

Abstract
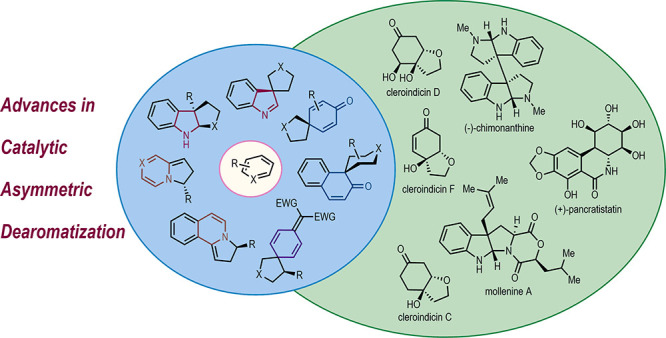
Asymmetric catalysis has been recognized as the most enabling strategy for accessing chiral molecules in enantioenriched forms. Catalytic asymmetric dearomatization is an emerging and dynamic research subject in asymmetric catalysis, which has received considerable attention in recent years. The direct transformations from readily available aromatic feedstocks to structurally diverse three-dimensional polycyclic molecules make catalytic asymmetric dearomatization reactions of broad interest for both organic synthesis and medicinal chemistry. However, the inherent difficulty for the disruption of aromaticity demands a large energy input during the dearomatization process, which might be incompatible with the conditions generally required by asymmetric catalysis. In this Outlook, we will discuss representative strategies and examples of catalytic asymmetric dearomatization reactions of various aromatic compounds and try to convince readers that by overcoming the above obstacles, catalytic asymmetric dearomatization reactions could advance chemical sciences in many respects.
Short abstract
Recent advances in catalytic asymmetric dearomatization provide diverse polycyclic molecular scaffolds, novel mechanistic insights, and unprecedented disconnection strategies for total synthesis.
Introduction
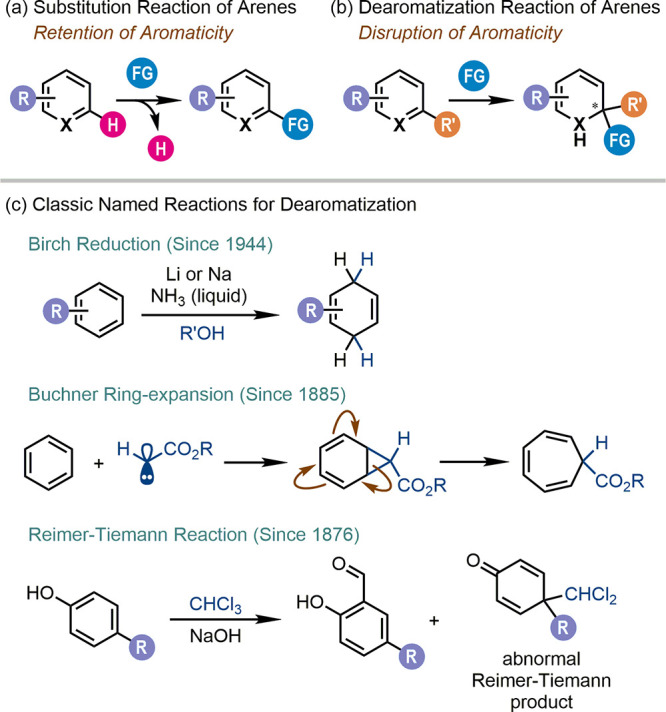
Since the discovery of benzene by Michael Faraday in 1825,1 the research and application of aromatic compounds in both the academic and industrial levels have been contributing to the development of humankind for almost two centuries. As bulk and fundamental chemical feedstocks, aromatic compounds play a prominent role in organic synthesis. However, as a result of “aromaticity”, the extraordinary stability caused by the delocalization of the π-electrons,2 aromatic compounds mainly participate in substitution reactions, where a hydrogen atom on the aromatic ring is replaced by a functional group; yet, its aromaticity is not disrupted (Scheme 1a).3 On the other hand, dearomatization is another general but relatively underdeveloped type of transformation of aromatic compounds where a functional group is added to the aromatic ring, leading to the permanent loss or significant decrease of its aromaticity (Scheme 1b). Historically, Birch reduction,4 Buchner ring-expansion,5 and the Reimer–Tiemann reaction6 of para-substituted phenols were among the rare examples of named reactions for dearomatization (Scheme 1c), which were usually operated under harsh conditions or with narrow substrate scopes. Notably, the nucleophilic addition7 and hydrogenation reactions8 of aromatic compounds can be promoted by chiral catalysts. Besides, enzyme-catalyzed transformations are well-known for dearomatization reactions, which are exemplified by the arene cis-dihydroxylation promoted by arene dioxygenase enzymes.9
Scheme 1. (a, b) General Reaction Types of Arenes and (c) Classic Named Reactions for Dearomatization.
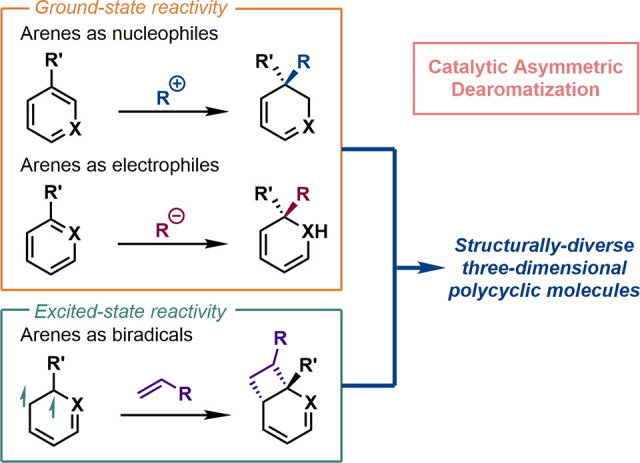
The preparation of chiral molecules in enantioenriched forms is of great importance in synthetic chemistry,10 pharmaceutical industry,11 and materials science.12 Among various available methods to this end, homogeneous asymmetric catalysis is probably the most efficient and diverse one.13 The great achievements in this area were acknowledged by the Nobel Prize in Chemistry in 2001 to Knowles, Noyori, and Sharpless in honor of their contributions for the development of asymmetric hydrogenation and oxidation reactions, respectively.14 One of the most active directions in asymmetric catalysis in recent years is to push the limit of functional groups compatible with diverse transformations to those traditionally regarded as “inert” ones. In this regard, catalytic asymmetric dearomatization (CADA) reactions15 have emerged as a powerful synthetic strategy in the past decade, which makes various aromatic units reactive functionalities for asymmetric synthesis.
The most distinctive feature of catalytic asymmetric dearomatization reactions is the potential for exploring previously untouched chemical spaces.16 They not only provide alternative retrosynthetic strategies to access known polycyclic molecules but also serve as indispensable tools to forge novel molecular scaffolds with diverse and unprecedented topologies. Particularly, the increased levels of saturation resulting from dearomatization, and of stereoisomerism led by the incorporation of new stereogenic centers, make the libraries of products of catalytic asymmetric dearomatization reactions appealing in the drug-discovery process.
However, multiple challenges associated with catalytic asymmetric dearomatization reactions need to be addressed. In general, the extraordinary stability of aromatic compounds makes the dearomatization process thermodynamically unfavorable. Therefore, many successful dearomatization reactions are usually coupled with the irreversible formation of a strong carbon–carbon, carbon–hydrogen, or carbon–heteroatom bond to compensate the energetic uphill required by the disruption of aromaticity. Meanwhile, achieving high stereochemical control during the dearomatization process is another challenging task. The design and development of enabling chiral catalytic systems is a key solution to reduce the energetic barriers of dearomatization and at the same time to distinguish between the subtle diastereomeric transition states.
