

Abstract
The sulfosugar sulfoquinovose (SQ) is produced by essentially all photosynthetic organisms on Earth and is metabolized by bacteria through the process of sulfoglycolysis. The sulfoglycolytic Embden–Meyerhof–Parnas pathway metabolizes SQ to produce dihydroxyacetone phosphate and sulfolactaldehyde and is analogous to the classical Embden–Meyerhof–Parnas glycolysis pathway for the metabolism of glucose-6-phosphate, though the former only provides one C3 fragment to central metabolism, with excretion of the other C3 fragment as dihydroxypropanesulfonate. Here, we report a comprehensive structural and biochemical analysis of the three core steps of sulfoglycolysis catalyzed by SQ isomerase, sulfofructose (SF) kinase, and sulfofructose-1-phosphate (SFP) aldolase. Our data show that despite the superficial similarity of this pathway to glycolysis, the sulfoglycolytic enzymes are specific for SQ metabolites and are not catalytically active on related metabolites from glycolytic pathways. This observation is rationalized by three-dimensional structures of each enzyme, which reveal the presence of conserved sulfonate binding pockets. We show that SQ isomerase acts preferentially on the β-anomer of SQ and reversibly produces both SF and sulforhamnose (SR), a previously unknown sugar that acts as a derepressor for the transcriptional repressor CsqR that regulates SQ-utilization. We also demonstrate that SF kinase is a key regulatory enzyme for the pathway that experiences complex modulation by the metabolites SQ, SLA, AMP, ADP, ATP, F6P, FBP, PEP, DHAP, and citrate, and we show that SFP aldolase reversibly synthesizes SFP. This body of work provides fresh insights into the mechanism, specificity, and regulation of sulfoglycolysis and has important implications for understanding how this biochemistry interfaces with central metabolism in prokaryotes to process this major repository of biogeochemical sulfur.
Short abstract
Sulfoglycolysis is a pathway for metabolism of the sulfosugar sulfoquinovose. We illuminate how sulfoglycolytic enzymes bind and turnover their substrates, and identify a new transiently produced intermediate, sulforhamnose.
Introduction
Global sulfur cycling involves the interconversion of inorganic, gaseous, and organic forms of the element.1 While there are innumerable biosulfur species on Earth, just a handful comprise the majority of sulfur in the biosphere, namely, the amino acids cysteine and methionine, the osmolytes dimethylsulfoniopropionate (DMSP)2 and dimethylsulfoxonium propionate,3 and the sulfosugar sulfoquinovose (SQ).4 Global production of SQ is estimated to be 1010 tonnes per annum, comprising up to half of all biosulfur,5 and it is produced by essentially all photosynthetic organisms. SQ exists as the carbohydrate headgroup of the plant and cyanobacterial sulfolipid sulfoquinovosyl diacylglycerol (SQDG) within the photosynthetic membranes of photoautotrophs.4 SQDG is believed to primarily enter the environment through decomposition of photosynthetic tissues and herbivory, whereupon it becomes available to environmental and intestinal bacteria.6−10 The catabolism of SQ, a process termed sulfoglycolysis,6−8 is therefore of great relevance to understanding the biogeochemical sulfur cycle.
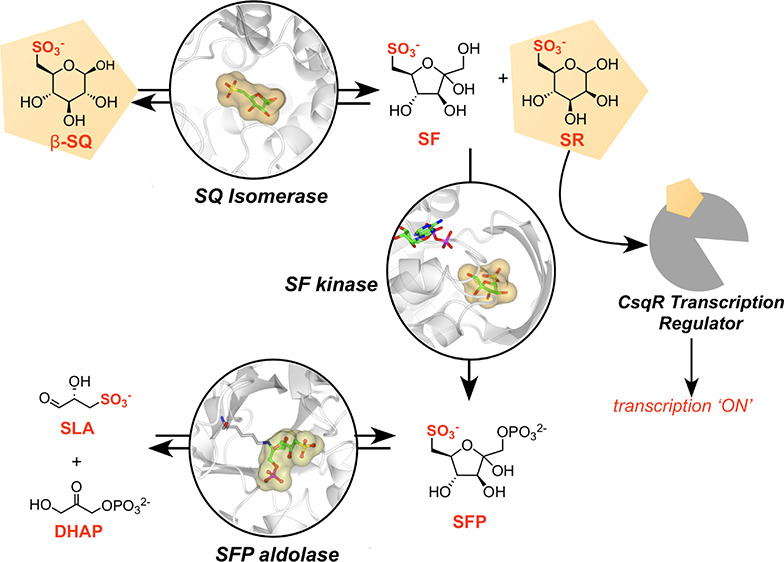
The first sulfoglycolytic pathway discovered comprises a variant of the classical Embden–Meyerhof–Parnas glycolysis pathway and is termed the sulfoglycolytic Embden–Meyerhof–Parnas (sulfo-EMP) pathway (Figure 1a).7 It was initially described in Escherichia coli and involves the importation of SQ and its glycosides (which undergo intracellular hydrolysis to liberate SQ), catabolic enzymes to produce dihydroxyacetone phosphate (DHAP) and sulfolactaldehyde (SLA), a reductase to convert SLA to dihydroxypropanesulfonate (DHPS), and a permease to export DHPS from the cell.7 The pathway is encoded by a 10-gene cluster (ompL and yihO-W, where yihW was renamed csqR) that encodes a transcriptional regulator (CsqR, formerly YihW);11 a sulfolipid porin (OmpL) and two transmembrane permeases (YihO, YihP); a sulfoquinovosidase for cleavage of SQ glycosides (YihQ);12,13 an SQ mutarotase (YihR);14 three core sulfoglycolytic enzymes: SQ-sulfofructose (SF) isomerase (YihS), SF kinase (YihV), and SF-1-phosphate (SFP) aldolase (YihT); and finally SLA reductase (YihU),15 which forms DHPS for export (Figure 1b). The three core sulfoglycolytic enzymes YihS, YihV, and YihT perform roles analogous to three enzymes involved in upper glycolysis: glucose-6-phosphate (G6P) isomerase, phosphofructose kinase (PFK), and fructose bisphosphate (FBP) aldolase, respectively. However, nothing is known about the specificity of these sulfoglycolytic enzymes for intermediates from sulfoglycolysis versus glycolysis. Metabolic flux through the glycolytic pathway is tightly regulated with the most important control exerted upon PFK, with allosteric activation and inhibition by assorted cellular metabolites such as adenosine monophosphate (AMP), adenosine diphosphate (ADP), adenosine triphosphate (ATP), fructose 6-phosphate (F6P), FBP, phosphoenolpyruvate (PEP), and citrate.16 It is unknown whether similar metabolites control flux through the sulfoglycolytic pathway.
Figure 1.

The Embden–Meyerhof–Parnas sulfoglycolysis pathway in E. coli. (a) Proposed biochemical pathway for metabolism of SQGro. Lipase cleavage of acyl groups in SQDG occurs in the environment. (b) Sulfo-EMP operon showing binding sites for the CsqR transcriptional repressor. SQDG, sulfoquinovosyl diacylglycerol; SQGro, α-sulfoquinovosyl glycerol; SQ, sulfoquinovose; SF, 6-deoxy-6-sulfofructose; SR, 6-deoxy-6-sulforhamnose; SFP, 6-deoxy-6-sulfofructose-1-phosphate; SLA, sulfolactaldehyde; DHPS, 2,3-dihydroxypropanesulfonate; DHAP, dihydroxyacetone phosphate; NADH, nicotinamide adenine dinucleotide, reduced form. Formation of SQGro is achieved by lipase action on sulfoquinovosyl diacylglycerol.
