

Abstract
Heat shock protein 70 (Hsp70) is a potent antiapoptotic agent. Here, we tested whether it directly regulates renal cell survival and organ function in a model of transient renal ischemia using Hsp70 knockout, heterozygous, and wild-type mice. The kidney cortical Hsp70 content inversely correlated with tubular injury, apoptosis, and organ dysfunction after injury. In knockout mice, ischemia caused changes in the activity of Akt and glycogen synthase kinase 3-β (kinases that regulate the proapoptotic protein Bax), increased active Bax, and activated the proapoptotic protease caspase 3. As these changes were significantly reduced in the wild-type mice, we tested whether Hsp70 influences ischemia-induced apoptosis. An Hsp70 inducer, geranylgeranylacetone, increased Hsp70 expression in heterozygous and wild-type mice, and reduced both ischemic tubular injury and organ dysfunction. When administered after ischemia, this inducer also decreased tubular injury and organ failure in wild-type mice but did not protect the knockout mice. ATP depletion in vitro caused greater mitochondrial Bax accumulation and death in primary proximal tubule cells harvested from knockout compared with wild-type mice and altered serine phosphorylation of a Bax peptide at the Akt-specific target site. In contrast, lentiviral-mediated Hsp70 repletion decreased mitochondrial Bax accumulation and rescued Hsp70 knockout cells from death. Thus, increasing Hsp70 either before or after ischemic injury preserves renal function by attenuating acute kidney injury.
Keywords: acute kidney injury, apoptosis, mitochondria, proximal tubule
Heat shock protein 70 (Hsp70) is a potent antiapoptotic protein that works by simultaneously interrupting several steps in the cell death pathway. Induction of Hsp70 by nonlethal insults in vitro or in vivo is associated with acquired cytoprotection.1,2 Enhanced Hsp70 expression in transgenic mice protects the heart and brain from ischemia by an unknown mechanism(s).3–7 Furthermore, direct injection of Hsp70-containing adenovirus into the myocardium before ischemia preserves contractile function.8 In the kidney, indirect evidence suggests that Hsp70 inhibits ischemic acute kidney injury (AKI). The Brown Norway rat, a species with relatively abundant renal Hsp70 expression, is resistant to ischemic AKI.9 In addition, nonspecific agents that induce heat stress proteins reduce ischemic tissue injury, partly by inducing the expression of Hsp70.10–12 Although previous evidence is suggestive, a direct role for Hsp70 in ischemic AKI has not been demonstrated and the mechanism of Hsp70-mediated protection in ischemic tissue injury is controversial.
Recent evidence suggests that Hsp70 targets mitochondria,13 a key site for integrating survival and death signal events regulated by members of the B-cell lymphoma 2 (BCL2) protein family. In the intact kidney as well as in renal epithelial cells subjected to ATP depletion, BCL2 proteins mediate apoptosis by causing mitochondrial membrane injury, resulting in the release of caspase-dependent and caspase-independent factors.14–21 Bax and Bcl2 (proto-type pro- and anti-apoptotic BCL2 proteins, respectively) are the primary mediators of survival after insults that target mitochondria.22,23 Although other BCL2 members likely have ‘supportive roles’ in cell death, Bax and Bcl2 determine the apoptotic ‘set point.’24 Furthermore, maneuvers directed at one or both of these BCL2 proteins alter stress-induced cell death in a predictable fashion.25–29 Several laboratories, including our own, have shown that Hsp70 regulates members of the BCL2 family. Specifically, Hsp70 inhibits the activation of Bax30,31 and preserves Bcl2,32,33 a Bax antagonist during cell stress. By preventing both Bax activation and preserving Bcl2, Hsp70 could promote cell survival.
Bax activation itself is a multistep (and hence regulable) process that involves sequential checkpoints: conformational change that exposes the N-terminal 6A7 epitope, oligomerization, mitochondrial translocation, and clustering at the outer membrane.34–37 Bax activation is elegantly regulated by stress kinases including Akt38 and glycogen synthase kinase 3β (GSK3β).39 Stress induces 6A7 epitope exposure either by Bax ser184 de-phosphorylation, normally regulated by Akt,38 or by Bax ser163 phosphorylation, regulated by GSK3β.38–40 Considerable evidence suggests that ser184 is a key Bax regulatory site. Constitutive Bax targeting to the mitochondrial surfaces results from either a single substitution at this site by valine or alanine (Bax-S184V, Bax-S184A) as well as by ser184 deletion (Bax-deltaS184).14,41 None of these Bax mutants is inactivated by Akt and each is highly toxic to mitochondria. However, ser163 phosphorylation by GSK3β is also sufficient to activate Bax and induce apoptosis.38–40 Interestingly, both ischemia in vivo and ATP depletion in renal cells in vitro (as well as in other tissues) simultaneously inactivate Akt and activate GSK3β.14,42 These events might be expected, as Akt normally inactivates GSK3β. When Akt inactivation is coupled with GSK3β activation, site-specific serine phosphorylation events promote Bax activation and translocation, outer mitochondrial membrane injury, and apoptotic cell death.38,39,43
In this study, we propose that Hsp70 promotes proximal tubule epithelial cell survival, maintains tubular structure, and preserves organ function after acute ischemia caused by bilateral renal pedicle occlusion (RPO). Specifically, we hypothesized that Hsp70 reduces Bax activation and mitochondrial injury after ischemia by modulating the activity of Akt and GSK3β. Our results show that Hsp70 promotes renal epithelial cell survival and preserves organ function after ischemia partly by inhibiting proapoptotic changes in the activity of Akt and GSK3β. Furthermore, we attempted to confirm that protection by geranylgeranylacetone (GGA) is causally linked to Hsp70 induction and is an effective maneuver for preserving renal function administered before or after an ischemic insult.
RESULTS
Hsp70.1 and Hsp70.3 gene deletion suppresses renal Hsp70 expression
Immunoblot analysis of Hsp70 in renal cortical homogenates confirmed relatively high expression in Hsp70+/+ mice, intermediate expression in Hsp70 +/− mice, and no detectable Hsp70 in Hsp70−/− mice (Figure 1). Relative Hsp70 expression levels in the renal cortex were 100, 45±9, and 0% for Hsp70+/+, Hsp70+/−, and Hsp70−/− mice, respectively (n = 6). Developmental defects were not detected in any of these mice (data not shown).
Figure 1 |. Heat shock protein 70 (Hsp70) expression in mouse genotypes.

Immunoblot analysis of Hsp70 expression in renal cortical homogenates harvested from Hsp70.1/3 wild-type (Hsp70+/+), heterozygote (Hsp70+/−), and homozygote (Hsp70−/−) C57BL/6J mice; this blot is representative of six separate studies; densitometric analysis appears in Results.
Effect of Hsp70 on renal function and survival after ischemia
To assess the importance of Hsp70 as a regulator of ischemic AKI, Hsp70+/+, Hsp70 +/−, and Hsp70−/− mice were subjected to an intermediate duration of bilateral RPO sufficient to induce renal dysfunction. After 25 min of RPO, blood urea nitrogen (BUN) and creatinine increased in all three genotypes in the following order: Hsp70−/− >Hsp70+/− >Hsp70+/+, indicating that the degree of renal impairment is inversely related to Hsp70 content (Figure 2a). In addition, 100% of Hsp70−/− and 25% of Hsp70+/− mice died within 72 h after ischemia, whereas all Hsp70+/+ mice survived. As a result of this variable susceptibility to injury, the duration of pedicle occlusion was adjusted to achieve renal dysfunction in each Hsp70 genotype in subsequent studies (that is, 21–40 min).
Figure 2 |. Effect of heat shock protein 70 (Hsp70) deficiency or Hsp70 induction by geranylgeranylacetone (GGA) on postischemic renal function.
