

Abstract
Low molecular weight synthetic peptides have been demonstrated to be effective catalysts for an increasingly wide array of asymmetric transformations. In many cases, these peptide-based catalysts have enabled novel multifunctional substrate activation modes and unprecedented selectivity manifolds. These features, along with their ease of preparation, modular and tunable structures, and often biomimetic attributes make peptides well-suited as chiral catalysts, and of broad interest. Many examples of peptide-catalyzed asymmetric reactions have appeared in the literature since the last survey of this broad field in Chemical Reviews (Chem. Rev. 2007, 107, 5759–5812). The overarching goal of this new review is to provide a comprehensive account of the numerous advances in the field. As a corollary to this goal, we survey the many different types of catalytic reactions, ranging from acylation to C–C bond formation, in which peptides been successfully employed. In so doing, we devote significant discussion to the structural and mechanistic aspects of these reactions that are perhaps specific to peptide-based catalysts and their interactions with substrates and/or reagents.
Graphical Abstract

1. Introduction
Mimicry of biological systems has spawned an enormous amount of research. Under the banner of “biomimetic chemistry,” scientists have synthesized countless compounds that resemble biological molecules in some way, reproducing aspects of both structure and function.1 Function itself may be considered from several perspectives. On the one hand, a small molecule that mimics a partner in a binding event, such as a protein-protein interaction,2 constitutes a significant achievement in molecular design at the interface of chemistry and biology, with a particular emphasis on thermodynamics. On the other hand, mimicking bond-forming reactions is a parallel thrust, and expands the challenge to include the kinetic details of the respective transition states.3
In this latter context, low molecular weight peptides have played a significant role in terms of both establishing new fundamental concepts and contributing useful catalysts for powerful, important, and often challenging reactions. While the study of peptide-based catalysis may well have begun in search of analogies between synthetic catalysts and enzymes,4 it quickly became clear that many of the mechanistic features underlying effective catalysis by small molecules and enzymes were similar. For example, the reliance on noncovalent interactions is now a theme throughout the field of asymmetric catalysis, and is one that is routinely identified in studies of both small molecule catalysts and enzymes.5 The search for peptide-based catalysts in the context of synthetically useful transformations has also created synergy with the broad field of organic synthesis.6
The exploration of peptide catalysis has also provided particular opportunities for heuristic comparison to enzymes, given the shared reliance on amino acid sequences to deliver folded, three-dimensional structures and arrays of cooperative functional groups for substrate activation.7,8 These synthetic peptides have provided taut springboards for the investigation of conformationally dynamic—if not “flexible”—catalysts, which are now ever more under study.9 Moreover, peptide catalysts have both enabled novel, multifunctional substrate activation modes and addressed selectivity challenges in unique stereochemical contexts, spanning point, axial, and conformation-dependent chirality. In the process, numerous peptides have now been shown to be effective catalysts for an increasingly wide array of important asymmetric transformations from the perspective of synthetic utility, including many that are not extant in biochemistry. These attributes, along with their ease of preparation, tunable structures, and often biomimetic characteristics, have made peptides well-suited as chiral catalysts and contributed to the growing interest in their application. Indeed, features of oligopeptides now routinely appear in partially peptidic, and even non-peptidic, catalyst designs of very broad mechanistic and functional scope. Examples abound in organocatalytic systems,10–12 as well as in organometallic chemistry, wherein the peptidic functionalities serve as ligands on transition metal complexes.13
Many highly creative and synthetically useful examples of peptide-catalyzed asymmetric reactions have appeared in the literature since our group’s first Chemical Reviews article on the subject.8 The overarching goal of this new review—thirteen years later—is to provide as comprehensive an accounting of the numerous advances in the field as is possible. As a corollary to this goal, we survey the many different types of asymmetric reactions, ranging from C–C bond forming reactions to halogenation, in which peptides been successfully employed. In so doing, we devote significant discussion to the structural and mechanistic aspects of these reactions that perhaps began as specific to peptide-based catalysts and their interactions with substrates and/or reagents, but which are now appearing more broadly in many other catalytic scaffolds. Our goal has been to compliment the other, more focused reviews of peptide-based catalysts that have recently appeared.14–16
Because of the explosive growth in the field, boundary conditions are necessary in order to limit this review to a tractable length. Although many exemplary reactions catalyzed by single amino acids and their derivatives have been reported to date, we intend to focus primarily on oligopeptides consisting of 2–20 amino acids, where both secondary structure and multifunctional activation modes likely contribute to selective catalysis. In select cases, we do opt to discuss amino acid-catalyzed reactions, when the discussion illuminates an instructive concept. Additionally, since we have recently reviewed the topic of site-selective functionalization of complex molecules,17,18 this current review focuses solely on catalytic, enantioselective reactions mediated by synthetic peptides. Our literature survey endeavors to span the years of 2007–2019.
2. Oxidation
The most common asymmetric oxidation mediated by peptides prior to 2007, and overall one of the most well studied peptide-catalyzed reactions, was the Juliá–Colonna epoxidation (Section 2.1.1). Since then, numerous additional reports improving both the efficiency of the transformation and studying the mechanism have been disclosed. Additionally, a substantial amount of recent work has focused on the development of new and more powerful peptide-based catalysts for epoxidations based on aspartyl (Asp) peracids (Section 2.1.2) and dioxiranes (Section 2.1.3). Numerous other reports highlighted that the sequences of these Asp-based catalysts could be reprogrammed to facilitate Baeyer–Villiger oxidations (Section 2.2), Trp oxidations, and N-oxidation of pyridines (Section 2.3.1). Finally, various reports have also highlighted the utility of other peptides as catalysts for oxidations of alcohols (Section 2.3.2) and carbonyl α-positions (Section 2.3.3).
2.1. Epoxidation
2.1.1. Juliá–Colonna Epoxidation.
Since its initial development in the 1980s, few peptide catalyzed reactions have received as much attention as the Juliá–Colonna epoxidation.19,20 A subclass of Weitz–Scheffer epoxidation, Juliá–Colonna reactions involve the nucleophilic epoxidation of electron-deficient olefins with aqueous hydrogen peroxide in the presence of both base and a poly(amino acid) catalyst (Figure 1a). In lieu of traditional stepwise peptide synthesis, these catalysts are normally synthesized through polymerization in the presence of an initiator amine and a sufficient amount of N-carboxyanhydride derived from the desired amino acid (Figure 1b). The epoxidation reactions are traditionally performed in a triphasic mixture consisting of both water and organic solvent, along with solid-supported peptide catalysts.
Figure 1.
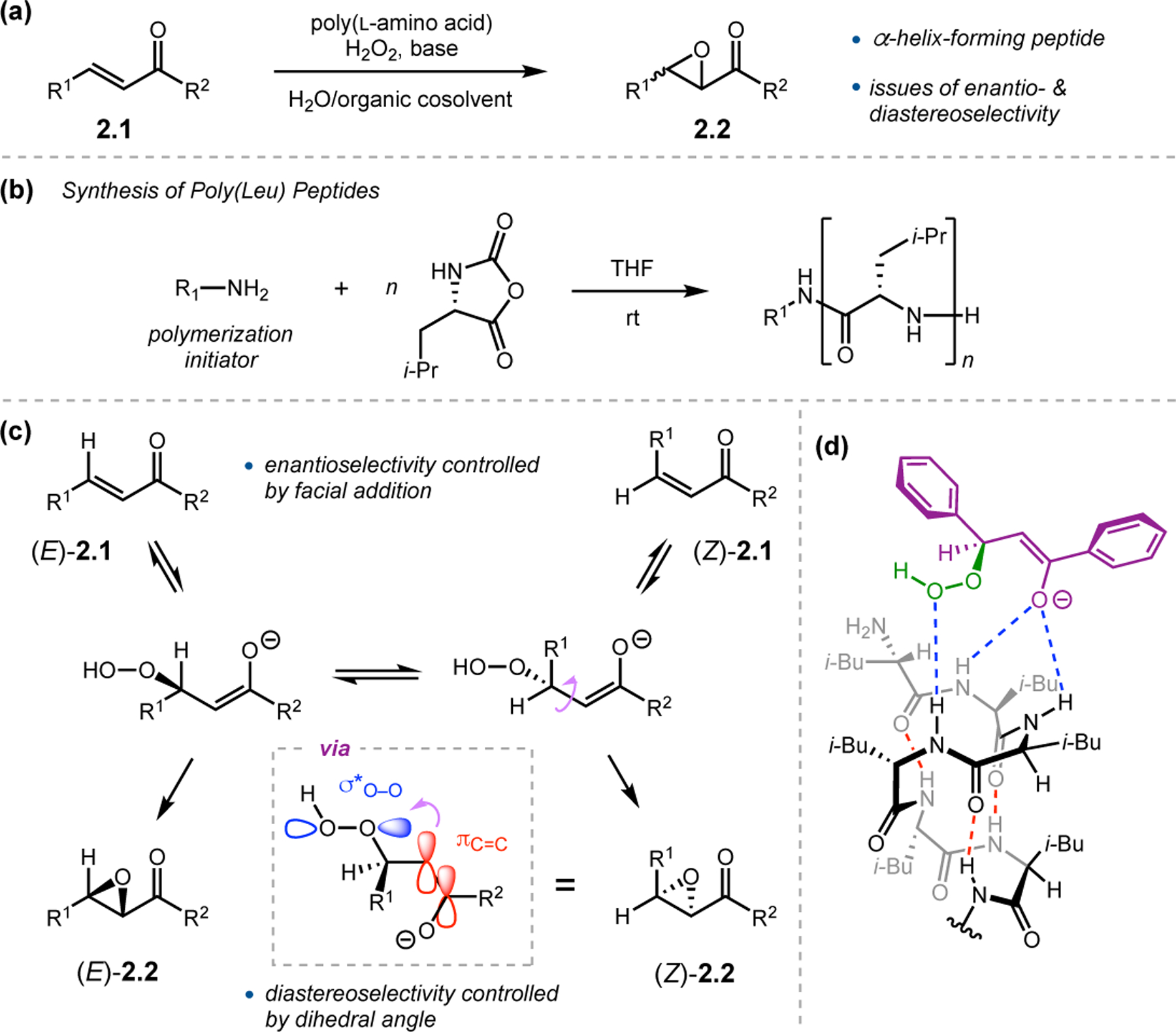
(a) Juliá–Colonna epoxidation. (b) Synthesis of polypeptides by polymerization with N-carboxyanhydrides. (c) Analysis of reaction mechanism and intermediates. (d) Proposed model for stereoselectivity.21
Analysis of the reaction mechanism in epoxidations of chalcone derivatives (2.1)—the most common substrate for this transformation—reveals a complicated picture (Figure 1c).21,22 Beginning either with stereopure 2.1 or a mixture of both Z- and E-alkenes, nucleophilic addition of peroxide occurs, possibly with assistance from the peptide catalyst. While reversible, the facial specificity of this addition is enantiodetermining for the β-position. The C–C bond adjacent to the peroxy group in the resulting intermediate can freely rotate to facilitate orbital overlap of the peroxy species with either face of the enolate π-bond, leading to either diastereomer of epoxide 2.2. Diastereoselectivity is hence determined by the orientation of this dihedral angle. In assessing how peptides can control the selectivity in both of these critical steps, the most common catalysts are purported to adopt α-helical conformations, such as poly(Leu) peptides.21,23 In this structure, the four N-terminal NH groups are not engaged in intrachain, helix-forming H-bonds and thus make excellent handles for H-bond donation to substrates. A putative model containing the critical noncovalent interactions associated with the peptide (red) and between the peptide and substrates (blue), was proposed to account for the observed enantioselectivity (Figure 1d).
Given the high enantioselectivity afforded by these peptides, numerous reports since 2007 have focused on advancing this system through (1) improvement over the often challenging triphasic (water/organic/solid) reaction conditions, (2) further development of more selective catalysts, (3) expansion of substrate scope, and (4) mechanistic analysis.
One aspect of the Juliá–Colonna epoxidation that has been the subject of extensive study is catalyst design. A complication of the earlier conditions for this reaction is that the peptide catalysts often formed a voluminous gel upon workup, making extractions difficult and impeding reuse. A workaround for this issue has been to synthesize solid-supported polypeptides, which are easier to separate.
In continuing to make more powerful catalysts that (1) have a lower molecular weight and (2) are easier to reuse, Yang, Tang, and co-workers applied concepts from phase transfer catalysis toward these epoxidations.24 Efforts in phase transfer catalysis have previously been undertaken with regards to the Juliá–Colonna epoxidation, specifically in the use of additives to increase the concentration of peroxide in the organic layer.25 Building on this approach, the authors synthesized poly(Leu) peptides with an imidazolium-containing initiator to form peptide 2.3a (Figure 2a). When comparing this catalyst to the poly(Leu) peptide synthesized with n-butylamine as an initiator (2.3b) in the epoxidation of 2.1, imidazolium-containing 2.3a provided epoxide 2.2 in substantially higher yield and enantioselectivity (Figure 2b). Furthermore, catalysts were easily re-isolated from reactions, and reusable with minimal perturbation to enantioselectivity. Reaction times even decreased after the first reaction with a batch of catalysts.
Figure 2.
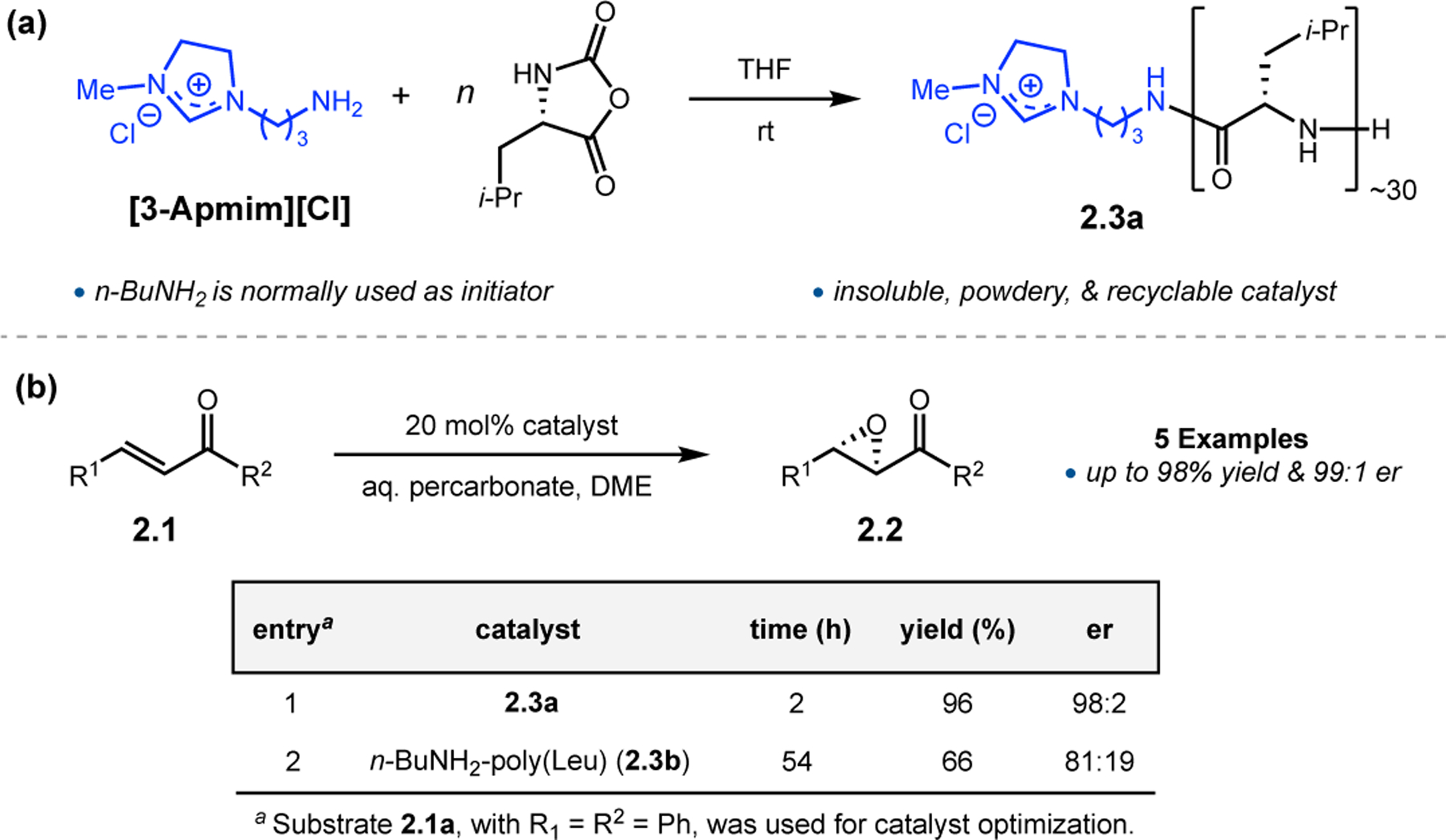
(a) Synthesis of imidazolium-containing poly(Leu) catalyst. (b) Epoxidation reactions.24
While helical polypeptides have dominated the field of Juliá–Colonna epoxidation since its inception, limited work has focused on incorporating other peptide structural elements within this framework. Kudo and co-workers had previously developed peptide catalysts for transfer hydrogenations (see Section 3.1) consisting of a Pro-containing β-turn attached to a poly(Leu) chain supported on resin. The N-terminal Pro-residue operates via iminium catalysis to activate enals toward reduction with Hantzsch ester, while the β-turn provides a good chiral environment for asymmetric induction. The poly(Leu) chain was later added to increase the hydrophobicity of the catalyst for reactions in aqueous media. However, given the known efficacy of poly(Leu)-based peptides in epoxidation reactions, the authors further studied their catalysts in this context.26 Specifically, the aqueous epoxidation of enals via iminium catalysis was examined, which is similar, yet mechanistically distinct, from the classic Juliá–Colonna epoxidation of enones. Commencing with a previously developed catalyst for transfer hydrogenation, peptide 2.6a was found to afford epoxide 2.5 in a good er of 14:86, albeit with low conversion (Figure 3a, entry 1). Removing the first N-terminal Pro residue and replacing the α-aminoisobutyric acid (Aib) residue with 1-aminocyclohexane carboxylic acid (Achc, 2.6b) enabled an increase in conversion to 77% with similar enantioselectivity (entry 2). Deletion of the two Trp-containing residues resulted in reduction of the observed er to 79:21, indicating these positions play an essential role in enantioinduction (entry 3). Given the importance of these positions, the authors postulated that the inclusion of very bulky pyrenylalanine (Ala(1-Pyn)) might increase the enantioselectivity, and indeed catalysts 2.6d and 2.6e provided 2.5 in up to 97:3 er (entries 4–5). Finally, the α-helical forming poly(Leu) chain on the C-terminus was confirmed to be important for catalyst activity, as variation in this sequence resulted in lower ers and conversions.
Figure 3.
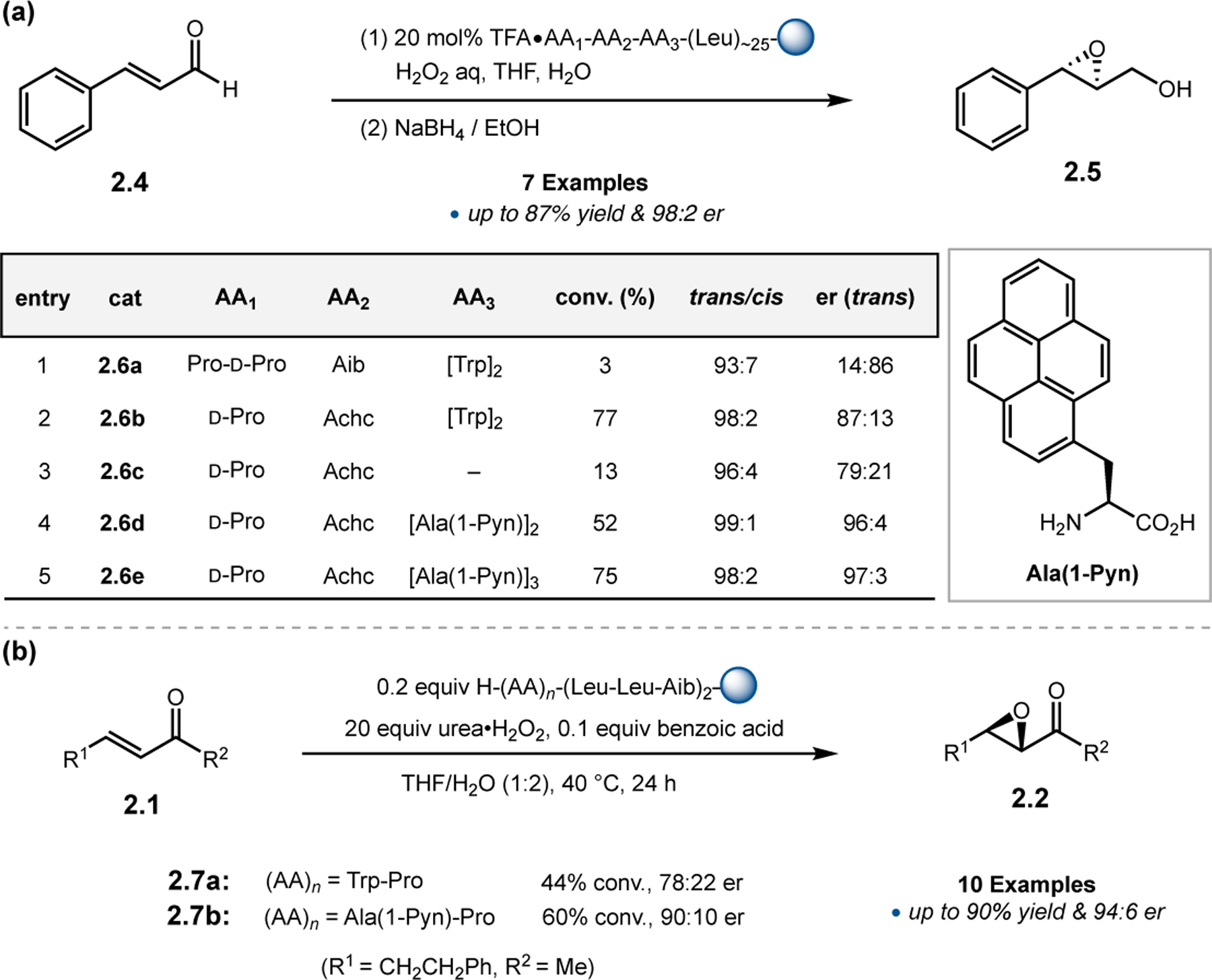
(a) Epoxidations of enals mediated by Pro-containing peptides with both β-turn and α-helix design elements. (b) Epoxidations of enones mediated by free N-terminal arene-containing residues.26,27
Given the success in the context of asymmetric epoxidation of enals, Kudo and co-workers next applied their system toward the oxidation of enones (Figure 3b).27 In this case, secondary amines are too sterically hindered to activate ketones via iminium catalysis, so the authors turned to primary amine-based catalysts.28 Specifically, given the observed effect of the aromatic residues near the N-terminus in epoxidations of 2.4, the authors designed new catalysts with N-terminal aromatic residues (2.7a & 2.7b). Either Trp or Ala(1-Pyn) residues were appended to a Pro for structural rigidity, followed by a 310-helix forming Leu-Leu-Aib repeating sequence. While Trp-containing 2.7a provided epoxide 2.2 in low conversion and er, Ala(1-Pyn)-containing 2.7b afforded 2.2 with 90:10 er and 60% conversion. Strikingly, the absolute stereochemistry of 2.2 is reversed compared to reactions mediated by poly(Leu) catalysts, despite both containing only l-amino acids.
Further efforts to render peptide catalysts more easily separable from reaction mixtures were accomplished by Segarra and co-workers, who reported the immobilization of poly(Leu)-containing peptides on hydrotalcite (Figure 4).29 Synthetic polypeptides were immobilized in less than 30 min under ultrasonic conditions. These nanohybrid materials did not require any pre-activation time, provided rapid and selective epoxidations, and were easily reutilized in subsequent reactions without change in activity.
Figure 4.
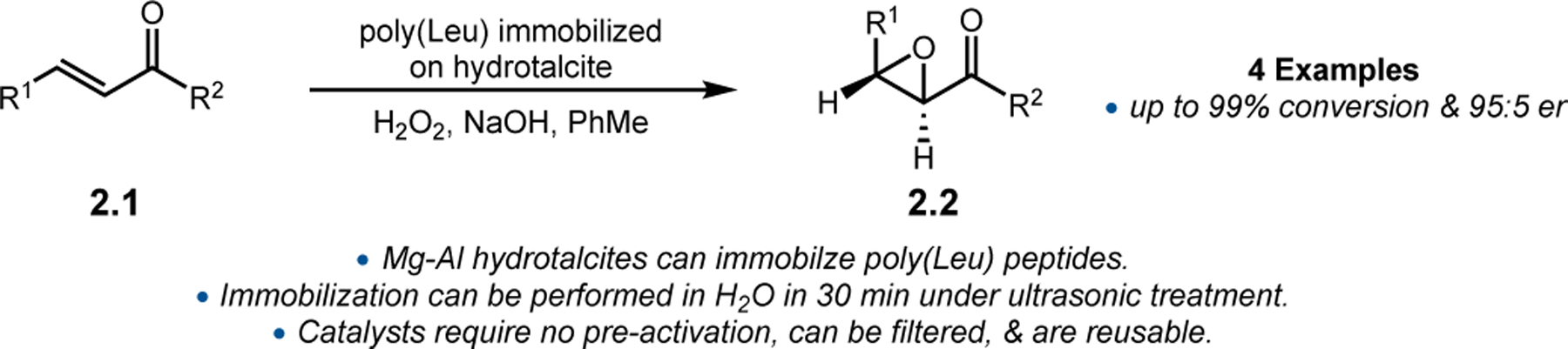
Juliá–Colonna epoxidations mediated by poly(Leu) peptide immobilized on hydrotalcite.29
In addition to solid-supported catalysts, recent work has focused on the development of de novo solution-phase peptides with a variety of intriguing design features.
Given the hypothesis that Juliá–Colonna catalysts predominantly exist as helices, Kurihara and co-workers attempted to stabilize this secondary structure by stapling the peptide backbone (Figure 5).30–32 Intrachain stapling between residues positioned adjacent to each other within a helical scaffold, such as the i and i+4 positions, has been shown to stabilize that conformation.33 As such, allylated l/d-Ser derivatives were incorporated into short peptide chains (2.8) and subjected to ring-closing metathesis and subsequent olefin hydrogenation to access all four diastereomers of stapled peptide 2.9 (Figure 5a). Spectroscopic analysis of these peptides was performed to assess the different structures of the unstapled (2.8) and stapled (2.9) variants of each stereoisomer. Overall, seven of the eight peptides were observed to adopt 310-helical conformations, indicating that stapling did not affect the structures of three of the four diastereomers of 2.8. However, stapling of 310-helical (3R,7R)-2.8 altered the conformation of (3R,7R)-2.9 to an α-helix, an observation later confirmed by X-ray crystallography. Intriguingly, when applying all eight of these peptide catalysts (2.8 & 2.9) toward the epoxidation of 2.1a, α-helical (3R,7R)-2.9 was the lone stapled peptide that provided 2.2a in substantially higher er compared to the uncyclized catalyst (3R,7R)-2.8. This catalyst also achieved the highest overall er (Figure 5b, entries 7 & 8). This study highlights how even subtle changes to peptide or helical type can have a substantial impact on catalyst selectivity.
Figure 5.
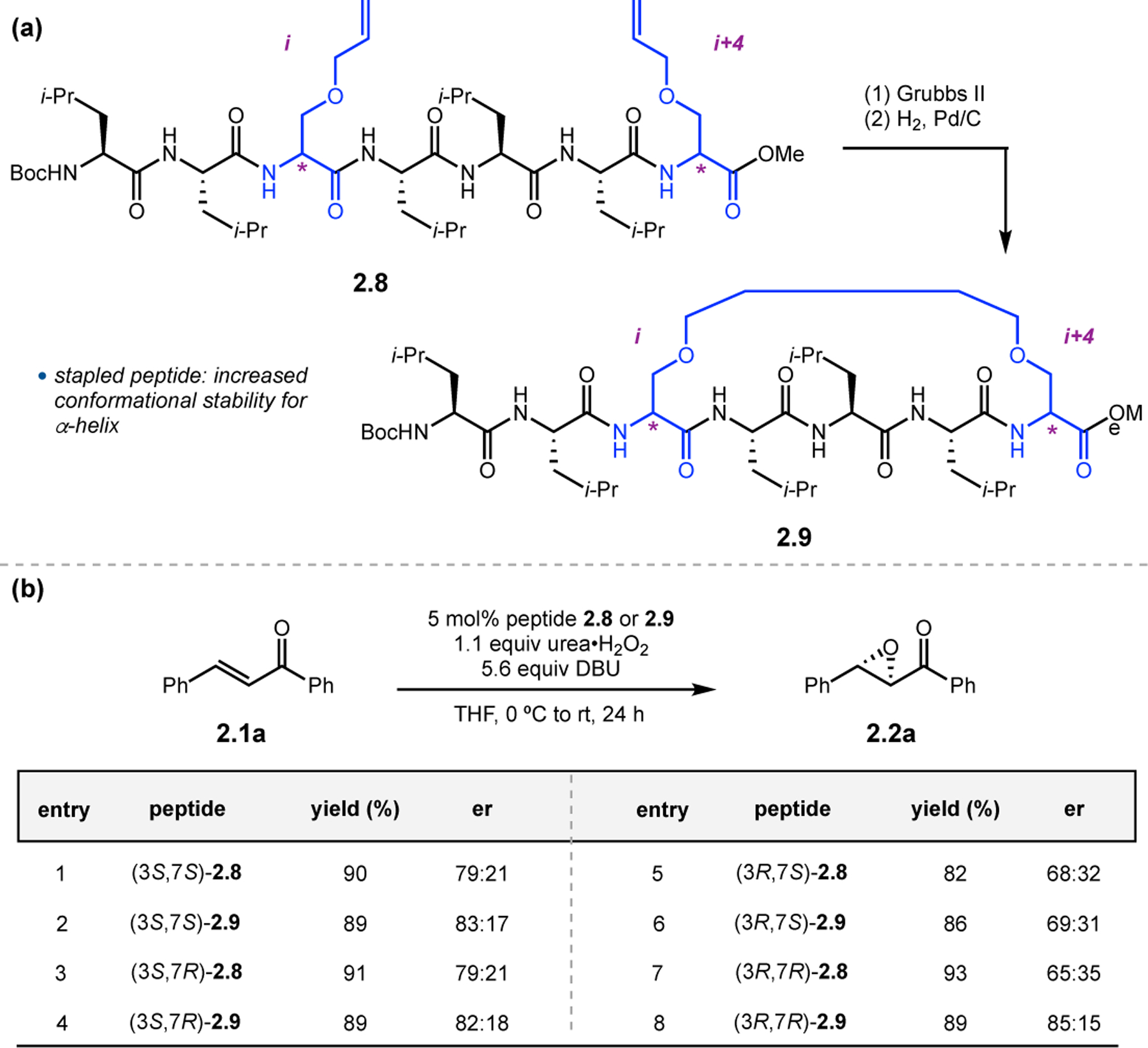
(a) Stapling of short peptides in order to stabilize helical conformations. (b) Epoxidations utilizing stapled and unstapled peptides.30
Alternatively, Tanaka and co-workers chose to stabilize α-helical conformations by including α,α-disubstituted residues in a poly(Leu) peptide.34 Residues such as the cycloleucine (Cle) derivatives in Figure 6a are known to induce the formation of α-helices within (Leu-Leu-Xaa) units. While myriad peptides were synthesized and assayed in the epoxidation of 2.1, general trends are presented here. Peptide length was shown to have a significant effect on selectivity, as the er rose from 66:34 to 99:1 by increasing the length of catalyst from six residues in 2.10a to twelve in 2.10c (entries 1–3). Furthermore, most N-terminal Boc-containing peptides provided low ers (entries 4 & 5). Finally, the choice of α,α-disubstituted residue had a significant effect on selectivity. Compared to catalysts 2.10 and 2.11, peptides of the 2.12 set, which contain the (S)-3-methoxy Cle residue, provided high er even at unoptimized lengths and with Boc groups (entries 6–9). Crystallographic analysis of high performing peptide 2.10c revealed an α-helix (Figure 6b). Further evidence that this conformation appears to be important for achieving high enantioselectivity in Juliá–Colonna epoxidations.
Figure 6.
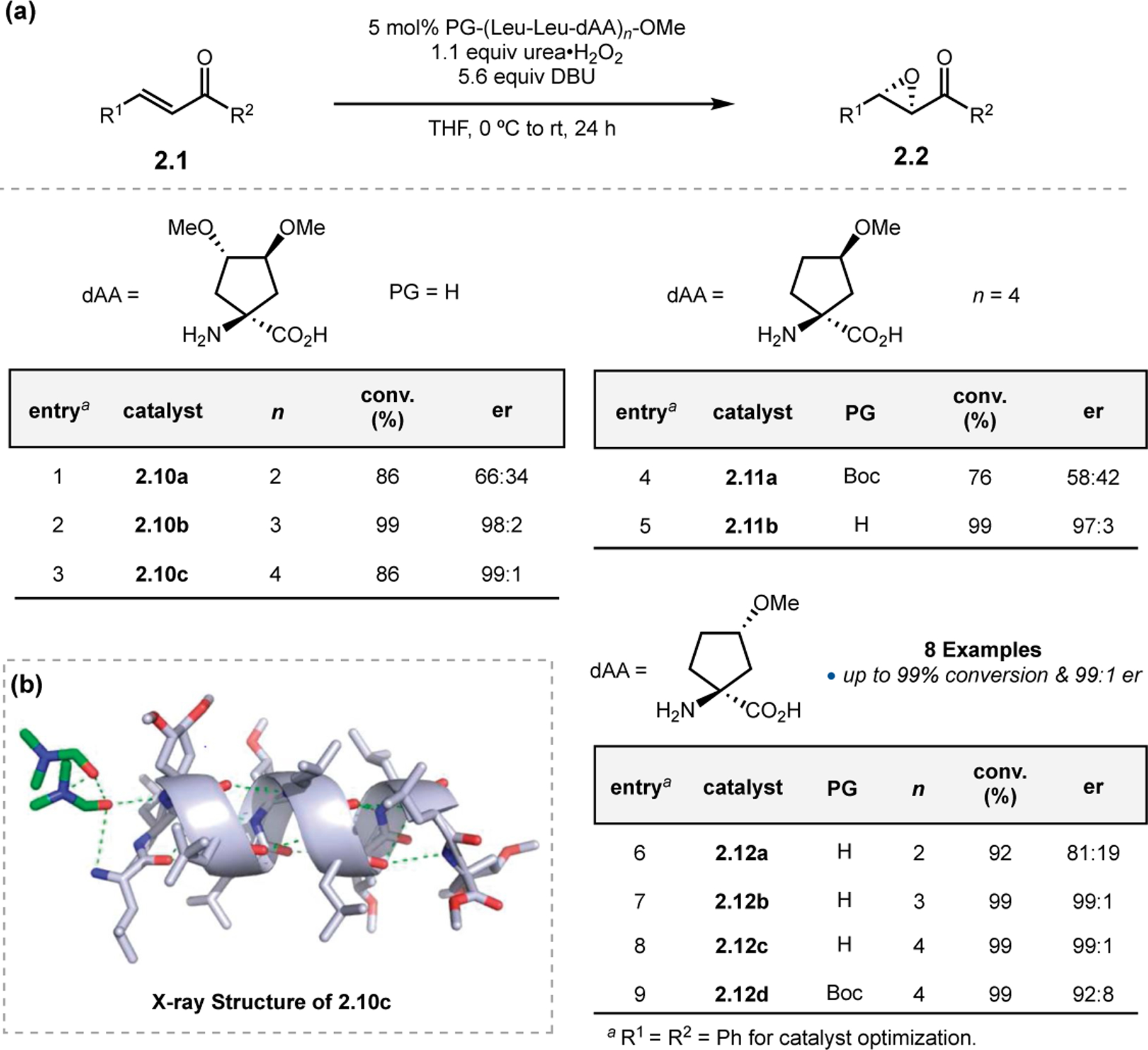
(a) Epoxidation with peptide containing helical forming cyclic residues. (b) Crystal structure of peptide 2.10c. Adapted with permission from ref. 34. Copyright 2010 American Chemical Society.
In contrast to larger, helical forming peptides, Voyer and co-workers explored the use much smaller cyclic dipeptides (Figure 7).35 Known for a variety of other asymmetric transformations, these minimal peptides have never been applied toward epoxidation.36 Despite the substantial reduction in size compared to the helical polypeptides, diketopiperazine 2.13a was able to achieve 85:15 er in the epoxidation of 2.1. The relative stereochemistry of the dipeptide was essential for enantioselectivity, as 2.13c provided racemic product. Furthermore, a linear dipeptide variant (2.14) only achieved epoxidations in 58:42 er, implying that cyclization was required for high catalytic activity. While conversions were generally low in reactions performed in hexanes, they could be accelerated in protic solvents, albeit at expense of enantioselectivity.
Figure 7.
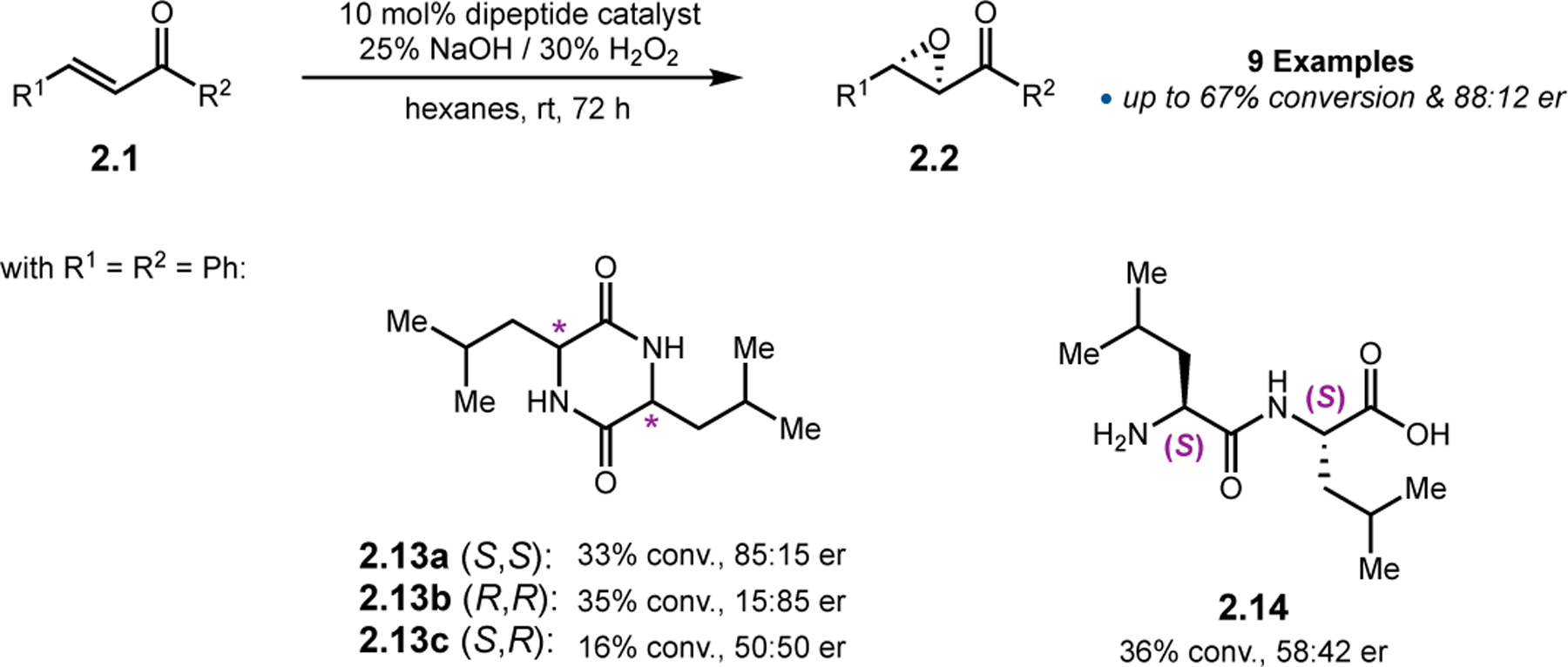
Juliá–Colonna epoxidations mediated by cyclic dipeptides.35
While the effects of variations to peptide catalysts have been studied extensively, one challenge still facing Juliá–Colonna epoxidations is the paucity of methods for the fast and controllable synthesis of longer helical peptides.
Leigh and co-workers addressed this issue in their report on alternative, faster strategies for the construction of poly(amino acid) chains.37 Central to their approach was a rotaxane-based molecular machine appended with a short peptide chain (Figure 8a).38 The macrocycle is threaded onto a mono-dispersed oligomeric track, on which are attached various amino acid phenyl esters. As the rotaxane moves along the linear chain, it collects all residues blocking its path and incorporates them onto the growing peptide chain. As such, a one-barrier [2]rotaxane (2.15) was synthesized (Figure 8b). The ring was appended with a protected trimer sequence (Cys-Gly-Gly) on which to grow the peptide. This ring (blue) was blocked on one side by a phenyl ester (green) on the linear chain (black), which contains the next amino acid building block to incorporate onto the rotaxane ring. Attached to the linear chain is a polymer precursor containing the leucine building blocks, and this was synthesized via copolymerization with styrene and p-leucyloxystyrene (purple). On average, the molecular machines contained approximately 52 styrene units, and 6 Leu units.
Figure 8.
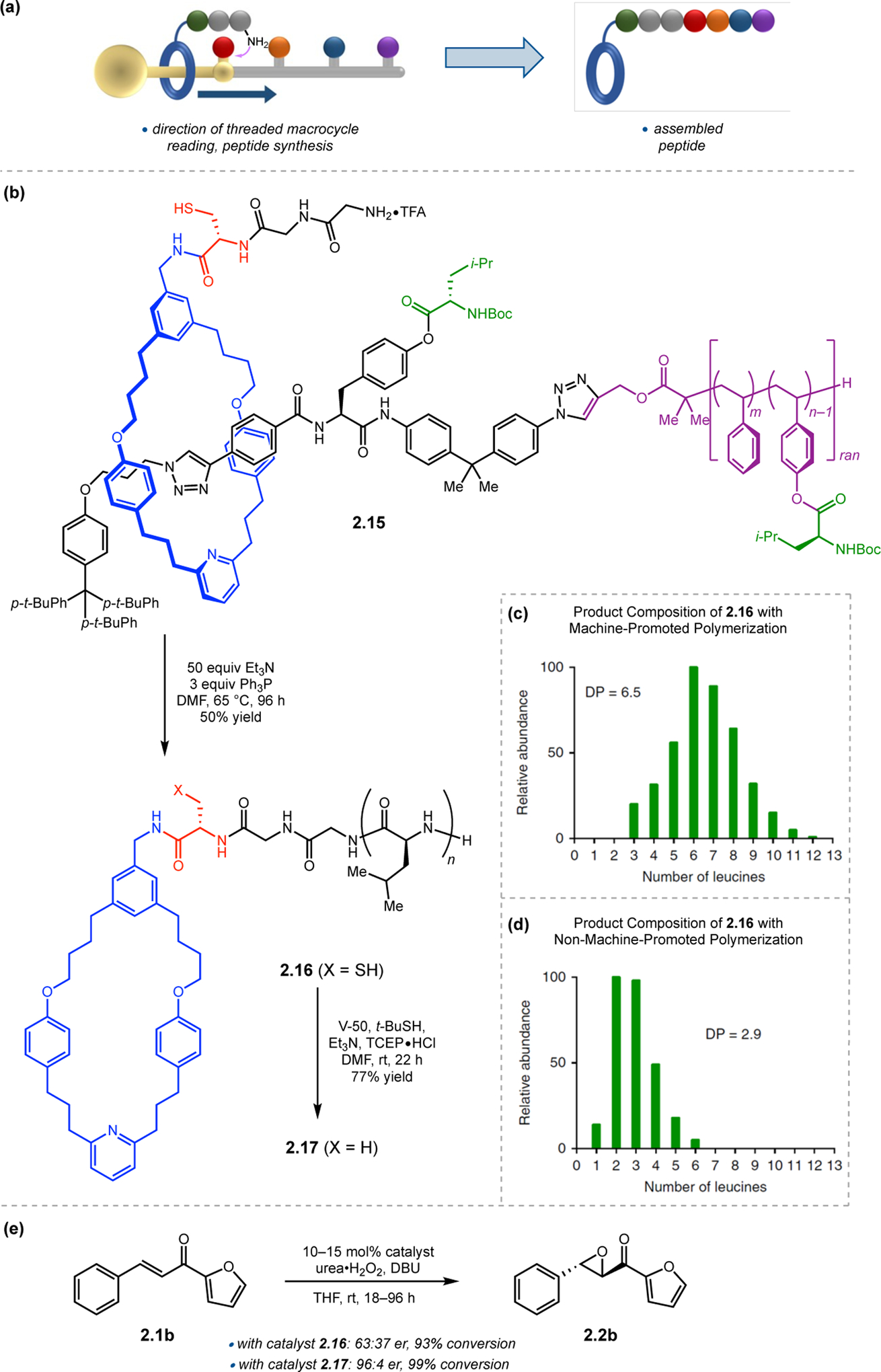
(a) Cartoon depicting the synthesis of a polypeptide with a rotaxane-based molecular machine. (b) Synthesis of a poly(Leu) peptide with a rotaxane-based molecular machine. (c & d) The degree of polymerization (DP) with and without the molecular machine approach. (e) Epoxidations mediated by the rotaxane-based poly(Leu) catalysts. Adapted with permission from ref. 37. Copyright 2017 Nature Publishing Group.
To facilitate chain-walking peptide synthesis, 2.15 was treated with triethylamine at 65 °C in DMF (Figure 8b). The reaction proceeds first via native chemical ligation onto a Cys appended to the rotaxane (red), followed by transfer to the N-terminal amino acid. After reacting with the phenyl ester, the rotaxane ring can walk over the unprotected phenol. Upon reacting with all the phenyl esters, macrocycle-containing peptide 2.16 is eventually released from the linear chain, which contains on average 6.5 Leu residues incorporated (Figure 8c). Alternatively, as a control, the peptide synthesis was performed without the molecular machine, with a mimic of the barrier leucine phenyl ester, and only 2.9 couplings were performed under similar conditions, highlighting the importance of the constrained rotaxane as a driving force for chain elongation (Figure 8d). In applying these catalysts toward the epoxidation of 2.1b, catalyst 2.16 was found to provide minimal levels of enantioselectivity (Figure 8e). The Cys in 2.16 was found to oxidize to a sulfonic acid under the reaction conditions, and this is hypothesized to disrupt the α-helical structure of the peptide. Peptide 2.16 was subjected to reductive radical desulfurization to produce 2.17, which provided epoxide 2.2b in an er of 96:4. Molecular machines hence could be a useful method for the synthesis of polypeptide catalysts.
Hernández and co-workers explored alternative techniques to accomplish polymerization using a combination of enzymatic catalysis with the aid of mechanochemical synthesis (Figure 9a).39 Proteases have previously been shown to function as efficient amino acid polymerization catalysts in solution. However, issues of oligopeptide solubility often limit the degree of polymerization (DP) due to intermediate product precipitation.40 A workaround that the authors explored in this study was to facilitate chain growth with ball milling (Figure 9b). This type of milling involves the grinding of solid reagents in a rotating cylindrical tube containing one or numerous freely moving balls, and its utility in synthesis has recently gained traction.41 Under near solvent-free conditions, a combination of powdered protease papain and sodium carbonate was found to mediate the polymerization of various amino acids with good chain lengths (Figure 9a). Notably, the protein is stable to this mechanochemical technique. After optimization of conditions with Phe 2.18a, 99% yield of peptide could be obtained with a DP of 8.0 using hydrated sodium carbonate under planetary ball milling conditions (entry 1). Switching to anhydrous base resulted in erosion of both yield and DP, and this observation was made for Leu 2.18b as well (entries 2–3). While water did not play as great a role in governing the polymerization yields for small residues, such as Gly 2.18c, the DP was substantially lowered with anhydrous base (entries 4–5). The authors hypothesize that the addition of water could create a hydrophobic pocket that attracts the amino acid residues closer to the enzyme active site (Figure 9c). This effect is more pronounced for more lipophilic residues such as Phe and Leu as opposed to Gly. Performing epoxidations on 2.1 with enzymatically and mechanosynthesized 2.18a resulted in excellent yields and enantioselectivities (Figure 9d).
Figure 9.
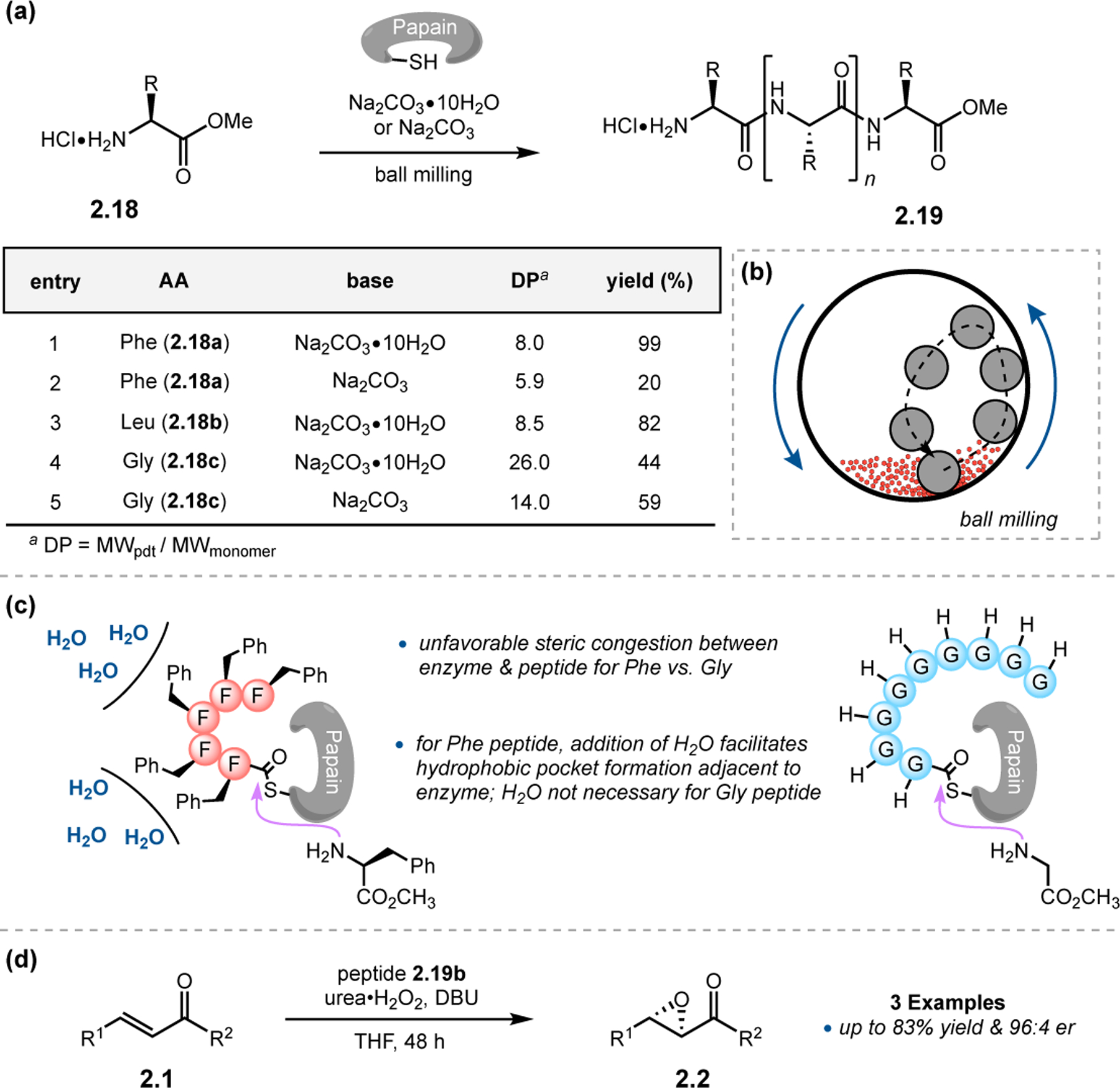
(a) Polymerization of amino acids mediated by the protease papain utilizing mechanosynthesis. (b) Cartoon representation of simple ball milling. (c) Water is beneficial for many lipophilic amino acids, indicating that a hydrophobic effect could be operative. (d) Epoxidations with mechanosynthesized peptides.39
Finally, to combat the normal limitation of performing Juliá–Colonna epoxidations under triphasic conditions (water/organics/solid-supported catalyst), Voyer and co-workers developed purely aqueous conditions (Figure 10a).42 The pH of the medium plays an important role in both er and yield. Yields are normally higher under more basic conditions, albeit at the expense of enantioselectivity (entries 1–3). At pH 8, when the catalytic loading was increased to 10 mol%, yields as high as 97% could be achieved (entry 4). Given that H-bonding interactions are often disrupted in pure water, the authors proposed that a hydrophobic effect between 2.1 and catalyst may be required to obtain enantioselectivity in this reaction setup. MD simulations revealed that the sidechains of poly(Leu) peptide 2.20 could form a hydrophobic pocket adjacent to the α-helix (Figure 10b). Lipophilic chalcones are presumed to bind to the catalyst in these chiral clefts.
Figure 10.
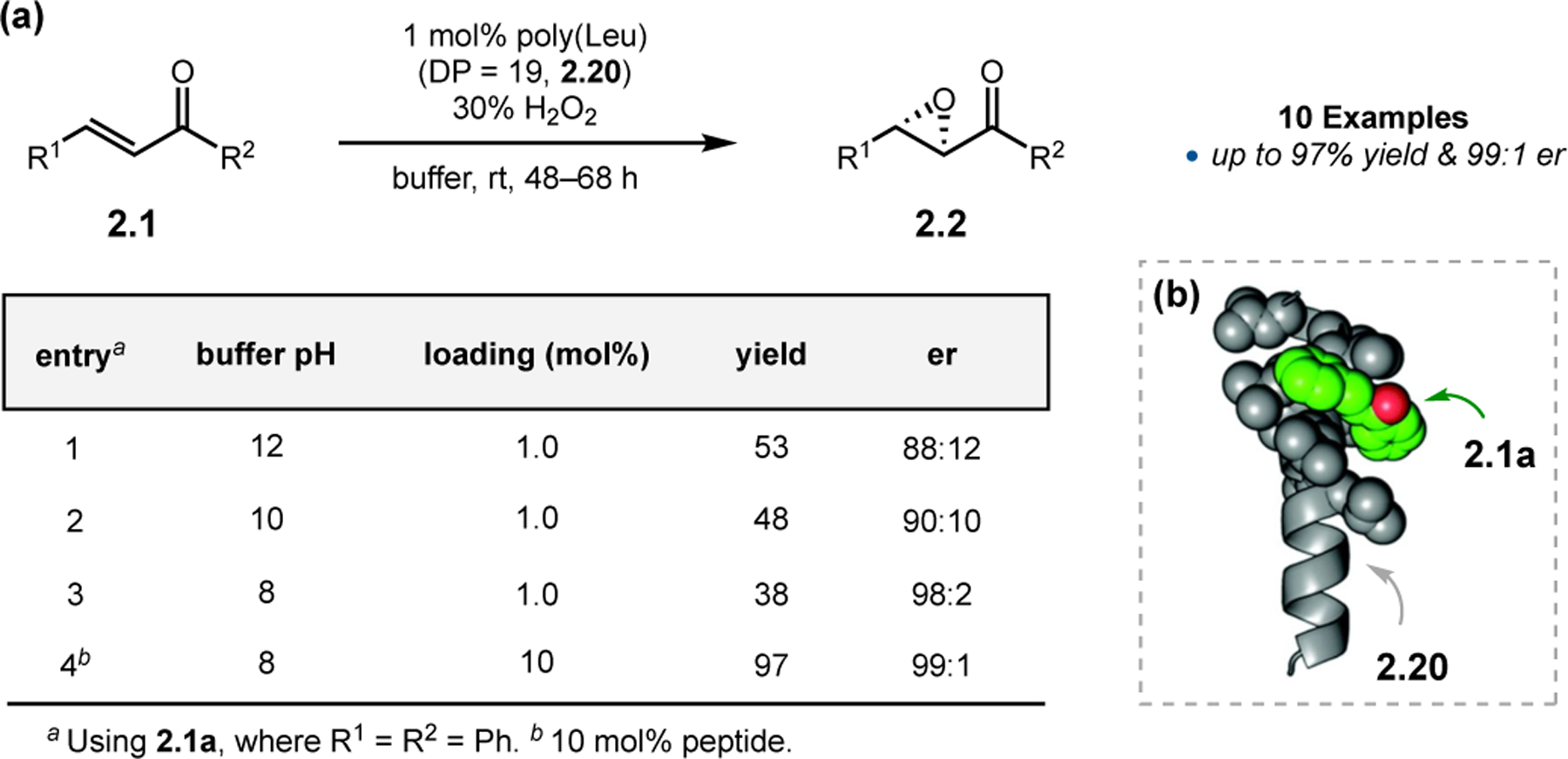
(a) Juliá–Colonna epoxidations in pure water. (b) Mechanistic model centered on a hydrophobic cavity formed from the Leu sidechains of the α-helix. Adapted with permission from ref. 42. Copyright 2017 Royal Society of Chemistry.
2.1.2. Aspartyl Peracid-Based Epoxidations.
Orthogonal to the use of poly(Leu) catalysts for nucleophilic Juliá–Colonna epoxidations of chalcone derivatives, our group sought to develop a complementary peptide-based catalytic system functioning by an alternative electrophilic epoxidation mechanism.43 While Juliá–Colonna epoxidations rely on stereocontrolled nucleophilic additions of peroxides to electron-deficient olefins, our initial approach centered on the generation of a transient, catalytically-active peracid species (C, Figure 11a), mirroring the widely used peracid oxidants, such as m-CPBA.44 We envisioned that this peracid could be formed via carbodiimide activation of an aspartic acid residue in the presence of 30% aqueous H2O2. There were many potential challenges associated with this catalytic approach, including (1) depletion of DIC due to interception by other compounds and (2) formation of off-cycle diacyl compounds (C′). To combat these issues, DMAP was added to accelerate both acyl transfer of B to C and the breakdown of diacyl peroxide C′ to peracid C.45 Under these conditions, Asp-containing peptides were able to epoxidize allylic carbamates 2.21 with high conversions to 2.22 (Figure 1b). Substrate 2.21 was chosen specifically to facilitate noncovalent interactions between the catalyst and substrate. Evaluation of a minimal library of peptide catalysts led to tripeptide 2.23, which provided up to 96:4 er in epoxidations of 2.21.
Figure 11.
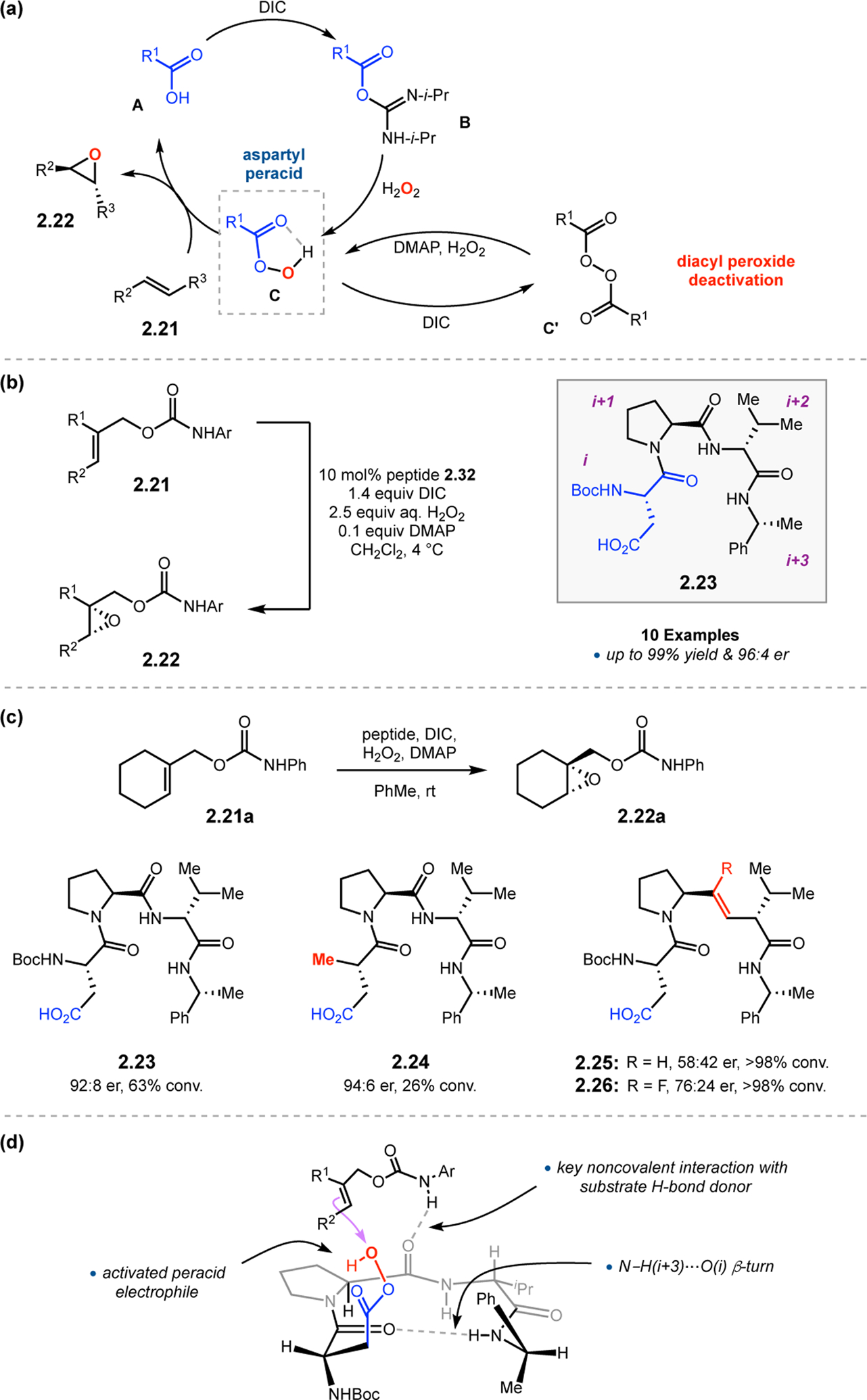
(a) Putative catalytic cycle for the generation of electrophilic aspartyl peracid species. (b) Epoxidation of 2.21 with aspartyl peracid-based peptides. (c) Functional analysis of catalyst 2.23 through synthesis of peptidomimetic analogues. (d) Hypothesized transition state for enantioselective epoxidations.43,46
To study the nature of the putative H-bonding interactions between 2.21 and peptide 2.23, the catalyst was subjected to various point mutations in which key H-bond donors and acceptors were deleted (Figure 11c).46 Tripeptide 2.23 was proposed to adopt a type II β-turn,47 and it was crucial to this study that this overall secondary structural motif remained largely unaltered despite significant functional changes. It was hypothesized that the N-terminal Boc group of 2.23 could be deleted without affecting catalyst structure since it was not expected to be involved in intramolecular H-bonding. Indeed, catalyst 2.24 afforded epoxide 2.22a with similar levels of enantioselectivity as parent catalyst 2.23, albeit with lower conversion. A greater challenge was how to appropriately substitute the loop region amide for a non-H-bonding functional group, since it is such an integral part of the β-turn backbone structure. One approach for the isosteric replacement of amide bonds is using alkenes.48 Alkene-containing peptide 2.25 was found to give significantly lower enantioselectivity than 2.23. However, further structural analysis revealed that 2.25 exhibited substantially more conformational flexibility than 2.23. While unfunctionalized alkenes may not be capable of fully mimicking characteristics of the polarized nature of amides, fluoroalkenes were proposed to serve this purpose.49 Indeed, peptide 2.26 was shown to primarily adopt a single conformation in solution that is quite similar to that of 2.23. While fluoroalkene-containing 2.26 provided 2.22a with lower er than parent catalyst 2.23, its efficacy was vastly improved over alkene-containing catalyst 2.25. Within the type II β-turn framework, it is possible that the i+1 C=O of the loop amide is essential for H-bonding with the carbamate of 2.21a (Figure 11d). The structural similarity between 2.23 and 2.26 could imply that the C–F bond of 2.26 is a competent, yet significantly inferior, H-bond acceptor.
These results highlight the utility of peptide-based catalysts biased toward β-turn conformations, as their programmable secondary structures could allow for selective reactions with many different substrate classes. However, while the modular peptide framework and the availability of amino acids provides nearly limitless potential for the discovery of unique catalyst sequences and structures, the rapid discovery of high-performing catalysts that are matched with specific substrates can be challenging. Analogous to the field of enzyme-mediated transformations, it is often difficult to predict the folded secondary structures of specific peptide sequences50 and which peptide structures will provide high levels of selectivity for particular transformations or substrates.51
Building upon precedent in combinatorial catalyst library screening,52,53 our group sought to develop an analogous high-throughput method for the discovery of competent oxidation catalysts.54 We envisioned that this approach could facilitate the identification and optimization of catalyst sequences for enantioselective transformations of small molecules (e.g., 2.21a), as well site-selective modifications of more complex molecules like farnesol (2.27, Figure 12a).55,56 Specifically, a one-bead-one-catalyst (OBOC) screening platform was chosen, as it had previously been effective in developing highly selective, peptide-based acylation catalysts.57
Figure 12.
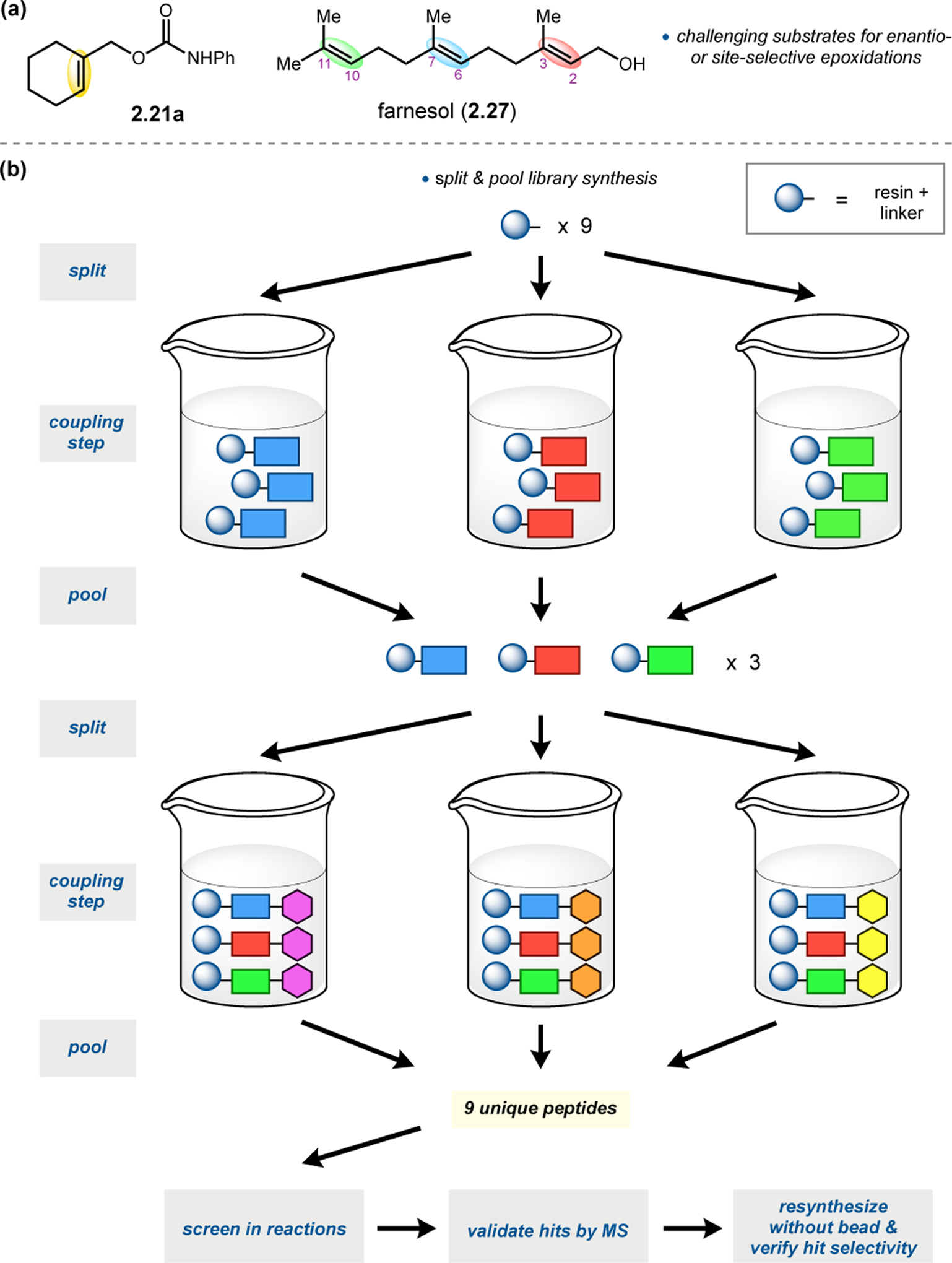
(a) The prediction of peptide sequences that are matched to perform selective chemistry on new substrates remains challenging. (b) Split-and-pool one-bead-one-catalyst library synthesis, screening, and identification.54
The workflow for OBOC synthesis, screening, and identification of hit sequences is presented in Figure 12b, which follows a split-and-pool method.58 Beginning with resin-bound precursors having an appropriate linker, individual beads can be split into various reaction vessels and coupled to various amino acid residues. To generate diversity, these beads are then pooled together, followed by random splitting and further coupling. In the simple example presented in Figure 12b, with two coupling steps each with three unique amino acids, nine theoretical dipeptides are possible. Moreover, this method offers control over the identities of amino acids at each position. For example, if a Pro residue was proposed to be essential at i+1, all beads could be programmed to contain a Pro or Pro-like derivative at that position. After synthesizing large arrays of on-bead peptide libraries, each bead can be manually sorted into individual reaction vessels and screened for activity using GC or HPLC to assess conversion and enantioselectivity. High-performing catalysts can then be sequenced via partial Edman degradation-mass spectrometry.59 As such, it is essential that all amino acids utilized in the synthesis of the library have different molecular weights in order to discriminate between mass fragments. Upon identification of hit sequences, the peptides are resynthesized and evaluated in solution. It is often the case that peptide-based catalysts perform notably better in solution, implying that the solid-support can interfere in enantioinduction. This means that even small increases in selectivity observed on-bead are often amplified in solution.60 As a proof of concept, an on-bead library of catalysts for the epoxidation of 2.21a was synthesized and screened using the OBOC method.54 Of the 512 peptide sequences synthesized, the highest performing catalyst (Boc-Asp-Pro-d-Val-Tyr(t-Bu)-linker-bead) was quite similar in structure to peptide 2.23 that was previously developed for the for epoxidation of 2.21a,43 differing only at the i+3 position.
Having validated the OBOC peptide library screening platform, we next sought to expand its utility in more challenging reaction paradigms, such as the site-selective epoxidation of complex molecules.55,56 Farnesol (2.27) contains three different alkenes that are expected to exhibit similar reactivity patterns. Because the only notable difference between these olefins is their relative distance from the terminal alcohol, it is particularly difficult to achieve the differentiation required for site-selective modification. In nature, enzymes often overcome site-selectivity issues by taking advantage of well-defined binding pockets to interact with substrates in a precisely controlled manner.61 For example, squalene epoxidase is able to selectively oxidize one alkene of the three chemically distinct alkenes in squalene.62 This catalyst-controlled site-selectivity has been a challenging paradigm to translate to small molecule catalysts.17 Our group has held that peptide catalysis may offer a solution, as diverse peptide sequences could tune the reactivity and selectivity of a conserved catalytic residue through the formation of specific secondary structures and precise noncovalent interactions with complex substrates.
Inspired by the selectivity exhibited by squalene epoxidase,62,63 our group sought to apply aspartic acid-containing peptides toward the site-selective epoxidation of farnesol (Figure 13a).55 While m-CPBA delivered an unselective mixture of epoxide products (Figure 13b, entry 1), initial screening revealed that peptide 2.31 selectively targets the 2,3-alkene to provide 2.28 in a 4:1:1 ratio over 6,7-epoxide 2.29 and 10,11-epoxide 2.30 (entry 2). A peptide library biased toward sequence 2.31 was synthesized using the OBOC method by coupling approximately 50% of the beads with the parent residue at each position. From this library, peptide 2.32 was discovered, which exhibited an increased 2,3-selectivity of 8.2:1.0:1.3 (entry 3). The Pro-Asn(Trt) diad was retained from parent sequence 2.31, highlighting its importance to selectivity. A second library now biased toward 2.32 led to the identification of two even more selective catalysts, 2.33a and 2.34a (entries 4–5). Upon validation of these on-bead sequences in solution, their site-selectivities increased to >100:1:1 (entries 6–7). We hypothesized that highly site-selective catalysts might also be highly enantioselective, since the best catalysts would presumably form specific networks of noncovalent interactions with substrates. Indeed, catalyst 2.34b was able to perform the site-selective epoxidation at the 2,3-position of farnesol with an er of 93:7 (entry 7). Further study of this peptide revealed that it was a highly selective in the epoxidation of allylic alcohols, providing high levels of enantioselectivity over a range of substrates 2.35 (Figure 13c). The alcohol was hypothesized to serve as a directing group for the functional group-rich backbone of the peptide, facilitating reaction at the adjacent alkene.
Figure 13.
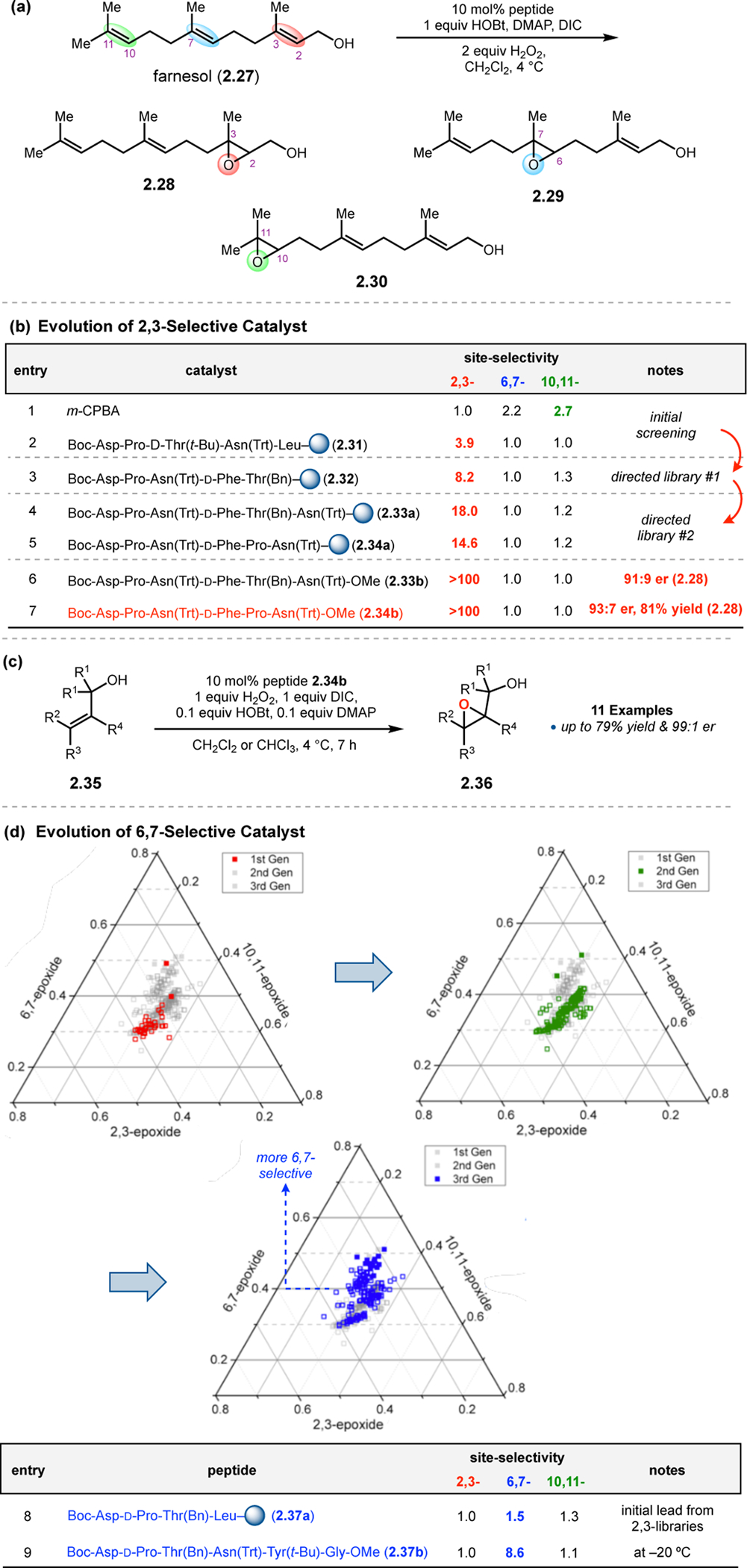
(a) The epoxidation of farnesol 2.27 could provide up to six epoxide products, posing challenges in site-selectivity and enantioselectivity. (b) Biased libraries for selective 2,3-epoxidation revealed hit catalyst 2.34b. (c) Peptide 2.34b can function as a highly selective catalyst in the epoxidation of numerous allylic olefins. (d) Evolution of a 6,7-selective farnesol epoxidation catalyst.55,56 Ternary plots show the positional selectivity, with 6,7-selective catalysts toward the top. Reproduced with permission from ref. 56. Copyright 2014 American Chemical Society.
Up to this point, peptides appeared to favor alcohol-directed 2,3-epoxidations of farnesol. However, our group sought to identify different peptide-based catalysts that could selectively to deliver the other farnesyl epoxides 2.29 and 2.30.56 An initial hint of selectivity in favor of the 6,7-alkene came with catalyst 2.37b (Figure 13d, entry 8). Based on this initial hit, three subsequent libraries of extended peptide sequences were synthesized, and the data were presented in the ternary plots shown in Figure 13d. These plots represent the relative ratios of the 2,3-, 6,7-, and 10,11-epoxide products. 6,7-Selective catalysts appear closer to the top vertex of the graph. The first library produced mostly non-selective catalysts or peptides that still favored 2,3-epoxidation (plot 1), though it did generate a high performing catalyst with a Thr(Bn) residue at the i+2 position. Two subsequent biased libraries (plots 2 and 3) revealed hit catalyst 2.37b, which provided a 1.0:8.6:1.1 ratio of products favoring 6,7-epoxide 2.29 upon validation in solution (entry 9). It is noteworthy that this rapid enhancement of 6,7-selectivity was enabled by on-bead library screening based on a lead sequence with very modest positional selectivity (2.37a).
In order to determine the origins of catalyst-controlled selectivity, NMR studies (in CDCl3) were performed to assess the solution structures of peptides 2.34b64 and 2.37b.56 Structural analysis of peptides can often be challenging due to their inherent conformational flexibility. One approach to overcome this issue is to use a combination of NMR and computational methods to predict an ensemble of possible structures, thereby modeling catalyst dynamics in solution. Two-dimensional ROESY spectra were recorded, and the through-space contacts were assigned, integrated, and used to calculate inter-proton distances. These NMR-derived distances were then used as restraints to calculate the most plausible structures.65 Both peptides were found to adopt multiple conformations in solution, but key structural features of these ensembles emerged. While the termini of 2.34b were quite flexible in solution, a central N–H(i+5)•••O=C(i+3) γ-turn H-bond was retained in most structures (Figure 14a). A key N–H(i+3)•••O=C(i) β-turn H-bond dominated the structures of 2.37b (Figure 14b). Because numerous, discrete conformations of 2.34b were consistent with the NMR data, it was difficult to propose one particular active conformer. However, since this peptide is highly selective for 2,3-epoxidation of 2.27, it likely interacts with the adjacent allylic alcohol via H-bonding (Figure 14c). On the other hand, given the more consistent β-turn conformation of 2.37b, it is plausible that the allylic alcohol binds much further away from the aspartyl peracid, possibly through H-bonds with N–H(i+2) and the sidechain of Thr(Bn), placing the 6,7-alkene in closer proximity to the peracid (Figure 14d). While elucidation of the exact catalytically active conformation of minimal peptides remains challenging, NMR and computational tools exist that enable ensembles of potential structures to be considered.
Figure 14.
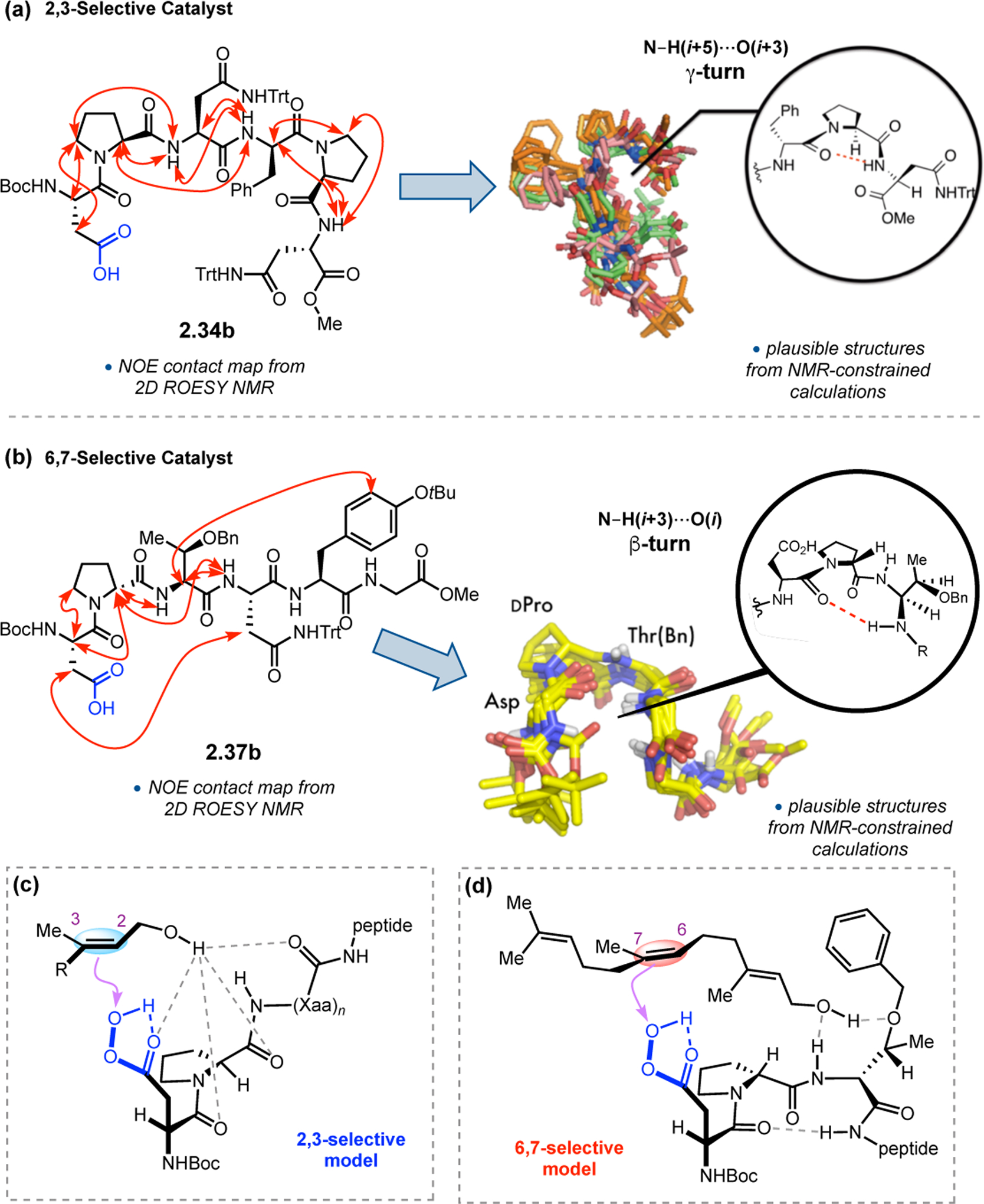
(a) NMR and computational structural analysis of peptide 2.34b. Adapted with permission from ref. 64. Copyright 2014 Royal Society of Chemistry. (b) NMR-constrained computational structural analysis of peptide 2.37b. Reproduced with permission from ref. 56. Copyright 2014 American Chemical Society. (c) Proposed model for the 2,3-selective epoxidation of farnesol with 2.34b. (d) Proposed model for the 6,7-selective epoxidation of farnesol with 2.37b.
Given the effectiveness of Asp-containing peptides for both enantio- and site-selective epoxidations, this catalytic strategy has been applied in the total synthesis of natural products.66,67 While oxidized indoles are a prodigious class of natural products, efforts to effect catalyst-controlled, enantioselective indole oxidations have suffered from complications associated with chemoselectivity and the propensity for product rearrangement. To address these issues, our group applied Asp-based peptides toward selective synthesis of 3-hydroxy-indolenines in collaboration with the Movassaghi Group.66 We hypothesized that, upon initial epoxidation of indole 2.38 with aspartyl peracids, subsequent rearrangement would result in 2.39 (Figure 15a). Upon optimization of reaction conditions, evaluation of a small library of Asp-containing catalysts resulted in the discovery of pentapeptide 2.40, which afforded a wide-range of 3-hydroxy-indolenines 2.39 with er values up to 95:5. Having developed a highly efficient catalyst, we turned our attention toward substrates 2.41 as a means to address issues of site- and diastereoselectivity (Figure 15b), which can become quite important in the context of total synthesis and complex molecule functionalization. While reactions in the presence of an achiral carboxylic acid revealed a subtle substrate-controlled selectivity favoring the (R)-stereochemistry of the oxidized indole (69:31 dr), peptide 2.40 enhanced this selectivity to 92:8 dr (entries 1–2). Interestingly, using ent-2.40 provided a mismatched case and negligible diastereoselectivity (entry 3).
Figure 15.
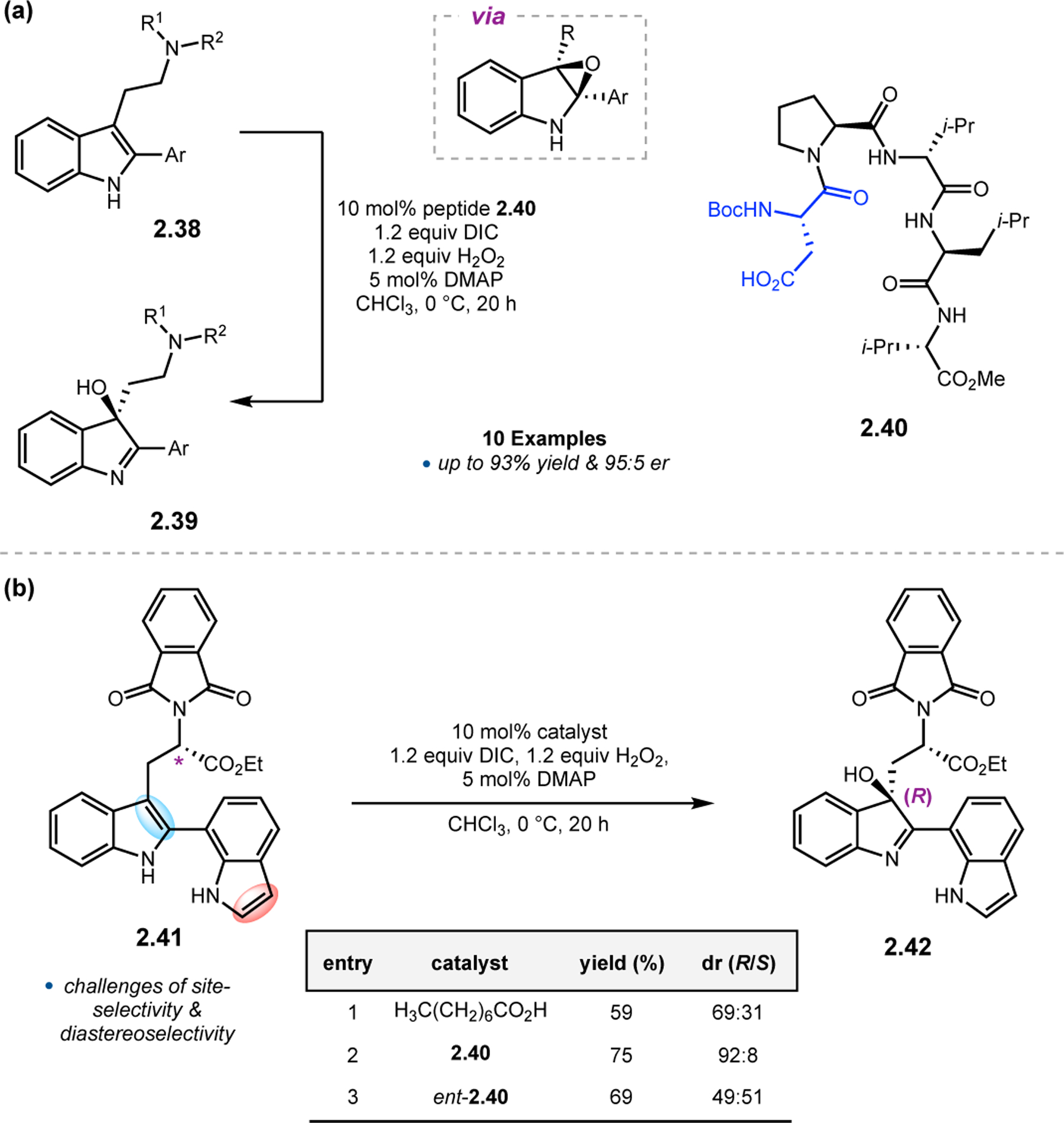
(a) Enantioselective peptide-based oxidations of achiral indole 2.38. (b) Diastereo- and site-selective oxidations of indole 2.41 with peptides.66
Similar selective indole oxidations were pursued in the total synthesis of citrinalin B and cyclopiamine B by a team of researchers led by the Sarpong Group.67 Cyclopiamine B (2.43) could be accessed through an indole-to-spirooxindole rearrangement of 2.44, which itself could arise from indole 2.45 (Figure 16a). When oxaziridines (2.49–2.51) were utilized to oxidize the C3 position of indole 2.45, the desired alcohol diastereomer 2.46 was not obtained as the major product, suggesting that the inherent facial selectivity of O-atom delivery favors diastereomer 2.47 (Figure 6b). Rearranged spirocycle 2.48 was also formed, though unfortunately this compound was epimeric to the desired spirocycle 2.43. These results spurred a campaign to develop reagents or catalysts that could overturn this inherent selectivity. Asp-containing peptides were particularly promising given that the acetamide of 2.45 could function as a directing group to enable oxidation from the desired face of the molecule. Indeed, following evaluation of a focused catalyst library, peptide 2.52 was found to provide the desired oxidized indole 2.46 in 83% yield. Unfortunately, further rearrangement of 2.46 to the desired spirooxindole was not observed, as pseudoindoxyl 2.53 was the isolated product. In the end, a different synthetic route was pursued, which did not require the peptide-controlled indole oxidation to access cyclopiamine B.
Figure 16.
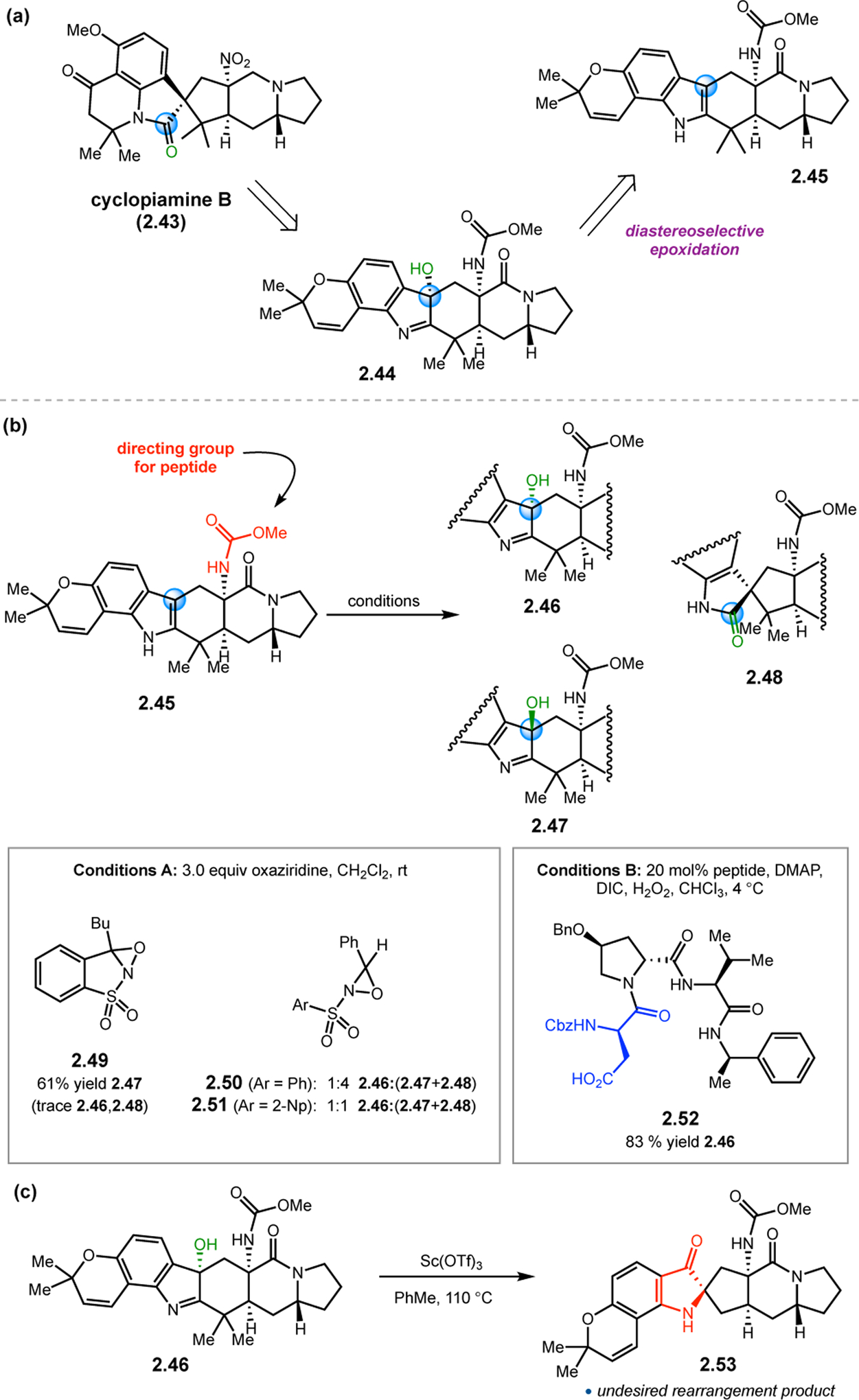
(a) Initial retrosynthesis of 2.43 proceeded through a key, diastereoselective indole oxidation of 2.45. (b) Catalyst 2.52 overturned the inherent substrate-derived selectivity and provided 2.46, syn to the acetamide motif. (c) Rearrangement of 2.45 provided pseudoindoxyl 2.53, instead of the desired spirooxindole 2.43.67
2.1.3. Trifluoromethyl Ketone-Derived Dioxiranes for Epoxidations.
In addition to peracids, dioxiranes have also been successfully employed as O-atom transfer catalysts in selective epoxidation reactions. In particular, Shi and co-workers developed fructose-based ketone 2.54, which can be converted into a dioxirane upon treatment with mild oxidants in situ (Figure 17a).68 This dioxirane is a highly effective epoxidizing agent known for delivering high levels of enantioselectivity over a broad range of substrates. As such, our group sought to investigate whether transient dioxiranes could be generated within a peptidic framework and used to effect enantioselective olefin epoxidation reactions.69 Trifluoromethyl ketone-containing 2.55 was chosen as the catalytically active motif, as these electron deficient ketones have been shown to form highly reactive dioxirane species under mild conditions.70 Chiral ketone 2.55 could be prepared in a short synthetic sequence starting with a chiral oxazolidinone and subsequently attached to peptide sequences using established amide coupling methods. A focused library of catalysts was evaluated in the epoxidation of 2.56 to 2.57 using hydrogen peroxide as the stoichiometric oxidant (Figure 17b). Peptide 2.58a emerged as the lead catalyst, affording up to 91:9 er over a range of unfunctionalized olefin substrates with no additional H-bonding functionality.
Figure 17.
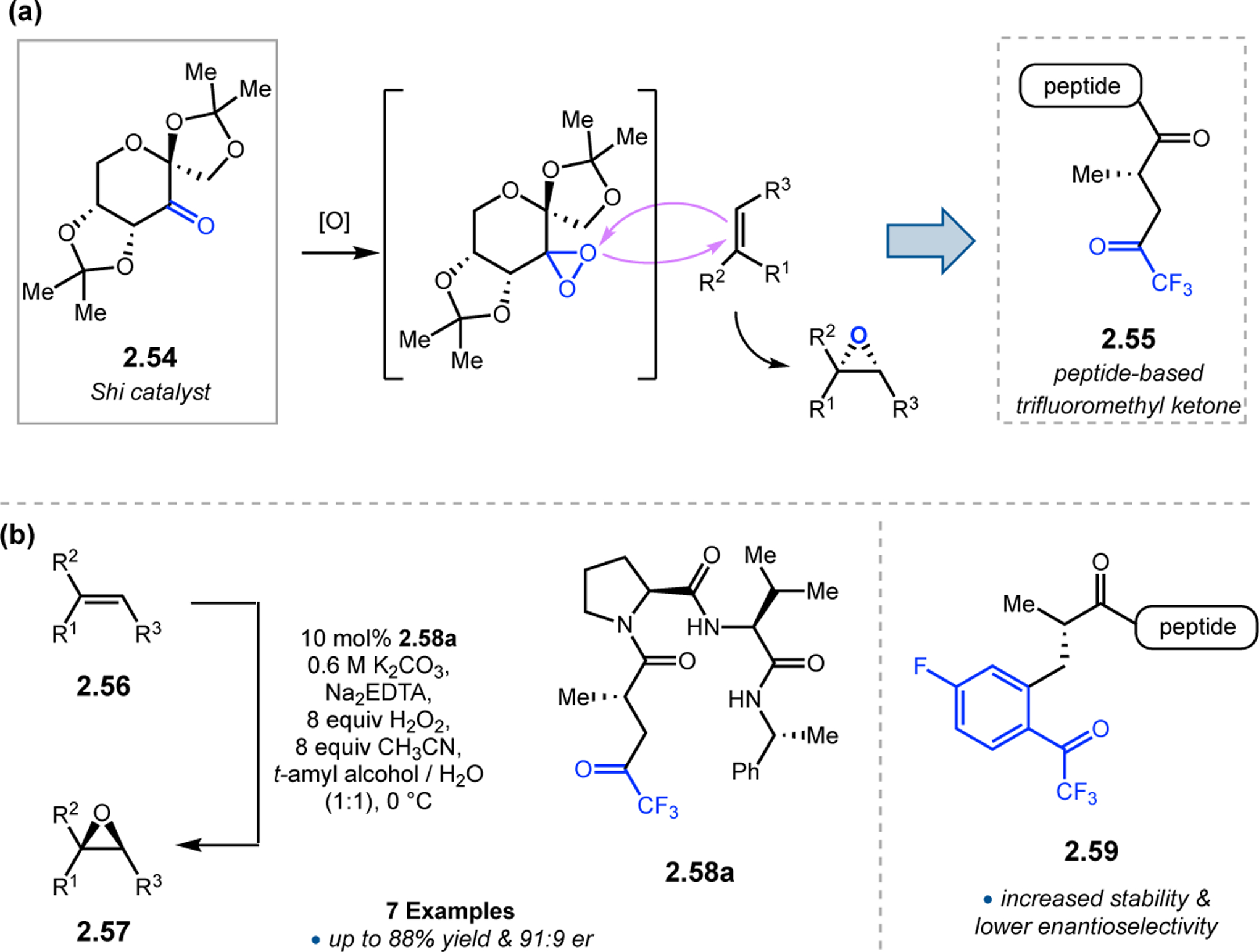
(a) Dioxirane-based epoxidation catalysts. (b) Epoxidation of 2.56 with trifluoromethyl ketone-containing peptide catalysts.69,71
Further optimization of the catalytic residue was pursued, specifically targeting aryl trifluoromethyl ketones like 2.59.71 This class of compounds could allow for additional tuning of the ketone electronic via modification of the arene para-position, such as with fluorine.72 Synthesis of the aryl ketone monomer could be accomplished in a short sequence, originating from serine-like derivatives. Incorporation of a methyl group at the α-position of 2.59 was required, given that traditional protected amino acid residue could cyclize onto the ketone and deactivate the catalyst. Elongated peptide catalyst containing 2.59 provided a range of epoxidized products with excellent levels of conversion, albeit with low enantioselectivities. More decisive secondary interactions with the substrates will be needed in future generations of catalysts.
2.2. Baeyer–Villiger Oxidation
While epoxidation has received significant attention in the field of asymmetric catalysis, asymmetric Baeyer–Villiger (BV) oxidations have traditionally been more difficult to accomplish.73 Enantioselectivity in epoxidation reactions is achieved via discrimination of the enantiotopic alkene faces, which is a fairly common selectivity paradigm in asymmetric catalysis. However, the situation is markedly different in BV oxidations of symmetrical ketones. Following initial addition of the peroxide/peracid nucleophile to form the Criegee intermediate, enantioselectivity is determined solely by the dihedral angle of the C–C–O–O bond, which dictates which C–C bond will migrate (Figure 18a).74 This type of catalyst-control over a single torsion is quite challenging to achieve. However, numerous catalysts have recently emerged that overcome this inherent obstacle.
Figure 18.
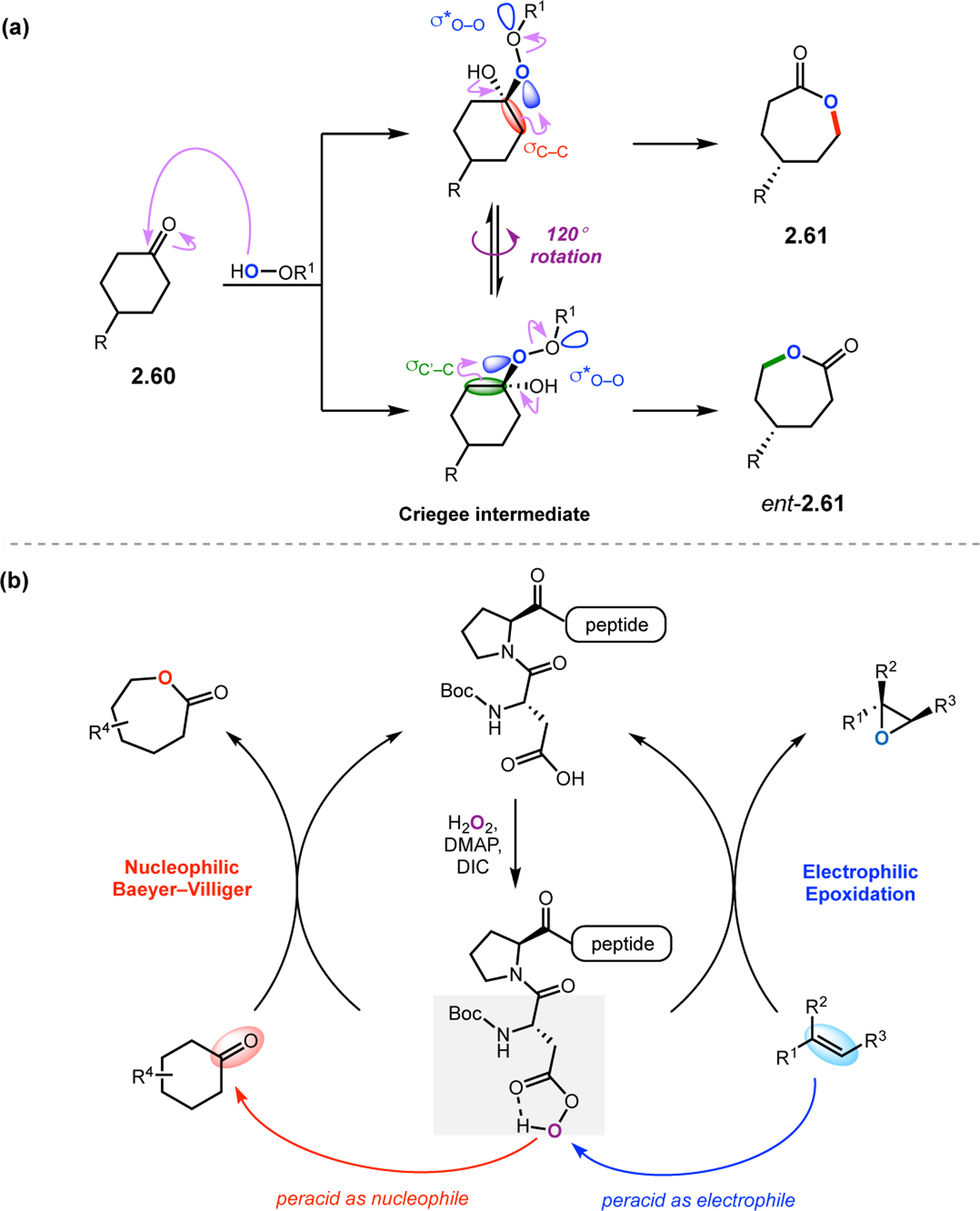
(a) The mechanism of Baeyer–Villiger (BV) oxidation: nucleophilic addition of the peracid species followed by 1,2-migration of an adjacent C–C bond. (b) A conserved, peptide-based aspartyl peracid catalyst could function as both a nucleophile in BV oxidations and as an electrophile in epoxidations.75
Building on previous peptide-mediated epoxidations with Asp-containing peptides, our group sought to expand the utility of these catalysts into BV oxidations (Figure 18b). Owing to the similarities between the catalytically active Asp-derived peracid and m-CPBA, which is known to exhibit diverse patterns of reactivity, it seemed plausible that Asp-containing peptides might also function through alternative mechanistic paradigms.75 In this sense, BV oxidations would also require the peracid as a key moiety in the catalytic cycle, but it would need to function as a nucleophilic oxidant, in contrast to its role in alkene epoxidations, providing an excellent testing ground for the versatility of these peptides.
Our group set out to apply these aspartyl peracids to BV oxidations (Figure 19),75 shortly after the initial epoxidation was reported.43 Unfortunately, the previously optimized conditions for epoxidation provided only low conversions to lactone 2.61a (13%, Figure 19a). It was hypothesized that a slow rate of addition of peracid C to the ketone substrate slowed the reaction, allowing the peracid species to dimerize and form off-cycle diacyl peroxide C′ (Figure 19c). When the DIC was introduced slowly to limit the amount of excess peracid species, yields were increased to 76%. At this stage, it was hypothesized that increasing the acidity of the catalyst could have an advantageous effect on reaction rates. Indeed, pentafluorobenzoic acid catalyst 2.63 (pKa = 1.74) outcompeted 2.62 (pKa ≈ 4.4) in the BV oxidation of 2.60b to lactone 2.61b using batch-wise delivery of DIC (Figure 19b). However, despite the low reactivity of Asp-derived catalyst 2.62, it remained plausible that incorporation of this monomer into an extended peptide sequence would enhance reaction rates via multifunctional substrate activation. Indeed, evaluation of Boc-Asp-Pro-d-Val-Val-(R)-α-Mba (Mba = methylbenzylamine) with substrate 2.60b (R1 = Ph, R2 = OH) under the slow addition protocol provided product with 70:30 er, a promising start for future studies.
Figure 19.
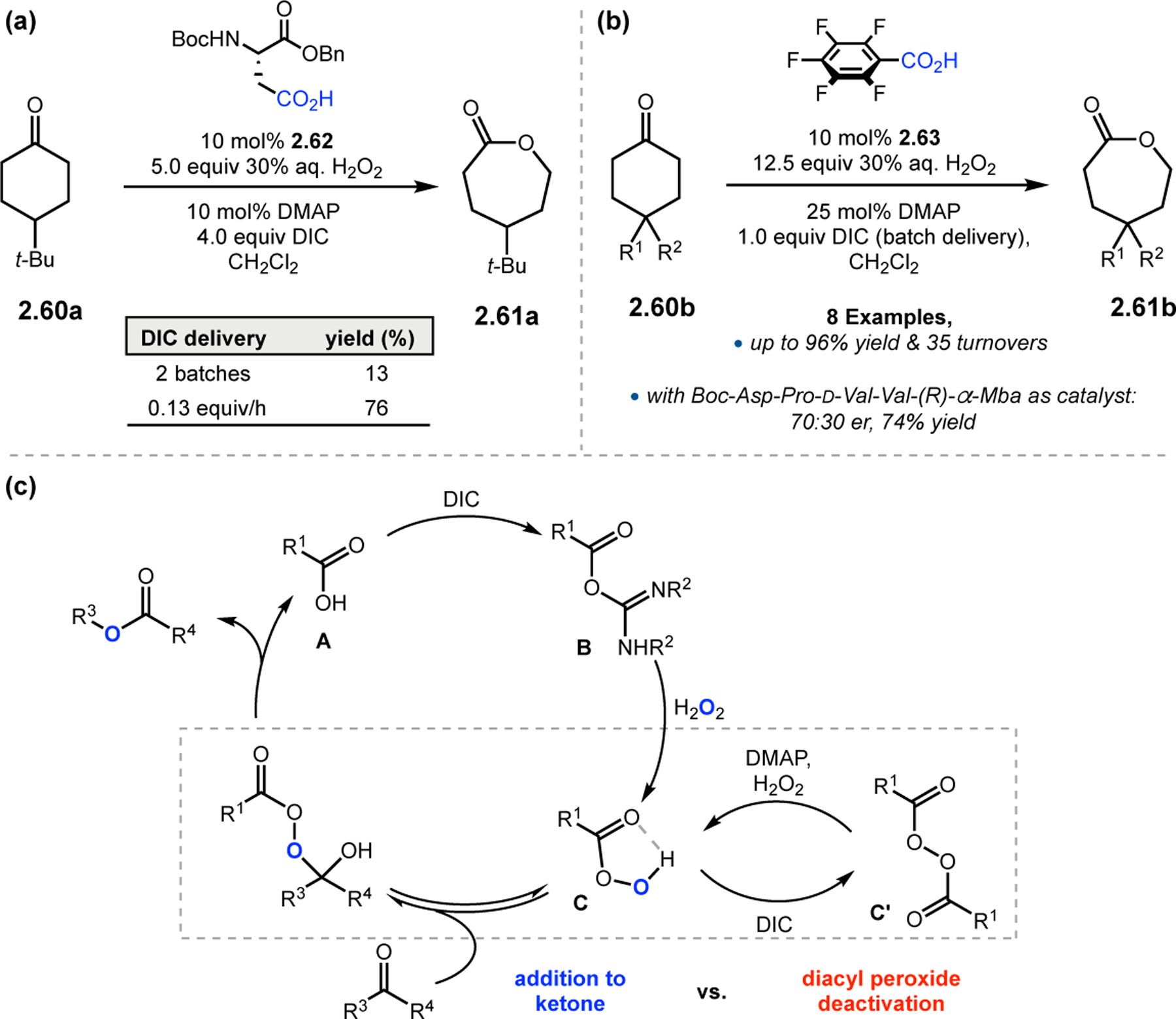
(a) BV oxidations with monomeric Asp catalyst. (b) BV oxidations with perfluorinated benzoic acid. (c) Mechanism of BV oxidation, with key off-cycle intermediate inhibiting higher conversions.75
In choosing a new substrate class to maximize peptide/substrate interactions, compounds like 2.64, which contain an amide directing group at the 3-position, were evaluated in peptide-catalyzed BV reactions (Figure 20).60 In these kinetic resolutions, the relative rates of reaction for each ketone enantiomer determine the ultimate enantioselectivity of the product, and migration of either adjacent C–C bond now creates an opportunity to control regioselectivity. Typically, the most electron-rich bond migrates. However, it is plausible that catalyst control could reverse the inherent migratory aptitude through precise control of the C–C–O–O dihedral angle.
Figure 20.
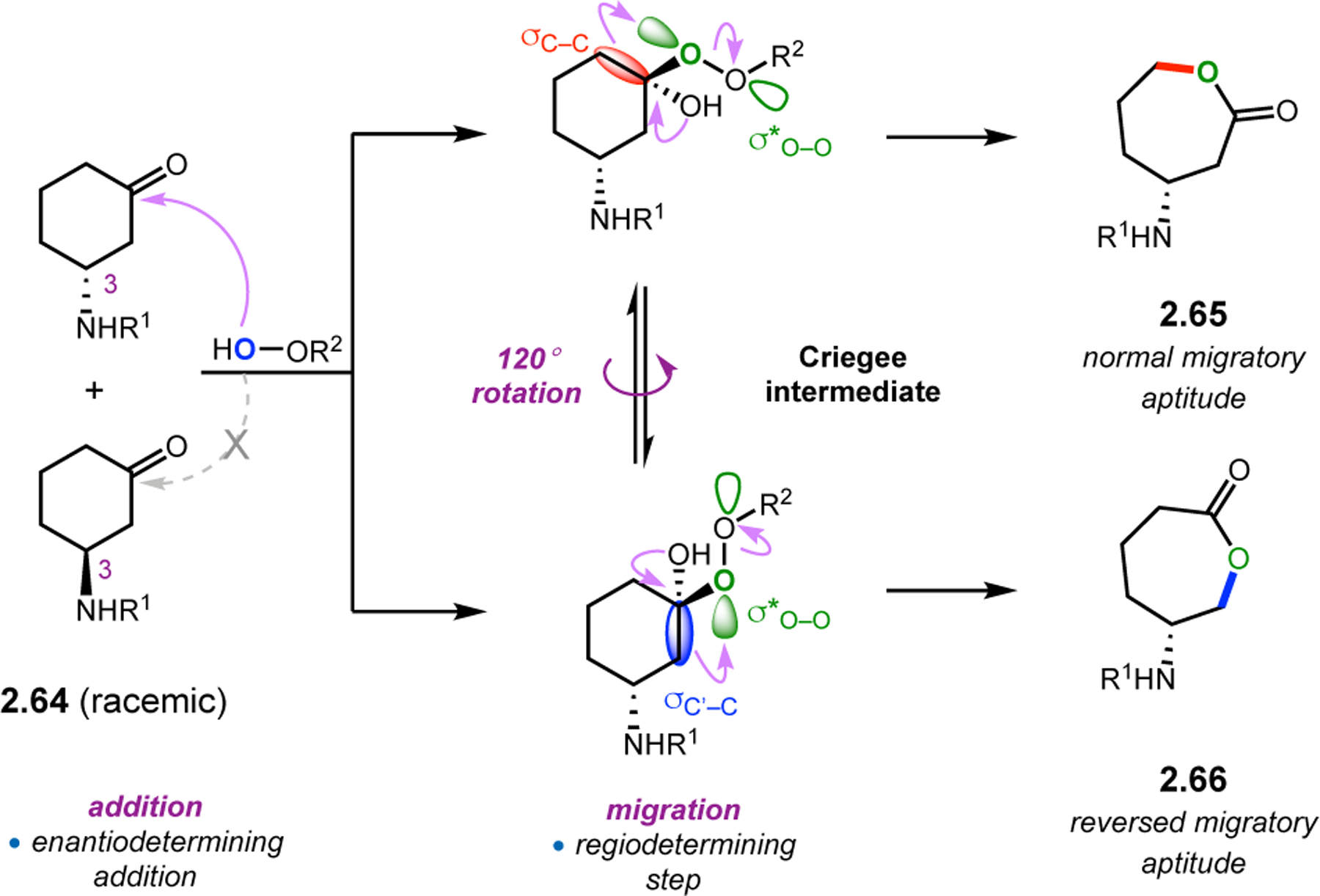
Mechanistic analysis of kinetic resolution of 2.64 via BV oxidation. In this case, rotation of the peracid dihedral results in the formation of regioisomeric products.60
Initially, incorporation of Asp into various peptide sequences that were selective in epoxidations and other reactions were not effective in BV oxidations of 2.64. As such, a library of on-bead catalysts was synthesized using the OBOC method (see Section 2.1.2, Figure 12). The library was biased to have a Pro residue at the i+1 position, as we have observed that this design element is essential for enantioinduction in many cases with substantial variation to the remainder of the sequence (Figure 21a). Ketone substrate 2.64a was initially used in catalyst screening, as its 2-naphthyl chromophore facilitated reaction analysis. Evaluation of the catalyst library led to the identification of peptide 2.68b, the resin-cleaved variant of 2.68a, which mediated the BV kinetic resolution of 2.64a to lactone 2.65a with a krel of 1.7, a promising starting point for subsequent optimization.
Figure 21.
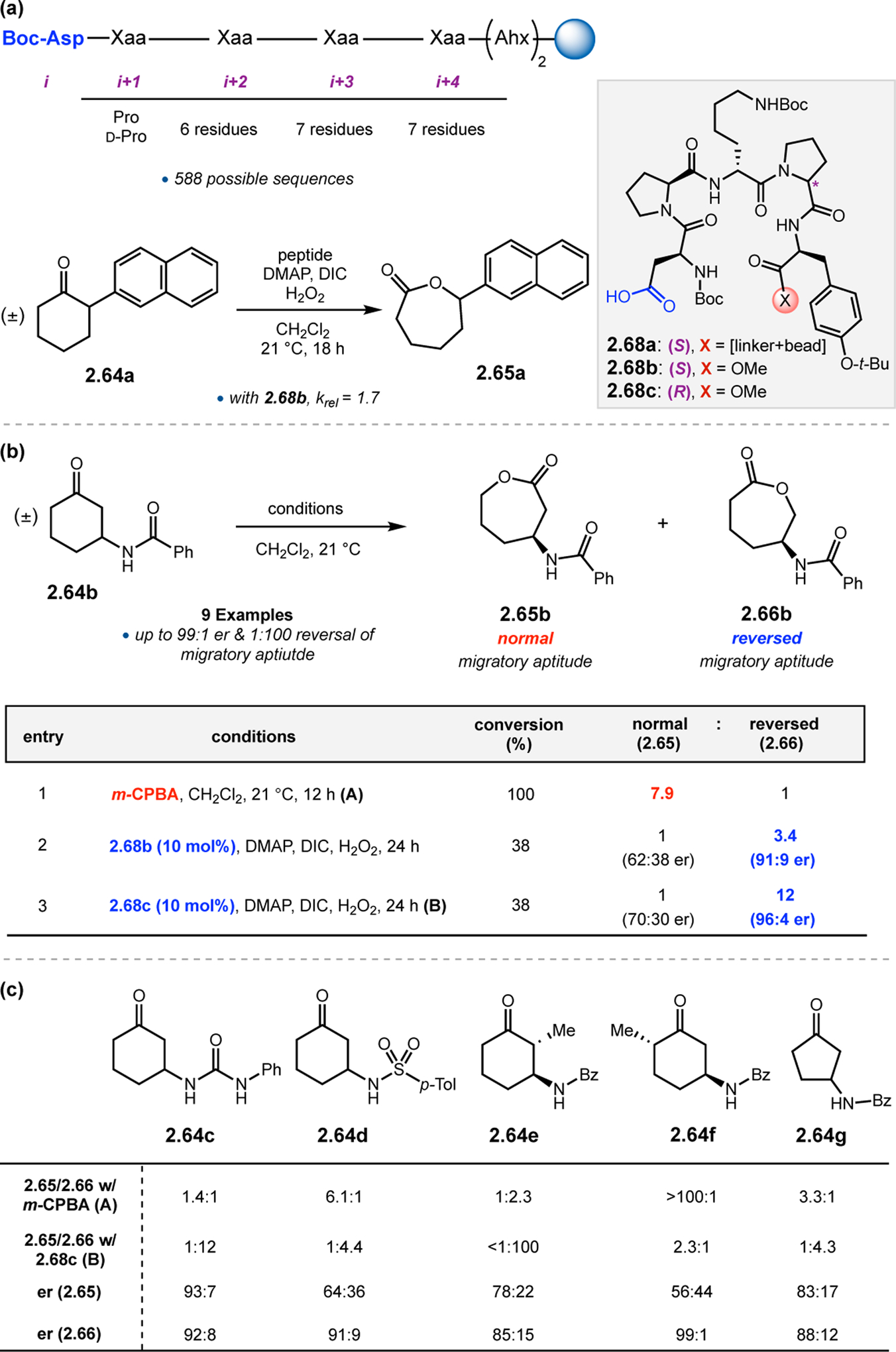
(a) On-bead screening of Asp-containing peptide catalysts for the BV oxidation of 2.66. (b) Solution-phase screening of hit Asp peptides for the BV oxidation of 2.64a. (c) Selected substrate scope entries highlights the general tendency of 2.68c to reverse the inherent migratory aptitude.60
This catalyst was next tested on parent substrate 2.64b (Figure 21b). The inherent preference for migration of this substrate in oxidations with m-CPBA is the normal migratory mode by a 7.9:1 ratio (entry 1). Alternatively, catalyst 2.68b provided reversed product 2.66b, in a 1:3.4 ratio and 91:9 er (entry 2). This catalyst-controlled overturning of the preferred migratory aptitude was further enhanced with minimal peptide optimization. Epimeric catalyst 2.68c delivered 2.66b in 1:12 ratio and 96:4 er (entry 3). Alteration of the 3-amido group of 2.64 revealed a continued preference for the reversed migration product using catalyst 2.68c (Figure 21c); 2.66e was isolated in <1:100 ratio. Using m-CPBA, 2.64f was converted to normal product 2.65f in >100:1. Catalyst 2.68c was able to reduce this large migratory preference to 2.3:1 with an er of 99:1.
On-bead screening has been extremely valuable for the identification and optimization of highly selective catalysts for a variety of oxidation reactions, including epoxidation and BV oxidation. Many of these catalysts, including 2.68c, possessed never-before-studied sequences. As such, it was unclear at the outset what secondary structural attributes, if any, peptide 2.68c would exhibit under the reaction conditions. The solution structure of 2.68c was examined using NMR spectroscopy.76 Through-space NOE contacts were used to generate distance restraints for structural calculations in CNS to generate an ensemble of structures,65 the lowest energy of which was subjected to DFT optimization (Figure 22). Two critical H-bonds were found in the ground state structure of 2.68c: (1) a seven-membered ring N–H(Tyr)•••O=C(d-Pro) H-bond indicative of a γ-turn, and (2) an H-bond between the sidechain N–H of Lys and its backbone carbonyl. Furthermore, it is apparent that the active Asp sidechain is nearby the Lys residue in space. Moreover, NMR structural analyses of 1:1 mixtures of peptide 2.68c with (1) a mismatched substrate enantiomer (S,R)-2.64e and (2) the favored product (R,S)-2.66e revealed subtle, yet noticeable, differences between these complexes and the structure of 2.68c alone. More telling is that the structures of the two complexes themselves were extremely similar. By comparison, the 1:1 mixture of 2.68c and matched substrate (R,S)-2.64e elicited significant changes in the catalyst structure. Most notably, the Lys sidechain was displaced from its ground-state H-bond with the backbone carbonyl. It is possible that this reorganization facilitates substrate docking or perhaps the substrate induces this change to a more catalytically active conformation. Overall, these data illuminate the precise nature of the noncovalent interactions between matched substrates and peptide 2.68c.
Figure 22.
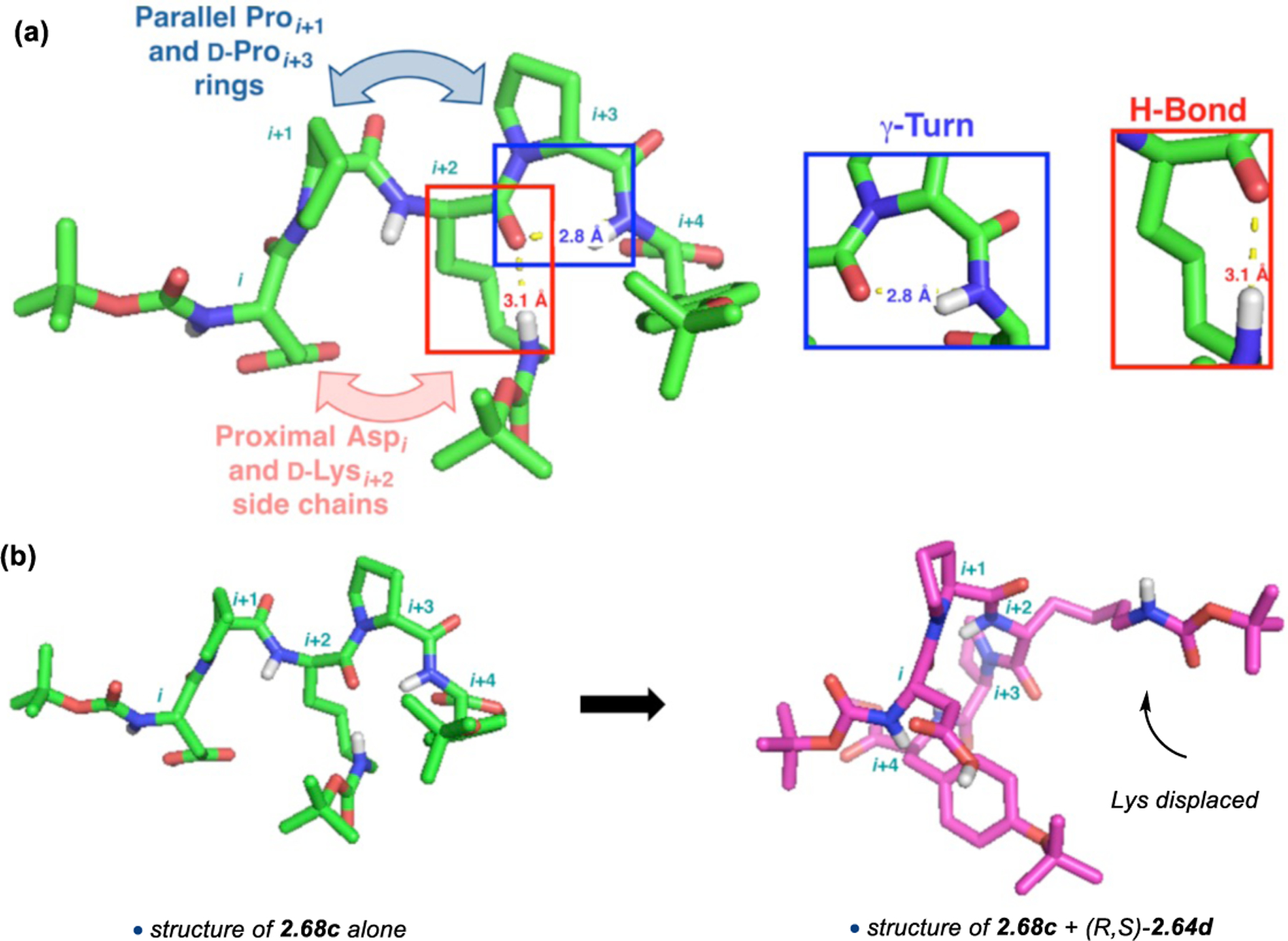
(a) Ground state structure of 2.68c. (b) Structure of 2.68c with an equimolar amount of (R,S)-2.64e Adapted with permission from ref. 76. Copyright 2016 American Chemical Society.
While BV oxidation catalyst 2.68c and epoxidation catalyst 2.34b share the same catalytic Asp residue, the reactive peracid intermediate functions as a nucleophile in the former case and an electrophile in the latter (Figure 23a). The tuning of a conserved reactive species for diverse transformations is a hallmark of enzymatic catalysis,77 which is normally accomplished through evolutionary alteration to the proteins’ three-dimensional structure. Our group wondered whether the distinct sequences of peptides 2.68c and 2.34b could control aspartyl peracid reactivity toward complex substrates in which epoxidation and BV oxidation are viable reaction pathways.78 Substrate trans-2.69, which contains a 3-amidocyclohexanone motif for BV oxidation and an allylic alcohol for epoxidation, was chosen to explore these questions (Figure 23b). Potential oxidation products include the lactones from normal (2.70) and reversed (2.71) migration, two mono-epoxide diastereomers (2.72 & 2.73), and numerous over-oxidation products. Reactions in the presence of m-CPBA produced an unselective mixture of products with a slight preference for epoxides 2.72 and 2.73 (1:1 ratio) over lactones 2.70 and 2.71 (entry 1). The preferred BV migratory aptitude favored the normal lactone over the reversed in a 2.9:1 ratio. On the other hand, epoxidation catalyst 2.34b provided substantial rate enhancement for its matched epoxide product 2.72 in >100:1:1 chemoselectivity and >99:1 diastereoselectivity (entry 2). The BV oxidation catalyst 2.68c was initially found to give low selectivity in the reaction of trans-2.69, suggesting that the catalyst was mismatched with the enantiomer of 2.69 that was synthesized. However, peptide ent-2.68c was found to provide good conversion to a mixture of products favoring lactones 2.70 and 2.71 in a 12.5:1 ratio over epoxides 2.72 and 2.73 (entry 3). The catalyst also overturned the inherent migratory preference of the substrate in favor of reversed lactone 2.71 in >50:1 ratio. Interestingly, a larger amount of water was found to be productive for BV selectivity, potentially by disrupting the H-bonding interactions between the catalyst and the allylic alcohol that might direct epoxidation (Figure 23c), as with peptide 2.34b (Figure 23d). Overall, this study further demonstrated the concept that minimal peptides can tune the reactivity of a shared catalytic residue through precise outer-sphere interactions with substrates, with control over not only stereochemical issues, but also through divergent functional group selection.
Figure 23.
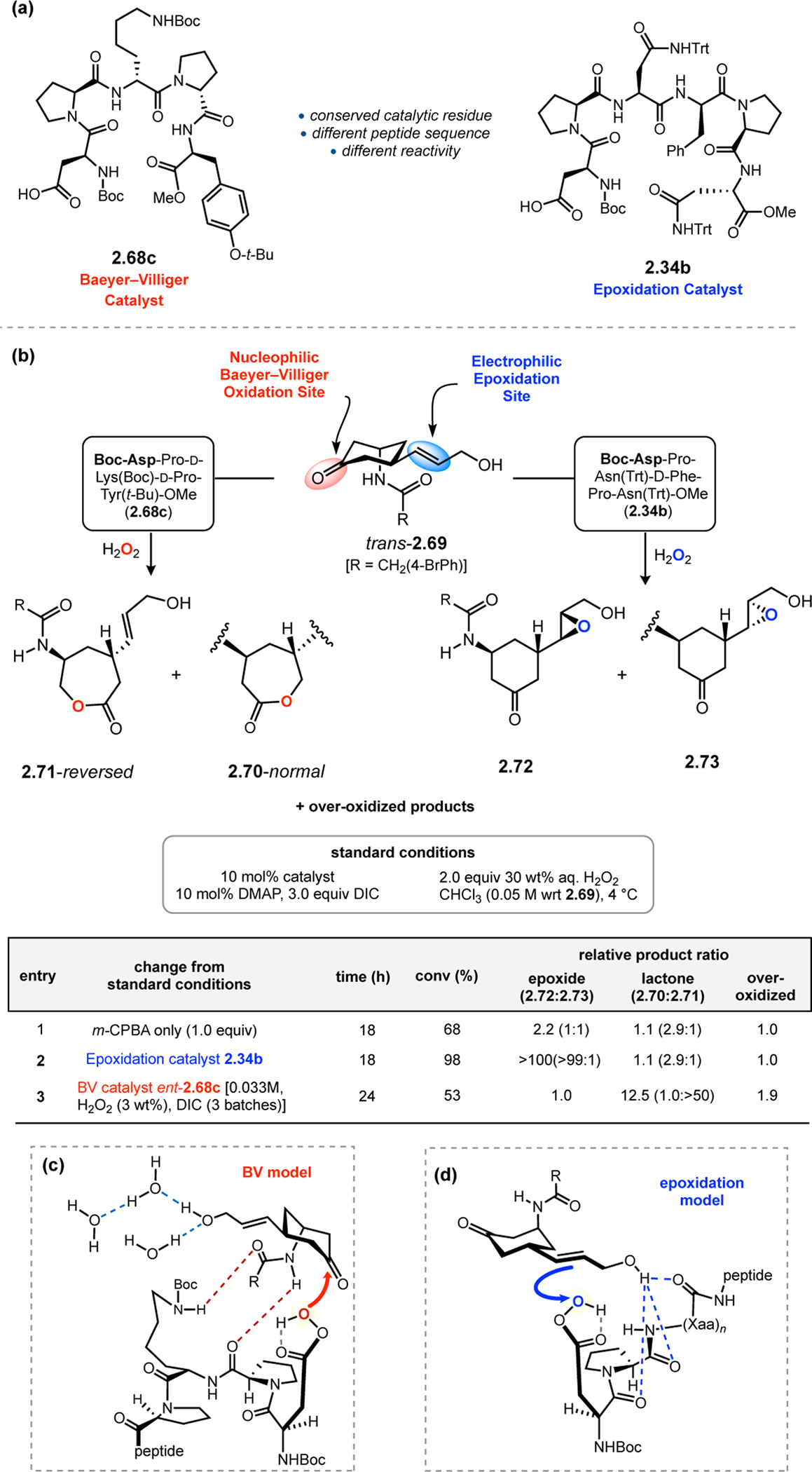
(a) Asp-containing peptide catalysts for BV oxidation and epoxidation. (b) Dual substrate trans-2.69, containing both a 3-amidocyclohexanone motif for BV oxidation and an allylic alcohol for epoxidation. While achiral catalysts provide limited selectivity, catalysts 2.68c and 2.34b facilitated their matched reactivities. (c) Model for selective BV oxidation with ent-2.68c. (d) Model for selective epoxidation with 2.34b.78
The utility of Asp-containing peptides was further explored in BV oxidations of ketone substrates that were not addressed by catalyst 2.68c. Unfunctionalized ketone substrate 2.74 was of particular interest, as the absence of any obvious H-bonding functionality would require the development of complementary catalyst scaffolds. In collaboration with the Anslyn Group, we pursued on-bead peptide screening to address this challenge, and two moderately selective catalysts were discovered (2.76a & 2.77a, Figure 24a).79 These two catalysts provided opposite absolute configurations of product 2.75, even though each contained only l-amino acids. In order to model putative catalyst–substrate interactions, exact stereochemical assignments of these products were required. One avenue for obtaining this information is using circular dichroism (CD). To facilitate the determination of absolute configuration using CD, it would be desirable to incorporate the products into zinc(II) complexes. Upon methanolysis of 2.75 to alcohol 2.78, TREN-like ligands were added to facilitate dynamic assembly of zinc(II) complexes, such as 2.79 (Figure 24b). These complexes exhibit characteristic Cotton effects, in which the intensity of the CD signal varies linearly with the ee of the appended alcohol. Furthermore, the sign of the signal correlates with either the M or P helical twist of the pyridine ligands around the zinc center, which is governed by the absolute configuration of the chiral alcohol. We expected that this method would not only be effective at assessing the absolute stereochemical configuration of products, but also as an alternative, rapid assay for the evaluation of catalyst selectivity. Catalysts discovered in the on-bead assay were re-synthesized in solution and screened in the BV oxidation of 2.74. The resulting lactone products 2.75 were then assayed for er using traditional HPLC methods. Lactones 2.75 were subsequently ring-opened, converted to Zn-complexes 2.79, and assayed for er using CD. The measurements recorded using both methods were quite close over a range of high and low performing catalysts, and measurements by CD reduced the time cost from ~30 min by HPLC to a few seconds per sample. Furthermore, the absolute configuration of products of both 2.76b and 2.77b were determined by CD to be (2R,6S) and (2S,6R), respectively. It appears that the identity of the i+2 position is essential for this inversion of enantioselectivity, as catalysts with Leu produced products with the (2R,6S) configuration, whereas catalysts incorporating Ser(t-Bu) at this position favor (2S,6R)-2.75. Further development of this CD assay is currently underway, including studies on the nature of the pyridine ligands utilized to form the zinc complexes.80
Figure 24.