In prokaryotes, the gluconeogenesis pathway uses two enzymes of upper glycolysis (G6P-F6P isomerase, FBP aldolase) and the pathway-specific enzyme fructose 1,6-bisphosphatase to drive the reverse of upper glycolysis and support the biosynthesis of G6P, which enters the pentose phosphate pathway (PPP) and cell wall biosynthesis.17 In contrast, the sulfo-EMP pathway is catabolic and is presumed to operate exclusively in the forward direction and does not provide key intermediates for gluconeogenesis such as F6P and G6P. Sulfoglycolytic cells must therefore utilize the traditional gluconeogenesis pathway to satisfy demands for cell wall biosynthesis and the PPP.10 Since these pathways must operate in tandem, it is presumably important that the sulfoglycolytic enzymes exhibit high selectivity for sulfosugar intermediates and limited activity on the analogous glycolytic intermediates to ensure the chemical unidirectionality of each pathway and avoid futile cycles.
In this work, we illuminate how the three core enzymes of the sulfo-EMP pathway recognize the unique sulfonate groups of their substrates and highlight key features of their catalytic mechanisms. We show that YihS catalyzes the interconversion of SQ, SF, and the previously unknown metabolite sulforhamnose (SR), and provide evidence that SR, as well as SQ, can act as a transcriptional derepressor through CsqR. We demonstrate the activation and inhibition of YihV by common cellular metabolites, revealing that this enzyme acts as an important control point for regulating flux through the sulfoglycolytic pathway. Importantly, we also show that this triad of enzymes exhibits high selectivity for sulfoglycolytic intermediates, ensuring sulfoglycolysis and gluconeogenesis can operate in tandem.
Results
SQ-SF Isomerase Also Produces Sulforhamnose
YihS was identified as the SQ-SF isomerase in the E. coli sulfo-EMP pathway by Schleheck and co-workers.7 It is classified as a member of Pfam database family PF07221,18 which includes enzymes that catalyze the epimerization of the α carbon in acyclic aldoses (the Lobry de Bruyn-Alberta van Ekenstein reaction). We chose to revisit the activity of E. coli YihS by incubating this protein with SQ in phosphate buffered saline, heat-inactivating the enzyme, and then exchanging the sample into deuterated solvent for 1H NMR analysis. This analysis revealed that YihS produced two new products (Figure 2a), which were not produced in the absence of enzyme. One product was identified as SF through comparison with authentic material.19 Resonances for the second new compound did not match either anomer of SQ or SF, particularly singlets in the 1H NMR spectrum at δ 4.86 and 5.10 ppm, and in the 13C NMR spectrum at δ 74.6 and 71.7 ppm. Further insight into the structure of the unknown product was obtained by conducting an additional experiment on the reaction of SQ with EcYihS, this time in buffered D2O. Under these conditions, the anomeric signals for SQ collapsed from a doublet to a singlet, consistent with H/D exchange at C2, and the signals for the unknown in the 13C NMR spectrum changed to triplets shifted slightly upfield, consistent with incorporation of deuterium (I = 1). Prior to the discovery that the sulfo-EMP operon encodes for sulfoglycolysis, Itoh et al. reported that recombinantly produced EcYihS can interconvert mannose, fructose, and glucose.20 We therefore speculated that the unknown compound could be the C2-epimer of SQ, namely, sulforhamnose (SR). Comparison of chemically synthesized SR with the mixture of products obtained from the reaction of EcYihS and SQ provided a good match for the unknown compound.
Figure 2.

Characterization and crystal structure of SQ isomerase YihS. (a) 1H NMR spectrum (in D2O) showing the equilibrium mixture resulting from isomerization of SQ by EcYihS, and reference spectra for pure SQ, SF, and SR. (b) Time course plot for the EcYihS-catalyzed equilibration of SQ to SF and SR. Substrate depletion plot obtained by HPLC was normalized to final composition determined by 1H NMR analysis (SF, 30.2; SR, 21.3; SQ, 48.5%). (c) Proposed mechanism of EcYihS-catalyzed isomerization of SQ to produce SF and SR. (d) Overall fold of SeYihS monomer showing location of active site. (e) Crystal structure of SeYihS·SF showing the sulfonate pocket. Ribbon diagram of protein backbone is depicted in blue, and SF and side chains of active site residues are shown in cylinder format. Electron density corresponds to the 2Fo – Fc map (in blue) at levels of 1.5σ. (f) Cartoon of ligand binding pocket of SeYihS·SF complex depicting hydrogen bonding interactions with active site residues.
To confirm that SQ, SF, and SR are in equilibrium, we individually treated each compound with EcYihS until equilibrium was established. 1H NMR spectra of each reaction revealed identical mixtures, providing evidence that all three compounds are substrates of EcYihS (Figure S1). At equilibrium under these experimental conditions, the ratios of the three sulfosugars were SF/SR/SQ = 30:21:49. We established an HPLC-MS assay for the YihS-catalyzed isomerization reaction using a ZIC-HILIC column to follow the time course for equilibration of SQ to the mixture of sulfosugars (Figure 2b). The normalized plot for SQ depletion shows that formation of SF and SR is coincident with consumption of SQ. These data are consistent with an enzymatic mechanism that involves the formation of acyclic SQ and enzyme-catalyzed deprotonation of C2 to form a 1,2-enediol (Figure 2c). Protonation at C1 then forms SF, protonation at C2 from the “bottom” face forms SR, while protonation on the “top” face at C2 regenerates SQ.
As the sulfo-EMP pathway is a catabolic process with the isomerization of SQ to SF the most physiologically relevant, we chose to investigate the kinetics of this process in greater detail. EcYihS exhibited Michaelis–Menten parameters for formation of SF from SQ, with kcat = (7.90 ± 0.27) × 101 s–1, KM = 1.89 ± 0.28 mM, and kcat/KM = (4.17 ± 0.51) × 104 M–1 s–1 (Figure S2). Comparison of this data with that for d-mannose reported by Itoh and co-workers20 shows that SQ is 178-fold better as a substrate for EcYihS (in terms of kcat/KM). Under similar conditions, no activity toward G6P (10 mM) was detected (as previously reported by Itoh and co-workers20), demonstrating that EcYihS can discriminate between a 6-phosphate and sulfonate and that no activity is expected for YihS on glycolysis intermediates.
SQ exists as a mixture of α- and β-anomers, which are interconverted by SQ mutarotase.14 It is unknown if EcYihS acts on a single anomer or both. The mechanism utilized by EcYihS to catalyze the isomerization of SQ to SR and SF also results in H/D exchange of the proton at C2 of SQ when the reaction is performed in D2O. This leads to the 1H NMR signal for the anomeric proton of SQ changing from a doublet to a singlet. We therefore monitored the multiplicity of H1 in the two SQ anomers as a function of time in the presence of EcYihS. The time course 1H NMR spectra showed a rapid collapse of the doublet corresponding to H1 of β-SQ, and a slower collapse of the doublet corresponding to H1 of α-SQ (Figure S3). These data are consistent with stereospecific H/D exchange at C2 of β-SQ, demonstrating that the enzyme preferentially acts on β-SQ. We interpret the slow H/D exchange responsible for the conversion of the doublet of H1 of α-SQ to a singlet to be a result of spontaneous mutarotation from the deuterated β-anomer (2-D-β-SQ), though we cannot discount a low rate of catalysis by EcYihS. As an added control, this experiment was repeated in the presence of the SQ mutarotase HsSQM,14 which increases the rate of interconversion of α-SQ and β-SQ. This revealed simultaneous collapse of anomeric doublets for both α-SQ and β-SQ. Together these experiments confirm that the mechanism for H/D exchange of α-SQ is primarily by mutarotation of β-SQ, which is the substrate of EcYihS. This study also demonstrates the functional significance of the SQ mutarotase in sulfoglycolysis: it ensures that the α-SQ product of the sulfoquinovosidase YihQ can be efficiently utilized by the SQ-SF isomerase YihS, which has a clear preference for β-SQ and little-to-no activity on α-SQ.