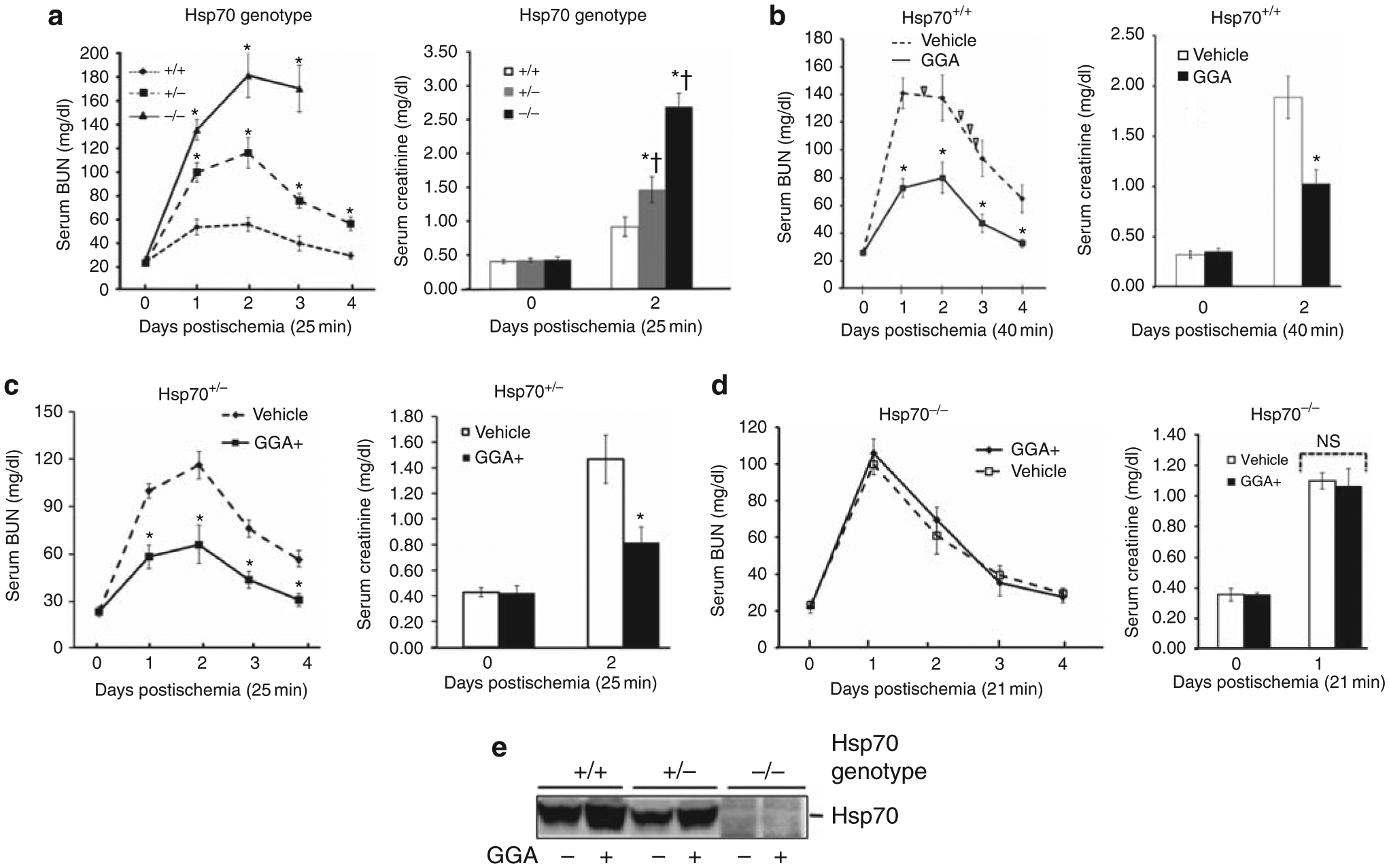
(a) Serum blood urea nitrogen (BUN) and creatinine at baseline and after 25 min of bilateral renal pedicle occlusion (RPO) in Hsp70+/+, Hsp70+/−, and Hsp70−/− mice; *P<0.05 vs Hsp70+/+; †P<0.05 vs Hsp70+/+ after ischemia, n = 6. (b) Serum BUN and creatinine at baseline and after 40 min of bilateral RPO in Hsp70+/+ mice with and without pretreatment with GGA; ∇ indicates animal death; *P<0.05 GGA vs vehicle alone; n = 6. (c) Serum BUN and creatinine levels at baseline and after 25 min of bilateral RPO in Hsp70+/− mice with and without GGA pretreatment; *P<0.05 for GGA vs vehicle alone; n = 6. (d) Serum BUN and creatinine levels at baseline and after 21 min of bilateral RPO in Hsp70−/− mice with and without pretreatment with GGA; NS P>0.05 for GGA vs vehicle; n = 6. (e) Immunoblot analysis of Hsp70 content in renal cortical homogenates harvested from Hsp70+/+, Hsp70+/−, and Hsp70−/− mice after treatment with GGA (400 mg/kg) or vehicle treatment; GGA increased Hsp70 expression only in Hsp70+/+ and Hsp70+/− mice; densitometric analysis appears in Results. GGA, 400 mg/kg, was orally administered twice at 18 and 2 h before injury.
Effect of GGA-mediated Hsp70 induction on ischemic AKI
Hsp70+/+ mice required more prolonged ischemia (35–40 min) in order to achieve reversible AKI. In these Hsp70+/+ mice, pretreatment with GGA, an Hsp70 inducer, significantly decreased BUN on days 1–4 after 40 min of RPO and reduced serum creatinine on day 2, the peak time of renal dysfunction (Figure 2b). Half of the vehicle-treated Hsp70+/+ mice died, whereas all GGA-treated mice survived. In Hsp70+/− mice, GGA also reduced the severity of renal dysfunction after 25 min of RPO, a sufficient duration to cause reversible AKI associated with a fivefold increase in BUN and 3.5-fold increase in serum creatinine (Figure 2c). Preischemia treatment with GGA significantly decreased BUN on days 1–4 after injury and reduced creatinine by 50% in Hsp70+/− mice. Interestingly, RPO for only 25 min caused severe AKI and death in all Hsp70−/− mice (data not shown). To assess the potential efficacy of GGA in Hsp70−/− mice, the duration of RPO was shortened to 21 min. Compared with vehicle alone, GGA failed to improve either BUN on days 1–4 or reduce peak serum creatinine on day 1 in postischemic mice that lack Hsp70 (Figure 2d). GGA significantly increased renal cortical Hsp70 expression (Figure 2e) by 196±14% and 104±14% in Hsp70+/+ and Hsp70+/− animals, respectively (P<0.05; n = 6), but failed to increase its expression in Hsp70−/− mice (P>0.05; n = 4).
Effect of Hsp70 and GGA on renal histology after ischemia
No significant morphological differences between Hsp70+/+ and Hsp70−/− mice were detected in renal cortical tissue sections at baseline (Figure 3a, panels A vs B). After 25 min of RPO and 24 h of reperfusion, however, less tubular injury was observed in the renal cortex of Hsp70 +/+ vs Hsp70−/− mice (Figure 3a, panels C vs D). Compared with Hsp70+/+ mice, Hsp70−/− animals exhibited extensive cortical damage with tubular dilation, epithelial cell detachment, and mild tubulointerstitial infiltration by inflammatory cells. Most surviving tubular epithelial Hsp70−/− cells evidenced cytoplasmic swelling. Although evidence of renal epithelial cell necrosis was lacking in all tissue sections, many Hsp70−/− tubular cells showed nuclear chromatin condensation indicative of apoptosis. In the medulla, the tubular lumen was filled with hyaline material known to result from the loss of tubular cells from more proximal nephron segments. Before ischemia, virtually no difference in histological score was observed between Hsp70+/+ and Hsp70−/− mice (data not shown). However, after ischemia, the mean renal injury score was 2.8 in Hsp70−/− mice but only 1.3 in Hsp70+/+ mice (P<0.01 between groups; Table 1). GGA did not alter baseline tubular morphology in either Hsp70+/+ or Hsp70−/− mice subjected to sham ischemia (Figure 3b, panels A vs B). In Hsp70+/+ mice, brief RPO failed to cause marked tubular injury in the presence or absence of GGA (Figure 3b, panels A vs C). In contrast, 25 min of ischemia caused significant tubular injury in Hsp70−/− mice with or without GGA (Figure 3a panel D vs panel D of Figure 3b, and Table 1). To confirm the efficacy of GGA, Hsp70+/+ mice were subjected to 40 min of RPO, an insult sufficient to produce marked tubular injury and renal dysfunction in these animals. Compared with vehicle alone, pretreatment with GGA effectively prevented tubular injury (Figure 3c, panel A vs B).
