

Abstract

Pulse-dipolar EPR is an appealing strategy for structural characterization of complex systems in solution that complements other biophysical techniques. Significantly, the emergence of genetically encoded self-assembling spin labels exploiting exogenously introduced double-histidine motifs in conjunction with CuII-chelates offers high precision distance determination in systems nonpermissive to thiol-directed spin labeling. However, the noncovalency of this interaction exposes potential vulnerabilities to competition from adventitious divalent metal ions, and pH sensitivity. Herein, a combination of room-temperature isothermal titration calorimetry (ITC) and cryogenic relaxation-induced dipolar modulation enhancement (RIDME) measurements are applied to the model protein Streptococcus sp. group G. protein G, B1 domain (GB1). Results demonstrate double-histidine motif spin labeling using CuII-nitrilotriacetic acid (CuII–NTA) is robust against the competitor ligand ZnII–NTA at >1000-fold molar excess, and high nM binding affinity is surprisingly retained under acidic and basic conditions even though room temperature affinity shows a stronger pH dependence. This indicates the strategy is well-suited for diverse biological applications, with the requirement of other metal ion cofactors or slightly acidic pH not necessarily being prohibitive.
As the complexity of biomolecular assemblies implicated in health and disease has increased, so too has interest in pulse-dipolar EPR (PD-EPR) as a robust strategy for solution-state structural characterization of proteins1,2 and nucleic acids3,4 in the nanometer distance regime.5,6 PD-EPR is a powerful tool that complements X-ray crystallography, NMR, cryo-EM, and Förster Resonance Energy Transfer (FRET) data by providing structural insight without the need for crystallization, size limitation, or structurally perturbative labels. Hence, PD-EPR has been applied to study conformational equilibria,7 oligomerization degree,8,9 complexation events,10−12 and competing structural models.13
Pairs of paramagnetic moieties are commonly introduced into diamagnetic systems of interest using thiol-based site-directed spin labeling.14 Cysteine residues are typically covalently modified, as for the nitroxide R1 side chain (Figure 1a top). This strategy is suboptimal in systems containing essential cysteine residues, nonpermissive to post-translational reduction. However, CuII-based genetically encodable self-assembling spin labels using double-histidine motifs have emerged as an alternative labeling strategy.15,16 Additionally, the bipedal mode of CuII-chelate attachment at the double-histidine motif (Figure 1a bottom) results in significantly improved precision and accuracy in the distance domain. CuII-nitrilotriacetic acid (CuII–NTA) spin labeling of double histidine motifs for PD-EPR has been applied successfully to enzymes,17 metalloproteins,18 and nucleoprotein complexes.19
Figure 1.

Spin label structures and the GB1 constructs used in this work. (a) MTSL nitroxide conjugated to a cysteine residue, resulting in the R1 side chain (top) and CuII–NTA coordinated to a double-histidine motif (bottom). (b) Cartoon representation of the I6R1/K28H/Q32H GB1 construct, with the R1 nitroxide and CuII–NTA spin labels shown in stick representation. (c) Cartoon representation of the K28H/Q32H GB1 construct, with the CuII–NTA shown in stick representation.
Despite this success, optimization of the spin-labeling approach is nontrivial, because the noncovalency of the interaction predisposes sensitivity to variations in binding affinity, although CuII–NTA labeling may be easier to interpret using molecular dynamics simulations.20 For instance, different buffer conditions influence the double-histidine motif labeling efficiency with CuII–NTA.21 Furthermore, while the influence of pH upon formation of CuII-chelates has been characterized by CW-EPR previously,22 current literature has not shown how pH variations influence binding at the double-histidine motif, particularly under cryogenic temperatures. Similarly, current literature has not addressed competition for double-histidine motif sites by adventitious divalent metal ions, and so warrants investigation. In the current study, Streptococcus sp. Group G. protein G, B1 domain (GB1) constructs I6R1/K28H/Q32H (Figure 1b) and K28H/Q32H (Figure 1c) were used as biological model systems, in CuII-nitroxide relaxation-induced dipolar modulation enhancement (RIDME)23 pseudotitrations,10,24 and isothermal titration calorimetry (ITC) measurements, respectively. Building on our previous work that demonstrated high concentration sensitivity and was reflective of the temperature regime wherein the equilibrium dynamics are frozen-out,24 here we establish the use of pulse dipolar EPR for competitive binding assays, and the pH dependence of the equilibrium.