Salmonella enterica possesses a sulfo-EMP operon syntenic to that from E. coli, and the crystal structures of wild-type S. enterica YihS (PDB ID: 2AFA) and EcYihS (PDB ID: 2RGK) were previously solved prior to determination of their physiological function.20 We revisited the catalytic activity of SeYihS to demonstrate that it also catalyzed the interconversion of SQ, SF and SR (Figure S4). To elucidate what interactions are made between YihS and the sulfonate moiety of its sulfosugar substrates, the inactive mutant SeYihS-H248A was produced for structural studies. Size exclusion chromatography-multiangle light scattering (SEC-MALS) revealed that SeYihS exists as a hexamer in solution, although an asymmetric elution peak and marginal decrease in molecular weight may signify some dissociation under experimental conditions (Figure S5). This protein was cocrystallized with SF and X-ray diffraction techniques used to determine the structure of the SeYihS-H248A·SF product complex, which existed as a crystallographic dimer-of-trimers (Table S1, Figure S5), commensurate with the observation of hexamers in solution by SEC-MALS.
As previously observed, the overall fold of YihS comprises an (α6/α6)-barrel scaffold displaying close similarity to N-acyl-d-glucosamine epimerases (Figure 2d).20 Clear density for a furanose sulfosugar was seen in all six chains that allowed modeling of β-SF in a highly compact active site. Overlaying the SeYihS-H248A·SF structure with the ligand-free wild-type SeYihS structure (2AFA.pdb) gave an rmsd of 0.7 Å over 496 residues, with the catalytic residues His248 and His383 positioned on opposite sides of the C1 hydroxyl group of bound SF, and shows that the active site architecture remains largely unchanged (Figure S6). However, upon binding SF, two loops surrounding the active site undergo reorganization. The loop comprising residues 228–242 moves toward the ligand pocket such that the side-chain of Phe239 reorients and projects into the active site. A previously unstructured, flexible loop 370–377 now adopts an ordered conformation and interacts with residues from loop 228–242, such that Trp375 makes π-stacking interactions with Arg238. This cation-π interaction between the residues in two separate loops encloses the pocket to form the active site.
SF is buried in a deep pocket surrounded by bulky residues on either side (Phe239, Trp316 and Trp51) (Figure 2e,f). Sugar hydroxyls make several hydrogen-bonding interactions with polar residues. The sulfonate is accommodated by a network of hydrogen-bonding interactions, with one of the sulfonate oxygens hydrogen bonding with Gln379 (2.8 Å), a second oxygen with Gln362 (3.1 Å) and an ordered water molecule (3.1 Å), and the third oxygen forming a salt bridge with Arg55 (2.8 Å), which also hydrogen bonds to the endocyclic oxygen of SF (3.1 Å). The observation of SF binding to YihS as the β-anomer does not definitely show which anomer is produced by the enzyme. In order to understand the ease with which SF can anomerize, we measured its unidirectional rate of uncatalyzed anomerization using an NMR-based chemical exchange spectroscopy method at equilibrium previously used to determine the mutarotation rate of SQ.14 At pD 7.5 and 25 °C, β-SF mutarotates to α-SF with k = 0.026 s–1, corresponding to t1/2 = 26 s (Figure S7) (the corresponding data for mutarotation of β-SQ to α-SQ are k = 3.87 × 10–5 s–1, t1/2 = 299 min). The rate was unaffected by addition of a SQ-specific mutarotase from Herbaspirillium seropedicae. This rapid rate of mutarotation suggests facile interconversion of SF anomers in cellulo.
Sulforhamnose Is a Transcription Factor Derepressor
The observation that SR is a product and substrate of SQ isomerase raised questions as to its possible role in sulfoglycolysis: is it simply an unproductive intermediate that is ultimately isomerized to SF and consumed, or does it play a role in gene regulation, akin to the role that allolactose plays in regulating the lac operon?21 The gene csqR within the SQ-utilizing gene cluster of E. coli encodes a DeoR-family transcription factor (TF) that has been demonstrated to bind inside the intergenic spacer between the yihUTS operon and the yihV gene, and in doing so represses expression of sulfoglycolytic genes (Figure 1b).11 SQ and sulfoquinovosyl glycerol (SQGro) are derepressors for CsqR binding to DNA, as demonstrated using gel-shift analyses with DNA sequences that encompass the CsqR binding sites and a reporter assay.11 We used this same gel-shift assay to assess whether SR can influence the CsqR-DNA interaction. Purified CsqR was used to probe dsDNA encompassing the yihUV intergenic region using a polyacrylamide gel-shift assay. Disappearance of the DNA resulted from high levels of cooperative binding of CsqR along the probe DNA that prevents the polymeric CsqR–DNA complexes from entering the gel during electrophoresis (Figure 3a). Titration with increasing [SQ] leads to dissociation of CsqR from its DNA complexes, releasing free DNA that could be detected within the gel, in agreement with the previous report.11 Titration with increasing [SR] also led to dissociation of CsqR-DNA complexes. However, completely free DNA was not observed; rather, higher molecular weight complexes were formed that decreased in size at higher [SR], indicating that SR is a weaker mediator of the CsqR–DNA binding interaction.
Figure 3.

Sulforhamnose is a derepressor of the transcriptional repressor CsqR. (a) PAGE gel shift assay showing effect of SQ and SR on binding of CsqR to a DNA probe of the yihUV intergenic region. Effector concentrations, lanes 1–8: 0, 0, 5, 10, 25, 50, 75, 100 mM. DNA was stained with GelRed and imaged. (b) Representative AFM observations of the effect of SQ and SR on binding of CsqR to yihUV intergenic DNA probe. All images cover an area of 250 × 250 nm.
To gain further insight into the effect of SQ, SQGro, and SR on the transcription regulator CsqR, two constructs were designed based on the predicted domain boundaries using InterPro Classification (Figure S8a): full-length CsqR, which contains two distinct domains, the winged helix-turn-helix HTH DNA binding domain (DBD, residues 2–66) and the DeoR effector binding domain (EBD, residues 80–260), and a truncated EBD-CsqR construct comprised of only the EBD. Binding studies using a temperature unfolding assay with purified EBD-CsqR and different sulfosugars as effector molecules gave improved stability only with SQ (ΔTm = 4.6 °C at 10 mM), with no change of Tm noted for SQGro or SR at 10 mM (Figure S8b).
To provide further insight into the effect of SQ, SQGro, and SR on CsqR-DNA binding, we imaged CsqR-DNA interactions by atomic force microscopy (AFM). Purified full-length CsqR was mixed with the yihUV probe in the presence of increasing concentrations of SQ or SR, and subjected to AFM imaging under the same conditions as employed in a previous study (Figure 3b).11 Titration with increasing concentrations of SQ caused a reduction in the size of the aggregates of CsqR–DNA complexes, at low concentrations of 0.5 and even 0.05 mM. By contrast, titration of CsqR–DNA aggregates with SR did not result in dissipation of aggregates at 0.05 mM, limited dissipation at 0.5 mM, and with complete dissociation observed only at 5.0 mM. Collectively, these data suggest that SR is a transcription inducer with a lower potency than SQ.
