

Abstract
Glioblastoma (GBM) is a lethal form of primary brain tumor in human adults. The impact of tumor-intrinsic alterations is not exclusively confined to cancer cells but can also be extended to the tumor microenvironment (TME). Glioblastoma-associated macrophages/microglia (GAMs) are a prominent type of immune cells that account for up to 50% of total cells in GBM. Emerging evidence suggests that context-dependent GBM-GAM symbiotic interactions are pivotal for tumor growth and progression. Here, we discuss how specific genetic alterations in GBM cells affect GAM biology and, reciprocally, how GAMs support GBM progression. We hypothesize that understanding context-dependent GBM-GAM symbiosis may reveal the molecular basis of GBM tumorigenesis, and lead to novel candidate treatment approaches aiming to improve GBM patient outcomes.
Keywords: Glioblastoma, Macrophages, Microglia, Crosstalk, Symbiosis, Heterogeneity
GBM Genetic Alterations and Associations with the TME
Cancer has been recognized as a genetic disease for more than a century, as driven by either the activation of oncogenes or the inactivation of tumor suppressor genes (TSGs), triggering tumor formation and progression [1]. In-depth studies on cancer genomics have yielded comprehensive atlases of oncogene and TSG alterations for specific types of cancer, which can reveal specific cancer vulnerabilities (Box1). Glioblastoma (GBM) is the most aggressive and highly lethal form of primary brain tumor in human adults [2, 3], and the current standard of care offers only modest survival benefit to GBM patients [4–6]. Genomic profiling of patient tumors has led to GBM subclassification based on gene alterations affecting core signaling pathways: specifically, those associated with P53/ARF/MDM2, receptor tyrosine kinase (RTK)/RAS/PI3K/PTEN and RB/CDKN2A alterations [4, 7–9]. These subclassifications have motivated clinical trials for testing targeted therapies, such as those against RTK signaling [4]. Regrettably, attempts to inhibit aberrant signaling that results from these gene alterations have not been successful in improving GBM patient outcomes. The underlying basis for these failures relates to the inherent cellular heterogeneity (see Glossary) of GBM that ensures the survival of cancer cells, irrespective of treatments [4, 10]. Glioma stem cells (GSCs) are subpopulations of cells endowed with stem cell properties (e.g., self-renewing capacity and tumor-propagating potential), which have been shown to promote GBM intratumoral heterogeneity [11, 12].
Box 1. Personalized Medicine for Cancer Patients.
Discoveries of synthetic lethality and oncogene addiction have revealed specific cancer vulnerabilities that can be exploited for treatment with vulnerability-targeted agents [68, 74–76]. For example, treatment with inhibitors targeting epidermal growth factor receptor (EGFR) or poly ADP ribose polymerase (PARP) provides significant benefits to cancer patients with an EGFR mutation [77, 78] or a BRCA mutation [78–80], respectively. Unfortunately, many gene alterations (including alterations of oncogenes and tumor suppressor genes) have yet to reveal druggable targets. Recent discoveries with new conceptual approaches (e.g., synthetic essentiality and collateral lethality) suggest that targeting lethal gene-gene interactions within cancer cells may provide a means to identify novel cancer therapeutic targets [75, 81, 82]. Now emerging, is the concept of lethal gene-gene interactions between cancer cells and the TME [83]. Together, such lethal gene-gene interactions suggest opportunities for novel treatment approaches.
The impact of aberrant cancer-associated signaling is not limited to cancer cells, but also extends to normal cells of the tumor microenvironment (TME) [13]. The function and composition of the GBM TME are influenced by cancer cell-intrinsic signaling pathways and secreted factors [13–16]. Reciprocally, the TME can promote GBM progression by modulating multiple cancer biologic properties, including cell proliferation, survival, migration, and immune surveillance [5, 17]. Among the TME, glioblastoma-associated macrophages/microglia (GAMs) constitute the most abundant cell population, and account for up to 50% of total live cells in the GBM tumor mass [17, 18]. GAMs are composed of multiple subpopulations including bone-marrow-derived macrophages (BMDMs, hereafter referred to as macrophages) and brain-resident microglia (hereafter referred to as microglia) (Box 2) [18, 19]. GAMs can also be classified as polarized toward pro-inflammatory and ‘alternatively activated’ phenotypes (Box 3), which exhibit anti-tumor/immune-stimulatory and pro-tumor/immunosuppressive effects, respectively [20]. However, it is highly likely that GAMs are composed of heterogeneous subpopulations that are not polarized to either state with variable immune functional capabilities, such as antigen presentation, phagocytosis, and tumor supportive functions [21]. A study of a large cohort of GBM patients has shown that tumors with mutations in PTEN, epidermal growth factor receptor (EGFR) and neurofibromin 1 (NF1) are significantly enriched with GAMs [22]. With respect to the transcriptomic classification of GBM, the most biologically aggressive mesenchymal subtype is enriched for alterations in PTEN, TP53, NF1, and RB1 [23], and is highly enriched with immunosuppressive macrophages [24]. Transcriptional regulatory network analyses have also revealed that the master regulators and their target genes for GAMs in mesenchymal GBM are associated with genetic aberrations in NF1 and PTEN/PI3K/mTOR/AKT pathways [25]. Together, these findings support a relationship in which GBM cells and GAMs can influence each other under specific genetic backgrounds.
Box 2. Identity of macrophages and microglia in GBM.
Glioblastoma-associated macrophages/microglia (GAMs) are composed of two subpopulations, including bone-marrow-derived macrophages (BMDMs, hereafter referred to as macrophages) and microglia [17, 18]. Distinguishing these two populations is complicated due to the lack of specific macrophage or microglia markers [18, 84]. Combining Cx3cr1GFP/WT;Ccr2RFP/WT knock-in mice with a genetically engineered mouse model (Pdgfb-driven glioma) or GL261 model has allowed to distinguish macrophages from microglia in tumors, in which macrophages account for approximately 85% of GAMs in these glioma models [18]. However, a growing body of evidence suggests that the ratio between macrophages and microglia in GBM is dependent on the methods used to quantify them, as well as on tumor genetic backgrounds and subtypes. For example, macrophage abundance in GL261 tumors is significantly enhanced in the irradiation bone marrow transplantation (IR-BMT) lineage tracing model compared to the Flt3:Cre;Rosa26:mTmG lineage tracing model [84]. Based on genetic lineage tracing in murine models, transcriptional profiling followed by functional studies have revealed that CD49D is specifically absent in microglia, and can distinguish microglia from macrophages in mouse and human GBM tumors [84]. Mutations in isocitrate dehydrogenase 1 and 2 (IDHmut) are generally observed in low-grade glioma, and GBM patients with IDHmut have a significantly better prognosis than those with wild-type (WT) IDH [85]. Analysis of glioma patient samples have demonstrated that IDHmut tumors harbor more microglia and fewer macrophages relative to IDH WT tumors [85, 86]. With regard to GBM tumor subtypes, macrophage and microglia signatures have been reported to be highly enriched in GBM patients with mesenchymal and neural subtypes, respectively [84].
Box 3. Pro-inflammatory and alternatively activated macrophages.
Macrophages are highly plastic cells consisting of distinct phenotypes, including a “classically activated” pro-inflammatory phenotype and an “alternatively activated” anti-inflammatory phenotype, which can be activated by specific factors or cytokines [87, 88]. For example, macrophages are polarized toward a pro-inflammatory phenotype upon stimulation with lipopolysaccharide, IFNγ or TNFα, which in turn produce pro-inflammatory cytokines, such as interleukin 1 beta (IL-1β), IL-12, and IL-23, to kill cancer cells and microorganisms. Instead, macrophage alternative polarization can be triggered by exposure to IL-4, IL-13 or IL-10, which in turn produces anti-inflammatory factors, such as IL-10, transforming growth factor beta 1 (TGF-β), C-C motif ligand 17 (CCL17), and CCL22, to promote tumor progression, angiogenesis and tissue repair [87–89]. These two phenotypes have been widely used to define macrophage subpopulations in cancer and immunology; however, this dichotomy is facing challenges due to the fact that: i) macrophages can respond to the dynamic tumor microenvironment in vivo; ii) both phenotypes often coexist in tumors, and iii) phenotype specific macrophage markers are still missing [90, 91]. However, accumulating evidence demonstrates that glioblastoma-associated macrophages/microglia (GAMs) are usually biased toward an alternatively activated phenotype, promoting tumor progression and creating an immunosuppressive microenvironment [19]. Consequently, reprogramming GAMs toward a pro-inflammatory phenotype has been reported as an effective antitumor strategy in certain models. For example, treatments with colony stimulating factor 1 receptor (CSF-1R) inhibitor BLZ945, angiopoietin 2/ vascular endothelial growth factor (Ang-2/VEGF) bispecific antibodies, and oncolytic viruses G47Δ expressing murine IL-12) can skew GAMs toward a pro-inflammatory phenotype, and extend survival in GBM mouse and PDX models [92–94]. Moreover, this strategy can synergize with immunotherapies (e.g., anti-PD1 antibody (Ab) or anti-CTLA4 Ab therapy) to extend survival in CT2A and 005 GSC GBM mouse models [94]. In addition, clinical evidence further suggests that anti-PD1 Ab therapy resistance is often observed in GBM patients with high numbers of alternatively activated GAMs, and PTEN deletion/mutation [95].