In this Outlook, we would like to give a personal perspective on this dynamic research field with a few representative examples in which aromatic compounds participate in asymmetric dearomatization reactions as nucleophiles, electrophiles, and excited state biradicals (Scheme 2). Selective applications of asymmetric dearomatization reactions in total synthesis will also be covered. Rather than being a comprehensive review, this Outlook will focus on how advances in catalytic asymmetric dearomatization reactions impact the research of organic chemistry with innovative mechanistic understanding, expanded chemical space, and transformative synthetic routes toward complex target molecules.
Scheme 2. General Strategies for Catalytic Asymmetric Dearomatization Reactions.
Arenes as Nucleophiles
In the most-studied type of catalytic asymmetric dearomatization reaction, electron-rich arenes react with an appropriately tethered electrophile, leading to various spirocyclic molecules. Particularly, the reactions involving the electrophilic π-allyliridium species catalyzed by a chiral Ir-complex have exhibited general synthetic potential and a broad scope.17
In 2010, our group reported the first Ir-catalyzed intramolecular asymmetric allylic dearomatization reaction (Scheme 3).18 In the presence of a catalyst consisting of [Ir(cod)Cl]2 and Me-THQphos (R,Ra)-L1, tryptamine-derived allylic carbonates 1 were converted smoothly to chiral six-membered-ring spiroindolenines 2 in up to 95% yield. Two contiguous stereogenic centers including a quaternary one were established with excellent stereochemical control (up to >99:1 dr and 96% ee). This reaction mode was recently extended to bis(indol-3-yl) substituted allylic carbonates 3.19 The enantioselective desymmetrization of 3 was realized under slightly modified conditions, allowing the exclusive formation of chiral six-membered-ring spiroindolenines decorated with an additional indole ring (4) in up to 99% yield and 99% ee. Notably, the core structure of 2 is related to the orally active growth hormone secretagogue MK-677 (ibutamoren) and other bioactive molecules.20
Scheme 3. Synthesis of Six-Membered-Ring Spiroindolenines via Ir-Catalyzed Asymmetric Allylic Dearomatization Reactions.

When the linkage between the indole core and the allylic carbonate in the substrates was shortened by one methylene group, the synthesis of chiral five-membered-ring spiroindolenines was also achieved (Scheme 4). The asymmetric allylic dearomatization of indol-3-yl allylic carbonates 5 by an Ir-catalyst derived from the Feringa phosphoramidite (S,S,Sa)-L3 led to spiroindolenines 6, whose imine moiety was reduced in situ by NaBH3CN to afford the corresponding spiroindolines 7 in up to 95% yield, 13:1 dr, and 98% ee.21 Alternatively, when racemic indol-3-yl methanamine-derived allylic carbonates (±)-8 were subjected to the same reaction conditions, three diastereoisomers of five-membered-ring aza-spiroindolenines 9a–c were delivered in high enantiopurity (up 98% ee).22 To be noted, in all the above syntheses of chiral spiroindolenines, the absolute configuration of the allylic stereogenic center was dominated by the chiral Ir-catalyst, while the usually high facial selectivity for the prochiral nucleophiles should be attributed to the structurally well-defined intramolecular cyclization transition states.
Scheme 4. Synthesis of Five-Membered-Ring Spiroindolenines via Ir-Catalyzed Asymmetric Allylic Dearomatization Reactions.

The most intriguing reactivity of the chiral spiroindolenines is their stereoselective ring-expansive migration (Scheme 5). When treated with a catalytic amount of tosylic acid (30 mol %), five-membered-ring spiroindolenines 6 underwent allyl migration, affording tetrahydrocarbazoles 7 in up to 92% yield. Interestingly, this allyl migration was highly stereoretentive, with es values of up to 99% [es = (eeproduct/eesubstrate) × 100%], and the absolute configuration at the allylic position remained unchanged during the migration.21 Comprehensive mechanistic studies revealed that the allyl migration proceeded through a “three-center–two-electron (3c–2e)”-type transition state (TS1). The attractive interaction between the positively charged allyl moiety and the electron-rich indole ring guaranteed the stereoretentive nature of the migration process.23 It was also found that if two potential migratory groups were available for a spiroindolenine, the one with the stronger ability to stabilize positive charge was more reactive. In addition, the activity of spiroindolenines toward ring-expansive migration was also influenced by other stereogenic centers in the molecule. Therefore, the treatment of the three diastereoisomers of five-membered-ring aza-spiroindolenines 9a–c with tosylic acid provided significantly varied outcomes. The iminium migration of 9c was finished within 1 min at room temperature, while the similar reaction of 9b required 12 h. Both reactions delivered tetrahydro-β-carboline cis-11 in high yields and es values. In contrast, 9a remained intact in the presence of tosylic acid even at 50 °C for 12 h. However, with stronger acid (saturated HCl in THF), 9a underwent ring-expansive migration with the configuration of the iminium carbon partially reversed.22 Guided by these mechanistic insights, a one-pot asymmetric allylic dearomatization/ring-expansive iminium migration sequence of allylic carbonate 12 was realized with the Ir-catalyst derived from BHPphos (R)-L4, which furnished tetrahydro-β-carboline 13 in 74% yield and 94% ee. The N-Bn methanamine tether that was originally attached to the C3 position of the indole ring finally moved to the C2 position. The proposed spiroindolenine intermediate was observed by in situ IR spectroscopy experiments.24
Scheme 5. Stereoselective Ring-Expansive Migration of Spiroindolenines.

Mechanistically, the enantioselective formation and ring-expansive migration of five-membered-ring spiroindolenines are closely related to asymmetric Pictet–Spengler reactions.25 On the basis of the systematic studies on the chemistry of spiroindolenines, we demonstrated the relationship of the electronic properties of the substrates, reaction pathways, and stereochemistry of asymmetric Pictet–Spengler reactions by density functional theory (DFT) calculations and Born–Oppenheimer molecular dynamics (BOMD) simulations.26 A unified two-dimensional mechanistic spectrum with two limiting conditions was proposed and successfully applied in the rational designs of a series of asymmetric transformations of spiroindolenines beyond classic Pictet–Spengler reactions.27
The scope of Ir-catalyzed asymmetric allylic dearomatization reactions could be extended to a variety of fused bicyclic (hetero)aromatic compounds, including naphthols,28 (iso)quinolines,29 benzoxazoles, benzothiazoles, and benzimidazoles,30 etc. However, in most cases, the aromaticity of only one aromatic ring was perturbed, while the other, usually a phenyl ring, remained intact. It was believed that the restoration of the aromaticity of this phenyl ring might be a key compensating factor to the unfavorable dearomatization process. In this regard, the simultaneous weakening of the aromaticity of two consecutive aromatic rings was an ambitious challenge in the area of catalytic asymmetric dearomatization reactions. In 2018, we disclosed an Ir-catalyzed intramolecular asymmetric allylic amination of hydroxyquinoline-derived allylic chlorides 14 (Scheme 6).31 The deprotonation of the hydroxyl group promoted the nucleophilicity of the nitrogen atom of 14, which facilitated the desired asymmetric allylic amination reactions. The utilization of an N-heterocyclic carbene ligand derived from a chiral triazolium salt (S)-L5 permitted the high yields (up to 99%) and excellent enantiopurity (up to 97% ee) of cyclic conjugated enone products 15. Theoretical analyses including NICS(1)_ZZ (the ZZ tensor component of the nuclear independent chemical shift values at the points 1 Å above the ring centers) and multicenter bond indices confirmed that the aromaticity of both rings of the quinoline substrates decreased significantly in this reaction.
Scheme 6. Ir-Catalyzed Asymmetric Allylic Amination with 5-Hydroxyquinoline and 6-Hydroxyisoquinoline Derivatives.