SF Kinase Activity Is Regulated by Sulfoglycolytic and Central Metabolites
YihV is a member of the Pfam pfkB carbohydrate kinase family22 and is an ATP-dependent kinase that mediates phosphoryl transfer from ATP to SF to give SFP. We established an HPLC-MS/MS assay for EcYihV (Figure S9) using a ZIC-HILIC column and chemically synthesized SF substrate (Figure S10).19 Initially, we measured the kinetics for production of SFP. Under conditions of constant SF (1.0 mM) and varying ATP in the presence of MgCl2, EcYihV exhibited Michaelis–Menten kinetics with kcat 3.1 ± 0.2 s–1, KM = 1.0 ± 0.2 mM, and kcat/KM = 3.2 ± 0.8 mM s–1 (Table 1, Figure S11). Conversely, under conditions of constant ATP (1.0 mM) and varying SF, EcYihV exhibited weak substrate inhibition, with an estimated KI value for SF of 8 mM. The KI value was sensitive to ATP concentration, and at [ATP] = 0.1 mM, dropped to 0.3 mM. As phosphofructokinase is a critical allosteric control point for glycolysis, we investigated whether EcYihV is sensitive to a range of metabolites from sulfoglycolysis (SQ and SLA), glycolysis (F6P, FBP, DHAP, and PEP), or central metabolism (citrate, representing the Krebs cycle and ADP). This revealed that EcYihV is strongly inhibited by ADP and is activated by SQ, SLA, and DHAP (through effects on KM) and by F6P, FBP, PEP, and citrate (through effects on kcat/KM) (Figure 4a, Table 1). Taken together, these data suggest that EcYihV is an important control point for flux through the sulfo-EMP pathway.
Table 1. Kinetic Parameters for EcYihV in the Presence and Absence of Cellular Metabolites.
| variablea substrate | additive | kcat (s–1) | KM (mM) | KI (mM) | kcat/KM (s–1 mM–1) |
|---|---|---|---|---|---|
| ATPb,c | 3.1 ± 0.2 | 1.0 ± 0.2 | 3.2 ± 0.8 | ||
| SF | 1.2 ± 0.2 | 1.3 ± 0.3 | 8.5 ± 2.8 | 0.9 ± 0.4 | |
| SFd | 0.30 ± 0.5 | 0.3 ± 1.1 | |||
| SF | SQ | 0.7 ± 0.1 | 0.6 ± 0.2 | 16 ± 9 | 1.2 ± 0.4 |
| SF | SLA | 0.77 ± 0.13 | 0.60 ± 0.19 | 6.4 ± 2.7 | 1.3 ± 0.6 |
| SF | DHAP | 1.4 ± 0.3 | 0.61 ± 0.24 | 3.0 ± 1.4 | 2.3 ± 1.4 |
| SFc | citrate | 3.2 ± 0.4 | 1.6 ± 0.5 | 2.0 ± 0.9 | |
| SF | PEP | 1.9 ± 0.2 | 0.11 ± 0.04 | 4.4 ± 1.5 | 17.7 ± 8.6 |
| SF | F6P | 2.5 ± 1.2 | 0.97 ± 0.7 | 2.6 ± 1.9 | 2.5 ± 3.1 |
| SF | FBP | 1.2 ± 0.1 | 0.04 ± 0.01 | 5.8 ± 1.5 | 27.8 ± 10.8 |
| SFc | ADP | 0.14 ± 0.01 | 0.06 ± 0.03 | 2.3 ± 1.3 |
Reactions were conducted in 200 μL volume in 25 mM BTP buffer (pH 7.5), 25 mM KCl, 5 mM MgCl2, 0.1 mg/mL BSA, 36.6 nM EcYihV at 30 °C, for 60 min. [ATP] = 1.0 mM, and kinetic data were analyzed using a substrate inhibition kinetic model, unless otherwise noted. Additives were at 10 mM.
[SF] = 1.0 mM.
Kinetic parameters obtained by analysis using the Michaelis–Menten equation.
[ATP] = 0.1 mM.
Figure 4.

Crystal structures of EcYihV SF kinase. (a) Kinetic plots showing effect of metabolites on EcYihV-catalyzed phosphorylation of SF to SFP at [ATP] = 1.0 mM. (b) (Left) EcYihV dimer in complex with AMPPNP·Mg in open and closed conformations is shown in ribbon with the two subunits shown in coral and blue. Each subunit is composed of two-domain architecture with α/β nucleotide binding domain and β-sheet “lid” domain. (Right) Lid domains of the dimer form a β-clasp dimerization motif that serves both structural and catalytic roles. (c) Close-up view of EcYihV·AMPPNP·Mg showing nucleotide binding site in the closed conformation. (d) EcYihV·SFP complex structure and active site interactions with bound SFP product molecule. (e) Close-up view of EcYihV·ADP·Mg·SF active site showing hydrogen bonding interactions in a quaternary complex. Backbone and carbon atoms of subunits A and B are shown in coral and blue, respectively, and ADP, AMPPNP, SF, and SFP are shown in cylinder format. Electron density corresponds to the 2Fo – Fc and in blue at levels of 1σ for c–e. (f) Cartoon of ligand binding pocket of EcYihV·ADP·Mg·SF complex depicting hydrogen bonding interactions with active site residues.
We next examined whether EcYihV could catalyze phosphorylation of F6P. Incubation of EcYihV with 1 mM F6P, ATP, and MgCl2 did not lead to the formation of FBP, showing that this enzyme is specific for SF and indicating that no cross-talk exists between sulfoglycolysis and the EMP glycolysis or gluconeogenesis pathways.
To delineate the specific interactions with substrates and products, three crystal structures of EcYihV were obtained in complex with substrate SF and AMPPNP or ADP as ATP analogues, as well as product SFP (Tables S1–S2, Figure 4b–f). EcYihV forms a dimeric assembly, and SEC-MALS analysis of EcYihV revealed a mixture of solution states with the major peak corresponding to a dimer (>60%) and a minor (13%) subpopulation corresponding to a tetrameric ensemble (Figure S9). The overall structure of YihV kinase displays a two-domain architecture comprising major α/β nucleotide binding domain and β-sheet serving as a “lid” domain covering the active site. The orthogonal packing of eight β strands within the lid domains of the two subunits form a “β-clasp” dimerization motif, a distinctive feature previously seen in members of the ribokinase superfamily that includes tagatose-6-phosphate kinase (TPK) and the minor isozyme from the glycolytic pathway, F6P kinase (PfkB).23−25 Close hydrophobic contacts and reciprocal interactions are observed at the dimer interface where residues from the opposite subunit in a β-clasp motif protrude into the active site, which is likely to be crucial for both catalysis and structural stability.
Cocrystallization with the non-hydrolyzable ATP analogue AMPPNP resulted in the EcYihV·AMPPNP·Mg complex showing a surface-accessible nucleotide binding site (Figure 4b,c). Density for AMPPNP bound in an anti-conformation with hydrated Mg ions was present in all four subunits in the structure. YihV subunits were observed in “open” and “closed” conformations due to interdomain rotation. In the open conformation, the γ-phosphate of AMPPNP is hydrogen bonded to the backbone amide of Gly243 (2.7 Å) and a water molecule (2.5 Å) and is present within 5 Å distance of conserved catalytic Asp244 (Figure S13), whereas in the closed conformation the γ-phosphate moves closer to the substrate cleft and achieves an appropriate distance for phosphoryl transfer. This closed conformation of YihV most likely denotes an inactive conformation that would prevent binding of substrate. Analysis using the DynDom program showed the β-clasp domain rotates 31 degrees about four hinge bending regions, reflective of dynamic domain movements associated with binding of substrates (Figure S14). Considering the regulatory role of this enzyme in the pathway, these dynamic movements may be promoted by binding of ligands at an allosteric site(s), thereby driving transitions between productive and unproductive conformations.
