

Abstract
Gene editing with the CRISPR-Cas9 system could revolutionize hematopoietic stem cell (HSC)-targeted gene therapy for hereditary diseases, including sickle cell disease (SCD). Conventional delivery of editing tools by electroporation limits HSC fitness due to its toxicity; therefore, efficient and non-toxic delivery remains crucial. Integrating lentiviral vectors are established for therapeutic gene delivery to engraftable HSCs in gene therapy trials; however, their sustained expression and size limitation preclude their use for CRISPR-Cas9 delivery. Here, we developed a Cas9 protein delivery non-integrating lentiviral system encoding guide RNA and donor DNA, allowing for transient endonuclease function and inclusion of all editing tools in a single vector (all-in-one). We demonstrated efficient one-time correction of the SCD mutation in the endogenous βs-globin gene up to 42% at the protein level (p < 0.01) with the Cas9 protein delivery non-integrating lentiviral all-in-one system without electroporation. Our findings improve prospects for efficient and safe genome editing.
Keywords: lentiviral vector, protein delivery, genome editing, CRISPR-Cas9, sickle cell disease, gene correction, non-integrating vector
Graphical abstract
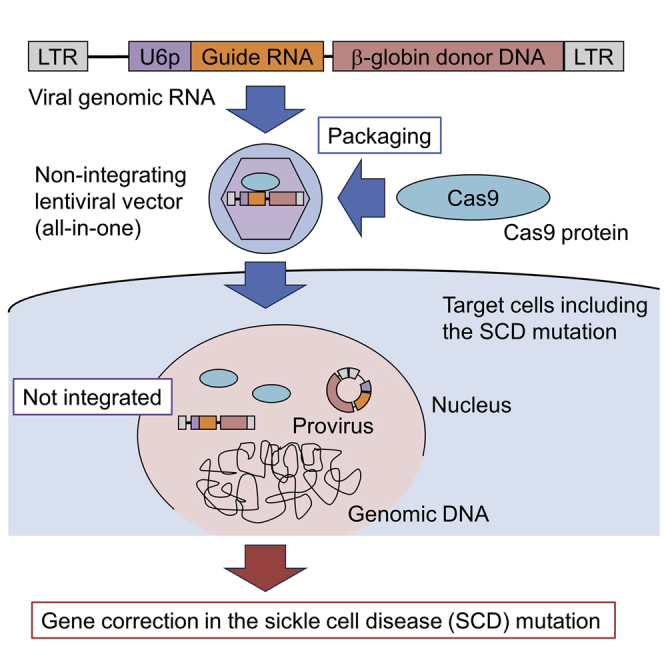
Uchida et al. develop a Cas9 protein delivery non-integrating lentiviral system encoding guide RNA and donor DNA, allowing for transient endonuclease function and inclusion of all editing tools in a single vector (all-in-one). One-time correction of the sickle cell disease mutation is demonstrated by this non-integrating lentiviral delivery without electroporation.
Introduction
Gene correction is an ideal gene therapy strategy for hereditary diseases, including hemoglobinopathies such as sickle cell disease (SCD). Proof of concept for gene addition therapy, in which a therapeutic gene is inserted to compensate for a missing or defective gene, has been established in clinical trials for various disorders affecting hematopoietic stem cells (HSCs). Since the initial gene therapy trials in severe combined immunodeficiency patients in the 1990s, tremendous improvements have been made in the collection, transduction, and editing of HSCs, particularly in the establishment of efficient human immunodeficiency virus type 1 (HIV-1)-based lentiviral gene delivery systems, which can achieve stable, long-term expression of therapeutic genes among the progeny of recipient CD34+ cells.1, 2, 3, 4, 5, 6, 7 Recently, description of the CRISPR-CRISPR-associated protein 9 (Cas9) system and its development as a tool for site-specific DNA breakage and gene correction (when accompanied with donor DNA) has generated intense interest in not just inserting therapeutic genes but also correcting the underlying mutations responsible for hereditary disorders.8, 9, 10, 11, 12
The CRISPR-Cas9 system relies upon the combination of Cas9 endonuclease, which cleaves DNA, and a guide RNA sequence, which is custom-designed to recognize and target a specific gene for cleavage by Cas9. When homologous donor DNA is also provided, CRISPR-Cas9-based site-specific DNA breakage followed by homology-directed repair (HDR) has been shown to greatly improve gene correction compared to donor DNA-only transfection, including correction of the SCD mutation in the β-globin gene (HBB: c.20A>T, βs-globin).13, 14, 15, 16, 17, 18 One barrier to realizing this system’s potential for HSC correction is that electroporation, the most common method for delivering guide RNA and Cas9 protein, reduces cell viability,13 and a reduction in cell viability is particularly problematic in disorders such as sickle cell disease in which harvesting sufficient HSCs for manufacturing is difficult.19 The toxicity associated with electroporation is indeed what initially spurred the development of alternative viral-vector-based delivery systems, which can now be used to reliably and efficiently modify CD34+ HSCs capable of long-term engraftment, as demonstrated in several animal models and clinical trials.1, 2, 3, 4, 5, 6, 7,20, 21, 22, 23
Given that lentiviral vector systems have been successfully used for gene addition to CD34+ HSCs in clinical trials, they represent a promising method for delivery of gene correction components with the potential for more rapid translation to the clinic. Both guide RNA and Cas9 can be encoded in a single lentiviral genome, allowing for DNA breakage.24,25 However, permanent lentiviral vector integration into the genome raises the possibility of genotoxicity due to continuous expression of Cas9 endonuclease, and insertional mutagenesis remains a potential concern when using integrating lentiviral vectors; thus, vector systems that deliver without integration would be preferable.6,26,27 In addition, the large-sized Cas9 DNA (4.3 kb) limits efficient lentiviral packaging, which likely reduces transduction efficiency, and, due to the size limit, donor DNA sequence cannot be included in the lentiviral genome encoding both guide RNA and Cas9. Therefore, in this study, we developed a Cas9 protein delivery system with non-integrating lentiviral vectors (NILVs) encoding both guide RNA and donor DNA that allows for efficient one-time gene correction of the SCD mutation (all-in-one for HDR), given that gene modification with the lentiviral vector system has been proved to yield genetically modified HSCs capable of long-term engraftment. In addition, we developed a versatile enhanced green fluorescent protein-to-enhanced yellow fluorescent protein (GFP-to-YFP) gene conversion assay that could be adapted for studying gene correction using other methods or in other diseases.
Results
Efficient GFP-to-YFP gene conversion with Cas9 protein delivery lentiviral vectors encoding guide RNA and donor DNA
As a control for efficient delivery and stable gene expression, we first used an integrating HIV-1-based lentiviral vector system to deliver the CRISPR-Cas9-based gene-editing tools, including guide RNA and Cas9 endonuclease.20,21,28 To eliminate the requirement of Cas9 DNA integration, we hypothesized that fusion proteins between Cas9 and Cyclophilin A (CypA) would allow for Cas9 packaging in lentiviral particles for delivery, since CypA is capable of binding to lentiviral capsids (Figure S1A).29 We designed two Cas9-CypA fusion proteins: CypA-Cas9 (N-terminal-CypA-Cas9-C-terminal) and Cas9-CypA (N-terminal-Cas9-CypA-C-terminal), and lentiviral vectors were prepared to encode a GFP-targeting guide RNA with the fusion proteins. Following transduction of a GFP-positive stable human erythroleukemia (HEL) cell line (encoding a single copy of the GFP gene) with GFP-targeting guide RNA vectors containing Cas9 fusion proteins (Figure S1B), GFP-positive percentages (%GFP) were reduced with both CypA-Cas9 and Cas9-CypA protein delivery vectors (GFP-negative 36%–42%, p < 0.01), compared to an untransduced control as well as a guide RNA-alone vector control (Figure S1C). The GFP disruption level was comparable to that obtained using a guide RNA-Cas9 integrating vector (GFP-negative 52%). These data demonstrate that Cas9-CypA fusion proteins are delivered with lentiviral vector particles and have sufficient endonuclease function to efficiently induce a GFP DNA break.