Increasing evidence underscores the myriad interactions and intertwined roles of cancer cells and GAMs in GBM biology. In this opinion article, we highlight and discuss how the alterations of specific TSGs and oncogenes in GBM cells affect GAM biology (including migration, adhesion and polarization), and reciprocally, how GAMs support GBM growth and progression. We propose to discuss context-dependent GBM-GAM symbiotic interactions, therapeutic potential via targeting GBM-GAM symbiosis, and future perspectives. We hypothesize that these emerging insights not only expand our understanding of GBM tumorigenesis, but might also provide a roadmap for the development of novel putative therapeutic strategies that disrupt this context-dependent circuit in GBM.
The Influence of TSG Alterations on GAM Biology
Factors secreted by GBM cells in the TME are known to recruit and polarize GAMs [20]. Increasing evidence shows that GBM cell-derived factors are regulated by specific TSG alterations (Figure 1). PTEN is a TSG that acts in opposition to PI3K signaling in GBM, and PTEN is mutated and/or deleted in about 30–40% of GBMs [8, 23]. The Cancer Genome Atlas (TCGA) analyses and immunohistochemical staining in GBM patient tissue microarrays (TMAs) have revealed that PTEN mutations are significantly associated with increased intratumoral macrophages, but have no effect on microglia [24]. These findings strongly suggest that PTEN deficiency in GBM cells might specifically trigger macrophage infiltration. This hypothesis is further supported by in vitro and in vivo (including mouse and human GBM models) studies showing that depletion of PTEN in PTEN-WT GBM increases macrophage recruitment, whereas overexpression of PTEN in PTEN-deficient GBM has opposite effects [24]. Mechanistically, PTEN deficiency in GBM cells activates SRC and AKT pathways, which, in turn, activates yes-associated protein 1 (YAP1) signaling [24]. YAP1 is a transcriptional co-activator that binds to the promoter of lysyl oxidase (LOX) and upregulates LOX expression in GBM cells [24]. LOX is a secreted enzyme that can be internalized by macrophages through the β1 integrin receptor, which, in turn, causes reactive oxygen species, hydrogen peroxide, to promote macrophage migration via activation of PYK2 signaling [24]. Moreover, in PTEN-null, but not PTEN-WT GBM models (e.g., human and mouse orthotopic GBM models established in SCID and C57BL/6 mice), LOX inhibition suppresses tumor macrophage infiltration, and increases the survival of tumor-bearing mice [24]. However, activation of PI3K/AKT signaling in human GBM cells and GSCs has been shown to promote GBM progression via activation of other signaling pathways, such as the WNT/β-catenin pathway (e.g., increasing GBM cell proliferation and GSC self-renewal, inhibiting GBM cell apoptosis, and inducing GSCs differentiation into endothelial-like cells relative to controls) [26, 27]. For example, GSCs in GBM can secrete WNT‐induced signaling protein 1 (WISP1) to promote GAM survival and GSC stemness through activation of the α6β1 integrin/AKT pathway in vitro and in GBM patient-derived xenograft (PDX) models [28]. Inhibition of the WNT/β-catenin/WISP1 pathway by shRNA knockdown (KD), or pharmacological inhibitor carnosic acid in human GSCs, suppresses GBM growth and extends survival through a mechanism that involves reduced numbers of intratumoral GAMs and disrupted GSC maintenance in NSG mouse models [28]. Collectively, these findings suggest that PTEN/PI3K/AKT signaling is essential for producing soluble factors (e.g., LOX and WISP1) in human or mouse GBM cells that recruit and maintain GAMs via activation of macrophage β1 integrin signaling. These data not only reveal some of the molecular basis underlying an interplay between PTEN-deficient GBM cells and GAMs, but also highlight a synthetic lethal interaction between mutation/deletion of PTEN and suppression of macrophage chemokines. This in turn, might offer synthetic lethal approaches to target GBM-GAM symbiosis in PTEN-null GBM.
Figure 1. Inactivation of tumor suppressors in GBM cells that affect GAM biology.
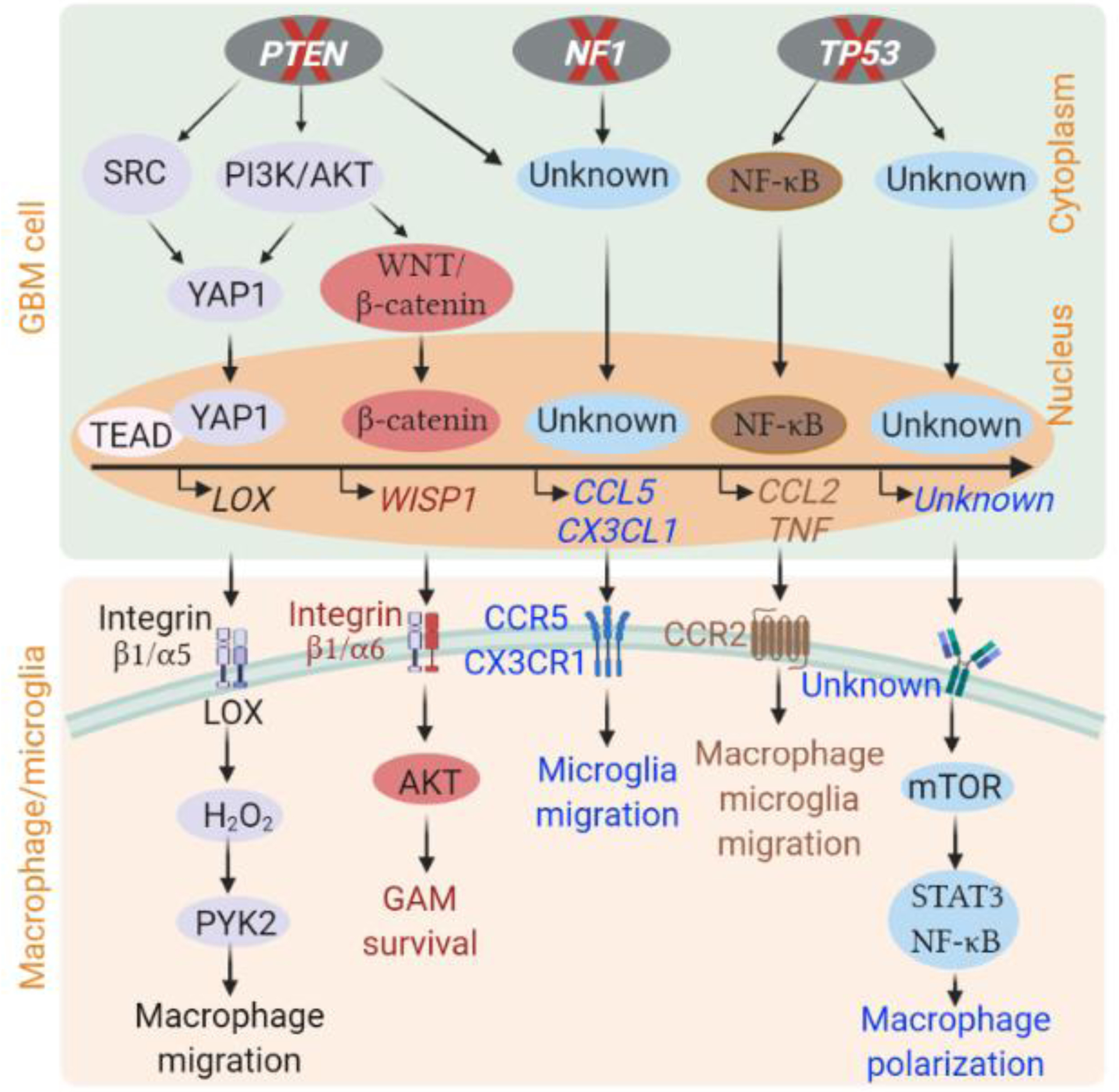
Inactivation of tumor suppressor genes (e.g., PTEN, NF1 and TP53) in GBM cells can increase the expression and secretion of various soluble factors through activation of distinct signaling pathways, which in turn recruit macrophages and microglia into the GBM tumor microenvironment, and then polarize them toward an alternatively activated phenotype in mouse and human GBM models [24, 28, 30, 32, 36]. The detailed signaling pathways and soluble factors involved in these processes are indicated. Unknown factors or signaling pathways are shown as unknown. This figure was created using BioRender (https://biorender.com/).