In the EcYihV·SFP binary complex, the two subunits in a dimer pair are both in a closed conformation (Figure S12). SFP bound at the cleft between the nucleotide domain and β-barrel motif, showing that binding of the sulfonate ligand induces interdomain rotation and subsequent closure. The EcYihV·SFP complex revealed a sulfonate subpocket where one of the sulfonate oxygens of SFP is hydrogen bonded to Nε of Arg138 (2.8 Å) and Asn109 (3.2 Å), and the second sulfonate oxygen forms a salt bridge with the in-trans Lys27 (2.9 Å), which projects into the active site from the β3 strand of other subunit in the dimer pair and makes reciprocal interactions with Asp162 present at 3 Å (Figure 4d). Lys27 also forms hydrogen bonds with the C1 hydroxyl and ring oxygen of SFP, each present at 2.9 Å.
A compact dimer pair is seen for a dead-end complex of EcYihV with SF and ADP·Mg, formed by closure of the active site (Figure 4e,f). In this complex, the sugar is deeply sequestered at the edge of the interdomain cleft, and the ADP molecule (with a penta-hydrated Mg ion coordinated to α-phosphate) is bound close to the substrate phosphorylation site. The C1 hydroxyl of SF is hydrogen bonded to catalytic base Asp244 (2.7 Å) and Lys27 (3.0 Å), with the substrate positioned for phosphoryl transfer. The remaining sugar hydroxyls make several hydrogen bonding interactions with other active site residues suggesting SF binding facilitates domain closure. Specifically, the C2 hydroxyl is hydrogen bonded to Asp244 (2.6 Å), Asp13 hydrogen bonds C3 and C4 hydroxyls each at 2.5 Å, and the C4 hydroxyl makes an additional hydrogen bond to Ser95 (2.7 Å). The hydrogen-bonding network at the sulfonate binding site is similar to the EcYihV·SFP structure above, wherein two of the sulfonate oxygens interact with Nε of Arg138 (2.8 Å) and Asn109 (3.2 Å) and the in-trans Lys27 (2.7 Å). The third sulfonate oxygen hydrogen bonds with a bound water molecule that in turn interacts with the backbone carbonyl of Tyr28 and another water molecule.
An ordered sequential Bi–Bi mechanism is reported for ribokinases, with sugar binding interactions driving domain closure that precedes ATP binding.23−25 Nano differential scanning fluorimetry (nanoDSF) studies using EcYihV in the presence of SF and ADP resulted in a Tm shift of 6 °C concurrent with binding of the sugar (and only a 1 °C shift seen upon binding ADP) indicating ligand-induced conformational stabilization of the kinase (Figure S15). These data complement the structural details revealed in the complexes reported here, showing that domain movements upon SF binding result in a closed, functional state, and ATP can access the active site through a surface groove (Figures S14–S15). This may avoid premature hydrolysis of the γ-phosphate of ATP, as reported in ribokinases.25−27
Given the close functional relationship of SF kinase (YihV) and PfkB and TPK, sequence- and structure-based analysis of their binding sites was undertaken. Two conserved motifs have been described in these phosphosugar kinases, the TR and GXGDXX motifs, responsible for substrate binding and catalysis, respectively.28,29 Sequence alignment of PfkB from E. coli and TPK from Staphylococcus aureus with SF kinase showed that the GXGDXX motif comprising the catalytic aspartate D244 (EcYihV numbering) is conserved in all three subfamilies (Figure S16). Comparison of the closed conformation of EcYihV with TPK and PfkB revealed that overall fold is highly conserved in these sugar kinases: with PfkB (3N1C.pdb), rmsd 2.6 over 248 residues; with TPK (2JG1.pdb), rmsd 2.2 over 265 residues (Figure S17). Finally, close inspection of the 6-phospho versus 6-sulfonate binding sites highlighted key differences consistent with the chemical structures of bound sugars (Figure S18). In PfkB and TPK, Arg88 present within the substrate binding TR motif is part of the conserved RRS triad for recognition of the 6-phosphate moiety. The substrate-specific TR motif and the consensus RRS triad are absent in SF kinases. Instead, a KRN sulfonate recognition triad (K27-R138-N109) was identified in the SFP and SF complexes, which is conserved in annotated YihV kinases (Figures S16 and S18).
SFP Aldolase
SFP aldolase catalyzes retro-aldol cleavage of SFP to DHAP and SLA. Schleheck and co-workers detected the formation of DHAP and SLA in a coupled system containing SQ and recombinantly produced YihS, YihV, and YihT, supplemented with ATP and MgCl2.7 Here, we established an LCMS-MS assay for direct analysis of the reaction catalyzed by YihT. Recombinantly expressed YihT from E. coli was incubated with chemo-enzymatically synthesized SFP;19 however, we could not detect the formation of DHAP or SLA under a range of conditions. Instead, we recombinantly expressed YihT from Salmonella enterica, which shares 87% similarity (Figure S19). Incubation of recombinant SeYihT with SFP revealed the formation of SLA and DHAP. The enzyme exhibited Michaelis–Menten kinetics with kcat = 47.7 ± 2.4 s–1, KM = 3.57 ± 0.42 mM, and kcat/KM = 13 ± 2 mM s–1 (Figure 5a). Under similar conditions, no activity was noted on FBP (1 mM).
Figure 5.

Kinetics, reversibility, and structure of SeYihT SFP aldolase. (a) Michaelis–Menten and Lineweaver–Burk plots (inset) for SeYihT-catalyzed conversion of SFP to SLA and DHAP, analyzed for DHAP. (b) Mass spectrum mirror plot comparison of the product ion scans of the product of the reaction of SeYihT incubated with SLA and DHAP, and independently synthesized SFP. (c) Overview of the SeYihT protein showing location of substrate binding site. (d) Close-up view of SeYihT·SFP active site with Lys193 engaged in a Schiff base with the sulfosugar. Electron density in blue corresponds to 2Fo – Fc contoured at 1.2σ. (e) Close-up view of SeYihT·DHAP active site with Lys193 engaged in a Schiff base with DHAP. Electron density in blue corresponds to 2Fo – Fc map contoured at 1σ. (f) Cartoon of ligand binding pocket of SeYihT·SFP complex depicting hydrogen bonding and electostatic interactions with active site residues.
Benson reported that SFP can be generated from SLA and DHAP by an unspecified aldolase.30 We revisited this observation by incubating recombinantly expressed SeYihT with chemically synthesized SLA and commercial DHAP. A product with identical hplc retention time and mass spectrometric fragmentation pattern to authentic SFP was obtained, demonstrating the reversibility of the YihT-catalyzed reaction (Figure 5b).
Fructose bisphosphate aldolases are classified into Class I and II enzymes based on their mechanism of action. Class I aldolases utilize an active-site lysine to form a Schiff base with the substrate, whereas prokaryotic and fungal Class II FBP aldolases are dependent on divalent metal ions for their activity. Class I aldolases are commonly found in algae, protozoa, plants, and animals and adopt homotetrameric active forms; archaeal and bacterial enzymes utilize similar mechanisms but have low sequence similarity and are distinguished from their eukaryotic counterparts through self-association as multimers exhibiting tetrameric to decameric quaternary structures, leading to their subgrouping into Class Ia.31,32 In order to reveal structural features of SFP aldolase, the X-ray crystal structures of YihT from E. coli and Salmonella enterica were obtained with 2 and 12 molecules in an asymmetric unit (Figure S20), respectively; however, SEC-MALS showed YihT exists as a homotetramer in solution (Figure S19). YihT displays an overall (α/β)8-barrel architecture similar to class I fructose bisphosphate aldolases (Figure 5c, Table S2).33 A DALI search using YihT against the RCSB PDB library gave annotated bacterial class I aldolases as closest structural neighbors. These included tagatose-1,6-bisphosphate aldolases from Streptococcus mutans (PDB ID: 3IV3 with DALI z score of 31.8, rmsd 2.4 and 21% sequence ID) and from Streptococcus porcinus (PDB ID: 5HJL with DALI z score of 31.7, rmsd 2.2 and 21% sequence ID), as well as fructose bisphosphate aldolase from Slackiahelio trinireducens (PDB ID: 4MOZ with DALI z score of 22.9, rmsd 2.5 and 15% sequence ID), indicating high structural similarity despite low sequence ID.
