

SUMMARY:
Human pluripotent stem cells show considerable promise for applications in regenerative medicine including the development cell replacement paradigms for the treatment of Parkinson’s disease. Protocols have been developed to generate authentic midbrain dopamine (mDA) neurons capable of reversing dopamine-related deficits in animal models of Parkinson’s disease. However, the generation of mDA neurons at clinical scale suitable for human application remains an important challenge. Here we present an mDA neuron derivation protocol based on a two-step WNT signaling activation strategy that improves expression of midbrain markers such as EN1 while minimizing expression of contaminating posterior (hindbrain) and anterior (diencephalic) lineage markers. The resulting neurons exhibit molecular, biochemical and electrophysiological properties of mDA neurons. Cryopreserved mDA neuron precursors can be successfully transplanted into 6OHDA lesioned rats to induce recovery of amphetamine-induced rotation behavior. The protocol presented here is the basis for clinical grade mDA neuron production and preclinical safety and efficacy studies.
Keywords: Directed differentiation, human embryonic stem cells, human induced pluripotent stem cells, neural patterning, WNT signaling, midbrain development, transplantation, Parkinson’s disease, cell therapy, preclinical study
Graphical Abstract
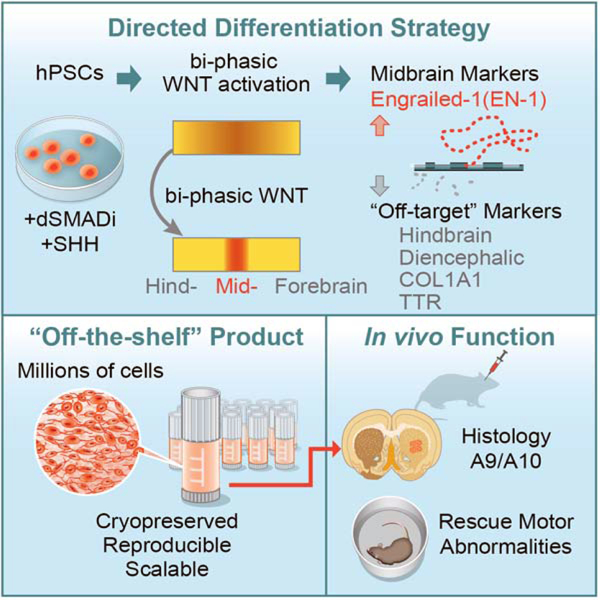
eTOC:
Studer, Tabar and colleagues present a midbrain dopamine neuron differentiation protocol that uses bi-phasic WNT activation to optimize the induction of midbrain identity and to avoid inappropriate neural and non-neural contaminants. The resulting cryopreserved, off-the-shelf product induces functional recovery in Parkinsonian rats and may be suitable for human translational use.
INTRODUCTION:
The use of pluripotent stem cells in regenerative medicine has moved closer to clinical trials for several disorders in the brain or other organ systems (Blau and Daley, 2019; Fox et al., 2014; Tabar and Studer, 2014). The development of a pluripotent-based cell therapy for Parkinson’s disease has been a particular focus given the extensive history of dopamine neuron grafting using fetal tissue sources. Human fetal midbrain dopamine neurons are capable of long-term engraftment in Parkinsonian patients for up to 24 years (Li et al., 2016). While there have been questions about the robustness of the clinical results that can be achieved using human fetal tissue grafts in PD (Barker et al., 2013), some studies report impressive long-term outcomes in at least a subset of patients. Those include the near complete restoration of physiological dopamine levels in the putamen of grafted patients as assessed by PET imaging and discontinuation of L-Dopa treatment (Kefalopoulou et al., 2014). In addition to the question as to how such remarkable results can be achieved more consistently, there is a consensus that realizing the potential of cell-based therapies in Parkinson’s disease will require a readily accessible and scalable human dopamine neuron source.
Human pluripotent stem cells (hPSC) have emerged as the currently most promising source of cells for mDA neuron replacement in PD with both human embryonic stem cell (hESC) and human induced pluripotent stem cell (hiPSC) at early stages of clinical testing (Barker et al., 2017; Parmar et al., 2020; Schweitzer et al., 2020). Robust engraftment of in vitro hPSC-derived mDA neurons in mouse, rat or monkey models of PD required the development of floor plate derived midbrain dopamine neuron differentiation protocols (Doi et al., 2014; Kikuchi et al., 2017; Kirkeby et al., 2012; Kriks et al., 2011; Sundberg et al., 2013; Xi et al., 2012). All of those protocols are based on the combined action of an activator of WNT signaling, typically the GSK3- inhibitor CHIR99021, in combination with a strong activation of SHH signaling to trigger midbrain floor plate induction and neurogenic conversion into midbrain DA neurons (Kriks et al., 2011). However, there are considerable differences among the various protocols in both the timing and concentration of CHIR99021 as well as in the use and timing of FGF8 as an additional inducer of midbrain dopamine neuron identity (Kim et al., 2020).
Recent studies suggest that expression of the floor plate marker FOXA2 in combination with the midbrain progenitor marker LMX1A is not sufficient to define mDA neuron precursor identity, but also marks cells in the adjacent anterior, subthalamic precursor cell region, giving rise to glutamatergic rather than dopaminergic neurons (Kee et al., 2017). Treatment with FGF8 following midbrain/diencephalic floor plate induction has been proposed as a strategy to refine the patterning of floor plate precursor towards midbrain domain (Kirkeby et al., 2017). Those findings are in agreement with earlier work reporting late FGF8 treatment (following floor plate induction), as beneficial for human primate-based mDA neuron induction (Xi et al., 2012). However, FGF8 treatment can induce expression of caudal markers beyond the midbrain boundary including HOXA2 (Kee et al., 2017; Kirkeby et al., 2017) and other hindbrain markers, a finding compatible with the role of FGF8 during early mid/hindbrain development (Liu et al., 2003). Therefore, FGF8 treatment needs to be carefully titrated as to avoid contamination with either hindbrain or other proliferating precursor cell populations.
Here we present an mDA neuron patterning strategy that is based on a bi-phasic WNT signaling activation, which avoids the use of extrinsic FGF8 and which triggers the robust and consistent induction of midbrain markers such as engrailed-1 (EN1) by day 11 of differentiation as compared to previous protocols (Kriks et al., 2011). The use of EN1 knockout hPSC lines demonstrates that enhanced mDA neuron differentiation under those conditions is dependent, at least in part, on EN1. The resulting mDA neurons show robust differentiation and functional properties in vitro. Furthermore, cryopreserved mDA neurons, generated via the bi-phasic (CHIR-boost) WNT activation, were transplanted into the adult hemiparkinsonian rat model resulting in functional recovery. The robustness of patterning hPSCs towards mDA neurons makes this protocol suitable for translational applications.
RESULTS:
Chir-boost treatment for the induction of mDA and repression of non-mDA markers.
Previous mDA neuron induction protocols have used a broad range of CHIR99021 concentrations to trigger midbrain identity (Gantner et al., 2020; Kirkeby et al., 2012; Kriks et al., 2011; Xi et al., 2012; Xiong et al., 2020). An important variable in those studies is the base media composition as one of the varying components of the medium. “Standard” Chir concentrations for mDA neuron differentiation depend on whether a fully defined (0.4–1µM Chir) or KSR-based (3µM Chir) medium is used. Knockout-serum replacement (KSR), contains lysophosphatidic acid (LPA) which has been shown to reduce the potency of CHIR99021 by dampening the levels of WNT signaling (Blauwkamp et al., 2012). Another concern is the observation that induction of midbrain markers such as EN1 can vary quite dramatically across differentiations, even when using an identical concentration (0.7 µM) of CHIR99021 (Kirkeby et al., 2017) in each of the experiments. Such batch-to-batch variability in the efficiency of midbrain induction, sensitive to very small changes in CHIR99021 concentrations, can lead to frequent contamination with hindbrain or diencephalic fates respectively. FGF8b has been proposed to induce or stabilize midbrain marker expression (Kirkeby et al., 2017; Kriks et al., 2011; Xi et al., 2012). However, FGF8b exposure at early stages of mDA patterning may not significantly improve robustness of midbrain marker expression (Kriks et al., 2011) while treatment at later differentiation stages appears to enhance or stabilize EN1 levels but can induce contaminating hindbrain makers and may promote the growth of undesired mesenchymal-like cell types.
WNT signaling plays temporally distinct roles in early CNS and midbrain development including an early, dose-dependent anterior-to-posterior patterning effect to drive default forebrain identity towards diencephalic, midbrain and hindbrain fates (Kiecker and Niehrs, 2001; Nordstrom et al., 2002), and a later region-specific effect of WNT signaling on promoting OTX2+ midbrain vs FGF8-mediated GBX2+ hindbrain identity (Liu and Joyner, 2001). Therefore, we tested the hypothesis that biphasic activation of WNT signaling during neural differentiation of hPSCs may trigger induction of midbrain/hindbrain identity at low Chir concentrations, mimicking the dose-dependent caudalization mediated by WNT signaling at early developmental stages, followed by locking in midbrain rather than hindbrain fate at high Chir concentrations (Chir-boost), mimicking the high WNT1 levels expressed at the anterior (OTX2+) border of the midbrain/hindbrain boundary. We used an initial concentration of 0.7 µM CHIR99021, similar to the concentrations used in previous studies under KSR-free media conditions (Kirkeby et al., 2012; Xi et al., 2012) (Figure 1A; Low Chir). To mimic the later role of WNT signaling, we optimized the timing and concentration of the “Chir boost” (Figure S1) and observed that an earlier Chir boost decreases forebrain contamination (PAX6 and NKX2.1) and increases the midbrain marker EN1 (Figure S1A, B). However, applying the Chir boost at day 3 dramatically induced NKX6.1 (a non mDA progenitor marker; data not shown). Also, Chir boosting to levels higher than 7.5 µM triggered dose-dependent cell death (Figure S1C, D). Based on these results, we used a Chir-boost concentration of 7.5 µM starting from day 4 of differentiation (Figure 1A; Boost Chir). As a control condition, we exposed cells to 7.5 µM throughout the neural induction process (Figure 1A; High Chir). We observed that traditional midbrain floor plate markers such as FOXA2 and LMX1A remain unaffected when comparing Low vs Boost Chir (Figure 1B–E, Figure S1B, S3B,3C). In contrast, robust induction of EN1 and concomitant suppression of PAX6 (a forebrain marker) or suppression of NKX2.2 (a diencephalic and hindbrain marker) was dependent on boosting CHIR99021 levels (Figure 1B–E). Time course mRNA expression analysis confirmed induction of comparable levels of FOXA2, LMX1A, and OTX2 in Low versus Boost Chir treated cells, while Boost Chir triggered increased EN1 levels and suppressed PAX6 and NKX2–2 induction (Figure S2A). In contrast, exposure to continued “High Chir” levels triggered loss of OTX2 but induced the expression of hindbrain markers such as HOXA2 and HOXB1 (Figure 1B). In addition to analyzing changes in gene expression by qRT-PCR analysis, we quantified the percentages of EN1, PAX6 and FOXA2+ cells at day 11 of differentiation (Figure 1D, Figure S1F), and we performed RNAseq, which confirmed a dosedependent upregulation of EN1 in response to increasing CHIR boost levels with increased levels of AXIN2, and suppression of NKX2.2, NKX2.1 and PAX6 (Figure 1E).