CCL2, CC chemokine ligand 2; CCL5, CC chemokine ligand 5; CCR2, C-C chemokine receptor type 2; CCR5, C-C chemokine receptor type 5; CX3CL1, C-X3-C motif chemokine ligand 1; CX3CR1, C-X3-C motif chemokine receptor 1; GAM, glioblastoma-associated macrophage/microglia; GBM, glioblastoma; LOX, lysyl oxidase; mTOR, mammalian target of rapamycin; NF-κB, Nuclear factor kappa B; PI3K, phosphoinositide 3-kinase; PYK2, proline-rich tyrosine kinase 2; STAT3, signal transducer and activator of transcription 3; TNFα, tumor necrosis factor alpha; WISP1, Wnt1-inducible signaling pathway protein-1; YAP1, yes-associated protein 1.
NF1 is a TSG that is mutated or deregulated in approximately 40% of the mesenchymal subtype of GBM and in 16% of all GBM patients [8, 29]. In addition to cancer cell intrinsic effects, tumor development in the context of NF1 loss has been reported to be regulated by steroid hormones in the CMV-CreERT2;Nf1flox/-;ROSA26 neurofibromas mouse model, thereby suggesting that tumor NF1 status can influence the TME [29]. Indeed, Nf1-deficient GSCs isolated from tumors of the Nf1 genetically engineered mouse model (Nf1flox/neo; GFAP-Cre mice) on a C57BL/6 background produce chemokines C-X3-C motif ligand 1 (CX3CL1) and C-C motif ligand 5 (CCL5), thus recruiting microglia into the TME, and this effect is further amplified by loss of Pten in GSCs isolated from Nf1flox/neo; Ptenflox/wt; GFAP-Cre mice [30]. TCGA data shows that GBM patients with NF1 abnormalities are associated with higher expression of intratumoral GAM-specific genes, such as ITGAM (integrin subunit alpha m; aka, CD11B) and AIF1 (allograft inflammatory factor 1; aka, IBA1), relative to patients with wild-type (WT) NF1 [31]. Additional support characterizing GBM cell NF1 status as a possible determinant of intratumoral GAM was offered by a study in which conditioned medium from NF1 shRNA KD human GBM cells increased the recruitment of macrophages and microglia relative to conditioned medium from shRNA control cells in a transwell migration assay [31]. Together, these findings suggest that loss of NF1 in GBM cells might promote intratumoral GAM infiltration. Evidently, further studies are needed, and these might also reveal some of the molecular mechanisms underlying GAM infiltration in human GBM. Accordingly, in this regard, GBM PDX models might lead to the development of novel synthetic lethal approaches to target GBM-GAM symbiosis in NF1-deficient GBM.
Another TSG alteration that affects GBM cellular composition is that of TP53. TP53 mutation is frequent in human GBM, and in addition to its effects on cell cycling and apoptosis [8, 32], tumor expression of mutant p53 increases the expression of CCL2 and tumor necrosis factor alpha (TNFα) via activation of nuclear factor kappa B (NF-κB) signaling in human GBM cells (e.g., U87 and 19NS) [32]. The increased CCL2 and TNFα, in turn, can recruit macrophages and microglia into the GBM TME [32]. The GL261 glioma mouse model in C57BL/6 mice harbors Trp53 mutant tumor cells [33]; these can communicate with GAMs via the release of extracellular vesicles (EVs) and soluble factors [34, 35]. For example, GL261 cell-derived EVs contain micro-RNA miR-21 that can increase microglia proliferation [34]. GL261 cells additionally produce secreted phosphoprotein 1 (SPP1) as a chemokine to trigger Iba1+ macrophage tumor infiltration via integrin αvβ5 signaling in tumors [35]. Moreover, results from several genetically engineered GBM mouse models (e.g., Ntv-a;Pdgfb;shTrp53, Pten−/−;Trp53−/− and Pten−/−;Trp53−/−;Idh1R132H;Pdgfb) demonstrate that GSCs with Trp53 deficiency can activate mTOR signaling in microglia, thus skewing microglia into an alternatively activated phenotype via activation of signal transducer and activator of transcription 3 (STAT3) and NF-κ signaling [36]; this in turn, suggests a potential therapeutic strategy involving inhibition of the mTOR/STAT3/NF-κB pathway in Trp53-deficient GBM. In addition to modulating GAM migration and polarization, Trp53 deficiency might also affect the phagocytic activity of GAMs. For instance, Trp53 and Sparc co-deficiency in mouse models promotes phagocytosis of glioma cells by GAMs, and induces decreased animal survival [37]. Together, these findings highlight the key role of p53 mutations in influencing GAM tumor infiltration, polarization, and phagocytic activity in GBM human and mouse models.
Consequently, we posit that there are significant symbiotic interactions between GAMs and GBM cells harboring deficiency of PTEN, NF1 and TP53, and suggest that targeting this context-dependent GBM-GAM symbiosis might reveal synthetic lethality. However, further studies are needed to design synthetic lethal approaches to target GBM-GAM symbiosis for specific TSG-deficient GBM (e.g., PTEN, NF1 or TP53). In addition, it will be relevant to determine whether this GBM-GAM symbiosis exists in GBMs that harbor a deficiency in other TSGs, such as CDKN2A and CDKN2B, as this might offer additional candidate personalized therapeutic strategies.
Role of Oncogene Alterations on GAM Biology
In addition to TSGs, the activation and amplification of oncogenes can also play an important role in modulating GAM infiltration and polarization in GBM (Figure 2). Among these is EGFR amplification and activation, which is a hallmark of GBM, and presents in about 60% of human GBM [8, 38–40]. TCGA analyses and immunohistochemical staining using patient TMAs have revealed that EGFR activation correlates positively with intratumoral macrophages in GBM [40, 41]. Mechanistically, EGFR activation in GBM cells can modulate the expression and activity of a variety of factors that are important for macrophage adhesion, infiltration, and polarization. For example, carbonic anhydrase IX (CAIX) mRNA and protein expression amounts are highly upregulated in human GBM cells (e.g., U87 and U251) under hypoxic conditions, and are associated with poor patient prognosis [42]. Moreover, EGFR activation plays a role in CAIX upregulation via stabilization of hypoxia-inducible factor 1-alpha in human GBM cells that in turn, promotes macrophage adhesion and polarization toward an alternatively activated phenotype [42]. EGFR activation can additionally increase TNFα-induced vascular cell adhesion molecule-1 (VCAM-1) expression in human (U251) and mouse (ALTS1C1) GBM cells via a P38/STAT3 pathway-dependent mechanism, and increased VCAM-1 promotes macrophage adhesion [43]. These macrophages conversely stimulate GBM cell proliferation, invasion, and further production of TNFα, thus forming a positive feedback loop between GBM cells and macrophages [43]. In addition to TNFα, stimulation of EGFR with EGF in human GBM cells (e.g., U251, D54 and A172) can activate protein kinase C epsilon type (PKCε) and NF-κB pathways, in turn upregulating VCAM-1 to promote macrophage adhesion, and thus increase GBM cell invasiveness in vitro [40]. Notably, half of EGFR-amplified human GBM tumors harbor an EGFR truncating mutation (EGFRvIII) [38]. Co-expression of EGFR and EGFRvIII in human GBM cells (e.g., U87 and A172) activates KRAS, and activated KRAS increases CCL2 expression, which, as noted above, can recruit CD68+ macrophages and TMEM119+ microglia into the GBM TME of GBM mouse models established in BALB/C nu/nu mice [44]. The above findings provide novel insights into EGFR-dependent GAM regulation, and offer potential therapeutic targets for GBM patients that specifically harbor EGFR amplifications or mutations.