We then designed an all-in-one Cas9 protein delivery NILV encoding both GFP-targeting guide RNA and YFP donor DNA, which includes all essential editing tools for GFP-to-YFP gene conversion without integration of the vector genome (Figure 1A). GFP-to-YFP conversion can model gene correction, since strong sequence homology exists between GFP and YFP genes, and their fluorescent signals can be separated by flow cytometry. Inclusion of an intact YFP gene strongly reduced vector titers in the Cas9 protein delivery all-in-one vector (p < 0.01), while several silent mutations into the guide RNA target site in the YFP gene allowed for efficient all-in-one vector preparation (Figure 1B), demonstrating that de-targeting from guide RNA to donor DNA is important for efficient lentiviral preparation in all-in-one vectors. We transduced a GFP-positive stable HEL cell line with the all-in-one NILV using the Cas9 fusion proteins (Figure 1C) and observed a significant reduction in GFP positivity (GFP and YFP double negative ∼25%, p < 0.01) as well as conversion to YFP positivity (YFP 14%–19%, p < 0.01) with both Cas9 fusion protein delivery vectors, compared to an untransduced control as well as no Cas9 control (Figure 1D). Similar levels of GFP DNA breakage and GFP-to-YFP gene conversion were obtained between Cas9 fusion protein delivery vectors (slightly lower GFP DNA breakage) and a Cas9 gene integrating vector, as well as between all-in-one NILVs with Cas9 fusion protein delivery (slightly higher GFP-to-YFP gene conversion) and integrating guide RNA vectors with Cas9 fusion protein delivery (Figure 1D). These data demonstrate that the Cas9 protein delivery NILV system encoding both guide RNA and donor DNA (all-in-one) allows for one-time gene conversion with similar efficiency as an integrating vector system.
Figure 1.

Cas9 protein delivery is allowed by guide RNA-encoding vectors
(A) Hypothetical genome editing model for Cas9 protein delivery with an all-in-one non-integrating lentiviral vector (NILV) encoding both enhanced green fluorescent protein (GFP)-targeting guide RNA and enhanced yellow fluorescent protein (YFP) donor DNA. The Cas9 protein was used with or without cyclophilin A (CypA) fusion for lentiviral packaging (CypA-Cas9 or Cas9-CypA). (B) Lentiviral titers at escalating doses of Cas9 fusion protein plasmids, evaluated by YFP expression in HeLa cells. Several silent mutations were added to the YFP gene to escape from GFP guide RNA targeting. (C) A GFP-to-YFP gene-conversion model using a GFP-positive human erythroleukemia (HEL) cell line (including a single copy of GFP gene) transduced with Cas9 protein delivery guide RNA/donor DNA vectors (all-in-one). GFP DNA breakage (GFP-YFP-), GFP-to-YFP gene conversion (GFP-YFP+), and false-positive conversion (GFP+YFP+) were evaluated by GFP- and YFP-positive percentages (% GFP and YFP). (D) GFP-to-YFP gene conversion using lentiviral Cas9 protein delivery with integrating guide RNA vectors or all-in-one NILVs at multiplicity of infection (MOI) 5, evaluated by flow cytometry 9 days post-transduction. (E) GFP-to-YFP gene conversion using all-in-one NILVs with CypA-Cas9 and Cas9-CypA fusion proteins as well as Cas9 protein alone (without CypA fusion), evaluated by flow cytometry 10 days post-transduction. (F) GFP DNA breakage 7 days post-transduction, evaluated by Surveyor nuclease assay using integrating or non-integrating guide RNA vectors (not encoding donor DNA) with protein delivery among CypA-Cas9, Cas9-CypA, and Cas9 without CypA fusion (intact: 0.8 kb, DNA break: 0.3 kb and 0.5 kb). (G) The all-in-one vector was separated into a GFP-targeting guide RNA vector and YFP donor DNA vector. Cas9 protein was delivered with either guide RNA vector or donor DNA vector. (H) GFP-to-YFP gene conversion using (1) guide RNA vector and Cas9 protein delivery donor DNA vector, and (2) Cas9 protein delivery guide RNA vector and donor DNA vector, evaluated by flow cytometry 9 days post-transduction. LTR, long terminal repeat; U6p, U6 promoter; Mp, murine stem cell virus promoter; gRNA, guide RNA. Values: mean ± standard error. All experiments were performed in triplicate except (F) (single run).
To evaluate whether the delivered Cas9 protein has transient nuclease activity, we added a guide RNA/donor DNA NILV without the Cas9 protein in a GFP-positive cell line 6 days after transduction of a Cas9 protein delivery all-in-one NILV (Figure S2). %YFP in GFP+ HEL cells was temporally elevated after NILV transduction at day 0 as well as day 6, and it plateaued 15 days after 1st transduction. No increases in GFP DNA breakage or GFP-to-YFP conversion were observed in additional guide RNA/donor DNA transduction at day 6 of lentiviral Cas9 protein delivery, demonstrating that Cas9 function was lost over the short term.
Cas9 protein delivery is enabled by presence of a guide RNA sequence on the lentiviral vector genome
To further investigate Cas9 protein delivery in lentiviral vectors, we compared Cas9-CypA fusion proteins to Cas9 protein alone (without CypA fusion) in the GFP-to-YFP gene conversion model in a GFP+ HEL cell line with all-in-one NILV transduction (Figure 1E). Surprisingly, both Cas9 fusion proteins and Cas9 protein alone (without CypA fusion) allowed for significant GFP breakage (double negative 20%–24%, p < 0.01) as well as efficient GFP-to-YFP conversion (YFP 13%–18%, p < 0.01), compared to an untransduced control. We confirmed efficient GFP DNA breaks from all Cas9 protein delivery guide RNA vectors (not encoding donor DNA) with and without CypA fusion in a GFP-positive cell line by Surveyor nuclease assay (Figure 1F). GFP-to-YFP gene conversion with a Cas9 protein delivery all-in-one vector (without CypA fusion) was confirmed by polymerase chain reaction (PCR) as well as DNA sequencing (Figure S3). In quantitative PCR (qPCR) analysis, lower GFP vector copy number (VCN) (p < 0.05) and higher YFP VCN (p < 0.01) were detected in GFP-to-YFP gene conversion with an all-in-one vector, as compared to an untransduced control (Figure S4). Total VCNs (1.0–1.2) in GFP-to-YFP gene-converted cells were similar to the VCN 1.0 in the untransduced control (not significant). In addition, to reduce false-positive YFP expression from the NILV without GFP-to-YFP conversion, a promoter for YFP expression was removed from the all-in-one vector, but we still observed GFP-to-YFP gene conversion with lentiviral Cas9 protein delivery (Figure S5). These data demonstrate that Cas9 protein can be delivered with lentiviral vector particles without CypA fusion, allowing for GFP-to-YFP gene conversion.
We hypothesized that the unfused Cas9 protein is delivered by binding to the guide RNA sequence encoded in the lentiviral RNA genome, since Cas9 endonuclease functions to bind guide RNA for site-specific DNA breakage. To investigate this hypothesis, we split the all-in-one NILV (encoding both GFP-targeting guide RNA and YFP donor DNA) into separate guide RNA and donor DNA vectors, and each NILV was prepared with or without Cas9 protein (Figure 1G). Using the GFP-to-YFP conversion model in a GFP+ HEL cell line, we evaluated (1) Cas9 protein delivery all-in-one vector encoding both guide RNA and donor DNA, (2) guide RNA vector and donor DNA-encoding Cas9 protein delivery vector, and (3) guide RNA-encoding Cas9 protein delivery vector and donor DNA vector. We observed more efficient GFP DNA breakage and GFP-to-YFP gene conversion with the all-in-one vector (YFP 24% and double negative 35%) and guide RNA-encoding Cas9 protein delivery vector (YFP 14% and double negative 36%), compared to the donor DNA-encoding Cas9 protein delivery vector (YFP 7% and double negative 11%) and an untransduced control (YFP 2% and double negative 0%) (Figure 1H). These data demonstrate that the Cas9 protein is predominantly delivered with guide RNA-encoding lentiviral vectors. Low-level gene editing with donor DNA vector-mediated Cas9 protein delivery suggests that small amounts of Cas9 protein might be non-specifically delivered with lentiviral particles.