Figure 3 |. Effect of pretreatment with geranylgeranylacetone (GGA) on ischemic renal injury.

(a) Representative tissue sections from Hsp70+/+ and Hsp70−/− mice after sham renal pedicle occlusion (RPO) vs 25 min of bilateral RPO followed by 24 h of reperfusion; original magnification × 400. (b) Typical injury observed in Hsp70+/+ and Hsp70−/− mice after 25 min of RPO with 24 h of reperfusion with and without GGA pretreatment. (c) Effect of GGA pretreatment on renal histology in Hsp70+/+ mice, 24 h after 40 min of RPO; GGA, 400 mg/kg, was administered twice at 18 and 2 h before ischemia; tissues were stained with hematoxylin and eosin; original magnification ×400.
Table 1 |.
Tubular injury score
| Postischemic injury | Hsp70+/+ | Hsp70−/− | Hsp70−/− plus GGA |
|---|---|---|---|
| Tubular dilation | 1 + | 2–3+ | 2–3+ |
| Vacuolar changes | 0–1+ | 2+ | 2 |
| Brush border loss | 2+ | 3+ | 3+ |
| Cell detachment | 0–1+ | 2+ | 2+ |
| Nuclear condensation | 0 | 2 | 2+ |
| Intratubular casts | 0 | 3+ | 3+ |
| Interstitial edema | 1+ | 2+ | 2+ |
| Leukocyte infiltration | 0–1+ | 2+ | 2+ |
| Capillary edema | 0 | 2+ | 2+ |
Abbreviations: GGA, geranylgeranylacetone; Hsp70, heat shock protein 70. Semiquantitative grading of whole kidney cross-sections after hematoxylin and eosin (H&E) staining. Histological injury score did not differ between any of the above groups before 25 min of ischemia (data not shown). More prolonged ischemia was required to assess the efficacy of GGA on tubular injury in Hsp70+/+ mice (see Figures 2b and 7a).
Antiapoptotic role of Hsp70
The effect of renal Hsp70 expression on apoptosis, a potential cause of ischemia-induced tubular injury and renal dysfunction, was assessed. At baseline, the content of active Bax (a primary cause of renal epithelial cell mitochondrial membrane injury and apoptosis) was similar in the renal cortical homogenates harvested from Hsp70+/+, Hsp70+/−, and Hsp70−/− mice (Figure 4a). Bax activation during ischemia and reperfusion, characterized by a conformational change in the amino-terminus, occurred to the greatest degree in Hsp70−/− mice and least in Hsp70+/+ mice. In contrast, total Bax did not change under any experimental condition in these three groups. Loss of immunoreactive Bcl2 was most remarkable in the renal cortices of Hsp70 +/− and Hsp70−/− mice during perfusion. Densitometric analysis confirmed a significant, stepwise increase in the content of active Bax and a parallel decrease in total Bcl2 in Hsp70−/− and Hsp70+/− vs Hsp70+/+ mice (P<0.01 for Hsp70−/− vs Hsp70+/+ mice; P<0.05 for Hsp70+/− vs Hsp70−/− mice). As a result of these reciprocal changes in active Bax and total Bcl2, a marked increased in the Bax/Bcl2 ratio was observed (Figure 4b). This stepwise increase in the Bax/Bcl2 ratio in Hsp70+/+, Hsp70+/−, and Hsp70−/− mice was associated with a parallel increase in caspase 3 activation in the renal cortex (Figure 4c). Compared with Hsp70+/+mice, active caspase 3 increased by 548±58% and 1597±67% in the renal cortex of Hsp70+/− and Hsp70−/− animals, respectively (P<0.05 vs Hsp70+/+; n = 3).
Figure 4 |. Apoptotic markers after renal ischemia.

(a) Heat shock protein 70 (Hsp70), active and total Bax, and B-cell lymphoma 2 (Bcl2) content in renal cortical homogenates of Hsp70+/+, Hsp70+/−, and Hsp70−/− mice at baseline, immediately after 25 min of bilateral renal pedicle occlusion (RPO), and after 60 min of reperfusion; β-actin loading control (bottom panel). (b) Densitometric analysis of active Bax (6A7), Bcl2, and the Bax/Bcl2 ratio from at least four separate experiments; mean active Bax and Bcl2 values, obtained after 60 min of reperfusion, were used to calculate the Bax/Bcl2 ratio in each group; *P<0.05 vs Hsp70−/−; n = 4. (c) Active (‘cleaved’) caspase 3 content in renal cortical homogenates after 60 min of reperfusion; the immunoblot shown is representative of three separate studies; densitometric analysis of caspase 3 data appears in Results.
To confirm that Hsp70 per se inhibits Bax and promotes survival, active Bax was colocalized with a mitochondrial marker before and after ATP depletion in cultured primary proximal tubules cells harvested from Hsp70−/− mice in the presence and absence of exogenous Hsp70. At baseline, virtually no active Bax was detected in cells infected with lentivirus containing either empty vector or Hsp70 (Figure 5a, upper panels). After stress, however, active Bax colocalized with a mitochondrial marker in Hsp70−/− cells (Figure 5a, bottom panels). In Hsp70−/− cells, lentiviral-induced expression of Hsp70 to a level similar to that observed in Hsp70+/+ cells markedly inhibited mitochondrial Bax accumulation and rescued the cells from death caused by ATP depletion (P<0.01 vs empty vector; Figure 5b). Neither Hsp70 expression nor infection with empty lentivirus altered baseline cell survival (data not shown).
Figure 5 |. Effect of heat shock protein 70 (Hsp70) on mitochondrial Bax translocation and survival in primary renal epithelial cells harvested from Hsp70−/− mice.