Figure 2. Activation of oncogenes in GBM cells that affect GAM biology.
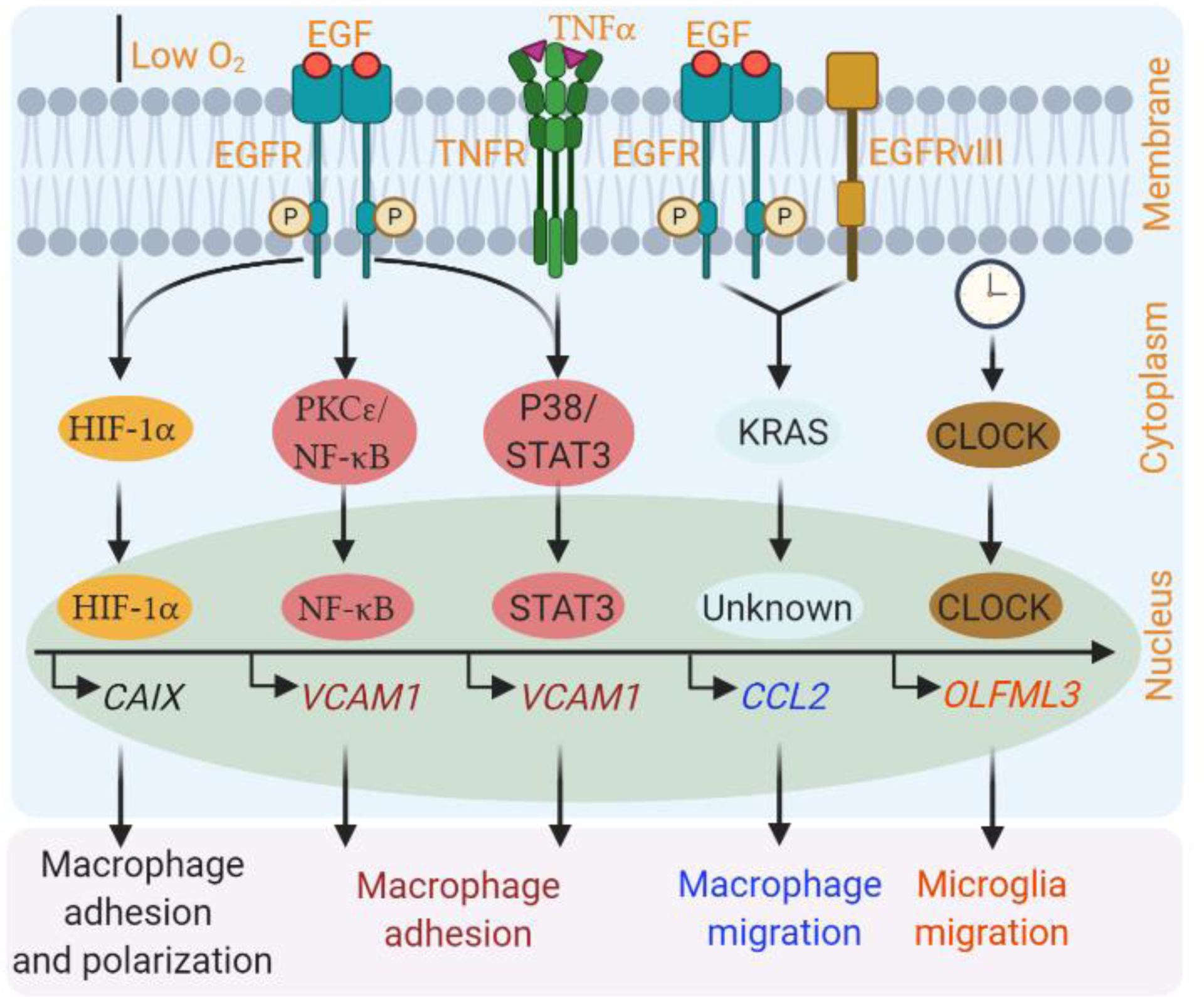
Activation of oncogenes (e.g., EGFR and CLOCK) in GBM cells can increase macrophage adhesion, migration and alternative polarization, as well as microglia migration through distinct mechanisms. For example, EGFR is required for hypoxia-induced activation of HIF-1α/CAIX axis in human GBM cells (e.g., U87 and U251), which can promote macrophage adhesion and alternative polarization [42]. In addition, EGFR is essential for EGF-induced activation of the PKCε/NF-κB pathway, and TNFα-induced activation of the P38/STAT3 axis in human and mouse glioma cells, which in turn, upregulate VCAM1 to increase macrophage adhesion [43]. EGFR can cooperate with EGFRvIII to upregulate KRAS, which in turn upregulates CCL2 to recruit macrophages. How CCL2 is regulated by KRAS in human GBM cells is still unknown [44]. CLOCK can transcriptionally upregulate chemokine OLFML3 in GSCs, which in turn recruits microglia into the GBM TME in mouse and human models [48]. This figure was created using BioRender (https://biorender.com/).
CAIX, carbonic anhydrase IX; CCL2, CC chemokine ligand 2; HIF-1α, EGF, Epidermal growth factor; EGFR, EGF receptor; EGFRvIII, EGFR variant III; GAM, glioblastoma-associated macrophage/microglia; GBM, glioblastoma; hypoxia-inducible factor 1-alpha; NF-κB, Nuclear factor kappa B; OLFML3, olfactomedin-like 3; PDX, patient-derived xenograft; PKCε, protein kinase C epsilon type, STAT3, signal transducer and activator of transcription 3; TNFα, tumor necrosis factor alpha; VCAM1, vascular cell adhesion molecule 1.
From another angle, the circadian rhythm is an important regulatory system that plays a pivotal role in regulating cancer cell proliferation, metabolism, and DNA repair [45–47]. Circadian locomotor output cycles kaput (CLOCK) is a key circadian regulator, which can function as an oncogene or TSG, based on TME factors [48]. In GBM, CLOCK is amplified in ~5% of human GBM cases and functions as an oncogene [48]. Inhibition of CLOCK by shRNA KD or pharmacological inhibitor SR9009 in human GBM PDX lines (e.g., T3565, T387, GSC272, and GSC20) reduces GSC self-renewal due to its effects on cell metabolism [48, 49]. Unbiased profiling studies have shown that high cancer cell stemness correlates with increased immunosuppressive activities in human GBM, including enhancing the GAM component [50], suggesting a potential role of stemness regulation in triggering GAM infiltration in GBM. Examination of TCGA GBM datasets revealed that a microglia signature (but not a macrophage signature) was enriched in CLOCK-high relative to CLOCK-low GBM patients [48]. Furthermore, high expression of CLOCK in human (e.g., GSC272 and GSC20) and mouse (e.g., QPP7) GSCs has been shown to recruit CX3CR1+ microglia into GBM tumors [48]. Mechanistically, this is presumed to occur with CLOCK upregulating the expression of olfactomedin-like protein 3 (OLFML3) in GSCs, which serves as a chemokine for microglia [48]. Inhibition of CLOCK by shRNA KD or pharmacological inhibitor SR9009 in human PDX (e.g., GSC272, GSC20, T387, and T3565 in SCID and NSG mice) and mouse (e.g., CT2A in C57BL/6 mice) orthotopic models can increase survival by reducing GSC stemness and microglia tumor infiltration relative to controls [48, 49]. Similarly, tumor-bearing mouse survival was also enhanced upon shRNA KD of OLFML3 in the GSC272 mouse model [48]. These data suggest that blockade of the GSC-microglia interplay via inhibition of OLFML3 might be a potentially promising therapeutic strategy in CLOCK-high GBM. However, the mechanism by which OLFML3 induces microglia tumor infiltration remains unknown. Future studies might identify druggable mediators of OLFML3’s effect on microglia, which might thereby expand the number of putative therapeutic targets in CLOCK-high GBM.