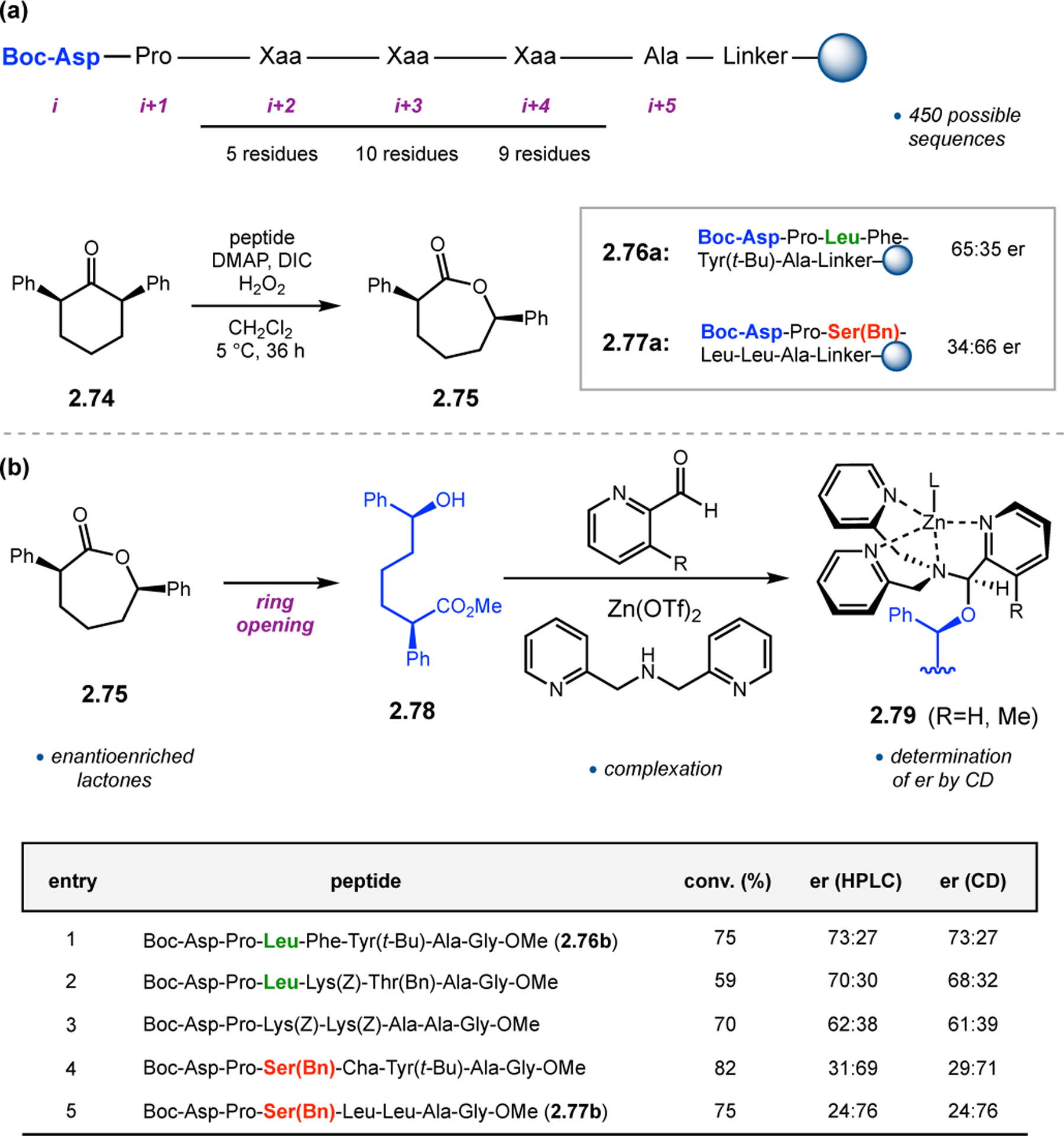
(a) On-bead peptide library screening for the BV oxidation of 2.74 revealing two peptides that give opposite senses of asymmetric induction. (b) Coordination of chiral, ring opened products to zinc(II) pyridyl complexes for analysis of absolute stereochemistry and enantioselectivity by CD.80
While most of the advances in peptide-mediated BV oxidation have relied on aspartyl peracids, new classes of Brønsted acid catalysts have recently emerged to provide an alternative approach. BINOL-derived chiral phosphoric acids (CPAs), which are well known to mediate a wide variety of transformations,81,82 have recently been extended toward BV oxidation.83 Unlike Asp-containing peptides, which react via a transiently generated peracid to enable asymmetric BV oxidations, CPAs function through a Brønsted acid catalytic mechanism to activate ketones for outer-sphere attack by hydrogen peroxide and subsequently facilitate selective bond migration of the Criegee intermediate (Figure 25).84 Inspired by this complementary mechanistic paradigm, our group set out to develop a new class of peptide-based BV oxidation catalysts based using phosphothreonine (pThr) as a Brønsted acidic residue.85 These pThr-containing peptides (i.e., phosphopeptides) had been previously demonstrated in enantioselective reduction chemistry (see Section 3),86,87, and we hoped to expand this approach into the realm of oxidation.
Figure 25.
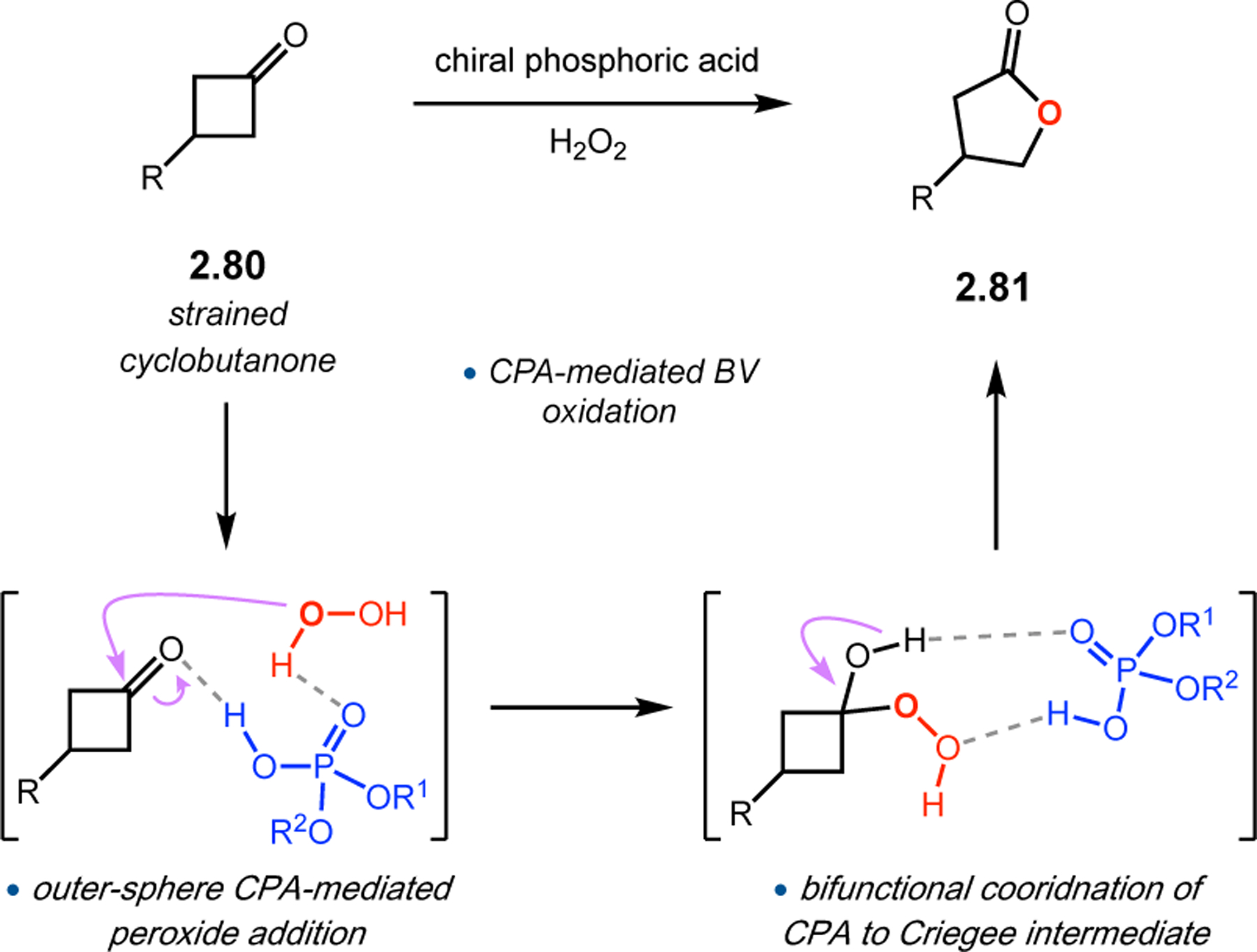
Putative mechanism of BV oxidation with CPAs, which activate the ketone and Criegee intermediate via outer-sphere interactions.
The phosphopeptide strategy was tested in the BV oxidation of cyclobutanones 2.80, which contain a pendent directing group to facilitate catalyst–substrate interactions, using aqueous hydrogen peroxide as the oxidant (Figure 26a).85 Evaluation of various pThr-containing catalysts led to the discovery that an additional directing group installed at the i+3 position of the peptide, namely an acetyl-protected l-diaminopropionic acid (Dap) residue (2.82), was highly advantageous for enantioselectivity. This effect was especially pronounced for substrates containing H-bond donor functionality (e.g., 2.80a–e), as Dap(Ac)-containing 2.82 provided lactones 2.81 with higher yields and ers than Phe-containing 2.83. On the other hand, substrates lacking a pendent H-bond donor group (e.g., 2.80f–h) were processed more effectively by catalyst 2.83. NMR structural studies of peptides 2.82 and 2.83 revealed that both catalysts adopt a β-hairpin structure in solution. Since the enhanced selectivities exhibited by the Dap(Ac)-containing catalyst do not stem from the formation of a unique secondary structure, it is plausible that the primary function of this residue is to nucleate additional noncovalent interactions with the substrate. These observations led to a proposed model for enantioinduction, wherein the phosphoric acid moiety of pThr engages with the Criegee intermediate while the Dap(Ac) side chain H-bonds with the substrate directing group (Figure 26b).
Figure 26.
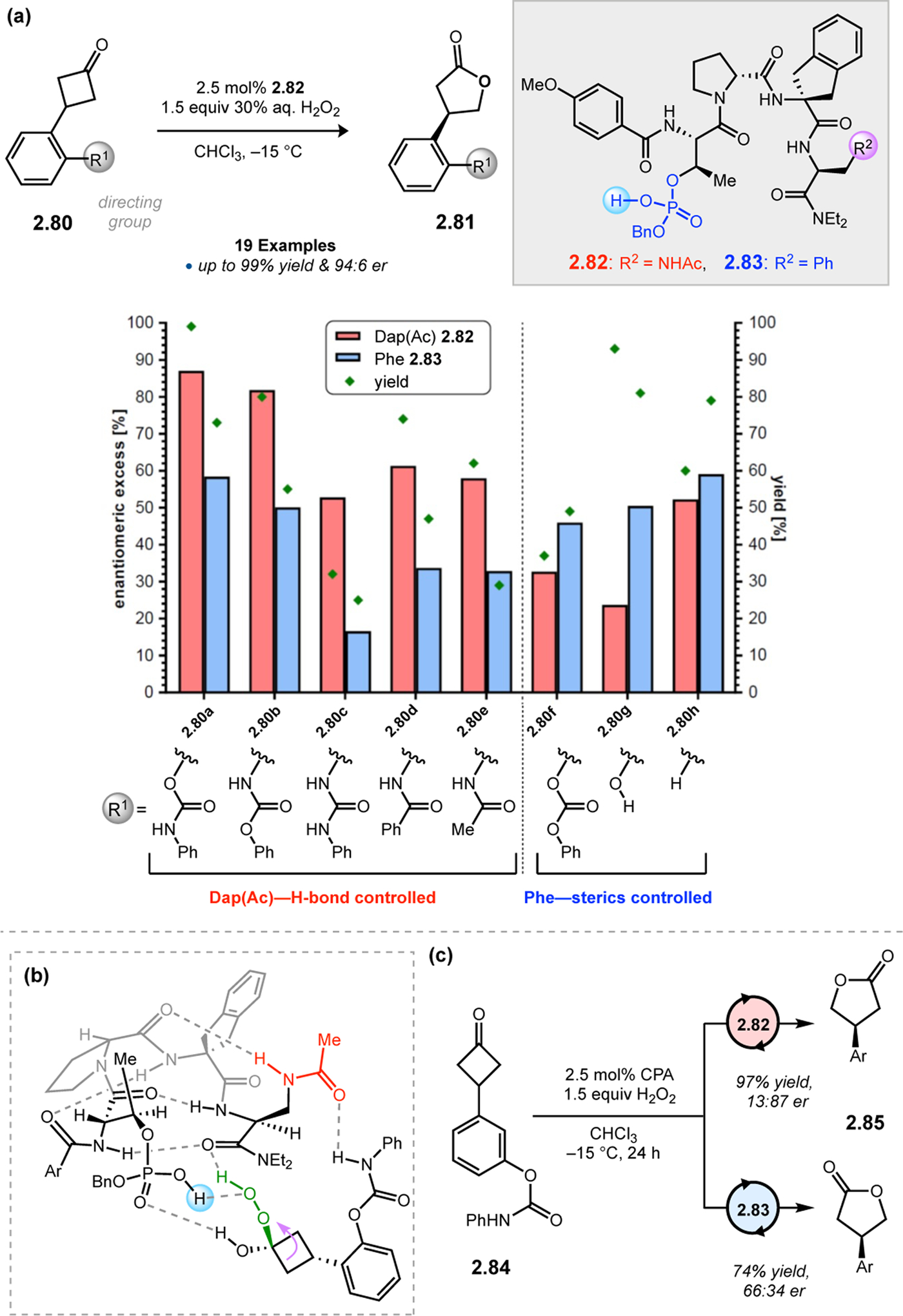
(a) Phosphopeptides as catalysts for the BV oxidation of cyclobutanones 2.80 with aqueous H2O2. A key Dap(Ac) residue at the i+3 position facilitates noncovalent interactions with the substrate. (c) Reversal in the sense of asymmetric induction upon switching from an ortho- (2.80) to a meta-directing group (2.84).85
The high degree of specificity in the interactions between peptide 2.82 and its substrates was further probed using cyclobutanone 2.84, in which the carbamate directing group is installed at the meta-position (Figure 26c). While Phe-containing peptide 2.83 processes 2.84 with moderate er on par with what was measured for 2.80a (79:21 vs. 66:34), Dap(Ac)-containing 2.82 provides lactone 2.85 with the opposite absolute stereochemistry relative to 2.80a (94:6 vs. 13:87 er). These data show that it is possible to observe enantiodivergence without altering a single stereocenter in the peptide catalysts, as Dap(Ac) and Phe are both l-amino acids. It is also striking that peptide 2.82 can process such similar substrates with highly enantiodivergent outcomes solely based on the position of a remote directing group. These phenomena speak to the central challenge of asymmetric BV oxidations, as catalysts must be able to control subtle torsional angle differences within the Criegee intermediate to produce enantiomeric products (Figure 20). Minimal peptide catalysts appear capable of addressing these challenges through the construction of precise noncovalent interactions with substrates.
In an analogous approach, Imada and co-workers employed isoalloxazines inspired by flavin-dependent monooxygenase enzymes to enable biomimetic oxidation reactions (Figure 27a).88 FAD functions as a cofactor in numerous classes of oxidations in vivo, often through the generation of a hydroperoxyflavin with molecular oxygen (C, Figure 27b).89 By embedding isoalloxazine units at the N-termini of short peptide-sequences, the authors were able to harness FAD-like reactivity in a synthetic context. Potential peptide sequences to be appended to the isoalloxazine were evaluated using DFT computations, which led to the incorporation of Pro at the i+1 position in order to favor a γ-turn structure and thereby orient the peptide scaffold nearby the catalytically active moiety. Acidic functionality was also incorporated at the i+3 position to interact with the intermediate peroxy species. Polymer-supported peptide 2.86 was found to be competent in both sulfide and BV oxidations. In oxidations of sulfide 2.87 to sulfoxide 2.88, catalyst 2.86 was utilized in tandem with oxygen and hydrazine, the latter of which functions as the terminal reductant for catalyst regeneration (Figures 2.27b–c). Inclusion of hydrazine is thought to activate the peroxy species toward nucleophilic attack by the sulfide through H-bonding (Figure 27b). This hypothesis was corroborated through the generation of a Hammett plot, which suggested that electron-rich sulfides were more reactive (Figure 27c). Peptide 2.86 was also able to mediate the BV oxidation of 2.89 using Zn as the terminal reductant (Figure 27d). In these competition experiments, oxidants such as m-CPBA inherently favored sulfide oxidation while 2.86 showed exceptional chemoselectivity for BV oxidation (Figure 27e). This chemoselectivity is possibly due to H-bonding between the i+3 carboxylic acid and the peroxy species to enhance its nucleophilicity (Figure 27b). Hence, subtle variation of reaction conditions enables access to distinct reaction mechanisms of a conserved hydroperoxyflavin species. Although no enantioselectivity was reported in these particular oxidation reactions, this novel flavin-containing peptide catalyst platform opens the door for future development of asymmetric reactions.
Figure 27.
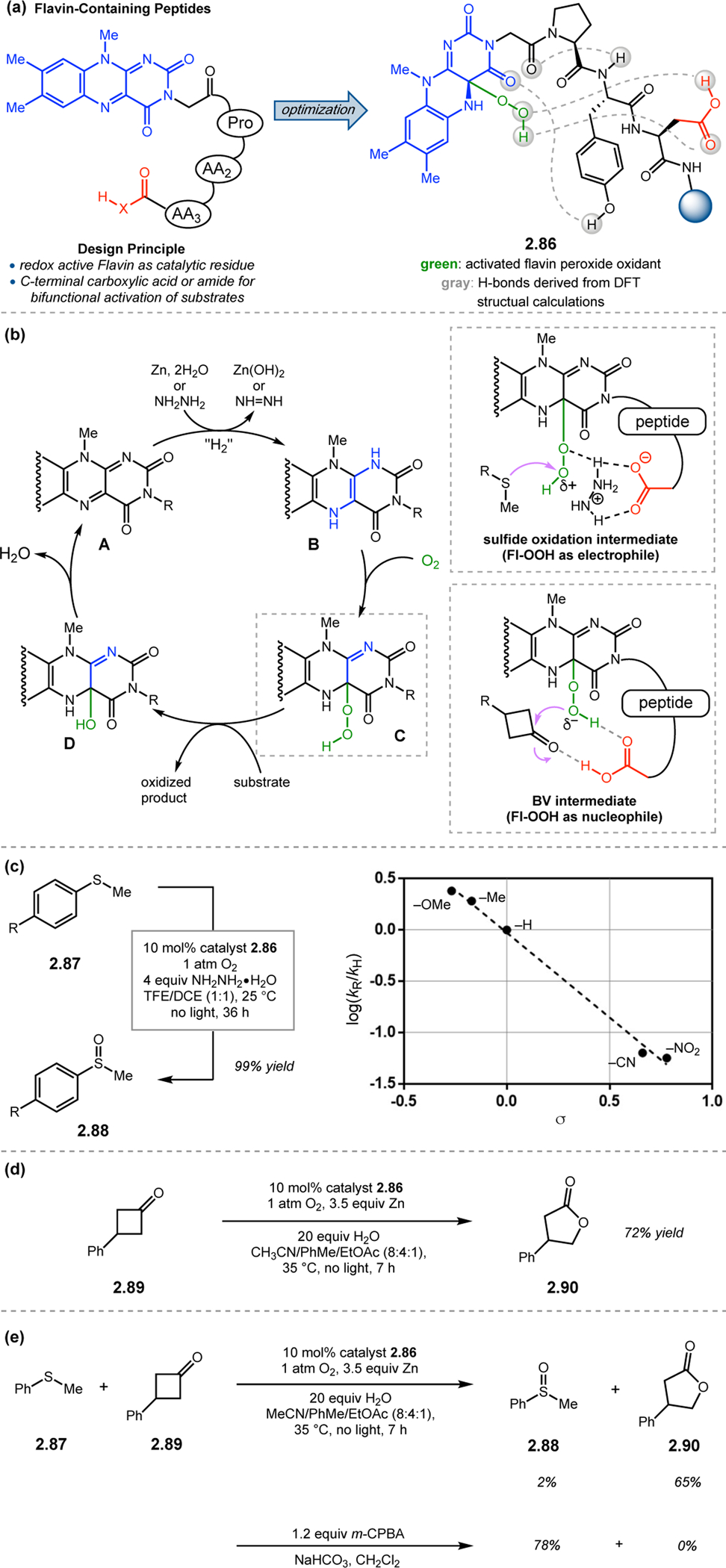
(a) Flavin-embedded peptides as oxidation catalysts, with a specifically designed secondary point of contact at the i+3 position for further intra- and intermolecular interactions. (b) Proposed mechanistic cycle for flavin-based oxidations. (c) Flavin catalyzed sulfide oxidation. (d) Flavin catalyzed BV oxidation. (e) Competition experiment revealed that flavin-based peptides are more matched to facilitate BV oxidation than sulfide oxidation.88
2.3. Additional Oxidations
Beyond epoxidation and BV reactions, peptide-based catalysts have also been developed to mediate a number of other oxidative transformations, including the N-oxidation of pyridines (Section 2.3.1), alcohol oxidations (Section 2.3.2), and α-functionalization reactions (Section 2.3.3).
2.3.1. N-Oxidation of Pyridines.
Asp-Containing peptides have been shown to function as powerful catalysts for various asymmetric transformations. The ease of generating the active aspartyl peracid moiety in situ has enabled peptides containing this catalytic residue to be applied in epoxidations (Section 2.1.2)43 and Baeyer–Villiger oxidations (Section 2.2).75 Notably, the aspartyl peracid can be encoded to function as either a nucleophilic or an electrophilic oxidant by tuning the peptide sequence in which it is embedded.78 This renders the aspartyl peracid moiety highly general and capable of diverse reactivity.
Given the range of reported peracid-mediated oxidations, our group sought to program the Asp-containing peptides for new reactions, specifically in the construction of decorated, chiral pyridines (Figure 28). While these heterocycles are ubiquitous in natural product and drug scaffolds, methods for their enantioselective syntheses remain challenging.90 One common strategy for functionalizing pyridines uses N-oxidation to generate an electrophilic pyridine N-oxide species that is highly activated toward nucleophilic addition reactions. In 1983, the Sharpless Group demonstrated that Ti-based complexes can catalyze the kinetic resolution of tertiary amines via N-oxidation.91 Building upon these precedents, our group envisioned a scenario wherein Asp-containing peptides could oxidize pyridines to the corresponding N-oxides through the aspartyl peracid scheme.92 Complex substrates exhibiting multiple conformations or containing various oxidizable functionality would pose issues of site- and stereoselectivity that could serve as a testing ground for this new paradigm in catalyst-controlled selective oxidations.
Figure 28.
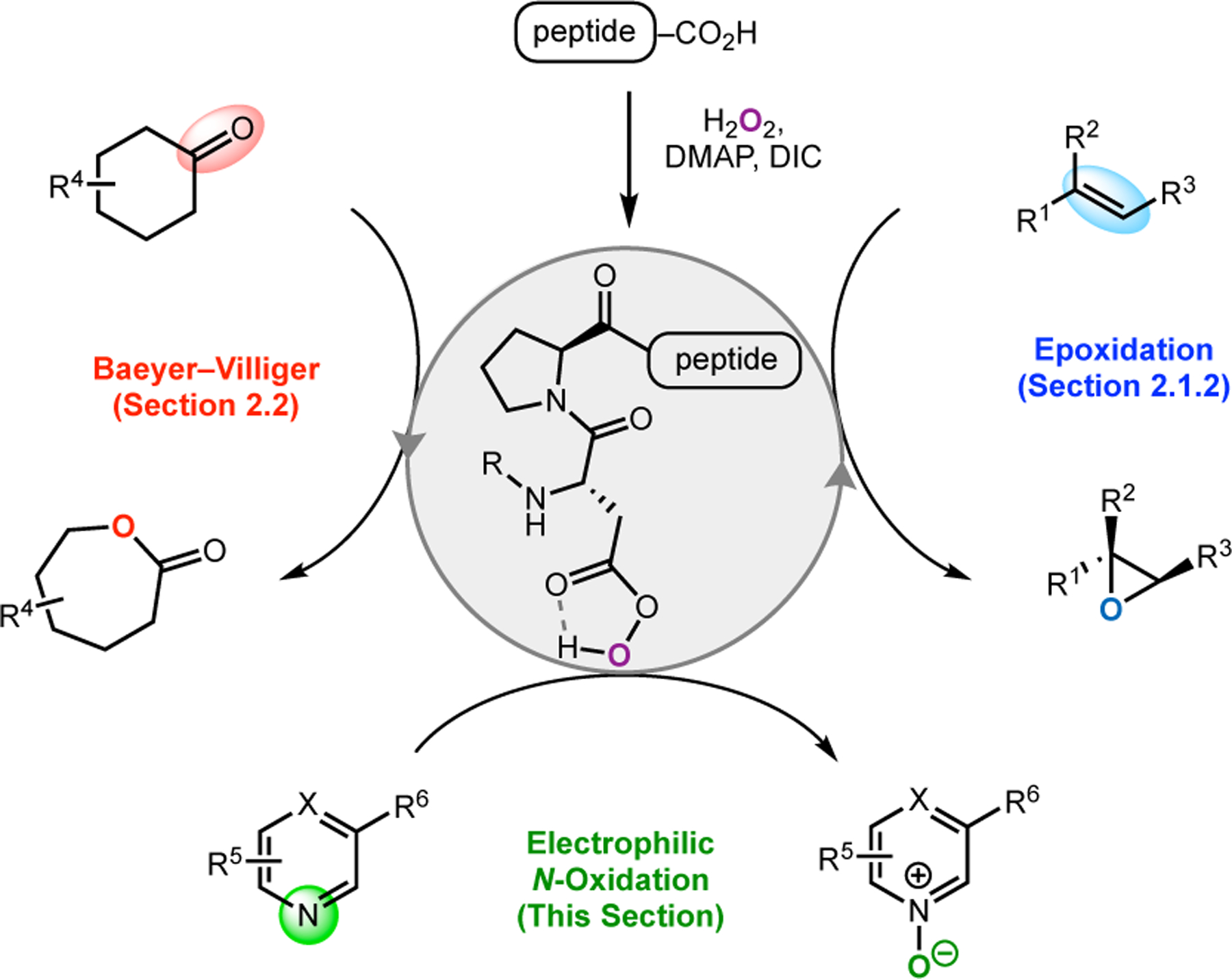
Asymmetric reactions mediated by aspartyl peracid-based peptides: epoxidations, BV oxidations, and most recently, N-oxidations of pyridines.
Bis(pyridine) 2.91a was chosen as a test system for enantioselective N-oxidation, as it contains a prochiral pivalamide directing group to maximize the potential for noncovalent interaction with the catalyst (Figure 29a).92 Effective desymmetrization of 2.91a would require remote asymmetric induction in order to differentiate the enantiotopic pyridines that are separated from each other and the prochiral center by great distances. Various peptides were evaluated in the desymmetrization of 2.91a that derived from either a combinatorial OBOC library or rationally designed β-turn-biased sequences. Both approaches yielded initial hit results, providing pyridine mono-N-oxide 2.92a in as high as 74:26 er, and β-turn-containing peptide 2.94 was chosen for further study. Most mutations of the i+1, i+2, and i+3 positions of 2.94 resulted in lower observed selectivities. Catalysts with an additional residue at the i–1 position were substantially more selective, and the best catalyst, phenylglycine (Phg)-containing 2.95, provided 2.92a with an er of 86:14. This catalyst was tolerant of variable substitution at the C6 position of 2.91, delivering the corresponding products 2.92 with up to 99:1 er (Figure 29b). Although 2.95 was also able to address substrates with C5-functionality, C2-substitution led to racemic products.
Figure 29.
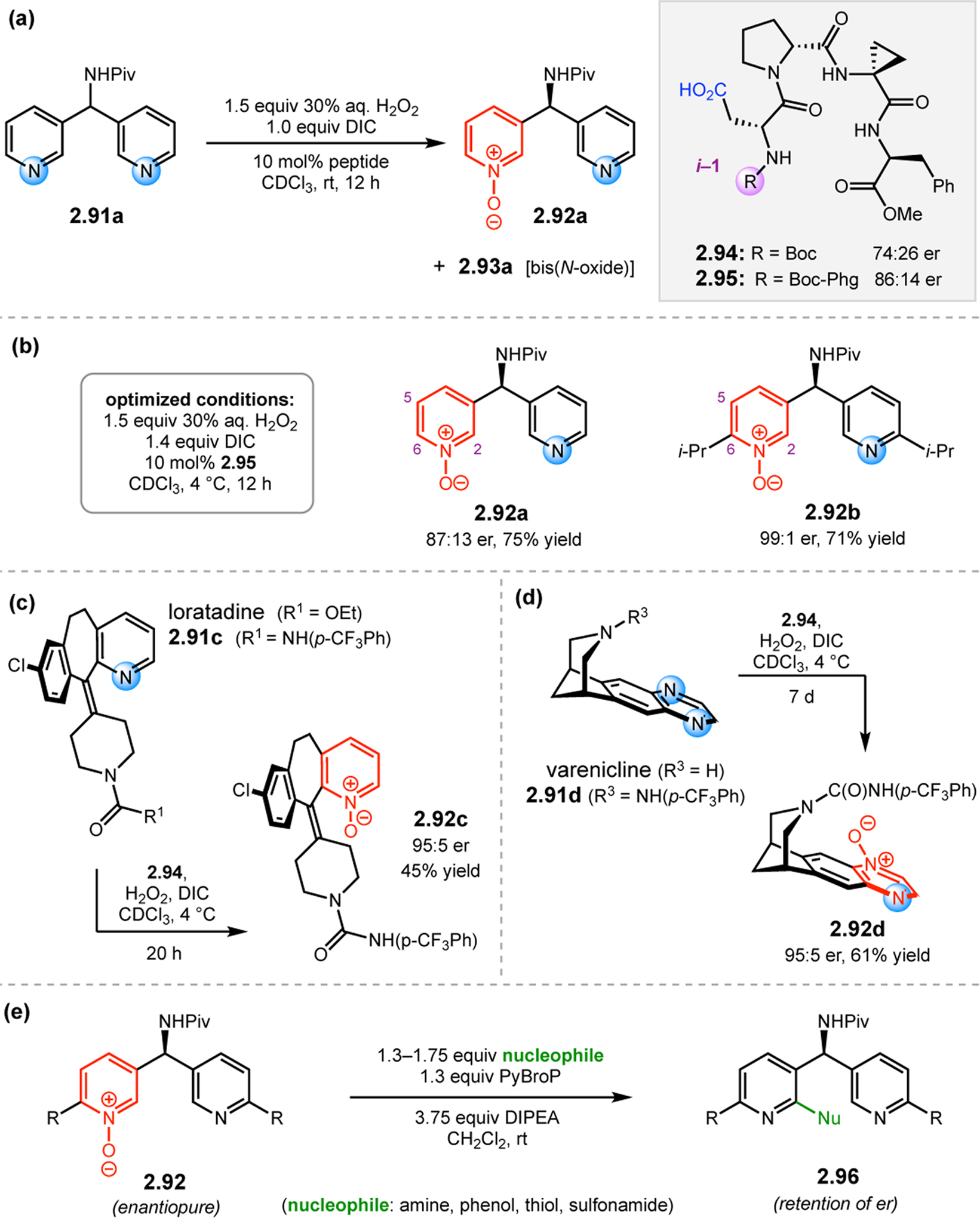
(a) Desymmetrization of bis(pyridine) 2.91a to the corresponding mono-N-oxide 2.92a catalyzed by Asp-containing peptide 2.95. (b) Optimal conditions and selected substrates for the 2.95-catalyzed desymmetrizing N-oxidation. (c) Enantioselective synthesis of loratadine derivative 2.91c, which freely interconverts between two helically chiral conformations and is locked upon 2.94-catalyzed N-oxidation. (d) Desymmetrizing N-oxidation of varenicline catalyzed by 2.94. (e) The N-oxidized bis(pyridine)s could be further functionalized with retention of enantiopurity.92
Upon developing an enantioselective N-oxidation catalyst, we sought to examine its potential in the synthesis of medicinally relevant derivatives. Loratadine is the active ingredient in the antihistamine Claritin®, which exists in rapid equilibrium between two enantiomeric, helically chiral conformations.93 This scenario provided the possibility for a peptide-catalyzed dynamic kinetic resolution (DKR) if N-oxidation were to effectively inhibit racemization. As such, peptide 2.94 was indeed able to effectively convert loratadine derivative 2.91c, in which urea directing group was incorporated in place of the native ethyl carbamate, to N-oxide 2.92c in 45% yield and 95:5 er (Figure 29c). Additionally, varenicline, the active ingredient in smoking cessation aid Chantix®, contains a symmetrical quinoxaline ring,94 which we hypothesized to be an excellent candidate for a peptide mediated desymmetrization. While oxidations of this more electron deficient heterocycle were slower, varenicline derivative 2.91d was successfully transformed into N-oxide 2.92d in 95:5 er and 61% yield (Figure 29d). Finally, to showcase the broad synthetic utility of enantiopure N-oxidized pyridines, a variety of nucleophiles were added to bis(pyridine) 2.92 under previously reported conditions.95 Stereoretentive amination, etherification, thioetherification, and sulfonamidation reactions were straightforward, highlighting the power of this platform in the site- and stereoselective functionalization of medicinally relevant N-heterocycles.
2.3.2. Alcohol Oxidation.
A common strategy for the development of new asymmetric catalysts is to embed a known reactive species into a chiral backbone. Ward, Kirshenbaum, and co-workers sought to generate a chiral variant of the known oxidant, TEMPO,96 by embedding the persistent radical species within an oligopeptide framework. The authors studied whether the incorporation of TEMPO into these foldamers, or oligopeptide sequences of non-proteinogenic amino acids that are known to adopt specific secondary structures, would produce a new class of oxidation catalyst.97 Specifically, they chose to investigate peptoids derived from N-substituted glycine residues that are known to exhibit helical structures in which the helix sense is controlled by the absolute configuration of the phenethylamine substituents (Figure 30a).98 A library of peptoid-based TEMPO catalysts was synthesized and screened in the oxidative kinetic resolution of compound 2.100, given the well documented activity of TEMPO as a general oxidant for secondary alcohols (Figure 30b). In the presence of external peptoid 2.98, TEMPO-derivative 2.97 provided negligible selectivities. However, TEMPO-embedded catalyst 2.99 was notably more effective, giving a krel of 4.6 in the kinetic resolution of 2.100. Further catalyst screening revealed that it was preferable to embed the TEMPO motif at the N-terminus of the peptoid, as selectivities were eroded upon moving the TEMPO moiety into the center of the sequence. Analysis of the structure of 2.99 revealed that the steric environment near TEMPO is quite hindered, leaving a small cleft accessible for substrate binding.
Figure 30.
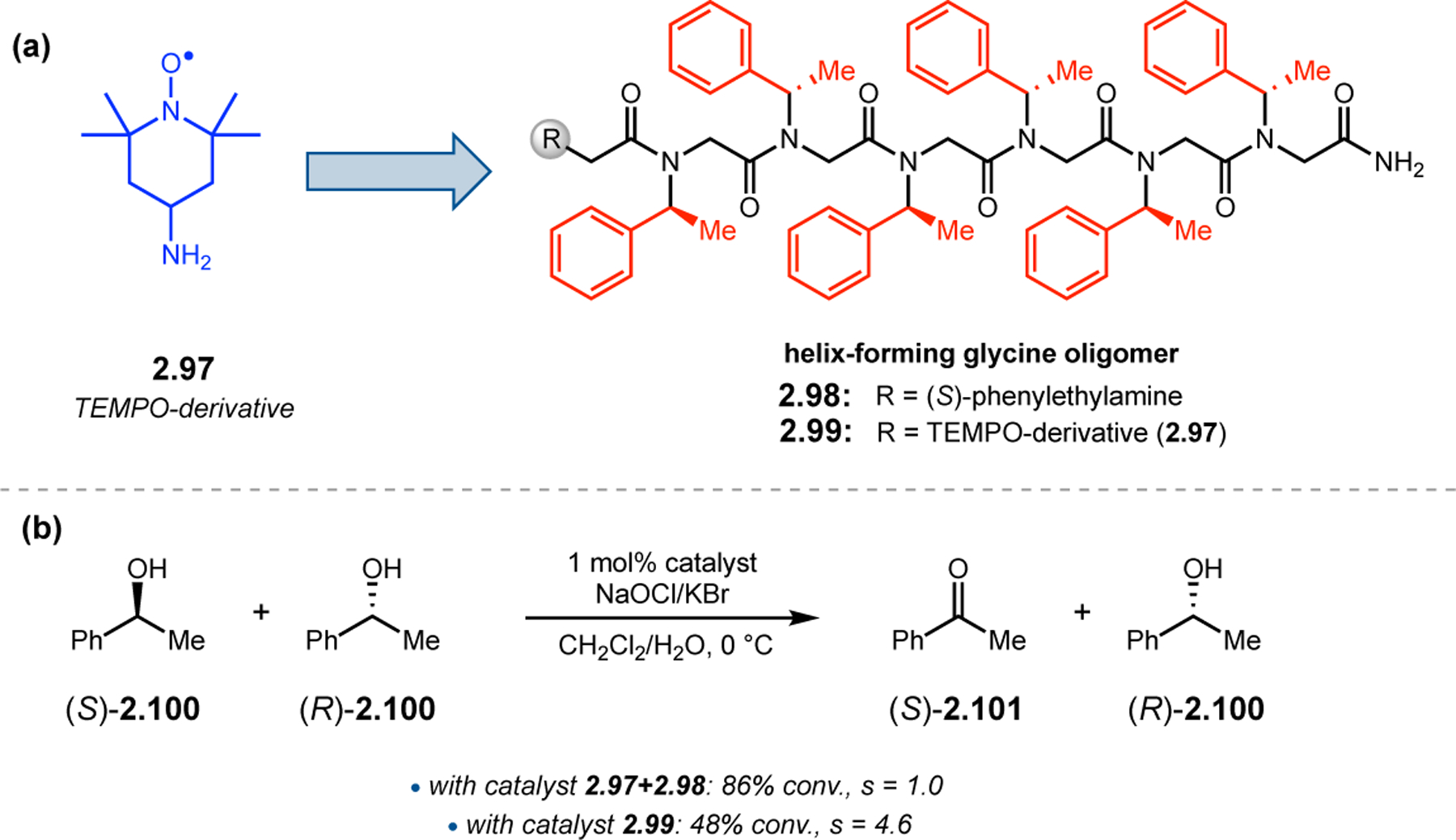
(a) Incorporation of a TEMPO catalyst into a helix-forming peptoid sequence. (b) Examination of peptoid-based catalysts in oxidative kinetic resolutions of secondary alcohols.97
2.3.3. α-Functionalization of Aldehydes.
Alternatively, Kudo and co-workers focused on controlling the reactivity of TEMPO through the addition of external peptide catalysts.99–101 They specifically targeted α-oxyaminations of aldehydes,102 which has previously been reported with TEMPO in the presence of chiral imidazolidinone catalysts.103 Upon condensation of the imidazolinone onto the aldehyde, single-electron oxidation of the incipient enamine resulted in formation of a planar radical cation, which can undergo enantioselective C–O bond formation with TEMPO. The Kudo Group sought to develop a variant of this reaction with a new class of Pro-containing peptides in aqueous conditions.
The use of Pro as a catalytic residue for the activation of substrates is among the critical advances in the field of organocatalysis.11 Specifically, Pro and other chiral secondary amines are able to condense onto aldehydes and ketones, activating the carbonyl compounds as either (1) iminium ions for stereocontrolled nucleophilic attack or (2) enamines for stereoselective addition to electrophiles. Pro-containing peptides have featured prominently in these reaction paradigms through the years.14,15
The Kudo group has pioneered the development of Pro-containing catalysts like 2.104 for a variety of reactions in aqueous conditions.16 Peptide 2.104 contains both a β-turn element (d-Pro-Aib-Trp) known to bolster asymmetric induction and a poly(Leu) chain to provide a hydrophobic environment for attracting organic substrates when water is used as a solvent. First developed in 2009 for reductions (Section 3.1),104 the authors sought to expand the catalysts’ utility into enamine-type activation mechanisms, such as α-oxidation (Figure 31a). Indeed, conditions were developed utilizing both Fe(II)100 and Cu(I) salts101 to facilitate SET from the activated enamines, with molecular oxygen as the terminal oxidant. Selectivities of up to 97:3 er were achieved.
Figure 31.
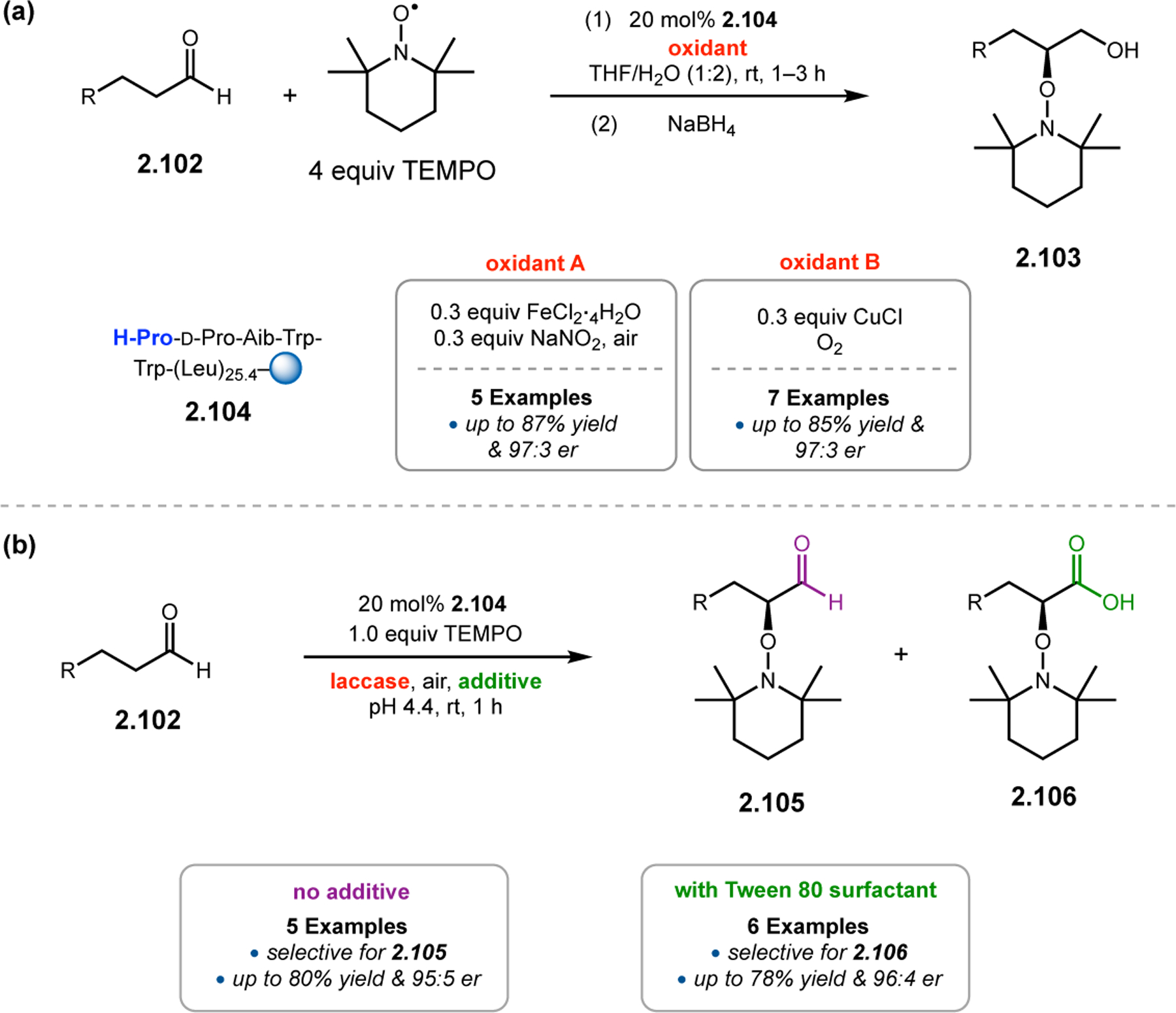
(a) Pro-mediated α-oxidation of aldehydes with TEMPO. (b) Combination of peptide-based catalyst and enzymatic oxidant.99–101
Upon developing efficient conditions for α-oxidations in water, Kudo and co-workers further optimized the reaction conditions with environmentally friendly reagents.101 Laccase is a copper-containing oxidase enzyme that has emerged as a potential candidate for diverse applications in synthetic chemistry.105 However, it has yet to be reported within the context of aldehyde α-functionalization. Given the excellent activities afforded by peptide 2.104 in aqueous conditions, laccase appeared to be suitably matched for use with an enzymatic cocatalyst in water (Figure 31b). Following optimization of the reaction conditions, two products predominated the reaction mixtures, aldehyde 2.105 and over-oxidized carboxylic acid 2.106. It was found that the product distribution could be controlled by the addition of a nonionic surfactant, Tween 80. The presence of Tween 80 resulted in an acceleration of the aldehyde to carboxylic acid oxidation by laccase. Overall, this method demonstrates the possibility of dual catalysis using short peptides and enzymes together to achieve highly selective α-oxidations of aldehydes with up to 96:4 er.
Enamine activation by Pro-based peptide catalysts have also been reported in α-amination reactions of aldehydes 2.107 using azodicarboxylate 2.108 (Figure 32).106,107 Kudo and co-workers further explored the utility of recyclable, on-bead peptides for the formation of amino alcohols 2.109.108 While Pro alone linked to the resin produced only a moderate er of 19:81 (entry 1), extending the peptide chain to include a β-turn forming motif (d-Pro-Aib-Xaa) provided higher enantioselectivities favoring the opposite enantiomer (entries 2–4). Indeed, catalyst 2.110 delivered 2.109 in 96:4 er (entry 4). The absolute sense of asymmetric induction is controlled by the configuration of the N-terminal Pro residue; using the l-Pro variant of 2.110 provides 2.109 in 5:95 er favoring the opposite enantiomer compared to 2.110. While recycling the freebase of catalyst 2.110 resulted in a reduction in yields, the TFA salt of 2.110 could be reused up to 10 times with minimal perturbation to product yields and ers.
Figure 32.
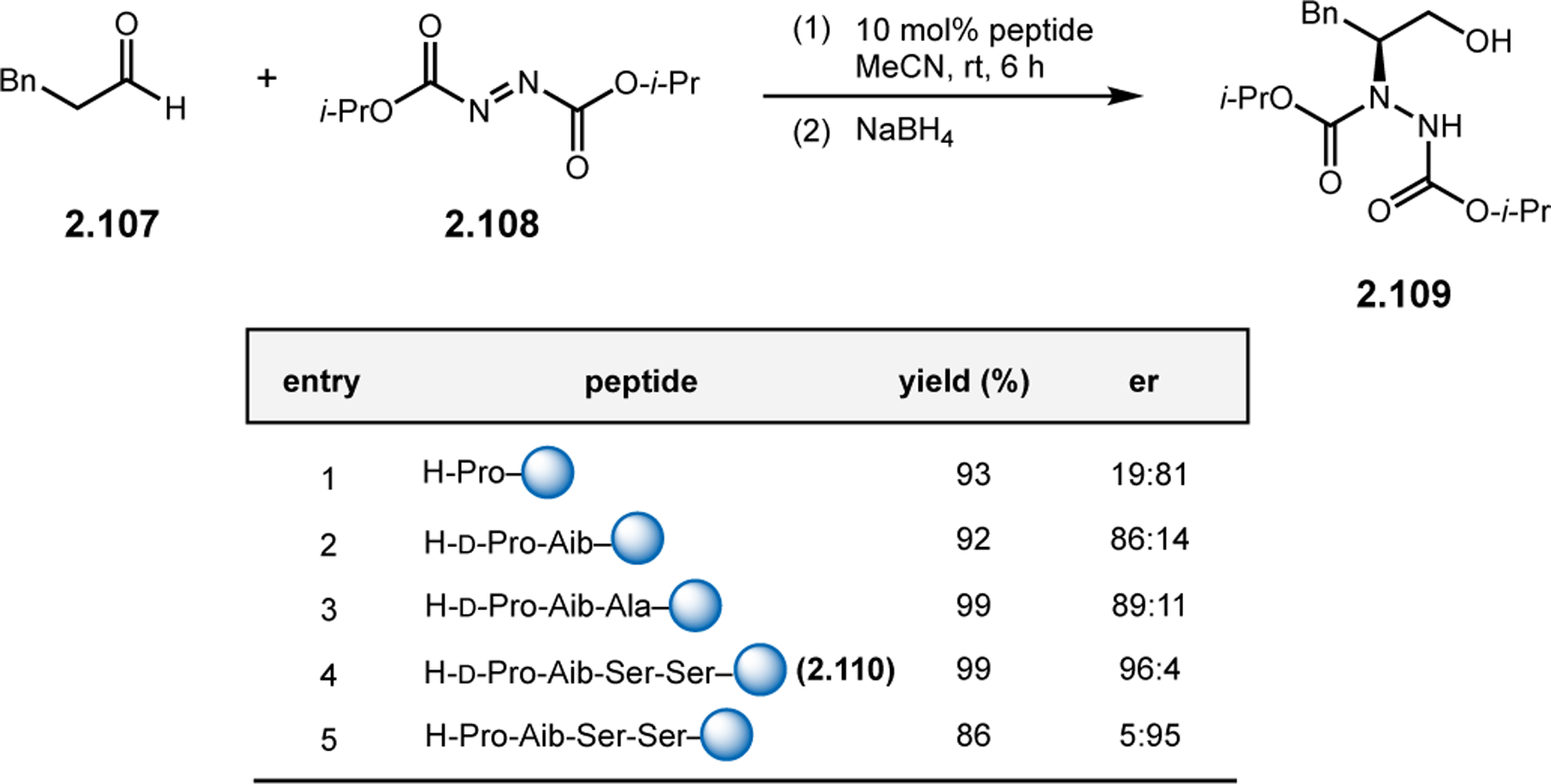
Development of a resin-bound, Pro-based peptide catalyst for the α-amination of aldehydes.108
Expanding upon the use of solid-supported peptides as catalysts for α-functionalizations of aldehydes, Fülöp and co-workers developed a continuous-flow type process to produce α-aminated products in a high-throughput manner (Figure 33).109 A library of minimal peptide catalysts containing Pro at the N-terminus and a variety of carboxylic acid-containing residues embedded into the sequences were evaluated in the α-oxidation of aldehyde 2.111 with azodicarboxylate 2.112. Peptide 2.115 was able to provide amino alcohols 2.114 with high er values both in batch and flow reaction setups. Upon optimization of the flow parameters, including flow rate, residence time of reactants with the catalyst bed, reagent concentrations, and pressure, a highly efficient protocol was developed, allowing for the formation of 2.113 in up to 99:1 er on gram-scale. The immobilized peptide column was found to be durable, with no signs of decrease in er or efficiency even after 20 consecutive hours of use.
Figure 33.
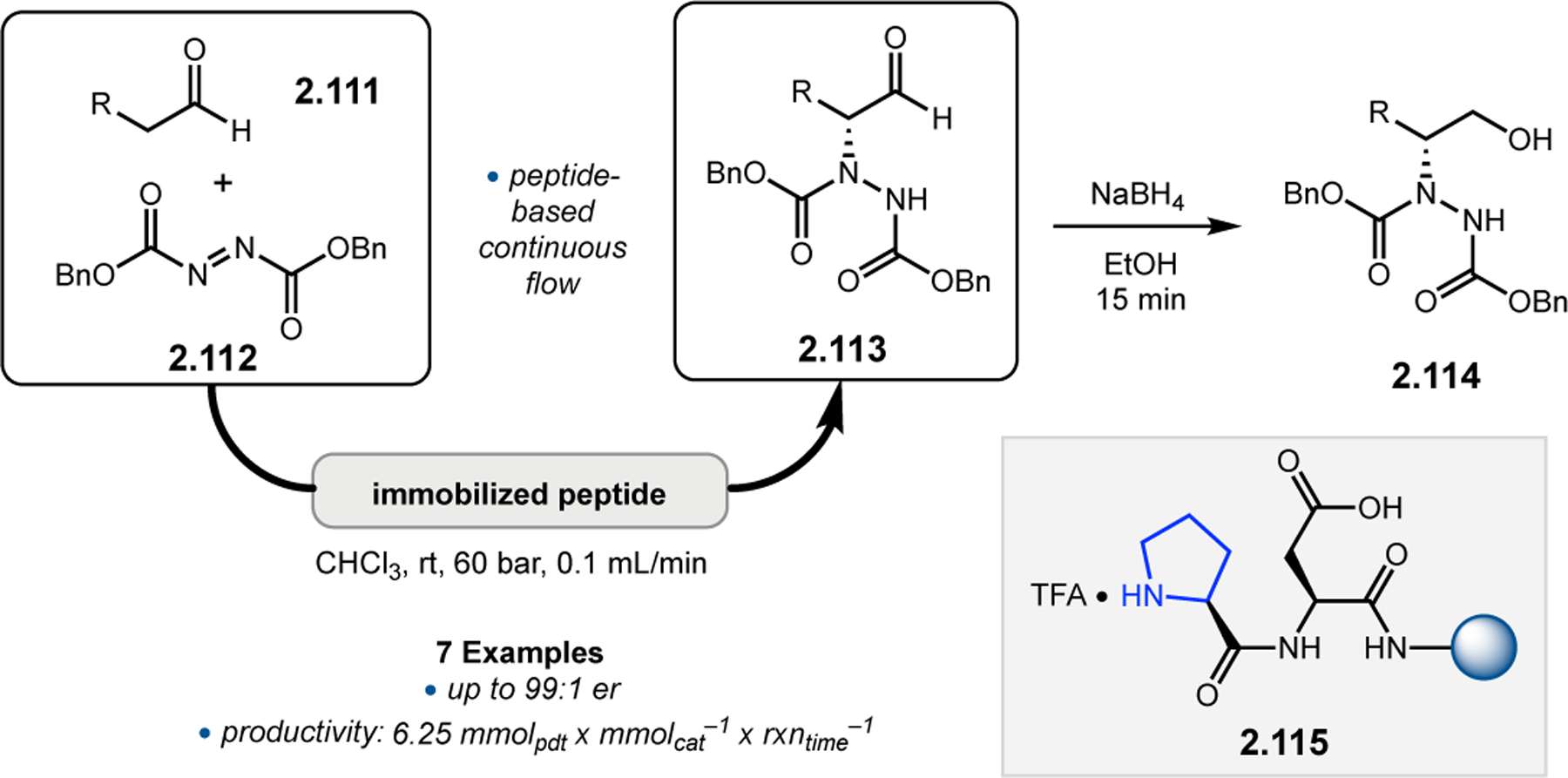
Development of a flow process, featuring solid-supported proline-containing peptides, for α-amination of aldehydes.109
3. Reduction
Prior to 2007, peptide-based catalysts had been demonstrated only sporadically in asymmetric reductions of various functional groups. Among the earliest contributions were electrochemical reductions, wherein polypeptide-coated electrodes were employed to enable asymmetric induction during electron transfer events.110–113 Since then, numerous advances have been made in the application of both solution-phase and resin-supported peptide catalysts toward the reductions of ketones, imines, quinolines, and conjugated alkenes, often with high levels of enantiocontrol. In particular, three classes of peptides have emerged as excellent platforms for such processes, including (1) amine-containing peptides via iminium catalysis, (2) phosphothreonine (pThr)-containing peptides for Brønsted acid catalysis, and (3) various peptide-based ligands in metal-based reduction systems (Section 11.2).
3.1. Transfer Hydrogenation
Proline is one of the most robust and versatile catalytic residues for peptide-based asymmetric catalysis.11 In particular, Pro-containing peptides are well known to form chiral iminium ions following condensation onto aldehydes, which can then be intercepted by a variety of nucleophiles with enantiofacial selectivity governed by the peptide catalyst. Building on the seminal advances from the MacMillan114 and List115 Groups, which revealed that secondary amine catalysts could enable enantioselective reductions of enals with Hantzsch ester (HEH) via iminium catalysis, Kudo and co-workers reported an aqueous, peptide-catalyzed variant in 2008 (Figure 34).116 Peptide-mediated reductions with HEH are a fascinating class of reactions, as both reaction partners are biomimetic in nature—peptide-based catalysts are often modeled on certain features of enzymes, while the dihydropyridine-based reductant mimics one of nature’s most prolific redox cofactors, NAD(P)H.117
Figure 34.
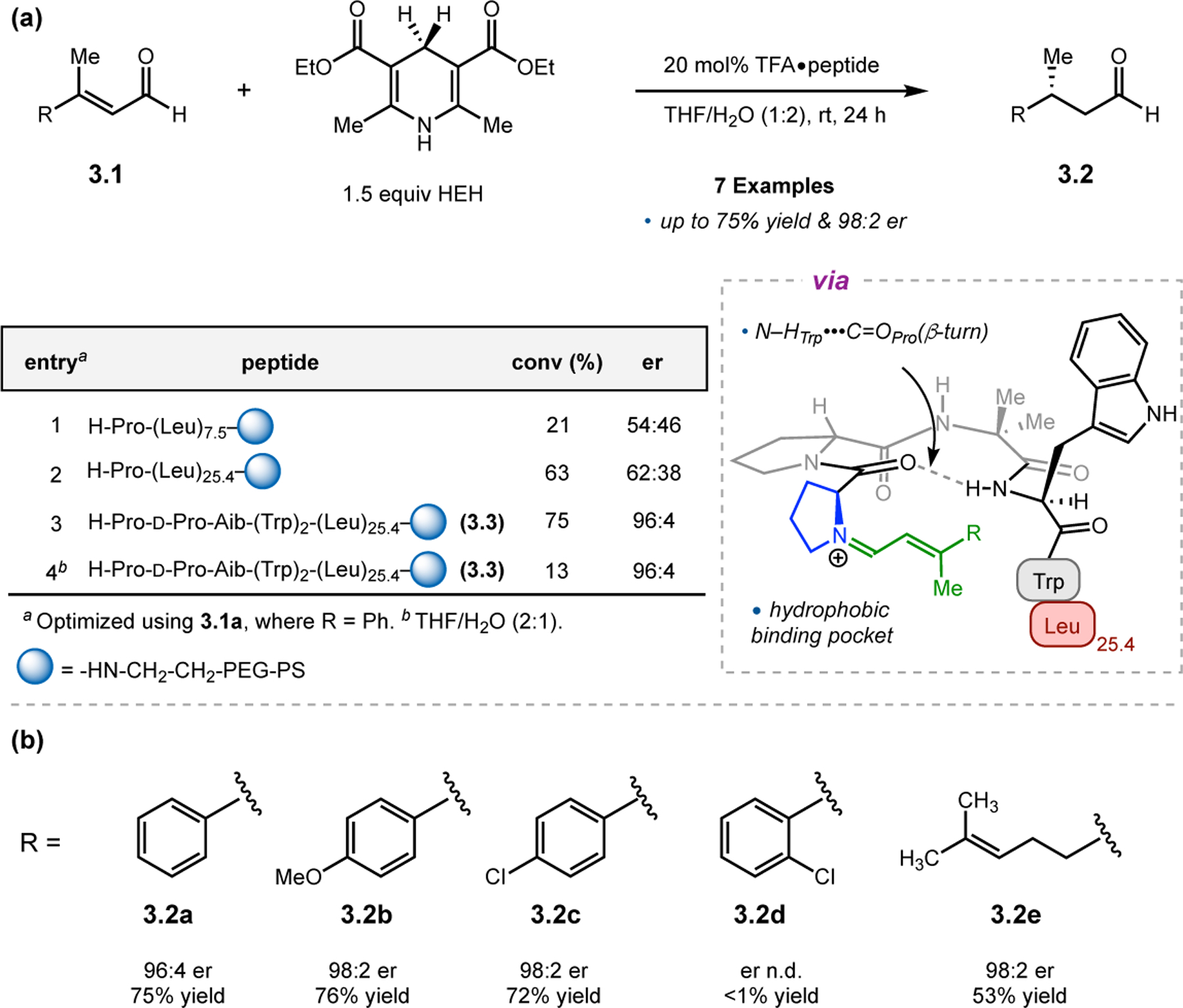
(a) Reduction of enals 3.1 with HEH using proline-containing peptide catalysts. Catalysts required a β-turn motif to achieve high selectivity, as well as a hydrophobic poly(Leu) chain to give high conversion. (b) Selected substrate scope entries.116,119
In that context, the authors began with the reduction of enal 3.1 to aldehyde 3.2 in THF/H2O (1:2 v/v) solvent mixtures using resin-bound, Pro-containing peptide catalysts (Figure 34a).116 However, Pro alone did not provide meaningful levels of conversion to 3.2.118 The authors posited that the incorporation of a hydrophobic environment near the Pro residue would be required to attract the lipophilic substrate and reductant to catalyst binding pocket. A key aspect of this design principle was to employ peptide-based catalysts in which the N-terminal, catalytically active Pro residue was linked to the resin through a helical poly(Leu) chain. While catalysts with short poly(Leu) domains delivered 3.2 in 21% conversion and 54:46 er (entry 1), reactivity and selectivity was enhanced to 63% conversion and 62:38 er upon increasing the average length to ~25.4 Leu units (entry 2)—sufficiently long to form a stable, helical secondary structure. Although this design feature seemed to address the issue of low reactivity, product enantioselectivities were still quite low. As such, peptide 3.3 was devised, in which a β-turn inducing sequence, d-Pro-Aib-(Xaa)2, was inserted between the catalytic Pro residue and poly(Leu) chain.119 Using this catalyst, 3.2 was formed in 75% conversion and with 96:4 er (entry 3), suggesting that the β-turn motif is able to provide excellent levels of asymmetric induction. To highlight putative hydrophobic effects exhibited by this catalyst, decreasing the amount of water to 2:1 THF/H2O resulted in only 13% conversion to 3.2 using peptide 3.3. A variety of enals 3.1 were addressed by the catalyst, except for sterically hindered compounds, such as ortho-chloro-containing 3.1d (Figure 34b).
Having developed highly enantioselective enal reductions using peptide 3.3, Kudo and co-workers next probed whether these Pro-containing catalysts could be applied toward more complex substrates, such as α,β,γ,δ-unsaturated aldehyde 3.4 (Figure 35).120 In addition to enantioselectivity, substrates like 3.4 pose inherent challenges in regioselectivity, namely the control of initial 1,4-reduction to deliver γ,δ-unsaturated aldehyde 3.5 over 1,6-reduction that could afford achiral α,β-unsaturated 3.6 or saturated aldehyde 3.7. Utilizing pyrrolidine as a simple achiral standard, 76% of 1,4-addition product 3.5 was obtained (entry 1). However, catalyst 3.3 predominantly gave 1,6-addition products 3.6 and 3.7, an example of catalyst-controlled reversal of inherent regioselectivity (entry 2). In that scenario, peptide 3.3 delivered 3.7 in 94:6 er. Further optimization of the β-turn-inducing sequence revealed that electron-rich Trp residues enhanced the er of 3.7 to up to 99:1 (entry 3), and variation of the i+2 position from Aib to Achc increased conversion to this product to 87% (entries 4). Truncation of the α-helical poly(Leu) sequence from 25 to 6 residues resulted in increased conversion and enantioselectivity, revealing peptide 3.8 as the optimal catalyst (entry 5), which was found to be effective over a range of substrates.
Figure 35.
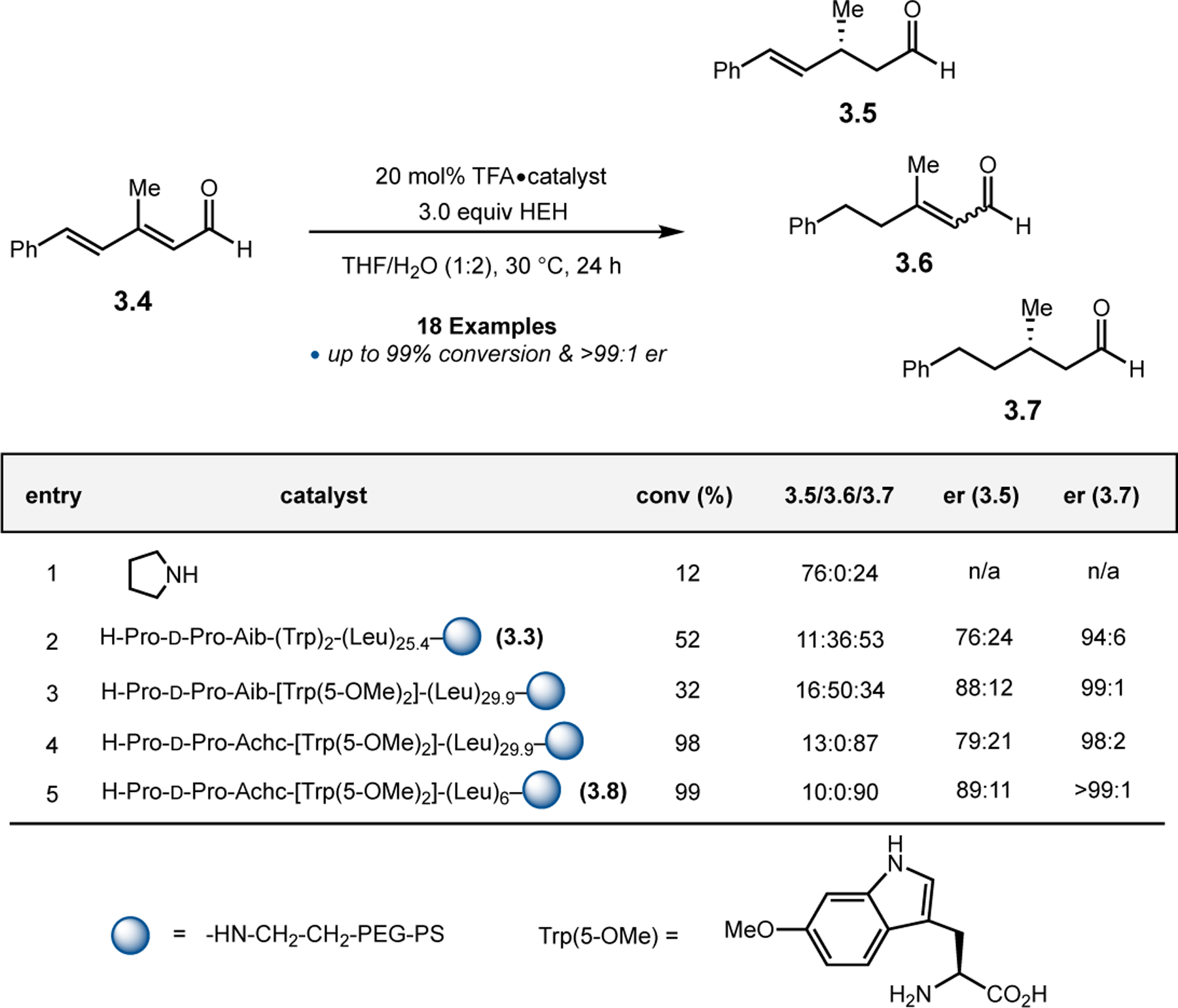
Enantio- and regioselective reduction of α,β,γ,δ-unsaturated aldehyde 3.4 with Pro-containing catalysts.120
Kudo and co-workers further applied these Pro-containing catalysts in the kinetic resolution of planar-chiral ferrocene complexes via asymmetric reduction of racemic, enal-containing 3.9 (Figure 36a).121 While β-turn-containing catalysts lacking an appended hydrophobic chain provided (Rp)-3.10 with a modest krel of 2.3 (entry 1), incorporation of a (Leu-Leu-Aib)2 unit increased the krel to 4.0 (entry 2). Unlike previous catalysts, such as 3.3 (Figure 34) and 3.8 (Figure 35), which relied on an α-helical polyleucine chain, the (Leu-Leu-Aib)2 motif is believed to form a 310 helix. Further optimization of this catalyst revealed that alterations to the i+4 position played an essential role in selectivity, with methyl-protected homoserine (Hse(Me)) increasing the krel to 11.7 (3.11, entry 3). While the effect of this residue is not entirely understood, the β-turn is thought to position Hse(Me) adjacent to the catalyst-bound substrate. This orientation potentially facilitates secondary interactions between the iminium cation and the oxygen lone pairs of Hse(Me), which may inhibit particular approach trajectories of the HEH reductant (Figure 36a). This methodology was further expanded to address symmetrical, bis(enal)-containing ferrocene compounds, such as 3.12 (Figure 36b).122 In this case, both mono-reduced 3.13 and the product of intramolecular cyclization (3.14) were possible. The choice of an acidic additive proved essential to controlling the product ratio. Using 3.11 as the catalyst with substoichiometric HCl as the additive, 3.13 was obtained with 87:13 er in a 99:1 ratio over 3.14 (entry 1). Using two equivalents of benzoic acid instead, peptide 3.11 now favored 3.14 in a 73:27 ratio, with products 3.13 and 3.14 being isolated with 97:3 er and 93:7 er, respectively (entry 2).
Figure 36.
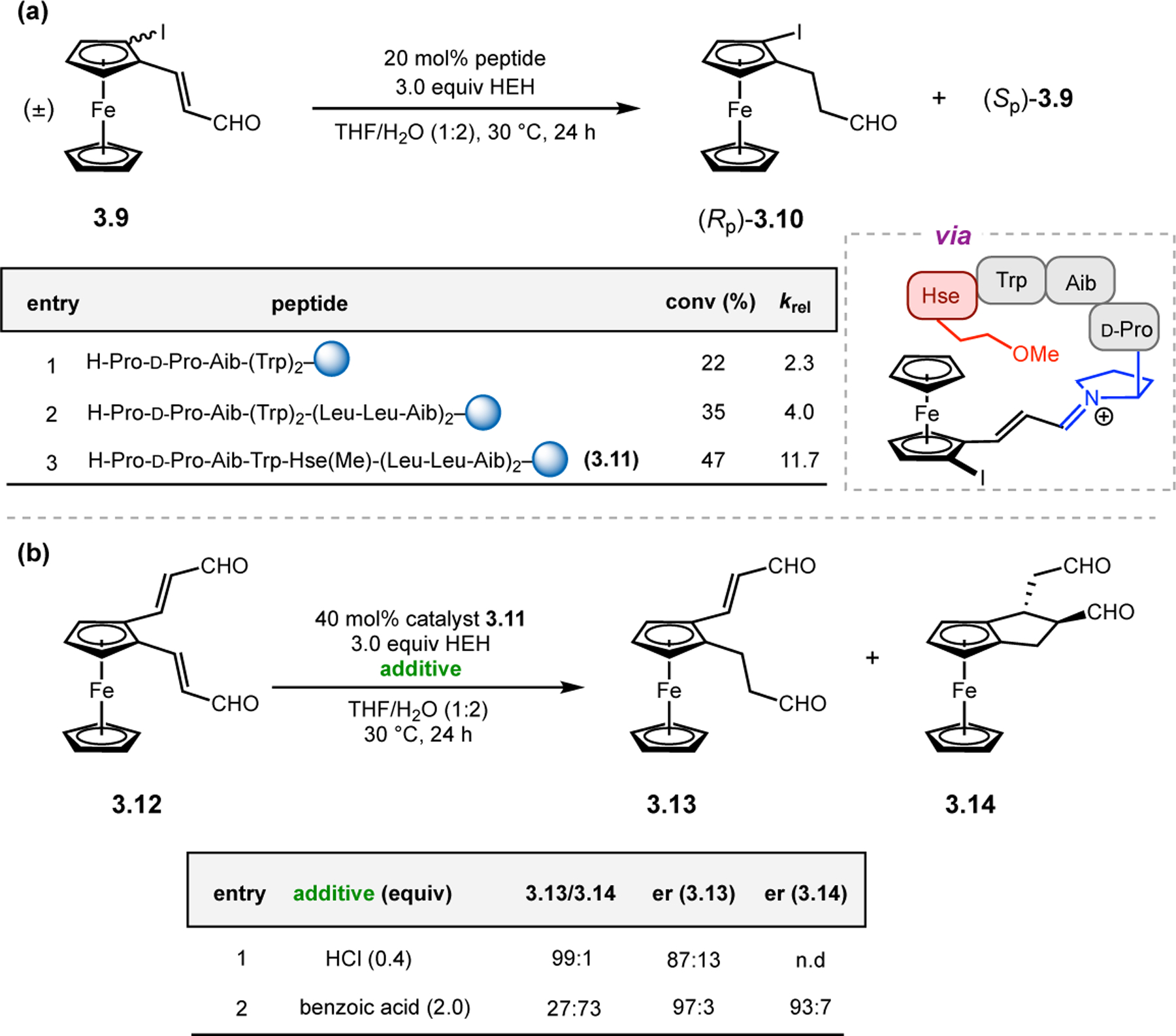
(a) Kinetic resolution of enal-containing ferrocene complexes with Pro-based peptide catalysts. (b) Desymmetrization of bis(enal) containing ferrocene complexes.121,122
In a complementary approach to iminium activation by Pro-containing catalysts, Brønsted acid catalysts have also been reported to mediate Hantzsch ester-based reductions. In 2004, the Akiyama123 and Terada124 Groups independently reported the development of a new BINOL-derived chiral phosphoric acid (CPA). CPAs are now considered privileged catalysts, and they have since been reported to mediate a vast array of transformations,82 including reductions.125–128 However, efforts to develop alternative catalyst architectures have been slow. The C2-symmetry of the CPA scaffold has been proposed to be essential for effective catalysis, as this property makes tautomerization of the phosphoric acid proton between the two phosphate oxygens degenerate, which would otherwise provide distinct diastereomeric species with potentially different catalytic activities.
In pursuit of an alternative to the archetypal CPA scaffold, our group postulated that short-sequence peptides could serve as a new family of chiral Brønsted acid catalysts. In this case, catalyst rigidity would perhaps not be necessary, as noncovalent interactions between the flexible peptide backbone and substrates could enable high degrees of selectivity. Moreover, the modularity and tunability of the peptide sequence would enable the development of catalysts with highly diverse structures and functions. Our first forays into this realm began in 2015, when we identified and developed pThr as a catalytic residue that could be easily embedded into peptidic scaffolds to enable Brønsted acid catalytic activation modes (Figure 37a).86 Although pThr is a well-known product of post-translational modification that is central to the regulation of cell function, it has never been reported as part of a catalytic apparatus in nature.129
Figure 37.
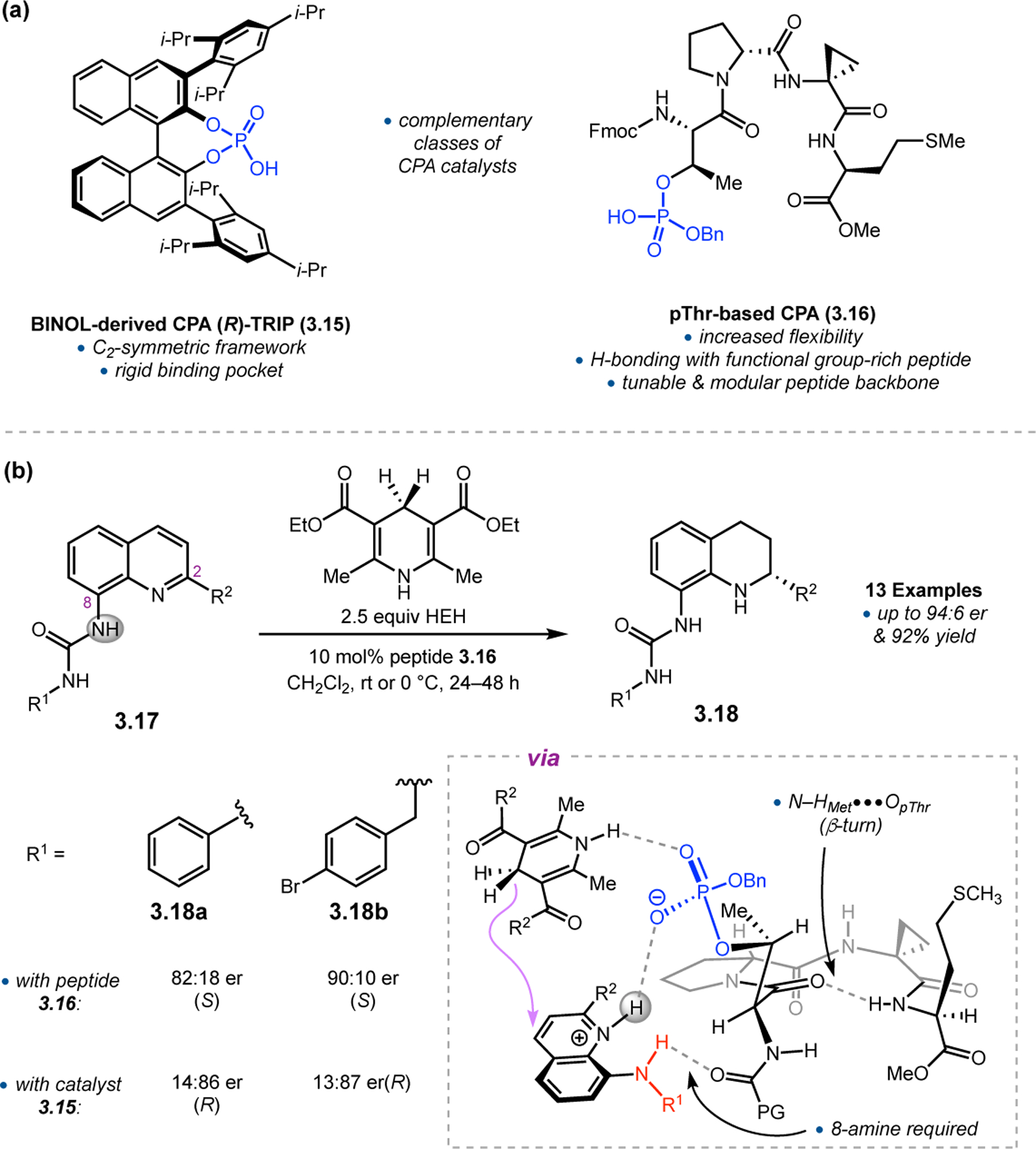
(a) Two complementary classes of chiral phosphoric acid (CPA) catalysts: BINOL-derived (3.15) and peptide-based (3.16) CPAs. (b) The transfer hydrogenation of 8-aminoquinolines with pThr-based catalysts.86
Our efforts to develop enantioselective pThr-based catalysts began with studies on the HEH-mediated transfer hydrogenation of quinolines (Figure 37b).86 While minimally substituted quinolines were reduced with high conversions, no enantioselectivity was observed. However, incorporation of a directing group handle on the quinoline C8-position (e.g., 3.17), which was presumed to facilitate noncovalent interactions between catalyst and substrate, greatly improved the efficacy of the peptide-mediated reduction to tetrahydroquinoline 3.18. Evaluation of a small library of peptides containing an N-terminal pThr residue revealed that a central d-Pro-Acpc dyad (Acpc = 1-aminocyclopropane carboxylic acid) at the i+1 and i+2 positions was critical for obtaining high levels of selectivity. Further optimization of the i+3 residue led to the identification of Met-containing phosphopeptide 3.16 (Figure 37a), which mediated the transfer hydrogenation of 8-aminoquinolines 3.17 in good yields and up to 94:6 er (Figure 37b). These selectivities were comparable to those afforded by a known BINOL-derived CPA, (R)-TRIP (3.15, Figure 37a). NMR studies showed that, despite the increased flexibility of peptide-based catalyst 3.16 as compared to 3.15, high selectivities could be achieved through: (1) the incorporation of critical secondary structural elements into the peptide, in this case a β-turn motif that is stabilized by a 10-membered ring N–H(i+3)•••O(i) H-bond, and (2) secondary interactions between the catalyst and substrate directing group, specifically the 8-amino group of 3.17 (Figure 37b). These results demonstrate the difference between pThr-peptides and BINOL-derived CPAs, which achieve selectivity through distinct activation mechanisms.
Given the high selectivities achieved by both BINOL-derived and pThr-based CPAs despite their significant differences in design and mechanism, our group sought to further probe the activities of these catalysts in more complex transformations. For example, the reduction of bis(quinoline) 3.19 would pose interesting selectivity challenges, as two sequential transfer hydrogenations can occur on one substrate (Figure 38a).130 Following initial formation of both enantiomers of mono-reduced 3.20, further reduction can result in a mixture of both chiral (3.21) and meso (3.22) products. The use of two chirality-generating steps in tandem is a strategy for enhancing the enantioselectivity of the overall process, as governed by Horeau’s Principle.131 Under ideal conditions, with the assumption that the catalyst reduces 3.19 to 3.20 and 3.20 to 3.21 with the same levels of selectivity, the final er of doubly reduced 3.21 can be predicted by squaring the er of the first reduction.
Figure 38.
(a) Reduction of bis(quinoline) 3.19 with both BINOL-derived CPA ent-3.15 and peptide-based CPA 3.16. (b) Overview of this process when both catalysts are added together. Catalyst ent-3.15 is matched for the first reduction, while catalyst 3.16 is matched for the second.130
Applying pThr-containing catalyst 3.16 to the reduction of 3.19, mono-reduced 3.20 was produced with 67:33 er (s1 = 2.0, Figure 38a, entry 1),130 notably lower than previously observed for the reduction of quinoline 3.17 (Figure 37b). Based on this result, the theoretical er of the di-reduced product is 81:19 (s12 = 4.1), yet catalyst 3.16 produced 3.21 with 95:5 er (s2 = 19). This result indicates that 3.16 is substantially more selective in the second reduction step than in the first. Alternatively, BINOL-derived catalyst ent-3.15 performed the first reduction with 86:14 er, yet only produced di-reduced 3.21 with 87:13 er (entry 2), implying this catalyst is matched preferably in the first reduction.
Given the complementarity of catalysts ent-3.15 and 3.16, reductions of 3.19 were performed in the presence of both catalysts in one pot, with the expectation that each catalyst would selectively facilitate its matched step. Indeed, using the catalysts together, both 3.20 and 3.21 were produced with high enantioselectivities of 83:17 er and 94:6 er, respectively, which is comparable with the values each catalyst was able to achieve separately (entry 3). These observations suggest that both catalysts indeed exerted control over their matched steps (Figure 38b). To further test this hypothesis, catalyst ent-3.15 was substituted with (R)-TRIP (3.15) in these one-pot reactions (entry 4). While 3.15 maintained control over the first step with the opposite sense of enantioinduction, delivering (R)-3.20 in 28:72 er, peptide 3.16 produced (S,S)-3.21 as the major di-reduced product, reversing the absolute stereochemistry for the product of the second step (63:37 er). This phenomenon is a rare instance of two co-catalysts with the same catalytic motif (a phosphoric acid) acting in tandem in a one-pot process. By appending different chiral scaffolds to a phosphoric acid (e.g., BINOLs vs. peptides), each catalyst is matched for a different substrate—BINOL-derived CPAs for bis-quinoline 3.19 and peptide-based CPAs for mono-reduced 3.20. These results speak to the potential complementarity of BINOL-derived and peptide-based CPAs in complex settings, including situations where they might be employed in situ and in one-pot.
While divergent selectivities were observed between different CPA catalysts for reductions, our group had also developed π-methylhistidine (Pmh)-based catalysts that exhibit distinct reactivity profiles based solely on the appended peptide sequence.130 In 1999, Pmh-containing peptide 3.23 was shown to mediate the kinetic resolution of racemic amino alcohol 3.25 via acylation with acetic anhydride (Figure 39a).132–134 Additionally, Pmh-containing peptide 3.24 was found to selectively phosphorylate myo-inositol derivative 3.26 at the 1-hydroxyl group.135,136 In analogy to the concept explored using CPAs to perform the reductions of bis(quinoline) 3.21 (Figure 38), we had shown that whether each of these catalysts 3.23 and 3.24 could perform their matched reactions when all the reagents and catalysts were added together in one pot (Figure 39b).130 Inclusion of both substrates 3.25 and 3.26, acylation and phosphorylation reagents, and catalysts 3.23 and 3.24 in one reaction could result in myriad of mono- and poly-acylated and phosphorylated products and the corresponding stereoisomers. Despite this complexity, the acylated amino alcohol 3.27 and phosphorylated inositol derivative 3.28 were the primary products of this reaction, and each was formed with good enantioselectivity. The only other significant product observed was 3.29, in which the unreactive enantiomer of 3.25 was phosphorylated. These results demonstrated the sequence-dependent reactivity Pmh-containing peptides 3.23 and 3.24 for acylation and phosphorylation of targeted substrates and foreshadowed our more recent findings with CPAs. This peptide-encoded reactivity is presumably the result of precise outer-sphere interactions between the peptides and their substrates.
Figure 39.
(a) Two π-methylhistidine (Pmh)-containing peptides previously found to mediate asymmetric acylations (3.23) and phosphorylations (3.24), respectively. (b) One-pot acylation and phosphorylation of 3.25 and 3.26 by both Pmh catalysts, synthesizing their respective, desired products with high fidelity and stereoselectivity.130
3.2. Reductive Amination
The utility of pThr-embedded peptides for HEH-mediated reductions was further explored in the reductive amination of 3-amidocyclohexanones 3.30 with p-anisidine (Figure 40a).87 Both the cis- and trans-isomers of amine product 3.31 were possible in this case, providing enantio- and diastereoselectivity challenges for the peptide-based catalysts to address. Upon confirming the inherent trans-selectivity of the reaction with achiral diphenyl phosphate as the catalyst (DPP, 69:31 dr, entry 1), peptide 3.16, the lead catalyst for quinoline reduction,86 was evaluated in this new context. Unfortunately, 3.16 gave nearly racemic products (entry 2). Extensive screening of the peptide sequence revealed that bulky, l-amino acids at the i+2 position, such as Val, provided cis-3.31 with up to 83:17 er (entry 3). However, further increases in enantioselectivity were difficult to achieve.
Figure 40.
(a) Reductive amination of 3-amidocyclohexanones 3.30. (b) Kinetic analysis and proposed transition state of the reaction.87
Given that the protected pThr monomer alone was able to provide fair levels of enantioselectivity in cis-3.31 (75:25 er, entry 4), we suspected that changes to the catalytic residue could have a more substantial impact on catalyst selectivities. Indeed, changing the N-terminal protecting group from Fmoc to the more electron-rich p-methoxybenzamide and further tailoring the peptide sequence led to the identification of 3.32, which produced cis-3.31 in 93:7 er (entries 5–6). Peptide 3.32 was also modestly more cis-selective than DPP (51:49 vs. 69:31 dr, entries 6 vs. 1). NMR studies revealed that the electron-rich benzamide enabled the formation of a 10-membered ring N–H(i+2)•••O(i–1) H-bond, defining a β-turn that is shifted over one residue with respect to the secondary structure of peptide 3.16. Alternatively, BINOL-derived CPA ent-3.15 yielded higher dr and er for trans-3.31 (entry 7). This complementarity was further investigated using kinetic studies (Figure 40b). While ent-3.15 operates via a kinetic resolution regime, in which the (S)-enantiomer of 3.30 reacts more rapidly and with higher selectivity than the other; peptide 3.32 performs a parallel kinetic resolution, in which both enantiomers are converted to different products with high selectivity in each—(R)-3.30 to cis-3.31 and (S)-3.30 to trans-3.31.
The complementarity of peptide-based Brønsted acid catalysts compared to BINOL-derived CPAs is further explored in Section 11
4. Group Transfer
Group transfer reactions, such as phosphorylation and acylation, are not only key steps in biosynthetic and regulatory pathways in vivo, but they are also highly useful and often strategic transformations in synthetic chemistry. As such, a wide variety of catalytic group transfer methods have been developed through the years, many of which take advantage of Lewis basic activation modes. In the context of polyfunctional molecules, selective group transfer reactions can provide direct access to functionalized targets without recourse to protecting group manipulations. While this sort of selective transformation is routine for enzymes, it presents a greater challenge for small molecule catalysts. Drawing upon inspiration from nature’s group transfer catalysts, synthetic peptide catalysts have been developed for asymmetric group transfer reactions in recent years, including phosphorylation (Section 4.1), sulfonylation (Section 4.2), carbamoylation (Section 4.3), and acylation (Section 4.4). Many of these reactions rely on a Pmh residue embedded within the peptide framework to enable Lewis base catalytic pathways. However, catalytic residues based on tetrazoles and pyridines have also been developed. The advent of these new catalytic systems has also been coupled to mechanistic studies using experimental and computational techniques.
4.1. Phosphorylation
Protein phosphorylation is the most common post-translational modification, which is crucial for regulating protein function.137,138 In nature, histidine-dependent kinases utilize an active site His residue to mediate phosphorylation events via Lewis base catalysis. Inspired by the reactivity and selectivity exhibited by various kinases, small peptides have been designed and developed to catalyze phosphate group transfer reactions of both natural and synthetic small molecule substrates, as well as complex natural products.17 Similar to histidine-dependent kinases, these biomimetic peptide catalysts typically rely on Pmh as the catalytically active residue. However, alternative reaction strategies have also been developed utilizing tetrazolylalanine (Atz).
In 2001, our group first demonstrated that short-sequence Pmh-embedded peptides could catalyze asymmetric phosphorylation reactions. Using either peptide 4.3135 or β-turn-biased 4.4,139 myo-inositol derivative 4.1 could be desymmetrized to afford either 4.2 (65% yield, >98% ee) or ent-4.2 (56% yield, >98% ee) with excellent levels of enantioselectivity (Figure 41a). Though both peptides contain the same absolute configuration of the catalytically active Pmh residue, 4.3 and 4.4 provide enantiodivergent outcomes in this phosphorylation reaction, likely due to their different secondary structures. These early studies indicated that peptide-catalysts are well-suited for the enantioselective synthesis of biologically relevant inositol monophosphates.
Figure 41.
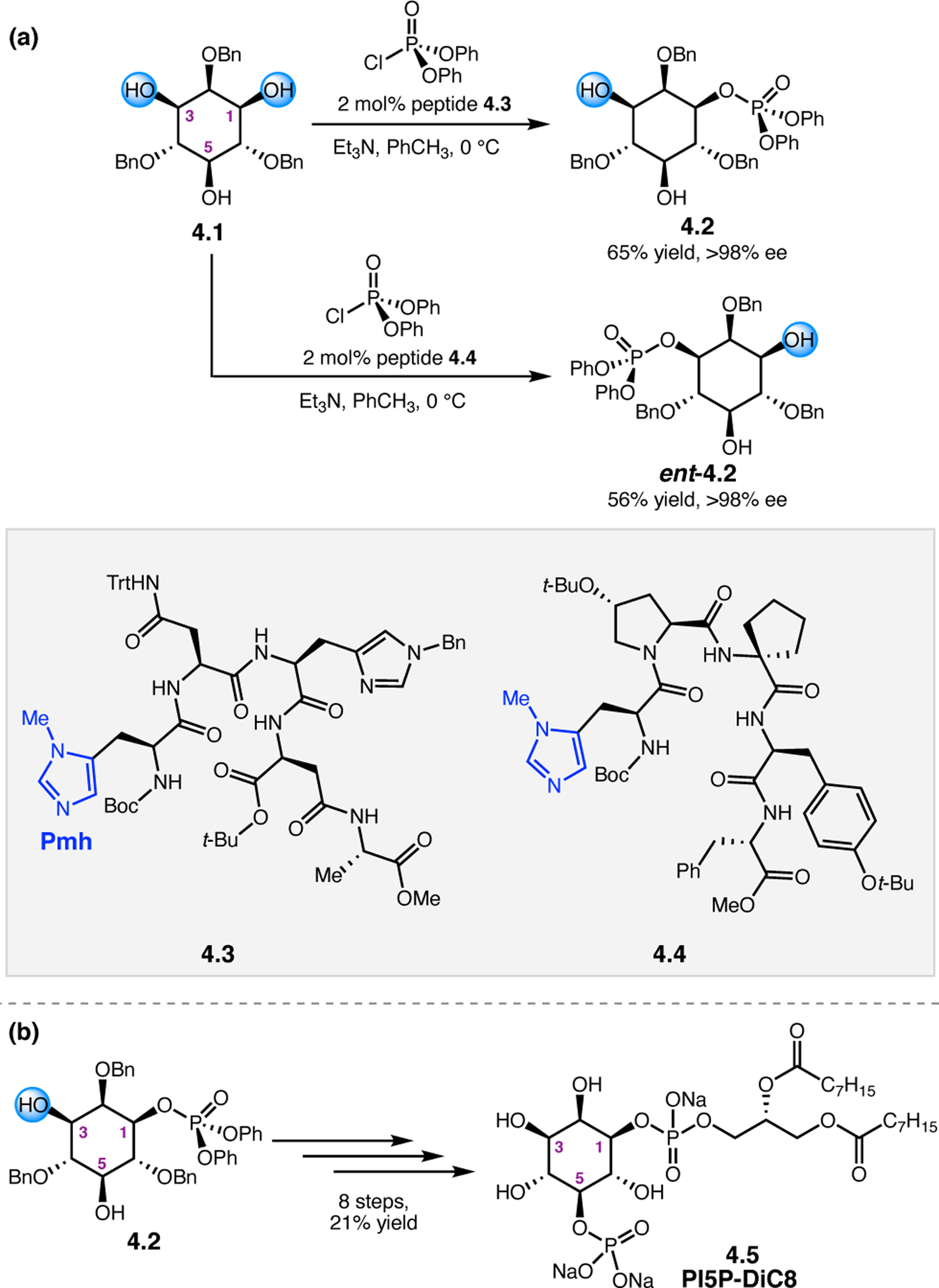
(a) Desymmetrization of myo-inositol derivative 4.1 using Pmh-containing peptides 4.3 or 4.4 to provide each enantiomer of monophosphate 4.2 with high levels of enantioselectivity.135,139 (b) This strategy was employed in the total synthesis of PI5P-DiC8 4.5.141
Among the monophosphates of inositol, phosphatidylinositol-5-phosphate (PI5P) has gained attention for its role in regulating cellular processes.140 However, investigations of PI5P had been limited, perhaps due to a lack of biological or synthetic availability. To address this challenge, our group developed a rapid and efficient total synthesis of PI5P derivative 4.5 using our previously developed strategy for the desymmetrization myo-inositol 4.1 (Figure 41a).141 We began with a large-scale synthesis of 4.2 using peptide 4.3 to catalyze the enantioselective phosphorylation. This material was then converted to PI5P derivative 4.5 in 21% yield from 4.2 over 8 synthetic steps (Figure 41b). Alternatively, peptide 4.4 could be employed to synthesize ent-4.2, which was ultimately converted into ent-4.5, the total synthesis of which had not been previously reported. Facile access to 4.5 and ent-4.5 enabled further studies of the biological roles that these inositol monophosphates play. Moreover, the rapid and efficient total synthesis of PI5P analogs highlights the utility of Pmh-embedded peptide catalysts both for asymmetric phosphate group transfer and in target-oriented synthesis.
A similar strategy was applied to the syntheses of l,l- and l,d-di-myo-inositol-1,1′-phosphate (DIP, 4.6), which has been identified as a major intracellular solute in several hyperthermophiles, acting as a stabilizer of enzyme activity under high temperature conditions (Figure 42).142 The stereochemical configuration of naturally occurring DIP has been an ongoing topic of discussion in the literature, as there have been reports implicating both chiral l,l-4.6 and meso l,d-4.6 isomers.143–145 Given our previous use of Pmh-containing peptides for the desymmetrization of myo-inositol derivative 4.1, we proposed that peptides like 4.3 and 4.4 could be used to synthesize both DIP isomers selectively. Access to both diastereomers of 4.6 could help to shed light on the question of which isomer is indeed the naturally occurring one.
Figure 42.
Two diastereomers of di-myo-inositol-1,1′-phosphate (DIP; 4.6).142
Starting from ent-4.2, which was synthesized using peptide 4.4 according to the conditions reported in Figure 41a, 4.7 can be accessed in 73% yield over 3 steps. Efforts were then directed toward identifying reaction conditions to promote the selective coupling of 4.7 with 4.1 (Figure 43). Achiral nucleophilic catalysts, such as NMI, established the degree of inherent substrate control during the formation of DIP adducts l,d-4.8 and l,l-4.8 in a 4:1 ratio (entry 1). Using peptide 4.4, a modest reversal of the intrinsic selectivity was observed, favoring l,l-4.8 in a 1:1.25 ratio (entry 2). Upon evaluating 20 other catalysts, peptides 4.9 and 4.10 were identified as highly selective for l,d-4.8 (13:1) and l,l-4.8 (1:2.5), respectively (entries 3 & 4). Although Figure 43 reports the product ratio as the relative amounts of l,d-4.8 and l,l-4.8, this reaction is actually more stereochemically complex. A diastereomeric mixture of phosphoryl chlorides—which are epimeric at the stereogenic P-center—is prepared in situ from 4.7 and then coupled with 4.1 to afford the desired products. This further highlights the challenges associated with reaction optimization and perhaps makes the results achieved by peptides 4.9 and 4.10 all the more remarkable.
Figure 43.

Examination of catalysts for the selective synthesis of both l,l- and l,d-di-myo-inositol-1,1′-phosphate derivatives 4.8.142
With each individual isomer of 4.8 in hand, these compounds were globally deprotected to form the sodium salts of l,d- and l,l-4.6 (Figure 42). The differential biological activity of each isomer was then assessed.142 Thermal stability studies indicated that the sodium salts of both l,l-4.6 and l,d-4.6 were similarly effective. In most cases, biological targets are tuned to interact with a specific isomer of a substrate. However, this scenario represents a potentially unusual example in which there is virtually no specificity for one DIP isomer over the other.
At this point, the peptide-catalyzed desymmetrizing phosphorylation of myo-inositol derivative 4.1 had been well established as a reliable method to differentiate the enantiotopic alcohols at the 1- and 3-positions.139 However, efforts to use a peptide-based strategy to discriminate between the enantiotopic 4- and 6-hydroxyls in analogous myo-inositol derivative 4.11 had remained an unmet challenge (Figure 44); over 150 Pmh-containing peptides were screened to no avail. In 2010, our group reported a related approach to address the desymmetrization of 4.11 that capitalizes on the known ability of tetrazoles to mediate phosphoramidite transfer reactions.146 Whereas Pmh-containing peptides mediate the transfer of P(V) groups through an activated phosphorylimidazolium intermediate (Figure 44a), we proposed that Atz would mediate P(III) group transfer chemistry, possibly providing access to an alternate mechanistic pathway that would enable the desymmetrization of 4.11 (Figure 44b). This mechanism would begin with protonation of the phosphoramidite reagent by Atz and subsequent displacement of the amine ligand to afford an active tetrazolylated phosphoramidite intermediate A for P(III) group transfer to 4.11. In this scenario, an amine scavenger is required to facilitate catalyst turnover.
Figure 44.
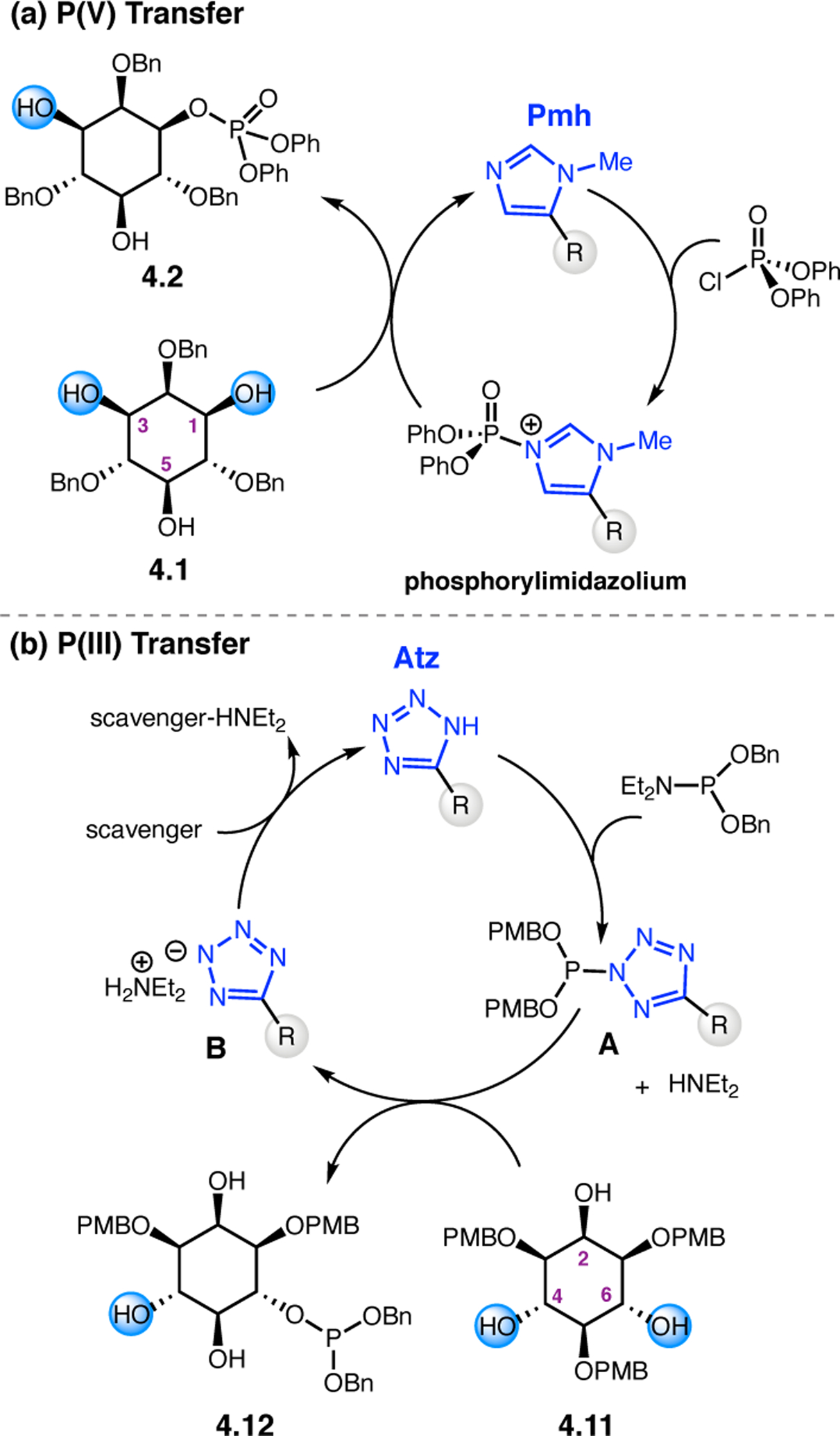
Mechanism of (a) Pmh-based catalysis of P(V) transfer vs. (b) Atz-containing peptide catalysis of P(III) transfer.146
As such, Atz was synthesized from β-cyanoalanine and tributyltin azide via a [3+2] cycloaddition and subsequently incorporated into a library of peptides using conventional coupling methods. These Atz-containing peptides were screened in the desymmetrization of 4.11 in the presence of dibenzyl-N,N-diethylphosphoramidite using 10 Å molecular sieves to scavenge diethylamine. A workup with aqueous hydrogen peroxide was used to convert initially formed phosphite 4.12 to the corresponding phosphate 4.13. Evaluation of the peptide library led to the identification of 4.15, which delivered 4.13 in 71% yield and 85:15 er. Notably, 4.15 shares several constituent amino acids with highly selective P(V)-transfer catalyst 4.4, namely the tert-butyl-protected hydroxyproline (Hyp) residue at i+1 and the C-terminal diad of benzylic residues. When (±)-4.13 was subjected to the 4.15-catalyzed phosphorylation conditions, a significant krel of 34.5 was measured en route to the bis(phosphate) byproduct, suggesting that secondary kinetic resolution contributes, at least in part, to the observed er in the desymmetrization reaction.
We were able to leverage this observation to our advantage in order to obtain highly enriched 4.13 to be used in the enantioselective total synthesis of d-myo-inositol-6-phosphate 4.14 (Figure 45). Following initial desymmetrization of 4.11, enantioenriched 4.13 (85:15 er) was re-subjected to the peptide 4.15-catalyzed phosphoramidite transfer conditions on gram-scale. Following oxidative workup, monophosphate 4.13 was isolated in 52% yield and >98:2 er, as the minor enantiomer was selectively converted into the symmetrical bis(phosphate). Monophosphate 4.13 was then globally deprotected via dissolving metal reduction to yield 4.14, a biologically relevant phosphorylated metabolite, in nearly quantitative yield.
Figure 45.
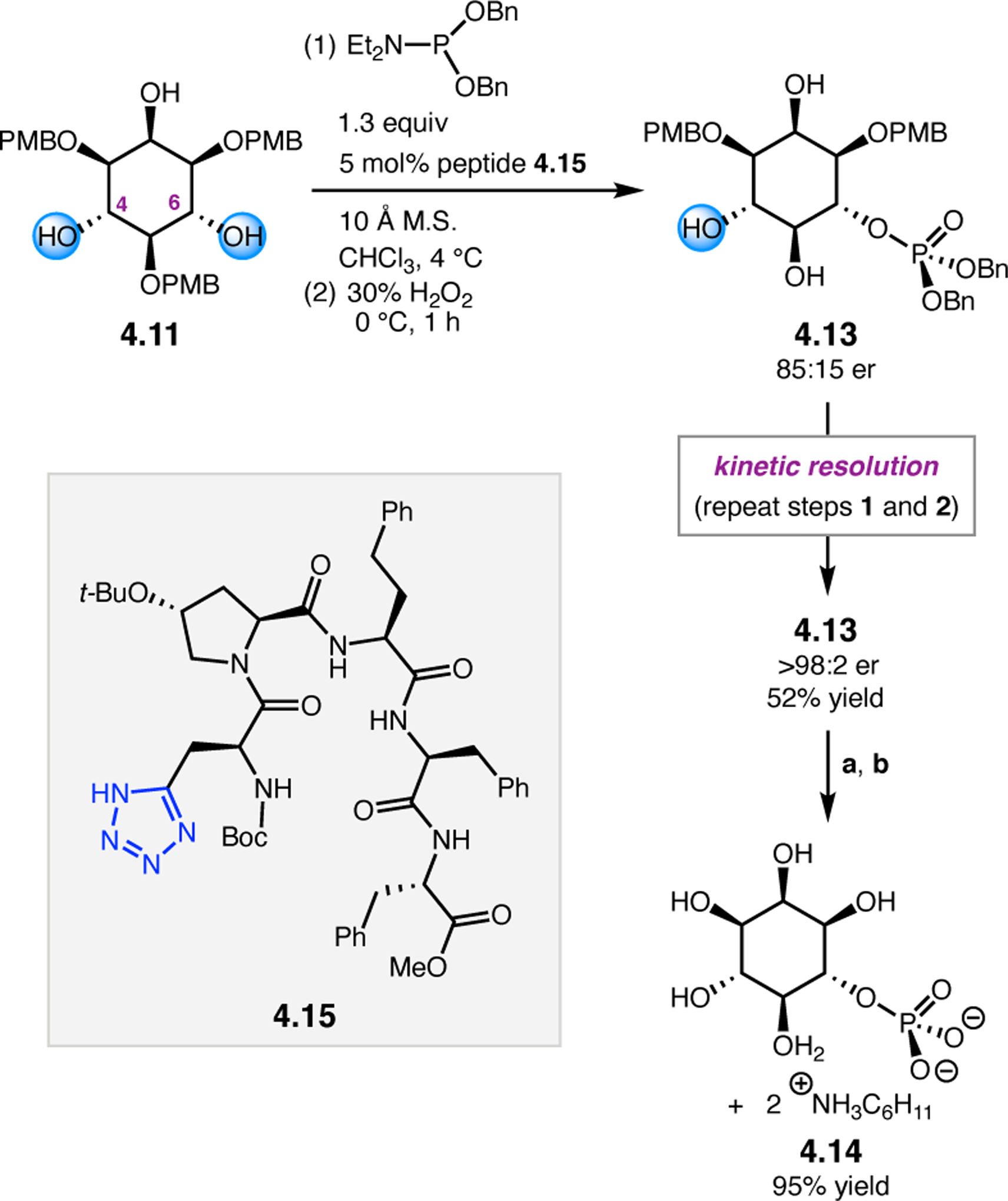
Peptide 4.15-catalyzed P(III) group transfer to myo-inositol derivative 4.11 followed by kinetic resolution of 4.13 and subsequent deprotection to afford highly enantioenriched d-myo-inositol-4-phosphate 4.14 on gram scale. Conditions: (a) NH3, Na, −78 °C; (b) DOWEX 50WX8-500, C6H11NH2.146
4.2. Sulfonylation
Although Pmh-containing peptides had been used to mediate a number of group transfer reactions with alcohols, such as phosphorylation (Section 4.1) and acylation (Section 4.4), the selective transfer of sulfur-based groups was less thoroughly explored and limited to sulfinylation.147 To address this, our group developed a method for the peptide-catalyzed, enantioselective sulfonylation of meso-1,3-diols (Figure 46).148 Reaction optimization was performed using the familiar myo-inositol derivative 4.1 as the substrate. Among the sulfonylating reagents assessed, p-nitrobenzenesulfonyl chloride (p-NsCl) was found to be optimal. Using sodium bicarbonate as the base, no racemic background reaction was detected in the absence of a catalyst, yet good conversion to mono-sulfonylated inositol 4.16 was observed under Pmh catalysis. Thus, a series of peptide-based catalysts were evaluated for their ability to desymmetrize 4.1 via Pmh-mediated sulfonylation. Pentapeptide 4.4, which was previously identified as highly enantioselective in the desymmetrizing phosphorylation of 4.1 (Figure 41),139 delivered 4.16 with a modest er of 67.5:32.5. After screening several Pmh-containing catalysts, tetrapeptide 4.18, which shares the same β-turn-inducing Hyp(t-Bu)-Cle sequence (Cle = cycloleucine) as pentapeptide 4.4, was identified as the lead catalyst, providing 4.16 in 76% yield and 97:3 er. Among the 10 substrates examined under these conditions, those based on the inositol framework were processed by 4.18 with excellent yields and enantioselectivities, while cis-cyclohexane-1,3-diol led to racemic product. Glycerols were also effective substrates, and the degree of enantioselectivity observed was dependent on the relative stereochemistry of the substituents. Taken together, these data suggest that 4.16 is better able to differentiate the enantiotopic hydroxyl groups in meso-1,3-diol substrates with certain conformational preferences.
Figure 46.
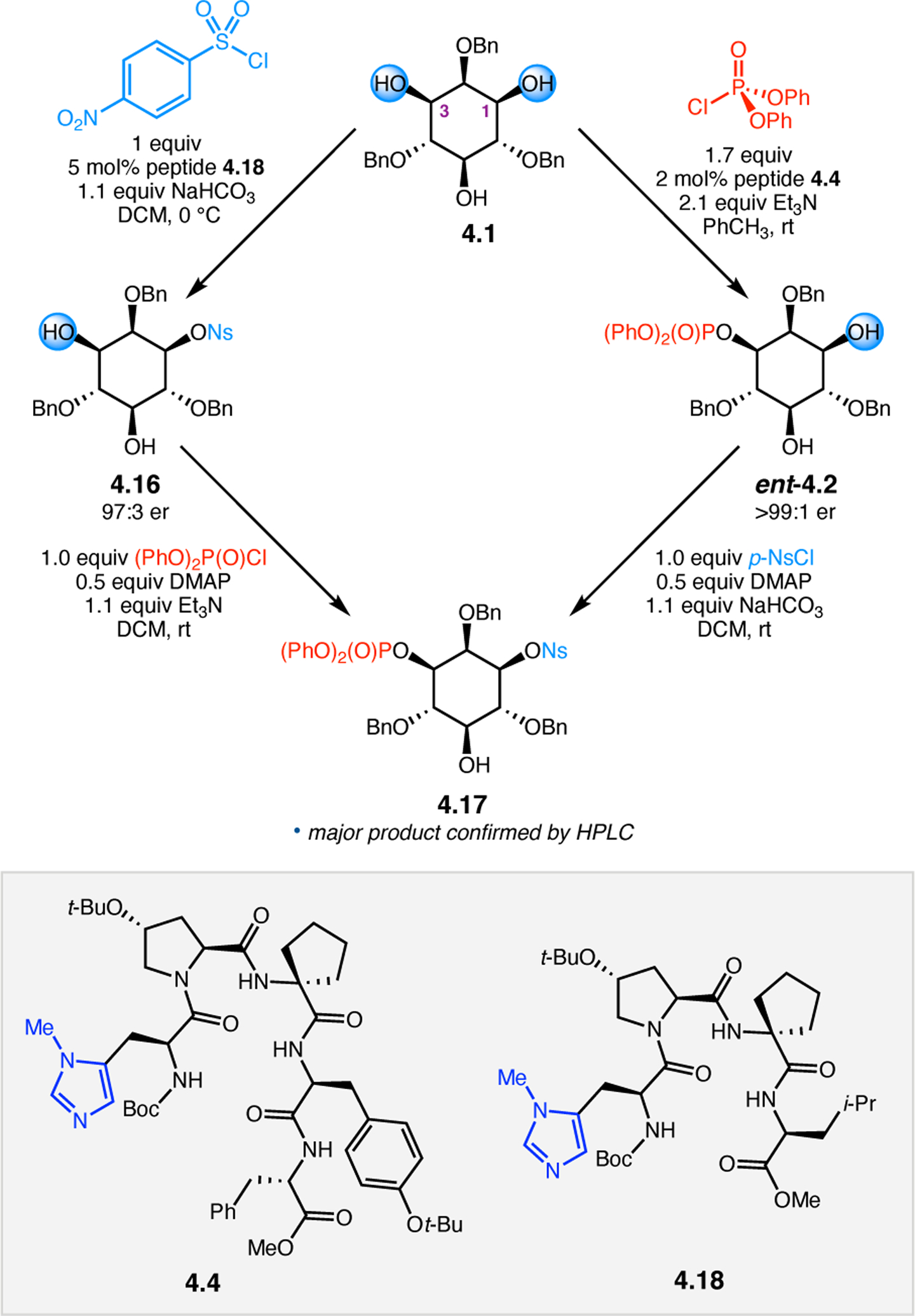
Comparison between Pmh-peptide catalyzed sulfonylation and phosphorylation.148
Although sulfonylation catalyst 4.18 and phosphorylation catalyst 4.4 share similar sequences, these peptides interact with myo-inositol 4.1 in unexpectedly different ways (Figure 46). When 4.1 was subjected to the phosphorylation conditions with peptide 4.4, phosphoryl group transfer occurred selectively at the 3-hydroxyl, affording ent-4.2 in >99:1 er. Conversely, under the 4.18-catalyzed sulfonylation conditions, the hydroxyl group at the 1-position was selectively sulfonylated to provide 4.16 in 97:3 er. However, when ent-4.2 and 4.16 were subsequently subjected to the sulfonylation and phosphorylation conditions, respectively, both species converge to the same enantiomer of enantiomer of mono-phosphorylated, mono-sulfonylated 4.17 as confirmed by HPLC retention times. These data show that 4.4 and 4.18 indeed target enantiotopic hydroxyl groups of 4.1 despite their sequence homology, suggesting that these two peptides operate via disparate catalytic activation modes. While it is tempting to conclude that this stems from differences in secondary structure, it is also possible that mechanistic changes as a function of the electrophile, stoichiometric base, or solvent give rise to this unexpected enantiodivergence. As is so often the case with peptide-based catalysts, seemingly minor differences in sequence and reaction conditions can lead to large perturbations in reactivity and selectivity.
4.3. Carbamoylation
In addition to group transfer reactions of alcohols, Pmh-containing peptide catalysts have also been used to mediate the enantioselective carbamoylation of formamides and thioformamides 4.19 as an entry into chiral secondary amine derivatives 4.20 (Figure 47).149 Chiral amine building blocks are highly valuable in synthetic chemistry. However, the ability to use group transfer kinetic resolutions to access chiral amines has presented a longstanding challenge to the field. Prior to this work, non-enzymatic approaches using small molecule catalysts under mild conditions were fairly limited.150–154 Since then, however, several methods have been developed for the synthesis of optically enriched amines employing small molecule catalysts.155 Combining the benefits of enzymatic and small molecule catalysis, our group sought to employ Pmh-containing peptides to catalyze the kinetic resolution of (thio)formamides 4.19 via selective Boc group transfer from di-tert-butyldicarbonate (Boc2O). The Pmh-containing peptide was anticipated to initially transfer a Boc group to the (thio)carbonyl group of 4.19. This kinetically more favorable O- or S-acylated intermediate I would subsequently rearrange to deliver the more thermodynamically stable imide 4.20. Enantioenriched 4.20 could then be selectively deprotected to deliver chiral amine building blocks.
Figure 47.
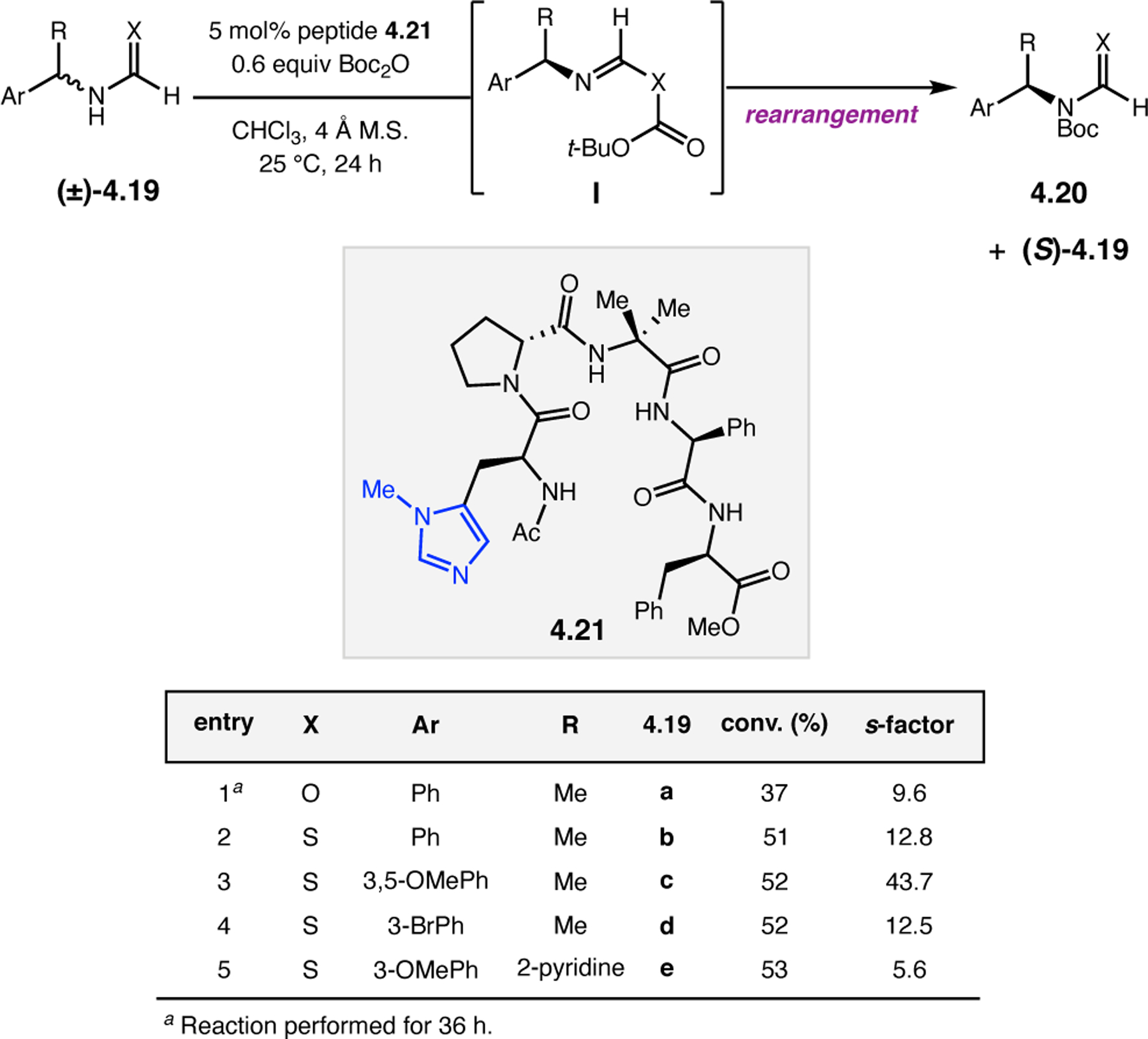
Peptide 4.21-catalyzed kinetic resolution of (thio)formamides (±)-4.19.149
Catalyst optimization led to the discovery of β-hairpin-biased peptide 4.21, which yielded formamide 4.19a with an s-factor of 9.6 at 37% conversion (Figure 47, entry 1). We hypothesized that thioformamides would be more reactive both toward initial S-acylation and subsequent S-to-N acyl shift. Indeed, thioformamide 4.19b substantially was processed by 4.21 with an s-factor of 12.8 at 51% conversion (entry 2). When examining the reaction scope of thioformamides, electron rich aryl substituents promoted higher s-factors than electron poor ones, as exemplified by 4.19c and 4.19d, respectively (entries 3 & 4). The largest s-factor was obtained with the highly electron rich thioformamide 4.19c, with an s-factor of 43.7. N-Heterocycle-containing 4.19e was also tolerated under the reaction conditions, albeit with a reduced s-factor of 5.6 (entry 5). In this case, it is possible that the pyridine moiety of 4.19e catalyzes a competing Boc group transfer process that erodes the selectivity, though different interactions with peptide 4.21 are also possible.
We further demonstrated the utility of these enantioenriched thioformamide products for the synthesis of chiral amine building blocks (Figure 48). Cleavage of the Boc group from 4.20b using trifluoroacetic acid, followed by S-to-O exchange led to the formation of formamide 4.20a in 82% yield. Additionally, Boc-protected 4.22 could be accessed in 81% yield via oxidative hydrolysis of the thioformamide group. These results further highlight the ability of Pmh-containing peptides to deliver synthetically useful products that are difficult to access by other means.
Figure 48.
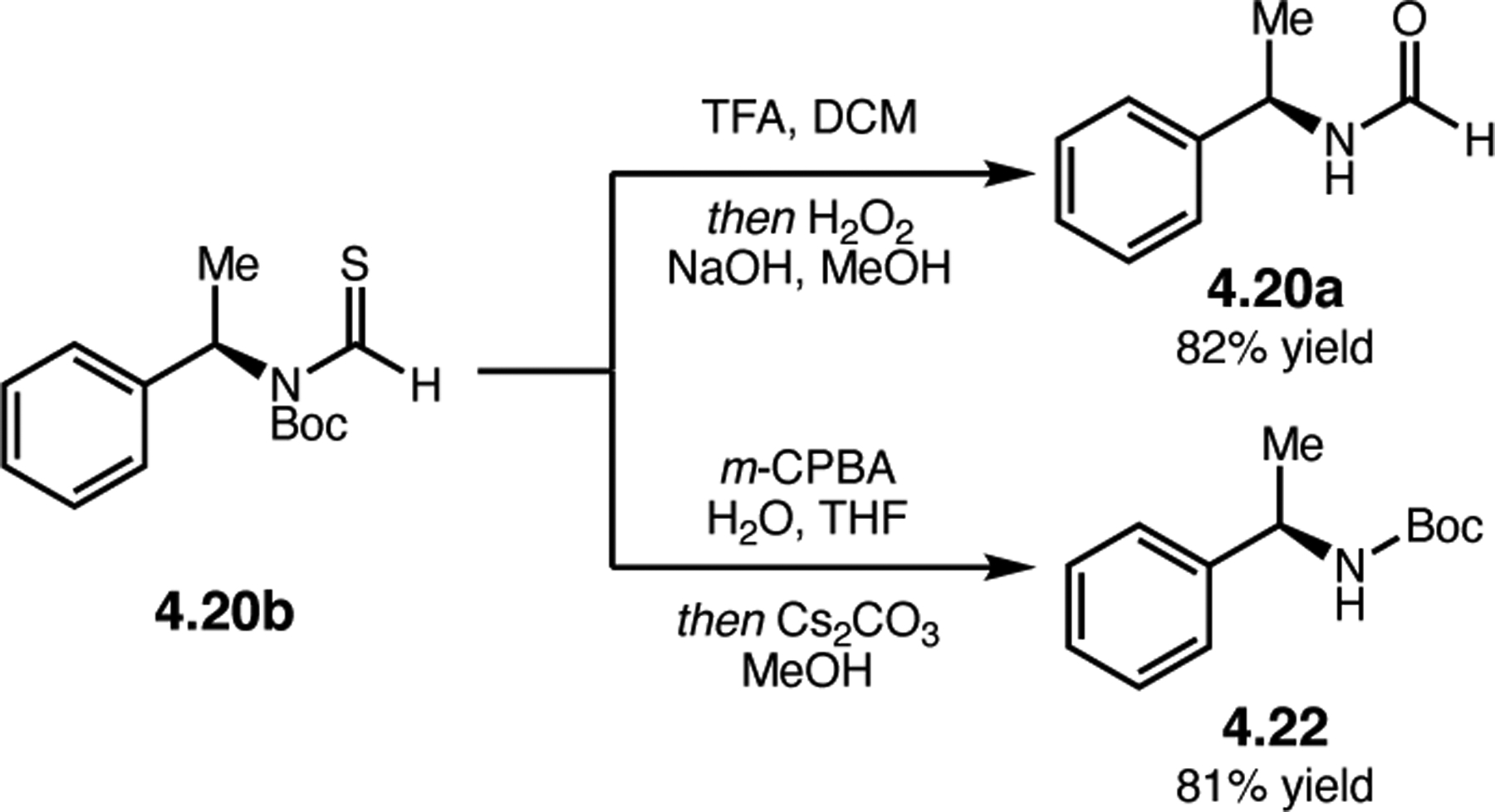
Derivatization of thioformamide 4.20b.149
4.4. Acylation
Since the initial reports on the use of Pmh-containing peptides for the enantioselective acylation of amino alcohols,6,132 the scope of acylation reactions amenable to Pmh-embedded peptide catalysis has grown significantly. While the earliest studies of peptide-catalyzed asymmetric acylation reactions were discussed in our previous review,4 this Section reports on the more recent advances in this area that amount to a significant expansion of scope and mechanistic understanding.
In collaboration with Merck in 2006, we reported the first example of a peptide-catalyzed remote desymmetrization reaction.156,157 Prior to this report, remote asymmetric induction was often thought to require enzymes in order to differentiate enantiotopic groups separated by great distances.158 However, Pmh-containing peptide 4.25 was able to desymmetrize diarylmethane bis(phenol) 4.23a to mono-acylated 4.24a in 80% yield and 97.5:2.5 er (Figure 49). In the context of this pharmaceutically relevant scaffold,159 peptide 4.25 was able to discriminate between enantiotopic hydroxyl groups that are separated from one another by nearly a full nanometer (9.9 Å) and from the pro-stereogenic center by 5.8 Å.
Figure 49.
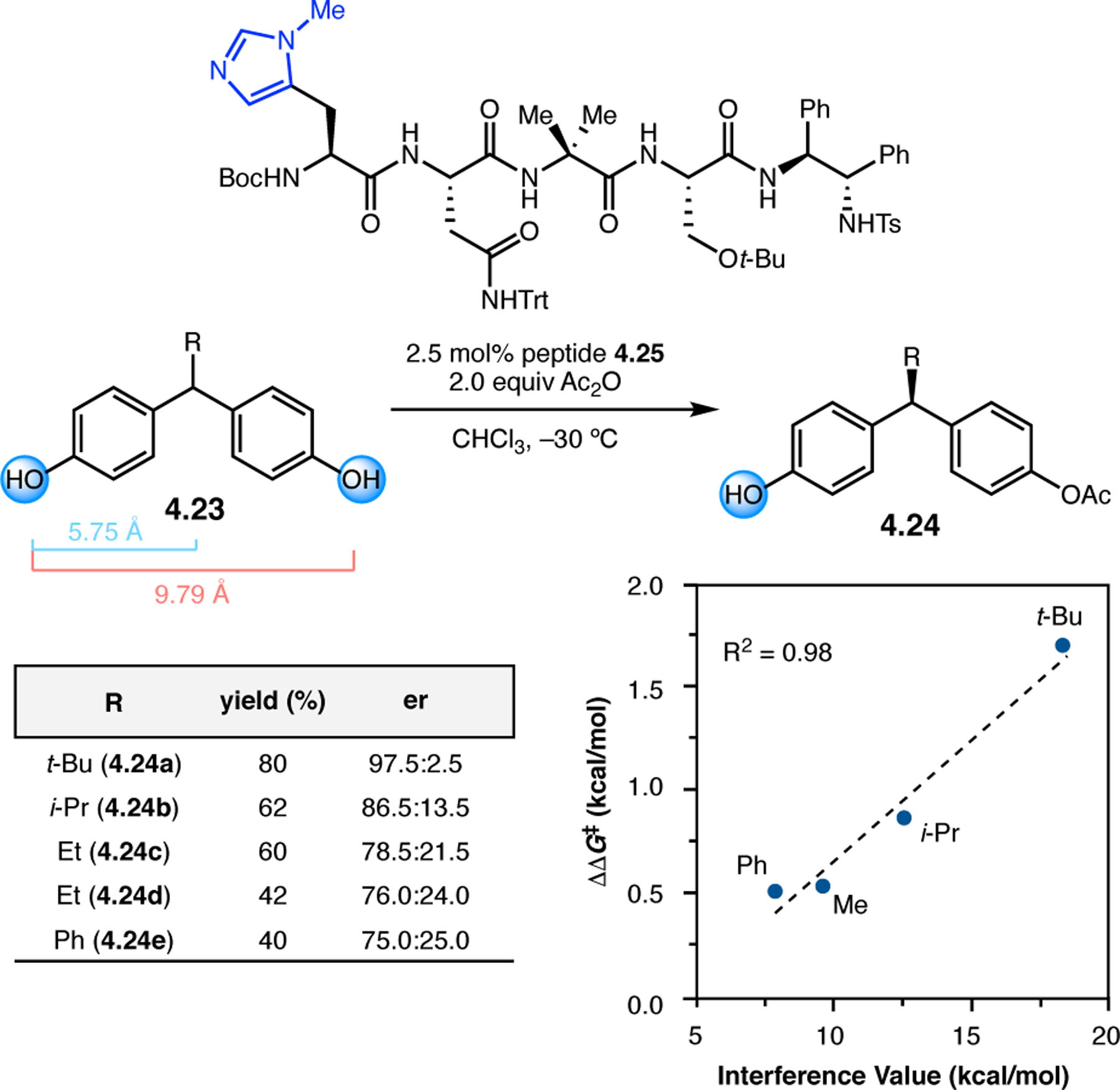
Peptide-catalyzed remote desymmetrization of bis(phenol) 4.23, noting the correlation between the observed enantioselectivity and the steric demand of the substituent R.160
While examining the scope of this remote desymmetrization reaction, a trend between the steric demand of the prochiral substituent of 4.23 and the enantioselectivity was observed. In collaboration with the Sigman Group, we employed physical organic tools to investigate the factors that govern selectivity in this peptide-catalyzed reaction.160 Linear free-energy relationships were used to correlate the observed enantioselectivity (ΔΔG‡) with various steric parameters, including Sternhell interference energies (Figure 49).161 A steep linear dependence on the steric demand of the prochiral substituent was identified, with bulky substituents, such as tert-butyl, delivering the best enantioselectivities. It was reasoned that bulky substituents could give rise to conformational preferences, such as differences in the propeller-like twisting of the diarylmethane scaffold, that might enable the discrimination of the enantiotopic arenes. In a follow-up study, the Sigman Group was able to further elaborate on the source of selectivity in this reaction using multidimensional parameterization techniques.162,163 In this study, the authors were able to identify correlations between the measured enantioselectivity and DFT-computed vibrational frequencies of the arene rings of 4.23. These new data suggested that peptide 4.25 is sensitive to both steric and electronic substituent effects in diarylmethane 4.23.
In addition to the early reports from our group, highly significant contributions in the arena of Pmh-containing peptide-mediated acylation reactions have been made by the Schreiner Group in recent years.164–166 Unique to the Pmh-containing peptides developed by Schreiner and co-workers is the presence of the non-proteinogenic amino acid γ-aminoadamantanecarboxylic acid (AGly, Figure 50). This residue can be synthesized in one step from the corresponding carboxylic acid via direct C–H bond amidation using acetonitrile as the nitrogen nucleophile.167 Incorporation of this rigid, hydrophobic residue was designed to diminish catalyst self-association (e.g., dimerization or folding), enable low catalyst loadings, and facilitate solubility in nonpolar organic solvents.
Figure 50.
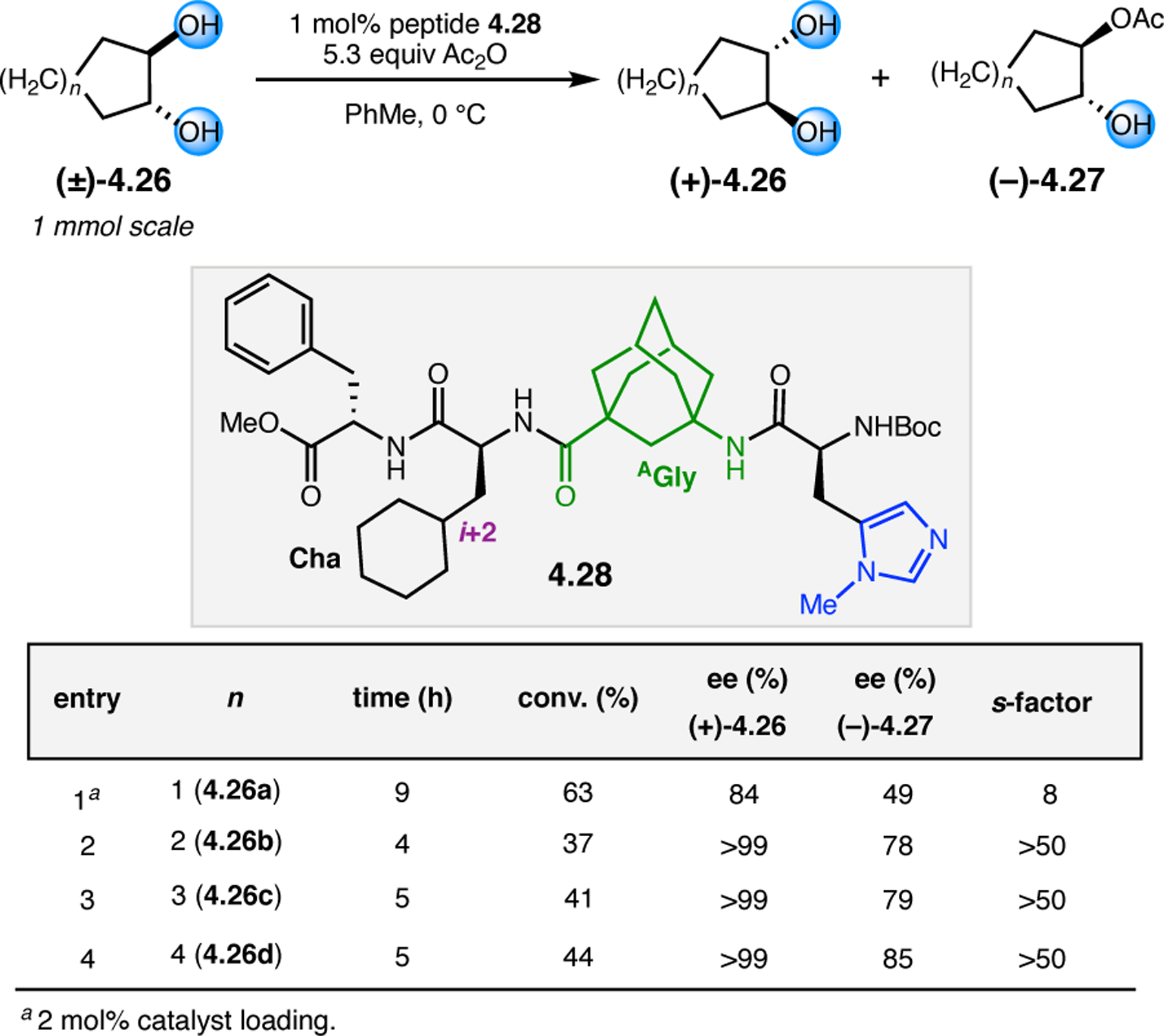
Kinetic resolution of trans-cycloalkane-1,2-diols 4.26 with tetrapeptide 4.28.164
Using this class of peptide catalysts, Schreiner and co-workers first pursued the kinetic resolution of simple trans-cycloalkane-1,2-diols 4.26 via acylation (Figure 50), a reaction which had posed significant challenges for methodology development at the time, even with enzymatic catalysts.164,166 After evaluating several lipophilic amino acids at the i+2 position, ultimately 4.28, bearing cyclohexylalanine (Cha), was selected as the lead catalyst. In toluene as solvent with a mere 1 mol% catalyst loading, 4.28 was able to achieve excellent selectivities in the acylation of trans-1,2-diols 4.26 with ring sizes ranging from 5–8 members (entries 1–4). While six- (4.26b), seven- (4.26c), and eight-membered rings (4.26d) were highly selective in this transformation with s-factors > 50 (entries 2–4), trans-cyclopentane-1,2-diol 4.26a was the exception, which was only modestly selective with an s-factors of 8 (entry 1). Low selectivity with this substrate is likely due to the need for minimal amounts of dichloromethane co-solvent to overcome the poor solubility of 4.26a in toluene, and the selectivity was found to be quite sensitive to the reaction medium. Though linear diols were also examined, peptide 4.28 achieved significantly higher levels of selectivity with cyclic diols.166
The authors turned to computations in order to shed light on the origins of enantioselectivity in the kinetic resolution of 4.26 with peptide 4.28.164 Molecular dynamics simulations (MMFF) were used to search for low energy conformations of the active N-acylimidazolium intermediate of 4.28. Regardless of the starting geometry, all of the lowest-energy conformers placed the i+2 cyclohexylalanine (Cha) residue in close proximity to the acylimidazolium moiety (Figure 51). The authors used the low-energy conformations of acylated 4.28 to propose a transition state model, which evokes an H-bond between the carbonyl of the i+2 residue with the nonreacting diol and hydrophobic interactions between Cha and the cycloalkane of 4.26 to induce high selectivity in favor of the experimentally observed (−)-enantiomer of 4.27. This model was further supported by NMR studies performed by Thiele and co-workers.168 Examination of peptide 4.28 alone using NOE- and residual dipolar coupling (RDC)-based methods revealed an ensemble of four major conformers, which resemble the “pocket-like” structure shown in Figure 51. Upon addition of (−)-4.27b, intermolecular NOE contacts between the substrate and catalyst could be detected using advanced NMR methods, including a pure shift EASY ROESY experiment developed by the authors. Notably, NOE contacts between the cyclohexyl groups of the substrate and peptide were observed (blue arrow, Figure 51), which further supports the importance of hydrophobic interactions for achieving high enantioselectivity in this reaction.