Figure 1. CHIR-boost effect on the induction of mDA and repression of non-mDA markers.
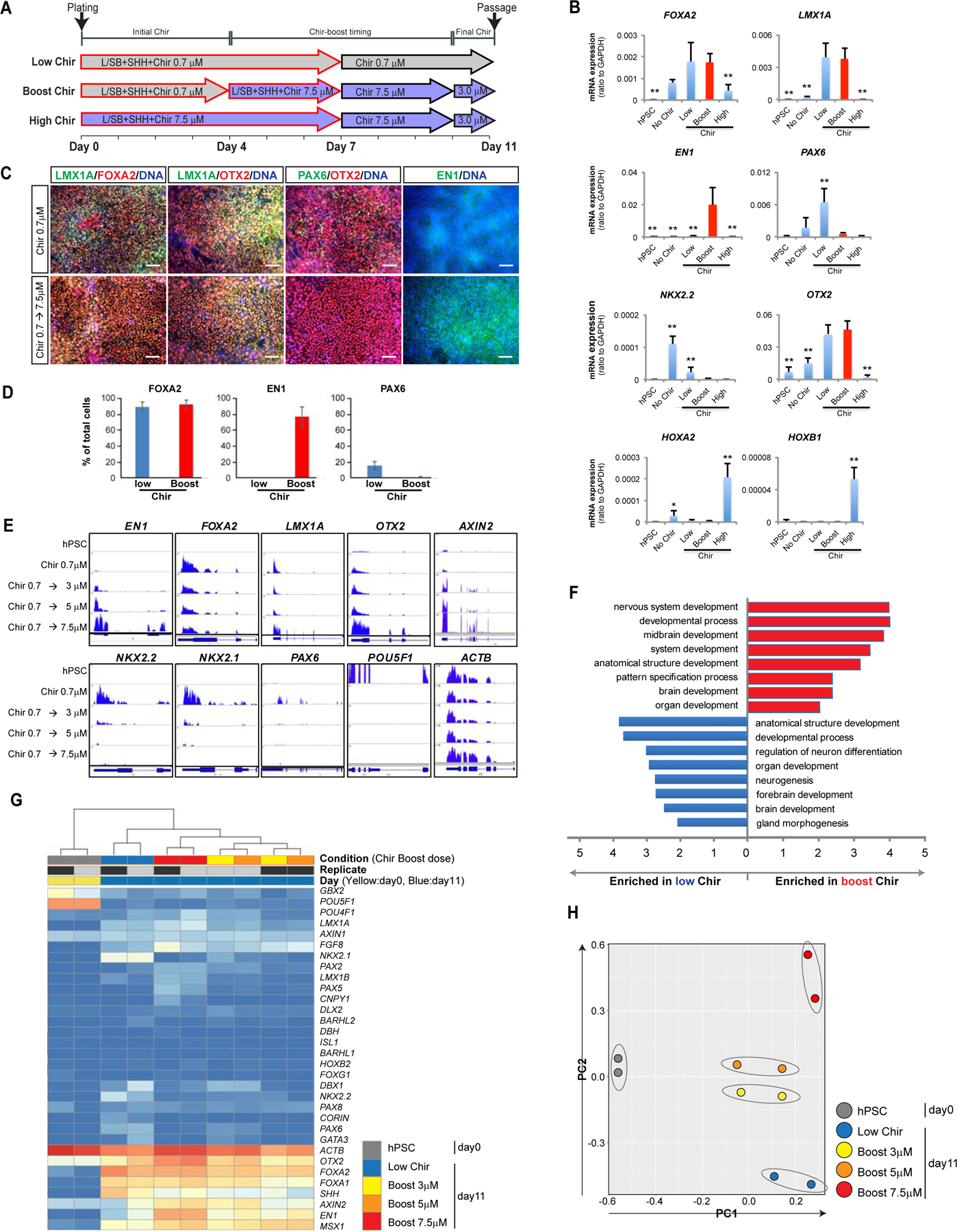
(A) Schematic illustration of the Low Chir, Boost Chir and High Chir culture conditions tested. (B) qRT-PCR analysis of hPSC-derived cells at day 11, Boost condition at 7.5µM. (C) Immuno-fluorescent staining of Low- and Boost- CHIR treated cells at day 11 of differentiation. Scale bars = 100 µm. (D) Quantification of the percentage of FOXA2+, EN1+ and PAX6+ cells at day 11 of differentiation following low Chir or Boost Chir treatment. (E) RNA sequencing data for key midbrain (upper row) and non-midbrain contaminating markers (lower row) are represented as tracks on human genome using IGV (integrative genomics viewer) (IGV). Chir 0.7µM is applied from day 0 to day 4, Chir boost dose from day 4 to day 10 and finally 3µM Chir from D10 (see timeline marked in (A)). Comparison of different concentrations of CHIR-boosting (day 4–10) measured at day 11. (F) Gene ontology (GO) analysis from RNA-sequencing between Low- and Boost- Chir conditions at day 11. (G) Heat map view of selected gene sets from RNA sequencing of hPSC and mDA differentiated cells with different CHIR-boosting at day 11. The unit of the color in each gene are log(TPM+1). Blue is low transcript while red color is high transcript. (H) PCA analysis of selected mDA genes from RNA-sequencing among hPSC and different CHIR-boost conditioned cells at day 11.
We next performed chromatin immunoprecipitation (ChIP-seq) for histone modifications including H3K4me3 and H3K27me3. Those results indicated that CHIR-boost promotes a more open chromatin structure at an EN1 locus based on increased H3K4me3 and decreased H3K27me3 levels. In contrast, analysis of the NKX2–2 and PAX6 loci showed the opposite pattern with decreased H3K4me3 and increased H3K27me3 levels under CHIR-boost conditions (Figure S2B). Gene ontology analysis, heatmap of differentially expressed transcripts and PCA analysis all supported the notion that CHIR boost conditions lead to the enrichment of midbrain transcripts and depletion of forebrain transcripts and transcripts of other contaminating populations (Figure 1F–H and Figure S2C).
Suppression of subthalamic fate in Boost CHIR is dependent on EN1
We next addressed whether Chir boost conditions suppress contaminating diencephalic fates including the expression of subthalamic nucleus markers (Kee et al., 2017; Nouri and Awatramani, 2017). Interestingly, CHIR-boost conditions repressed diencephalic and subthalamic precursor markers in a dose-dependent manner (Figure 2A and Figure S3A). An alternative strategy to suppress diencephalic fates is treatment with FGF8, known to convert diencephalic into midbrain fates in the developing chick embryo (Martinez et al., 1999) and to suppress diencephalic markers in hESC-derived mDA precursors (Kee et al., 2017; Kirkeby et al., 2017). Interestingly, Boost CHIR induced expression of endogenous FGF8 transcript and additional markers known to be involved in the FGF8 regulatory loop during early midbrain development (Ye et al., 2001) such as PAX2 and 5 (Figure 2B). Several of the transcripts induced by Boost Chir have been previously reported to predict mDA neuron engraftment potential in xenografting assays (Kirkeby et al., 2017). As an independent molecular assessment of the changes induced by Boost CHIR, ChIP-Seq at genes regulating diencephalic versus midbrain/hindbrain identity showed reduced levels of H3K4me3 at DBX1 and BARHL2 loci under Boost Chir conditions without an obvious change in the levels of H3K27me3. In contrast, at FGF8, PAX2, and PAX5 loci, H3K4me3 levels were increased in parallel to decreased levels of H3K27me3 in Boost versus Low Chir conditions (Figure 2C, D).
Figure 2. Suppression of subthalamic fate in Boost CHIR is dependent on EN1.
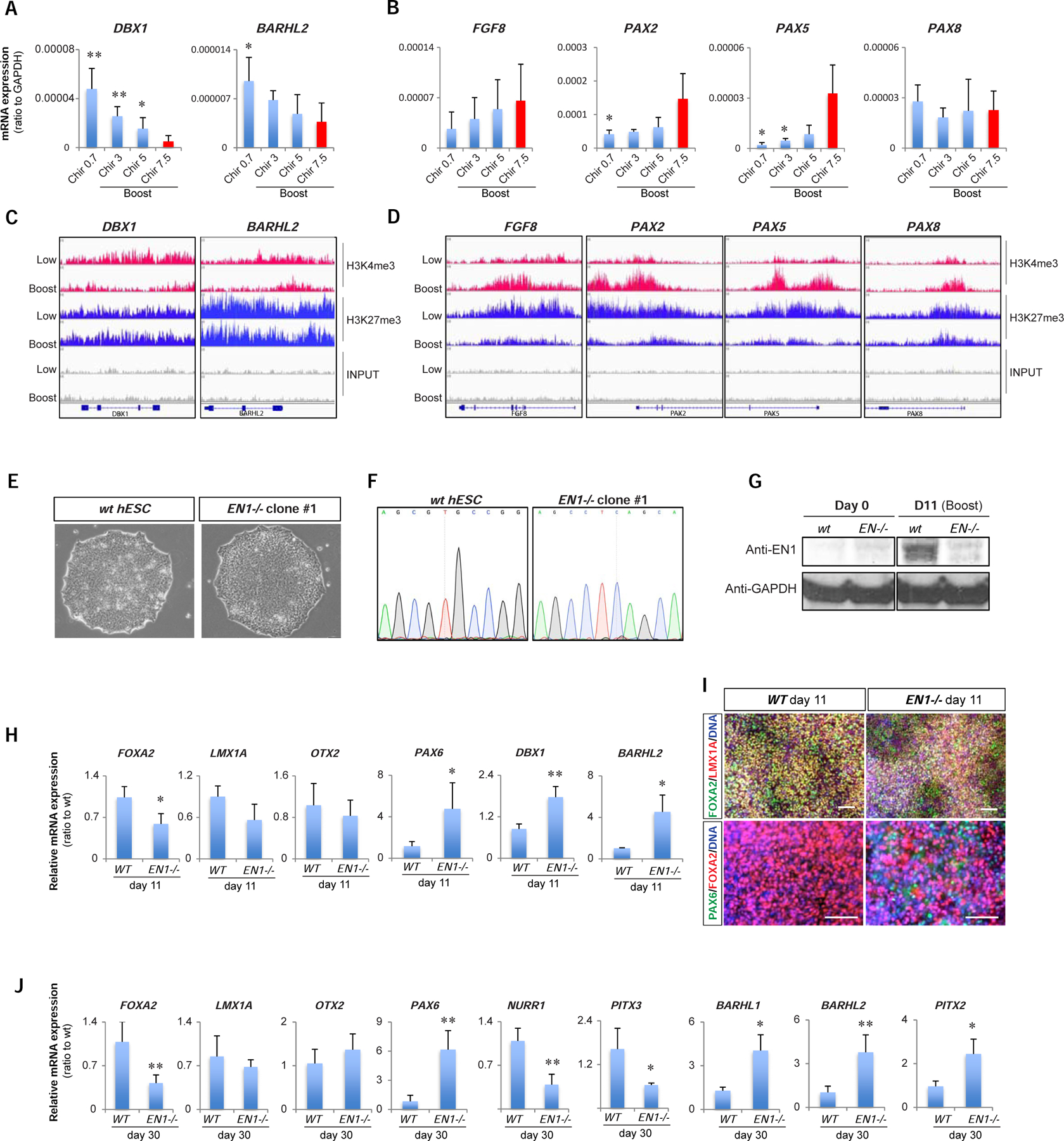
(A, B) qRT-PCR of subthalamic nucleus markers (DBX1 and BARHL2; A) and FGF8 related genes (FGF8, PAX2, PAX5, and PAX8; B) among different CHIR-boost treated cells at day 11. (C-D) IGV view of ChIP-sequencing data for H3K4me3 and H3K27me3 between Low- and Boost-CHIR treated mDA differentiated cells at day 11 at the loci of subthalamic nucleus markers (DBX1 and BARHL2; C) and FGF8 related genes (FGF8, PAX2, PAX5, and PAX8; D). (E) hPSC morphology of wild-type (WT) and EN1 knockout (EN1−/−) clones. (F) Sanger sequencing chromatograms comparing WT and EN1−/− hPSC clones. (G) Western-blotting of EN1 and GAPDH between day 0 and day 11 mDA differentiated cells from WT and EN1−/− hPSCs. (H, I) qRT-PCR analysis (H) and immunofluorescent staining (I) of day 11 mDA differentiated cells from WT and EN1−/− hPSCs. Scale bars = 100 µm. (J) qRT-PCR analysis of day 30 of mDA neuron differentiation from WT and EN1−/− hPSCs.