Taken together, these findings highlight the notion that oncogene alterations (e.g., EGFR and CLOCK amplifications and/or mutations) in GBM cells can change the properties of GAMs (e.g., migration and polarization), which might in turn contribute to promoting GBM progression, atleast from what has been gauged from mouse models. Although further studies are needed, these findings provide a rationale for developing personalized therapeutic strategies that target specific oncogene alteration-mediated GBM-GAM symbiosis (e.g., EGFR and CLOCK). Moreover, these findings provide a framework for the discovery of druggable targets in GBM that harbor specific alterations of other important oncogenes, such as PI3K, cyclin-dependent kinase 4 (CDK4), and platelet-derived growth factor receptor alpha (PDGFRA).
Impact of GAMs on GBM Progression
Once macrophages and microglia have infiltrated into the TME, they are educated by GBM cells [20]. The inverse is also true: i.e., recruited GAMs can reciprocally promote GBM progression [20] (Figure 3). A growing body of evidence indicates that GAMs promote GSC self-renewal, and stemness is known to be important for sustained tumor growth and resistance to therapy in GBM mouse models and patient samples [5, 25]. Mechanistically, GAMs can regulate GSC self-renewal by secreting stemness-supporting factors, including heparin-binding EGF-like growth factor, IL-12, IL-1β and CCL8 [51–53], as well as lipocalin 2, hepatocyte growth factor, vascular endothelial growth factor and IL-6 [54] in mouse and human GBM models. Furthermore, recent studies have demonstrated that pleiotrophin (PTN) is expressed and secreted by GAMs, and PTN helps sustain GSC stemness through its receptor PTPRZ1, which activates AKT signaling in GBM PDX lines [55]. shRNA KD of PTN in alternatively activated macrophages impairs their ability to promote GSC growth, and inhibition of GSC-associated PTPRZ1 dramatically impairs GSC maintenance and tumorigenic potential in human PDX GBM models, such as T4121 and T0912 models in SCID mice [55]. GAMs that are skewed toward an alternatively activated phenotype appear to be essential for maintaining GSC populations in GBM mouse models and patients [17, 56]. Reprogramming of GAMs toward an pro-inflammatory phenotype using vitamin B3 or amphotericin B not only impairs the stemness and tumorigenicity of human and mouse GSCs in vitro and in vivo, but can also sensitize brain tumors to chemotherapy in PDX models (e.g., BT048 and BT53M) [57, 58]. In addition to macrophages, microglia can also contribute to GSC maintenance in GBM. For instance, when microglia are stimulated by CD8+ T cell-derived CCL4, they secrete CCL5 that can increase GSC stemness by activating CD44/AKT/GSK3β/CREB signaling in GSCs isolated from Nf1flox/mut;GFAP-Cre mice [59]. Together, these findings suggest that GAMs are a component of the GSC niche, which can sustain GSC properties via secretion of a variety of stemness-supporting factors.
Figure 3. Impact of glioblastoma-associated macrophages/microglia (GAMs) on glioblastoma (GBM) progression.
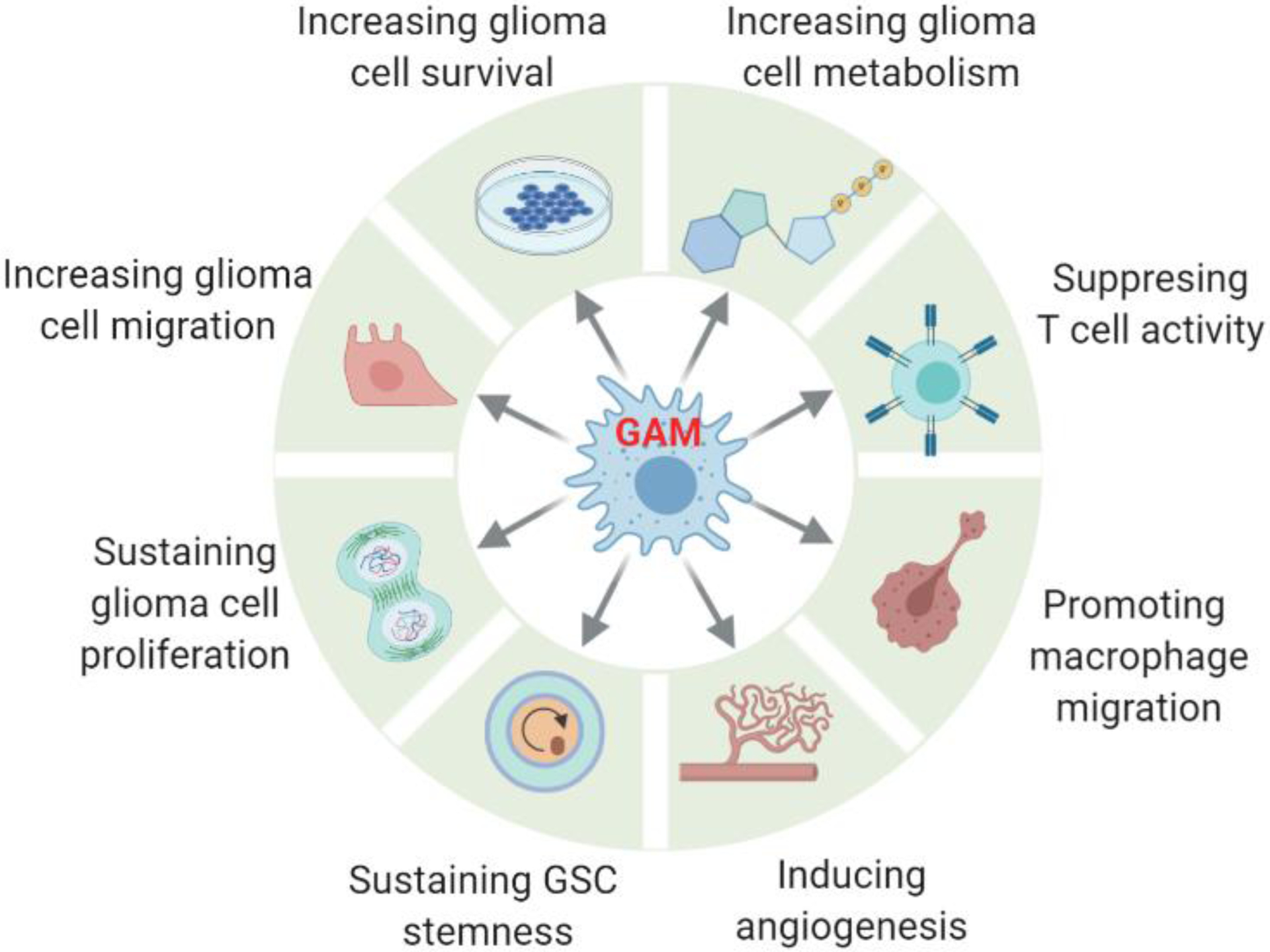
Once infiltrated into the GBM tumor, GAMs contribute to GBM progression by promoting glioma stem cell (GSC) stemness, GBM cell proliferation, survival, altered metabolism, and migration, as well as suppressing CD4+ and CD8+ T cell activity, stimulating angiogenesis, and recruiting additional macrophages in mouse and human GBM models. This figure was created using BioRender (https://biorender.com/).