Figure 51.
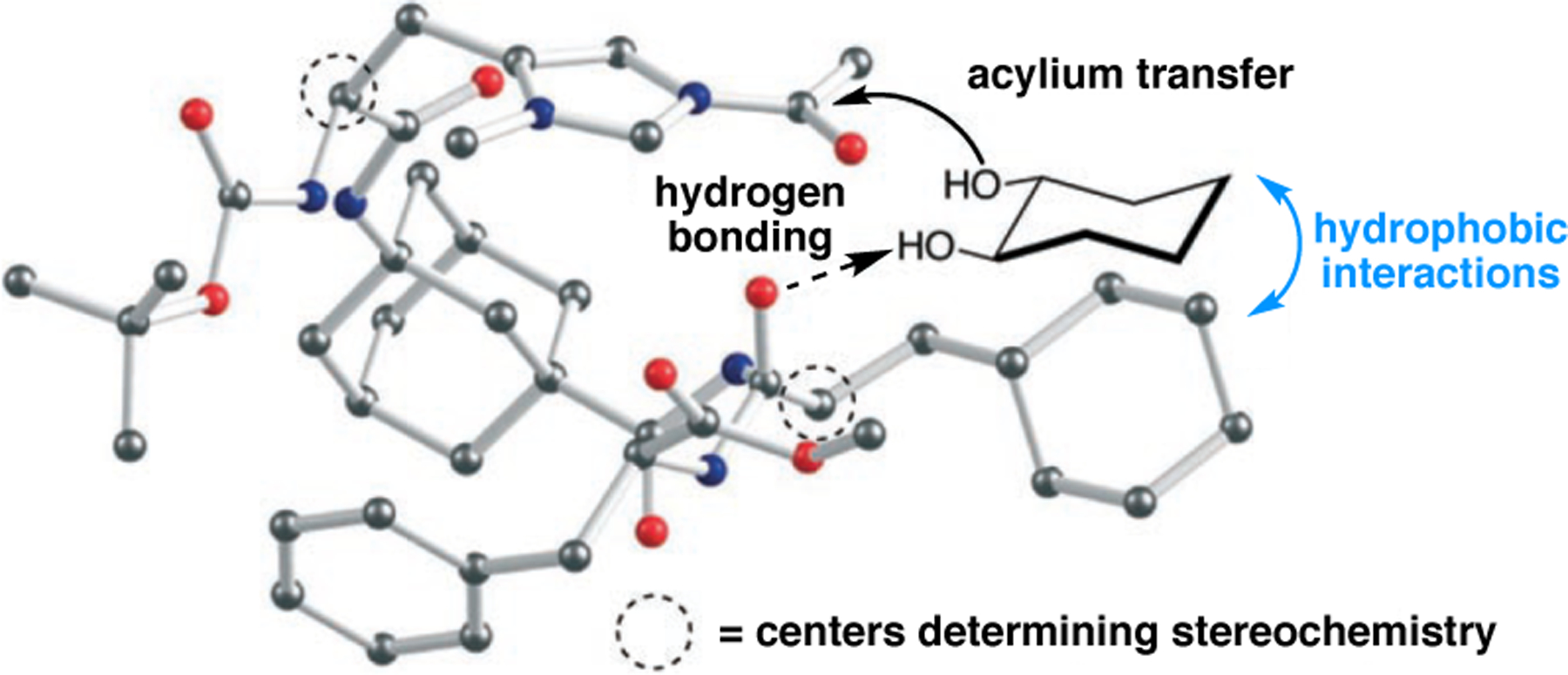
Model for the enantioselective acylation of trans-cycloalkane-1,2-diols 4.26 in the chiral “pocket” of acylated peptide 4.28. Hydrogens are omitted for clarity. Adapted with permission from ref. 164. Copyright 2011 Wiley.
In a subsequent computational study by Sunoj and co-workers, the mechanism of the peptide 4.28-catalyzed acylation of trans- (4.26b) and cis-cyclohexane-1,2-diols (4.29a) was examined in more depth.169 Although cis-diols were not reported in Schreiner’s original paper,164 the authors studied this substrate class computationally. For geometry optimizations, the most crucial parts of the reacting system—the diol and acylated Pmh residue—were described using density functional theory (B3LYP/6-31G*), while the peptide backbone was treated as a lower layer using the semiempirical PM3 method. When considering the mechanism of these acylation reactions, a complex between acylated 4.28 and the diol substrate is first formed, and then stereoselective acyl group transfer occurs within this chiral environment. Thus, the inclusion complex between different stereoisomers of the diols (either 4.26b or 4.29a) and acylated-peptide (4.28) was first examined (Figure 52). The lowest-energy complex for trans-diol 4.26b leading to a (1R, 2R)-configuration (Complex I)—the observed enantiomer of the product—shows a stabilizing H-bonding interaction between the carbonyl of the i+2 Cha residue and the nonreacting hydroxyl group of 4.26b. This type of interaction was in agreement with the model proposed by Schreiner and co-workers (Figure 51).164 While Complex I, leading to (1R, 2R)-4.27b, was most stable for trans-4.26b, Complex II with a (1R,2S)-orientation was the most preferred for cis-4.29a. This reinforces the notion of a proclivity toward selective chiral recognition of each diol by the peptide, which produces the desired chiral environment for enantioselective acyl group transfer.
Figure 52.
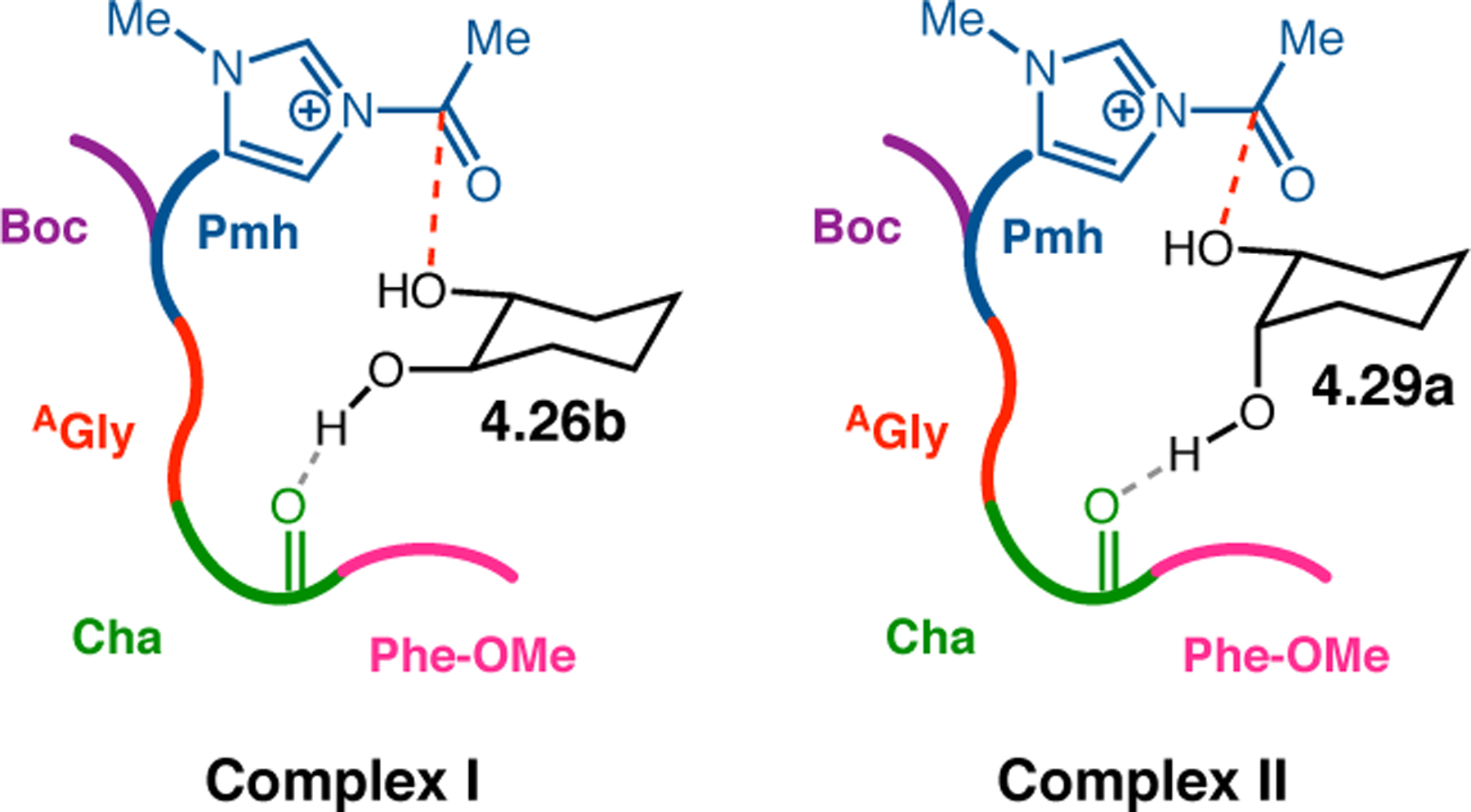
Lowest-energy computed complexes between acylated-peptide 4.28 and trans-(1R,2R)-4.26b (Complex I, left) and cis-(1R,2S)-4.29a (Complex II, right). Computations were performed at the B3LYP/6-31G*//ONIOM2(B3LYP/6-31*:PM3) level of theory.169
Next, the transition states (TSs) for the acyl group transfer step were identified.169 The two lowest-energy transition states of each enantiomer of trans-diol 4.26b with acylated peptide 4.28 are shown in Figure 53. As anticipated, TS 4.1 with the (1R,2R)-enantiomer of 4.26b is the lowest conformer, which leads to the experimentally observed product 4.27b.164 Comparing the geometrical features of TS 4.1 and TS 4.2 further emphasizes the crucial role of the stabilizing H-bond between the nonreacting hydroxyl of 4.26b and the i+2 carbonyl of peptide 4.28. The distances between the acyl group and the Pmh nitrogen and diol oxygen also suggest a product-like TS for this acyl transfer reaction. Across the various TSs for this step, the conformation of the peptide, particularly the orientation of cyclohexyl group of the Cha residue, exhibited significant deviations (e.g., TS 4.1 vs. TS 4.2, Figure 53). The effects of the peptide conformation and noncovalent interactions between the acylated-peptide and diol culminate in a significant energy separation between the diastereomeric acyl transfer TSs to render this acylation selective for the trans-(1R,2R)-4.26b. Although the stereochemical outcome was correctly predicted, the calculated enantiomeric excess of 4.27b (>99% ee) was significantly higher than the experimental outcome (Figure 50, entry 2).
Figure 53.
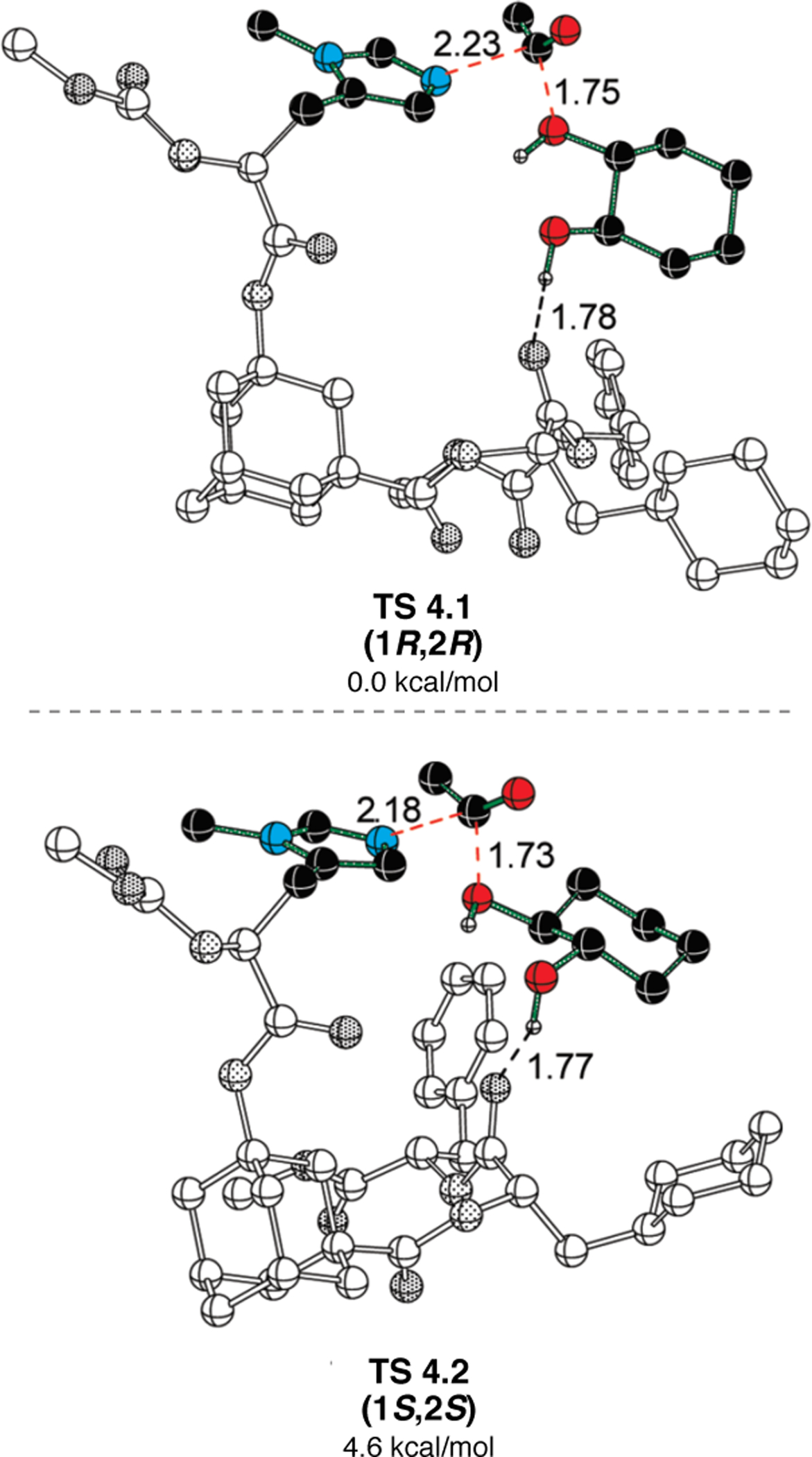
Two lowest-energy transition states for acyl transfer from 4.28 to trans-diol 4.26b leading to each enantiomer of 4.27b. Only select hydrogens are shown for clarity. Lighter gray and darker colored shadings, respectively, represent the lower (PM3) and higher (B3LYP) layers in the ONIOM2 partitioning. Adapted with permission from ref. 169. Copyright 2009 American Chemical Society.
For the desymmetrizing acylation of cis-diol 4.29a, this computational method predicted that the (1R,2S)-enantiomer would give the lowest-energy TS, with a ΔΔG‡ of 4.3 kcal/mol between the diastereomeric TSs.169 Such a large energy difference indicates that peptide 4.28 should also be able to acylate cis-1,2-diols 4.29, affording (1R,2S)-4.30. Overall, this in-depth computational study of the kinetic resolution of trans- and cis-1,2-diols further highlights the bifunctional nature of Pmh-based catalysts, which not only serve as nucleophilic catalysts, but also govern selectivity through peptide secondary structure, noncovalent interactions, and chiral recognition.
Fulfilling the computational predictions by Sunoj and co-workers,169 the desymmetrization of meso, cis-1,2-diols (4.29) using the same peptide (4.28) was later developed by Schreiner and co-workers.170 The reaction conditions used for the kinetic resolution of trans-diols 4.26 (Figure 50) translated well to cis-diols 4.29, with only minor optimization required. Although exogenous base is not required in this reaction, the inclusion of 5.3 equivalents of N,N-diisopropylethylamine (DIPEA) lead to significantly faster reaction rates and increased selectivity; the molar equivalency of acetic anhydride was adjusted to match that of the base. Under these conditions, peptide 4.28 successfully desymmetrized meso-vicinal diols 4.29 (Figure 54), achieving up to 94:6 er with 4.29a at −40 °C (entry 1). Further cooling the reaction mixture to −55 °C increased the enantioselectivity of 4.30a to 95:5 er but decreased the yield to 61%. Both cyclic and linear meso diols provided >90:10 er, with the exception of meso-hydrobenzoin 4.29e (entry 5) and heteroatom-containing diol 4.29f (entry 6).
Figure 54.
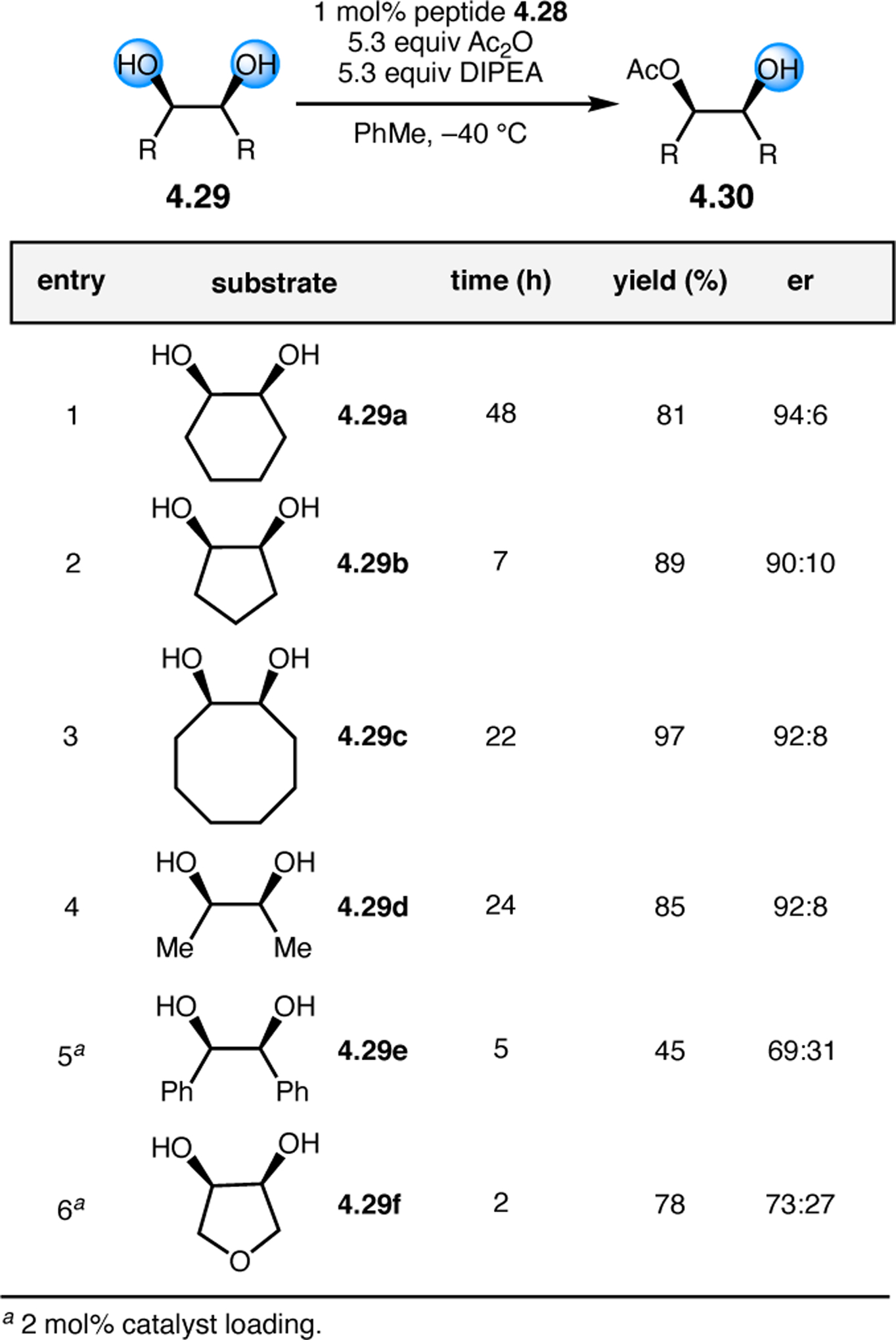
Desymmetrization of meso, cis-Diols 4.29 using peptide 4.28.170
The ability to achieve high enantioselectivity with meso-diols 4.29 is challenging, due in part to the intramolecular acyl group migration that can occur in 4.30, which effectively racemizes the product. Indeed, the authors noted that partial racemization of 4.30 was detected upon reaction workup. To combat this problem, a subsequent oxidation of the remaining alcohol was developed to convert 4.30 into α-hydroxyketones 4.31 (Figure 55).170 After exploring various oxidants, TEMPO and m-CPBA were selected as co-oxidants for in situ oxidation of stereochemically labile 4.30a to afford 4.31a in 70% yield and 94:6 er. This protocol was applied to several other meso-1,2-diols 4.29 without significant perturbation to the enantioselectivity after oxidation.
Figure 55.
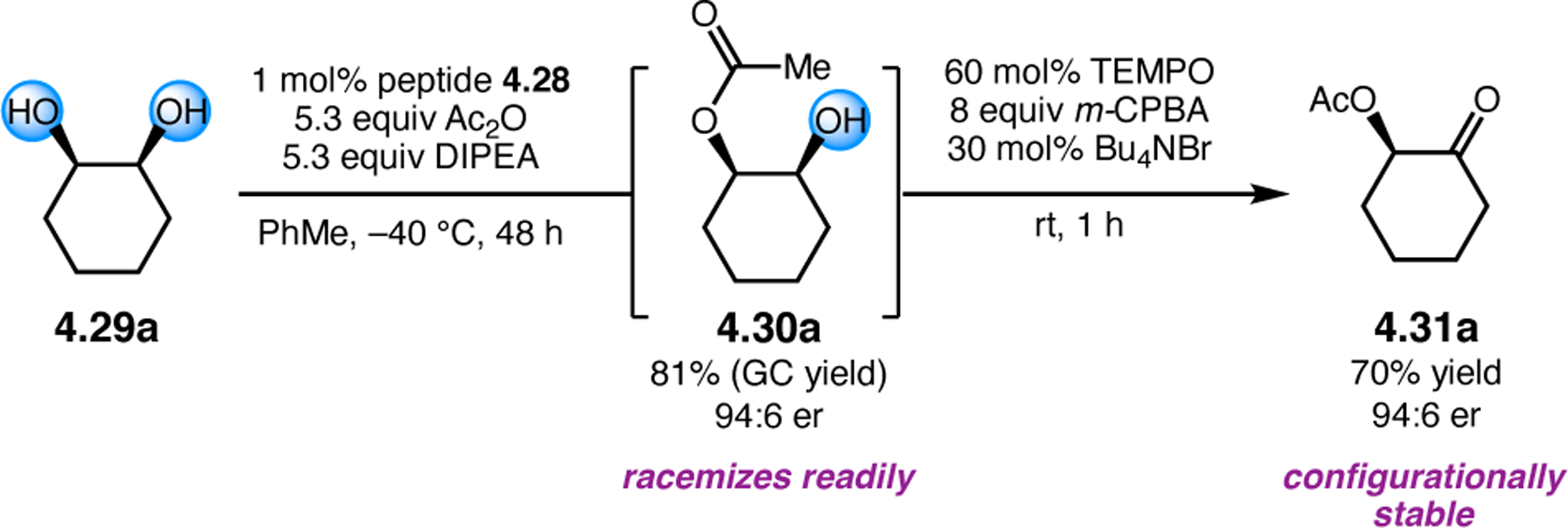
Peptide 4.28-catalyzed acyl transfer followed by in situ oxidation to produce configurationally stable 4.31a.170
This one-pot enantioselective acylation/oxidation protocol was later advanced to use a single multicatalyst to perform both the enantioselective acylation and subsequent alcohol oxidation (Figure 56a).171 This represents the first report of an organocatalytic, enantioselective tandem reaction mediated by a single catalyst containing orthogonal catalytic moieties. Several multicatalysts were synthesized and evaluated, varying the location of the TEMPO moiety relative to the Pmh residue. The best results were obtained using peptide 4.32, whose structure and sequence most closely resemble 4.28, which was previously optimized for acyl transfer.164,170 An overlay of the lowest-energy conformations of the two acylated catalysts 4.28 and 4.32 obtained using molecular force-field analysis highlights their structural similarities (Figure 56b).
Figure 56.
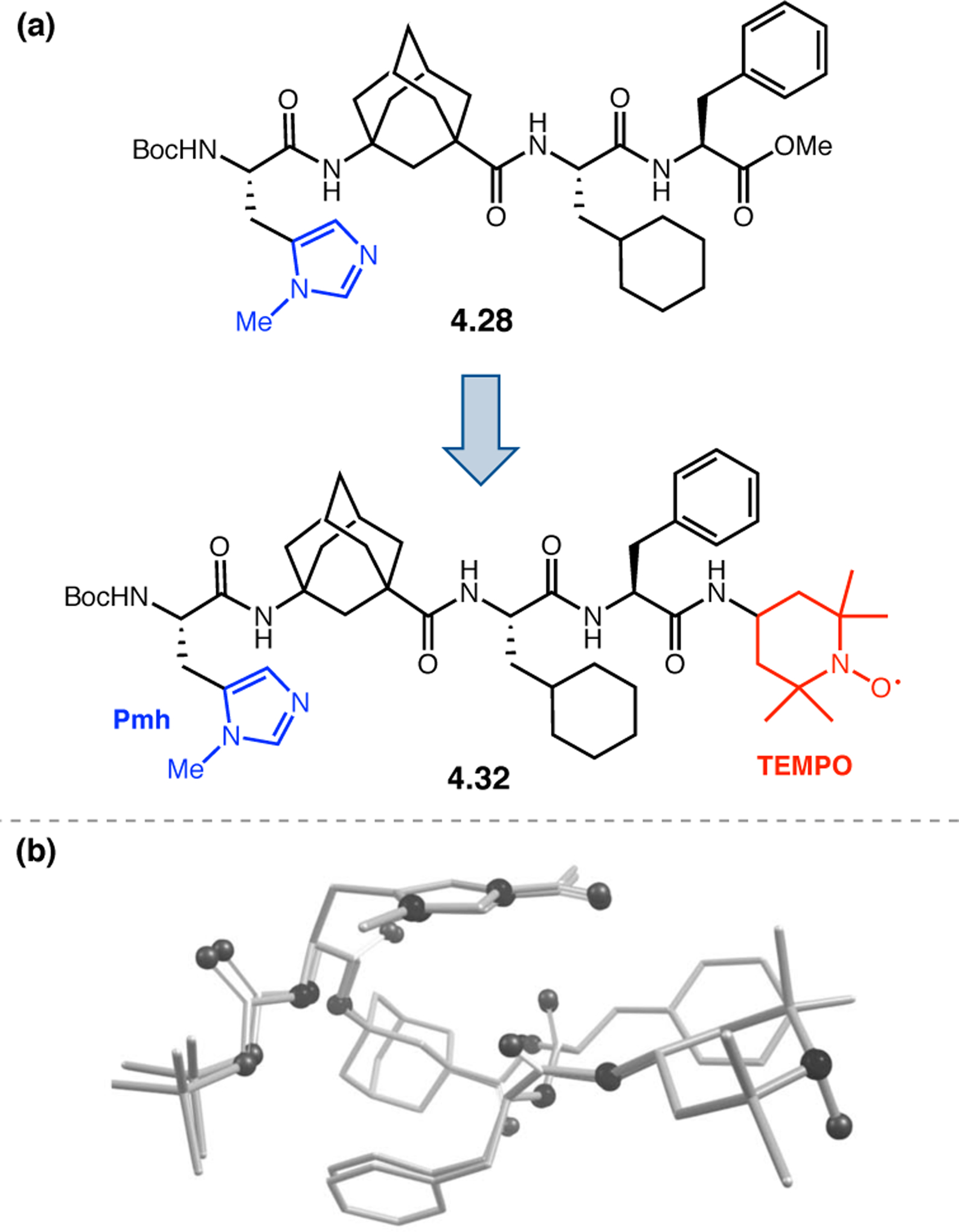
(a) Peptidic multicatalyst 4.32 designed from the sequence of lead catalyst 4.28 for asymmetric acyl-transfer. (b) An overlay (molecular force-field analysis) of both acylated-peptides is shown below. Adapted with permission from ref. 171. Copyright 2011 Wiley.
A comparison between both approaches for the acylation/oxidation of meso-diols, which proceed either by concurrent tandem desymmetrization/oxidation accomplished with multicatalyst 4.32 (blue) or peptide 4.28-catalyzed desymmetrization followed by one-pot TEMPO oxidation (red; Figure 57).170,171 With most substrates, yields and selectivities were slightly reduced using multicatalyst 4.32. However, using 4.29b, higher enantioselectivity was achieved (87:13 er) with peptide 4.32 rather than with the two-step approach using 4.28 followed by TEMPO (82:18 er, entry 2). The incorporation of the TEMPO moiety within the peptide sequence increased the oxidation activity as compared to TEMPO alone. This is reflected in the milder oxidation conditions employed when using multicatalyst 4.32. In this way, the multicatalyst approach offers some benefit over the two-step route to access 4.31. This proof-of-principle study demonstrates the first organocatalytic multicatalyst reaction using short oligopeptides with two orthogonal catalytic moieties. The Schreiner Group has since developed several other peptidic multicatalyst systems that are discussed below.
Figure 57.
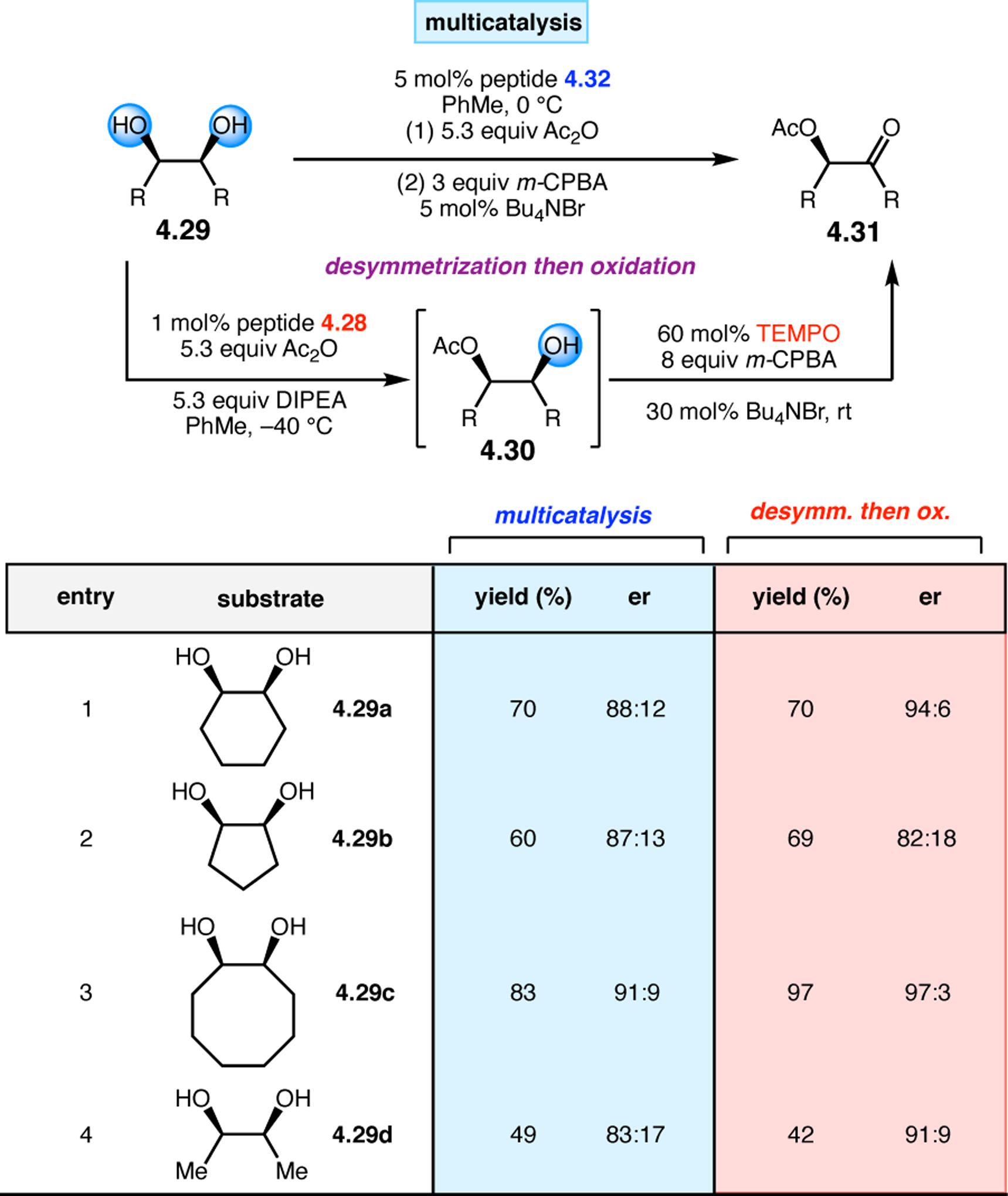
Concurrent tandem desymmetrization/oxidation of meso-diols 4.29 using multicatalyst 4.32171 as compared to a two-step approach with peptide 4.28.170
In addition to performing acetylation of trans-1,2-diols 4.26 using acetic anhydride, the Schreiner Group developed a Steglich-type protocol, which relies on the use of a carboxylic acids rather than anhydrides (Figure 58).172 Using carboxylic acids is often advantageous, as some anhydrides may be unstable or not readily available. In the presence of DIC, the carboxylic acid can be converted into an O-acyl isourea intermediate, which can then further react to form a symmetrical anhydride. A Lewis basic catalyst can then deliver this acyl group to kinetically resolve trans-1,2-diols such as 4.26b. By using a Pmh-embedded peptide catalyst, this report represents the first enantioselective Steglich-type esterification of alcohols.
Figure 58.
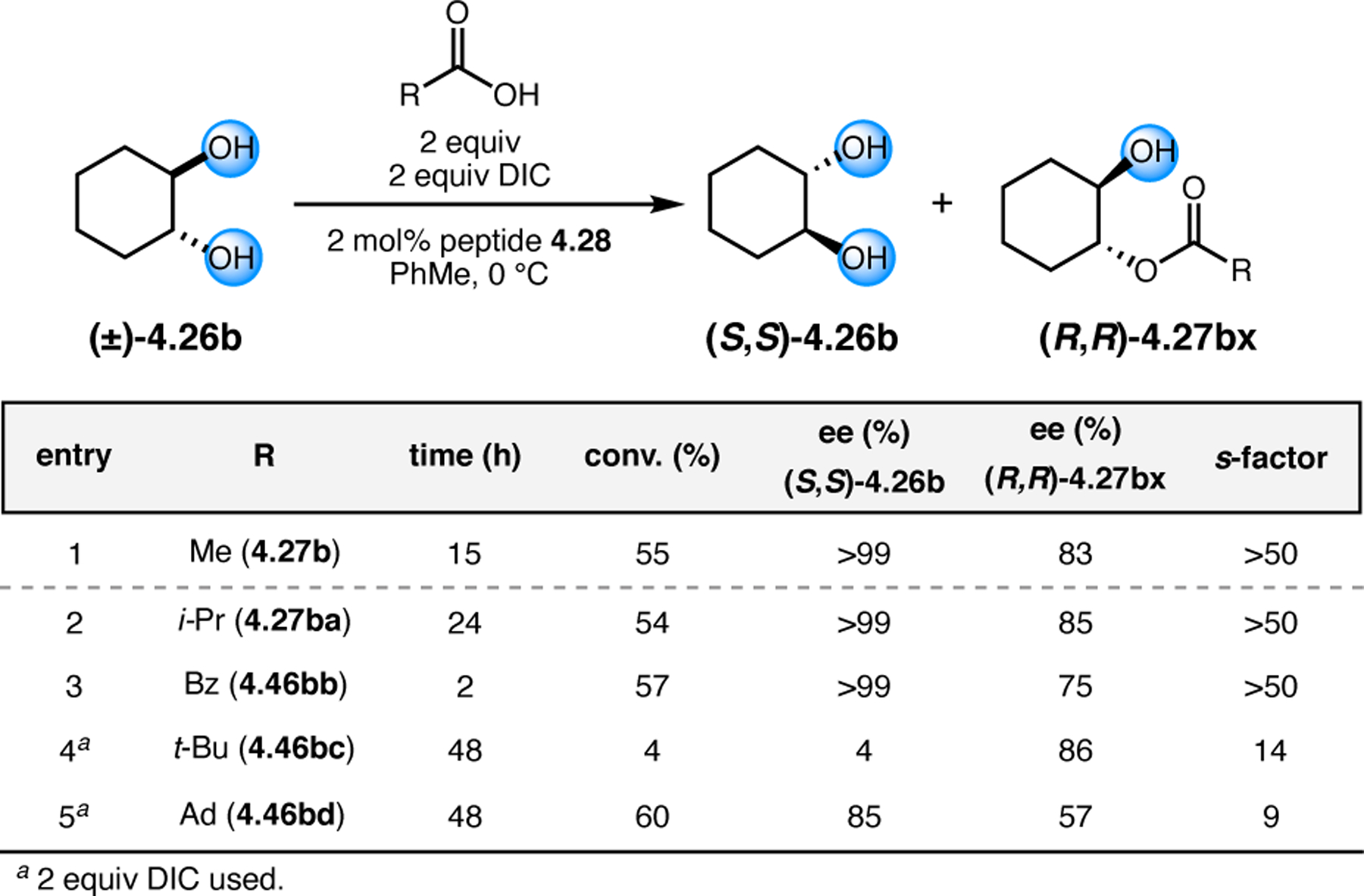
Acylation of trans-cyclohexane-1,2-diol 4.26b peptide 4.28–catalyzed Steglich esterification.172
Peptide 4.28, which had been optimized for acylation with acetic anhydride,164 delivered monoacetylated 4.27b in higher conversion (55 %) and selectivity (83% ee) using acetic acid rather than acetic anhydride (Figure 58, entry 1 vs. Figure 50, entry 2). Using diol 4.26b, 10 carboxylic acids were examined, selected examples of which are shown in Figure 58. s-Factors >50 were achieved for the kinetic resolution of trans-cyclohexane-1,2-diol 4.26b with propionic and phenylacetic acids, as well (entries 2 & 3). Sterically hindered acids, such as tert-butyl and adamantane carboxylic acids, resulted in significantly lower s-factors (entries 4 & 5). Overall, this Pmh-mediated Steglich esterification strategy greatly improves the scope of this method from simple acetylation to general acylation.
Schreiner and co-workers expanded this method for enantioselective acyl group transfer to diol 4.26 by developing conditions for the oxidative esterification of aldehydes (Figure 59).173 Using aldehydes is advantageous given that these compounds are typically more soluble in organic solvents, easier to purify, and more reactive than anhydrides. In this approach, an aldehyde can be activated by a sacrificial carboxylic acid and subsequently oxidized to form a mixed anhydride in situ. This species can then be selectively transferred to the diol via Pmh-containing peptide catalysis, analogously to the previously reported methods. Conditions optimization was first performed using TEMPO as the oxidation catalyst and peptide 4.28 to mediate the selective acyl group transfer. Various carboxylic acids were evaluated to determine the ideal aldehyde activator for in situ anhydride formation. 4-Nitrobenzoic acid was found to be optimal for both conversion into and enantioselectivity of monoacylated 4.27be. This two-catalyst method was applied to various trans-cycloalkane-1,2-diols 4.26 and aldehydes, achieving up to 81% ee of 4.26 and 88% ee of mono-acylated product 4.27 (s-factor = 39).
Figure 59.
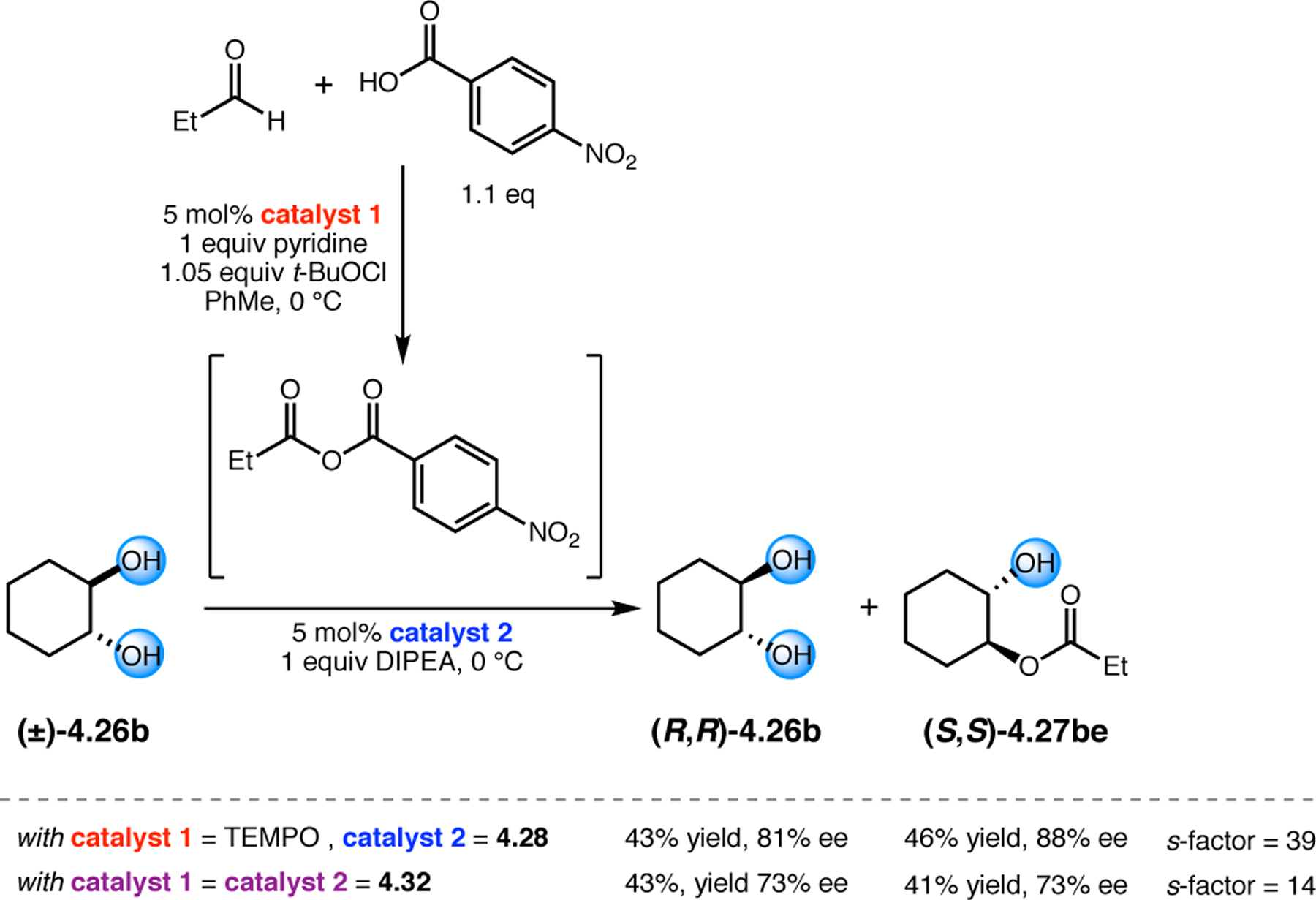
Kinetic resolution of (±)-4.26b via oxidative esterification of aldehydes.173
With conditions optimized using a two-step approach, peptidic multicatalyst 4.32 was employed in the one-pot, tandem, enantioselective oxidative esterification of 4.26b. Though the yields were comparable, the enantioselectivities obtained for 4.26b (73% ee) and 4.27be (73% ee; s-factor =14) were slightly lower than with the two-step protocol. By enabling the use of aldehydes over anhydrides, Schreiner and co-workers further expanded the scope of peptide-mediated acyl transfer to trans-1,2-diols 4.26 and demonstrated the power of peptidic multicatalysts to perform two different transformations with a single peptide.
In 2015, the protocol for the tandem oxidative esterification reaction of trans-1,2-diols 4.26 with multicatalyst 4.32 was further improved to use alcohols as acyl synthons (Figure 60).174 Alcohols are superior acyl equivalents to anhydrides, acids, and aldehydes owing to their high stability and commercial accessibility. Similar to their strategy using aldehydes,173 Schreiner and co-workers proposed that multicatalyst 4.32 could be used to first oxidize the alcohol in situ to the corresponding carboxylic acid with its TEMPO moiety along with co-oxidant m-CPBA. The acid could then be activated with DIC to provide a mixed anhydride that could then be transferred to various trans-1,2-diols 4.26 via the Pmh-residue of 4.32. Using this three-step, one-pot strategy with multicatalyst 4.32, higher yields and selectivities were obtained for recovered diols 4.26 and mono-acylated products 4.27 compared to the two-step procedure starting with aldehydes (Figure 59). Figure 60 shows a selection of the substrate scope, achieving up to 97% ee for 4.26 and 87% ee for 4.27. Alcohols with smaller steric bulk proved most successful in this transformation (e.g., 4.27b vs. 4.27be vs. 4.27bf). The ring size of the diol did not significantly affect enantioselectivity (4.26b vs. 4.26d), and the use of acyclic diols, such as 4.26e, was also possible with this route.
Figure 60.
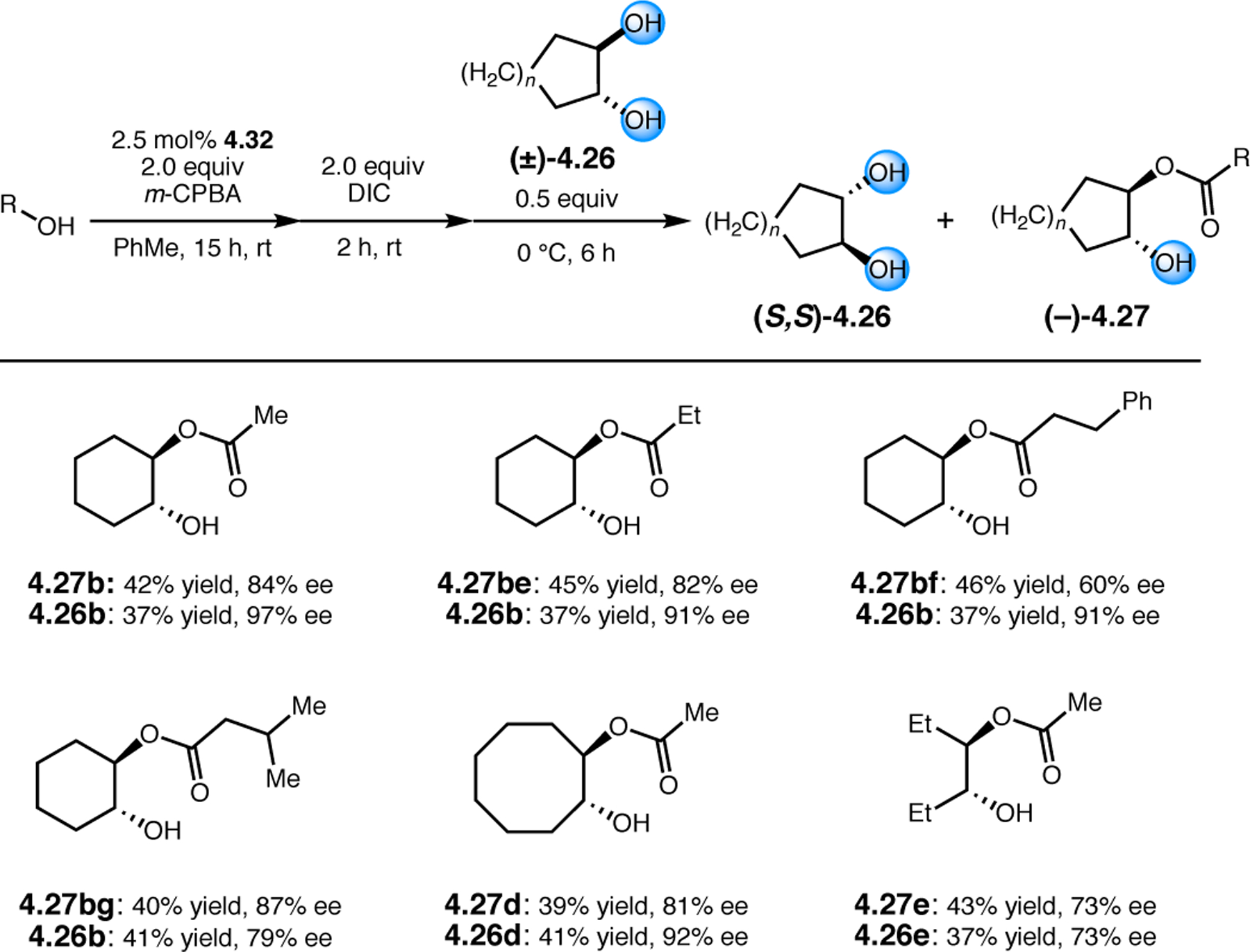
Kinetic resolution of trans-diols (4.26) using various alcohols as acyl synthons with peptidic multicatalyst 4.32.174
Instead of employing trans-1,2-diols 4.26 as the starting material for enantioselective acylation, Schreiner and co-workers developed a method to generate the requisite diol in situ starting from simple alkenes using a one-pot multicatalyst approach (Figure 61).175 This strategy relies on a sequence of catalytic epoxidation, epoxide-opening with water as the nucleophile, and kinetic resolution of the resulting racemic diol via enantioselective, peptide-catalyzed acylation. While streamlined, some challenges associated with this strategy included competitive catalyst oxidation, or interference of epoxide side products in the following steps. Therefore, each step in this sequence was optimized separately. Beginning with meso-epoxides 4.33, the last two steps of this sequence were examined using peptide-based multicatalyst 4.34 in a one-pot reaction. This peptidic salt combines the ability of sulfuric acid to catalyze the racemic epoxide opening via Brønsted acid catalysis with that of the Pmh residue to serve as a nucleophilic catalyst for enantioselective acylation. Peptide 4.34—the bisulfate salt analog of the highly efficient acylation catalyst 4.28164—was found to be an efficient catalyst for both the racemic epoxide opening with water and the subsequent enantioselective acyl group transfer, providing s-factors of up to 48 (entries 1–4). Moreover, this reaction can be scaled up to 1 mmol with minimal perturbations to selectivity (entry 2).
Figure 61.
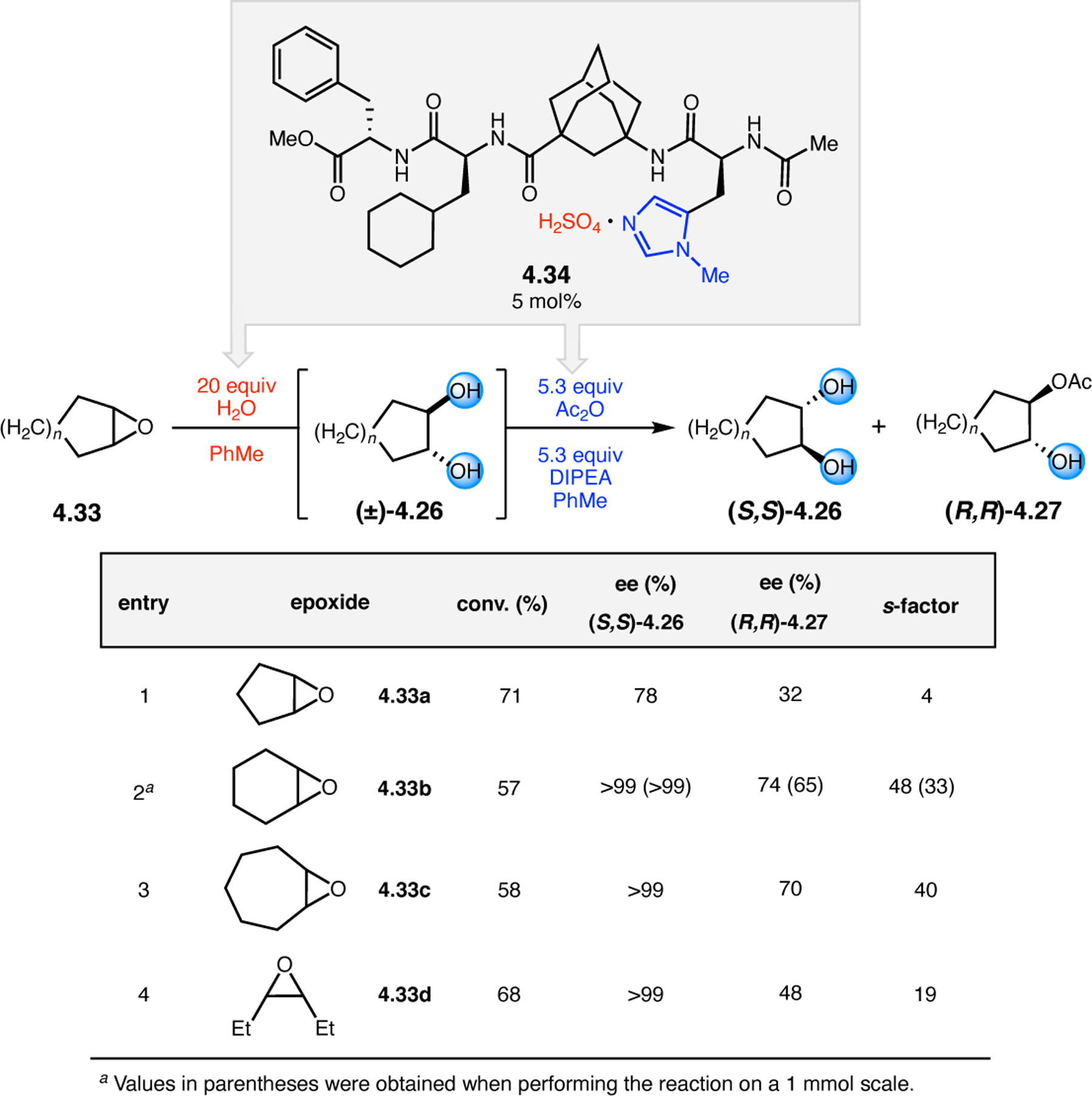
Peptide salt 4.34-catalyzed epoxide opening to trans-1,2-diols 4.26 and subsequent kinetic resolution via acylation.175
With a two-step strategy in hand, Schreiner and co-workers were then able to extend this system to the desired three-step, one-pot reaction beginning with alkenes (Figure 62).175 Several multicatalysts containing β-aspartate for epoxidation43 and Pmh for enantioselective acylation were synthesized, varying the spacer length between the two catalytic moieties. Multicatalyst 4.35 was identified as the lead catalyst for epoxidation and enantioselective acylation. Under the optimized conditions, hydrazine bisulfate was found to be the lead Brønsted acid catalyst for epoxide opening. Several alkenes were transformed into diol 4.26 in situ, then kinetically resolved to the mono-acylated product 4.27 with s-values of up to 26.
Figure 62.
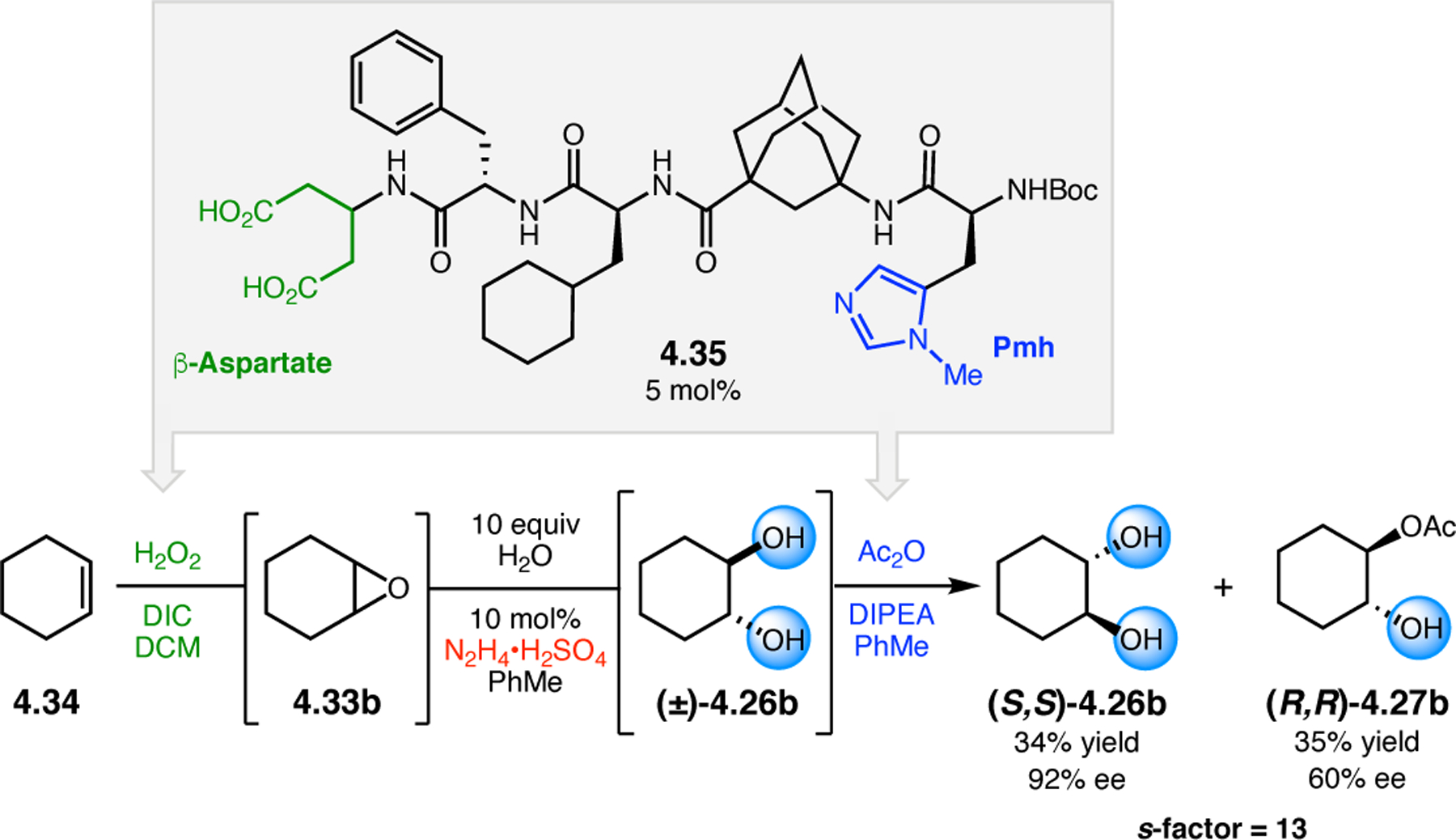
Epoxidation, ring-opening, and subsequent acylation mediated by peptide 4.35 and hydrazine bisulfate leading to enantioenriched mono-acylated diol 4.27b and trans-diol 4.26b from alkene 4.34.175
The mechanism of this one-pot, three-step transformation catalyzed by 4.35 was later investigated using electrospray ionization mass spectrometry in a collaboration between the Schreiner and Schrader Groups (Figure 63).176 The initial epoxidation of cyclohexene 4.34 with multicatalyst 4.35 was examined first. During this step, DIC activates the β-aspartate residue via O-acyl isourea intermediate I, which can either release diisopropyl urea and water to produce anhydride II or rearrange to the off-cycle N-acylurea III. Intermediate II can further react with hydrogen peroxide to give the catalytically active peracid IV, which can convert cyclohexene 4.34 to cyclohexene oxide 4.33b. A potential side product of the incorporation of hydrogen peroxide is catalyst oxidation to form V. Due to the short lifetime of some of the reactive intermediates, an online microflow reactor and MS methods were used to identify and study these species in detail.
Figure 63.
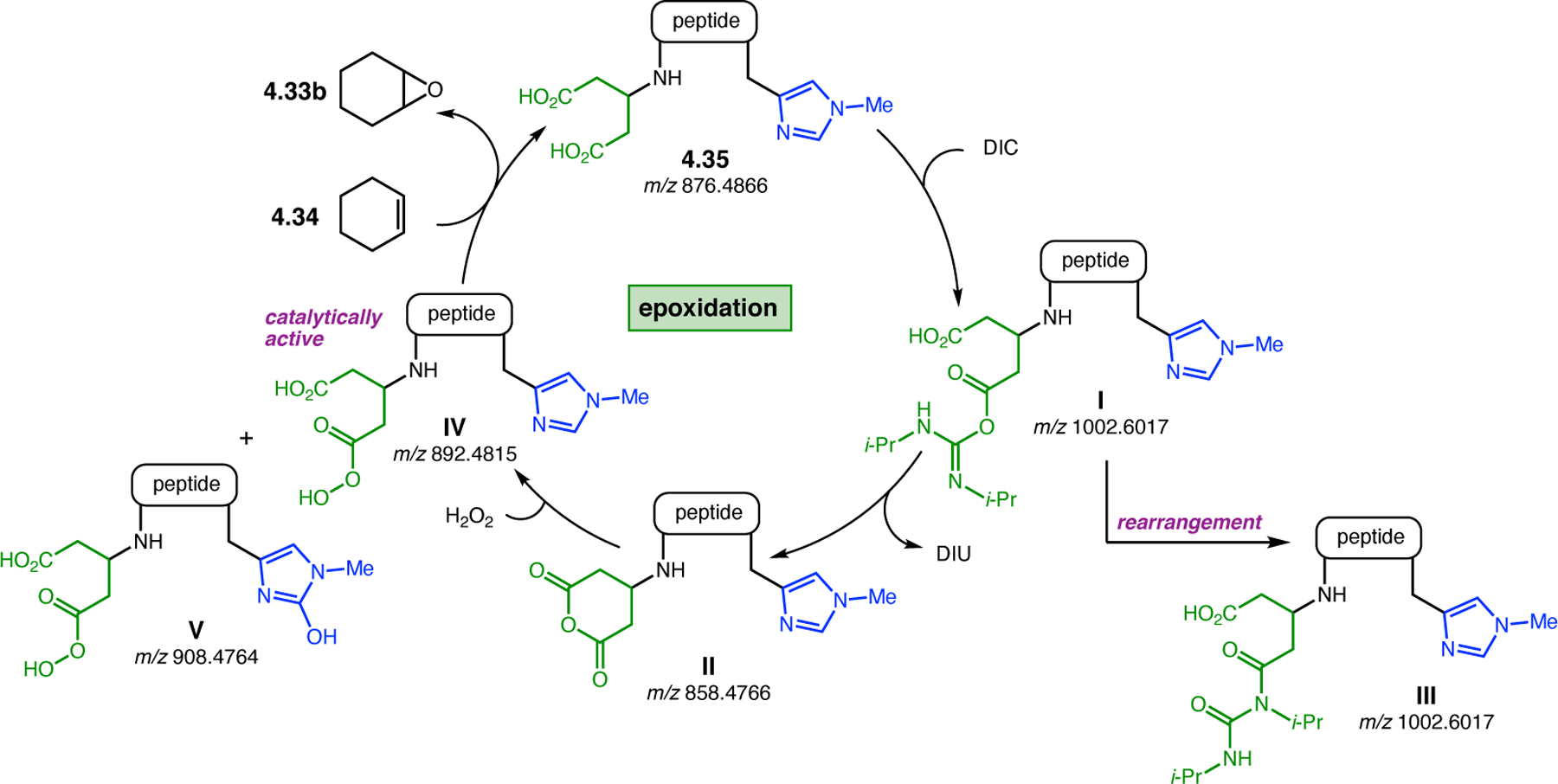
Catalytic cycle for the epoxidation of cyclohexene (4.34) with multicatalyst 4.35.176
Using these techniques, species I–V were identified as intermediates of the epoxidation of 4.34 (Figure 63). While rearrangement product III was observed, the molecular ion was present in such a low level that it was deemed fairly irrelevant to catalysis. Under multicatalysis conditions with 4.35, only 35% of mono-acetylated 4.27b was obtained when starting from cyclohexene (Figure 62), yet when starting from epoxide 4.33b with catalyst 4.34, 57% yield of 4.27b was obtained (Figure 61).175 The diminished activity of multicatalyst 4.35 was suspected to be caused by oxidation of the peptide. Indeed, intermediate V, in which the Pmh-moiety has been oxidized, was identified via MSn experiments.
The next catalytic step investigated was the enantioselective acylation of racemic diol 4.26 (Figure 64).176 To simulate the one-pot reaction conditions, 4.35 was subjected to both acetic anhydride and hydrogen peroxide. It was found that not only the Pmh catalytic moiety is activated by acetic anhydride, but also the β-aspartate residue, providing the catalytically active intermediate VI, confirmed via high resolution MSn experiments. Under the one-pot conditions, several other species were detected, including II and VII, all of which may influence the enantioselectivity of acyl transfer. This hypothesis is rooted in the observation that 4.35 provides lower selectivities as compared to the originally developed acylation catalyst 4.28.164,175 Overall, the use of high-resolution mass spectroscopy was valuable for the identification of ephemeral reaction intermediates and potentially deleterious side products found in these one-pot tandem epoxidation/epoxide opening/ acylation reactions.
Figure 64.
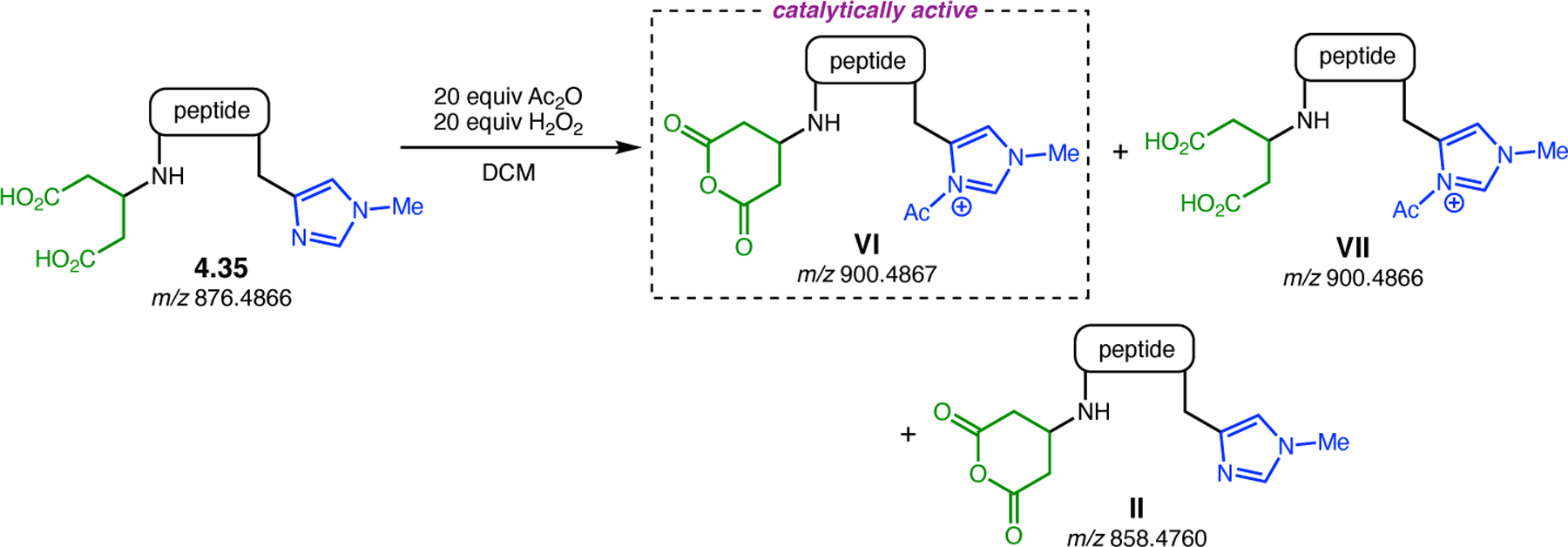
Activation of Pmh- and β-Asp residues in multicatalyst 4.35 for acylation of (±)-4.26.176
In addition to the many contributions from the Schreiner Group, Qu and co-workers were able to modify previously reported Pmh-containing peptide 4.38a for the enantioselective acylation of amino alcohols 4.36 (Figure 65).177 In our original report, tetrapeptide 4.38a was able to catalyze the kinetic resolution of (±)-4.36, affording enantioenriched (1S,2S)-4.36 in 98% ee and acetylated product 4.37 in 73% ee, with a krel of 28 (entry 1).132 High selectivity in this transformation can be attributed, in part, to the β-hairpin conformation of 4.38a. Chen and Qu hypothesized that incorporation of a thioamide—in which the carbonyl oxygen of the i+1 Pro residue is replaced with sulfur—in peptide 4.38a might improve the enantioselectivity of this kinetic resolution. Thioamide replacement in peptides can give rise to several consequences, such as increased acidity of the thioamide N–H, increased rotational barrier about the peptide C–N bond, and decreased conformational flexibility of the surrounding residues.178–183 Incorporation of a thioamide at the i+2 position or a sulfonamide at the N-terminus was expected to strengthen the intramolecular H-bonds reinforcing the β-hairpin secondary structure. Alternatively, a thioamide could be inserted at the i+1 or i+2 positions. The substrate was proposed to interact with 4.38a via H-bonding with the i+2 amide N–H. Modification of this position to a thioamide could improve the H-bond donor ability of the peptide, and thereby improve the enantioselectivity.
Figure 65.
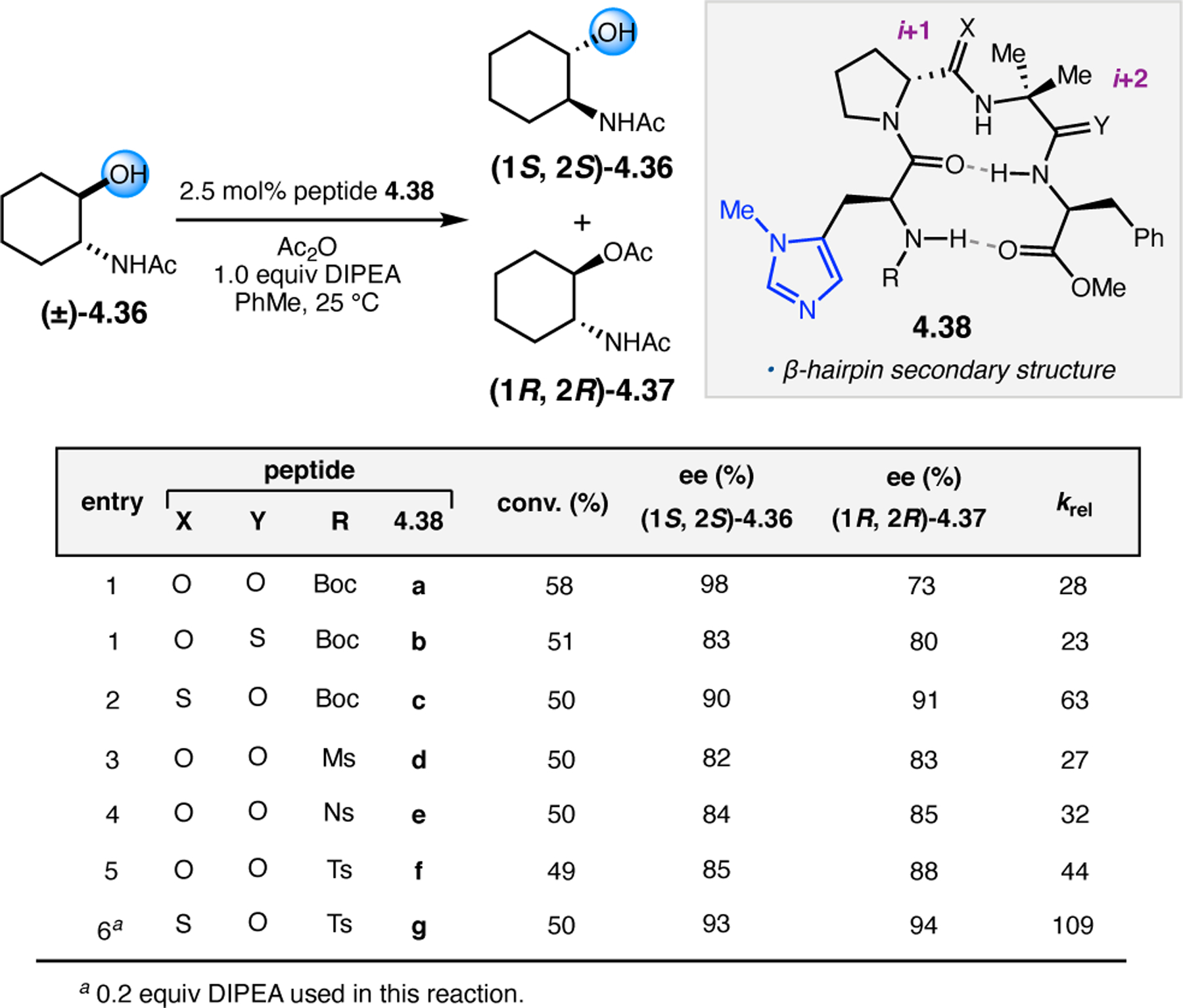
Kinetic resolution of (±)-4.36 with various β-hairpin-biased tetrapeptides 4.38.177
Thus, Qu and co-workers synthesized several derivatives of peptide 4.38 varying the location of the thioamide modification.177 The kinetic resolution of (±)-4.36 with peptide 4.38b, containing a thioamide substitution at the i+2 position, resulted in a diminished krel of 23 (Figure 61, entry 2). However, incorporation of the thioamide at i+1 (4.38c) resulted in a significant enhancement in selectivity, with krel reaching 63 (entry 3). Tetrapeptides modified at the N-terminus (4.38d–f) gave higher selectivity than that of 4.38a, with an upward trend from Ms to Ns to Ts (entries 3–5). Combining the benefits of the N-terminal Ts-protecting group and the i+1 thioamide, peptide 4.38g provided the highest selectivity in this reaction, with a krel of 109 using only 0.2 equivalents of DIPEA (entry 6). Using peptide 4.38g, 8 additional 1,2-amino alcohols were examined, with 4.36 providing the highest selectivity. The secondary structures of modified peptides 4.38 were studied using NMR and FT-IR techniques. Examination of the NHs of lead peptide 4.38g by FT-IR indicated a tight first interstrand H-bond, suggesting that incorporation of the thioamide at i+1 position promotes a more rigid β-hairpin conformation. Though Ns-containing 4.38e bears the most acidic N–H, which is often correlated with strong interstrand H-bonds, it did not provide the highest stereoselectivities in this asymmetric acyl transfer reaction. This may support the notion that some degree of flexibility is an important feature for asymmetric peptide catalysis and also highlights that acidity and H–bond strength are not always correlated in a simple way.184
Mechanistic studies on the peptide-mediated kinetic resolution of (±)-4.36 have also been reported by the Liao, Santoro, and Himo Groups using DFT calculations (Figure 66).134 The reaction was treated as proceeding through two sequential steps: (1) acylation of peptide 4.38a with acetic anhydride to form intermediate I via TS 4.3, and (2) acyl transfer from I to 4.36 with a concerted proton transfer from 4.36 to the acetate ion (TS 4.4). The authors focused on modeling the second step of the reaction with tetrapeptide 4.38a, as this is the process that confers asymmetric induction. Even with the assumption that the peptide adopts a stable β-hairpin structure, the Pmh-residue can still freely rotate about bonds A, B, and C of intermediate I, generating numerous conformations to consider in TS 4.4. Among the conformations of I, the re- and si-face attack of I by 4.36, and each enantiomer of 4.36, 48 distinct transition states were considered, each of which was optimized using DFT with the B3LYP functional.
Figure 66.
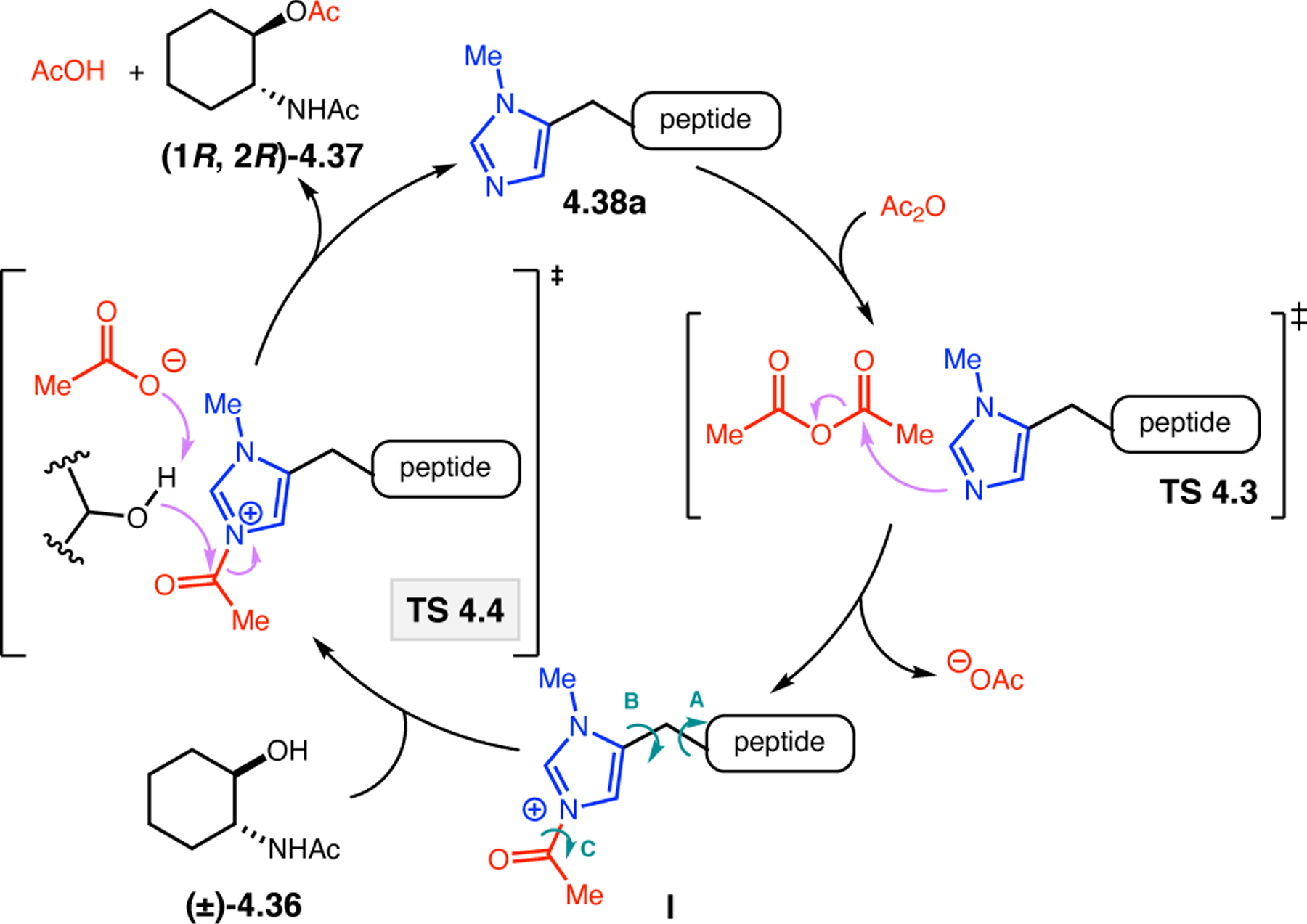
General reaction mechanism for the acylation of (±)-4.36 catalyzed by peptide 4.38a.134
The 48 transition states were categorized into three groups depending on the interaction mode between peptide intermediate I, amino alcohol 4.36, and the acetate ion. The lowest energy conformation of each mode of TS 4.4 with experimentally favored (1R,2R)-4.36 is depicted in Figure 67. In TS 4.4A, the N–H of the i+2 Aib residue of I forms an H-bond to the carbonyl of the 4.36 acetamide. Alternatively, the Aib N–H of I can H-bond with the acetate ion (TS 4.4B). In TS 4.4C, no H-bond is formed between the peptide backbone and 4.36. All of the TSs show the acetamide N–H of 4.36 H-bonding with the carbonyl of the acetylated Pmh-residue in I. Overall, the lowest energy TS leading to the favored (1R, 2R)-enantiomer of 4.37 is TS 4.4A, in which the peptide backbone H-bonds to the acetamide carbonyl of 4.3, in accordance with, but expanding in detail and precision, the model that we originally proposed.185
Figure 67.
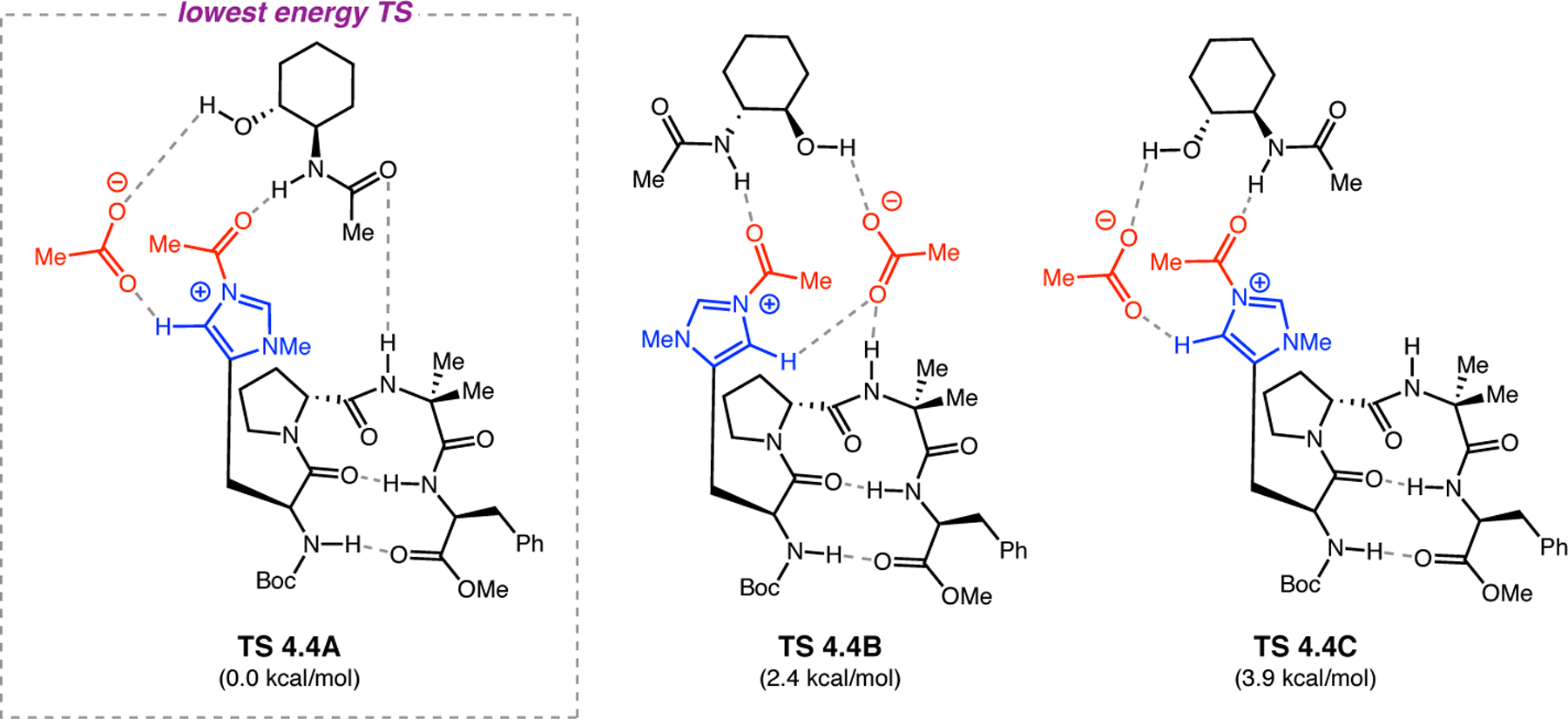
Lowest-energy transition states for the three interaction modes of intermediate I with (1R, 2R)-4.36.134
The lowest-energy diastereomeric TSs leading to each enantiomer of 4.37 are shown in Figure 68.134 As described by the authors, the favored enantiomer of 4.37 engages in noncovalent interactions with 4.38a as represented in TS 4.4A (Figure 68a). Alternatively, the lowest energy TS leading to disfavored (1S, 2S)-4.37 proceeds via TS 4.4B (Figure 68b), in which the N–H of the i+2 residue H-bonds to the acetate anion. This indicates that mode A of TS 4.4 offers a more stabilizing network of noncovalent interactions. Experimental studies show the relative acylation rate of each enantiomer to be 28 (Figure 61, entry 1),132 which converts to 2.0 kcal/mol. Therefore, the calculations (ΔΔG‡ = 2.6 kcal/mol) are in reasonable agreement with the experimental findings.
Figure 68.
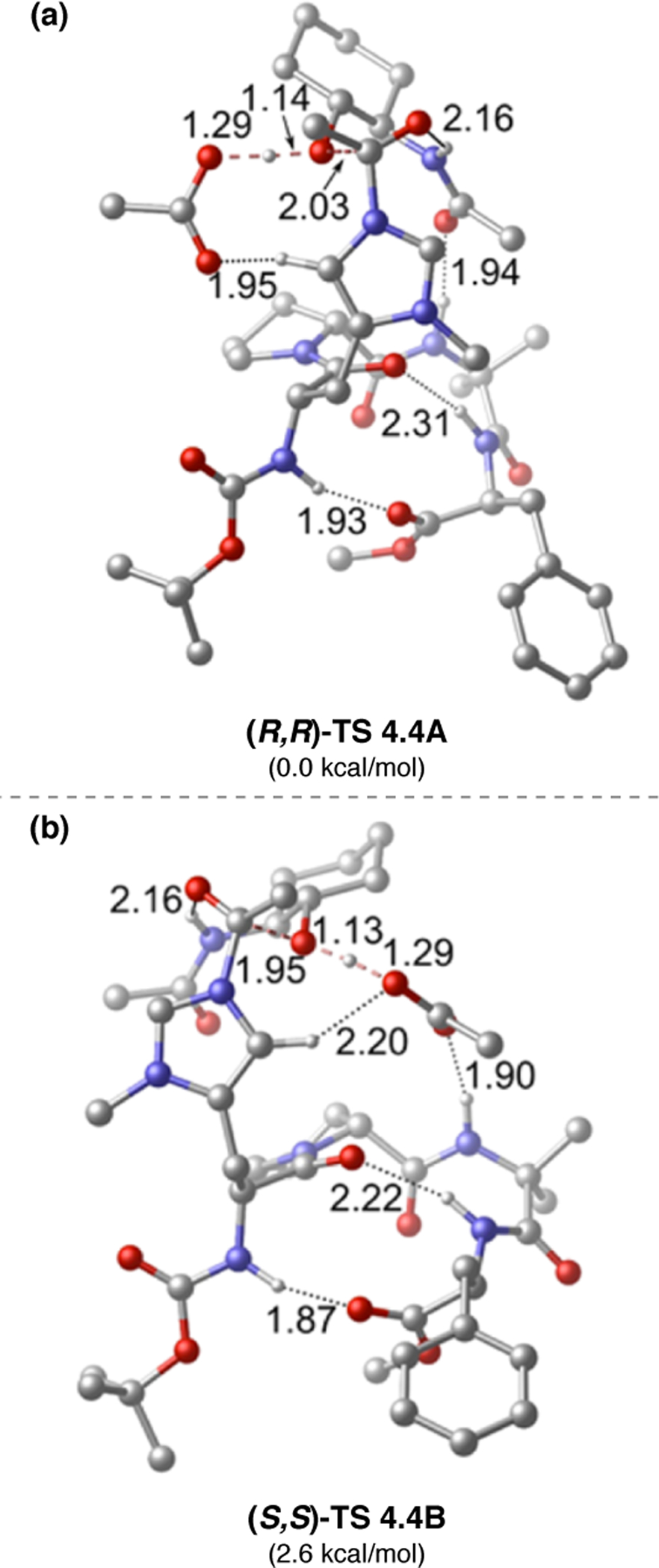
Lowest-energy transition states of (a) TS 4.4 with (1R, 2R)-4.36 and (b) (1S, 2S)-4.36. Adapted with permission from ref. 134. Copyright 2016 American Chemical Society.
Another type of acylation reaction is the Dakin–West (DW) reaction, which was first disclosed in 1928 as a method to access α-acylamido ketones, such as 4.40, from α-amino acids, such as, 4.39 (Figure 69).186,187 In the presence of a base at elevated temperatures, carbon dioxide is evolved from 4.39 en route to the α-acylamido ketone product, providing a thermodynamic driving force for this multistep process. Though several advances have been made to this reaction since 1928,188 it was not rendered asymmetric until 2016, when Schreiner and co-workers reported the first enantioselective DW reaction using Pmh-containing peptide catalysts.189
Figure 69.
Proposed mechanism of the peptide 4.41-catalyzed Dakin–West reaction of (±)-4.39.189
The authors envisioned that a Pmh-containing peptide catalyst could serve multiple roles along the putative DW reaction pathway (Figure 69).189 Amino acid 4.39 is first converted to the mixed anhydride, followed by cyclodehydration to oxazolone intermediate I. Subsequent acetylation of I affords oxazole II, which can undergo a Steglich rearrangement to form α-acyl oxazolone intermediate III. Alternatively, enolate formation and subsequent C-acetylation of I can also produce III directly. Acid mediated ring-opening of III affords β-ketoacid IV, which undergoes decarboxylation to form enolate V. Enantioselective protonation of V produces α-acylamido ketone 4.40. Performing the DW reaction enantioselectively is particularly challenging, as it requires a stereoselective proton transfer in the presence of stoichiometric acid within a rather complex multistep reaction framework. Moreover, the Pmh-catalyst would be required to serve a dual role as a Lewis base for acyl transfer and Brønsted base in the decarboxylative protonation step.
When screening reaction conditions, the authors found that addition of N,Nʹ-dicyclohexylcarbodiimide (DCC) improved the reaction by (1) facilitating cyclization to I, (2) acting as an auxiliary base in the deprotonation steps, and (3) promoting the conversion of the acetic acid byproduct back into the anhydride. Importantly, the byproduct formed from using DCC—dicyclohexyl urea (DCU)—cannot act as a base in the final decarboxylation step and thereby minimizing racemic background reactivity. Peptide 4.41 was identified as the lead catalyst for this transformation. The strikingly better performance of tripeptide 4.41 in the asymmetric DW reaction compared to that of tetrapeptide 4.28—which was successfully employed in previous acyl group transfer reactions165—may be a result of the location of the Pmh moiety as C-terminal residue in this case, leading to more efficient association with 4.39. With peptide 4.41, 10 α-amino acids 4.39 were examined in this transformation, achieving up to 58% ee in the ketone products 4.40. Some products could be recrystallized up to 84% ee.
Schreiner and co-workers also investigated the mechanism of this reaction using DFT calculations.189 The exact nature of the decarboxylative step is not clear, however, asymmetric induction is believed to stem from the deprotonation of IV, release of carbon dioxide, and subsequent enantioselective protonation of enolate V. Possible complexes between enolate V and protonated peptide 4.41 were therefore computed. The lowest-energy complex, shown in Figure 70, reveals the close proximity (1.94 Å) of the α-carbon of V to the protonated Pmh residue. This computed complex leads to the observed (S)-enantiomer, which is favored by 2.1 kcal/mol over the complex leading to the (R)-product. The observed selectivity may result from shielding of one face of enolate V by its own sidechain and the Cha residue of 4.41, inhibiting non-selective protonation by acetic acid in solution. Comparing various substrates, the higher selectivity observed for N-acyl amino acids bearing aliphatic sidechains (e.g., 4.39) likely originates from attractive dispersion interactions between the Cha residue of catalyst 4.41 and the side chain of enolate V.190
Figure 70.
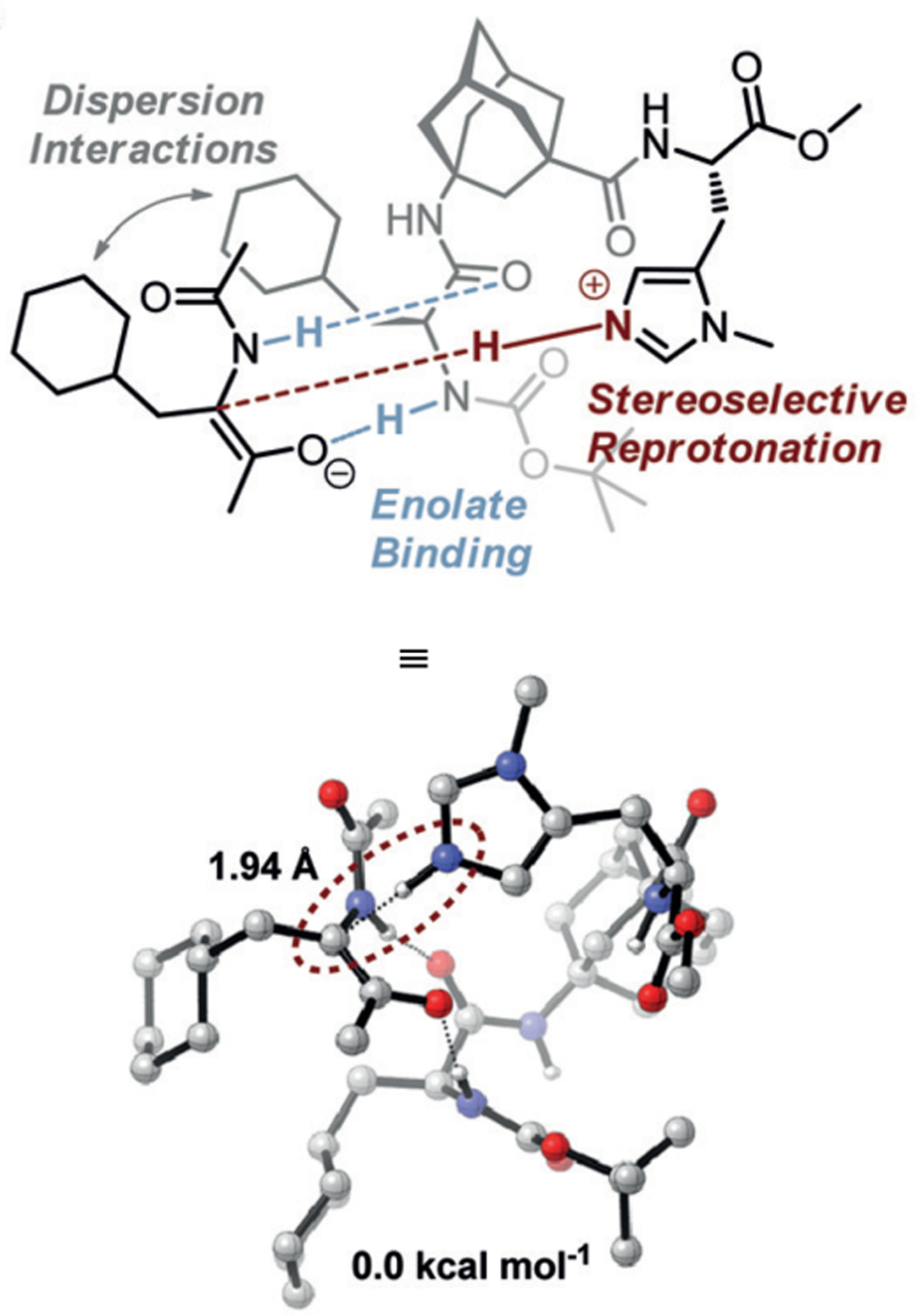
B3LYP-D3 (BJ)/6–31+G(d,p) optimized structures representing the lowest-energy complex of protonated peptide 4.41 with enolate V. Key noncovalent interactions are highlighted. Reproduced with permission from ref. 189. Copyright 2016 Wiley.
In addition to Pmh-containing catalysts, advances have been made in asymmetric acylation using peptides bearing pyridine derivatives as the catalytic residue.165 The 4-dimethylaminopyridine (DMAP)-191,192 and pyrrolidinopyridine (PPY)-type193,194 catalysts have found wide use as nucleophilic catalysts for acyl group transfer reactions in organic synthesis. Combing the modularity of peptides with the robustness of DMAP as a catalyst, Mandai, Suga and co-workers developed a robust synthetic strategy to synthesize peptide-tethered DMAPs 4.43 from aldehyde 4.42 and α-amino acids using an Ugi multicomponent reaction (Figure 71).195 Using this strategy, diastereomerically pure peptides 4.43a and 4.43b were obtained in 51 and 37% yield, respectively. Peptides 4.43 were then examined as chiral catalysts in the asymmetric Steglich rearrangement of acyloxyindoles 4.44 to oxindoles 4.45.196
Figure 71.
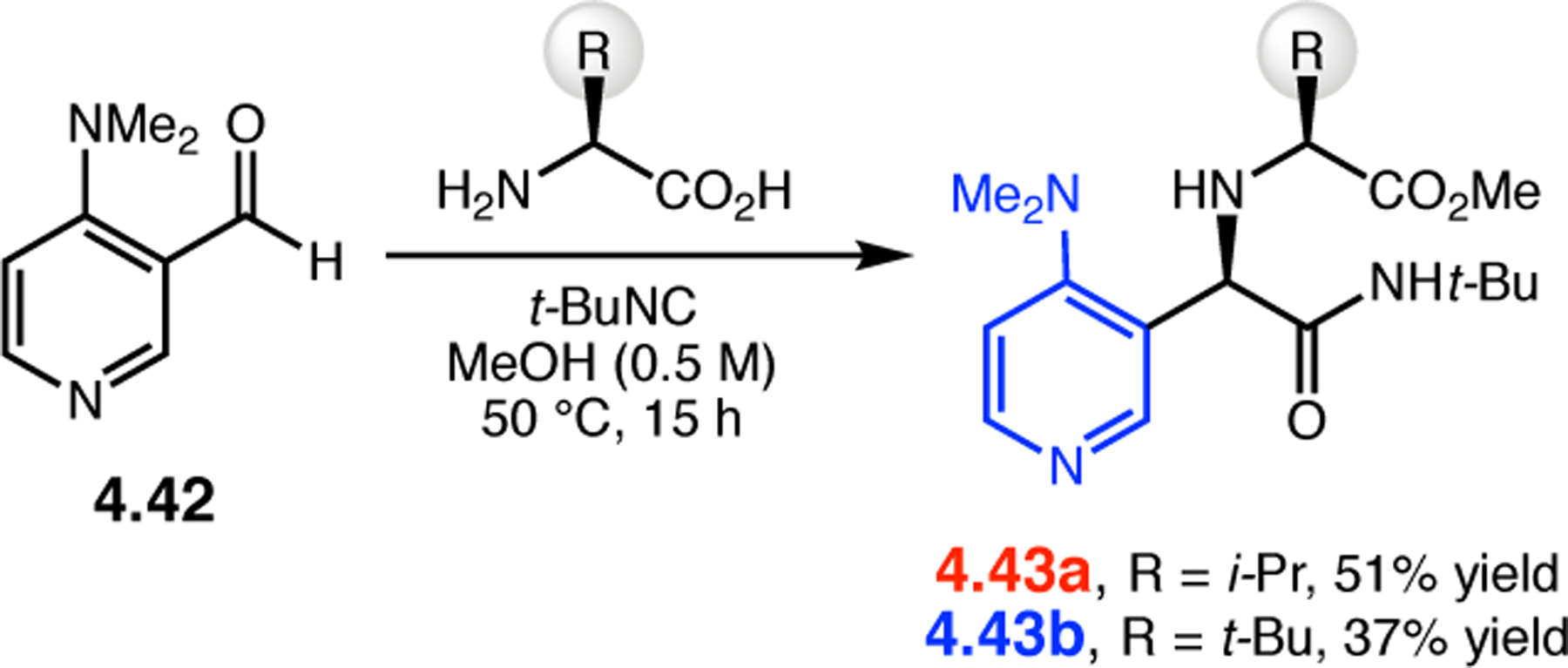
Synthesis of diastereomerically pure DMAP derivatives 4.43 via a one-pot, Ugi multicomponent reaction.195
Impressively, all 13 acyloxyindoles 4.44 examined were processed by peptides 4.43 with exceptional yields of >98% and enantioselectivities of >92:8 er (Figure 72).196 For the majority of substrates, Val-containing catalyst 4.43a tended to provide higher selectivities than the corresponding tert-leucine (Tle)-containing catalyst 4.43b. Acyloxyindoles 4.44a–d proceeded efficiently, achieving 92:8 to 99:1 er of the desired products (entries 1–4). Additionally, 3-aryl-(entry 5) or 3-heteroaryl-substituted oxindoles (entry 6) gave equally excellent enantioselectivities. Mechanistically, the Steglich rearrangement is believed to proceed through the formation of an ion-pair between the pyridinium cation of peptide 4.43 and the oxindole enolate derived from 4.44, followed by C–C bond formation to give the C-acyl oxindole 4.55. TSs to C–C bond formation for the ion-pair complex between acylated 4.43b and the enolate of 4.44b were calculated using DFT at the B3LYP/6-31G(d) level of theory. Schematic representations of the lowest energy TSs that deliver (R)- and (S)-4.45b are shown in Figure 73. (R)-TS 4.5 may be disfavored due to destabilizing steric repulsion between the tert-butyl of the Boc group in 4.43b and the phenylene group of 4.44b. The energy difference between the two transition states (ΔΔG‡) was calculated to be 2.99 kcal/mol, which is consistent with the experimentally obtained enantioselectivity of 99:1 er (Figure 72, entry 2).
Figure 72.
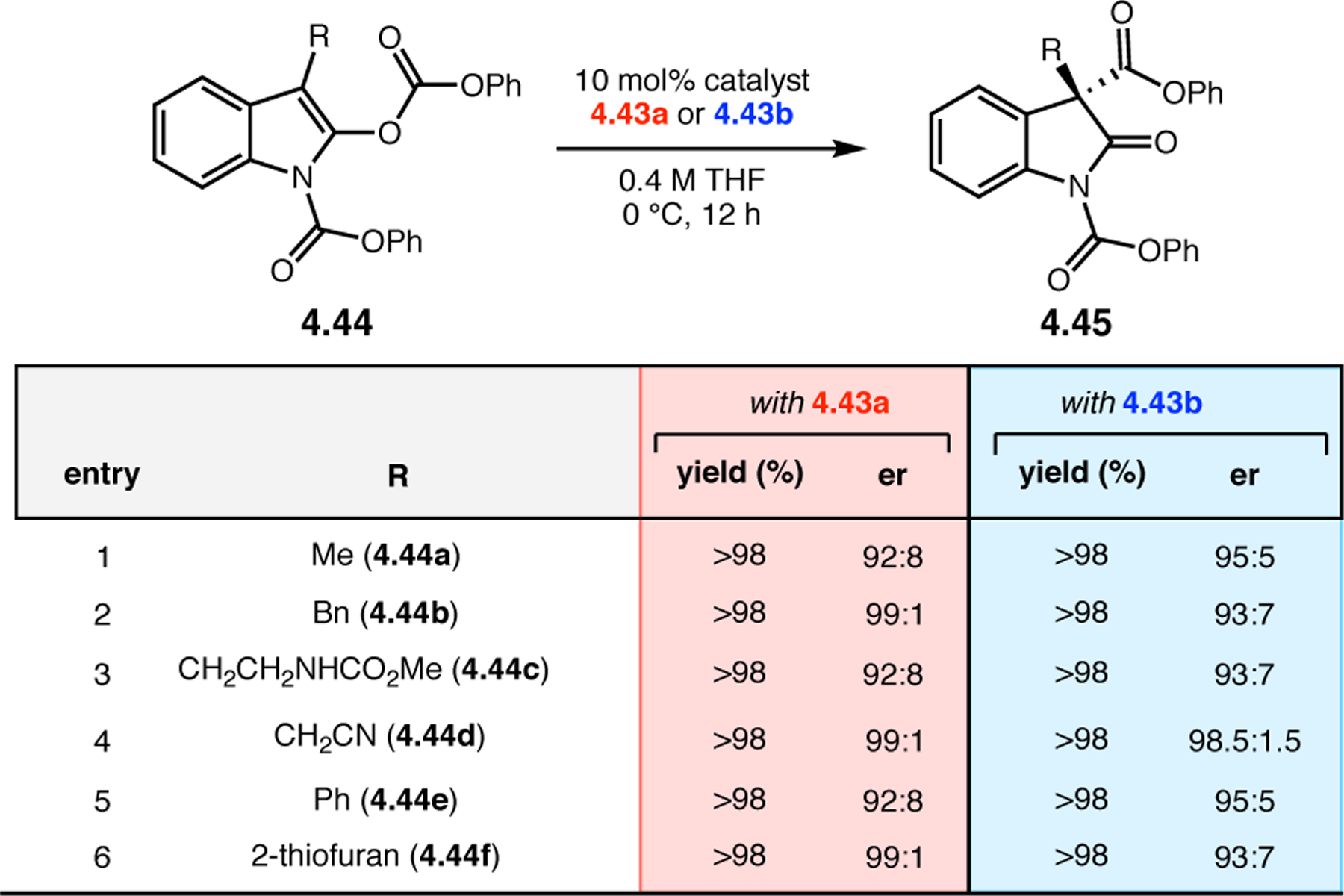
Enantioselective Steglich rearrangement of various O-acylated oxindole derivatives 4.44 using catalysts 4.43a or 4.43b.196
Figure 73.
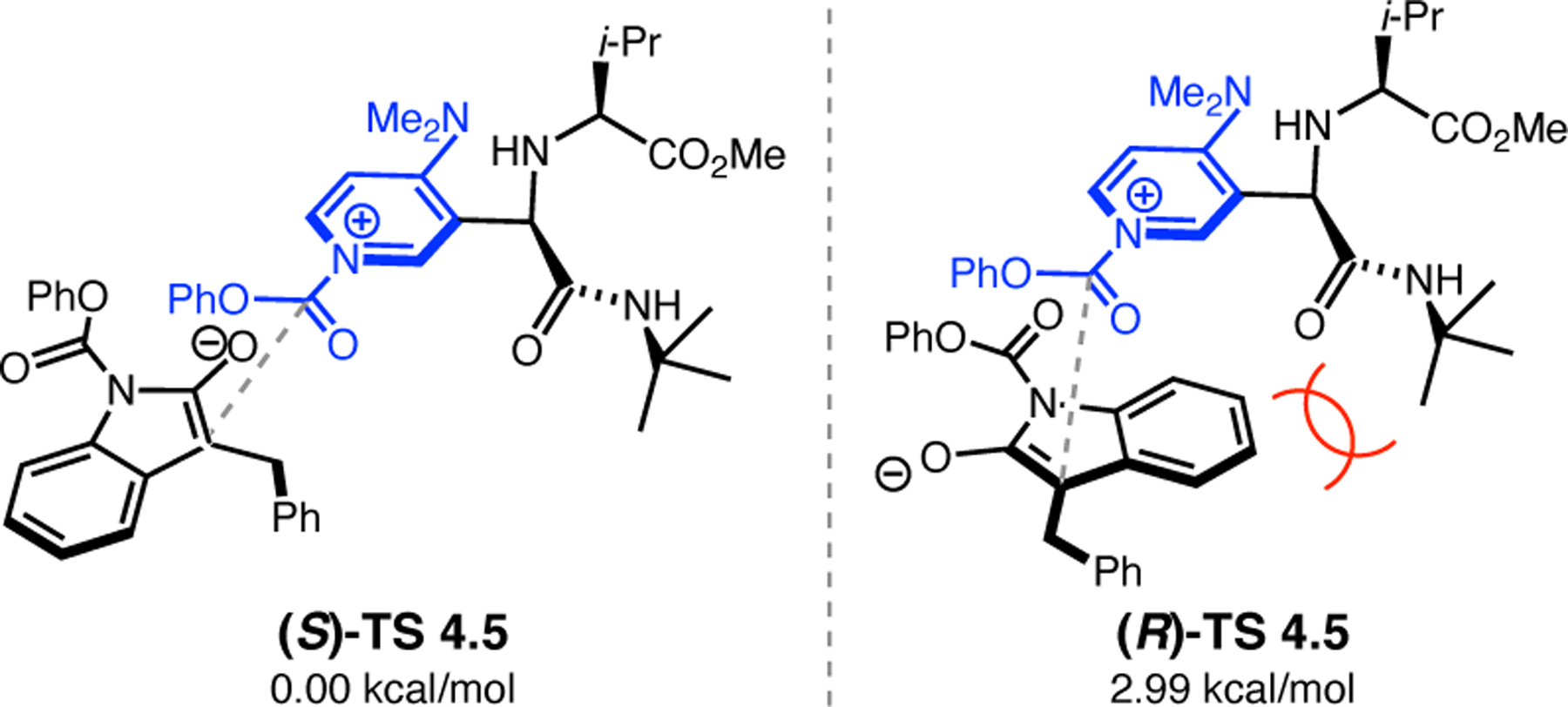
Schematic models of the energetically favorable transition states leading to (S)- and (R)-4.45b.196
Using another type of pyridine-containing peptide, Kawabata and co-workers employed PPY-embedded, C2-symmetric catalysts to promote the desymmetrization of meso-diols via acylation (Figure 74).197 The desymmetrization of meso-diols 4.29 through acylation had been well-studied by Schreiner and co-workers using Pmh-containing peptides,165 providing a standard to which this new catalyst archetype may be compared. Various C2-symmetric, chiral PPY-based peptides 4.46 were prepared from 5-oxo-Pro in several synthetic steps (Figure 74a)197,198 and subsequently evaluated in the enantioselective desymmetrization of 4.29a with isobutyric anhydride (Figure 74b). Chiral catalysts containing aromatic residues, such as l- (4.46a) or d-Trp (4.46b) and l- (4.46c) or d-(2-naphthyl)alanine (2-Nal, 4.46d) were first synthesized and tested (entries 1–4). Peptide 4.46a had proven to be a highly efficient catalyst in the regioselective acylation of glucopyranoses.198 As such, it was the first peptide to be employed in this system, delivering mono-acylated 4.30ab in 85% and 73% ee (entry 1). In screening the other aromatic catalysts, the highest selectivity was achieved with d-2-Nal-containing 4.46d, affording 4.30ab in 77% yield and 83% ee (entry 4). The absolute configuration of 4.30ab was unaffected by the configuration of the amino acid residue, though the extent of the enantioselectivity was indeed impacted (entries 1 vs. 2 & 3 vs. 4). Aliphatic residues, such as Val (4.46e) and Leu (4.46f) were also examined (entries 5 & 6), but neither was as selective as 4.46d, though Leu-containing 4.46f was quite similar overall. Lastly, achiral pendent groups were assessed, including Gly-containing 4.46g and N-hexyl 4.46h (entries 7 & 8). Interestingly, 4.46h delivered 4.30ab in 75% yield and 87% ee, suggesting that pendent residues with chiral sidechains were not necessary for selectivity in the context of these C2-symmetric PPY-based catalysts.
Figure 74.
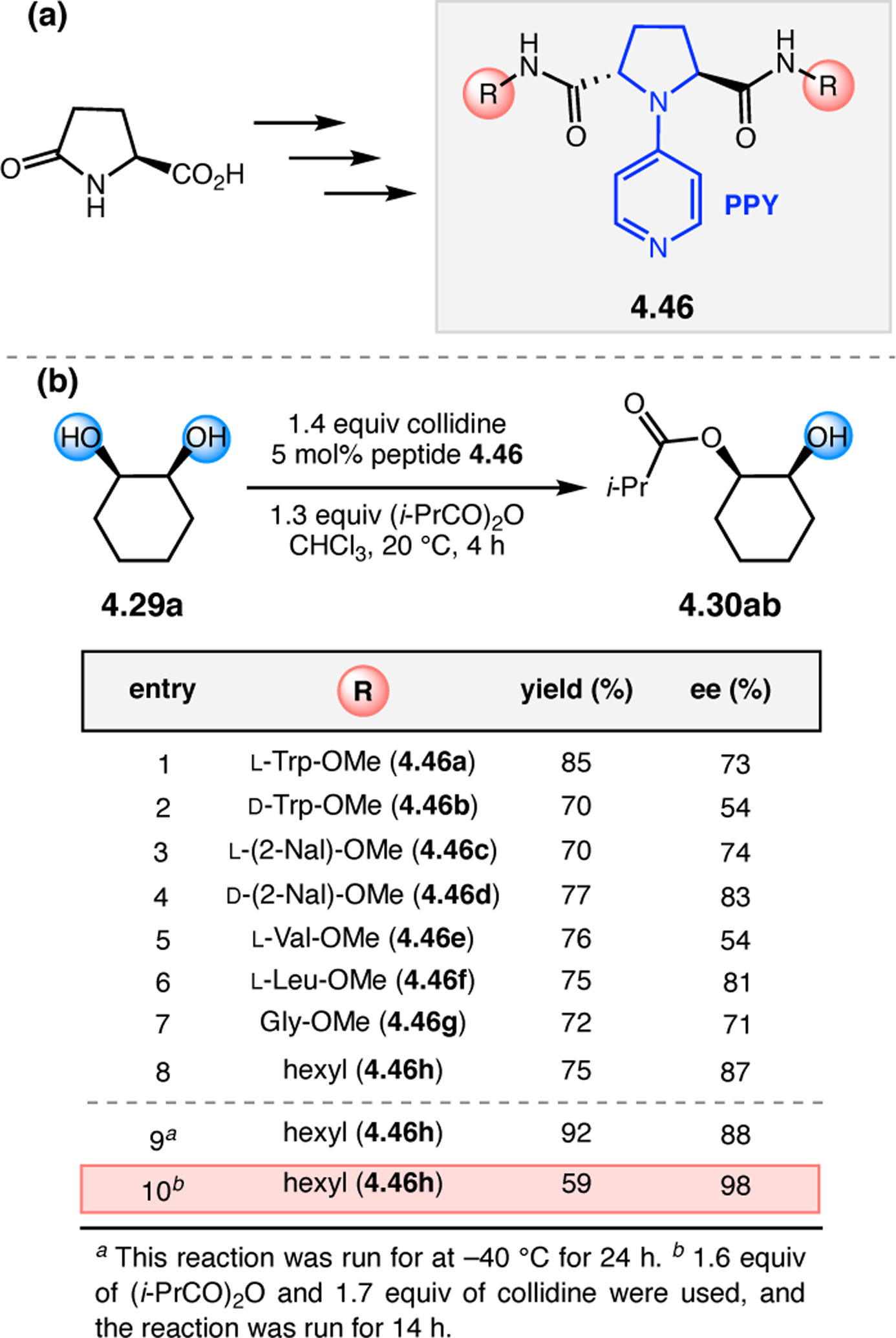
(a) Synthesis of chiral, C2-symmetric PPY catalysts 4.46 for (b) the asymmetric desymmetrization of meso-diol 4.29.197
With optimal catalyst 4.46h in hand, reaction conditions such as solvent, temperature, time, equivalents of anhydride, and catalyst loading were investigated.197 Lowering the reaction temperature to −40 °C, 4.30ab was produced in 92% yield and 88% ee after 24 h (Figure 74, entry 9). The highest selectivity of 98% ee was achieved by increasing the equivalents of collidine (entry 10). The increased ee of mono-acylated 4.30ab is associated with an attendant increase in the amount of diacylated material, eroding the yield to 59%. This suggests that the ee of 4.30ab is amplified to some degree by a secondary kinetic resolution en route to the symmetrical diacylated byproduct. To confirm this suspicion, racemic 4.30ab was subjected to the reaction conditions, affording (1R,2S)- 4.31ab in 67% ee with an s-factor of 7.7. The authors proposed two potential stereochemical models for the 4.46-catalyzed acylation of 4.29a (Figure 75). In one model, the amide carbonyls of 4.46 formed H-bonds with the non-reacting alcohol of 4.29a. The other model invokes the opposite antipode of H-bonding between 4.46 and 4.29a, wherein the amide N–H of 4.46 engages with the non-reacting alcohol of 4.29a. Indeed, when the amide of 4.46h is methylated or replaced with an ester linkage, 4.30ab is produced in only 13% ee. Though the authors prefer the former model, both transition state assemblies remain possible.
Figure 75.
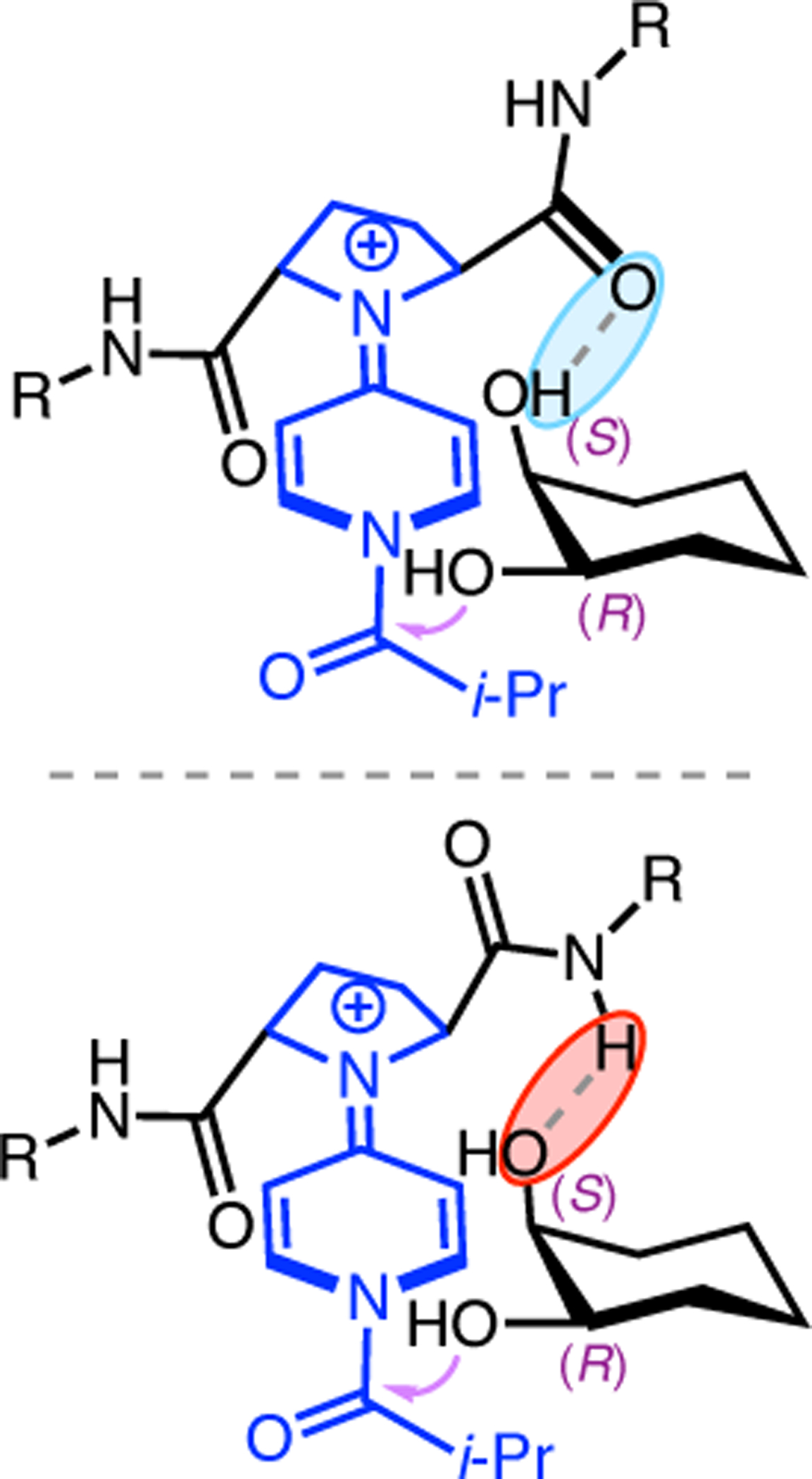
Two possible models for the transition state assembly in the asymmetric acylation of 4.29a catalyzed by 4.46h.197
Another variant of PPY-containing peptide catalysts based on general scaffold 4.49 was developed by Hunter and co-workers for the enantioselective acylation of racemic sec-alcohols 4.47 with isobutyric anhydride (Figure 76).199 Molecular modeling was used as a tool to facilitate catalyst optimization for the kinetic resolution of alcohol 4.47a via acylation. The first PPY-catalyst tested contained a Leu-Trp-OMe sidechain (4.49a). The Trp residue was incorporated in an attempt to engage 4.47a in π-stacking interactions with the N-acylated pyridinium intermediate, as shown in model 4.50. This was hypothesized to encourage facial discrimination of the pyridine ring as a means of achieving asymmetric induction in the acyl group transfer. A Leu residue was added upstream of Trp to improve catalyst solubility and further facilitate discrimination of one of the pyridine faces. Using 4.49a as an initial catalyst for enantioselective acylation, a low, yet nonnegligible, s-value of 5.3 was measured (entry 1).
Figure 76.
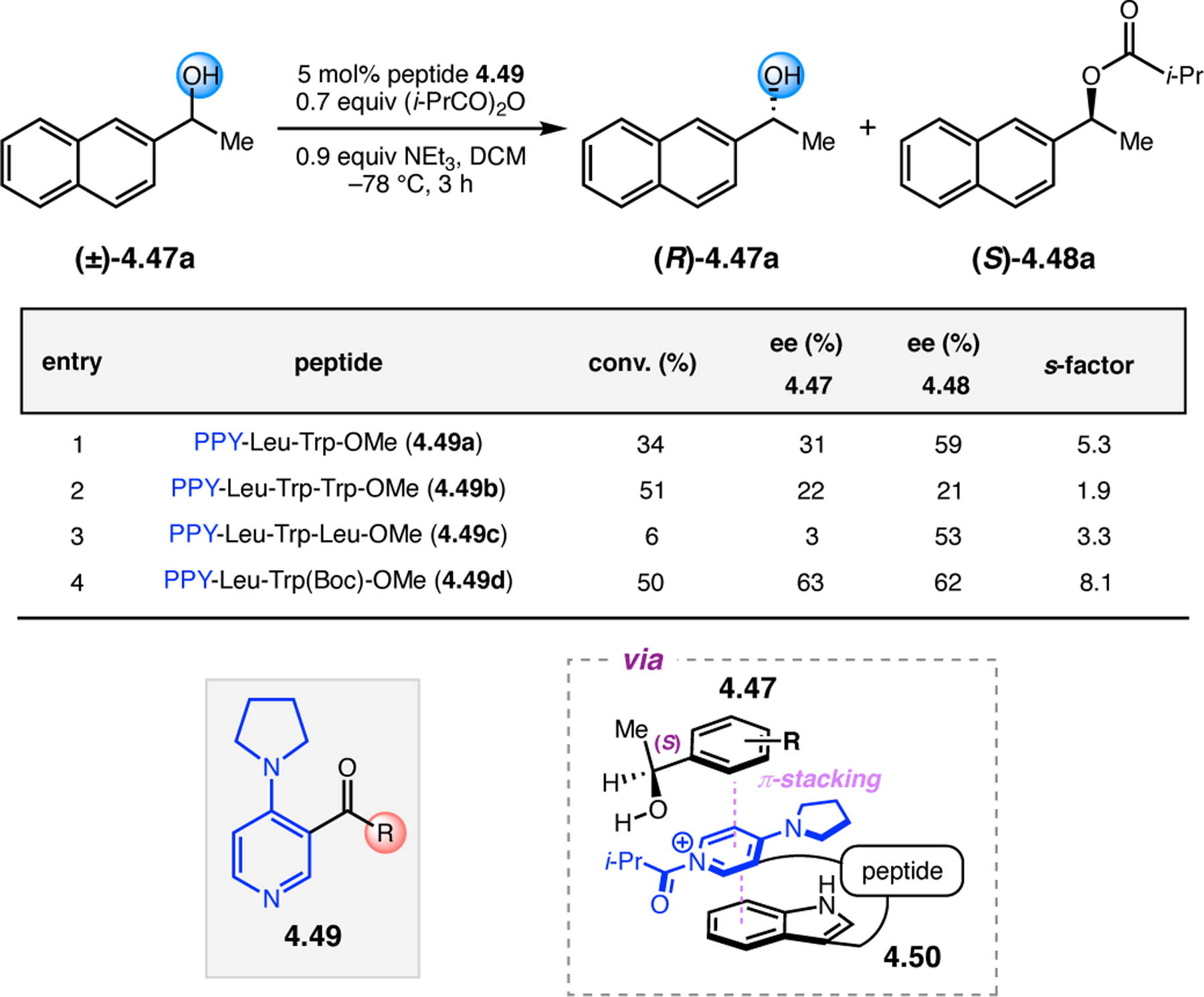
The kinetic resolution of racemic sec-alcohol 4.47a catalyzed by PPY-containing peptides 4.49.199
Computational modeling of the N-acyl intermediate 4.50 was then employed to identify potential structural elements for improvement. An initial set of 1,000 conformers of 4.50a were generated and optimized using molecular mechanics (MMFF94). A subset of the 100 lowest-energy conformers were then further optimized via quantum mechanical methods (HF-3c). From these 100 structures, the 10 lowest-energy conformers were refined using DFT calculations (B3LYP-D3(BJ)/def2-SVP), and the lowest-energy conformer identified is shown in Figure 77a. In this structure, the peptide chain curves underneath the pyridinium ring, favoring attack of alcohol 4.47 from the opposite re-face. A potential H-bond is also observed between the indole N–H of Trp and the carbonyl of the catalytic residue (2.335 Å). The aromatic pyridinium and Trp sidechain appear too far apart to for a stabilizing π-stacking interaction. NMR studies further supported the calculated structure.
Figure 77.
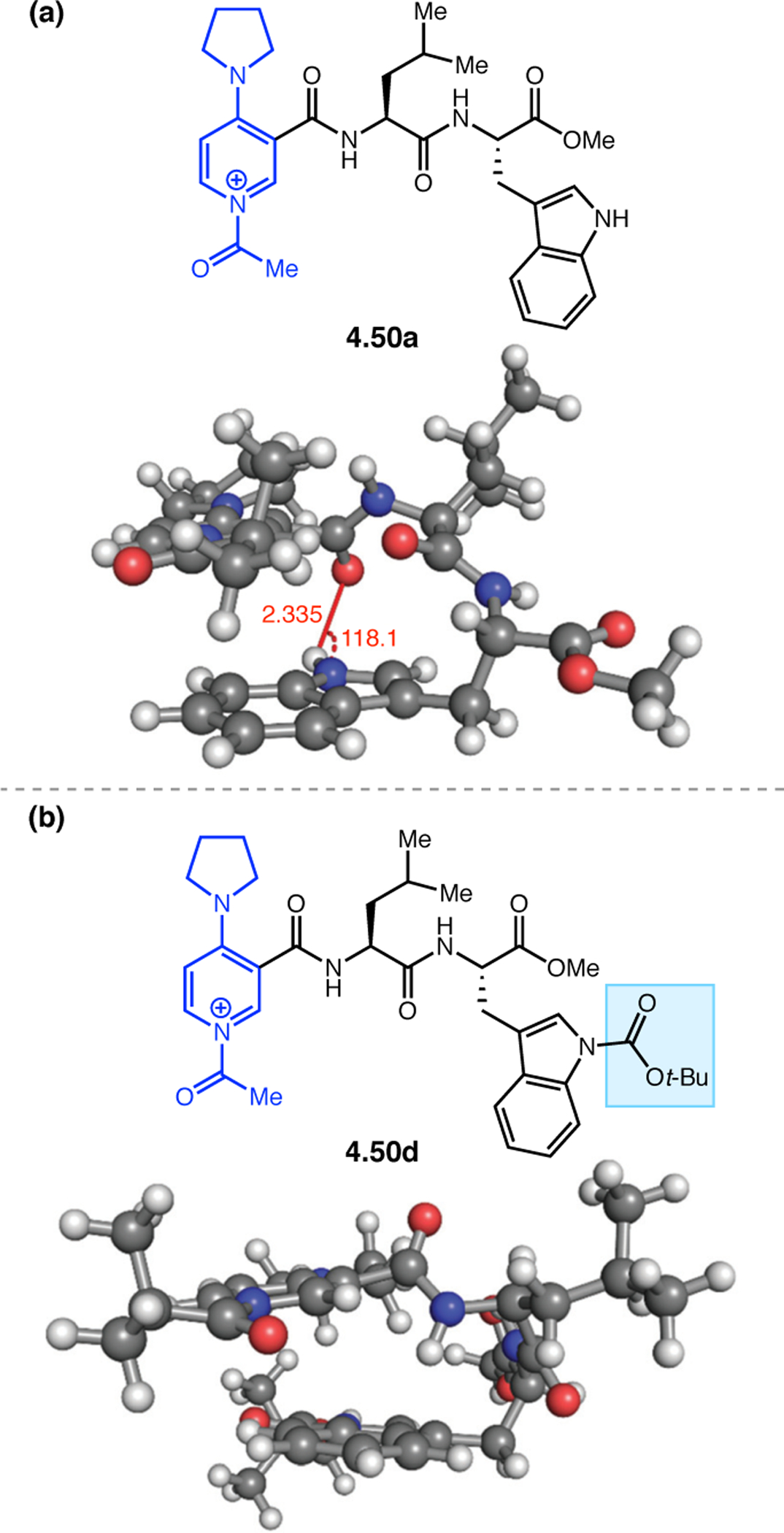
Lowest-energy conformers of (a) 4.50a and (b) 4.50d. Adapted with permission from ref. 199. Copyright 2016 Royal Society of Chemistry.
Based on the computed structure, the authors decided to extend the catalyst from a di- to tripeptide. Thus, peptides 4.49b and 4.49c were synthesized and evaluated in the kinetic resolution of 4.47a (Figure 76, entries 2 & 3). Unfortunately, the s-values for these tripeptides were even lower than those of dipeptide 4.49a. Once again, Hunter and co-workers turned to modeling for a solution. The optimized structure of 4.50a also indicated that functionalization of the indole ring of Trp could be a fruitful avenue. Before synthesizing a new catalyst, the structure of 4.50d was calculated using the same method as described previously. The lowest energy conformer of 4.50d is shown in Figure 77b. As noted with 4.50a, the peptide chain once again coils underneath the pyridinium ring. However, with 4.50d, the N-acyl group is in an s-cis conformation, which was shown to be a more favorable orientation. Most notably, introduction of the Boc group into the dipeptide resulted in a shift of the Trp indole ring to lay more in-line with the pyridinium ring. In this configuration of 4.50d, a π-stacking interaction between Trp and the pyridinium ring should be possible.
Given the promising results of modeling 4.50d, peptide 4.49d was synthesized and tested in the enantioselective acylation of 4.47a, achieving the highest s-factor yet of 8.1 (Figure 76, entry 4). With dipeptide 4.49d in hand, the reaction was performed with several other sec-alcohols 4.47 (Figure 78). 1-Phenylethanol 4.47b achieved a slightly higher s-factor of 8.9 compared to 1-(2-naphthyl)ethanol 4.47a. Increased steric bulk on the aryl ring by inclusion of methyl groups, as in 4.47c, resulted in the highest overall s-value of 10.8 (entry 2). Electron-donating groups, such as para-methoxy in 4.47d (entry 3), were preferred over electron-withdrawing substituents, like para-nitro in 4.47e (entry 4). A preference for electron-donating substituents also supports the importance of π-stacking interactions governing selectivity, with increased electron density in the arene of 4.47 favoring a tighter association with the electron-deficient pyridinium ring of 4.50.
Figure 78.
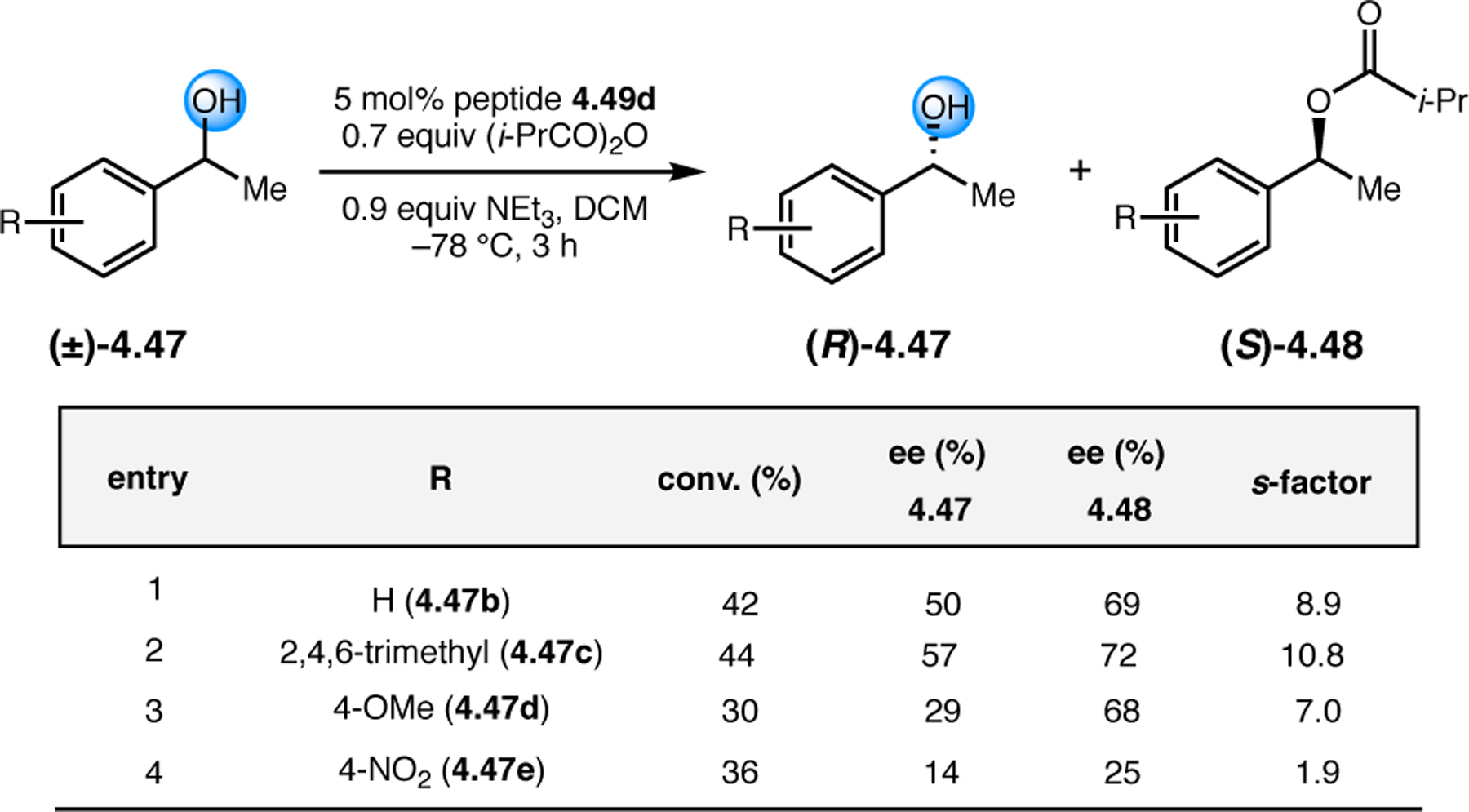
Kinetic resolution of various racemic sec-alcohols 4.47 catalyzed by peptide 4.49d.199
5. Nucleophilic Additions to Carbonyls & Epoxides
The addition of nucleophiles to carbonyls and epoxides are two of the most fundamental and useful transformations in organic synthesis. Countless asymmetric carbonyl addition and epoxide-opening methodologies have been reported to date,200 and peptide-based catalysts have played an important role in the development of this area. This Section summarizes the recent reports of peptide-catalyzed 1,2-additions to aldehydes (Sections 5.1 & 5.2) and esters (Section 5.3), wherein the nucleophiles are simple, commercially available reagents. Reactions of enols and enolates are reserved for subsequent sections (Sections 6–8). A peptide-catalyzed meso-epoxide opening is also summarized in Section 5.4.