More efficient genome editing with Cas9 protein delivery lentiviral vectors including multiple guide RNA sequences
Reasoning that a guide RNA expression cassette can produce many copies of guide RNA but deliver only one Cas9 protein per guide RNA, we constructed vectors containing multiple copies of the same GFP-targeting guide RNA sequence (2×, 4×, 6×, and 9×) in the all-in-one GFP-to-YFP gene conversion vector (Figure 2A). These vectors included additional tandem guide RNA sequences with no additional promoter, permitting Cas9 protein binding without additional transcription; thus, we have termed these additional sequences “pseudo guide RNA.” Insertion of multiple pseudo guide RNAs reduced lentiviral titers with Cas9 protein delivery (Figure 2B), and the reduction of lentiviral titers with multiple pseudo guide RNAs was enhanced by addition of a Cas9 protein (Figure S6), suggesting that the acceptable Cas9 protein amount per vector particle is limited. The addition of multiple pseudo guide RNAs resulted in a strong increase of DNA breakage (double negative 46%–83%) and a slight increase in GFP-to-YFP gene conversion (YFP 5%–17%) in a GFP+ HEL cell line, compared to the all-in-one vector without pseudo guide RNA addition (YFP 9% and double negative 33%) and an untransduced control (YFP 0% and double negative 0%) (Figure 2C). In addition, we evaluated Cas9 protein amounts delivered by all-in-one vectors with or without 4× pseudo guide RNA. In western blot analysis of the vector pellets, greater amounts of Cas9 protein were detected in both all-in-one vectors compared to a donor DNA vector without guide RNA (p < 0.01) (Figure S7), and Cas9 protein amounts were increased 1.4-fold by addition of 4× pseudo guide RNA (Figure S7). These data demonstrate that adding guide RNA sequences to the all-in-one vector allows for greater amounts of lentiviral Cas9 protein delivery and thus DNA breakage; however, additional donor DNA may be required to improve gene conversion. Based on this hypothesis, we simply increased the amounts (from MOI 5 to MOIs 10, 25, and 50) of the all-in-one NILV (Figure 2D; Figure S8), and, strikingly, this resulted in not only an increase in GFP DNA breakage (double negative 43%–51%) but also more efficient GFP-to-YFP gene conversion (YFP 14%–38%), compared to MOI 5 transduction (YFP 7% and double negative 27%) and an untransduced control (YFP 0% and double negative 0%). These data demonstrate that more efficient gene conversion in the NILV system can result from enhanced Cas9 protein delivery as well as greater amounts of donor DNA sequence.
Figure 2.

More efficient genome editing with Cas9 protein delivery lentiviral vectors including multiple guide RNA sequences
(A) Design of lentiviral vectors encoding GFP-targeting guide RNA derived from a U6 promoter, multiple tandem copies (2×, 4×, 6×, and 9×) of pseudo guide RNA (the same GFP-targeting guide RNA with no additional promoter), and YFP donor DNA with Cas9 protein delivery. (B) Lentiviral vector titers for guide RNA/donor DNA (all-in-one) NILVs with Cas9 protein delivery including multiple pseudo guide RNA. (C) GFP-to-YFP gene conversion using these multiple pseudo guide RNA vectors at MOI 5, evaluated by flow cytometry 20 days post-transduction in a GFP+ HEL cell line. (D) GFP-to-YFP gene conversion at escalating MOIs (5, 10, 25, and 50) of guide RNA/donor DNA (all-in-one) NILV with Cas9 protein delivery without multiple pseudo guide RNA 14 days post-transduction. Values: mean ± standard error. All experiments were performed in triplicate.
βs-to-β-globin gene correction with Cas9 protein delivery NILVs encoding guide RNA and donor DNA
We then switched from the GFP-to-YFP gene conversion to a therapeutic model of SCD gene correction. To select an optimal guide RNA for both βs-globin gene targeting and Cas9 protein delivery, we evaluated several guide RNAs (BG1, BG3, BG4, BG5, BG8, and SG11) (Table S1) targeting the βs-globin gene within exon 1 (including the SCD mutation) and intron 1 (Figure 3A), which were validated for efficient DNA targeting with Cas9-integrating lentiviral vectors in K562 cells (Figure S9). Of these guide RNAs, the SG11 guide RNA is the only one that targets the SCD mutation site in the βs-globin gene. To evaluate βs-globin gene breakage, we designed a chimeric gene between βs-globin (exons 1–2 with the SCD mutation) and YFP, which can be expressed within a single reading frame. βs-globin/YFP-expressing K562 cells were transduced with βs-globin-targeting guide RNA vectors with Cas9 gene integration or Cas9 protein delivery (Figure 3B). βs-globin/YFP knockdown was observed with all integrating guide RNA/Cas9 vectors (p < 0.01) compared to an untransduced control, while only BG3, BG5, BG8, and SG11 guide RNAs resulted in βs-globin/YFP knockdown with Cas9 protein delivery NILVs (p < 0.05). These data demonstrate that not all guide RNAs optimized by Cas9-integrating vectors can allow for efficient gene editing with Cas9 protein delivery NILVs. Different features of the guide RNA may affect Cas9 protein delivery, compared to DNA targeting.
Figure 3.

Efficient βs-to-β-globin gene correction with a guide RNA-encoding NILV with Cas9 protein delivery
(A) Design of several guide RNAs (BG1, BG3, BG4, BG5, BG8, and SG11) targeting β-globin gene with the sickle cell disease (SCD) mutation (βs-globin). The SG11 guide RNA only targets the SCD mutation site. (B) DNA breakage of βs-globin/YFP chimeric gene (βs-globin exons 1–2 and YFP, which can express on a single reading frame) using βs-globin-targeting guide RNA/Cas9 integrating vectors as well as βs-globin-targeting guide RNA NILVs with Cas9 protein delivery, evaluated by YFP expression in flow cytometry 4 days post-transduction in K562 cells. (C) Design of NILVs encoding SG11 guide RNA with Cas9 protein delivery and/or β-globin donor DNA. (D) An endogenous βs-globin gene editing model using an immortalized erythroid cell line including the SCD mutation (sickle HUDEP-2 cells) transduced with gene editing vectors at MOI 25. (E) DNA breakage in the βs-globin gene 8 days post-transduction in sickle HUDEP-2 cells, evaluated by Surveyor nuclease assay (intact: 0.8 kb, DNA break: 0.4 kb and 0.4 kb). (F) βs-to-β-globin gene correction at the DNA level 7–8 days after erythroid differentiation in gene-corrected sickle HUDEP-2 cells, evaluated by targeted deep sequencing. (G and H) βs-to-β-globin gene correction at the protein level, evaluated by reverse-phase high-performance liquid chromatography (RP-HPLC) 7–19 days after erythroid differentiation. (I) Cell counts in gene-corrected cells 7–19 days after erythroid differentiation, evaluated by trypan blue staining. ΔEx3, 5′-side partial β-globin exon 3 without stop codon; Ex2, β-globin exon 2; Ex1, β-globin exon 1; BGp, β-globin promoter; HDR, homology-directed repair. Values: mean ± standard error. (B) was performed in triplicate; (E) was performed in single run; and (F), (H), and (I) were performed 2–4 times.
We selected the SG11 guide RNA targeting the SCD mutation site for further analysis. We transduced immortalized erythroid cells including the SCD mutation (sickle HUDEP-2 cells) with a SG11 guide RNA NILV with Cas9 protein delivery (Figures 3C and 3D). Endogenous βs-globin DNA breakage was detected in Cas9 protein delivery guide RNA NILVs with or without 4× pseudo guide RNA by Surveyor nuclease assay (Figure 3E) and qPCR (p < 0.01) (Figure S10), compared to an untransduced control. Unexpectedly, more efficient genome editing in the βs-globin gene was observed with the Cas9 protein delivery SG11 guide RNA vector without additional pseudo guide RNA (56%) compared to that with 4× pseudo guide RNA (24%), maybe due to the lower vector titer (less efficient transduction) with the 4× pseudo guide RNA vector.