We next compared neural patterning markers following biphasic Chir activation (Boost Chir) versus biphasic low Chir followed by early FGF8b treatment. Biphasic Chir activation resulted in higher EN1 induction at day 11 than either lowChir or low Chir followed by FGF8b treatment (Figure S3B). At day 30 of differentiation, biphasic low Chir followed by late FGF8b treatment, mimicking an alternative strategy to induce EN1 expression and to suppress diencephalic fates (Kirkeby et al., 2017; Xi et al., 2012), resulted in robust EN1 levels comparable to Chir-boost, but triggered a significant increase in COL1A1, SMA, and SIX1 (Figure S3C), genes characteristic of non-neural contaminants such as the perivascular fibroblast-like cell population identified in mDA neuron grafts in vivo (Tiklova et al., 2020). Another contaminant reported in some mDA neuron grafts are choroid plexus epithelial cells (Doi et al., 2020), characterized the expression of TTR. TTR expression was suppressed in Boost Chir treatment (Figure S3C). Therefore, bi-phasic CHIR activation achieves high levels of EN1 induction without inducing non-neural contaminants such as COL1A1+ and TTR+ cells.
Given the broad set of changes triggered by Boost CHIR, we next asked which of those changes could be functionally linked to midbrain precursor cell specification. One key candidate is EN1, a transcriptional regulator that was dramatically upregulated by Boost CHIR treatment and a gene previously shown to rescue WNT1−/− midbrain phenotypes when expressed under control of the WNT1 regulator elements (Danielian and McMahon, 1996). We established two independent EN1−/− PSC knockout clones by CRISPR/Cas9-based targeting triggering a frameshift mutation resulting in the near complete loss of EN1 protein expression (Figure 2E–G). Using the Boost CHIR conditions in both isogenic control and EN1−/− knockout lines demonstrated that lack of EN1 results in significant increases in DBX1 and BARHL2 expression suggesting that EN1 contributes to suppressing diencephalic and subthalamic precursors fates. EN1−/− cells further showed increased expression of the anterior marker PAX6 (Figure 2H, I) mimicking the results obtained previously for wild-type hPSCs under low CHIR conditions. The partial loss of midbrain identity in EN1−/− cells was corroborated at day 30 of differentiation by the decreased expression of mDA neuron markers NURR1 and PITX3 and the increased levels of subthalamic neuron markers such as PITX2 (Figure 2J).
In vitro maturation and functional characterization of CHIR-boost treated mDA neurons
Gene expression analysis from RNA-seq study confirmed the progressive differentiation and maturation of Boost CHIR treated precursors into mDA neurons by day 30 of differentiation (Figure 3A–E and Figure S4A). Further maturation to day 60 of differentiation showed a time dependent increase of more mature mDA neuron markers including PITX3, ADCYAP1, CHRNA4, GIRK2 and SNCA (Figure 3F). The ability of Boost CHIR treated cultures to trigger more mature neuronal and synaptic marker expression was confirmed by immunocytochemistry (Figure 3G, H). To characterize electrophysiological properties of hPSC-derived mDA neurons at different stages of maturation, we conducted patch-clamp recording at day 40, 60, and 75 post differentiation. For these experiments, mDA neurons were plated onto a monolayer of rat cortical astrocytes, as described previously (Rayport et al., 1992). Comparison of the basal neuronal membrane properties revealed that cells at day 60 versus 75 showed comparable properties while younger neurons displayed a more positive resting membrane potential (Figure 3I; day 40: −34.9 ± 3.7 mV, n=14; day 60: −49.1 ± 1.8 mV, n=20; day 75: −49.1 ± 1.6 mV, n=24, P < 0.001 for day 40 vs both day 60 and day 75 by one-way ANOVA). Furthermore, day 40 cells showed a downward shift on the I-V curve (Figure 3J), and higher input resistance compared to day 60 and 75 mDA neurons (Figure 3K, day 40: 2,170 ± 504.2 MΩ; day 60: 964.2 ± 92.7 MΩ; day 75: 968.7 ± 80.8 MΩ, P < 0.001 for day 40 vs both day 60 and day 75 by oneway ANOVA). These values are high compared to the input resistance of rodent DA neurons (Grace and Onn, 1989; Rayport et al., 1992), but similar to previously published values for human mDA neurons (Ganat et al., 2012; Yan et al., 2005). Similarly, plasma membrane capacitance and thus cell surface area were much smaller in younger neurons (Figure 3L, day 40: 17.0 ± 0.9 pF; day 60: 53.0 ± 4.0 pF; day 75: 61.8 ± 4.2 pF, P < 0.001 for day 40 vs both day 60 and day 75 by one-way ANOVA). DA neurons showed spontaneous activity that we categorized into three types: silent, moderate spiking (<10Hz) and burst spiking (>10Hz) (Figure 3M). At day 40, 43% were silent and 57% showed moderate activity (Figure 3N). Whereas the proportion of silent types decreased in more mature mDA neurons (day 60: 20%; day 75: 21%), other types of activity patterns became more predominant (moderate spiking: day 60: 50%; day 75: 58%; burst spiking: day 60: 30%; day 75: 21%). Additionally, neurons at day 40 had much wider spontaneous action potentials (AP) and smaller afterhyperpolarization than neurons at DIV 60 (Figure 3O). In response to depolarizing step currents, neurons at day 60 and 75 showed multiple APs and spike-frequency adaptation, whereas only a single AP was observed in day 40 neurons, regardless of the amount of injected current (Figure 3P, Q). Furthermore, day 60 neurons expressed membrane currents typical of mature neurons, including HCN (Ih), KCNQ (M), fast inwards sodium and slow outwards rectifier potassium currents (Figure S4B–D). Because younger cells were vulnerable to current/voltage injections, we could not analyze their membrane currents. Taken together, mDA neurons at day 40 displayed “immature” electrophysiological properties, including high input resistance, low membrane capacitance, wide APs and an inability to maintain AP generation at depolarizing potentials. In contrast, electrophysiological characteristics of day 60 and 75 neurons were similar and reminiscent of those reported for rodent midbrain DA neurons (Grace and Onn, 1989; Rayport et al., 1992). Additionally, by HPLC analysis day 60 neurons were able to synthesize and release DA neurotransmitter in a stimulation and Ca2+-dependent manner (Figure 3R).
Figure 3. In vitro maturation and functional characterization of CHIR-boost treated mDA neurons.
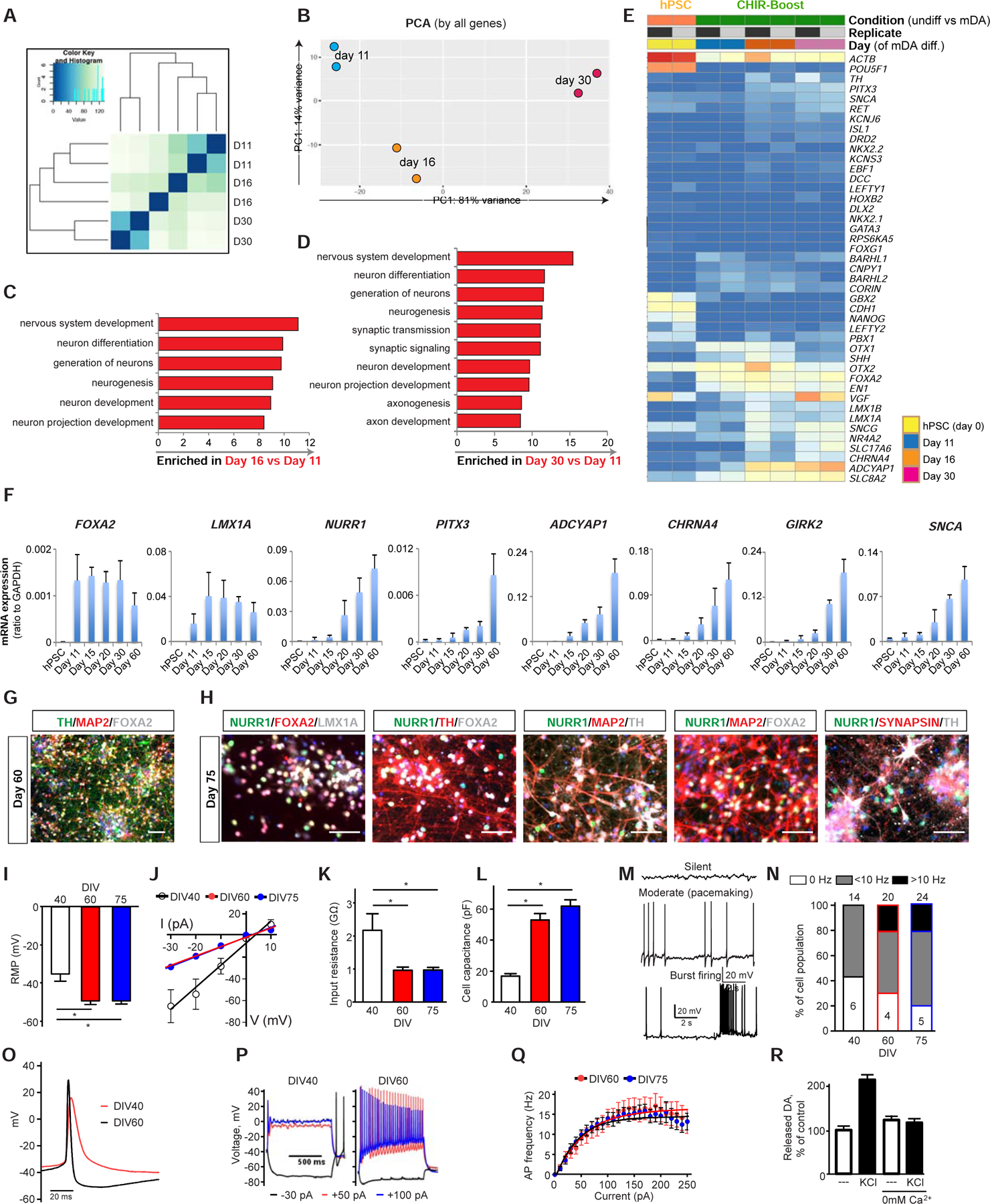
(A, B) Sample distance plot (A) and PCA analysis (B) for RNA expression among different time points of mDA differentiated cells using CHIR-Boost. (C, D) GO analysis from RNA-sequencing within day 11 versus day 16 (C) and day 11 versus day 30 (D) mDA differentiated cells. (E) Heat map of selected gene sets from RNA-sequencing at different time points of mDA differentiation. The unit of the color in each gene are log(TPM+1). Blue is low transcript while red color is high transcript. (F) qRT-PCR of mDA markers during mDA differentiation of hPSC using CHIR-boost protocol. (G, H) Immunofluorescent staining of mDA markers of day 60 (G) and day 75 (H) mDA differentiated cells. Scale bars = 100 µm. (I-Q) Electrophysiological properties of hPSC- derived mDA neurons. (I-L) Neurons at day 40 displayed higher resting membrane potentials (RMP) (I), steeper current-voltage dependence (J), higher input resistance (K) and lower membrane capacitance (L) than neurons at day 60 and 75 (* - p<0.001 by one-way ANOVA; n=14 for DIV 40, n=20 for day 60 and n=24 for day 75). (M) Representative traces of spontaneous neuronal activity recorded in cell-attached mode. (N) Percentages of cells displaying different types of spontaneous firing. Neurons at day 40 were either silent or fired at relatively slow frequency (< 10 Hz), while at day 60 and 75 neurons showed more variable spiking types. Total number of cells is shown on top of each bar graph, while numbers of cells displaying each activity type is shown inside the bars. (O) Examples of spontaneous action potential of day 40 and 60 mDA neurons. (P) Representative voltage traces elicited by somatic step current injections (−30, +50 and +100 pA for 1s) from day 40 and day 60 mDA neurons. (Q) Dependence of the number of action potentials on injected current for day 60 and day 75 neurons. (R) HPLC measurements of evoked DA release from day 60 mDA neurons following stimulation with 40 mM KCl. Zero mM Ca2+ saline was used to inhibit stimulation-dependent exocytosis.