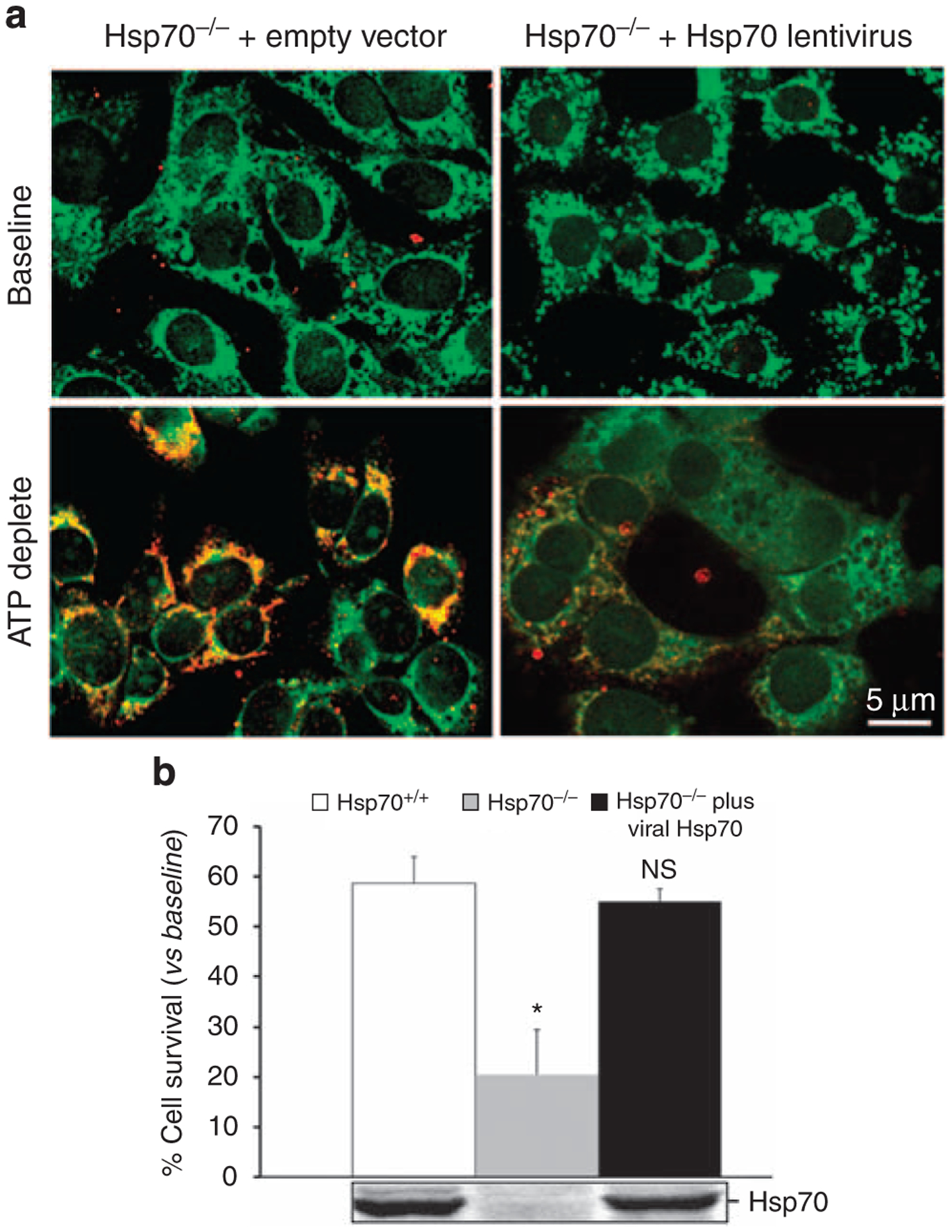
(a) Localization of active Bax (that is, 6A7 epitope exposed; shown in red) and mitochondria (using MitoTracker Green FM) in control (‘empty vector’) and Hsp70-expressing (‘Hsp70 lentivirus’) proximal tubule cells in primary culture at baseline and 6 h following 1 h of ATP depletion; colocalization of the two probes appears as an orange–yellow color; virtually no active Bax was detected at baseline; results are representative of six separate studies. (b) Effect of Hsp70 expression on survival after 1 h of ATP depletion and 6 h of recovery in primary proximal tubule cells harvested from Hsp70+/+ and Hsp70−/− mice in the presence of lentivirus virus containing either empty vector or human Hsp70 (Hsp70). *P<0.05; NS, not significant. Data are mean±s.e; n = 7; immunoblot analysis shows Hsp70 content in cell lysates.
Effect of Hsp70 on Akt and GSK3β: Bax regulators
As Akt and GSK3β regulate conformational Bax activation, the effect of Hsp70 expression on the activity of these two kinases was examined after ischemia in vivo. Basal-level Akt activity (as measured by p-ser473 Akt content) was similar in Hsp70+/+ and Hsp70−/− renal cortical homogenates (Figure 6a). Compared with Hsp70−/− mice, Hsp70+/+ cortical homogenates exhibited less Akt inactivation and more rapid kinase reactivation (that is, greater p-ser473 Akt content) during recovery from ischemia. Similarly, active GSK3β (that is, p-ser9 GSK3β content) was lower in Hsp70+/+ vs Hsp70−/− homogenates during and after stress. Compared with Hsp70−/− mice, Akt activity was 10% higher during ischemia and nearly 75% higher during reperfusion in Hsp70+/+ cortical homogenates (P<0.05 for both time points). GSK3β activity was 36% lower during ischemia and 60% lower during recovery in Hsp70+/+ cortical homogenates (P<0.05; for both time points vs Hsp70−/−; Figure 6b). As we recently reported GSK3β site-specific Bax phosphorylation after ATP depletion,42 only Akt-mediated phosphorylation of Bax ser184 was examined in a cell-free assay using a synthetic Bax peptide substrate (Figure 6c). Compared with ATP-replete cells, incorporation of 32[P] into the ser184 residue was markedly reduced in immunoprecipitates harvested from cultured primary proximal tubules cell cells subjected to ATP depletion (P<0.05). These data confirm that ATP depletion inactivates Akt and reduces Bax ser184 phosphorylation, thereby promoting Bax activation.
Figure 6 |. Effect of heat shock protein 70 (Hsp70) expression on Akt-glycogen synthase kinase 3β (GSK3β) activity after renal ischemia and effect of stress on Bax serine phosphorylation.

(a) Steady-state content of active (p-ser473) and total Akt, inactive (ser9) and total GSK3β, and β-actin in renal cortical homogenates obtained from Hsp70+/+, Hsp70+/−, and Hsp70−/− mice at baseline, immediately after 25 min of bilateral renal pedicle occlusion (RPO), or after 30, 60, or 120 min of reperfusion. (b) Densitometric assessment of active Akt and inactive GSK3β at 30 min after bilateral RPO; *P<0.05 vs sham control at baseline; n = 6 separate experiments. (c) Effect of ATP depletion on 32[P] incorporation into a synthetic Bax peptide containing the Akt-specific phosphorylation site (ser184); *P<0.05 vs baseline; n = 3.
Treatment of ischemic AKI with GGA
As Hsp70 induction by GGA before ischemia ameliorated tubular injury and organ failure, we hypothesized that GGA effectively treats ischemic AKI. To test this hypothesis, the severity of ischemia was reduced to 35 min and GGA was administered twice to Hsp70+/+ mice only after ischemia. Compared with vehicle alone, GGA treatment resulted in lower BUN levels, more rapid renal recovery, and increased renal cortical Hsp70 content postischemia (Figure 7a). GGA treatment also preserved tubular morphology after injury (Figure 7b) and improved animal survival. Of eight vehicle-treated Hsp70 +/+ animals, four (50%) died within 72-h postischemia. In contrast, none of the GGA-treated Hsp70+/+ animals died.
Figure 7 |. Effect of postischemic geranylgeranylacetone (GGA) treatment on renal function and histology after renal pedicle occlusion (RPO).