Very recently, we realized the intermolecular version of this reaction and uncovered an unprecedented phenomenon in asymmetric catalysis, namely, time-dependent enantiodivergent synthesis (Scheme 6).32 The asymmetric allylic amination reactions between hydroxyisoquinolines 16 and racemic tert-butyl allylic carbonates [(rac)-17, 2 equiv] were promoted by a chiral Ir-complex derived from Carreira-type (phosphoramidite, olefin) ligand (S)-L6. Interestingly, each enantiomer of the desired products 18 could be obtained in high yields and enantiopurity when the reactions were quenched at different reaction times [(R)-18, up to 78% yield, 99% ee for 9–11 h; (S)-18, up to 80% yield, 94% ee for 5–10 min]. Systematic mechanistic investigations revealed that four independent transformations, allylic amination of (S/R)-17 with hydroxyisoquinolines 16 and allylic etherification of (S/R)-18 with methanol, proceeded in the presence of the same chiral Ir-catalyst. The appropriate permutation of individual reaction rates was crucial for achieving enantiodivergent synthesis of 18 with the reaction time as a key parameter.
Arenes as Electrophiles
The asymmetric nucleophilic addition to electron-deficient (hetero)aromatic compounds constitutes another important category of dearomatization reactions, with the Reissert reaction being the most famous example.7 In this section, we highlight some recent contributions on the catalytic asymmetric dearomative cyclization reactions of 3-nitroindoles initiated by nucleophilic additions at the C2 position of the indole ring.
In 2014, the Trost group reported one example of Pd-catalyzed asymmetric dearomative cyclization of N-phenylsulfonyl 3-nitroindole 19 with trimethylenemethane (TMM)-donor 20 (Scheme 7).33 The Pd-TMM complex formed in situ from Pd(dba)2, chiral phosphoramidite (R,R,Ra)-L3, and 20 underwent a formal [3 + 2] cyclization with 19, leading to 21 in quantitative yield with 66% ee. Inspired by this pioneering work, our group achieved stereodivergent dearomative [3 + 2] cyclization of a series of 3-nitroindoles 22 with racemic 2-vinyloxiranes (rac)-23 catalyzed by a Pd-complex derived from [Pd(η3-C3H5)Cl]2 and a novel PHOX ligand (S)-L7 (Scheme 7).34 The reaction started with the oxidative addition of (rac)-23 with the Pd-catalyst, which resulted in a zwitterionic species that underwent the dearomative cyclization with 22. Interestingly, the diastereoselectivity of the reactions was significantly influenced by the solvent. The two stereogenic centers on the indoline ring of the target products always adopted the cis configuration (3aR and 8aR when R′ ≠ H) due to the ring strain, while the absolute configuration at the allylic position was the opposite in toluene [24 (3S), up to 99% yield, 95:5 dr, 88% ee] or acetonitrile [24′ (3R), up to 98% yield, 93:7 dr, 98% ee]. Finally, Hammett analyses and ESI-MS experiments suggested varied rate-determining steps in the two reaction systems, namely, the first addition to 3-nitroindole in toluene and the second addition to π-allylpalladium moiety in acetonitrile, respectively.
Scheme 7. Pd-Catalyzed Asymmetric Dearomatization of 3-Nitroindoles.

Almost at the same time, the groups of Shi,35 Wang,36 and Ding and Hou37 independently reported the Pd-catalyzed asymmetric dearomative cyclization [3 + 2] reactions of 3-nitroindoles with vinyl cyclopropanes [(rac)-26 and (rac)-29] and vinyl aziridines [(rac)-32], respectively (Scheme 7). Although different chiral Pd-complexes, derived from chiral phosphoramidite ligand [(R,R,Sa)-L8], BOX ligands (L9, and L10), or bisphosphine ligand (L11), were identified as the optimal catalysts in each cases, these reactions all proceeded in similar sequences, accessing densely substituted chiral cyclopenta[b]indolines (27, 30, and 30′) or pyrroloindolines (33 and 33′) with high yields and good stereochemical controls. It should be noted that in the reactions of vinyl aziridines, the relative configurations of the major products were different in the two reaction systems. Therefore, the stereodivergent syntheses of these pyrroloindolines could be achieved when each enantiomer of L10 and L11 was applied.
Besides, the groups of Arai,38 and Stanley39 realized the Cu-catalyzed asymmetric dearomative [3 + 2] cyclization reactions of 3-nitroindoles with azomethine ylides. Yuan and co-workers40 reported the corresponding [3 + 2] and [4 + 2] reactions of 3-nitroindoles with 3-isothiocyanato oxindoles or Nazarov reagents by quinine-based chiral bifunctional thiourea/thiocarbamate catalyst, or chiral Zn-complex. Notably, the asymmetric dearomative cyclization reactions could also be extended to 2-nitrobenzofurans.41
Recently, chiral phosphine-catalyzed dearomative [3 + 2] cyclization reactions between 3-nitroindoles 34 and allenoates 35 were reported by the groups of Zhang42 and Lu43 independently (Scheme 8). In this reaction design, the addition of chiral phosphines P1 or P2 to allenoates generated the key zwitterionic species, which subsequently attacked 3-nitroindoles with its α-terminus. After the second C–C bond-formation between the C3-position of the indole ring and the γ-terminus, and the extrusion of the phosphine catalyst, the desired cyclopenta[b]indoline products 36 were delivered in good yields with high enantioselectivity in both cases. Besides, Wang, Guo and co-workers realized asymmetric dearomative [3 + 2] cyclization reactions of 2-nitrobenzofurans by applying similar strategies.44
Scheme 8. Phosphine-Catalyzed Asymmetric Dearomatization of 3-Nitroindoles.

Arenes as Excited State Biradicals
Visible-light-promoted [2 + 2] reactions via an energy transfer mechanism have recently witnessed considerable attention from the synthetic chemistry community.45 Particularly, mediated by a suitable photosensitizer, aromatic compounds in excited states can be generated and display distinctive reactivity compared with the original ground-state molecules. It has been proven that, if appropriately decorated, the arenes in excited states can be further activated by chiral Lewis acid catalysts, which allows the asymmetric dearomatization reactions with external unsaturated functionalities via [2 + 2] cycloaddition.
In 2018, Meggers, Baik, and co-workers reported the first visible-light-promoted asymmetric dearomatization reactions (Scheme 9).46 2-N-Acylpyrazole benzofurans 37 were identified as the suitable substrate for the coordination to the chiral Lewis acid Δ-RhS. The resulting complex reached its first singlet excited state (S1) under the irradiation of blue LEDs. The subsequent intersystem crossing led to the corresponding triplet excited state (T1), which reacted with external olefins 38, furnishing asymmetric dearomative [2 + 2] cycloaddition reactions. After the hydrolysis of the N-acylpyrazole moiety and the methyl esterification, the desired dearomatized products 39 were obtained in high yields (up to 93%) and good regio- and enantioselectivities (up to 99% ee). DFT calculations revealed a stepwise cyclization between the 1,2-biradical of the T1 state arenes and the olefins. The origin of regioselectivity was well-explained based on the resonance stabilization for the unpaired electron by the neighboring aryl or carbonyl groups. A working model was also proposed, accounting for the experimentally observed absolute configuration of the major products.
Scheme 9. Visible-Light-Promoted Asymmetric Dearomatization Reactions.

The Bach group achieved significant progress on visible-light-promoted asymmetric cycloaddition reactions (Scheme 9).47 Guided by the UV/vis spectra of phenanthrene-9-carboxaldehyde in the presence of variable amounts of EtAlCl2, it was confirmed that the coordination to a Lewis acid would induce a bathochromic shift of the π–π* transition of 40 (from 316 to 387 nm). Therefore, the utilization of a chiral Lewis acid catalyst would promote the asymmetric photocycloaddition of 40 under long wavelength irradiation by inhibiting the racemic background reactions of uncoordinated substrates. This reaction design was successfully executed using various alkenes including 41 as the partners in the presence of chiral oxazaborolidine C1 and visible light (λ = 457 nm). The desired products 42 were delivered smoothly in up to 93% yield with 96% ee.