5.1. Hydro- & Silylcyanation of Aldehydes.
One of the earliest contributions to the field of peptide catalysis dates to 1981, when Inoue and co-workers reported the enantioselective hydrocyanation of benzaldehydes catalyzed by a cyclic dipeptide, or diketopiperazine.36 As one of the first highly selective, organocatalytic transformations,201 this reaction triggered a cascade of excellent new applications and mechanistic studies through the early 2000s, which was covered extensively in our previous review.4 However, the mechanistic picture has undergone continuous exploration and enhancement in recent years. Below, Section 5.1.1 updates the mechanistic model for this important transformation, and Section 5.1.2. summarizes an alternate peptide-catalyzed strategy for the 1,2-addition of cyanide to benzaldehydes.
5.1.1. Hydrocyanation of Aldehydes.
Under the conditions originally reported by Inoue, cyclo[l-Phe-l-His] 5.3 catalyzes the highly enantioselective hydrocyanation of benzaldehyde 5.1a to the corresponding cyanohydrin 5.2a in 97% yield and 97% ee (Figure 79).202 High levels of selectivity are only possible in this reaction with nonpolar solvents, such as toluene, making these reactions heterogeneous. Moreover, Danda and co-workers reported the observation of enantioselective autoinduction, in which the ee values increased with higher conversion to the (R)-product.203 The stoichiometry of 5.3 to (R)-5.2a to in the active catalyst was found to be approximately 1:1. Kinetic analyses showed that the reaction is second-order in 5.3, suggesting that the catalytically active species is dimeric in solution.203,204 Three limiting models have been proposed for this transformation (A–C, Figure 79), none of which are consistent with all of the data. Model C notably employs a catalytic dimer, in which one imidazole activates 5.1a as an H-bond donor, while the other delivers the nucleophile.205
Figure 79.
Asymmetric hydrocyanation of benzaldehyde 5.1a to cyanohydrin 5.2a catalyzed by diketopiperazine 5.3 originally reported by Inoue and co-workers.36,202 Three limiting models for asymmetric induction (A, B, & C) have been proposed.
In this context, Houk and co-workers performed a computational study in 2009 aimed at providing a unified mechanistic picture of the 5.3-catalyzed asymmetric hydrocyanation reaction.206 Their study began with an assessment of achiral model systems consisting of imidazole(s), hydrogen cyanide, and methanol was intended to approximate the incorporation of product 5.2a into the active catalyst. Three preliminary DFT findings informed their initial model: (1) in the presence of two methanol molecules, the barrier for isomerization of H–CN to CN–H was only about 1 kcal/mol in the gas phase; (2) imidazolium was determined a more effective H-bond donor for aldehyde activation than the peptidic core of 5.3; and (3) transition states with a 2:2:1 stoichiometry of imidazole/methanol/HCN were lower in energy. Together, these data led to TS1 (Figure 80), which was the lowest energy transition state located in an assessment of multiple possibilities with different ratios of methanol and HCN.
Figure 80.
Computed structures of TS1 and D1 led to the transition state models TS2 and TS3, which provide a basis for stereoselectivity in the 5.3-catalyzed hydrocyanation of benzaldehyde 5.1a. B3LYP/6-31G* geometries and energies reported. Adapted in part with permission from reference 206. Copyright 2009 American Chemical Society.
To account for the dimeric nature of the active catalyst, the authors used MM simulations to search for low-energy dimer conformations beginning from three initial arrangements—one head-to-head and two different head-to-tail—the outputs of which were optimized using DFT. All three converged to head-to-tail dimer D1 as the global minimum (Figure 80), in which the two 5.3 amides are engaged in reciprocal N–H•••O H-bonds and the imidazole sidechains H-bond with one another and the peptide core. To derive transition states for the enantioselective hydrocyanation of 5.1a, features of both TS1 and D1 were combined, replacing a methanol equivalent with (R)-5.2a to account for the known product complexation, and the resulting structures were optimized to transition states at −20 °C. The result was TS2 and TS3, wherein one imidazole activates 5.1a through H-bonding while the other imidazolium delivers the cyanide nucleophile (Figure 80). TS2, which leads to the experimentally observed (R)-cyanohydrin, is favored over (S)-selective TS3 by 1.8 kcal/mol, in good agreement with the observed enantioselectivity (ΔG‡ = 2.1 kcal/mol). This stabilization predominantly results from an extended edge-to-face π-interaction network involving the Phe sidechain of 5.3 and the phenyl groups of both 5.1a and (R)-5.2a. These interactions are largely absent in (S)-selective TS3.
Danda and co-workers reported that adding catalytic quantities of (R)-5.2a to the reaction results in constant, high levels of selectivity over the course of the reaction, whereas asymmetric autoinduction is observed when the opposite enantiomer is included.203 The authors re-optimized the transition states including (S)-5.2a as the additive and found that (R)-selective TS4 and (S)-selective TS5 now only differ by 1.1 kcal/mol, resulting in reduced selectivity (Figure 81). Additionally, TS4 was found to be about 1 kcal/mol higher in energy than TS2 due to differences in the π-interaction network. Thus, the (R)-5.2a•5.3 complex is expected to outcompete the (S)-complex, which is consistent with Danda’s observations.
Figure 81.
Computed structures of TS4 and TS5. B3LYP/6–31* geometries and energies reported. Adapted in part with permission from reference 206. Copyright 2009 American Chemical Society.
Taken together, these computational data provide a more unified model for enantioinduction in this important, peptide-catalyzed reaction. Thematically, these studies reinforce that excellent catalytic activity is made possible by the ability of peptide 5.3 to engage in productive noncovalent interactions with its substrate.
5.1.2. Silylcyanation of Aldehydes.
An alternate peptide-catalyzed method for the cyanation of aldehydes was reported in 2012 by Kudo and co-workers.207 Drawing inspiration from the Juliá–Colonna epoxidation (see Section 2.1),26 their strategy was to employ helical polypeptides to mediate the enantioselective cyanosilylation of benzaldehyde 5.1a to TMS-protected cyanohydrin 5.4a (Figure 82). Catalyst platform 5.5 was designed as a PEG-PS-supported poly(l-Leu) sequence, which is known to adopt an α-helical conformation once a critical chain length is met. The authors chose to use a solid support for ease of synthesis and handling and to facilitate solubility. Moreover, much like in the Juliá–Colonna epoxidation, a free N-terminus was proposed to be crucial to catalysis in this cyanosilylation reaction.
Figure 82.
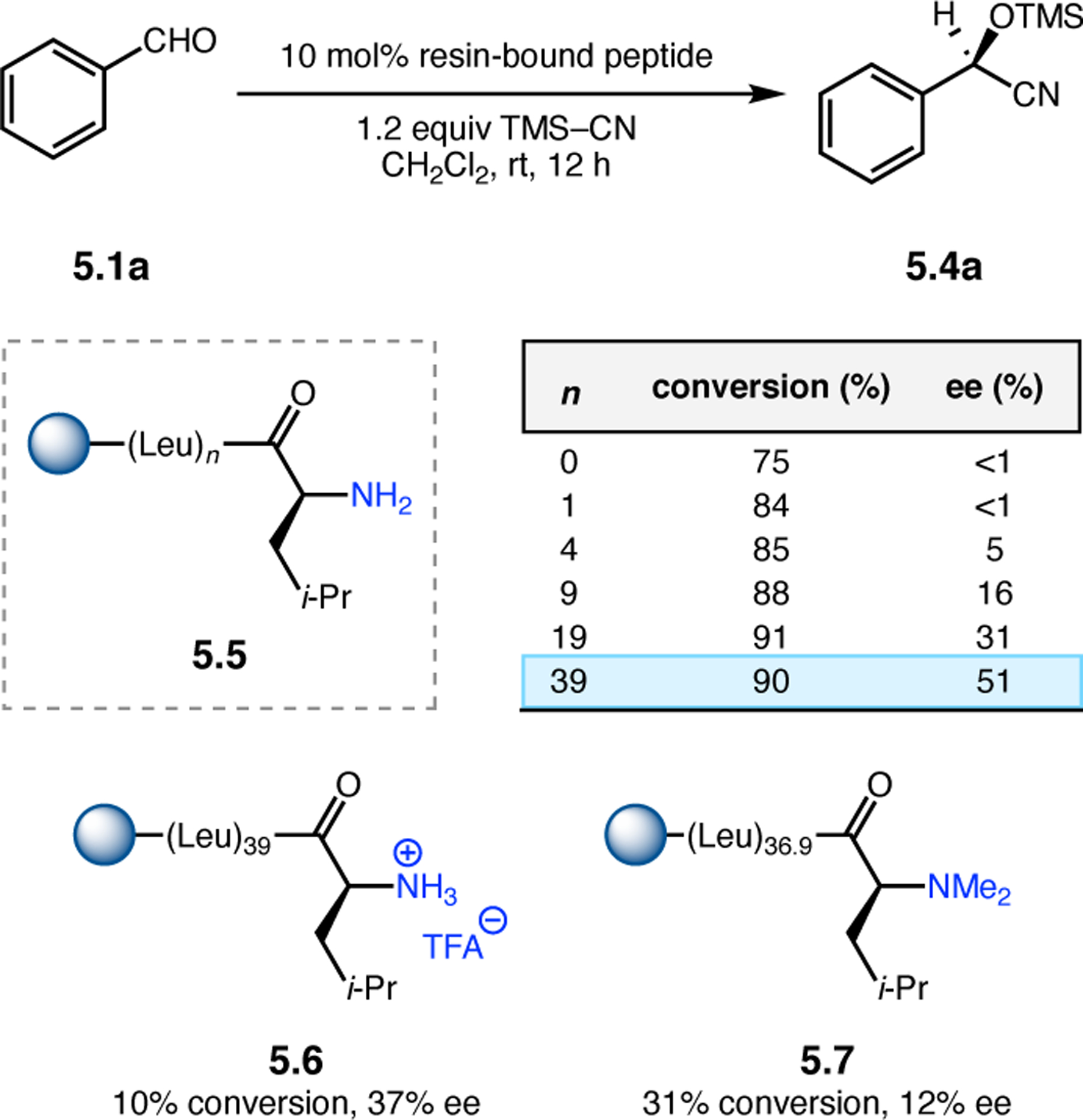
Optimization of the asymmetric cyanosilylation of benzaldehydes catalyzed by resin-bound helical polypeptides 5.5.207
An investigation of catalysts 5.5 with varying poly(l-Leu) chain lengths revealed the importance of the helical structure to both reactivity and selectivity (Figure 82). Short sequence peptides (n < 5) provided reasonable conversions to 5.4a with very low levels of ee. Of particular note, a single resin-bound Leu residue provides racemic product 5.4a, suggesting that the point chirality of this amino acid alone is unable to induce asymmetry. Low, yet measurable ee values were obtained with chain lengths of 10–20 Leu residues, but the highest observed enantioselectivities required longer poly(Leu) sequences (n > 35). Using 40-mer 5.5, TMS-protected cyanohydrin 5.4a was obtained in 90% conversion and 51% ee. Poly(l-Leu) sequences were uniquely suited to this reaction, as poly(l-Ala) and poly(l-Glu(Bn)) provided reasonable conversion with low ee values. The authors used TFA-salt 5.6 and dimethylamine 5.7 as control peptides to probe the effect of the N-terminus. Both peptides exhibit low reactivity and selectivity compared to 5.5, suggesting that a free amino group is required to achieve tractable levels of enantioselectivity.
A key difference between this poly(Leu)-catalyzed aldehyde cyanosilylation and the classical Juliá–Colonna epoxidation is that, in this reaction, good conversion can be achieved independent of the chain length, while useful ee values are only possible with longer sequences. The opposite is true in the Juliá–Colonna reaction; good enantioselectivity can be achieved with only a few amino acids, but increasing the chain length increases yields.208 This is likely a manifestation of the different reaction mechanisms and modes of asymmetric induction.
Lowering the reaction temperature to −20 °C, peptide 5.5 was able to deliver 5.4a in quantitative conversion and 62% ee (compared to 90% conversion and 51% ee at ambient temperature). These conditions were then used to investigate the substrate scope of this reaction (Figure 83). On preparative scale, isolated yields and enantioselectivities of 5.4 were recorded following hydrolysis of the TMS ether and subsequent O-acetylation. Peptide 5.5 addressed a modest scope of benzaldehydes 5.1 using 1.2–2.0 equivalents with good yields and ee values ranging from 31–62%.
Figure 83.
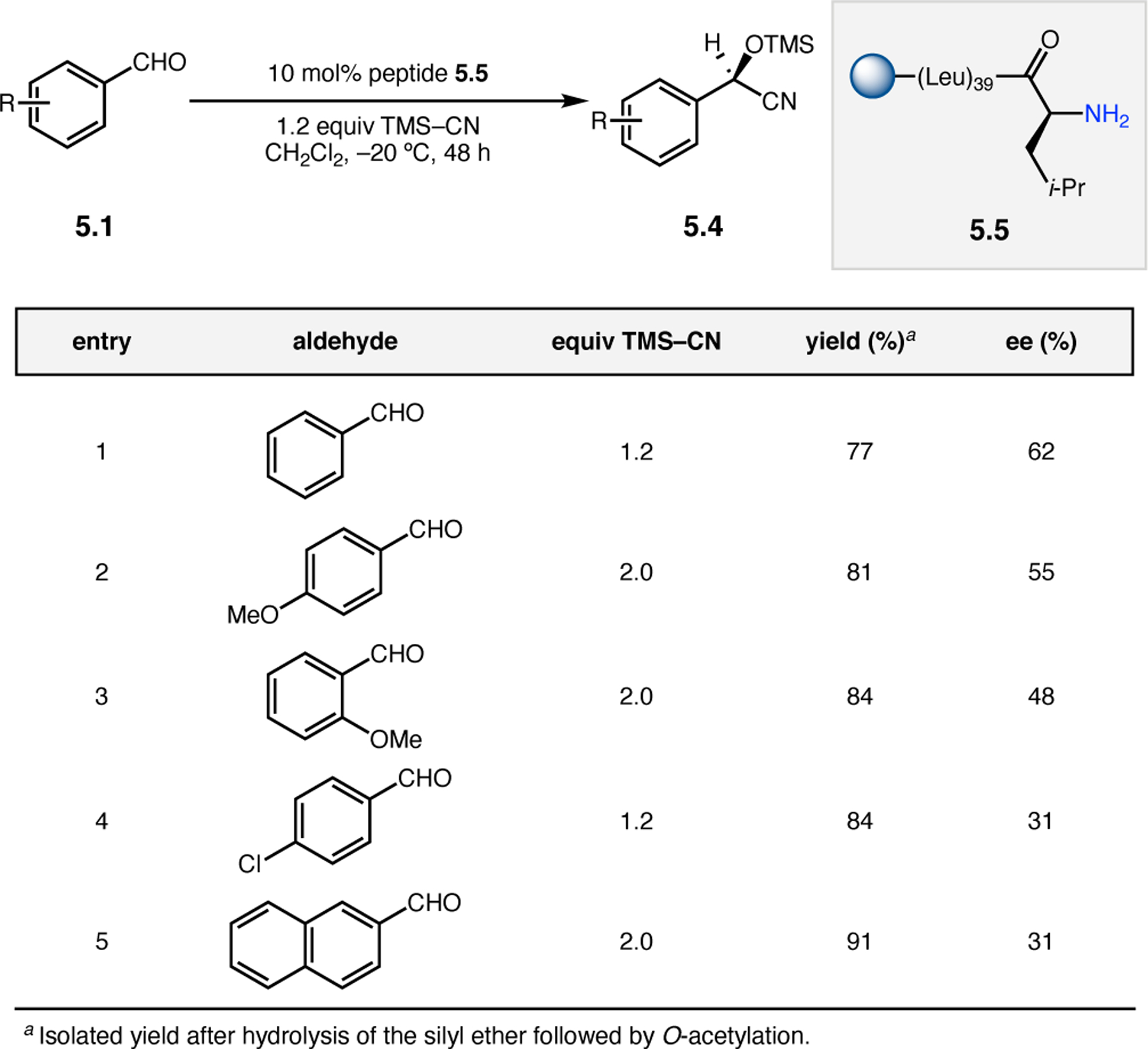
Scope of the 5.5-catalyzed asymmetric cyanosilylation of benzaldehydes 5.1.207
5.2. Organozinc Additions to Aldehydes.
Since the 1990s, short sequence peptides have been widely applied as chiral ligands for various asymmetric organometallic additions to carbonyl compounds.209,210 An archetypal scaffold for these ligands is a dipeptide sequence capped on the N-terminus with a Schiff base moiety for metal chelation. Originally reported by Inoue and co-workers for the Ti-catalyzed, asymmetric hydrocyanation of aldehydes,211 the impactful expansion of this general ligand framework was pioneered by Hoveyda, Snapper, and co-workers throughout the 2000s, which led to the development of highly enantioselective organometallic 1,2-212–214 and 1,4-addition215,216 reactions. These methodologies helped to establish the dipeptide-based ligand as a reliable and robust means of achieving asymmetric induction in challenging scenarios.
Building off of this body of work, Park and co-workers reported a peptide-catalyzed method for the enantioselective alkylation of aldehydes with dimethylzinc.217 Their strategy was to optimize a dipeptide scaffold—without an N-terminal Schiff base—that is able to chelate to and activate dimethylzinc for addition to prochiral aldehydes, such as 5.8, providing access to enantioenriched secondary alcohols 5.9 (Figure 84). All of the peptides examined adhered to the general sequence R2-Xaa-Pro-OMe (5.10), in which the N-terminus was either mono- or dialkylated. The catalyst library was evaluated in the addition of dimethylzinc to benzaldehyde 5.8a in toluene at ambient temperature.
Figure 84.
Optimization of a dipeptide-based ligand for the asymmetric addition of dimethylzinc to benzaldehyde 5.8a.217
When Xaa was d-Phg, the degree of alkylation at N-terminus had an interesting effect on enantioselectivity (Figure 84, entries 1–3). While the monobenzyl amine variant delivered 5.9a in 35% yield and 53:47 er (entry 1), N-benzyl-N-methyl amine gave a notable boost in yield to 85% with equal and opposite enantioselectivity (entry 2). However, the dibenzylamine-containing peptide returned 5.9a in 85% yield and 88:12 er, restoring (and amplifying) the sense of asymmetric induction (entry 3). These data suggest that the reaction is quite sensitive to N-terminal substitution. The effect of the C-terminal ester was probed next (entries 3–6), and selectivity was found to decrease significantly with increasing steric bulk of the ester substituent. The tert-butyl ester also provided an enantiodivergent outcome (entry 6). Ligands containing d-Phe (entry 7) and d-Ala (entry 8) as the N-terminal residue were modestly less selective than the d-Phg variant (entry 3), but the Gly variant delivered nearly racemic product (entry 9). Epimerizing the N-terminal residue to l-Ala improved the er to 92:8 at the expense of some yield (entry 10), and l-Leu was similarly reactive but less selective (entry 11). Benzyl-protected l-Asp was found to be optimal at this position, providing 5.9a in 90% yield and 94:6 er (entry 12). The yield and er could be further improved by modification of the N-terminal dibenzylamine (entries 13–15), with bulky 3,5-di-tert-butylbenzyl variant 5.10a delivering the product in 95% yield and 97:3 er (entry 15).
Having identified 5.10a as the leading ligand, the authors proceeded to examine the substrate scope for this transformation. Peptide 5.10a was effective in the dimethylzinc addition to various benzaldehydes 5.8, providing secondary alcohols (S)-5.9 in 65–94% yield and 94:6 to 96:4 er (Figure 85). The authors also demonstrated that excellent yield and er could be obtained with only 5 mol% catalyst loading (entry 2).
Figure 85.
Scope of the 5.10a-catalyzed, asymmetric organozinc addition to benzaldehydes 5.8.217
5.3. Ester Hydrolysis & Alcoholysis
Lipases are an important class of hydrolytic enzymes that have been widely employed in asymmetric synthesis,218 even on industrial scale.219 These enzymes often leverage a catalytic triad of Ser, His, and Asp residues that are precisely oriented in the active site to enable the rapid and highly selective hydrolysis and alcoholysis of esters.220,221 This mechanistic framework has inspired efforts to develop synthetic, small molecule catalysts that expand upon the native activity and substrate scope of lipases. Peptide-based catalysts are well-suited for these goals due to their ability to fold into well-defined secondary structures, wherein catalytically active residues are oriented in close proximity to one another and to H-bond donor/acceptor groups of the peptide backbone. This Section summarizes recent campaigns to design peptide-based catalysts that mimic lipase activity.
5.3.1. Solvolysis of Formylphenyl Esters.
In 2016, Kudo and co-workers devised a strategy to develop synthetic catalysts with lipase-like hydrolytic activity.222 They envisioned a scenario in which a bifunctional oligopeptide containing catalytic Pro and His residues could mediate the hydrolysis of formylphenyl esters 5.11 to the corresponding carboxylic acid 5.12 and 4-formylphenol (Figure 86). Building upon their previous work in Michael addition chemistry (see Section 7.2), they proposed that the N-terminal Pro residue could potentially engage ester 5.11 through the reversible formation of an iminium ion intermediate. This intermediate would subsequently be hydrolyzed by the C-terminal His residue via either a Lewis base or Brønsted base activation mechanism.
Figure 86.
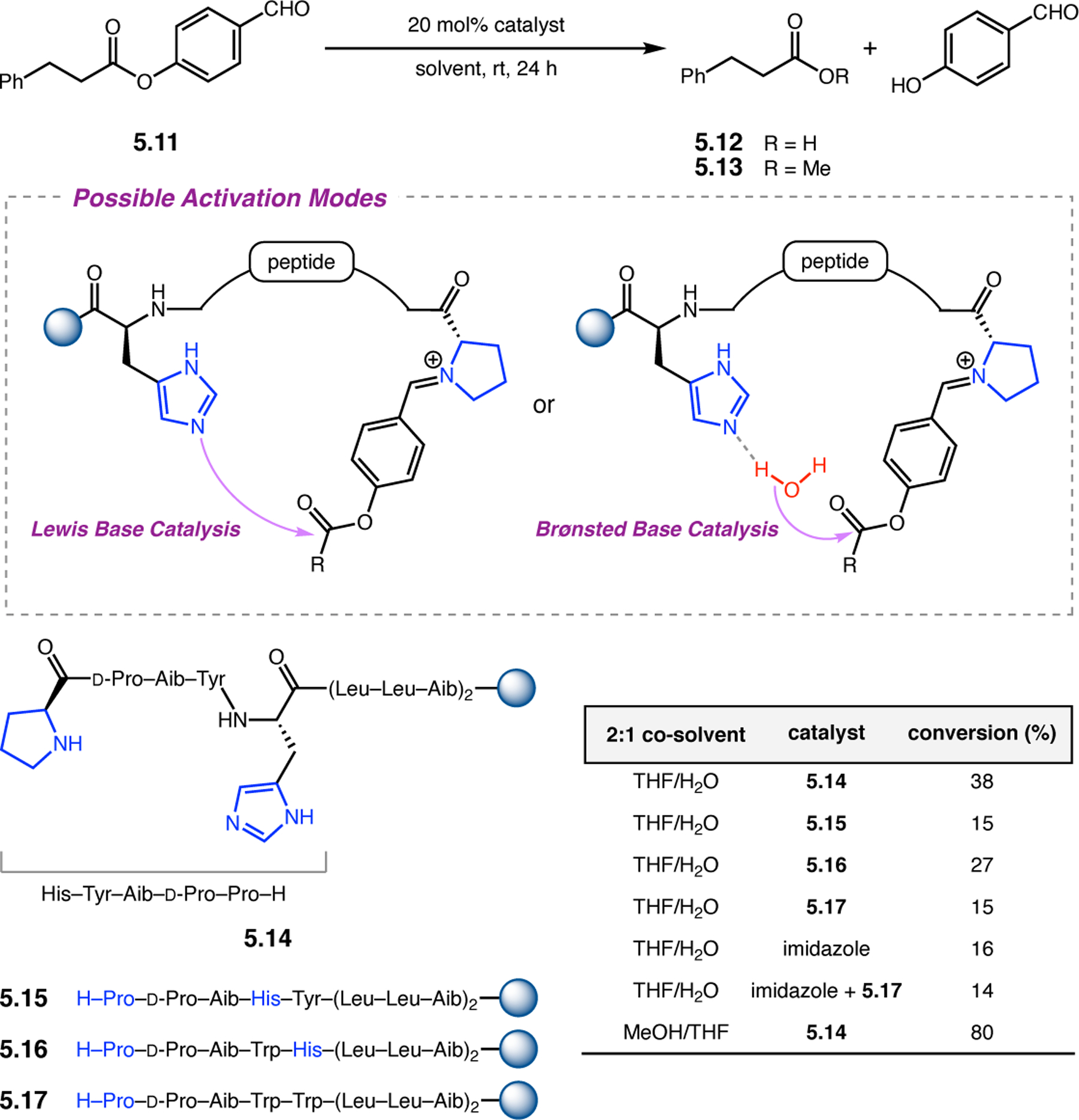
Development and optimization of a resin-bound, bifunctional peptide catalyst for the hydrolysis of formylphenyl esters 5.11.222
This mechanistic hypothesis was investigated using resin-bound peptides in which the active His and Pro residues were interspersed with a β-turn biased sequence of three amino acids.223 The catalytic pentapeptide was attached to the ambiphilic resin through a helical linker. Following some initial optimization, the authors identified peptide 5.14, which was able to catalyze the hydrolysis of 5.11 in 38% conversion (Figure 86). The precise spacing of the Pro and His residues was found to be important, as peptide 5.15, in which the Pro and His are one-residue closer together, was notably less active. Moreover, substituting out the Tyr residue of 5.14 proved modestly detrimental to catalysis. For example, Trp-containing 5.16 delivered carboxylic acid 5.12 in 27% conversion. Lacking a His residue, peptide 5.17 was significantly less reactive, providing only 15% conversion to the products. Including 20 mol% of imidazole along with 5.17 provided no advantage over the reaction catalyzed by imidazole alone, suggesting that the peptide-based catalyst requires both the Pro and His residues in order to meaningfully accelerate the reaction. Additionally, the 4-cyano variant of ester 5.12 exhibited low reactivity, providing evidence in support of the putative iminium ion intermediate. Replacing the water co-solvent with methanol resulted in increased reactivity, as peptide 5.14 delivered methyl ester 5.13 in 80% conversion.
Recognizing the lipases are principally used for hydrolytic kinetic resolutions of esters, the authors sought to demonstrate the viability of their approach in the kinetic resolution of 2-formylphenyl ester 5.18 (Figure 87). Preliminary results using catalyst 5.14 under the methanolytic conditions led to a very modestly selective kinetic resolution, in which methyl ester 5.19 was formed in 74% conversion and 7% ee, with 5.18 being enriched to 11% ee. These initial results establish proof-of-principle that alcoholytic reactions can be mediated by small molecule peptide catalysts and set the stage for further optimization.
Figure 87.
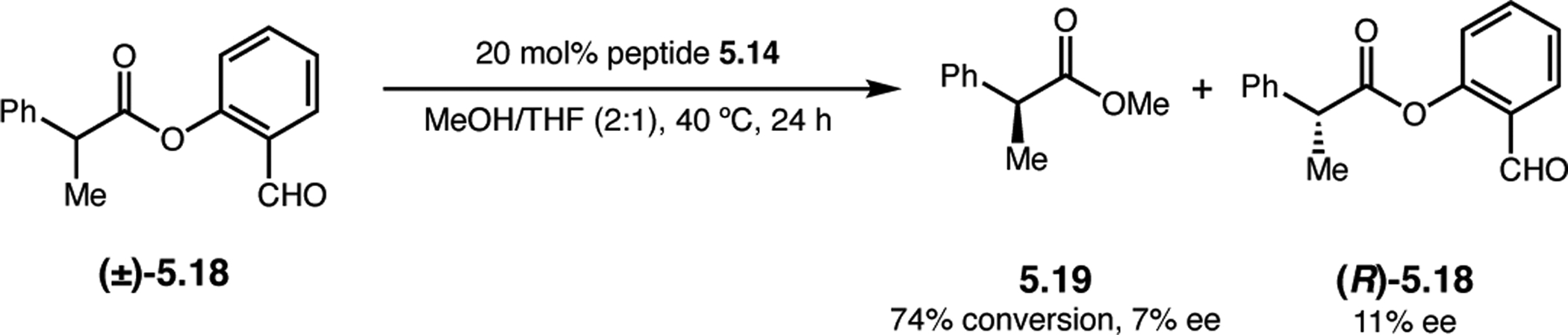
Preliminary results for a hydrolytic kinetic resolution.222
5.3.2. Ring-Opening DKR of Oxazolones.
Oxazol-5(4H)-ones, such as 5.20, are versatile synthetic building blocks due to their multifaceted reactivity profiles that provide access to a variety of useful heterocyclic frameworks.224 These cyclodehydrated amino acid derivatives have anomalously acidic α-C–H bonds (pKa ~ 9), which is due to aromatic stabilization of the oxazole enolate conjugate base (5.21, Figure 88). This elevated acidity allows for rapid, autocatalytic racemization in solution, making oxazolones 5.20 prime candidates for ring-opening dynamic kinetic resolution (DKR) reactions, a process that provides access to enantioenriched α-amino acid derivatives 5.22. Various catalyst types have been employed in alcoholytic DKRs of oxazolones, ranging from transition metal complexes225 and organocatalysts226,227 to lipase enzymes.228 Peptide-based catalysts have also been developed for this purpose.
Figure 88.
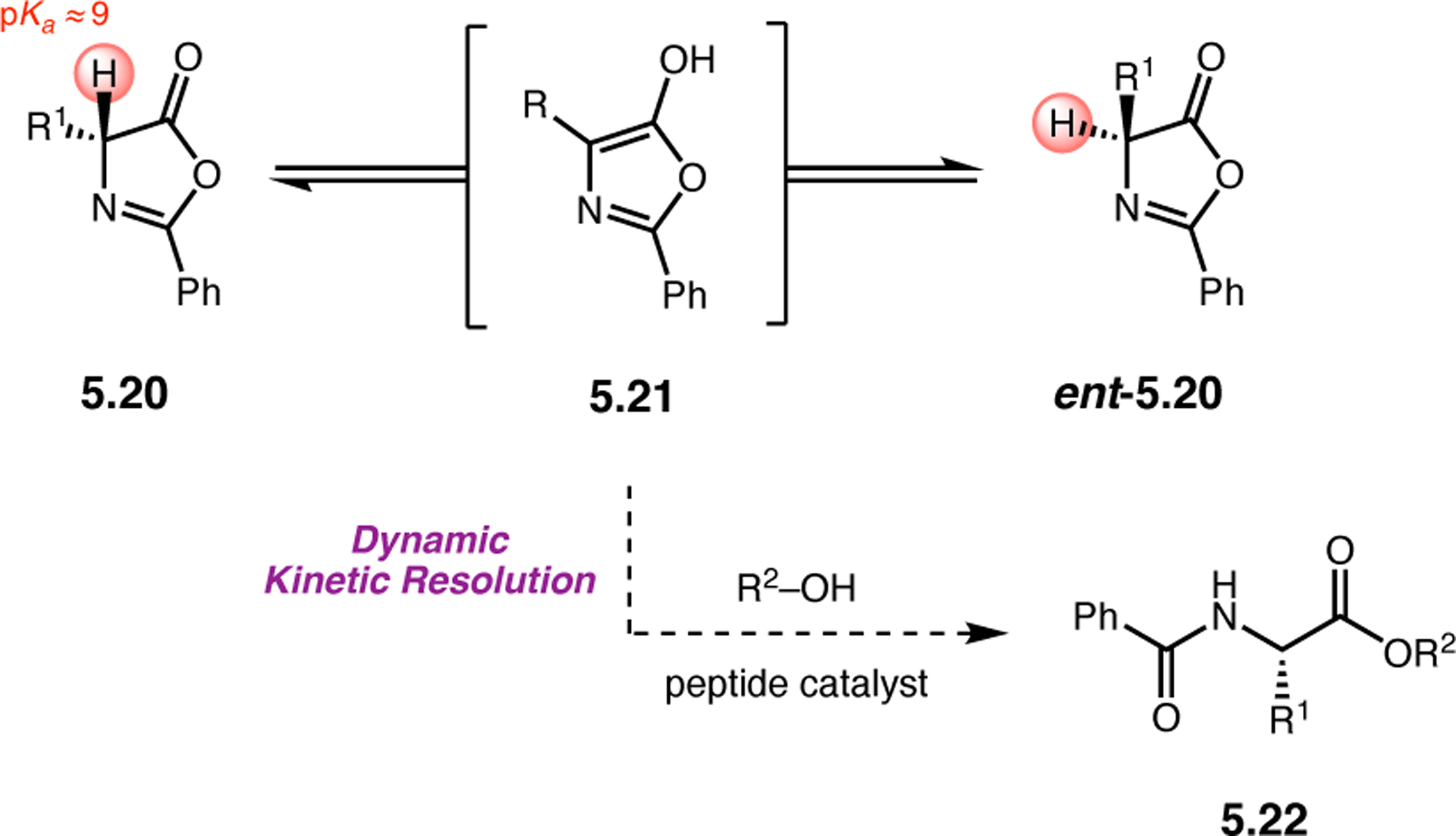
Peptide-catalyzed, alcoholytic ring-opening DKR of oxazol-5(4H)-ones 5.20.
In one of the earlier examples of an oxazolone ring-opening DKR, Hua and co-workers reported that diketopiperazine 5.3, the lead catalyst in Inoue’s benzaldehyde hydrocyanation methodology (see Section 5.1),36 was also able to catalyze the conversion of racemic oxazolone 5.20a to protected Phe-derivative 5.22a (Figure 89).229 In the absence of any chiral additive, 5.3 provided only very low levels of enantioinduction in this reaction. Based on mechanistic studies of the 5.3-catalyzed hydrocyanation of benzaldehyde suggesting that the active catalyst could be a complex of 5.3 and cyanohydrin 5.2,203 the authors postulated that a chiral additive might enhance the selectivity of 5.3 in the ring-opening reaction of 5.20a. Indeed, using 2 equivalents of diisopropyl-l-tartrate as an additive, 5.3 delivered (S)-5.22a in quantitative conversion and 27–39% ee depending on the nature of the alcohol nucleophile. While only modestly selective, this study provided a foothold for future developments.
Figure 89.
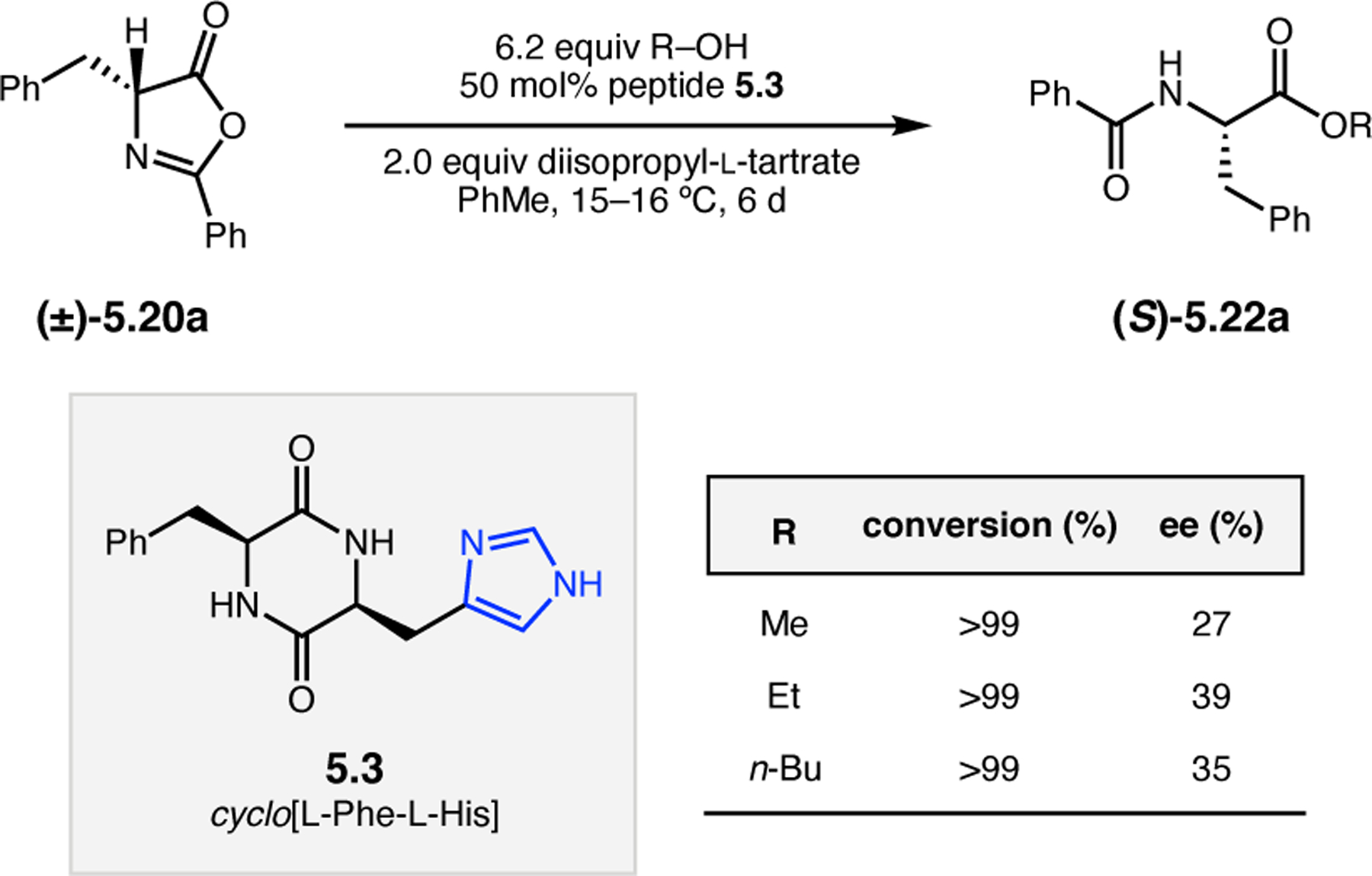
Diketopiperazine-catalyzed, alcoholytic ring-opening DKR of oxazolone 5.20a.229
In 2014, our group reported a highly selective methanolytic DKR of oxazolones 5.20 catalyzed by tertiary amine-containing tetrapeptides.230 The catalyst library was designed around general scaffold 5.23, in which a β-dimethylaminoalanine (Dmaa) residue is embedded at the N-terminus of a well-defined β-turn secondary structure (Figure 90). This general arrangement provided the possibility for a multifunctional activation mechanism, wherein the tertiary amine side chain of Dmaa engages and selectively delivers methanol to one enantiomer of (±)-5.20, while the peptide backbone stabilizes charge buildup on the substrate through noncovalent interactions.
Figure 90.
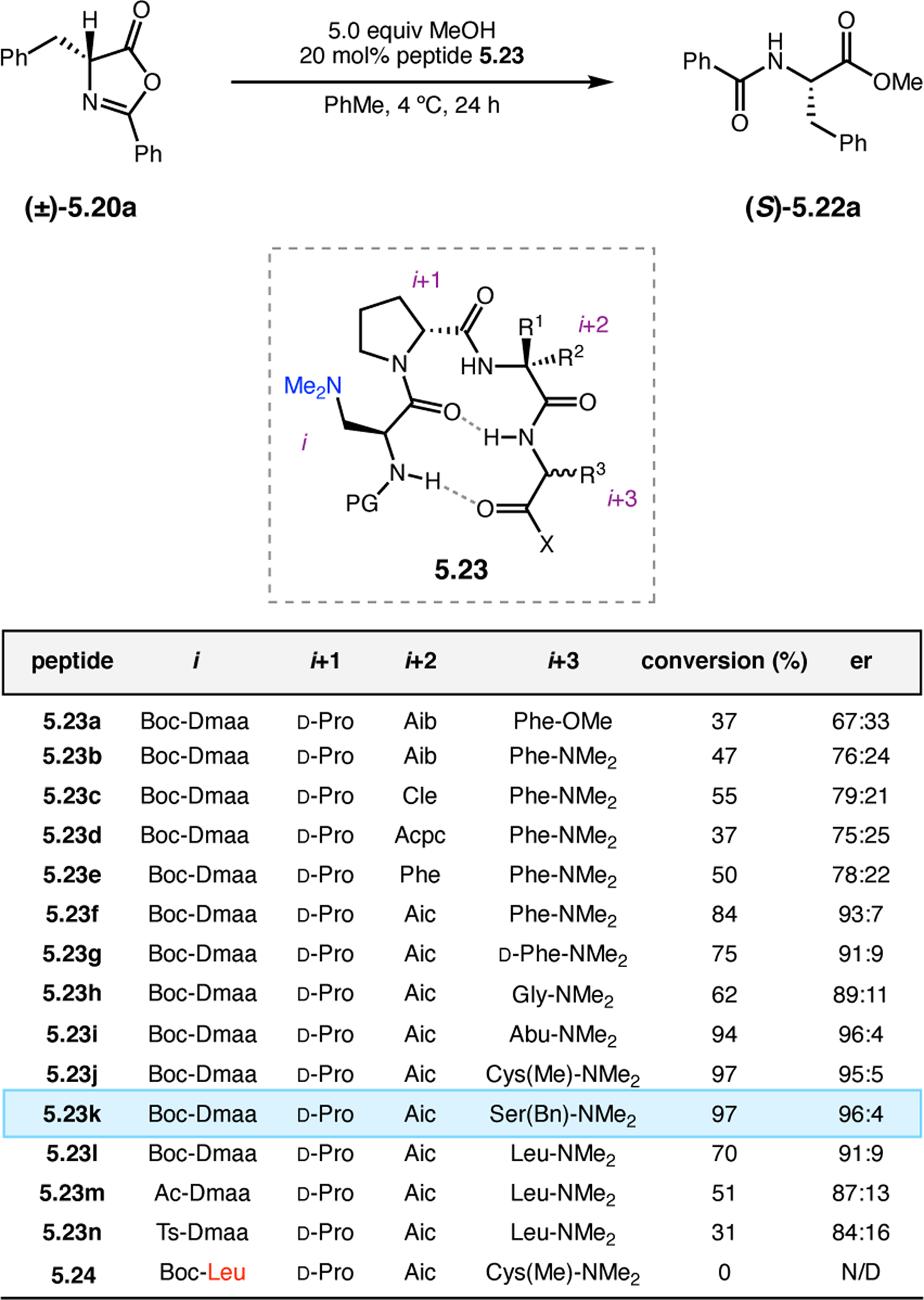
Optimization of a β-turn biased peptide catalyst for the methanolytic ring-opening DKR of oxazol-5(4H)-one 5.20a.230
The library consisted of 14 Dmaa-containing peptides biased toward a type IIʹ β-turn by incorporation of a central d-Pro-Xaa sequence motif (e.g., 5.23, Figure 90).223 These catalysts were evaluated in the ring-opening DKR of oxazolone 5.20a in the presence of 5 equivalents of methanol. Peptide 5.23a, which possesses an archetypal d-Pro-Aib β-turn sequence and a Phe residue at the i+3 position, delivered 5.22a in 37% conversion and 67:33 er. Changing the C-terminal cap to a dimethyl amide in 5.23b led to a modest improvement in conversion to 47% and a notable boost in selectivity to 74:26 er. This is possibly due to the increased H-bond acceptor ability of the amide relative to the ester, making the 14-membered ring β-hairpin H-bond more favorable. In an assessment of the effect of the i+2 residue, 2-aminoindane-2-carboxamide (Aic)-containing 5.23f stood out as significantly more reactive and selective than the other variants (5.23b–e), providing 5.22a in 84% conversion and 93:7 er. Exchanging the i+3 Phe with d-Phe in 5.23g only slightly eroded the yield and er, and an evaluation of other i+3 residues (5.23f–l) led to the identification of Ser(Bn)-containing 5.23k, which delivered 5.22a in 97% conversion and 96:4 er. In an attempt to further strengthen the β-hairpin H-bond, different N-terminal protecting groups were assessed (5.23l–n), but none were as effective as Boc. Lastly, peptide 5.24, in which the Dmaa residue is replaced with Leu—its C-isostere—did not catalyze the methanolysis reaction.
A sequence truncation study was performed to examine the effect of the lead catalyst’s residues on the reactivity and selectivity observed in the methanolysis of 5.20a (Figure 91). Compared to tetrapeptide 5.23k, tripeptides 5.25 were notably less reactive and selective. However, secondary amide-containing 5.25b was significantly more reactive and selective than its tertiary amide counterpart 5.25a, providing 5.22a in 62% conversion and 86:14 er—a value on par with some of the less selective tetrapeptides. This perhaps speaks to the importance of the 10-membered ring β-turn H-bond to catalysis, as 5.25a is able to nucleate that interaction while 5.25a cannot. Dipeptides 5.26 provided only modestly enantioenriched product in low conversion, while Dmaa monomer 5.27 was essentially inactive. It is noteworthy that 5.27 provides very low levels of selectivity in favor of the opposite enantiomer of 5.22a, suggesting that the secondary structure of higher-order peptides overturns the inherent preference of the catalytic monomer.
Figure 91.
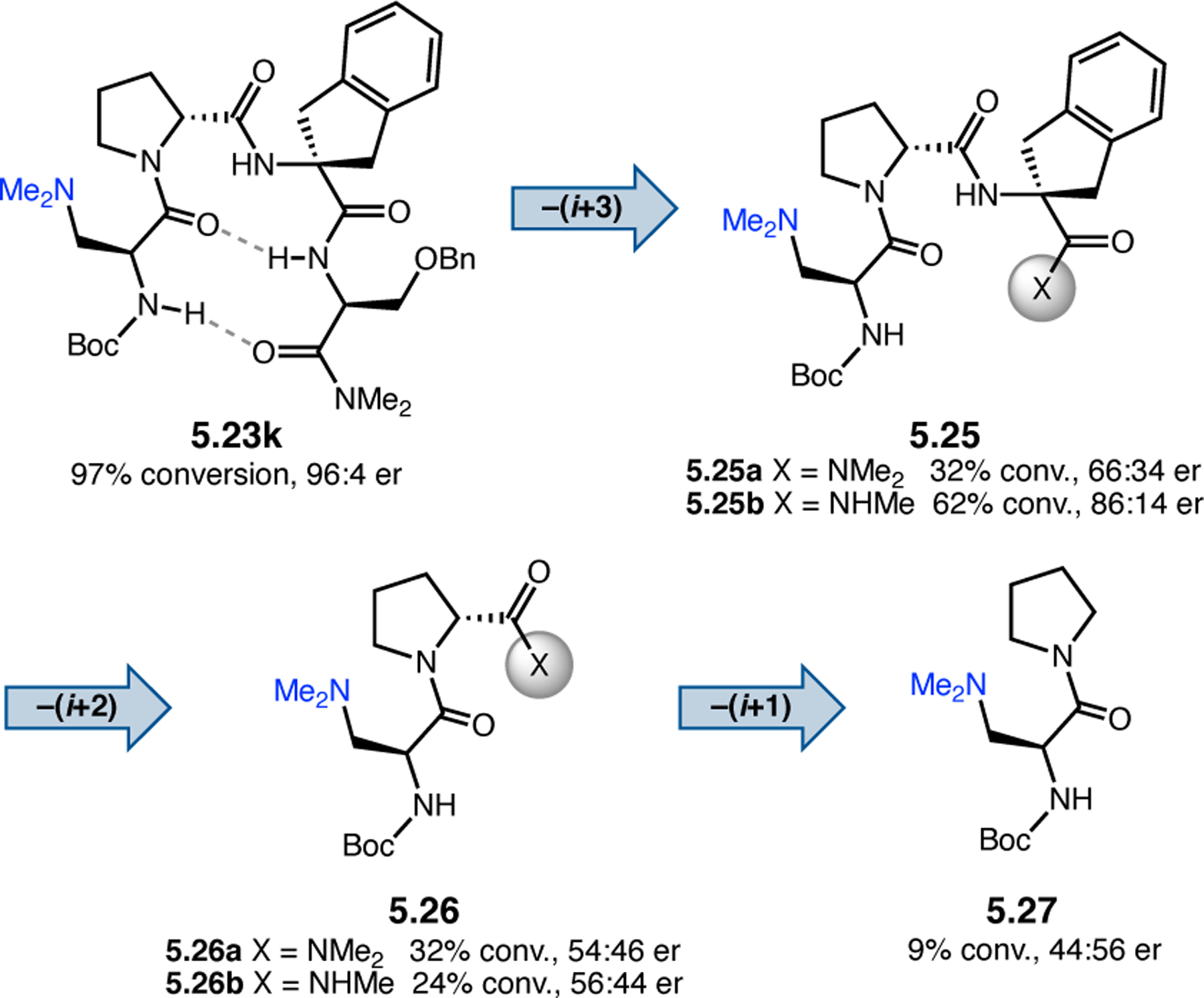
Sequence truncation study showing the effect of each residue on the reactivity and selectivity in the methanolytic DKR of 5.20a under the conditions described in Figure 90.230
Peptide 5.23k was able to address a scope of oxazolones 5.20 bearing variable substitution at the α-position (Figure 92). Benzylic and heterobenzylic substrates (entries 1–7) were processed by 5.23k with excellent yields (78–93%) and ers (88:12 to 98:2). Electron rich benzylic oxazolones (entry 6) were more selective than their electron-poor congeners (entry 7). Compared to 5.20a (entry 1), benzhydryl substitution led to only a modest decrease in selectivity to 90:10 er with a significant drop in yield to 19% (entry 8), while homologating the side chain by one methylene unit led to decreases in both yield and er (entry 9). Ester substitution at the β-position was well tolerated (entry 10), but branched alkyl substrates were generally less reactive and selective benzylic ones (entries 11–12). Yields and er values decreased with increasing substitution at both the β- (entry 11 vs. 13) and γ-positions (entry 12 vs. 14).
Figure 92.
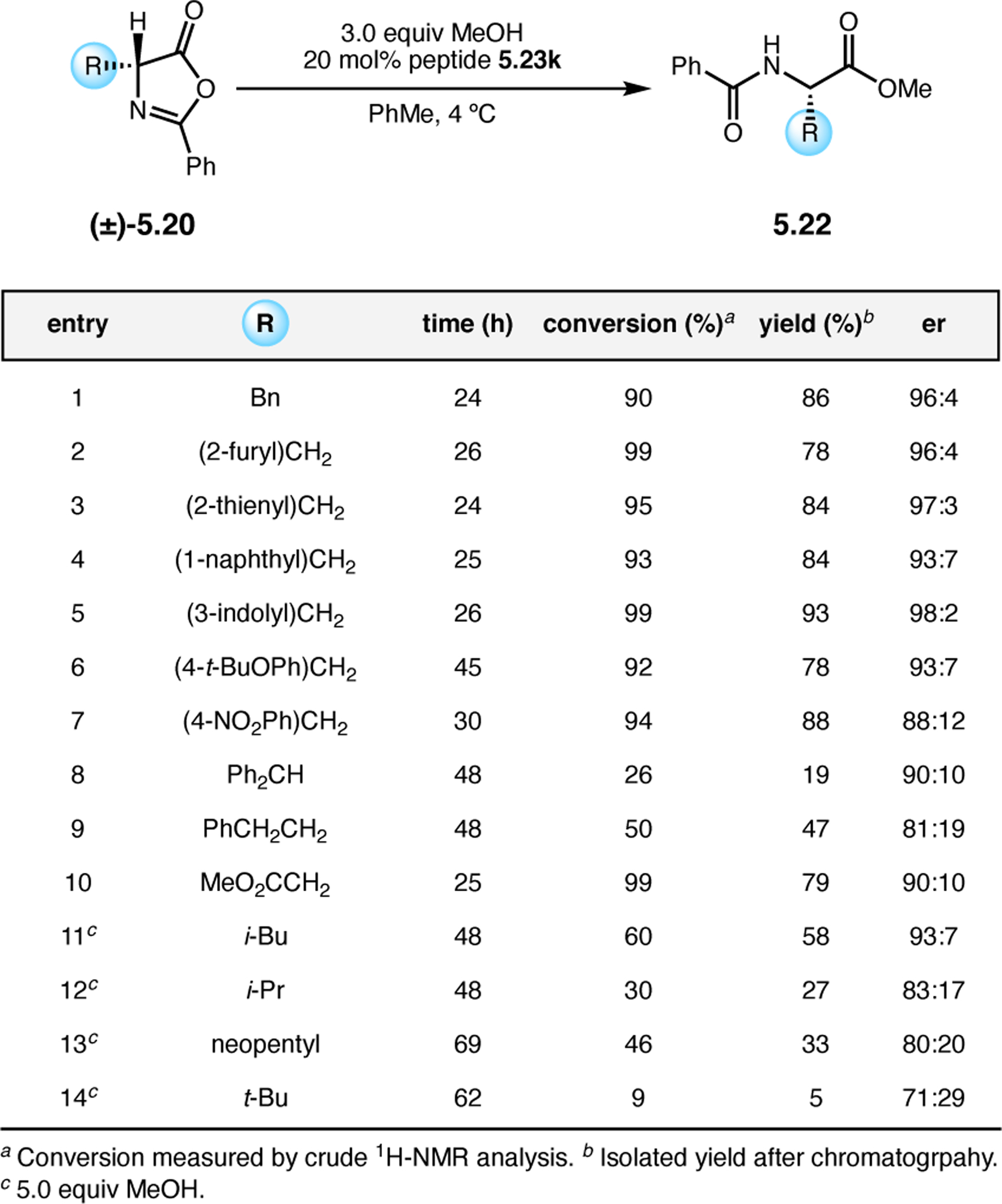
Scope of the methanolytic DKR of oxazolones 5.20 catalyzed by 5.23k.230
Based on the peptide-optimization data, substrate trends, and preliminary mechanistic experiments, a self-consistent stereochemical model was proposed that accounts for the observed absolute configuration of methyl esters 5.22 (Figure 93). In this model, the Dmaa sidechain of 5.23k delivers methanol to selectively to the re-face of (S)-5.20a, as the peptide stabilizes the transition state leading to the zwitterionic tetrahedral intermediate through charge-stabilizing H-bonds. Putative edge-to-face π-stacking between the oxazolone α-substituent and the i+2 Aic residue perhaps accounts for the catalyst’s preference for benzylic substrates. The convergence of these noncovalent interactions to both activate the nucleophile and stabilize the intermediate is reminiscent of enzymatic oxyanion holes.231 In that sense, 5.23k is proposed to operate through a biomimetic mechanism.
Figure 93.
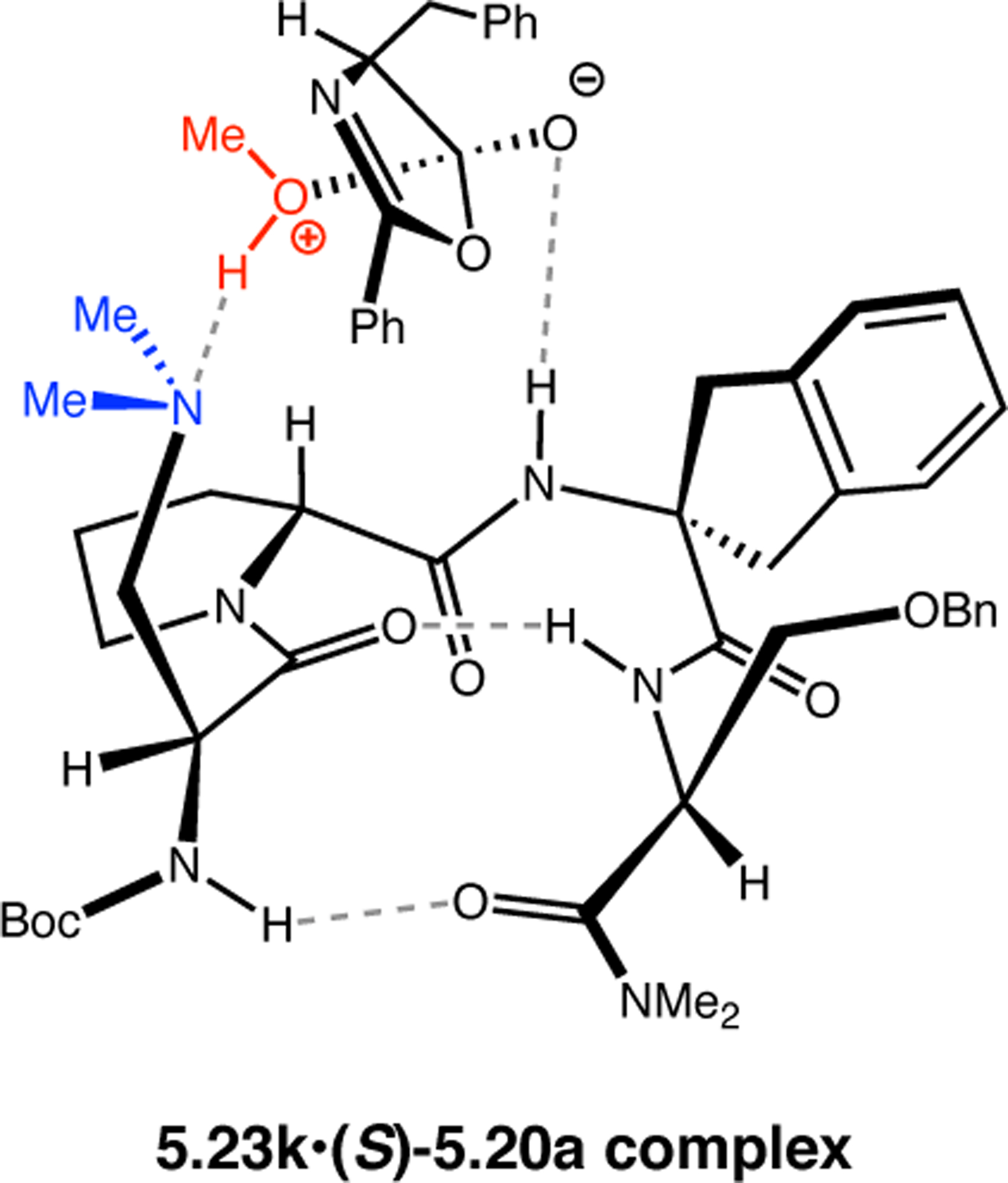
Stereochemical model for the 5.23k-catalyzed methanolysis of (S)-5.20a.230
5.4. meso-Epoxide Opening
Asymmetric ring-opening reactions of epoxides are valuable methodologies that provide access to chiral, enantioenriched alcohol derivatives, which themselves are important synthetic building blocks. These methods generally fall into two strategic categories: (1) kinetic resolutions of racemic epoxides, or (2) enantioselective ring-openings of meso-epoxides.232 Many examples of each strategy have been reported to date that implement a variety of catalyst types and architectures. While peptide-based catalysts have track record in epoxidation (see Section 2.1), peptide-mediated epoxide opening reactions have only recently been developed.
In 2014, Chimni and co-workers reported a peptide-catalyzed, asymmetric ring-opening reaction of meso-stilbene oxides 5.28 with N-phenylpiperazine 5.29 to access chiral amino alcohols 5.30 (Figure 94).233 The general catalyst design centered on short, β-turn forming sequences capped with an N-terminal thiourea moiety. In that way, the authors intended to capitalize on the demonstrated ability of thioureas to activate electrophiles through H-bonding interactions,234 as well as the folded secondary structures of peptides to provide a robust chiral environment for asymmetric induction. They proposed a model wherein the N-terminal thiourea engages the epoxide, while the peptide scaffold activates the amine nucleophile via H-bonding with backbone carbonyls (X in Figure 94).
Figure 94.
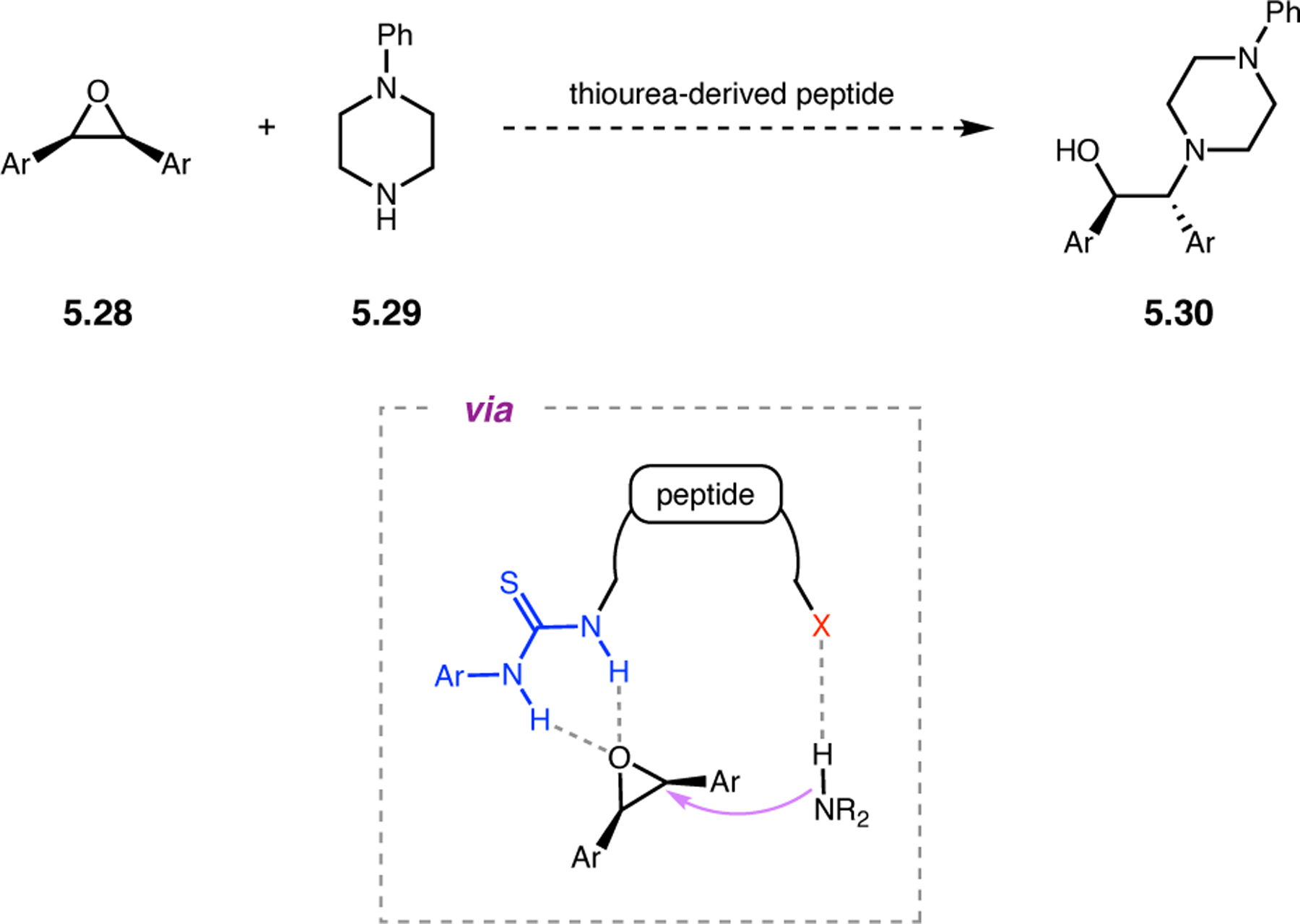
Thiourea-derived peptide catalysts for the asymmetric ring-opening of meso-stilbene oxides.233
The authors evaluated a modest library of peptides biased toward β-turn conformations by incorporation of either l-Pro-d-Pro15,235 or d-Pro-Gly236 turn motifs. The former sequence emerged as the front-runner, and optimization of the i residue led to the identification of thiourea-embedded tripeptide 5.31a, which delivered amino alcohol 5.30 (Ar = Ph) in 89% yield and 79% ee (entry 1, Figure 95). Peptide 5.31a effectively catalyzed the ring-opening of a small scope of stilbene oxides 5.28 using piperazine 5.29. Compared to the parent substrate (entry 1), substitution of Cl or Br at the meta-position of the phenyl rings was tolerated in terms of yield, but selectivity decreased notably to 50–52% ee (entries 2 & 3). Substitution at the ortho-position of the aryl groups in 5.28 was not tolerated (entry 4), nor was nitro group incorporation anywhere on the phenyl ring (entries 5–7). The authors attribute this to competitive binding of the nitro group by the thiourea of 5.31a. Piperidine, N-methyl piperazine, N-benzyl piperazine were also poorly selective in this reaction, providing the corresponding amino alcohol product in <10% ee.
Figure 95.
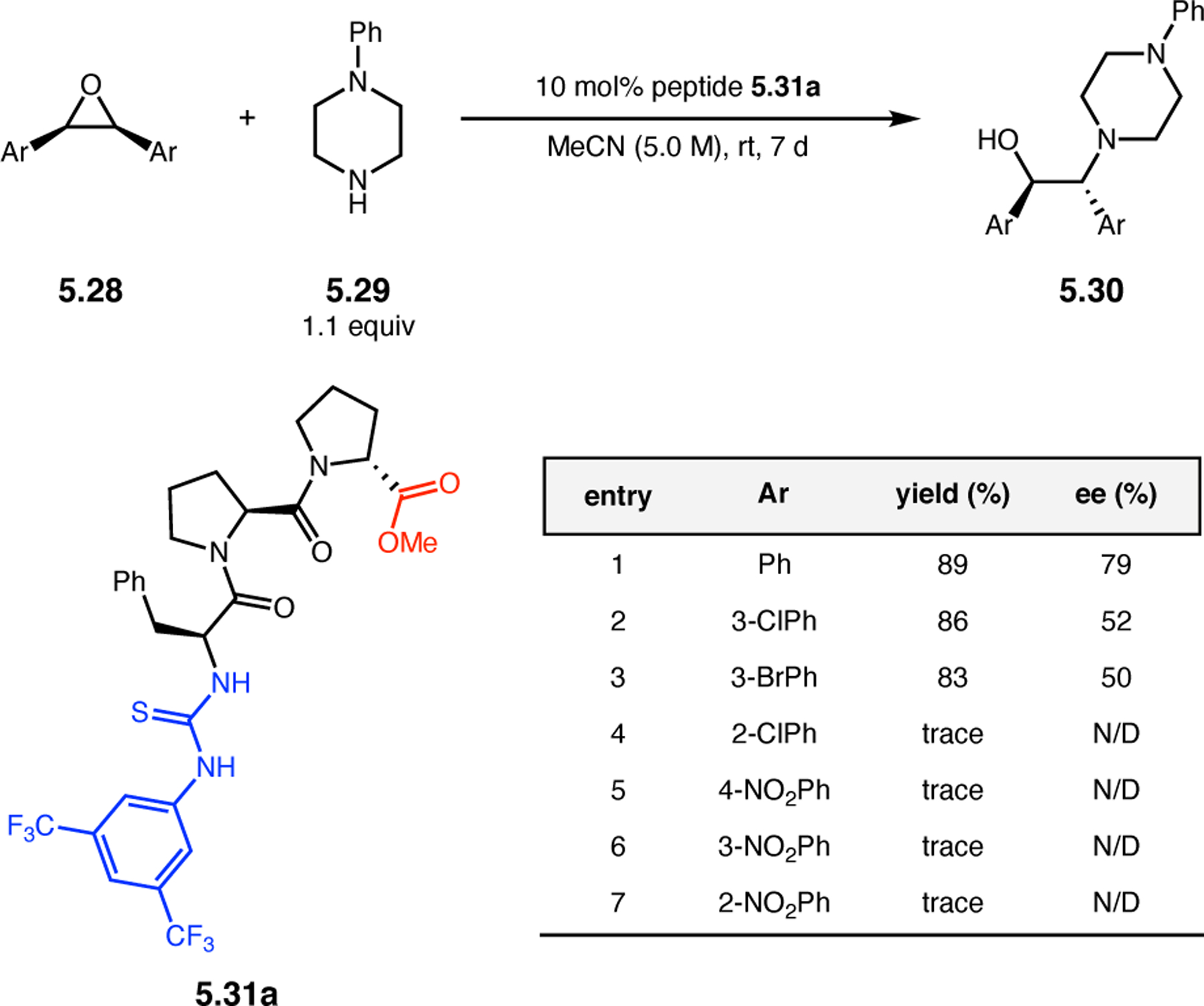
Asymmetric ring-opening of stilbene oxides 5.28 with piperazine 5.29 catalyzed by thiourea-functionalized peptide 5.31a.233
A series of epimeric catalysts were assessed under the standard epoxide-opening conditions in order to examine the effect of each stereogenic center of peptide 5.31a on the observed enantioselectivity (Figure 96). Compared to the lead catalyst, converting the C-terminal residue from d-Pro to l-Pro in 5.31b led to a notable decrease in ee from 79% to 22% (entry 1 vs. 2), suggesting that this Pro residue plays a role in the selective transition state. Peptide 5.31c, in which the central Pro residue is epimerized from d to l, gave a somewhat less drastic drop in ee to 33%. Notably, the Phe-epimer 5.31d was found to deliver 5.30a in 52% ee favoring the opposite enantiomer, which suggests that the configuration of the N-terminal Phe residue largely controls the selectivity in this reaction. These data show that the various diastereomers of 5.31 form different secondary structures that exhibit different degrees of selectivity in the epoxide opening.
Figure 96.
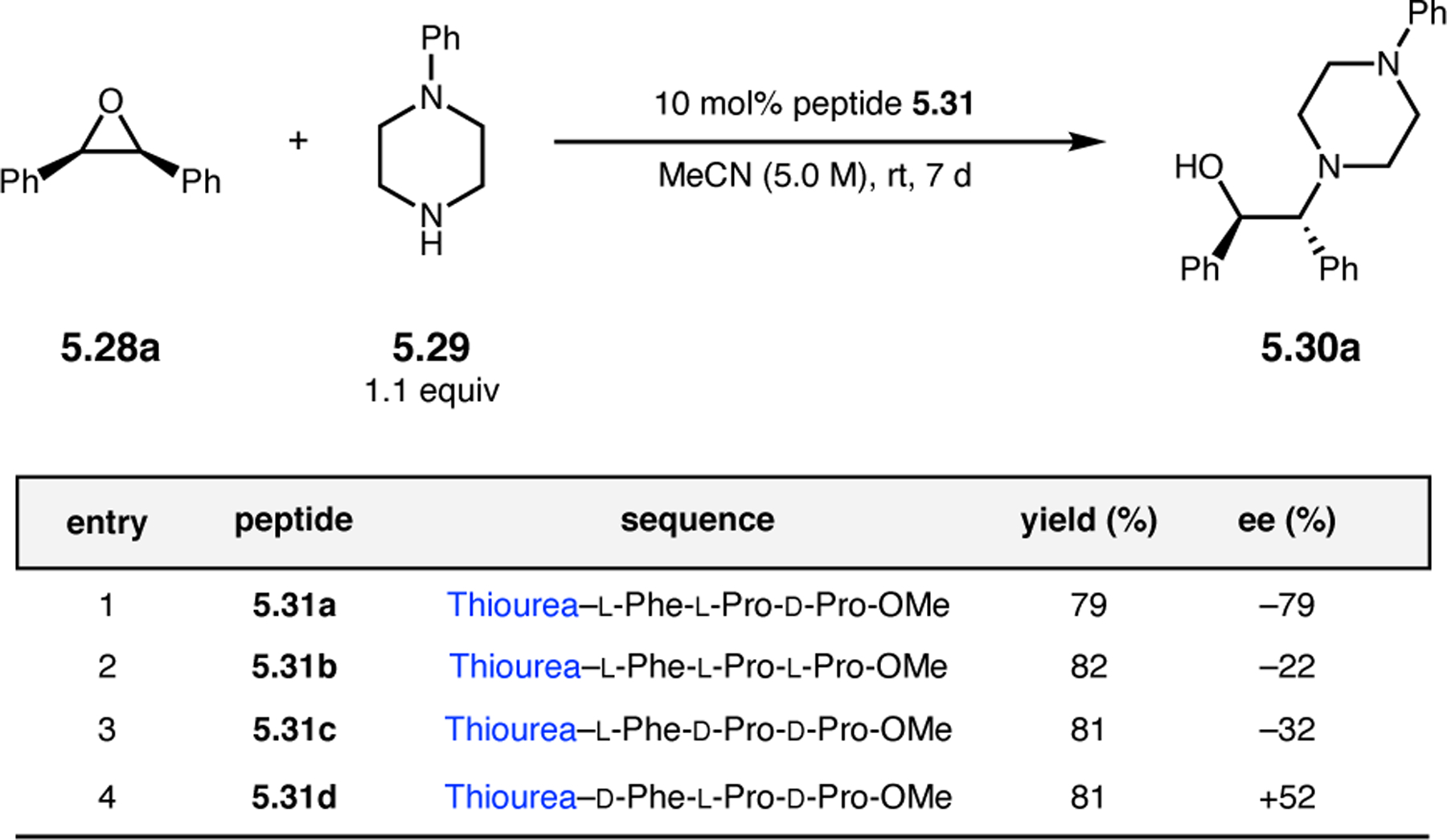
Effect of catalyst epimerization and the observation of enantiodivergence.233
6. Aldol Reaction
The aldol reaction is a powerful C–C bond-forming process, and when it is performed with control of regio-, diastereo-, and enantioselectivity, it is able to streamline the synthesis of complex molecules. Historically, asymmetric aldol reactions relied on the use of stoichiometric chiral reagents or auxiliaries. Although many of these are reliable approaches, they often require additional synthetic steps to install or remove the chiral control elements and generate stoichiometric byproducts. Therefore, catalytic variants of this powerful transformation have undergone intense development. More recently, a large body of literature on catalytic asymmetric aldol reactions employing metal-based or organocatalysts have been reported.237 Among them, the Pro-catalyzed aldol reaction is best connected to peptide-based catalysis.238 Despite the many advances in this area, certain challenges still remain unmet, such as reducing catalyst loading, increasing catalytic efficiency, improving stoichiometry of substrates, and expanding the substrate scope. To address these issues, there has been efforts to develop new peptide-based catalysts for asymmetric aldol reactions.
Many peptide-based catalysts for aldol reactions can be generally categorized into two different classes (Figure 97). In the most common design, Pro—a venerable catalyst for the aldol reaction in its own right—is positioned at the N-terminus of a peptidic scaffold, availing its free pyrrolidine moiety for condensation onto aldehydes and ketones. While extensively studied, mechanistic analyses of the Pro-catalyzed aldol will not be considered here, as the focus is on more elaborated peptide-catalyzed processes.239–243 The other major class of catalysts is inspired by type I aldolase, an enzyme that catalyzes aldol reaction in vivo using a catalytically-active Lys residue in the active site.244 These catalysts typically bear a free primary amino group at the N-terminus. Both of these catalyst classes are designed to engage their substrates via enamine catalysis. As detailed below, the modular nature of peptides provides easy access to diverse Pro- and aldolase-like catalysts with tunable lengths, secondary structures, and functional groups, which together can be exploited to engage in precise noncovalent interactions with substrates. These characteristics provide opportunities to tailor and expand the scope of the aldol reaction. Additionally, replacement of the α-carboxylate group of simple Pro-based catalysts, for example, with peptide backbones is a means of suppressing known potential off-cycle processes, such as formation of oxazolidinones.241
Figure 97:
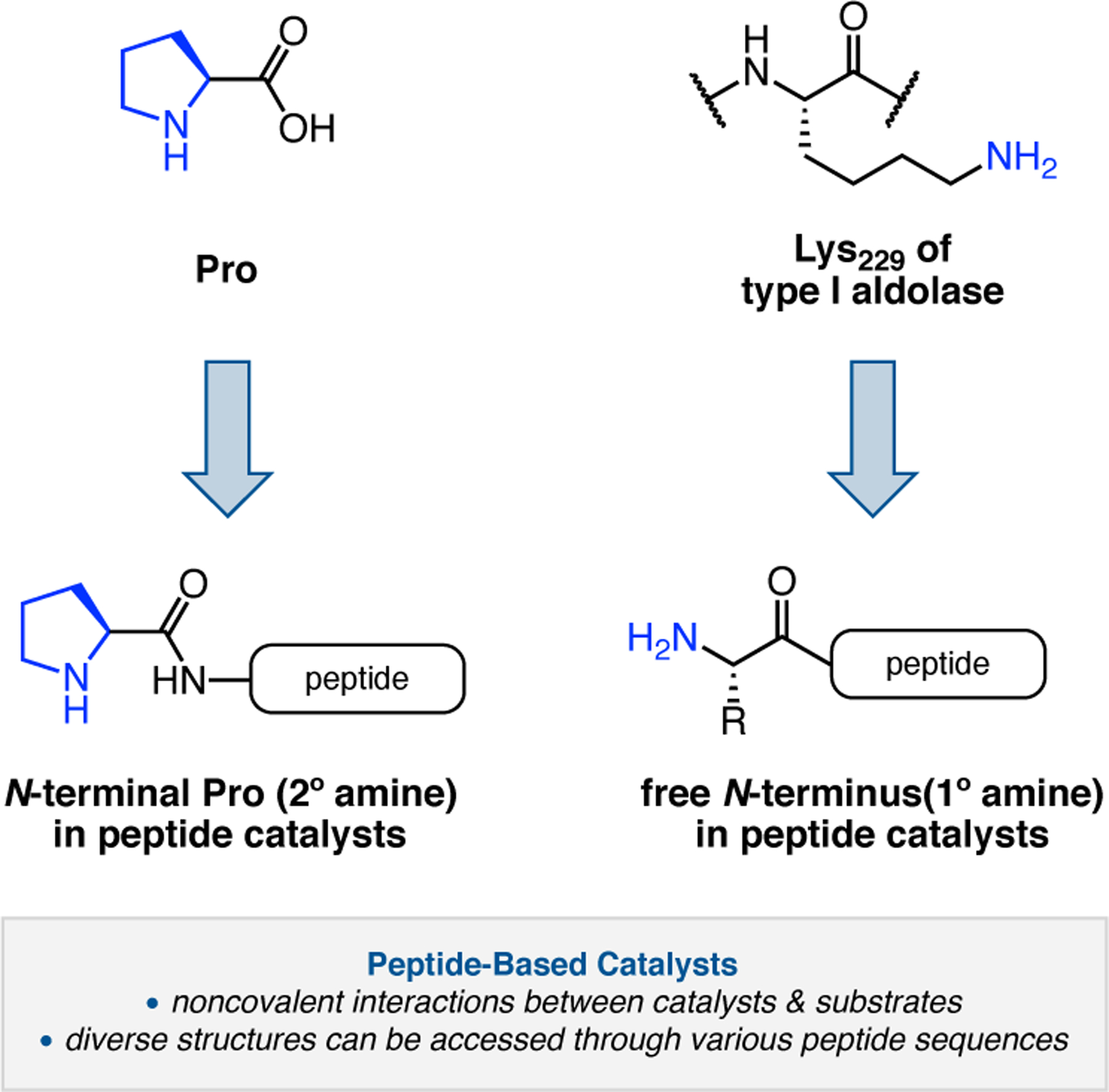
Inspirations for the design of amine-based peptide catalysts.
The focus of this Section is to present a comprehensive overview of amine-containing peptide catalysts for asymmetric aldol reactions. In lieu of presenting a detailed substrate scope for each catalyst, pertinent issues in catalyst concepts and design are discussed. For those reactions catalyzed by a single amino acid or its derivatives, prior reviews provide the necessary insight.237,238
6.1. Enantio- & Diastereoselective Aldol Reactions Using Amine-Based Peptide Catalysts
To assist in the discovery of new peptide catalysts, highly electrophilic acceptors, such as 4-nitrobenzaldehyde, and simple donors, such as acetone and cyclohexanone, that are often used in large excess, have been employed as benchmark substrates. The new catalysts that have been developed in recent years are structurally quite diverse, as their designs were inspired by various attributes including known amino acid sequences in enzymes, protein secondary structures, and solid-supported materials. All of these peptides were found to be effective catalysts for asymmetric aldol reactions between 4-nitrobenzaldehyde and acetone (Figure 99) and cyclohexanone (Figure 100) with varying degrees of reactivity and selectivity. For diastereoselective aldol reactions, most peptide catalysts are selective for the anti-diastereomer, much like in the corresponding Pro-catalyzed reactions.
Figure 99:
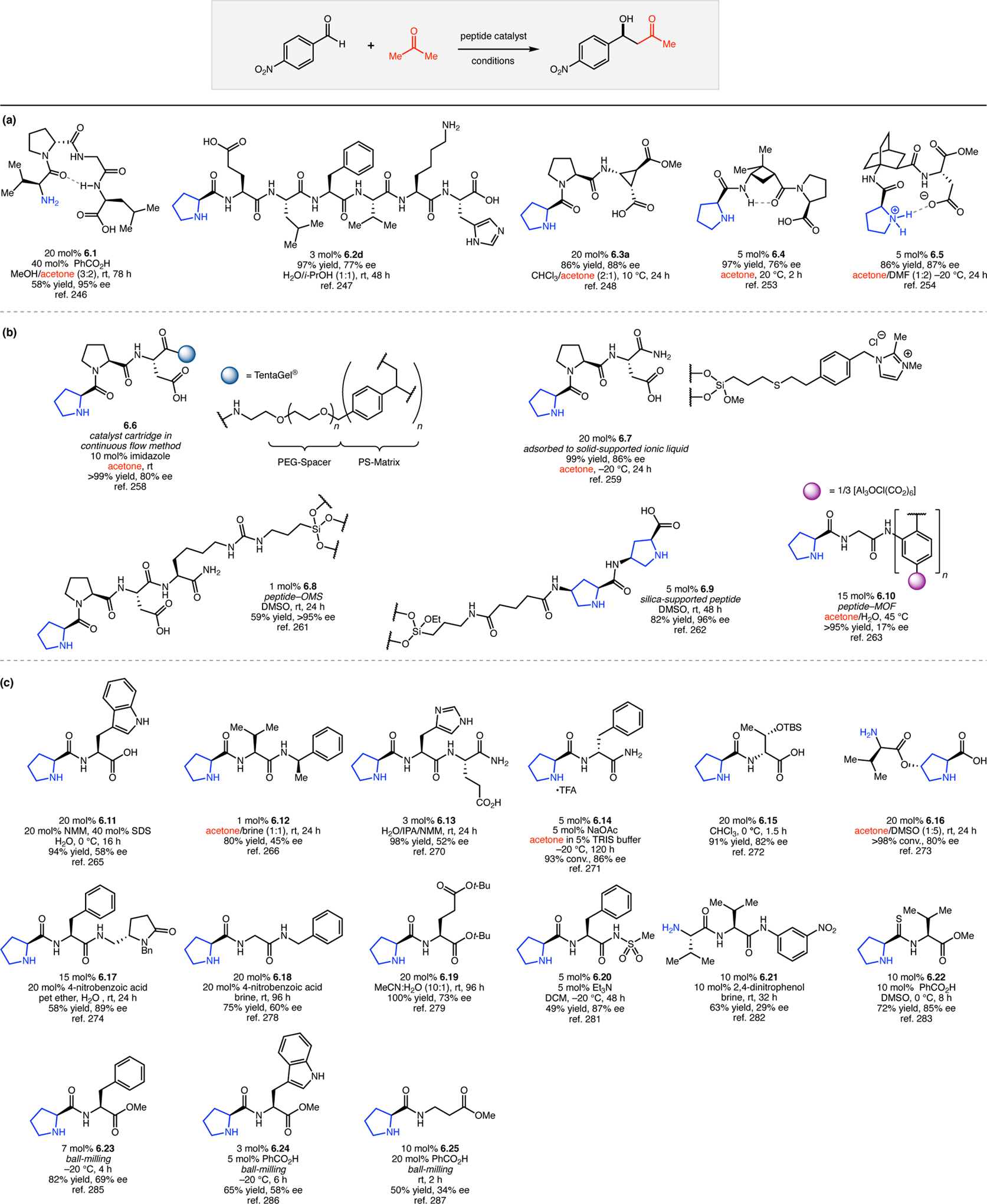
Peptide catalysts for the asymmetric aldol reaction between 4-nitrobenzaldehyde and acetone: (a) Enzyme/protein-inspired or geometrically constrained peptide catalysts; (b) Solid-supported catalysts; and (c) various di/tri-peptide catalysts.
Figure 100:
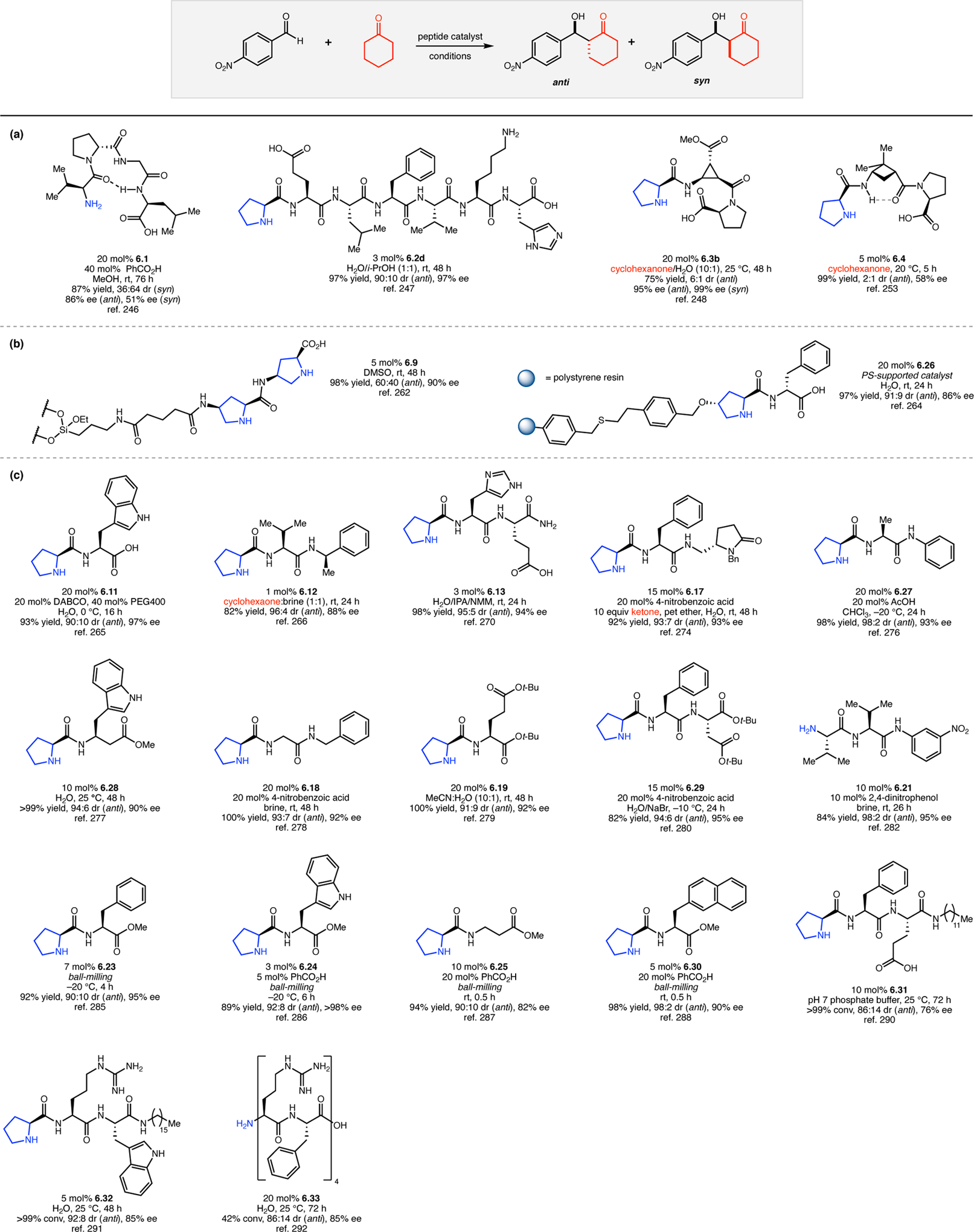
Peptide catalysts for the asymmetric aldol reaction between 4-nitrobenzaldehyde and cyclohexanone: (a) Enzyme/protein-inspired or geometrically constrained peptide catalysts; (b) Solid-supported catalysts; (c) Various di/tri-peptide catalysts.
Inspired by enzymes, many catalyst designs are centered on the ability of peptides to form defined secondary structures or stable geometries. The β-turn is a common secondary structure found within proteins47 that has proven particularly fruitful in achieving asymmetric induction.245 Da and co-workers used the β-turn secondary structure as a key feature in the design of their catalyst that bears an N-terminal primary amine.246 After evaluation of multiple tetrapeptides, peptide 6.1 was identified as the lead catalyst (Figure 99a). Incorporation of the central d-Pro-Gly sequence was intended to bias the structure toward a β-turn conformation, which was supported by CD and NOESY NMR structural studies in methanol, the solvent for the reaction. Additionally, truncation studies of peptide 6.1 revealed that the corresponding di- and tripeptides were far less selective catalysts, delivering the aldol product in < 31% ee. It is likely that the shorter peptides do not form a stable β-turn under the reaction conditions, especially in H-bond competitive solvents, like methanol. Furthermore, the authors identified that the presence of a C-terminal carboxylic acid is crucial for obtaining high levels of enantioselectivity. Although high catalyst loadings were used (20 mol% of 6.1 and 40 mol% of benzoic acid co-catalyst), the product was obtained in 58% yield and 95% ee. The same catalyst worked fairly well using cyclohexanone as the nucleophile (Figure 100a). Interestingly, the syn-diastereomer was modestly favored (64:36 dr for syn) using 6.1, which provided the aldol product in 87% total yield and 86% ee in the major syn-diastereomer. The minor anti-diastereomer was also formed with a moderate ee of 51%.
Using a more biomimetic approach, Rahman and co-workers designed a peptide catalyst for asymmetric aldol reactions based on the active sites of aldo-keto reductases (AKRs, PDB = 1VBJ), which bear a conserved Lys residue (Figure 98).247 Although enzymes often achieve high selectivities under mild reaction conditions, they often suffer from limitations in scope. To address this issue, the authors targeted peptide-based catalysts as a means of emulating the activity of AKRs in a more tunable catalytic scaffold. Initially, two 18-mers (6.2a & 6.2b) and one 16-mer (6.2c) were evaluated in the reaction of 4-nitrobenzaldehyde with cyclohexanone in aqueous media (entries 1–3). Using 6.2a, the aldol product was isolated in 88% yield with an exceptional dr of 99:1 and moderate enantioselectivity of 63% ee (entry 1). Significant improvements to both yield (95%) and enantioselectivity (86% ee) were observed when 6.2b was employed as the catalyst (entry 2). On the other hand, peptide 6.2c showed diminished yield (67%) and enantioselectivity (39% ee) compared to the 18-mers (entry 3). Following truncation studies and an investigation into the importance of a free Lys residue, octapeptide 6.2d bearing an N-terminal Pro residue was discovered. This catalyst afforded the highest observed yield (97%) and stereoselectivity (90:10 dr for anti, 97% ee) of the catalysts examined (entry 4; also shown in Figure 100a). Furthermore, 6.2d provided high yield (97%) and good enantioselectivity (77%) in the aldol reaction with acetone (Figure 99a). The reusability of 6.2d was also demonstrated by recovering and reusing this catalyst up to 10 times (83% yield, 76% ee vs. entry 4).
Figure 98:
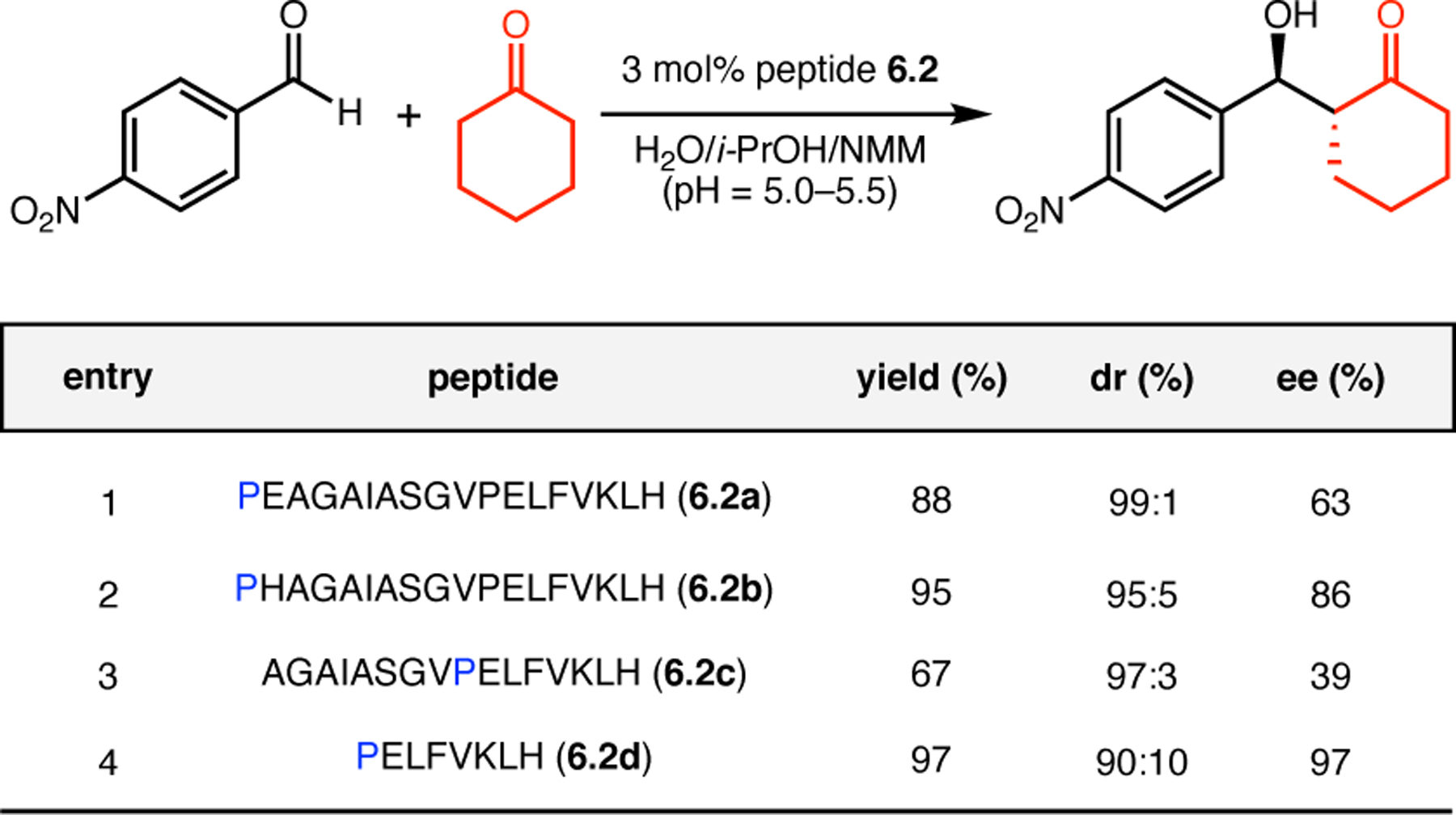
Aldol reaction catalyzed by aldo-keto reductase mimics.247
Additionally, there have been many efforts to use non-proteinogenic amino acids to promote a rigid secondary structure in peptide-based catalysts. Reiser and co-workers integrated conformationally restricted β-amino acids, specifically β-aminocyclopropylcarboxylic acid (β-Acc), into short peptide sequences in order to stabilize turn-like secondary structures.248,249 As such, β-Acc was incorporated into either the central or terminal positions of various N-terminal Pro-containing peptides. For the reaction between acetone and 4-nitrobenzaldehyde, catalyst 6.3a, which contains β-Acc as the C-terminal residue, gave the aldol product in 86% yield and 88% ee (Figure 99a). In an aqueous media, peptide 6.3b, a constitutional isomer of 6.3a in which the β-Acc residue was moved to the central position of the tripeptide, delivered the aldol product in 75% yield and 6:1 dr favoring the anti-diastereomer (Figure 100a). Both diastereomers were isolated with excellent levels of enantioselectivity (95% ee for anti & 99% ee for syn).
Illa, Branchadell, Ortuño, and co-workers synthesized short peptides bearing a central cis-cyclobutane γ-amino acid (cis-γ-Cbaa), which is derived from (−)-verbenone.250,251 As observed in their previous study on cis-γ-Cbaa-containing foldamers, these catalysts (e.g. 6.4) are designed to form a seven-membered N–H(i+1)•••O(i+1) H-bond across the central cyclobutane framework (Figure 99a).252 The intramolecular H-bond contributes to the high rigidity of the peptide catalyst, which is supported by NMR and computational studies. Upon examination of various diastereomers of cis-γ-Cbaa-bearing peptides in the benchmark acetone aldol reaction, peptide 6.4 emerged as the lead catalyst, providing the aldol product it 97% yield and 76% ee in only 2 hours (Figure 99a).253 For the reaction with cyclohexanone, peptide 6.4 was found to be notably less selective. In this case, the aldol product was obtained in 99% and modest selectivity (2:1 dr favoring anti, 58% ee, Figure 100a). However, the catalyst was ineffective towards electron-rich 4-methoxybenzaldehyde and gave no conversion.
Calmès and co-workers also sought to use conformationally restricted residues to nucleate turn structures in peptide-based catalysts.254 The authors incorporated a conformationally constrained 2-aminobicyclo[2.2.2]octane carboxylic acid (Aboc) residue into short peptide sequences. This bicyclic amino acid was prepared previously by the authors using an asymmetric Diels–Alder reaction,255 and it was demonstrated to induce a β-turn secondary structure when embedded at the central position of a tripeptide sequence.256 Using Aboc residues, the authors prepared a number of di- and tripeptides bearing N-terminal Pro residues for enamine catalysis. In general, tripeptides with heterochirality were more efficient than their corresponding homochiral diastereomers. With 5 mol% of the lead catalyst 6.5, the aldol product between acetone and 4-nitrobenzaldehyde was obtained in 86% yield and 87% ee (Figure 99a). As they hypothesized, an X-ray crystal structure of 6.5 revealed a β-turn conformation, wherein the N-terminal pyrrolidine and the C-terminal carboxylate groups were in close proximity, possibly interacting through H-bonding. The authors reasoned that this geometric arrangement of the two termini of the peptide allows for selectivity in the peptide-catalyzed aldol reaction.
Solid-phase peptide synthesis (SPPS) is a powerful and practical way of preparing peptides, and as such there are number of reports detailing the development of solid-supported peptide-based catalysts. This approach offers a number of advantages, including ease of synthesis and catalyst recycling. Such benefits represent an atom-economical, effective solution to the recurring requirement of high catalyst loadings in peptide-catalyzed aldol reactions.
Fülöp and co-workers developed a highly recyclable, solid-supported peptide-catalyzed continuous flow (CF) process. Inspired by Wennemers’ tripeptidic aldol reaction catalyst,257 the authors synthesized 6.6 on TentaGel® resin via SPPS, which they directly immobilized onto a CF cartridge, obviating the need for any purification steps (Figure 99b).258 With the benchmark substrates, this CF method can generate the aldol product in quantitative conversion with 80% ee, and the catalyst cartridge can be recycled up to 20 times while retaining efficiency and selectivity. Such CF process could be a useful tool for large-scale and sustainable catalytic reactions.
Gruttadauria and co-workers also utilized Wennemers’ previously reported aldol catalyst 6.7 in developing recyclable materials.259 The peptide catalyst (H–Pro-Pro-Asp–NH2) was adsorbed onto the surface of silica gel functionalized with 1,2-dimethyl-imidazolium salts (Figure 99b). In the first use of the supported catalyst in the aldol reaction of 4-nitrobenzaldehyde and acetone, the β-hydroxyketone product was obtained in 99% yield and 86% ee, comparable results to those obtained under homogeneous conditions. However, both yield and enantioselectivity diminished over the course of a few subsequent runs with the recycled catalysts (38% yield, 83% ee).
Ordered mesoporous silicas (OMSs) can be covalently functionalized with large biomolecules, including enzymes.260 Utilizing known trialkoxysilane chemistry that functionalizes OMSs, Mehdi, Subra, and co-workers developed a method for synthesizing hybrid peptide-OMS materials.261 They installed a trialkoxysilane group onto the C-terminus of Wennemers’ aldol catalyst 6.7 via a urea linker (Figure 99b), and this peptide-silane building block was used to directly graft a non-functionalized silica matrix to give the hybrid peptide-OMS catalyst 6.8. Following a brief optimization of the reaction conditions, this hybrid bioorganic material proved to be a highly selective catalyst, providing the aldol product in >95% ee, albeit with lower reactivity (59% yield) than in the corresponding homogeneous system.257 The hybrid peptide-OMS could be recovered from the reaction mixture by a simple filtration step. However, the use of recycled OMS in aldol reaction was not demonstrated.
Wang and co-workers developed a silica-supported, non-canonical diproline catalyst 6.9, which gave high yield (82%) and enantioselectivity (96% ee) in the aldol reaction between acetone and benzaldehyde (Figure 99b).262 With cyclohexanone as the substrate, 6.9 also delivered the aldol product in high yield (98%) and with good diastereo- (60:40 dr favoring anti) and enantioselectivity (90% ee) in the major diastereomer (Figure 100b). Other catalysts containing a range of Pro units were examined, but they were less selective than the dipeptide catalyst 6.9. It was demonstrated that this catalyst can be recycled up to 5 times while preserving the observed yield and enantioselectivity.
Furthermore, Canivet and co-workers demonstrated the first example of functionalizing metal-organic-frameworks (MOFs) with peptides using microwave-assisted amide bond formation.263 First, MOFs bearing 2-aminoterephthalates were coupled to the C-terminus of the desired peptide sequence. Then the N-terminal Boc protecting group is removed under microwave/thermal conditions to afford the functionalized MOFs 6.10 (Figure 99b). As a proof-of-concept, the MOFs were employed as catalysts for the aldol reaction between 4-nitrobenzaldehyde and acetone. Despite the low enantioselectivity of 17% ee, the product was obtained in near quantitative yield.
Gruttadauria and coworkers utilized a thiol-ene coupling reaction between mercaptomethyl functionalized polystyrene resin and a styrene-containing dipeptide to prepare solid-supported aldol catalyst 6.26 (Figure 100b).264 The desired peptide sequence was synthesized using a trans-4-styrenyl-Hyp. After the evaluation of various dipeptides and stereoisomers, the supported catalyst bearing a Hyp-d-Phe sequence gave the highest yield (97%), diastereoselectivity (91:9 dr favoring anti), and enantioselectivity (86% ee for anti) in the aldol reaction between cyclohexanone and 4-nitrobenzaldehyde under aqueous conditions. It was shown that the catalyst can be reused four times with reproducible results. Also, an electron-rich aromatic aldehyde underwent the 6.26-catalyzed aldol reaction to give the corresponding product in high diastereo- and enantioselectivity (Figure 101b).
Figure 101:
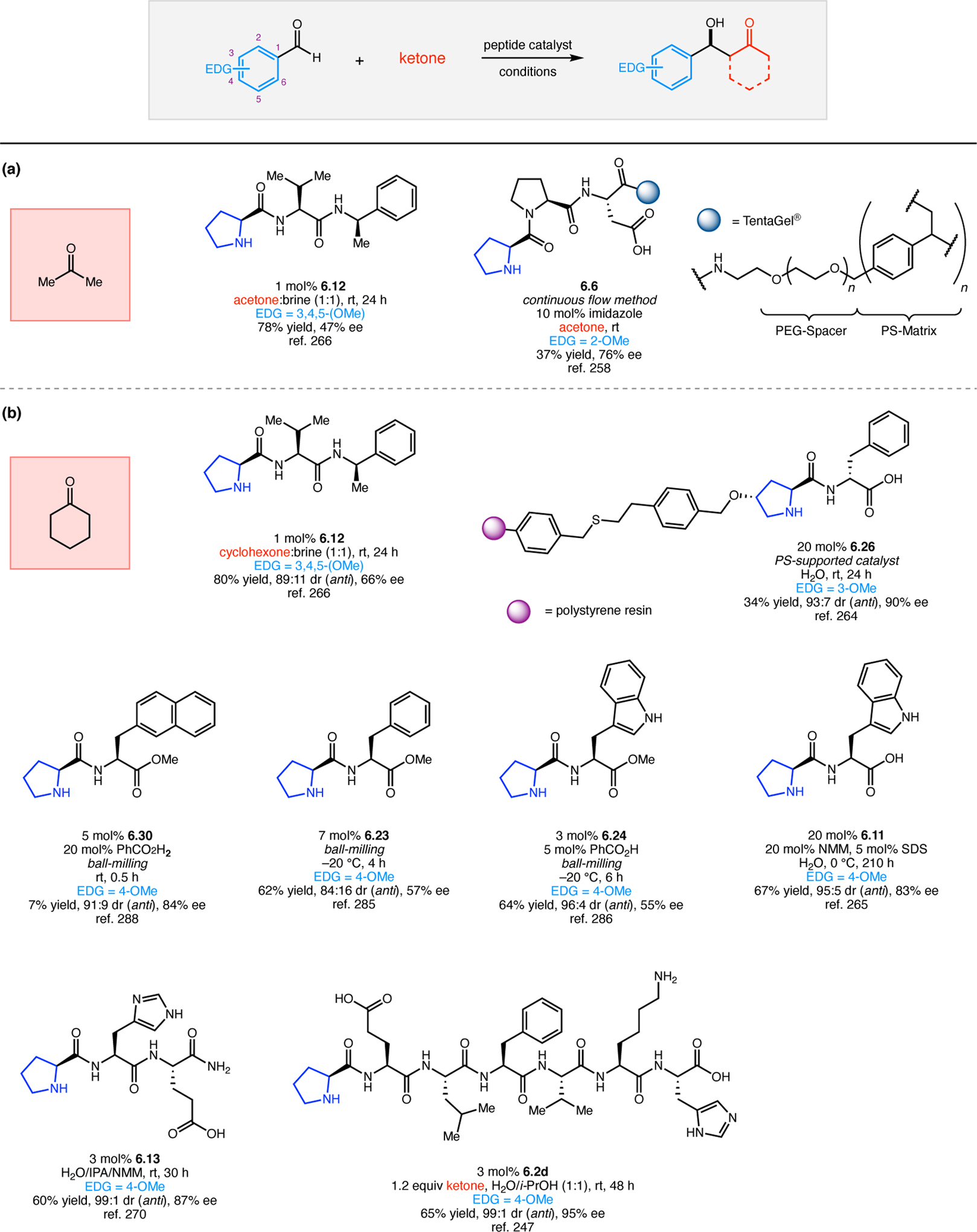
Peptide catalysts for the asymmetric aldol reaction between electron-rich benzaldehydes and (a) acetone and (b) cyclohexanone.
To date, the majority of new peptide-based catalysts for aldol reactions are two or three residues in length. Since a carboxylic acid moiety of an amino acid—Pro in many cases—can be easily modified through well-established amide bond forming methods, large libraries of diverse catalysts can be accessed (Figures 6.2c & 6.3c).
Li and co-workers showed that globally unprotected dipeptide 6.11 (H- Pro-Trp-OH) can be used to catalyze the aldol reaction of simple ketones with various aldehydes in water.265 Due to the aqueous media, the presence of a surfactant (e.g., SDS) was proven to be essential for reactivity, as only trace product was observed when the reagent was omitted. A high yield of 94% and a good enantioselectivity of 58% ee were obtained in the reaction of 4-nitrobenzaldehyde with acetone (Figure 99c). For aldol reaction with cyclohexanone, corresponding aldol product was obtained in 93% yield, 90:10 dr in favor of the anti-isomer, and 97% ee (Figure 100c). This peptide-based catalyst was able to address a range of aldehyde substrates, including electron-rich aromatic (Figure 101b) and aliphatic aldehydes (Figure 103b), and it was also effective using an unsymmetrical aliphatic ketone (Figure 109). In many cases, the aldol products were obtained with enantioselectivities of up to 97% ee.
Figure 103:
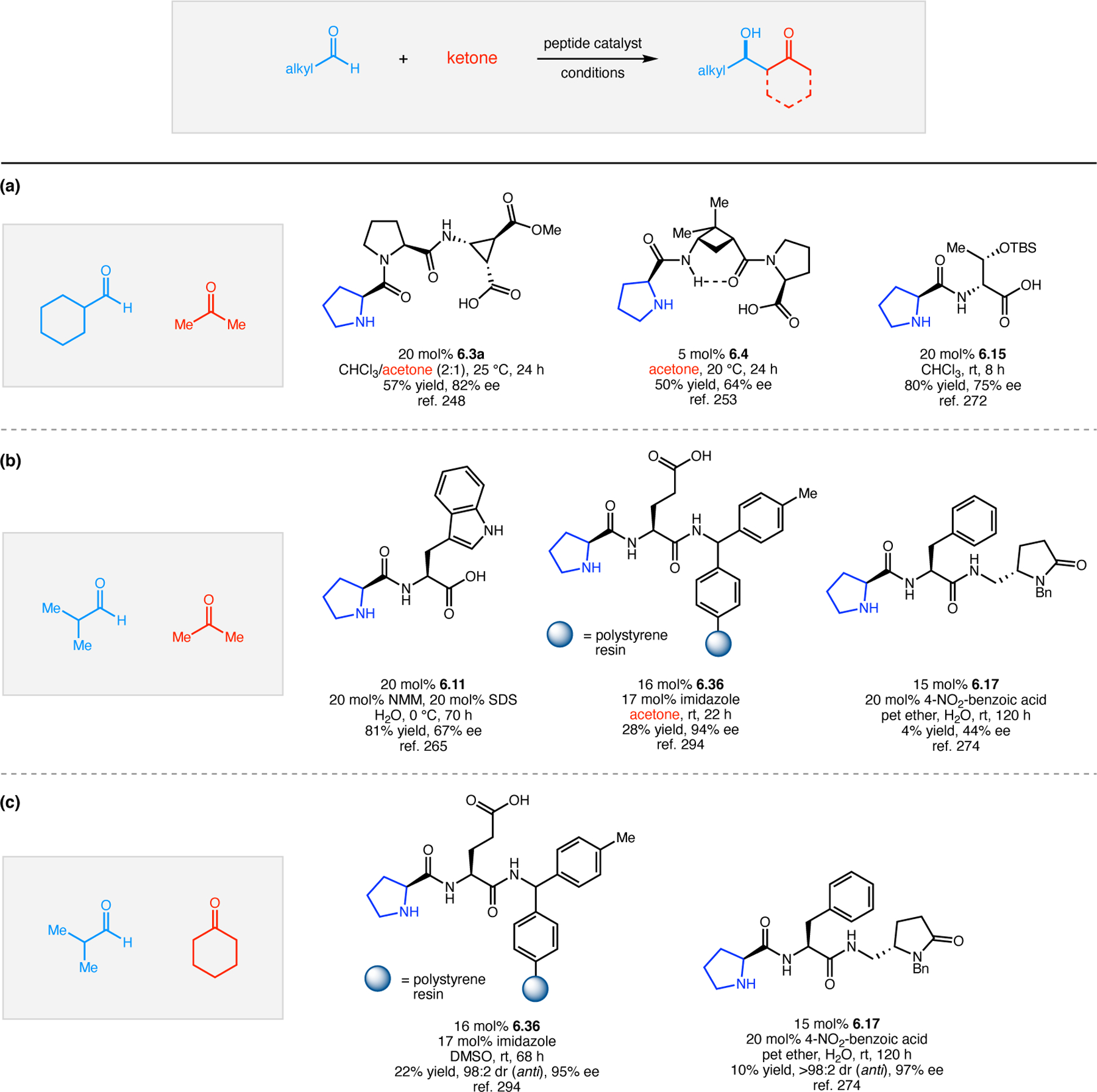
Peptide catalysts for the asymmetric aldol reaction between (a) cyclohexanecarboxaldehyde and acetone, (b) iso-butyraldehyde and acetone, and (c) iso-butyraldehyde and cyclohexanone.
Figure 109:
Divergence in site-selective, asymmetric aldol reactions catalyzed by short peptides.265,246
Organocatalyzed asymmetric aldol reactions often require high catalyst loadings—typically 10–30 mol%—in order to achieve synthetically useful levels of enantioselectivity. However, Zhang, Wang and co-workers reported that dipeptide catalyst 6.12 is an effective catalyst at only 1 mol% loading.266 Under the optimized conditions, peptide 6.12 delivered the aldol product of 4-nitrobenzaldehyde and acetone in 80% yield and 45% ee (Figure 99c). During the course of catalyst optimization, it was found that the (R)-α-methylbenzyl amide C-terminal cap was crucial for enantioselectivity, as peptides having the (S)-variant or an achiral amide group led to lower enantioselectivity in the product. Moreover, the authors discovered that performing the reaction in brine was also key to success. When the reaction was performed in water, the enantioselectivity decreased significantly to 7.5% ee. The authors proposed that this observation may be due to the previously reported “salting-out” effect.267–269 The authors further demonstrated that this catalytic system was highly effective for both electron-deficient and electron-rich benzaldehydes. In the aldol reaction between 4-nitrobenzaldehyde and cyclohexanone, 6.12 afforded the product in 82% yield and 96:4 dr in favor of the anti-isomer, which was enriched to 88% ee (Figure 100c). The reactions between an electron-rich benzaldehyde and acetone (Figure 101a) or cyclohexanone (Figure 101b) also provided impressive yields and good selectivities using peptide 6.12 (see Section 6.2).
In an alternate catalyst design strategy, Rahman and co-workers prepared tripeptides containing residues with polar sidechains (Tyr, Glu, or His) in order to exploit their ability to engage in H-bonding interactions with substrates.270 Evaluating their library, they identified His-bearing catalyst 6.13, which gave the aldol product of 4-nitrobenzaldehyde and acetone in 98% yield and 52% ee (Figure 99c). High stereoselectivities were observed in the reaction with cyclohexanone under the same conditions, in which the product was isolated in 98% yield, 95:5 dr favoring the anti-product, and 94% ee (Figure 100c). The authors speculate that aldehyde may be activated by the imidazole via H-bonding in the aldol transition state.
Andreu and co-workers showed that simple dipeptide 6.14 (as the TFA salt) can catalyze highly selective acetone aldol reactions (up to 86% ee) when used in combination with co-catalytic zinc or sodium acetate (Figure 99c).271 Deprotonation of 6.14•TFA by the acetate base is thought to reveal the active amine catalyst. Furthermore, it is proposed that those metal cations could act as Lewis acids to activate aldehydes toward aldol addition of the enamine nucleophile. These metal cations were shown to greatly enhance conversion relative to the corresponding reactions lacking them.
In 2009, Chandrasekhar and co-workers discovered that dipeptide 6.15 is an effective catalyst for the acetone aldol reaction, providing the β-hydroxyketone product in 91% yield and 82% ee (Figure 99c).272 While the C-and N-termini were unprotected, the lead catalyst employed a TBS-ether on the Thr sidechain. Despite using different conditions and being reported by different authors, peptides 6.14 and 6.15—both of which possess the same stereodiad of l-Pro-d-Xaa—are remarkably consistent in terms of their efficacy and selectivity in the benchmark acetone aldol reaction.
By comparison, Al-Momani has explored an alternative catalytic design based upon sidechain functionalized Hyp residues.273 Catalyst 6.16, in which a Val residue is coupled to the γ-hydroxyl group of Hyp, was identified as the lead catalyst, providing the acetone aldol product in 98% conversion and 80% ee (Figure 99c). Although there is no experimental support, it is speculated that enamine formation occurs at the secondary amine site rather than at the primary amine of the pendent Val ester.
Moutevelis-Minakakis and co-workers developed C-terminal pyrrolidinone-containing peptide 6.17,274 in which the lactam motif was derived from glutamic acid.275 When the catalyst was employed in asymmetric aldol reactions with acetone using co-catalytic 4-nitrobenzoic acid, the product was obtained in 58% yield and 89% ee (Figure 99c). Higher yields and selectivities were observed in the corresponding cyclohexanone aldol reaction, in which 6.17 delivered the product in 92% yield favoring the anti-diastereomer in 93:7 dr and 93% ee (Figure 100c).
Upon examination of various N-terminal Pro-based dipeptide catalysts, the Peng Group found that it was important to fine-tune the sidechain sterics of the second residue.276 For instance, Ala-containing peptide 6.27 gave the best selectivity in the cyclohexanone aldol reaction (Figure 100c, 98:2 dr favoring anti, 93% ee), whereas residues with bulkier sidechains, such as Val, Leu, and Ile, delivered to lower ee values (79%, 83%, and 90% ee, respectively). Interestingly, when Gly was incorporated at the second residue, only trace amount product was observed. Furthermore, both electron-rich and electron-poor substituents on the C-terminal aniline resulted in lower yields and selectivities.
In addition to α-amino acids, β-amino acids have been utilized in the design of peptide-based catalysts. Nisco and co-workers prepared and examined various Pro-β3-amino ester catalysts for diastereo- and enantioselective aldol reactions under aqueous conditions.277 The authors found that peptide 6.28, which possesses a β3-Trp-OMe residue with a free indole sidechain, gave the best results in the aldol reaction of 4-nitrobenzaldehyde and cyclohexanone, providing the product in >99% yield, 94:6 dr favoring anti, and 90% ee (Figure 100c). A π–π interaction might play a role in organizing the transition state.
The Kokotos Group has developed numerous highly selective peptides to date, including 6.18 and 6.19 (Figures 6.2c & 6.3c) and 6.29 (Figure 100c).278–280 These catalysts all possess an N-terminal Pro residue for enamine catalysis but vary with respect to their lengths and residue composition. Using both acetone and cyclohexanone as a substrate with 4-nitrobenzaldehyde, these catalysts work well in an aqueous media to generate the corresponding aldol products in up to quantitative yield with stereoselectivities up to 94:6 dr favoring anti and 95% ee.
One common strategy used to enhance the efficiency of peptide-based catalysts is to increase the acidity of its amide N–H bonds. It is often proposed that this modification will enable stronger H-bonding interactions between the aldehyde substrate and the peptide backbone, which can effectively increase the electrophilicity of the aldehyde partner.
For example, Kokotos and co-workers showed that the presence of a C-terminal methanesulfonamide group in dipeptide 6.20 greatly improved the enantioselectivity of the aldol reaction between 4-nitrobenzaldehyde and acetone from 34% ee to 77% ee (Figure 99c).281 The authors proposed that the increase in the enantioselectivity can be attributed to the increased H-bond donor ability of the C-terminal residue. Upon further optimization of the reaction temperature, the aldol product was obtained in 49% yield with 87% ee.
Zhang and co-workers identified Val-Val-based dipeptide 6.21 as a catalyst for asymmetric aldol reactions.282 Using 10 mol% of 2,4-dinitrophenol as a co-catalyst, they observed that peptides bearing a C-terminal 3-nitroaniline group exhibited increased stereocontrol. Although low enantioselectivity was observed in the acetone aldol (Figure 99c), the reaction with cyclohexanone was highly selective, providing the aldol product in 84% yield, 98:2 dr favoring anti, and 95% ee (Figure 100c).
Compared to the corresponding native amide-based peptides, the thioamide-containing catalyst 6.22 developed by Li and co-workers gave higher yield (72% vs. 59%) and enantioselectivity (85% vs. 35%) in the direct aldol reaction of 4-nitrobenzaldehyde with acetone (Figure 99c).283 Thioamides are stronger H-bond donors than the corresponding amides. As such, Pro thioamide-containing peptide 6.22 is thought to be a more effective catalyst due to its enhanced ability to activate the aldehyde reaction partner through H-bonding,180 although additional mechanistic studies are required. Notably, reduction of the C-terminal methyl ester of 6.22 to a primary alcohol led to a further increase in enantioselectivity to 95% ee (not shown).
High Speed Ball-Milling (HSBM) has been utilized as an environmentally friendly, mechanochemical tool for performing asymmetric aldol reactions under solvent-free conditions. This area was pioneered by the Bolm Group in 2007 in the context of Pro catalysis.284 Using a related mechanochemical technique, Juaristi and co-workers have found that a series of dipeptides 6.23–6.25 (Figure 99c) can also be used to catalyze asymmetric aldol reactions with good yields (50–82%) and enantioselectivities (34–69% ee).285–287 For the catalysts bearing aromatic sidechains (e.g., 6.23 & 6.24), the authors have speculated that noncovalent π-π interactions between the aromatic motif and the aromatic aldehyde could contribute to the high enantioselectivities observed. In comparison to the reactions with acetone under ball-milling conditions, catalysts 6.23–6.25 and 6.30 mediated highly diastereoselective aldol reactions with cyclohexanone as the donor, delivering the aldol products in up to 98% yield, 98:2 dr favoring anti, and >98% ee (Figure 100c). Much like in the solution phase reactions, anti-diastereoselectivity was also observed under these solvent-free conditions.286–288
Self-assembled fibrillar networks of organic molecules play important roles in the localization of reactants within cellular environments.289 Inspired by such supramolecular phenomena, Ashkenasy, Escuder, and co-workers developed a short peptide 6.31, which possesses a long alkyl chain at the C-terminus, that was shown to self-assemble into high aspect ratio aggregates and hydrogels.290 They observed that these aggregates are able to catalyze the aldol reaction of 4-nitrobenzaldehyde with cyclohexanone in aqueous media, providing the product in high yield (>99% conversion), 86:14 dr favoring anti, and 76% ee (Figure 100c), whereas non-aggregated analogues gave notably diminished reactivity (32% yield). The authors proposed that the long, hydrophobic alkyl chain of the catalyst may function as a surfactant to help solubilize substrates in the aqueous media. Moreover, the authors also used 6.31 to catalyze the self-aldol reaction of TBDPS-protected 2-hydroxyacetaldehyde to afford a tetrose derivative in 84% yield, 90:10 dr favoring anti, and 82% ee (not shown).
Using a similar self-assembly approach, Alves and co-workers designed an amphiphilic tripeptide catalyst 6.32, which possesses Arg and Trp residues for hydrophilicity and a C-terminal n-hexadecyl chain for lipophilicity.291 This peptide was reported to catalyze the cyclohexanone aldol reaction in excellent yield and 92:8 dr favoring the anti-isomer with 85% ee (Figure 100c). Various imaging techniques (small-angle X-ray scattering and cryo-TEM), as well as molecular dynamics simulations, supported the hypothesis that 6.32 forms micelles under the aqueous reaction conditions. The same research group later demonstrated that oligopeptide 6.33 can self-assemble in an aqueous medium, and this supramolecular assembly can mediate the cyclohexanone aldol reaction with moderate yield (42% conversion) and good stereoselectivity (86:14 dr favoring anti, 85% ee).292
6.2. Expansion of Substrate Scope
In addition to 4-nitrobenzaldehyde, other electron-deficient aromatic aldehydes, such as those functionalized with halogen, nitrile, and trifluoromethyl substituents, are used in the vast majority of peptide-catalyzed asymmetric aldol reactions. This is presumably due to their greater electrophilicity and increased propensity toward nucleophilic addition. In comparison, only a handful of examples using electron-rich aromatic and aliphatic aldehydes have been reported to date. The peptide-based catalysts that are able to address these more challenging aldol substrates are presented below in Figures 101 and 103, respectively. Additional examples of peptide-catalyzed aldol reactions employing aldehydes other than benzaldehyde and its simple derivatives are also presented below.
Zhang and Wang showed that peptide 6.12 effectively catalyzed aldol reactions using 3,4,5-trimethoxybenzaldehyde as the electrophile.266 Using only 1 mol% of 6.12 under mild conditions, the acetone aldol product was obtained in 78% yield and 47% ee (Figure 101a), comparable to the results obtained with 4-nitrobenzaldehyde (see Figure 99c). Peptide 6.12 was similarly reactive and slightly more selective in the corresponding reaction with cyclohexanone, delivering the product in 80% yield favoring the anti-diastereomer in 89:11 dr and 66% ee (Figure 101b).
The reduced reactivity of electron-rich benzaldehydes has also been observed under continuous flow and ball-milling conditions. Although Fülöp’s solid-supported peptide catalyst 6.6 was proven to be highly recyclable and robust in aldol reactions with 4-nitrobenzaldehyde (see Figure 99b), the corresponding reaction with 2-methoxybenzaldehyde was significantly lower yielding, providing only 37% yield of the aldol product in 76% ee (Figure 101a).258 Similarly, Gruttadauria’s recyclable polystyrene resin-supported catalyst 6.26 was also less reactive with 3-methoxybenzaldehyde and cyclohexanone, as the product was obtained in only 34% yield favoring the anti-diastereomer in 93:7 dr and 93% ee (Figure 101b).264 Moreover, under solvent-free HSBM conditions, peptide catalyst 6.30 afforded only 7% yield of the product in the aldol reaction between 4-methoxybenzaldehyde and cyclohexanone (Figure 101b).288 Despite the very low yield, a good selectivity was observed, as the product was obtained in 91:9 dr favoring the anti-isomer and 84% ee. Yet, in other HSBM methods with different peptide catalysts, such as 6.23 and 6.24, much improved yields of 62–64% were observed, complimented by good anti-selectivities of 84:16–96:4 dr and enantioselectivities up to 57% ee (Figure 101b).285,286 However, these ee values are significantly lower than those measured for the reaction with 4-nitrobenzaldehyde (see Figures 6.2c & 6.3c)
Spawning from the continuous efforts to expand the scope of peptide-catalyzed, asymmetric aldol reactions, a few peptides have been reported that provide both high yields and stereoselectivities with electron-rich benzaldehyde substrates. For example, Li’s dipeptide 6.11 was an effective catalyst for the aldol reaction of 4-methoxybenzaldehyde and cyclohexanone, affording 67% yield of the anti-product in 95:5 dr and 83% ee, although long reaction times were required (Figure 101b).265 Perhaps some of the most stereoselective examples were reported by the Rahman Group. The His-bearing tripeptide 6.13 worked quite well with 4-methoxybenzaldehyde, delivering the product in 60% yield, 99:1 dr favoring the anti-isomer, and 87% ee.270 Moreover, their enzyme-inspired catalyst 6.2d, a globally unprotected heptapeptide, essentially afforded a single stereoisomer in the aldol reaction between 4-methoxybenzaldehyde and only 1.2 equivalents of cyclohexanone as the anti-product was isolated in 65% yield, 99:1 dr and 95% ee (Figure 101b).247
Inspired by dehydratases, Da and co-workers devised a complementary method for generating enantioenriched aldol products via dehydrative kinetic resolution of racemic bhydroxyketones (Figure 102).293 Interestingly, the authors found that peptide 6.1, the optimal catalyst from their asymmetric aldol reaction (see Figure 99a), was also the most effective in this context. As mentioned above, catalytic aldol reactions of electron-rich aldehydes often give low yields and enantioselectivities. On the other hand, dehydrative kinetic resolutions were particularly effective for these electron-rich substrates. For example, the ortho-methoxy substituted (±)-6.34 undergoes 6.1-catalyzed dehydration with an s factor of 21.2 to provide the enantioenriched 6.34 in 45% isolated yield and 91% ee (Figure 102a). Enone byproduct (E)-6.35 was also isolated as a single geometric isomer in 48% yield. Based on preliminary studies, the authors proposed a mechanism that requires both termini of 6.1 to be unprotected, enabling a bifunctional activation mechanism; the Pro condenses onto the ketone to first form imine I en route to enamine II, which subsequently dehydrates to leave α,β-unsaturated imine III (Figure 102b). Rehydration and cleavage from the peptide generates 6.35 and turns over the catalyst.
Figure 102:
(a) Dehydrative kinetic resolution of hydroxy ketones (b) Proposed catalytic cycle.293
Examples of peptide-catalyzed direct aldol reactions with aliphatic aldehydes are scarce (Figure 103). Alkyl aldehydes are often problematic substrates due to their propensity to undergo self- or cross-aldol reactions. To combat this issue, aldehydes that are non-enolizable or would lead to less reactive enamine intermediates have been used. Commonly, α,α-disubstituted aldehydes are employed. Peptides with rigid conformations, such as 6.3a and 6.4, have been shown fairly effective as catalysts for the asymmetric aldol reaction between cyclohexanecarbaldehyde and acetone, providing the β-hydroxyketone product in 57% and 50% yield and 82% ee and 64% ee, respectively (Figure 103a).248,253 In 2009, Chandrasekhar and co-workers used simple dipeptide 6.15 to catalyze the same aldol reaction to give a fairly high yield of 80% and a good selectivity of 75% ee (Figure 103a).272
iso-Butyraldehyde has been also used as an electrophile in asymmetric aldol reactions, and a handful of peptide-based catalysts have been used to mediate its reaction with acetone and/or cyclohexanone with varying degrees of efficacy. Notably, Bartók and co-workers developed peptide 6.36, which gave 94% ee using acetone as the donor substrate (Figure 103b) and 98:2 dr (anti) and 95% ee using cyclohexanone (Figure 103c), albeit with <30% yield in each case.294 The authors observed effects of catalyst-lengths on enantiodivergence, which is described in Section 6.3 below (Figure 114). Moutevelis-Minakakis and co-workers also applied their C-terminal pyrrolidinone-containing peptide 6.17 in an asymmetric aldol reaction of iso-butyraldehyde with acetone (Figure 103b).274 However, the desired aldol product was produced in only 4% yield and 44% ee. Using cyclohexanone as the donor, a similarly low yield of 10% yield was obtained, but the product was formed in >98:2 dr favoring the anti-isomer in 97% ee (Figure 103c).
Figure 114:
Aldol reactions catalyzed by di- and tripeptides that provide enantiodivergent outcomes.262
Beyond using traditional aldehydes as substrates, Kudo and co-workers have focused on expanding the scope of these asymmetric aldol reactions by employing masked or unconventional acceptors. Employing acetals, the authors developed the first enantioselective, successive acid- and base-catalyzed reactions in a single reaction vessel using resin-bound peptide catalysts to mediate the process (Figure 104).295 This method builds upon their first report of a polymer-supported peptide catalyst for asymmetric aldol reactions performed under aqueous conditions.118 Using commercially available Amberlite IR-120 resin (H+ form) in tandem with PEG-PS-bound tripeptide 6.3, acetal 6.37 was hydrolyzed to 2-nitrobenzaldehyde, which then underwent a 6.39-catalyzed aldol reaction with acetone to afford β-hydroxyketone 6.38 in one-pot, 89% NMR yield, and 73% ee. The authors demonstrated the reusability of the resin-supported catalyst, which can be easily separated from the reaction mixture by filtration and subsequently regenerated by drying. Even after six uses, the catalytic activity of 6.39 was largely retained.
Figure 104:
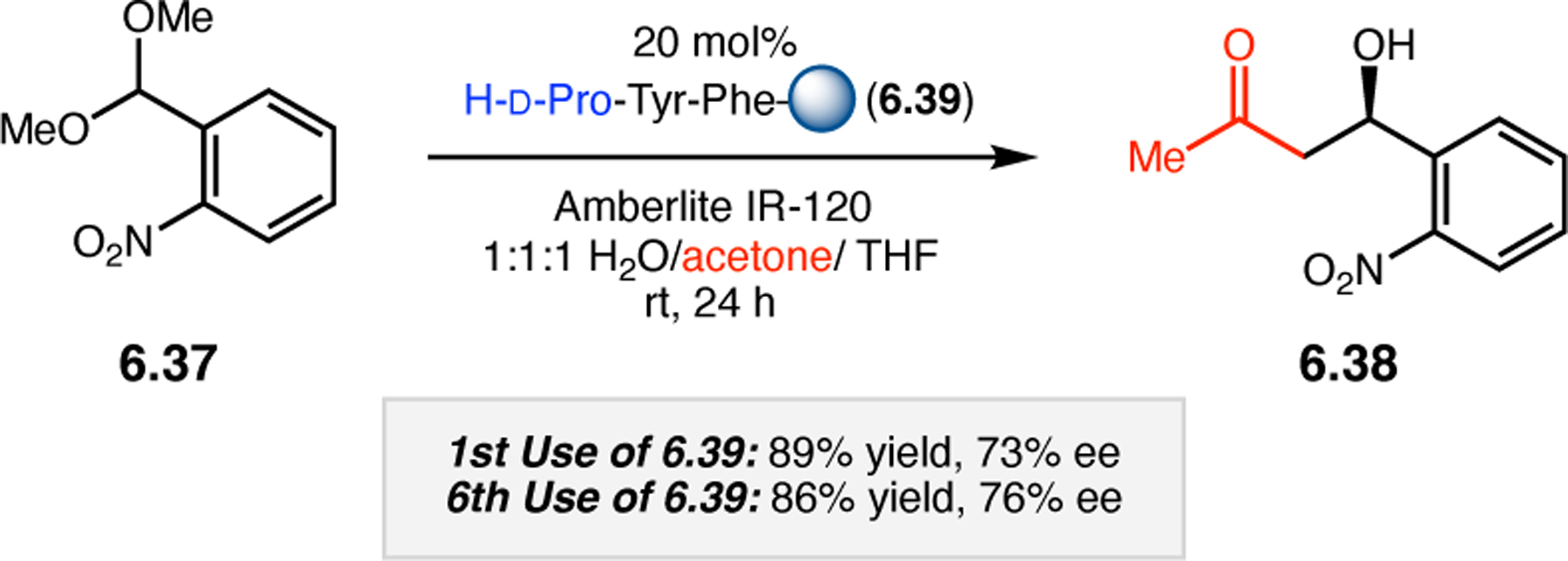
One-pot tandem aldol reaction using solid-supported acid and base catalysts showcasing the reusability of the catalysts.295
Kudo and co-workers also used resin-bound peptide-based catalysts to explore a tandem oxidation/direct aldol strategy, which could improve reaction outcomes for chemically unstable aldehydes and for those that are only commercially available in the alcohol oxidation state (Figure 105).296 Using a similar strategy to their previous report,295 the authors developed two resin-supported catalysts to mediate each step of the process. Together with a Cu(I) catalyst, TEMPO-derived peptide 6.41 would mediate the oxidation of benzylic alcohol 6.40 to the corresponding benzaldehyde.297 Pre-treatment of 6.41 with a Cu(I) salt and 2,2′-bipyridine ligand was necessary to achieve suitable yields in the oxidation step. A Pro-containing peptide would then catalyze the direct aldol reaction between 2-nitrobenzaldehyde and acetone via enamine catalysis to provide β-hydroxyketone 6.38. Modest optimization of their previously reported PEG-PS-bound tripeptide led to 6.42—the sidechain-protected variant of 6.39 (Figure 104)—which was found to be a more active catalyst under the reaction conditions. Employing the pre-adsorption method, the one-pot, tandem oxidation/aldol sequence was performed under the optimized reaction conditions to afford 6.38 in 78% yield and 87% ee (Figure 105), a marked improvement in selectivity over the previous report.295 This system also lends itself to catalyst recycling, as peptide 6.42 maintained its activity over eight cycles. TEMPO-containing 6.41 was removed via filtration after the alcohol oxidation to prevent decomposition of the aldol catalyst. Peptide 6.42 was then added to the reaction mixture along with EDTA to deactivate any remaining Cu species.