In the SeYihT structure, density for a single sulfate ion in the proposed substrate binding pocket was observed in all 12 chains at occupancies 0.8–1 (Figure S21). The sulfate ion is located close to conserved Lys193, which is proposed to be involved in Schiff base formation. Substrate-soaking experiments at saturating concentrations of SFP resulted in trapping of the Schiff base adduct of SeYihT (Figure 5c–d, Table S3). Clear, contiguous density (at an occupancy of 0.8) was observed for a hexose covalently attached to Lys193 within two of the subunits of the tetrameric protein structure representing SFP in an open-chain conformation. In the other two subunits, there was evidence of in-crystal cleavage of the C3–C4 bond of the substrate, and a new Schiff base formed with a DHAP molecule could be modeled, consistent with the reaction catalyzed by this enzyme (Figure 5e, Figure S22). The SFP fragment bound to Lys193 allowed unambiguous identification of both phosphate and sulfonate pockets within SeYihT. The phosphate oxygens interact with Ser226, Ser227, and Arg253 as described in the SeYihT·SO42– complex above. One sulfonate oxygen forms an electrostatic interaction with Arg253 (2.7 Å), and the other two sulfonate oxygens make hydrogen bonding interactions with two bound water molecules present at a distance of 2.7 and 2.8 Å (Figure 5f). These residues are conserved across annotated SFP aldolases and are proposed to comprise a sulfonate binding pocket that defines this subclass of enzymes (Figure S23).
Discussion
The sulfo-EMP pathway allows metabolism of the widespread sulfosugar SQ and its glycosides. The pathway shares a similar series of steps with the EMP pathway, with 6-sulfonate taking the place of phosphate. This similarity raises obvious questions about whether the sulfo-EMP pathway can act on intermediates in the EMP pathway or if the two pathways are functionally segregated. Kinetic analysis reveals that the SQ-SF isomerase has no detectable activity on G6P, the SF kinase has no activity on F6P, and the SFP aldolase has no activity on FBP, under conditions where robust enzymatic activity on their namesake substrates can be detected. This selectivity is likely to be of significance under conditions where SQ is the sole substrate for growth, in which case gluconeogenesis is required to supply F6P for the PPP and cell wall biosynthesis, and where reversal of flux is required. Among these steps, high selectivity for SF kinase to act solely on SF and not F6P is likely to be the most critical, as gluconeogenesis requires a switch from PFK to FBP phosphatase to change directionality from catabolism to anabolism.
Using X-ray crystallography, we obtained three-dimensional (3D) structures of all three enzymes and through complexes with SF or SFP defined sulfonate binding pockets that show how these enzymes recognize their sulfosugar substrate. In all three enzymes, the sulfonate pocket was lined with positively charged residues (for SQ isomerase: Arg55-Gln379-Gln362; for SF kinase: Lys27-Asn109-Arg138; for SFP aldolase: Arg253-Ala26(H2O)), which in every case includes an Arg residue to balance the charge. These data complement X-ray data for 3D structures of sulfoquinovosidase and SLA reductase, which also exhibit selectivity for their sulfonate substrates and possess well-defined and conserved sulfonate binding pockets. While the EcSQase binding pocket is comprised of Arg301-Trp304-Tyr508(H2O),13 the SLA reductase binding pocket lacks a direct interaction with an Arg; rather, it is comprised of Asn174-Ser178 and the backbone amide of Arg123.15
Transcription factors (TFs) involved in transcriptional regulation of the genes for metabolism are often controlled through allosteric interactions with metabolites. Previously, regulation of CsqR activity by both SQGro and SQ (sulfoquinovose) was identified.11 Here we demonstrate that SR is also a regulator of CsqR-DNA binding, inducing the dissociation of repressor CsqR from its regulatory target DNA. The level of CsqR inactivation will therefore potentially be controlled depending on the level of three metabolites, SQGro, SQ, and SR, with the induction level of sulfo-EMP operon depending on the affinity of each inducer ligand to CsqR. In E. coli K-12, regulation of TF activity by multiple metabolites has been found for TFs such as allantoin repressor (AllR) by allantoin and glyoxylate,34 cysteine B (CysB) by O-acetyl-l-serine and thiosulfate,35 glycine cleavage A (GcvA) by glycine and purine,36 purine repressor (PurR) by hypoxanthine and guanine,37 and pyrimidine utilization regulator (RutR) by uracil and thymine.38 As in the case of CsqR, the activity and target selectivity of tyrosine repressor (TyrR) is controlled by three metabolites, phenylalanine, tryptophan, and tyrosine,39 with the level of TF activity dependent on the intracellular concentrations of all three metabolites.
The role of SF kinase in sulfoglycolysis is analogous to that of PFK in glycolysis. PFK is widely recognized as a central regulatory step in carbohydrate metabolism in most organisms and has complex allosteric properties. These effects help regulate distribution of G6P into the PPP pathway, cell wall synthesis, the production of carbohydrate storage molecules such as glycogen, maltose, and trehalose, the channeling of PEP into the citric acid cycle and fatty acid synthesis, as well as ensuring ATP production is managed when in surplus. We showed modulatory effects on SF kinase by a range of sulfoglycolytic and central metabolites, including substrate inhibition by SF, strong inhibition by ADP, and activation by SQ, SLA, F6P, FBP, PEP, DHAP, and citrate. Substrate inhibition is widely recognized as a control strategy to maintain steady pathway flux even in the presence of large fluctuations in substrate concentration,40 which may help to limit variation in flux through sulfoglycolysis as environmental concentrations of SQ and SQGro fluctuate. Because the wiring of cellular metabolism and pathway yields under conditions of sulfoglycolysis differ from glycolysis, significant differences in regulation are to be expected. Notably, while SF kinase is inhibited by ADP and not ATP, E. coli PFK-1 and -2 are inhibited by ATP, while only PFK-1 is activated by ADP and inhibited by ATP.41,42 While sulfoglycolysis and glycolysis both yield PEP, unlike glycolysis, sulfoglycolysis does not produce G6P, and thus there may not be the same requirement for ATP/ADP control over the pathway to manage distribution of substrate into catabolic and anabolic pathways, as for G6P in glycolysis. Instead, an important branchpoint post-sulfoglycolysis is likely to be distribution of PEP into gluconeogenesis and central metabolism (citric acid cycle and fatty acid synthesis). Activation by citrate, PEP, FBP, and F6P may assist in maintaining flux into these anabolic and catabolic pathways. Additionally, it may be beneficial for glycolytic-EMP and sulfo-EMP pathways to be regulated in distinct manners, which could allow them to operate in parallel if SQ is available together with glucose, providing greater metabolic flexibility, and allowing the regulation of cellular energy metabolism as the ATP yield of the two pathways differ.
Conclusions
The present work provides the first detailed biochemical and structural analysis for the three core enzymes of the E. coli sulfo-EMP pathway: SQ isomerase, SF kinase, and SFP aldolase. Collectively, these data demonstrate kinetic selectivity for the core sulfoglycolytic enzymes for sulfoglycolytic intermediates over the corresponding intermediates in glycolysis. This selectively arises from conserved sulfonate binding pockets in each enzyme. The effect of this selectivity is to allow functional segregation of this pathway from glycolysis/gluconeogenesis, preventing futile cycling, and it highlights the specific structural adaptations that have led to the evolution of this pathway. The present work provides a roadmap to conduct more detailed bioinformatic analyses of this pathway in complex microbial communities and to understand how sulfoglycolysis contributes to bacterial metabolism.