To optimize donor DNA vectors encoding a normal β-globin gene, we designed NILVs containing various sizes of the β-globin donor DNA sequence (0.3 kb, 0.4 kb, 0.7 kb, 1.7 kb, and 2.0 kb), which was terminated upstream of the stop codon to prevent vector-derived globin expression and included several silent mutations to escape from SG11 guide RNA targeting (Figure S11A). Immortalized erythroid cells including the SCD mutation (sickle HUDEP-2 cells) were transduced with a SG11 guide RNA NILV with Cas9 protein delivery as well as the β-globin donor NILVs. After erythroid differentiation of edited cells, βs-to-β-globin gene correction was detected at the protein level for all donor vectors with up to 31.5% of gene correction using 1.7 kb β-globin donor DNA as well as minimal cell toxicity (Figures S11B and S11C), suggesting that more efficient gene correction results from large-sized donor DNA encoded in NILVs. Donor-size-dependent gene correction was also observed with an integrating guide RNA vector with Cas9 protein delivery and these donor DNA NILVs, resulting in 24%–43% gene correction and 57%–76% insertions or deletions (indels) at the DNA level (p < 0.01 by qPCR) and 50%–91% β-globin production at the protein level with slight reduction of cell counts (0.5- to 0.7-fold) (Figure S12).
We selected the 1.7 kb β-globin donor DNA to produce Cas9 protein delivery NILVs encoding both SG11 guide RNA and β-globin donor DNA (all-in-one), which were prepared in two vector constructs of 5′-guide RNA-donor DNA-3′ and 5′-donor DNA-guide RNA-3′. We observed 27%–34% of βs-to-β-globin gene correction (HDR, p < 0.05) and 28%–34% of indel (p < 0.05) in sickle HUDEP-2 cells at the DNA level (by targeted deep sequencing) in all Cas9 protein delivery gene correction NILVs (Figure 3F) with undetectable off-target editing in the δ-globin gene (Figure S13), which has high homology to the β-globin sequence and was reported as a major off-target site in β-globin editing.30 Efficient βs-to-β-globin gene correction was observed at the protein level in the Cas9 protein delivery SG11 guide RNA NILV along with the β-globin donor DNA NILV (34.2%, p < 0.01) as well as Cas9 protein delivery all-in-one NILVs encoding both SG11 guide RNA and β-globin donor DNA (34.5%, p < 0.05 and 41.9%, p < 0.01, respectively) (Figures 3G and 3H; Figure S14). Similar cell counts were obtained between lentiviral gene-corrected cells (0.9- to 1.0-fold) and no transduction control (Figure 3I). When we evaluated cell toxicity between NILV delivery and electroporation in immortalized erythroid cells, similar cell counts were observed between lentiviral gene correction and no editing control; however, electroporation-mediated delivery strongly reduced cell counts (0.2- to 0.5-fold) as compared to no editing control (Figure S15), suggesting that unlike electroporation, gene correction with lentiviral Cas9 protein delivery is well tolerated, with minimal cell toxicity. These data demonstrate that the Cas9 protein delivery NILV system allows for β-globin DNA breakage and βs-to-β-globin gene correction.
Discussion
New genome-editing technologies have the potential to allow us to develop a gene-correction therapy for various hereditary diseases, including SCD. Electroporation is the current standard delivery method for editing tools;15, 16, 17, 18 however, toxicity to target cells remains a concern, including for HSCs.31, 32, 33 Adeno-associated virus (AAV) vectors can be used for larger-size donor DNA delivery, but electroporation is still employed for efficient guide RNA and Cas9 protein delivery.13 Lentiviral vector systems were developed for safe and efficient delivery of therapeutic genes, as well as long-term engraftment of gene-modified HSCs, as demonstrated in animal models and clinical trials.1, 2, 3, 4, 5, 6, 7,20,21,28 Given the proven safety and efficacy of lentiviral systems for gene therapy without toxicity to engrafting HSCs, lentiviral delivery systems adapted for genome editing have the potential for rapid translation to the clinic. Preliminary lentiviral gene-editing systems encoded guide RNA and Cas9 without donor DNA;24,25 therefore, it can result in long-term Cas9 expression with potential genotoxicity by lentiviral integration, and it allows for DNA breakage but not HDR due to no inclusion of donor template DNA. Therefore, in this study, we developed an “all-in-one” Cas9 protein delivery NILV system encoding guide RNA and donor DNA. We obtained up to ∼50% DNA breakage and up to ∼30% gene conversion (HDR) with an all-in-one NILV designed to induce GFP-to-YFP gene conversion (Figures 1 and 2). The Cas9 protein delivery NILVs also allowed for efficient SCD gene correction (up to ∼40% at the protein level) in the endogenous βs-globin gene with minimal cell toxicity (Figure 3; Figure S11) in cell lines. Our Cas9 protein delivery NILVs result in efficient gene/protein delivery, which should allow for long-term engraftment of gene-edited HSCs without genotoxicity by insertional mutagenesis.
We first used an integrating lentiviral vector system to deliver both the guide RNA and Cas9 gene, since this is the most established method for efficient gene delivery into engraftable HSCs (Figure 1; Figure S1). Lentiviral vector integration allows for long-term gene expression; however, persistent expression of Cas9 endonuclease can increase the risk of genotoxicity in target cells. Therefore, we developed a Cas9 protein delivery NILV system, which abrogates Cas9 gene integration by replacing the Cas9 gene with the Cas9 protein and does not require integration of the entire vector genome. In addition, this Cas9 protein delivery system eliminates the requirement for a large Cas9 gene (4.3 kb) in the lentiviral genome, which creates space to include both guide RNA and donor DNA within a single vector genome (all-in-one allowing for HDR) and should allow more efficient gene/protein delivery due to its small-sized genome. In our Cas9 protein delivery NILV system, site-specific DNA breakage was induced short term (<6 days, demonstrated in Figure S2), which is sufficient for gene correction but possibly reduces DNA damage in target cells. Cas9 protein activity is reportedly limited to within ∼24 h,34 which should be independent of delivery methods.
We initially hypothesized that Cas9-CypA fusion allows for protein delivery in lentiviral vector particles, since CypA binds to lentiviral capsid as an innate immune factor.29 We demonstrated that Cas9-CypA fusion proteins can be delivered with lentiviral vector particles without compromising endonuclease function, allowing for genome editing (Figure 1; Figure S1). Surprisingly, when investigating the mechanism of lentiviral Cas9 protein delivery, we found that CypA fusion is not required for Cas9 protein. We then hypothesized that Cas9 protein can be delivered by binding to the guide RNA sequence encoded in the lentiviral RNA genome, since Cas9 protein functions to bind guide RNA for site-specific DNA breakage.8 We found that inclusion of the guide RNA sequence is important for both GFP-to-YFP gene conversion and GFP DNA break with Cas9 protein delivery lentiviral vectors (Figure 1). Additional guide RNA sequences enhanced both GFP DNA breakage (Figure 2) and the Cas9 protein amount packaged per vector (Figure S7). Interestingly, a background Cas9 protein signal was detected in a donor vector control (Figure S7), possibly because small amounts of Cas9 proteins were non-specifically packaged in lentiviral particles, and the residual Cas9 protein produced in transfected 293T cells was contaminated in concentrated vector pellets. In contrast, the Cas9 plasmid used for vector preparation should be contained in concentrated vector solutions;35 however, the residual plasmids in vector pellets should not produce Cas9 proteins, since 0.22 μm filtration and high-speed ultracentrifugation (25,000 rpm) break most cells that are required for plasmid DNA to produce mRNA and proteins. These data demonstrate that the Cas9 protein is predominantly delivered with guide RNA-encoding lentiviral vectors, potentially binding to guide RNA sequences on the lentiviral RNA genome.