Reproducibility and suitability of protocol for generating cryopreserved, “off the shelf” mDA cell product
Next, we examined whether the Boost-Chir protocol is suitable to generate consistent batches of mDA neurons with appropriate marker expression avoiding expression of markers related to contaminating lineages. We established a limited gene-set panel of 42 genes (41 genes plus ACTB). To address consistency of marker expression across independent differentiations, we present data as normalized cycle threshold (Ct) values (Note: higher Ct values correspond to lower gene expression levels and vice versa). We observed highly consistent gene expression patterns for 16 independent differentiation runs measured at day 10, day 16 and day 20 of differentiation (Figure 4A). We also validated the Boost Chir differentiation protocol in independent human ESCs such as MEL1 and human iPSC lines such as J1 (Figure S5A–D). To facilitate the use of mDA neurons for translational use, it is desirable to have a cryopreserved “off-the-shelf” product to allow for extensive and repeated product testing prior to clinical use (Barker et al., 2017). In fact, an “off-the-shelf” approach may enable pre-clinical and ultimate clinical testing from the same cryopreserved batch of cells. Inconsistent batch-to-batch performance is a problem in the field that may have contributed to past clinical failures in neural transplantation (Temple and Studer, 2017). We compared various reagents for cryopreservation of mDA neuron precursors using a controlled rate freezer, and we determined mDA neuron precursor viability following thawing. STEM-CELLBANKER™ yielded high post-thaw viability and produced cultures with the expected neuronal morphology, and with no obvious difference in marker expression before and after freezing (Figure 4B, C). High viability was maintained for many hours post thawing and following loading and extrusion of cells through a stereotactic injection cannula to mimic the transplantation procedure (Figure 4D, E).
Figure 4. Development of the clinically compatible “off the shelf” mDA cell product.
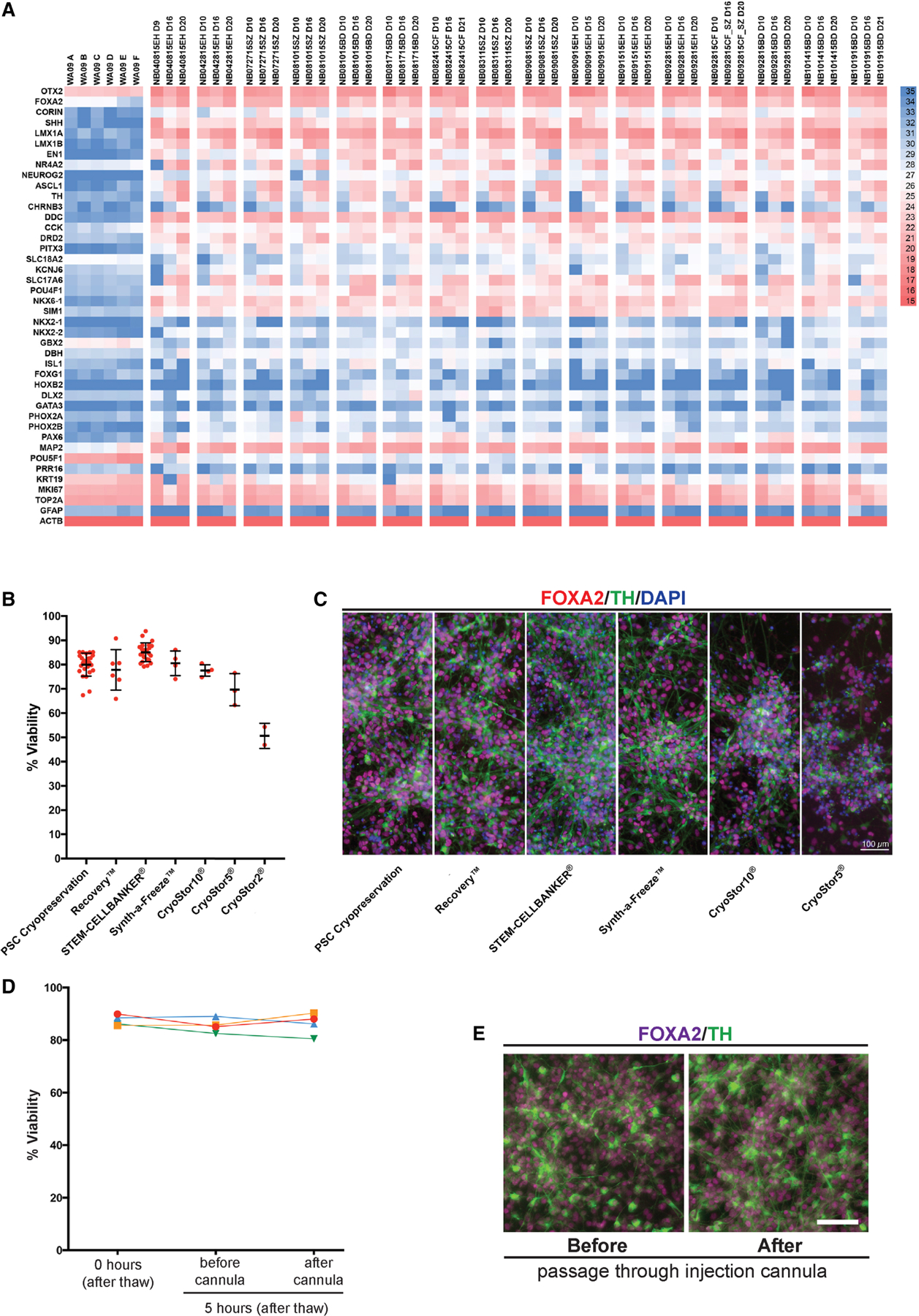
(A) Quantitative RT-PCR analysis using gene-set panel of 42 genes for 16 independent mDA differentiation over time showing derivation of a consistent mDA cell product with the comparable marker expression. “WA09” are H9-hESCs (day 0), and each differentiation shows a day 9 or 10 (D9 or D10), a day 16 (D16), and a day 20 or 21 (D20 or D21) of mDA differentiated cells from hESCs in Chir-Boost condition. The color at each gene is Cq value. Genes with high expression levels have low Cq values while genes with low expression have high values. (B) Viability of cryopreserved midbrain dopamine neurons post thawing in multiple commercial cryopreservation reagents. Cell survival was measured by AOPI system (Nexcelom Bioscience). (C) Immuno-fluorescent staining of mDA cells with FOXA2 (red), TH (green) and DAPI (blue). Cells were analyzed after 5-days of further mDA differentiation (day 21) post thawing the day 16 mDA product. The reagents used for each cell freezing is shown in the bottom. Scale bar = 100 µm. (D) Viability was assessed at 0 hours and in cells kept on ice for 5 hours each before/after passage through the injection cannula across 4 independent experiments. (E) Immuno-fluorescence staining of FOXA2 and TH for mDA cells, which were differentiated for 5 additional days from (D; Cells were on ice for 5 hours prior to passage through the cannula Scale bar = 100 µm.
In vivo survival and function of cryopreserved mDA neuron grafts in murine host
To determine the in vivo potential for “off-the-shelf” mDA neuron precursors, we transplanted mDA neuron precursors into adult 6OHDA lesioned rat striatum. Cells were cryopreserved on day 16 of differentiation. Upon thawing and confirmation of viability at > 80% (Nexcelom Bioscience), cells were injected at 450 × 103 cells/animal into the striatum of adult immunocompromised (NIH-Foxn1rnu) rats via stereotactic surgery, as described previously (Kriks et al., 2011). Following pilot studies to assess short-term survival in unlesioned hosts (Figure S5E), we performed long-term studies (Sham, n=4; mDA, n=5) in 6OHDA unilateral lesioned rats. Grafted animals showed a time-dependent recovery of amphetamine-induced rotational asymmetry as compared to sham-treated, vehicle solution injected animals (Figure 5A). Histology at 5.5 months after transplantation demonstrated survival of human mDA neurons as characterized by the co-expression of human nuclear antigen (hNA) and tyrosine hydroxylase (TH) (Figure 5B). Stereological analysis revealed the presence of 9,173± 2,576 TH/hNA positive cells and the graft volume was 6.22 +/− 1.77 mm3 (mean+/−SEM). Extensive TH+ fibers were observed emanating from graft core extending far into the host striatum and co-expressing human specific cytoplasmic marker SC121 or NCAM (Figure 5C, D, Figure S5F). Among the hNA+ cell population within the graft there were very few (< 1%) of the cells expressing serotonin (5HT, Figure 5E). The very low percentage of serotonin+ cells in those grafts may be advantageous compared to the higher percentages of serotonin+ cells reported for human fetal grafts, as serotonin+ neurons have been linked to an increased risk for triggering graft induced dyskinesia (Politis et al., 2010). We also observed a small number of glial cells as characterized by the expression of human specific GFAP while the percentage of Ki67+ proliferating cells was < 1% at 5.5 months. We further performed additional in vivo analyses on Chir-boost graft at 6 months post-transplantation in mice. The presence of TH+ cells expressing GIRK2, a widely used A9 markers, is 65% of TH% cells while the percentage of TH+ cells expressing the A10 marker CALB1 is 23% (Figure 5F, G). We also analyzed another A9 mDA neuron subtype marker, ALDH1A1, and found that most ALDH1A1+ cells are co-positive with TH and GIRK2 (Figure 5H). We further characterized mDA subtype identity based on neuron morphology. GIRK2+/TH+ mDA neurons were larger and more angular and often multipolar while CALB1+/TH+ mDA neurons were comparatively smaller and rounder (Figure 5G). Finally, the distribution pattern of CALB1+ vs ALDH1A1+ mDA neurons was also distinct with CALB1+ somas commonly located in the center of the graft whereas ALDH1A1+ somas primarily located at the graft periphery (Figure S5G). Fiber extension from ALDH1A1+ neurons could be densified within the striatum (using a human specific ALDH1A1 antibody) projecting to dorso-lateral and -medial regions of the striatum (Figure 5I).