Applications in Total Syntheses
Catalytic asymmetric dearomatization reactions have been applied in the total syntheses of complex molecules and provided unprecedented retrosynthetic disconnection strategies. The asymmetric dearomative cyclizations of tryptamine derivatives are probably the most widely studied methods for the construction of the key pyrroloindoline skeletons in diverse indole-based natural products.48 In this regard, MacMillan and co-workers have made pioneering contributions and completed the total syntheses of (−)-(debromo)flustramine B, (−)-diazonamide A, hodgkinsine, and a series of related natural products using iminium catalysis or Cu-catalyzed asymmetric arylation with iodonium salts as the key steps.49
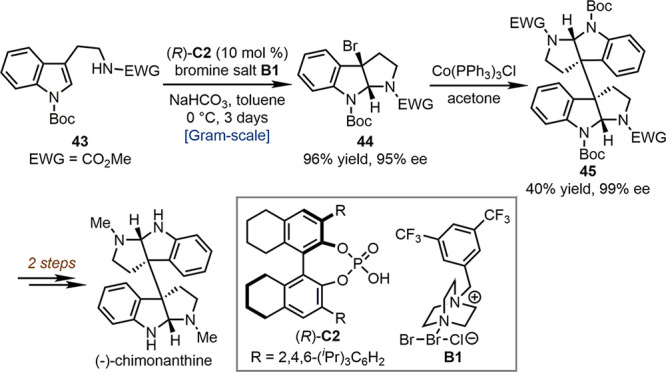
In 2013, Xie, Lai, Ma, and co-workers developed a highly enantioselective dearomative bromocyclization reaction and applied this reaction in the total synthesis of (−)-chimonanthine (Scheme 10).50 The reaction of tryptamine derivative 43 with electrophilic brominating reagent B1 in the presence of a chiral phosphoric acid (R)-C2 afforded bromo-substituted pyrroloindoline 44 on a gram scale (96% yield, 95% ee). Subsequently, 44 underwent Co-catalyzed dimerization leading to the pyrroloindoline dimer 45. The enantiopurity of 45 was improved to 99% ee probably due to the resolution during the dimerization process. Finally, the total synthesis of (−)-chimonanthine was completed after two steps of functional group manipulations.
Scheme 10. Total Synthesis of (−)-Chimonanthine by the Asymmetric Dearomative Bromocyclization Reaction.
In 2018, our group developed Pd-catalyzed asymmetric dearomative prenylation reactions.51 With various C3-substituted indole derivatives as the substrates, this reaction enantioselectively installed a prenyl group or related isoprenoids at the C3 position of the indole ring with the concomitant cyclization of the side chain, leading to prenylated pyrroloindolines and related molecules. The synthetic potential of this reaction was exemplified by the expedient syntheses of a series of natural products containing the prenylated pyrroloindoline structural core (Scheme 11). For example, the reaction of 6-bromo-substituted tryptamine derivative 46 and prenyl carbonate 47 proceeded in the presence of Pd-catalyst derived from chiral phosphoramidite ligand Allylphos (R)-L12. The corresponding product 48 was obtained in 95% yield with 94% ee. The N-prenylation of 48 generated 49, which was a known precursor for (−)-debromoflustramine B. Besides, simple two-step functional-group manipulations of 49 delivered (−)-flustramine B efficiently (90% yield). Starting from another chiral functionalized pyrroloindoline product 50, the total synthesis of (−)-pseudophrynaminol was achieved by reducing the N-CO2Me group and removing the silyl protecting group of a primary alcohol. Notably, this asymmetric dearomative prenylation reaction permitted the structure revision and facile synthesis of mollenine A. The treatment of prenylated pyrroloindoline 52, which was generated from Boc-l-Trp-OMe, with TMSI led to the release of free secondary amine in 53 (83% yield). The subsequent condensation with l-leucic acid furnished the corrected structure of mollenine A (59% yield). In addition, mollenine A could also be obtained in gram scale (82% yield) from l-Trp-l-leucic acid 54 and prenyl carbonate 47 under the standard conditions of the asymmetric dearomative prenylation reaction. In this protocol, three chemical bonds were formed in a highly ordered manner.
Scheme 11. Application of Pd-Catalyzed Asymmetric Dearomative Prenylation Reactions in Total Syntheses.

Enantioselective transformations of the molecules obtained from dearomatization are another important strategy that has been employed in natural product syntheses. In 2010, our group reported chiral phosphoric acid-catalyzed intramolecular oxo-Michael addition reactions for the desymmetrization of cyclohexadienones.52 Using this reaction as a key step, the asymmetric synthesis of cleroindicins was realized (Scheme 12). The oxidative dearomatization of commercially available 4-(2-hydroxyethyl)phenol 55 by oxone delivered cyclohexadienone 56. Subsequently, the desymmetrization of this molecule by intramolecular oxo-Michael addition in the presence of chiral phosphoric acid (S)-C3 was achieved, leading to bicyclic enone 57 in 80% ee. The treatment of 57 with Triton B and aluminum amalgam promoted intramolecular epoxidation and reduction, yielding cleroindicin D (27% yield from 56). Besides, the reduction of the hydroperoxyl group in 57 by P(OPh)3 generated cleroindicin F (80% ee, 57% yield from 56). Further hydrogenation of the enone moiety of cleroindicin F with Pd/C furnished cleroindicin C in 94% yield with 81% ee.
Scheme 12. Asymmetric Syntheses of Cleroindicins based on Desymmetrization of Cyclohexadienones.

Sarlah and workers systematically studied the dearomatization of plain aromatics (benzene, naphthalene, tetracene, etc.) with N-methyl-1,2,4-triazoline-3,5-dione (MTAD) as an arenophile under visible light. The reaction afforded the arene-arenophile adduct which could be trapped under low temperature by olefin-like transformation or transition-metal-catalyzed amino functionalization reactions.53 This reaction found broad applications in the total syntheses of polycyclic natural products (Scheme 13).54 For example, treating the arene-arenophile adduct with a chiral Ni-complex derived from PHOX ligand L13 and the aryl Grignard reagent generated trans-1,2-carboamination products 58 with high enantiopurity (96% ee) on a >10 g scale. The two-step oxidation of the resulting 1,4-diene moiety of 58 delivered 59. The subsequent urazole reduction and Co-catalyzed carbonylative coupling furnished (+)-7-deoxypancratistatin, from which a formal C–H oxidation led to (+)-pancratistatin. Notably, the synthesis of trans-1,2-carboamination products 58 could be upgraded to >20 g scale without erosion of yield and enantioselectivity with reduced loading of Ni-precursor and L13. With this reaction as the first step, the total syntheses of (+)-lycoricidine and (+)-narciclasine could be finished in six steps on a >8 g scale and seven steps on a gram scale, respectively.
Scheme 13. Synthetic Applications of Arenophile-Mediated Dearomative Functionalization.
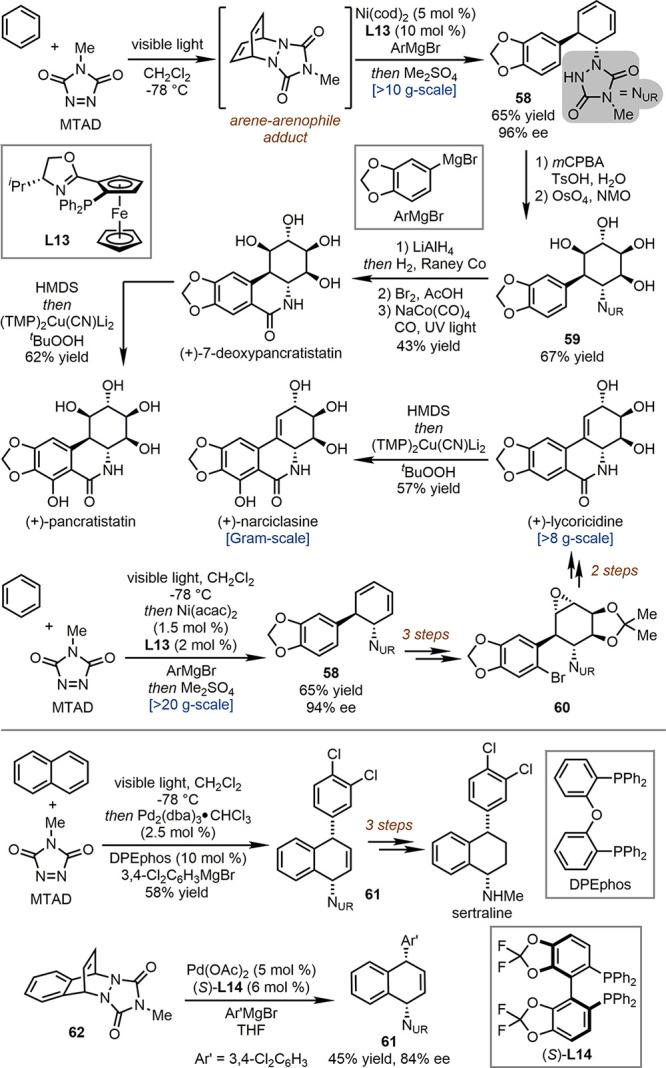
Sarlah and co-workers also developed Pd-catalyzed dearomative syn-1,4-carboamination of naphthalenes with the aryl Grignard reagent.55 Product 61 could be readily transformed to sertraline in three steps (Scheme 13). It was confirmed that the isolated arene-arenophile adduct 62 underwent an enantioselective ring-opening reaction in the presence of a Pd-catalyst derived from chiral bisphosphine ligand DIFLUORPHOS (S)-L14. Compound 61 could be obtained in 45% yield with 84% ee.