In addition, we used a NILV system for Cas9 protein delivery, resulting in transient gene expression and reducing the risk of genotoxicity by insertional mutagenesis; however, lower transgene expression was observed in NILVs, compared to integrating vectors.36,37 We used a D64V integrase-deficient NILV system for guide RNA expression and Cas9 protein delivery, which allows for transient transgene expression (Figure S16) and minimal elevation of VCN 14 days post-transduction (Figure S4), and, surprisingly, Cas9 protein delivery NILVs allowed for efficient GFP DNA breakage as well as GFP-to-YFP gene conversion, at levels similar to Cas9 protein delivery integrating vectors (Figure 1). These data demonstrate that guide RNA amounts derived from NILVs are sufficient for genome editing with lentiviral Cas9 protein delivery.
We used the GFP-to-YFP gene conversion to model gene correction in our early experiments due to its relative ease of interpretation. The GFP fluorescent signal is useful for lentiviral optimization in gene therapy research due to its specific and sensitive detection, which is more reliable than PCR-based DNA analysis.35 In addition, strong homology between GFP and YFP allows for fair evaluation due to similar DNA size, fluorescence intensity, and immunogenicity. While auto-fluorescence in transduced cells might result in false positives in GFP and YFP, we also demonstrated βs-to-β-globin gene correction at the protein level with Cas9 protein delivery NILVs.
After establishing Cas9 protein delivery NILVs in the GFP-to-YFP gene conversion model, we focused on editing the βs-globin gene (including the SCD mutation) to develop a therapeutic strategy for SCD. We performed a screening of βs-globin-targeting guide RNAs in both integrating guide RNA/Cas9 vectors and Cas9 protein delivery NILVs encoding guide RNA (Figure 3), demonstrating that suitable guide RNA sequences for lentiviral Cas9 protein delivery may be different from those required for efficient DNA targeting. An optimal guide RNA (SG11) allowed for robust DNA breakage in the endogenous βs-globin gene with Cas9 protein delivery NILVs (Figure 3). We also optimized the size of normal β-globin gene (1.7 kb) in donor DNA NILVs, which is larger than the commonly used size (∼0.1 kb) for electroporation, and larger sizes of donor DNA enhanced gene correction in the endogenous βs-globin gene along with the Cas9 protein delivery guide RNA NILV (Figure S11). The Cas9 protein delivery system allowed for all-in-one NILVs encoding both SG11 guide RNA and 1.7 kb β-globin donor DNA due to no requirement of Cas9 gene (4.3 kb), and efficient βs-to-β-globin gene correction was achieved with Cas9 protein delivery all-in-one NILVs (Figure 3). Interestingly, fetal hemoglobin levels were increased by β-globin editing in sickle HUDEP-2 cells (Figure S14), probably since indels of the β-globin gene could generate thalassemia-like HUDEP-2 cells, and low proliferation of thalassemia-like cells might result in biological selection of fetal hemoglobin-producing HUDEP-2 subclones.38 These data demonstrate that the Cas9 protein delivery NILV system allows for efficient βs-to-β-globin gene correction in cell lines and warrants further optimization in engrafting HSCs.
Electroporation-related toxicity may in part explain why gene correction in primary CD34+ HSCs remains challenging, evidenced by only low-level long-term engraftment of gene-converted CD34+ cells (with HDR) in recent attempts in our non-human primate model despite efficient gene conversion rates in input cells.39 Based on persistent engraftment of gene-modified CD34+ cells in lentiviral gene therapy trials, this lentiviral Cas9 protein delivery should be applicable for engraftable CD34+ cells; however, further optimization will be required in this population for efficient gene correction with lentiviral Cas9 protein delivery. Recently, enhancement of HDR was demonstrated by 53BP1 inhibition,40,41 which may be useful for the lentiviral gene correction. In addition, Gag-Cas9 fusions allowed for vesicular stomatitis virus G protein (VSVG) particle-based delivery including guide RNA, resulting in targeted DNA breakage.42 Another VSVG particle-based Cas9 delivery system utilized a fused dimerization domain, which can bind to an engineered membrane-associated protein (Gesicle, Takara Bio, Shiga, Japan). These systems do not include a viral RNA genome (donor DNA) as well as reverse transcriptase, and therefore do not allow for gene correction. Potentially, Gag-Cas9 fusion proteins could be combined with our lentiviral Cas9 delivery system to improve Cas9 protein delivery and gene correction, since a Gag-Cas9 fusion was also reported in a murine leukemia virus (MLV) system including the viral genome.43 Zinc finger nucleases (ZFNs) have previously been used for genome editing with NILV delivery;44,45 however, design of ZFNs is more difficult and editing efficiency is likely less efficient than with the CRISPR-Cas9 system. In addition, nanoparticle-based delivery is also available for genome editing, but it is further from clinical translation than lentiviral delivery.46
In summary, we developed a Cas9 protein delivery system with NILVs, which resulted in efficient DNA breakage and genome correction (HDR) in SCD. Our Cas9 protein delivery system allows for efficient one-time gene correction without electroporation using an all-in-one NILV encoding both guide RNA and donor DNA. Our findings improve the prospects for safe delivery and efficient genome editing.
Materials and methods
HIV-1-based lentiviral vector preparation with Cas9 protein delivery
Self-inactivating lentiviral vectors were prepared by co-transfection of 293T cells (American Type Culture Collection [ATCC], Manassas, VA, USA) with 6 μg of Gag/Pol plasmid, 2 μg of Rev/Tat plasmid, 2 μg of VSVG envelope plasmid, and 10 μg of HIV-1 vector genome plasmid, including a guide RNA expression cassette derived from a U6 promoter (Addgene plasmid #43860, kindly provided by Dr. Keith Joung), cultured in Dulbecco’s modified Eagle’s medium (Corning, Tewksbury, MA, USA) containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific, Waltham, MA, USA).47,48 Lentiviral vectors were filtered (0.22 μm, MilliporeSigma, St. Louis, MO, USA) to remove transfected 293T cells and 100-fold concentrated by ultracentrifugation (25,000 rpm in an SW28 rotor for 1 h, Optima XE-90 ultracentrifuge, Beckman Coulter Life Sciences, Indianapolis, IN, USA) to remove soluble molecules.35 Lentiviral titers were evaluated by YFP-positive percentages or VCNs in HeLa cells (ATCC), as previously described.28,35,48 NILVs were prepared with an integrase-defective Gag/Pol (D64V) plasmid.36 For Cas9 protein delivery, we added 4 μg (2–8 μg) of Cas9 plasmid on the co-transfection step during vector preparation, which expresses a codon-optimized Streptococcus pyogenes Cas9 with 3× FLAG and 2× nuclear localization signals (Addgene plasmid #42230, kindly provided by Dr. Feng Zhang) under the control of a cytomegalovirus enhancer/chicken β-actin (CAG) promoter.49 The CypA-Cas9 and Cas9-CypA fusion genes were designed by ligation of two genes after deleting both the start codon (ATG) and stop codon (TAA) in the ligation site to express on a single reading frame. The YFP donor DNA vector encoded a YFP expression cassette under the control of murine stem cell virus promoter (Mp),28 which is almost the same as the GFP target vector except a 9-base difference between GFP and YFP genes (193C>T, 195G>C, 196A>G, 197C>G, 205G>C, 217A>G, 218G>C, 610A>T, and 611C>A) as well as 5-base silent mutations in the YFP gene (150C>A, 153C>A, 156C>A, 165C>T, and 171C>A) to escape from GFP guide RNA targeting while preserving the YFP amino acid sequence (Figure S17). Human β-globin donor DNA vectors encoded both β-globin promoter (0.1 kb, 0.3 kb, or 0.7 kb) and β-globin gene (exon 1 only [0.1 kb], exons 1–2 [0.4 kb], or exons 1–3 [1.3 kb]) with silent mutations (27G>A, 28T>A, 29C>G, 30T>C, 33C>A, and 36T>G) in reverse orientation but did not include the locus control region, stop codon, and polyadenylation signals, which are required for globin expression from the vector construct.22
Genome editing with Cas9 protein delivery lentiviral vectors
The GFP-positive HEL cell line includes one copy of lentiviral vector genome (3.6 kb) encoding a GFP expression cassette under the control of Mp,20 in which the same vector backbone was utilized as the YFP donor DNA vector (Figure S17), cultured in Roswell Park Memorial Institute 1640 Medium (Corning) containing 10% FBS. GFP DNA was edited by a GFP-targeting guide RNA (5′-GGG CAC GGG CAG CTT GCC GG-3′), of which the target site has an appropriate protospacer adjacent motif (PAM) of NGG (Table S1).50 GFP-to-YFP conversion was performed with the GFP-targeting guide RNA and/or YFP donor DNA vectors. GFP and/or YFP expression was evaluated by flow cytometry (FACSCalibur, Becton Dickinson, East Rutherford, NJ, USA) (Figure S8), and GFP positive in the untransduced control was normalized to 100%.
