

Abstract
Phosphatidylinositol-3-kinases (PI3Ks) are lipid kinases that phosphorylate phosphatidylinositol 4,5-bisphosphate to generate a key lipid second messenger, phosphatidylinositol 3,4,5-bisphosphate. PI3Kα and PI3Kγ require activation by RAS proteins to stimulate signaling pathways that control cellular growth, differentiation, motility and survival. Intriguingly, RAS binding to PI3K isoforms likely differ, as RAS mutations have been identified that discriminate between PI3Kα and PI3Kγ, consistent with low sequence homology (23%) between their RAS binding domains (RBDs). As disruption of the RAS/PI3Kα interaction reduces tumor growth in mice with RAS- and epidermal growth factor receptor driven skin and lung cancers, compounds that interfere with this key interaction may prove useful as anti-cancer agents. However, a structure of PI3Kα bound to RAS is lacking, limiting drug discovery efforts. Expression of full-length PI3K isoforms in insect cells has resulted in low yield and variable activity, limiting biophysical and structural studies of RAS/PI3K interactions. This led us to generate the first RBDs from PI3Kα and PI3Kγ that can be expressed at high yield in bacteria and bind to RAS with similar affinity to full-length PI3K. We also solved a 2.31 Å X-ray crystal structure of the PI3Kα-RBD, which aligns well to full-length PI3Kα. Structural differences between the PI3Kα and PI3Kγ RBDs are consistent with differences in thermal stability and may underly differential RAS recognition and RAS-mediated PI3K activation. These high expression, functional PI3K RBDs will aid in interrogating RAS interactions and could aid in identifying inhibitors of this key interaction.
Keywords: RAS, Phosphoinositide 3-kinase (PI3K), protein/protein interactions, cell signaling, cancer
Graphical Abstract
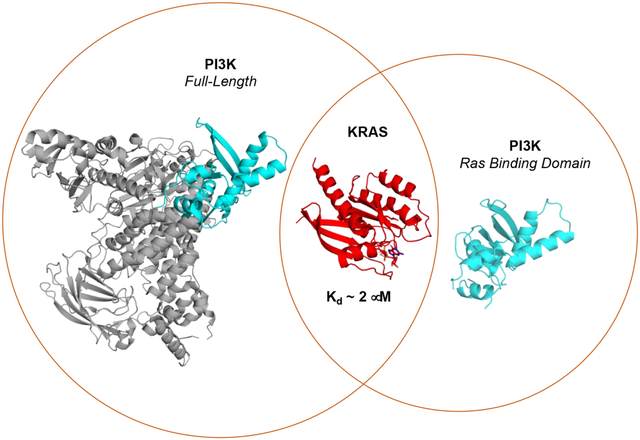
Introduction
Phosphatidylinositol-3-kinases (PI3Ks) are a family of lipid kinases that mediate a host of cellular functions, including cellular growth, differentiation, motility and survival. Upon activation, Class I PI3Ks (α, β, δ, and γ) convert the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) to a key lipid second messenger, phosphatidylinositol 3,4,5-triphosphate (PIP3). The product, PIP3, can activate multiple targets, including AKT, to promote cell survival, growth, proliferation and metabolism [1].
Class I PI3Ks are obligate heterodimers that consist of a catalytic subunit (p110) and a regulatory subunit. While the p110 subunit contains the kinase domain that catalyzes production of PIP3, the regulatory subunit interacts with the catalytic subunit to downregulate substrate access and kinase activity [2]. There are several classes and isoforms of PI3K, but only three Class I PI3Ks, PI3Kα, PI3Kγ, and PI3Kδ, have been shown to be activated by RAS subfamily proteins [3–7]. While PI3Kα and PI3Kβ are ubiquitously expressed, PI3Kγ and PI3Kδ isoforms are predominantly found in leukocytes [8]. Activation of these PI3K isoforms requires binding of receptor tyrosine kinases (RTKs) or heterotrimeric G-protein βγ subunits to the regulatory subunit to release inhibitory interactions with the catalytic subunit in conjunction with RAS binding to the catalytic domain [9].
While RAS binding to the p110 catalytic subunit of PI3Kα, δ, and γ isoforms promote kinase activation, how RAS binds and activates the PI3Kα is poorly understood. RAS proteins are the founding members of the RAS superfamily of GTPases. The three isoforms (HRAS, KRAS, and NRAS) function as highly regulated molecular switches that cycle between inactive GDP- and active GTP-bound states. GTP binding causes a conformational change in RAS that confers higher affinity binding to downstream effectors, including PI3Ks. This in turn promotes effector activation and downstream signaling [10]. Oncogenic point mutations that occur in roughly 30% of cancers cause constitutive activation of RAS and its downstream targets, including RAF kinases and PI3Ks, resulting in deregulated growth control. PI3Kα is also frequently mutated or amplified in several common cancers, such as breast, ovarian, colon, endometrial, gastric, cervical and lung cancer [11]. Of note, several studies have shown that disruption of the RAS/PI3Kα interaction in RTK and RAS-driven cancers has resulted in improved survival of tumor-bearing mice in both lung and skin cancer models, a reduction in tumor initiation and growth, and tumor-induced angiogenesis [12–15]. While these studies suggest that targeting the RAS/PI3Kα binding interface may be a clinically effective approach, a clear strategy has not been established as a structure of the KRAS/PI3Kα complex is lacking. The low sequence homology (23%) between the RBDs of PI3Kα and PI3Kγ makes it difficult to extrapolate known RAS/PI3Kγ binding determinants to that of PI3Kα. Moreover, RAS mutants (G12R, E37G and Y40C) have been identified that show differential binding and activation of PI3Kα and PI3Kγ isoforms [16–18]. These observations suggest that the interaction of RAS with these PI3K isoforms is distinct.
Class I PI3Ks are large heterodimers that are difficult to generate for biophysical and structural studies, as they require baculovirus-mediated insect cell expression, and result in low yield and variable activity. Truncated PI3K constructs that only contain the RBD of PI3K can be expressed in bacteria; however, these constructs show greatly reduced affinity compared to the full-length PI3K and require an affinity or fluorescent tag to maintain solubility [4,19–22]. To help overcome the obstacles associated with studying KRAS/PI3Kα interactions, we designed novel, soluble RBDs for both PI3Kα and PI3Kγ to facilitate biochemical and structural characterization of RAS/PI3K interactions. Both constructs can be expressed at high yield in E. coli and retain GTP-dependent binding to KRAS similar to their full-length counterparts. We also solved a 2.3 Å X-ray crystal structure of the truncated PI3K-RBD. While the structure is similar to the RAS binding domain in the context of the full-length p110 subunit, intriguingly, structural differences are observed between the PI3Kα and PI3Kγ RBDs, consistent with differences in thermal stability observed between these two isoforms. The observed structural differences between the PI3K isoforms likely underly differential RAS recognition and RAS-mediated PI3K activation. Our structural and biophysical characterization of these novel PI3K RBDs validate their use in delineating differential binding interactions between RAS, PI3Kα, and PI3Kγ, and provides a platform for the development of inhibitors that ablate this key interaction.
Results
Designing novel PI3K RBDs as tools to investigate RAS/PI3K interactions
In an effort to improve recombinant expression of the PI3Kα-RBD, we inspected the full-length crystal structure [23] to identify a minimal globular domain. Shown in Fig. 1 is the 3D structure and individual domains of the PI3K catalytic subunit (p110α) in complex with the N-terminal and inter-SH2 domains (niSH2) of the regulatory p85α subunit [23]. All five domains of p110α are shown, which consist of the adapter binding domain, the RBD, C2, helical domain and kinase domain. A commonly referenced GST-PI3Kα-RBD construct colored in red (Fig. 1), shows reduced binding relative to full-length PI3K and requires GST fusion to retain solution stability [4]. As the construct contains unstructured termini that likely de-stabilize the protein, these regions were excluded from our PI3Kα-RBD construct. The truncated RBD domain we constructed is colored in cyan (Human PI3Kα residues: 157–300; Fig. 1). To improve parity when comparing isoforms of PI3K RBDs, a truncated RBD construct was also generated for PI3Kγ, based on an alignment of this minimal PI3Kα-RBD within the full-length structure of PI3Kγ complexed to HRASG12V (PDB ID: 1HE8). Although these two class I PI3Ks share significant structural similarity (1.0 Å RMSD of 80 Cα atoms) and are of similar length, they differ significantly in primary sequence. Structure-based sequence alignment of the PI3Kα and PI3Kγ RBDs (Fig. 1B) revealed that the amino acid sequence of these two RBDs are only 23% identical. Notably, key residues within the PI3Kγ-RBD critical for RAS binding (Fig. 1B, red stars), F221, T232, K251 and K255, are distinct from the PI3Kα-RBD.
Figure 1.