Figure 105:
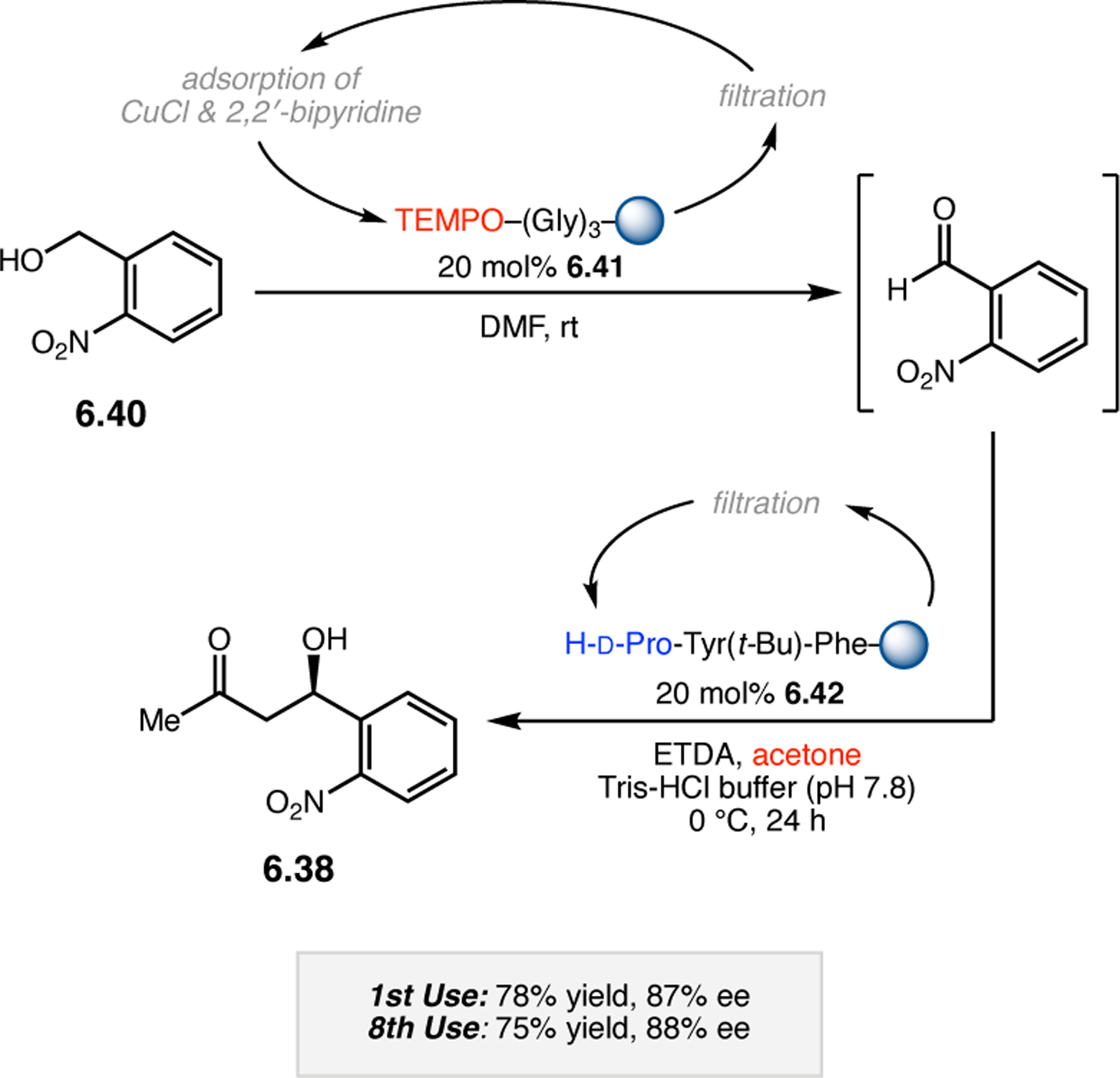
One-pot, tandem oxidation/aldol reaction using resin-supported catalysts that may be reused up to eight times.296
Using previously identified peptide 6.39 (Figure 104) and its enantiomer (ent-6.39), Kudo and co-workers developed a strategy for the kinetic resolution of racemic ansa cyclophanes (i.e., cyclophanes with an aliphatic bridge) via a sequential aldol/retro-aldol process (Figure 106).298 Planar-chiral cyclophanes have been used as sources of chirality in asymmetric catalysis and in the synthesis of functional materials.299,300 However, limited methods for the enantioselective synthesis of ansa cyclophanes have been reported, presumably due to the flexibility of the aliphatic bridge.301 Peptide catalysis proved a viable solution to this unmet challenge. Each step of the tandem process was optimized individually. The initial aldol reaction of ansa cyclophane 6.43 with acetone catalyzed by 6.39 selectively delivered β-hydroxyketone (Sp, S)-6.44 in 93:7 dr and 67% ee, enriching starting material 6.43 to 19% ee (entry 1). The ansa chain was hypothesized to block si-face attack that would produce the (Rp)-isomer about the planar-chiral cyclophane. This aldol product with two elements of chirality—a chiral plane and a chiral center—could then be resolved itself via a retro-aldol reaction catalyzed by ent-6.39, in which minor aldol product (Rp,S)-6.44 was preferentially converted back into the starting aldehyde, thus increasing the enantiopurity of both (Sp,S)-6.44 and 6.43. Using this two-step kinetic resolution strategy, aldol product (Sp,S)-6.44 was successfully obtained in 37% yield and with an enhanced ee of 82%; aldehyde 6.43 was also recovered in 38% yield and 46% ee, and the minor diastereomer (Rp,S)-6.44 was produced in 4% yield and 95% ee (entry 2). The reusability of the resin-supported peptide catalysts was tested, and the selectivity was found to diminish only slightly after two reuses (entry 3). Substrates with an ansa chain of 8 residues and sulfur-linkages were also tested in this reaction, yielding similar results (entries 4 & 5).
Figure 106:
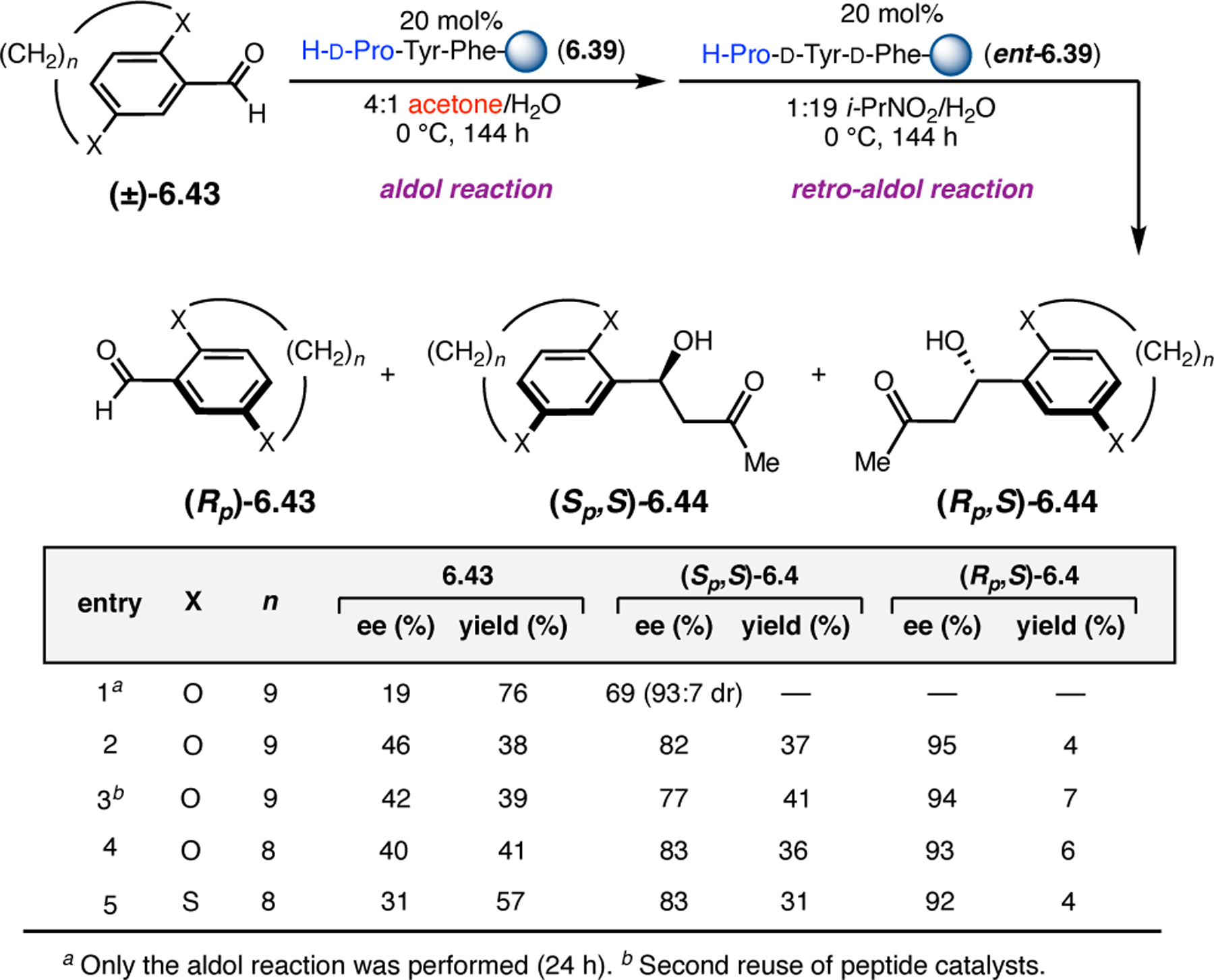
Kinetic resolution of ansa cyclophanes by aldol and retro-aldol reactions.298
Various ketones have been also used as acceptors in peptide-catalyzed direct aldol reactions (Figure 107). Reiser and co-workers demonstrated that β-Acc-containing tripeptide 6.3b catalyzed the Robinson Annulation of 6.45 to the Wieland–Miescher ketone (6.46) in 88% yield and 92% ee (Figure 107a).248 These results are comparable to the best-known examples in the literature.302
Figure 107:
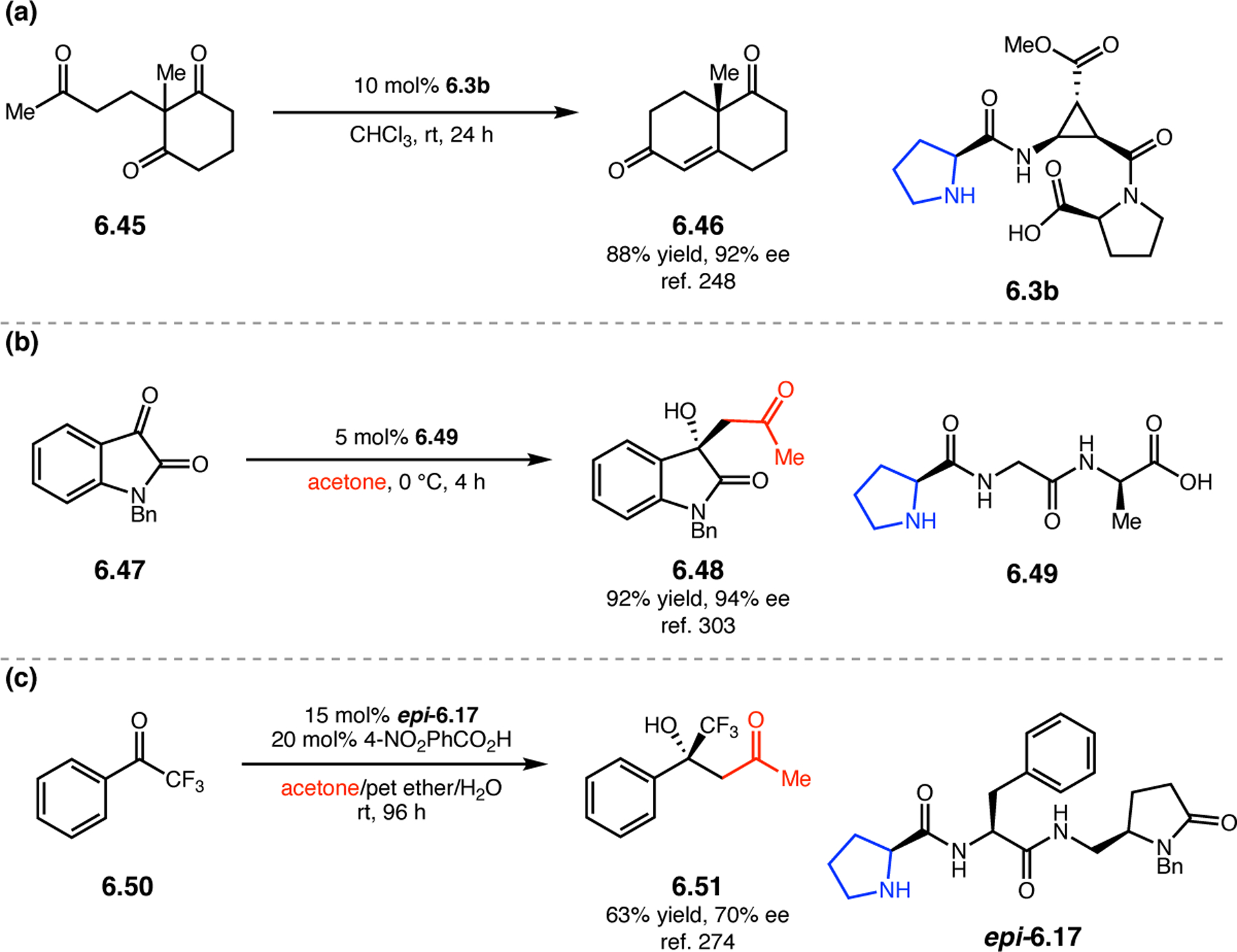
Peptide-catalyzed asymmetric aldol reactions using various ketone acceptors.
Intermolecular direct aldol reactions of more electron deficient ketones are also known. Kohari and co-workers reported the asymmetric direct aldol reaction of isatins like 6.47 with acetone to afford functionalized products 6.48 (Figure 107b).303 Upon evaluation of various tripeptide catalysts, it was apparent that both a d-configured residue and a free carboxylic acid at the C-terminus were essential for achieving high levels of enantioselectivity. Using peptide catalyst 6.49, in which the catalytically active Pro and d-Ala are interspersed with a Gly residue, β-hydroxyketone 6.48 was obtained in 92% yield and 94% ee. Moreover, Moutevelis-Minakakis and co-workers also utilized an electron-deficient ketone, trifluoroacetophenone 6.50, which delivered 6.51 in 63% yield and 70% ee using pyrrolidinone-containing catalyst epi-6.17 (Figure 107c).274 Interestingly, this peptide is the C-terminal epimer of the catalyst that provided optimal results in the aldol reactions of 4-nitrobenzaldehyde (see Figure 99c) and iso-butyraldehyde (Figure 103c) with acetone.
6.3. Stereo- & Site-Divergence in Asymmetric Aldol Reactions
As the diversity of peptide-based catalyst libraries continues to increase, hints of the next directions that could expand the scope of the asymmetric aldol reaction beyond benchmark substrates are beginning to emerge. For example, when using an unsymmetrical aliphatic ketone as an aldol nucleophile, an additional level of control must be considered due to the presence of multiple enolizable sites that could lead to a mixture of β-hydroxyketone products (Figure 108). Fully catalyst-controlled site-, diastereo-, and enantioselectivity are goals that are clearly becoming within reach.
Figure 108:
Potential future directions for catalytic direct aldol reactions.
Divergence in site-selectivity has been observed in asymmetric aldol reactions using different catalytic conditions (Figure 109). For instance, in the aldol reaction between 4-nitrobenzaldehyde and methyl iso-butyl ketone, Li and co-workers observed complete branch site-selectivity when dipeptide 6.11 was used as the catalyst. Despite the long reaction time of 180 h, 6.11 delivered branched product 6.52a in 67% yield, 95:5 dr favoring anti, and 83% ee for the major diastereomer.265 However, in the same reaction, Da’s β-turn-biased peptide 6.1 solely produced the linear isomer with a high 93% ee, albeit in only 24% yield after 240 h.246 In addition to differences in secondary structure, peptides 6.11 and 6.1 differ in the degree of substitution on the catalytically active amine: 6.11 employs the secondary amine of its N-terminal Pro, while 6.1 uses an N-terminal primary amine of Val. As such, these data show that it is possible to achieve site-divergence by modifying the catalyst sequence. In that sense, the modularity of peptide-based catalysts relative to other organocatalytic scaffolds is particularly beneficial.
In another example, Peng observed that both branched and linear β-hydroxyketone products were formed in the peptide 6.27-catalyzed aldol reaction between 4-nitrobenzaldehyde and 2-butanone (Figure 110).276 Branched product 6.53a was obtained in 54% yield, 90:10 dr in favor of the anti-isomer (96% ee), while linear product 6.53b was formed in only 36% yield and with 53% ee. On the other hand, using 2-pentanone as the nucleophile, which is only a single methylene unit longer than 2-butanone, only linear product 6.54b was formed in 69% yield and 39% ee. No branched product 6.54a was observed in this reaction. The authors speculated that two enolizable sites on the ketone substrates could lead to two different chair-like transition states, wherein the size of the ketone substituents would affect the degree of site-selectivity. Although this may explain the apparent substrate-controlled site-selectivity observed for this particular catalytic system, the examples from Figure 109 may suggest that such selectivity could also be achieved in a catalyst-controlled manner.
Figure 110:
Substrate-influenced site-selectivity in asymmetric aldol reaction.276
Diastereoselective direct aldol reactions that operate via enamine-catalysis have often been shown to favor the anti-diastereomer as the major product. Indeed, most of the peptide-based catalysts discussed in Section 6.2 above were anti-selective (see Figures 100, 101, & 103). However, it is sometimes possible to achieve catalyst-controlled diastereodivergence as a function of catalyst structure. For example, in the aldol reaction of 4-nitrobenzaldehyde with 2-hydroxyacetone, β-turn-containing peptide 6.1 gave 6.55 in quantitative yield, 74:26 dr favoring anti, and 87% ee in the major diastereomer (Figure 111).246 The use of co-catalytic (S)-BINOL was found to be beneficial for catalysis, but nearly identical diastereo- and enantioselectivities were obtained using either enantiomer of BINOL. Notably, replacement of the BINOL additive with benzoic acid switched the sense of diastereoselectivity to slightly favor the syn-diastereomer in 58:42 dr. By comparison, Tomasini and co-workers observed significant syn-selectivity in the same reaction under the control of peptide catalyst 6.56 (Figure 111).304 In this case, syn-6.55 was obtained in 98% yield, 83:17 dr, and 88% ee. Interestingly, only branched products were observed in these reactions. Again, catalysts 6.1 and 6.56 differ in their structures and in the nature of the catalytically active amine, both of which could give rise to the observed preferences in diastereoselectivity.
Figure 111:
Divergence of diastereoselectivity in peptide-catalyzed, asymmetric aldol reactions.246,304
Enantiodivergence has also been observed in peptide-mediated aldol reactions. Messerer and Wennemers discovered a unique reversal in the enantioselectivity of a peptide-catalyzed aldol reaction solely by changing the reaction medium (Figure 112).305 β-Turn-biased tripeptide 6.58 was previously identified as an effective catalyst for direct aldol reactions.257,306 To improve the solubility of 6.58, peptide 6.59 was synthesized, which bears a long-chain N-alkyl amide rather than a primary amide. This peptide produced comparable results to 6.58 in the original aldol reaction, indicating that the alkyl chain did not affect the catalytic properties, only the solubility. The catalytic properties of 6.59 were assessed in the aldol reaction between cyclohexanone and 4-nitrobenzaldehyde (Figure 112). Although the aldol product was generally obtained in 1:2 dr favoring the anti-isomer, the solvent had a remarkable effect on observed enantioselectivity. In the absence of water, both DMSO and methanol solvents resulted in the formation of (R,S)-6.57 in 55% and 58% ee, respectively (entries 1 & 4). However, adding of as little as 2.5 vol% of water to DMSO resulted in a reversal in the absolute configuration of the product, favoring (S,R)-6.57 in 42% ee (entry 2). Larger quantities of water increased the enantioselectivity up to 69% ee with 10% water in DMSO (entry 3). This trend was also observed in methanol solvent; as the amount of water increased, the enantioselectivity of (S,R)-6.57 increased up to 20% ee (entries 5 & 6).
Figure 112:

Aldol reaction between cyclohexanone and 4-nitrobenzaldehyde showcasing a reversal in enantioselectivity with 6.46 in different solvents.305
To gain a better understanding of the source of this enantiodivergence, the authors recorded CD spectra of 6.59 in methanol, water, and mixtures of the two solvents (Figure 113). The spectra of 6.59 revealed remarkable conformational differences in methanol as compared to water, suggesting that the switch in the enantioselectivity of this aldol reaction is due to a conformational change from one β-turn conformation to another that has the opposite twist sense.
Figure 113:
CD spectra of 6.59 in methanol (blue), 10% H2O in methanol (red), 50% H2O in methanol (green), and water (orange). Reproduced with permission from ref. 305. Copyright 2011 Thieme.
Bartók and co-workers also noted an interesting case of enantiodivergence when using solid-supported di- versus tri-peptides as catalysts in asymmetric aldol reactions (Figure 114).294 In all the reactions examined, dipeptide 6.36, which is bound to polystyrene resin through a methyl benzhydryl amine (MBHA) linker, delivered the (R)-configured aldol product (entries 1 & 3). However, under otherwise identical reaction conditions, tripeptide 6.60 reliably favored the (S)-aldol product (entries 2 & 4). Imidazole was included as a co-catalyst in these reactions to suppress the formation of side-products, and it was shown not to influence the stereochemical outcomes. To determine whether the resins had any effect on the enantioselectivity outcomes, the same reactions of 4-nitrobenzaldehyde and acetone were performed using solution-phase variants of 6.36 (Pro-Glu-NH2) and 6.60 (Pro-Pro-Glu-NH2). These soluble catalysts provided the (R)-product in 32% ee (25% conversion) and the (S)-product in 42% ee (5% conversion), respectively (compare to entries 1 & 2). These data suggest that the MBHA linker and resin may exert some influence over the reactivity and selectivity of the aldol reaction. In order to better understand the source of enantiodivergence in these peptide-catalyzed reactions, DFT calculations were performed on the enamine intermediates, which were modeled using related aldol catalysts, H-Pro-Asp-MHBA and H-Pro-Pro-Asp-MBHA. The relative energies of the diastereomeric enamine-aldehyde adducts were compared. With respect to tripeptide 6.60, formation of the (S)-enantiomer was favored by 0.52 kcal/mol. However, dipeptide 6.36 favored the (R)-enantiomer by 0.82 kcal/mol.
Taken together, all of these reports show that peptide catalysis is a viable approach for controlling stereochemical outcomes in many direct aldol reactions. Initially discovered in now straightforward reactions, the venerable Pro-catalyzed transformation is expanding into ever more complicated situations.
7. Conjugate Addition
The prowess of enamine catalysis for the aldol reaction (Section 6) has now also been demonstrated explosively in reactions involving conjugate addition. In the mechanism proposed by List and Houk for Pro catalysis, proton transfer between the carboxylic acid moiety of the catalyst and the substrate is critical for nucleating catalyst–substrate interactions.307,308 In this regard, peptide-based catalysts have proven particularly well-suited for engaging conjugate acceptor substrates, as the increase in distance between the C-terminal carboxylic acid and N-terminal Pro residue provides space to accommodate the additional two atoms present in conjugate acceptor substrates (Figure 115). This Section summarizes recent advances in peptide-catalyzed conjugate addition reactions, with a particular emphasis on catalyst structural and mechanistic features. Additions of aldehyde- and ketone-derived nucleophiles into activated olefin acceptors (Section 7.1) is treated separately from the conjugate addition of other C-based nucleophiles, such as indoles and nitronates (Section 7.2).
Figure 115.
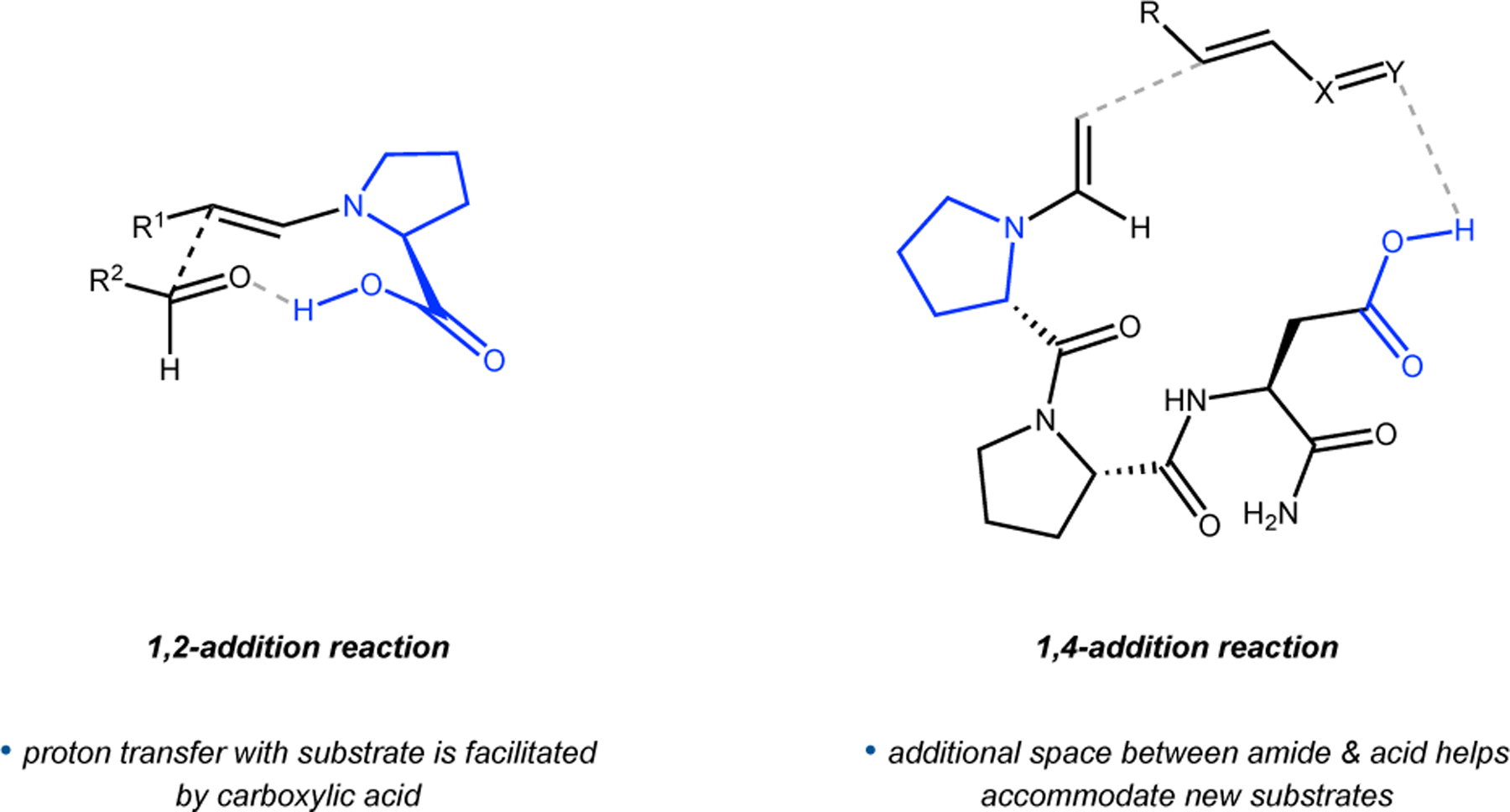
Pro-catalyzed 1,2-addition reaction (i.e., aldol) compared to a peptide-catalyzed 1,4-addition reactions.
7.1. Conjugate Additions of Aldehydes & Ketones to Activated Olefin Acceptors
Recognizing the increased distance requirements of 1,4- relative to 1,2-additions, Wennemers and co-workers hypothesized that β-turn-biased tripeptides, which had previously been shown to be effective catalysts for asymmetric aldol reactions,257 might also be well suited for conjugate addition reactions (Figure 116).309 Molecular modeling indicated that approximately 3 Å of additional space between the secondary amine and carboxylic acid moieties is present in the lowest-energy structure of tripeptide 7.4 (H-Pro-Pro-Asp-NH2, Figure 116a) compared to the Pro monomer (Figure 116b), which could accommodate the extra two atoms present in conjugate acceptors. Peptide-catalyzed conjugate addition reactions of aldehydes into nitroolefins had previously been demonstrated and was thus selected as a model reaction.4 Indeed, these tripeptides proved highly effective in the reactions of aldehydes 7.1 with nitroolefins 7.2, providing γ-nitroaldehyde products 7.3 in excellent yields, diastereoselectivities, and enantioselectivities at catalyst loadings as low as 1 mol% (Figure 116c). Of the four possible diastereomers 7.4–7.7 (entries 1–4), it was found that the d-Pro-Pro-Asp stereotriad of 7.7 was the most selective, providing syn-7.3 in 25:1 dr and 97:3 er (entry 4). It is notable that related chiral amine catalysts typically require catalyst loadings of 10–20 mol% in order to achieve similar results. Studies of the primary sequence revealed that while the N-terminal Pro stereogenic center controls the enantioselectivity, the remaining stereocenters on the catalyst must be matched in order to achieve high catalytic performance.
Figure 116.
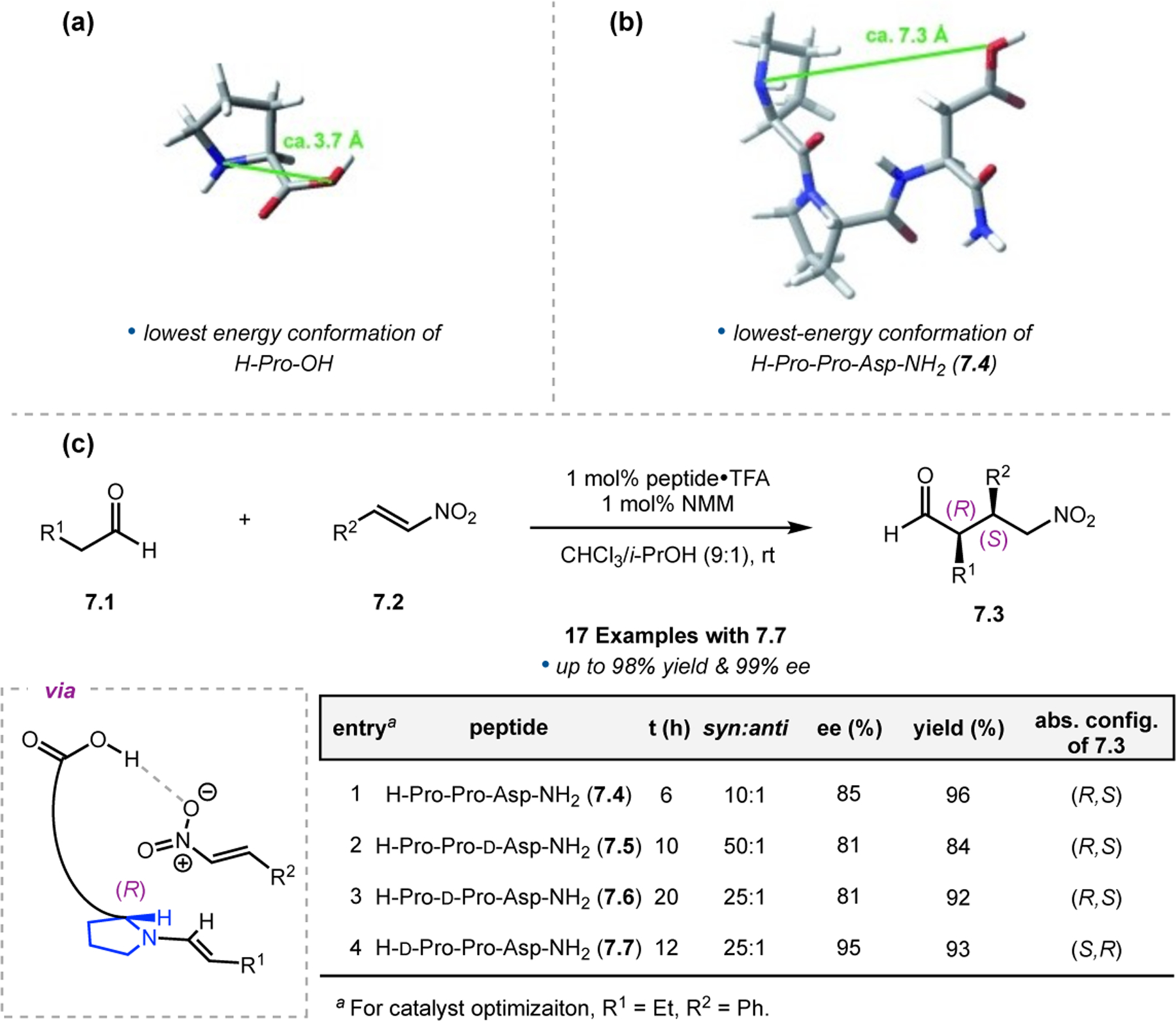
Lowest-energy conformations of (a) H-Pro-OH and (b) H-Pro-Pro-Asp-NH2 (7.4) calculated using MacroModel 8.0. (c) Peptide-catalyzed conjugate addition of aldehydes 7.1 into nitroolefins 7.2, which is proposed to proceed through an enamine intermediate and is assisted by judicious placement of a protic sidechain. Reproduced in part from with permission from ref. 309. Copyright 2008 Wiley.
Following this initial study, the Wennemers Group investigated conjugate additions of aldehydes 7.1 into terminal nitroolefins 7.8 to provide γ-nitroaldehydes 7.9, which in turn could be converted into monosubstituted γ2-amino acids (Figure 117).310 The previously identified peptide 7.7 was found to be an effective catalyst for this transformation, as well. However, significant rate enhancement and increases in the observed enantioselectivity were observed by simply exchanging the C-terminal Asp residue for a Glu residue, which bears an additional methylene (7.10, entry 2). While such conjugate addition reactions are often plagued by the formation of homoaldol side-reactivity, this byproduct was not observed using catalyst 7.10. Additionally, only a minimal excess (1.5 equiv) of aldehyde relative to nitroolefin was required to achieve high levels of reactivity. With slightly higher catalyst loadings (3 mol%), substrates with bulky substituents were tolerated, such as isopropyl and tert-butyl groups.
Figure 117.
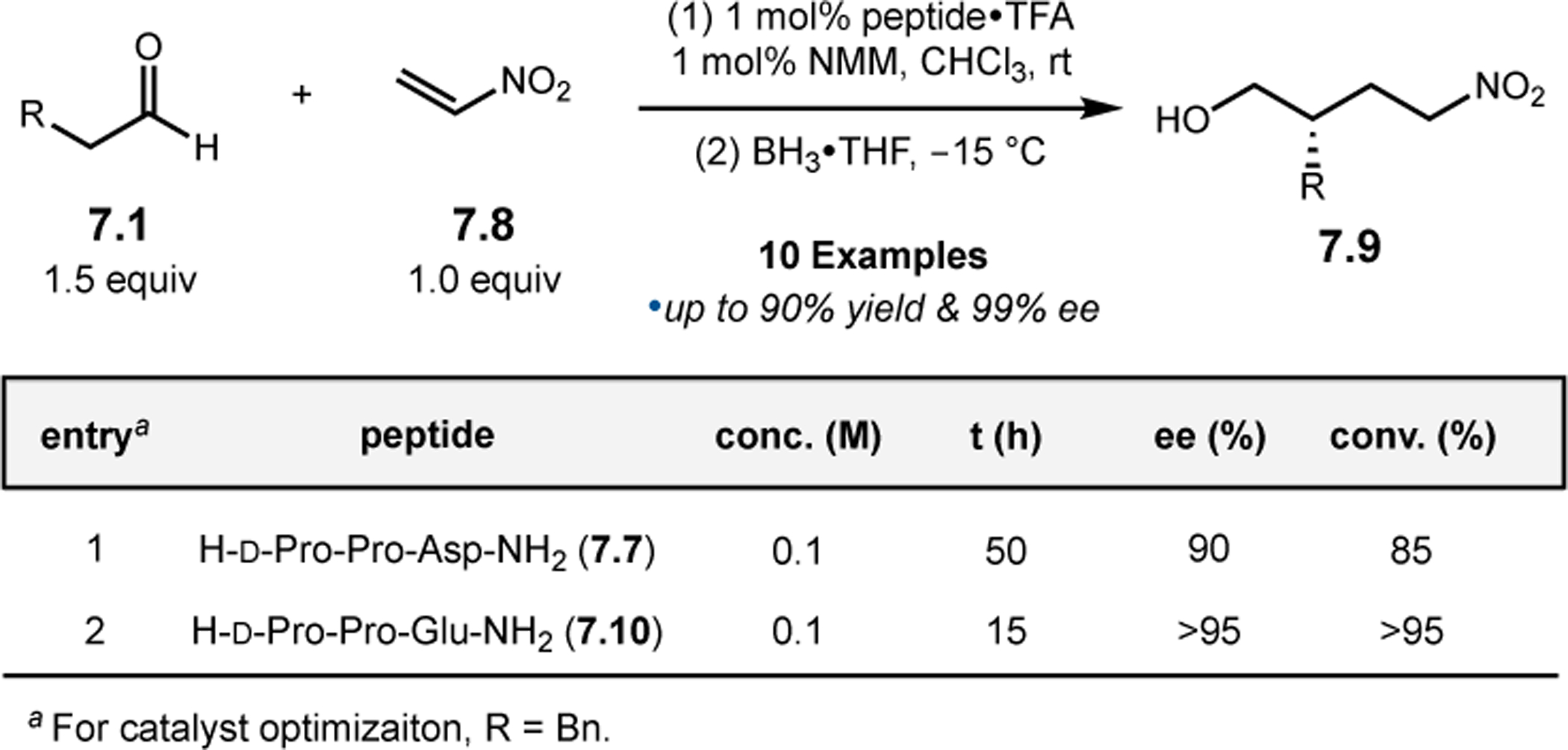
Conjugate addition of aldehydes 7.1 into nitroolefins 7.8 lacking substitution at the β-position.310
A more in-depth interrogation of the C-terminal residue in the peptide-catalyzed conjugate addition of 7.11 into 7.12 revealed that the enantioselectivity of the reaction was relatively insensitive to the identity of the C-terminal functional group (Figure 118, entries 1–3).311 Conversely, when the identity of the sidechain carboxylate was altered to an amide (entry 4), significant reductions in both conversion and enantioselectivity were observed, suggesting that this carboxylic acid plays a critical role in substrate coordination and orientation. While the insertion of an additional methylene unit to obtain Glu from Asp was found to be beneficial to both yield and selectivity (entry 5), addition of a third methylene within the sidechain residue decreased the observed selectivity (entry 6).
Figure 118.

Optimization of the C-terminal residue of tripeptides in the asymmetric conjugate addition of aldehydes into nitroolefins.311
Moreover, the effect of water on the reactivity of this system was assessed by comparing “standard” and “dry” reaction conditions, in which water had been rigorously excluded (Figure 119a).312 It was found that reaction times could be substantially shortened under the dry conditions (entries 1 vs. 2). Additionally, at longer reaction times, substantially lower catalyst loadings could be used (0.1 mol%) with only a minor drop in enantioselectivity (entry 3). Studies of the fundamental steps revealed that, at higher concentrations, the reaction displays a zero-order dependence with respect to butanal 7.11, indicating a steady state is reached between the aldehyde and the catalyst, presumably to form an enamine intermediate (Figure 119b, green line). This suggests that enamine formation is not rate-determining. When using the dry conditions (red line), this steady state is reached at a lower concentration. When additional water is intentionally included (blue line), the steady state is never reached. Kinetic studies revealed that reactions conducted under the anhydrous conditions proceeded rapidly, while reactions with added water were relatively sluggish (Figure 119c).
Figure 119.
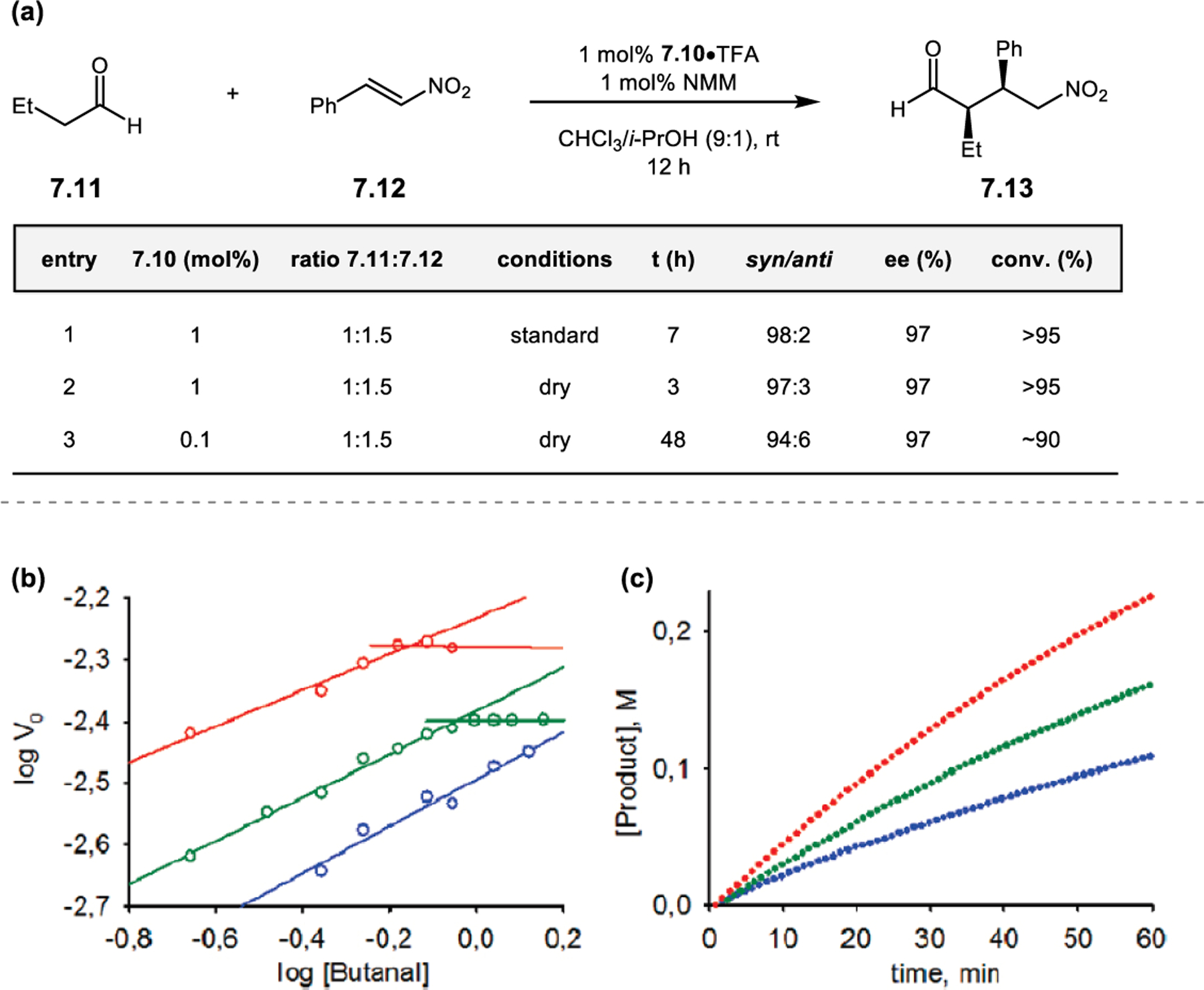
(a) Conjugate additions between n-butanal 7.11 and nitrostyrene 7.12 under different conditions. Under the “dry” conditions, anhydrous reagents and solvents were used in contrast to the standard conditions. (b) Initial rates vs. concentration of 7.11 under various conditions in the conjugate addition of 7.11 to 7.12 (green = standard conditions, red = anhydrous conditions, and blue = 10 mol% excess of water). (c) Influence of water on rates of product formation. Reproduced in part from with permission from ref. 312. Copyright 2010 American Chemical Society.
While an enamine mechanism for this transformation is widely accepted, it had not been unambiguously determined for peptide catalyzed reactions. The groups of Wennemers and Pfaltz employed ESI-MS in order to rigorously investigate the presence of an enamine intermediate, (Figure 120).313 According to the principle of microscopic reversibility, if the C–C bond-forming step was rate-determining, then the related C–C bond cleavage step of the reverse reaction should be governed by the same transition states. As such, the difference in energy between transition states should be the same, which governs the enantioselectivity of a transformation. The reverse C–C bond cleavage step requires the addition of peptide to form an iminium ion. Therefore, if the forward reaction proceeds through an iminium/enamine mechanism, the enantioselectivity of both the forward and the reverse process should be equal. By using a mixture of quasi-enantiomeric substrates 7.14 that vary at the 4-position of the aryl ring, enantioselectivity can be measured rapidly by quantitative MS analysis. Indeed, the mass distribution between enamine 7.16a and 7.16b reflected the measured ee values for the forward transformation catalyzed by peptide 7.10. The performance of related catalysts that lack an acidic group (e.g., 7.17), suggest that C–C bond formation is only rate determining in cases where rapid protonation is facilitated by an optimally positioned acidic group.
Figure 120.
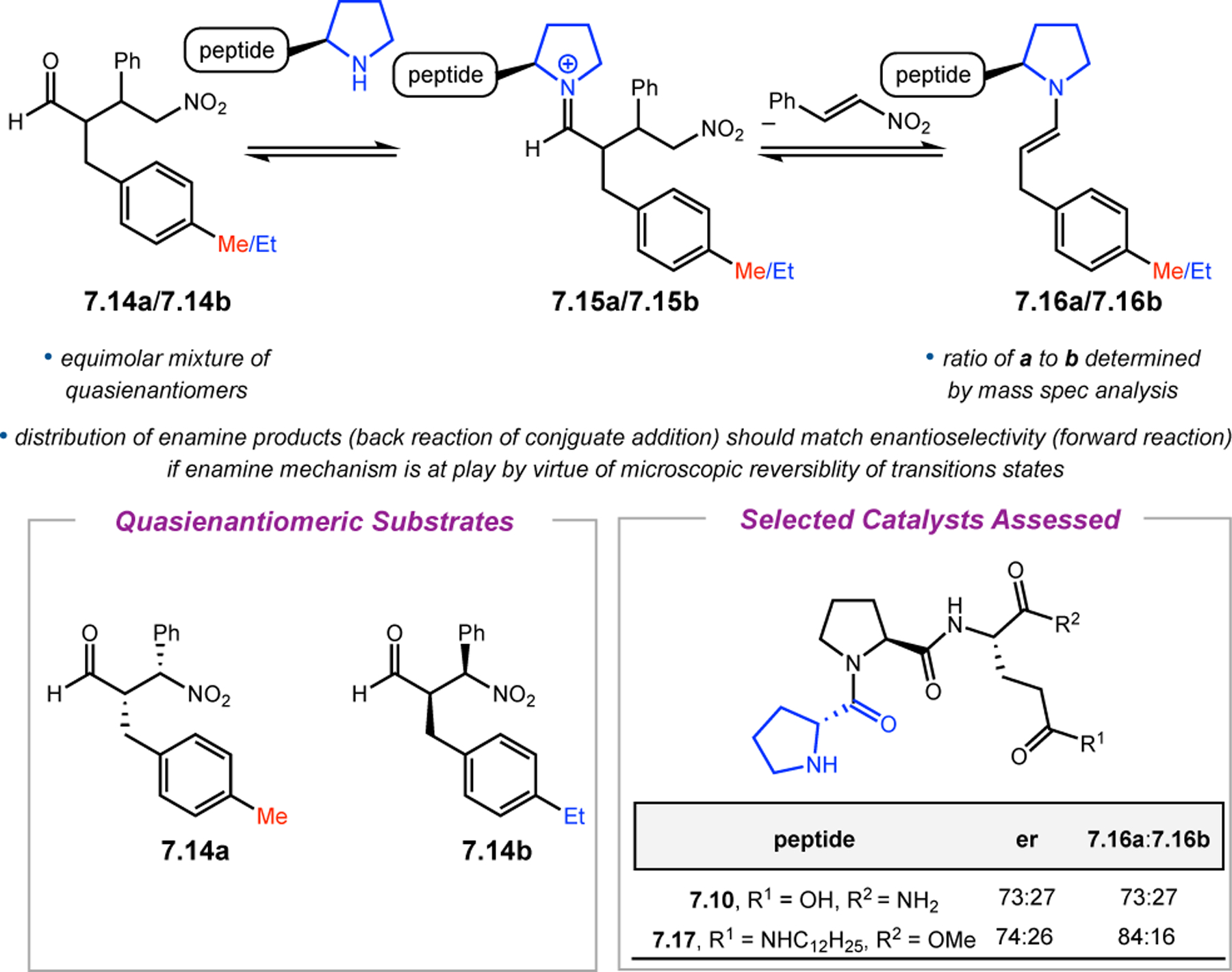
Strategy for verifying an enamine mechanism by employing an ESI-MS study of the reverse-reaction using mass-labeled quasi-enantiomeric conjugate addition products.313
The Wennemers Group has also demonstrated that the ability to synthesize peptides on-resin allows for the facile development of immobilized catalysts, which can be recycled multiple times.314 In principle, organic catalysts are particularly attractive when immobilized, as they tend to be moisture and air stable. These peptide-based catalysts for conjugate addition reactions have been shown to be highly effective when immobilized on solid support, allowing for the isolation of conjugate addition products in high yield and stereoselectivity after simple filtration of the catalyst and removal of volatiles (Figure 121a). In this system, catalysts were able to be recycled at least 30 times without diminished reactivity or selectivity (Figure 121b). Similarly, immobilized peptide catalysts have also been successfully employed in flow reaction setups.315
Figure 121.
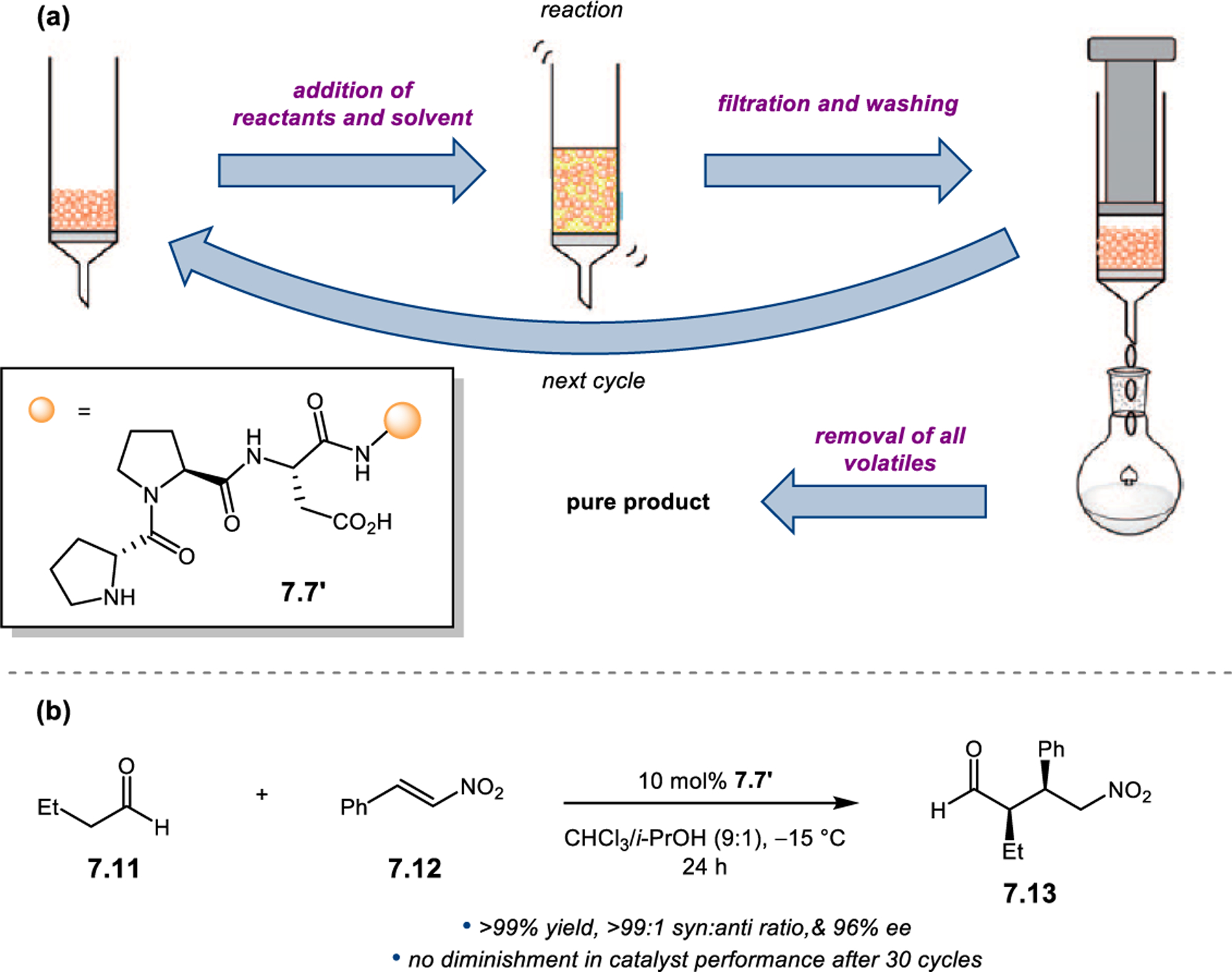
(a) Strategy for conducting reactions with tripeptide catalysts suspended on resin. (b) Performance of the proposed strategy in the asymmetric conjugate addition of aldehyde 7.11 into nitroolefin 7.12. Reproduced in part from with permission from ref. 314. Copyright 2011 Wiley.
Additionally, Wennemers and co-workers have also explored the behavior of these trimeric catalysts under aqueous reaction conditions.316 When lead catalyst 7.10 was appended with long alkyl chains at the C-terminus and on the Pro sidechain, peptide catalysts 7.18 and 7.19, respectively, were found to form emulsions in aqueous media due to the hydrophobic effect (Figure 122). Despite the negative effect of water in these conjugate additions (vide supra), these emulsions were found to efficiently catalyze the conjugate addition of 7.1 into 7.2 with catalyst loadings as low as 3 mol% (entries 2 & 3). Catalysts lacking these modifications did not emulsify in aqueous media, and diminished catalytic efficiency was observed as a result (entry 1). The rate of reactivity for these transformations was found to depend not only on the reactivity of the nitroolefin, but also on the ability of the peptide to effectively form an emulsion in water.
Figure 122.
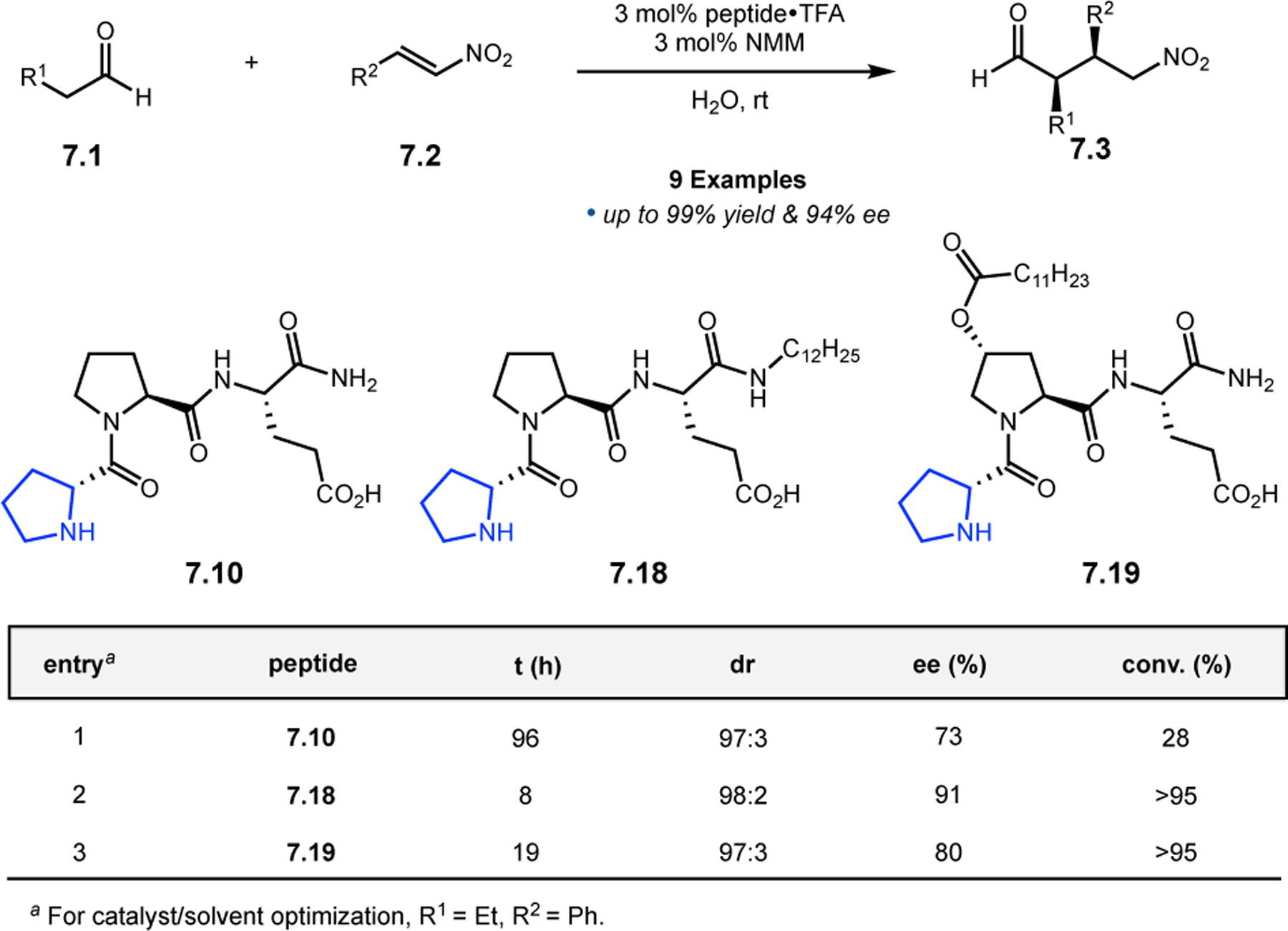
Application of tripeptide catalysts with added alkyl chains (e.g., 7.18 & 7.19) to promote micelle formation in asymmetric conjugate addition reactions under aqueous conditions.316
Encouraged by these promising results, the Wennemers Group further expanded their studies to include conjugate additions of α,β-disubstituted nitroolefins 7.20 (Figure 123a), which have historically proven to be particularly challenging substrates.317 The presence of α-substitution not only provides additional steric hindrance, but it also distorts the planarity between the aryl β-substituent and the double bond, hampering conjugation and deactivating the substrate toward 1,4-addition. Furthermore, the formation of an additional stereogenic center in these reactions must be controlled in order to achieve a highly selective transformation. After examining several variations of parent catalyst 7.10, peptides 7.22 and 7.23 were identified as particularly effective. These peptides contain a Pro-Pro sequence instead of the d-Pro-Pro motif that had proven effective in the previously studied reactions. It is notable that of the four possible diastereomers of γ-nitroolefin 7.21 (without considering their enantiomers), significantly different distributions are observed when catalysts 7.22 and 7.23 are employed as opposed to pyrrolidine or Pro (Figure 123b, entries 3 & 4 vs. 1 & 2). In fact, upon examining the distribution of 7.21a and 7.21c, which are epimers at the α-position, opposite epimers are favored in the case of peptide- versus non-peptide-catalyzed reactions, indicating that the protonation step is also under catalyst control. It is also notable that catalysts 7.22 and 7.23 favor the same product diastereomer (7.21a) despite having different stereotriads; 7.23 has the opposite configuration at the C-terminal residue.
Figure 123.
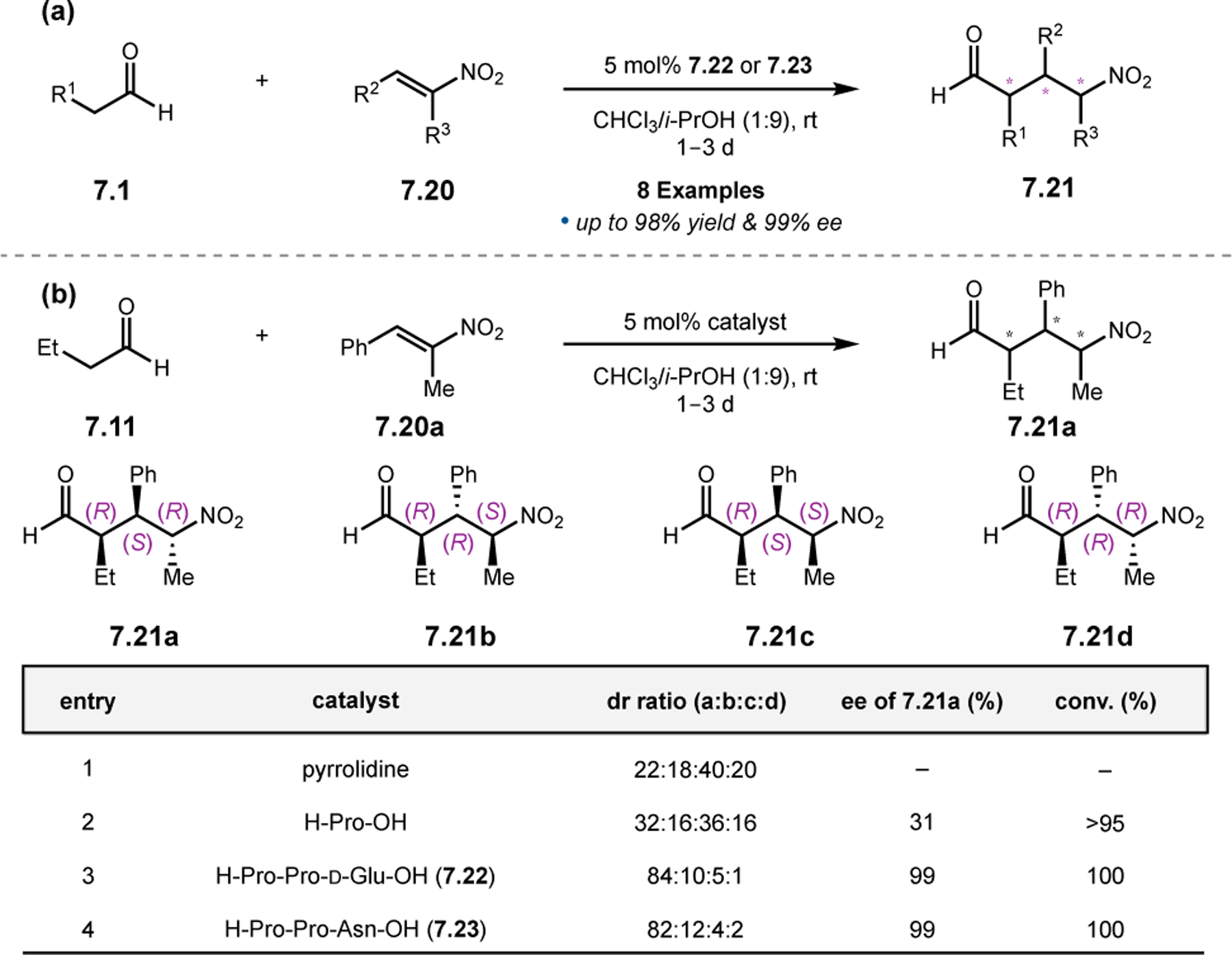
(a) Conjugate addition of aldehydes 7.1 to nitroolefins 7.20 bearing both α- and β-substitution catalyzed by peptides 7.22 and 7.23. (b) Influence of catalysts on the distribution of diastereomers 7.21a–d in the conjugate addition of aldehyde 7.11 into nitroolefin 7.20a bearing both α- and β-substitution. Both peptide catalysts 7.22 and 7.23 substantially alter the intrinsic diastereomeric preference.317
Despite great successes in the conjugate addition of aldehydes into α,β-disubstituted nitroolefins, sterically congested β,β-disubstituted nitroolefins, such as 7.24, proved to be more challenging initially (Figure 124).318 Evaluation of the previously explored peptides resulted in low conversions to the desired product 7.25 and produced large amounts of the undesired homoaldol product (entry 1). It was found that substituting out the C-terminal carboxylic acid and amide of peptide 7.10 with aryl groups dramatically increased conversion (entries 2 & 3). This observation was in contrast to earlier studies, which had identified that carboxylic acid functionality as critical for selective catalysis. Additionally, new solvent conditions were required for high reactivity, with tert-butanol ultimately emerging as the optimal solvent (entry 4). Under these conditions, peptide 7.27 afforded γ-nitroaldehyde 7.25, which possesses an all-carbon quaternary stereocenter, in 96% conversion, 6:1 dr, and 94% ee. Further mechanistic studies of this reaction are presented below.
Figure 124.

Optimization of tripeptides for the conjugate addition of aldehydes 7.1 into sterically hindered β,β-disubstituted nitroolefins 7.24 to generate an all-carbon quaternary stereogenic center.318
Seeking to expand the scope of peptide-catalyzed, asymmetric conjugate additions, the Wennemers Group examined the addition of acetophenone and related ketones 7.28 to dicyanoolefins 7.29 (Figure 125).319 Under the optimized conditions, catalyst 7.10 was found to be effective without the need for further catalyst optimization, despite the well-known challenges in iminium ion formation between secondary amines and ketones. In order to overcome low reactivity, however, 20 mol% of the catalyst was used, and an excess of ketone 7.28 (5.0 equiv) was required. Under these conditions, 7.10 delivered ketones 7.30 in up to 90% yield and 88:12 er.
Figure 125.
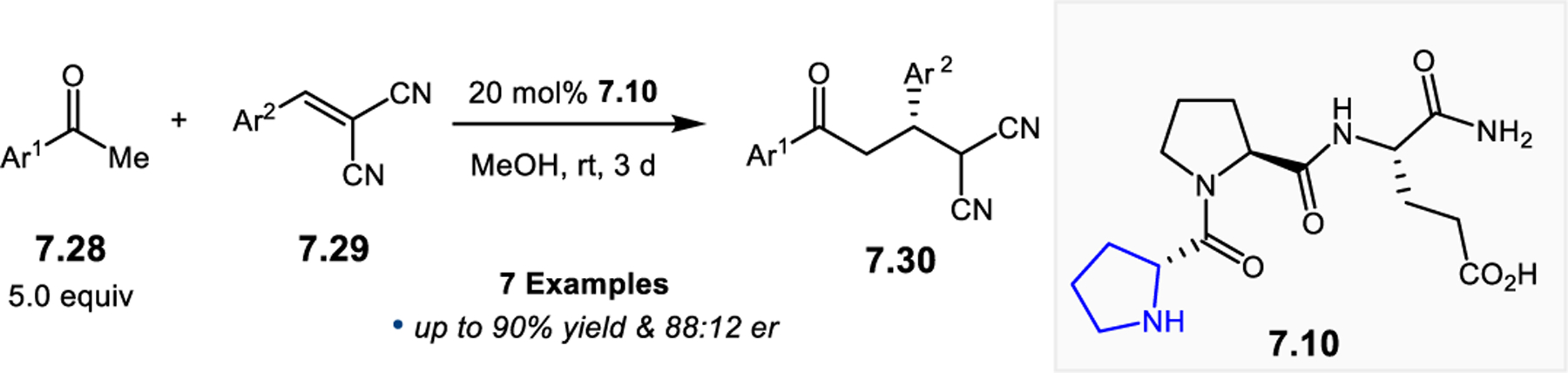
Conjugate addition of acetophenone into dicyanoolefins catalyzed by a tripeptide 7.10 containing a secondary amine.319
The Wennemers Group reasoned that, given the highly tunable nature of peptide scaffolds and their previous successes in their studies of related conjugate additions, maleimide (7.32) could also serve as a potential conjugate acceptor (Figure 126).320 After screening previously reported peptides with limited success (Figure 126a, entries 1 & 2), it was found that the incorporation of additional amide functionalities at the C-terminus (entries 3 & 4) proved to be effective in promoting both reactivity and enantioselectivity in the reaction of aldehydes 7.31 with 7.32. Lead catalyst 7.35 delivered the succinimide product 7.33 in 93% conversion, 86:14 dr, and 98% ee under the optimized conditions. It was proposed that H-bonding, rather than protonation, is critical for accelerating the C–C bond forming pathway. In fact, when maleimide 7.36 (which lacks an H-bond donor) was subjected to the reaction conditions, the product was obtained with high enantioselectivity, but in low yield (Figure 126b), which suggests an H-bonding interaction with the maleimide helps to increase reactivity.
Figure 126.
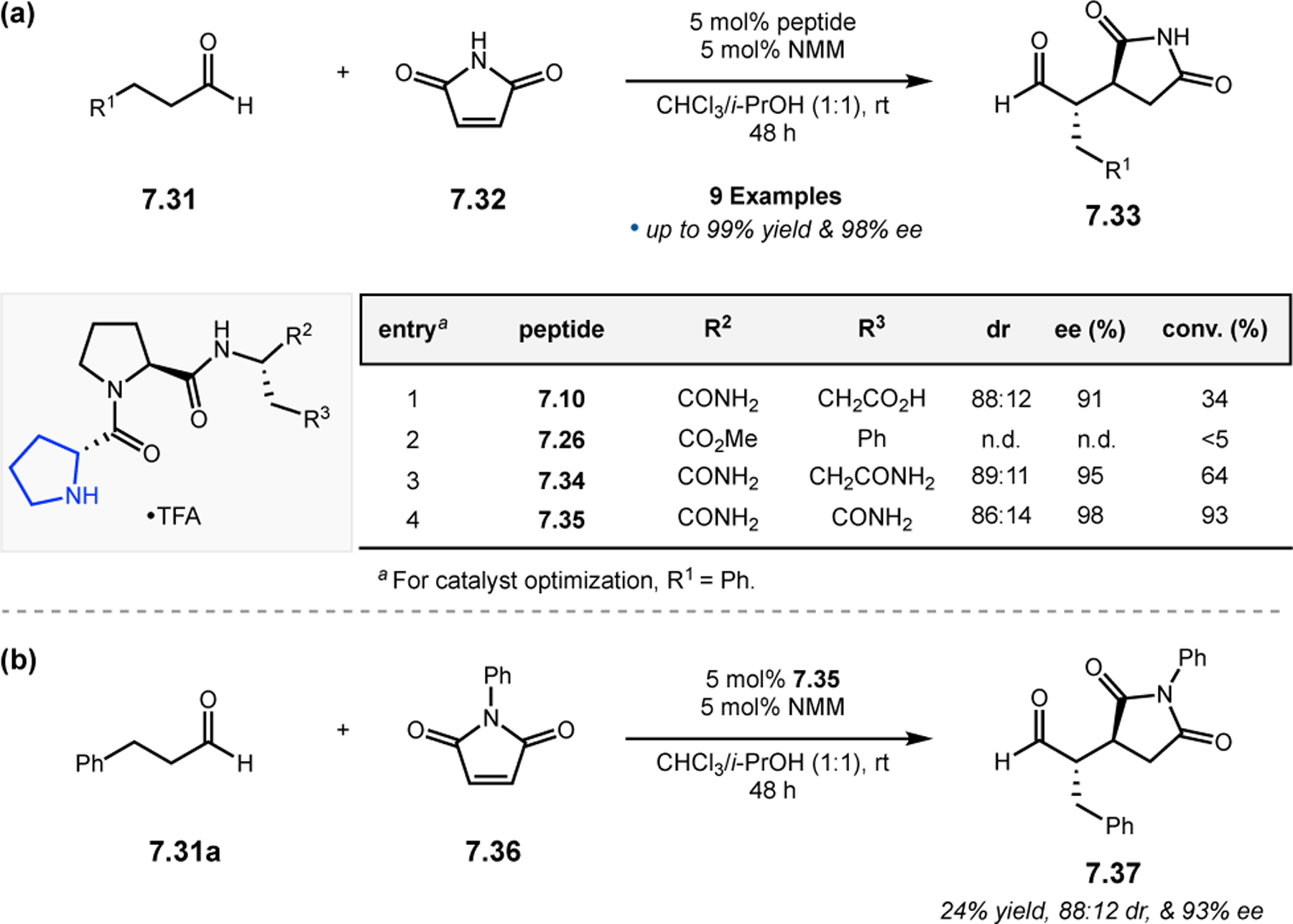
(a) Optimization of tripeptide-based catalysts for the asymmetric conjugate addition of aldehydes 7.31 into maleimide 7.32. (b) Low-yielding conjugate addition into N-phenyl protected maleimide.320
While many of the lead peptides identified over the course of these studies contain a d-Pro-Pro motif, subtle differences in the C-termini led to drastically different catalytic performances. In order to gain insight into how structural differences between the peptides might impact catalytic function, X-ray crystal structures of peptides 7.7, 7.27, and 7.35 were obtained and analyzed (Figure 127a).321 An overlay of these structures reveals that they adopt relatively similar conformations with respect to the d-Pro-Pro turn motif, with some variation being observed in the ring pucker of the i+1 Pro residue (Figure 127b). A significant difference between these three peptides, however, is the presence or absence of an intramolecular H-bond between the C- and N-termini. This H-bond, or lack thereof, was dependent on the identity of the C-terminal residue. While all three peptides adopt a similar β-turn structure in the solid state, the turn conformations do vary as a result of this C- to N-terminal interaction. Additionally, the presence of an n→π* interaction322 between the C-terminal d-Pro carbonyl and the adjacent Pro carbonyl (Figure 127c) is present in peptides 7.7 and 7.27 as evidenced by Bürgi–Dunitz trajectories (95° < θ < 125°) and significantly shortened Oi–1 to Ci distances. This interaction is not present in 7.35, which instead is engaged in an intramolecular H-bond between the C-terminal amide and N-terminal Pro. While these studies provide valuable insight into the conformation of peptide catalysts, it was recognized that more in-depth studies of the solution-phase structure of the peptides would be required for understanding catalytically relevant species.
Figure 127.
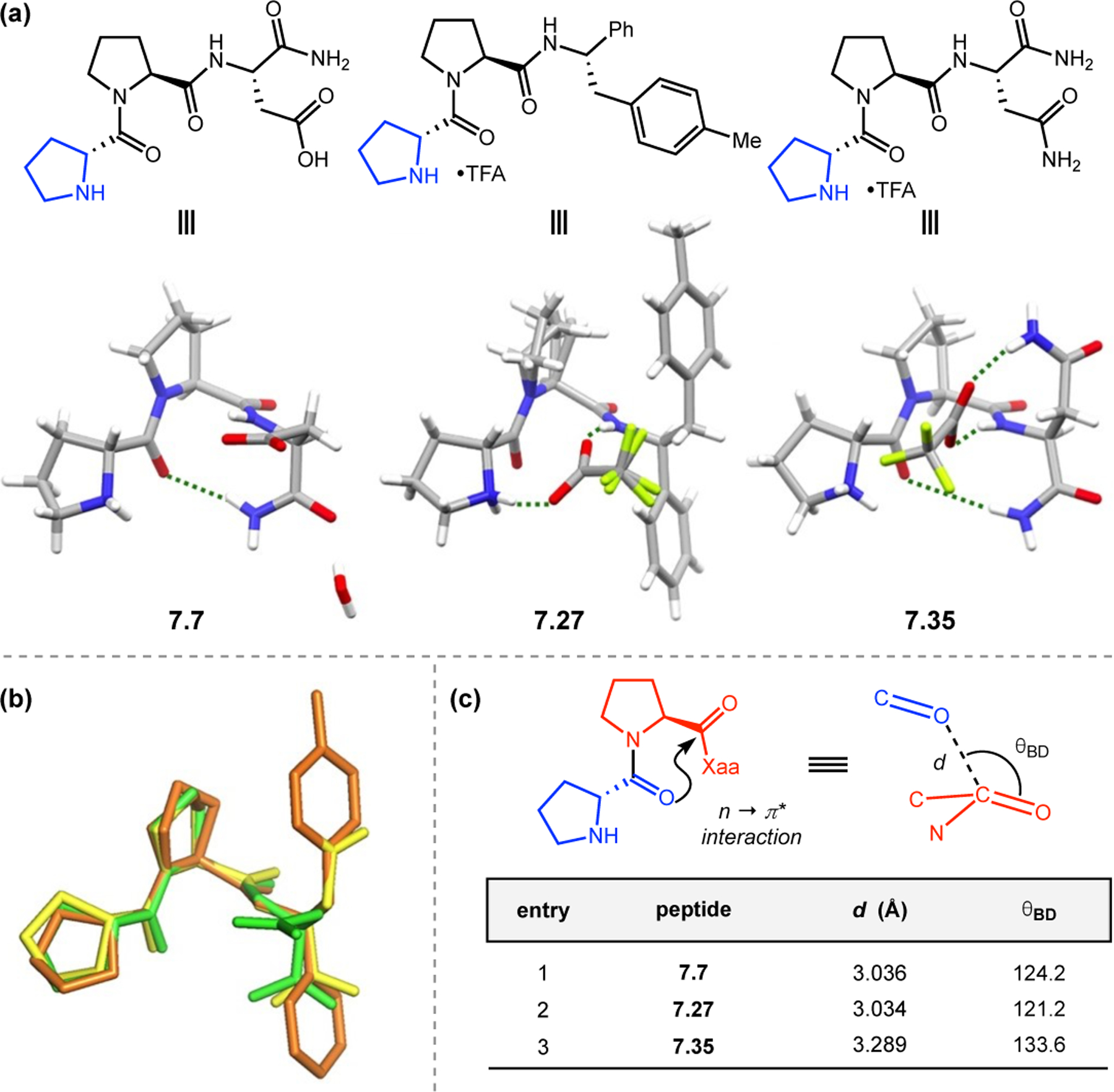
(a) Crystal structures of three optimized catalysts for conjugate addition reactions with various olefin acceptors. (b) Overlay of the three crystal structures for 7.7 (green), 7.27 (orange), and 7.35 (yellow) with H-atoms and TFA counterion removed for clarity. (c) The n→π* interactions suggested by the distance between the carbonyl O and the adjacent carbonyl C, as well as the approximation of the Bürgi–Dunitz angle. Reproduced in part with permission from ref. 321. Copyright 2011 Wiley.
The n→π* interaction present in the crystal structures of 7.7 and 7.27 has been the study of several solution-phase studies by the Wennemers Group. This interaction is of particular interest, as it has been shown to influence the trans/cis equilibrium (Kt/c) of Pro amides and to impact the structures of peptides and proteins.323,324 In light of this, Wennemers and co-workers hypothesized that such interactions may influence the catalytic performance of tripeptide-based catalysts. Accordingly, peptides with different i+1 Pro analogs of various ring sizes were examined for their trans/cis ratio in solution (Figure 128).325 NMR analysis was used to measure Kt/c, which was found to be highly correlated with enantioselectivity in the addition of aldehyde 7.11 to nitroolefin 7.12. It is noteworthy that, upon examining different solvent mixtures, peptides 7.38, 7.10, and 7.39 exhibited different trans/cis ratios, and the catalyst that displayed the highest Kt/c would vary depending on the medium. However, the enantioselectivity consistently displayed a linear relationship with the percentage of peptide adopting the trans conformation. It was confirmed that the Kt/c of the unfunctionalized peptide is correlated with the Kt/c of the reactive enamine intermediate 7.10-En that is relevant to catalysis. This trend was found to be true for conjugate addition reactions involving maleimides as well.
Figure 128.
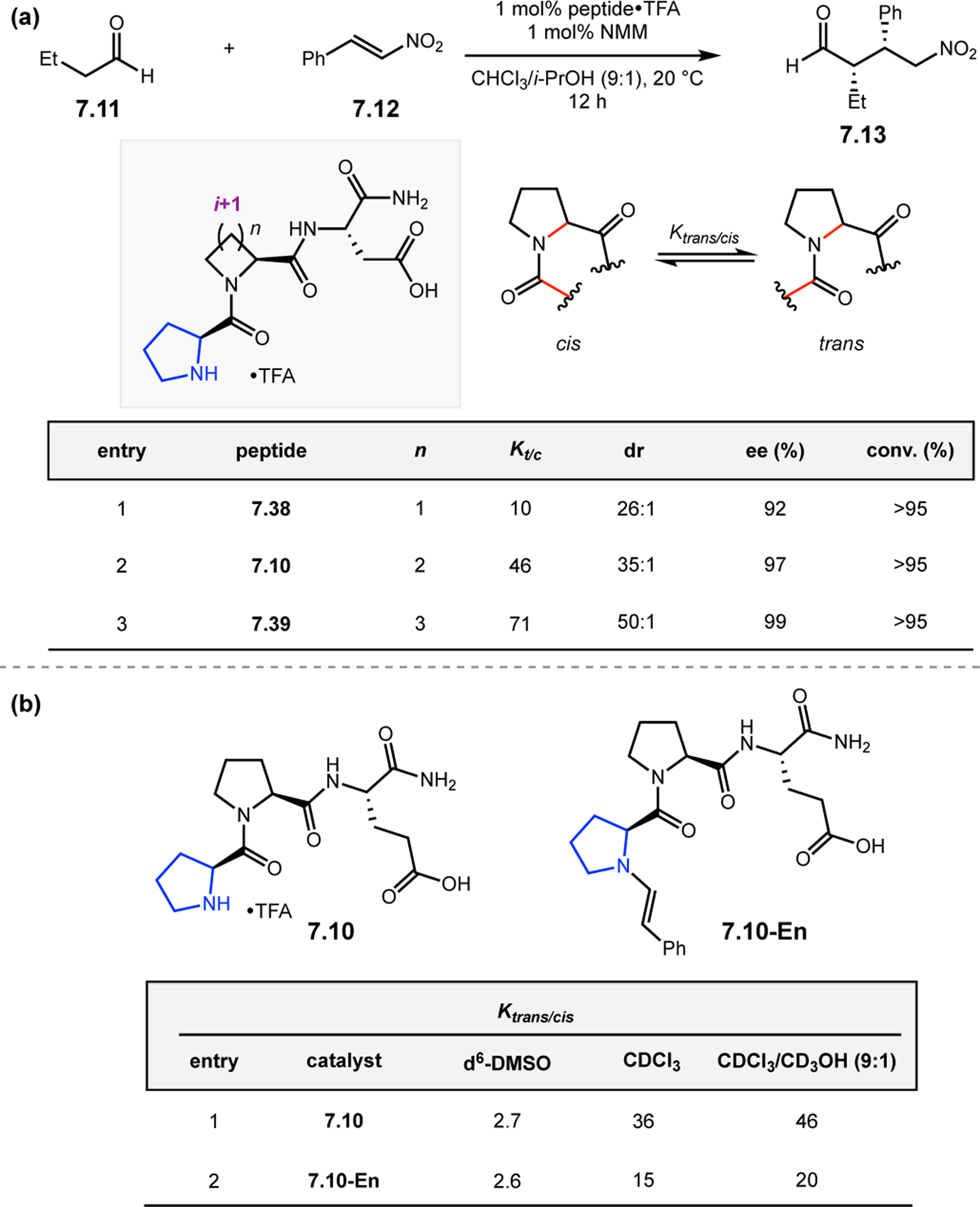
(a) Influence of ring size on the trans/cis ratio for the amide bond of the central Pro residue and its relation to enantioselectivity in the conjugate addition reactions. (b) Kt/c values for catalyst 7.10 and its enamine form 7.10-En in different solvents.325
Substituents on the pyrrolidine ring of the i+1 Pro have also been shown to affect the trans/cis ratio in peptide catalysts.326 4R-Substituted pyrrolidine rings typically favor the Cγ-exo ring pucker, in contrast to unsubstituted pyrrolidine rings, which favor the Cγ-endo ring pucker. Accordingly, peptides with this pyrrolidine substitution adopt a β-turn conformation that lacks the stabilizing n→π* interaction that is present in other peptides. Lower selectivity is observed in reactions catalyzed by these peptides, which is consistent with previous studies on trans/cis ratios (Figure 129, entries 2 & 3 vs. 1). In contrast, 4S-substituted pyrrolidine rings, which favor the Cγ-endo ring pucker have an inverse relationship to Kt/c. It is proposed that the substituent points directly towards the cavity of the catalyst and overrides the selectivity afforded by the higher trans/cis ratio (entries 4–6).
Figure 129.
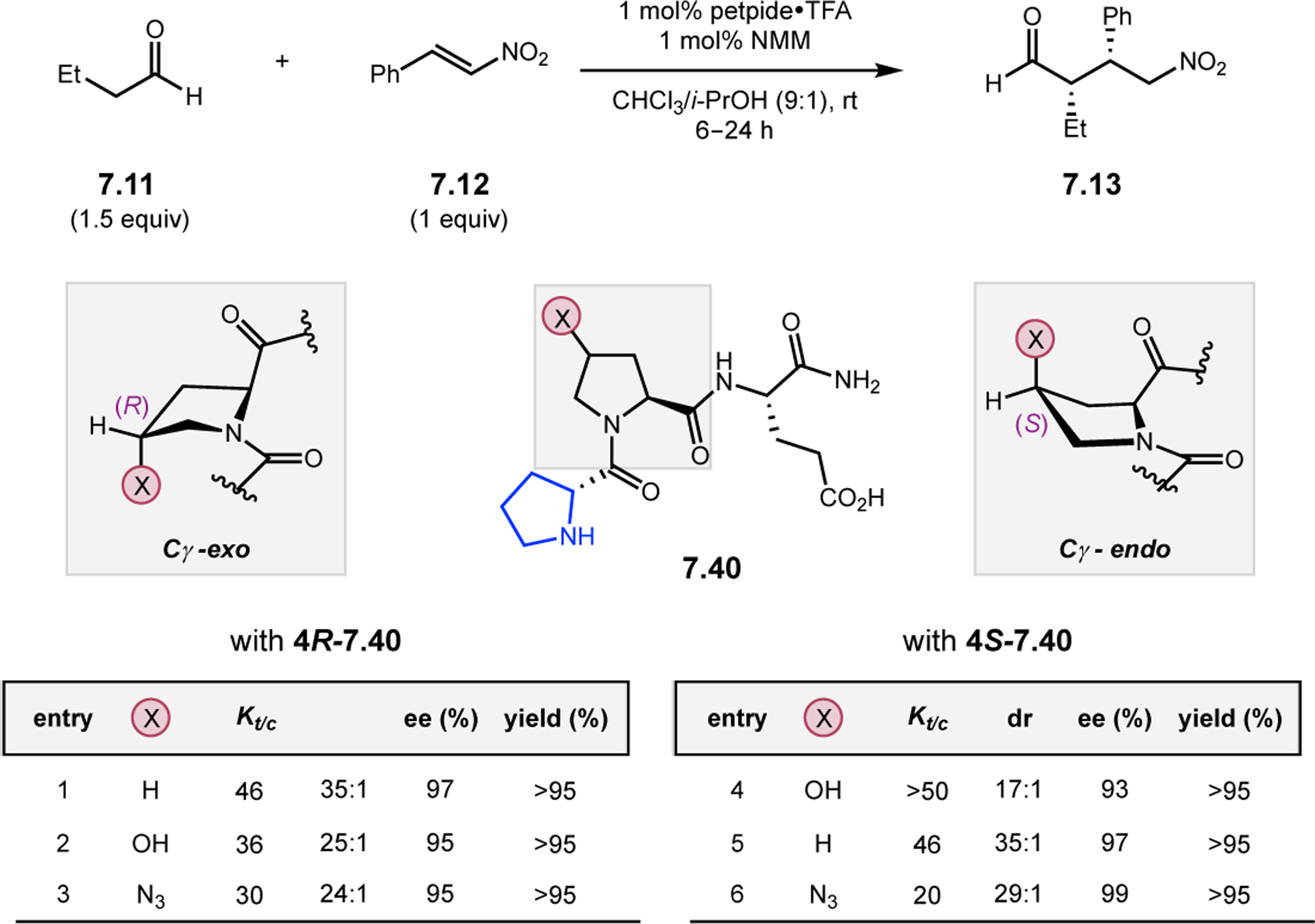
Effects of substitution on the ring pucker of the i+1 Pro residue and its influence on enantioinduction in the asymmetric conjugate additions of aldehydes 7.11 into nitroolefins 7.12.326
Additional studies had identified that trans/cis ratios are influenced not only by an n→π* interaction, but also by the net dipole moment of the peptide molecule (Figure 130).327 DFT calculations revealed that the Boltzmann-averaged dipole moment of model compound 7.42, which contains a C-terminal dimethylamide, is significantly higher for the cis conformation than that of the trans (Δμ). This is in stark contrast to methyl esters 7.41, in which both amide isomers have very similar dipole moments. As such, the variability in Kt/c as a function of solvent polarity is much greater for amide 7.42, wherein the trans conformer is greatly favored in non-polar solvents, such as chloroform and dioxane. NMR analysis of peptide 7.7, which also contains a dialkylamide motif adjacent to the N-terminal Pro residue (i.e., the i+1 Pro), in DMSO-d6 and a 9:1 mixture of CDCl3/CD3OD revealed that a greater trans/cis ratio is indeed observed in the more non-polar solvent mixture. Reactions conducted in these solvents reflected this difference in the enantioselectivity of the reaction.
Figure 130.
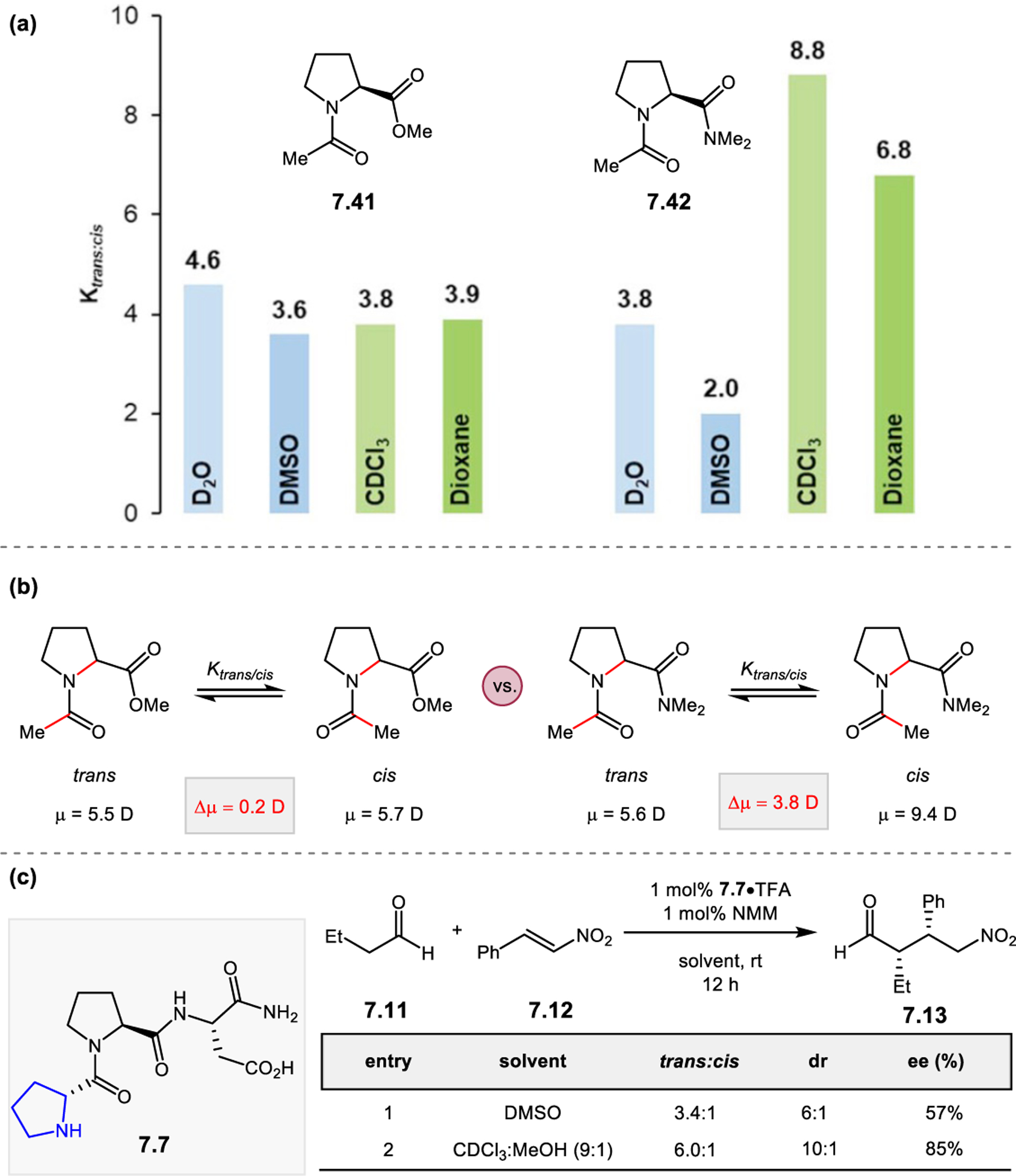
(a) Ktrans/cis values for 7.41 and 7.42 in various solvents. (b) Computed Boltzmann-averaged dipole moments of trans and cis conformers for compounds 7.34 and 7.35. (c) Solvent influence on trans/cis ratios of peptide 7.7 and its effect of enantioselectivity. Ratios were determined by NMR analysis in DMSO-d6 and a 9:1 mixture of CDCl3:CD3OD. Reproduced in part from with permission from ref. 327. Copyright 2015. Royal Society of Chemistry.
In order to assess the potential structures of catalytically relevant species, detailed NMR studies were conducted on peptide catalyst 7.10, providing insight into the structure in solution, which is not apparent from previous studies analyzing crystal structures.328 Using a combination of HSQC, ROESY, TOCSY, DQF-COSY, and HMBC methods, the solution phase structure of peptide 7.10 was found to be consistent with a type I β-turn (Figure 131a).47 Notably, several changes were observed in the NOE interactions of enamine derivative 7.10-En, wherein some interactions were notably stronger or weaker, suggesting a relaxation of the β-turn structure (Figure 131b). When explored computationally, it was found that the lowest energy structures of both 7.10 and 7.10-En showed significant overlap in the backbone suggesting a β-turn (Figure 131c). However, four conformations were found to exist within 2 kcal/mol of this ground state for 7.10-En, suggesting a highly flexible structure (Figure 131d). The noted shifts in NOE interactions were in good agreement when these four structures were considered. It was proposed that upon formation of the enamine, a stabilizing H-bonding interaction with the C-terminal carboxylate is weakened, greatly increasing flexibility, which could help accommodate the incoming nitroolefin.
Figure 131.
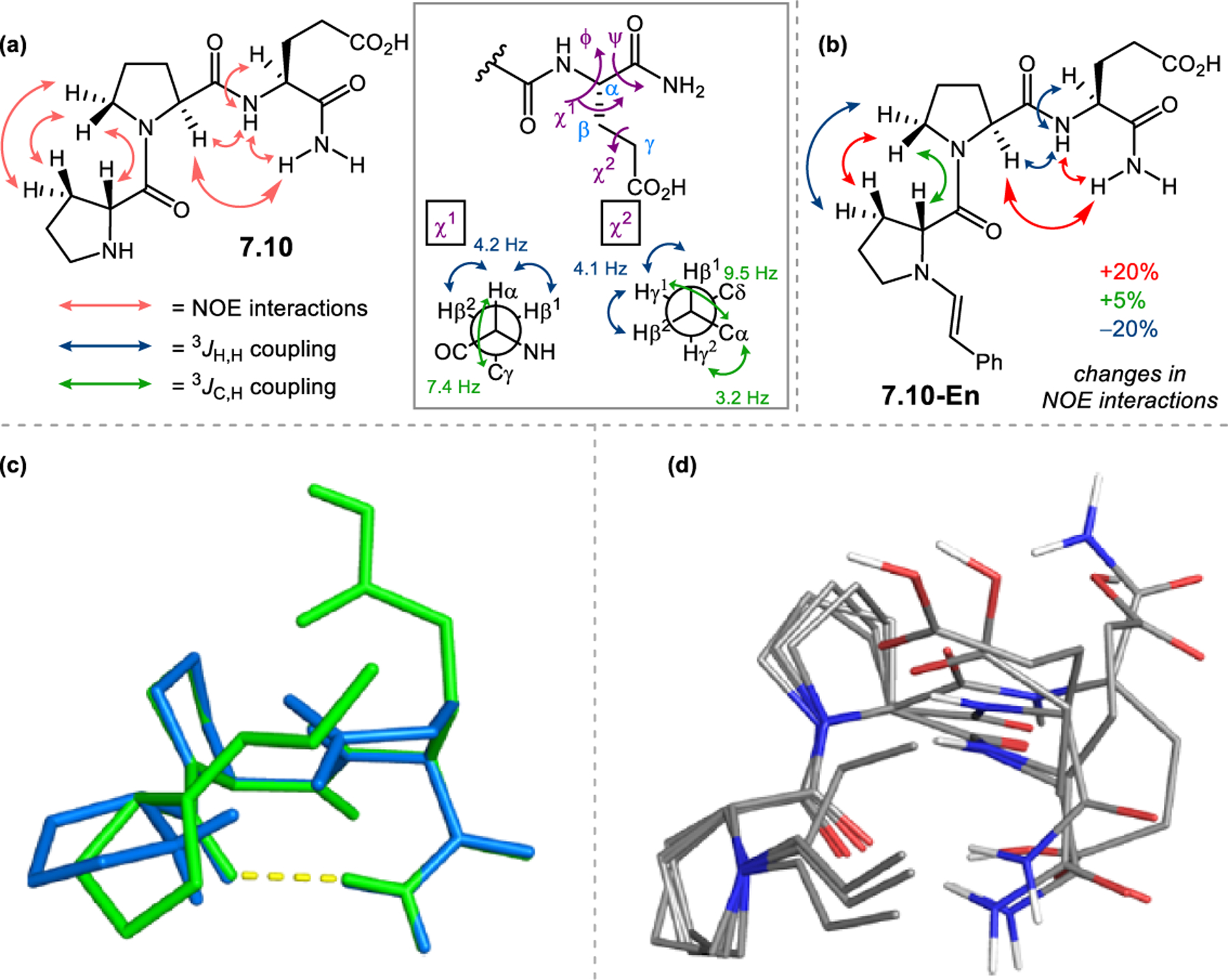
(a) Selected NOE, 3JH,H coupling, and 3JC,H coupling interactions for catalyst 7.10. (b) Variations in NOE interactions between 7.10 and 7.10-En. (c) Overlay of lowest energy DFT structures 7.10 (blue) and 7.10-En (green) (d) Overlay of four calculated structures within 2 kcal/mol of lowest energy structure of 7.10-En. Reproduced in part from with permission from ref. 328. Copyright 2018 American Chemical Society.
Taking these mechanistic studies into account, the Wennemers Group proposed the following catalytic cycle (Figure 132). The ground state of the catalyst primarily adopts a type I β-turn (7.43), which undergoes reversible condensation with an aldehyde substrate. The resulting enamine 7.44 has increased structural flexibility that may assist in accepting a nitroolefin during the rate-determining C–C bond-forming step. Nitronate intermediate 7.45 is then rapidly protonated by an appropriately positioned acidic group before releasing the product through hydrolysis of iminium ion 7.46.
Figure 132.
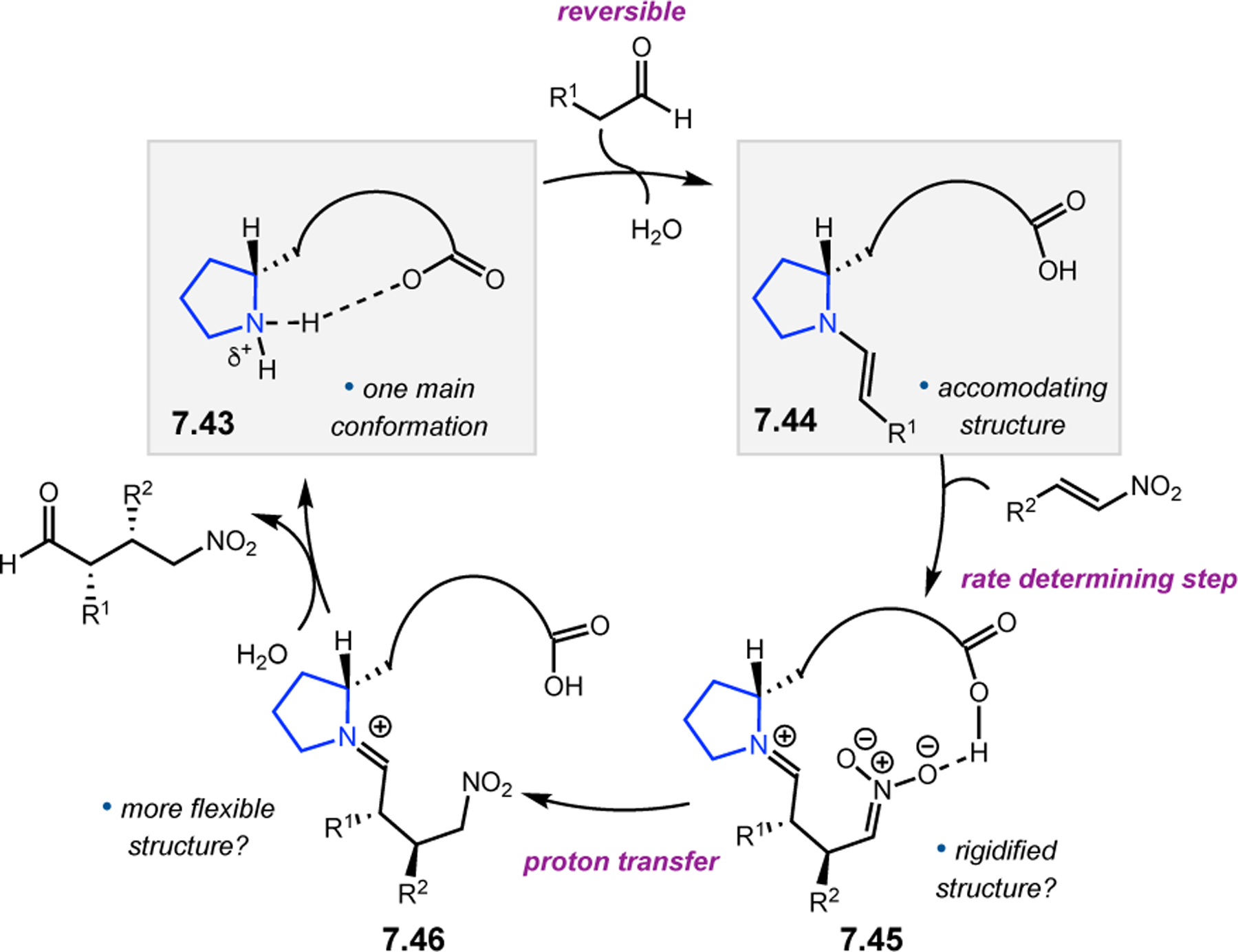
Proposed catalytic cycle for the peptide-catalyzed, asymmetric conjugate addition of aldehydes to nitroolefins considering all previous mechanistic studies.328
While venerable peptide coupling protocols have enabled the rapid synthesis of peptide-based catalysts,329 other strategies have emerged for the synthesis of diverse peptide scaffolds. For instance, the groups of Orru and Paixao have demonstrated that 4-component Ugi reactions can be employed to generate tripeptide catalysts similar to those explored by Wennemers and co-workers (Figure 133a).330 In several publications, Orru recognized that peptide 7.49 resembled trimeric peptides that Wennemers had previously disclosed. When employed as a catalyst in the same asymmetric addition of aldehyde 7.47 to nitroolefins 7.2, high enantioselectivity was observed in γ-nitroaldehyde 7.48 (Figure 133b). Paixao and co-workers further utilized a 4-component Ugi reaction to generate various structurally diverse trimeric peptides.331 Upon assessing 13 different catalysts in the reaction between 7.1 and 7.2, peptides 7.50 and 7.51 emerged as reasonably selective catalysts (Figure 133c). A conformational search of both catalysts using Monte Carlo molecular mechanics revealed that 673 conformations of 7.50 existed within 5 kcal/mol of one another. In contrast, 7.51 exhibited only 162 conformations that were within 5 kcal/mol. Computationally optimized structures of the 7.51 enamine revealed that the cyclohexyl group provides significant shielding on one face of this intermediate. Ultimately, 7.51 was found to be the more selective catalyst, providing 7.3 in up to 93% yield, 97:3 dr, and 98% ee in a series of conjugate addition reactions. In later studies, Paixao demonstrated that immobilized versions of these peptides could be easily formed by using silicon appended substrates and demonstrated the efficacy of these catalysts in flow chemistry setups.332
Figure 133.
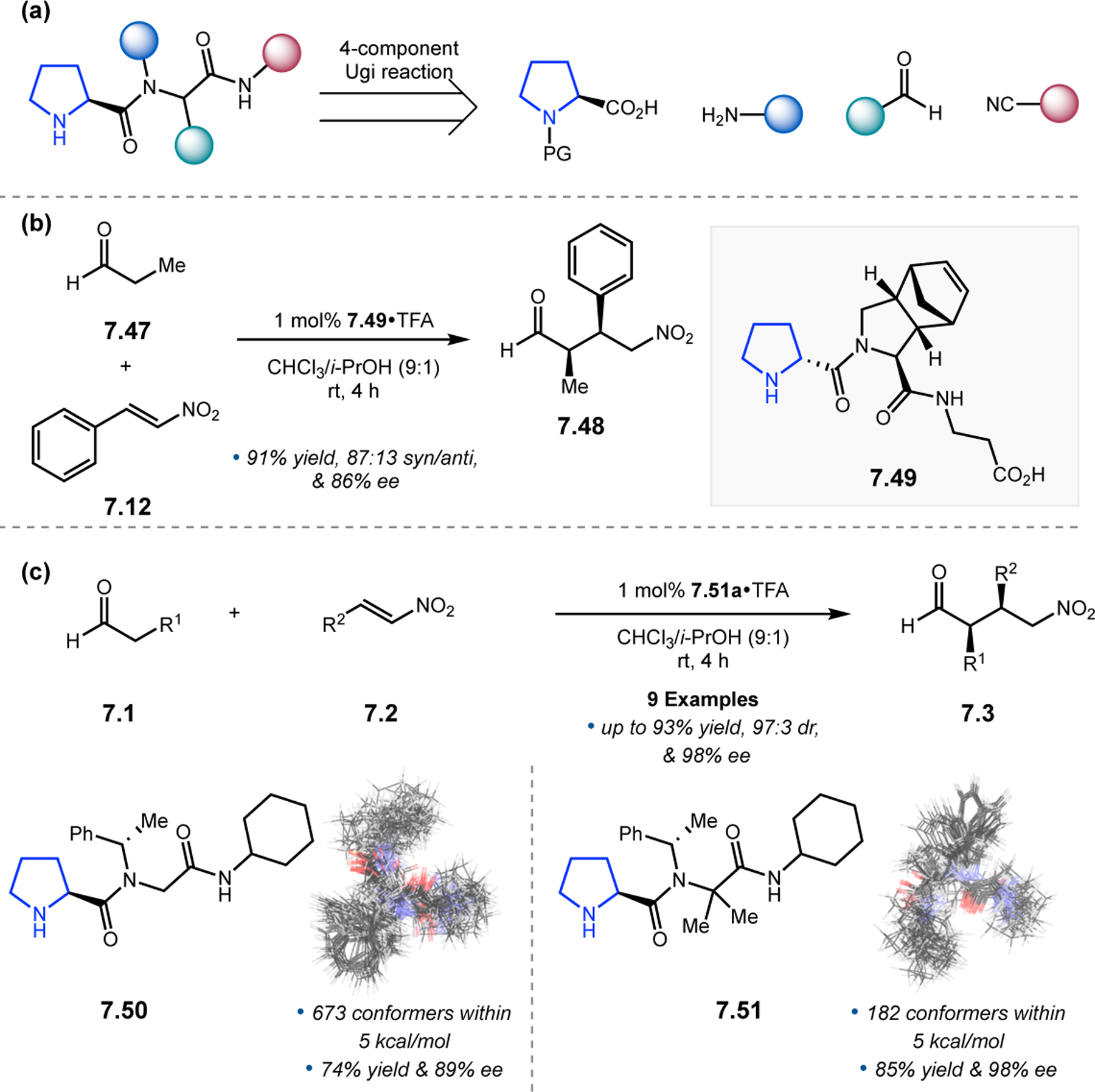
(a) Strategy for the synthesis of diverse peptide catalysts scaffolds via a four-component Ugi reaction. (b) Asymmetric conjugate addition of aldehyde 7.47 into nitroolefin 7.2 catalyzed by peptide 7.49.330 (c) Diversity oriented approach pursued by Paixao and co-workers to develop catalysts for asymmetric conjugate addition reactions and a comparison of the number of low energy conformations of peptides 7.50 and 7.51. Reproduced in part from with permission from ref. 331. Copyright 2013 American Chemical Society.
Lecouvey and co-workers explored trimeric peptide scaffolds that contain a phosphonic acid on the C-terminal residue as catalysts in the asymmetric conjugate addition of aldehydes 7.1 into nitroolefins 7.2 (Figure 134).333 Due to the multiple potential protonation states of the catalyst, it was envisioned that enamine formation could be favored without the introduction of external base to deprotonate the N-terminal Pro residue. Reactivity and selectivity were found to be influenced by the protonation state of the catalyst. Fully protonated peptide 7.52 was ineffective as a catalyst, while fully deprotonated 7.53 delivered full conversion in 24 h, providing 7.3 in 85:15 dr and 87:13 er. Mono deprotonated 7.54, however, was notably more reactive and selective. Using this catalyst, full conversion was achieved within 20 h, and 7.3 was obtained in 91:9 dr and 93:7 er. These catalysts were found to be easily recyclable through aqueous extraction and could be reused up to 10 times with only a slight decrease in enantioselectivity. Intriguingly, the opposite enantiomer of the product was obtained compared to what Wennemers and co-workers observed, presumably as a function of the phosphonic versus the carboxylic acid functional group on the C-terminal residue sidechain.
Figure 134.
Asymmetric conjugate addition of aldehydes 7.1 to nitroolefins 7.2 catalyzed by tripeptides bearing a bifunctional phosphonic acid moiety at the C-terminal residue.333
Tsogoeva and co-workers had previously demonstrated that short peptides were capable of catalyzing conjugate addition reactions between enone substrates and nitroalkanes.334 Expanding upon these results, they demonstrated that dipeptide H-Pro-Phe-OH can catalyze the conjugate addition reactions of ketones 7.55 to nitroolefins 7.2 under completely aqueous conditions (Figure 135).335 Judicious choice of base was required in order to achieve high reactivity, with NaOH ultimately emerging as the optimal choice. It was proposed that H-bonding between the Pro carbonyl and water increases the acidity of the corresponding amide proton enough for it to act as a H-bond donor to the nitroolefin.
Figure 135.
Asymmetric conjugate addition of aldehydes 7.1 into nitroolefins 7.2 catalyzed by H-Pro-Phe-OH under aqueous conditions.335
In light of the contributions to the field of nitroolefin conjugate additions by the Wennemers Group, Piarulli and co-workers studied whether diketopiperazines could function in a similar capacity in the reaction of aldehyde 7.11 with nitroolefin 7.12 (Figure 136).336 A series of diastereomeric diketopiperazines 7.57–7.60 were synthesized in order to explore the stereochemical relationships within the catalyst. Intriguingly, the catalytic behavior of 7.57 was matched with 7.59, both of which have an appended d-Pro residue, whereas those appended with l-Pro demonstrated lower selectivity. This trend was found to be true across multiple substrates.
Figure 136.
Asymmetric conjugate addition of aldehydes into nitroolefins catalyzed by bifunctional diketopiperazine.336
7.2. Conjugate Additions of Nucleophiles to Unsaturated Aldehydes & Ketones
Substantial contributions have been made by Kudo and co-workers in the area of peptide-catalyzed conjugate additions by exploiting many of the same design strategies featured in their studies of aqueous oxidations and transfer hydrogenations (see Sections 2 & 3). Catalysts explored for these transformations typically involved: (1) a β-turn region, which provides a robust chiral environment suitable for asymmetric catalysis; (2) an α-helical region, which provides a hydrophobic pocket in aqueous media and has been shown to greatly enhance reaction rates; and (3) a solid support resin. These strategies were found to be highly transferable and effective in accelerating the Friedel–Crafts-type alkylations of indoles under fully aqueous conditions (Figure 137).337 In fact, reactions conducted in the absence of water, with THF as the sole solvent, failed to provide any product. The authors proposed that rate acceleration is achieved by enhanced hydrophobic interactions between the peptide and its substrates as a consequence of the aqueous media. Fine-tuning of the β-turn region of 7.64 revealed that the stereocenters of the two Trp residues had a large impact on enantioselectivity in the reaction between enal 7.61 and indole 7.62, and that an l-Trp-l-Trp stereochemical array was required for optimal enantioselectivity (Figure 137a, entries 1–3). Using catalyst 7.64a, alkylated indole 7.63 was obtained in 85% yield and 88% ee (entry 1), while ee values <45% were obtained with d-Trp-containing peptides 7.64b and 7.64c. Furthermore, it was observed that a poly(Leu) linker sequence had a high propensity to form an α-helical structure. Consistent with their hypothesis, the hydrophobic pocket formed in this region was required for both high reactivity and enantioselectivity (entries 4–7). Upon further examination, peptide 7.64f, which contains a related Leu-Leu-Aib triad that promotes a 310-helix rather than an α-helix further enhances selectivity, provided 7.63 in 92% conversion and 91% ee (entry 7).338 Techniques such as IR, ROESY, and CD confirmed the helicity of such structures in aqueous media. Taking advantage of the compatibility of catalyst 7.64f with water, a one-pot sequential alkylation, oxidation sequence was realized, wherein the oxidation was facilitated by the oxidizing enzyme, laccase.339 This synthetic sequence was enabled by the viability of peptides in aqueous media.
Figure 137.
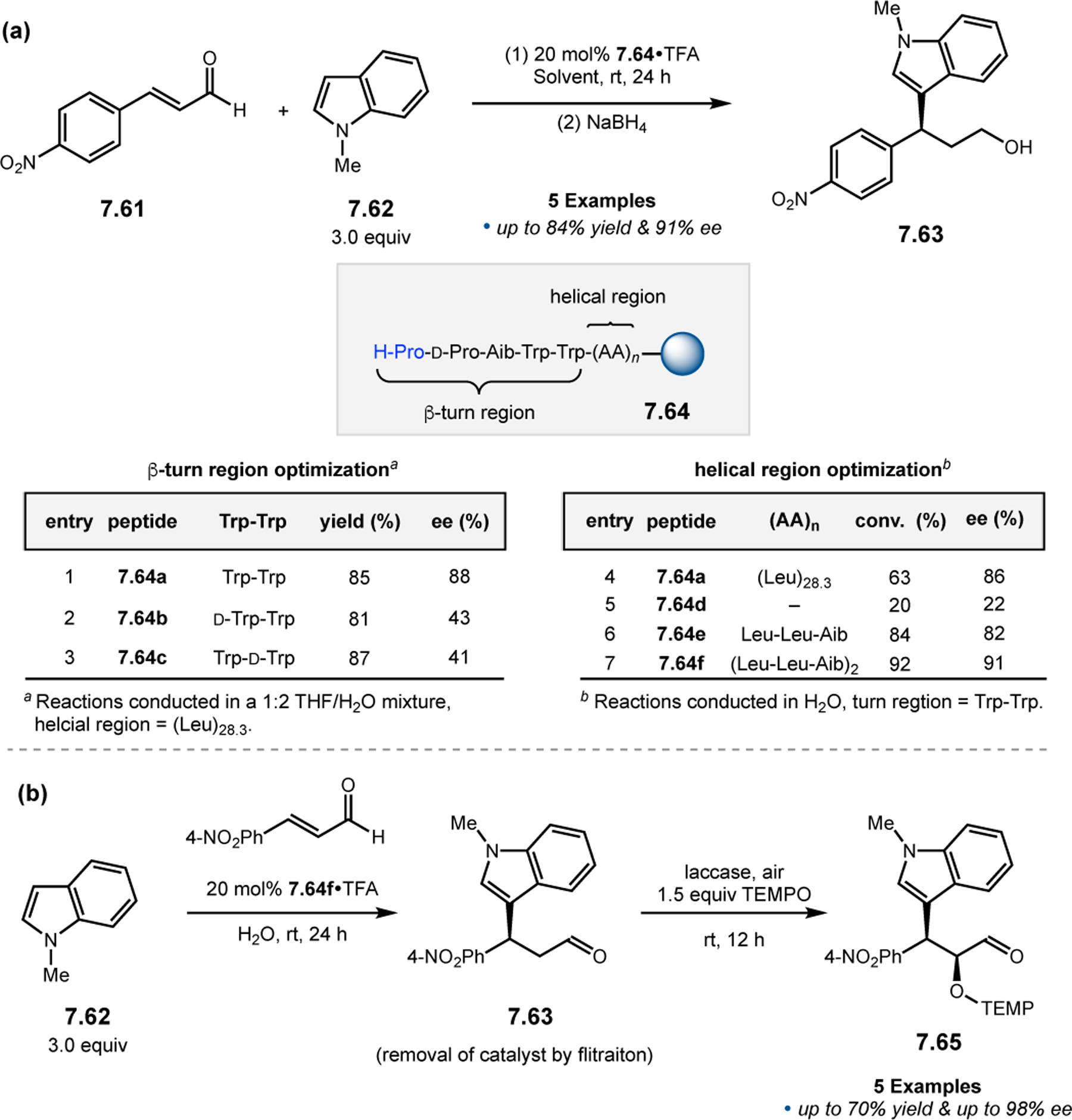
(a) Peptide-catalyzed asymmetric Friedel–Crafts-type addition of indole 7.62 into unsaturated aldehydes 7.62 in aqueous media.338 (b) One-pot sequential Friedel-crafts-type alkylation and oxidation catalyzed by 7.64f and laccase.339
Catalyst 7.64f also proved competent in the asymmetric conjugate addition of nitromethane (7.67) into β,β-disubstituted α,β-unsaturated aldehydes 7.66, although low yields were observed in reactions conducted in H2O (Figure 138a, entry 1).340 Upon examining related solvent mixtures (entries 2–5), it was found that mixtures of methanol and water were optimal (entry 5) relative to reactions conducted in methanol alone (entry 4). This suggests that hydrophobic interactions were crucial for the efficacy of this transformation, as well. Under these conditions, 7.64f delivered γ-nitroaldehyde 7.68a in 47% yield and 91% ee (entry 5). The hydrophobic helical region was shown to affect both conversion and enantioselectivity (entry 5 vs. 6), and reactivity could be enhanced by using α-helical, poly(Leu)-containing peptide 7.69, which provided 7.68a in 54% yield and 96% ee. This result stands in contrast to the authors’ previous work on the Friedel–Crafts reaction, wherein the 310-helical linkers were found to be superior catalysts. Under the optimized conditions, a variety of substrates containing all-carbon quaternary centers could be synthesized with high levels of enantioselectivity (Figure 138b).
Figure 138.
(a) Catalyst and solvent optimization for the conjugate addition of nitromethane (7.67) to unsaturated aldehyde 7.66a. (b) Asymmetric conjugate addition of nitromethane 7.67 into β,β-disubstituted α,β-unsaturated aldehydes 7.66 catalyzed by peptide 7.69 in aqueous media.340
While peptides such as 7.64f and 7.69 are effective catalysts for conjugate additions into enals, which easily form iminium ions upon condensation with secondary amines, Kudo and co-workers anticipated that catalysts with primary amine-containing catalytic resides would be better suited for enone substrates (Figure 139).28 A series of resin-bound catalysts containing various unprotected N-terminal residues were assessed in the addition of nitromethane (7.67) into enones 7.70 (entries 1–5). Ultimately, peptide 7.75, which contains an N-terminal diad of Trp resides and a 310-helix-promoting linker region, was identified as the lead catalyst, as it afforded product 7.71 in 93% yield and 91% ee (entry 4). Notably, peptide 7.76, which contains an α-helical linker, was ineffective as a catalyst (entry 5).
Figure 139.
Asymmetric conjugate addition of nitromethane (7.67) into α,β-unsaturated ketones 7.70 catalyzed by peptides containing unprotected N-termini.28
Similar to the enantioselective reduction of α,β,γ,δ-unsaturated aldehydes via transfer hydrogenation previously reported (Section 3.1),120 the Kudo Group demonstrated that selectivity could also be imparted in the related conjugate addition of 2-naphthalenethiol (7.78) into α,β,γ,δ-unsaturated aldehydes 7.77 (Figure 140).341 When imidazolidinone catalyst 7.80 was employed, product 7.79, which is obtained from two sequential 1,6- and 1,4-conjugate additions, was formed as a nearly racemic mixture for each possible diastereomer. The authors examined peptide 7.64f due to its successful application in previous conjugate addition reactions (vide supra). Indeed, when 7.64f was employed, the ratio of the three possible products greatly favors 7.79c, with modest enantioselectivity detected for both possible diastereomers. The identity of the counter ion was found to greatly influence conversion in this reaction. Moreover, resin-bound 7.64f could be recycled three times with essentially no decrease in catalytic performance.
Figure 140.
Asymmetric conjugate addition of 2-naphthalenethiol 7.78 into α,β,δ,γ-unsaturated aldehydes 7.77.341
During their investigations into the kinetic resolution of metallocenes 7.81 via transfer hydrogenation (Section 3.1), Kudo and co-workers also explored conjugate additions of nitromethane (Figure 141).122 Using peptide 7.83—the same catalyst that was used to catalyze the transfer hydrogenation of 7.81—a high krel was observed, providing the conjugate addition product (Rp)-7.82 in >99:1 dr and 95% ee (Figure 141a). However, this catalyst proved ill-suited in catalyzing the related conjugate addition of nitromethane into [2.2] paracyclophane derivatives 7.84 (Figure 141b). The authors reasoned that a less sterically hindered catalyst lacking a β-turn-region and containing a primary amine as opposed to a secondary amine might be more suitable in this transformation, as the catalyst must engage a relatively hindered ketone rather than an aldehyde. After screening a variety of N-terminal residues, it was found that residues with linear sidechains generally performed well, such as Hse(Me). Additionally, the 310-helix-inducing triad of Leu-Leu-Aib was crucial for promoting reactivity. Ultimately, the desired kinetic resolution was achieved with a krel of 39 using peptide 7.86, which delivered (Sp,R)-7.85 in 41% yield and 90% ee; the starting material was also recovered in 53% yield and 63% ee. It was proposed that H-bonding between the amide backbone and the nucleophile helps to direct reactivity.
Figure 141.
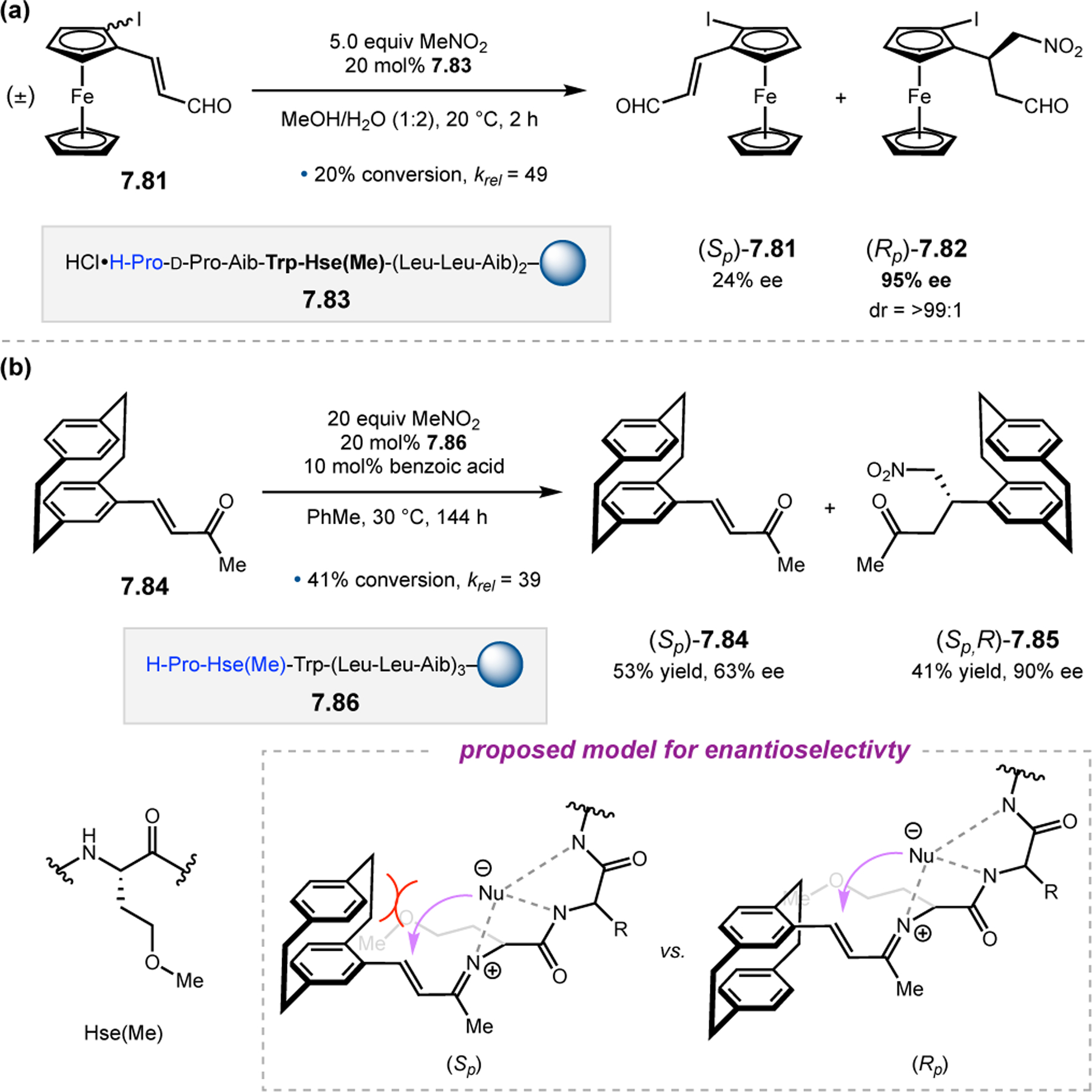
(a) Kinetic resolution of metallocene complexes via peptide 7.83-catalyzed conjugate addition of nitromethane into α,β-unsaturated aldehydes 7.81. (b) Kinetic resolution of paracyclophane derivatives 7.84 via peptide 7.86-catalyzed conjugate addition.122
In addition to reactions in aqueous media, several transformations have also been reported in completely organic solvents, such as the conjugate addition of vinyl boronic acids 7.88 into α,β-unsaturated aldehydes 7.87 (Figure 142).342 In analogy to previous studies, the presence of a helical motif linking the solid support to the catalytically active Pro residue was found to be important for reactivity and selectivity compared to simple monomers and dimers (entries 1 & 2 vs. 3 & 4). It was found that helices comprised of Leu and Ala residues were most effective in this particular transformation (entries 3 & 4). It is noteworthy that Leu and Ala are the amino acid monomers with the highest helical propensity, and that sequences containing other residues performed notably worse in the conjugate addition reaction. Peptide 7.91 delivered hemiacetal 7.90 in 99% conversion and 91% ee. NMR titration experiments, as well as IR studies, confirmed the helical conformation of peptide 7.91 in solution.
Figure 142.

Asymmetric conjugate addition of boronic acids 7.88 into γ-hydroxy-α,β-unsaturated aldehydes 7.87.342
Although diverse peptide libraries can be rapidly synthesized on solid support via split and pool methods, analysis of hundreds of reactions can become rate-limiting for project progress. Our Group and the Wennemers Groups had previously addressed this issue by using strategies involving (1) fluorescent reporter molecules attached to substrates or (2) reactions in which the substrate is covalently bound to the peptide catalyst.343,344 However, these strategies were incompatible with the conjugate addition reactions studied by the Kudo Group, and a new approach was required in order to accelerate the screening process. Recognizing that the fundamental steps of Pro-catalyzed conjugate additions involve the formation of an iminium ion, Kudo and co-workers reasoned that reduction of this intermediate would produce a resin bound conjugate addition product (Figure 143a).345 Using dye-modified malonate 7.92 as conjugate addition substrate, this strategy could be used to covalently append a dye molecule to a catalytically active peptide. Upon washing away excess dye, beads that remained colored would identify peptides with high reactivity, and presumably high enantioselectivity, which would help to prioritize reactions for sequencing by mass spectroscopic techniques. Initial screening efforts focused on introducing diversity at the fourth and fifth positions in order to keep the helical and β-turn regions intact (Figure 143b). Both Trp-Trp and Phe-His motifs were found with a high frequency using this approach. It was determined that the conjugate addition of a His sidechain at the fifth position was highly reversible, and could contribute to an increase in the ratio of the enal taken up by the resin (Figure 143c). Catalysts containing this His residue typically demonstrated higher rates of reactivity in addition to increased enantioselectivity. A more in-depth screen that included the helical region of identified peptides 7.101 and 7.102, which despite sharing a similar N-terminal residue, afford opposite enantiomers of the product (Figure 143d).346 Both peptides contain a His residue at the seventh residue. These new peptides were found to be even more effective in entrapping substrate onto the resin, possibly via the mechanism described in Figure 143c.
Figure 143.
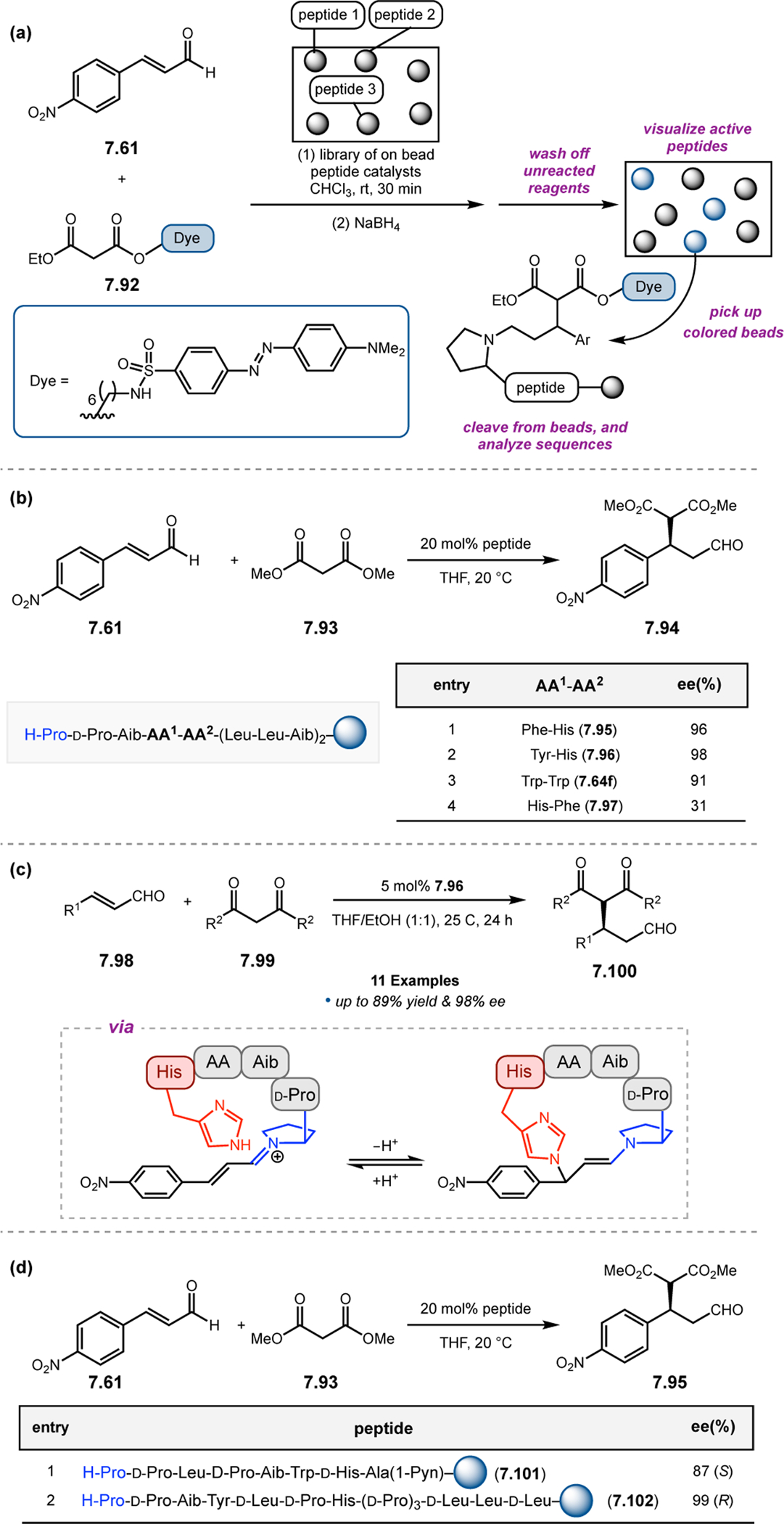
(a) Strategy for the visualization and identification of strongly active catalysts via immobilization of dye molecules to on-bead peptide catalysts. This facilitates rapid screening of peptide catalysts for the conjugate addition of malonate nucleophiles, such as 7.92, into α,β-unsaturated aldehyde like 7.61. (b) Selected enantioselectivity values for high performing catalysts in the reaction of aldehyde 7.61 with dimethyl malonate (7.93). (c) Optimized conditions for asymmetric conjugate addition of 7.99 into enals 7.98 and proposed model for the efficacy of His residues at the 5th position. (d) Catalysts identified after further screening that demonstrate divergent enantioselectivity in the reaction of 7.61 with 7.93.
8. Other C–C Bond-Forming Reactions
Reactions that form C–C bonds are at the very heart of synthetic and biological chemistry, and a plethora of methods have been developed to forge these highly important bonds with control over stereochemical outcomes. Asymmetric C–C bond-forming reactions permeate multiple sections of this Review. Thus far, classical additions of C-centered nucleophiles into carbonyl compounds have been discussed (Section 5), as well as aldol (Section 6) and Michael addition reactions (Section 7). Moreover, more modern photochemical and transition metal-catalyzed methods for C–C bond formation will be treated in subsequent sections (Sections 9 & 11, respectively). This Section is dedicated to the other asymmetric, peptide-catalyzed C–C bond-forming reactions that do not easily fit into the other categories, such as cyclopropanation (Section 8.1), Morita–Baylis–Hillman (Section 8.2), and Rauhut–Currier reactions (Section 8.3). These are also very powerful transformations that have been widely utilized in organic synthesis through the years. Recent examples of peptide-catalyzed asymmetric variants of these important C–C bond-forming reactions are discussed below with a particular emphasis on mechanistic study, which has received significant attention over the past decade.
8.1. Cyclopropanation
Despite being the most strained cycloalkane ring-system, cyclopropanes are widely found in natural products and pharmaceuticals, and they also serve as useful synthetic intermediates for subsequent transformations.347–350 Thus, the development of stereoselective methods for the construction of cyclopropanes is of particular interest and value to the field. One of the most widely used methods for constructing three-membered carbocycles is the Simmons–Smith cyclopropanation, in which a Zn species inserts into diiodomethane to form a reactive organozinc carbenoid capable of converting alkenes into cyclopropanes.351,352 Asymmetric variants of the Simmons–Smith reaction that utilize chiral auxiliaries, reagents, or catalysts typically rely on the presence of a directing group in the alkene substrate. Coordination of a heteroatom in the alkene substrate to the Zn reagent not only enhances the reaction rate, but also improves stereocontrol. Therefore, prior to 1998, there were only two reports of an enantioselective cyclopropanation of unfunctionalized olefins, achieving <4% ee.353,354 However, using a chiral alcohol to coordinate to the organozinc species, Shi and co-workers were able to cyclopropanate an unfunctionalized olefin with 51% ee.355
This method was later improved using dipeptide 8.3a to mediate the asymmetric Simmons–Smith type cyclopropanation of styrene 8.1 (Figure 144).356 By adding ethyl methoxyacetate (EMA) as an achiral additive, this reaction was rendered catalytic, decreasing the loading of 8.3a from 1.25 equivalents to 25 mol%.357 Under these conditions 8.3a delivered the cyclopropane 8.2 in 87% yield and 89% ee. In a subsequent study, Shi and co-workers explored the mechanism of this peptide-catalyzed transformation with 8.1.358 In contrast to the optimized catalyst 8.3a, no conversion was observed using N-methylated dipeptide 8.3b. This suggested that the free N–H of 8.3a is crucial for reactivity in this cyclopropanation reaction. The authors probed the mechanism further by subjecting peptide 8.3a to the cyclopropanation reaction conditions sequentially and characterizing the intermediates (Figure 145). Initial treatment of 8.3a with diethylzinc evolves ethane gas and generates intermediate I, in which Zn coordinates to both the amide nitrogen and the carbonyl oxygen of the Val residue. This intermediate can then react with diiodomethane at lower temperatures to afford carbenoid intermediate II. The structures of these intermediates were assigned using NMR data coupled with an X-ray crystal structure of a related dipeptide.
Figure 144.
Dipeptide 8.3-catalyzed cyclopropanation of 8.1 to afford 8.2.357,358
Figure 145.