Measurements were first performed in the presence of the model competitor ligand, ZnII–NTA, which was chosen because (i) it is a weak ligand for double-histidine motifs compared to CuII–NTA and (ii) it is diamagnetic, so it does not contribute to the detected EPR signal. An EPR silent competitor ligand is desirable because analysis of pseudotitration data is simplified (see Supporting Information (SI) section 1.6). Room temperature ITC data (Figure 2a) fitted to a one-site model where binding stoichiometry could vary indicated a dissociation constant (KD) of 513 μM. The binding affinity was extrapolated to 235 K (i.e., the temperature at which the binding equilibrium is found to freeze out in our samples, such that diffusional processes cease, meaning our EPR data reflect equilibria at 235 K),24 to determine the influence of the competitor ligand upon double-histidine loading efficiency with CuII–NTA under PD-EPR conditions. Importantly, throughout this work the temperature is determined by the self-consistency between EPR and ITC under the assumption that the enthalpy change (ΔH) is temperature independent when extrapolating binding affinities to cryogenic temperatures. While the individual assumptions are not necessarily well met, the extrapolation to 235 K using room temperature binding enthalpies is in good agreement with experimental values24 even though this might be rooted in a cancellation of errors.
Figure 2.
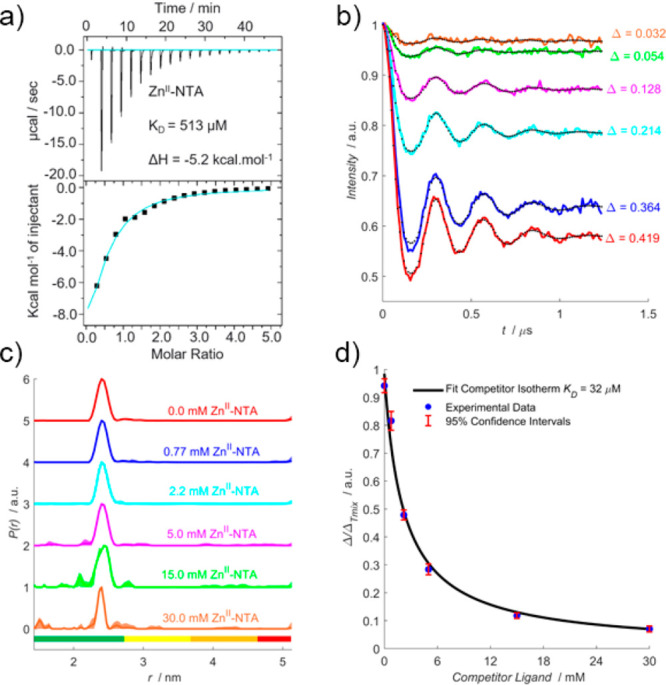
ZnII–NTA competitor RIDME pseudotitration. (a) ITC data performed at 298 K, 800 μM 28H/32H GB1 titrated against 12 mM ZnII–NTA. (b) RIDME dipolar evolution functions, in absence (red) and presence of 0.77 mM (blue), 2.2 mM (cyan), 5.0 mM (magenta), 15 mM (green), and 30 mM (orange) ZnII–NTA, with the corresponding fits shown in dotted black. Modulation depths (Δ) are indicated. (c) Validated RIDME distance distributions, corresponding to the dipolar evolution functions shown in (b). The color scheme is the same in (b) and (c). The concentrations of ZnII–NTA are indicated. Color bars represent reliability ranges (green: shape reliable; yellow: mean and width reliable; orange: mean reliable; red: no quantification possible). (d) A univariate fit of the competitor dissociation constant (32 μM) is shown in solid black. Experimental points are shown as the blue scatter, and 95% confidence intervals are shown as the red error bars.