Figure 5. In vivo survival and function of cryopreserved mDA neuron grafts in murine host.
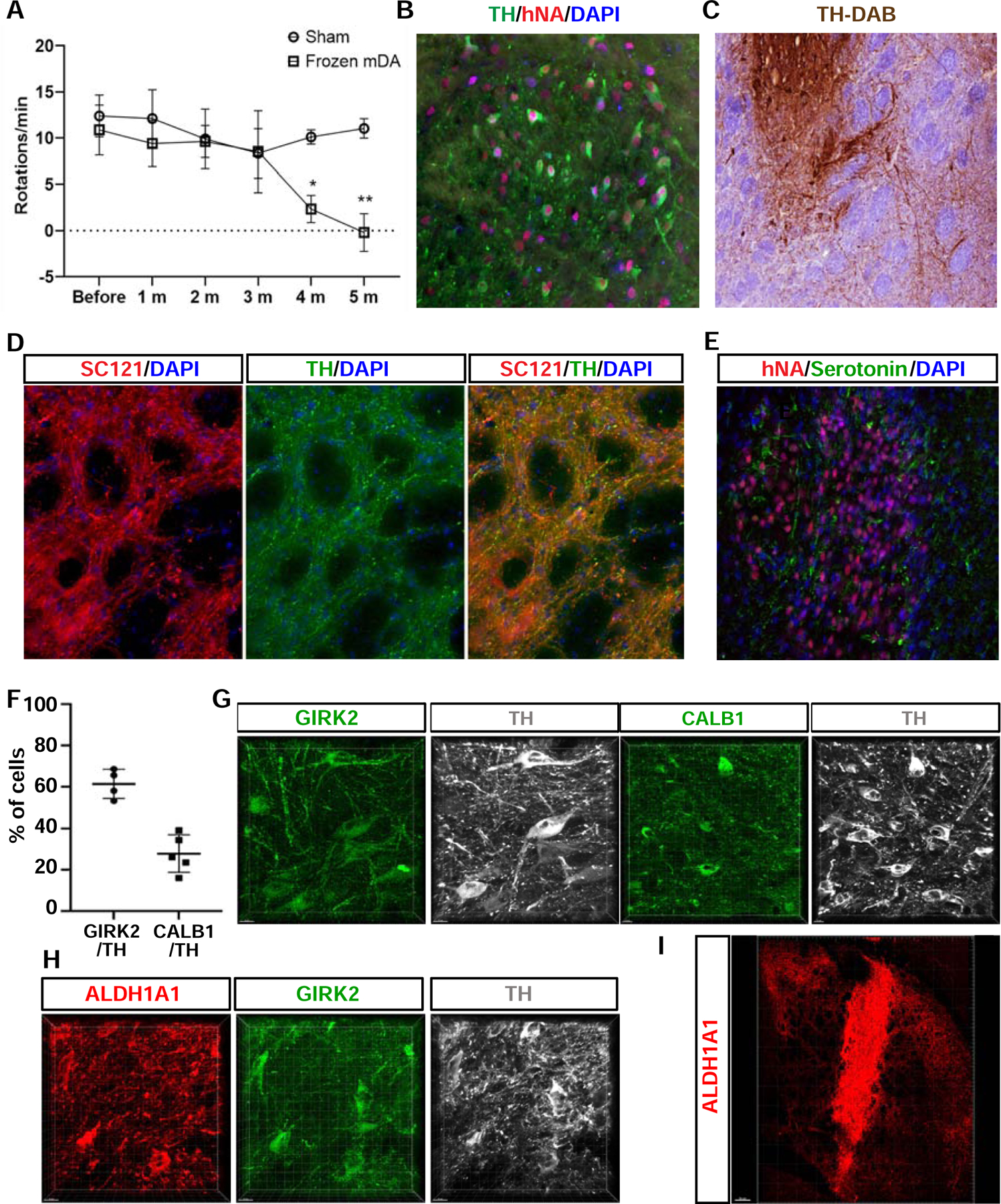
(A) Amphetamine induced rotation analysis of 6OHDA-lesioned rats comparing sham treated (vehicle solution, n=4) versus cryopreserved mDA neuron grafts (n=5). Data are represented as mean ± standard deviation (SD). (B) TH and hNA expression at 5.5 months after transplantation confirming human and mDA neuron identity. (C) TH-DAB staining at 5.5 month after grafting indicating robust axonal outgrowth from graft core. (D) Neurite outgrowth from grafts at 5.5 month showing expression of SC121 (human specific cytoplasmic marker) to confirm human identity of TH+ fibers at distances away from graft core. (E) Serotonin (5-HT) expression in grafted region at 5.5 month after transplantation showing the near absence of 5--HT+ neurons. (F - I) Characterization of mDA neuron subtype markers for Chir-Boost protocol at 6 months post transplantation in unlesioned NSG mice. (F) Quantification of the percentage of grafted TH+ cells expressing GIRK2 or CALB. (G, H) Representative confocal microscope mage of mDA neurons co-labeled with subtype markers. (I) Representative image of fiber outgrowth of the ALDH1A1+ mDA neurons from the graft. *P<0.05, **P<0.01. Scale bars = 100 µm in (B-E), 15 µm in (G, H), and 200 µm in (I).
DISCUSSION:
Our study is focused on developing a protocol for mDA neuron derivation from hPSCs that yields midbrain marker expression such as expression of EN1 in a robust and consistent manner and a protocol that is suitable for large scale manufacturing and the generation of an off-the shelf GMP product for cell transplantation. We report that boosting of CHIR exposure during a narrow differentiation window results in greatly enhanced EN1 expression levels, and that EN1 itself is a critical mediator of improved mDA neuron specification from hPSCs under Boost CHIR conditions.
An interesting question is whether for future iterations of the protocol the Boost CHIR condition should be combined with FGF8 treatment at later stages of differentiation. While we demonstrate that the robust induction of EN1 does not require extrinsic FGF8 exposure, it is conceivable that FGF8 treatment is important for maintenance rather than induction in our protocol. Preliminary data suggest that EN1 levels do decrease following midbrain floor plate induction at day 11 to the time point of mDA neurogenesis, though EN1 is maintained at those levels even in postmitotic mDA neurons. It will be intriguing to compare the performance of mDA neurons at distinct levels of EN1 expression to see whether engrailed levels affect A9 versus A10 like behavior or mDA neuron survival (Simon et al., 2001) or potentially other mDA neuron features in vitro and in vivo (for review (Rekaik et al., 2015)). However, for clinical translation a major concern is to avoid the presence of contaminating cell populations that may be promoted by FGF8 exposure such as posterior and potentially mesenchymal-like fates.
While our Boost CHIR cells yield GIRK2+ cells in vivo with a smaller proportion of Calb2+ TH+ cells, future studies will be required to further improve our ability to conclusively define and manipulate the ratio of A9 versus A10 mDA neurons in vitro and in vivo. Another area of future improvement is the development of strategies to address the limited initial survival of transplanted mDA neurons. While mDA neuron show long-term survival of 5.5 months in our current study and up to 24 years in human fetal dopamine neuron grafts (Li et al., 2016), most published studies report that initial mDA neuron survival is less than 10% of the grafted cells. While the proposed translational applications will simply inject larger cell numbers to compensate for initial cell loss post grafting, improved mDA neuron survival would allow the grafting of lower cell numbers.
In conclusion, our optimized derivation protocol yields functional DA neuron from hPSCs in vitro and survival of mDA neurons in vivo with improved EN1 expression while minimizing contamination with undesired cell types in a consistent manner. Those conditions should be suitable for producing a scalable, off-the-shelf mDA neuron product, compatible for human translation. Accordingly, based on this protocol, we have established detailed standard operating procedures (SOPs) for manufacturing at scale. We have produced cryopreserved mDA neuron batches for pre-clinical testing under GLP-conditions, as detailed further in the accompanying article (Piao et al. 2020) towards use in a first-in-human clinical study in PD patients.
LIMITATION OF STUDY
Our study provides the basis for the clinical-grade protocol moving towards human clinical trials (as detailed in the accompanying Piao et al. manuscript). The main features of the protocol include: i) suitability for clinical translation (GMP manufacturing and Cryopreservation, Scalability, and Reproducibility); ii) optimized AP patterning to avoid anterior (diencephalic) and posterior (hindbrain) fates, iii) lack of non-neural contaminants such as COL1A1 perivascular fibroblasts and TTR choroid plexus epithelial cells. In future studies, it will be important to further demonstrate the reproducibility of achieving long-term functional recovery across many independent batches of mDA neurons produced for a given hPSC line and produced across independent lines. Such data are critical for dose-finding in clinical trials and for validating in vitro potency assays which are a requirement by regulatory agencies when a product moves from early stage trials to market approval and commercial use. The development of reliable in vitro potency assays can complement in vitro molecular and functional characterizations, as performed in the current study, to enable comparisons across mDA neuron protocols and cell lines. In the current study, we demonstrate functionality of our cells in vivo by amphetamine-induced recovery, as our study was focused on developing a GMP-compatible protocol for the subsequent preclinical animal work. The amphetamine rotation assay is very robust when applied properly and correlates well with extent of mDA neuron loss and loss of striatal innervation. However, additional behavioral assays (e.g. cylinder, stepping or corridor assays) would be helpful in studying mDA neuron physiology and functional integration, and should be included in future studies aimed at comparing the potency of various mDA neuron differentiation protocols. Comparative in vivo studies are challenging as some mDA protocols may need to be optimized for each hPSC line prior to use, and several protocols are currently not suitable for cryopreservation. Accordingly, such studies would have to either compare cryopreserved against fresh cell products, which may complicate interpretations, or use fresh cells throughout despite the goal of using an off-the-shelf, cryopreserved product for eventual clinical use.