Treatment of dipeptide 8.3a with diethylzinc followed by diiodomethane.358
The cyclopropanation of 8.1 was then tested using peptide-zinc complex II, which had been synthesized according to Figure 145. After 24 h, complex II had only delivered 7% conversion of 8.1 into 8.2, although with 83% ee. Upon adding one equivalent of Zn(CH2I)2 to II, conversion into 8.2 was fully restored to 89%, and the ee increased to 90%. This result suggests that Zn(CH2I)2 is necessary for catalyst turnover. One hypothesis for the role of Zn(CH2I)2 is that it may decompose to ZnI2, which has been demonstrated to facilitate cyclopropanation in other systems.359–361
Taking all of these mechanistic results together, the authors proposed two possible catalytic cycles (Figure 146). Once intermediate II is generated by deprotonation with diethylzinc and subsequent halogen exchange with diiodomethane, it can be activated by XZnI to form either complex III or IV. If intermediate III is produced (blue cycle), this species can cyclopropanate 8.1 directly to form olefin 8.2 and intermediate IV. Transmetalation with Zn(CH2I2) regenerates III, closing the catalytic cycle. Alternatively, intermediate IV can act as a chiral Lewis acid to activate XZnCH2I, forming V (red cycle). This species can then cyclopropanate 8.1 to regenerate intermediate IV. Overall, this study sheds light on the interactions between dipeptides and organozinc reagents that ultimately give rise to a highly enantioselective cyclopropanation reaction.
Figure 146.

Two potential mechanisms for the dipeptide 8.3a-catalyzed cyclopropanation of 8.1.358
Following the earlier works of MacMillan,362,363 Arvidsson,364 and Ye,365 Kudo and coworkers sought to use resin-supported, N-terminal Pro-containing peptides to catalyze the asymmetric cyclopropanation of α,β-unsaturated aldehydes 8.4 with sulfur ylides (Figure 147).366 This strategy invokes an iminium catalytic activation mechanism to produce 1,2,3-trisubstituted cyclopropanes bearing three contiguous stereogenic centers. Cinnamaldehyde 8.4a was used as a model substrate for screening. Although chloroform was initially tested as the reaction medium, it was found that the reaction rate could be significantly accelerated (3 vs. 24 h) using aqueous conditions (THF/H2O, 2:1). Furthermore, this solvent system enabled in situ generation of the requisite sulfur ylide using sodium bicarbonate as the base rather than pre-forming the ylide, which tends to be unstable.
Figure 147.
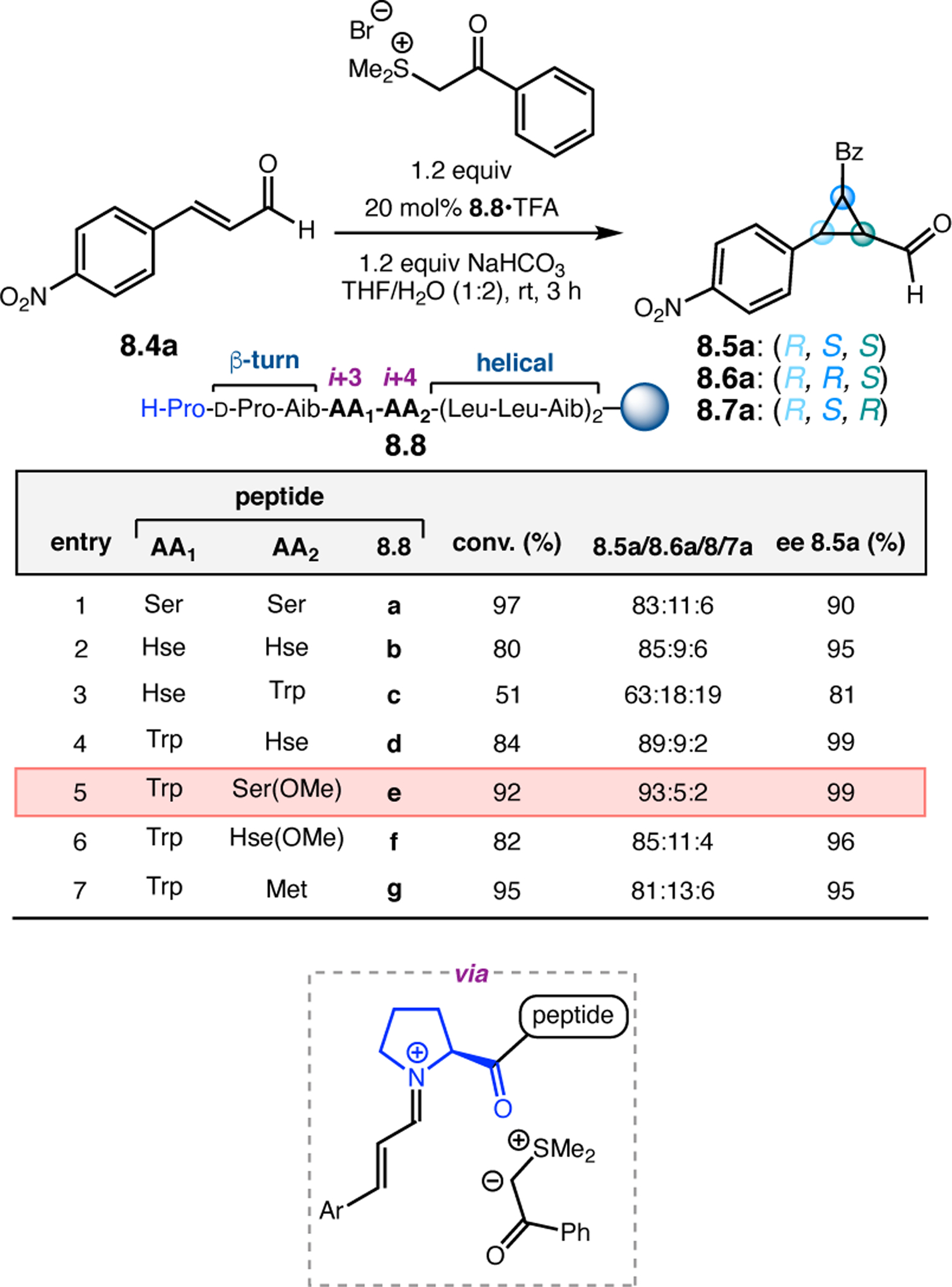
Peptide catalyst optimization for the cyclopropanation of α,β-unsaturated aromatic aldehyde 8.4a.366
The authors then evaluated several resin-supported peptides 8.8 as secondary amine catalysts for the cyclopropanation of 8.4a with an acetophenone-derived sulfonium ion (Figure 147). These peptides were designed to include both a β-turn motif, induced by incorporation of a central d-Pro-Aib sequence, and a helical section linker. The authors proposed that the β-turn motif is crucial for controlling enantioselectivity, while the hydrophobic helical portion could stabilize the peptide in the aqueous reaction medium.338 The i+3 and i+4 residues, which were hypothesized to be important for noncovalent interactions with aldehyde 8.4, were first examined pairwise. It was found that residues containing a hydroxyl group, such as Ser and Hse, gave promising diastereoselectivities (entries 1 & 2). When the i+4 residue was replaced with Trp, both the diastereo- and enantioselectivity decreased significantly (entry 3). However, incorporation of Trp at i+3 provided a boost in selectivity (entry 4). With Trp at i+3, the position of the oxygen atom and the length of the chain at the i+4 position was then assessed (entries 4–7). It was found that Ser(Me)-containing 8.8e gave the best overall stereoselectivity, providing 92% conversion to 8.5a in 93:5:2 dr and 99% ee (entry 5). While the role of the sidechain ether functionality has not been fully elucidated, the authors propose an attractive interaction between the ether O-atom and the sulfonium salt, which may increase the selectivity for three-membered ring formation via transition state stabilization.
With peptide 8.8e in hand, various α,β-unsaturated aldehydes 8.4 were investigated in this reaction (Figure 148, entries 1–5). Both electron-rich and electron-poor α,β-unsaturated aryl aldehydes consistently afforded 8.5 in high yields (>84%), diastereoselectivities (>94:3:3), and enantioselectivities (99%). The reusability of the resin-supported catalyst was also examined with enal 8.4e. No significant decrease in catalytic activity was observed up through the fifth reuse of the catalyst, in which yield decreased slightly from 87% to 83%, yet enantioselectivity of 8.5e remained constant at 99% ee (entry 5).
Figure 148.
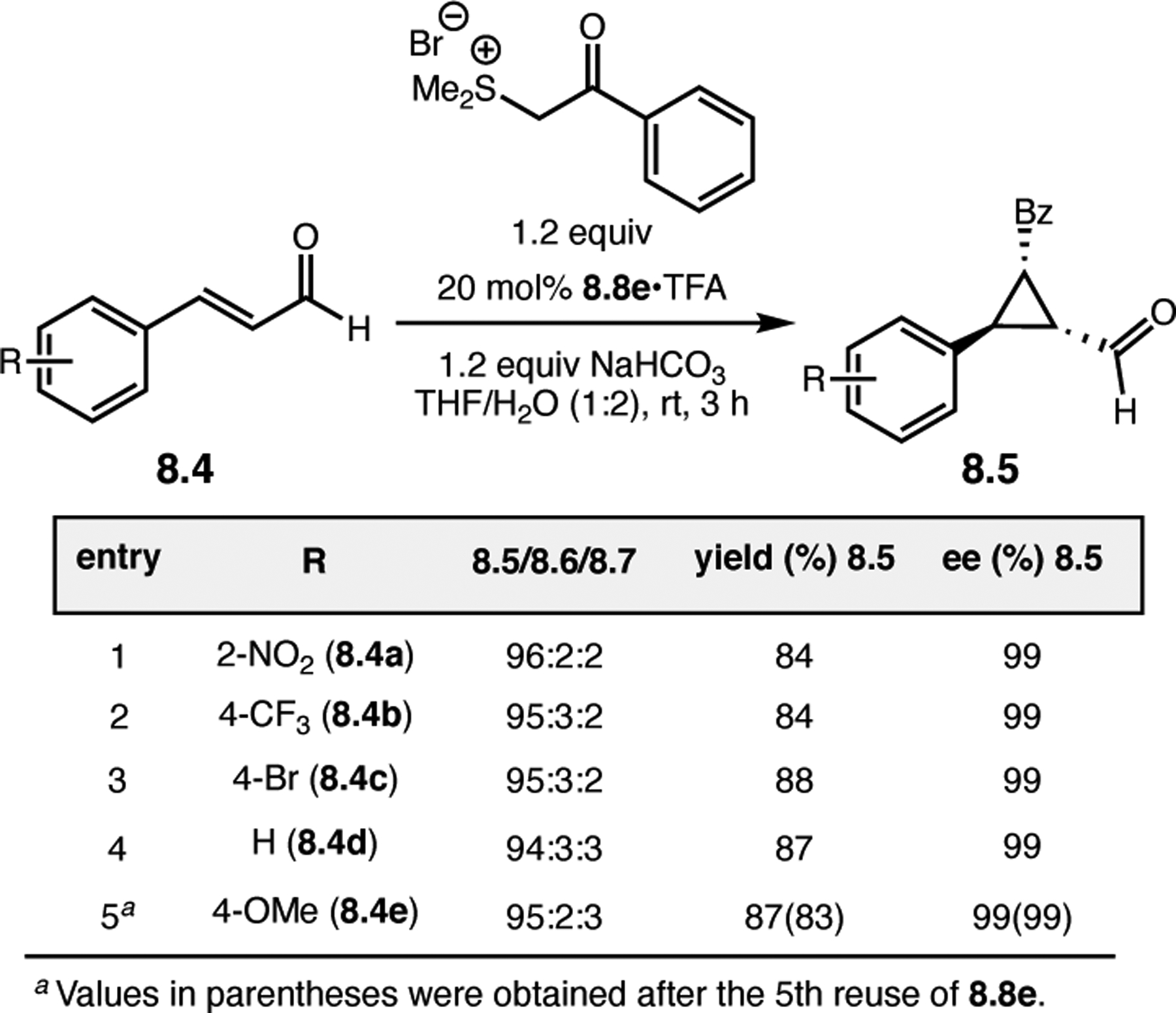
Selected substrate scope for the asymmetric cyclopropanation of 8.4 using peptide 8.8e.366
8.2. Morita–Baylis–Hillman Reaction
Since its discovery over 50 years ago, the asymmetric Morita–Baylis–Hillman (MBH) reaction has become a useful method for forging C–C bonds with control over the newly formed alcohol- or amine-bearing stereogenic center.367 Broadly defined, MBH reactions operate under Lewis base catalytic mechanisms, often employing tertiary amine- or phosphine-based catalysts to mediate the coupling of an α,β-unsaturated carbonyl compound with an aldehyde or imine. The nucleophilic catalysts are thought to undergo 1,4-addition with the α,β-unsaturated substrate, activating it as a transient enolate equivalent for addition into the aldehyde or imine electrophile. Proton transfer and conjugate elimination then restores the catalyst to its active form.
When considering the reaction between allenoates 8.9 and imines 8.10, two possible reaction pathways lead to two different types of products (Figure 149). Using a chiral phosphine catalyst, an asymmetric formal [3+2] cycloaddition can occur between 8.9 and 8.10 to generate dihydropyrroles 8.13.368–370 Alternatively, 8.9 and 8.10 can react in a classical aza-MBH fashion to yield allenoates 8.11. Employing a chiral phosphine catalyst, Lu and co-workers had previously indicated the imine N-substituent on 8.10 could influence the product distribution, with tosylates favoring 8.13 and carbamates preferentially proceeding through the MBH reaction.371 When coupling allenoates 8.9 with enones, our group observed that the product distribution could be affected by catalyst type.372 In this system, phosphines preferred to catalyze the formal cycloaddition, while amines tended to favor the aza-MBH reaction. Therefore, our group hypothesized that peptidic amine catalysts could offer complementary reactivity to the phosphine catalysts previously employed for the coupling of 8.9 and 8.10.373 This strategy would provide access to enantioenriched allenic amines 8.11, representing the first enantioselective aza-MBH reaction of its class.
Figure 149.
Two potential modes of reactivity in the Lewis base-catalyzed reactions of allenoates 8.9 and imines 8.10.373
Reaction optimization included both tertiary amine and phosphine catalysts to mediate the coupling of allenoate 8.9a with imine 8.10a (Figure 150).373 Using triphenyl phosphine led to 57% isolated yield of the aza-MBH product 8.11aa (entry 1). However, N-heterocyclic catalysts, such as DMAP and protected Pmh monomer only afforded trace product (entries 2 & 3). Pyridine and the protected 3-pyridylalanine (3-Pal) monomer, however, proved to be quite effective catalysts for this transformation, forming 8.11aa in 75% and 71% yield, respectively (entries 4 & 5).
Figure 150.
Catalyst optimization for the aza-MBH reaction of allenoate 8.9a and imine 8.10.373
With promising levels of conversion using Pal, a library of Pal-embedded peptides designed to adopt β-turn secondary structures was synthesized, as this motif had proven to be highly effective in peptide-catalyzed asymmetric transformations previously (entries 6–10). The library was biased by strategic incorporation of a central Pro-Aib sequence motif, which is known to induce β-turn conformations in solution.223 While peptide 8.12a, which contains a Pro residue at the i+1 position, gave detectable enantioenrichment in 8.11aa (56:44 er, entry 6), d-Pro-containing 8.12b provided a significant increase in enantioselectivity to 78:22 er (entry 7). Using peptide 8.12b, the N-protecting group of imines 8.10 was then evaluated. Exchanging ethoxycarbonyl group of 8.10a for a benzyl group (8.10b) fully ablated reactivity (entry 8). However, N-benzoylated imine 8.10c proved to be more reactive and selective than carbamate 8.10a, affording 8.11ac in 94% yield and 84:16 er (entry 9). Exchanging the C-terminal protecting group of the peptide from a methyl ester in 8.12b to a dimethyl amide in 8.12c resulted in a further increase in er to 92:8 (entry 10).
With catalyst 8.12c in hand, the substrate scope was examined for this aza-MBH reaction (Figure 151). With benzyl allenoate 8.9a, aryl imines with electron donating substituents at both the ortho- (8.10d) and para- (8.10e) positions were well tolerated (entries 1 & 2). Moreover, 8.11ae could be recrystallized up to 98.5:1.5 er without significant loss in yield (entry 2). Imines with halide substitution on the arene, such as 8.10f, were marginally less effective in terms of yield and enantioselectivity (entry 3). Once again, recrystallization could be used to increase enantioselectivity of 8.11af to 96.5:3.5 er. Allenoate 8.9b was also a highly effective coupling partner with imine 8.10c, with optimal yields and selectivity obtained at 0 °C (entries 4 vs. 5). Under these enhanced reaction conditions, a more expansive substrate scope was examined, (entries 5–9). Once again, electron rich-aryl imines 8.10e and 8.10g were processed with high enantioselectivities of 92.5:7.5 er (entries 6 & 7). Naphthyl-substituted imine 8.10h was also coupled with 8.9b to deliver 8.11bh in 66% yield with 92:8 er (entry 8). Aliphatic imines, such as 8.10i, were also tolerated in this reaction, albeit with reduced yield and enantioselectivity (42% yield, 80.5:19.5 er; entry 9).
Figure 151.
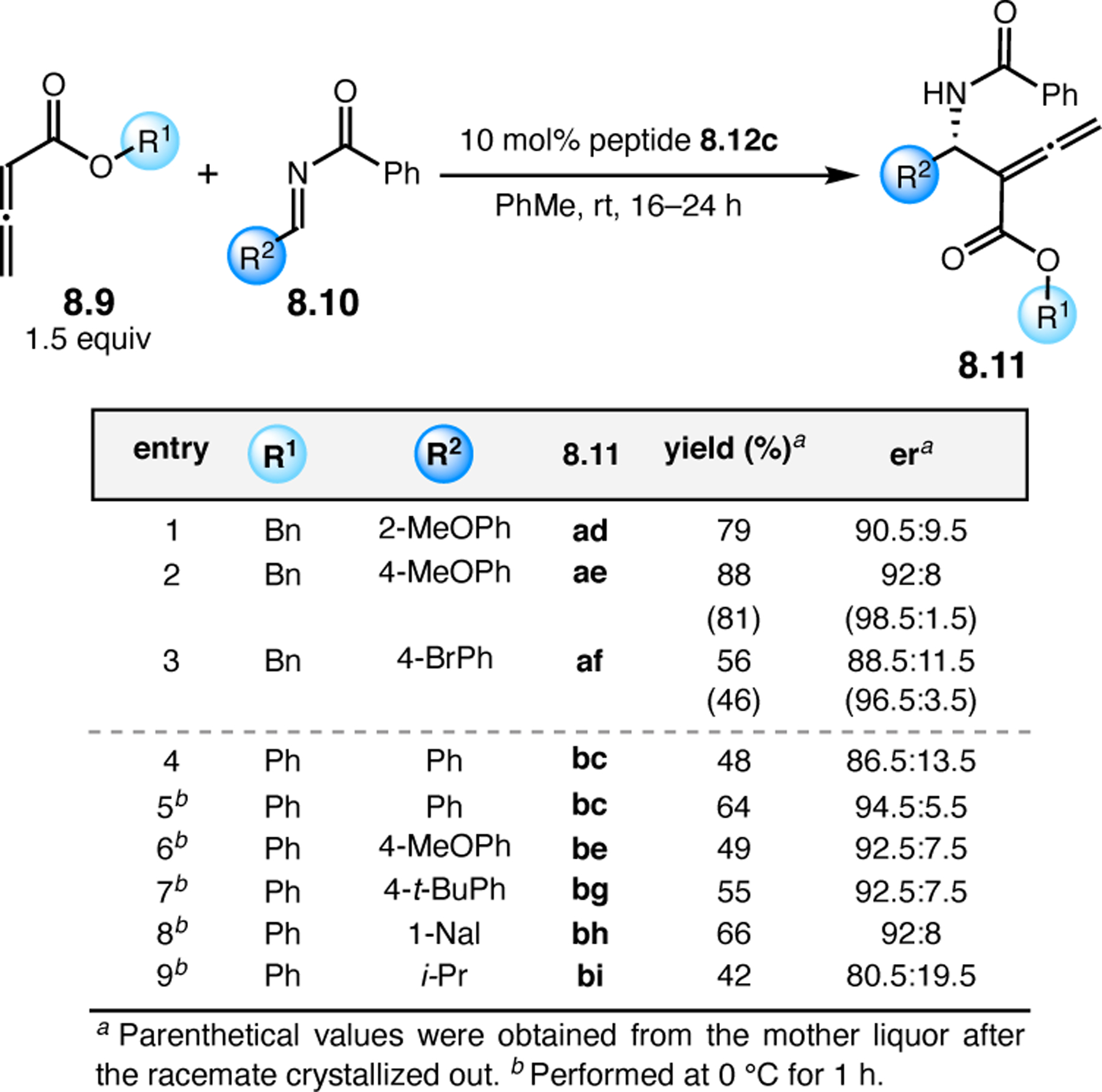
Scope of the peptide-catalyzed aza-MBH reaction between allenoates 8.9 and imines 8.10.373
In a subsequent report, our group performed an in-depth mechanistic study of this peptide-catalyzed aza-MBH reaction using various kinetics experiments.374 A series of initial rate studies revealed that the reaction between 8.9a and 8.10c is first order in both allenoate and peptide 8.12c and zero order with respect to the imine. Compared to other MBH and aza-MBH reactions, the kinetic order of the imine 8.10c in this peptide-catalyzed reaction is unusual.367 This may be attributed to the possibility that the rates were measured at catalyst saturation in imine, though that concentration would have to be lower than NMR titration experiments suggested. We then sought to identify the rate determining step (RDS). Comparing allenoate 8.9a to its α-deuterated variant, a small KIE of 1.08 was measured, suggesting that proton transfer is likely not rate-determining, also in contrast to previously studied aza-MB reactions.367 Since the kinetic data indicated that imine 8.10c is not involved in the RDS, catalyst addition to allenoate 8.9a was proposed to be rate determining in this case. To further establish if this mechanism is unique to the peptide-catalyzed reaction, analogous kinetics experiments were performed using pyridine as the catalyst rather than 8.12c. In this case, a nearly identical allenoate KIE was observed (kH/kD = 1.03). However, the pyridine-catalyzed reaction was significantly slower than the 8.12c-catalyzed variant, suggesting that the peptide is better suited to activate the allenoate for coupling with the imine. Based on these data, a mechanism for this transformation was proposed (Figure 152). First, the Lewis basic pyridine moiety of the peptide adds into allenoate 8.9 at the β-position to form zwitterionic intermediate I in what may be the RDS. Intermediate I then reacts with imine 8.10 to yield intermediate II through the formation of a C–C bond. Proton transfer gives enolate intermediate III, which undergoes elimination via an E1cB mechanism to release the peptide-based catalyst and deliver product 8.11.
Figure 152.
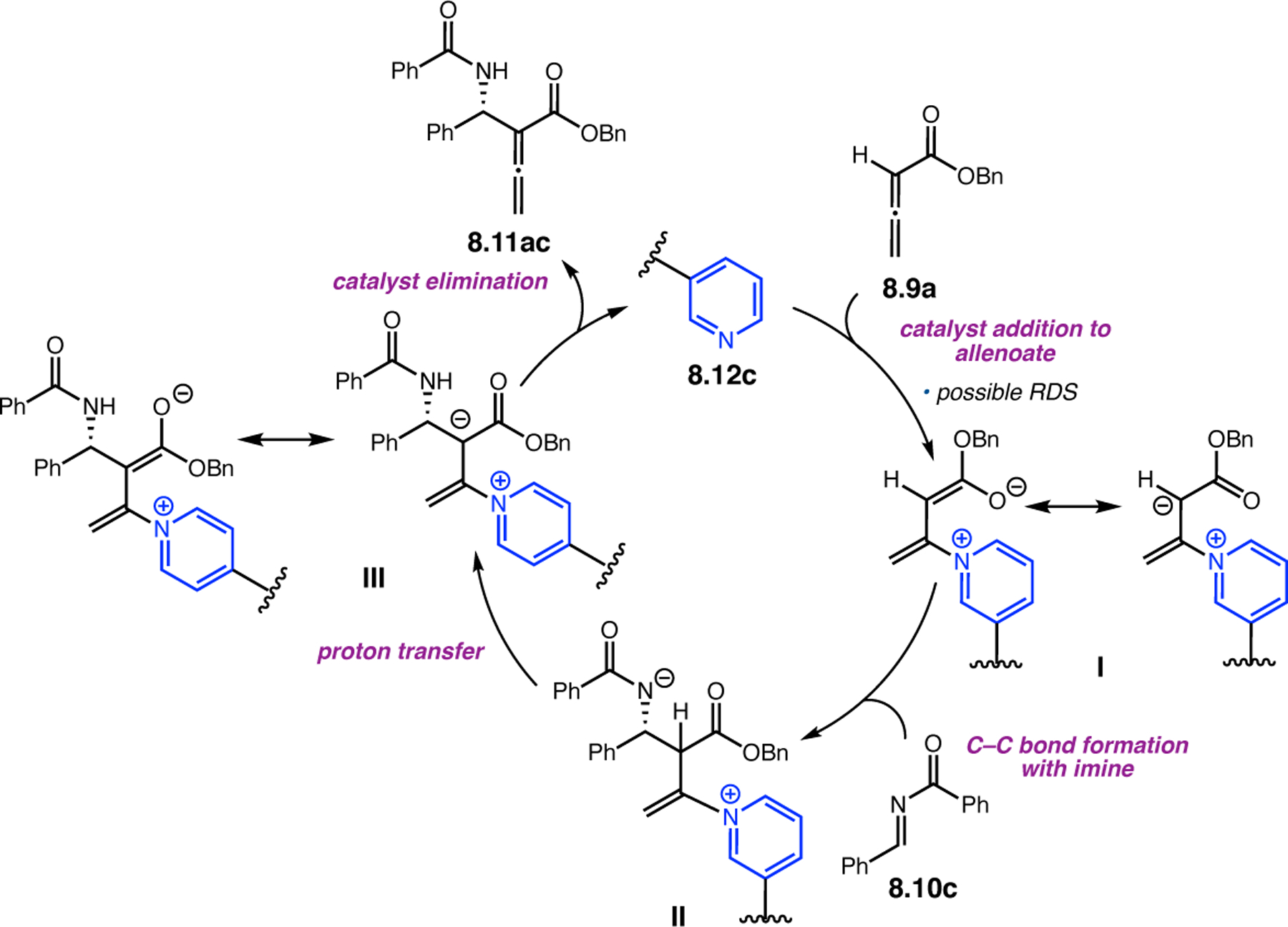
Proposed mechanism for the peptide-catalyzed aza-MBH reaction.374
In 2014, our group extended this Pal-containing peptide-catalyzed aza-MBH reaction to the coupling of allenoates 8.14 with N-acylimines 8.10 to deliver products 8.15 that possess two different stereochemical elements—a stereogenic center and an axis of chirality (Figure 153).375 Adding to both the challenge and intrigue of this transformation, the peptide-based catalyst must be able to exert control over both stereogenic elements in order to achieve a highly selective process. Initially, the previously identified catalyst 8.12c and conditions were tested in the reaction of chiral, racemic allenoate 8.14a.373 Under these conditions, 8.15ac was produced in 67% yield, with modest diastereo- and enantioselectivities of 1:1.2 dr and 38.5:61.5 er (entry 1). An additional site for H-bonding between allene 8.14 and the peptide was added in an effort to enhance selectivity. Indeed, using anilide-substituted allenoate 8.14b, 85% of 8.15bc was isolated in just 1 h with an improved dr of 1:2.7 (entry 2). Replacing the amide of 8.14b with an ester, as in 8.14c, eroded the dr to 1:1.2, further supporting the importance of an H-bond donor at this position (entry 3).
Figure 153.
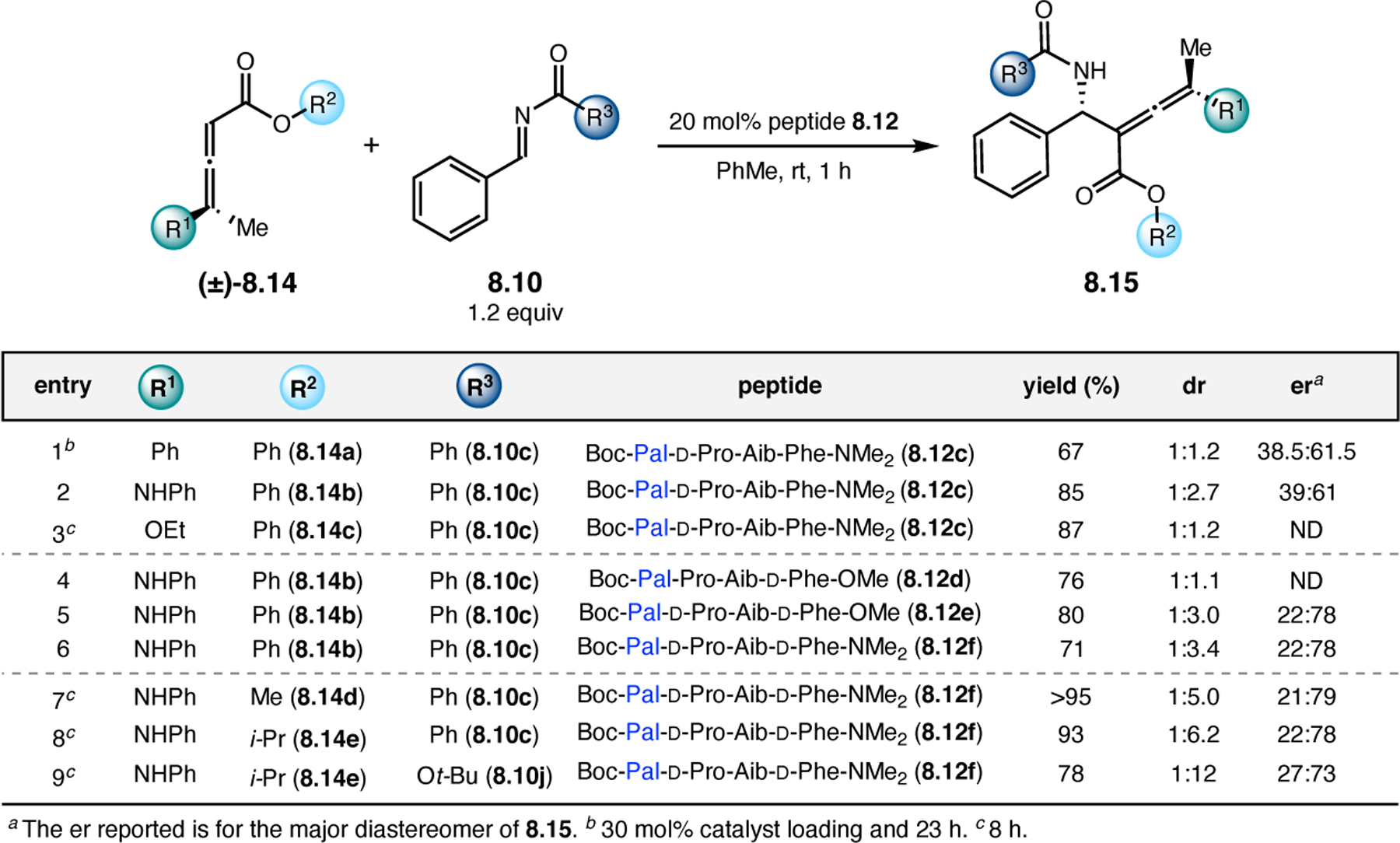
Optimization of the peptide 8.12-catalyzed coupling of allenoates (±)-8.14 with imines 8.10.375
Using allene 8.14b with imine 8.10c, analogs of peptide 8.12c were examined (Figure 153, entries 4–6). A matched and mismatched case of stereochemistry at the i+1 and i+3 positions was observed. With l-Pro at i+1 and d-Phe at i+3 (8.12d), diastereoselectivity decreased even further to 1:1.1 dr (entry 4). However, employing peptide 8.12e, which contains d-configured residues at both i+1 and i+3, 8.15bc was obtained in 1:3.0 dr and 22:78 er (entry 5). Exchange of the C-terminal protecting group of the peptide from a methyl ester to a dimethylamide in 8.12f provided a slight boost in dr to 1:3.4 with the same level of enantioselectivity (entry 6). Having achieved substantial dr and er in this transformation using peptide 8.12f, the effects of altering the allenic ester substituent of 8.14 were then investigated. Exchange of the phenyl ester for aliphatic groups improved dr, with the bulkier isopropyl ester 8.14e giving the highest dr of 1:6.2 (entries 7 & 8). With respect to the imine, sterically bulkier carbamate 8.10j further improved the dr to 1:12 (entry 9).
Several additional peptides were examined in the aza-MBH reaction between 8.14e and 8.10j in pursuit of higher enantioselectivity values. Ultimately, tetrapeptide 8.12g, bearing a trans-4-hydroxy-l-proline (Hyp) residue at i+1 and 2-aminoindane-2-carboxylic acid (Aic) at i+2, delivered 8.15ej in 92% yield, 1:16 dr, and 88:12 er (Figure 154, entry 1). Electron-donating substituents on the imine aryl ring had a negligible effect on selectivity, while electron-deficient aryl imine 8.10k provided marginally decreased selectivities of 1:10 dr and 85.5:14.5 er (entry 2). Tetrasubstituted allene 8.15ek could be recrystallized up to 98:2 er. This aza-MBH reaction was reasonably sensitive to substitution at the γ-position of allene 8.14, as 8.14f required a longer reaction time to isolate 8.15fj in 70% yield with reduced dr of 1:11 and er of 71.5:28.5 (entry 3). The reaction between allenic thioester 8.14g and Cbz-protected imine 8.10l was also examined, producing 8.15gl in 90% yield, 1:42 dr, and 94:6 er (entry 4). Increased steric bulk at the γ-position of thioester 8.13h also resulted in a significant loss of both reactivity and selectivity, producing 8.15hm in 58% yield with 1:5.8 dr and 75.5:24.5 er (entry 5).
Figure 154.
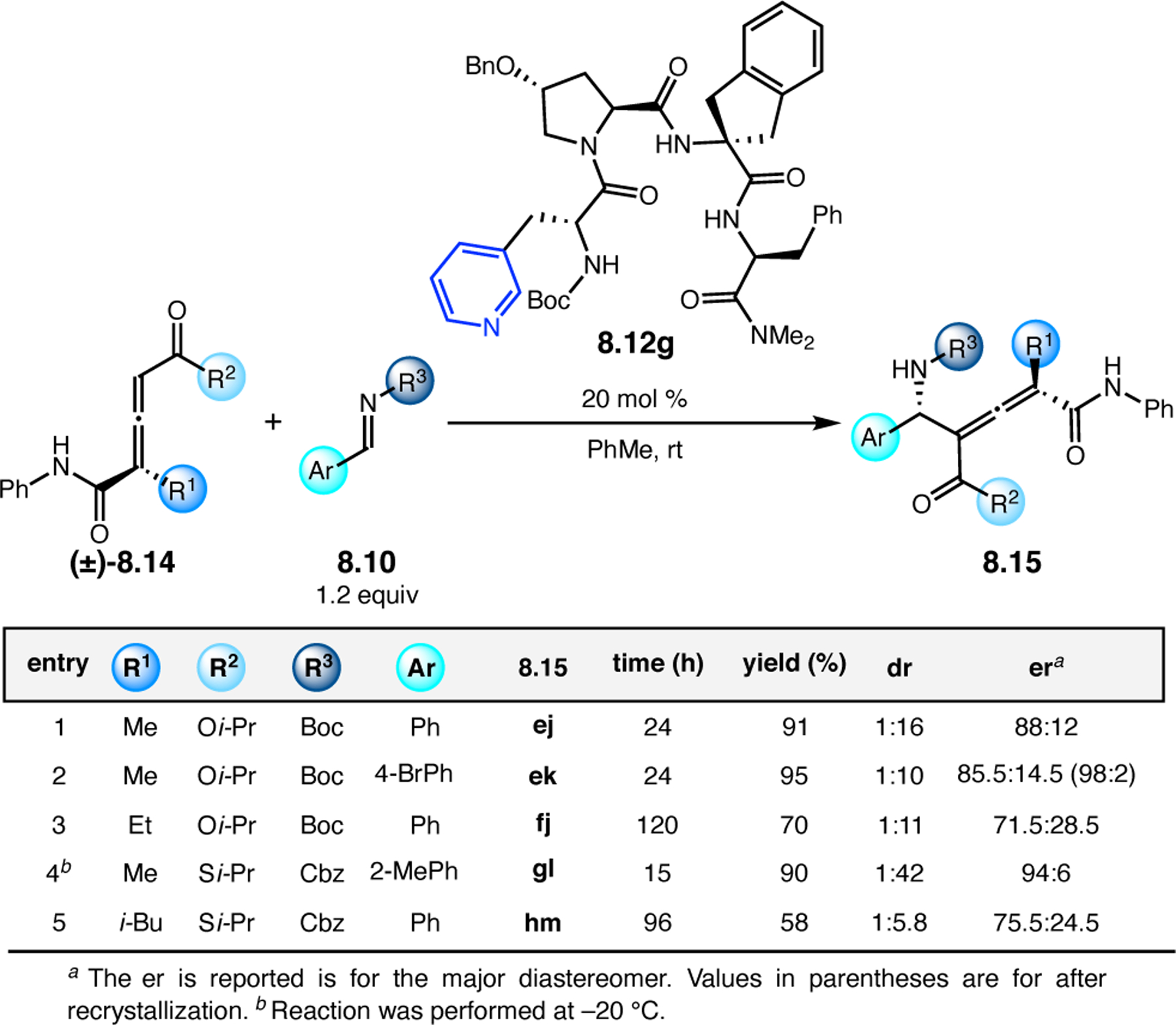
Selected scope for the peptide 8.12g-catalyzed coupling of allenoates 8.14 and imines 8.10.375
Several kinetics experiments were performed to elucidate the mechanism of this interesting aza-MBH variant.375 We first sought to define the role of the peptide as either a Brønsted or Lewis base catalyst. Hydrogen/deuterium-exchange studies were performed with a representative allenoate using D2O or MeOD-d4 as the deuteron source in the presence of various bases. The rates of H/D-exchange with inorganic bases, such as potassium carbonate and potassium hydroxide, under phase-transfer conditions were orders of magnitude greater than with any tertiary amine bases examined. Of the amine bases, the rate of exchange with peptide 8.12g was significantly faster than pyridine, quinuclidine, and triethylamine, all of which are expected to be more basic than 8.12g from a pKa standpoint. These data suggest that different bases may operate via alternate catalytic mechanisms. Additional kinetics studies were conducted to determine the RDS of this peptide-catalyzed transformation. The reaction was found to be first order in both allenoate 8.14e and peptide 8.12g. The rate dependence on imine 8.10j was very small at synthetically relevant concentrations and could be interpreted as close to zero order, consistent with imine saturation. Again, no primary KIE was observed for the proton transfer step. These results mirror those obtained in our previous study (Figure 152),374 which support catalyst addition to the allenoate as the RDS.
These studies culminated in the proposed mechanism depicted in Figure 155, in which peptide 8.12g acts as a Lewis base catalyst. First, 8.12g adds into (±)-8.14e to form zwitterionic intermediate I, which may exist in several isomeric forms (e.g., IA & IB). Interconversion of these intermediates effectively ablates the axial chirality of allenoate 8.14e. While this step may be rate-determining, it is most likely not stereodetermining. Imine 8.10j can then react with intermediate I to form intermediate II, thus establishing the α-amine stereocenter. At least four isomers of intermediate II (A–D) may exist, with catalyst control over the distribution of these species. Subsequent α-proton transfer followed by catalyst elimination defines the chiral axis of 8.15ej and determines the dr.
Figure 155.
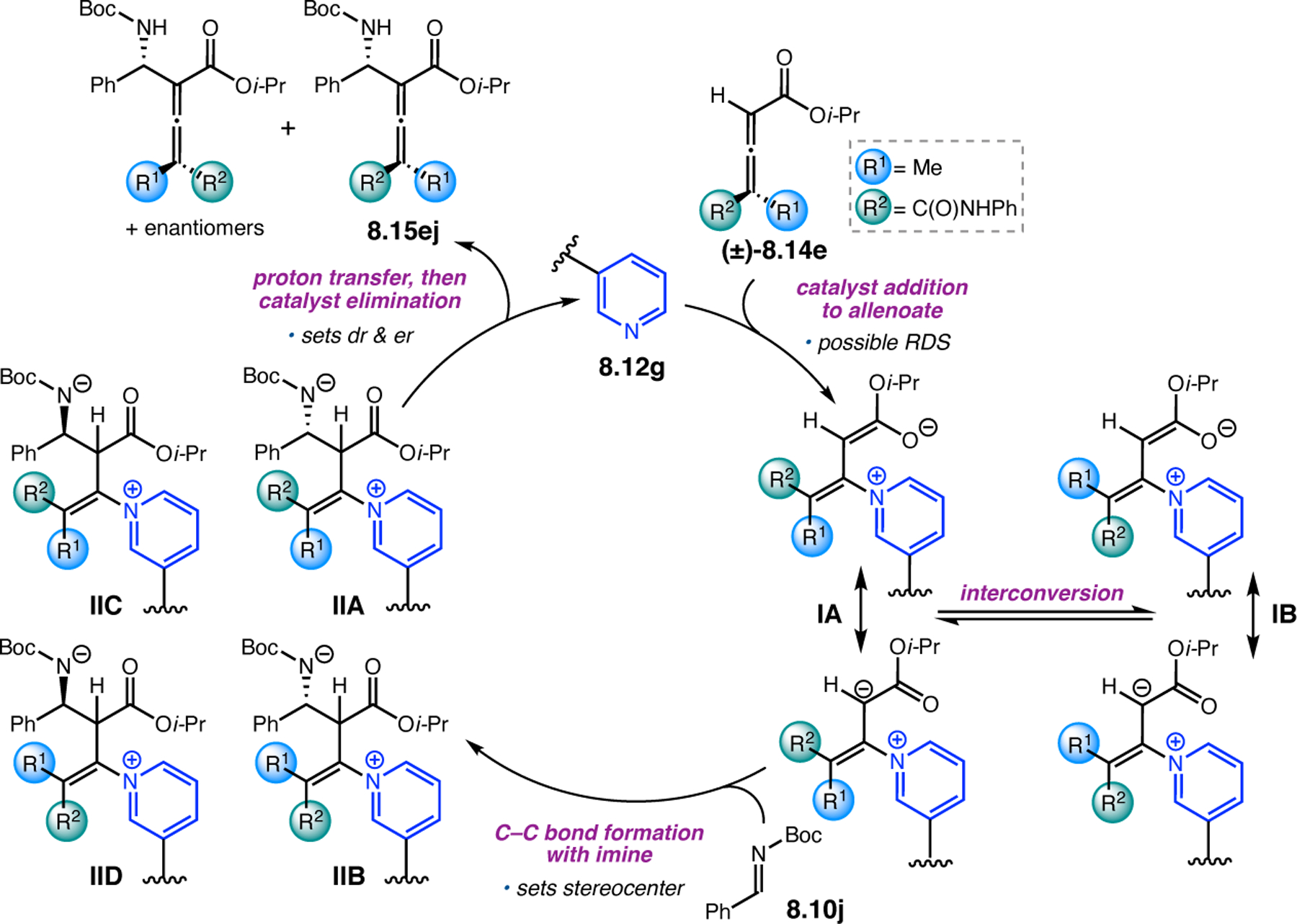
Proposed mechanism for the peptide 8.12g-catalyzed aza-MBH reaction of allenoate 8.14e and imine 8.10j.375
NMR experiments were performed in order to shed light on the mode of activation in this peptide-catalyzed reaction. Spectra recorded of peptide 8.12g alone show multiple signatures of a β-turn secondary structure. However, addition of 1 equivalent of allenoate 8.14e under otherwise identical conditions produced several changes in the 1H-NMR resonances of 8.12g, consistent with the formation of a ground state complex. Taking these data into consideration, a potential binding model was constructed (Figure 156). In intermediate I, the peptide (gray) is proposed to maintain a β-turn secondary structure while H-bonding to the N–H of allene 8.14e through the C-terminal carbonyl. Allenes lacking this N–H generally resulted in low selectivity. As such, we believe this moiety is crucial for effective catalyst–substrate docking and orientation. The i+2 N–H of the peptide may also form an H-bond with N-acylimine 8.10j, facilitating stereoselective C–C bond formation.
Figure 156.
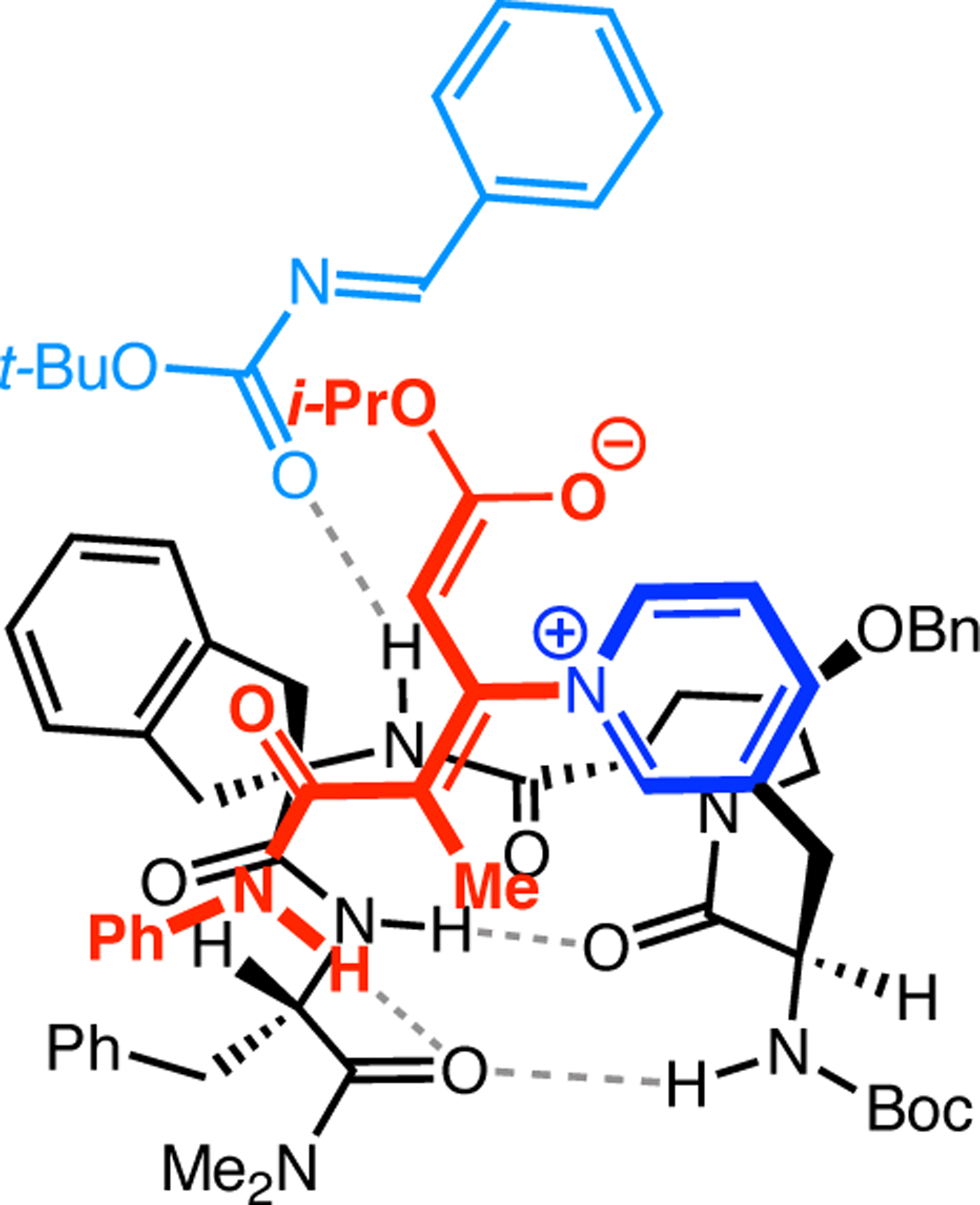
Speculative binding model for the addition reaction between intermediate I and imine 8.10j leading to intermediate II.375
8.3. Rauhut–Currier Reactions
Related to the MBH reaction, the Rauhut–Currier (RC) reaction—also termed the vinylogous MBH—involves the coupling of two α,β-unsaturated carbonyl compounds under Lewis base catalysis to deliver dicarbonyl products through the formation of a C–C bond between the α- and β-positions of the Michael acceptor substrates. While it was first disclosed in 1963, it was not until 2007 that an RC reaction was rendered asymmetric.376,377
Though Cys is an exceedingly common nucleophilic catalyst for biochemical reactions in vivo owing to the nucleophilicity of the sidechain thiol, this proteinogenic residue had not yet been utilized in small molecule Lewis base catalysis for synthetically relevant transformations. In 2007, our group reported the first enantioselective RC reaction that was enabled by Cys catalysis (Figure 157a).378,379 We chose to investigate this process in the context of the cycloisomerization of bis(enone) 8.16 to carbocycle 8.17, and while Cys-containing peptides were initially targeted, it was found that protected Cys monomer 8.18 was quite reactive and selective on its own. Following careful optimization of the reaction parameters (e.g., base, solvent, temperature, & additives), stoichiometric 8.18 was able to deliver 8.17 in 70% yield and 95% ee using potassium tert-butoxide as the base in a solvent mixture of water and acetonitrile at −40 °C. Using 20 mol% of 8.18, the cycloisomerization of 8.16 proceeded in 75% yield and 92% ee, but decreasing the loading further led to a steep decline in the reaction yields. Several additional bis(enone) substrates were examined, affording carbocyclic products in 54–73% yield and 67–95% ee at 100 mol% loading of 8.18.
Figure 157.
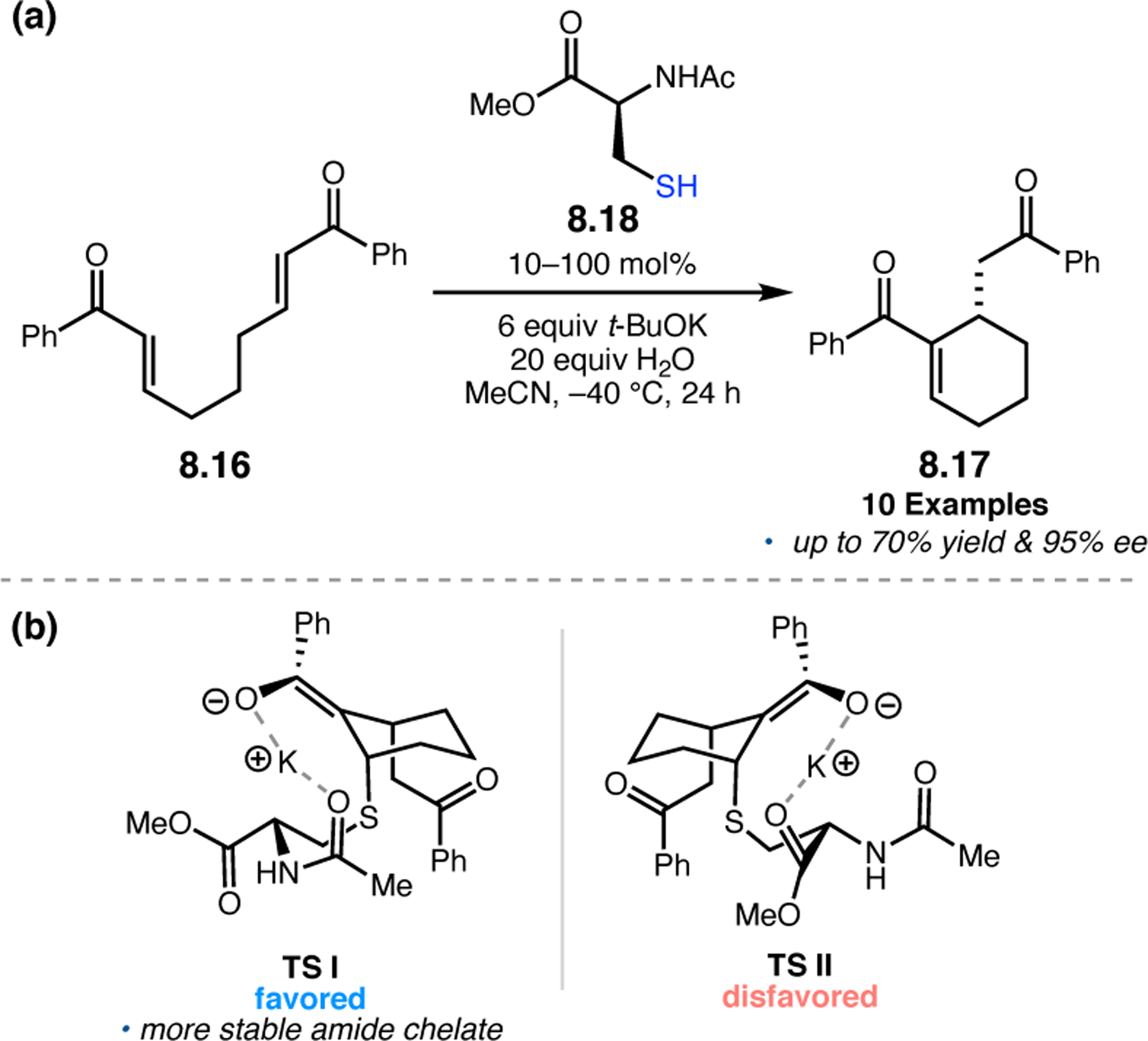
(a) Cys-catalyzed, intramolecular Rauhut–Currier reaction of bis(enone) 8.16 and (b) proposed model for selectivity.378,379
Following reaction optimization, mechanistic aspects of this Cys-catalyzed transformation were explored in order to shed light on the nature of asymmetric induction. To frame these studies, a plausible catalytic cycle was proposed (Figure 158). The Cys catalyst 8.18 is first deprotonated to generate thiolate I, which reversibly adds into 8.16 to deliver intermediate II. This covalent catalyst–substrate adduct subsequently undergoes an intramolecular Michael addition to afford III. Irreversible proton transfer and subsequent catalyst elimination yields 8.17 and regenerates active thiolate I. Two observations informed this mechanistic proposal. First, when 18-crown-6 was included in the reaction, the reaction was very sluggish and cyclic enone 8.17 was not the major product, but rather a deconjugated isomer of 8.17 predominated. This suggested that potassium ion was important in the selective reaction pathway. Second, when the reaction was run to low conversion, 8.18 delivered 8.17 in 29% yield and 95% ee, but the remainder of the mass balance consisted of five diastereomers of the protonated form of intermediate III. When this covalent complex was oxidized to the sulfone and eliminated under irreversible conditions, 8.17 was obtained in only 40% ee. This suggested that the proton transfer and catalyst elimination steps are both rate- and stereodetermining. Based on these data, a stereochemical model was proposed (Figure 157b). Important for achieving high levels of enantioselectivity could be a stabilizing potassium ion chelate between the carbonyl of the N-acyl protecting group of 8.18 and the enolate of intermediate III.
Figure 158.
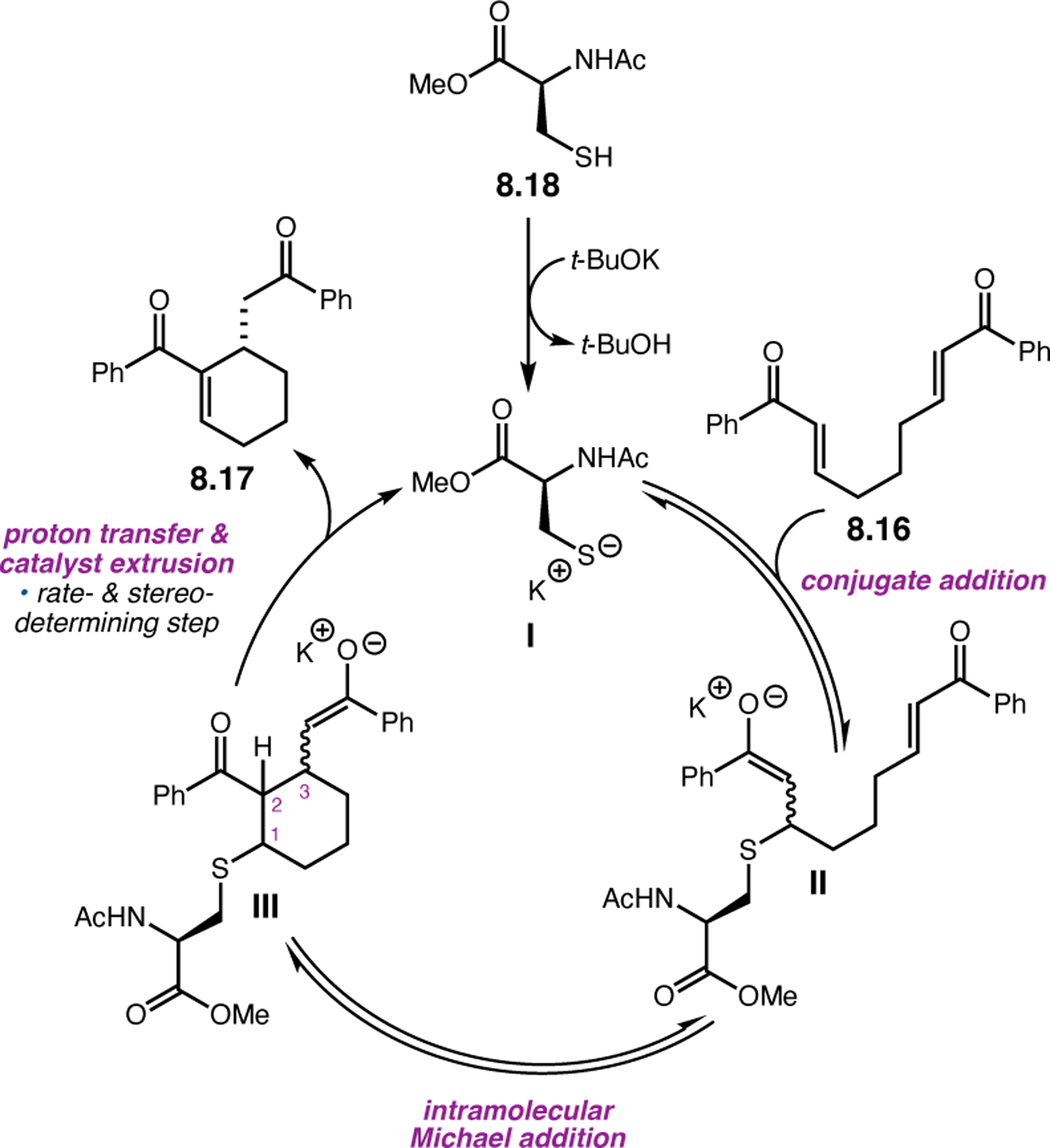
Proposed mechanism for the Cys-catalyzed intramolecular RC reaction.378,379
Houk and co-workers then probed the reaction mechanism further using DFT calculations.380 The computational data supported that deprotonation/thiolate elimination is rate determining, and it proceeds through an E1cB mechanism. This process is comprised of two steps: (1) α-deprotonation and (2) catalyst extrusion to form 8.17 and regenerate thiolate I (Figure 158). Since catalyst elimination can only occur when the Cys moiety is in an axial orientation, the number of reasonable stereoisomers of III was reduced from eight to four. Regardless of the stereoisomer, the deprotonation step was found to have a higher activation barrier than the subsequent catalyst elimination step, consistent with an E1cB mechanism. During reaction optimization, it became apparent that the catalyst was sensitive to the equivalents of water added and the nature of the base employed. Therefore, the effects of the counterion and water on this E1cB process were studied computationally.
Transition states with zero, one, or two water molecules and either potassium, sodium, or no countercation were optimized. A synergistic effect between the potassium counterion and the presence of water was identified, and the lowest energy transition states for the α-deprotonation step were obtained when two water molecules and one potassium cation were included (Figure 159); one water was proposed to stabilize the hydroxide anion that deprotonates III at the α-position, and the other could interact with the enolate. The potassium cation was found to orient the hydroxide anion proximally to the site of deprotonation. TS II, which leads to the experimentally observed (S)-enantiomer of 8.17, was favored by about 0.8 kcal/mol (79.5:20.5 er). Experimentally, the addition of one equivalent of water at room temperature resulted in 71:29 er, and three equivalents led to 78.5:21.5, quite similar to the computed energy difference.379 Houk and co-workers propose that TS II may be favored due to the pre-organization of the potassium cation and the hydroxide anion and the more favorable, planar orientation of cysteine.
Figure 159.
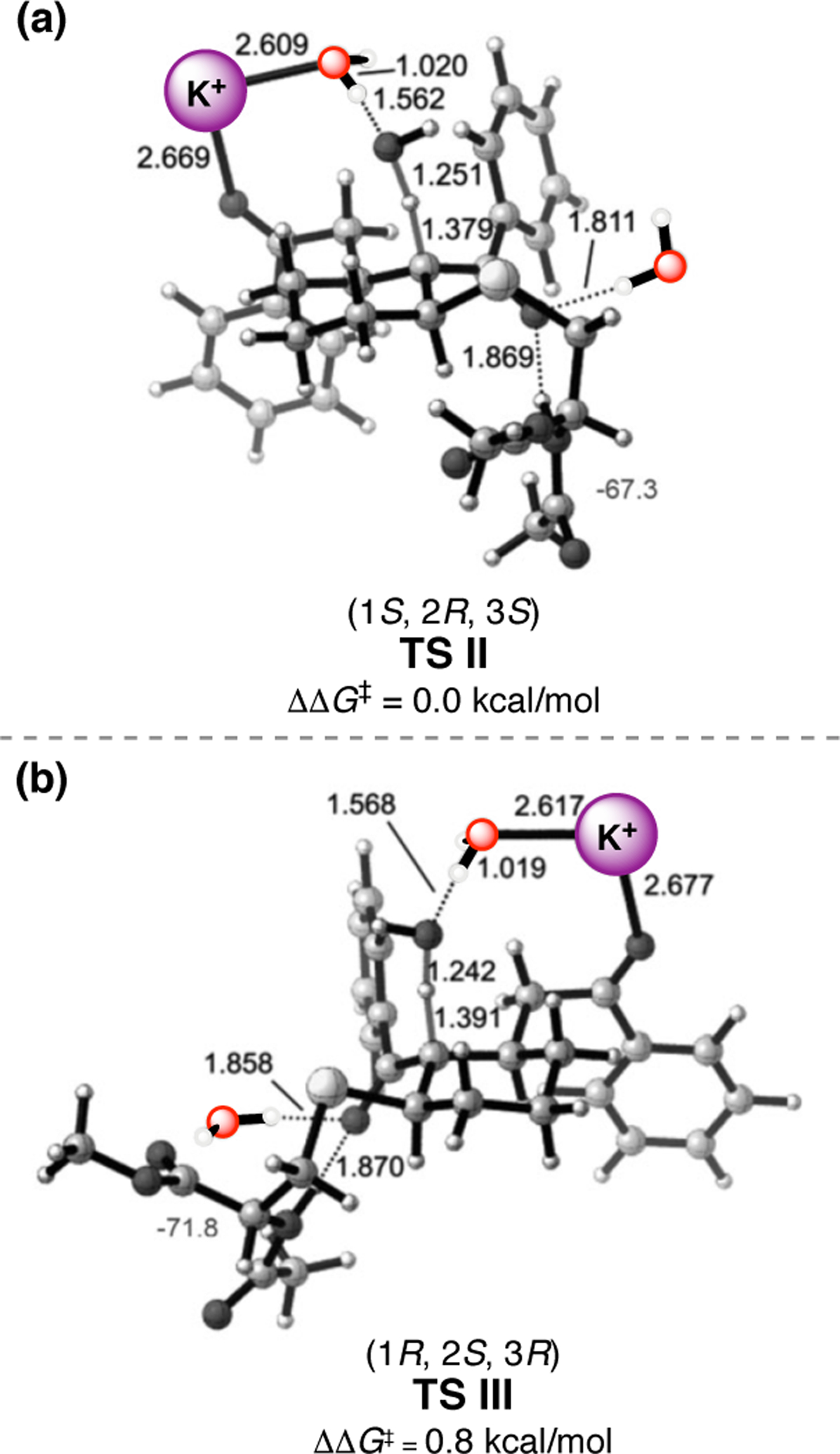
Optimized transition states for the α-deprotonation step of the E1cB process for (1S, 2R, 3S) and (1R, 2S, 3R) enantiomers of intermediate III including potassium counterion and two explicit water molecules. Distances and angles are denoted in Å and degrees, respectively. Adapted with permission from ref. 380. Copyright 2013 Wiley.
The enantioselective, cysteine-catalyzed RC reaction was also applied in the asymmetric total synthesis of Sch-642305 (8.21, Figure 160),381 which has been shown to exhibit promising anti-HIV activity.382 As noted previously, Cys residues within enzymes are known to catalyze a number of biological reactions, but an enzymatic, cysteine-mediated RC reaction had not yet been identified. However, we wondered if a cysteine-dependent enzyme might be capable of mediating the ring contraction of 8.19 to 8.20 en route to 8.21. Thus, we proposed a Cys-catalyzed ring contraction process as a quasi-biomimetic strategy to rapidly access natural product 8.21.
Figure 160.
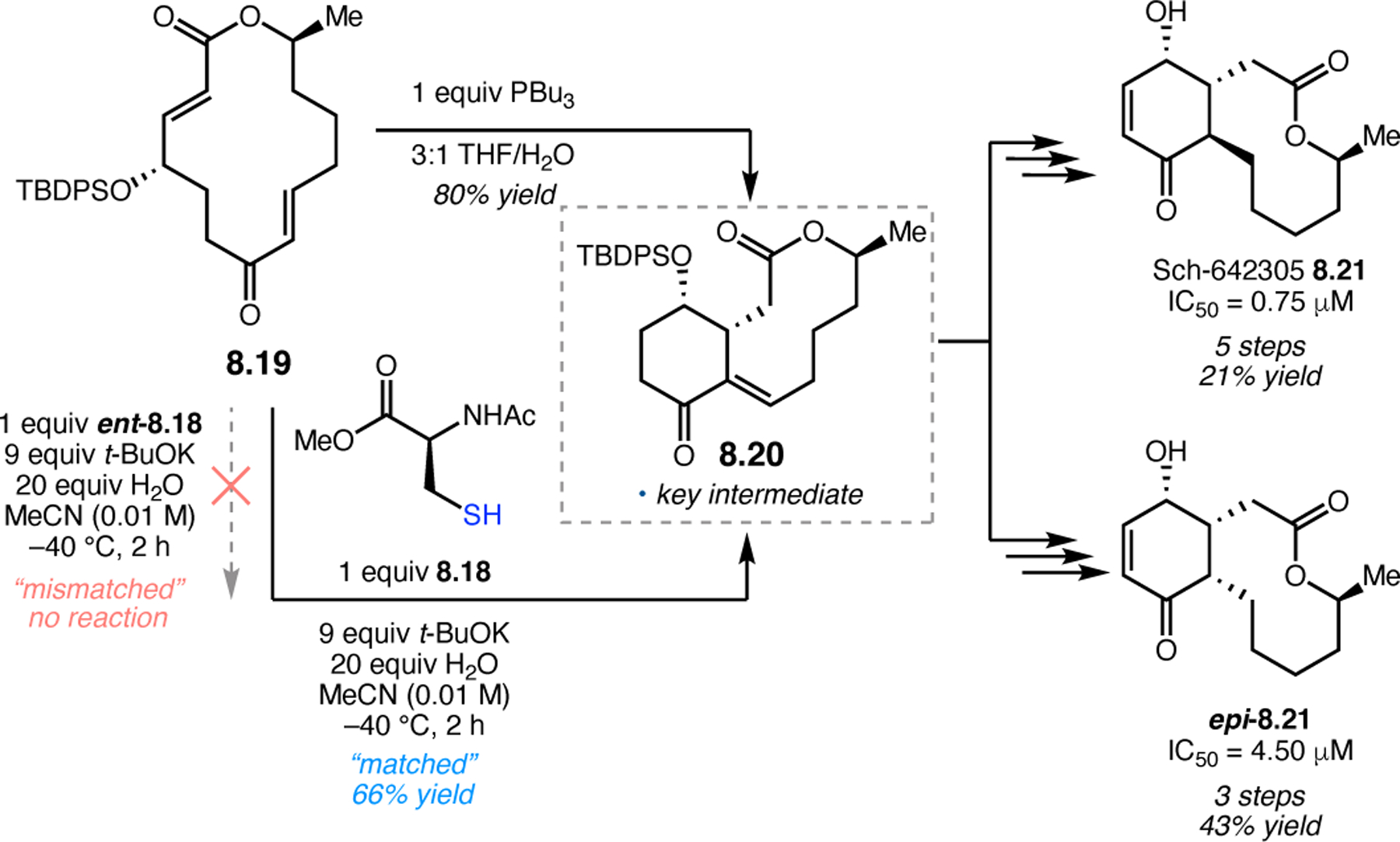
Substrate controlled Cys-catalyzed RC ring contraction leading to Sch-642305 (8.21) and epi-Sch-642305 (epi-8.21).381
Macrocycle 8.19 was synthesized in order to study the key Cys-mediated RC reaction (Figure 160). Tributyl phosphine was initially employed as a catalyst, affording 8.20 in 80% yield and >30:1 dr. This implied that the substrate may indeed govern selectivity in this transformation. Reaction optimization with Cys 8.18 culminated in the delivery of 8.20 in 66% yield. Under the same conditions, yet with ent-8.18 as the catalyst, the desired product was not detected. These results imply that a matched/mismatched situation exists between 8.19 and catalyst 8.18. Moreover, when achiral isobutyl thiol was used as a putative catalyst, the RC reaction product 8.20 was not obtained. Instead, a mixture of thiol conjugate addition products was observed, suggesting that 8.18 is uniquely apt as an RC catalyst in this context.
With compound 8.20 in hand, natural product 8.21 could be accessed in 21% yield over five steps. Additionally, 8.20 could be funneled to epi-8.21 in 43% yield over 3 steps. These compounds, along with other structural analogs, were tested against HIV-1 in MT-2 human T-cells. Notably, natural product 8.21 exhibited significantly higher activity (IC50 = 0.75 μM) than the four other analogs tested.
The stereochemical dependence of this RC reaction stimulated an experiment to determine whether 8.20 could be produced via catalyst- rather than substrate-control. Thus, macrocycle 8.23, lacking any stereogenic centers, was tested in this RC reaction with catalyst 8.18 (Figure 161). After 24 h with 7.5 equivalents of exogenous base, 8.24 was produced in 25% yield and 82.5:17.5 er. Studies aimed at further improving these results are currently ongoing.
Figure 161.
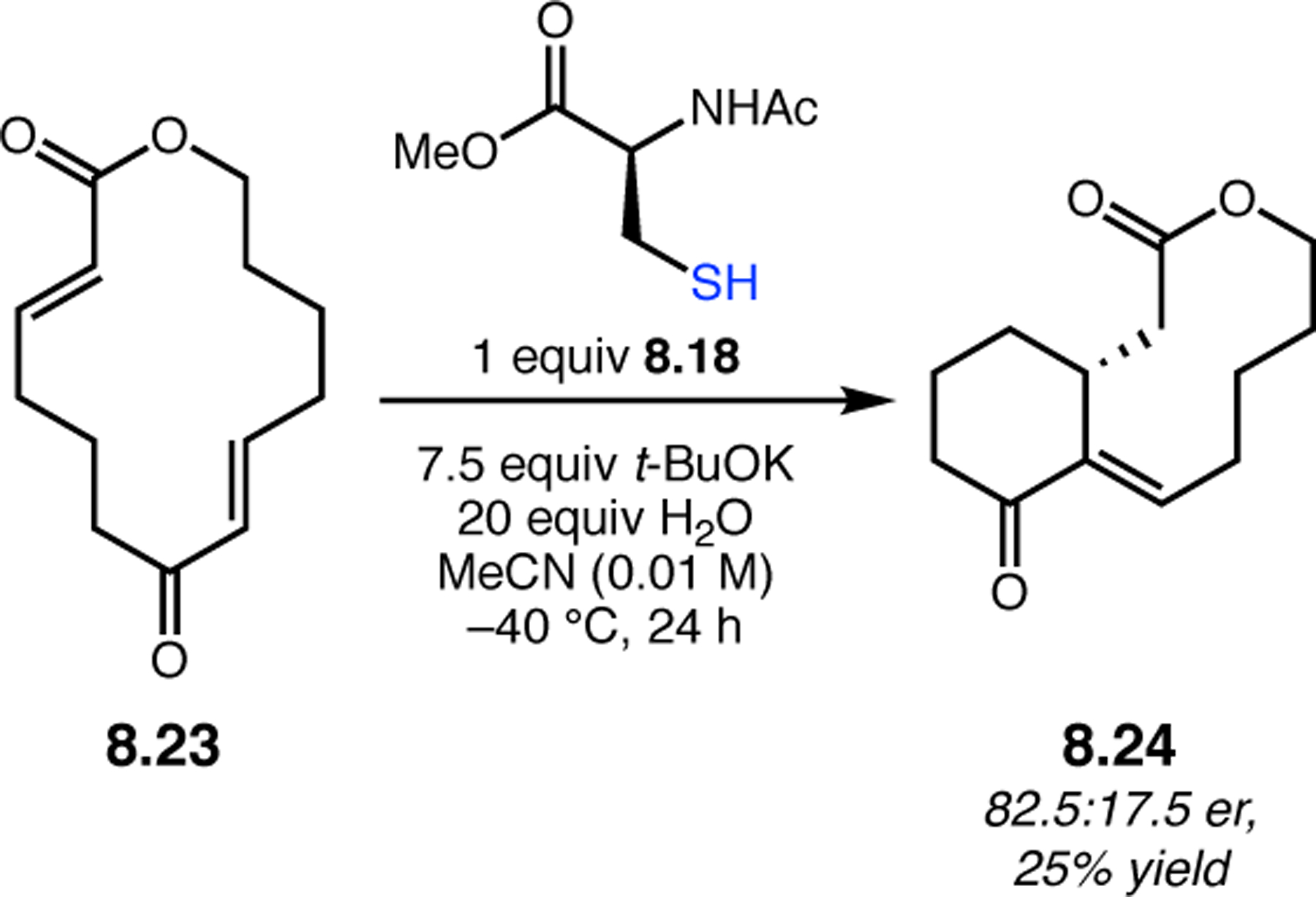
Catalyst 8.18-controlled enantioselective RC ring contraction of 8.23.381
9. Photochemical Radical Reactions
Photochemical reactions often involve high-energy free radical intermediates, which are difficult to control using catalytic asymmetric approaches.383,384 While many advances have been made on this front, peptide catalysis remained largely unexplored. This may have been due to perceived challenges associated with their conformational flexibility, which might amplify the already daunting task of asymmetric induction with radical species. Moreover, the noncovalent interactions that peptides typically exploit to organize transitions states in selective reactions may have been anticipated to be weak or inoperative with neutral, open-shell intermediates. Despite these liabilities, highly enantioselective photochemical reactions utilizing peptide-based catalysts have indeed been demonstrated in recent years.
Two of these methodologies were enabled by the strategic use of cysteine as a catalytic residue. Enzymes are known to use active-site cysteine residues to mediate both polar and radical transformations in vivo.385,386 Cysteine proteases are an example of the former, which use nucleophilic cysteine thiolates to catalyze amide bond hydrolysis.385 On the other hand, ribonucleotide reductases enlist cysteine residues in the active site as HAT catalysts that assist in the deoxygenation of ribonucleotides to deoxyribonucleotides.387 Following the initial report of cysteine as a nucleophilic catalyst for asymmetric Rauhut–Currier reactions (Section 8.3),378,379 our group became interested in targeting the radical reactivity of cysteine-containing peptides. This catalytic platform was first assessed in the context of covalent thiyl radical catalysis (Section 9.1) and was later explored in enantioselective HAT chemistry governed by putative noncovalent interactions (Section 9.2).
A third recent strategy for peptide-catalyzed radical chemistry employs peptidic ligands on rare-earth metals to induce asymmetry in a dual photoredox-/Lewis acid-catalyzed [2+2] cycloaddition (Section 9.3). Among the various “privileged chiral scaffolds” examined,388 peptide-based ligands were uniquely effective in promoting this transformation.
9.1. Vinylcyclopropane Ring-Opening/Cycloadditions
Although the reactivity patterns of thiyl radicals have been extensively studied in a host of synthetically useful transformations (e.g. HAT, additions to C=C bonds, etc.), very few examples of asymmetric, thiyl-catalyzed reactions exist to date.389,390 Early examples reported by Roberts and co-workers used sugar-derived thiols as chiral, polarity-reversal HAT catalysts391 for modestly enantioselective alkene hydrosilylation chemistry.392,393 In 2014, Maruoka and co-workers reported an asymmetric vinyl cyclopropane (VCP) ring-opening/cycloaddition cascade reaction catalyzed by chiral thiophenols.394 In this process, the chiral thiyl was presumed to directly engage the substrate in covalent catalysis, implementing a steric blocking strategy to deliver elaborated cyclopentane products with excellent diastereo- and enantioselectivities. Given the demonstrated thiyl radical reactivity of cysteines in vivo,386 as well as the challenges associated with the synthesis and diversification of chiral thiophenol-based organocatalysts, a peptide-mediated process seemed a viable extension.
Our group explored the VCP ring-opening/cycloaddition cascade reaction as a means to probe the hitherto unprecedented radical reactivity of cysteine in asymmetric catalysis.395 The overall strategy was to employ disulfide-bridged peptide dimers as thiyl radical precursors that would enable the formal radical cycloaddition of VCPs 9.1 and acceptor olefins 9.2 to yield cyclopentane derivatives 9.3 with high levels of selectivity (Figure 162). Mechanistically, UV-photoexcitation of the cysteine disulfide (i.e., cystine) homolytically cleaves the S–S bond, providing two equivalents of active thiyl radical catalyst. The thiyl then adds to the C=C bond of 9.1, resulting in covalent complex I following rapid cyclopropane ring-opening. Addition of electron-deficient radical I to alkene 9.2 results in heteroatom-stabilized radical intermediate II. Subsequent 5-exo-trig cyclization and catalyst elimination delivers 9.3 with diastereo- and enantioselectivity governed by interactions between the peptide and its covalently bound substrate.
Figure 162.
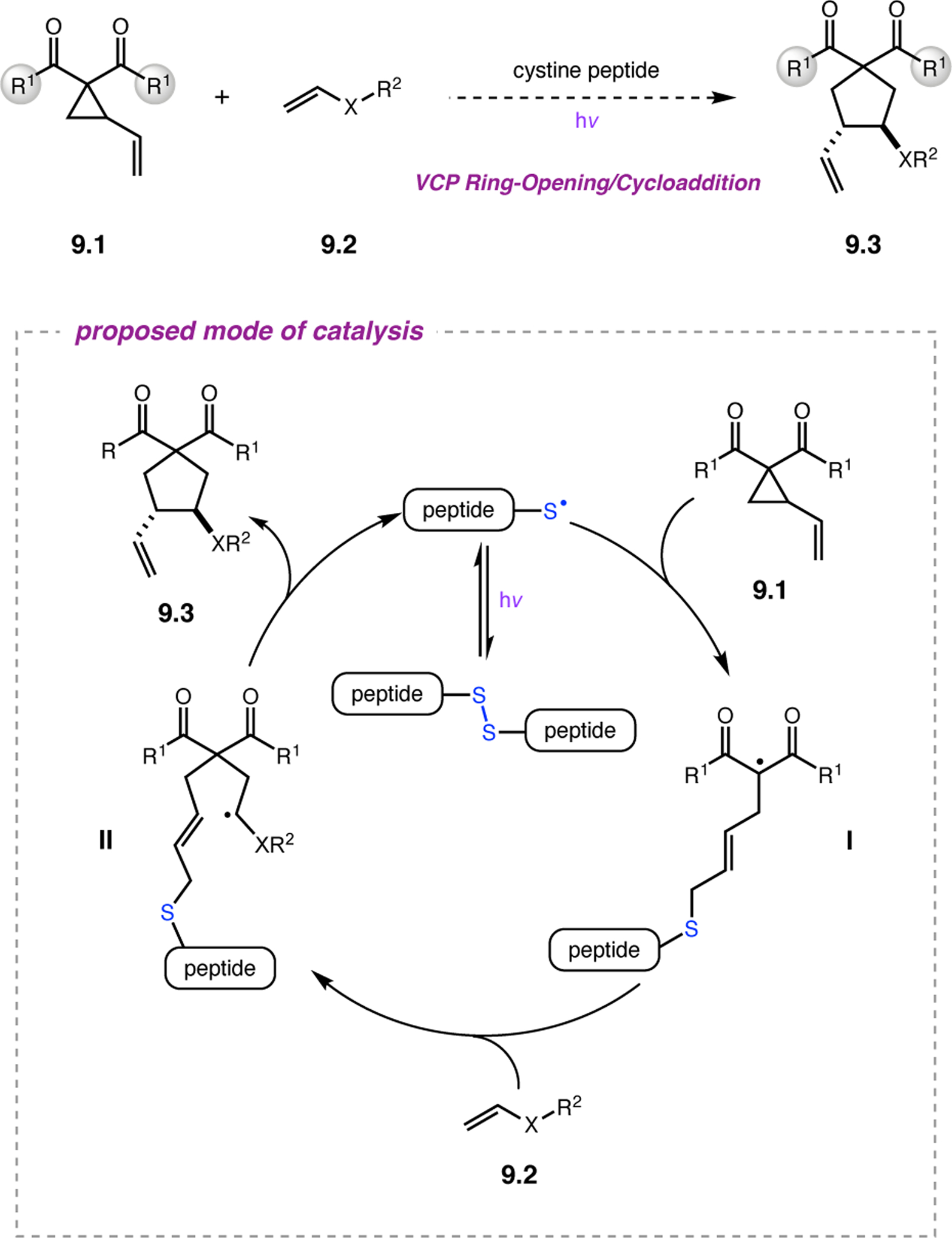
General strategy and mechanistic proposal for the peptide-catalyzed VCP ring-opening/cycloaddition cascade enabled by cysteine thiyl radicals.395
Preliminary results showed that the diester VCP 9.1a used by Matsubara and co-workers was an ineffective substrate in peptide-catalyzed reactions (Figure 163). Multiple cystine-containing peptides were examined, and while good reactivity was often observed, the reactions were not enantioselective. However, the analogous diamide VCP 9.1b was modestly selective under the influence of cystine-containing peptides. Using β-turn-biased peptide 9.4, tert-butyl vinyl ether 9.2, and exposure to UV light, diamide 9.1b was converted to a nearly 1:1 mixture of cis- and trans-9.3b, with the cis-isomer being enriched to 38:62 er.
Figure 163.
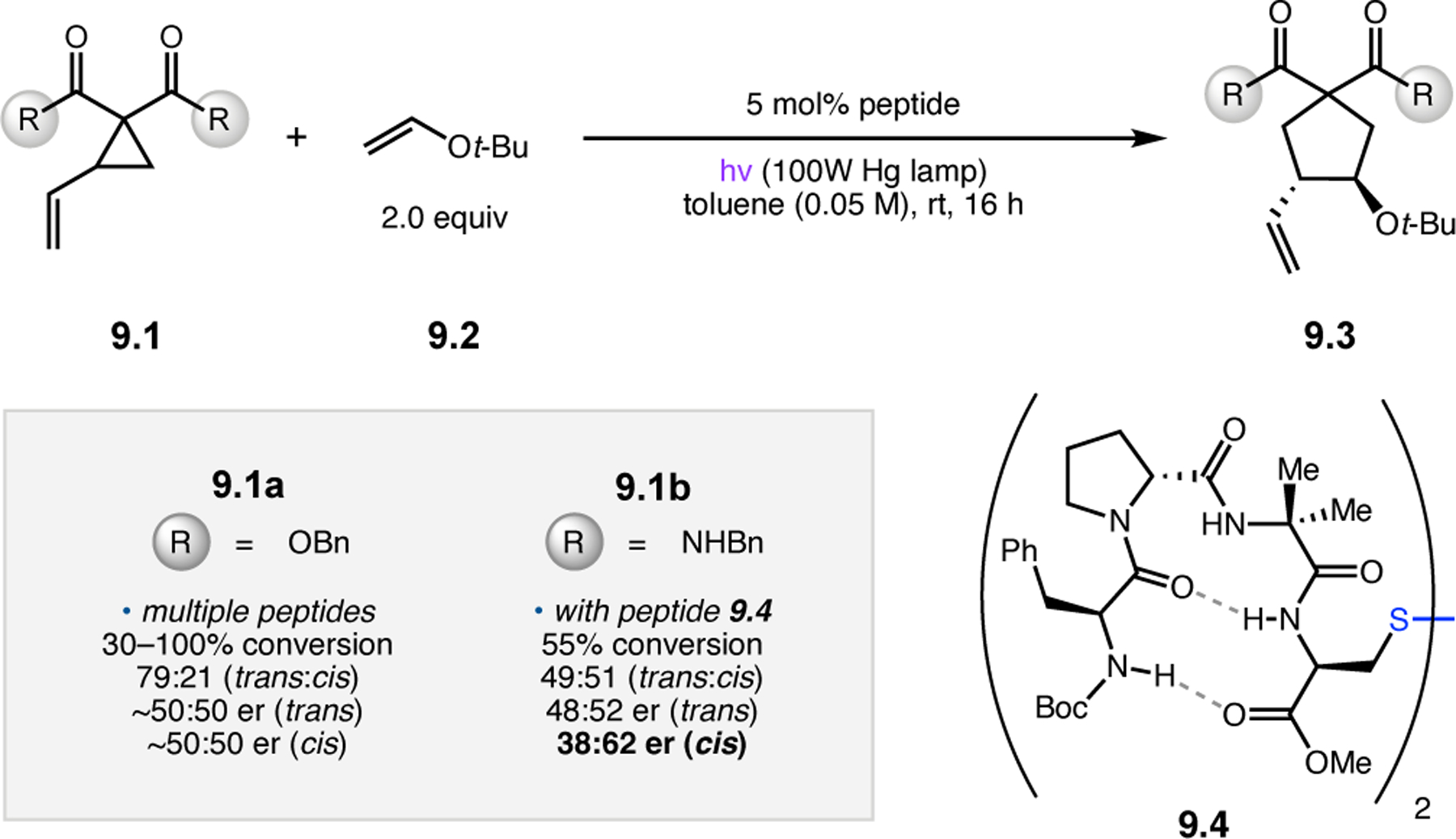
Initial results establishing proof-of-principle with amide-functionalized VCPs 9.1.395
Catalyst optimization was carried out for the reaction of VCP 9.1c with tert-butyl vinyl ether under continuous UV irradiation (Figure 164). Known catalyst scaffolds 9.4 and 9.5 were not selective in this context, suggesting that these legacy structural motifs were not effectively engaging the diamide substrate. Reasoning that the spin density of key intermediates I and/or II (Figure 162) was distal to the peptide, scaffold 9.6 was conceived, wherein the i+1 Pro residue is functionalized at the γ-position in an attempt to extend the chiral pocket and improve asymmetric induction. Indeed, peptide 9.6a delivered 9.3c in quantitative conversion and notably improved er for both diastereomers—80:20 er for the major trans isomer and 57:43 for the minor cis isomer.
Figure 164.
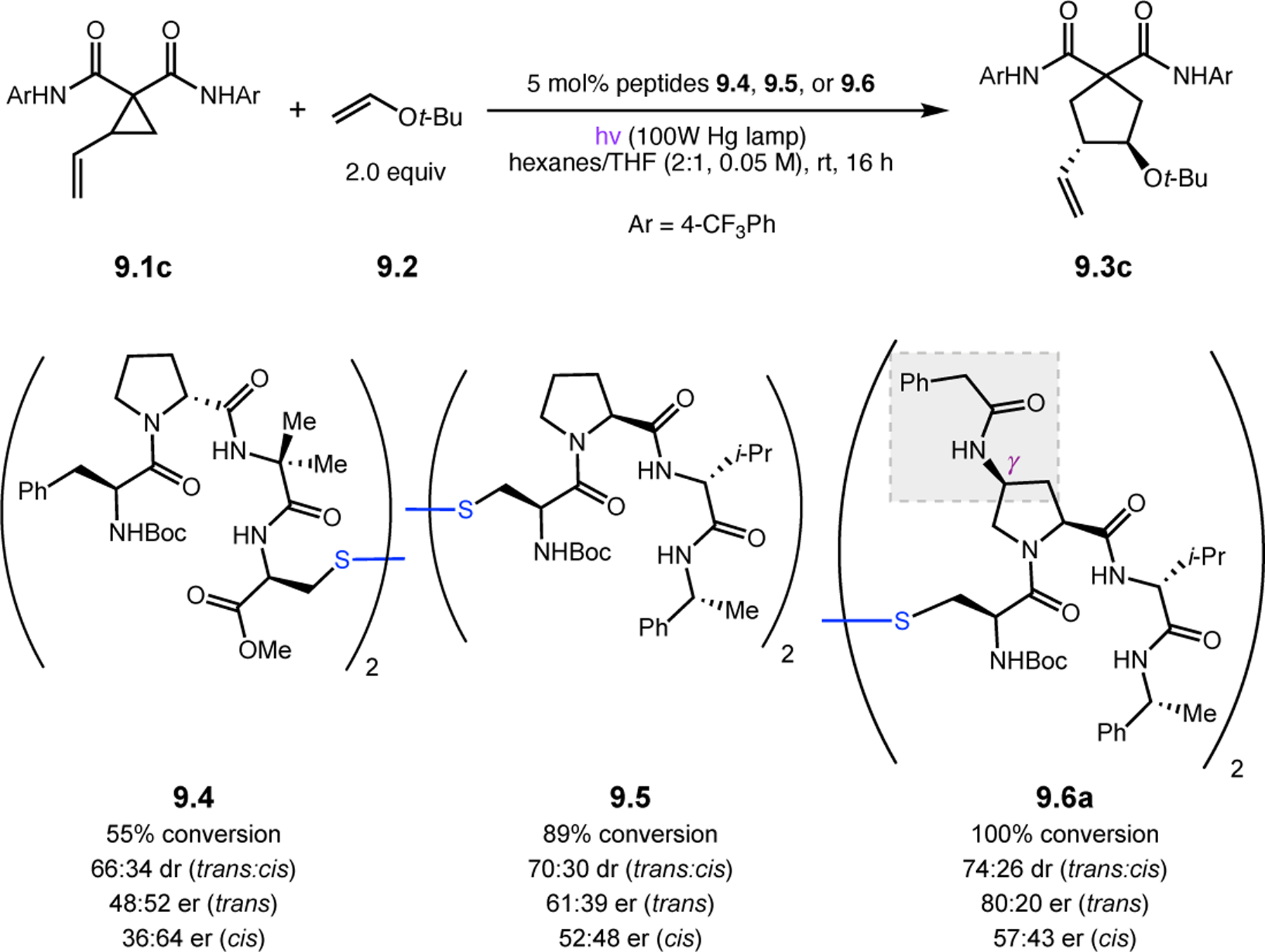
Peptide optimization for the VCP ring-opening/cycloaddition cascade reaction. Scaffold 9.6 led to significant improvements in enantioselectivity compared to the other archetypal peptides 9.4 and 9.5.395
Modification of the Pro γ-substituent provided another accessible handle for catalyst optimization (Figure 165). While the pivalamide led to further improvements in er for both diastereomers (9.6b), both tosyl- (9.6c) and phenyl urea-protected amines (9.6d) were significantly inferior catalysts. γ-Arylated derivatives were similarly poor in the reaction of 9.1c with tert-butyl vinyl ether (9.6e–g). A Hammett analysis of various γ-benzamide-containing peptides showed that enantioselectivity was not sensitive to electronic perturbations. However, er did correlate with the steric size of the γ-amine protecting group. Bulky amide groups provided the requisite boost in enantioselectivity, and peptide 9.6h was chosen as the lead catalyst, providing the major trans-9.3c in 90:10 er and minor cis-9.3c in 75:25 er.
Figure 165.
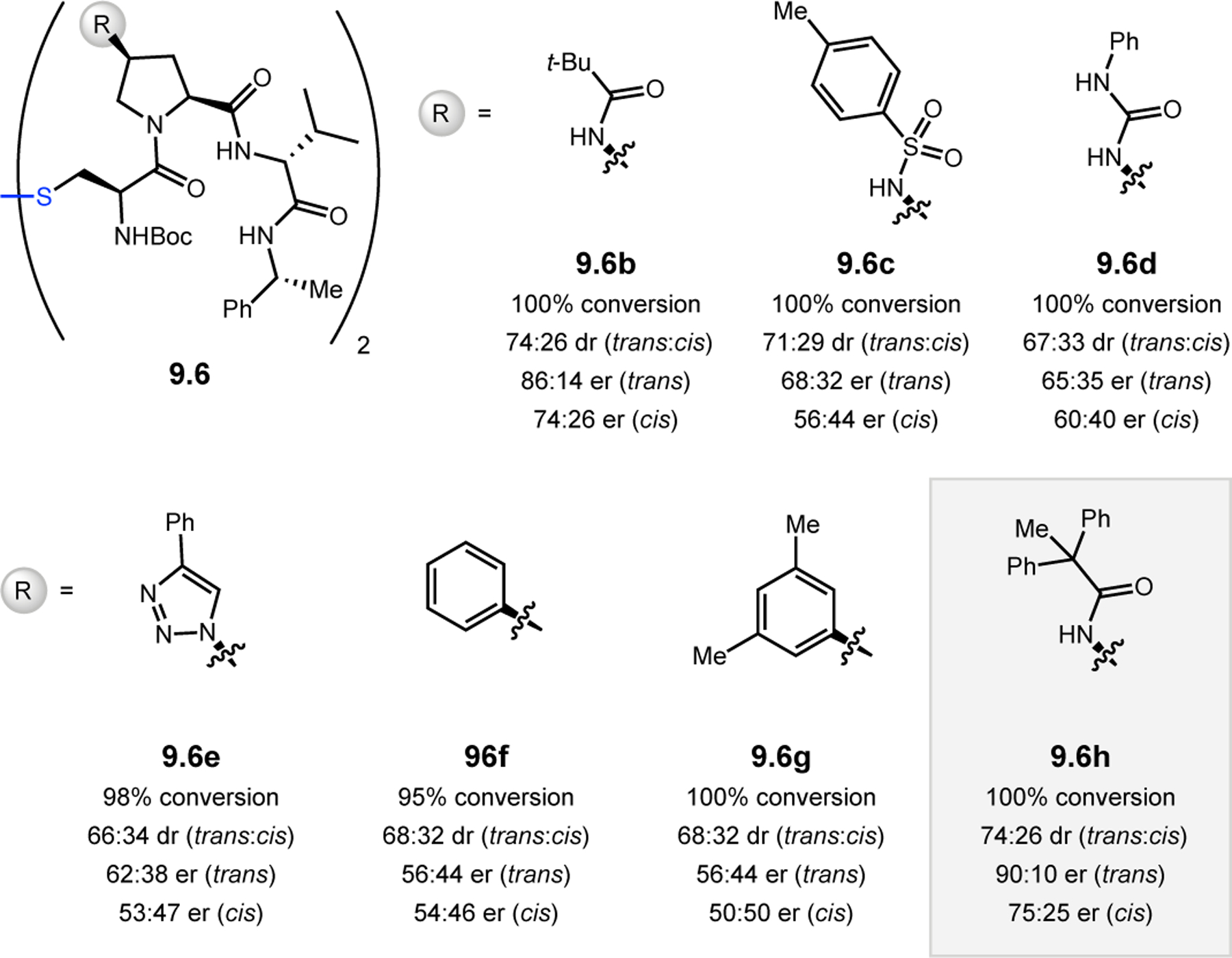
Modulation of the i+1 Pro γ-substituent led to the identification of lead catalyst 9.6h. Results are presented for the reaction of VCP 9.1c under the conditions described in Figure 164.395
Peptide 9.6h was found to address a reasonable scope with respect to both VCPs 9.1 and acceptor olefins 9.2 (Figure 166). Diastereoselectivity was found to be intrinsic to each VCP substrate, and the major trans diastereomer was always formed with higher levels of enantioselectivity. In general, electron-deficient amides were more selective in the reaction (entries 3 & 6–9 vs. entries 1 & 4–5). This provides some evidence in support of H-bonding between the VCP substrate and the catalyst. With regard to the acceptor olefin, only electron-rich alkenes were converted to the corresponding cyclopentane products 9.3d–i with good to excellent er values.
Figure 166.
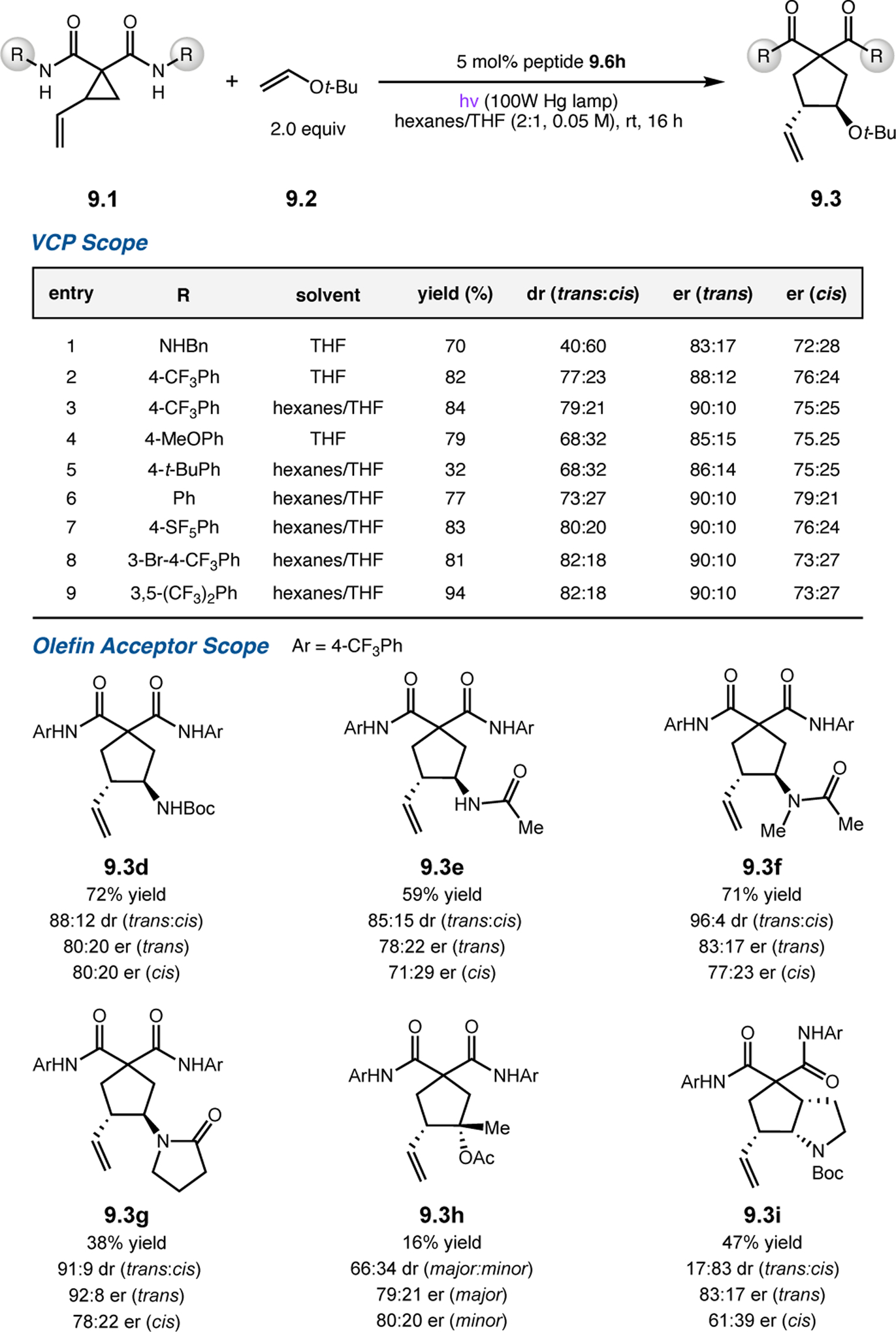
Substrate scope of the 9.6h-catalyzed VCP ring-opening/cycloaddition cascade reaction.395
A model was proposed for the 9.6-catalyzed ring-opening/cycloaddition cascade reaction of VCP 9.1c (Figure 167). In the model, an amide N–H of the VCP substrate engages in an H-bond with the i+1 carbonyl of peptide 9.6h. The combination of this key noncovalent interaction with the steric blocking effect of the Pro γ-substituent requires that radical addition to the acceptor olefin occur from opposite side, such that the t-butoxy group is oriented away from the γ-substituent. This putative arrangement is consistent with the absolute configuration of trans-9.3 as determined by X-ray crystallography.
Figure 167.
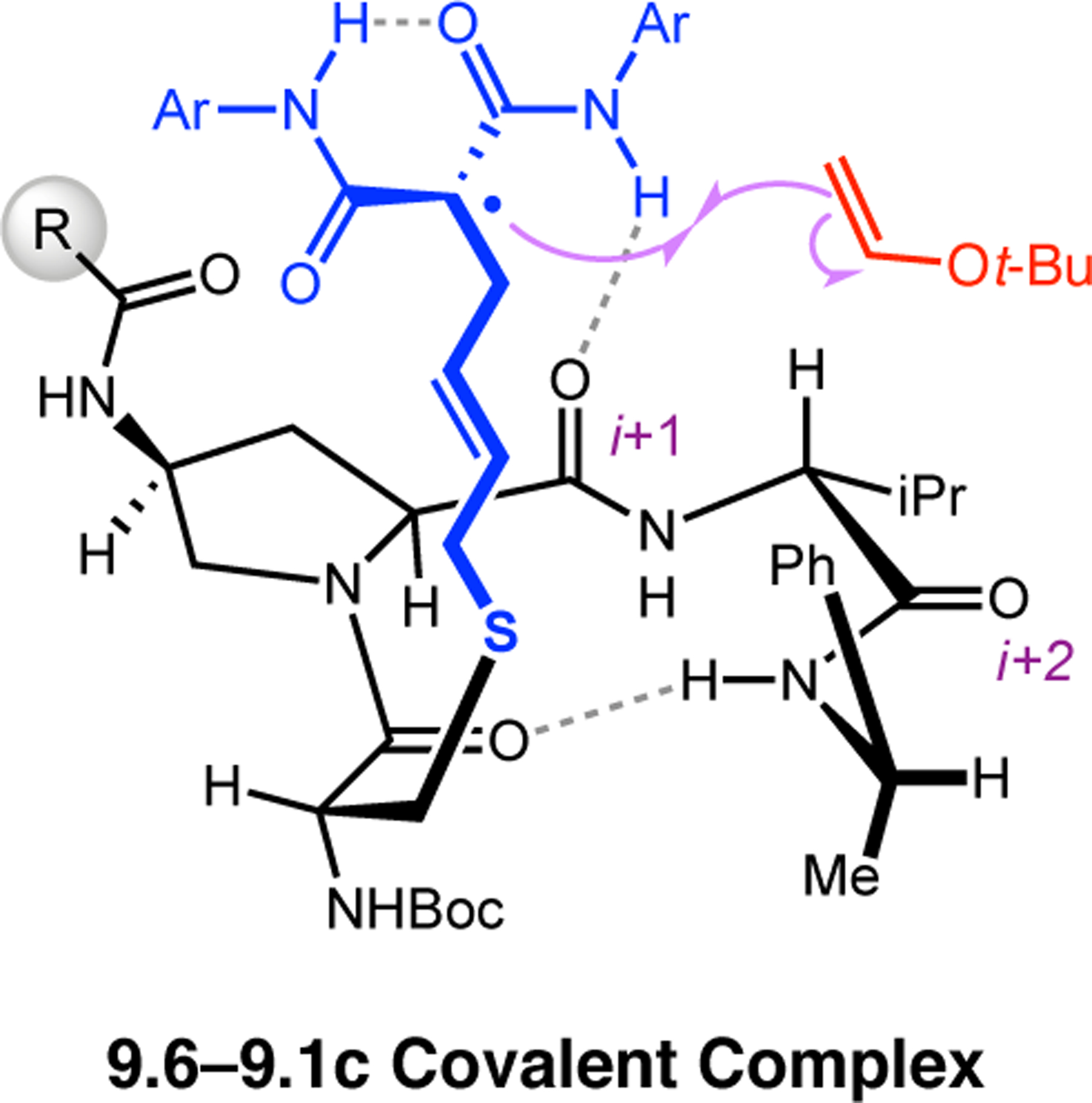
Proposed model for the 9.6-catalyzed VCP ring opening/cycloaddition cascade reaction.395
9.2. Photoredox Deracemization
An underexplored avenue for asymmetric synthesis is deracemization, or the direct conversion of a racemate into a single enantiomer of the same compound (Figure 168).396 This process may occur, in principle, when a chiral catalyst converts each enantiomer of the starting material into a common, achiral intermediate at different rates, enriching the reaction mixture in the slow-reacting isomer. Subsequent non-selective back-reactions from the achiral intermediate can then return both enantiomers of the starting material, which reenter the cycle. As these steps are repeated iteratively, the reaction mixture may become enriched in one enantiomer, such that synthetically useful ee values can be obtained. Seminal examples by Turner, Toste, and Zhou implement sequential redox steps to achieve deracemization.397–399
Figure 168.
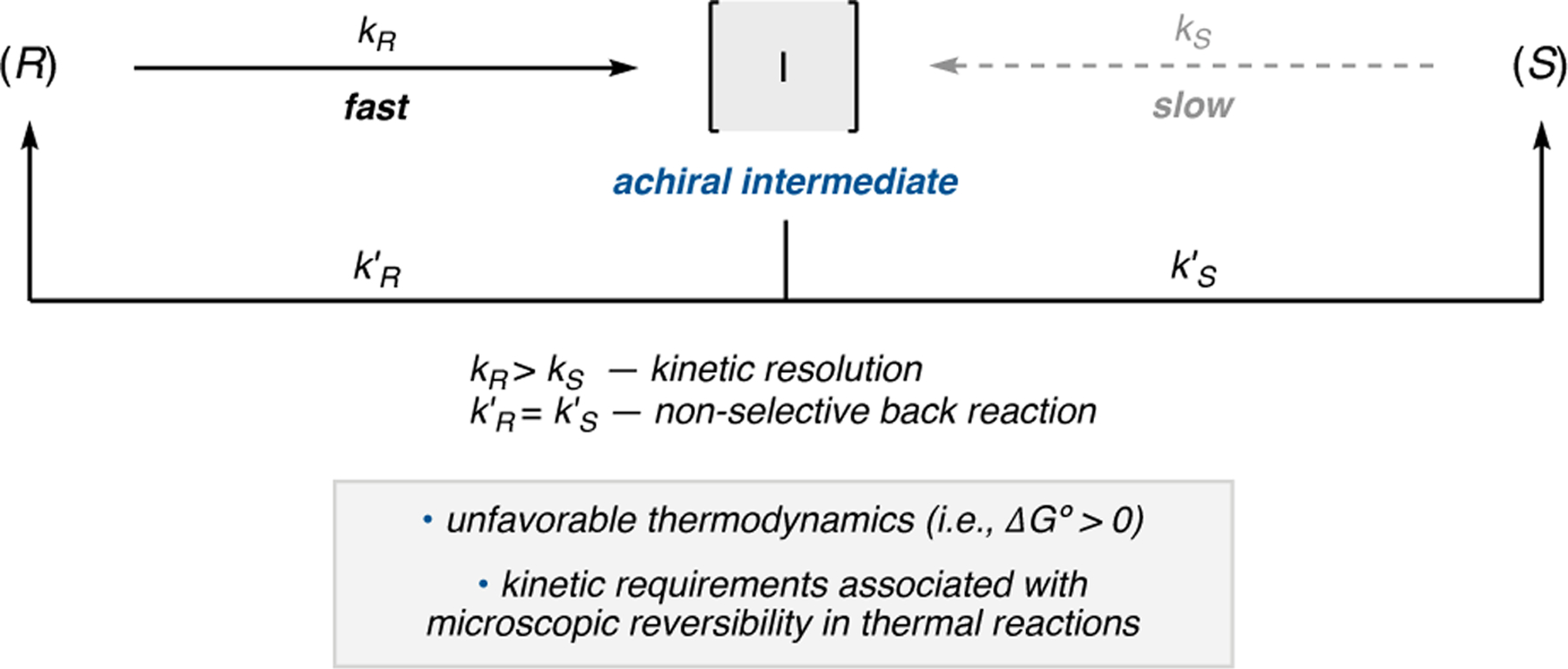
General conception of and challenges associated with cyclic deracemization strategies.396
Although conceptually quite straight forward, fundamental challenges associated with deracemization methodologies have contributed to their relative scarcity in the literature. First, the conversion of a two-state racemate to a one-state (i.e., enantiopure) system makes these processes thermodynamically uphill (ΔG° = +0.42 kcal/mol). From a kinetic standpoint, the elementary steps that destroy and create stereochemistry must occur through distinct mechanisms in order to observe deracemization. If these steps are microscopic reverses of one another, then an equilibrium mixture—that is, a racemate—is inevitable. Chemoselectivity is also a critical challenge in deracemization, as the reagents that effect the stereoablative and stereogenic steps are often incompatible (e.g., oxidants and reductants).
In collaboration with the Knowles Group, we recently reported a light-driven deracemization method for the direct optical enrichment of cyclic urea substrates 9.7 (Figure 169).400 The conversion of (±)-9.7 to enantioenriched (R)-9.7 is jointly mediated by an Ir(III)-based photosensitizer, a chiral BINOL-derived Brønsted base, and a cysteine-containing peptide as an HAT catalyst. Together, these three catalysts act to break and reform the stereogenic α-C–H bond of the substrate in a sequence of electron-transfer (ET), proton transfer (PT), and HAT steps. This process is driven solely by the absorption of visible photons and uses no stoichiometric reagents and produces no waste byproducts.
Figure 169.

Deracemization of cyclic ureas 9.7 jointly mediated by an Ir(III)-based photocatalyst, a chiral Brønsted base, and a cysteine-containing peptide catalyst.400
The proposed deracemization mechanism begins with non-selective single-electron oxidation of (±)-9.7 by the excited state of the Ir(III) photoredox catalyst,401 which then provides both enantiomers of radical cation 9.7+• in an equal ratio (Figure 170). These reactive intermediates are then kinetically resolved via enantioselective deprotonation by the chiral phosphate base, delivering achiral α-amino radical 9.8. This PT step is proposed to enrich the reaction mixture in the slower reacting enantiomer (R)-9.7+•, which reverts to (R)-9.7 through charge recombination with the reduced Ir(II) state of the photocatalyst. Enantioselective HAT catalyzed by a cysteine-containing peptide then converts 9.8 into either (R)- or (S)-9.7 depending on the sense asymmetric induction. In the matched case, the peptide-catalyzed HAT forms (R)-9.7, causing the reaction mixture to further enrich in that isomer. The resulting peptidic thiyl is then reduced and protonated to restore the three catalytic components to their active states. Unlike other reported deracemization methods,397–399,402 this process involves two mechanistically distinct, enantioselective steps—PT that ablates chirality and HAT that re-establishes it. This provides the unique opportunity to observe multiplicative enantioselectivity outcomes, wherein the observed er is the product of the individual ers of both the PT and HAT steps (erobs = erPT • erHAT). Within this framework, two modestly selective steps could give rise to a highly selective overall transformation.
Figure 170.
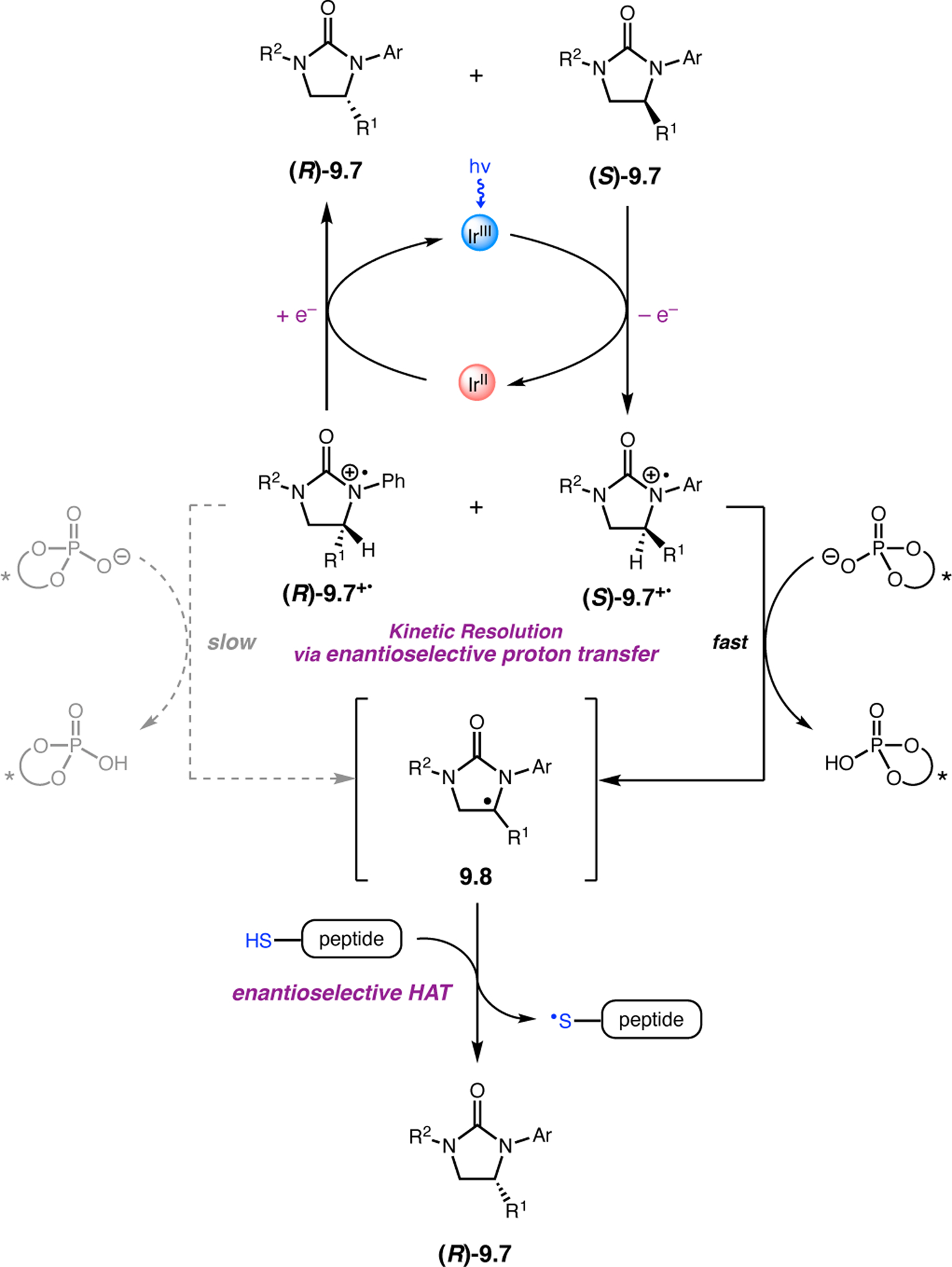
Proposed mechanism for the deracemization of urea 9.7. The cysteine containing peptide-based catalyst would mediate the enantioselective HAT step.400
Optimization of this deracemization process began with a separate evaluation of each enantioselective step using amide-functionalized urea (±)-9.7a as the model substrate and [Ir(dF(CF3)ppy)2(bpy)]PF6 as the photocatalyst (Figure 171). Pyrene-substituted BINOL phosphate 9.9 emerged from base screening as the most selective PT catalyst. Using thiophenol as an achiral HAT catalyst, 9.9 was able to deracemize 9.7a in 84% recovered yield and 79:21 er (entry 1). Addition of molecular sieves improved both the yield and selectivity, providing (R)-9.7a in 92% yield and 86:14 er (entry 2).
Figure 171.
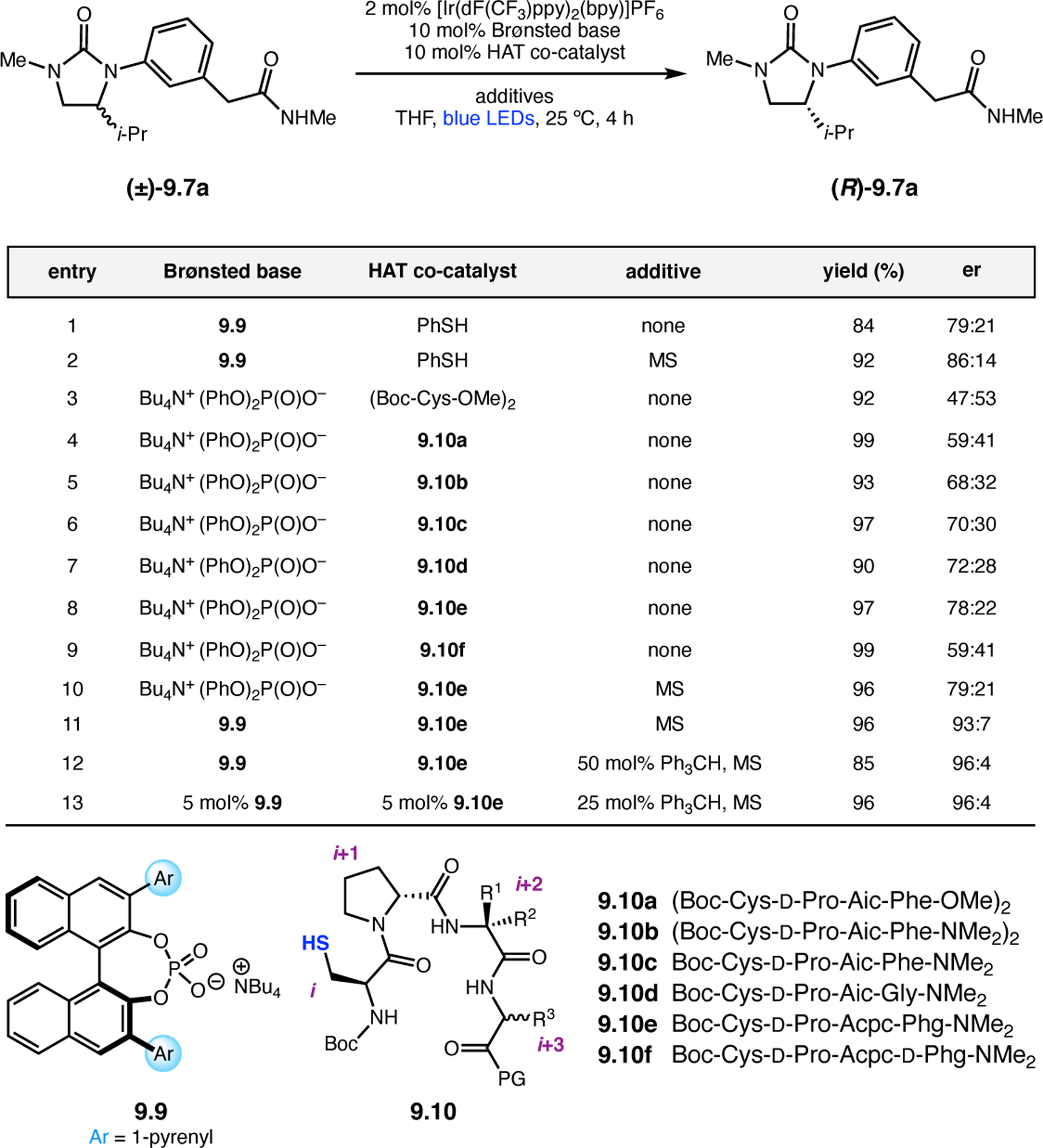
Optimization of the light-driven deracemization of urea 9.7. The enantioselective PT and HAT steps were mediated by chiral BINOL-derived phosphate 9.9 and cysteine-containing tetrapeptides 9.10, respectively.400
Cysteine-containing peptides were then evaluated as HAT catalysts using an achiral phosphate as the base (Figure 171). While protected cystine returned 9.7a in 92% yield and 47:53 er (entry 3), disulfide-bridged tetrapeptide 9.10a, which was biased toward a β-hairpin conformation with its central d-Pro-Aic sequence, provided a modest boost in selectivity to 59:41 er in favor of the opposite enantiomer (entry 4). This suggests that the secondary structure of 9.10a can overturn the inherent enantiopreference of the catalytically active HAT residue. Substituting the methyl ester for a dimethyl amide C-terminal cap in 9.10b resulted in a notable increase in selectivity (entry 5), as did reducing disulfide 9.10b to the corresponding thiol 9.10c (entry 6). Changing the i+3 residue from Phe in 9.10c to Gly in 9.10d led to an increase in er at the expense of some yield (entry 7). However, peptide 9.10e, which contained Acpc and Phg (phenylglycine) residues at the i+2 and i+3 positions, respectively, deracemized 9.7a in 97% yield and 78:22 er (entry 8). Interestingly, selectivity was significantly reduced with epimeric, d-Phg-containing 9.10f (entry 9). The selectivity of 9.10e was slightly better when molecular sieves were included (entry 10).
Using both catalysts 9.9 and 9.10e under the optimized conditions, urea 9.7a was returned in 96% yield and 93:7 er (Figure 171, entry 11), proving that two modestly selective catalysts can, together, perform a highly effective deracemization in the proposed mechanistic framework. However, the theoretical er based on the product of erPT (entry 2) and erHAT (entry 10) is 96:4 er, slightly higher than what was observed experimentally (entry 11). It was reasoned that the thiyl radical of peptide 9.10e could induce some degree of racemization via HAT from the α-C–H bond of (R)-9.7a. This adventitious side reactivity was suppressed by addition of 50 mol% triphenylmethane as an H-atom buffer, and the theoretical er of 96:4 er was obtained (entry 12). Using 5 mol% of 9.9 and 9.10e and 25 mol% of triphenylmethane, 9.7a was effectively deracemized in 96% isolated yield and 96:4 er (entry 13).
Together, phosphate 9.9 and peptide 9.10e were able to effectively deracemize an array of urea substrates with variable substitution on both the core and pendent amide functionality (Figure 172). Various α-amino substituents (9.7a–e) and urea N-substituents (9.7f–h) were accommodated by the dual catalyst system with excellent yields and ers. Likewise, perturbations to the pendent secondary amide led to only minor changes in yield and er (9.7i–n). However, tertiary amide 9.7o was significantly less selective. Investigating each of the enantioselective steps separately, peptide 9.10e was found to process 9.7o with 77:23 er, while phosphate 9.9 returned only 58:42 er. This indicates that the secondary amide moiety is crucial in the enantioselective PT step. Assessed separately, the enantioselective HAT step was relatively invariant across all substrates, suggesting that 9.10e effectively associates with the urea core and likely induces asymmetry through stabilizing noncovalent interactions along the HAT reaction coordinate. Based on previous data, it is possible that the i+2 amide N–H of 9.10e forms an H-bond with the urea carbonyl of 9.7 in the selective transition state.
Figure 172.
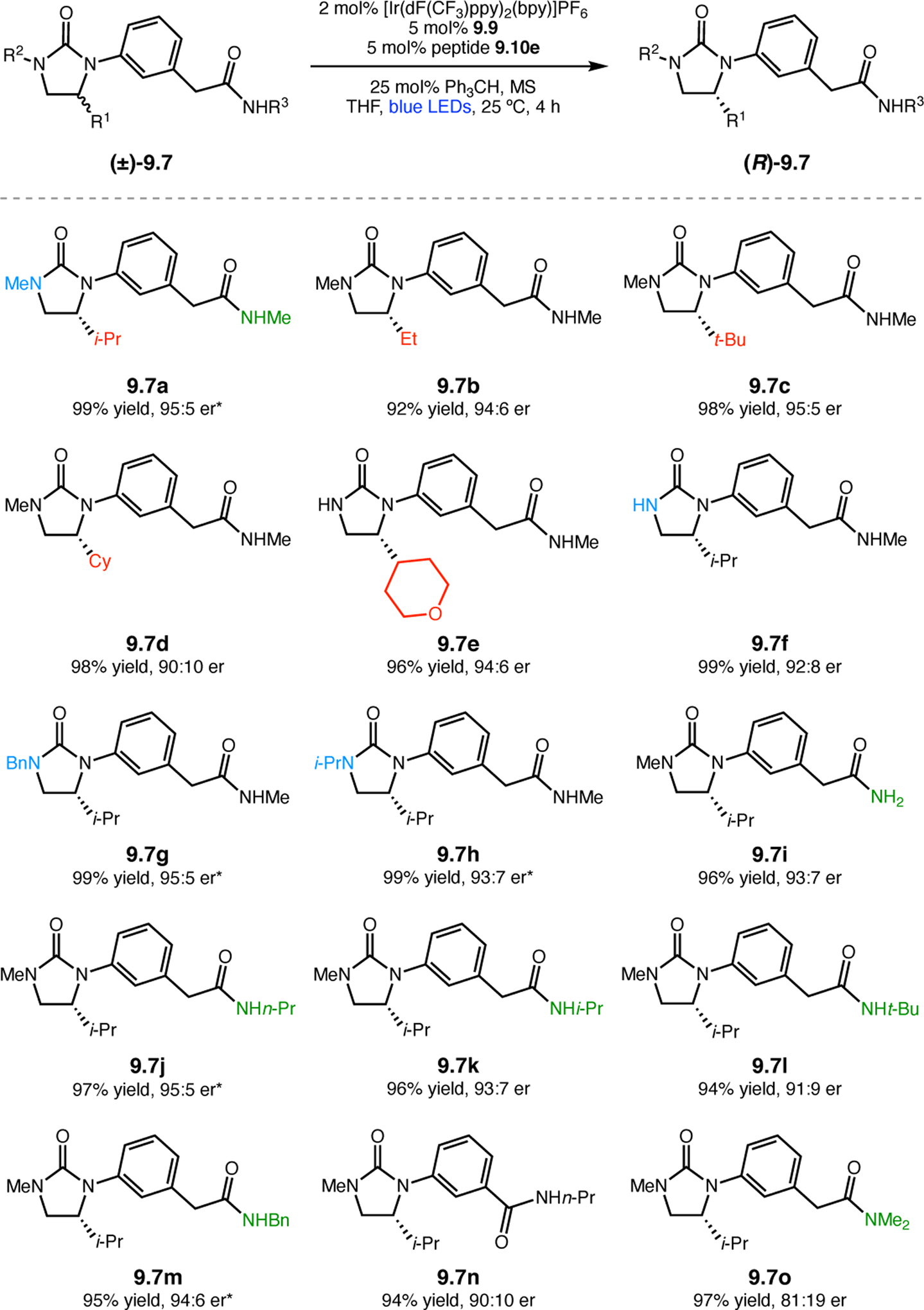
Scope of the light-driven deracemization of urea 9.7. Entries marked with an asterisk (*) indicate a 12 h reaction time.400
In addition to providing the proof-of-principle for this light-driven, dually enantioselective deracemization process, this study provided a proving ground for Cys-containing peptides as selective HAT catalysts. Examples of enantioselective HAT reactions are quite rare,383,403 possibly owing to the inherent difficulty of controlling the trajectory of H-atom delivery to a reactive intermediate. Peptide-based catalysts have proven to be a viable solution and may be particularly useful as this area is likely to expand.
9.3. Photoredox [2+2] Cycloadditions
In 2014, Yoon and co-workers reported a photochemical [2+2] cycloaddition of enones 9.11 and 9.12 to decorated cyclobutanes 9.13 employing a dual photoredox/Lewis acid catalytic strategy (Figure 173).404 Previous works by the Yoon group showed that Lewis acid coordination to enones can enable single-electron reduction events that are otherwise unfavorable.405,406 In this study, the authors sought to investigate the possibility of enantioselective, intermolecular [2+2] photocycloadditions by including a chiral ligand for the Lewis acid catalyst. The proposed catalytic cycle begins with photoexcitation of a Ru(II)-based photocatalyst to its triplet excited state.401 Oxidative quenching of the excited-state Ru(II) species by a tertiary amine generates a reduced Ru(I) species and the amine radical cation. Alone, the Ru(I) species (E° = −1.3 V vs. SCE) is insufficient to reduce enone 9.11 (E° = −1.4 V vs. SCE) to the corresponding radical anion. However, Lewis acid coordination to 9.11 makes this SET step feasible, resulting in radical intermediate 9.14, which subsequently undergoes a formal [2+2] cycloaddition with 9.12 and one-electron oxidation to form cyclobutanes 9.13.
Figure 173.
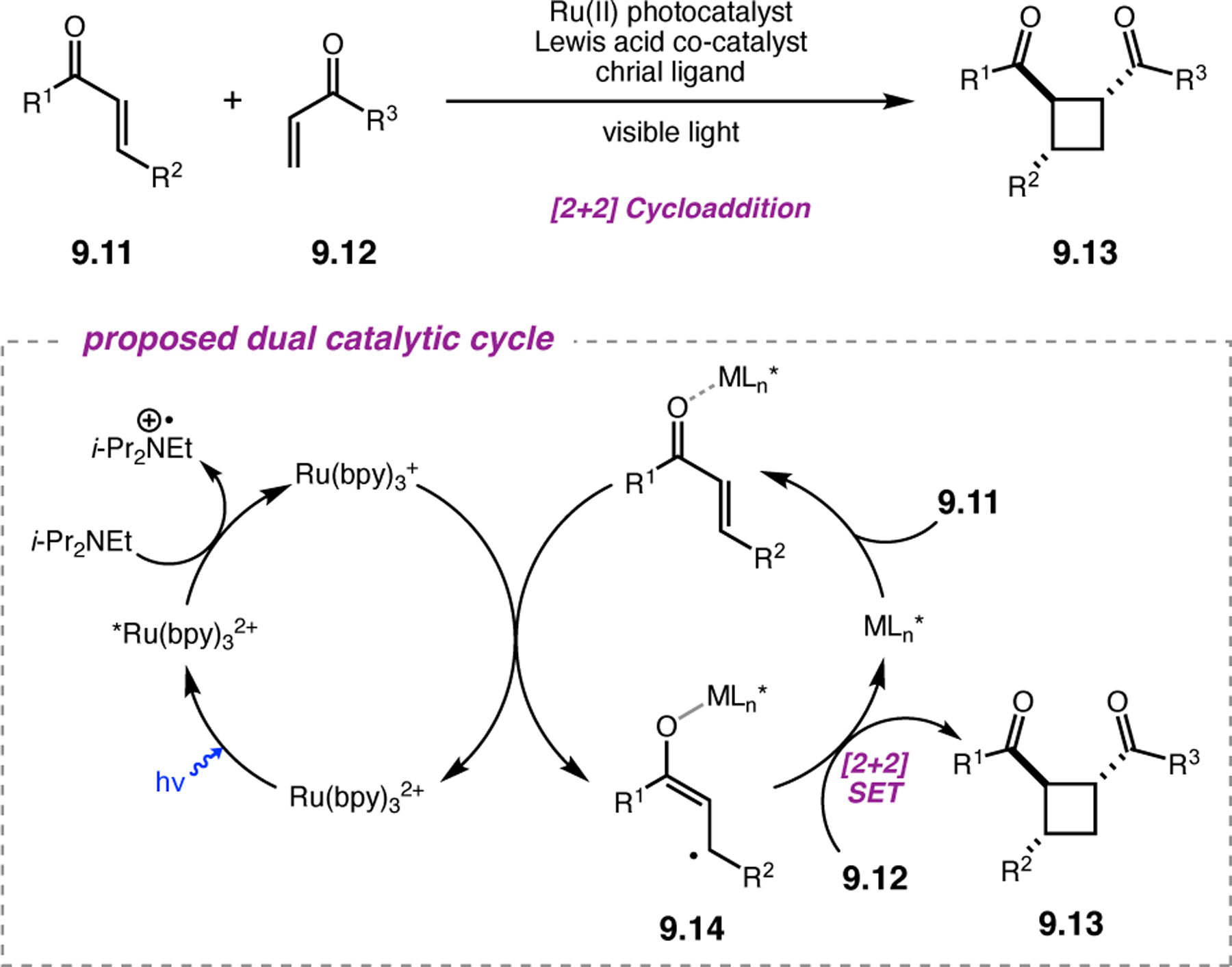
Dual catalytic strategy for the photochemical [2+2] cycloaddition of enones 9.11 and 9.12 to deliver substituted cyclobutanes 9.13 with high levels of diastereo- and enantioselectivity.404
Their studies began with an evaluation of chiral ligands that have proven highly selective in a host of different reactions through the years (Figure 174).388 A preliminary evaluation of different Lewis acids revealed that lanthanide triflates were the most effective in the photocycloaddition of 9.11a and methyl vinyl ketone 9.12a using 5 mol% of Ru(bpy)3Cl2 as the photocatalyst. In the absence of a chiral ligand, Gd(OTf)3 provided cyclobutane 9.13a in 32% yield as a 3:1 mixture of 1,2-trans- and cis-diastereomers (entry 1). BINOL 9.15 (entry 2), TADDOL 9.16 (entry 3), and PyBOX 9.17 (entry 4) were ineffective ligands for this transformation, providing yields and drs that were no better than without ligand. However, dipeptide 9.18 delivered the [2+2]-cycloadduct in 56% yield and 6:1 dr favoring trans-9.13a, which was enriched to 21% ee (entry 5). These Schiff base-functionalized dipeptides were reported in the 2000s by Hoveyda and co-workers, who found them to be highly selective in a variety of enantioselective organometallic reactions (see Section 5.2).209,407
Figure 174.
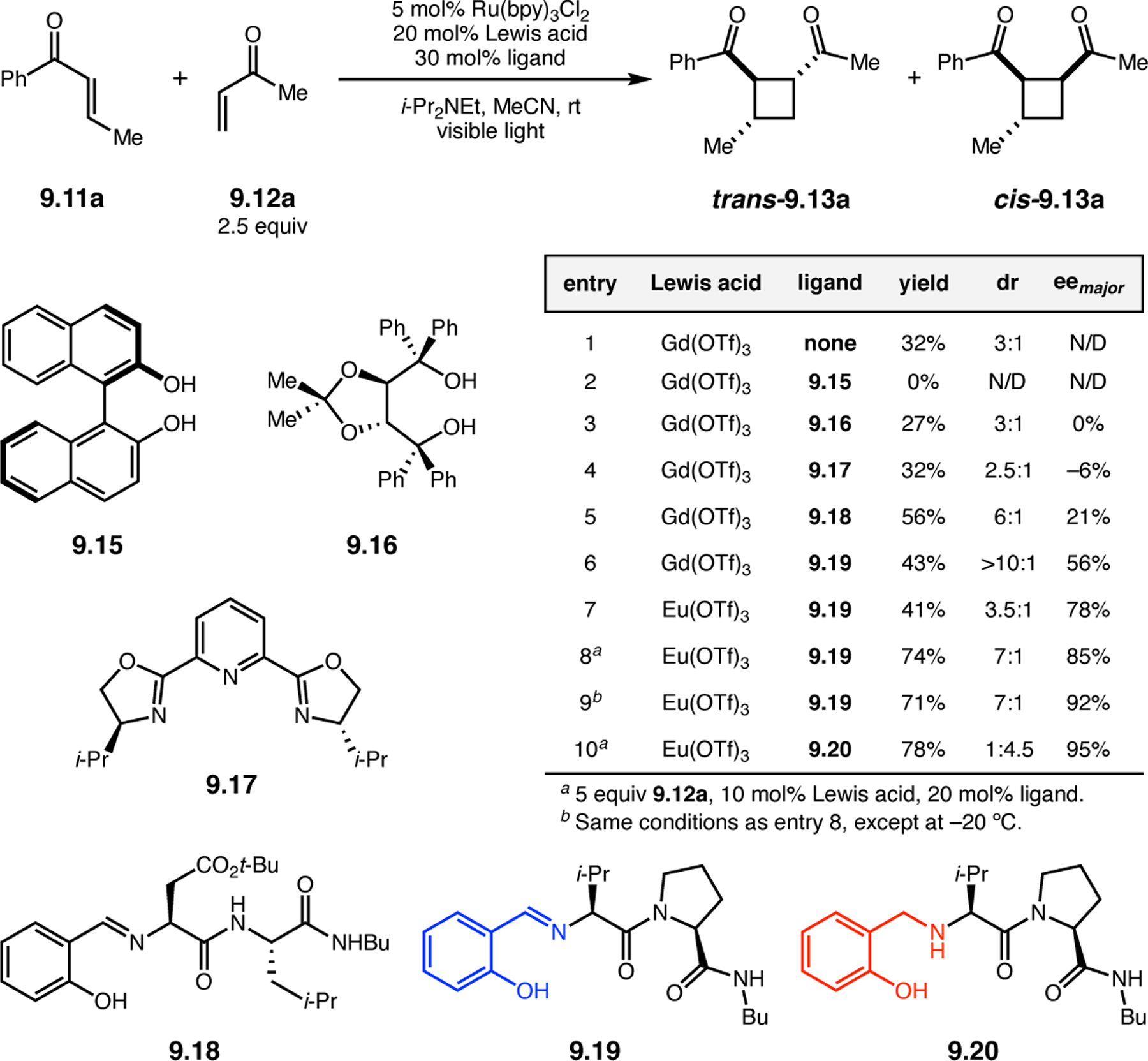
Optimization of the Lewis acid and chiral ligand led to the identification of peptides 9.18 and 9.19, which provided diastereodivergent results in the [2+2] cycloaddition of 9.11a with 9.12a.404
This result spurred a combinatorial screen of dipeptide sequences, which led to Pro-containing 9.19 that gave 43% yield of trans-9.13a (>10:1 dr) in 56% ee (entry 6). Substituting Eu(OTf)3 as the Lewis acid provided a noteworthy boost in ee to 78% at the expense of dr (entry 7). Increasing the equivalents of 9.12a and reducing the loadings of Eu(OTf)3 and 9.19 led to a significant improvements in yield, dr, and ee (entry 8), and −20 °C provided the highest observed ee of 92% (entry 9). Control experiments showed that the reaction completely shuts down in the absence of light, photocatalyst, Lewis acid, and DIPEA base. Moreover, enantioselectivity is independent of Eu(OTf)3/9.19 loading, suggesting the absence of a racemic background reaction.408
Interestingly, diastereodivergence was observed using peptide 9.20, in which the imine functionality is reduced to an amine. Even though this modification occurs to an achiral portion of the catalyst scaffold, the selectivity switches to favor cis-9.13a (1:4.5) in 95% ee, even at ambient temperature (entry 10). This result possibly points to a change in the ligand conformation that overrides the intrinsic trans-selectivity of the transformation.