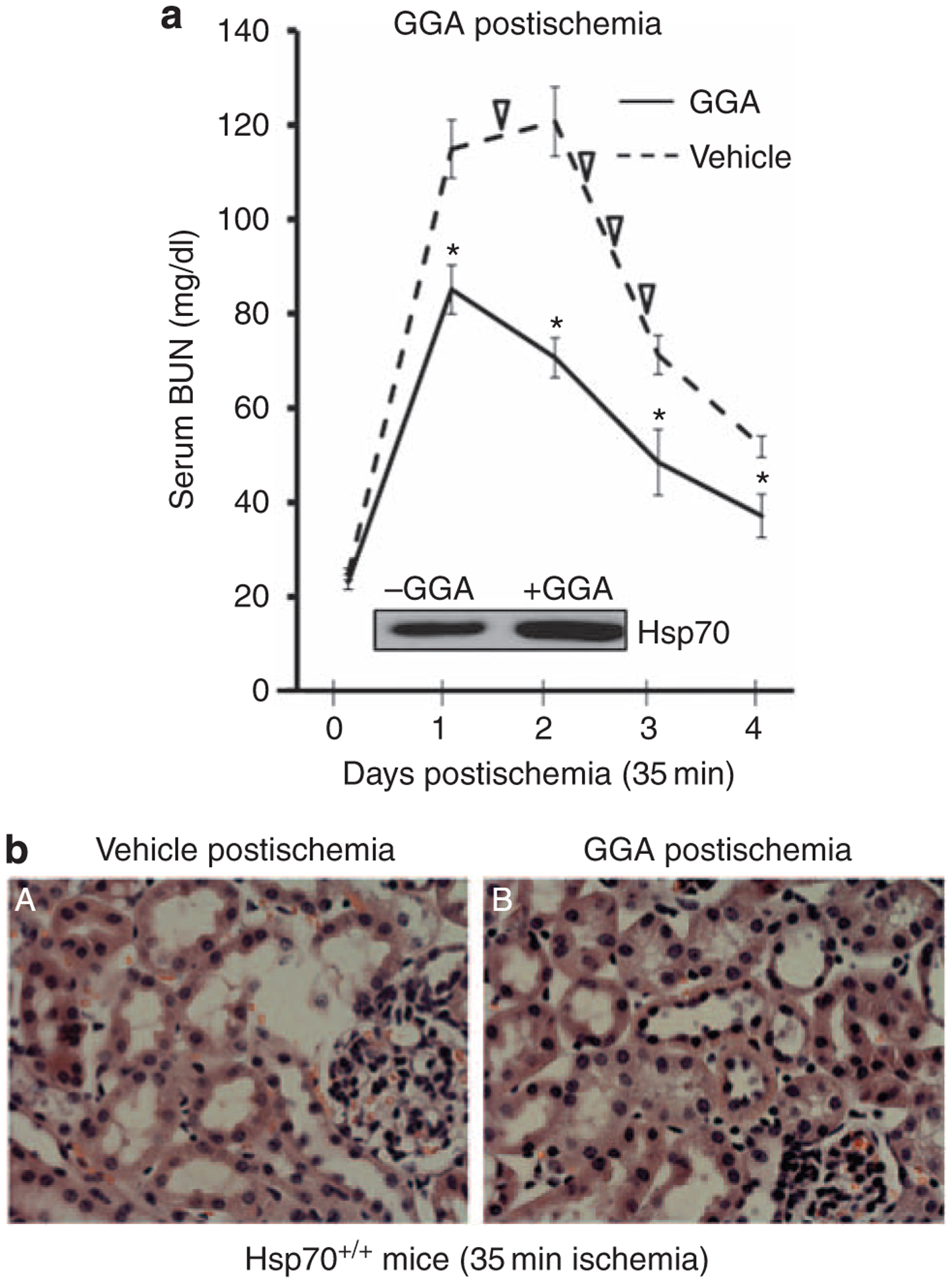
(a) Serial blood urea nitrogen (BUN) at baseline and on days 1–4 in Hsp70+/+ mice subjected to 35 min of bilateral RPO. Mice were treated with GGA (800 mg/kg intraperitoneal (i.p.) was administered immediately after 35 min of bilateral RPO and again after 18 h of recovery) vs vehicle alone; ∇ indicates animal death; inset shows the effect of GGA postischemia on heat shock protein 70 (Hsp70) content; *P<0.05 for GGA treatment vs vehicle alone; n = 6. (b) Representative renal histology in the presence of vehicle vs GGA; tissue sections stained with hematoxylin and eosin; original magnification ×400.
DISCUSSION
In this study, both endogenous and induced expressions of Hsp70 in the murine kidney inhibit apoptosis, an important contributor to ischemic AKI. How does Hsp70 reduce mitochondrial membrane injury and apoptosis in renal epithelial cells? As it does not interact with Bax,30,44 we hypothesized that Hsp70 mediates events upstream of conformational Bax activation (characterized by exposure of the 6A7 epitope, Hetz et al.45). In our study, renal ischemia caused a transient, 80% decrease in Akt activity (that is, reduced ser473 Akt content; Figure 6b), and a 50% increase in GSK3β activation (that is, decreased ser9 GSK3β content; Figure 6b). We reported identical changes in Akt and GSK3β activity in renal epithelial cells after ATP depletion in vitro.15,42 Interestingly, Akt inactivation results in ser9-GSK3β de-phosphorylation and activation, biochemically linking these two kinases. During ischemic stress, loss of Akt activity precedes conformational Bax activation (Figure 4a), Bax translocation to mitochondria (Figure 5a), and activation of caspase 3 (caused by cytochrome c;46 Figure 4c), associated with decreased cell survival (Figure 5b), marked tubular injury (Figure 3), and loss of organ function (Figure 2a and b). In renal cell immunoprecipitates, Akt inactivation during stress results in serine184 de-phosphorylation of a Bax peptide (Figure 6c). Similarly, GSK3β activation during renal ischemia promoted ser163 phosphorylation of a Bax synthetic peptide, connecting GSK3β activation with Bax activation in renal cells.42 In that report, direct manipulation of GSK3β using constitutively active and GSK3β-specific RNA interference produced reciprocal changes in Bax activation and renal cell death after ATP depletion, whereas RNA interference-mediated Bax suppression prevented the toxic effects of the constitutively active GSK3β mutant during stress, confirming that Bax is a major target of activated GSK3β.42 In the present study, Hsp70−/− mice exhibited significantly greater GSK3β activation (as well as more prolonged Akt inactivation) during ischemia and reperfusion than wild-type mice (Figure 4a and b). Taken together, these results support the hypothesis that stress-induced changes in Akt and GSK3β alter kinase-specific Bax serine phosphorylation, resulting in conformational Bax activation, Bax translocation to mitochondria, and cell death.
By altering the activation of both Akt and GSK3β, we propose that Hsp70 regulates key BCL2 proteins, apoptosis, and renal cell injury during ischemic AKI. In fact, a stepwise decrease in renal Hsp70 expression resulted in a stepwise increase in both Bax activation and Bcl2 loss in cortical homogenates (Figure 4b). Activation of Bax coupled with loss of Bcl2 markedly increased the active Bax/Bcl2 ratio after renal ischemia (Figure 4b) by 28-fold in Hsp70−/− mice, 8-fold in Hsp70 +/− mice, and by only 2.5-fold in Hsp70+/+ mice. This stepwise increase in the active Bax/Bcl2 ratio was associated with a parallel increase in caspase 3 activation (Figure 4c). Interestingly, loss of Bcl2, described as the ‘nail in the apoptotic coffin,’ results from its degradation by caspase 3, resulting in the conversion of Bcl2 from an anti-to a pro-apoptotic protein.47 Our in vitro data substantiate the role of Hsp70 as an antiapoptotic mediator. As observed after ischemia in vivo, proximal tubule cells isolated from Hsp70−/− mice are more sensitive to ATP depletion-induced cell death (Figure 5b). This increased sensitivity to death is completely reversed by repletion of Hsp70 to the level detected in wild-type mice (Figure 5b), confirming that Hsp70 is critical for renal epithelial cell survival after stress.
Although Bax activation persists in the postischemic period in Hsp70+/− and Hsp70−/− mice (Figure 4a) and is a potential Hsp70 target, other cytoprotective mechanisms may be important. Hsp70 also increases cyclin D1, a key regulator of cell proliferation,48 an important determinant of renal recovery.49,50 Alternatively, by inhibiting caspase 3 activation,51 Hsp70 could reduce the inflammation that impairs postischemic organ recovery. This hypothesis is supported by the observation that greater leukocyte infiltration (Table 1), a hallmark of tissue inflammation, is observed in Hsp70−/− mice.
Although a role for inducible stress proteins in regulating survival kinases has been reported, the mechanism by which Hsp70 regulates Akt, GSK3β, and Bax activation is unknown. Hsp27, a potent antiapoptotic protein with similar cytoprotective effects as Hsp70, increases the activity of phosphatidylinositol 3-phosphate kinase, the regulatory enzyme upstream of Akt, GSK3β,52 and Bax activation. As Hsp70 indirectly inhibits JNK (c-jun-N-terminal kinase) by preventing a JNK-specific phosphatase from inactivation during stress,53 it is also possible that Hsp70 acts on the phosphatases that regulate Akt and GSK3β. At present, it is difficult to study the effect of Hsp70 on Bax serine phosphorylation because of the absence of phospho-specific antibodies and the presence of multiple serine sites with opposing effects on Bax activity.
Resistance to ischemic AKI varied in a ‘dose-dependent’ manner that positively correlated with renal Hsp70 expression. In Hsp70−/− mice, 21 min of ischemia time was sufficient to produce moderate, reversible AKI (Figure 2d). For Hsp70 heterozygotes, the required clamp time increased to 25 min (Figure 2c). In contrast, Hsp70 +/+ mice needed 35–40 min of clamp time to cause AKI (Figures 2b and 7a). GGA, a nontoxic drug currently used to treat human peptic ulcer disease, induced Hsp70, protected tubular structure, and preserved organ function in both wild-type and Hsp70 heterozygote mice (Figure 2b and c). However, GGA failed to protect Hsp70 knockout mice, indicating that this agent requires Hsp70 to afford renoprotection (Figure 2d). Remarkably, GGA reduced the severity of ischemic AKI by 40–60% even when given during reperfusion (Figure 7a).