In addition to maintaining GSC populations, GAMs can influence multiple GBM biologic properties, including proliferation, survival and migration [20, 60]. The influence may occur through a variety of mechanisms that include the secretion of soluble factors, the release of exosomes, as well as cell-to-cell contact. For example, GAMs secrete IL-6 and IL-1β that can enhance 3-phosphoinositide-dependent protein kinase 1 (PDPK1)-mediated activation of phosphoglycerate kinase 1 (PGK1) [61], as well as PI3K/PKCδ-mediated activation of glycerol-3-phosphate dehydrogenase (GPD2) [62], respectively, in GBM cells (such as U87 and U251). These activations, in turn, can promote GBM cell glycolysis and proliferation [61, 62]. Inhibition of PDPK1/PGK1 (shRNA KD of PGK1, or PDPK1 inhibitor OSU-03012 treatment) or PI3K/PKCδ/GPD2 (shRNA KD of PRKCD or GPD2) axes in GBM cells, or neutralization of macrophage-derived IL-6 or IL-1β, can attenuate GAM-associated effects on tumor cell glycolysis, proliferation, and tumorigenesis in human (e.g., U87 and U251) and mouse (e.g., GL261) models [61, 62]. In addition to cytokines, GAMs also secrete SPP1 [24], lysophosphatidic acid [63], and transforming growth factor (TGF)-β1 [64], and produce exosomes containing arginase-1 [65], to promote GBM biologic activities (e.g., proliferation, survival, and migration). These pro-tumor effects can also be achieved through a GAM-to-GBM contact mechanism that involves GAM-induced upregulation of PDGFRB in GBM cells [66]. Lastly, GAMs can promote GBM growth and progression via indirect mechanisms, which may include the recruitment of additional macrophages [35], the suppression of CD4+ and CD8+ T cell infiltration, the effector activity of these cells [35, 36, 67], as well as an increase in angiogenesis [24]. In summary, these studies suggest that GAMs may contribute to GBM progression by regulating several cancer hallmarks, which include increasing GBM cell survival, metabolism, migration and proliferation, promoting macrophage infiltration and angiogenesis, sustaining GSC stemness, and suppressing antitumor T cell activity (Figure 3). In addition, these findings highlight the therapeutic potential of targeting GAMs during the symbiotic interactions between GAMs and GBM cells. However, further studies are needed to characterize whether these GAM effects are context-dependent (including species-dependent). Ideally, these forward-looking investigations might lead to the development of novel candidate personalized therapies to treat GBM.
Concluding Remarks
While therapeutic strategies involving the identification of synthetic lethality or inhibiting crucial oncogene addiction have achieved success in treating some types of cancer [68], such approaches have yet to impact GBM patient outcomes. This could also be said for immunotherapeutic approaches that have been tested in treating GBM, but have not succeeded, This opinion article has specifically highlighted the molecular circuitry between GBM cells, with specific genetic backgrounds, and the GAM component of the TME. Our current understanding suggests a context-dependent GBM-GAM symbiosis that appears to be essential for sustained tumor growth and progression, but which may inform the development of personalized therapy in GBM (Figure 4).
Figure 4. Context-dependent glioblastoma (GBM)-macrophage/microglia symbiosis.
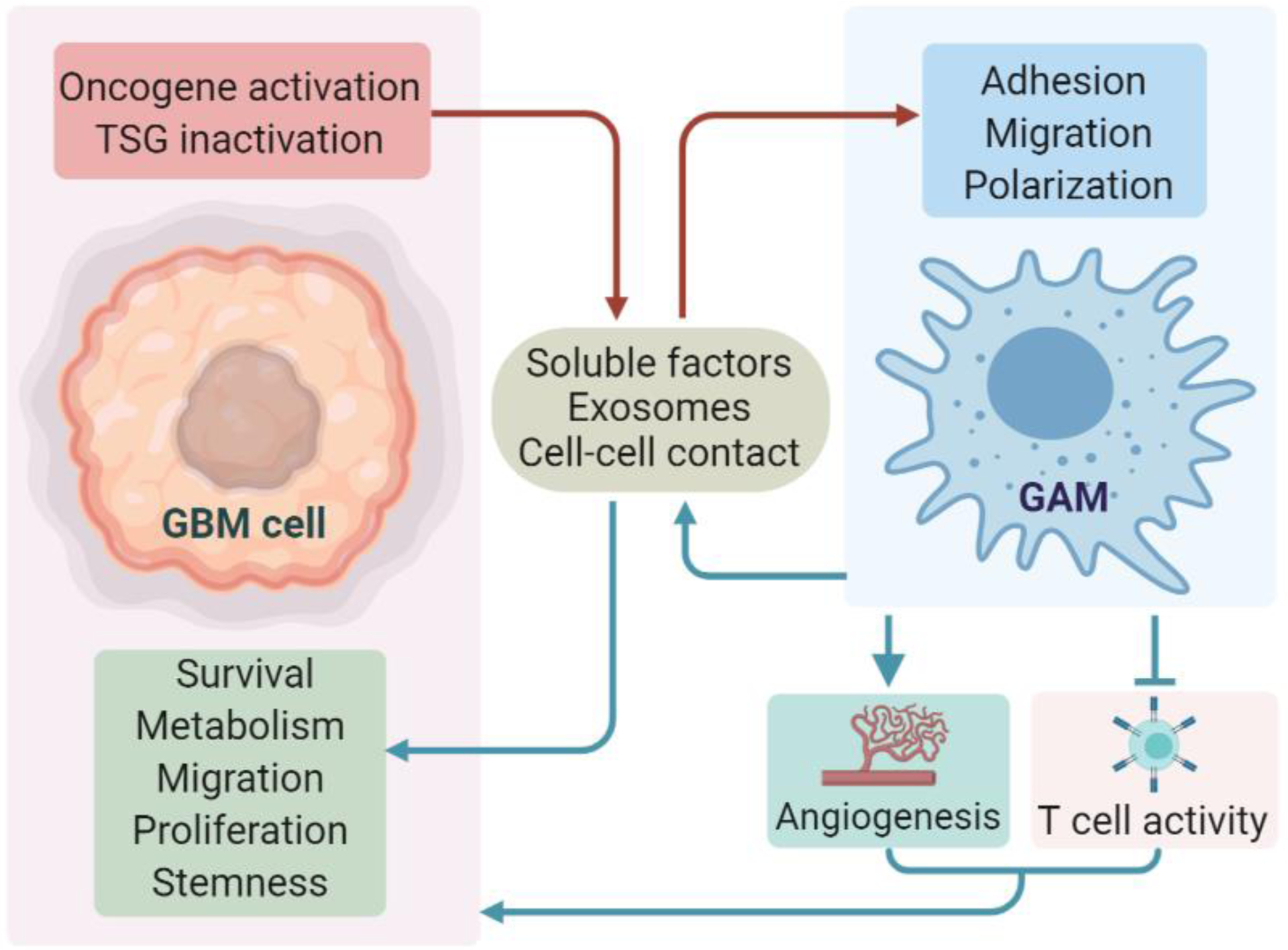
Inactivation or activation of specific tumor suppressor genes (TSGs) or oncogenes in GBM cells can regulate the adhesion, migration, and polarization of macrophages and microglia via secretion of soluble factors and exosomes, or through a cell-to-cell contact mechanism in mouse and human GBM models. Reciprocally, such glioblastoma-associated macrophages/microglia (GAMs) can promote GBM cell survival, proliferation, metabolism, migration, and self-renewal. GAMs can also promote GBM progression via indirect mechanisms (e.g., stimulating angiogenesis, and suppressing CD8+ and CD4+T cell activity). This figure was created using BioRender (https://biorender.com/).
Although our knowledge of the role of GBM-GAM crosstalk in tumorigenesis has increased in the past few years, multiple open questions remain regarding the molecular mechanisms underlying this symbiosis, and how we might target this crosstalk, especially when taking into consideration the unique genetic features of individual GBM types (see Outstanding Questions). Absent from the text above is a discussion of one of the hallmark features of GBM: intratumoral heterogeneity [69–72]. The heterogeneity and dynamic plasticity of GBM cells, as well as the heterogeneity of GAMs (e.g., macrophage vs. microglia and pro-inflammatory vs. alternatively activated or their overlapping phenotypes) highlight the challenges in identifying patient-specific GBM-GAM circuits that might be therapeutically accessible. Single-cell RNA sequencing (scRNA-seq) may provide insights regarding patient-specific targets. With respect to pre-clinical research, an emerging alternative is to grow GBM tumors as ex vivo organoids, which may better recapitulate certain tumor features [72, 73]. GBM organoid and macrophage/microglia cocultures could serve as a powerful model system to study the molecular circuits underlying GBM-GAM symbiosis under specific genetic backgrounds, and to test therapeutic agents targeting their crosstalk.
Outstanding Questions.
Among the different genetic alterations, which ones are the drivers regulating GAM recruitment and polarization? Answering this question might enable key elements of GBM-GAM symbiosis and associated therapeutic strategies.
What are the specific differences and similarities of GBM-GAM symbiosis in GBM -- in terms of different genetic alterations, or in terms of the same genetic backgrounds at different tumor stages? Can we achieve successful clinical outcomes in GBM patients by blocking GBM-GAM symbiosis under specific genetic backgrounds? The definition of these issues might help to develop personalized therapies for GBM patients.