Acknowledgments
This work was supported by the Australian Research Council (DP180101957, DP210100233), the Leverhulme Trust (RPG-2017-190), MEXT Cooperative Research Program of Network Joint Research Center for Materials and Devices (AI and TS), the National Health and Medical Research Council of Australia (GNT1139546 and GNT1139549), and the Royal Society for the Ken Murray Research Professorship to G.J.D. We acknowledge support from The Walter and Eliza Hall Institute of Medical Research; the Australian Cancer Research Fund; and a Victorian State Government Operational Infrastructure support grant. We acknowledge the staff of the Diamond Light Source (U.K.) for provision of IO3, IO4, and IO4-1 beamline facilities (proposal numbers mx-13587, mx-18598, and mx-24948). We thank Noriyuki Kodera (Kanazawa Univ.) for fabricating the electron beam deposited tips of AFM, and Christopher Bengt, Joe Joiner, and Prof. David Vocadlo for constructive advice.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01285.
Additional figures (Figures S1–S23) and tables (Tables S1–S3) including X-ray data, structures, enzyme kinetics, NMR analysis of products, and protein purification and characterization. Methods describing protein expression, purification, biophysical characterization, and crystallography; enzymatic reactions, gel shift, and atomic force microscopy, and methods for chemical synthesis of sulforhamnose (and associated NMR data) (PDF)
Accession Codes
The coordinate files and structure factors have been deposited in the Protein DataBank (PDB) with accession numbers 7AG4, 7AGH, 7AG6, 7AGK, 7AG1, 7AG7, and 7NE2.
Author Contributions
G.J.D, A.I., and S.J.W conceived the project. G.J.D, E.D.G.-B., A.I. and S.J.W designed the experiments. M.S., Y.J., and J.L. performed molecular biology and protein chemistry. P.A., R.E., and S.J.W. conducted enzyme kinetics and analyzed results. P.A., J.M., and R.E. synthesized substrates/reagents. M.S., J.Y., and G.J.D. conducted X-ray crystallography and analyzed results. T.S., M.N., and A.I. conducted CsqR experiments and analyzed results. M.S., S.J.W., and G.J.D. wrote the manuscript with input from the other authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Canfield D.; Farquhar J.. The global sulfur cycle. In Fundamentals of Geobiology; Knoll A. H., Canfield D. E., Konhauser K. O., Eds.; Blackwell Publishing Ltd, 2012; pp 49–64. [Google Scholar]
- Dickschat J. S.; Rabe P.; Citron C. A. The chemical biology of dimethylsulfoniopropionate. Org. Biomol. Chem. 2015, 13, 1954–1968. 10.1039/C4OB02407A. [DOI] [PubMed] [Google Scholar]
- Thume K.; Gebser B.; Chen L.; Meyer N.; Kieber D. J.; Pohnert G. The metabolite dimethylsulfoxonium propionate extends the marine organosulfur cycle. Nature 2018, 563, 412–415. 10.1038/s41586-018-0675-0. [DOI] [PubMed] [Google Scholar]
- Goddard-Borger E. D.; Williams S. J. Sulfoquinovose in the biosphere: occurrence, metabolism and functions. Biochem. J. 2017, 474, 827–849. 10.1042/BCJ20160508. [DOI] [PubMed] [Google Scholar]
- Harwood J. L.; Nicholls R. G. The plant sulpholipid - a major component of the sulphur cycle. Biochem. Soc. Trans. 1979, 7, 440–447. 10.1042/bst0070440. [DOI] [PubMed] [Google Scholar]
- Frommeyer B.; Fiedler A. W.; Oehler S. R.; Hanson B. T.; Loy A.; Franchini P.; Spiteller D.; Schleheck D. Environmental and Intestinal Phylum Firmicutes Bacteria Metabolize the Plant Sugar Sulfoquinovose via a 6-Deoxy-6-sulfofructose Transaldolase Pathway. iScience 2020, 23, 101510. 10.1016/j.isci.2020.101510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denger K.; Weiss M.; Felux A. K.; Schneider A.; Mayer C.; Spiteller D.; Huhn T.; Cook A. M.; Schleheck D. Sulphoglycolysis in Escherichia coli K-12 closes a gap in the biogeochemical sulphur cycle. Nature 2014, 507, 114–117. 10.1038/nature12947. [DOI] [PubMed] [Google Scholar]
- Felux A. K.; Spiteller D.; Klebensberger J.; Schleheck D. Entner-Doudoroff pathway for sulfoquinovose degradation in Pseudomonas putida SQ1. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E4298–4305. 10.1073/pnas.1507049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck H.; Kerim K.; Jessica L.; Anna B.; Alexander F.; Karin D.; Benjamin F.; Craig H.; Thomas R.; Nicolai K.; Nicola S.; David S.; Alexander L. Sulfoquinovose is a select nutrient of prominent bacteria and a source of hydrogen sulfide in the human gut. Res. Square 2021, 10.21203/rs.21203.rs-49676/v21201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Epa R.; Scott N. E.; Skoneczny D.; Sharma M.; Snow A. J. D.; Lingford J. P.; Goddard-Borger E. D.; Davies G. J.; McConville M. J.; Williams S. J. A Sulfoglycolytic Entner-Doudoroff Pathway in Rhizobium leguminosarum bv. trifolii SRDI565. Appl. Environ. Microbiol. 2020, 86, e00750. 10.1128/AEM.00750-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T.; Yamamoto K.; Nakano M.; Watanabe H.; Schleheck D.; Ishihama A. Regulatory role of CsqR (YihW) in transcription of the genes for catabolism of the anionic sugar sulfoquinovose (SQ) in Escherichia coli K-12. Microbiology 2019, 165, 78–89. 10.1099/mic.0.000740. [DOI] [PubMed] [Google Scholar]
- Abayakoon P.; Jin Y.; Lingford J. P.; Petricevic M.; John A.; Ryan E.; Wai-Ying Mui J.; Pires D. E. V.; Ascher D. B.; Davies G. J.; Goddard-Borger E. D.; Williams S. J. Structural and Biochemical Insights into the Function and Evolution of Sulfoquinovosidases. ACS Cent. Sci. 2018, 4, 1266–1273. 10.1021/acscentsci.8b00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speciale G.; Jin Y.; Davies G. J.; Williams S. J.; Goddard-Borger E. D. YihQ is a sulfoquinovosidase that cleaves sulfoquinovosyl diacylglyceride sulfolipids. Nat. Chem. Biol. 2016, 12, 215–217. 10.1038/nchembio.2023. [DOI] [PubMed] [Google Scholar]
- Abayakoon P.; Lingford J. P.; Jin Y.; Bengt C.; Davies G. J.; Yao S.; Goddard-Borger E. D.; Williams S. J. Discovery and characterization of a sulfoquinovose mutarotase using kinetic analysis at equilibrium by exchange spectroscopy. Biochem. J. 2018, 475, 1371–1383. 10.1042/BCJ20170947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Abayakoon P.; Lingford J. P.; Epa R.; John A.; Jin Y.; Goddard-Borger E. D.; Davies G. J.; Williams S. J. Dynamic Structural Changes Accompany the Production of Dihydroxypropanesulfonate by Sulfolactaldehyde Reductase. ACS Catal. 2020, 10, 2826–2836. 10.1021/acscatal.9b04427. [DOI] [Google Scholar]