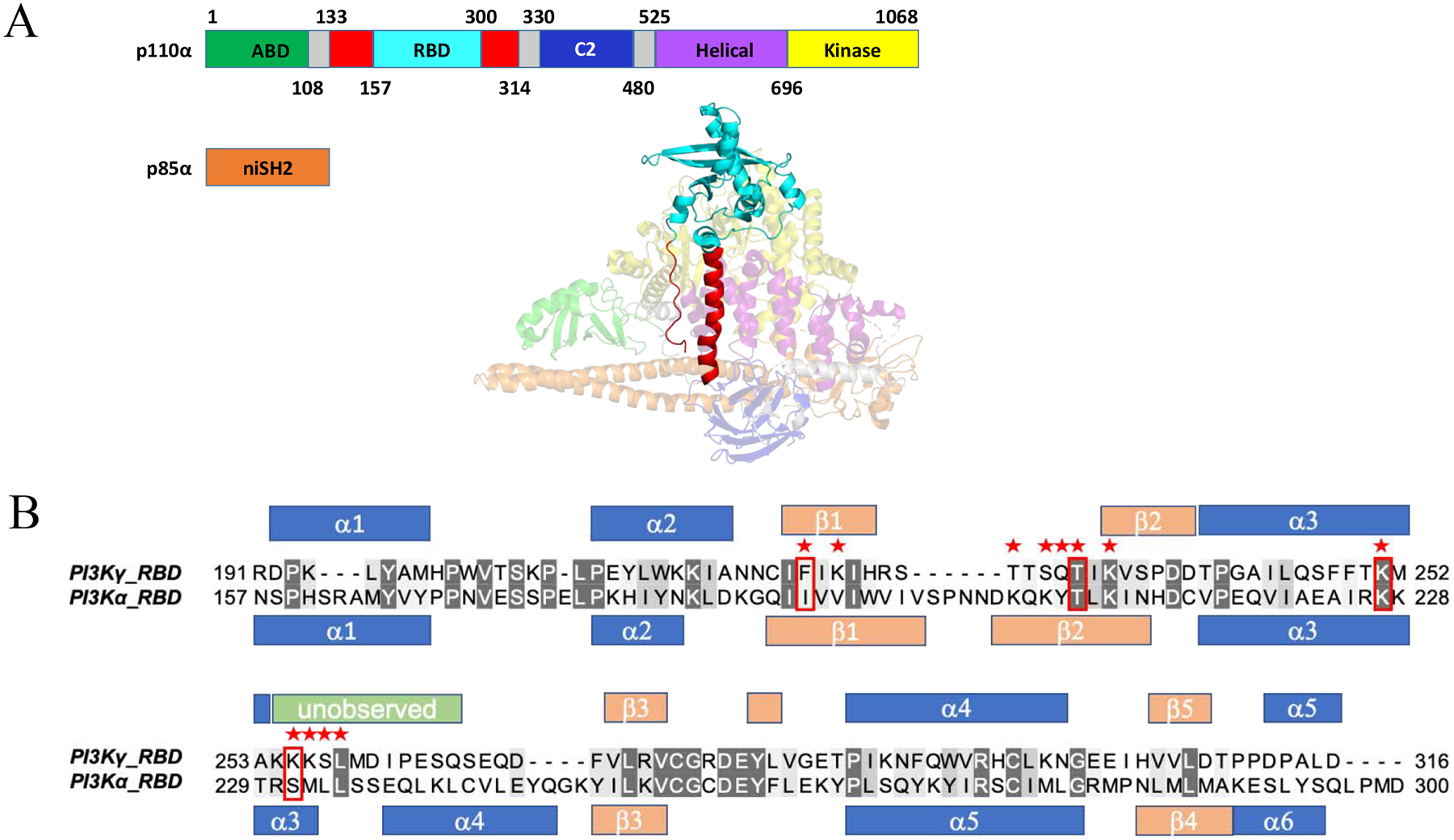
Generation of PI3Kα and PI3Kγ RAS binding domains (RBD) for high level bacterial expression. (A) Domains within the catalytic (p110α) and regulatory subunit (p85α). The adaptor binding domain (ABD) is shown in green, RBD in red (Rodriguez-Viciana et al, 1996), truncated RBD in cyan (Residues 157–300), C2 domain in blue, helical domain in purple and helical domain in yellow, with linker regions in gray. The N-terminal iSH2 domain within the regulatory subunit is shown in orange (PDB ID: 3HHM). (B) Structure-based sequence alignment for both the PI3Kγ (PDB ID: 1HE8) and truncated PI3Kα (PDB ID: 3HHM) RBDs created using PyMol {DeLano, 2002 #422}. Red stars indicate residues in PI3Kγ that contact HRASG12V in the HRASG12V/PI3Kγ crystal structure (PDB ID: 1HE8) and red boxes indicate the four residues shown to be critical for HRASG12V/PI3Kγ binding. Secondary structural elements are shown for the RBDs (α: α-helix in blue; β: β-strand in peach) of p110γ (PDB ID: 1E8X) and p110α (PDB ID: 3HHM). Unobserved residues in the p110α RBD (PDB ID: 3HHM) (Res. 254–265) are shown in green.
Novel PI3K RBDs bind RAS in a GTP-dependent manner
Both the PI3Kα-RBD and PI3Kγ-RBD domains were cloned into a pCDB24 bacterial expression vector, recombinantly expressed at high levels and subsequently purified to obtain ~15 mg of protein per liter. Given their high-level expression, we next evaluated binding of the PI3K RBDs to the KRAS GTPase. We conducted isothermal titration calorimetry (ITC) measurements, as this approach allows assessment of binding affinity, stoichiometry and thermodynamics. As shown in Fig. 2, results obtained from ITC experiments verified that both PI3K RBDs bound to KRASWT loaded with the non-hydrolysable GTP analog, GMPPCP (guanosine-5′-[(β,γ)-methyleno]triphosphate). KRASWT-GMPPCP bound to the PI3Kα-RBD (Fig. 2A) with a Kd of 2.19±0.48 μM and to the PI3Kγ-RBD (Fig. 2B) with a Kd of 2.29±0.38 μM. These binding affinities are similar to previously reported values for full-length PI3K [16,18]. The interaction is GTP-dependent, as binding of the PI3K RBDs is not detectable for KRASWT loaded with guanosine diphosphate (GDP) (Fig. 2A–D, Table 2).
Figure 2.
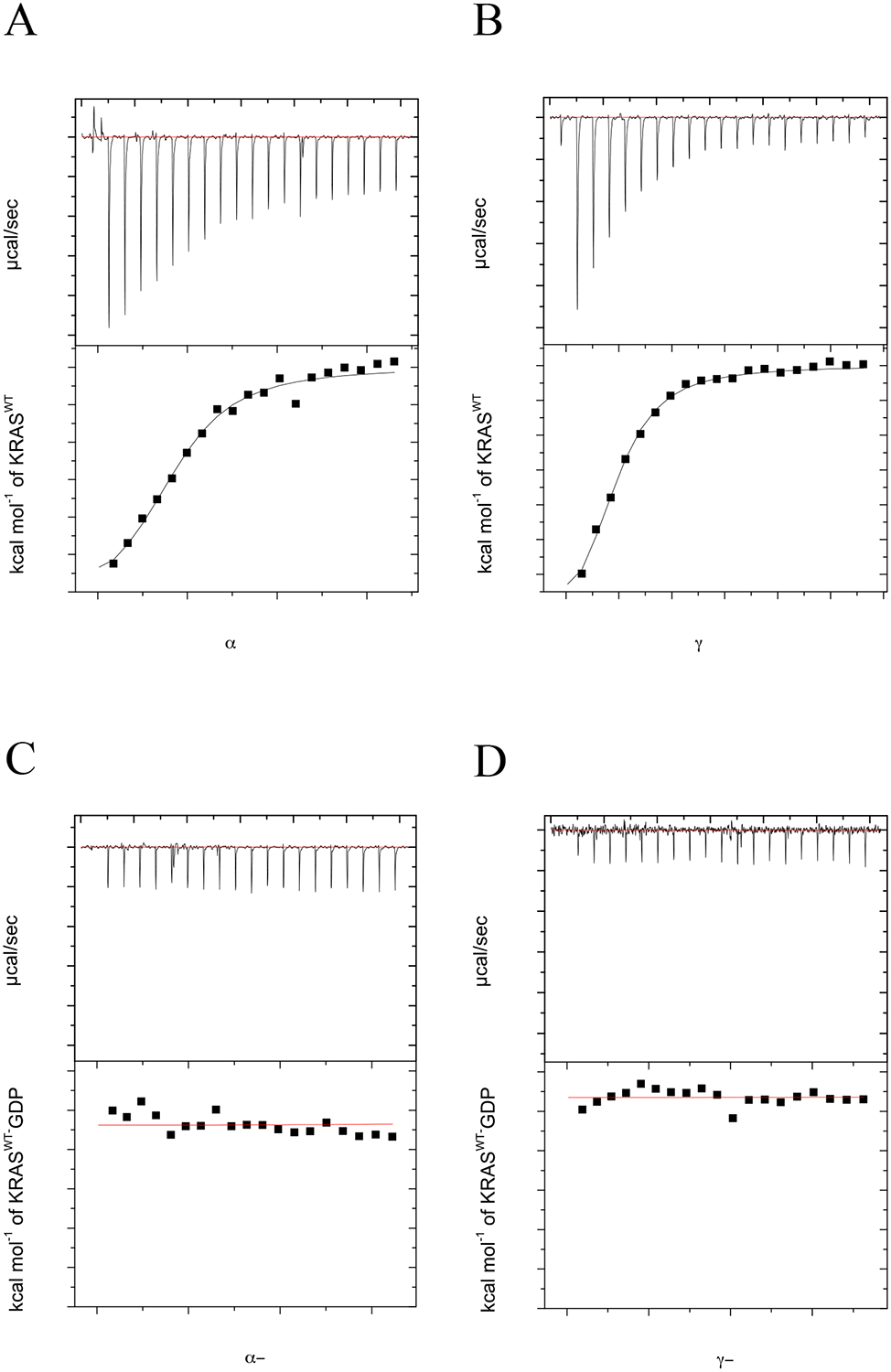
KRAS binds to both PI3Kα and PI3Kγ RBDs in a GTP-dependent manner. ITC experiments were performed with either 12 μM PI3Kα or PI3Kγ RBD in the presence of varying concentration of KRASWT (0–215 μM) at 25°C. Isothermograms of (A) KRASWT-GMPPCP injected into a cell containing PI3Kα-RBD, (B) KRASWT-GMPPCP injected into a cell containing PI3Kγ-RBD, (C) KRASWT-GDP injected into a cell containing PI3Kα-RBD and (D) KRASWT-GDP injected into a cell containing PI3Kγ-RBD. Top panel shows titration curve obtained for 20 automatic injections, 2 μL each, of KRASWT. The bottom panel shows the integrated curve showing experimental (■) points and the best fit ( — ). All data was processed and curve fitting was used with a single site model in Origin 7.0. Data is representative of N=3 for KRASWT-GMPPCP experiments and N=2 for KRASWT-GDP experiments. Binding stoichiometry (N), Kd, enthalpy (ΔH) and entropy (ΔS) values were calculated in Origin 7.0 and reported as the mean ± std. dev. in Table 2.
Table 2.