Finally, our study supports a role for apoptosis in the decline in renal function after ischemia. In contrast, morphological evidence of cell necrosis was not observed in any of the kidney tissue sections after ischemia. Although ischemic AKI is clinically synonymous with acute tubular necrosis ‘ATN,’ this simplistic ‘structure-function paradigm’ has been challenged.54–56 In human renal biopsies, the severity of ischemic ‘ATN’ fails to predict renal function, the need for renal replacement therapy, or the likelihood of organ recovery either in native kidneys or transplant allografts.54 In contrast, ischemia-induced renal dysfunction has been shown to correlate with the degree of apoptosis, at least in rodents.55 Although apoptosis was first reported after renal ischemia in humans nearly 20 years ago,57 its contribution to renal failure has been difficult to quantify.58 This is partly because of the stochastic nature of apoptosis, the evanescent nature of apoptotic cells, and the insensitivity of current assays.58 Despite these limitations, recent evidence strongly suggests that BCL2 protein-mediated mitochondrial membrane injury, caspase activation, and apoptosis contribute to renal epithelial cell death and organ failure.15,16,21,33,42,59–61 The present observation that Hsp70, an established antiapoptotic protein, preserves organ function adds to mounting evidence that apoptosis contributes to ischemic AKI. To date, many treatment strategies have failed in human trials despite their initial success in animals, partly because of their untoward side effects. This study opens the door to potential treatment of ischemic AKI by showing that a nontoxic agent already in human use induces Hsp70 in mammalian cells, limits apoptosis, and preserves both organ structure and function even after the insult has occurred.
MATERIALS AND METHODS
Reagents
All chemical and reagents were obtained from Sigma-Aldrich (St Louis, MO) unless otherwise indicated.
Animals
Congenic wild-type (Charles River Laboratory, Wilmington, MA) and Hsp70−/− mice41 were bred to produce Hsp70−/−, Hsp70+/−, and Hsp70+/+ mice at the Boston University animal core facility and were maintained under specific pathogen-free conditions. In mice, disruption of both Hsp70.1 and Hsp70.3 genes in embryonic stem cells is required to completely prevent Hsp70 expression.62 Speed congenics was utilized to transfer the deletion mutation into the C57BL/6J mouse strain (Washington University Mouse Genetics Core, St Louis, MO). Mice were tested through N5 and had 97.4% conversion. All procedures were performed under conditions in accordance with the guidelines set by the National Institutes of Health, the Institutional Animal Care and Use Committee of Boston Medical Center.
GGA administration
GGA (Wako Industries, cat no. 20–1573, Osaka, Japan) was emulsified with 0.5% gum arabic and 0.008% tocopherol in distilled water and was administered by gavage using a disposable, flexible feeding tube. As control, the same components was prepared and emulsified but without GGA (vehicle). Experimental mice received GGA (400 mg/kg p.o.), 18 and 2 h before surgery, whereas control received vehicle alone. To assess GGA as a potential treatment for AKI, GGA was administered twice, immediately after 35 min of renal ischemia and again 18 h later (800 mg/kg, intraperitoneal per dose). Control animals were given an identical volume of vehicle only.
Kidney ischemia–reperfusion
Six-week-old mice weighing 20–22 g were used to perform ischemia–reperfusion studies under standardized conditions as recently described.42 Mice were anesthetized with intraperitoneal injection of tribromoethanol (250 mg/kg body weight). Mice were placed onto a sterile, warmed operating table and kidneys were exposed by a midline incision. To control the severity of injury, both renal arteries and veins were occluded for 25, 35, or 40 min with nontraumatic microaneurysm clamps. After clamp removal, restoration of blood flow was visually confirmed for 1 min. The abdomen was closed using interrupted sutures. Sham-operated mice received identical surgical procedures except that the micro-vascular clamps were not applied to the renal artery. For signal transduction and histological studies, mice were killed at 0, 0.25, 1, and 24 h after ischemia. Renal function studies (see below) were serially performed for up to 4 days after injury, using blood drawn from the lateral tail vein.
BUN and serum creatinine
Upon completion of the in vivo experiments, a whole-blood sample was harvested either from the tail (BUN) or aorta (creatinine). Serum BUN and creatinine levels were measured using QuantiChrom BUN and or Creatinine assay kit (BioAssay System, Hayward, CA)
Histology
Kidney tissue was fixed with 10% formaldehyde, embedded in paraffin, and cut into 8 μm sections before staining with hematoxylin and eosin reagent. Histological scores were calculated by a single blinded observer (RB) from 50 randomly selected cortical tubules with visible basement membranes on cross-section using a 0–3 semiquantitative score (with ‘0’ representing normal structure and ‘3’ representing severe injury) as previously reported.42
Kidney tissue protein extraction
Renal cortical tissue was added into prechilled RIPA buffer (Boston BioProducts, Boston, MA) containing a 1% protease inhibitor cocktail designed for mammalian tissue extracts and then homogenized. Samples were incubated for 30 min at 4 °C and centrifuged at 16,000 g at 4 °C for 15 min to pellet the tissue debris. The supernatant was stored at −70 °C. Protein concentrations were determined by a colorimetric protein assay (Bio-Rad, Hercules, CA) using protein standards.
Primary cell culture
Four-week-old mice were killed by overdose with inhaled CO2 using established protocols. Immediately after killing, the kidneys were removed and maintained at 4 °C (on ice). The cortex was collected, minced, and then digested in a solution with collagenase for 60 min at 37 °C. The collagenase was neutralized with calf serum and the digested cortex was washed twice with buffered F12 medium. Cells were harvested and characterized as proximal tubule in origin as described previously by our laboratory63,64 and were maintained in a 50:50 (vol/vol) mixture of Dulbecco’s modied Eagle’s medium (DMEM)/Ham’s F12 medium containing insulin (5 mg/l), hydro-cortisone (50 nM), and apotransferrin (500 mg/l) and were cultured at 37 °C for 5–7 days in a 5% CO2 incubator.
Lentiviral-mediated Hsp70 expression
Lentivirus containing full-length human Hsp70 cDNA or empty lentivirus was generated and renal epithelial cells were infected as recently described by our laboratory.42 Cells were exposed to virus for 16 h at a multiplicity of infection of 6. Hsp70 expression was confirmed in cell lysates by immunoblot analysis 24 h after removal of the virus.
Cell stress
ATP depletion, an established model of renal ischemia,31,46,65–67 was induced by exposing cells to glucose-free DMEM that contained (in mmol/l): adenosine, 1; allopurinol, 0.2; and rotenone, 10 (a mitochondrial complex I inhibitor), at 37 °C in the presence of 5% CO2 as previously reported.67 Cells were rinsed three times with glucose-free DMEM followed by DMEM containing the above metabolic inhibitors for 60–90 min at 37 °C with 5% CO2. Cell recovery was initiated by replacing the medium with DMEM containing (in mmol/l): adenosine, 1; allopurinol, 0.2; and heptanoic acid, 1 (to bypass complex I inhibition). These maneuvers resulted in a 90% reduction in cell ATP content during the period of metabolic stress and a rapid increase in ATP during recovery.67
Immunoblot analysis
Aliquots (30 g) of kidney homogenates were separated on 10% polyacrylamide gel and transferred to a polyvinylidene fluoride membrane. After blocking with dry milk, membranes were incubated with antibodies directed against Hsp70 (C92F3A-5; Stressgen, Ann Arbor, MI), active Bax (YTH-6A7; Trevingen, Gaithersburg, MD) total Bax (YTH-5B7; Trevingen), Bcl2 (Cell Signaling, cat no. 2876), Akt (cat no. 9272), p-ser473 Akt (cat no. 4085), GSK3β (cat no. 9332), p-ser9 GSK3β (cat no. 9336), or active caspase 3 (cat no. 9661), 5% bovine serum albumin in Tris-buffered saline overnight at 4 °C. Membranes were washed, incubated with secondary antibody conjugated to horseradish peroxidase, and then rewashed. Positive bands were detected by chemiluminescence technology using the G:Box gel documentation and analysis system (Fuji 4000, Tokyo, Japan). As a loading control, each membrane was probed with an anti-β-actin antibody.