What are the specific regulatory and functional differences between macrophages and microglia in GBM? scRNA-seq studies might help to address this question. This knowledge might allow to identify specific macrophage- or microglia-targeted therapies for GBM.
What signaling pathways are essential for regulating GBM-GAM symbiosis? Such knowledge might help to identify novel therapeutic targets.
Can scRNA-seq identify new subpopulations and/or states of GBM cells, macrophages and microglia, as well as new GBM-GAM interactions? Answering this question might inform novel elements of GBM-GAM symbiosis.
Can organoid cultures mimic the in vivo tumor microenvironment during GBM-GAM symbiosis? This knowledge might allow the design of translational preclinical studies targeting GBM-GAM symbiosis.
Does GBM-GAM symbiosis affect T cell-mediated immune responses and immunotherapies? If so, how? This can have key implications for developing novel strategies to improve antitumor efficiency from immunotherapies in GBM patients.
Is there a context-dependent GBM-myeloid derived suppressor cells (MDSCs) symbiosis in GBM? Similar to macrophages, MSDCs originate from the bone marrow, have heterogeneous subpopulations, and can play an important role in GBM tumorigenesis. MDSCs can also differentiate into macrophages in the tumor microenvironment. Answering this question might help define context-dependent GBM-MDSC symbiosis in GBM.
Altogether, our current knowledge of the molecular GBM-GAM symbiosis and its functional impact on GBM progression is still at an early stage. Recent studies have provided valuable information showing that GBM-GAM interactions are of fundamental importance to the biologically aggressive characteristics of GBM. We anticipate that future studies may lead to a detailed understanding of context-dependent GBM-GAM symbiotic interactions, and may offer a roadmap for the development of novel putative anti-cancer therapeutic strategies that disrupt this dynamic circuitry.
Highlights.
Symbiotic Glioblastoma-Macrophage/microglia (GBM-GAM) interactions reveal synthetic lethality in glioblastoma (GBM) harboring a deficiency in a specific tumor suppressor gene (e.g., PTEN, NF1 or TP53).
Cancer-cell-intrinsic activation of oncogenes (e.g., EGFR and CLOCK) can shapes a pro-tumor immune response by modulating GAM biology.
GBM-GAM symbiosis can contribute to GBM progression by promoting GSC stemness, GBM cell proliferation, survival, migration, as well as by suppressing T-cell-mediated immune response in mouse and PDX models.
Characterizing GBM-GAM symbiosis might reveal personalized therapeutic targets. For example, LOX and CLOCK inhibition can impair tumor progression and GAM infiltration, specifically in PTEN-deficient and CLOCK-high GBM, in mouse and PDX models, respectively.
Acknowledgments:
This work was supported in part by NIH R00 CA240896 (to P.C.), NIH P50CA221747 (to P.C., Brain cancer SPORE CEP Award) and Northwestern University start-up funds (to P.C.).
Glossary
- Alternatively activated macrophages
skewed towards an immunosuppressive and pro-tumor phenotype in cancer
- Bone marrow-derived macrophages (BMDMs)
here, infiltrating macrophages in GBM; originate from the bone marrow
- Brain-resident microglia
specialized macrophages in the brain that originate from progenitors seeding the embryonic yolk
- Cellular heterogeneity
fundamental property of biological systems that covers different aspects of cells, ranging from genetic diversity to cell-to-cell variability; driven by stochastic molecular interactions involved in all cellular processes
- Circadian rhythm
an organism’s internal clock that regulates the sleep-wake cycle and repeats on each rotation every 24 hours (e.g. in humans)
- Extracellular vesicles (EVs)
lipid bilayer-delimited particles that are naturally secreted by cancer cells and/or stromal cells in the tumor
- Hypoxia-inducible factor 1-alpha
the master transcriptional regulator of cellular response to hypoxia
- Lysyl oxidase (LOX)
a copper-dependent amine oxidase that plays a key role in the biogenesis of connective tissue matrices by crosslinking collagen and elastin, and in regulating macrophage migration
- Oncogene addiction
dependency of tumor cells on a single oncogene to maintain their pro-tumor activity
- Organoids
(Tumor organoid)3D multicellular in vitro tissue construct that can mimic its corresponding in vivo tumor microenvironment; it can be used to study aspects of tumor biology in a tissue culture dish
- Patient-derived xenograft (PDX)
cancer patient-derived xenograft model that can reflect properties of the original patient tumors
- Phagocytosis
process where phagocytes such as macrophages engulf particles, debris, or other cells
- Polarization
macrophage activation into distinct phenotypes characterized by e.g., pro-inflammatory and anti-inflammatory gene expression profiles
- Single-cell RNA sequencing (scRNA-seq)
technology that can provide the expression profiles of individual cells, thus offering a better understanding of phenotypes, states, and functions of an individual cell in the context of its microenvironment
- Stemness
molecular process underlying the fundamental stem cell properties of self-renewal and generation of differentiated cells
- GBM-GAM symbiosis
symbiotic interaction where GBM cells and GAMs benefit from each other via secretion of soluble factors and vesicles, or through a cell-to-cell contact mechanism
- Synthetic lethal interaction
relationship between two functional entities (e.g., genes) where loss of either one is viable, while loss of both entities induces cell death
- Synthetic lethality
a strategy based on synthetic lethal interactions
- The Cancer Genome Atlas (TCGA)
landmark cancer genomics program characterizing over 20,000 primary cancer and matched normal human samples across 33 cancer types, including GBM
- Tumor microenvironment (TME)
stromal components of a tumor, including the surrounding immune cells, blood vessels, fibroblasts, as well as signaling molecules and extracellular matrix
- Glioblastoma-associated macrophages/microglia (GAMs)
macrophages and microglia infiltrated in GBM that promote tumor progression and exhibit immunosuppressive functions
- Yes-associated protein 1 (YAP1)
key factor in the Hippo pathway; functions as a transcriptional regulator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wellenstein MD and de Visser KE (2018) Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 48 (3), 399–416. [DOI] [PubMed] [Google Scholar]
- 2.Geraldo LHM et al. (2019) Glioblastoma Therapy in the Age of Molecular Medicine. Trends Cancer 5 (1), 46–65. [DOI] [PubMed] [Google Scholar]
- 3.Zanders ED et al. (2019) Therapy for glioblastoma: is it working? Drug Discov Today 24 (5), 1193–1201. [DOI] [PubMed] [Google Scholar]
- 4.Dunn GP et al. (2012) Emerging insights into the molecular and cellular basis of glioblastoma. Genes & Development 26 (8), 756–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hambardzumyan D and Bergers G (2015) Glioblastoma: Defining Tumor Niches. Trends Cancer 1 (4), 252–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khosla D (2016) Concurrent therapy to enhance radiotherapeutic outcomes in glioblastoma. Ann Transl Med 4 (3), 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennan CW et al. (2013) The somatic genomic landscape of glioblastoma. Cell 155 (2), 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research, N. (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455 (7216), 1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng H et al. (2008) Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harb Symp Quant Biol 73, 427–37. [DOI] [PubMed] [Google Scholar]
- 10.Patel AP et al. (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344 (6190), 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gimple RC et al. (2019) Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev 33 (11–12), 591–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prager BC et al. (2020) Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 6 (3), 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quail DF and Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19 (11), 1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen S et al. (2001) PTEN controls tumor-induced angiogenesis. Proc Natl Acad Sci U S A 98 (8), 4622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parsa AT et al. (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 13 (1), 84–8. [DOI] [PubMed] [Google Scholar]
- 16.Zerrouqi A et al. (2012) P14ARF inhibits human glioblastoma-induced angiogenesis by upregulating the expression of TIMP3. J Clin Invest 122 (4), 1283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quail DF and Joyce JA (2017) The Microenvironmental Landscape of Brain Tumors. Cancer Cell 31 (3), 326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z et al. (2017) Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res 77 (9), 2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pathria P et al. (2019) Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol 40 (4), 310–327. [DOI] [PubMed] [Google Scholar]
- 20.Wei J et al. (2020) Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro Oncol 22 (2), 180–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabrusiewicz K et al. (2016) Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 1 (2), e85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C et al. (2017) Tumor Purity as an Underlying Key Factor in Glioma. Clin Cancer Res 23 (20), 6279–6291. [DOI] [PubMed] [Google Scholar]