Despite the progress in mDA neurons differentiation, there is room for further improvement beyond the current study, such as the possibility of grafting specific mDA neuron subtypes (A9 mDA neurons or specific subtypes within the A9 compartment). In future studies, mDA subtype characterization by GIRK2, CALB and ALDH1A1 immunohistochemistry should be complemented with advanced multiplex approaches such as nucSeq or spatial transcriptomics to define detailed subtype identities (Poulin et al., 2020) among grafted mDA neurons and to compare relative proportions of mDA neuron subtypes across differentiation protocols. Other developments may involve improving in vivo mDA neuron survival and enhancing our ability to control the transition from mDA neuron precursor to fully mature postmitotic mDA neuron stage, advances that can further improve the reliability and safety of mDA neuron grafting in the future.
STAR* METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Lorenz Studer (studerl@mskcc.org).
Materials Availability
Cell lines generated and used in this study are available upon reasonable request from the Lead Contact.
Data and Code Availability
The RNA and ChIP sequencing data generated in this paper are uploaded to GEO with accession number GEO: GSE162884.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human pluripotent stem cells [hPSCs; WA09 (H9; 46XX) and MEL1 (46XY)], EN1 knockout H9 hPSCs, and J1 human induced PSC (MRC5), which was previously published in Miller et al., (2015), were grown onto Vitronectin (VTN-N, Thermo Fisher #A14700) coated dishes with Essential 8 media (Life Technologies #A1517001). hPSCs were passaged every 4–5 days by EDTA, and passage 35–55 hPSCs were used for the experiments. For EN1 knockout in hPSCs, guide RNA was predicted with a top score from the CRISPR design tool (http://crispr.mit.edu). Sequence of sgRNA for EN1 knockout was 5-AGCGATGGAGACAGCGTGC-3, and cloned into a CAG-Cas9 2A-GFP U6-sgRNA vector (Addgene, PX458) according to the published instruction (Ran et al., 2013). 5ug of plasmid was transfected to H9 hPSCs using Nucleofector (Lonza Kit V using the B-016 program). After 48h later, GFP expressed cells were FACS sorted using a BD FACS Aria III in the MSKCC Flow Cytometry core facility followed by growing clonally. Each colony was picked manually, genomic DNA was extracted, and validated EN1 knockout by DNA Sanger sequencing from amplified PCR product of the target region. PCR primers for this are 5-GCCGAGCATGGAAGAACA-3 and 5-CGGGTTCCCAGCTTTAGAC-3. All cell lines are cultured at 37°C with 5% CO2 and routinely tested for mycoplasma.
In vivo Animal studies
Transplantation of hPSC-derived mDA neurons into nu/nu rat and NSG mice
All procedures were performed following NIH guidelines and were approved by the local Institutional Animal Care and Use Committee (IACUC), the Institutional Biosafety Committee (IBC) and the Embryonic Stem Cell Research Committee (ESCRO). Female NIH nude (NIH-Foxn1rnu) rats were purchased from Taconic Biosciences. The animals were acclimated for at least five days to laboratory conditions before the procedures.
6-OHDA lesioning at 6–8 weeks old rats and cell transplantation were performed as described in Kriks et al., 2001. For cell transplantation, cells (450 000 cells/rat, 150 000/µl) were stereotaxic injected into right striatum at two deposit sites (1.5 µl/site) (AP: +1.0, ML: −2.5mm; VL: −4.7 and −4.4 mm; toothbar set at −2.5) of rat. Sham group received vehicle solution instead. For mouse studies, 6–8 weeks old NSG (NOD-SCID IL2Rgc−/−) mice (Jackson Laboratory) were used, and a total of 2 µl cells (200,000/mouse) were injected at the speed of 0.5 µl/min into the dorsal striatum (AP +0.5, ML −1.8, DV −3.4 from dura) with the aid of stereotactic apparatus and electrical pump (Boston Scientific) to drive the syringe.
Amphetamine-induced rotation test
Amphetamine-induced rotation test were performed before transplantation and once in a month after transplantation until 5 months post grafting. The rats were injected intraperitoneally of D-Amphetamine (Sigma, 5mg/kg). After 10 minutes, the rotation behavior was recorded 40 minutes and the total rotates were automatically counted by Ethovision XT 11.5 (Noldus Information Technology Inc., USA). The data were presented as (Ipsilateral-contralateral) rotates per minute.
METHOD DETAILS
Directed differentiation into midbrain dopamine neurons (mDA)
hPSCs were dissociated into single cells using Accutase (Cell Technologies, #AT104), and plated at 400K cells/cm2 onto Geltrex (Life Technologies, #A1413201) coated dishes with Neurobasal (Life Technologies)/N2(Stem Cell Technologies)/B27(Life Technologies) media containing 2mM L-glutamine, 500ng/ml SHH C25II (R&D systems #464-SH), 250nM LDN (Stemgent # 04-0074-02), 10µM SB431542 (R&D systems #1614), 0.7µM CHIR99021 (R&D systems #4432), and 10uM Rock inhibitor (Y-27632, R&D systems #1254), which represents day 0 of differentiation, and cultured until day 3 without Rock inhibitor from day 1. On day 4, cells were exposure to different concentration of CHIR 0.7, 3, 5, and 7.5 µM until day 10. On day 7, LDN, SB, and SHH were withdrawn. On day 10, media was changed to Neurobasal/B27/L-Glu supplemented with BDNF (brain-derived neurotrophic factor, 20ng/ml; R&D #248-BD), ascorbic acid (0.2 µM, Sigma #4034), GDNF (glial cell line-derived neurotrophic factor, 20 ng/ml; Peprotech # 450–10), TGFβ3 (transforming growth factor type β3, 1 ng/ml; R&D #243-B3), dibutyryl cAMP (0.5 mM; Sigma #4043), and CHIR 3 µM. On day 11, cells were dissociated using Accutase and replated under high cell density (800K cells/cm2) on polyornithine (PO; 15 µg/ml)/ laminin (1 µg/ml)/ fibronectin (2 µg/ml) coated dishes in mDA differentiation media [(NB/B27/L-Glu, BDNF, ascorbic acid, GDNF, dbcAMP, and TGFβ3 until day 16 with adding DAPT (10 µM R&D #2634)] from day 12. On day 16, cells were dissociated and plated as same procedure of day 11 and cultured until day 25 using mDA differentiation media. On day 25, cells were dissociated using Accutase and replated under low cell density (200K~300K cells/cm2) in mDA differentiation media until the desired experiments. For the cryopreservation of mDA precursor neurons, day16 mDA differentiated cells were treated with Accutase for 20–30 minutes, washing, detached, single cells, and pelleting. Cell pellets were resuspended at a cell density of 8 million cells/mL of STEM-CELLBANKER™. Controlled rate freezer (ThermoFisher) was used to cryopreserve cell product.
Immunohistochemistry
Cells were fixed in 4% paraformaldehyde (PFA) (Affymetrix #MFCD00133991) in DPBS for 15 min at room temperature. Cells were subsequently washed with DPBS. Then samples were permeabilized with 0.5% Triton X100 and blocked with 2% BSA in DPBS. The samples were subsequently incubated with primary antibody overnight at 4°C. The next day, after washing with DPBS, the samples were incubated with secondary antibody conjugated with Alexa Fluor 488- 555-, or 647- (Thermo Fisher) diluted at 1:400 in 2% BSA (DPBS) for 1 hour at room temperature. Then the samples were washed with DPBS and count-stained with 4′, 6-diamidino-2-phenylindole (DAPI) (Sigma, #D9542). Images were visualized using an Olympus and Zeiss inverted fluorescence microscope. Mouse and chicken anti-MAP2 (1:1500, Sigma and 1:2000, Abcam), rabbit and mouse anti-TH (1:500, PelFreez and 1:1000, Immunostar), goat anti-FOXA2 (1:200, R&D), Rabbit anti-LMX1A (1:1500, Abcam), Goat anti-OTX2 (1:1000, Neuromics), rabbit and mouse anti-PAX6 (1:500, Covance and 1:200, BD-Biosciences), mouse and rabbit anti-EN1 (1:50, DSHB and 1:200 Invitrogen), goat anti-ALDH1A1 (1:250, Santa Cruz), rabbit anti-GIRK2 (1:400, Almonte), rabbit anti-CALB1 (1:2000 Swant), and mouse anti-NURR1 (1:1500, Perseus Proteomics) were used for immuno-fluorescent staining. Donkey anti- mouse, goat, rabbit or chicken secondary antibodies conjugated with Alexa-Fluor-488, Alexa-Fluor-555 or Alexa-Fluor-647 fluorophore (1:400, Life technologies) were used. Nuclei were counterstained by DAPI.
Western blotting
Cultured cells were collected and lysed with 2X Laemmli Sample Buffer (Bio-Rad, #161–737). After protein quantification using BCA protein assay kit (Pierce, #23228), same amount of proteins from samples were loaded and separated by NuPAGE™ 4–12% Bis-Tris Protein Gel (Invitrogen, #NP0322BOX) using NuPAGE™ MES SDS Running Buffer (Invitrogen, #NP0060). Proteins were electrophoretically transferred to a nitrocellulose membrane using NuPAGE™ Transfer Buffer (Invitrogen, #NP0006) with 20% Methanol. Then membranes were blocked in 5% skim milk (TBS-T) for 1 hour at room temperature and incubated primary antibodies overnight at 4°C. After washing with TBS-T, secondary mouse or rabbit antibodies conjugated to horseradish peroxidase were incubated for 1 hour at room temperature. After three times washing, developing the signals was performed by using an enhanced chemiluminescence (ECL) detection kit (PerkinElmer, #NEL104001WA).
RNA extraction and Real-time qRT-PCR
Total RNAs from samples were isolated with TRIzol (Qiagen) using the Direct-zol™ RNA MiniPrep kit (Zymo Research, #R2052). 1ug of RNA was used to generate cDNA using the iScript™ Reverse Transcription Supermix (BioRad, #170–8841). Real-time qRT-PCR was performed using the SSoFAST EvaGreen Mix (BioRad) in a BioRad CFX96 Thermal Cycler. All reactions were performed according to the manufactured protocol. Primer sequences are listed below, and some primers were obtained from Qiagen (Quantitect Primer assays). Results were normalized to GAPDH. Primer sequences are listed in Table S1.
RNA-sequencing
RNA-seq library preparation was performed at the MSKCC Integrated Genomics Operation Core Facility. Libraries were sequenced on an Illumina HiSeq 2500 platform with 50bp paired end reads. Sequencing data was filtered for quality filtered and adapter sequences were removed using Flexbar (v.2.2) (Dodt et al., 2012) and aligned to hg19 using STAR aligner (v.2.4.2a) (Dobin et al., 2013). On average, we obtain ~50M reads per sample with >97% mapped reads. Gene read coverage was generated using FeatureCounts (v.1.4.2) (Liao et al., 2014) using GENCODE annotation (v19) (Harrow et al., 2012). Differential gene expression was performed using DESeq2 (v. 1.12.4) (Love et al., 2014) and annotated using biomaRt package (v. 2.28) (Durinck et al., 2009).