We designed a chimeric gene between βs-globin (exon 1 including the SCD mutation, intron 1, and exon 2) and YFP, which can be expressed on a single reading frame by removing splicing signals and stop codons. A K562 erythroleukemia cell line (ATCC) was transduced with a lentiviral vector encoding the βs-globin/YFP chimeric gene under the control of Mp, and the βs-globin/YFP-positive cells were edited by several βs-globin-targeting guide RNA sequences (Table S1), cultured in Iscove’s modified Dulbecco’s medium (IMDM, Corning) containing 10% FBS. All target sites from each guide RNA have an appropriate PAM (NGG). The endogenous βs-globin gene in a human immortalized erythrocyte cell line (HUDEP-2 cells, provided by Drs. Yukio Nakamura and Ryo Kurita, RIKEN Tsukuba Branch, Ibaraki, Japan) including the SCD mutation was edited with the βs-globin-targeting guide RNA (SG11: 5′-GTA ACG GCA GAC TTC TCC AC-3′), cultured in proliferation media based on StemSpan Serum-Free Expansion Medium (STEMCELL Technologies, Vancouver, BC, Canada) containing 50 ng/mL stem cell factor (SCF, R&D Systems, Minneapolis, MN, USA), 3 U/mL erythropoietin (EPO, AMGEN, Thousand Oaks, CA, USA), 1 μM dexamethasone (VETone, Boise, ID, USA), and 1 μg/mL doxycycline (Thermo Fisher Scientific).51 The gene-corrected cells were further differentiated for hemoglobinization using IMDM-based differentiation media containing 50 ng/mL SCF, 3 U/mL EPO, 40 ng/mL insulin (Lilly, Indianapolis, IN, USA), 0.4 mg/mL transferrin (MilliporeSigma), 2 U/mL heparin (Sandoz, Holzkirchen, Germany), 1 μg/mL doxycycline, and human type AB plasma (Rhode Island Blood Center, Providence, RI, USA), and globin protein production was evaluated by reverse-phase high-performance liquid chromatography (RP-HPLC)52,53 and hemoglobin electrophoresis.22,52 Cell counts were evaluated by Countess II Automated Cell Counter (Thermo Fisher Scientific) with trypan blue staining.
In addition, we performed electroporation (4D-Nucleofector X Kit S, Lonza, Basel, Switzerland) 1 day after transduction (MOI 25) of immortalized erythrocyte cells including the SCD mutation to deliver SG11 modified guide RNA (200 pmol, Synthego, Menlo Park, CA, USA), Cas9 protein (30 μg, QB3 MacroLab, University of California, Berkeley, CA, USA), and/or β-globin donor single-strand DNA (0.1 kb, 300 pmol, Integrated DNA Technologies, Coralville, IA, USA).
PCR
The GFP-to-YFP converted gene (PCR product: 1,057 bp) and YFP donor DNA vector (PCR product: 667 bp) were separately detected by nested PCR using external primers (GFP recombination F1: 5′-TCA GAC CCA CCT CCC AAC CC-3′ and GFP recombination R1: 5′-CAC TGC AGG CCG TAG CCG-3′) and internal primers (GFP recombination F2: 5′-GGG ACC GAG CTC AAG CTT C-3′ and GFP recombination R2: 5′-GCA CAG GCA GCT TTC CTG TT-3′). The non-edited GFP gene cannot be expanded in these primers, since these reverse primers were specific for the YFP gene, with silent mutations to escape from GFP guide RNA targeting.
qPCR-based single-nucleotide polymorphism (SNP) genotyping and VCN analysis
The SCD mutation (HBB: c.20A>T) and intact β-globin sequence were separately detected by qPCR-based SNP genotyping using sickle FWD4 primer: 5′-GG CAG AGC CAT CTA TTG CTT AC-3′, sickle REV2 primer: 5′-CCA ACT TCA TCC ACG TTC ACC-3′, sickle FAM probe (SCD mutation): 5′-FAM-CTG ACT CCT GTG GAG AA-3′, sickle VIC (intact β-globin) 5′-VIC-CTG ACT CCT GAG GAG AA-3′, and sickle reference (for all β-globin sequences): 5′-Cy5-CCT CAA ACA GAC ACC AT-3′. The SCD mutation and intact β-globin signals were standardized by the sickle reference, and the percentages of SCD mutation and intact β-globin were measured by comparison to a βs-globin control (100% of SCD mutation) and an intact β-globin control (100% of intact β-globin), respectively. The indel (DNA breakage) percentages were calculated by the equation: indel (%) = reference (100%) − SCD mutation (%) − intact β-globin (%). VCNs were evaluated by specific probe/primers targeting GFP, YFP, self-inactivating long-terminal repeat (SIN-LTR), or an HIV-1 packaging signal (LV2), as previously described.20,35,48
Surveyor nuclease assay
Both GFP and βs-globin DNA breaks were evaluated by Surveyor Mutation Detection Kits (Integrated DNA Technologies; Coralville, IA, USA) according to company protocol. The target sites for genome editing were expanded by nested PCR using external primers (GFP Surveyor F1: 5′-GCC CTC AGC AGT TTC TAG AGA ACC-3′ and GFP Surveyor R1: 5′-GTC CAT GCC GAG AGT GAT CCC-3′) and internal primers (GFP Surveyor F2: 5′-AGT TCG CTT CTC GCT TCT GTT C-3′ and GFP Surveyor R2: 5′-GAA CTC CAG CAG GAC CAT GTG-3′) in the GFP gene, as well as external primers (LBGpF: 5′-GAC GCA GGA AGA GAT CCA TCT AC-3′ and BGEx2R: 5′-CTC AGG ATC CAC GTG CAG CTT G-3′) and internal primers (LBGpF2: 5′-CAA TAT GCT TAC CAA GCT GTG ATT CC-3′ and BGEx2R2: 5′-GCT CAC TCA GTG TGG CAA AGG-3′) in the βs-globin gene. The PCR products from edited genes and intact genes were mixed, denatured, and annealed. After digestion with a Surveyor endonuclease, indels (mismatch) were detected as smaller DNA bands in electrophoresis.