Summary and Perspectives
The progress in catalytic asymmetric dearomatization reactions in recent years has exerted great influence on organic chemistry. The dearomatization processes are now no longer the “forbidden zone” for versatile chemical synthesis. On the contrary, the exploration of previously untouched chemical spaces brings about numerous opportunities for advancing the chemical sciences in multiple respects. As showcased by the examples discussed in this Outlook, the energetically unfavorable disruption of aromaticity can be readily compensated by irreversible formation of carbon–carbon, carbon–hydrogen, or carbon–heteroatom bonds. In addition, asymmetric dearomatization processes deliver polycyclic skeletons with increased saturation and stereochemical complexity. The dearomatized products with structural diversity might serve as the novel candidates for drug discovery. Mechanistically, dearomatized intermediates have been proposed for many classic organic reactions. Overlooking such dearomatization pathways might lead to incorrect structure assignment for the products. In this regard, deep understanding of the dearomatized species and manipulating their reactivities would undoubtedly contribute to the development of novel synthetic methods. Finally, the asymmetric dearomatization reactions employ aromatic systems, traditionally regarded as “inert” structural units, as reactive functional groups, thus providing innovative retrosynthetic plans for complex molecules.
However, there are still significant challenges that should be overcome for further development in this area. Currently, some certain types of “activated” arenes such as indoles, phenols, pyridines, etc. are generally utilized. In contrast, the direct catalytic asymmetric dearomative transformations of “non-activated” arenes such as benzenes, naphthalenes, etc. are rather limited.56 Notably, several examples of asymmetric hydrogenation of this kind of arenes have been reported.57 To break through this predicament, one probably needs very reactive reaction partners that are generated under mild conditions and well embedded in a subtle chiral environment. To this end, we expect that the implementation of modern catalytic technologies, such as visible-light-catalysis,58 electrocatalysis,59 nanocatalysis, etc., can revolutionize the seminal dearomatization reactions discussed in the Introduction by bringing about their catalytic asymmetric variants. In addition, the known enzyme-promoted asymmetric dearomatization of nonactivated arenes might provide an alternative solution by stimulating de novo design of small molecule catalysts that mimic relevant enzymes or by improving the performance of such enzymes employing directed evolution technology. We are quite confident that the dynamic research on catalytic asymmetric dearomatization reactions will push the frontier and shape the future of synthetic chemistry.
Acknowledgments
We thank the National Natural Science Foundation of China (21772219, 21821002, and 22031012), Science and Technology Commission of Shanghai Municipality (18QA1404900 and 18JC1411302), Chinese Academy of Sciences (XDB20000000), and Youth Innovation Promotion Association (2017302) of CAS for generous financial support for our works presented in this review. S.-L.Y. acknowledges the support from the Tencent Foundation through the XPLORER PRIZE.
The authors declare no competing financial interest.
References
- Kaiser R. Bicarburet of Hydrogen”. Reappraisal of the Discovery of Benzene in 1825 with the Analytical Methods of 1968. Angew. Chem., Int. Ed. Engl. 1968, 7, 345–350. 10.1002/anie.196803451. [DOI] [Google Scholar]
- a Schleyer P. v. R.; Jiao H. What is aromaticity?. Pure Appl. Chem. 1996, 68, 209–218. 10.1351/pac199668020209. [DOI] [Google Scholar]; b Randić M. Aromaticity of Polycyclic Conjugated Hydrocarbons. Chem. Rev. 2003, 103, 3449–3606. 10.1021/cr9903656. [DOI] [PubMed] [Google Scholar]; c Hua Y.; Zhang H.; Xia H. Aromaticity: History and Development. Chin. J. Org. Chem. 2018, 38, 11–28. 10.6023/cjoc201709009. [DOI] [Google Scholar]
- a Astruc D., Ed. Modern Arene Chemistry: Concepts, Synthesis, and Applications; Wiley-VCH, 2002. [Google Scholar]; b Mortier J., Ed. Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds; Wiley-VCH, 2015. [Google Scholar]
- For seminal reports:; a Birch A. J. Reduction by dissolving metals. I. J. Chem. Soc. 1944, 430–436. For selected reviews: 10.1039/jr9440000430. [DOI] [Google Scholar]; b Birch A. J. The Birch reduction in organic synthesis. Pure Appl. Chem. 1996, 68, 553–556. 10.1351/pac199668030553. [DOI] [Google Scholar]; c Heravi M. M.; Fard M. V.; Faghihi Z. Recent Applications of Birch Reduction in Total Synthesis of Natural Products. Curr. Org. Chem. 2015, 19, 1491–1525. For selected recent examples: 10.2174/1385272819666150608220335. [DOI] [Google Scholar]; d Peters B. K.; Rodriguez K. X.; Reisberg S. H.; Beil S. B.; Hickey D. P.; Kawamata Y.; Collins M.; Starr J.; Chen L.; Udyavara S.; Klunder K.; Gorey T. J.; Anderson S. L.; Neurock M.; Minteer S. D.; Baran P. S. Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-Ion Battery Chemistry. Science 2019, 363, 838–845. 10.1126/science.aav5606. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chatterjee A.; König B. Birch-Type Photoreduction of Arenes and Heteroarenes by Sensitized Electron Transfer. Angew. Chem., Int. Ed. 2019, 58, 14289–14294. 10.1002/anie.201905485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For seminal reports:; a Buchner E.; Curtius T. Synthesis of β-keto esters from aldehydes and diazoacetic acid. Ber. Dtsch. Chem. Ges. 1885, 18, 2371–2377. 10.1002/cber.188501802118. [DOI] [Google Scholar]; For selected reviews:; b Lebel H.; Marcoux J.-F.; Molinaro C.; Charette A. B. Stereoselective Cyclopropanation Reactions. Chem. Rev. 2003, 103, 977–1050. 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]; c Reisman S. E.; Nani R. R.; Levin S. Buchner and Beyond: Arene Cyclopropanation as Applied to Natural Product Total Synthesis. Synlett 2011, 2011, 2437–2442. 10.1055/s-0031-1289520. [DOI] [Google Scholar]; d Ford A.; Miel H.; Ring A.; Slattery C. N.; Maguire A. R.; McKervey M. A. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. 10.1021/acs.chemrev.5b00121. [DOI] [PubMed] [Google Scholar]