ChIP-sequencing
Chromatin immune-precipitation (ChIP) for H3K27me3 (Millipore, #07–449) and H3K4me3 (Abcam, #ab8580) from each sample were performed using SimpleChIP® Plus Enzymatic Chromatin IP Kit (Cell signaling Tech, #9005) according to the instructions of the manufacturer. ChIP-sequencing library was generated at the MSKCC Integrated Genomics Operation Core Facility. Libraries were sequenced on an Illumina HiSeq 2500 platform with 50bp paired end reads. Generated each FASTQ files are processed to remove any adapter sequences at the end of the reads using cutadapt (v1.6). The files are then mapped using the BWA mapper (bwa mem v0.7.12). After mapping the SAM files are sorted and read group tags are added using the PICARD tools. After sorting in coordinate order, the BAM’s are processed with PICARD MarkDuplicates. Peak calling is then doing using the MACS program (Version 2).
Electrophysiological recordings
Patch-clamp electrophysiological recording were performed on hPSC-derived mDA neurons plated on a monolayer of rat cortical astrocytes, as described previously (Rayport et al., 1992). Recording were conducted at day 40, 60, and 75 on randomly selected neurons at room temperature in a Tyrode’s solution containing (in mM): 119 NaCl, 3 KCl, 10 glucose, 2 CaCl2, 1.2 MgCl2-6 H2O, 3.3 HEPES, and 2.7 HEPES-Na+ salt (pH 7.4, 270 mOsm). For whole-cell patch-clamp studies, borosilicate glass pipettes (G150F-4, Warner Instruments) with a tip resistance of 3–4 MΩ were pulled on a P-97 Flaming-Brown micropipette puller (Sutter Instruments) and filled with (in mM): 115 K-gluconate, 20 KCl, 10 HEPES, 2 MgCl2, 2 ATP-Mg, 2 ATP-Na2 and 0.3 GTP-Na, (pH 7.25, ~280 mOsm). Neurons were visualized under a 40x water immersion objective using Olympus BX51W1 microscope (Olympus), and recording were performed with an Axopatch 700B amplifier (Molecular Devices) and digitized at 10 kHz with ITC-18 (HEKA Instruments Inc). Data were acquired using WinWCP software (John Dempster, University of Strathclyde, UK). In each cell, input resistance (measured by −100 pA, 1s hyperpolarizing pulse), resting membrane potential and spontaneous firing frequency were monitored throughout the recording. Current-voltage relationship and evoked action potentials were measured by injecting a 1s long somatic current from −30 to +20 pA in +10 pA increments and from 0 to +250 pA in +10 pA increments, respectively. To measure HCN currents, cells were held at −50 mV in voltage clamp mode and hyperpolarizing voltage steps were applied from −70 to −160 mV. KCNQ currents were measured at −30 mV holding potential with −30 to −70 mV hyperpolarizing voltage range. Sodium and slow potassium currents were induced by a depolarizing voltage step from 0 to +110 mV. Data analysis and statistics were performed using Clampfit (Molecular Devices) and GraphPad Prism (GraphPad software). Data are presented as mean ± SEM.
HPLC
For DA measurement experiments, mDA neurons were plated onto PO/laminin/fibronectin coated 24-well plates in 5 × 105 cells on day 25 and used between day 60 and day 75. HPLC with electrochemical detection (HPLC-EC) was done as previously described (Pothos et al., 1996). Briefly, prior to supernatant collection, cells were incubated in fresh DMEM: F12 + N2 media for 30 min. After exposure to either Tyrode’s saline alone or supplemented with high KCl (40 mM, Sigma) for 10 min at room temperature, supernatant was collected and immediately mixed with perchloric acid (0.1 M final concentration) to deproteinize the sample and prevent dopamine auto-oxidation. Samples were sonicated at room temperature for 10 min, centrifuged at 10,000 g for 5 min), stored at −80˚C and analyzed within the following two weeks by reverse phase HPLC-EC. Cells in each sample were collected to normalize for protein content. DA concentrations in each group of samples were normalized to the levels in the corresponding control group; data are shown as averaged normalized values from 2 independent experiments.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-PAX6 | Biolegend | Cat#901301; RRID: AB_256503 |
| Mouse monoclonal anti-Tyrosine Hydroxylase, clone LNC1 | Millipore | Cat#MAB318; RRID:AB_2201528 |
| Rabbit polyclonal anti-LMX-1 | Millipore | Cat#AB10533; RRID: AB_10805970 |
| Goat Anti-OTX2 | Neuromics | Cat#GT15095-100; RRID: AB_2157174 |
| Chicken polyclonal anti-MAP2 | Abcam | Cat# ab5392; RRID:AB_2138153 |
| Rabbit polyclonal anti-TH | PelFreez Biologicals | Cat#P40101-150; RRID:AB_2617184 |
| Mouse monoclonal anti-Ki-67 antigen (clone MIB-1) | Agilent | Cat# M724001-2, RRID:AB_2631211 |
| Goat polyclonal anti-FOXA2 | R&D Systems | Cat# AF2400; RRID:AB_2294104 |
| Mouse monoclonal anti-NURR1 | Perseus Proteomics | Cat# PP-N1404-00; RRID:AB_2251476 |
| Mouse monoclonal anti-EN1 | DSHB | Cat#4G11; RRID: AB_528219 |
| Rabbit polyclonal anti-Calbindin D-28k | Swant | Cat#CB38; RRID: AB_2721225 |
| Mouse monoclonal anti-STEM121 | Takara Bio Inc | Cat#AB-121-U-050; RRID: AB_2632385 |
| Goat anti-ALDH1A1 | Santa Cruz Biotechnology | Cat#sc-22588; RRID:AB_2289311 |
| Rabbit polyclonal anti-Synapsin I | Sigma-Aldrich | Cat#S193; RRID:AB_261457 |
| Rabbit polyclonal anti-Serotonin (5-HT) | Sigma-Aldrich | Cat#S5545; RRID: AB_477522 |
| Rabbit polyclonal anti-GIRK2 | Alomone labs | Cat#APC-006; RRID:AB_2040115 |
| Mouse monoclonal anti-human nuclei | Millipore | Cat#MAB1281; RRID: AB_94090 |
| Rabbit polycolnal anti-EN1 | Thermo Fisher Scientific | Cat#PA5–84917; RRID:AB_2792066 |
| Rabbit polyclonal anti-H3K27me3 | Millipore | Cat#07–449; RRID:AB_310624 |
| Rabbit polyclonal to Histone H3 (tri methyl K4) | Abcam | Cat#ab8580; RRID:AB_306649 |
| HRP-linked donkey anti–rabbit IgG | GE Healthcare | Cat# NA934; RRID:AB_772206 |
| HRP-linked anti-Goat IgG (H+L) | Invitrogen | Cat# A15999; RRID:AB_2534673 |
| HRP-linked sheep anti–mouse IgG | GE Healthcare | Cat: NA931; RRID:AB_772210 |
| AlexaFluor Donkey Anti-Goat 488 | Thermo Fisher Scientific | Cat#A-11055; RRID: AB_2534102 |
| AlexaFluor Donkey Anti-Goat 568 | Thermo Fisher Scientific | Cat#A-11057; RRID: AB_142581 |
| AlexaFluor Donkey Anti-Goat 647 | Thermo Fisher Scientific | Cat#A-21447; RRID: AB_141844 |
| AlexaFluor Donkey Anti-Rabbit 488 | Thermo Fisher Scientific | Cat#A-21206; RRID: AB_141708 |
| AlexaFluor Donkey Anti-Rabbit 555 | Thermo Fisher Scientific | Cat#A-31572; RRID: AB_162543 |
| AlexaFluor Donkey Anti-Rabbit 647 | Thermo Fisher Scientific | Cat#A-31573; RRID: AB_2536183 |
| AlexaFluor Donkey Anti-Mouse 488 | Thermo Fisher Scientific | Cat#R37114; RRID: AB_2556542 |
| AlexaFluor Donkey Anti-Mouse 555 | Thermo Fisher Scientific | Cat#A-31570; RRID: AB_2536180 |
| AlexaFluor Donkey Anti-Mouse 647 | Thermo Fisher Scientific | Cat#A-21235; RRID: AB_141693 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Vitronectin (VTN-N) | Thermo Fisher Scientific | A14700 |
| Trizol | Thermo Fisher Scientific | 15596026 |
| Recombinant human FGF8b | R&D | 423-F8 |
| Accutase | Innovative Cell Technologies | AT104–500 |
| 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | 15575–020 |
| L-Glutamine (100X) | Thermo Fisher Scientific | 25030–081 |
| Penicillin Streptomycin | Thermo Fisher Scientific | 15140–122 |
| Essential 8 (E8) | Thermo Fisher Scientific | A1517001 |
| Neurobasal | Life Technologies | 21103–049 |
| N2 supplement B | Stem Cell Technologies | 7156 |
| B27 | Life Technologies | 12587–010 |
| Y-27632 (ROCKi) | R&D | 1254 |
| SB431542 (SB) | R&D | 1614 |
| LDN193189 (LDN) | Stemgent | 04–0074 |
| CHIR99021 | R&D | 4432 |
| SHH C25II | R&D | 464-SH |
| brain-derived neurotrophic factor (BNDF) | R&D | 248-BD |
| ascorbic acid (AA) | Sigma | 4034 |
| dibutyryl cAMP (cAMP) | Sigma | 4043 |
| glial cell line-derived neurotrophic factor (GDNF) | Peptrotech | 450–10 |
| transforming growth factor type β3 (TGFβ3) | R&D | 243-B3 |
| DAPT | R&D | 2634 |
| Poly-L-Ornithine (PO) | Sigma Aldrich | P3655 |
| Mouse Laminin I (LAM) | R&D | 3400–010-1 |
| Fibronectin (FN) | Thermo Fisher Scientific | 356008 |
| Geltrex | Life Technologies | A1413201 |
| STEM-CELLBANKER™ | Amsbio | 11890 |
| 4% paraformaldehyde | Affymetrix | MFCD00133991 |
| 4′, 6-diamidino-2-phenylindole (DAPI) | Sigma | D9542 |
| Critical Commercial Assays | ||
| RNA MiniPrep kit | Zymo Research | R2052 |
| SsoFast EvaGreen® Supermix | Bio-Rad | 172–5202 |
| iScript Reverse Transcription Supermix | Bio-Rad | 170–8841 |
| SimpleChIP® Plus Enzymatic Chromatin IP Kit | Cell signaling Tech | 9005 |
| BD Perm/Wash Buffer | BD Biosciences | 554723 |
| BCA protein assay kit | Pierce | 23228 |
| Deposited Data | ||
| RNA-Seq | This study | GSE162884 |
| ChIP-Seq | This study | GSE162884 |
| Experimental Models: Cell Lines | ||
| Human: H9 (WA-09) hESC line | WiCell Research Institute | NIHhESC-10–0062 |
| MEL-1 hESC line | Stem Cells Ltd | NIHhESC-11–0139 |
| MRC5 (J1) iPSC line | MSKCC Stem Cell Core | Miller et al., 2013 |
| Oligonucleotides | ||
| See Table S1 | ||
| Recombinant DNA | ||
| PX458 Cas9-GFP | Addgene | Addgene: 48138 |
| Software and Algorithms | ||
| DESeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R | https://cran.r-project.org/ | N/A |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2 |
| STAR aligner (v.2.4.2a) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| FLEXBAR (v.2.2) | (Dodt et al., 2012) | https://github.com/seqan/flexbar |
| FeatureCounts (v.1.4.2) | (Liao et al., 2014) | http://bioinf.wehi.edu.au/featureCounts/ |
| MACS | (Zhang et al., 2008) | http://liulab.dfci.harvard.edu/MACS/ |
| CRISPR design tool | (http://crispr.mit.edu) | N/A |
| FIJI – ImageJ | (Schindelin et al., 2012) | https://fiji.sc/ |
| FlowJo 9 | http://www.flojo.com | N/A |
| Integrative Genomics Viewer (IGV) | Robinson et al., 2011 | http://software.broadinstitute.org/software/igv/ |
| Picardtools (version 2.9.5) | , Broad Institute | http://broadinstitute.github.io/picard/ |
HIGHLIGHTS:
Scalable and reproducible mDA neuron differentiation via biphasic WNT activation
EN1 is required to mediate effects of biphasic WNT activation
Protocol minimizes diencephalic, hindbrain and non-neural contaminants
Cryopreserved mDA precursors yield A9 mDA neurons in vivo and rescue PD-rat model
Acknowledgements:
We thank members of the Studer and Tabar lab for discussions on the manuscript. RNA-seq and ChIP-seq library preparation and sequencing were performed at the Sloan Kettering integrated genomics core. Flow cytometry was performed by Kiran Ramnarine at the Stem Cell Core of MSKCC. The work was supported in part by a contract C028503 from NYSTEM and by the core grant P30CA008748 from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
L.S. is a scientific co-founder and consultant and V.T. is a founding investigator and consultant of Bluerock Therapeutics Inc. M.J.T and S.I. are employed by BlueRock Therapeutics Inc. L.S., S.K., S.I. and M.J.T. are inventors of a patent WO2016196661A1 filed by Memorial Sloan Kettering Cancer Center on the methods described in this study.