The corresponding RIDME pseudotitration was performed at 1 μM protein concentration in the presence of 10 μM CuII–NTA (to ensure quantitative loading at the double histidine motif prior to addition of competitor ligand (see SI section 1.6)) and varying ZnII–NTA concentrations. Importantly, the dipolar evolution functions (Figure 2b) and distance distributions (Figure 2c) show that, in all cases, the expected peak at ∼2.5 nm is retrieved as the only significant feature following data validation. The fitted competitor KD value (32 μM) is within 2-fold of that determined from ITC when extrapolated to 235 K (48 μM) (Figure 2d). This suggests that CuII–NTA is robust against the ZnII–NTA competitor ligand in vast excess, >1000-fold, even at low μM protein concentrations. Additionally, this benchmarks quantitation of CuII-nitroxide RIDME modulation depths for remotely determining binding affinities of EPR silent ligands, in a competition assay format.
Next, the influence of pH upon double-histidine motif loading efficiency with CuII–NTA was investigated by measuring ITC and RIDME at pH 6.4. Since only deprotonated histidine residues can coordinate CuII–NTA, it follows that binding affinity should decrease under acidic conditions. Indeed, room-temperature ITC performed at pH 5, below the approximate pKA of solvent-exposed histidine,25 shows negligible binding (see SI section 2.3), and measurements at pH 6.4, fitted to a one-site model, indicated a 20-fold reduction in affinity compared to previous work24 (Figure 3a). Extrapolating ΔH to 235 K suggested a binding affinity of ∼4 μM.
Figure 3.
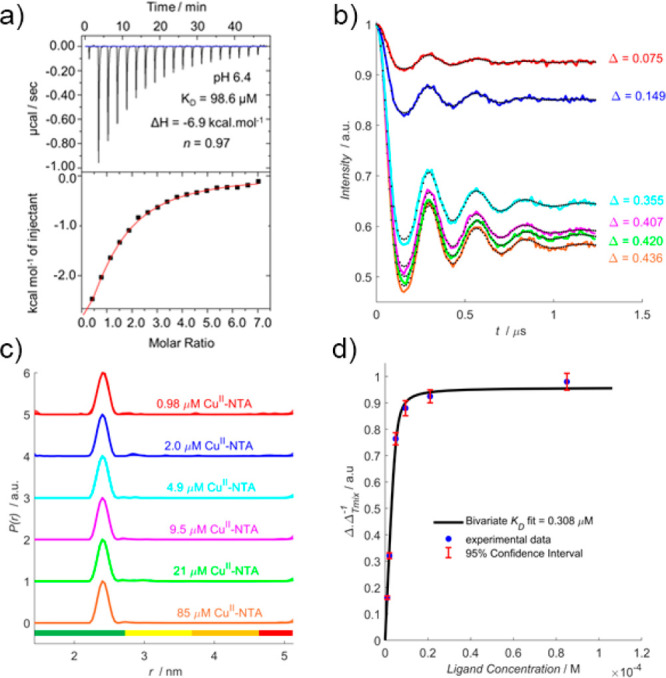
pH 6.4 RIDME pseudotitration. (a) ITC data performed at 298 K, 75 μM K28H/Q32H GB1 titrated against 2 mM CuII–NTA. (b) RIDME dipolar evolution functions, with the corresponding fits shown in dotted black. Modulation depths (Δ) are indicated. (c) Validated RIDME distance distributions, corresponding to the dipolar evolution functions shown in (b). The color scheme is the same in (b) and (c). The concentrations of CuII–NTA are indicated. (d) A bivariate fit of the dissociation constant (0.31 μM) is shown in solid black. Experimental points are shown as the blue scatter, and 95% confidence intervals are shown as the red error bars.
A RIDME pseudotitration was performed at 5 μM protein concentration to validate the room-temperature ITC prediction of reduced affinity under PD-EPR conditions. Significantly, the dipolar evolution functions (Figure 3b) show CuII–NTA binding is only marginally reduced at lower pH, with 1 equiv of CuII–NTA saturating ∼70% of available double-histidine motifs. This is further borne out by the fitted dissociation constant (Figure 3d), 0.31 μM compared to 0.14 μM in previous work at pH 7.4.24 The affinity reduced by only 2-fold, indicating that the influence of pH upon double-histidine motif loading may be attenuated at lower temperatures. A possible explanation is that histidine protonation is endothermic,26 driving the equilibrium toward the deprotonated state at lower temperatures, compensating for reduced pH and facilitating double-histidine loading. Importantly, this would also imply significantly tighter binding at higher pH, where histidine deprotonation is already favored.