Akt-mediated Bax serine184 phosphorylation
Akt-mediated phosphorylation of a synthetic Bax peptide was performed as previously described for GSK3β.42 In brief, Akt was immunoprecipitated from 400 μg cell protein by constant mixing with 5 μg of an anti-Akt monoclonal antibody and protein A-sepharose for 2 h at 4 °C. The resulting immunoprecipitates were added to a kinase assay wash buffer, resuspended in kinase assay mixture (pH 7.2) to which a cAMP-dependent protein kinase inhibitor peptide (IP20), 100 Ci [γ−32P] ATP (PerkinElmer Life Sci., Boston, MA) and a synthetic Bax-specific peptide (FVAGVLTASL-TIWKKMG) that flanks the serine184 (shown in bold) were added. The assay was terminated after 30 min of incubation at 30 °C by spotting onto P81 ion-exchange paper. The paper was washed four times in 0.6% phosphoric acid and bound radioactivity was quantified by scintillation counting. A commercially available Akt phosphorylation substrate (Upstate Biotech, Lake Placid, NY) was used to optimize the reaction conditions.
Cell viability
In this injury model, ATP depletion causes apoptotic cell death without necrosis,31,33,46,65,68 permitting the use of cell survival measurements to estimate apoptosis in this model. Cell viability was assayed using a modified colorimetric technique that is based on the ability of live cells to convert 3-(4,5 dimethylthiazol)-2,5-diphenyl tetrazolium bromide (MTT), a tetrazolium compound, into purple formazan crystals as previously reported.69 The number of surviving cells is expressed as a percentage of viable control cells detected at baseline.
Statistical analysis
Data were analyzed using the Statview statistics program (Microsoft, Redmond, WA). Significant differences between groups were determined by either paired (Student’s t-test) or unpaired observations (analysis of variance using Scheffe’s test), with values of P<0.05 considered significant.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health research grants (RO-1) DK-53387 (SCB), DK-52898 (JHS), AG025286 (CRH), a James A. Scherbenske Award from the American Society of Nephrology (SCB), and an ARC award from the Evans Center for Interdisciplinary Research at Boston University. None of the authors has any significant primary financial arrangements with commercial companies that produce or sell products that are the subject of the studies contained in this manuscript, or with competitors of such companies.
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.Kabakov AE, Budagova KR, Latchman DS et al. Stressful preconditioning and HSP70 overexpression attenuate proteotoxicity of cellular ATP depletion. Am J Physiol Cell Physiol 2002; 283: C521–C534. [DOI] [PubMed] [Google Scholar]
- 2.Trost SU, Omens JH, Karlon WJ et al. Protection against myocardial dysfunction after a brief ischemic period in transgenic mice expressing inducible heat shock protein 70. J Clin Invest 1998; 101: 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uchida S, Fujiki M, Nagai Y et al. Geranylgeranylacetone, a noninvasive heat shock protein inducer, induces protein kinase C and leads to neuroprotection against cerebral infarction in rats. Neurosci Lett 2006; 396: 220–224. [DOI] [PubMed] [Google Scholar]
- 4.Fudaba Y, Tashiro H, Ohdan H et al. Efficacy of HSP72 induction in rat liver by orally administered geranylgeranylacetone. Transpl Int 2000; 13: S278–S281. [DOI] [PubMed] [Google Scholar]
- 5.Kelly S, Bieneman A, Horsburgh K et al. Targeting expression of hsp70i to discrete neuronal populations using the Lmo-1 promoter: assessment of the neuroprotective effects of hsp70i in vivo and in vitro. J Cereb Blood Flow Metab 2001; 21: 972–981. [DOI] [PubMed] [Google Scholar]
- 6.Marber MS, Mestril R, Chi SH et al. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest 1995; 95: 1446–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsumori Y, Hong SM, Aoyama K et al. Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab 2005; 25: 899–910. [DOI] [PubMed] [Google Scholar]
- 8.Belke DD, Gloss B, Hollander JM et al. In vivo gene delivery of HSP70i by adenovirus and adeno-associated virus preserves contractile function in mouse heart following ischemia-reperfusion. Am J Physiol Heart Circ Physiol 2006; 291: H2905–H2910. [DOI] [PubMed] [Google Scholar]
- 9.Basile DP, Donohoe D, Cao X et al. Resistance to ischemic acute renal failure in the Brown Norway rat: a new model to study cytoprotection. Kidney Int 2004; 65: 2201–2211. [DOI] [PubMed] [Google Scholar]
- 10.Lu A, Ran R, Parmentier-Batteur S et al. Geldanamycin induces heat shock proteins in brain and protects against focal cerebral ischemia. J Neurochem 2002; 81: 355–364. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki S, Maruyama S, Sato W et al. Geranylgeranylacetone ameliorates ischemic acute renal failure via induction of Hsp70. Kidney Int 2005; 67: 2210–2220. [DOI] [PubMed] [Google Scholar]
- 12.Chen CC, Liu ZM, Wang HH et al. Effects of ulinastatin on renal ischemia-reperfusion injury in rats. Acta Pharmacol Sin 2004; 25: 1334–1340. [PubMed] [Google Scholar]
- 13.Jayakumar J, Suzuki K, Sammut IA et al. Heat shock protein 70 gene transfection protects mitochondrial and ventricular function against ischemia-reperfusion injury. Circulation 2001; 104: I303–I307. [DOI] [PubMed] [Google Scholar]
- 14.Gupta S, Li S, Abedin MJ et al. Effect of Notch activation on the regenerative response to acute renal failure. Am J Physiol Renal Physiol 2009; 298: F209–F215. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Havasi A, Gall JM et al. Beta-catenin promotes survival of renal epithelial cells by inhibiting Bax. J Am Soc Nephrol 2009; 20: 1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brooks C, Wei Q, Cho SG et al. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 2009; 119: 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolfs TG, de Vries B, Walter SJ et al. Apoptotic cell death is initiated during normothermic ischemia in human kidneys. Am J Transplant 2005; 5: 68–75. [DOI] [PubMed] [Google Scholar]
- 18.Chiang-Ting C, Tzu-Ching C, Ching-Yi T et al. Adenovirus-mediated bcl-2 gene transfer inhibits renal ischemia/reperfusion induced tubular oxidative stress and apoptosis. Am J Transplant 2005; 5: 1194–1203. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Wei Q, Wang CY et al. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem 2004; 279: 19948–19954. [DOI] [PubMed] [Google Scholar]
- 20.Kelly KJ, Sutton TA, Weathered N et al. Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. Am J Physiol Renal Physiol 2004; 287: F760–F766. [DOI] [PubMed] [Google Scholar]
- 21.Castaneda MP, Swiatecka-Urban A, Mitsnefes MM et al. Activation of mitochondrial apoptotic pathways in human renal allografts after ischemiareperfusion injury. Transplantation 2003; 76: 50–54. [DOI] [PubMed] [Google Scholar]
- 22.Gupta S, Kass GE, Szegezdi E et al. The mitochondrial death pathway: a promising therapeutic target in Diseases. J Cell Mol Med 2009; 13: 1004–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galluzzi L, Kroemer G. Mitochondrial apoptosis without VDAC. Nat Cell Biol 2007; 9: 487–489. [DOI] [PubMed] [Google Scholar]
- 24.Korsmeyer SJ, Shutter JR, Veis DJ et al. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol 1993; 4: 327–332. [PubMed] [Google Scholar]
- 25.Scorrano L, Oakes SA, Opferman JT et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003; 300: 135–139. [DOI] [PubMed] [Google Scholar]
- 26.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647–656. [DOI] [PubMed] [Google Scholar]
- 27.Er E, Oliver L, Cartron PF et al. Mitochondria as the target of the pro-apoptotic protein Bax. Biochim Biophys Acta 2006; 1757: 1301–1311. [DOI] [PubMed] [Google Scholar]
- 28.Sorenson CM. Bcl-2 family members and disease. Biochim Biophys Acta 2004; 1644: 169–177. [DOI] [PubMed] [Google Scholar]