The substrate scope of the dual photoredox- and Lewis acid-catalyzed [2+2] cycloaddition was evaluated with both ligands 9.19 and 9.20 (Figure 175). The scope was found to be general for both trans-selective 9.19 and cis-selective 9.20. Various aryl ketones were well-tolerated by both peptidic ligands (entries 1–8), as were substitutions at the enone β-position (entries 1 & 9–11). Overall, yields and ee values tended to be modestly improved with peptide 9.20, whereas dr values were generally higher with imine-containing 9.19.
Figure 175.
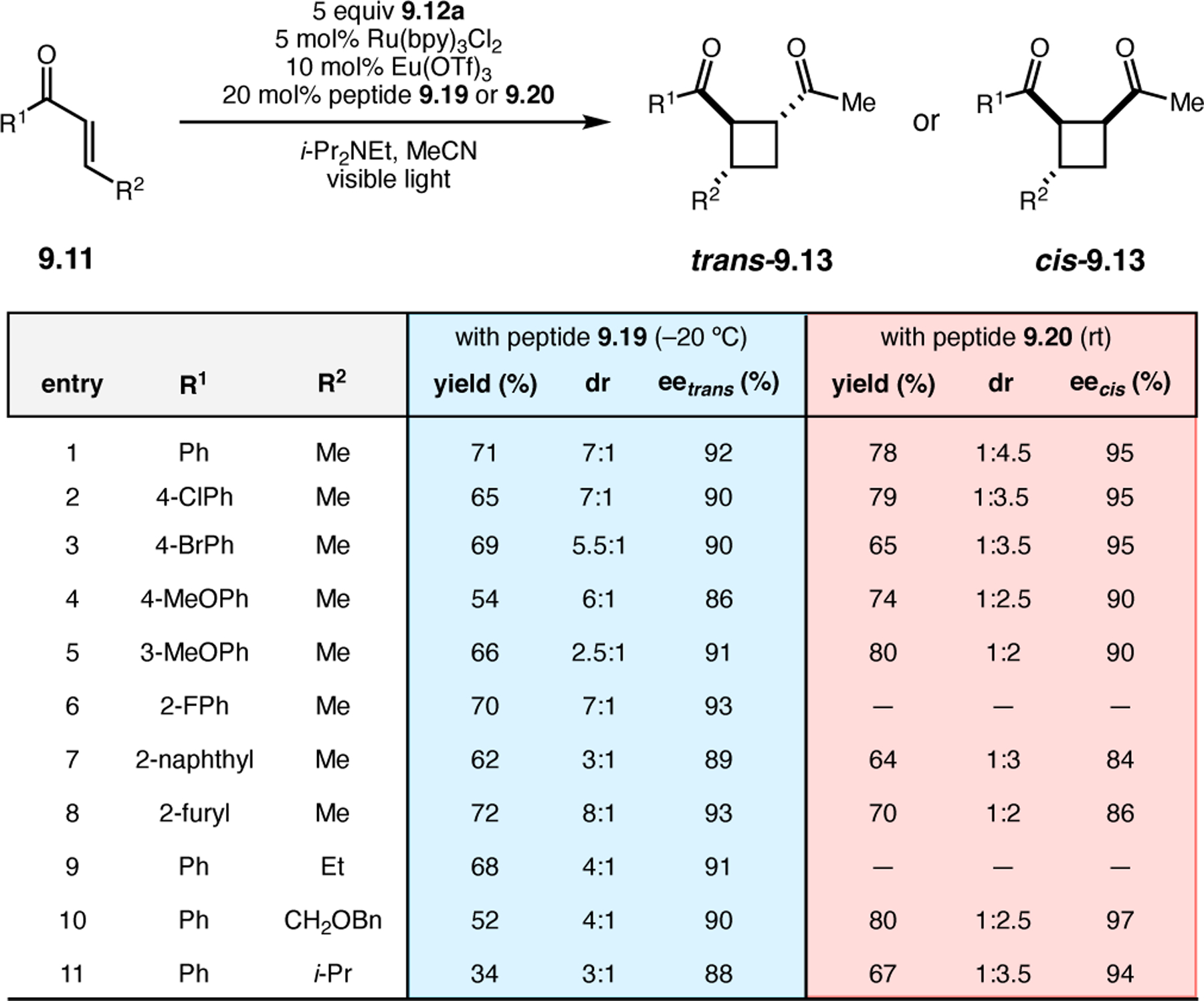
Scope of the dual catalytic [2+2] photocycloaddition of enones 9.11 and 9.12a using both peptides 9.19 and 9.20.404
10. Halogenation
A wide variety of asymmetric halogenation methodologies have been developed over the past three decades, ranging from α-halogenations of carbonyl compounds409,410 and olefin halofunctionalizations411,412 to dihalogenation reactions of allylic alcohols.413 In recent years, the application of peptide-based catalysts in this area has led to a significant expansion of scope and mechanistic understanding. Not only have peptides been employed as catalysts for venerable halolactonization reactions (Section 10.3), but they have also enabled novel asymmetric SEAr chemistries that presented interesting selectivity challenges. Of particular note is the ability of these peptidic catalysts to achieve remote asymmetric induction in compounds where the pro-stereogenic element is distal from the reaction site (Sections 10.1 & 10.2).245 In this Section, we summarize the recent peptide-catalyzed, asymmetric bromination methods with a particular emphasis on the specific attributes of peptide-based catalysts that make them well-suited to address intriguing stereochemical problems.
10.1. Atroposelective Electrophilic Aromatic Bromination
Previous examples of asymmetric SEAr methods largely follow a mechanistic paradigm wherein an arene nucleophile adds into a prochiral electrophile, with enantiofacial discrimination governed by the catalyst–electrophile complex.414–416 However, the rapidly growing interest in enantiopure atropisomers—compounds that exhibit axial chirality by virtue of hindered rotation about a single bond—for natural products,417 pharmaceuticals,418 and catalysis,81,419 provided an impetus for the development of direct atroposelective SEAr reactions.
In that regard, our group envisaged a scenario in which one enantiomer of a chiral, rapidly racemizing biaryl phenol (e.g., 10.1) could undergo selective electrophilic aromatic bromination via preferential interaction with a peptide-based catalyst (Figure 176). This would enable the DKR of 10.1 if the following criteria are met: (1) racemization of 10.1 is fast relative to SEAr; (2) the product is configurationally stable under the reaction conditions; and (3) the peptide-based catalyst is able to engage in remote asymmetric induction, as the phenol group is distal from the axis of chirality. Mono-ortho-substituted biaryls, such as 10.1, are known to have low enantiomerization barriers (ΔΔG‡ ~ 7 kcal/mol), which all but ensured rapid racemization under the reaction conditions. However, we anticipated that two ortho-bromination events would be necessary to lock the configuration of the product. In order to meet this requirement and also avoid complicated mixtures of poly-brominated product isomers, we decided to pursue the tribromination of 10.1. The biaryl tribromide products (e.g., 10.2) were expected to have enantiomerization barriers exceeding 30 kcal/mol, which is sufficiently high to avoid in situ racemization. Lastly, we hypothesized that incorporation of a suitable directing group on the opposite ring (X, Figure 176) would facilitate multidentate interactions with the peptide catalyst and provide a more robust handle for enantio-discrimination in the stereodetermining transition state.
Figure 176.
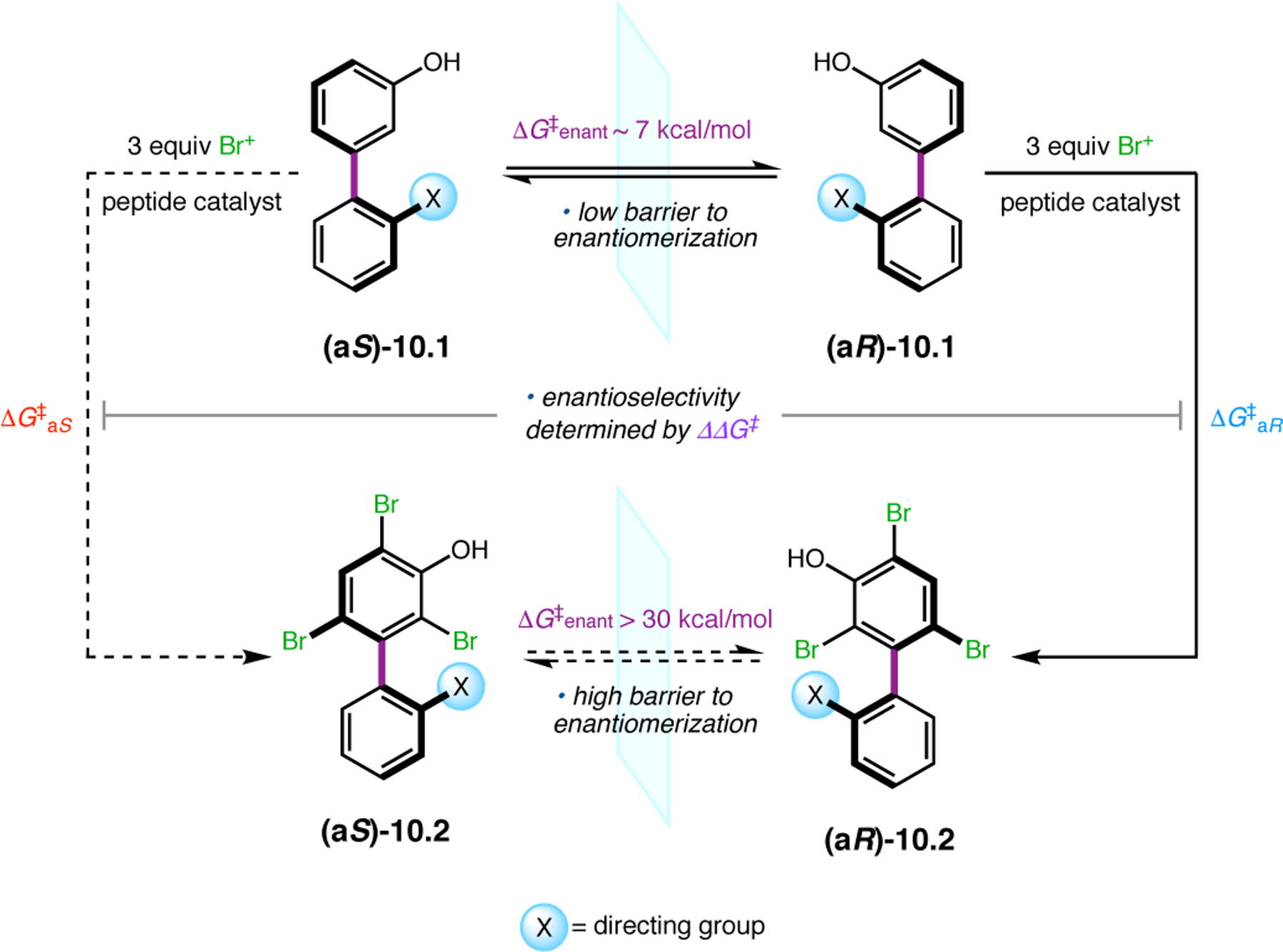
DKR of biaryl compounds via atroposelective electrophilic aromatic bromination.
Using this general DKR strategy, our group has been able to develop highly atroposelective, peptide-catalyzed bromination reactions of biaryl (Section 10.1.1), non-biaryl (Section 10.1.2 & 10.1.3), and heterobiaryl (Section 10.1.4) atropisomers. Unique peptide catalysts were optimized for each substrate class, and multifunctional binding and activation mechanisms were proposed to be crucial for achieving high levels of enantioselectivity in each case. Moreover, the tribrominated products are able to be regioselectively functionalized using robust cross-coupling protocols to access high value derivatives that often have drug-like properties.
Prior to this body of work, most reported examples of catalytic, atroposelective methods took advantage of transition metal-mediated cross-coupling reactions to forge biaryl C–C bonds with stereocontrol over the nascent axis.420 However, a conceptually similar DKR strategy had been developed by Bringmann and co-workers, which involved the atroposelective ring opening of biaryl lactones with various chiral nucleophiles.421 These strained lactones racemize rapidly due to ground state destabilization, but the ring-opened products are configurationally stable ortho-tetrasubstituted biaryls, which are especially difficult to access using asymmetric cross-coupling methods. Since then, catalytic versions of this reaction have recently been reported.422 A single catalytic, atroposelective DKR example had been reported by Murai and co-workers prior to our initial report.423 This method employed a chiral Rh(I)-complex to effect a C–H/olefin coupling via a modestly atroposelective C–H activation event. These pioneering studies informed many aspects of our atroposelective DKR platform.
10.1.1. Atroposelective Bromination of Biaryls.
In 2010, our group successfully implemented the atroposelective DKR strategy in the peptide-catalyzed bromination of biaryls 10.1 (Figure 177).424 Our catalyst design was centered around an N-terminal, tertiary amine-containing catalytic residue, β-dimethylaminoalanine (Dmaa), which was intended to nucleate complexation with 10.1 through H-bonding/ion pairing interactions. Indeed, high levels of selectivity were only observed when the directing group on scaffold 10.1 was a carboxylic acid; pendent ester, amide, and nitro groups resulted in poor to modest enantioinduction. Tripeptide 10.3 emerged from sequence optimization studies as the lead catalyst. Under the optimized reaction conditions employing N-bromophthalimide (NBP) as the brominating reagent and acetone as a polar additive, 10.3 was able to address a reasonable scope of biaryls 10.1 with variable substitution on the benzoic acid moiety. Tribromides 10.2 were isolated in up to 85% yield and 97:3 er (entries 1–9). Even a pyrrole-containing substrate was selectively brominated by 10.3 in 77% yield and 85:15 er when four equivalents of NBP were used (entry 10). The absolute configuration of the product was assigned as (aR) for tribromide 10.2a (R = H) using single crystal X-ray crystallography.
Figure 177.
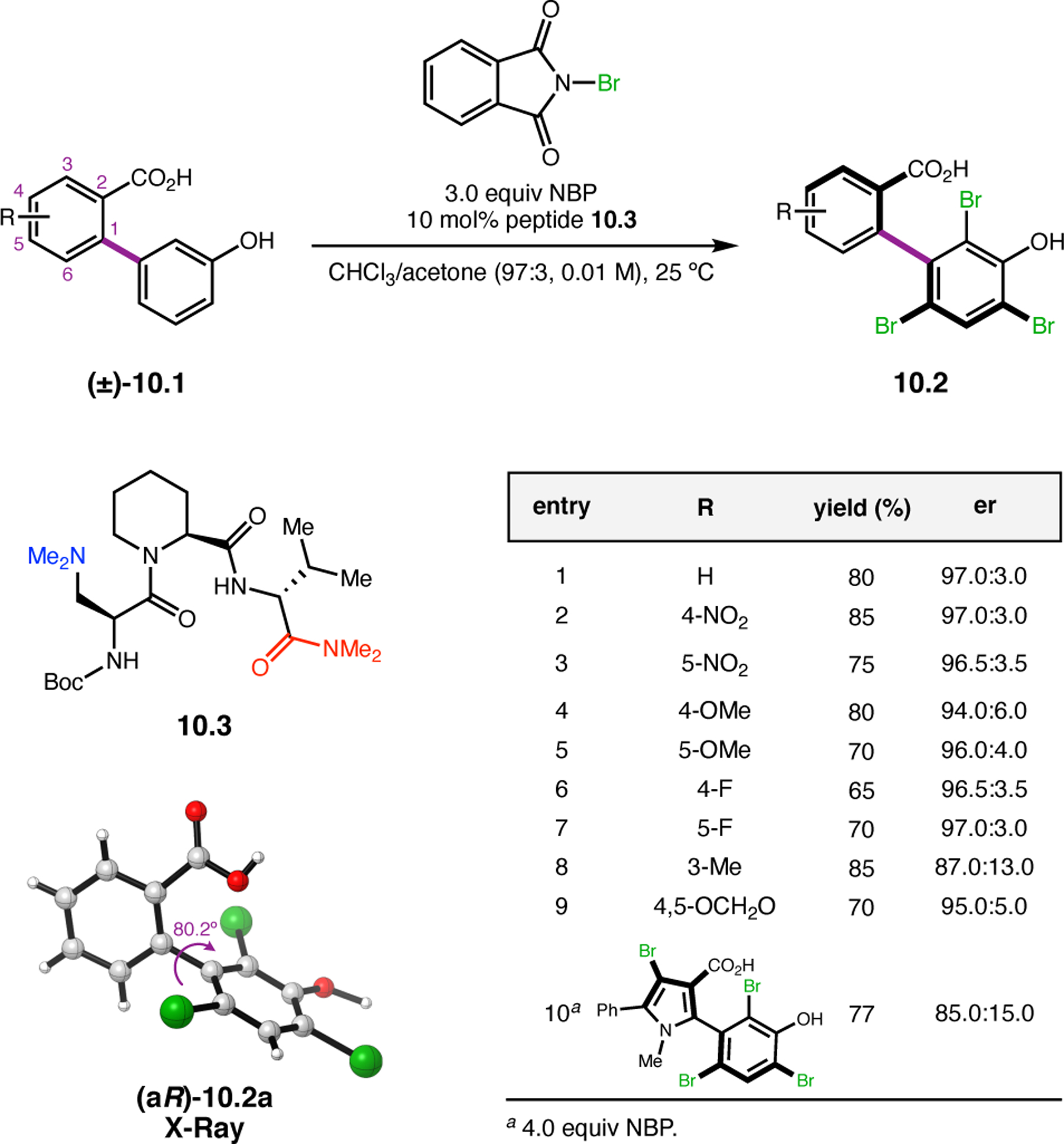
Tripeptide 10.3-catalyzed atroposelective bromination of biaryls 10.1.424
A series of control reactions were performed in order to glean which elements of tripeptide 10.3 were essential for catalysis (Figure 178). In the absence of a catalyst, background bromination of 10.1a was slower and non-selective; only 15% of 10.2a was isolated after 18 hours, with the remainder of the mass balance consisting of mono- and dibrominated isomers. The yield of 10.2a improved to 31% when 10 mol% of DIPEA was employed as a mimic of the Brønsted basic Dmaa residue. However, a boost to 91% yield was observed using catalytic (±)-Boc-Val-NMe2 to simulate the C-terminus of 10.3. Taken together, these data suggest that the C-terminal d-Val residue of 10.3 contributes more substantially to bromination catalysis, perhaps through bromenium ion delivery by the tertiary amide moiety. Lewis basic carbonyl-type functionality are known to accelerate halogenation reactions in other contexts.425
Figure 178.
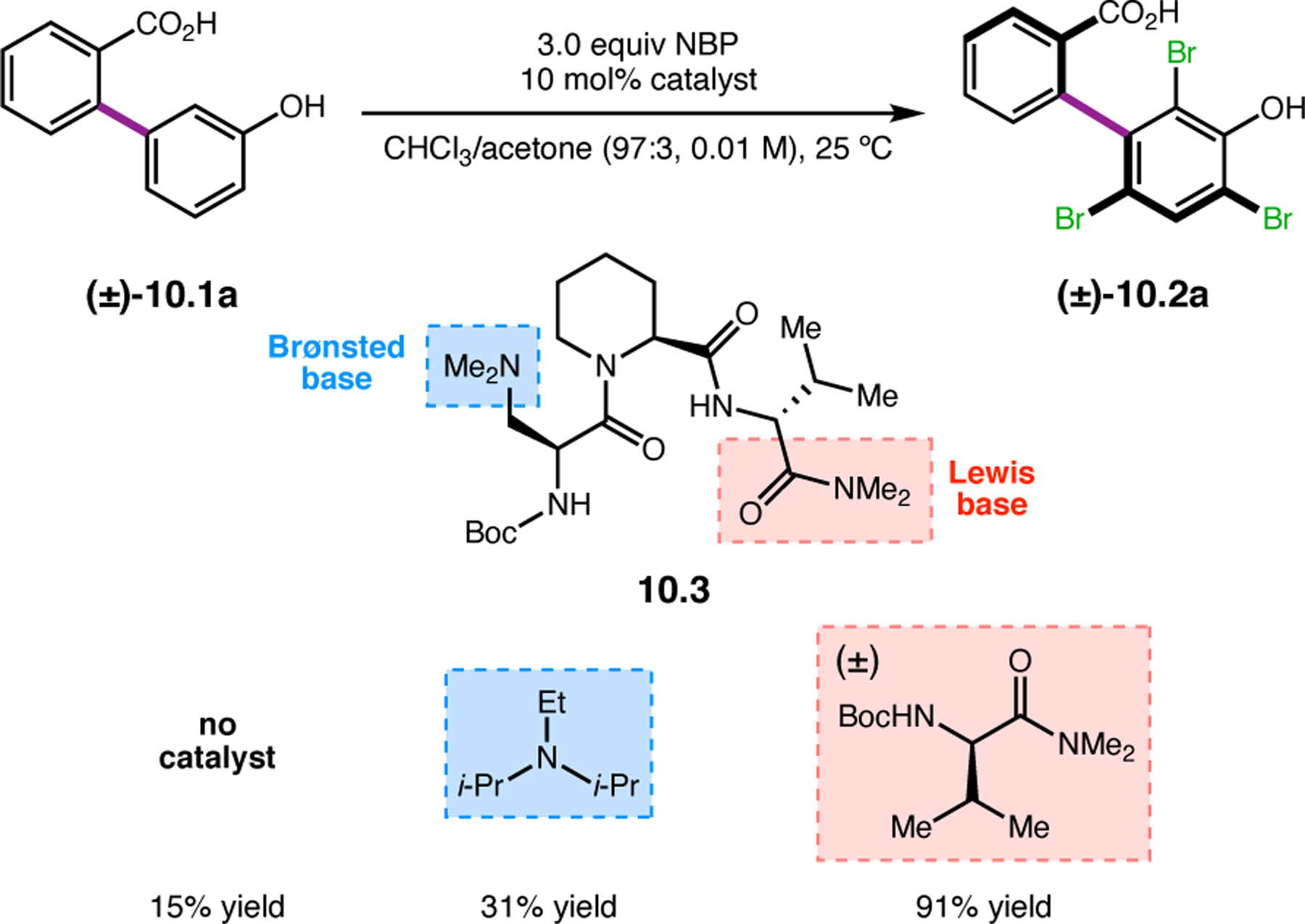
Racemic control reactions to investigate the elements of peptide 10.3 that are crucial for catalysis.424
Based on all of the data, we proposed a self-consistent binding and activation model was proposed that accounts for the observed absolute configuration of product 10.2 (Figure 179). Proton transfer from 10.1a to 10.3 allows for ion pairing/H-bonding between the carboxylate of 10.1a and the Dmaa residue of 10.3. This salt bridge formation may nucleate additional multidentate H-bonding interactions between the backbone amides of 10.3 and the phenol moiety 10.1a. This possibly leaves the C-terminal tertiary amide of 10.3 available for bromenium ion transfer, though no direct evidence for such an interaction has been obtained to date. In collaboration with the Johnson Group, we used cryogenic infrared spectroscopy426 to study the nature of the ground state 10.3•10.1a complex.427 The results of this study were quite consistent with the proposed model.
Figure 179.
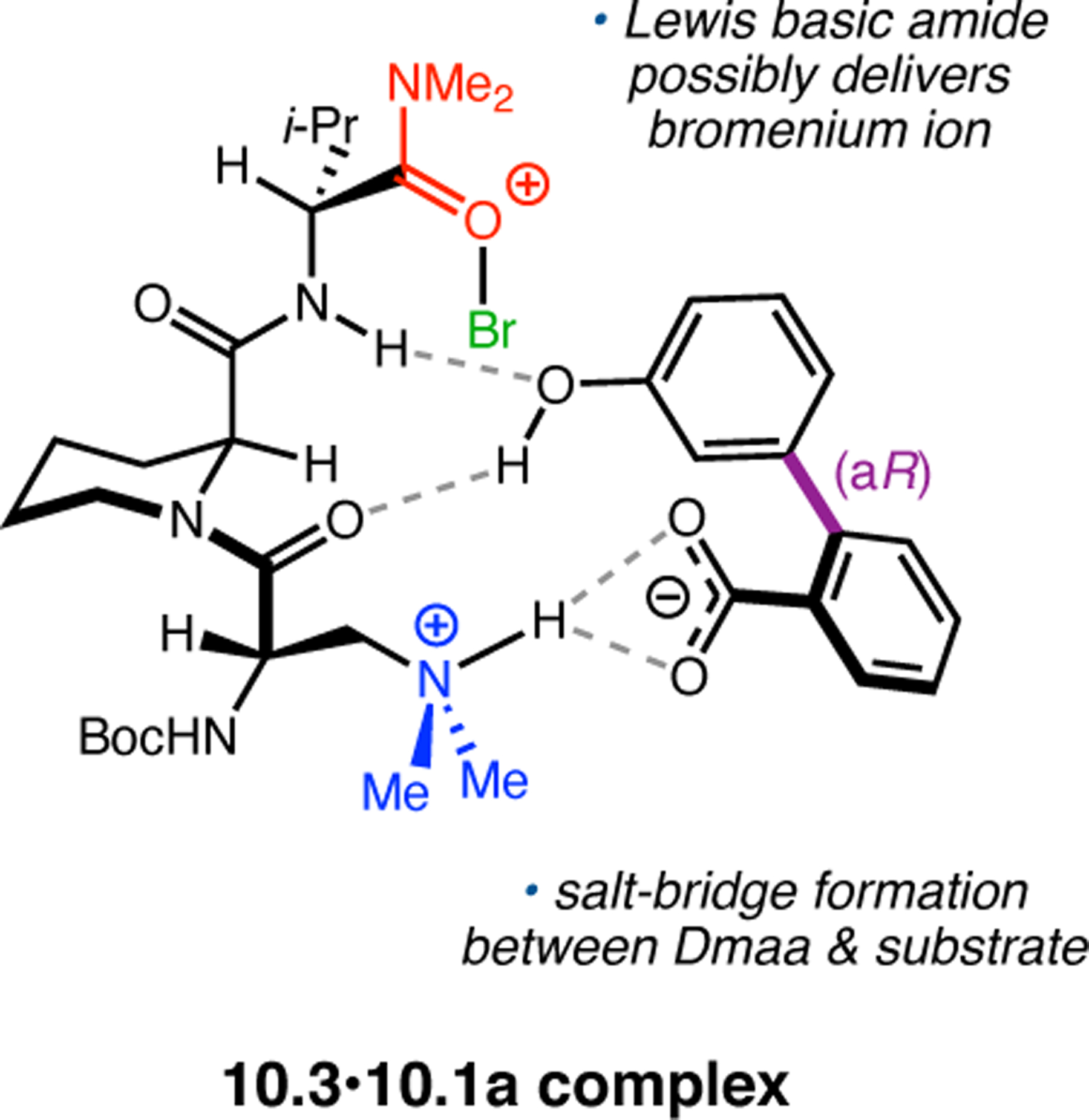
Proposed binding and activation model for the 10.3-catalyzed bromination of 10.1a.424,427
To demonstrate the synthetic utility of these tribrominated products, we showed that protected tribromides 10.4 can undergo sequential cross-coupling reactions with no erosion of configurational integrity (Figure 180).428 This “A-B-C” coupling strategy capitalizes on the steric and electronic differentiation of each individual bromide to enable regioselective functionalization. The A-coupling selectively targets the bromide para to the chiral axis to provide C–C-, C–N-, C–O-, and C–H-coupled products 10.5 in 45–70% yield. Next, the B-coupling allows for the functionalization of the bromide para to the methoxy group, delivering derivatives 10.6 in 37–65% yield. The final C–Coupling functionalizes the most sterically encumbered bromide to provide tris-functionalized products 10.7 in 70–80% yield under slightly more forcing conditions. Differentiated pentaaryls are accessible in virtually enantiopure form (99:1 er) using this strategy.
Figure 180.
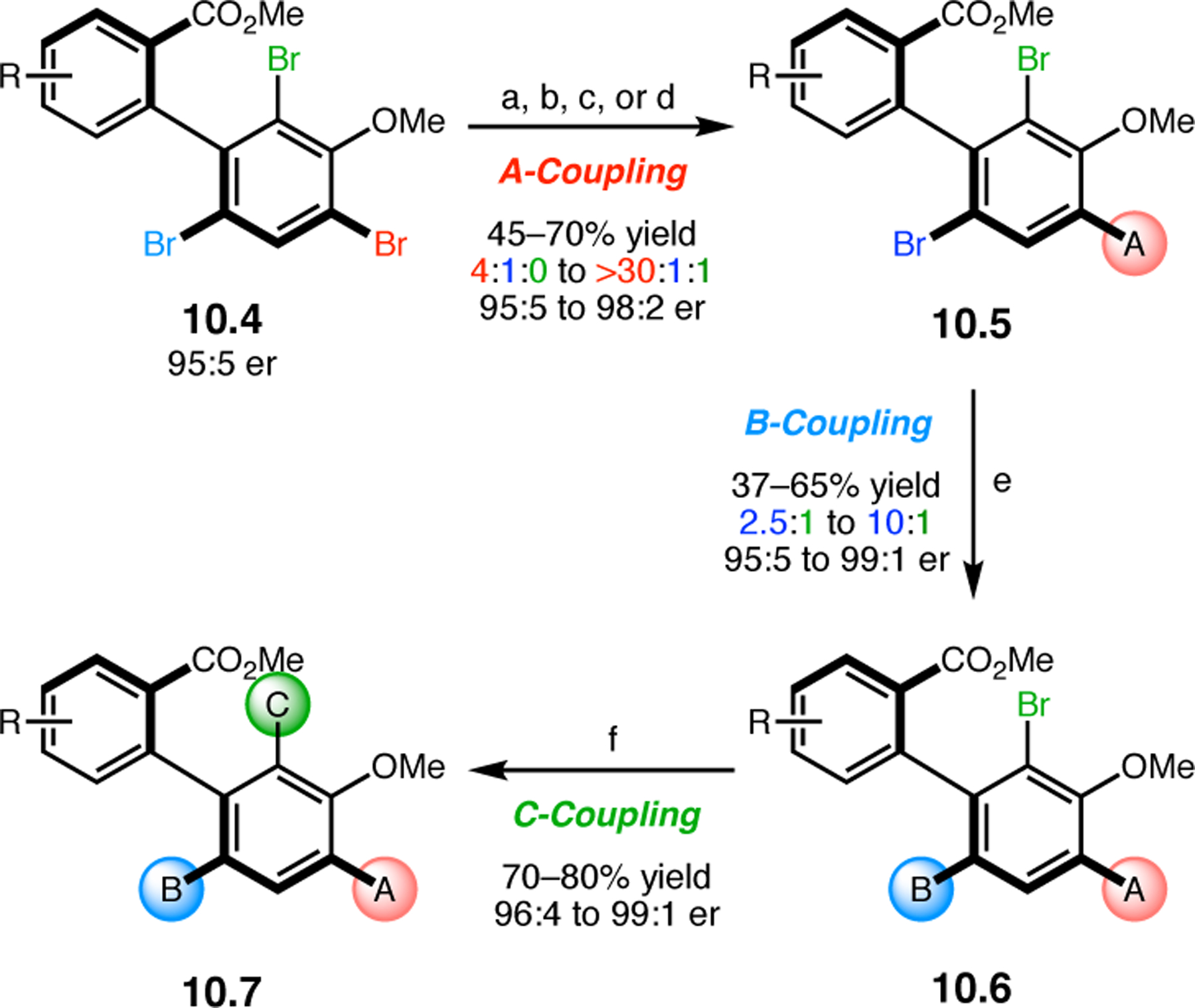
Sequential “A-B-C” cross-coupling of 10.4 enables the synthesis of highly functionalized derivatives 10.7. Conditions: (a) 1 equiv aryl MIDA-boronate, 10 mol% Pd(PPh3)4, 8 equiv K3PO4, THF/H2O (5:1, 0.1 M), 65 °C, 14 h; (b) 1 equiv benzylamine, 10 mol% Pd(OAc)2, 20 mol% rac-BINAP, PhMe (0.1 M), 100 °C, 18 h; (c) 1.2 equiv phenol, 10 mol% Pd(OAc)2, 13 mol% t-Bu2XPhos, 2 equiv K3PO4,PhMe (0.3 M), 100 °C, 14 h; (d) 1 equiv NaBH4, 5 mol% Pd(OAc)2, 5.5 mol% rac-BINAP, 1.5 equiv TMEDA, THF (0.25 M), 50 °C, 14 h; (e) 1 equiv aryl MIDA-boronate, 10 mol% Pd2(dba)3, 10 mol% (S)-BINAP, 8 equiv K3PO4, THF/H2O (4:1, 0.1 M), 65 °C, 14 h; (f) 1 equiv aryl boronic acid, 5 mol% Pd(PPh3)4, THF/2 M aq. Na2CO3 (2:1, 0.1 M), 100 °C, μw irradiation, 1 h.428
10.1.2. Atroposelective Bromination of Tertiary Benzamides.
Our group expanded the general atroposelective DKR strategy to include non-biaryl atropisomers with our 2013 report of the peptide-catalyzed, asymmetric bromination of tertiary benzamides 10.8 (Figure 181).429 Axially chiral benzamide motifs have appeared frequently in pharmaceutical compounds exhibiting a range of biochemical functions.418,430,431 In-depth studies by Mannschreck,432 Clayden,433 and others on the atropisomeric properties of benzamides like 10.8 and 10.9 provided crucial insight into the challenges associated with the atroposelective bromination of these compounds. Namely, the amide N-substituents would have to be sufficiently large so as to ensure the configurational stability of the tribromide products 10.9. We hypothesized that this would also provide the added benefit of minimizing background tertiary amide-catalyzed bromination. Preliminary results showed that the N,N-diisopropyl benzamide motif indeed provided improved yields and selectivities over the corresponding piperidinyl and piperazinyl benzamides in the peptide-catalyzed bromination reaction.
Figure 181.
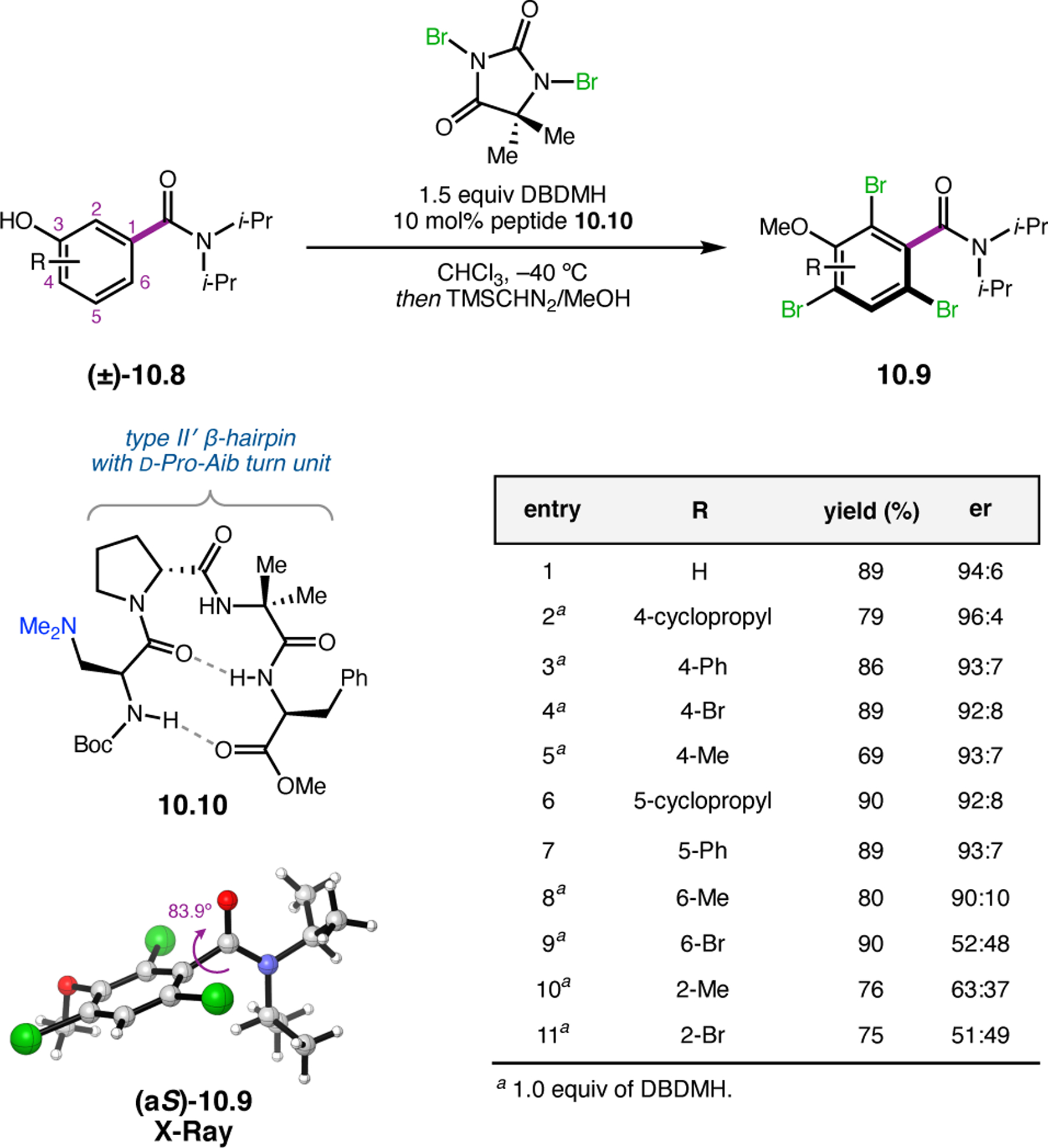
Peptide 10.10-catalyzed, atroposelective bromination of tertiary benzamides 10.8.429
Sequence optimization led to the discovery of Dmaa-containing tetrapeptide 10.10 as the lead catalyst for this transformation. Using 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) as the brominating reagent, 10.10 delivered tribromide 10.9a (R = H) in 89% yield and 94:6 er favoring the (aS)-configuration (Figure 181, entry 1). The secondary structure of 10.10 was biased toward a type IIʹ β-hairpin by incorporation of the central d-Pro-Aib sequence, a feature that proved to be important for selectivity in the bromination of 10.8. Interestingly, this same sequence (i.e., Boc-Xaa-d-Pro-Aib-Phe-OMe) emerged as highly effective in a variety of mechanistically distinct reactions.4,6
Peptide 10.10 selectively brominated a variety of substituted benzamides 10.8 under the optimized conditions. Variable substitution at the 4- and 5-positions was well-tolerated (Figure 181, entries 2–7), while ortho-substituents presented a more complicated scenario. At the 6-position, methyl substitution was processed selectively by 10.10 (entry 8), whereas a pre-installed bromide completely ablated the selectivity (entry 9). The observed disparity is possibly due to differences in the enantiomerization barriers; the ortho-bromides are expected to decrease the rate of racemization relative to ortho-methyl groups due to their increased demand,161 which would decrease the efficacy of the DKR. On the other hand, neither methyl nor bromide substituents were tolerated at the 2-position (entries 10–11). A possible rationale for this observation was gleaned from LC/MS analyses of crude, low-conversion reaction mixtures (Figure 182). In the absence of a catalyst and when 10 mol% of triethylamine was employed, monobromination of 10.8a at both the 4- and 6-positions was observed at low conversion, leading to monobromides 10.11a and 10.12a, respectively, in a ratio favoring 10.12a. However, only 10.13a was observed in the presence of 10.10, suggesting that the peptide selectively brominates 10.8a at the 2-position. These data suggest that 10.10 overturns the inherent regioselectivity of the reaction by promoting monobromination at the most sterically encumbered position. Moreover, if the first bromination is stereodetermining, then 2-substituted benzamides are expected to provide poor selectivities in the 10.10-catalyzed reaction, which is consistent with the substrate data (Figure 181, entries 10–11).
Figure 182.
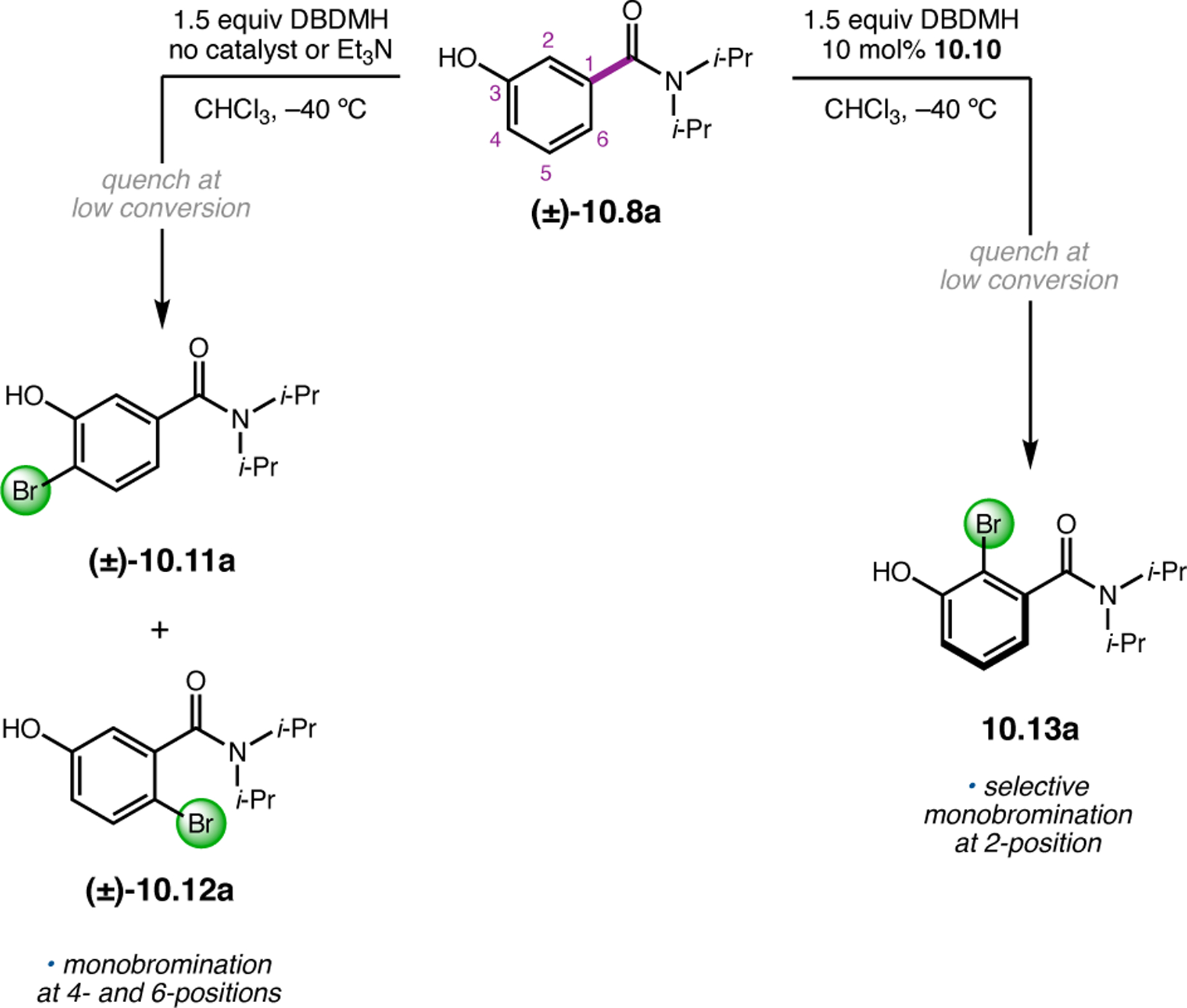
LC/MS studies of reaction mixtures at low conversion under various catalytic conditions.429
Based on NMR studies of the ground state catalyst-substrate complex, we proposed a model for binding and enantioinduction that involves multi-point docking between 10.10 and 10.8a (Figure 183). H-Bonding between the tertiary amine of 10.10 and the phenol of 10.8a is presumed to nucleate catalyst–substrate association and activate the phenol ring toward SEAr by making it more phenolate-like. An additional H-bonding interaction between the substrate and the N–H of the i+2 Aib residue holds 10.8a in the (aS)-configuration with the 2-position oriented toward the peptide scaffold. If one of the Lewis basic amides of 10.10 indeed delivers bromenium to 10.8a, then this model is also consistent with the predicted site of stereodetermining monobromination.
Figure 183.
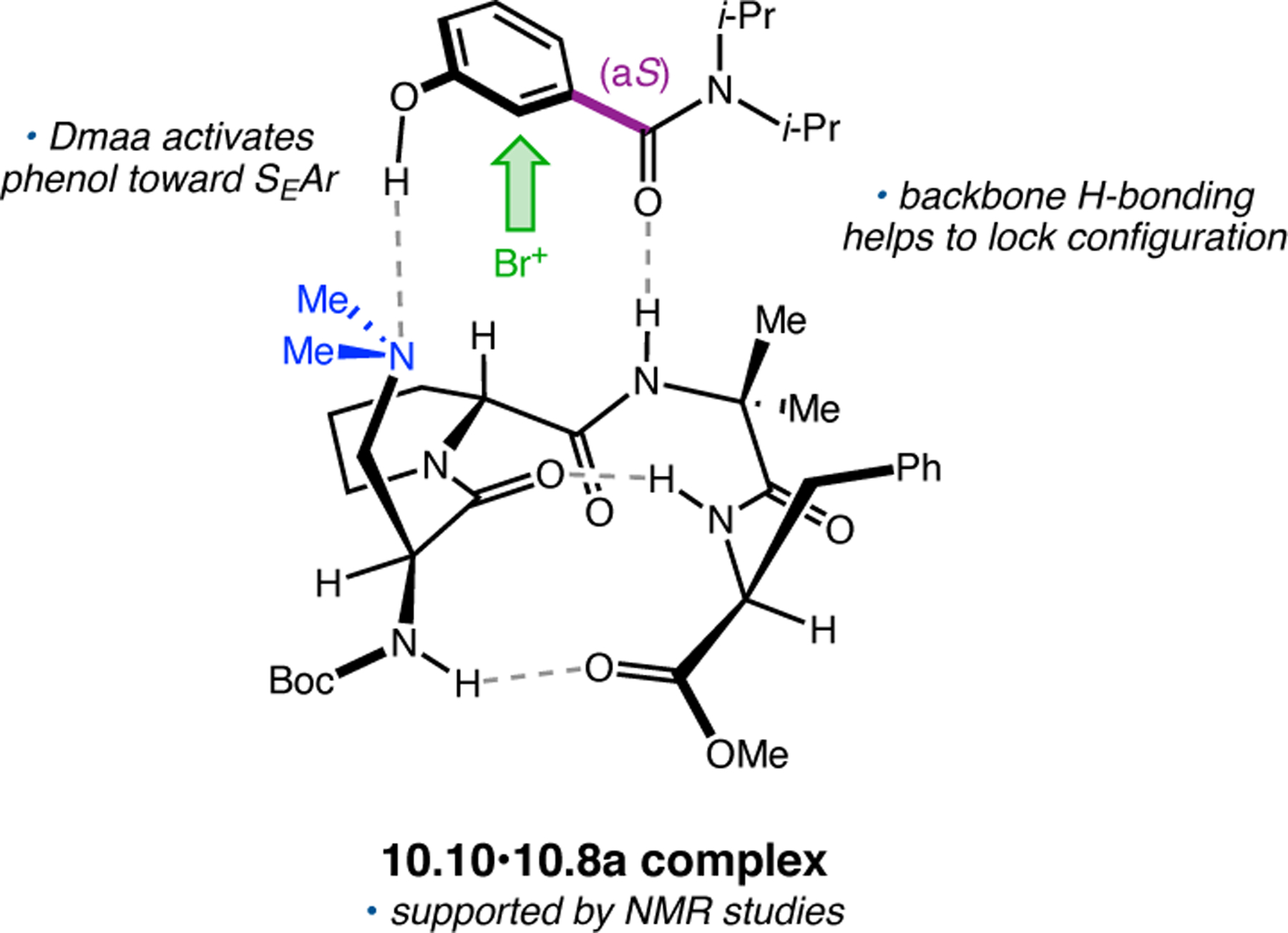
Binding and activation of benzamide 10.8a by peptide 10.10.429
Given the apparent pharmaceutical importance of this class of compounds, we next demonstrated that tribromides 10.9 could be regioselectively derivatized en route to functional, drug-like molecules (Figure 184a).434 Selective cross-coupling of the para-bromide provided monofunctionalized compounds 10.14 in 61–80% yield and with little to no erosion of er (ΔΔG‡ ~ 28 kcal/mol). ortho-Lithiation of the 2-bromide followed by electrophile trapping led to products 10.15 in good yields and up to 99:1 er following recrystallization. The utility of these derivatives was demonstrated by utilizing phosphine 10.15a as a chiral ligand for a Pd-catalyzed, enantioselective Tsuji–Trost-type allylation (Figure 184b). Remarkably, 10.16 was converted into allylated malonate 10.17 in 93% yield and 96:4 er.
Figure 184.
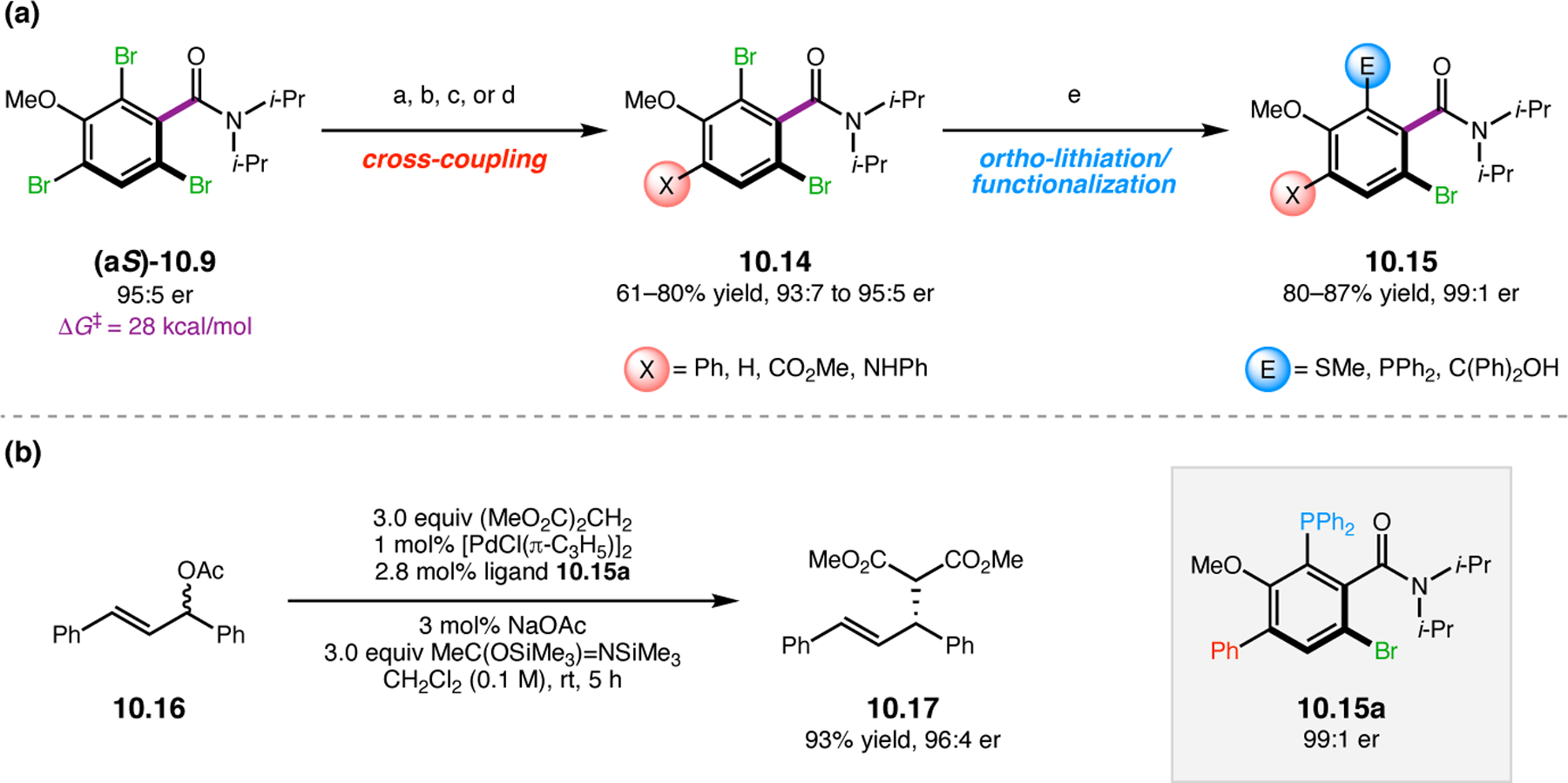
(a) Regioselective derivatization of tribromides 10.9 using a cross-coupling/ortho-lithiation sequence. Conditions: (a) 1 equiv phenyl boronic acid, 5 mol% Pd(PPh3)4, 8 equiv K3PO4, THF/H2O (5:1, 0.1 M), 50 °C, 20 h; (b) 1.05 equiv NaBH4, 5 mol% Pd(OAc)2, 5.5 mol% rac-BINAP, 1.5 equiv TMEDA, THF (0.25 M), 50 °C, 17 h; (c) 1 atm CO, 10 mol% Pd(OAc)2, 10 mol% Xant-Phos, 10 equiv Et3N, MeOH (0.05 M), 60 °C, 16 h; (d) 1.1 equiv aniline, 5 mol% Pd2(dba)3, 10 mol% Xant-Phos, 2.5 equiv NaOt-Bu, THF (0.25 M), 50 °C, 3.5 h; (e) 1.1 equiv n-BuLi, THF (0.1 M), −78 °C; then 1.2 equiv (i) (MeS)2, (ii) Ph2P–Cl, or (iii) Ph2C=O, 10 min. (b) Implementation of the derivatized product 10.15 as a chiral ligand in enantioselective Tsuji–Trost-type allylation.434
As an extension of this methodology, we next pursued the atroposelective bromination of benzamides such as 10.18 that possess two stereogenic axes—one defined by the CAr–C(O) bond (red) and the other about the N–C(O) bond of the differentially N,N-disubstituted amide (blue, Figure 185).435 Tribromination of 10.18 has the potential to deliver up to four stereoisomers of 10.19. We hypothesized that, if both axes were configurationally stable following tribromination, it may be possible to optimize a unique, peptide-based catalyst that would provide selective access to each diastereomeric product and enable the fully catalyst-controlled, stereodivergent synthesis of 10.19.
Figure 185.
Spontaneous enantioenrichment following 10.10-catalyzed bromination of two-axis substrate 10.18.435
We began by importing the optimized conditions (Figure 181) from the bromination of benzamides 10.8 into this system. In the absence of a catalyst, bromination of 10.18 initially provided a 40:60 trans/cis mixture of (±)-10.19 which slowly equilibrated to 76:24 over 72 hours at ambient temperature. DFT computations confirmed that, while the barrier to rotation about the CAr–C(O) axis is sufficiently high as to be configurationally locked following tribromination, rotation about the amide N–C(O) axis is accessible, albeit slow, at ambient temperature. Under the control of tetrapeptide 10.10, tribromide 10.19 was initially obtained with a similar trans/cis ratio of 43:57, but each amide isomer was processed with different enantioselectivity; cis-10.19 was processed by 10.10 with an er of 88:12 favoring the (aS)-isomer, while trans-10.19 was characterized by a lower er value of 66:34. However, this kinetically controlled mixture of diastereomers 10.19 changed over time as the trans/cis ratio converged on its equilibrium value of 76:24. Over the 72 hour equilibration period, the er of trans-10.19 spontaneously increased to 79:21 as the er of cis-10.19 eroded to the same value. These findings marked a rare example of spontaneous enantioenrichment, which was made possible by the ability of peptide 10.10 to differentially process the amide isomers of benzamide 10.18.
10.1.3. Atroposelective Bromination of Amido-Tropolones.
Murelli and co-workers recently reported the atroposelective bromination of amide-substituted α-hydroxytropolone derivative 10.20 (Figure 186).436 α-Hydroxytropolones are potent inhibitors of several medicinally relevant metalloenzymes,437 including HIV reverse transcriptase RNase H, a promising target for the development of HIV therapeutics.438 Like the analogous benzamides, amido-tropolones such as 10.20 are axially chiral about the CAr–C(O) bond. However, these compounds are expected to exhibit higher enantiomerization barriers owing to the wider internal bond angles of the seven-membered tropolone ring that exacerbate strain in the coplanar rotational transition state. DFT calculations show that amido-tropolone 10.20 has an enantiomerization barrier of 16.9 kcal/mol versus 13.0 kcal/mol for benzamide 10.21, which corresponds to roughly a 750-fold difference in half-life. This effect is even more pronounced in the monobromide products; tropolone 10.22 has an enantiomerization barrier of 31.2 kcal/mol versus 24.2 kcal/mol in benzamide 10.23—a 160,000-fold difference in rotational half-life. Binding simulations suggest that some α-hydroxytropolone derivatives preferentially dock with the RNase H domain of HIV RT in a single atropisomeric form.439 Therefore, the development of single-enantiomer ahydroxytropolone-based drugs could lead to enhanced potency and selectivity for an array of metalloenzyme targets.
Figure 186.
Peptide 10.22-catalyzed bromination of tropolone 10.20. Compared to the corresponding benzamides, amido-tropolones have notably higher rotational barriers.436
In order to test this hypothesis, the authors developed a peptide-catalyzed bromination of 10.20, providing access to configurationally stable monobromide 10.22.436 Following assessment of a series of Dmaa-containing peptide catalysts with sequences biased toward type IIʹ β-turns, tetrapeptide 10.24 was identified as the lead catalyst, providing 10.22 in 50% yield and 75:25 er. Enantioselectivity was proposed to originate from binding and activation in analogy to that of complex 10.10•10.8a (Figure 183), wherein the Dmaa residue engages with the free O–H of 10.20 while the peptide backbone H-bonds with the amide group. Moreover, selective monobromination of 10.20 was possible in this case due to the electronically deactivated nature of the tropolone ring, which may also contribute to the overall lower reactivity observed in this system. While preliminary, this result proves that the peptide-catalyzed DKR strategy is viable for synthesizing enantioenriched α-hydroxytropolone derivatives bearing a functional handle for additional modification and SAR study. Further optimization of this transformation is currently underway in the Murelli Group.
10.1.4. Atroposelective Bromination of 3-Arylquinazolinones.
In 2015, our group reported the atroposelective bromination of 3-arylquinazolin-4(3H)-ones (e.g., 10.25), a class of heterobiaryl compounds that exhibit axial chirality about the anilide N–CAr bond.440 The atropisomeric quinazolinone nucleus is an important pharmacophore that frequently appears in a range of biologically active molecules, from recreational hallucinogens to anti-cancer drugs.441 Many such 3-arylquinazolinone-based drugs are known to engage in enantiomer-specific binding with their biological targets, and access to single enantiomer compounds could enable the development of more potent and/or selective drugs. In that vein, we envisioned that peptide-catalyzed, atroposelective bromination could be an effective means of accessing enantiopure 3-arylquinazolinone tribromides that are primed for late-stage derivatization and SAR study.
Our investigations began with the optimization of a peptide-based catalyst for the atroposelective conversion of quinazolinone 10.25a into tribromide product 10.26a using NBS as the brominating agent (Figure 187). Under these conditions, product yields were always near-quantitative, and so enantioselectivity was used as the sole determinant for catalyst selection. Examination of an initial library of 21 Dmaa-containing tetrapeptides 10.27, all of which were biased toward a type IIʹ β-hairpin geometry by incorporating a central d-Pro-Xaa sequence element, revealed several interesting trends, including a particularly striking effect of the i+2 residue. Comparing a homologous series of i+2 variants (Figure 187, blue), we observed that Acpc-containing 10.27h delivered 10.26a in 88:12 er, significantly higher than the Aib- (10.27c, 70:30 er), Cle- (10.27g, 70:30 er), l-Phe- (10.27i, 70:30 er), and Aic-containing (10.27j, 61:39 er) homologs. This effect was tentatively attributed to a beneficial effect of the wider i+2 main-chain angle (τi+2) of Acpc-containing 10.27h relative to the more acute values expected in the other homologs. Having identified a suitable i+2 residue, the catalyst sequence was fine-tuned by modification of the i+3 residue. Alkyl-substituted residues were found to be more selective than Phe- or Gly-derivatives, and we ultimately arrived at Leu-containing peptide 10.27q as the lead catalyst (Figure 187, red), which provided 10.26a in 92:8 er. Further sequence optimization did not lead to noticeable improvements in selectivity.
Figure 187.
Optimization of a peptide-based catalyst for the atroposelective bromination of 3-arylquinazolinone 10.25a, highlighting the effect of the i+2 residue.440
Seeking further understanding of the structural attributes that contribute to high selectivity, a truncation study was performed, wherein residues were sequentially removed from the C-terminus of lead peptide 10.27q (Figure 188). For tripeptides 10.28, while methyl ester 10.28a and dimethylamide 10.28b provided modest selectivities in the bromination of quinazolinone 10.25a under the screening conditions, N-methyl amide 10.28c remained a viable catalyst, furnishing 10.26a in 82:18 er. This observation was attributed to the ability of 10.28c to maintain a β-turn secondary structure through the formation of the crucial N–H(i+3)•••O(i) H-bond. A similar trend was observed in the dipeptides 10.29 following truncation of the Acpc residue. Only N-methyl amide 10.29c provided any significant selectivity (71:29 er), while methyl ester 10.29a and dimethyl amide 10.29b both delivered nearly racemic product. Thus, having an N–H bond at the i+2 position available for H-bonding with the substrate is likely important for selective catalysis, although formation of a γ-turn secondary structure via an N–H(i+2)•••O(i) H-bond could also contribute to the elevated selectivity of dipeptide 10.29c. Interestingly, Dmaa-monomer 10.30 provides modest selectivity (46:54 er) in favor of the opposite enantiomer, suggesting that secondary structural attributes overturn the inherent selectivity of the catalytically active residue.
Figure 188.
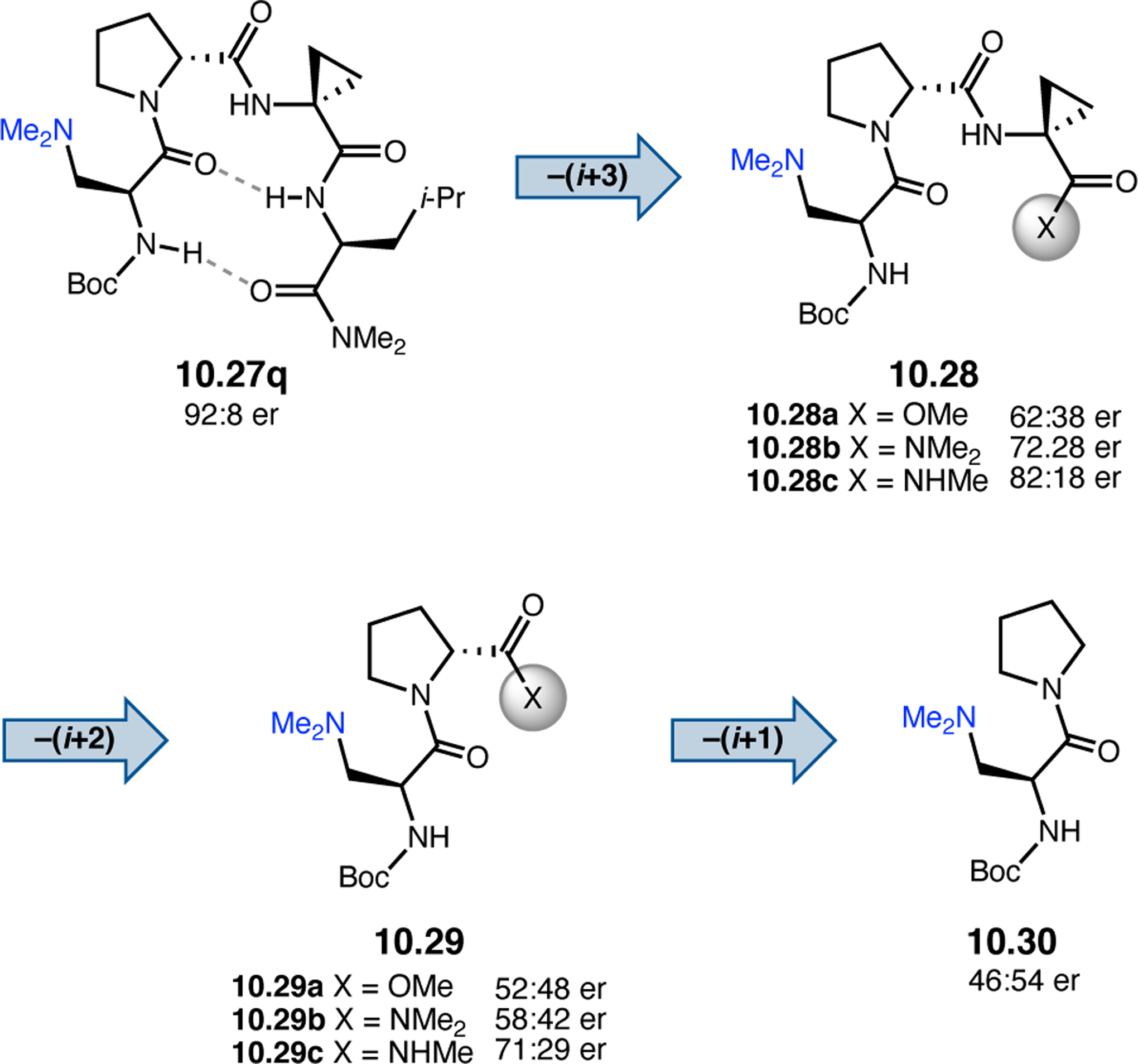
Sequence truncation reveals the structural attributes key for selectivity in the bromination of quinazolinone 10.25a under the conditions described in Figure 187.440
DFT computations showed that the enantiomerization barrier of 10.25a is 19 kcal/mol, which corresponds to a half-life of approximately seven seconds at ambient temperature. Compared to the other substrates we have examined in the context of atroposelective bromination, namely biaryls 10.1 (ΔG‡ ≈ 7 kcal/mol) and benzamides 10.8 (ΔG‡ ≈ 10 kcal/mol), these quinazolinones are expected to racemize significantly slower, likely as a consequence of the shorter N–CAr bond defining the chiral axis. Thus, we reasoned that slow delivery of NBS could result in higher levels of enantioselectivity by permitting more complete re-racemization of 10.25a on the extremely rapid bromination time scale, thereby enabling a more efficient DKR. Indeed, syringe pump addition of NBS over three hours in the presence of peptide 10.27q led to an energetically significant boost in the er of 10.26a from 92:8 to 97:3 er (Figure 189, entry 1). X-Ray crystallography showed that 10.27q delivered 10.26a in the (aS)-absolute configuration.
Figure 189.
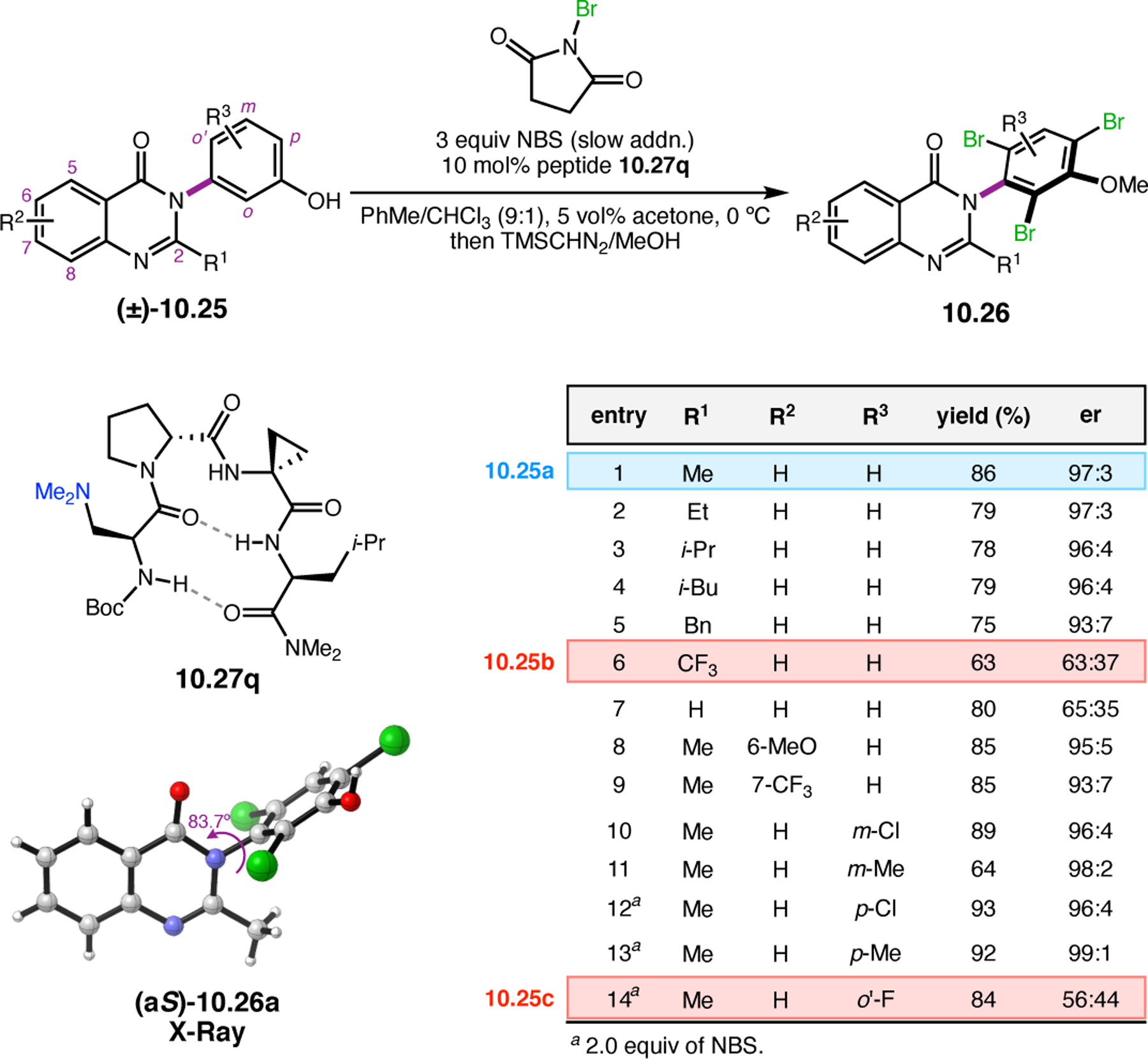
Peptide 10.27q-catalyzed, atroposelective bromination of quinazolinones 10.25.440
Under the optimized reaction conditions, tetrapeptide 10.27q was found to address a reasonably broad scope of quinazolinones 10.25 (Figure 189). Alkyl substitution at the 2-position of 10.25 was well tolerated (entries 1–5), as were electronically diverse substituents on the benzo moiety (entries 8–9). Additionally, substrates containing methyl and chloro substituents at the meta- and para-positions (relative to the chiral axis) of the phenol ring were also processed by 10.27q with high levels of enantioselectivity (entries 10–13). In fact, para-methyl 10.26 was isolated in 92% yield and 99:1 er (entry 13). However, three of the substrates examined performed rather poorly in the atroposelective bromination reaction (entries 6, 7, and 14). While the 2-trifluoromethyl group of 10.25b was presumed to negatively influence catalyst-substrate assembly via unfavorable electrostatic interactions, the other two substrates likely underperformed due to rotational barrier-related issues. For example, the preinstalled ortho-fluoro substituent of 10.25c was proposed to significantly reduce the racemization rate of this substrate, thereby precluding it from effective DKR. Indeed, 10.26c was formed in 84% yield and only 56:44 er (entry 14), and DFT computations determined the enantiomerization barrier of 10.25c to be over 26 kcal/mol. However, when the reaction with 10.25c is run to low conversion using only 0.5 equivalents of NBS, 10.26c can be isolated in 14% yield and 93:7 er, suggesting that compounds like these are suitable kinetic resolution substrates.
Mechanistically, low-conversion LC/MS studies revealed that peptide 10.27q overturns the inherent regioselectivity of the enantiodetermining step by promoting monobromination at the ortho-position, as was observed in the bromination of benzamides 10.8. Monobromination at the least sterically encumbered para-position is observed in triethylamine-catalyzed reaction. Moreover, due to the inherently high N–CAr rotational barriers of the quinazolinone system, the ortho-monobromide of 10.25a was able to be isolated with 98:2 er. This unambiguously demonstrates that ortho-monobromination is the enantiodetermining step in the 10.27q-catalyzed reaction.
NMR complexation experiments were also performed in order to study catalyst-substrate association in solution. Relative to their individual spectra acquired under identical conditions, the 1H-NMR spectrum of a 1:1 mixture of 10.25a and 10.27q showed a number of chemical shift perturbations consistent with intermolecular association. For example, the N–H(i+2) resonance shifted downfield by over 1 ppm, suggesting that this amide engages the substrate in H-bonding, likely with the carbonyl group of the quinazolinone core structure. NOESY experiments also revealed a number of intermolecular NOEs between 10.25a and 10.27q, a relatively uncommon finding for systems involving weak association (Figure 190). The distances ascribed to the NOEs allowed for the development of a precise, self-consistent binding and activation model (10.27q•10.25a complex) that predicts the site of enantiodetermining monobromination, as well as the observed (aS) product configuration. Again, association is likely nucleated by H-bonding between the phenol of 10.25a and the Dmaa sidechain of 10.27q, with backbone H-bonding from the N–H(i+2) further stabilizing the complex.
Figure 190.
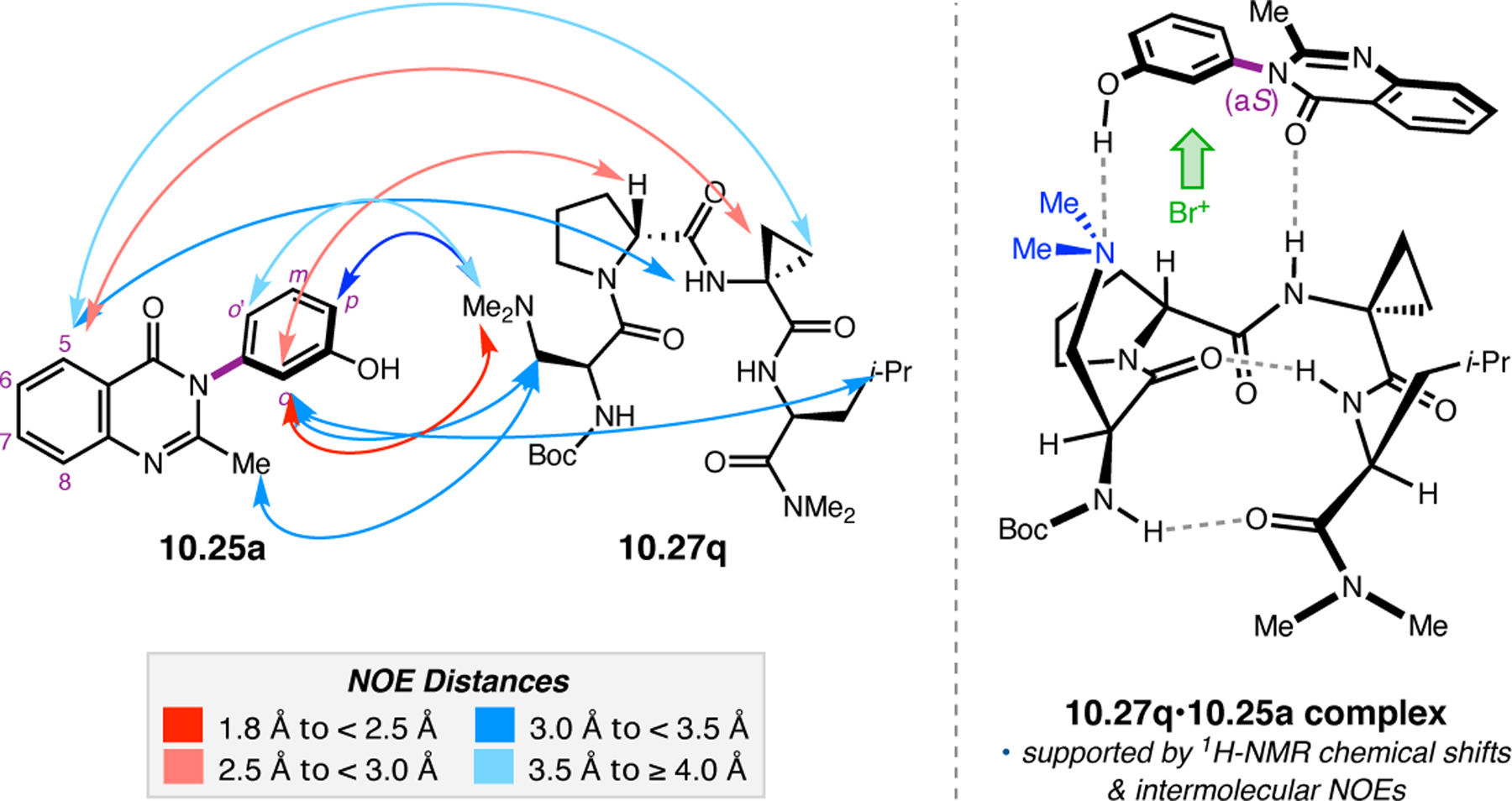
A model for binding and activation of quinazolinone 10.25a by peptide 10.27q was proposed on the basis of NMR complexation experiments (10 mM in C6D6, 25 °C, 600 MHz), which revealed a number of intermolecular NOEs.440
Due to the pharmaceutical relevance of these compounds, we also demonstrated that tribromides 10.26 could be regioselectively derivatized to access functionalized quinazolinones (Figure 191). Selective removal of the least sterically encumbered bromides could be achieved by careful hydrogenation of 10.26a, providing monobromide 10.31 with no erosion of er. DFT computations showed that 10.31 is configurationally stable with an enantiomerization barrier of over 35 kcal/mol. Suzuki–Miyaura cross-coupling of 10.31 delivered arylated derivatives 10.32 in 83–95% yield and with no enantioerosion. Alternatively, selective Buchwald–Hartwig amination targeted the para-bromide of 10.26a, furnishing amines 10.33 in 60–75% yield and in 96:4 er.
Figure 191.
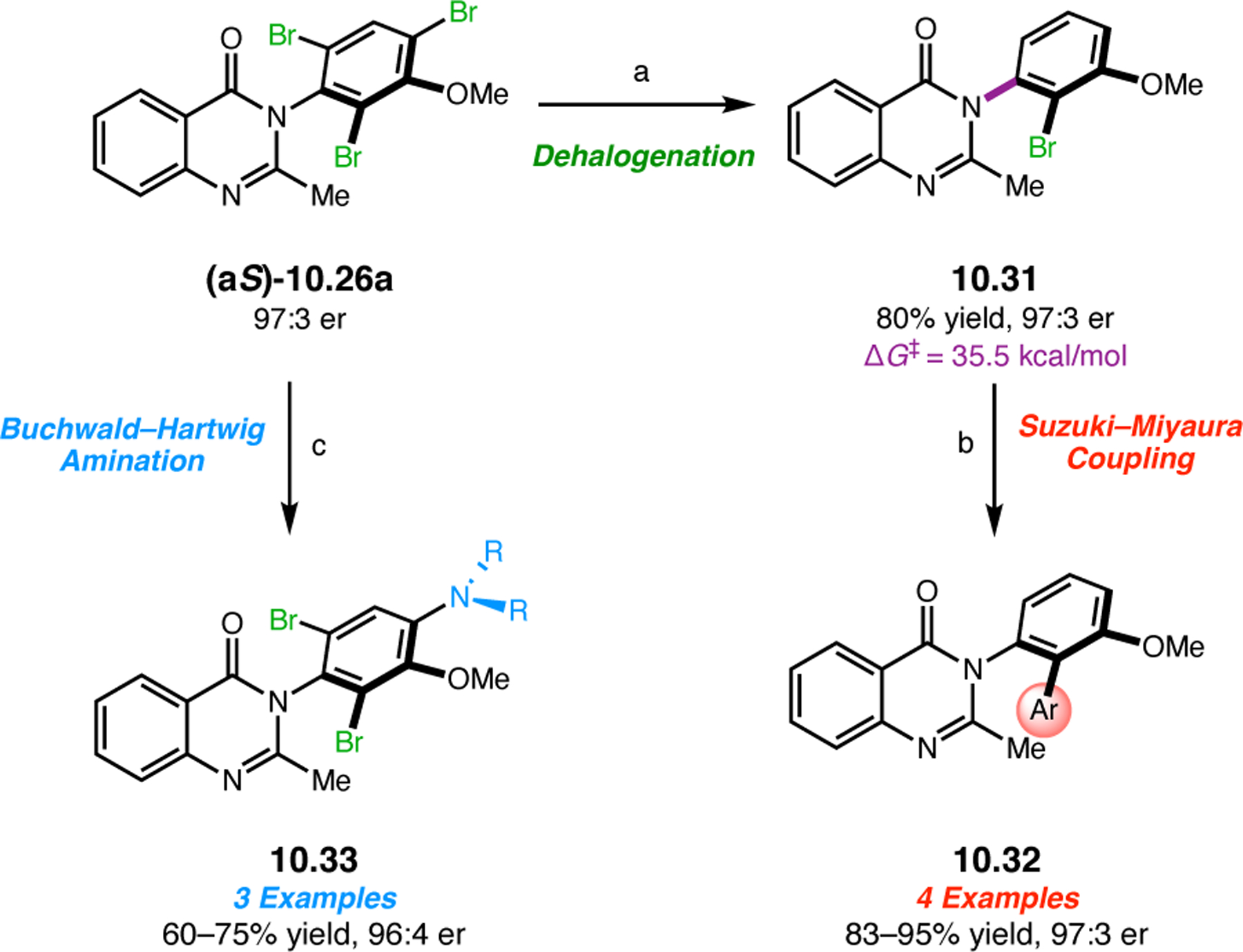
Regioselective derivatization of tribromides 10.26a using cross-coupling methodologies. Conditions: (a) 1 atm H2, 10 mass% Pd/C, MeOH (0.02 M), 12 h, 0 °C. (b) 2.0 equiv Ar–B(OH)2,10 mol% Pd2(dba)3, 20 mol% P(t-Bu)(Cy)2•HBF4, 4.0 equiv K3PO4, THF/H2O (3:1, 0.1 M), 45 °C, 24 h. (c) 2.0 equiv NHR2, 10 mol% Pd2(dba)3, 20 mol% rac-BINAP, 1.4 equiv NaOt-Bu, PhMe (0.1 M), 80 °C, 18 h.440
A follow-up study on the peptide-catalyzed quinazolinone bromination reported an expansion of the catalyst library in search of additional information on the relationship between structure and function.442 Interestingly, although C-terminal methyl esters were previously found to be inferior to dimethyl amides in the lower-selectivity regime (Figure 187, 10.27a–c), peptide 10.34a—the methyl ester variant of the original lead catalyst (10.27q)—was found to be a more reactive and selective catalyst (Figure 192, entries 1 vs. 2). Under the screening conditions, 10.34a delivered 10.26a in 95:5 er, even at only 1 mol% loading (entry 2). Recourse to the slow addition protocol provided a modest boost in selectivity to 97:3 er at 1 mol% of 10.34a. The origin of this increased catalyst efficacy by methyl ester substitution is currently uncertain, but the effect only manifests with Acpc at the i+2 position.
Figure 192.

Expansion of the catalyst library for the peptide-catalyzed bromination of quinazolinone 10.25a.442
Another effect that is seemingly unique to Acpc-containing peptides was identified that involves the substitution of l-Pro for d-Pro within an otherwise homologous sequence (Figure 192, entries 2–7). Epimerization of the catalyst in that way is expected to invert the turn sense from a so-called “mirror image β-turn” (e.g., type IIʹ) to a canonical β-turn (e.g., type II).47,223 We have often found previously that catalyst epimerization at the i+1 position results in selectivity favoring the opposite enantiomer with a significant reduction in the absolute er value.4 Such is the case here for the Aib- and Aic-containing peptides. The change from d-Pro-Aib (10.34b) to l-Pro-Aib (epi-10.34b) produced a drop in selectivity from 68:32 er to 45:55 er (entries 4 vs. 5). The change from d-Pro-Aic (10.34c) to l-Pro-Aic (epi-10.34c) followed suit (entries 6 vs. 7). However, the epi-10.34a maintained a high level of enantioselectivity in the opposite direction (11:89 er) relative to the d-Pro-Acpc-variant 10.34a (entry 2 vs. 3). This result may suggest that absolute enantiocontrol is largely dictated by the turn sense with relatively minor contribution from the flanking residues. Using the optimized slow addition protocol, peptide epi-10.34a delivers ent-10.26a in 78% yield and 9:91 er. This degree of enantiodivergence is notable and unprecedented for an epimeric catalyst.
In that same follow-up study, some preliminary structural data was reported for the lead quinazolinone bromination catalysts (10.27q and 10.34a).442 Three discrete conformations of 10.27q were observed in the solid state by X-ray crystallography (Figure 193). One crystal structure showed two conformations within the same asymmetric unit, both of which were characterized as type IIʹ β-hairpins based on the ϕ and ψ dihedral angles of the central two residues (i.e., the loop region)47 and presence of 10-membered N–H(i+3)•••O(i) and 14-membered N–H(i)•••O(i+3) intramolecular H-bonds (I and II). This was the designed secondary structure for the initial quinazolinone bromination catalyst library (see Figure 187). The primary difference between conformers I and II is the degree of backbone bending. An unexpected secondary structure was observed in a separate crystallographic sample of 10.27q. Conformer III also possessed the 10-membered ring N–H(i+3)•••O(i) β-turn H-bond observed in the designed conformers, but its loop region amide was inverted relative to I and II, allowing for a second 10-membered N–H(i+2)•••O(Boc) H-bond. This sequential double turn structure renders III prehelical in nature, with the central ϕ and ψ dihedrals resembling those of a type Iʹ β-turn.47 Interestingly, peptide 10.34a, which was found to be more active and selective than 10.27q, reliably adopted the prehelical conformation in the solid state. These unanticipated findings prompted an NMR study of these two peptides in solution. Two-dimensional NOESY spectra were acquired for each under identical conditions, and the cross-peak volumes were converted into distance restraints for structure simulations. The NOEs tabulated for 10.27q satisfied both the hairpin and prehelical geometries, and as such the simulated structure was intermediate between the two. However, peptide 10.34a predominantly adopted the prehelical type Iʹ β-turn conformation. Taken together, these results suggest that the most selective quinazolinone bromination catalysts can adopt multiple conformations, including secondary structures that were not designed at the outset.
Figure 193.
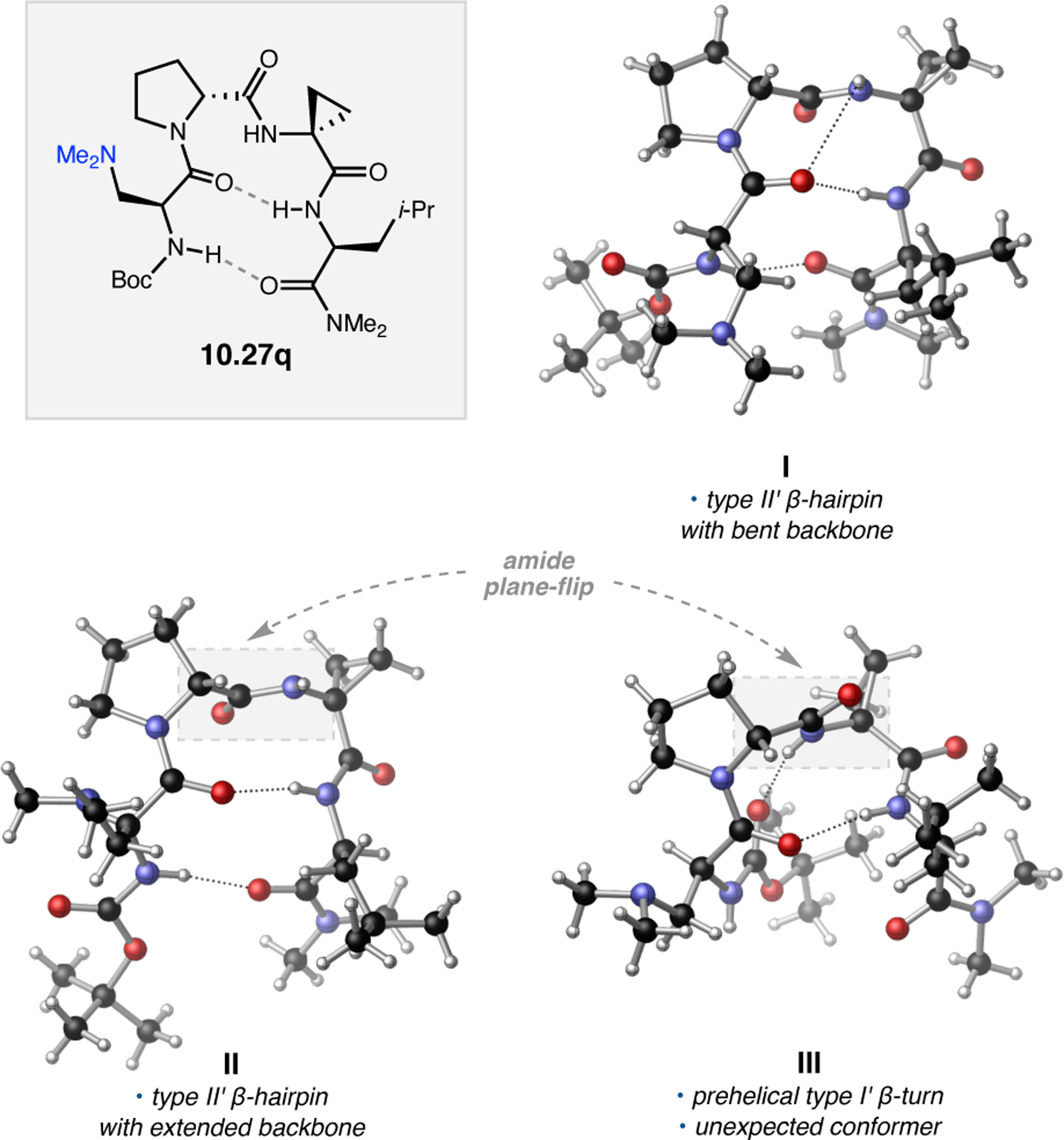
The three conformations of peptide 10.27q observed by X-ray crystallography. The X-ray structure of peptide 10.34a overlays closely with conformer III.442
In order to glean more information about the conformations accessible to these tetrapeptides and better understand how structure affects catalysis, we conducted an in-depth structural investigation of the bromination catalyst library.50 X-Ray crystal structures of 35 different peptides were obtained, giving rise to 53 total peptide conformations due to the occurrence of symmetry-independent molecules and polymorphs for multiple cases. Various structural parameters of these peptides were analyzed, including backbone dihedrals, side chain dihedrals, and H-bond geometries, among others, in search of information that might be relevant to catalyst design and function. In general, the peptides were found to be quite structurally diverse for a library that was biased toward a particular β-turn conformation. This structural diversity in the solid state often translated to solution phase dynamics, as many of the peptides showed NOE signatures of multiple limiting conformations, including type IIʹ β-hairpins like I and II, prehelical type Iʹ β-turns like III, and γ-turns. In many cases, the designed conformation was not predominant in solution.
One noteworthy aspect of this study came from the investigation of homologous i+2 variants of sequence 10.34 (Figure 194). While all of the catalysts were designed to adopt type IIʹ β-hairpins by incorporating a central d-Pro-Xaa sequence (where Xaa is a symmetrically disubstituted residue), only Aib-containing 10.34b exhibited this conformation in the solid state. All of the other members of this series (10.34a,c–g) existed in the unexpected type Iʹ prehelical conformation with very little backbone deviation. Most of the variation between these structures occurred at the C-termini and the i+3 sidechains (see structural overlay). Both high- and low-selectivity catalysts adopted the prehelical secondary structure in the solid state, and given the high degree of backbone similarity, it was unclear what factors might make certain sequences more selective, namely Acpc-containing 10.34a and Gly-containing 10.34g.
Figure 194.
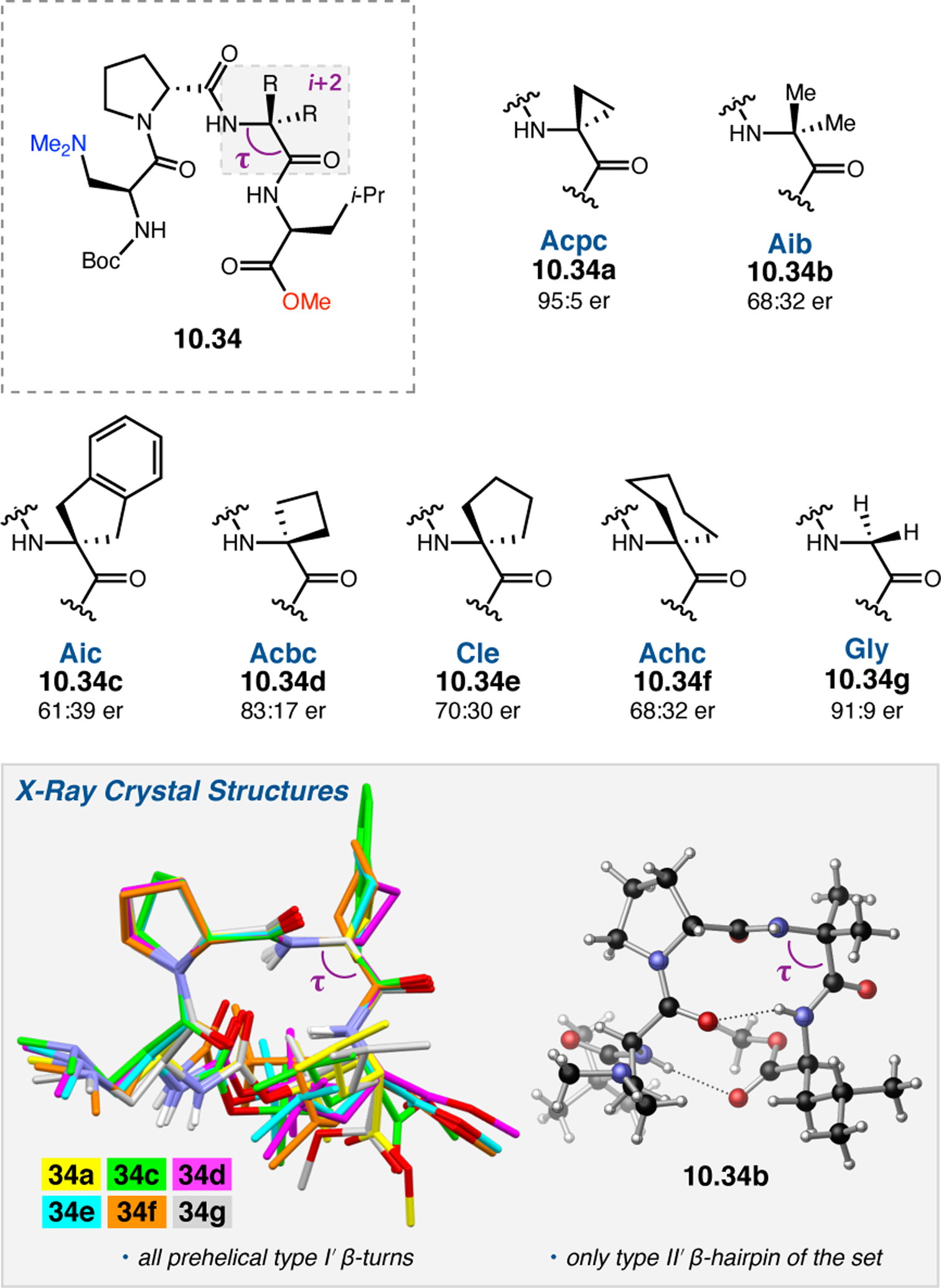
X-Ray crystal structures of the homologous i+2 variants of sequence 10.34. All but 10.34b adopted the unexpected prehelical conformation, including the most selective quinazolinone bromination catalysts. Enantioselectivity data are for the bromination of quinazolinone 10.25a to tribromide 10.26a under the conditions reported in Figure 192.50
A possible explanation emerged in the form of a linear correlation between the enantioselectivity measured in the peptide-catalyzed bromination of quinazolinone 10.25a and the main-chain angle of the catalyst i+2 residue (τi+2, Figure 195). While this feature was not designed into the catalyst library at the outset, the data showed that catalysts with wider τi+2 values tend to be more selective. In proteins, turn residues with wide τ values often have relaxed torsional energy profiles, making their respective secondary structures more structurally flexible.443 In that light, these data hinted at the possibility that the most selective quinazolinone bromination catalysts are also conformationally dynamic.9
Figure 195.
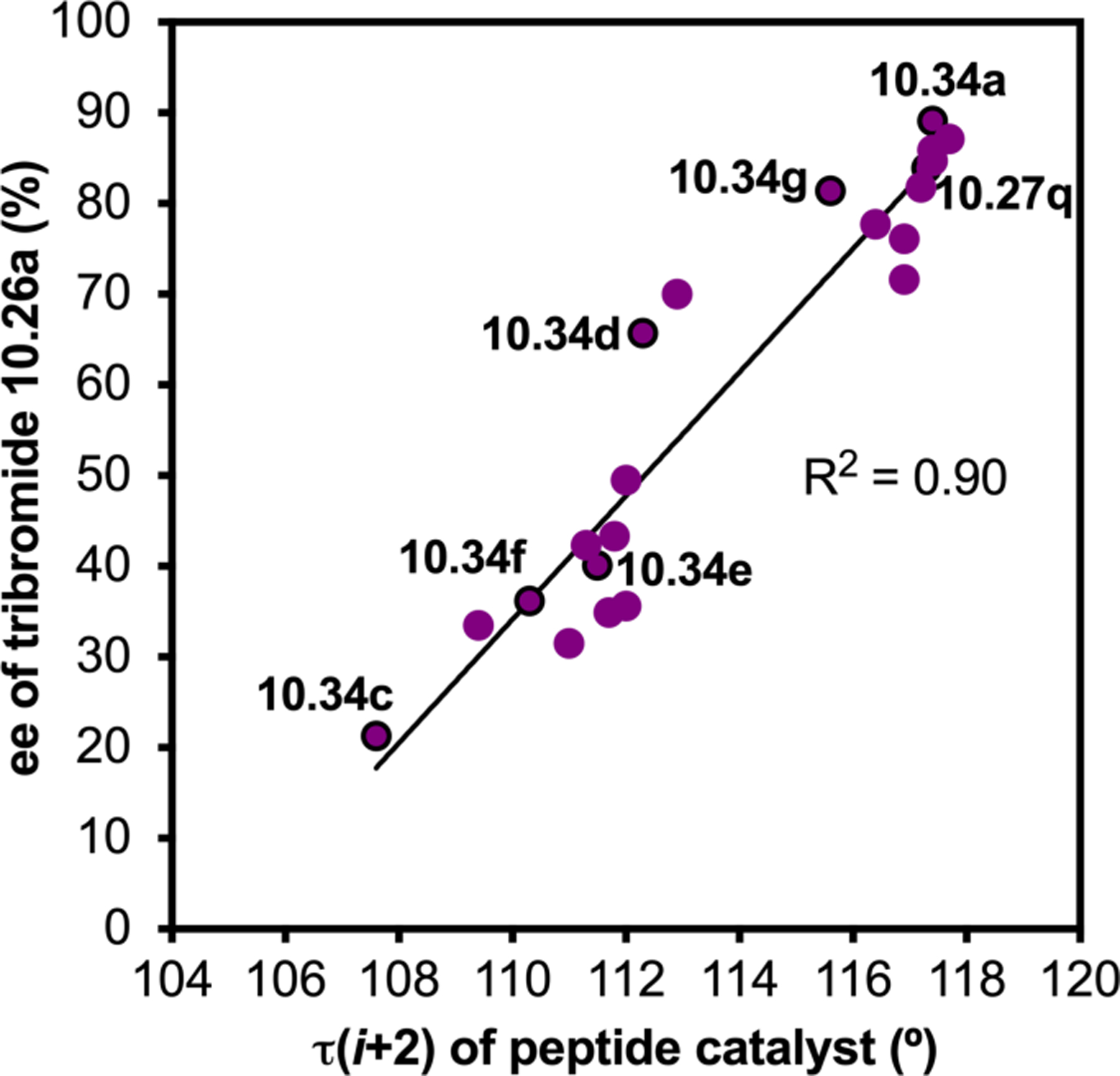
Correlation between enantioselectivity (ee) and catalyst τi+2. The homologous catalysts highlighted in Figure 194 are indicated on the plot. Enantioselectivity data are for the bromination of quinazolinone 10.25a to tribromide 10.26a under the conditions reported in Figure 192.50
Evidence in support of this hypothesis was obtained from solution-phase NMR studies of the same homologous i+2 series.50 Analysis of the NOESY spectra of peptides 10.34a–g revealed that most of the peptides were conformationally dynamic in solution, as determined by the presence of NOE signatures of two or more secondary structural archetypes (e.g., β-hairpin, prehelical β-turn, and γ-turn). The exception was 10.34b. This low-selectivity catalyst only showed NOEs consistent with the designed β-hairpin, mirroring the solid-sate observations (Figure 196). In contrast, highly selective catalysts like 10.34a and 10.34g predominantly adopted the prehelical conformation in solution, while also equilibrating between multiple minor secondary structures.
Figure 196.
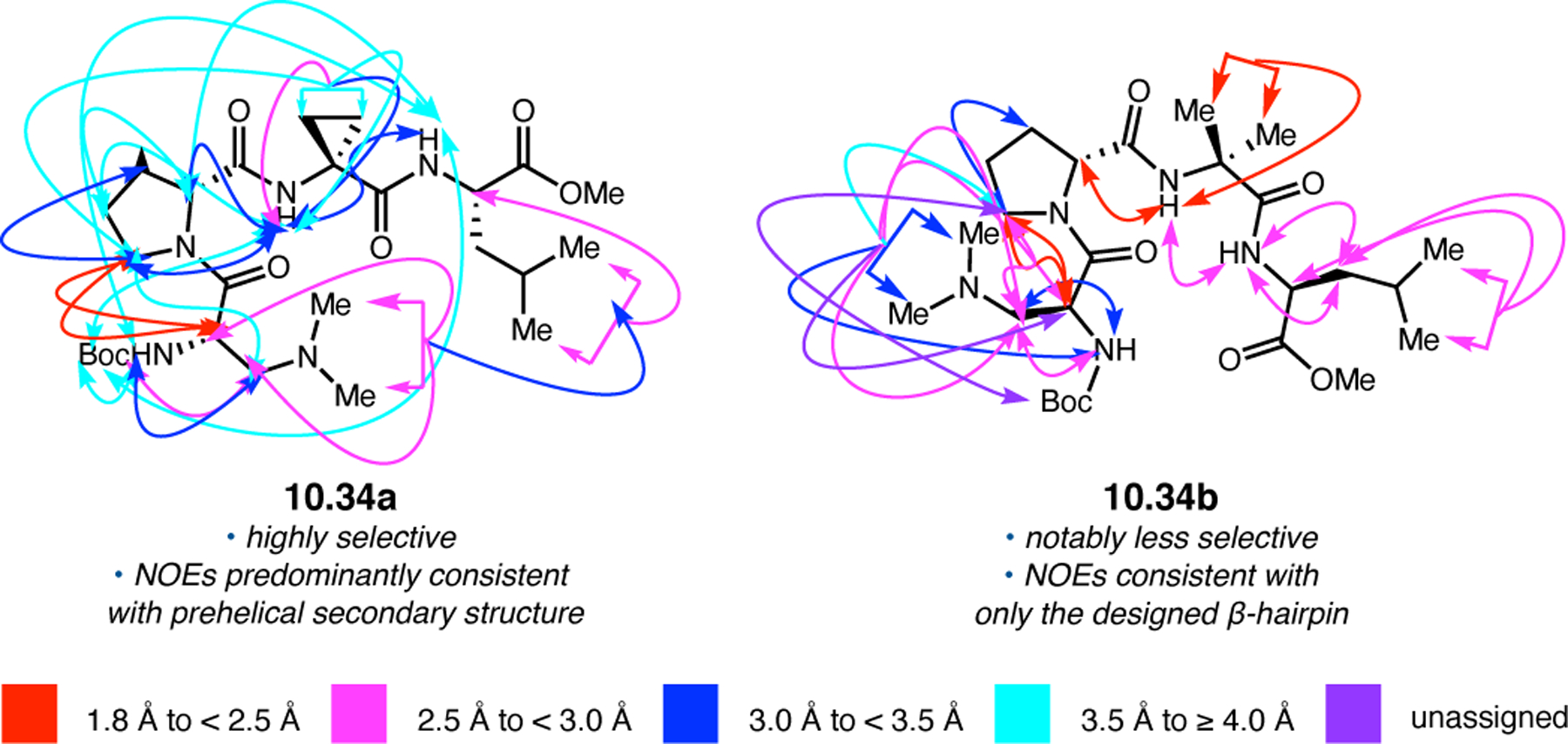
NOESY correlation diagrams for highly selective catalyst 10.34a and less selective catalyst 10.34b.50
The situation was different in the presence of quinazolinone substrates 10.25. When lead catalyst 10.27q was treated with an equimolar amount of 10.25a, its solution conformation changed from predominantly prehelical to the β-hairpin.50 However, when 10.27q was treated with an equivalent of quinazolinone 10.25b—a poorly performing substrate (see Figure 189)—the degree of change was notably less. In collaboration with the Jorgensen Group, high level molecular dynamics (MD) simulations were performed to model these NMR spectroscopic observations.444 Over the course of a 1600 ns simulation in explicit benzene solvent, 10.27q alone was found to exhibit the prehelical conformation 75% of time, while the remaining 25% consisted of the β-hairpin and other conformers (Figure 197a). Upon treatment with selective substrate 10.25a, the conformation of the peptide catalyst flipped to favor the β-hairpin 75% of the time. In contrast, the conformational landscape in the presence of 10.25b favored a hairpin-like conformation only 51% of the time. In both cases, the lowest energy binding modes very closely resembled the proposed model shown in Figure 190; the complex with 10.25b, however, was notably more dissociated than that of 10.25a (Figure 197b). These data seemed to imply that selective bromination substrates induce a more substantial conformational change than do less-selective ones in a manner analogous to conformational selection in enzymatic catalysis.
Figure 197.
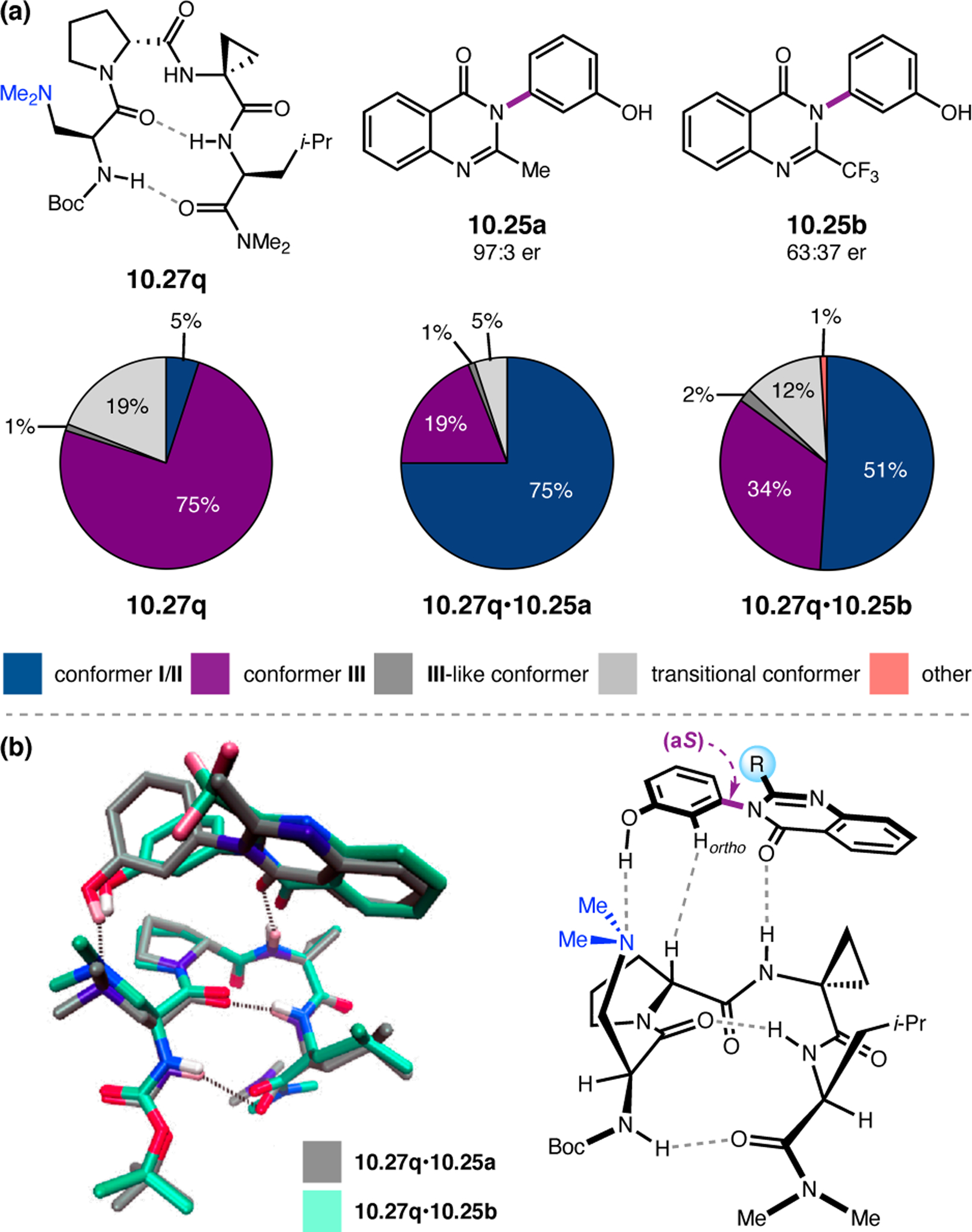
(a) Conformational landscape of peptide 10.27q alone and in the presence of 1.0 equiv of quinazolinone substrates 10.25a and 10.25b. (b) Lowest-energy binding modes showing key intermolecular interactions.444
While these NMR titration studies generated valuable information on the ground state catalyst-substrate complex, the findings do not necessarily provide a readout on the stereodetermining transition state of the peptide-catalyzed quinazolinone bromination reaction. Following the identification of the τi+2 correlation shown in Figure 195, we engaged in a collaboration with the Sigman Group to utilize multidimensional reaction analysis163 to identify catalyst parameters relevant to asymmetric induction.445 Features of both limiting catalyst conformations—the type IIʹ β-hairpin and the prehelical type Iʹ β-turn—correlated well with the measured enantioselectivity (ΔΔG‡) in the bromination of quinazolinone 10.25a (Figure 198). For the type IIʹ β-turns, the infrared stretching frequency and intensity of the loop region amide C=O and N–H bonds, respectively, likely read out on the importance of this amide in nucleating an H-bonding interaction with the quinazolinone substrate. For the prehelical conformers, τi+2 emerged as an important parameter, as well as the 10-membered β-turn H-bond strength measured through natural bond order (NBO) analysis. The Sterimol parameter L of the i+3 sidechain was found to be important in both models, suggesting that the steric effect of this residue is common to both peptide conformations. Ultimately, these findings imply that the enantioselectivity observed in the bromination of quinazolinones 10.25 may derive from a transition state ensemble wherein the catalyst interacts with the substrate from multiple conformational states.
Figure 198.
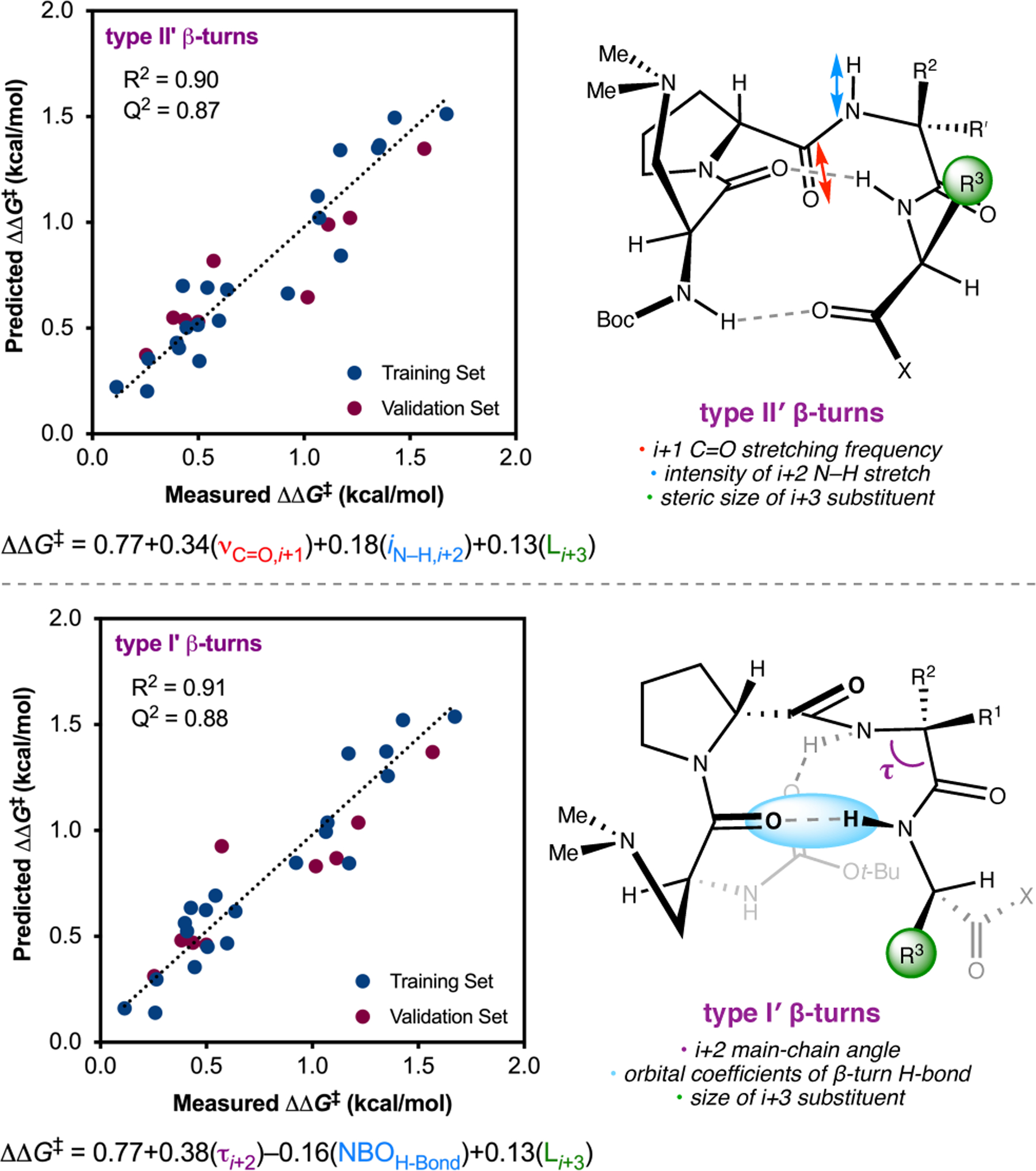
Multidimensional parameterization data shows that features of both limiting catalyst conformations correlate well with the observed enantioselectivity in the bromination of quinazolinone 10.25a.445
Taken together, this body of work on the atroposelective bromination of biaryls (Section 10.1.1), tertiary benzamides (Section 10.1.2), amido-tropolones (Section 10.1.3), and 3-arylquinazolinones (Section 10.1.4) has provided a viable and robust strategy for the synthesis of enantioenriched atropisomers. The tribromide products of these transformations are of significant value in medicinal chemistry due to the drug-like properties of the core atropisomeric structures and their demonstrated elaboration using reliable cross-coupling methods. Moreover, these works have contributed significantly to the fundamental understanding of peptide-catalyzed transformations and the degree to which concepts imported from structural biology can be reliably exploited to achieve selectivity in small-molecule catalysis.
10.2. Desymmetrizing Bromination of Diarylmethylamido Bis(phenol)s
In the atroposelective bromination reactions reviewed in Section 10.1, the peptide-based catalysts were able to impart enantioselectivity by effectively differentiating the enantiomeric orientations of a meta-phenol ring. While the crucial phenol OH group is somewhat distal from the axis of chirality in these systems, asymmetric induction becomes significantly more challenging when the reactive site or directing group is even further removed from the stereogenic element. Examples of remote asymmetric induction were once most commonly observed using enzymatic catalysts,158 which was intuitively comfortable since proteins are large enough and sufficiently well-organized to exert catalytic influence at a distance. In 2006, our group demonstrated that peptide-based catalysts can also achieve remote asymmetric induction in the highly enantioselective, desymmetrizing acylation of diarylmethane bis(phenol)s (Section 4.4),156 an important scaffold in pharmaceutical chemistry.159 Although other small molecule-catalyzed desymmetrizations of diarylmethane substrates have since been reported,446,447 peptides are particularly well-suited for this sort of “outer-sphere” selectivity due to their tunable lengths, high functional group density, and programmable secondary structures.245
In order to further demonstrate these concepts, our group set out to develop a peptide-catalyzed remote desymmetrization of diarylmethylamido bis(phenol)s 10.35 via electrophilic aromatic bromination (Figure 199).448 Given the emerging relevance of this scaffold in medicinal chemistry,159,449 access to enantioenriched monobromides 10.36 would provide a useful platform for late-stage functionalization and SAR. The enantiotopic phenols of 10.35 are separated from one another by 9.6 Å and from the prochiral center by 5.7 Å. A peptide-based catalyst might be able to differentiate the enantiotopic arenes of 10.35 over such long distances by taking advantage of attractive noncovalent interactions with the substrate. If the sequence were sufficiently long, a Dmaa-containing peptide could possibly interact with both phenols of 10.35 directly (e.g., mode 1). Alternatively, the Dmaa residue could preferentially activate one phenol toward electrophilic bromination, while the peptide backbone engaged the prochiral amido group in H-bonding (e.g., mode 2).
Figure 199.
General strategy for the desymmetrizing bromination of diarylmethylamido bis(phenol)s 10.35.448
Our studies focused on elaborated diarylmethylamido bis(phenol)s 10.35a, in which amido groups were pre-installed ortho to the enantiotopic phenol groups in order to minimize the formation of complicated polybrominated products.448 Catalyst optimization led to the identification of peptide 10.38, which was highly selective in the bromination of 10.35 using a precise amount of NBS (1.35 equiv) and carefully controlled conditions (Figure 200). Peptide 10.38 catalyzed the conversion of 10.35a to a 69:31 mixture of mono- and dibrominated products 10.36a and 10.37a (entry 1). Monobromide 10.36a was isolated in 66% yield and 95:5 er. X-Ray crystallography confirmed the (R)-configuration of 10.36a. Under the same conditions, the reaction was slow and non-selective using peptide 10.39 (entry 2), in which the N-terminal Dmaa residue was replaced with its C-isostere—Leu. The reaction was similarly sluggish using triethylamine as a catalyst (entry 3) and with no added catalyst (entry 4). These results suggest that both the Dmaa residue and the peptide scaffold of 10.38 are crucial for organizing the transition state in the selective pathway.
Figure 200.
Results from the peptide-catalyzed desymmetrizing bromination of 10.35a demonstrating the importance of the Dmaa residue for selective catalysis.448
Because a significant amount of dibrominated material 10.37a was formed during the course of the reaction, the possibility existed that secondary kinetic resolution of monobromide 10.36a could be contributing significantly to the observed enantioselectivity. However, when authentically prepared (±)-10.36a was subjected to the reaction conditions constrained to low conversion using only 0.45 equivalents of NBS, a krel of 3.2 was measured at 37% conversion. This low value suggested that, while secondary resolution does contribute to some extent, the primary source of asymmetric induction is indeed enantiotopic arene differentiation.
During the course of catalyst optimization, an interesting case of enantiodivergence was noted (Figure 201). Whereas Acpc-containing 10.38 delivered monobromide 10.36a in 69% NMR yield and 95:5 er, its Aib homologue 10.10—the lead catalyst discovered for the atroposelective benzamide bromination discussed in Section 10.1.2—provided 46% yield modestly favoring the opposite enantiomer in 42:58 er (entry 1). Moreover, the degree of enantiodivergence was found to be substrate dependent, with Δ(ΔΔG‡) values ranging from 1.10–1.86 kcal/mol contingent upon nature of the amide substituents R and Rʹ (entries 2–5). Enantiodivergent outcomes in asymmetric catalysis are uncommon for non-enantiomeric catalysts. This case is particularly striking, because the two catalysts in question only differ by a 2 amu-perturbation at an achiral residue. As such, the observed enantiodivergence most likely stemmed from differences in the secondary structures of 10.38 and 10.10 and how those structures interact with 10.35.
Figure 201.
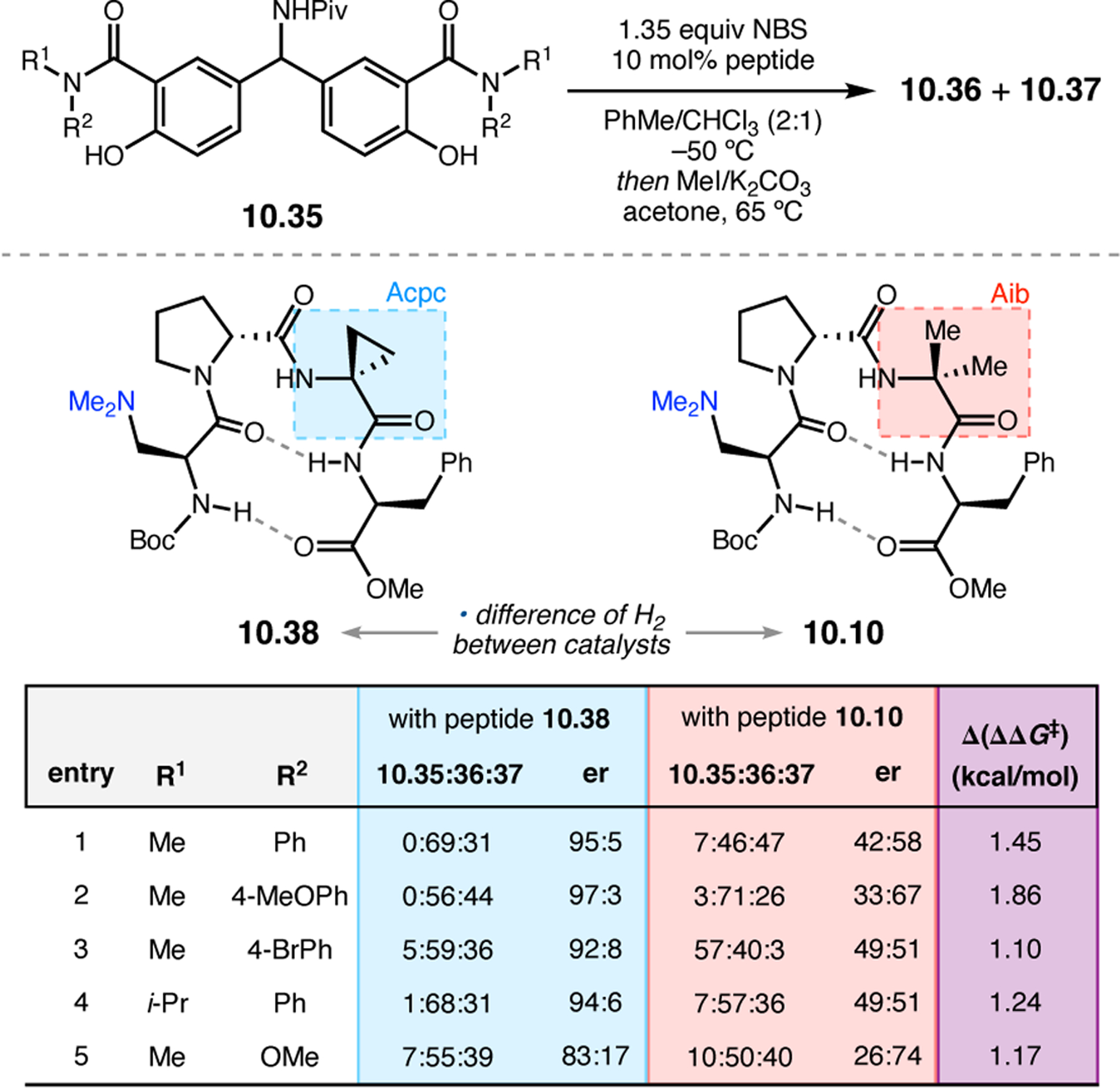
Enantiodivergence between catalysts 10.38 and 10.10 in the bromination of diarylmethylamido bis(phenol)s 10.35.448
A possible clue for this intriguing effect came from analysis of X-ray crystal structures of the catalyst library. As in the atroposelective quinazolinone bromination discussed in Section 10.1.4, a linear correlation between the measured ee of monobromide 10.36a and the main-chain angle of the catalyst i+2 residue (τi+2) was observed (Figure 202). Again, catalysts with wide τi+2 values delivered the highest ees, while values closer to tetrahedral (109.5°) gave modestly inverted selectivities. Based on the structural analyses we performed previously,50 wide-angle Acpc-containing catalysts like 10.38 were expected to primarily adopt prehelical type Iʹ β-turn structures in solution, while the narrower Aib-containing congeners like 10.10 were more likely to exist as type IIʹ β-hairpins (see Figure 193). A major difference between these two conformations is that the loop region amide is flipped, or oppositely oriented, which could give rise to the observed enantiodivergence.
Figure 202.
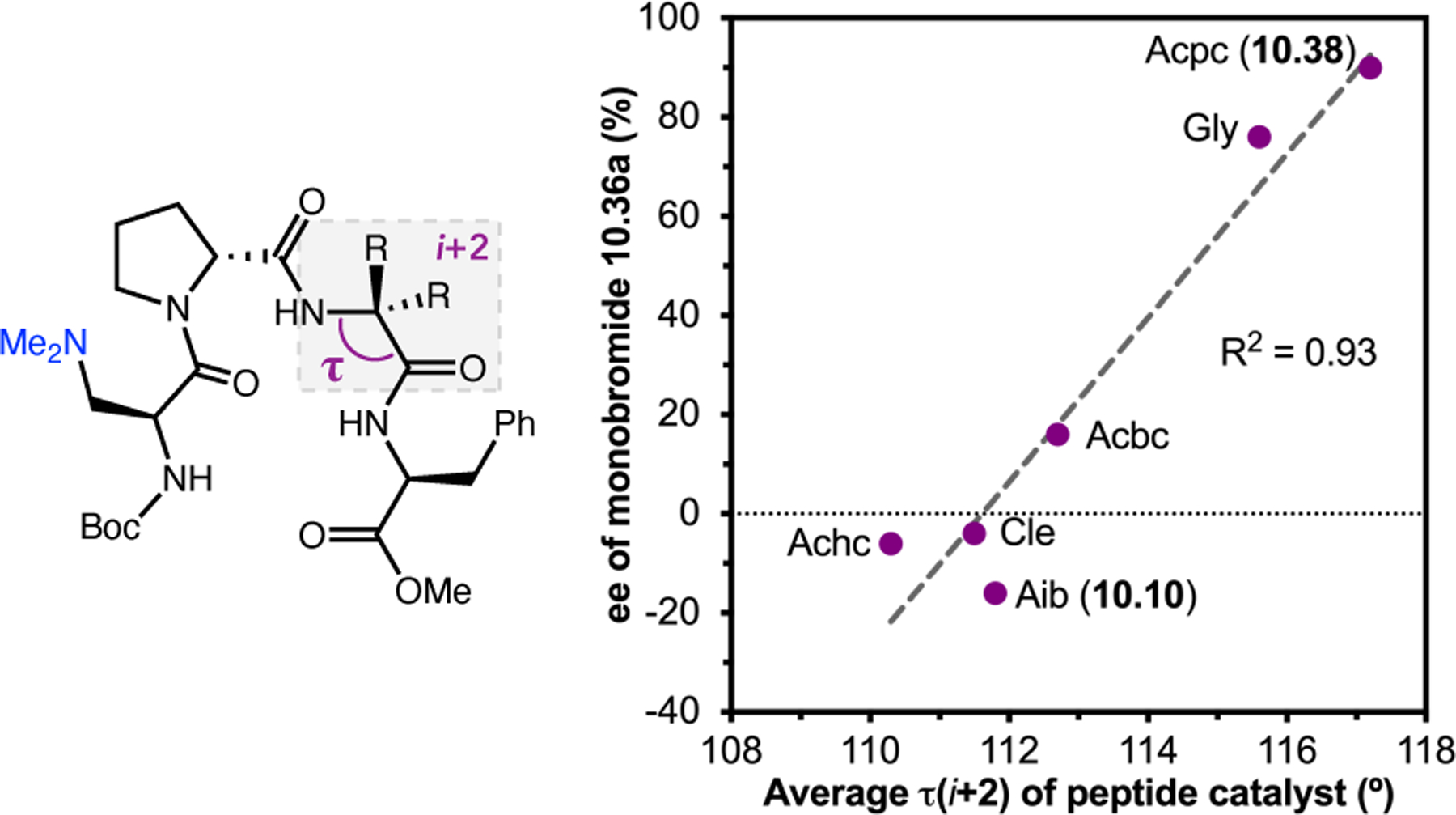
A plot of ee vs. catalyst τi+2 revealed a linear correlation for the desymmetrizing bromination of 10.35a under the conditions reported in Figure 201.448
Based on these concepts, we proposed models wherein diarylmethylamido bis(phenol) 10.35 interacts with peptide 10.38 from its likely prehelical conformation and with 10.10 from its β-hairpin structure (Figure 203). In both models, a key intermolecular H-bond between the tertiary amine of the catalyst Dmaa residue and a phenol O–H of 10.35 is proposed to activate the substrate toward SEAr. However, the oppositely oriented loop amides of 10.38 and 10.10 are proposed to invert the binding orientation of 10.35 through differential interaction with either antipode of the central pivalamide moiety.
Figure 203.
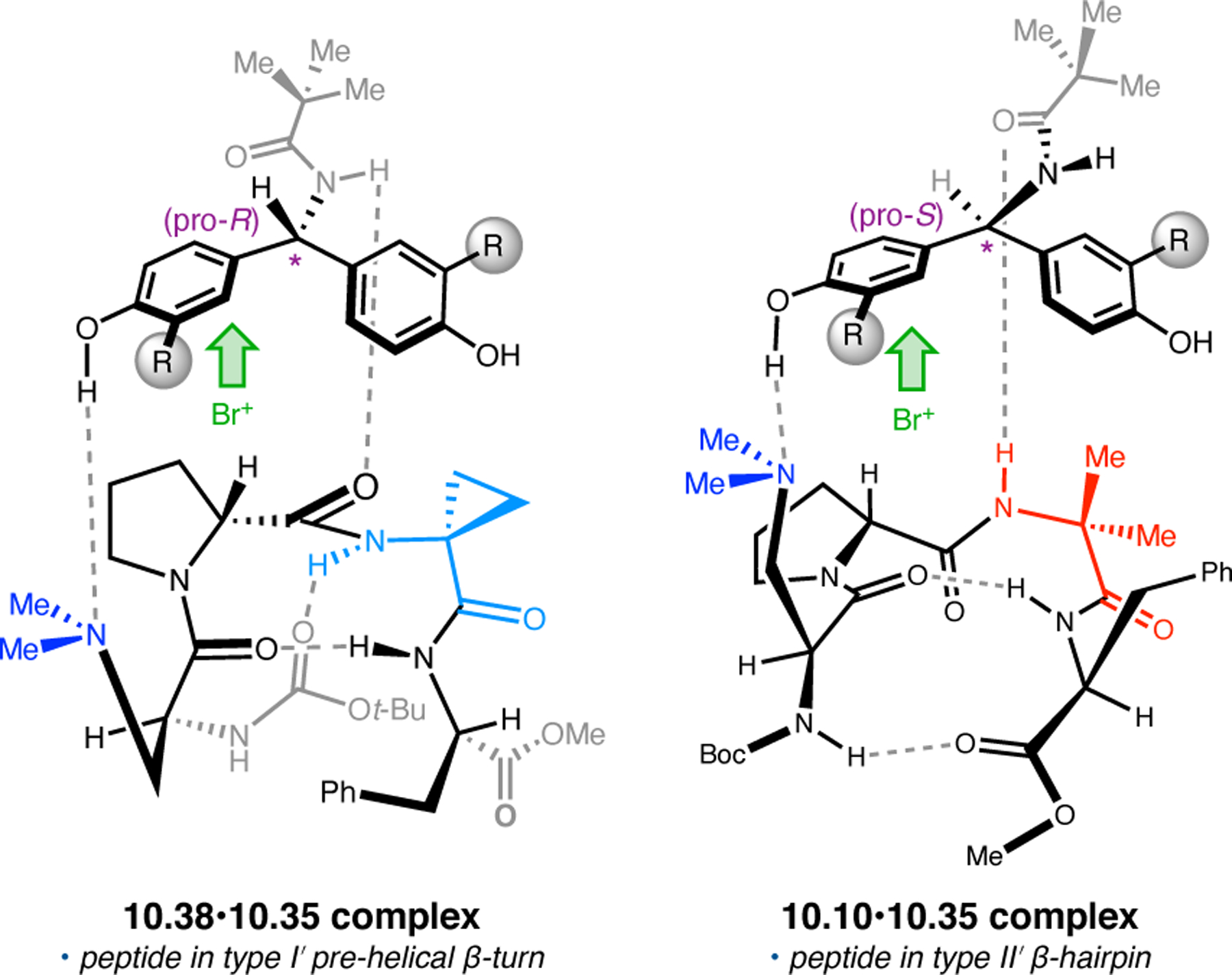
Differences in the solution conformations of 10.38 and 10.10 could possibly give rise to inverted binding and activation modes.448
This method provides access to highly enantioenriched diarylmethylamido bis(phenol) monobromides (e.g., 10.36) that can be reliably diversified using known cross-coupling protocols. A similar system was recently reported by Lewis and co-workers that employs a halogenase enzyme to enantioselectively chlorinate a diarylmethane bis(aniline) substrate with excellent levels of selectivity.450 On the other hand, the achievements of this study were made possible by the remarkable ability of a small molecule peptide to effectively differentiate enantiotopic phenols separated by long distances.