Targeted deep sequencing to evaluate β-globin gene editing
Efficiency of HDR (βs-to-β-globin gene correction) and indel was evaluated by targeted deep sequencing. We amplified human β-globin genome but not donor DNA sequence using BG external F (5′-TCT TCA ATA TGC TTA CCA AG CTG TG-3′) and BG external R (5′-CCC AAG AGT CTT CTC TGT CTC C-3′), then added partial P5 and P7 adapters (underlines) using hBG TargSeq F1 GW (5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT GTC AGG GCA GAG CCA TCT ATT G-3′) and hBG TargSeq R1 GW (5′-GAC TGG AGT TCA GAC GTG TGC TCT TCC GAT CTT CTA TTG GTC TCC TTA AAC CTG TCT TG-3′). Off-target δ-globin gene editing (indel) was analyzed by hDG TargSeq F1 GW (5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT AGG GAG GAC AGG ACC AGC AT-3′) and hDG TargSeq R1 GW (5′-GAC TGG AGT TCA GAC GTG TGC TCT TCC GAT CTC CCA GTT TCC ATT TGC CTC CTT G-3′). Deep sequencing was performed by GENEWIZ (South Plainfield, NJ, USA), and sequencing data were analyzed by a web-based software of CRISPResso2.54
Western blot analysis
After ultracentrifugation of lentiviral vectors, the vector pellets were resuspended in 1/100× volume of X-VIVO10 media (Lonza)22 and lysed in the same volume of SDS buffer (Sample Buffer, Laemmli 2× Concentrate, MilliporeSigma). The vector lysates (40 μL) were electrophoresed on Novex 4%–20% Tris-Glycine gel (Thermo Fisher Scientific) and transferred to 0.45 μm nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA).55 The membrane was blocked in Tris-buffered saline with 0.1% Tween-20 (TBST) and 3% blotting grade blocker (Bio-Rad) for 1 h and incubated with a 1:1,000 dilution of anti-FLAG antibody (clone M2, MilliporeSigma) overnight to detect Cas9 protein including FLAG tag. After washing three times in TBST for 5 min, the membrane was incubated with a 1:80,000 dilution of peroxidase-conjugated anti-mouse IgG antibody (polyclonal, MilliporeSigma) for 1 h. Anti-HIV-1 p24 Gag monoclonal antibody (1:1,000, clone #24-2, National Institutes of Health [NIH] AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases [NIAID], NIH, provided by Dr. Michael H. Malim) and peroxidase-conjugated anti-mouse IgG antibody were used to detect the HIV-1 capsid as an internal control for lentiviral vector.56 After washing three times in TBST, the blots were detected by ImageQuant LAS 4000 (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) and measured by ImageJ 1.51k (NIH, Bethesda, MD, USA).
Statistical analysis
Statistical analyses were performed using the JMP 14 software (SAS Institute, Cary, NC, USA). Two averages were evaluated with Student’s t test. The averages in various conditions were evaluated by Dunnett’s test (one-way ANOVA for a control) as well as Tukey’s honestly significant difference (HSD) test (one-way ANOVA among all groups) (Table S2). A p value of <0.01 or 0.05 was deemed significant. Standard error of the mean was shown as error bars in all figures. All experiments were performed in triplicate except RP-HPLC (single run), Surveyor nuclease assay (single run), and western blot analysis (duplicate).
Acknowledgments
This work was supported by the intramural research program of the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) at NIH. We thank Dr. Duck-Yeon Lee from the NHLBI Biochemistry Core for RP-HPLC analysis help. We published an international patent (WO 2017/059241) on April 6, 2017 and United States patent (US 2016/054759) on September 30, 2016.
Author contributions
N.U. designed the research, performed experiments, analyzed results, made the figures, and wrote the paper; C.M.D. performed experiments and wrote the paper; T.N. performed experiments; J.G. performed experiments; M.Y. performed experiments; J.D. performed experiments; Y.S. performed experiments; M.H. performed experiments; B.G. performed experiments; J.J.H.-M. designed the research; S.D. designed the research and performed experiments; and J.F.T. designed the research and wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.02.022.
Supplemental information
References
- 1.Aiuti A., Cattaneo F., Galimberti S., Benninghoff U., Cassani B., Callegaro L., Scaramuzza S., Andolfi G., Mirolo M., Brigida I. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 2.Aiuti A., Slavin S., Aker M., Ficara F., Deola S., Mortellaro A., Morecki S., Andolfi G., Tabucchi A., Carlucci F. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 3.Boztug K., Schmidt M., Schwarzer A., Banerjee P.P., Díez I.A., Dewey R.A., Böhm M., Nowrouzi A., Ball C.R., Glimm H. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010;363:1918–1927. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavazzana-Calvo M., Hacein-Bey S., de Saint Basile G., Gross F., Yvon E., Nusbaum P., Selz F., Hue C., Certain S., Casanova J.L. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 5.Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F., Down J., Denaro M., Brady T., Westerman K. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hacein-Bey-Abina S., von Kalle C., Schmidt M., Le Deist F., Wulffraat N., McIntyre E., Radford I., Villeval J.L., Fraser C.C., Cavazzana-Calvo M., Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 7.Ribeil J.A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 8.Horvath P., Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 9.Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R., Joung J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lattanzi A., Meneghini V., Pavani G., Amor F., Ramadier S., Felix T., Antoniani C., Masson C., Alibeu O., Lee C. Optimization of CRISPR/Cas9 Delivery to Human Hematopoietic Stem and Progenitor Cells for Therapeutic Genomic Rearrangements. Mol. Ther. 2019;27:137–150. doi: 10.1016/j.ymthe.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H., Kim J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 15.DeWitt M.A., Magis W., Bray N.L., Wang T., Berman J.R., Urbinati F., Heo S.J., Mitros T., Muñoz D.P., Boffelli D. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romero Z., Lomova A., Said S., Miggelbrink A., Kuo C.Y., Campo-Fernandez B., Hoban M.D., Masiuk K.E., Clark D.N., Long J. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019;27:1389–1406. doi: 10.1016/j.ymthe.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pattabhi S., Lotti S.N., Berger M.P., Singh S., Lux C.T., Jacoby K., Lee C., Negre O., Scharenberg A.M., Rawlings D.J. In Vivo Outcome of Homology-Directed Repair at the HBB Gene in HSC Using Alternative Donor Template Delivery Methods. Mol. Ther. Nucleic Acids. 2019;17:277–288. doi: 10.1016/j.omtn.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park S.H., Lee C.M., Dever D.P., Davis T.H., Camarena J., Srifa W., Zhang Y., Paikari A., Chang A.K., Porteus M.H. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47:7955–7972. doi: 10.1093/nar/gkz475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leonard A., Bonifacino A., Dominical V.M., Luo M., Haro-Mora J.J., Demirci S., Uchida N., Pierciey F.J., Jr., Tisdale J.F. Bone marrow characterization in sickle cell disease: inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br. J. Haematol. 2019;186:286–299. doi: 10.1111/bjh.15902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchida N., Hargrove P.W., Lap C.J., Evans M.E., Phang O., Bonifacino A.C., Krouse A.E., Metzger M.E., Nguyen A.D., Hsieh M.M. High-efficiency transduction of rhesus hematopoietic repopulating cells by a modified HIV1-based lentiviral vector. Mol. Ther. 2012;20:1882–1892. doi: 10.1038/mt.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchida N., Hsieh M.M., Hayakawa J., Madison C., Washington K.N., Tisdale J.F. Optimal conditions for lentiviral transduction of engrafting human CD34+ cells. Gene Ther. 2011;18:1078–1086. doi: 10.1038/gt.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uchida N., Hsieh M.M., Raines L., Haro-Mora J.J., Demirci S., Bonifacino A.C., Krouse A.E., Metzger M.E., Donahue R.E., Tisdale J.F. Development of a forward-oriented therapeutic lentiviral vector for hemoglobin disorders. Nat. Commun. 