GTP-bound KRASWT and oncogenic KRAS mutants bind with a similar affinity to the PI3K RBDs, while the T208D mutant in the PI3Kα-RBD does not show detectable binding. All values are for GMPPCP-bound KRAS, except where designated otherwise. Binding stoichiometry (N), Kd, enthalpy (ΔH), and TΔS values were calculated in Origin 7.0 and reported as the mean ± std. dev. as determined by three independent experiments. Gibbs free energy (ΔG) values were calculated using the Gibbs free energy equation and ITC-derived derived ΔH and ΔS values.
| RAS protein | Effector | n | Kd (μM) | ΔH (kcal/mol) | TΔS (kcal/mol/deg) | ΔG (kcal/mol |
|---|---|---|---|---|---|---|
| KRASWT | PI3Kα-RBD | 0.88±0.06 | 2.2±0.5 | −6.12±1.08 | 1.6±1.3 | −7.72 |
| KRASWT | PI3Kγ-RBD | 0.82±0.08 | 2.3±0.4 | −7.42±0.93 | 0.30±1.0 | −7.72 |
| KRASWT-GDP | PI3Kα-RBD | N/A | N/A | N/A | N/A | N/A |
| KRASWT-GDP | PI3Kγ-RBD | N/A | N/A | N/A | N/A | N/A |
| KRASG12V | PI3Kα-RBD | 1.07±0.14 | 2.3±0.5 | −3.82±0.37 | 4.11±0.3 | −7.93 |
| KRASG12V | PI3Kγ-RBD | 0.81±0.16 | 2.4±0.6 | −4.16±0.28 | 2.96±0.4 | −7.12 |
| KRASQ61H | PI3Kα-RBD | 1.06±0.17 | 4.0±0.5 | − 2.59±0.50 | 4.7±0.6 | −7.29 |
| KRASQ61H | PI3Kγ-RBD | 0.910±0.01 | 2.6±1.4 | −2.97±0.63 | 4.7±1.3 | −7.67 |
| KRASWT | PI3Kα-RBD(T208D) | N/A | N/A | N/A | N/A | N/A |
Structural characterization of the PI3K RBDs
After validating that our minimal PI3K RBDs bind to KRAS in a GTP-specific manner, we sought to structurally characterize the PI3K RBDs. Initially, we performed circular dichroism (CD), with detection at 222 nm, as a function of temperature to monitor the thermal unfolding of the PI3K RBDs. Results from these experiments revealed that the PI3Kα-RBD possesses a significantly higher thermal stability, with a melting temperature (Tm) of 53.8°C compared to 39.0°C for the PI3Kγ-RBD (Fig. 3A). Given the enhanced stability of the PI3Kα-RBD, we employed NMR spectroscopy to conduct preliminary characterization studies. We prepared 13C/15N-enriched PI3Kα-RBD and collected a 1H-15N 2D HSQC NMR spectrum as shown in Supp. Fig. 1. Enrichment with 15N allows detection of NH groups, including the peptide backbone, and provides a site-specific probe for every amino acid, with the exception of proline. The NMR spectrum shown in Supp. Fig. 1 is well dispersed, with ~82% of the backbone amides detectable. Moreover, the backbone amides display an average linewidth of 25.5 Hz and 13.7 Hz along the 1H and 15N dimension, respectively, indicative of a well folded monomeric protein (16.7 kDa). Given the quality of the NMR data, we sought structure determination by crystallography.
Figure 3.
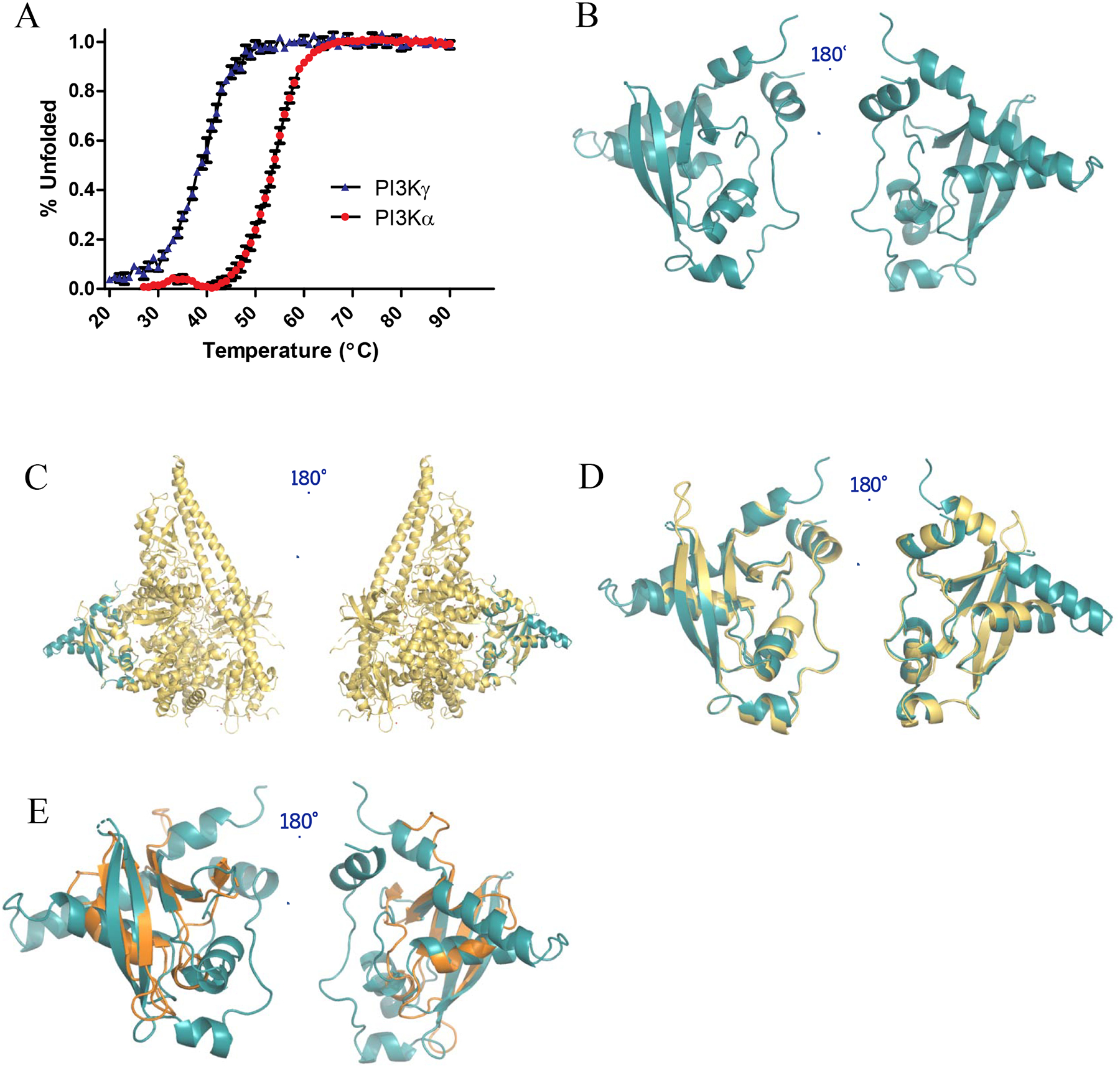
Structural characterization of PI3Kα-RBD. (A) CD thermal unfolding as quantified by the loss of mean residue ellipticity at 222 nm for 30 μM PI3Kα-RBD (red) and PI3Kγ-RBD (blue) over a temperature range of 20–90°C at pH 7.4. Data was normalized to the maximum and minimum ellipticity. Thermal denaturation curves and the midpoint of the thermal transition (Tm) were calculated by fitting the data using the Gibbs-Helmhotz equation. Results were graphed in GraphPad Prism and reported as the mean ± S.E. (error bars) (N=3 for PI3Kα, N=2 for PI3Kγ, each performed in duplicate). (B) X-ray crystallographic structure of the PI3Kα-RBD (residues 157–300) (PDB: 6VO7). The structure was refined to a resolution of 2.31 Å, with residual values of 19.65 % (Rwork) and 25.52 % (Rfree). The structure is complete, save for 4 unobservable residues in the flexible loop region between β-strands 1 and 2, and 16 unobservable residue sidechains. Superposition of (C) PI3Kα-RBD crystal structure (teal) onto the full length PI3Kα structure (PDB: 5XGH, yellow), (D) PI3Kα-RBD crystal structure (teal) overlayed with the RBD contained within the PI3Kα full-length crystal structure (PDB: 5XGH, yellow, correlated construct residues 157–300, Cα least squares quadratic (LSQ) RMSD 1.22 Å), (E) PI3Kα-RBD crystal structure (teal) overlayed with B-RAF-RBD (PDB: 3NY5, gold). The 4 β-strands align well between the B-RAF-RBD and PI3Kα-RBD, with similar orientations observed for the conserved helices adjacent to the beta sheets.
We were able to successfully obtain PI3Kα-RBD construct crystals in the C2221 space group with a single molecule in the asymmetric unit. The crystals refined to a resolution of 2.31 Å and the structure was refined with residual values of 19.68 % (Rwork) and 25.52 % (Rfree) (Fig. 3B). Additional X-ray data and refinement statistics for the PI3Kα-RBD are listed in Table 1. The X-ray diffraction data yielded a mostly complete structure, except for 4 unobservable residues (199–202) in a flexible loop region between β-strands 1 and 2 and 16 unobservable sidechains (S173, I197, V198, D203, K204, K206, E218, S231, S236, E237, K240, K249, E263, K264, E291 and M299). The PI3Kα-RBD crystal structure aligns well with the correlated residues of the previously deposited full-length PI3Kα structure (Fig. 3C,D) (PDB: 5XGH), with a Cα least squares quadratic RMSD value of 1.22 Å. Residues 228–247 were not present in the 5XGH molecular replacement search model, yet electron density maps fully supported building this region into our construct model. While these residues are absent in the full-length structure, the remainder of the RBD construct structure superposes well, except for the absence of RBD residues 199–200 in the flexible loop region between β-strands 1 and 2. Secondary structural element notation is provided in Fig. S1. The superposition of the PI3Kα-RBD crystal structure onto the BRAF-RBD crystal structure [24] is provided in Fig. 3E. The BRAF-RBD structure is smaller (11.26 kD compared to 16.78 kD for PI3Kα-RBD) and obtained at a slightly higher resolution (82 residues, 1.993 Å) than the PI3Kα-RBD (143 residues, 2.31 Å). The core RMSD for secondary structural alignment is 2.05 Å from 61 aligned residues, 4 gaps and 13% sequence identity. The PI3Kα-RBD has 33 N-terminal residues pre-alignment and 10 C-terminal residues post-alignment. Even though these RBD constructs are from different RAS effectors, their secondary structures align well, especially in the beta sheet region. The BRAF-RBD possesses an additional smaller β-strand, located between β3 and H5, that is not defined in the PI3Kα-RBD. Other differences between PI3Kα-RBD and the BRAF-RBD include a longer Helix 3 in the PI3Kα-RBD as well as the loop region between β1 and β2. Moreover, H4 and H6 are not present in the BRAF-RBD structure. Residues 202–204 following β3 are the only missing region in the BRAF-RBD, where the PI3Kα-RBD structure is missing residues 199–202 between β1 and β2.