To clarify the disparity between ITC and PD-EPR data
at pH 6.4,
room-temperature ITC was also performed at pH 8.4 (Figure 4a), fitted to a one-site model,
where a 20-fold increase in affinity was predicted (via improved thermodynamic favorability of binding) compared to previous
work. Another RIDME pseudotitration was performed at 2 μM protein
concentration, with dipolar evolution functions (Figure 4b) suggesting moderate improvement
in binding affinity. The fitted dissociation constant (Figure 4d) of 0.091 μM indicates
the binding affinity is approximately 2-fold higher than at pH 7.4,
consistent with observation at pH 6.4 that the influence of pH upon
binding affinity is attenuated with decreasing temperature. While
an endothermic protonation process would suggest much tighter binding
is to be anticipated at pH 8.4, consider that at this pH < 1% of
histidine  -nitrogen atoms should remain protonated.
This may explain why the relative increase in binding affinity is
smaller than expected, since the deprotonation is already driven toward
completion by the high pH.
-nitrogen atoms should remain protonated.
This may explain why the relative increase in binding affinity is
smaller than expected, since the deprotonation is already driven toward
completion by the high pH.
Figure 4.
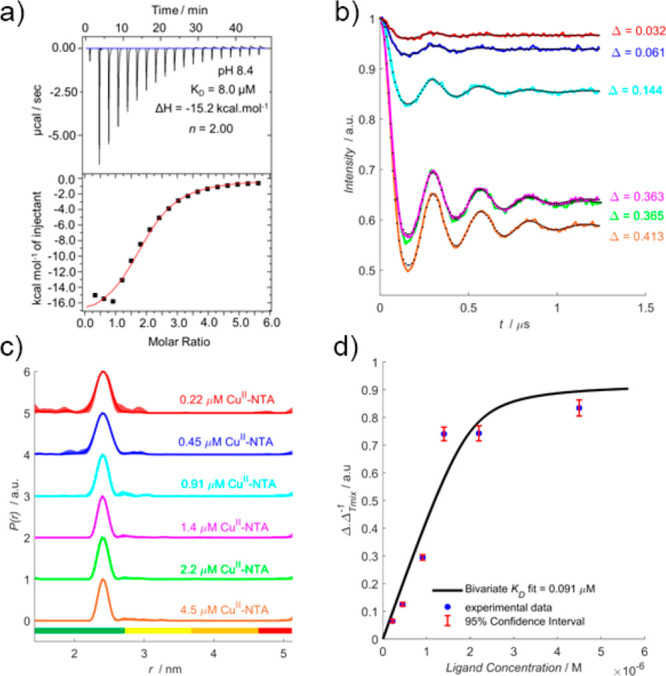
pH 8.4 RIDME pseudotitration. (a) ITC data performed at 298 K, 75 μM K28H/Q32H GB1 titrated against 2.5 mM CuII–NTA. (b) RIDME dipolar evolution functions, with the corresponding fits shown in dotted black. Modulation depths (Δ) are indicated. (c) Validated RIDME distance distributions, corresponding to the dipolar evolution functions shown in (b). The color scheme is the same in (b) and (c). The concentrations of CuII–NTA are indicated. (d) A bivariate fit of the dissociation constant (0.091 μM) shown in solid black. Experimental points are shown as the blue scatter, and 95% confidence intervals are shown as the red error bars.
While the data suggest that spin labeling and measurement at pH 8.4 will afford enhanced loading and sensitivity, it should be noted that the stoichiometry of binding is ∼2, compared to ∼1 at pH 6.4. This may arise from deprotonation of the protein surface that promotes nonspecific binding. This would explain the increased exothermic nature of the binding, if nonspecific or additional binding events contributed to the isotherm and would further inflate the binding affinity when extrapolated to cryogenic temperatures. However, the corresponding distance distributions (Figure 4c) do not contain additional peaks to support this hypothesis.