- Dunaway G. A. A review of animal phosphofructokinase isozymes with an emphasis on their physiological role. Mol. Cell. Biochem. 1983, 52, 75–91. 10.1007/BF00230589. [DOI] [PubMed] [Google Scholar]
- Hers H. G.; Hue L. Gluconeogensis and related aspects of glycolysis. Annu. Rev. Biochem. 1983, 52, 617–653. 10.1146/annurev.bi.52.070183.003153. [DOI] [PubMed] [Google Scholar]
- Finn R. D.; Mistry J.; Schuster-Böckler B.; Griffiths-Jones S.; Hollich V.; Lassmann T.; Moxon S.; Marshall M.; Khanna A.; Durbin R.; Eddy S. R.; Sonnhammer E. L. L.; Bateman A. Pfam: clans, web tools and services. Nucleic Acids Res. 2006, 34, D247–D251. 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abayakoon P.; Epa R.; Petricevic M.; Bengt C.; Mui J. W. Y.; van der Peet P. L.; Zhang Y.; Lingford J. P.; White J. M.; Goddard-Borger E. D.; Williams S. J. Comprehensive synthesis of substrates, intermediates and products of the sulfoglycolytic Embden-Meyerhoff-Parnas pathway. J. Org. Chem. 2019, 84, 2901–2910. 10.1021/acs.joc.9b00055. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Mikami B.; Hashimoto W.; Murata K. Crystal structure of YihS in complex with d-mannose: structural annotation of Escherichia coli and Salmonella enterica yihS-encoded proteins to an aldose-ketose isomerase. J. Mol. Biol. 2008, 377, 1443–1459. 10.1016/j.jmb.2008.01.090. [DOI] [PubMed] [Google Scholar]
- Jobe A.; Bourgeois S. lac Repressor-operator interaction. VI. The natural inducer of the lac operon. J. Mol. Biol. 1972, 69, 397–408. 10.1016/0022-2836(72)90253-7. [DOI] [PubMed] [Google Scholar]
- Bork P.; Sander C.; Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993, 2, 31–40. 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrell J. A.; Cameron A. D.; Mowbray S. L. Induced fit on sugar binding activates ribokinase. J. Mol. Biol. 1999, 290, 1009–1018. 10.1006/jmbi.1999.2938. [DOI] [PubMed] [Google Scholar]
- Sigrell J. A.; Cameron A. D.; Jones T. A.; Mowbray S. L. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 A resolution: insights into a new family of kinase structures. Structure 1998, 6, 183–193. 10.1016/S0969-2126(98)00020-3. [DOI] [PubMed] [Google Scholar]
- Park J.; Gupta R. S. Adenosine kinase and ribokinase-the RK family of proteins. Cell. Mol. Life Sci. 2008, 65, 2875–2896. 10.1007/s00018-008-8123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murillo-López J.; Zinovjev K.; Pereira H.; Caniuguir A.; Garratt R.; Babul J.; Recabarren R.; Alzate-Morales J.; Caballero J.; Tuñón I.; Cabrera R. Studying the phosphoryl transfer mechanism of the E. coli phosphofructokinase-2: from X-ray structure to quantum mechanics/molecular mechanics simulations. Chem. Sci. 2019, 10, 2882–2892. 10.1039/C9SC00094A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S.; Vivoli Vega M.; Harmer N. J. Carbohydrate Kinases: A Conserved Mechanism Across Differing Folds. Catalysts 2019, 9, 29. 10.3390/catal9010029. [DOI] [Google Scholar]
- Cabrera R.; Babul J.; Guixé V. Ribokinase family evolution and the role of conserved residues at the active site of the PfkB subfamily representative, Pfk-2 from Escherichia coli. Arch. Biochem. Biophys. 2010, 502, 23–30. 10.1016/j.abb.2010.06.024. [DOI] [PubMed] [Google Scholar]
- Miallau L.; Hunter W. N.; McSweeney S. M.; Leonard G. A. Structures of Staphylococcus aureus D-tagatose-6-phosphate kinase implicate domain motions in specificity and mechanism. J. Biol. Chem. 2007, 282, 19948–19957. 10.1074/jbc.M701480200. [DOI] [PubMed] [Google Scholar]
- Benson A. A.; Shibuya I. Sulfocarbohydrate metabolism. Fed. Proc. 1961, 20, 79. [Google Scholar]
- Lorentzen E.; Siebers B.; Hensel R.; Pohl E. Mechanism of the Schiff Base Forming Fructose-1,6-bisphosphate Aldolase: Structural Analysis of Reaction Intermediates. Biochemistry 2005, 44, 4222–4229. 10.1021/bi048192o. [DOI] [PubMed] [Google Scholar]
- Siebers B.; Brinkmann H.; Dörr C.; Tjaden B.; Lilie H.; van der Oost J.; Verhees C. H. Archaeal fructose-1,6-bisphosphate aldolases constitute a new family of archaeal type class I aldolase. J. Biol. Chem. 2001, 276, 28710–28718. 10.1074/jbc.M103447200. [DOI] [PubMed] [Google Scholar]
- Marsh J. J.; Lebherz H. G. Fructose-bisphosphate aldolases: an evolutionary history. Trends Biochem. Sci. 1992, 17, 110–113. 10.1016/0968-0004(92)90247-7. [DOI] [PubMed] [Google Scholar]
- Hasegawa A.; Ogasawara H.; Kori A.; Teramoto J.; Ishihama A. The transcription regulator AllR senses both allantoin and glyoxylate and controls a set of genes for degradation and reutilization of purines. Microbiology 2008, 154, 3366–3378. 10.1099/mic.0.2008/020016-0. [DOI] [PubMed] [Google Scholar]
- Kredich N. M. The molecular basis for positive regulation of cys promoters in Salmonella typhimurium and Escherichia coli. Mol. Microbiol. 1992, 6, 2747–2753. 10.1111/j.1365-2958.1992.tb01453.x. [DOI] [PubMed] [Google Scholar]
- Jourdan A. D.; Stauffer G. V. Mutational Analysis of the Transcriptional Regulator GcvA: Amino Acids Important for Activation, Repression, and DNA Binding. J. Bacteriol. 1998, 180, 4865. 10.1128/JB.180.18.4865-4871.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L. M.; Nygaard P. Identification of hypoxanthine and guanine as the co-repressors for the purine regulon genes of Escherichia coli. Mol. Microbiol. 1990, 4, 2187–2192. 10.1111/j.1365-2958.1990.tb00580.x. [DOI] [PubMed] [Google Scholar]
- Shimada T.; Hirao K.; Kori A.; Yamamoto K.; Ishihama A. RutR is the uracil/thymine-sensing master regulator of a set of genes for synthesis and degradation of pyrimidines. Mol. Microbiol. 2007, 66, 744–757. 10.1111/j.1365-2958.2007.05954.x. [DOI] [PubMed] [Google Scholar]
- Pittard A. J.; Davidson B. E. TyrR protein of Escherichia coli and its role as repressor and activator. Mol. Microbiol. 1991, 5, 1585–1592. 10.1111/j.1365-2958.1991.tb01904.x. [DOI] [PubMed] [Google Scholar]
- Reed M. C.; Lieb A.; Nijhout H. F. The biological significance of substrate inhibition: a mechanism with diverse functions. BioEssays 2010, 32, 422–429. 10.1002/bies.200900167. [DOI] [PubMed] [Google Scholar]
- Guixé V.; Babul J. Effect of ATP on phosphofructokinase-2 from Escherichia coli. A mutant enzyme altered in the allosteric site for MgATP. J. Biol. Chem. 1985, 260, 11001–11005. 10.1016/S0021-9258(17)39138-X. [DOI] [PubMed] [Google Scholar]
- Zheng R. L.; Kemp R. G. The mechanism of ATP inhibition of wild type and mutant phosphofructo-1-kinase from Escherichia coli. J. Biol. Chem. 1992, 267, 23640–23645. 10.1016/S0021-9258(18)35886-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.