Table 1.
X-ray data collection and refinement statistics for PI3Kα-RBD. Values in parentheses are for the highest resolution shell.
| PI3Kα RBD | |
|---|---|
| PDB Entry | 6VO7 |
| Data Collection | |
| Wavelength (Å) | 1.00 |
| Temperature (K) | 100 |
| Space Group | C2221 |
| Unit-cell parameters (Å) | |
| a | 71.55 |
| b | 83.91 |
| c | 63.99 |
| Unique reflections | 8,574 (414) |
| Completeness (%) | 98.8 (97.4) |
| Rmerge (%)b | 17.1 (83.2) |
| I/σ(I) | 21.0 (1.7) |
| Redundancy | 5.8 (5.0) |
| Refinement | |
| Resolution (Å) | 2.31 |
| Rwork (%)c | 19.68 (25.65) |
| Rfree (%)d | 25.52 (34.84) |
| No. of protein atoms | 1,109 |
| No. of solvent atoms | 26 |
| R.m.s.d from ideal geometrye | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 0.950 |
| Ramachandran plot (%)f | |
| Most favored region | 97.06 |
| Addl allowed region | 2.94 |
| Outliers | ----- |
Rmerge = ∑h∑i[|Ii(h) − 〈I(h)〉|/∑h∑iI(h)] × 100%, where Ii(h) is the ith measurement and 〈I(h)〉 is the weighted mean of all measurements of I(h).
Rwork = ∑|FO − FC|/∑FO × 100%, where FO and FC are the observed and calculated structure factors, respectively.
Rfree is the R factor for the subset (~9%) of reflections selected before and not included in the refinement.
Root-mean-square deviation.
The Ramachandran plot was created via MolProbity.
Oncogenic KRAS mutants bind PI3K similar to KRASWT, while T208D PI3Kα-RBD mutant abolishes RAS binding
As RAS mutations have been identified that differentially interact with PI3Kα and PI3Kγ, we conducted additional ITC studies to investigate binding of two oncogenic KRAS mutants to the two PI3K isoforms. We selected KRASG12V and KRASQ61H because they are among the most prevalent KRAS mutations found at the 12 and 61 positions in human cancer. Using ITC, we determined the affinity, stoichiometry and thermodynamics associated with these binding interactions. The PI3Kα and PI3Kγ RBDs bound GMPPCP-loaded KRASG12V with a Kd of 2.29±0.53 μM and 2.41±0.57 μM, respectively, and KRASQ61H with a Kd of 4.00±0.50 μM and 2.6±1.4 μM, respectively (Fig. 4A–D). As shown in Table 2, all binding affinities between the PI3K RBDs and KRASG12V and KRASQ61H are comparable to KRASWT-GMPPCP. We also performed an additional ITC experiment to further validate that our PI3Kα-RBD construct behaves similar to the full-length protein. Previous work investigating KRAS/PI3Kα interactions have used a point mutation in PI3Kα (T208D) to abrogate KRAS/PI3Kα binding [12]. This residue is also conserved in PI3Kγ and plays a key role in binding [18]. To evaluate whether our PI3Kα-RBD retains this crucial interaction, we performed ITC with PI3KαT208D-RBD and KRASWT-GMPPCP. As shown in Fig. 4E and Table 2, we did not observe detectable binding to KRASWT-GMPPCP.
Figure 4.
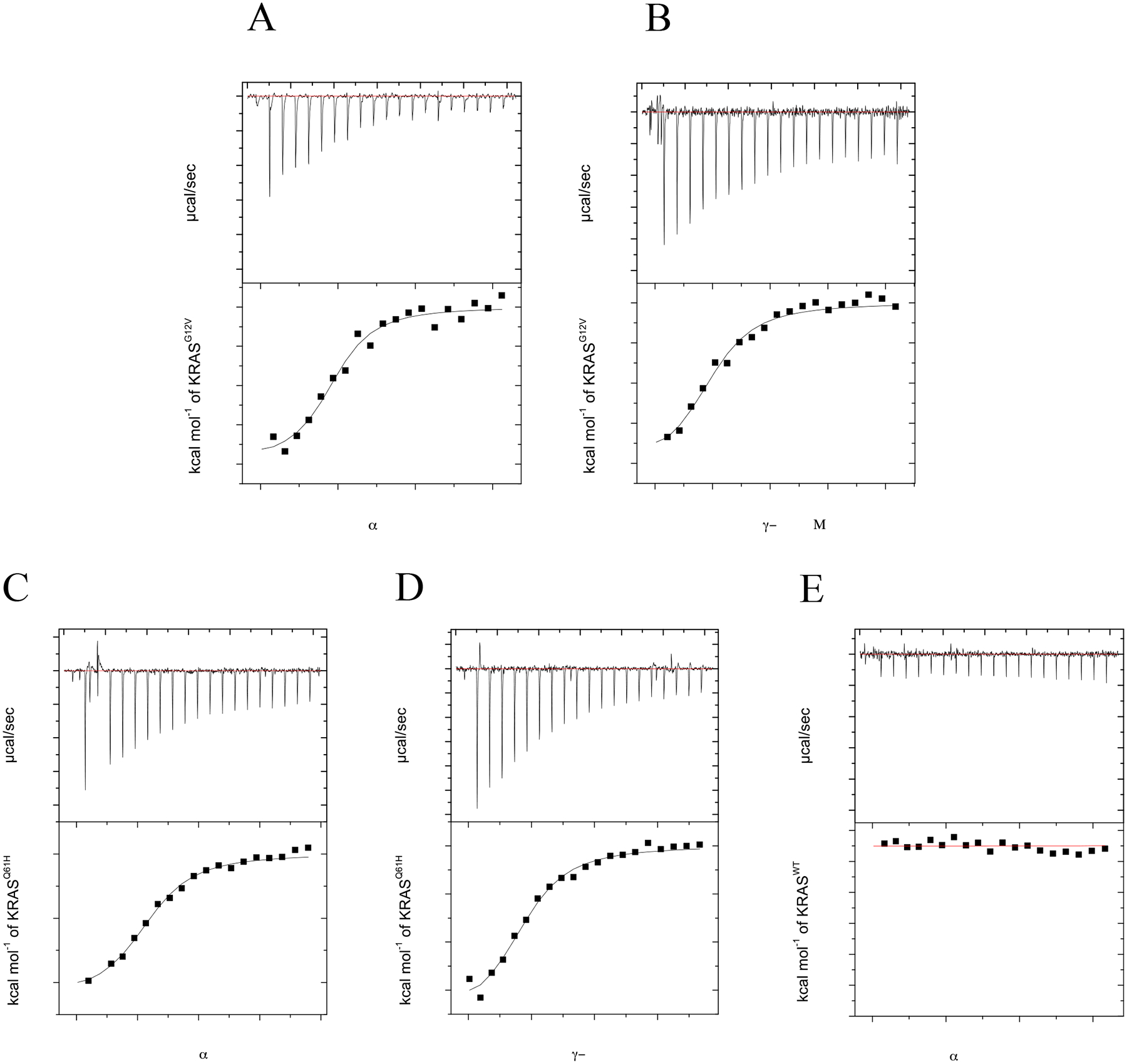
Oncogenic KRAS (G12V, Q61H)-GMPPCP shows similar binding to PI3Kα and PI3Kγ RBDs relative to KRASWT while binding to PI3Kα(T208D)-RBD is not detectable. ITC experiments were performed with either 12 μM PI3Kα or PI3Kγ RBD in the presence of varying concentration of KRAS (0–215 μM) at 25°C. Isothermograms of (A) KRASG12V-GMPPCP injected into a cell containing PI3Kα-RBD, (B) KRASG12V-GMPPCP injected into a cell containing PI3Kγ-RBD, (C) KRASQ61H-GMPPCP injected into a cell containing PI3Kα-RBD, (D) KRASQ61H-GMPPCP injected into a cell containing PI3Kγ-RBD and (E) KRASWT-GMPPCP injected into a cell containing PI3Kα(T208D)-RBD. Top panel shows titration curve obtained for 20 automatic injections, 2 μL each, of KRASWT. The bottom panel shows the integrated curve showing experimental (■) points and the best fit ( — ). All data was processed and curve fitting was used with a single site model in Origin 7.0. Data is representative of N=2 for all experiments. Binding stoichiometry (N), Kd, enthalpy (ΔH) and entropy (ΔS) values were calculated in Origin 7.0 and reported as the mean ± std. dev. in Table 2.
PI3Kα and PI3Kγ RBDs differ in secondary structure and stability
As the PI3Kγ-RBD displayed a greatly reduced stability via our CD thermal melt analysis (Fig. 3A), we examined known structures of the PI3K RBDs to provide an explanation for differences in stability between the two PI3K RBDs. Using existing crystal structures of full-length PI3Kα (Fig. 5A, cyan) and PI3Kγ (Fig. 5A, green) ([23] and [18] for PI3Kα and PI3Kγ, respectively), we identified two regions in both RBDs that differ. The first region consists of a flexible loop in PI3Kγ that is disordered in every PI3Kγ structure except the HRASG12V/PI3Kγ structure [18], which is bound to RAS. In PI3Kα, this region is more ordered and possesses helical secondary structure (Fig. 5A, yellow). The second region that differs corresponds to the N- and C- termini, which pack against each other with higher helical character in PI3Kα compared to PI3Kγ (Fig. 5A, orange) (Supp. Fig. 2).