Perhaps most significantly, these results clearly show that CuII–NTA binding affinity for double-histidine motifs is not strongly perturbed from the high nM concentration regime by fluctuations of pH between 6.4 and 8.4. Coupled with measurements in the presence of competitor ligand ZnII–NTA, findings support that CuII–NTA is a highly robust spin label when combined with α-helical double-histidine motifs. This is encouraging for the widespread application of double-histidine motifs in metalloproteins, or in systems where divalent metal cofactors are necessary. Additionally, the benchmarking of a competition assay using PD-EPR is particularly exciting because it allows remote detection of binding interactions with diamagnetic ligands and showcases investigation of competitor ligand binding at significantly reduced material, compared to more established methods like ITC. This will be promising in cases where paramagnetic ligand analogues are not available or cause structural perturbation. PD-EPR also has greater sensitivity than ITC, and the coupling of thermodynamic and structural information allows for the facile monitoring of nonspecific and competitor ligand interactions.27 Traditionally, monitoring competitive ligand binding has required expensive radio-labeling and judicious selection of appropriate isotopes.28,29 PD-EPR may complement these strategies, while obviating potential cost and safety considerations.
The research data supporting this publication can be accessed at 10.17630/d7138874-55dd-4874-a2e8-c026fbc0b67f.30
Acknowledgments
The authors thank Dr. Hassane El Mkami for help with EPR. J.L.W. is supported by the BBSRC DTP Eastbio. We thank the Leverhulme Trust for support (RPG-2017-214 and RPG-2018-397). This work was supported by equipment funding through the Wellcome Trust (099149/Z/12/Z) and BBSRC (BB/R013780/1). We gratefully acknowledge ISSF support to the University of St. Andrews from the Wellcome Trust.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.1c00211.
(I) Experimental Procedures: Construct Design, Expression and Purification; Pulse EPR Sample Preparation; Metal Chelate Spin Label Preparation; Mass Spectrometry; Pulse EPR Measurements; Competitive Binding Model; Isothermal Titration Calorimetry; UV–visible Spectroscopy. (II) Results and Discussion: Inversion Recovery Measurements; 5-pulse RIDME Measurements; Influence of Differential pH upon Double-Histidine Motif Affinity; Influence of Differential pH upon CuII–NTA Complex Formation; Optimization of CuII–IDA Complex Formation. (III) References. (PDF)
The authors declare no competing financial interest.
This letter was published ASAP on March 13, 2021. To correct an error caused by the publisher, the Acknowledgments section has been updated. This change has no impact to the research presented, and the letter was reposted ASAP March 18, 2021.
Supplementary Material
References
- Edwards D. T.; Huber T.; Hussain S.; Stone K. M.; Kinnebrew M.; Kaminker I.; Matalon E.; Sherwin M. S.; Goldfarb D.; Han S. Determining the Oligomeric Structure of Proteorhodopsin by Gd3+-Based Pulsed Dipolar Spectroscopy of Multiple Distances. Structure 2014, 22, 1677–1686. 10.1016/j.str.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Verhalen B.; Dastvan R.; Thangapandian S.; Peskova Y.; Koteiche H. A.; Nakamoto R. K.; Tajkhorshid E.; McHaourab H. S. Energy Transduction and Alternating Access of the Mammalian ABC Transporter P-Glycoprotein. Nature 2017, 543, 738–741. 10.1038/nature21414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann O.; Piton N.; Mu Y.; Stock G.; Engels J. W.; Prisner T. F. A PELDOR-Based Nanometer Distance Ruler for Oligonucleotides. J. Am. Chem. Soc. 2004, 126, 5722–5729. 10.1021/ja0393877. [DOI] [PubMed] [Google Scholar]