All other authors declare no conflict of interest.
REFERENCES:
- Barker RA, Barrett J, Mason SL, and Bjorklund A (2013). Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. The Lancet Neurology 12, 84–91. [DOI] [PubMed] [Google Scholar]
- Barker RA, Parmar M, Studer L, and Takahashi J (2017). Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 21, 569–573. [DOI] [PubMed] [Google Scholar]
- Blau HM, and Daley GQ (2019). Stem Cells in the Treatment of Disease. N Engl J Med 380, 1748–1760. [DOI] [PubMed] [Google Scholar]
- Blauwkamp TA, Nigam S, Ardehali R, Weissman IL, and Nusse R (2012). Endogenous Wnt signalling in human embryonic stem cells generates an equilibrium of distinct lineage-specified progenitors. Nature communications 3, 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, and McMahon AP (1996). Engrailed-1 as a target of the Wnt-1 signalling pathway in vertebrate midbrain development. Nature 383, 332–334. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt M, Roehr JT, Ahmed R, and Dieterich C (2012). FLEXBAR-Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms. Biology (Basel) 1, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi D, Magotani H, Kikuchi T, Ikeda M, Hiramatsu S, Yoshida K, Amano N, Nomura M, Umekage M, Morizane A, et al. (2020). Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nature communications 11, 3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi D, Samata B, Katsukawa M, Kikuchi T, Morizane A, Ono Y, Sekiguchi K, Nakagawa M, Parmar M, and Takahashi J (2014). Isolation of Human Induced Pluripotent Stem Cell-Derived Dopaminergic Progenitors by Cell Sorting for Successful Transplantation. Stem Cell Reports 2, 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox IJ, Daley GQ, Goldman SA, Huard J, Kamp TJ, and Trucco M (2014). Stem cell therapy. Use of differentiated pluripotent stem cells as replacement therapy for treating disease. Science 345, 1247391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganat YM, Calder EL, Kriks S, Nelander J, Tu EY, Jia F, Battista D, Harrison N, Parmar M, Tomishima MJ, et al. (2012). Identification of embryonic stem cell-derived midbrain dopaminergic neurons for engraftment. J Clin Invest 122, 2928–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner CW, de Luzy IR, Kauhausen JA, Moriarty N, Niclis JC, Bye CR, Penna V, Hunt CPJ, Ermine CM, Pouton CW, et al. (2020). Viral Delivery of GDNF Promotes Functional Integration of Human Stem Cell Grafts in Parkinson’s Disease. Cell Stem Cell 26, 511–526 e515. [DOI] [PubMed] [Google Scholar]
- Grace AA, and Onn SP (1989). Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. The Journal of neuroscience : the official journal of the Society for Neuroscience 9, 3463–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee N, Volakakis N, Kirkeby A, Dahl L, Storvall H, Nolbrant S, Lahti L, Bjorklund AK, Gillberg L, Joodmardi E, et al. (2017). Single-Cell Analysis Reveals a Close Relationship between Differentiating Dopamine and Subthalamic Nucleus Neuronal Lineages. Cell Stem Cell 20, 29–40. [DOI] [PubMed] [Google Scholar]
- Kefalopoulou Z, Politis M, Piccini P, Mencacci N, Bhatia K, Jahanshahi M, Widner H, Rehncrona S, Brundin P, Bjorklund A, et al. (2014). Long-term clinical outcome of fetal cell transplantation for Parkinson disease: two case reports. JAMA neurology 71, 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecker C, and Niehrs C (2001). A morphogen gradient of Wnt/beta-catenin signalling regulates anteroposterior neural patterning in Xenopus. Development 128, 4189–4201. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Morizane A, Doi D, Magotani H, Onoe H, Hayashi T, Mizuma H, Takara S, Takahashi R, Inoue H, et al. (2017). Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548, 592–596. [DOI] [PubMed] [Google Scholar]
- Kim TW, Koo SY, and Studer L (2020). Pluripotent Stem Cell Therapies for Parkinson Disease: Present Challenges and Future Opportunities. Front Cell Dev Biol 8, 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkeby A, Grealish S, Wolf DA, Nelander J, Wood J, Lundblad M, Lindvall O, and Parmar M (2012). Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep 1, 703–714. [DOI] [PubMed] [Google Scholar]
- Kirkeby A, Nolbrant S, Tiklova K, Heuer A, Kee N, Cardoso T, Ottosson DR, Lelos MJ, Rifes P, Dunnett SB, et al. (2017). Predictive Markers Guide Differentiation to Improve Graft Outcome in Clinical Translation of hESC-Based Therapy for Parkinson’s Disease. Cell Stem Cell 20, 135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Englund E, Widner H, Mattsson B, van Westen D, Latt J, Rehncrona S, Brundin P, Bjorklund A, Lindvall O, et al. (2016). Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci U S A 113, 6544–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu A, and Joyner AL (2001). Early anterior/posterior patterning of the midbrain and cerebellum. Annu Rev Neurosci 24, 869–896. [DOI] [PubMed] [Google Scholar]
- Liu A, Li JY, Bromleigh C, Lao Z, Niswander LA, and Joyner AL (2003). FGF17b and FGF18 have different midbrain regulatory properties from FGF8b or activated FGF receptors. Development 130, 6175–6185. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez S, Crossley PH, Cobos I, Rubenstein JL, and Martin GR (1999). FGF8 induces formation of an ectopic isthmic organizer and isthmocerebellar development via a repressive effect on Otx2 expression. Development 126, 1189–1200. [DOI] [PubMed] [Google Scholar]
- Nordstrom U, Jessell TM, and Edlund T (2002). Progressive induction of caudal neural character by graded Wnt signaling. Nat Neurosci 5, 525–532. [DOI] [PubMed] [Google Scholar]
- Nouri N, and Awatramani R (2017). A novel floor plate boundary defined by adjacent En1 and Dbx1 microdomains distinguishes midbrain dopamine and hypothalamic neurons. Development 144, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar M, Grealish S, and Henchcliffe C (2020). The future of stem cell therapies for Parkinson disease. Nat Rev Neurosci 21, 103–115. [DOI] [PubMed] [Google Scholar]
- Politis M, Wu K, Loane C, Quinn NP, Brooks DJ, Rehncrona S, Bjorklund A, Lindvall O, and Piccini P (2010). Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci Transl Med 2, 38ra46. [DOI] [PubMed] [Google Scholar]
- Pothos E, Desmond M, and Sulzer D (1996). L-3,4-dihydroxyphenylalanine increases the quantal size of exocytotic dopamine release in vitro. Journal of neurochemistry 66, 629–636. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Gaertner Z, Moreno-Ramos OA, and Awatramani R (2020). Classification of Midbrain Dopamine Neurons Using Single-Cell Gene Expression Profiling Approaches. Trends Neurosci 43, 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayport S, Sulzer D, Shi WX, Sawasdikosol S, Monaco J, Batson D, and Rajendran G (1992). Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology. The Journal of neuroscience : the official journal of the Society for Neuroscience 12, 4264–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekaik H, Blaudin de The FX, Prochiantz A, Fuchs J, and Joshi RL (2015). Dissecting the role of Engrailed in adult dopaminergic neurons--Insights into Parkinson disease pathogenesis. FEBS Lett 589, 3786–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer JS, Song B, Herrington TM, Park TY, Lee N, Ko S, Jeon J, Cha Y, Kim K, Li Q, et al. (2020). Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N Engl J Med 382, 1926–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HH, Saueressig H, Wurst W, Goulding MD, and O’Leary DD (2001). Fate of midbrain dopaminergic neurons controlled by the engrailed genes. The Journal of neuroscience : the official journal of the Society for Neuroscience 21, 3126–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg M, Bogetofte H, Lawson T, Jansson J, Smith G, Astradsson A, Moore M, Osborn T, Cooper O, Spealman R, et al. (2013). Improved cell therapy protocols for Parkinson’s disease based on differentiation efficiency and safety of hESC-, hiPSC-, and non-human primate iPSC-derived dopaminergic neurons. Stem Cells 31, 1548–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabar V, and Studer L (2014). Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nat Rev Genet 15, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple S, and Studer L (2017). Lessons Learned from Pioneering Neural Stem Cell Studies. Stem Cell Reports 8, 191–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiklova K, Nolbrant S, Fiorenzano A, Bjorklund AK, Sharma Y, Heuer A, Gillberg L, Hoban DB, Cardoso T, Adler AF, et al. (2020). Single cell transcriptomics identifies stem cell-derived graft composition in a model of Parkinson’s disease. Nature communications 11, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi J, Liu Y, Liu H, Chen H, Emborg ME, and Zhang SC (2012). Specification of midbrain dopamine neurons from primate pluripotent stem cells. Stem Cells 30, 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M, Tao Y, Gao Q, Feng B, Yan W, Zhou Y, Kotsonis TA, Yuan T, You Z, Wu Z, et al. (2020). Human Stem Cell-Derived Neurons Repair Circuits and Restore Neural Function. Cell Stem Cell. [DOI] [PMC free article] [PubMed]
- Yan Y, Yang D, Zarnowska ED, Du Z, Werbel B, Valliere C, Pearce RA, Thomson JA, and Zhang SC (2005). Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells 23, 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Bouchard M, Stone D, Liu X, Vella F, Lee J, Nakamura H, Ang SL, Busslinger M, and Rosenthal A (2001). Distinct regulators control the expression of the mid-hindbrain organizer signal FGF8. Nat Neurosci 4, 1175–1181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA and ChIP sequencing data generated in this paper are uploaded to GEO with accession number GEO: GSE162884.