- 29.Sharpe JC, Arnoult D, Youle RJ. Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta 2004; 1644: 107–113. [DOI] [PubMed] [Google Scholar]
- 30.Stankiewicz AR, Lachapelle G, Foo CP et al. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem 2005; 280: 38729–38739. [DOI] [PubMed] [Google Scholar]
- 31.Ruchalski K, Mao H, Li Z et al. Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J Biol Chem 2006; 281: 7873–7880. [DOI] [PubMed] [Google Scholar]
- 32.Lin C, Yin Y, Long F et al. Tissue-specific requirements of beta-catenin in external genitalia development. Development 2008; 135: 2815–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Knowlton AA, Christensen TG et al. Prior heat stress inhibits apoptosis in adenosine triphosphate-depleted renal tubular cells. Kidney Int 1999; 55: 2224–2235. [DOI] [PubMed] [Google Scholar]
- 34.Yethon JA, Epand RF, Leber B et al. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 2003; 278: 48935–48941. [DOI] [PubMed] [Google Scholar]
- 35.Annis MG, Soucie EL, Dlugosz PJ et al. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J 2005; 24: 2096–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nechushtan A, Smith CL, Lamensdorf I et al. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol 2001; 153: 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valentijn AJ, Metcalfe AD, Kott J et al. Spatial and temporal changes in Bax subcellular localization during anoikis. J Cell Biol 2003; 162: 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardai SJ, Hildeman DA, Frankel SK et al. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 2004; 279: 21085–21095. [DOI] [PubMed] [Google Scholar]
- 39.Linseman DA, Butts BD, Precht TA et al. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 2004; 24: 9993–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.del Mar Martinez-Senac M, Corbalan-Garcia S, Gomez-Fernandez JC. Conformation of the C-terminal domain of the pro-apoptotic protein Bax and mutants and its interaction with membranes. Biochemistry 2001; 40: 9983–9992. [DOI] [PubMed] [Google Scholar]
- 41.Hunt CR, Dix DJ, Sharma GG et al. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol 2004; 24: 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Z, Havasi A, Gall J et al. GSK3beta promotes apoptosis after renal ischemic injury. J Am Soc Nephrol 2010; 21: 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nechushtan A, Smith CL, Hsu YT et al. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J 1999; 18: 2330–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruchalski K, Mao H, Li Z et al. Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J Biol Chem 2006; 281: 7873–7880. [DOI] [PubMed] [Google Scholar]
- 45.Hetz C, Vitte PA, Bombrun A et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem 2005; 280: 42960–42970. [DOI] [PubMed] [Google Scholar]
- 46.Li F, Mao HP, Ruchalski KL et al. Heat stress prevents mitochondrial injury in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol 2002; 283: C917–C926. [DOI] [PubMed] [Google Scholar]
- 47.Cheng EH, Kirsch DG, Clem RJ et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 1997; 278: 1966–1968. [DOI] [PubMed] [Google Scholar]
- 48.Roue G, Pichereau V, Lincet H et al. Cyclin D1 mediates resistance to apoptosis through upregulation of molecular chaperones and consequent redistribution of cell death regulators. Oncogene 2008; 27: 4909–4920. [DOI] [PubMed] [Google Scholar]
- 49.Lieberthal W, Fuhro R, Andry CC et al. Rapamycin impairs recovery from acute renal failure: role of cell-cycle arrest and apoptosis of tubular cells. Am J Physiol Renal Physiol 2001; 281: F693–F706. [DOI] [PubMed] [Google Scholar]
- 50.Fiaschi-Taesch NM, Santos S, Reddy V et al. Prevention of acute ischemic renal failure by targeted delivery of growth factors to the proximal tubule in transgenic mice: the efficacy of parathyroid hormone-related protein and hepatocyte growth factor. J Am Soc Nephrol 2004; 15: 112–125. [DOI] [PubMed] [Google Scholar]
- 51.Wang YH, Knowlton AA, Li FH et al. HSP 72 expression enhances survival in adenosine triphosphate-depleted renal epithelial cells. Cell Stress Chaperones 2002; 7: 84–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Havasi A, Li Z, Wang Z et al. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem 2008; 283: 12305–12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gabai VL, Meriin AB, Yaglom JA et al. Suppression of stress kinase JNK is involved in HSP72-mediated protection of myogenic cells from transient energy deprivation. HSP72 alleviates the stress-induced inhibition of JNK dephosphorylation. J Biol Chem 2000; 275: 38088–38094. [DOI] [PubMed] [Google Scholar]
- 54.Rosen S, Heyman SN. Difficulties in understanding human ‘acute tubular necrosis’: limited data and flawed animal models. Kidney Int 2001; 60: 1220–1224. [DOI] [PubMed] [Google Scholar]
- 55.Kelly KJ, Plotkin Z, Dagher PC. Guanosine supplementation reduces apoptosis and protects renal function in the setting of ischemic injury. J Clin Invest 2001; 108: 1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly KJ, Plotkin Z, Vulgamott SL et al. P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. J Am Soc Nephrol 2003; 14: 128–138. [DOI] [PubMed] [Google Scholar]
- 57.Schumer M, Colombel MC, Sawczuk IS et al. Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am J Pathol 1992; 140: 831–838. [PMC free article] [PubMed] [Google Scholar]
- 58.Bonegio R, Lieberthal W. Role of apoptosis in the pathogenesis of acute renal failure. Curr Opin Nephrol Hypertens 2002; 11: 301–308. [DOI] [PubMed] [Google Scholar]
- 59.Plotnikov EY, Kazachenko AV, Vyssokikh MY et al. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int 2007; 72: 1493–1502. [DOI] [PubMed] [Google Scholar]
- 60.Jani A, Ljubanovic D, Faubel S et al. Caspase inhibition prevents the increase in caspase-3, −2, −8 and −9 activity and apoptosis in the cold ischemic mouse kidney. Am J Transplant 2004; 4: 1246–1254. [DOI] [PubMed] [Google Scholar]
- 61.Ortiz A, Justo P, Sanz A et al. Targeting apoptosis in acute tubular injury. Biochem Pharmacol 2003; 66: 1589–1594. [DOI] [PubMed] [Google Scholar]
- 62.Hampton CR, Shimamoto A, Rothnie CL et al. HSP70.1 and −70.3 are required for late-phase protection induced by ischemic preconditioning of mouse hearts. Am J Physiol Heart Circ Physiol 2003; 285: H866–H874. [DOI] [PubMed] [Google Scholar]
- 63.Borkan SC, Wang YH, Lieberthal W et al. Heat stress ameliorates ATP depletion-induced sublethal injury in mouse proximal tubule cells. Am J Physiol 1997; 272: F347–F355. [DOI] [PubMed] [Google Scholar]
- 64.Lieberthal W, Menza SA, Levine JS. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am J Physiol 1998; 274: F315–F327. [DOI] [PubMed] [Google Scholar]
- 65.Ruchalski K, Mao H, Singh SK et al. HSP72 inhibits apoptosis-inducing factor release in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol 2003; 285: C1483–C1493. [DOI] [PubMed] [Google Scholar]
- 66.Wang YH, Borkan SC. Prior heat stress enhances survival of renal epithelial cells after ATP depletion. Am J Physiol 1996; 270: F1057–F1065. [DOI] [PubMed] [Google Scholar]
- 67.Doctor RB, Bacallao R, Mandel LJ. Method for recovering ATP content and mitochondrial function after chemical anoxia in renal cell cultures. Am J Physiol 1994; 266: C1803–C1811. [DOI] [PubMed] [Google Scholar]
- 68.Wang YH, Knowlton AA, Li FH et al. Hsp72 expression enhances survival in adenosine triphosphate-depleted renal epithelial cells. Cell Stress Chaperones 2002; 7: 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sinha D, Wang Z, Ruchalski KL et al. Lithium activates the Wnt and phosphatidylinositol 3-kinase Akt signaling pathways to promote cell survival in the absence of soluble survival factors. Am J Physiol Renal Physiol 2005; 288: F703–F713. [DOI] [PubMed] [Google Scholar]