Figure 5.
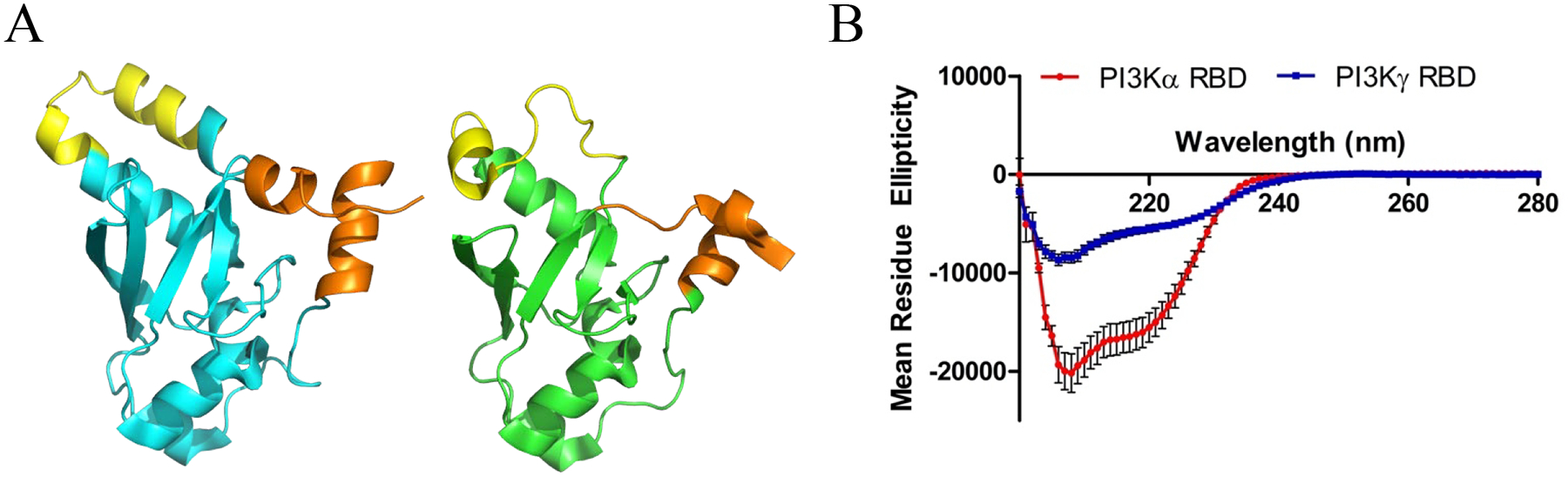
PI3Kα RBD possess higher helical content relative to the PI3Kγ-RBD. (A) Structures of the PI3Kα-RBD (cyan, left) and PI3Kγ (green, right), with proposed regions of differential structure colored in yellow and orange. The RBD domains were extracted from full-length PI3K structures (PDB ID: 3HHM & 1HE8 for PI3Kα and PI3Kγ, respectively. Two regions are highlighted in both PI3K RBDs, with one being a flexible loop in yellow in PI3Kγ (residues for PI3Kα = 229–244 and PI3Kγ = 253–268) and the terminal residues in orange (residues for PI3Kα = 157–167 & 290–300 and PI3Kγ = 191–198 & 309–316). (B) Circular dichroism (CD) spectral scans of PI3Kα (red) and PI3Kγ (blue) were collected at 20°C from 200–280 nm at pH 7.4. The PI3Kα RBD possesses 20.5% higher helical content relative to the PI3Kγ-RBD. Results are reported as the mean ± S.E. (error bars) (N=3 for PI3Kα, N=2 for PI3Kγ, each performed in duplicate).
To investigate differences in secondary structure between our truncated PI3Kα and PI3Kγ RBDs, we collected far UV-CD spectra and predicted secondary structure content. As shown in Fig. 5B, the PI3Kα-RBD shows higher helical content (35.1±4.1%) compared to the PI3Kγ-RBD (14.6±2.8%). The helical content is consistent with RBDs in the context of the full-length protein crystal structures (Supp. Table 1), suggesting that the termini of the PI3Kγ-RBD construct is unstructured. In contrast, calculations of secondary structure for the PI3Kα-RBD requires partial helicity at the termini to match the CD data. Combining our CD thermal stability and secondary structure analysis, our data indicates that the PI3Kα-RBD is more stable, likely due to enhanced helical structure relative to the PI3Kγ-RBD.
Discussion
Use of RBDs to Investigate RAS/PI3K Binding Determinants
Most effectors engage RAS using two types of ubiquitin-like domains, commonly referred to as RAS binding (RB) and RAS association (RA) domains [25]. Class I PI3Ks comprise key downstream effectors of RAS that contain an RBD within the catalytic subunit. RAS binding to PI3K via this domain plays an important role in kinase activation to promote PIP3 production [7,26,27]. Constitutive PIP3 production can occur in contexts where PI3Kα is mutated or amplified, which frequently occurs in breast, ovarian, colon, endometrial, gastric, cervical and lung cancer [11], or in RTK or RAS-driven cancers. Seminal studies in the RAS field have shown that introduction of two germline RBD mutations in mice (T208D/K227A) abolishes binding between RAS and PI3Kα, while the same mutations reduce oncogenic RAS-driven lung and skin tumorigenesis in mice and mouse embryonic fibroblasts MEFs [12] and causes regression of established lung tumors and long-term tumoristasis in mice [13]. Moreover, disrupting this key interaction reduces tumor growth, metastasis, and tumor-induced angiogenesis in both lung carcinoma and melanoma transplanted tumors [28], without causing toxicity. In addition, EGFR-mutant lung adenocarcinoma in mice containing the PI3KαT208D/K227A mutations inhibit tumor onset and cause significant regression of established tumors and increased survival [15]. This work highlights a route to target aberrant PI3K signaling without the use of PI3K inhibitors. While significant attention has been given to generating isoform specific PI3K inhibitors [29,30], PI3K inhibitors possess high cellular toxicity [31]. By targeting the RAS/PI3Kα interaction, a loss of RAS binding will attenuate PI3K activity, rather than eliminate it. While these studies suggest that targeting the RAS/PI3Kα binding interface may be a clinically effective strategy in RAS- and EGFR-driven cancers, a structure of the KRAS/PI3Kα complex is lacking and the low sequence homology between PI3Kα and PI3Kγ makes it difficult to correlate known RAS/PI3Kγ interactions to PI3Kα.
PI3K RBDs that can be expressed at high yield in bacteria have not been successfully generated [4,19–21], limiting characterization of RAS binding interactions and pharmacological approaches to targeting this key interaction. As RBDs for several RAS effectors have successfully been generated, we postulated that the PI3K RBDs require additional sequence elements outside of the core ubiquitin-like fold to stabilize the RBD. One example where a RAS effector utilizes these additional contacts to stabilize the ubiquitin fold can be seen in the NORE1A RBD, where an N-terminal α-helix and short β-strand form hydrophobic interactions with β1 and helix α2 [32]. Inspection of sequence conservation and 3D structures of PI3Ks led to the generation of truncated RBDs that express in high yield and bind to KRAS in a GTP-dependent manner. While our PI3K RBDs contain most determinants associated with RAS/PI3Kγ binding, a residue (E919) outside the RBD in the kinase domain of PI3Kγ, has been shown to make contact with residue 73 in RAS. However, this interaction does not appear to contribute significantly to the overall binding affinity, as mutation of this residue (E919K) in PI3Kγ did not affect binding [18].
Only four out of twelve amino acids in the PI3Kγ-RBD involved in RAS binding (T232, K234, K251, L258) are strictly conserved in the PI3Kα-RBD, with T251 and K232 shown to be absolutely critical for binding to RAS switch 1 residues D33, D38 and E37 [18]. Two other residues in PI3Kγ critical for RAS binding, F221 and K255, are not conserved between the two PI3K isoforms. The lack of sequence homology, 23% overall between the two RBDs, and the observation that KRASG12R fails to bind PI3Kα but retains binding to PI3Kγ suggests that binding determinants between these two PI3K isoforms differ [16]. Consistent with these observations, KRASY40C selectively binds PI3Kα but has severely attenuated binding to PI3Kγ [17,18]. Computational analysis of the KRAS/PI3Kα complex predicts that these two PI3K isoforms differentially engage RAS. Results from MD simulations indicate that while many RAS contacts remain in place, particularly for antiparallel β-sheet interactions upon binding with PI3Kα and PI3Kγ, new switch I and switch II contacts are predicted to form [33].
RAS Affinity for Novel PI3K RBDs is similar to p110
KRAS binding to full-length PI3Kα (Kd of 1.85±0.07 μM for GTP-KRASWT/PI3Kα (p110α) has been previously evaluated [16]. While a Kd is lacking for full-length PI3Kγ bound to RAS, a slightly truncated p110γ construct containing all known RAS binding determinates was reported to bind HRASG12V with a Kd of 2.9±0.4 μM [18]. Notably, our truncated PI3K RBDs show similar affinity to GMPPCP-loaded KRAS relative to these larger/full-length proteins. Moreover, both PI3K RBDs bind in a GTP-dependent manner and retain comparable Kd’s and ΔG values for the oncogenic KRASG12V and KRASQ61H mutants. In addition, our PI3KαT208D-RBD mutant failed to bind KRASWT as measured by ITC. This RBD mutant was previously shown to abolish KRAS/PI3Kα binding [12]. With our novel PI3K RBDs in hand, we then conducted biophysical and structural analyses to evaluate structural differences between the two isoforms.
Thermodynamics of KRAS/PI3K RBD Binding
As shown in Table 2, our ITC analysis indicates that binding of RAS to the PI3K RBDs is driven by both favorable enthalpy and entropy contributions. The PI3K RBDs are more structurally similar to the RALGDS-RBD [34] and bind with similar affinity to RAS relative to the CRAF-RBD [35]. However, RALGDS binding to RAS possesses a negative entropic contribution which is counteracted by a large negative change in enthalpy [36]. This may be due to release of water or conformational changes that restrict mobility upon complex formation.