2019;10:4479. doi: 10.1038/s41467-019-12456-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uchida N., Nassehi T., Drysdale C.M., Gamer J., Yapundich M., Demirci S., Haro-Mora J.J., Leonard A., Hsieh M.M., Tisdale J.F. High-Efficiency Lentiviral Transduction of Human CD34+ Cells in High-Density Culture with Poloxamer and Prostaglandin E2. Mol. Ther. Methods Clin. Dev. 2019;13:187–196. doi: 10.1016/j.omtm.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ortinski P.I., O’Donovan B., Dong X., Kantor B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. Methods Clin. Dev. 2017;5:153–164. doi: 10.1016/j.omtm.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hacein-Bey-Abina S., Garrigue A., Wang G.P., Soulier J., Lim A., Morillon E., Clappier E., Caccavelli L., Delabesse E., Beldjord K. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 28.Uchida N., Washington K.N., Hayakawa J., Hsieh M.M., Bonifacino A.C., Krouse A.E., Metzger M.E., Donahue R.E., Tisdale J.F. Development of a human immunodeficiency virus type 1-based lentiviral vector that allows efficient transduction of both human and rhesus blood cells. J. Virol. 2009;83:9854–9862. doi: 10.1128/JVI.00357-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colgan J., Yuan H.E., Franke E.K., Luban J. Binding of the human immunodeficiency virus type 1 Gag polyprotein to cyclophilin A is mediated by the central region of capsid and requires Gag dimerization. J. Virol. 1996;70:4299–4310. doi: 10.1128/jvi.70.7.4299-4310.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long J., Hoban M.D., Cooper A.R., Kaufman M.L., Kuo C.Y., Campo-Fernandez B., Lumaquin D., Hollis R.P., Wang X., Kohn D.B. Characterization of Gene Alterations following Editing of the β-Globin Gene Locus in Hematopoietic Stem/Progenitor Cells. Mol. Ther. 2018;26:468–479. doi: 10.1016/j.ymthe.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Tendeloo V.F., Willems R., Ponsaerts P., Lenjou M., Nijs G., Vanhove M., Muylaert P., Van Cauwelaert P., Van Broeckhoven C., Van Bockstaele D.R., Berneman Z.N. High-level transgene expression in primary human T lymphocytes and adult bone marrow CD34+ cells via electroporation-mediated gene delivery. Gene Ther. 2000;7:1431–1437. doi: 10.1038/sj.gt.3301252. [DOI] [PubMed] [Google Scholar]
- 32.Wu M.H., Liebowitz D.N., Smith S.L., Williams S.F., Dolan M.E. Efficient expression of foreign genes in human CD34(+) hematopoietic precursor cells using electroporation. Gene Ther. 2001;8:384–390. doi: 10.1038/sj.gt.3301393. [DOI] [PubMed] [Google Scholar]
- 33.Wu M.H., Smith S.L., Dolan M.E. High efficiency electroporation of human umbilical cord blood CD34+ hematopoietic precursor cells. Stem Cells. 2001;19:492–499. doi: 10.1634/stemcells.19-6-492. [DOI] [PubMed] [Google Scholar]
- 34.Kim S., Kim D., Cho S.W., Kim J., Kim J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uchida N., Evans M.E., Hsieh M.M., Bonifacino A.C., Krouse A.E., Metzger M.E., Sellers S.E., Dunbar C.E., Donahue R.E., Tisdale J.F. Integration-specific In Vitro Evaluation of Lentivirally Transduced Rhesus CD34(+) Cells Correlates With In Vivo Vector Copy Number. Mol. Ther. Nucleic Acids. 2013;2:e122. doi: 10.1038/mtna.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leavitt A.D., Robles G., Alesandro N., Varmus H.E. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J. Virol. 1996;70:721–728. doi: 10.1128/jvi.70.2.721-728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wanisch K., Yanez-Munoz R.J. Integration-deficient lentiviral vectors: a slow coming of age. Mol. Ther. 2009;17:1316–1332. doi: 10.1038/mt.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demirci S., Gudmundsdottir B., Li Q., Haro-Mora J.J., Nassehi T., Drysdale C., Yapundich M., Gamer J., Seifuddin F., Tisdale J.F., Uchida N. βT87Q-Globin Gene Therapy Reduces Sickle Hemoglobin Production, Allowing for Ex Vivo Anti-sickling Activity in Human Erythroid Cells. Mol. Ther. Methods Clin. Dev. 2020;17:912–921. doi: 10.1016/j.omtm.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uchida N., Li L., Nassehi T., Yapundich M., Gamer J., Drysdale C., Haro-Mora J.J., Demirci S., Bonifacino A., Krouse A. Preclinical Evaluation for Engraftment of Gene-Edited CD34+ Cells with a Sickle Cell Disease Mutation in a Rhesus Transplantation Model. Blood. 2019;134(Suppl 1):609. [Google Scholar]
- 40.Canny M.D., Moatti N., Wan L.C.K., Fradet-Turcotte A., Krasner D., Mateos-Gomez P.A., Zimmermann M., Orthwein A., Juang Y.C., Zhang W. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2018;36:95–102. doi: 10.1038/nbt.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jayavaradhan R., Pillis D.M., Goodman M., Zhang F., Zhang Y., Andreassen P.R., Malik P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019;10:2866. doi: 10.1038/s41467-019-10735-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montagna C., Petris G., Casini A., Maule G., Franceschini G.M., Zanella I., Conti L., Arnoldi F., Burrone O.R., Zentilin L. VSV-G-Enveloped Vesicles for Traceless Delivery of CRISPR-Cas9. Mol. Ther. Nucleic Acids. 2018;12:453–462. doi: 10.1016/j.omtn.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mangeot P.E., Risson V., Fusil F., Marnef A., Laurent E., Blin J., Mournetas V., Massouridès E., Sohier T.J.M., Corbin A. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019;10:45. doi: 10.1038/s41467-018-07845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lombardo A., Genovese P., Beausejour C.M., Colleoni S., Lee Y.L., Kim K.A., Ando D., Urnov F.D., Galli C., Gregory P.D. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 45.Hoban M.D., Cost G.J., Mendel M.C., Romero Z., Kaufman M.L., Joglekar A.V., Ho M., Lumaquin D., Gray D., Lill G.R. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gee P., Lung M.S.Y., Okuzaki Y., Sasakawa N., Iguchi T., Makita Y., Hozumi H., Miura Y., Yang L.F., Iwasaki M. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat. Commun. 2020;11:1334. doi: 10.1038/s41467-020-14957-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanawa H., Kelly P.F., Nathwani A.C., Persons D.A., Vandergriff J.A., Hargrove P., Vanin E.F., Nienhuis A.W. Comparison of various envelope proteins for their ability to pseudotype lentiviral vectors and transduce primitive hematopoietic cells from human blood. Mol. Ther. 2002;5:242–251. doi: 10.1006/mthe.2002.0549. [DOI] [PubMed] [Google Scholar]
- 48.Uchida N., Hanawa H., Dan K., Inokuchi K., Shimada T. Leukemogenesis of b2a2-type p210 BCR/ABL in a bone marrow transplantation mouse model using a lentiviral vector. J. Nippon Med. Sch. 2009;76:134–147. doi: 10.1272/jnms.76.134. [DOI] [PubMed] [Google Scholar]
- 49.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kurita R., Suda N., Sudo K., Miharada K., Hiroyama T., Miyoshi H., Tani K., Nakamura Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE. 2013;8:e59890. doi: 10.1371/journal.pone.0059890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uchida N., Demirci S., Haro-Mora J.J., Fujita A., Raines L.N., Hsieh M.M., Tisdale J.F. Serum-free Erythroid Differentiation for Efficient Genetic Modification and High-Level Adult Hemoglobin Production. Mol. Ther. Methods Clin. Dev. 2018;9:247–256. doi: 10.1016/j.omtm.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uchida N., Haro-Mora J.J., Fujita A., Lee D.Y., Winkler T., Hsieh M.M., Tisdale J.F. Efficient Generation of β-Globin-Expressing Erythroid Cells Using Stromal Cell-Derived Induced Pluripotent Stem Cells from Patients with Sickle Cell Disease. Stem Cells. 2017;35:586–596. doi: 10.1002/stem.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans M.E., Kumkhaek C., Hsieh M.M., Donahue R.E., Tisdale J.F., Uchida N. TRIM5alpha variations influence transduction efficiency with lentiviral vectors in both human and rhesus CD34(+) cells in vitro and in vivo. Mol. Ther. 2014;22:348–358. doi: 10.1038/mt.2013.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simon J.H., Carpenter E.A., Fouchier R.A., Malim M.H. Vif and the p55(Gag) polyprotein of human immunodeficiency virus type 1 are present in colocalizing membrane-free cytoplasmic complexes. J. Virol. 1999;73:2667–2674. doi: 10.1128/jvi.73.4.2667-2674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.