- For seminal reports:; a Reimer K.; Tiemann F. The effect of chloroform on phenolates. Ber. Dtsch. Chem. Ges. 1876, 9, 824–828. For selected reviews: 10.1002/cber.187600901247. [DOI] [Google Scholar]; b Wynberg H. The Reimer-Tiemann reaction. Chem. Rev. 1960, 60, 169–184. 10.1021/cr60204a003. [DOI] [Google Scholar]; c Wynberg H. The Reimer-Tiemann Reaction. Comp. Org. Synth. 1991, 2, 769–775. 10.1016/B978-0-08-052349-1.00048-2. [DOI] [Google Scholar]
- a Bull J. A.; Mousseau J. J.; Pelletier G.; Charette A. B. Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev. 2012, 112, 2642–2713. 10.1021/cr200251d. [DOI] [PubMed] [Google Scholar]; b Tsukano C.; Takemoto Y.. Dearomatization Reactions of Electron-Deficient Aromatic Rings. In Asymmetric Dearomatization Reactions; You S.-L., Ed.; Wiley-VCH, 2016; pp 247–278. [Google Scholar]
- a Zhou Y.-G. Asymmetric Hydrogenation of Heteroaromatic Compounds. Acc. Chem. Res. 2007, 40, 1357–1366. 10.1021/ar700094b. [DOI] [PubMed] [Google Scholar]; b Wang D.-S.; Chen Q.-A.; Lu S.-M.; Zhou Y.-G. Asymmetric Hydrogenation of Heteroarenes and Arenes. Chem. Rev. 2012, 112, 2557–2590. 10.1021/cr200328h. [DOI] [PubMed] [Google Scholar]; c Chen Z.-P.; Zhou Y.-G. Asymmetric Hydrogenation of Heteroarenes with Multiple Heteroatoms. Synthesis 2016, 48, 1769–1781. 10.1055/s-0035-1561622. [DOI] [Google Scholar]; d Mingat G.; Rueping M.. Organocatalytic Asymmetric Transfer Hydrogenation of (Hetero)Arenes. In Asymmetric Dearomatization Reactions; You S.-L., Ed.; Wiley-VCH, 2016; pp 33–68. [Google Scholar]; e Kuwano R.Transition-Metal-Catalyzed Asymmetric Hydrogenation of Aromatics. In Asymmetric Dearomatization Reactions; You S.-L., Ed.; Wiley-VCH, 2016; pp 69–102. [Google Scholar]
- Lewis S. E.Asymmetric dearomatization under enzymatic conditions. In Asymmetric Dearomatization Reactions; You S.-L., Ed.; Wiley-VCH. 2016; pp 279–346. [Google Scholar]
- a Christmann M., Braese S., Eds.; Asymmetric Synthesis: The Essentials, 2nd ed.; Wiley-VCH, 2008. [Google Scholar]; b Carreira E. M., Yamamoto H., Eds.; Comprehensive Chirality; Elsevier, 2012. [Google Scholar]; c Blaser H.-U., Schmidt E., Eds.; Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions; Wiley, 2004. [Google Scholar]
- McConathy J.; Owens M. J. Stereochemistry in drug action. Primary Care Companion J. Clin. Psychiatry 2003, 5, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang Y.; Xu J.; Wang Y.; Chen H. Emerging chirality in nanoscience. Chem. Soc. Rev. 2013, 42, 2930–2962. 10.1039/C2CS35332F. [DOI] [PubMed] [Google Scholar]; b Liu M.; Zhang L.; Wang T. Supramolecular chirality in self-assembled systems. Chem. Rev. 2015, 115, 7304–7397. 10.1021/cr500671p. [DOI] [PubMed] [Google Scholar]
- a Jacobsen E. N., Pfaltz A., Yamamoto H., Eds. Comprehensive Asymmetric Catalysis; Springer, 2000. [Google Scholar]; b Yoon T. P.; Jacobsen E. N. Privileged chiral catalysts. Science 2003, 299, 1691–1693. 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]; c Zhou Q.-L., Ed. Privileged Chiral Ligands and Catalysts; Wiley, 2011. [Google Scholar]
- a Knowles W. S. Asymmetric hydrogenations. Angew. Chem., Int. Ed. 2002, 41, 1998–2007. . [DOI] [PubMed] [Google Scholar]; b Noyori R. Asymmetric catalysis: science and opportunities. Angew. Chem., Int. Ed. 2002, 41, 2008–2022. . [DOI] [PubMed] [Google Scholar]; c Sharpless K. B. Searching for new reactivity. Angew. Chem., Int. Ed. 2002, 41, 2024–2032. . [DOI] [PubMed] [Google Scholar]
- a Zhuo C.-X.; Zhang W.; You S.-L. Catalytic asymmetric dearomatization reactions. Angew. Chem., Int. Ed. 2002, 51, 12662–12686. [DOI] [PubMed] [Google Scholar]; b Zheng C.; You S.-L. Catalytic Asymmetric Dearomatization by Transition-Metal-Catalysis: A Method for Transformations of Aromatic Compounds. Chem. 2016, 1, 830–857. 10.1016/j.chempr.2016.11.005. [DOI] [Google Scholar]; c You S.-L., Ed. Asymmetric Dearomatization Reactions; Wiley, 2016. [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- a Zhuo C.-X.; Zheng C.; You S.-L. Transition-Metal-Catalyzed Asymmetric Allylic Dearomatization Reactions. Acc. Chem. Res. 2014, 47, 2558–2573. 10.1021/ar500167f. [DOI] [PubMed] [Google Scholar]; b Cheng Q.; Tu H.-F.; Zheng C.; Qu J.-P.; Helmchen G.; You S.-L. Iridium-Catalyzed Asymmetric Allylic Substitution Reactions. Chem. Rev. 2019, 119, 1855–1969. 10.1021/acs.chemrev.8b00506. [DOI] [PubMed] [Google Scholar]
- Wu Q.-F.; He H.; Liu W.-B.; You S.-L. Enantioselective Construction of Spiroindolenines by Ir-Catalyzed Allylic Alkylation Reactions. J. Am. Chem. Soc. 2010, 132, 11418–11419. 10.1021/ja105111n. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zheng C.; You S.-L. Iridium-Catalyzed Asymmetric Allylic Dearomatization by a Desymmetrization Strategy. Angew. Chem., Int. Ed. 2017, 56, 15093–15097. 10.1002/anie.201708419. [DOI] [PubMed] [Google Scholar]
- Dean D. C.; Nargund R. P.; Pong S.-S.; Chaung L.-Y. P.; Griffin P.; Melillo D. G.; Ellsworth R. L.; Van Der Ploeg L. H. T.; Patchett A. A.; Smith R. G. Development of a High Specific Activity Sulfur-35-Labeled Sulfonamide Radioligand That Allowed the Identification of a New Growth Hormone Secretagogue Receptor. J. Med. Chem. 1996, 39, 1767–1770. 10.1021/jm960054c. [DOI] [PubMed] [Google Scholar]
- Wu Q.-F.; Zheng C.; You S.-L. Enantioselective Synthesis of Spiro Cyclopentane-1,3′-indoles and 2,3,4,9-Tetrahydro-1H-carbazoles by Iridium-Catalyzed Allylic Dearomatization and Stereospecific Migration. Angew. Chem., Int. Ed. 2012, 51, 1680–1683. 10.1002/anie.201107677. [DOI] [PubMed] [Google Scholar]
- Wu Q.-F.; Zheng C.; Zhuo C.-X.; You S.-L. Highly Efficient Synthesis and Stereoselective Migration Reactions of Chiral Five-Membered Aza-Spiroindolenines: Scope and Mechanistic Understanding. Chem. Sci. 2016, 7, 4453–4459. 10.1039/C6SC00176A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C.; Wu Q.-F.; You S.-L. A Combined Theoretical and Experimental Investigation into the Highly Stereoselective Migration of Spiroindolenines. J. Org. Chem. 2013, 78, 4357–4365. 10.1021/jo400365e. [DOI] [PubMed] [Google Scholar]