Additional Helical Content in PI3Kα-RBD Correlates with Higher Stability
One of the most striking differences with our novel PI3K RBDs is a significant difference in thermal stability. The PI3Kγ-RBD possesses a 14.8°C lower Tm relative to the PI3Kα-RBD. The difference in stability and secondary structure may be due to additional helical content and termini packing of the PI3Kα-RBD compared to the PI3Kγ-RBD. Helical content observed within the RBD domain of full-length PI3K crystal structures matched the CD measurements reported here for the truncated RBD constructs (Supp. Table 1), indicating that the more stable PI3Kα-RBD contains ~20% more helical content than PI3Kγ-RBD.
Based on full-length structures, differences in secondary structure are present at two locations within the PI3K isoforms (Fig. 5). The C-terminus of the PI3Kα-RBD contains a short helix that forms interactions with the N-terminus, whereas the PI3Kγ-RBD contains three proline residues that likely prevent secondary structure formation and helical packing with the N-terminus (Fig. 5A, orange, Supp. Fig. 2). Additionally, the yellow region indicated in Fig. 5A highlights a flexible loop in PI3Kγ, which is visible in the HRASG12V/PI3Kγ structure, but absent in the other 92 deposited PI3Kγ structures (Supp. Fig. 3). With RAS bound to PI3Kγ, the unstructured loop is stabilized and makes several key contacts with RAS. In contrast, the corresponding region in PI3Kα forms an additional helix, α4 (Supp. Fig. 4), which is only present in 27 of the 43 deposited PI3Kα structures (Supp. Fig. 3). Based on these findings, we postulate that the PI3Kγ-RBD is dependent on RAS complex formation to promote structural stability.
To investigate whether differences in stability observed for the two RBD constructs may also apply to RBD domains in the context of the full-length proteins, we conducted in silico analysis. Results from these analyses suggest PI3Kα may form more intramolecular contacts with other components of the p110 subunit compared to PI3Kγ. The interface size for the PI3Kα-RBD and the remainder of the protein is larger, and the interactions are stronger than those observed within the PI3Kγ crystal structure (Supp. Table 2). However, based on these predictions, the PI3Kα-RBD should be less stable from the full-length protein. Since this is not what our CD thermal data shows, we hypothesize that the contacts the PI3K RBDs makes with the rest of the protein do not significantly contribute to the PI3K RBDs stability in isolation. Therefore, the PI3Kγ-RBD might have other structural features that contribute to it being less stable than the PI3Kα-RBD. The untethered termini may differentially affect the two RBD isoforms resulting in reduction of secondary structure. While termini truncation may contribute to the reduced stability of PI3Kγ relative to PI3Kα, our analysis suggests that PI3Kγ stability is more dependent on RAS complex formation.”
Comparison of PI3K RBD Tertiary Structure with BRAF and RALGDS RBDs
Further comparison of the PI3Kα-RBD structure to the well-characterized BRAF-RBD and RALGDS-RBD shows many similarities, including the common ubiquitin-like fold and antiparallel β-sheet pairing with RAS. The PI3Kα-RBD differs from these RBDs by containing additional helices and one less β-sheet. In addition to the aforementioned α4 helix, additional helices in the PI3Kα-RBD can be found at the N-terminal (α1 and α7) and C-terminal (α6) end of the protein. These additional helices are outside the RAS binding region, except for α4, which packs against part of the ubiquitin-like fold responsible for key switch 1 contacts with RAS. The presence of α4 likely helps stabilize this part of the structure and leads to novel RAS contacts. The yellow helix described in Fig. 5A is also absent in the BRAF-RBD [24] and RALGDS-RBD [34].
PI3K RBDs May Facilitate RAS/PI3Kα Drug Discovery Efforts
One approach to reduce toxicity associated with PI3K kinase inhibitors is to decrease PI3K activity to levels that attenuate signaling. As RAS acts synergistically with RTKs to increase kinase activity (~30–200 fold compared to basal levels) [6,7], interfering with either of these activating agents may attenuate PI3Kα activity and toxicity. Identification of compounds that interfere with RAS/PI3Kα association represents an attractive approach, as disrupting RAS/PI3Kα complex formation using PI3Kα mutants defective in RAS binding has been shown to reduce tumorigenicity in RAS- and RTK-driven lung and skin cancer without causing toxicity[12–15]. Several promising drug discovery efforts aimed at disrupting RAS/effector protein-protein interactions (PPI) have recently been developed. A notable example includes allosteric KRASG12C inhibitors that target and stabilize the inactive GDP-bound form of RAS. These covalent inhibitors show efficacy with low toxicity and are in clinical trials, with Amgen 510 awaiting FDA approval for second-line treatment of patients with non-small cell lung cancer ([37–39]. Moreover, direct inhibitors of KRAS/effector PPI, including PPIN compounds, have recently been developed. The PPIN inhibitors interfere with effector association both in vitro and in vivo and inhibit downstream signaling of mutant RAS proteins in colorectal adenocarcinoma (DLD-1) cell lines [40]. Thus, characterizing binding determinants between wild-type and oncogenic RAS mutants with PI3Kα and PI3Kγ will provide insights into how PI3K isoforms differentially recognize RAS. This knowledge, in turn, will expediate drug discovery efforts that aim to inhibit the RAS/PI3Kα interaction, a key interaction in EFGR- and RAS-driven cancers. Due to difficulties associated with generating full-length PI3K from insect cells, our novel PI3K RBDs, which express at high yields in bacterial cell culture, will facilitate future structural and biophysical studies to characterize RAS interaction that may aid in development of RAS/PI3Kα inhibitors.
Materials and Methods
Recombinant Protein Expression
Human PI3Kα-RBD (residues 157–300), PI3Kγ-RBD (residues 193–316) and KRAS4B (residues 1–169, C118S) were subcloned into pCDB24 expression vectors containing an N-terminal 10-histidine tag and a SUMO cleavage site. In addition, wild-type human KRAS4B (residues 1–169, C118S) and KRAS-4BG12V and KRAS4BQ61H were subcloned into a pET21 vector that encodes an N-terminal 6-histidine tag and a TEV protease cleavage site. The PI3K RBDs and all of the KRAS plasmids were expressed in BL21 (DE3) RIPL cells and were grown at 37°C in Luria-Bertani (LB) medium. The cells were grown to an OD600 of ~0.5. The temperature was then reduced to 18°C for 30 minutes (min), protein expression induced with 500 μM Isopropyl β-D-1-thiogalactopyranoside (IPTG), and the cells grown for 16 hours (h) at 18°C. Cell pellets were obtained upon centrifugation at 4,500 x g for 30 min.
Protein Purification
PI3K RBDs
Following centrifugation, the PI3K RBDs pellets were resuspended in lysis buffer (50 mM HEPES, 200 mM NaCl, 20 mM Imidazole, and 10% glycerol, pH 8.0) and sonicated in the presence of a Roche protease inhibitor cocktail tablet (Roche Ref.# 05892970001). The PI3K cell lysates were subsequently centrifuged at 15,000 x g for 30 min. The supernatants were then purified using nickel-nitrotriacetic acid-agarose affinity (Ni-NTA agarose) chromatography. The supernatants were applied to the Ni-NTA agarose column, washed with 5 column volumes (CV) lysis buffer, 10 CV wash buffer (50 mM HEPES, 1 M NaCl, 30 mM Imidazole, 10% glycerol, 5mM β-mercaptoethanol (BME), pH 8.0) and again with 5 CV lysis buffer. The protein was eluted with 50 mM HEPES, 200 mM NaCl, 500 mM Imidazole, 5 mM BME and 10% glycerol, pH 8.0. The eluted protein was cleaved of its histidine tag using the Ubl-specific protease 1 (ULP1) from Saccharomyces cerevisiae during overnight dialysis in 50 mM HEPES, 500 mM NaCl, and 5 mM β-mercaptoethanol (BME), pH 8.0 at 4°C. Following dialysis, the tag was removed via the Ni-NTA agarose column. The PI3K RBDs were purified more than 95% by size exclusion chromatography (SEC) using a Sephadex S-75 (S-75) column and verified using SDS-PAGE analysis.
Wild-type and mutant KRAS4B
The pellet containing all KRAS proteins was resuspended in lysis buffer (50 mM HEPES, 150 mM NaCl, 20 mM imidazole, 5 mM MgCl2, 15 μM guanosine diphosphate (GDP), and 5mM BME, pH 7.75) and then sonicated. The supernatant was collected after centrifugation at 15,000 x g for 30 min and then applied to a Ni-NTA agarose column. The protein was washed with 5 CV lysis buffer, 10 CV wash buffer (50 mM HEPES, 750mM NaCl, 30 mM Imidazole, 5 mM MgCl2, 15 μM GDP, and 5mM BME, pH 7.75), and then 5 CV lysis buffer. The protein was eluted with 50 mM HEPES, 150 mM NaCl, 500 mM imidazole, 5 mM MgCl2, 15 μM GDP, and 5 mM BME, pH 7.75. Either the TEV protease or ULP1 was used to cleave the his-tag during overnight dialysis at 4°C in 50 mM HEPES, 150 mM NaCl, 5 mM MgCl2, 15 μM GDP and 5 mM BME. An S-75 size exclusion column was used to purify the protein to greater than 95% purity, as verified through SDS-PAGE analysis.
Circular Dichroism (CD) Thermal Melts and Spectral Scans
CD measurements were performed on a Jasco J-815 CD spectrometer using sparged 10mM KH2PO43−/K2HPO43−, pH 7.4 at 20°C. CD thermal melts were collected on both the PI3Kα and PI3Kγ RBDs using a 1-mm cuvette using 30 μM protein. Thermal melts were obtained at 222 nm, over a temperature range of 20–95°C, using a temperature increment of 1°C/min. The midpoint of the thermal transition (Tm) was obtained through non-linear fitting of the thermal denaturation using the Gibbs-Helmhotz equation and plotted as the mean ± SEM in GraphPad Prism. Wavelength scans were collected from 190–280 nm for both PI3K RBDs, with measurements taken every 1 nm.