- Babaylova E. S.; Malygin A. A.; Lomzov A. A.; Pyshnyi D. V.; Yulikov M.; Jeschke G.; Krumkacheva O. A.; Fedin M. V.; Karpova G. G.; Bagryanskaya E. G. Complementary-Addressed Site-Directed Spin Labeling of Long Natural RNAs. Nucleic Acids Res. 2016, 44, 7935–7943. 10.1093/nar/gkw516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G. The Contribution of Modern EPR to Structural Biology. Emerg. Top. Life Sci. 2018, 2, 9–18. 10.1042/ETLS20170143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanabria H.; Rodnin D.; Hemmen K.; Peulen T. O.; Felekyan S.; Fleissner M. R.; Dimura M.; Koberling F.; Kühnemuth R.; Hubbell W.; et al. Resolving Dynamics and Function of Transient States in Single Enzyme Molecules. Nat. Commun. 2020, 11, 1231. 10.1038/s41467-020-14886-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B.; Sikora A.; Cafiso D. S. Ligand Induced Conformational Changes of a Membrane Transporter in E. Coli Cells Observed with DEER/PELDOR. J. Am. Chem. Soc. 2016, 138, 1844–1847. 10.1021/jacs.5b13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagelueken G.; Ingledew W. J.; Huang H.; Petrovic-Stojanovska B.; Whitfield C.; ElMkami H.; Schiemann O.; Naismith J. H. PELDOR Spectroscopy Distance Fingerprinting of the Octameric Outer-Membrane Protein Wza from Escherichia Coli. Angew. Chem., Int. Ed. 2009, 48, 2904–2906. 10.1002/anie.200805758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode B. E.; Margraf D.; Plackmeyer J.; Dürner G.; Prisner T. F.; Schiemann O. Counting the Monomers in Nanometer-Sized Oligomers by Pulsed Electron-Electron Double Resonance. J. Am. Chem. Soc. 2007, 129, 6736–6745. 10.1021/ja065787t. [DOI] [PubMed] [Google Scholar]
- Giannoulis A.; Oranges M.; Bode B. E. Monitoring Complex Formation by Relaxation-Induced Pulse Electron Paramagnetic Resonance Distance Measurements. ChemPhysChem 2017, 18, 2318–2321. 10.1002/cphc.201700666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann K.; Giannoulis A.; Cordes D. B.; Slawin A. M. Z.; Bode B. E. Assessing Dimerisation Degree and Cooperativity in a Biomimetic Small-Molecule Model by Pulsed EPR. Chem. Commun. 2015, 51, 5257–5260. 10.1039/C4CC08656B. [DOI] [PubMed] [Google Scholar]
- Giannoulis A.; Ackermann K.; Spindler P. E.; Higgins C.; Cordes D. B.; Slawin A. M. Z.; Prisner T. F.; Bode B. E. Nitroxide-Nitroxide and Nitroxide-Metal Distance Measurements in Transition Metal Complexes with Two or Three Paramagnetic Centres Give Access to Thermodynamic and Kinetic Stabilities. Phys. Chem. Chem. Phys. 2018, 20, 11196–11205. 10.1039/C8CP01611A. [DOI] [PubMed] [Google Scholar]
- Theillet F. X.; Binolfi A.; Bekei B.; Martorana A.; Rose H. M.; Stuiver M.; Verzini S.; Lorenz D.; Van Rossum M.; Goldfarb D.; et al. Structural Disorder of Monomeric α-Synuclein Persists in Mammalian Cells. Nature 2016, 530, 45–50. 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- Hubbell W. L.; Altenbach C. Investigation of Structure and Dynamics in Membrane Proteins Using Site-Directed Spin Labeling. Curr. Opin. Struct. Biol. 1994, 4, 566–573. 10.1016/S0959-440X(94)90219-4. [DOI] [Google Scholar]
- Cunningham T. F.; Putterman M. R.; Desai A.; Horne W. S.; Saxena S. The Double-Histidine Cu2+-Binding Motif: A Highly Rigid, Site-Specific Spin Probe for Electron Spin Resonance Distance Measurements. Angew. Chem., Int. Ed. 2015, 54, 6330–6334. 10.1002/anie.201501968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Lawless M. J.; Rule G. S.; Saxena S. The Cu2+-Nitrilotriacetic Acid Complex Improves Loading of α-Helical Double Histidine Site for Precise Distance Measurements by Pulsed ESR. J. Magn. Reson. 2018, 286, 163–171. 10.1016/j.jmr.2017.12.005. [DOI] [PubMed] [Google Scholar]