- Zhuo C.-X.; Wu Q.-F.; Zhao Q.; Xu Q.-L.; You S.-L. Enantioselective Functionalization of Indoles and Pyrroles via an in Situ-Formed Spiro Intermediate. J. Am. Chem. Soc. 2013, 135, 8169–8172. 10.1021/ja403535a. [DOI] [PubMed] [Google Scholar]
- Stöckigt J.; Antonchick A. P.; Wu F.; Waldmann H. The Pictet-Spengler Reaction in Nature and in Organic Chemistry. Angew. Chem., Int. Ed. 2011, 50, 8538–8564. 10.1002/anie.201008071. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Xia Z.-L.; You S.-L. Unified Mechanistic Understandings of Pictet-Spengler Reactions. Chem. 2018, 4, 1952–1966. 10.1016/j.chempr.2018.06.006. [DOI] [Google Scholar]
- Zheng C.; You S.-L. Exploring the Chemistry of Spiroindolenines by Mechanistically-Driven Reaction Development: Asymmetric Pictet-Spengler-type Reactions and Beyond. Acc. Chem. Res. 2020, 53, 974–987. 10.1021/acs.accounts.0c00074. [DOI] [PubMed] [Google Scholar]
- a Cheng Q.; Wang Y.; You S.-L. Chemo-, Diastereo-, and Enantioselective Iridium-Catalyzed Allylic Intramolecular Dearomatization Reaction of Naphthol Derivatives. Angew. Chem., Int. Ed. 2016, 55, 3496–3499. 10.1002/anie.201511519. [DOI] [PubMed] [Google Scholar]; b Tu H.-F.; Zheng C.; Xu R.-Q.; Liu X.-J.; You S.-L. Iridium-Catalyzed Intermolecular Asymmetric Dearomatization of β-Naphthols with Allyl Alcohols or Allyl Ethers. Angew. Chem., Int. Ed. 2017, 56, 3237–3241. 10.1002/anie.201609654. [DOI] [PubMed] [Google Scholar]; c Shen D.; Chen Q.; Yan P.; Zeng X.; Zhong G. Enantioselective Dearomatization of Naphthol Derivatives with Allylic Alcohols by Cooperative Iridium and Brønsted Acid Catalysis. Angew. Chem., Int. Ed. 2017, 56, 3242–3246. 10.1002/anie.201609693. [DOI] [PubMed] [Google Scholar]
- Yang Z.-P.; Wu Q.-F.; Shao W.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Pyridines, Pyrazines, Quinolines, and Isoquinolines. J. Am. Chem. Soc. 2015, 137, 15899–15906. 10.1021/jacs.5b10440. [DOI] [PubMed] [Google Scholar]
- Yang Z.-P.; Zheng C.; Huang L.; Qian C.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Benzoxazoles, Benzothiazoles, and Benzimidazoles. Angew. Chem., Int. Ed. 2017, 56, 1530–1534. 10.1002/anie.201611056. [DOI] [PubMed] [Google Scholar]
- Yang Z.-P.; Jiang R.; Zheng C.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Alkylation of Hydroxyquinolines: Simultaneous Weakening of the Aromaticity of Two Consecutive Aromatic Rings. J. Am. Chem. Soc. 2018, 140, 3114–3119. 10.1021/jacs.8b00136. [DOI] [PubMed] [Google Scholar]
- Tu H.-F.; Yang P.; Lin Z.-H.; Zheng C.; You S.-L. Time-dependent Enantiodiver-gent Synthesis via Sequential Kinetic Resolution. Nat. Chem. 2020, 12, 838–844. 10.1038/s41557-020-0489-1. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Ehmke V.; O’Keefe B. M.; Bringley D. A. Palladium-Catalyzed Dearomative Trimethylenemethane Cycloaddition Reactions. J. Am. Chem. Soc. 2014, 136, 8213–8216. 10.1021/ja5044825. [DOI] [PubMed] [Google Scholar]
- Cheng Q.; Zhang F.; Cai Y.; Guo Y.-L.; You S.-L. Stereodivergent Synthesis of Tetrahydrofuroindoles via Pd-Catalyzed Asymmetric Dearomative Formal [3 + 2] Cycloaddition Reactions. Angew. Chem., Int. Ed. 2018, 57, 2134–2138. 10.1002/anie.201711873. [DOI] [PubMed] [Google Scholar]
- Sun M.; Zhu Z.-Q.; Gu L.; Wan X.; Mei G.-J.; Shi F. Catalytic Asymmetric Dearomative [3 + 2] Cycloaddition of Electron-Deficient Indoles with All-Carbon 1,3-Dipoles. J. Org. Chem. 2018, 83, 2341–2348. 10.1021/acs.joc.7b03259. [DOI] [PubMed] [Google Scholar]
- Zhang J.-Q.; Tong F.; Sun B.-B.; Fan W.-T.; Chen J.-B.; Hu D.; Wang X.-W. Pd-Catalyzed Asymmetric Dearomative Cycloaddition for Construction of Optically Active Pyrroloindoline and Cyclopentaindoline Derivatives: Access to 3a-Aminopyrroloindolines. J. Org. Chem. 2018, 83, 2882–2891. 10.1021/acs.joc.8b00046. [DOI] [PubMed] [Google Scholar]
- Suo J.-J.; Liu W.; Du J.; Ding C.-H.; Hou X.-L. Diastereo- and Enantioselective Palladium-Catalyzed Dearomative [3 + 2] Cycloaddition of 3-Nitroindoles. Chem. - Asian J. 2018, 13, 959–963. 10.1002/asia.201800133. [DOI] [PubMed] [Google Scholar]
- Awata A.; Arai T. PyBidine/Copper Catalyst: Asymmetric exo’-Selective [3 + 2] Cycloaddition using Imino Ester and Electrophilic Indole. Angew. Chem., Int. Ed. 2014, 53, 10462–10465. 10.1002/anie.201405223. [DOI] [PubMed] [Google Scholar]
- Gerten A. L.; Stanley L. M. Enantioselective dearomative [3 + 2] cycloadditions of indoles with azomethine ylides derived from alanine imino esters. Org. Chem. Front. 2016, 3, 339–343. 10.1039/C5QO00346F. [DOI] [Google Scholar]
- a Zhao J.-Q.; Zhou M.-Q.; Wu Z.-J.; Wang Z.-H.; Yue D.-F.; Xu X.-Y.; Zhang X.-M.; Yuan W.-C. Asymmetric Michael/Cyclization Cascade Reaction of 3-Isothiocyanato Oxindoles and 3-Nitroindoles with Amino-Thiocarbamate Catalysts: Enantioselective Synthesis of Polycyclic Spirooxindoles. Org. Lett. 2015, 17, 2238–2241. 10.1021/acs.orglett.5b00850. [DOI] [PubMed] [Google Scholar]; b Zhao J.-Q.; Wu Z.-J.; Zhou M.-Q.; Xu X.-Y.; Zhang X.-M.; Yuan W.-C. Zn-Catalyzed Diastereo- and Enantioselective Cascade Reaction of 3-Isothiocyanato Oxindoles and 3-Nitroindoles: Stereocontrolled Syntheses of Polycyclic Spirooxindoles. Org. Lett. 2015, 17, 5020–5023. 10.1021/acs.orglett.5b02489. [DOI] [PubMed] [Google Scholar]; c Yue D.-F.; Zhao J.-Q.; Chen X.-Z.; Zhou Y.; Zhang X.-M.; Xu X.-Y.; Yuan W.-C. Multiple Hydrogen-Bonding Bifunctional Thiourea-Catalyzed Asymmetric Dearomative [4 + 2] Annulation of 3-Nitroindoles: Highly Enantioselective Access to Hydrocarbazole Skeletons. Org. Lett. 2017, 19, 4508–4511. 10.1021/acs.orglett.7b02068. [DOI] [PubMed] [Google Scholar]
- a Cheng Q.; Zhang H.-J.; Yue W.-J.; You S.-L. Palladium-Catalyzed Highly Stereoselective Dearomative [3 + 2] Cycloaddition of Nitrobenzofurans. Chem. 2017, 3, 428–436. 10.1016/j.chempr.2017.06.015. [DOI] [Google Scholar]; b Zhao J.-Q.; Zhou X.-J.; Zhou Y.; Xu X.-Y.; Zhang X.-M.; Yuan W.-C. Diastereo- and Enantioselective Dearomative [3 + 2] Cycloaddition Reaction of 2-Nitrobenzofurans with 3-Isothiocyanato Oxindoles. Org. Lett. 2018, 20, 909–912. 10.1021/acs.orglett.7b03667. [DOI] [PubMed] [Google Scholar]