- 23.Verhaak RG et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17 (1), 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen P et al. (2019) Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 35 (6), 868–884 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sa JK et al. (2020) Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol 21 (1), 216–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu B et al. (2016) Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell 167 (5), 1281–1295 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuccarini M et al. (2018) The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes (Basel) 9 (2), 105–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tao W et al. (2020) Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat Commun 11 (1), 3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le LQ et al. (2009) Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell 4 (5), 453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo X et al. (2019) Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia. Neuro Oncol 21 (10), 1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q et al. (2017) Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 32 (1), 42–56 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ham SW et al. (2019) TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ 26 (3), 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szatmari T et al. (2006) Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci 97 (6), 546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abels ER et al. (2019) Glioblastoma-Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR-21. Cell Rep 28 (12), 3105–3119 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei J et al. (2019) Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. Journal of Clinical Investigation 129 (1), 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dumas AA et al. (2020) Microglia promote glioblastoma via mTOR-mediated immuno-suppression of the tumour microenvironment. EMBO J 39 (15), e103790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas SL et al. (2015) Loss of Sparc in p53-null Astrocytes Promotes Macrophage Activation and Phagocytosis Resulting in Decreased Tumor Size and Tumor Cell Survival. Brain Pathol 25 (4), 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heimberger AB et al. (2005) Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res 11 (4), 1462–6. [DOI] [PubMed] [Google Scholar]
- 39.Fan QW et al. (2013) EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 24 (4), 438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng Y et al. (2013) Epidermal growth factor (EGF)-enhanced vascular cell adhesion molecule-1 (VCAM-1) expression promotes macrophage and glioblastoma cell interaction and tumor cell invasion. J Biol Chem 288 (44), 31488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao Z and Guo D (2019) EGFR mutation: novel prognostic factor associated with immune infiltration in lower-grade glioma; an exploratory study. BMC Cancer 19 (1), 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang BR et al. (2020) CAIX Regulates GBM Motility and TAM Adhesion and Polarization through EGFR/STAT3 under Hypoxic Conditions. Int J Mol Sci 21 (16), 5838–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu YS et al. (2017) MiR-181b modulates EGFR-dependent VCAM-1 expression and monocyte adhesion in glioblastoma. Oncogene 36 (35), 5006–5022. [DOI] [PubMed] [Google Scholar]
- 44.An Z et al. (2018) EGFR Cooperates with EGFRvIII to Recruit Macrophages in Glioblastoma. Cancer Res 78 (24), 6785–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masri S and Sassone-Corsi P (2018) The emerging link between cancer, metabolism, and circadian rhythms. Nat Med 24 (12), 1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shafi AA and Knudsen KE (2019) Cancer and the Circadian Clock. Cancer Res 79 (15), 3806–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sulli G et al. (2019) Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer 5 (8), 475–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen P et al. (2020) Circadian Regulator CLOCK Recruits Immune-Suppressive Microglia into the GBM Tumor Microenvironment. Cancer Discov 10 (3), 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong Z et al. (2019) Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov 9 (11), 1556–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miranda A et al. (2019) Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A 116 (18), 9020–9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hide T et al. (2018) Oligodendrocyte Progenitor Cells and Macrophages/Microglia Produce Glioma Stem Cell Niches at the Tumor Border. EBioMedicine 30, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tabu K et al. (2020) Glioma stem cell (GSC)-derived autoschizis-like products confer GSC niche properties involving M1-like tumor-associated macrophages. Stem Cells 38 (8), 921–935. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X et al. (2020) CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab Invest 100 (4), 619–629. [DOI] [PubMed] [Google Scholar]
- 54.Yin J et al. (2020) ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat Commun 11 (1), 2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi Y et al. (2017) Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun 8, 15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen P et al. (2021) Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep 34 (1), 108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sarkar S et al. (2014) Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat Neurosci 17 (1), 46–55. [DOI] [PubMed] [Google Scholar]
- 58.Sarkar S et al. (2020) Control of brain tumor growth by reactivating myeloid cells with niacin. Sci Transl Med 12 (537), eaay9924. [DOI] [PubMed] [Google Scholar]
- 59.Guo X et al. (2020) Midkine activation of CD8(+) T cells establishes a neuron-immune-cancer axis responsible for low-grade glioma growth. Nat Commun 11 (1), 2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hambardzumyan D et al. (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19 (1), 20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y et al. (2018) Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol Cell 71 (2), 201–215 e7. [DOI] [PubMed] [Google Scholar]
- 62.Lu J et al. (2020) Tumor-associated macrophage interleukin-beta promotes glycerol-3-phosphate dehydrogenase activation, glycolysis and tumorigenesis in glioma cells. Cancer Sci 111 (6), 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.do Amaral RF et al. (2020) Microglial lysophosphatidic acid promotes glioblastoma proliferation and migration via LPA1 receptor. J Neurochem. [DOI] [PubMed] [Google Scholar]
- 64.Liu Y et al. (2019) An miR-340–5p-macrophage feedback loop modulates the progression and tumor microenvironment of glioblastoma multiforme. Oncogene 38 (49), 7399–7415. [DOI] [PubMed] [Google Scholar]
- 65.Azambuja JH et al. (2020) Arginase-1+ Exosomes from Reprogrammed Macrophages Promote Glioblastoma Progression. Int J Mol Sci 21 (11), 3990–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wallmann T et al. (2018) Microglia Induce PDGFRB Expression in Glioma Cells to Enhance Their Migratory Capacity. iScience 9, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takenaka MC et al. (2019) Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nature Neuroscience 22 (5), 729–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao D and DePinho RA (2017) Synthetic essentiality: Targeting tumor suppressor deficiencies in cancer. Bioessays 39 (8), 1700076–1700084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Puram SV et al. (2018) Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol Cell Oncol 5 (3), e1448244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bhaduri A et al. (2020) Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 26 (1), 48–63 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeCordova S et al. (2020) Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front Immunol 11, 1402–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jacob F et al. (2020) A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 180 (1), 188–204 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baker K (2018) Organoids Provide an Important Window on Inflammation in Cancer. Cancers (Basel) 10 (5), 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davies H et al. (2002) Mutations of the BRAF gene in human cancer. Nature 417 (6892), 949–54. [DOI] [PubMed] [Google Scholar]
- 75.Jerby-Arnon L et al. (2014) Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell 158 (5), 1199–1209. [DOI] [PubMed] [Google Scholar]
- 76.Bitler BG et al. (2015) Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med 21 (3), 231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lynch TJ et al. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350 (21), 2129–39. [DOI] [PubMed] [Google Scholar]
- 78.Lord CJ and Ashworth A (2017) PARP inhibitors: Synthetic lethality in the clinic. Science 355 (6330), 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bryant HE et al. (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 (7035), 913–7. [DOI] [PubMed] [Google Scholar]
- 80.Farmer H et al. (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434 (7035), 917–21. [DOI] [PubMed] [Google Scholar]
- 81.Dey P et al. (2017) Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 542 (7639), 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao D et al. (2017) Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 542 (7642), 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ashworth A et al. (2011) Genetic interactions in cancer progression and treatment. Cell 145 (1), 30–8. [DOI] [PubMed] [Google Scholar]
- 84.Bowman RL et al. (2016) Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep 17 (9), 2445–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klemm F et al. (2020) Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181 (7), 1643–1660 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Friebel E et al. (2020) Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 181 (7), 1626–1642 e20. [DOI] [PubMed] [Google Scholar]
- 87.Chen P et al. (2015) Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral nerve injury. Acta Neuropathol 130 (5), 605–18. [DOI] [PubMed] [Google Scholar]
- 88.Shapouri-Moghaddam A et al. (2018) Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233 (9), 6425–6440. [DOI] [PubMed] [Google Scholar]
- 89.Mantovani A et al. (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25 (12), 677–86. [DOI] [PubMed] [Google Scholar]
- 90.Martinez FO and Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Murray PJ et al. (2014) Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 41 (1), 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kloepper J et al. (2016) Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci U S A 113 (16), 4476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pyonteck SM et al. (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19 (10), 1264–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saha D et al. (2017) Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 32 (2), 253–267 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao J et al. (2019) Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 25 (3), 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]