- Lawless M. J.; Pettersson J. R.; Rule G. S.; Lanni F.; Saxena S. ESR Resolves the C Terminus Structure of the Ligand-Free Human Glutathione S-Transferase A1–1. Biophys. J. 2018, 114, 592–601. 10.1016/j.bpj.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble Jarvi A.; Cunningham T. F.; Saxena S. Efficient Localization of a Native Metal Ion within a Protein by Cu2+-Based EPR Distance Measurements. Phys. Chem. Chem. Phys. 2019, 21, 10238–10243. 10.1039/C8CP07143H. [DOI] [PubMed] [Google Scholar]
- Sameach H.; Ghosh S.; Gevorkyan-Airapetov L.; Saxena S.; Ruthstein S. EPR Spectroscopy Detects Various Active State Conformations of the Transcriptional Regulator CueR. Angew. Chem., Int. Ed. 2019, 58, 3053–3056. 10.1002/anie.201810656. [DOI] [PubMed] [Google Scholar]
- Bogetti X.; Ghosh S.; Gamble Jarvi A.; Wang J.; Saxena S. Molecular Dynamics Simulations Based on Newly Developed Force Field Parameters for Cu2+ Spin Labels Provide Insights into Double-Histidine-Based Double Electron-Electron Resonance. J. Phys. Chem. B 2020, 124, 2788–2797. 10.1021/acs.jpcb.0c00739. [DOI] [PubMed] [Google Scholar]
- Gamble Jarvi A.; Casto J.; Saxena S. Buffer Effects on Site Directed Cu2+-Labeling Using the Double Histidine Motif. J. Magn. Reson. 2020, 320, 106848. 10.1016/j.jmr.2020.106848. [DOI] [PubMed] [Google Scholar]
- Lawless M. J.; Ghosh S.; Cunningham T. F.; Shimshi A.; Saxena S. On the Use of the Cu2+-Iminodiacetic Acid Complex for Double Histidine Based Distance Measurements by Pulsed ESR. Phys. Chem. Chem. Phys. 2017, 19, 20959–20967. 10.1039/C7CP02564E. [DOI] [PubMed] [Google Scholar]
- Milikisyants S.; Scarpelli F.; Finiguerra M. G.; Ubbink M.; Huber M. A Pulsed EPR Method to Determine Distances between Paramagnetic Centers with Strong Spectral Anisotropy and Radicals: The Dead-Time Free RIDME Sequence. J. Magn. Reson. 2009, 201, 48–56. 10.1016/j.jmr.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Wort J. L.; Ackermann K.; Giannoulis A.; Stewart A. J.; Norman D. G.; Bode B. E. Sub-Micromolar Pulse Dipolar EPR Spectroscopy Reveals Increasing CuII-Labelling of Double-Histidine Motifs with Lower Temperature. Angew. Chem., Int. Ed. 2019, 58, 11681–11685. 10.1002/anie.201904848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. S.; Griffin J. H.; Schechter A. N. Nuclear Magnetic Resonance Titration Curves of Histidine Ring Protons. IV. The Effects of Phosphate and Sulfate on Ribonuclease. J. Biol. Chem. 1973, 248, 4305–4310. 10.1016/S0021-9258(19)43773-3. [DOI] [PubMed] [Google Scholar]
- Tanokura M.; Tasumi M.; Miyazawa T. NMR Study on the Protonation of Imidazole Ring of N-Acetyl-L-Histidine Methylamide, a Model for Histidine Residues Exposed toAqueous Solvent. Chem. Lett. 1978, 7, 739–742. 10.1246/cl.1978.739. [DOI] [Google Scholar]
- Bankston J. R.; DeBerg H. A.; Stoll S.; Zagotta W. N. Mechanism for the Inhibition of the CAMP Dependence of HCN Ion Channels by the Auxiliary Subunit TRIP8b. J. Biol. Chem. 2017, 292, 17794–17803. 10.1074/jbc.M117.800722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire J. J.; Kuc R. E.; Davenport A. P. Radioligand Binding Assays and Their Analysis. Methods Mol. Biol. 2012, 897, 31–77. 10.1007/978-1-61779-909-9_3. [DOI] [PubMed] [Google Scholar]
- Hulme E. C.; Trevethick M. A. Ligand Binding Assays at Equilibrium: Validation and Interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. 10.1111/j.1476-5381.2009.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wort J.; Arya S.; Ackermann K.; Stewart A. J.; Bode B. E., 2021, Pulse Dipolar EPR Reveals Double-Histidine Motif Spin-labelling is Robust Against Competitor Ions (Dataset); Dataset. University of St Andrews Research Portal; 10.17630/d7138874-55dd-4874-a2e8-c026fbc0b67f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.