Isothermal Titration Calorimetry (ITC)
For all ITC experiments, KRAS was either bound to guanine diphosphate (GDP) (Thermo Fisher Scientific #J61646) or a non-hydrolysable analog of GTP, guanosine-5′-[(β,γ)-methyleno]triphosphate GMPPCP (Sigma #M3509). For GDP studies, KRAS was purified in the presence of 15 μM GDP and then the protein was verified to only contain GDP via HPLC. To load KRAS with GMPPCP, KRAS proteins were loaded with five-fold excess guanine mono-phosphate (GMPPCP) (Sigma) in 20 mM HEPES (pH 8.0), 50 mM NaCl, 125 mM ammonium sulfate, 1 mM MgCl2, 1 mM ethylenediaminetetraacetic acid (EDTA), 100 μM ZnCl2 and then incubated for 48 h at 4°C in the presence of alkaline phosphatase agarose beads derived from cow intestine (Sigma). Following incubation, KRAS-GMPPCP protein was exchanged into 20 mM HEPES, 50 mM NaCl, 5 mM MgCl2, and 1 mM TCEP, which is the same buffer all ITC experiments were performed in. Nucleotide loading was then verified using HPLC. RAS proteins (~200–250 μM) were injected into the sample cell containing PI3K RBDs (~10–15 μM) using a MicroCal PEAQ-ITC (Malvern Paranalytical) or Microcal iTC200 (Malvern Paranalytical). The ITC experiment was performed at a 1:15–1:20 effector to RAS molar ratio at 25°C, stirring speed of 650 rpm, an initial delay of 120 seconds, with 19 2-μL injections spaced 180 seconds apart. To take into account heat of dilution, either the heat associated with the last few injections when saturation occurred was subtracted from all of the heat peaks (control subtraction) or the heat produced from KRAS injected into the sample cell containing only buffer was subtracted. All data was analyzed using a nonlinear least square algorithm and fit to a one-site model provided in Origin 7, the accompanying software for the ITC instruments.
NMR Analyses
To generate 13C/15N-enriched PI3Kα RBD, pCDB24 construct containing our gene of interest, was expressed in BL21(DE3)-RIPL E. coli bacterial strain in minimal medium [41] containing 1 g/L 15NH4Cl and 3g/L 13C-glucose (Cambridge Isotope Laboratories) as the sole source of nitrogen and carbon, respectively. The cells were grown at 37°C to an OD600 ~ 0.8 and induced with 500 μM IPTG followed by incubation at 18°C. After 22 h, the cells were harvested at 4 °C and 5000 x g and then purified as described above. 13C/15N-enriched PI3Kα protein (157 μM) was equilibrated in 20 mM HEPES (pH 6.8), 50 mM NaCl and 5 mM DTT containing 5% (v/v) D2O. Two-dimensional NMR 1H-15N HSQC spectrum of 13C/15N-labelled PI3Kα RBD was acquired on a Bruker Avance 850 MHz (19.97 T field strength) spectrometer at 25 °C, using a cryogenic (TCI) 5 mm triple-resonance probe equipped with z-axis gradient. NMR data was collected using a spectral width of 13 ppm and 33 ppm and complex points of 1024 and 256 along the 1H and 15N dimension, respectively. The NMR data was processed using NMRPipe [42] and the spectrum was visualized using SPARKY [43].
Computational Analysis
The PI3Kα RBD (residues 157–300) and PI3Kγ RBD (residues 193–316) sequences were selected for expression in vitro based on visual inspection of the full-length crystal structures (PDB ID: 3HHM and 1HE8, respectively). All images of molecular structures were created with PyMol [44]. The endpoints for the PI3Kα construct was selected based on the sequence that was expected to form a minimal globular unit. The PI3Kγ RBD was created based on this PI3Kα construct.
Amino Acid Occupancy
Crystal structures for PI3Kα and PI3Kγ were extracted from the Protein Data Bank (PDB) based on Uniprot Accession codes P42336 and P48736, respectively. A bash script was written to count the number of PDB files that contain a defined residue (i.e. sufficient electron density to resolve the residue) and the results were plotted with MS Excel.
Sequence Alignments
A structure-based sequence alignment for the RBD domains of PI3Kγ and PI3Kα (PDB ID: 3HHM and 1HE8, respectively) was created with PyMol. The alignment applied the BLOSUM62 substitution matrix and was followed by 10 cycles of iterative refinement of the Cα atoms with a RMSD of 0.97 Å for 80 residues. The sequence alignment was exported to clustalw format for image processing with Jalview [45]. Conservation scores were calculated based on the AMAS method [46].
Protein Crystallization and X-ray Diffraction Studies
The PI3Kα-RBD protein was brought to a concentration of 725 μM in crystallization buffer (20 mM HEPES, 50 mM NaCl, 3 mM (tris(2-carboxyethyl)phosphine) (TCEP), pH 7.4). Crystals were obtained through the sitting-drop vapor diffusion method. Using a 96-well plate set-up, three 0.3 μL drops were equilibrated against a reservoir volume of 30 μL. Initial PI3Kα RBD construct crystals were obtained from a mother liquor solution of 0.17M ammonium acetate, 0.085M sodium citrate HCl, and 25% w/v PEG4000, pH 5.6, equilibrated against an identical reservoir solution at 20°C in 7 days. As the crystals were of low quality, they were crushed and a 1:100 seed stock was used to perform random microseed matrix screening [47]. The final PI3Kα RBD crystal was obtained from a mother liquor of 0.7 M sodium citrate and 0.1 M Tris-HCl at pH 8.5, equilibrated against an identical reservoir solution at 20°C in 3 days. The crystals were cryo-protected by briefly dipping them in reservoir solution enhanced with 15% ethylene glycol and then flash frozen in liquid N2. Data were collected at 100 K on the SER-CAT ID22 beamline at the APS synchrotron facility, utilizing a wavelength of 1.00 Å. All data were scaled and integrated using HKL2000 [48], molecular replacement (MR) was performed with Phaser-MR [49] from the PHENIX [50] suite of programs. Truncation of the full-length PI3Kα (PDB ID: 5XGH) [51] to include only the residues of the PI3Kα-RBD construct design (residues 157–300) provided the search model for MR. Model building and manual placement of waters utilized COOT [52] and refinement was carried out using phenix.refine [53]. Alignment calculations were performed via COOT. X-ray data collection and refinement statistics are provided in Table 2.
Supplementary Material
Highlights.
KRAS selectively binds and activates different PI3K isoforms
PI3Kα is a key therapeutic target as mutations in PI3Kα are present in (~15–30%) of cancers
Novel, truncated PI3Kα/PI3Kγ constructs bind RAS in a GTP-dependent manner similar to full-length
Structural characterization of novel PI3Kα RAS binding domain reveals it retains similar structure as the full-length protein
Novel PI3K constructs will aid in characterizing RAS/PI3K interactions and drug discovery efforts
Acknowledgments
We thank all members of the Campbell laboratory for critical discussions and guidance. This study was supported in part by NCI PO1CA203657 (S.L.C.), Diversity Supplement Award 3P01CA203657-04S1 (N.G.M.), NIH R35 Grant GM131923 (B.K.) and NIH F32 GM136112 (D.T.)
SER-CAT and APS acknowledgement
Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID (or 22-BM) beamline at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions (see www.ser-cat.org/members.html), and equipment grants (S10_RR25528 and S10_RR028976) from the National Institutes of Health. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Macromolecular X-ray Crystallography Core/UNC Macromolecular Interactions Facility
This work was supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers
The coordinates & structure factors have been deposited in the Protein Data Bank under the accession number 6VO7.