- Wang H.; Zhang J.; Tu Y.; Zhang J. Phosphine-Catalyzed Enantioselective Dearomative [3 + 2]- Cycloaddition of 3-Nitroindoles and 2-Nitrobenzofurans. Angew. Chem., Int. Ed. 2019, 58, 5422–5426. 10.1002/anie.201900036. [DOI] [PubMed] [Google Scholar]
- Li K.; Gonçalves T. P.; Huang K.-W.; Lu Y. Dearomatization of 3-Nitroindoles by a Phosphine-Catalyzed Enantioselective [3 + 2] Annulation Reaction. Angew. Chem., Int. Ed. 2019, 58, 5427–5431. 10.1002/anie.201900248. [DOI] [PubMed] [Google Scholar]
- Yang X.-H.; Li J.-P.; Wang D.-C.; Xie M.-S.; Qu G.-R.; Guo H.-M. Enantioselective dearomative [3 + 2] cycloaddition of 2-nitrobenzofurans with aldehyde-derived Morita-Baylis-Hillman carbonates. Chem. Commun. 2019, 55, 9144–9147. 10.1039/C9CC04542B. [DOI] [PubMed] [Google Scholar]
- a Strieth-Kalthoff F.; James M. J.; Teders M.; Pitzer L.; Glorius F. Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. 10.1039/C8CS00054A. [DOI] [PubMed] [Google Scholar]; b Sherbrook E. M.; Yoon T. P.. Asymmetric Catalysis of Triplet-State Photoreactions. In Photochemistry; Protti S., Albini A., Protti S., Eds.; Royal Society of Chemistry, 2018; Vol. 46, pp 432–448. [Google Scholar]; c Zhou Q.-Q.; Zou Y.-Q.; Lu L.-Q.; Xiao W.-J. Visible-light-induced organic photochemical reactions via energy transfer pathways. Angew. Chem., Int. Ed. 2019, 58, 1586–1604. 10.1002/anie.201803102. [DOI] [PubMed] [Google Scholar]; d Strieth-Kalthoff F.; Glorius F. Triplet Energy Transfer Photocatalysis: Unlocking the Next Level. Chem. 2020, 6, 1888–1903. 10.1016/j.chempr.2020.07.010. [DOI] [Google Scholar]
- Hu N.; Jung H.; Zheng Y.; Lee J.; Zhang L.; Ullah Z.; Xie X.; Harms K.; Baik M.-H.; Meggers E. Catalytic Asymmetric Dearomatization by Visible-Light-Activated [2 + 2] Photocycloaddition. Angew. Chem., Int. Ed. 2018, 57, 6242–6246. 10.1002/anie.201802891. [DOI] [PubMed] [Google Scholar]
- Stegbauer S.; Jandl C.; Bach T. Enantioselective Lewis Acid Catalyzed ortho Photocycloaddition of Olefins to Phenanthrene-9-carboxaldehydes. Angew. Chem., Int. Ed. 2018, 57, 14593–14596. 10.1002/anie.201808919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C.; You S.-L. Catalytic Asymmetric Dearomatization Reaction Enabled Total Synthesis of Indole-based Natural Products. Nat. Prod. Rep. 2019, 36, 1589–1605. 10.1039/C8NP00098K. [DOI] [PubMed] [Google Scholar]
- a Austin J. F.; Kim S.-G.; Sinz C. J.; Xiao W.-J.; MacMillan D. W. C. Enantioselective organocatalytic construction of pyrroloindolines by a cascade addition-cyclization strategy: Synthesis of (−)-flustramine B. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5482–5487. 10.1073/pnas.0308177101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knowles R. R.; Carpenter J.; Blakey S. B.; Kayano A.; Mangion I. K.; Sinz C. J.; MacMillan D. W. C. Total synthesis of diazonamide A. Chem. Sci. 2011, 2, 308–311. 10.1039/C0SC00577K. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jamison C. R.; Badillo J. J.; Lipshultz J. M.; Comito R. J.; MacMillan D. W. C. Catalyst-controlled oligomerization for the collective synthesis of polypyrroloindoline natural products. Nat. Chem. 2017, 9, 1165–1169. 10.1038/nchem.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W.; Jiang G.; Liu H.; Hu J.; Pan X.; Zhang H.; Wan X.; Lai Y.; Ma D. Highly Enantioselective Bromocyclization of Tryptamines and Its Application in the Synthesis of (−)-Chimonanthine. Angew. Chem., Int. Ed. 2013, 52, 12924–12927. 10.1002/anie.201306774. [DOI] [PubMed] [Google Scholar]
- Tu H.-F.; Zhang X.; Zheng C.; Zhu M.; You S.-L. Enantioselective Dearomative Prenylation of Indole Derivatives. Nat. Catal. 2018, 1, 601–608. 10.1038/s41929-018-0111-8. [DOI] [Google Scholar]
- Gu Q.; Rong Z.-Q.; Zheng C.; You S.-L. Desymmetrization of Cyclohexadienones via Brønsted Acid-Catalyzed Enantioselective Oxo-Michael Reaction. J. Am. Chem. Soc. 2010, 132, 4056–4057. 10.1021/ja100207s. [DOI] [PubMed] [Google Scholar]
- a Okumura M.; Sarlah D. Arenophile-Mediated Dearomative Functionalization Strategies. Synlett 2018, 29, 845–855. 10.1055/s-0036-1591940. [DOI] [Google Scholar]; b Okumura M.; Sarlah D. Arenophile-Mediated Photochemical Dearomatization of Nonactivated Arenes. Chimia 2020, 74, 577–583. 10.2533/chimia.2020.577. [DOI] [PubMed] [Google Scholar]
- a Hernandez L. W.; Pospech J.; Klöckner U.; Bingham T. W.; Sarlah D. Synthesis of (+)-Pancratistatins via Catalytic Desymmetrization of Benzene. J. Am. Chem. Soc. 2017, 139, 15656–15659. 10.1021/jacs.7b10351. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bingham T. W.; Hernandez L. W.; Olson D. G.; Svec R. L.; Hergenrother P. J.; Sarlah D. Enantioselective Synthesis of Isocarbostyril Alkaloids and Analogs Using Catalytic Dearomative Functionalization of Benzene. J. Am. Chem. Soc. 2019, 141, 657–670. 10.1021/jacs.8b12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C.; Okumura M.; Zhu Y.; Hooper A. R.; Zhou Y.; Lee Y.-H.; Sarlah D. Palladium-Catalyzed Dearomative syn-1,4-Carboamination with Grignard Reagents. Angew. Chem., Int. Ed. 2019, 58, 10245–10249. 10.1002/anie.201905021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertjes W. C.; Southgate E. H.; Sarlah D. Recent advances in chemical dearomatization of nonactivated arenes. Chem. Soc. Rev. 2018, 47, 7996–8017. 10.1039/C8CS00389K. [DOI] [PubMed] [Google Scholar]
- Wiesenfeldt M. P.; Nairoukh Z.; Dalton T.; Glorius F. Selective Arene Hydrogenation for Direct Access to Saturated Carbo- and Heterocycles. Angew. Chem., Int. Ed. 2019, 58, 10460–10476. 10.1002/anie.201814471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura M.; Sarlah D. Visible-Light-Induced Dearomatizations. Eur. J. Org. Chem. 2020, 2020, 1259–1273. 10.1002/ejoc.201901229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv S.; Zhang G.; Chen J.; Gao W. Electrochemical Dearomatization: Evolution from Chemicals to Traceless Electrons. Adv. Synth. Catal. 2020, 362, 462–477. 10.1002/adsc.201900750. [DOI] [Google Scholar]