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT, The PI3K Pathway in Human Disease, Cell. 170 (2017) 605–635. 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Backer JM, The Regulation of Class IA PI 3-Kinases by Inter-Subunit Interactions, in: Rommel C, Vanhaesebroeck B, Vogt PK (Eds.), Phosphoinositide 3-Kinase Heal. Dis Vol. 1, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011: pp. 87–114. 10.1007/82_2010_52. [DOI] [Google Scholar]
- [3].Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J, Phosphatidylinositol-3-OH kinase direct target of Ras, Nature. 370 (1994) 527–532. 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- [4].Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J, Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation., EMBO J. 15 (1996) 2442–2451. 10.1002/j.1460-2075.1996.tb00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kurig B, Shymanets A, Bohnacker T, Prajwal, Brock C, Ahmadian MR, Schaefer M, Gohla A, Harteneck C, Wymann MP, Jeanclos E, Nürnberg B, Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110γ, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 20312–20317. 10.1073/pnas.0905506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Buckles TC, Ziemba BP, Masson GR, Williams RL, Falke JJ, Single-Molecule Study Reveals How Receptor and Ras Synergistically Activate PI3Kα and PIP3 Signaling, Biophys. J 113 (2017) 2396–2405. 10.1016/j.bpj.2017.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Siempelkamp BD, Rathinaswamy MK, Jenkins ML, Burke JE, Molecular mechanism of activation of class IA phosphoinositide 3-kinases (PI3Ks) by membrane-localized HRas, J. Biol. Chem 292 (2017) 12256–12266. 10.1074/jbc.M117.789263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Okkenhaug K, Vanhaesebroeck B, PI3K in lymphocyte development, differentiation and activation, Nat. Rev. Immunol 3 (2003) 317–330. 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- [9].Vadas O, Burke JE, Zhang X, Berndt A, Williams RL, Structural biology structural basis for activation and inhibition of class I phosphoinositide 3-kinases, Sci. Signal 4 (2011) 1–13. 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- [10].Erijman A, Shifman JM, RAS / Effector Interactions from Structural and Biophysical Perspective, (2016) 370–375. [DOI] [PubMed]
- [11].Thorpe LM, Yuzugullu H, Zhao JJ, PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting, Nat. Rev. Cancer 15 (2015) 7–24. 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J, Binding of Ras to Phosphoinositide 3-Kinase p110α Is Required for Ras-Driven Tumorigenesis in Mice, Cell. 129 (2007) 957–968. 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- [13].Castellano E, Sheridan C, Thin MZ, Nye E, Spencer-Dene B, Diefenbacher ME, Moore C, Kumar MS, Murillo MM, Grönroos E, Lassailly F, Stamp G, Downward J, Requirement for Interaction of PI3-Kinase p110α with RAS in Lung Tumor Maintenance, Cancer Cell. 24 (2013) 617–630. 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zelenay S, Murillo MM, Downward J, Lassailly F, Stamp G, Nye E, Castellano E, RAS interaction with PI3K p110α is required for tumor-induced angiogenesis, J. Clin. Invest 124 (2014) 3601–3611. 10.1172/jci74134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Murillo MM, Rana S, Spencer-Dene B, Nye E, Stamp G, Downward J, Disruption of the Interaction of RAS with PI 3-Kinase Induces Regression of EGFR-Mutant-Driven Lung Cancer, Cell Rep. 25 (2018) 3545–3553.e2. 10.1016/j.celrep.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, Anderson AO, Waters AM, Diehl JN, Papke B, Hodge RG, Klomp JE, Goodwin CM, DeLiberty JM, Wang J, Ng RWS, Gautam P, Bryant KL, Esposito D, Campbell SL, Petricoin EF, Simanshu DK, Aguirre AJ, Wolpin BM, Wennerberg K, Rudloff U, Cox AD, Der CJ, Atypical KRASG12R mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer, Cancer Discov. 10 (2020) 104–123. 10.1158/2159-8290.CD-19-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J, Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras, Cell. 89 (1997) 457–467. 10.1016/S0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- [18].Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL, Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma., Cell. 103 (2000) 931–43. http://www.ncbi.nlm.nih.gov/pubmed/11136978. [DOI] [PubMed] [Google Scholar]
- [19].Vargiu P, De Abajo R, Garcia-Ranea JA, Valencia A, Santisteban P, Crespo P, Bernal J, The small GTP-binding protein, Rhes, regulates signal transduction from G protein-coupled receptors, Oncogene. 23 (2004) 559–568. 10.1038/sj.onc.1207161. [DOI] [PubMed] [Google Scholar]
- [20].Nakhaeizadeh H, Amin E, Nakhaei-Rad S, Dvorsky R, Ahmadian MR, The RAS-effector interface: Isoform-specific differences in the effector binding regions, PLoS One. 11 (2016) 1–20. 10.1371/journal.pone.0167145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bery N, Cruz-Migoni A, Bataille CJ, Quevedo CE, Tulmin H, Miller A, Russell A, Phillips SE, Carr SB, Rabbitts TH, BRET-based RAS biosensors that show a novel small molecule is an inhibitor of RAS-effector protein-protein interactions, Elife. 7 (2018) 1–28. 10.7554/elife.37122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou Z-W, Ambrogio C, Bera A, Li Q, Li X-X, Li L, Son J, Gondi S, Li J, Campbell E, Jin H, Okoro J, Xu C-X, Janne P, Westover K, KRASQ61H preferentially signals through MAPK in a RAF dimer-dependent manner in non-small cell lung cancer, Cancer Res. (2020) canres.0448.2020. 10.1158/0008-5472.CAN-20-0448. [DOI] [PubMed] [Google Scholar]
- [23].Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B, Amzel LM, A frequent kinase domain mutation that changes the interaction between PI3Kα and the membrane, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 16996–17001. 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vorobiev L, Su S, Seetharaman M, Patel J, Xiao P, Ciccosanti R, Shastry C, Everett R, Nair JK, Acton R, Rost TB, Montelione B, Hunt GT, Tong JF, Crystal structure of the RBD domain of serine/theronine-protein kinase B-raf from Homo sapiens, RCSB PDB. (2010). [Google Scholar]
- [25].Patel M, Côté J-F, Ras GTPases’ interaction with effector domains: Breaking the families’ barrier., Commun. Integr. Biol 6 (2013) e24298. 10.4161/cib.24298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Suire S, Condliffe AM, Ferguson GJ, Ellson CD, Guillou H, Davidson K, Welch H, Coadwell J, Turner M, Chilvers ER, Hawkins PT, Stephens L, Gβγs and the Ras binding domain of p110γ are both important regulators of PI3Kγ signalling in neutrophils, Nat. Cell Biol 8 (2006) 1303–1309. 10.1038/ncb1494. [DOI] [PubMed] [Google Scholar]
- [27].Yang HW, Shin MG, Lee S, Kim JR, Park WS, Cho KH, Meyer T, Do Heo W, Cooperative Activation of PI3K by Ras and Rho Family Small GTPases, Mol. Cell 47 (2012) 281–290. 10.1016/j.molcel.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, Downward J, RAS interaction with PI3K p110α is required for tumor-induced angiogenesis, J. Clin. Invest 124 (2014) 3601–3611. 10.1172/JCI74134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X, Targeting PI3K in cancer: Mechanisms and advances in clinical trials 06 Biological Sciences 0601 Biochemistry and Cell Biology, Mol. Cancer 18 (2019) 1–28. 10.1186/s12943-019-0954-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Miller MS, Thompson PE, Gabelli SB, Structural determinants of isoform selectivity in pi3k inhibitors, Biomolecules. 9 (2019). 10.3390/biom9030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nunnery SE, Mayer IA, Management of toxicity to isoform α-specific PI3K inhibitors, Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 30 (2019) x21–x26. 10.1093/annonc/mdz440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Stieglitz B, Bee C, Schwarz D, Yildiz Ö, Moshnikova A, Khokhlatchev A, Herrmann C, Novel type of Ras effector interaction established between tumour suppressor NORE1A and Ras switch II, EMBO J. 27 (2008) 1995–2005. 10.1038/emboj.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang M, Jang H, Nussinov R, The structural basis for Ras activation of PI3Kα lipid kinase, Phys. Chem. Chem. Phys 21 (2019) 12021–12028. 10.1039/c9cp00101h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lan H, Franz H, Steven M, Sung-Hou K, Structural basis for the interaction of Ras with RalGDS, Nat. Struct. Biol 5 (1998). http://link.springer.com/10.1007/978-1-62703-429-6. [DOI] [PubMed] [Google Scholar]
- [35].Johnson CW, Reid D, Parker JA, Salter S, Knihtila R, Kuzmic P, Mattos C, The small GTPases K-Ras, N-Ras, and H-Ras have distinct biochemical properties determined by allosteric effects, J. Biol. Chem 292 (2017) 12981–12993. 10.1074/jbc.M117.778886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rudolph MG, Linnemann T, Grünewald P, Wittinghofer A, Vetter IR, Herrmann C, Thermodynamics of Ras/Effector and Cdc42/Effector Interactions Probed by Isothermal Titration Calorimetry, J. Biol. Chem 276 (2001) 23914–23921. 10.1074/jbc.M011600200. [DOI] [PubMed] [Google Scholar]
- [37].Kim D, Xue JY, Lito P, Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients, Cell. 183 (2020) 850–859. 10.1016/j.cell.2020.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, Lanman BA, Werner J, Rapaport AS, San Miguel T, Ortiz R, Osgood T, Sun JR, Zhu X, McCarter JD, Volak LP, Houk BE, Fakih MG, O’Neil BH, Price TJ, Falchook GS, Desai J, Kuo J, Govindan R, Hong DS, Ouyang W, Henary H, Arvedson T, Cee VJ, Lipford JR, The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity, Nature. 575 (2019) 217–223. 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- [39].Hallin J, Engstrom LD, Hargi L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, Burkard MR, Fell JB, Fischer JP, Vigers GP, Xue Y, Gatto S, Fernandez-Banet J, Pavlicek A, Velastagui K, Chao RC, Barton J, Pierobon M, Baldelli E, Patricoin EF, Cassidy DP, Marx MA, Rybkin II, Johnson ML, Ignatius SH, Lito P, Papadopoulos KP, Jänne PA, Olson P, Christensen JG, The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients, Cancer Discov. 10 (2020) 54–71. 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cruz-Migoni A, Canning P, Quevedo CE, Bataille CJR, Bery N, Miller A, Russell AJ, Phillips SEV, Carr SB, Rabbitts TH, Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 2545–2550. 10.1073/pnas.1811360116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cai M, Huang Y, Yang R, Craigie R, Clore GM, A Simple and Robust Protocol for High-Yield Expression of Perdeuterated Proteins in Escherichia Coli Grown in Shaker Flasks, J. Biomol. NMR 66 (2016) 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, NMRPipe: A multidimensional spectral processing system based on UNIX pipes, J. Biomol. NMR 6 (1995) 277–293. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- [43].Lee W, Tonelli M, Markley JL, NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy, Bioinformatics. 31 (2015) 1325–1327. 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].DeLano WL, The PyMOL Molecular Graphics System, Delano Sci. San Carlos (2002). [Google Scholar]
- [45].Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ, Jalview Version 2-A multiple sequence alignment editor and analysis workbench, Bioinformatics. 25 (2009) 1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Livingstone CD, Barton GJ, Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation., Comput. Appl. Biosci 9 (1993) 745–756. 10.1093/bioinformatics/9.6.745. [DOI] [PubMed] [Google Scholar]
- [47].Till M, Robson A, Byrne MJ, Nair AV, Kolek SA, Shaw Stewart PD, Race PR, Improving the success rate of protein crystallization by random microseed matrix screening, J. Vis. Exp (2013) 1–6. 10.3791/50548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Otwinowski Z, Minor W, Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol. 276 (1997) 307–326. [DOI] [PubMed] [Google Scholar]
- [49].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software, J. Appl. Crystallogr 40 (2007) 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, PHENIX: A comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr. Sect. D Biol. Crystallogr 66 (2010) 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yang X, Zhang X, Huang M, Song K, Li X, Huang M, Meng L, Zhang J, New Insights into PI3K Inhibitor Design using X-ray Structures of PI3Kα Complexed with a Potent Lead Compound, Sci. Rep 7 (2017) 1–7. 10.1038/s41598-017-15260-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot, Acta Crystallogr. Sect. D Biol. Crystallogr 66 (2010) 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD, Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallogr. Sect. D Biol. Crystallogr 68 (2012) 352–367. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Stride Web Interface, (n.d.).
- [55].Frishman D, Argos P, Knowledge-Based Protein Secondary Structure Assignment, Proteins Struct. Funct. Genet 23 (1995) 566–579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.