10.3. Asymmetric Bromolactonization
Halofunctionalization reactions of alkenes have been well established in the literature for over a century. These reactions typically proceed through the formation of a bridged haliranium ion followed by trapping with nucleophiles, allowing for the 1,2-difunctionalization of the olefin starting material.411,412,425 In many cases, formation of the haliranium ion intermediate renders these reactions stereospecific, making them quite valuable in organic synthesis. Olefin halolactonization, in particular, has found wide use as a reliable approach for the formation of lactone products that can be subsequently derivatized via displacement of the adjacent halide.451 However, catalytic asymmetric variants have been difficult to realize through the years.411 This is due, in part, to the challenge associated with olefin-to-olefin haliranium ion transfer, which ultimately leads to product racemization if the rate is competitive with that of nucleophilic addition. In this context, Borhan and co-workers became interested in developing asymmetric halolactonization methods. In 2010, they developed an enantioselective chlorolactonization protocol that employs a cinchona alkaloid-derived organocatalyst to mediate halenium ion transfer.452 This method provided the desired chlorolactone products in good yields and enantioselectivities up to 90% ee. The group soon expanded this methodology to address allylic amide substrates for the asymmetric synthesis of oxazoline and dihydrooxazine heterocycles depending on the cyclization mode.453
Building upon these works, the Borhan Group later pursued an alternative approach to catalytic, asymmetric halolactonization that would take advantage of hypervalent iodine(III) species generated in situ to mediate halenium ion transfer.454 The authors chose to investigate this strategy in the bromolactonization of 4-arylpentenoic acid 10.40 to the corresponding bromolactone 10.41 (Figure 204). They proposed to embed an ortho-iodobenzamide moiety within a peptide scaffold, which would oxidatively cyclize to form a bromoiodinane intermediate upon exposure to NBS. The peptidic bromoiodinane would then serve as a bromenium transfer catalyst. This concept was based on the known ability of simple ortho-iodobenzamides to accelerate non-stereoselective bromolactonization reactions via the bromoiodinane intermediate.455 By incorporating the iodoarene moiety within a well-defined peptidic framework, this process could be rendered asymmetric.
Figure 204.
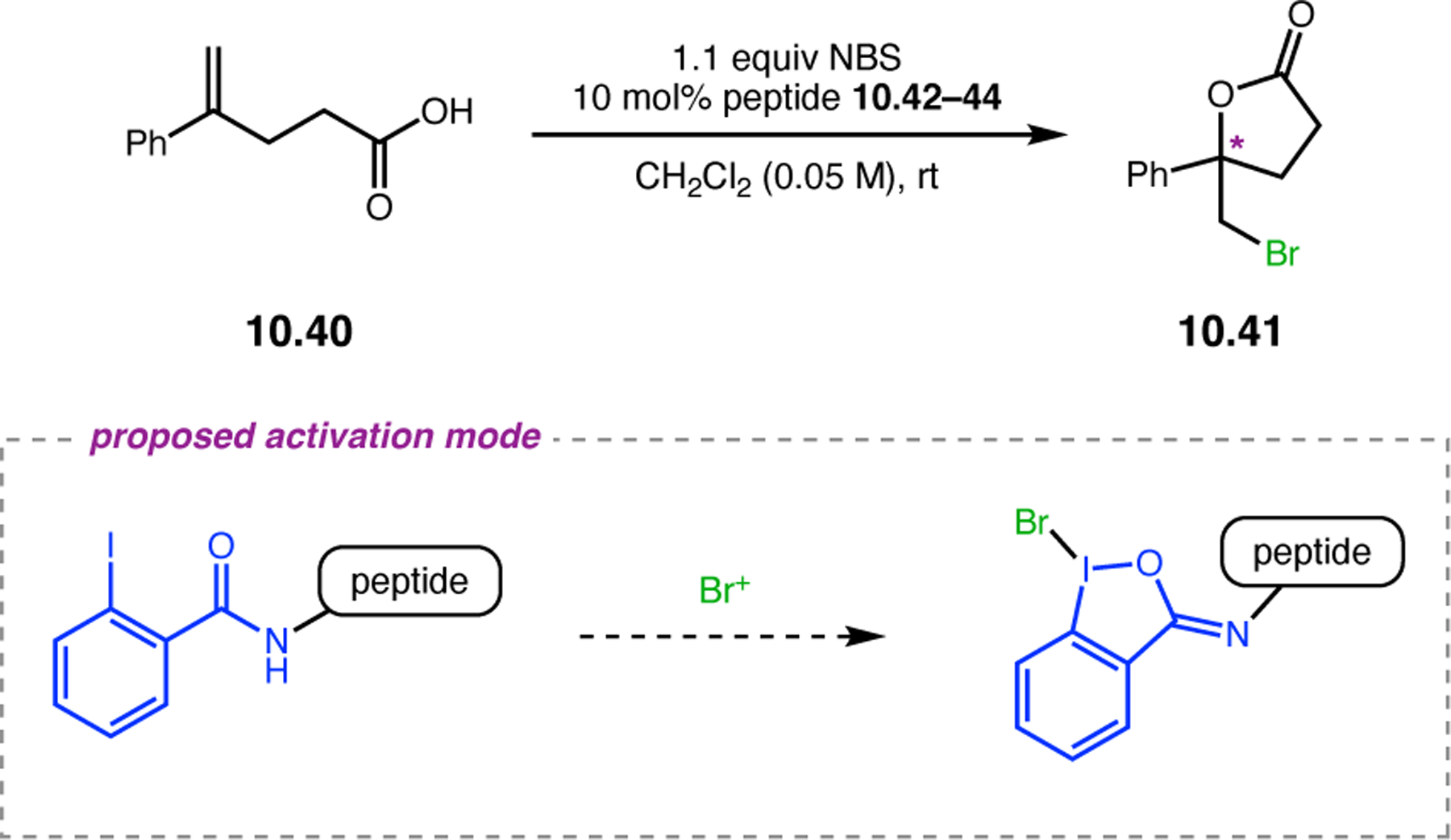
The general strategy for the peptide-catalyzed bromolactonization of pentenoic acid 10.40 to bromolactone 10.41 takes advantage of a bromoiodinane generated in situ to transfer bromenium ion to the olefin.454
The authors chose to investigate the bromolactonization of 10.40 using peptide-based catalysts adhering to three structural archetypes: (1) linear di- and tripeptides 10.42, (2) pentapeptides 10.43 biased toward β-turns by incorporation of a central Pro-d-Xaa sequence, and (3) symmetrically substituted 10.44 (Figure 205).454 While scaffolds 10.42 and 10.43 place the catalytically active moiety at the N-terminus of the peptides, the aryl iodide is centrally located on 10.44. The authors screened their catalyst library on microscale (50 μmol) and only used ee values as the basis for peptide evaluation. In total, the authors investigated a library of nearly 40 peptides. All of the 29 peptides 10.42 provided full conversion to bromolactone 10.41, and appreciable selectivity was only observed for tripeptides where R = t-Bu. The most selective catalyst of the set was tert-leucine-trimer 10.42a, which provided 10.41 in 14% ee under the screening conditions. The four β-turns 10.43 examined gave no appreciable selectivity, with 5% maximum ee observed. No selectivity was observed with any of the three symmetrical aryl iodides 10.44. Further sequence optimization did not yield further improvements. However, 10.41 could be obtained in quantitative conversion and up to 24% ee using 111 mol% of tripeptide 10.42a. The authors reasoned that this low “ceiling enantioselectivity” could be due to the high background rate and/or a low equilibrium population of the catalytically active bromoiodinane species.
Figure 205.
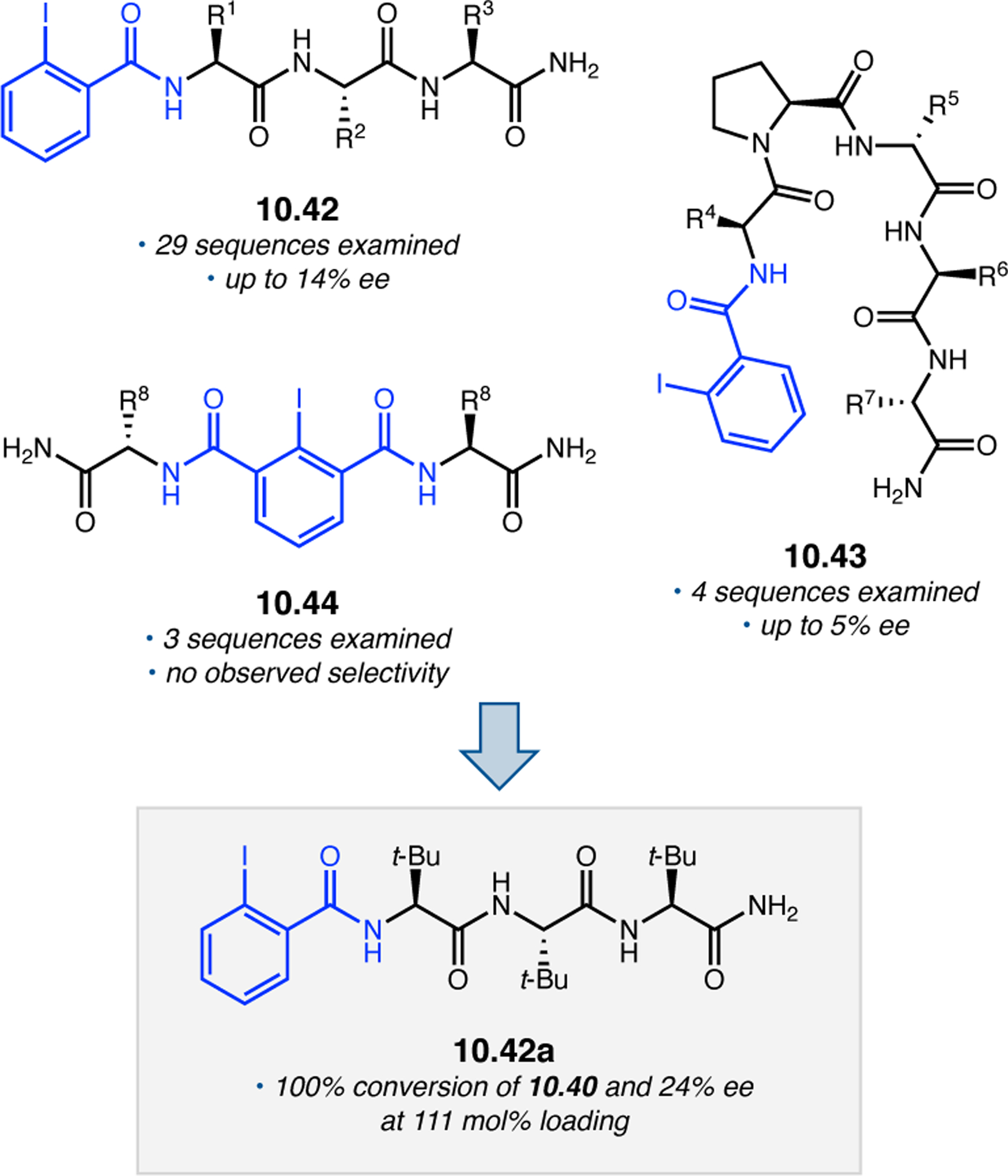
Key results from the evaluation of peptide scaffolds 10.42, 10.43, and 10.44 in the bromolactonization of pentenoic acid 10.40.454
While this approach ultimately did not deliver synthetically useful levels of ee in the bromolactonization of pentenoic acid 10.40, this study presented a novel and interesting approach to asymmetric halolactonization at a time where few examples of this type of transformation were reported. Since then, much progress has been made in this realm, but no further attempts using peptide-based catalyst scaffolds have been made.
11. Transition Metal- & Lewis Acid-Peptide Complexes in Asymmetric Catalysis
While enzymes have long been considered ideal catalysts with respect to the near perfect levels of chemo-, site-, and enantioselectivity that they are able to impart on their substrates, a number of valuable transition metal-catalyzed reactions exist solely within the realm of synthetic chemistry, including but not limited to hydroformylation, olefin metathesis, and cross-coupling among others. Conversely, while ligand design for transition metal catalysis has enabled a plethora of powerful transformations, enantioselectivity can be hard to achieve for particular reaction classes or substrates, in particular those that contain functional groups that may bind to metal centers. Additionally, many ligand classes are a difficult to derivatize, which often makes the tailoring of ligands for desired reactivity and selectivity outcomes quite challenging. The use of artificial metalloenzymes, wherein transition metal complexes are incorporated into the active site of an enzyme, has emerged as an extremely powerful solution.13,456–459 Additional approaches, however, are needed to combine the broad reactivity of organometallic complexes with the exquisite selectivity of enzymes. A strategy which has emerged at the interface between these two fields is the design of short peptide-based ligands, which can direct reactivity through noncovalent interactions with the substrate. The modular nature of peptide synthesis provides access to large amounts of chemical space, enabling the rapid generation of ligand derivatives. Elegant early applications of peptidic ligands in transition metal-catalyzed reactions include the pioneering work of Hoveyda, Snapper, and co-workers,214–216,460 Gilbertson and co-workers,461 as well as the resin-bound phosphine catalysts developed by Landis.462 This Section will specifically focus on the most recent advances in this area. Similarly, significant progress has been made in the application of single amino acid ligands in transition metal-catalyzed processes, such as the Cu-catalyzed cross-coupling reactions developed by Ma and co-workers463 and the C–H functionalization reactions reported by the Yu Group.464 In this Section, however, we have elected to focus only on examples employing ligands of two or more residues. Such peptide-based ligand systems have made significant recent contributions in several synthetically useful reaction classes, including allylation (Section 11.1), hydrogenation (Section 11.2), oxidation (Section 11.3), Lewis acid-catalyzed reactions (Section 11.4), Rh carbenoid-mediated insertions and cyclopropanations (Section 11.5), and cross-coupling (Section 11.6).
11.1. Allylation
The most common strategy for developing peptide-based ligands for transition metal catalysts is to incorporate a motif designed to coordinate the transition metal within the peptide sequence. In this scenario, the metal-ligating group directly interacts with the transition metal center, while the remainder of the peptide scaffold provides a (1) chiral environment for asymmetric induction through its secondary structure and (2) functional handles for orienting substrates through noncovalent interactions.
Many efforts have focused on exploring decorated phosphine ligands with peptide-based backbones. Seminal publications from Gilbertson and co-workers demonstrated that bidentate phosphine ligands, such as 11.3, could be applied as ligands in palladium-catalyzed asymmetric allylation reactions (Figure 206).465 This study employed SPPS in order to rapidly generate and assess 96 on-bead catalysts in the allylation of 11.1 with dimethylmalonate. It was found that catalysts with sequences that were expected to disrupt the designed β-turn conformation provided allylic malonate 11.2 with lower ee values, suggesting that peptide secondary structure played a role in governing the enantioselectivity of this transformation. Further examination revealed that α,α-disubstituted residues at the i+2 position, such as Aib, were detrimental to selectivity. While these types of residues are often strategically incorporated at the i+2 to favor β-turn formation,223 in this case they introduce steric bulk oriented toward the coordinated Pd, which was detrimental for catalysis. Ultimately, Pro-d-Val-containing bis(phosphine) 11.4 was identified as the lead ligand, providing 11.2 in 75% ee. This pioneering work laid the foundation for much of the research discussed in this Section by demonstrating the modularity and efficacy of peptides as ligands for transition metals.
Figure 206.
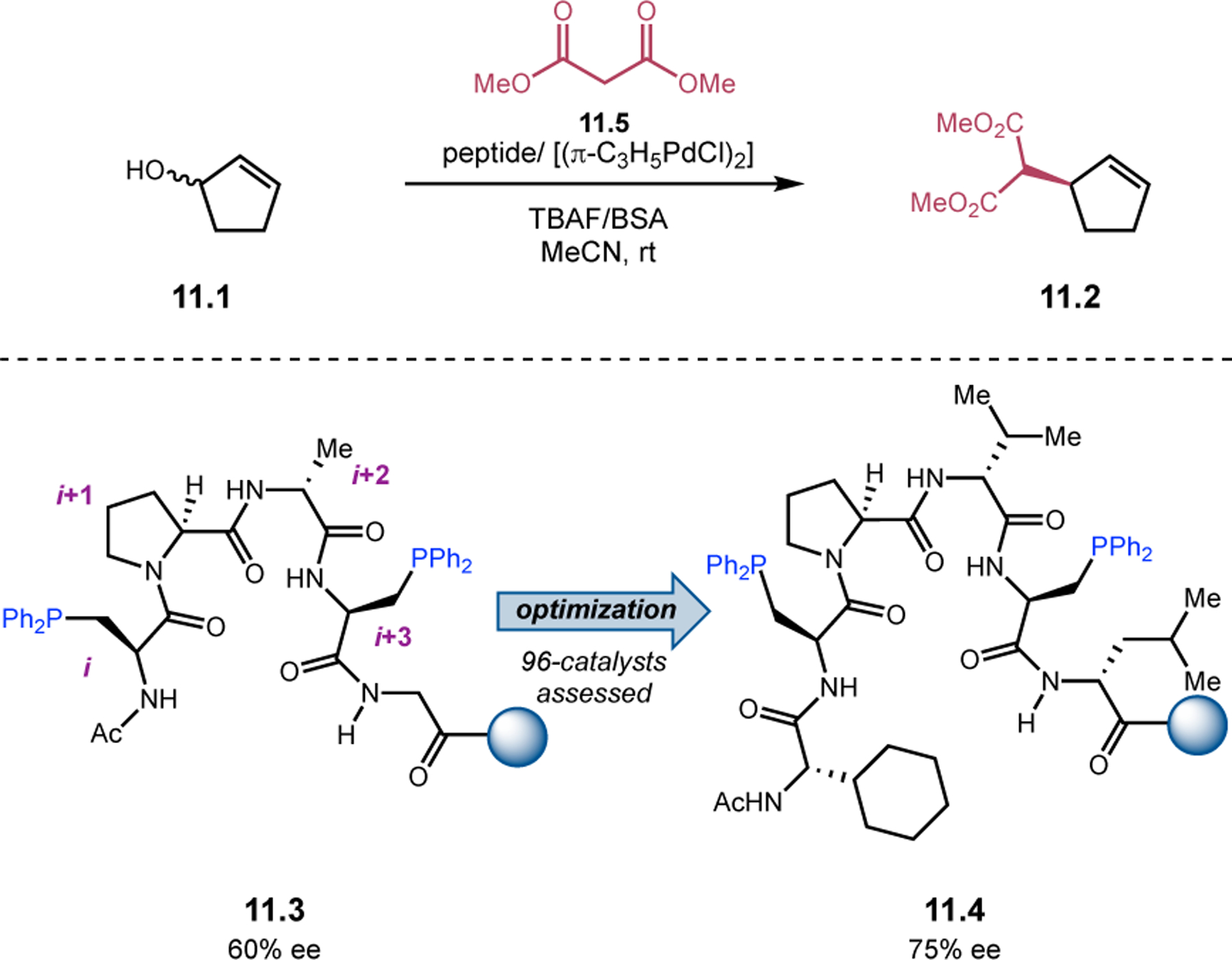
Asymmetric Pd-catalyzed allylation of dimethylmalonate with 11.1 using peptide-based phosphine ligands 11.3 and 11.4.465
More recently, Meldal and co-workers explored the benefits of modular peptide ligand synthesis in the context of Pd-catalyzed allylation reactions of malonate 11.5 and allylic acetate 11.6 (Figure 207).466 Recognizing that P,S-ligands had been previously shown to be effective in these types of reactions, a series of Cys(t-Bu)-containing peptides 11.8 was synthesized, in which a phosphine moiety was appended to the amide backbone adjacent to the thioether in an arrangement that was intended to favor chelation of Pd. Surprisingly, the configuration of allylic malonate 11.7 was found to invert not only when l-Cys(t-Bu) was replaced with d-Cys(t-Bu) (entries 1 & 2 vs. 3 & 4), but also when the number of methylene groups between the phosphine and amide backbone was changed (entries 1–4 vs. 5–8). This changes the palladacycle formed from a six-membered to a seven-membered ring. The complete reversal of enantioselectivity as the result of an additional methylene is challenging to predict a priori and emphasizes how rapid screening methods can lead to the discovery of efficient catalysts and unanticipated reactivity and selectivity outcomes. The most selective catalyst 11.8 for this reaction, which possesses d-Cys and l-Phe moieties, delivered 11.7 in 60% ee (entry 3).
Figure 207.
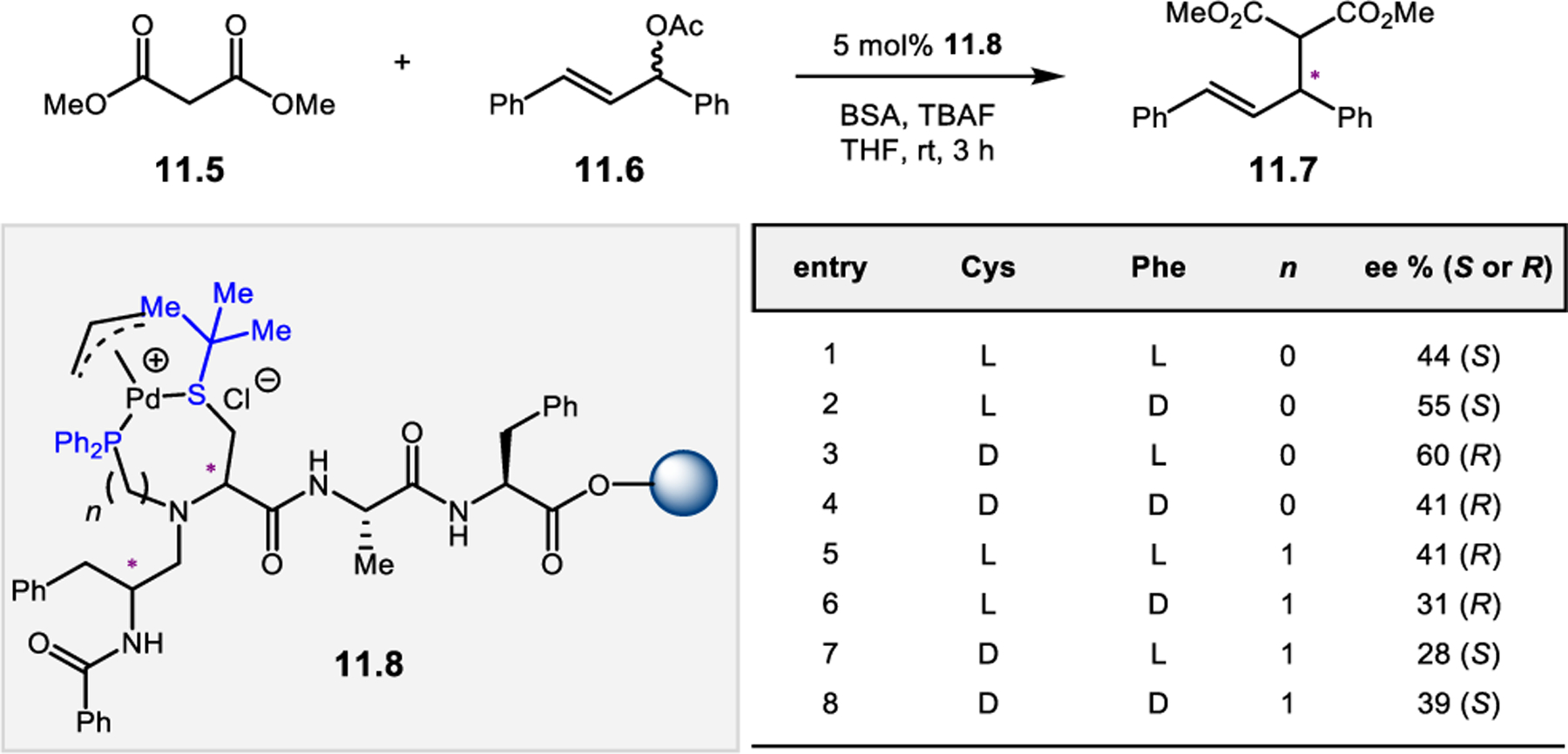
Application of peptide-based P,S-ligands 11.8 in the Pd-catalyzed allylation of malonate 11.5 with allylic acetate 11.6. Modular peptide synthesis enabled the assessment of many distinct stereochemical arrays.466
11.2. Hydrogenation
The self-assembly of subunits through complementary noncovalent interactions is an important process in nature that not only governs receptor–substrate association and protein folding, but it also enables the formation of macromolecular assemblies that are critical in biological catalysis. Inspired by this, Breit and co-workers have extensively explored the utility of spontaneous molecular assembly as a means for rapidly generating catalysts for asymmetric transformations. Their initial efforts were focused on developing peptide-based mimics of PlanePhos, a cyclophane-derived, planar-chiral phosphine ligand (Figure 208).467 While bidentate PlanePhos is a powerful ligand class, members of this family often require multistep syntheses.468 An alternative strategy would be to design a simple monodentate phosphite ligand that could be programmed to self-assemble into a π-stacked complex similar to that of PlanePhos in the presence of a transition metal (Figure 208a). Peptide chains appended to the arenes could serve this function, as their functional group-rich nature makes them strong candidates to facilitate ligand assembly through precise H-bonding networks.
Figure 208.
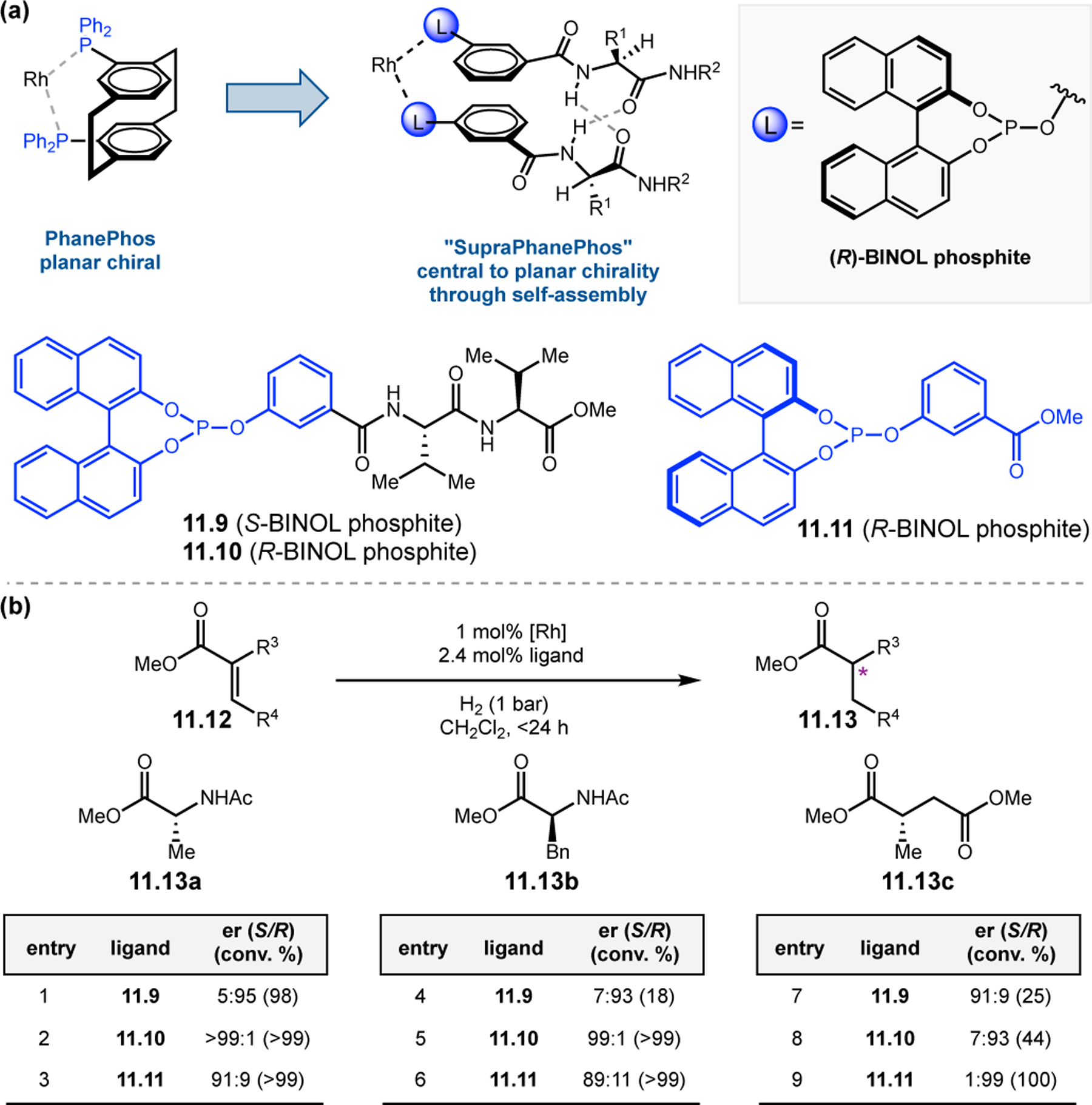
(a) Development of a peptide based “SupraPhanePhos” ligand. (b) Catalyst evaluation in the hydrogenation of α,β-unsaturated esters 11.12.467
Upon treatment of ligands 11.9 and 11.10, which contain both a BINOL-phosphite element and a peptide chain attached to the arene, with either cis-[Cl2Pt(cod)] or [Rh(cod)2]BF4, the desired homodimeric helical complexes formed.467 The structure of the Pt complex of 11.10 and its helical tail was confirmed by X-ray crystallography. However, NMR analysis revealed that while the arene stacking was stable in solution, H-bonding between peptide chains appeared to be dynamic. As a control, a Rh complex with ligand 11.11 that lacks the potential to self-assemble through H-bonding, was synthesized and evaluated. The three Rh complexes derived from ligands 11.9, 11.10, and 11.11 were next evaluated in the hydrogenation of α,β-unsaturated esters 11.12 (Figure 208b). The absolute configuration of products 11.13 was determined by the BINOL-phosphite moiety of the ligand. For 11.12a and 11.12b, the (S)-BINOL-derived catalyst led to (R)-11.13a,b (entries 1 & 4), while the (R)-BINOL catalyst diastereomer delivered (S)-11.13a,b selectively (entries 2–3 & 5–6). In the case of 11.12c, the (S)-BINOL catalyst provided the (S)-product 11.13c (entry 7), and the (R)-catalyst provided the (R)-product (entries 8 & 9). While subtle, the peptide chain does appear to be matched stereochemically with the (R)-BINOL-phosphite, as absolute enantioselectivities are higher for all substrates 11.12 with ligand 11.10 than 11.9. Intriguingly, catalyst 11.11, which does not have a peptide chain to anchor the ligand self-assembly, still provides excellent levels of enantioselectivity for all substrates and is most selective for 11.12c (entry 9).
Breit and co-workers further expanded upon this concept of self-assembly to probe the viability of developing heterodimeric ligands capable of spontaneously constructing β-sheets (Figure 209a).469 This phenomenon is not only of interest to the synthetic community as a means of generating PlanePhos mimics, but it is also relevant to the independent study of protein self-assembly in nature.470 As such, two classes of phosphine ligands, C-terminal peptidyl ligands (LC, 11.17–11.20) and N-terminal peptidyl ligands (LN, 11.21–11.23), were chosen that contained complementary amide functionality for matched H-bonding (Figure 209b). NMR and MS studies revealed that when LC and LN were added together without metal, they did not form a complex. However, upon addition of cis-[PtCl2(cod)] to a 1:1 mixture of both ligand classes, a coordination complex was observed to form. Despite the statistical probability of a 50:25:25 mixture of hetereodimeric complex to the two homodimeric complexes, heterodimer formation predominated in almost every case owing to the matched inter-peptide H-bonding. Further NMR and DFT studies confirmed the existence of the proposed β-sheet, with a non-traditional H-bond between the ortho-aryl C–H of LC and the pyridine of LN (Figure 209a). When these catalysts were applied in the hydroformylation of styrene 11.14, homodimeric complexes of both LC ligand 11.17 and LN ligand 11.22 were found to exert excellent control over the ratio of linear (L) and branched (B) products 11.15 and 11.16 (entries 1 & 2). However, the major branched aldehyde 11.15 was nearly racemic in both cases. Conversely, the heterodimeric complexes were notably more enantioselective at the expense of B/L ratio (entries 3–6). For example, the heterodimeric complex between 11.18 and 11.22 was found to deliver branched 11.15 in 87:13 B/L and 36% ee (entry 4).
Figure 209.
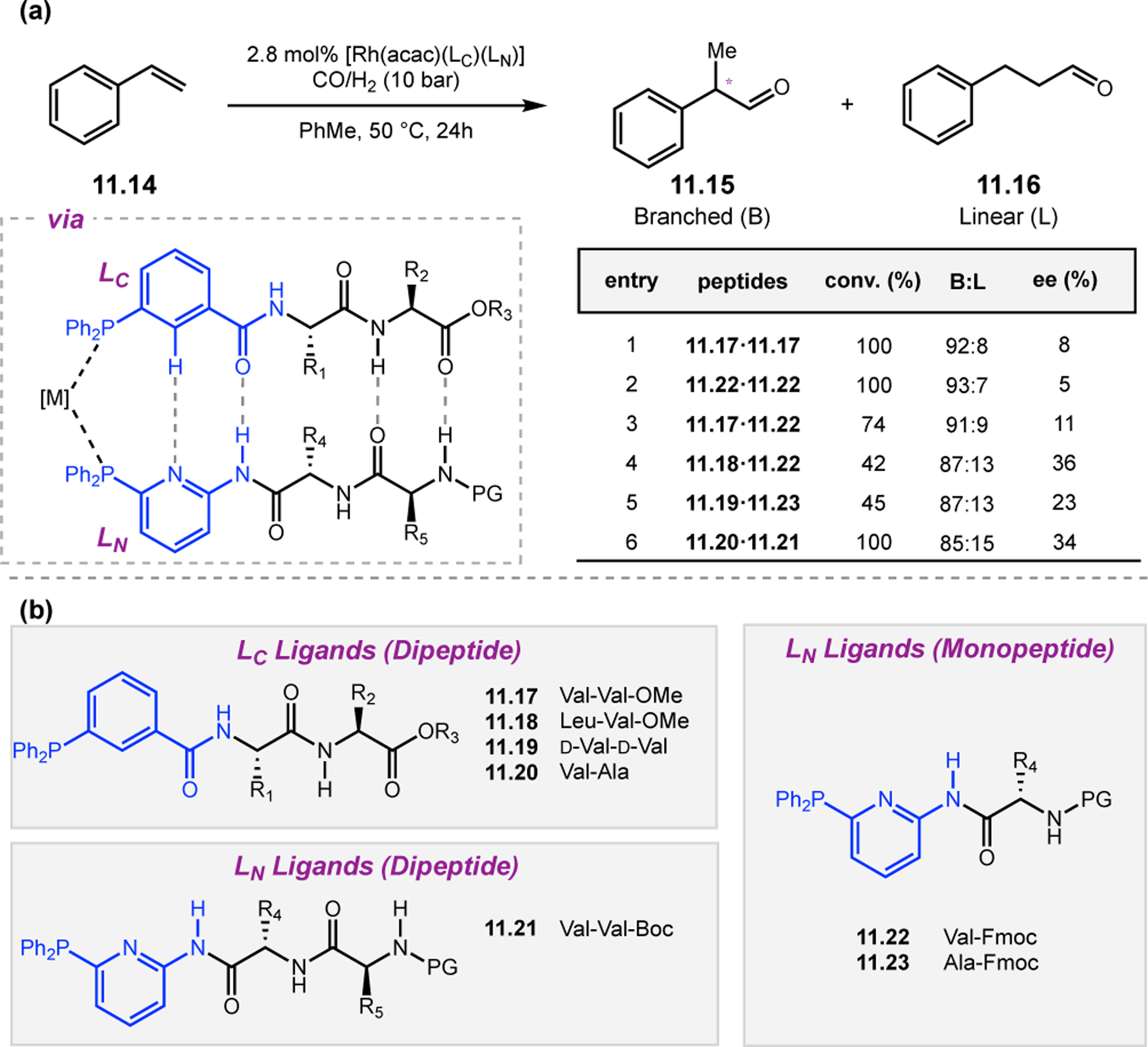
(a) Asymmetric hydroformylation of styrene 11.14 mediated by heterodimeric β-sheet-like self-assembled Rh complexes. (b) Design of various carbon linked (LC) and nitrogen linked (LN) ligands.469
In light of these publications from the Breit Group, Kirin and co-workers noted that the proposed interactions between the LC and LN ligands formed two 10-membered ring H-bonds, which is analogous to β-turn motifs (Figure 210).471 The effect of inter-strand H-bonding on ferrocene helicity had been previously studied by Herrick and co-workers,472,473 and it was found that the inter-strand H-bonding pattern within the sidechain is propagated through a “backdoor effect” to determine the helical chirality of the stacked aromatic rings (Figure 210a). The helical configuration of these ligands in turn governs the major product enantiomer observed in the hydrogenation of dehydroalanine derivative 11.12 (Figure 210b). In this way, remote chiral information is relayed to enforce the stereochemical outcome of the reaction.
Figure 210.
(a) Influence of peptide H-bonding patterns on the helical chirality of supramolecular phosphine structures. (b) Asymmetric, Rh-catalyzed hydrogenation of dehydroalanine 11.12 to afford 11.13. (c) Optimized ligand scaffolds that have been applied in the hydrogenation of 11.12.471,474,475
Keeping this mode of asymmetric induction in mind, a series of Rh catalysts were synthesized containing monodentate arylphosphine ligands similar to those explored by Breit,467 but with several key differences (Figure 210a). Initial studies focused on ligands containing para-arene substitution, which would render the system insensitive to 180° rotations about a single ligand. In combination with pendent l-amino acids, these para-substituted ligands induced P-helical chirality and ultimately provided the (R)-enantiomer of 11.13. Trimeric peptides with C-terminal amides were employed in order to maximize H-bonding interaction between strands (Figure 210c). Following optimization, peptide 11.24 was determined to be the premier ligand, affording 11.13 in >98% yield with 68% ee. In subsequent studies, the authors found that through incorporating additional H-bonding residues at the 3- and 5-positions of the phosphine ligand (e.g., 11.25), an increased number of H-bonding interactions could be promoted, further increasing enantioselectivity of 11.13 to 84% ee.474 It was generally observed that smaller peptide chains resulted in higher levels of enantioselectivity, while phosphine ligands containing longer peptide chains were less selective. Ultimately, peptide 11.25, which contains two Ala residues, was found to be the optimal ligand for the hydrogenation of 11.12. Consistent with their proposed model for “backdoor induction,” the observed (S)-enantiomer of the product was opposite to that observed with 4-substituted ligands, despite the fact that amino acids with the same sense of chirality were used in both cases. Finally, bidentate phosphines that were connected by various carbon-based linkers were explored (e.g., 11.26–11.28).475 It was found that as the rigidity of the linker increased, a concomitant increase in the enantioselectivity was generally observed, with 11.28 providing (R)-11.13 in >98% yield and 81% ee. It was proposed that this increased linker rigidity promoted H-bonding interactions between the two Ala residues, leading to a greater chirality transfer to the metal site.
In a similar way, the Neumann Group has taken advantage of the propensity of ambiphilic, anionic peptides, such as 11.29 and 11.30, to self-assemble upon treatment with cationic transition metal species (Figure 211).476,477 These peptides have been shown to self-assemble in water via coordination of the N-terminal phosphonate functionality with Ti(IV) to form macrocyclic complexes (Figure 211a). By incorporating a large number of hydrophobic residues, a porous material is formed with many chiral, hydrophobic pockets that are sequester organic reagents. This strategy was found to be highly effective for incorporating an achiral Ru complex into the chiral pocket, generating an molecular assembly capable of catalyzing the asymmetric hydrogenation of ketones, such as 11.31, to secondary alcohols, such as 11.32 (Figure 211b).477 In general, it was found that enantioselectivity correlated with the degree of hydrophobicity of the catalyst (entries 1–3), with Phg-containing 11.29 providing 11.32 in 70% ee when coupled with tetraphenyl Ru-complex c (entry 3). The same trend was observed with Leu-containing peptide 11.30 (entries 4–6), but the degree of enantioselectivity was generally lower. The authors also demonstrated that these peptide assemblies were able to encapsulate Mn-salen complexes to form chiral catalysts for the asymmetric epoxidation of styrenes 11.33 (Figure 211c).476 Peptide 11.29 effectively coordinated the Mn complex to generate assembly 11.29d, which delivered epoxides 11.34 and diols 11.35 in good yields and enantioselectivities over a modest scope (entries 1–4). In general, diols 11.35 were obtained with higher ee values.
Figure 211.
(a) Design strategy for the encapsulation of achiral catalysts in homochiral, peptide-based Ti-phosphonate assemblies in aqueous environments. (b) Asymmetric hydrogenation of acetophenone 11.31 catalyzed by achiral Ru complexes encapsulated by 11.29 and 11.30.477 (c) Asymmetric epoxidation/ring-opening of styrenes 11.33 catalyzed an achiral Mn-salen complex encapsulated by 11.29.476
In addition to the design of catalytic phosphine and phosphonate complexes that self-assemble, naturally occurring peptidic architectures have also been explored. For instance, a peptidic natural product, gramicidin S, adopts a β-hairpin conformation by virtue of its cyclic structure (Figure 212).478,479 Additionally, the two ornithine (Orn) residues orient their sidechains on the same face of the macrocycle, providing handles for functionalization with transition metal-coordinating groups. Using this parent scaffold, several peptidic arylphosphine ligands 11.36–11.39 were synthesized and examined in the asymmetric hydrogenation of dehydroalanine 11.12a. While, gramicidin-based ligands with ortho-arylphosphine (11.36) and alkylphosphine (11.39) linkages were inactive (entries 1 & 4), those with meta- (11.37) and para-linkages (11.38) to the Orn sidechains delivered 11.13a with modest enantioselectivities (entries 2 & 3). Intriguingly, ligands 11.37 and 11.38 were observed to be enantiodivergent, affording opposite enantiomers of 11.13a despite the fact that they contain identical stereoconfigurations. It was speculated that shortening the distance between the phosphine and the amide backbone may increase the enantioselectivity of the reaction by placing the metal center closer to the chiral information of gramicidin S.
Figure 212.
Application of bidentate phosphine ligands 11.36–11.39 derived from gramicidin S in the asymmetric hydrogenation of dehydroalanine 11.12a.478,479
11.3. Oxidation
Asp-containing peptides have found wide utility in peracid-mediated oxidations in recent years (Section 2.2).43,78 Based on this body of work, Costas and co-workers envisioned a complementary application of carboxylate-embedded peptides as ligands for organometallic oxidations (Figure 213).480 Fe-PDP complexes were first developed by White and co-workers in 2007 as highly selective C–H oxidation catalysts in the presence of hydrogen peroxide and acetic acid.481 The Costas Group later reported the more electron-rich Fe-PDPNMe2 complexes that were able to catalyze the epoxidations of enones with high levels of stereoselectivity.482 The authors hypothesized that carboxylate-containing peptides may serve as competent ligands for Fe-PDP, such that the selectivity afforded by the complex could be tuned via outer-sphere interactions rather than inner-sphere coordination (Figure 213a).480 This concept is fundamentally similar to how the selectivity of enzymatic Fe complexes is altered by virtue of the surrounding amino acid residues. It was found that when Fe-PDPNMe2 was paired with peptide 11.42, which contains a central d-Pro-Pro β-turn motif and a free C-terminus, high levels of selectivity could be achieved in the epoxidation of styrenes 11.40 via the carboxylate-coordinated Fe complex (Figure 213b). Epoxides 11.41 were obtained in up to 84% yield and 96:4 er under these conditions. While enantioselectivity was found to be governed by the configuration of the Fe-PDPNMe2 complex, the chirality of the peptide needed to be matched in order to ensure high levels of selectivity. Control experiments using acetic acid as a simple carboxylate ligand indicated that high selectivity is a function of not only the chiral metal center, but of the more remote stereocenters present on the peptide ligand 11.42 as well (Figure 213c).
Figure 213.
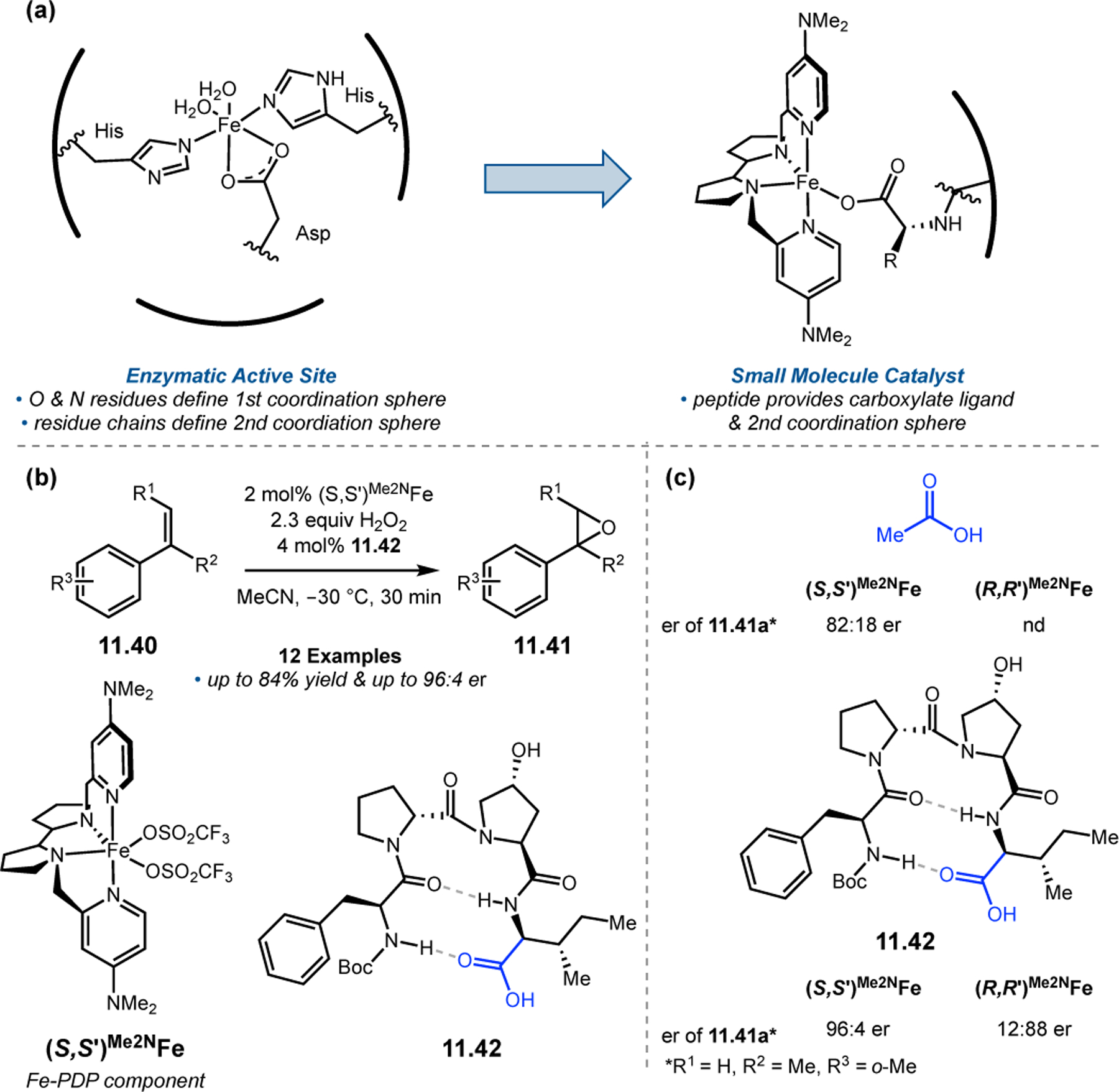
(a) Design of carboxylate-containing peptide ligands for Fe-based catalysts. (b) Asymmetric epoxidation of styrenes 11.40 mediated by Fe-PDPNMe2 and peptide ligand 11.42. (c) A comparison of enantioselectivity when acetic acid is used compared to peptide 11.42.480
11.4. Lewis Acid Catalysis
Multiple strategies have recently emerged for using peptides as chiral ligands in Lewis-acid catalyzed reactions. For example, Roelfes and co-workers took advantage of the secondary structural motifs present in bovine pancreatic polypeptide (bPP) to design a peptide-based ligand capable of coordinating Cu (Figure 214).483 The sequence of bPP is only 36 amino acids long, and its structure is comprised of a type II helix (residues 1–8), a β-turn (residues 9–12), and an α-helix (residues 13–31). These proteins naturally dimerize in water due to hydrophobic interactions to form an antiparallel homodimer (Figure 214a). It was thought that substitution of Tyr7 (shown as “N” in Figure 214a) for a Cu-coordinating residue, such as His or Pal, would be ideal, since this residue is found at the dimer interface such that a bidentate Cu complex could be formed upon dimerization via coordination with the “N” residue on both peptides. The authors therefore synthesized and designed a series of bPP mutants 11.43–11.47, in which Tyr7 is replaced with His, 4-Pal, & 3-Pal (Figure 214b). These ligands were then evaluated in Lewis acid-catalyzed reactions of enones 11.48 and 11.49 using Cu(II) salts (Figure 214c). In the Diels–Alder reaction of 11.48 with cyclopentadiene, peptide 11.45—the direct 3-Pal analog of bPP—delivered cycloadduct 11.50 in 73% conversion and 83% ee (entry 3). Peptide 11.46, in which the Asp10 residue is replaced with a Glu, was also found to be effective, providing 11.50 in quantitative conversion and 80% ee (entry 4). The His- and 4-Pal-containing peptides 11.43 and 11.44 were unselective in this transformation (entries 1 & 2), as was the triple bPP mutant 11.47 (entry 5). In the Cu-catalyzed conjugate addition of dimethylmalonate into 11.49, peptides 11.45 and 11.46 were again found to be selective, delivering the 11.51 in 66% and 86% ee, respectively, at good levels of conversion (entries 6 & 7). In this case, however, triple mutant 11.47 was similarly effective to 11.45. These data show that the identity of the coordinating residue has a profound impact on the selectivity of the peptidic Cu complex. In addition to providing a chiral environment for asymmetric induction, the dimerized peptides greatly accelerated reaction rates due to the effective increase in the concertation of reagents sequestered in the hydrophobic cavities.
Figure 214.
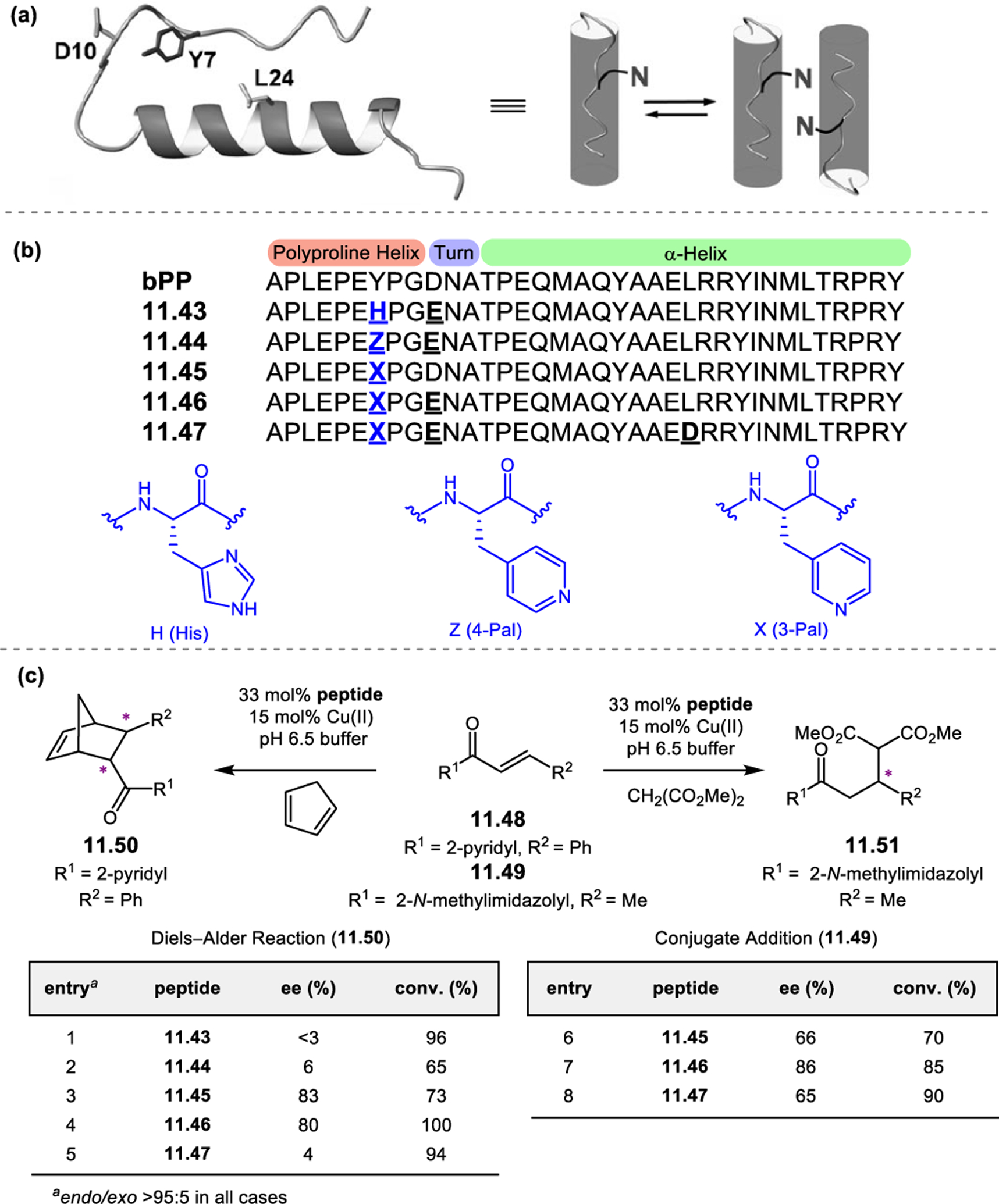
(a) Molmol representation of monomeric bPP and its dimerization driven by hydrophobic interactions. N represents a site for the introduction of a Cu-coordinating residue at the hydrophobic interface between the two monomers. (b) A comparison of ligand sequences and their relative positions in the secondary structure of bPP. (c) Asymmetric Diels-Alder and conjugate addition reactions of enones 11.48 and 11.49 catalyzed by Cu-bPP complexes in water. Reproduced in part from with permission from ref. 483. Copyright 2009 Wiley.
Herrmann and co-workers also examined the potential for small peptides to serve as chiral ligands in similar Lewis acid-catalyzed reactions (Figure 215).484 They reasoned that focusing on shorter peptide sequences would be generally more tractable than larger protein domains, facilitating the synthesis of small ligand libraries that could be evaluated in the Cu-catalyzed Diels-Alder reaction of enones 11.48. Inspired by the widespread occurrence of disulfide bridges in proteins and their influence on secondary structure, a series of cyclic peptides containing disulfide bridged Cys residues at the C- and N-termini were synthesized in order to enforce a rigid conformation within the peptide. These peptides could then ligate Cu(II) either via the free amine N-terminus or a His side-chain. While initial library screening identified selective His containing catalysts, ultimately 11.52 was found to be most selective, providing cycloadduct 11.50 in 85% ee. An Ala scan experiment was performed, wherein each individual residue is mutated to Ala in order to assess the contribution of each residue to selective catalysis. Surprisingly, the only mutation that significantly affected the reaction outcome was that of the achiral Gly residue just upstream of the N-terminal Cys; when this Gly was substituted for Ala, racemic 11.50 resulted. All of the other Ala variants provided only minor perturbations to the 85% ee result with 11.52. While these results are potentially consistent with N-terminal coordination of Cu(II), the exact site of binding was not rigorously determined. Truncation studies ultimately led to the identification of hexapeptides 11.53 and 11.54 as highly selective catalysts, which delivered 11.50 in 94% and 96% ee, respectively. Control experiments with linear variants of 11.53 and 11.54 showed no selectivity, confirming the efficacy of the cyclic structures. The scope of these ligands was then investigated in related Diels–Alder and Friedel–Crafts reactions (Figure 216). The authors focused on reactions of α,β-unsaturated 2-acylimidazole 11.55 rather than pyridine-containing 11.48 due to the challenges associated with removing pyridyl auxiliaries. Ligands 11.53 and 11.54 proved to be quite effective in both of these transformations, providing cycloadducts 11.56 in up to 99% conversion, 96:4 endo/exo ratio, and 99% ee (Figure 216a) and Friedel–Crafts products 11.57 in up to 99% conversion and 86% ee (Figure 216b).
Figure 215.
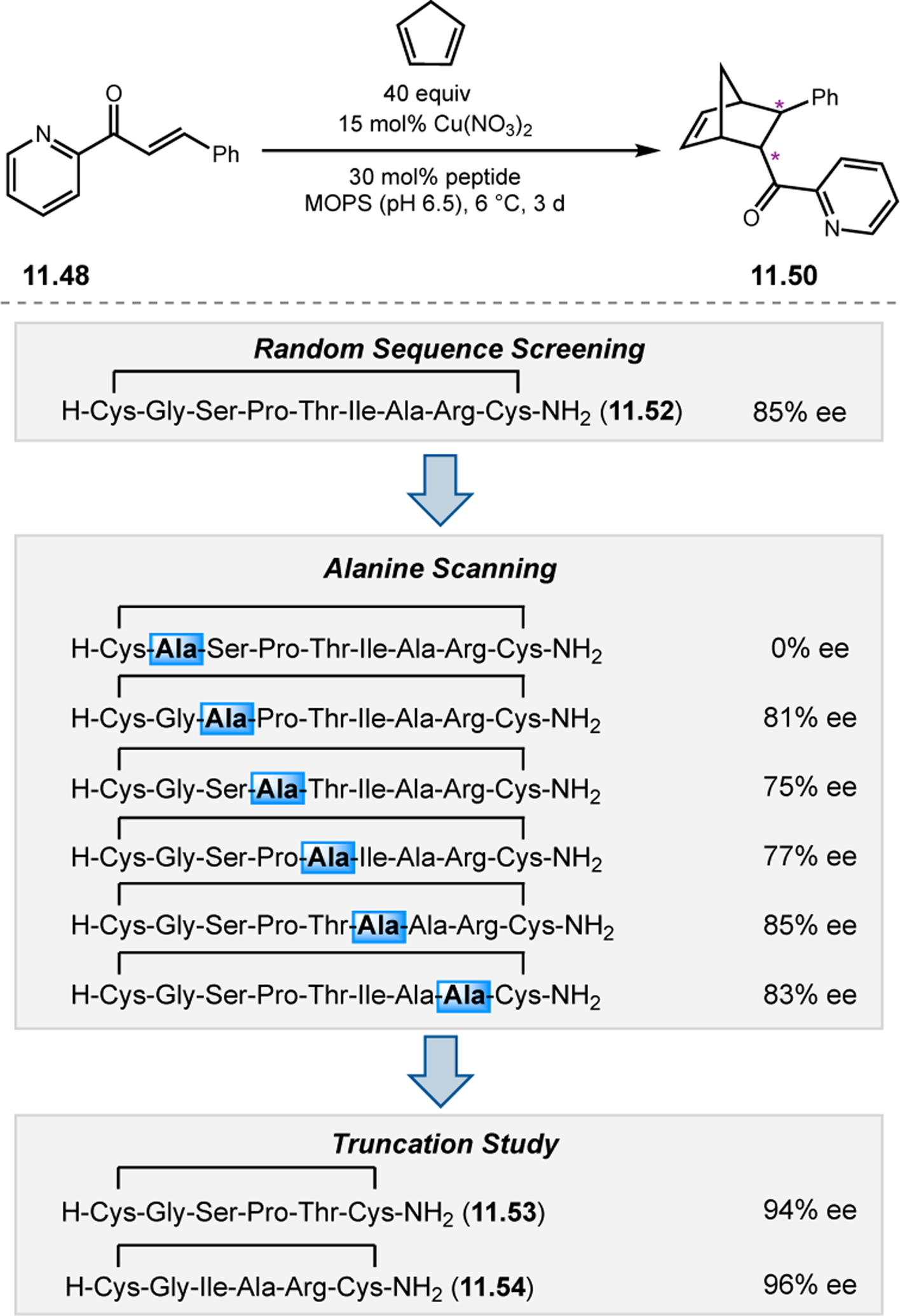
Optimization of Cu-coordinating peptides 11.52–11.54 in the asymmetric Diels–Alder reaction of enone 11.48. Ala scan and truncation experiments were used to study the peptides.484 MOPS = 3-(N-morpholino)propanesulfonic acid buffer.
Figure 216.

Application of peptides 11.53 and 11.54 in (a) asymmetric Diels-Alder reactions and (b) asymmetric Friedel–Crafts-like conjugate addition reactions of enones 11.55.484
A potentially powerful approach to the design of peptide-ligated transition metal catalysts is to take advantage of the natural metal binding domains found in enzymes. Human Ctr-1 is a membrane protein of 190 amino acids that serves as a Cu transporter. Within this protein, a short sequence of eight residues called Mets7 was known to bind well to Cu(I) and Pt(II).485 Recognizing this, Rimoldi and co-workers examined the efficacy of Mets7-Cu complexes as Lewis acid catalysts for Henry (nitroaldol) reactions of benzaldehyde 11.58 with nitromethane 11.59 (Figure 217).486 Native peptide 11.61a was found to adopt two major conformations using MD simulations, both of which were consistent with β-turn motifs (Figure 217b). However, this sequence exhibited a high degree of flexibility, and cluster analysis predicted that only 57% of the population would adopt either of these β-turn conformations at a given time. This structural heterogeneity may have resulted in lower levels of enantioselectivity in the Henry reaction, as the product was isolated in only 48% ee using 11.61a as the ligand. Recognizing that a more rigid structure may be better suited for asymmetric catalysis, modified peptide 11.61b was proposed, which possesses a nonnative β-turn-promoting motif consisting of a functionalized piperidine linked to a Pro residue that was expected to render the structure more rigid. Indeed, MD simulations predicted 91% of the population of 11.61b to occupy the two major β-turn conformations, and this ligand showed notable improvements in enantioselectivity, providing the nitroaldol product 11.60 in 75% ee at similar conversion.
Figure 217.

(a) Henry reaction of benzaldehyde 11.58 with nitromethane 11.59 used to study the application of peptides derived from the Crt-1 Mets7 motif. (b) The unmodified structure of Crt-1 Mets7 (11.61a) that naturally binds Cu and modified scaffold 11.61b, which is more conformationally rigid.486
11.5. Rhodium Carbenoid-Mediated Insertions & Cyclopropanations
The Ball Group has recognized that dirhodium tetracarboxylates are attractive transition metal complexes for incorporation into peptide scaffolds (Figure 218). Not only have they been shown to catalyze myriad carbenoid insertion reactions in water, but they have been found to be stable to carboxylate exchange in water at pH < 7.487 Previous work from the Ball Group had demonstrated that Rh(II) could be bound to peptides through Asp sidechains. These Rh-peptide catalysts were capable of site-selective carbenoid insertion reactions on complex peptide substrates.488 Emboldened by these successes, the authors began to explore the efficacy of Rh-peptide catalysts in asymmetric insertion reactions of small molecule substrates.489 Using the Si–H insertion of dimethylphenylsilane into methyl α-diazophenylacetates 11.62 as a model reaction (Figure 218a), they assessed a series of peptides containing either Glu or Asp at the i and i+4 positions so that both carboxylate functional groups would be displayed on the same face of the helical secondary structure (Figure 218b). Moreover, either monomeric or dimeric catalysts with respect to the peptide ligands were accessible from Rh-tetraacetate species. As such, monomer 11.64a, parallel dimer 11.64b, and anti-parallel dimer 11.64c were synthesized by complexation of peptide 11.64 with dirhodium tetraacetate. Evaluated in the silane insertion reaction of 11.62, the three Rh-peptide complexes exhibited different levels of enantioselectivity. Notably, anti-parallel dimer 11.64c was significantly more selective than the monomer and parallel isomer, providing silane product 11.63 in 45% ee. During ligand optimization, it was found that the anti-parallel isomer generally afforded higher levels of enantioselectivity than the other isomer across all 22 peptide sequences assessed. Modifications at residues adjacent to the Asp residues were particularly influential on the observed selectivity, presumably due to their proximity to the metal center. Ultimately, peptide 11.65 emerged from optimization as the lead ligand, providing silanes 11.63 in up to 97% yield and 99% ee. CD experiments confirmed that a helical conformation is achieved upon complexation with the dirhodium species.
Figure 218.
(a) Asymmetric Si–H insertion reaction of α-diazoacetates 11.62 with dimethylphenylsilane catalyzed by helical Rh-peptide complexes. (b) A comparison of the performance of monomeric Rh-peptide 11.64a, parallel dimeric complex 11.64b, and anti-parallel dimer 11.64c in the reaction of 11.62 with dimethylphenylsilane. (c) Peptide 11.65 was ultimately identified as the lead catalyst.489
While the parallel and anti-parallel isomers of the Rh-peptide dimers proved to be readily separable by chromatographic methods, identification proved to be more challenging, as both isomers showed similar NMR signatures, even at low temperatures, in many cases. Pyrene Excimer Fluorescence is a distance-sensitive technique that has been used as a tool in the study of peptide assembly. In this technique, fluorescence is only detected in situations where pyrene functionalities are in close proximity to one another, such that a π-stacking exciplex can be formed. In the context of dimeric Rh-peptide complexes, only the parallel configuration should exhibit fluorescence (Figure 219a). Given that the two isomers do not interconvert, even under forcing conditions, this technique was employed to unambiguously determine the orientation of isolated peptides.490 The superior performance of the anti-parallel peptide in carbenoid insertion reactions (Figure 218) is intriguing from the standpoint of molecular symmetry. While both dimeric isomers are C2-symmetric, the axis symmetry of anti-parallel 11.65-Pyr passes through both Rh atoms such that the metal centers are non-equivalent, allowing for multiple sites of catalysis (Figure 219b).
Figure 219.
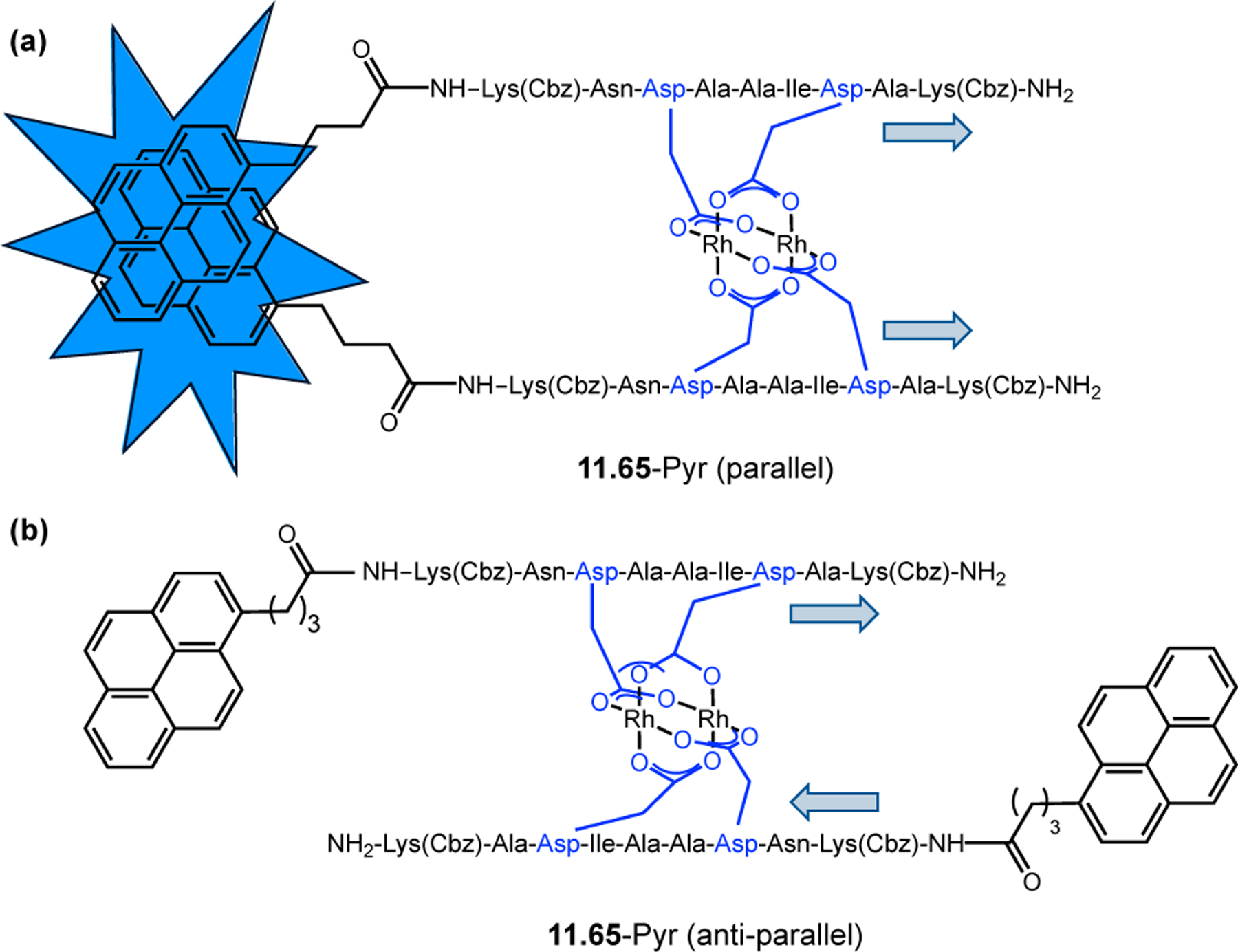
Catalyst 11.65 with appended pyrenes. While the (a) parallel isomer 11.65-Pyr that displays detectable fluorescence, (b) anti-parallel isomer 11.65-Pyr is not able to form an exciplex.490
Ball and co-workers have also applied similar Rh-peptide complexes in the asymmetric cyclopropanation of styrene derivatives (Figure 220).491 In order to accelerate the discovery of selective ligand sequences, a small library of peptides was synthesized on-bead (Figure 220a). Because it is challenging to synthesize dimeric peptide complexes on solid support, the authors elected to evaluate monomeric Rh-peptide complexes on-bead wherein two acetate ligands replaced a second peptide, and assumed that the selectivity of an optimized mono-peptide ligand would be improved upon conversion into a dimer complex with two peptides. However, while several dimeric peptides outperformed their monomeric, on-bead counterparts, no direct correlation was observed. Moreover, some peptides that had performed poorly on-bead demonstrated high enantioselectivities as the dimer complexes, suggesting that some viable candidates had been excluded using this method. However, the majority of unoptimized sequences were not effective catalysts. Following optimization, complex 11.68 was identified, which catalyzed the cyclopropanation of styrenes with α-diazophenylacetates 11.66 to deliver 11.67 in up to 95% yield and 95% ee over a modest scope (Figure 220b). Intriguingly, complex 11.69 also emerged from this screening strategy, delivering ent-11.67 in up to 94% yield and 97% ee. Peptides 11.68 and 11.69 are not enantiomers of one another, and yet they deliver enantiodivergent outcomes. Because these peptides are both comprised of only l-residues and differ only at residues flanking the active ligand motifs, it is possible that enantiodivergence stems from differences in secondary structure.
Figure 220.
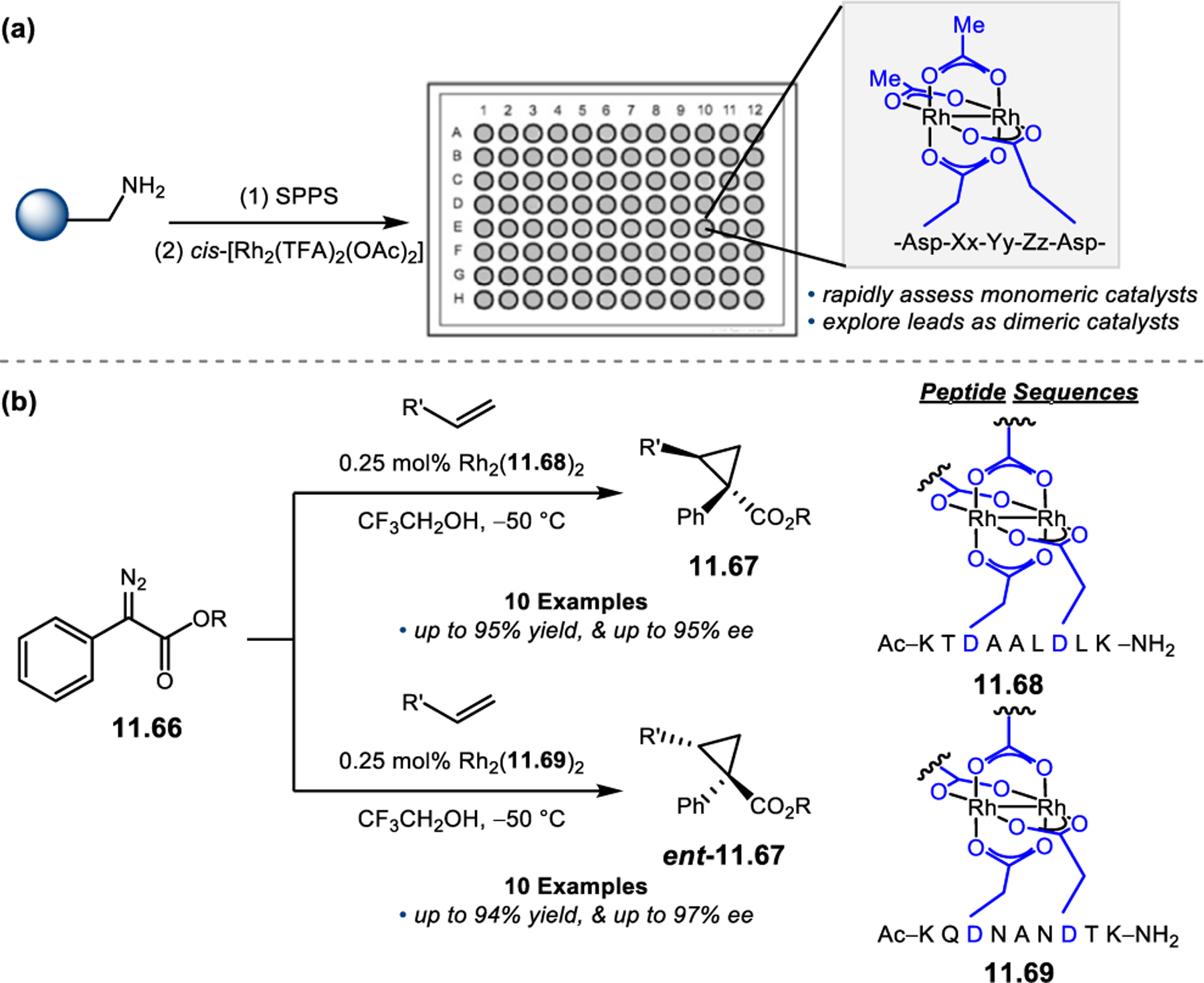
(a) Synthesis and screening of monomeric catalysts “on-bead” in order the rapidly identify selective sequences. (b) Application of optimized dimeric catalysts 11.68 and 11.69 in the asymmetric cyclopropanation of styrenes with α-diazophenylacetate 11.66, wherein divergent enantioselectivity is observed between the two peptides.491
In light of these results, the Ball Group next sought to develop monomeric Rh-peptide scaffolds that showed comparable reactivity and selectivity to the previously reported dimers and that would allow for on-bead screening of catalysts (Figure 220).492 A contributing factor to the poor performance of monomeric Rh-peptide complexes is the lack of symmetry, which leaves the two Rh atoms in chemically unique environments, each of which can catalyze the reaction with different levels of selectivity. In order to address this, the Ball and co-workers evaluated a library of peptides which contained adjacent His residues that could potentially coordinate to an exposed axial site of the Rh complex (Figure 221a). This was hypothesized to inactivate a less selective site of catalysis, which could improve the selectivity of the overall transformation. Following optimization, peptide ligand 11.70 was identified (Figure 2213b), which was found to be highly reactive and selective in the cyclopropanation of alkenes 11.71 with α-diazophenylacetates 11.66, providing cyclopropanes 11.72 in up to 99% yield and 99% ee over a modest scope (Figure 221c). In contrast to the previously optimized catalysts, this new His-containing peptide only showed high enantioselectivity as an on-bead catalyst as opposed to in solution. It was shown that axial binding of the His residue disrupts the helical structure of the peptide, which enables additional intermolecular His–Rh interactions with the more active Rh atom instead (Figure 2213a). While intermolecular His–Rh coordination keeps the helical structure intact, this results in the aggregation of the peptide catalysts, decreasing the enantioselectivity of the reaction. Conversely, on-bead catalysts have a limited ability to engage in intermolecular interactions, and do not aggregate. Ultimately, this strategy simultaneously simplified catalyst synthesis by removing the requirement to separate parallel and anti-parallel dimer isomers and enabled the rapid on-bead screening of catalysts, which led to the identification of more selective catalysts for this known cyclopropanation reaction.
Figure 221.
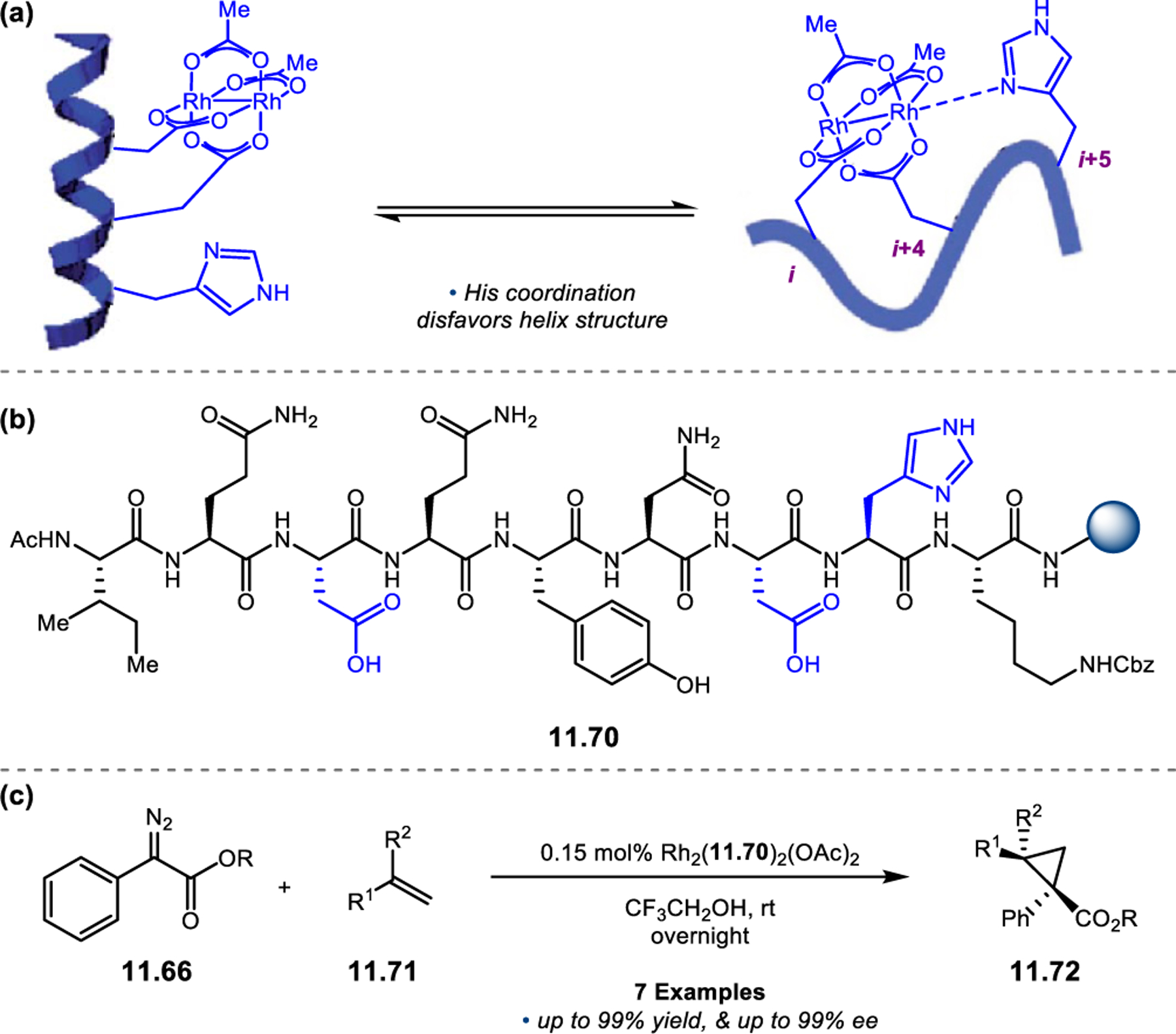
(a) Disruption of helicity and inactivation of one Rh-site via His coordination to an adjacent axial position on Rh. (b) Optimized peptide 11.70 for cyclopropanation that was identified form on-bead screening of monomeric Rh-peptide complexes. (c) Asymmetric cyclopropanation of olefins 11.71 with α-diazophenylacetates 11.66 catalyzed by resin-supported tripodal Rh-peptide complex 11.70.492
Section 11.6. Cross-Coupling
While examples of asymmetric cross-coupling are increasingly common in the literature, most reported examples involve bond formation directly at the newly formed stereogenic element.493–495 In contrast, cross-coupling reactions that establish chirality in the broader context of the molecule, as is the case of desymmetrization reactions, are relatively rare. In such transformations, chiral information needs to be relayed between the site of bond formation and the remote stereogenic element. This problem is further exacerbated when the catalyst is required to discriminate between remote enantiotopic sites, as is the case in desymmetrization reactions. Our group has recently detailed several reports wherein diarylmethine substrates, such as 11.73, are desymmetrized via Cu-catalyzed cross-coupling reactions to form C–C,496 C–O,497 and C–N bonds (Figure 222).498 Key to the success of these coupling reactions was the development of peptide-based ligands containing an N-terminal tetramethylguanidinylated Asp residue (TMG-Asp), which is linked to the peptide scaffold through its sidechain. This active ligand moiety is proposed to form a 5-membered N,O-chelate with the Cu(I) catalyst that facilitates bond formation.
Figure 222.
Desymmetrization of diarylmethine 11.73 via Cu-catalyzed C–C, C–O, and C–N cross-coupling reactions.496–498 Reactivity was enabled by peptide-based ligands containing TMG-Asp residues at the N-terminus, which are proposed to form a 5-membered metallocycle with the Cu(I) catalyst.
The TMG-Asp ligand moiety was originally developed for the cross-coupling of 11.73 with malonate nucleophiles to deliver C–C coupled products 11.74 (Figure 222).496 In order to relay stereochemical information between the prochiral center of 11.73 and the site of bond formation, additional recognition elements were required. It was hypothesized that a catalyst capable of interfacing with both termini of the substrate would enable close catalyst–substrate interactions and assist in the discrimination of the remote enantiotopic aryl bromides. The presence of a C-terminal carboxylate in peptide-based ligand 11.77 was found to improve enantioselectivity significantly. This carboxylate moiety was initially thought to interact with the distal site of 11.73 in a salt bridging interaction with the deprotonated trifluoroacetamide. In order to probe this hypothesis, a series of kinetic resolution experiments were performed on racemic substrates 11.80 (Figure 223). While racemic diarylmethine 11.80a, which lacks one of the aryl bromides, was processed with a moderate krel of 5.3, compound 11.80b lacking a trifluoroacetamide gave a high krel of 11.3, suggesting that the proposed salt bridging interaction was not operative in the selective desymmetrization pathway. Moreover, compound 11.80c, which lacks both bromide and trifluoroacetamide substituents on the distal arene, was also processed with a high krel of 10.3. To compliment these findings, substrates lacking aryl functionality altogether, such as 11.80d and 11.80e, afforded low krel values. Taken together, these data suggest that the aryl group itself, rather than any substituent, was required for strong catalyst–substrate interactions with peptide 11.77. As such, we ultimately proposed that the C-terminal carboxylate of the peptide interacted with the distal unfunctionalized terminus of the substrate through a cation-π interaction that is not possible with substrates 11.80d and 11.80e.
Figure 223.
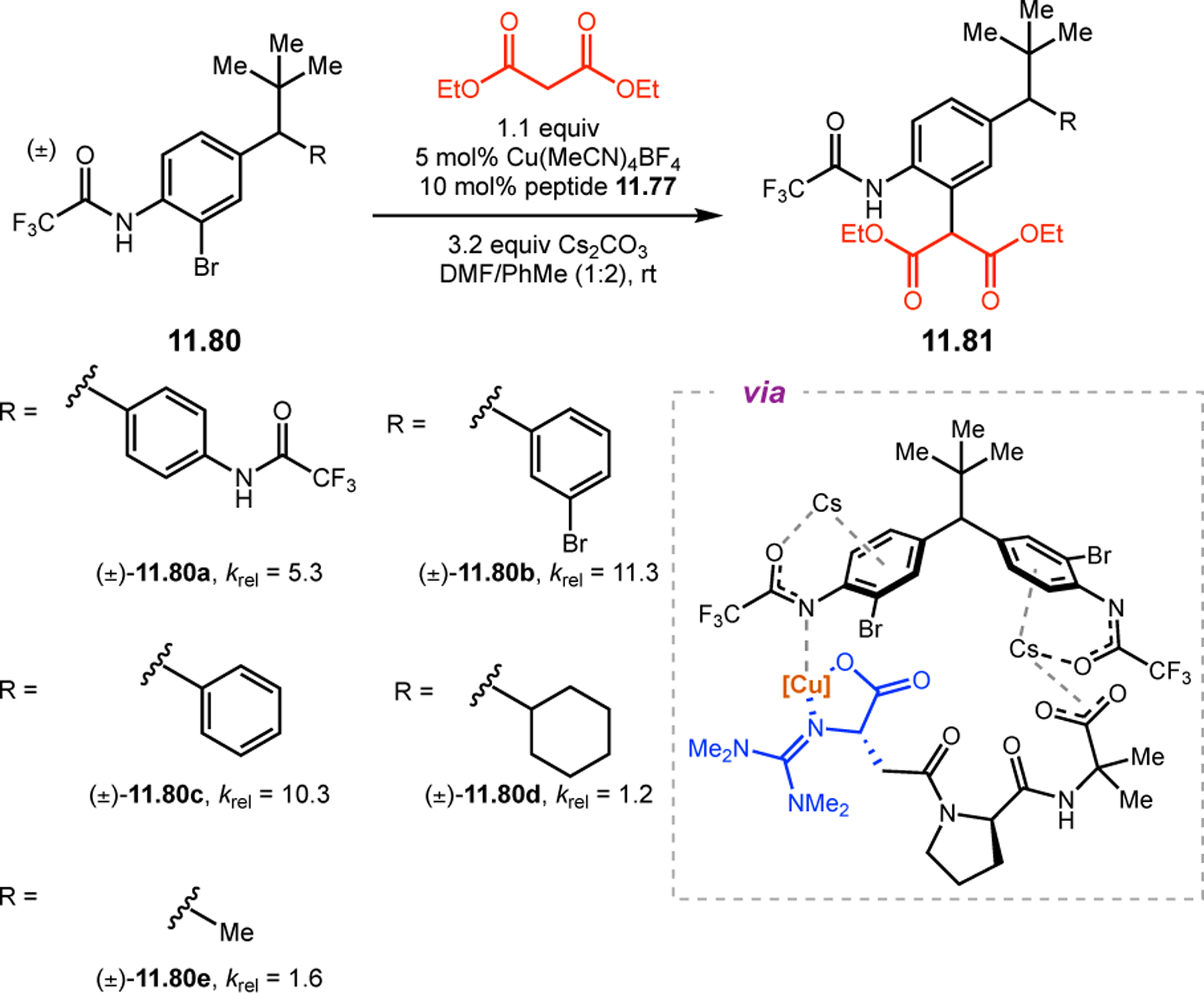
Kinetic resolutions of racemic substrates 11.80 informed a self-consistent model for catalyst–substrate interactions for the transformation depicted in Figure 222.496
During subsequent studies examining C–O bond formation using phenol nucleophiles to deliver diarylethers 11.75, stereochemical relationships within the peptide ligand were more rigorously explored (Figure 222). Although tetrapeptide 11.78—which contains four different residues—was found to be optimal with respect to yield and enantioselectivity, only the stereocenters at the i and i+1 positions were found to impact catalytic performance to a significant degree. Additionally, the i position was shown to be stereodetermining, as inverting this chiral center resulted in ent-11.75. In contrast, substitutions at other positions provided nearly identical results in most cases to those observed when 11.78 was employed. Using peptide ligand 11.78, a broad scope of diarylethers 11.75 could be accessed in up to 71% yield and 99:1 er. Notably, ortho-substituted phenols, a challenging class of substrates in other cross coupling methodologies, were viable nucleophiles in this Cu-peptide-mediated reaction. Furthermore, phenol substrates bearing benzylic alcohols as well as indole functionality were well tolerated, with exclusive coupling at the phenolic oxygen.
While exploring the viability of nitrogen-based nucleophiles in the desymmetrization of 11.73, it was found that both anilines and amines afforded the desired N-arylated product. However, due to the high nucleophilicity of the resulting diarylamine product, partial cyclization would occur for many substrates, affording the corresponding benzimidazoles 11.76 (Figure 222). As such, we elected to expose the crude reaction mixtures to acidic conditions in order to fully drive cyclization to completion. Under the optimized reaction conditions, a variety of anilines with diverse electronics and substitution patterns were found to be well tolerated by the newly optimized ligand 11.79, providing benzimidazoles 11.76 in up to 71% yield and >99:1 er. Similarly, benzylic, primary, and secondary amines were also found to be effective coupling partners. Intriguingly, upon acid-mediated cyclodehydration of the intermediate diarylaniline species containing sufficiently bulky ortho functionality, a chiral axis was formed as a result of hindered bond rotation about the C–N bond. We questioned if this process could be performed under catalyst control to selectively set an additional element of chirality within the same scaffold. Several pThr-containing peptide catalysts (see Section 3) were evaluated in the atroposelective cyclodehydration of diarylaniline 11.82 to benzimidazole 11.83 (Figure 224a). Peptide 11.84 emerged as highly selective for this transformation, providing 11.83 in 99% conversion and 96:4 er (entry 1). The configuration of the pThr β-stereocenter and the nature of the β-turn loop region was found to have influence the sense of asymmetric induction, as allothreonine-containing peptide 11.85 delivered ent-11.83 in 99% conversion and 17:83 er (entry 2). When this strategy was applied to enantioenriched diarylmethine 11.86, both diastereomers of product could be accessed with high diastereo- and enantioselectivity, under complete catalyst control (Figure 224b).
Figure 224.

(a) Atroposelective cyclodehydration of trifluoroacetamides 11.82 to benzimidazoles 11.83 catalyzed by pThr-containing peptides. (b) Catalyst-controlled diastereoselective cyclodehydration of enantioenriched 11.86.498
12. Conclusion & Outlook
Peptide-based catalysts now encompass many classes and have been applied to numerous reaction types. It seems inevitable that the field will continue to record impressive successes as this approach to catalysis is implemented in increasingly complicated reaction classes and substrate types. As a result, while opportunities for expansion seem easy to anticipate, it is more challenging to extract the broader lessons that these peptides may teach. For example, are we moving closer to an era in which it may be possible to design effective peptidic catalysts from first principles, or will a reliance on integrated design and screening approaches persist well into the future? The use of modern physical organic tools presents exciting opportunities to push the field toward a deeper understanding and, eventually, explicit molecular design.499,500 Yet, it also seems likely that at least some stochastic aspect of the catalyst discovery process will persist. In this sense, we are reminded of the evolutionary history of enzymes. While convergence on a finite number of protein folds supports catalysis of reactions in far greater number,77 it is also true that the nuances of catalyst variation within enzyme classes can be very great, and of course evolvable to the desires of humans directing evolution.53 Very likely, peptide-based catalysts—and maybe all classes of catalysts—will be subject to the same opportunities for convergence on a finite number of powerful scaffolds. However, adaptability to applications in the essentially infinite diversity presented by chemical space501 will continue to generate more catalysts for application-specific settings. In this sense, it is well to conclude on an optimistic note, celebrating the achievements of highly effective catalysts, including some that are privileged,388 while welcoming ever more catalysts into a less exclusive, but no less special club.
ACKNOWLEDGMENTS
We are grateful to the National Institute of General Medical Sciences of the National Institutes of Health (R35 GM132092) for current support of peptide-based catalysis research. A.J.M. is grateful to the NIGMS of the NIH for a postdoctoral fellowship (F32-GM128245) that supported early aspects of this work. C.R.S. thanks the NIH for a postdoctoral fellowship (F32-GM13307301). E.A.S. gratefully acknowledges financial support from the NSF Graduate Research Fellowship Program. We would also like to thank Carole Velleca for valuable assistance in preparing this manuscript.
AMINO ACID RESIDUE ABBREVIATIONS
- Aboc
2-aminobicyclo[2.2.2]octane carboxylic acid
- Acbc
1-aminocyclobutane carboxylic acid
- Acc
β-aminocyclopropylcarboxylic acid
- Achc
1-aminocyclohexane carboxylic acid
- Acpc
1-aminocyclopropane carboxylic acid
- AGly
γ-aminoadamantanecarboxylic acid
- Aic
2-aminoindane carboxylic acid
- Ala
alanine
- Ala(1-Pyn)
1-pyrenylalanine
- Arg
arginine
- Asn
asparagine
- Asp
aspartic acid
- Atz
tetrazolylylalanine
- Cbaa
cyclobutane γ-amino acid
- Cha
cyclohexylalanine
- Chg
cyclohexylglycine
- Cle
cycloleucine
- Cys
cysteine
- (Cys)2
cystine
- Dap
diaminopropionic acid
- Dmaa
dimethylaminoalanine
- Gln
glutamine
- Glu
glutamic acid
- Gly
glycine
- His
histidine
- Hse
homoserine
- Hyp
hydroxyproline
- Ile
isoleucine
- Leu
leucine
- Lys
lysine
- α-Mba
α-methylbenzylamine
- Met
methionine
- Nal
naphthylalanine
- Orn
ornithine
- 3-Pal
3-pyridylalanine
- Phe
phenylalanine
- Phg
phenylglycine
- Pip
pipecolic acid
- Pmh
π-methylhistidine
- Pro
proline
- pThr
phosphothreonine
- Ser
serine
- Thr
threonine
- Tle
tert-leucine
- TMG-Asp
tetramethylguanidinylated aspartic acid
- Trp
tryptophan
- Tyr
tyrosine
- Val
valine.
Biographies
Anthony J. Metrano was born in Boston and raised in North Attleborough, MA. He received a B.A. in chemistry and biology from the College of the Holy Cross (2011) and a Ph.D. in organic chemistry from Yale University (2017), where he was an NSF Graduate Research Fellow under the mentorship of Professor Scott J. Miller. His dissertation research focused on Brønsted base-containing peptide catalysts for dynamic kinetic resolution reactions. From 2017–2020, he was an NIH Postdoctoral Fellow in the Knowles Group at Princeton University, where he studied photoredox methods for olefin hydrofunctionalization utilizing a proton-coupled electron transfer strategy. He is currently a Senior Scientist in Medicinal Chemistry at AstraZeneca’s Oncology R&D site in Waltham, MA.
Alex J. Chinn began his chemical career at The College of William and Mary, where he received a B.S. in Chemistry. He went on to obtain a Ph.D. in chemistry from Yale University in 2019 under the guidance of Professor Scott J. Miller. While at Yale, his research focused on the design of peptide-based ligands and their application in asymmetric cross-coupling reactions. He is currently conducting postdoctoral research at Princeton University with Professor Abigail G. Doyle.
Christopher R. Shugrue is from Hartford, CT. He obtained a B.A. in chemistry from the College of the Holy Cross in 2013. Under the supervision of Professor Scott J. Miller, he received a Ph.D. in chemistry from Yale University in 2019, where he was also an NSF Graduate Research Fellow. He is currently an NIH Postdoctoral Fellow at Massachusetts Institute of Technology in the laboratory of Professor Bradley L. Pentelute.
Elizabeth A. Stone is from Rhinebeck, New York. She obtained a B.A. in chemistry and a minor in English from Connecticut College in 2016, where she conducted research in the laboratories of both Professor Timo V. Ovaska and Professor Stanton Ching. She is currently pursuing her Ph.D. in organic chemistry at Yale University as an NSF Graduate Research Fellow under the guidance of Professor Scott J. Miller.
Byoungmoo Kim received a B.Sc. in Biochemistry at the University of Waterloo, Canada, in 2008 and a Ph.D. in organic chemistry under the supervision of Professor Vy M. Dong at the University of Toronto and partly at the University of California, Irvine in 2014. In the Dong Group, he developed new stereoselective methods using rhodium catalysts. He then undertook a postdoctoral position in Professor Scott J. Miller’s research group at Yale University (2014–2019), where he developed new, chiral, peptidic ligands and discovered unique stereochemical phenomena. In the Fall of 2019, he started his independent career as an assistant professor in the Department of Chemistry at Clemson University. His group is interested in developing new synthetic methods for biomacromolecule functionalization.
Scott J. Miller is from Buffalo, NY. He received his B.A. (1989), M.A. (1989), and Ph.D. (1994) from Harvard University, working with Professor David Evans as an NSF Predoctoral Fellow. He was an NSF Postdoctoral Fellow at the California Institute of Technology with Robert Grubbs before joining the faculty at Boston College in 1996. Since 2006, he has been on the faculty at Yale University.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Breslow R Biomimetic Chemistry: Biology as an Inspiration. J. Biol. Chem 2009, 284, 1337–1342. [DOI] [PubMed] [Google Scholar]
- (2).Wells JA; McClendon CL Reaching for High-Hanging Fruit in Drug Discovery at Protein–Protein Interfaces. Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- (3).Jencks WP Catalysis in Chemistry and Enzymology; Dover Publications: New York, 1987. [Google Scholar]
- (4).Davie EAC; Mennen SM; Xu Y; Miller SJ Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev 2007, 107, 5759–5812. [DOI] [PubMed] [Google Scholar]
- (5).Knowles RR; Jacobsen EN Attractive Noncovalent Interactions in Asymmetric Catalysis: Links Between Enzymes and Small Molecule Catalysts. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 20678–20685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Miller SJ In Search of Peptide-Based Catalysts for Asymmetric Organic Synthesis. Acc. Chem. Res 2004, 37, 601–610. [DOI] [PubMed] [Google Scholar]
- (7).Gellman SH Foldamers: A Manifesto. Acc. Chem. Res 1998, 31, 173–180. [Google Scholar]
- (8).Miller SJ; Copeland GT; Papaioannou N; Horstmann TE; Ruel EM Kinetic Resolution of Alcohols Catalyzed by Tripeptides Containing the N-Alkylimidazole Substructure. J. Am. Chem. Soc 1998, 120, 1629–1630. [Google Scholar]
- (9).Crawford JM; Sigman MS Conformational Dynamics in Asymmetric Catalysis: Is Catalyst Flexibility a Design Element? Synthesis 2019, 51, 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dondoni A; Massi A Asymmetric Organocatalysis: From Infancy to Adolescence. Angew. Chem. Int. Ed 2008, 47, 4638–4660. [DOI] [PubMed] [Google Scholar]
- (11).MacMillan DWC The Advent and Development of Organocatalysis. Nature 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]
- (12).Doyle AG; Jacobsen EN Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev 2007, 107, 5713–5743. [DOI] [PubMed] [Google Scholar]
- (13).Deuss PJ; den Heeten R; Laan W; Kamer PCJ Bioinspired Catalyst Design and Artificial Metalloenzymes. Chem. Eur. J 2011, 17, 4680–4698. [DOI] [PubMed] [Google Scholar]
- (14).Wennemers H Asymmetric Catalysis with Peptides. Chem. Commun 2011, 47, 12036–12041. [DOI] [PubMed] [Google Scholar]
- (15).Lewandowski B; Wennemers H Asymmetric Catalysis with Short-Chain Peptides. Curr. Opin. Chem. Biol 2014, 22, 40–46. [DOI] [PubMed] [Google Scholar]
- (16).Akagawa K; Kudo K Development of Selective Peptide Catalysts with Secondary Structural Frameworks. Acc. Chem. Res 2017, 50, 2429–2439. [DOI] [PubMed] [Google Scholar]
- (17).Shugrue CR; Miller SJ Applications of Nonenzymatic Catalysts to the Alteration of Natural Products. Chem. Rev 2017, 117, 11894–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Giuliano MW; Miller SJ Site-Selective Reactions with Peptide-Based Catalysts. In Topics in Current Chemistry; Kawabata T, Ed.; Springer: Heidelberg, 2016; Vol. 372, pp 157–201. [DOI] [PubMed] [Google Scholar]
- (19).Bryliakov KP Catalytic Asymmetric Oxygenations with the Environmentally Benign Oxidants H2O2 and O2. Chem. Rev 2017, 117, 11406–11459. [DOI] [PubMed] [Google Scholar]
- (20).Zhu Y; Wang Q; Cornwall RG; Shi Y Organocatalytic Asymmetric Epoxidation and Aziridination of Olefins and Their Synthetic Applications. Chem. Rev 2014, 114, 8199–8256. [DOI] [PubMed] [Google Scholar]
- (21).Colonna S; Perdicchia D; Mauro ED Enantioselective Reactions Catalyzed by Synthetic Enzymes. A Model for Chemical Evolution. Tetrahedron: Asymmetry 2009, 20, 1709–1714. [Google Scholar]
- (22).Wakeham D; Crivoi DG; Medina F; Segarra AM; Rutland MW In-situ Study of Substrate–Catalyst Interactions in a Juliá–Colonna Epoxidation using Quartz Crystal Microbalance with Dissipation. J. Colloid Interface Sci 2016, 469, 263–268. [DOI] [PubMed] [Google Scholar]
- (23).Weyer A; Díaz D; Nierth A; Schlörer NE; Berkessel A The L-Leu Hexamer, a Short and Highly Enantioselective Peptide Catalyst for the Juliá–Colonna Epoxidation: Stabilization of a Helical Conformation in DMSO. ChemCatChem 2012, 4, 337–340. [Google Scholar]
- (24).Qiu W; He L; Chen Q; Luo W; Yu Z; Yang F; Tang J Imidazolium-Modified Poly(L-leucine) Catalyst: An Efficient and Recoverable Catalyst for Juliá–Colonna Reactions. Tetrahedron Lett 2009, 50, 5225–5227. [Google Scholar]
- (25).Geller T; Krüger CM; Militzer H-C Scoping the Triphasic/PTC Conditions for the Juliá–Colonna Epoxidation Reaction. Tetrahedron Lett 2004, 45, 5069–5071. [Google Scholar]
- (26).Akagawa K; Kudo K Asymmetric Epoxidation of α,β-Unsaturated Aldehydes in Aqueous Media Catalyzed by Resin-Supported Peptide-Containing Unnatural Amino Acids. Adv. Synth. Catal 2011, 353, 843–847. [Google Scholar]
- (27).Akagawa K; Hirata T; Kudo K Asymmetric Epoxidation of Enones by Peptide-Based Catalyst: A Strategy Inverting Juliá–Colonna Stereoselectivity. Synlett 2016, 27, 1217–1222. [Google Scholar]
- (28).Akagawa K; Suzuki R; Kudo K Development of a Peptide-Based Primary Aminocatalyst with a Helical Structure. Asian J. Org. Chem 2014, 3, 514–522. [Google Scholar]
- (29).Miranda R-A; Llorca J; Medina F; Sueiras JE; Segarra AM Asymmetric Epoxidation of Chalcone Catalyzed by Reusable poly-L-leucine Immobilized on Hydrotalcite. J. Catal 2011, 282, 65–73. [Google Scholar]
- (30).Yamagata N; Demizu Y; Sato Y; Doi M; Tanaka M; Nagasawa K; Okuda H; Kurihara M Design of a stabilized short helical peptide and its application to catalytic enantioselective epoxidation of (E)-chalcone. Tetrahedron Lett 2011, 52, 798–801. [Google Scholar]
- (31).Miller SJ; Blackwell HE; Grubbs RH Application of Ring-Closing Metathesis to the Synthesis of Rigidified Amino Acids and Peptides. J. Am. Chem. Soc 1996, 118, 9606–9614. [Google Scholar]
- (32).Blackwell HE; Sadowsky JD; Howard RJ; Sampson JN; Chao JA; Steinmetz WE; O’Leary DJ; Grubbs RH Ring-Closing Metathesis of Olefinic Peptides: Design, Synthesis, and Structural Characterization of Macrocyclic Helical Peptides. J. Org. Chem 2001, 66, 5291–5302. [DOI] [PubMed] [Google Scholar]
- (33).Walensky LD; Bird GH Hydrocarbon–Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem 2014, 57, 6275–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nagano M; Doi M; Kurihara M; Suemune H; Tanaka M Stabilized α-Helix-Catalyzed Enantioselective Epoxidation of α,β-Unsaturated Ketones. Org. Lett 2010, 12, 3564–2566. [DOI] [PubMed] [Google Scholar]
- (35).Bérubé C; Voyer N Biomimetic epoxidation in aqueous media catalyzed by cyclic dipeptides. Synth. Commun 2016, 46, 395–403. [Google Scholar]
- (36).Oku J-I; Inoue S Asymmetric Cyanohydrin Synthesis Catalysed by a Synthetic Cyclic Dipeptide. J. Chem. Soc., Chem. Commun 1981, 229–230. [Google Scholar]
- (37).De Bo G; Gall MAY; Kuschel S; De Winter J; Gerbaux P; Leigh DA An Artificial Molecular Machine that Builds an Asymmetric Catalyst. Nat. Nanotechnol 2018, 13, 381–385. [DOI] [PubMed] [Google Scholar]
- (38).Lewandowski B; De Bo G; Ward JW; Papmeyer M; Kuschel S; Aldegunde MJ; Gramlick PME; Heckmann D; Goldup SM; D’Souza DM; Fernandes AE; Leigh DA Sequence-Specific Peptide Synthesis by an Artificial Small-Molecule Machine. Science 2013, 339, 189–193. [DOI] [PubMed] [Google Scholar]
- (39).Ardila-Fierro KJ Crawford DE; Korner A; James SL; Bölm C; Hernandez JG Papain-Catalysed Mechanochemical Synthesis of Oligopeptides by Milling and Twin-Screw Extrusion: Application in the Juliá–Colonna Enantioselective Epoxidation. Green. Chem 2018, 20, 1262–1269. [Google Scholar]
- (40).Shoda S-I; Uyama H; Kadokawa J-I; Kimura S; Kobayashi S Enzymes as Green Catalysts for Precision Macromolecular Synthesis. Chem Rev 2016, 116, 2307–2413. [DOI] [PubMed] [Google Scholar]
- (41).Wang G-W Mechanochemical Organic Synthesis. Chem. Soc. Rev 2013, 42, 7668–7700. [DOI] [PubMed] [Google Scholar]
- (42).Bérubé C; Barbeau X; Lagüe P; Voyer N Revisiting the Juliá–Colonna Enantioselective Epoxidation: Supramolecular Catalysis in Water. Chem. Commun 2017, 53, 5099–5102. [DOI] [PubMed] [Google Scholar]
- (43).Peris G; Jakobsche CE; Miller SJ Aspartate-Catalyzed Asymmetric Epoxidations Reactions. J. Am. Chem. Soc 2007, 129, 8710–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kaur N; Kishore D Peroxy Acids: Role in Organic Synthesis. Synth. Commun 2014, 44, 721–747. [Google Scholar]
- (45).El-Faham A; Albericio F Peptide Coupling Reagents, More than a Letter Soup. Chem Rev 2010, 111, 6557–6602. [DOI] [PubMed] [Google Scholar]
- (46).Jakobsche CE; Peris G; Miller SJ Functional Analysis of an Aspartate-Based Epoxidation Catalyst with Amide-to-Alkene Peptidomimetic Catalyst Analogues. Angew. Chem. Int. Ed 2008, 47, 6707–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wilmot CM; Thornton JM Analysis and Prediction of the Different Types of β-Turn in Proteins. J. Mol. Biol 1988, 203, 221–232. [DOI] [PubMed] [Google Scholar]
- (48).Gante J Peptidomimetics—Tailored Enzyme Inhibitors. Angew. Chem. Int. Ed 1994, 33, 1699–1720. [Google Scholar]
- (49).Couve-Bonnaire S; Cahard D; Pannecoucke X Chiral Dipeptide Mimics Possessing a Fluoroolefin Moiety: A Relevant Tool for Conformational and Medicinal Studies. Org. Biomol. Chem 2007, 5, 1151–1157. [DOI] [PubMed] [Google Scholar]
- (50).Metrano AJ; Abascal NC; Mercado BQ; Paulson EK; Hurtley AE; Miller SJ Diversity of Secondary Structure in Catalytic Peptides with β-Turn-Biased Sequences. J. Am. Chem. Soc 2017, 139, 492–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Toste FD; Sigman MS; Miller SJ Pursuit of Noncovalent Interactions for Strategic Site-Selective Catalysis. Acc. Chem. Res 2017, 50, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Francis MF; Jacobsen EN Discovery of Novel Catalysts for Alkene Epoxidation from Metal-Binding Combinatorial Libraries. Angew. Chem. Int. Ed 1999, 38, 937–941. [DOI] [PubMed] [Google Scholar]
- (53).Arnold FH Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed 2018, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lichtor PA; Miller SJ One-Bead-One-Catalyst Approach to Aspartic Acid-Based Oxidation Catalyst Discovery. ACS Comb. Sci 2011, 13, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lichtor PA; Miller SJ Combinatorial Evolution of Site- and Enantioselective Catalysts for Polyene Epoxidation. Nat. Chem 2012, 4, 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Lichtor PA; Miller SJ Experimental Lineage and Functional Analysis of a Remotely Directed Peptide Epoxidation Catalyst. J. Am. Chem. Soc 2014, 136, 5301–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Copeland GT; Miller SJ Selection of Enantioselective Acyl Transfer Catalysts from a Pooled Peptide Library through a Fluorescence-Based Activity Assay: An Approach to Kinetic Resolution of Secondary Alcohols of Broad Structural Scope. J. Am. Chem. Soc 2001, 123, 6496–6502. [DOI] [PubMed] [Google Scholar]
- (58).Lam KS; Lebl M; Krchňak V The “One-Bead-One-Compound” Combinatorial Library Method. Chem. Rev 1997, 97, 411–448. [DOI] [PubMed] [Google Scholar]
- (59).Chait BT; Wang R; Beavis RC; Kent SBH Protein Ladder Sequencing. Science 1993, 262, 89–92. [DOI] [PubMed] [Google Scholar]
- (60).Romney DK; Colvin SM; Miller SJ Catalyst Control over Regio- and Enantioselectivity in Baeyer–Villiger Oxidations of Functionalized Ketones. J. Am. Chem. Soc 2014, 136, 14019–14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Denisov IG; Makris TM; Sligar SG; Schlichting I Structure and Chemistry of Cytochrome P450. Chem. Rev 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- (62).Van Tamelen EE; Heys JR Enzymic Epoxidation of Squalene Variants. J. Am. Chem. Soc 1975, 97, 1252–1253. [DOI] [PubMed] [Google Scholar]
- (63).Budt K-H; Vatele J-M; Kishi Y Terminal Epoxidation of Farnesate Attached to Helical Peptides. J. Am. Chem. Soc 1986, 108, 6080–6082. [DOI] [PubMed] [Google Scholar]
- (64).Abascal NC; Lichtor PA; Giuliano MW; Miller SJ Function-Oriented Investigations of a Peptide-Based Catalyst that Mediates Enantioselective Allylic Alcohol Epoxidation. Chem. Sci 2014, 5, 4504–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Brunger AT Version 1.2 of the Crystallography and NMR System. Nat. Protoc 2007, 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- (66).Kolundzic F; Noshi MN; Tjandra M; Movassaghi M; Miller SJ Chemoselective and Enantioselective Oxidation of Indoles Employing Aspartyl Peptide Catalysts. J. Am. Chem. Soc 2011, 133, 9104–9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Mercado-Marin EV; Garcia-Reynaga PG; Romminger S; Pimenta EF; Romney DK; Lodewyk MW; Williams DE; Andersen RJ; Miller SJ; Tantillo DJ; Berlinck RGS; Sarpong R Total Synthesis and Isolation of Citrinalin and Cyclopiamine Congeners. Nature 2014, 509, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Wang Z-X; Tu Y; Frohn M; Zhang J-R; Shi Y An Efficient Catalytic Asymmetric Epoxidation Method. J. Am. Chem. Soc 1997, 119, 11224–11235. [Google Scholar]
- (69).Romney DK; Miller SJ A Peptide-Embedded Trifluoromethyl Ketone Catalyst for Enantioselective Epoxidation. Org. Lett 2012, 14, 1138–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Wong OA; Shi Y Organocatalytic Oxidation. Asymmetric Epoxidation of Olefins Catalyzed by Chiral Ketones and Iminium Salts. Chem. Rev 2008, 108, 3958–3987. [DOI] [PubMed] [Google Scholar]
- (71).Featherston AL; Miller SJ Synthesis and Evaluation of Phenylalanine-Derived Trifluoromethyl Ketones for Peptide-Based Oxidation Catalysis. Bioorg. Med. Chem 2016, 24, 4871–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Limnios D; Kokotos CG 2,2,2-Trifluoroacetophenone: An Organocatalyst for an Environmentally Friendly Epoxidation of Alkenes. J. Org. Chem 2014, 79, 4270–4276. [DOI] [PubMed] [Google Scholar]
- (73).Zhou L; Liu X; Ji J; Zhang Y; Wu W; Liu Y; Lin L; Feng X Regio- and Enantioselective Baeyer–Villiger Oxidation: Kinetic Resolution of Racemic 2-Substituted Cyclopentanones. Org. Lett 2014, 16, 3938–3941. [DOI] [PubMed] [Google Scholar]
- (74).ten Brink G-J; Arends IWCE; Sheldon RA The Baeyer–Villiger Reaction: New Developments toward Greener Procedures. Chem. Rev 2004, 104, 4105–4123. [DOI] [PubMed] [Google Scholar]
- (75).Peris G; Miller SJ A Nonenzymatic Acid/Peracid Catalytic Cycle for the Baeyer–Villiger Oxidation. Org. Lett 2008, 10, 3049–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Abascal NC; Miller SJ Solution Structures and Molecular Associations of a Peptide-Based Catalyst for the Stereoselective Baeyer–Villiger Oxidation. Org. Lett 2016, 18, 4646–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Anantharaman V; Aravind L; Koonin EV Emergence of Diverse Biochemical Activities in Evolutionarily Conserved Structural Scaffolds of Proteins. Curr. Opin. Chem. Biol 2003, 7, 12–20. [DOI] [PubMed] [Google Scholar]
- (78).Alford JS; Abascal NC; Shugrue CR; Colvin SM; Romney DK; Miller SJ Aspartyl Oxidation Catalysts That Dial In Functional Group Selectivity, along with Regio- and Stereoselectivity. ACS Cent. Sci 2016, 2, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Giuliano MW; Lin C-Y; Romney DK; Miller SJ; Anslyn EV A Synergistic Combinatorial and Chiroptical Study of Peptide Catalysts for Asymmetric Baeyer–Villiger Oxidation. Adv. Synth. Catal 2015, 357, 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Lin C-Y; Giuliano MW; Ellis BD; Miller SJ; Anslyn EV From Substituent Effects to Applications: Enhancing the Optical Response of a Four-Component Assembly for Reporting ee Values. Chem Sci 2016, 7, 4085–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Terada M Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations. Synthesis 2010, 2010, 1929–1982. [Google Scholar]
- (82).Parmar D; Sugiono E; Raja S; Rueping M Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev 2014, 114, 9047–9153. [DOI] [PubMed] [Google Scholar]
- (83).Xu S; Wang Z; Zhang X; Zhang X; Ding K Chiral Brønsted Acid Catalyzed Asymmetric Baeyer–Villiger Reaction of 3-Substituted Cyclobutanones by Using Aqueous H2O2. Angew. Chem. Int. Ed 2008, 47, 2840–2843. [DOI] [PubMed] [Google Scholar]
- (84).Xu S; Wang Z; Li Y; Zhang X; Wang H; Ding K Mechanistic Investigation of Chiral Phosphoric Acid Catalyzed Asymmetric Baeyer–Villiger Reaction of 3-Substituted Cyclobutanones with H2O2 as the Oxidant. Chem. Eur. J 2010, 16, 3021–3035. [DOI] [PubMed] [Google Scholar]
- (85).Featherston AL; Shugrue CR; Mercado BQ; Miller SJ Phosphothreonine (pThr)-Based Multifunctional Peptide Catalysis for Asymmetric Baeyer–Villiger Oxidations of Cyclobutanones. ACS Catal 2019, 9, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Shugrue CR; Miller SJ Phosphothreonine as a Catalytic Residue in Peptide‐Mediated Asymmetric Transfer Hydrogenations of 8-Aminoquinolines. Angew. Chem. Int. Ed 2015, 54, 11325–11328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Shugrue CR; Featherston AL; Lackner RM; Lin A; Miller SJ Divergent Stereoselectivity in Phosphothreonine (pThr)-Catalyzed Reductive Aminations of 3-Amidocyclohexanones. J. Org. Chem 2018, 83, 4491–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Arakawa Y; Yamanomoto K; Kita H; Minagawa K; Tanaka M; Haraguchi N; Itsuno S; Imada Y Design of Peptide-Containing N5-Unmodified Neutral Flavins that Catalyze Aerobic Oxygenations. Chem. Sci 2017, 8, 5468–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Bruice TC Mechanisms of Flavin Catalysis. Acc. Chem. Res 2002, 13, 256–262. [Google Scholar]
- (90).Hill MD Recent Strategies for the Synthesis of Pyridine Derivatives. Chem. Eur. J 2010, 16, 12052–12062. [DOI] [PubMed] [Google Scholar]
- (91).Miyano S; Lu LDL; Viti SM; Sharpless KB Kinetic Resolution of Racemic β-Hydroxy Amines by Enantioselective N-Oxide Formation. J. Org. Chem 1983, 48, 3608–3611. [Google Scholar]
- (92).Hsieh S-Y; Tang Y; Crotti S; Stone EA; Miller SJ Catalytic Enantioselective Pyridine N-Oxidation. J. Am. Chem. Soc 2019, 141, 18624–18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Piwinski JJ; Wong JK; Chan TM; Green MJ; Ganguly AK Hydroxylated Metabolites of Loratadine: An Example of Conformational Diastereomers Due to Atropisomerism. J. Org. Chem 1990, 55, 3341–3350. [Google Scholar]
- (94).Rouden J; Lasne M-C; Blanchet J; Baudoux J (−)-Cytisine and Derivatives: Synthesis, Reactivity, and Applications. Chem. Rev 2013, 114, 712–778. [DOI] [PubMed] [Google Scholar]
- (95).Londregan AT; Jennings S; Wei L General and Mild Preparation of 2-Aminopyridines. Org. Lett 2010, 12, 5254–5257. [DOI] [PubMed] [Google Scholar]
- (96).Sheldon RA; Arends IWCE; ten Brink G-J; Dijksman A Green, Catalytic Oxidations of Alcohols. Acc. Chem. Res 2002, 35, 774–781. [DOI] [PubMed] [Google Scholar]
- (97).Maayan G; Ward MD; Kirshenbaum K Folded Biomimetic Oligomers for Enantioselective Catalysis. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 13679–13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Yoo B; Kirshenbaum K Peptoid Architectures: Elaboration, Actuation, and Application. Curr. Opin. Chem. Biol 2008, 12, 714–721. [DOI] [PubMed] [Google Scholar]
- (99).Akagawa K; Fujiwara T; Sakamoto S; Kudo K Efficient Asymmetric α-Oxyamination of Aldehydes by Resin-Supported Peptide Catalyst in Aqueous Media. Org. Lett 2010, 12, 1804–1807. [DOI] [PubMed] [Google Scholar]
- (100).Akagawa K; Fujiwara T; Sakamoto S; Kudo K One-Pot Sequential Alcohol Oxidation and Asymmetric α-Oxyamination in Aqueous Media Using Recyclable Resin-Supported Peptide Catalyst. Chem. Commun 2010, 46, 8040–8042. [DOI] [PubMed] [Google Scholar]
- (101).Akagawa K; Kudo K Peptide/Laccase Co-catalyzed Asymmetric α-Oxyamination of Aldehydes. Org. Lett 2011, 13, 3498–3501. [DOI] [PubMed] [Google Scholar]
- (102).Brown SP; Brochu MP; Sinz CJ; MacMillan DWC The Direct and Enantioselective Organocatalytic α-Oxidation of Aldehydes. J. Am. Chem. Soc 2003, 125, 10808–10809. [DOI] [PubMed] [Google Scholar]
- (103).Sibi MP; Hasegawa M Organocatalysis in Radical Chemistry. Enantioselective α-Oxyamination of Aldehydes. J. Am. Chem. Soc 2006, 129, 4124–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Akagawa K; Akabane H; Sakamoto S; Kudo K Asymmetric Transfer Hydrogenation in Aqueous Media Catalyzed by Resin-Supported Peptide Having a Polyleucine Tether. Tetrahedron: Asymmetry 2009, 20, 461–466. [DOI] [PubMed] [Google Scholar]
- (105).Witayakran S; Ragauskas AJ Synthetic Applications of Laccase in Green Chemistry. Adv. Synth. Catal 2009, 351, 1187–1209. [Google Scholar]
- (106).Franzén J; Marigo M; Fielenbach D; Wabnitz TC; Jørgensen KA A General Organocatalyst for Direct α-Functionalization of Aldehydes: Stereoselective C–C, C–N, C–F, C–Br, and C–S Bond-Forming Reactions. Scope and Mechanistic Insights. J. Am. Chem. Soc 2005, 127, 18296–18304. [DOI] [PubMed] [Google Scholar]
- (107).Kumaragurubaran N; Juhl K; Zhuang W; Bøgevig A; Jørgensen KA Direct L-Proline-Catalyzed Asymmetric α-Amination of Ketones. J. Am. Chem. Soc 2002, 124, 6254–6255. [DOI] [PubMed] [Google Scholar]
- (108).Tanaka T; Akagawa K; Mitsuda M; Kudo K Asymmetric α-Amination of Aldehydes by a Recyclable Resin-Supported Peptide Catalyst. Adv. Synth. Catal 2013, 355, 294–296. [Google Scholar]
- (109).Ötvös SB; Szloszár A; Mándity IM; Fülöp F Heterogeneous Dipeptide-Catalyzed α-Amination of Aldehydes in a Continuous-Flow Reactor: Effect of Residence Time on Enantioselectivity. Adv. Synth. Catal 2015, 357, 3671–3680. [Google Scholar]
- (110).Abe S; Nonaka T; Fushigama T Electroorganic Reactions on Organic Electrodes. 1. Asymmetric Reduction of Prochiral Activated Olefins on a Poly-L-Valine-Coated Graphite. J. Am. Chem. Soc 1983, 105, 3630–3632. [Google Scholar]
- (111).Abe S; Fuchigami T; Nonaka T Electrochemical Asymmetric Reduction of Prochiral Carbonyl Compounds, Oximes, and a gem-Dihalide on a Poly-L-Valine-Coated Graphite Electrode. Chem. Lett 1983, 12, 1033–1036. [Google Scholar]
- (112).Nonaka T; Abe S; Fuchigami T Electro-organic Reactions on Organic Electrodes. Part 2. Electrochemical Asymmetric Reduction of Citraconic and Mesaconic Acids on Optically-active Poly(Amino Acid)-coated Electrodes. Bull. Chem. Soc. Jpn 1983, 56, 2778–2783. [Google Scholar]
- (113).Abe S; Nonaka T Durable Chiral Graphite Electrodes Modified with Poly(L-Valine) and Poly(N-Acryloyl-L-Valine Methyl Ester). Chem. Lett 1983, 1541–1542. [Google Scholar]
- (114).Ouellet SG; Walji AM; MacMillan DWC Enantioselective Organocatlaytic Transfer Hydrogenation Reactions using Hantzsch Esters. Acc. Chem. Res 2007, 40, 1327–1339. [DOI] [PubMed] [Google Scholar]
- (115).Yang JW; Hechavarria Fonseca MT; List B A Metal-Free Transfer Hydrogenation: Organocatalytic Conjugation Reduction of α,β-Unsaturated Aldehydes. Angew. Chem. Int. Ed 2004, 43, 6660–6662. [DOI] [PubMed] [Google Scholar]
- (116).Akagawa K; Akabane H; Sakamoto S; Kudo K Organocatalytic Asymmetric Transfer Hydrogenation in Aqueous Media Using Resin-Supported Peptide Having a Polyleucine Tether. Org. Lett 2008, 10, 2035–2037. [DOI] [PubMed] [Google Scholar]
- (117).Zheng C; You SL Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors. Chem. Soc. Rev 2012, 41, 2498–2518. [DOI] [PubMed] [Google Scholar]
- (118).Akagawa K; Sakamoto S; Kudo K Direct Asymmetric Aldol Reaction in Aqueous Media using Polymer-Supported Peptide. Tetrahedron Lett 2005, 46, 8185–8187. [Google Scholar]
- (119).Akagawa K; Akabane H; Sakamoto S; Kudo K Asymmetric Transfer Hydrogenation in Aqueous Media Catalyzed by Resin-Supported Peptide Having a Polyleucine Tether. Tetrahedron: Asymmetry 2009, 20, 461–466. [DOI] [PubMed] [Google Scholar]
- (120).Akagawa K; Sen J; Kudo K Peptide-Catalyzed Regio- and Enantioselective Reduction of α,β,γ,δ-Unsaturated Aldehydes. Angew. Chem. Int. Ed 2013, 52, 11585–11588. [DOI] [PubMed] [Google Scholar]
- (121).Akiyama M; Akagawa K; Seino H; Kudo K Peptide-Catalyzed Kinetic Resolution of Planar-Chiral Metallocenes. Chem. Commun 2014, 50, 7893–7896. [DOI] [PubMed] [Google Scholar]
- (122).Akagawa K; Akiyama M; Kudo K Peptide-Catalyzed Desymmetrization of an Achiral Ferrocenyl Compound to Induce Planar Chirality. Eur. J. Org. Chem 2015, 3894–3898. [Google Scholar]
- (123).Akiyama T; Itoh J; Yokota K; Fuchibe K Enantioselective Mannich-Type Reaction Catalyzed by a Chiral Brønsted Acid. Angew. Chem. Int. Ed 2004, 43, 1566–1568. [DOI] [PubMed] [Google Scholar]
- (124).Uraguchi D; Terada M Chiral Brønsted Acid-Catalyzed Direct Mannich Reactions via Electrophilic Activation. J. Am. Chem. Soc 2004, 126, 5356–5357. [DOI] [PubMed] [Google Scholar]
- (125).Rueping M; Sugiono E; Theissmann T; Bolte M Enantioselective Brønsted Acid Catalyzed Transfer Hydrogenation: Organocatalytic Reduction of Imines. Org. Lett 2005, 7, 3781–3783. [DOI] [PubMed] [Google Scholar]
- (126).Hoffmann S; Seayad AM; List B A Powerful Brønsted Acid Catalyst for the Organocatalytic Asymmetric Transfer Hydrogenation of Imines. Angew. Chem. Int. Ed 2005, 44, 7424–7427. [DOI] [PubMed] [Google Scholar]
- (127).Storer RI; Carrera DE; Ni Y; MacMillan DWC Enantioselective Organocatalytic Reductive Amination. J. Am. Chem. Soc 2006, 128, 84–86. [DOI] [PubMed] [Google Scholar]
- (128).Rueping M; Antonchick AP; Theissmann T A Highly Enantioselective Bronsted Acid Catalyzed Cascade Reaction: Organocatalytic Transfer Hydrogenation of Quinolines and their Application in the Synthesis of Alkaloids. Angew. Chem. Int. Ed 2006, 45, 3683–3686. [DOI] [PubMed] [Google Scholar]
- (129).Yaffe MB; Elia AEH Phosphoserine/Phosphothreonine-Binding Domains. Curr. Opin. Cell Biol 2001, 13, 131–138. [DOI] [PubMed] [Google Scholar]
- (130).Shugrue CR; Sculimbrene BR; Jarvo ER; Mercado BQ; Miller SJ Outer-Sphere Control for Divergent Multicatalysis with Common Catalytic Moieties. J. Org. Chem 2019, 84, 1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Harned AM From Determination of Enantiopurity to the Construction of Complex Molecules: The Horeau Principle and its Application in Synthesis. Tetrahedron 2018, 74, 3797–3841. [Google Scholar]
- (132).Copeland GT; Jarvo ER; Miller SJ Minimal Acylase-like Peptides. Conformational Control of Absolute Stereospecificity. J. Org. Chem 1998, 63, 6784–6785. [DOI] [PubMed] [Google Scholar]
- (133).Jarvo ER; Copeland GT; Papaioannou N; Bonitatebus PJ; Miller SJ A Biomimetic Approach to Asymmetric Acyl Transfer Catalysis. J. Am. Chem. Soc 1999, 121, 11638–11643. [Google Scholar]
- (134).Liao R-Z; Santoro S; Gotsev M; Marcelli T; Himo F Origins of Stereoselectivity in Peptide-Catalyzed Kinetic Resolution of Alcohols. ACS Catal 2016, 6, 1165–1171. [Google Scholar]
- (135).Sculimbrene BR; Miller SJ Discovery of a Catalytic Asymmetric Phosphorylation through Selection of a Minimal Kinase Mimic: A Concise Total Synthesis of d-Myo-Inositol-1-Phosphate. J. Am. Chem. Soc 2001, 123, 10125–10126. [DOI] [PubMed] [Google Scholar]
- (136).Sculimbrene BR; Morgan AJ; Miller SJ Nonenzymatic Peptide-Based Catalytic Asymmetric Phosphorylation of Inositol Derivatives. Chem. Commun 2003, 1781−1785. [DOI] [PubMed] [Google Scholar]
- (137).Ardito F; Giuliani M; Perrone D; Troiano G; Muzio LL The Crucial Role of Protein Phosphorylation in Cell Signaling and its Use as Targeted Therapy (Review). Int. J. Mol. Med 2017, 40, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Ubersax JA; Ferrell JE Jr. Mechanisms of Specificity in Protein Phosphorylation. Nat. Rev. Mol. Cell. Biol 2007, 8, 530–541. [DOI] [PubMed] [Google Scholar]
- (139).Sculimbrene BR; Morgan AJ; Miller SJ Enantiodivergence in Small-Molecule Catalysis of Asymmetric Phosphorylation: Concise Total Syntheses of the Enantiomeric d-myo-Inositol-l-Phosphate and d-myo-Inositol-3-Phosphate. J. Am. Chem. Soc 2002, 124, 11653–11656. [DOI] [PubMed] [Google Scholar]
- (140).Pendaries C; Tronchère H; Racaud-Sultan C; Gaits-Iacovoni F; Coronas S; Manenti S; Gratacap M-P; Plantavid M; Payrastre B Emerging Roles of Phosphatidylinositol Monophosphates in Cellular Signaling and Trafficking. Adv. Enzyme Regul 2005, 45, 201–214. [DOI] [PubMed] [Google Scholar]
- (141).Kayser-Bricker KJ; Jordan PA; Miller SJ Catalyst-Dependent Syntheses of Phosphatidylinositol-5-Phosphate–DiC8 and its Enantiomer. Tetrahedron 2008, 64, 7015–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Longo CM; Wei Y; Roberts MF; Miller SJ Asymmetric Syntheses of l,l- and l,d-Di-myo-Inositol-1,1′-Phosphate and Their Behavior as Stabilizers of Enzyme Activity at Extreme Temperatures. Angew. Chem. Int. Ed 2009, 48, 4158–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Rodrigues MV; Borges N; Henriques M; Lamosa P; Ventura R; Fernandes C; Empadinhas N; Maycock C; da Costa MS; Santos H Bifunctional CTP:Inositol-1-Phosphate Cytidylyltransferase/CDP-Inositol:Inositol-1-Phosphate Transferase, the Key Enzyme for Di-myo-Inositol-Phosphate Synthesis in Several (Hyper)Thermophiles. J. Bacteriol 2007, 189, 5405–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Rodionov DA; Kurnasov OV; Stec B; Wang Y; Roberts MF; Osterman AL Genomic Identification and in Vitro Reconstitution of a Complete Biosynthetic Pathway for the Osmolyte Di-Myo-Inositol-Phosphate. Proc. Natl. Acad. Sci. USA 2007, 104, 4279–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Van Leeuwen SH; van der Marel GA; van Boom JH; Hensel R Synthesis of LL-Di-Myo-Inositol-1,1′-Phosphate: A Novel Inositol Phosphate from Pyrococcus woesei. Recl. Trav. Chim. Pays Bas 1994, 113, 335–336. [Google Scholar]
- (146).Jordan PA; Kayser-Bricker KJ; Miller SJ Asymmetric Phosphorylation Through Catalytic P(III) Phosphoramidite Transfer: Enantioselective Synthesis of d-myo-Inositol-6-Phosphate. Proc. Natl. Acad. Sci. USA 2010, 107, 20620–20624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Evans JW; Fierman MB; Miller SJ; Ellman JA Catalytic Enantioselective Synthesis of Sulfinate Esters Through the Dynamic Resolution of tert-Butanesulfinyl Chloride. J. Am. Chem. Soc 2004, 126, 8134–8135. [DOI] [PubMed] [Google Scholar]
- (148).Fiori KW; Puchlopek ALA; Miller SJ Enantioselective Sulfonylation Reactions Mediated by a Tetrapeptide Catalyst. Nat. Chem 2009, 1, 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Fowler BS; Mikochik PJ; Miller SJ Peptide-Catalyzed Kinetic Resolution of Formamides and Thioformamides as an Entry to Nonracemic Amines. J. Am. Chem. Soc 2010, 132, 2870–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Arp FO; Fu GC Kinetic Resolutions of Indolines by a Nonenzymatic Acylation Catalyst. J. Am. Chem. Soc 2006, 128, 14264–14265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Arai S; Chemie SB-L; Fu GC Kinetic Resolution of Amines by a Nonenzymatic Acylation Catalyst. Angew. Chem. Int. Ed 2001, 40, 234–236. [DOI] [PubMed] [Google Scholar]
- (152).Birman VB; Jiang H; Li X; Guo L; Uffman EW Kinetic Resolution of 2-Oxazolidinones via Catalytic, Enantioselective N-Acylation. J. Am. Chem. Soc 2006, 128, 6536–6537. [DOI] [PubMed] [Google Scholar]
- (153).Arnold K; Davies B; Hérault D; Whiting A Asymmetric Direct Amide Synthesis by Kinetic Amine Resolution: A Chiral Bifunctional Aminoboronic Acid Catalyzed Reaction Between a Racemic Amine and an Achiral Carboxylic Acid. Angew. Chem. Int. Ed 2008, 47, 2673–2676. [DOI] [PubMed] [Google Scholar]
- (154).De CK; Klauber EG; Seidel D Merging Nucleophilic and Hydrogen Bonding Catalysis: An Anion Binding Approach to the Kinetic Resolution of Amines. J. Am. Chem. Soc 2009, 131, 17060–17061. [DOI] [PubMed] [Google Scholar]
- (155).Kreituss I; Bode JW Catalytic Kinetic Resolution of Saturated N-Heterocycles by Enantioselective Amidation with Chiral Hydroxamic Acids. Acc. Chem. Res 2016, 49, 2807–2821. [DOI] [PubMed] [Google Scholar]
- (156).Lewis CA; Chiu A; Kubryk M; Balsells J; Pollard D; Esser CK; Murry J; Reamer RA; Hansen KB; Miller SJ Remote Desymmetrization at Near-Nanometer Group Separation Catalyzed by a Miniaturized Enzyme Mimic. J. Am. Chem. Soc 2006, 128, 16454–16455. [DOI] [PubMed] [Google Scholar]
- (157).Lewis CA; Gustafson JL; Chiu A; Balsells J; Pollard D; Murry J; Reamer RA; Hansen KB; Miller SJ A Case of Remote Asymmetric Induction in the Peptide-Catalyzed Desymmetrization of a Bis(Phenol). J. Am. Chem. Soc 2008, 130, 16358–16365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).García-Urdiales E; Alfonso I; Gotor V Enantioselective Enzymatic Desymmetrizations in Organic Synthesis. Chem. Rev 2005, 105, 313–354. [DOI] [PubMed] [Google Scholar]
- (159).Ameen D; Snape TJ Chiral 1,1-Diaryl Compounds as Important Pharmacophores. Med. Chem. Commun 2013, 4, 893–15. [Google Scholar]
- (160).Gustafson JL; Sigman MS; Miller SJ Linear Free-Energy Relationship Analysis of a Catalytic Desymmetrization Reaction of a Diarylmethane-Bis(Phenol). Org. Lett 2010, 12, 2794–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Bott G; Field LD; Sternhell S Steric Effects. A Study of a Rationally Designed System. J. Am. Chem. Soc 1980, 102, 5618–5626. [Google Scholar]
- (162).Milo A; Bess EN; Sigman MS Interrogating Selectivity in Catalysis Using Molecular Vibrations. Nature 2014, 507, 210–214. [DOI] [PubMed] [Google Scholar]
- (163).Sigman MS; Harper KC; Bess EN; Milo A The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res 2016, 49, 1292–1301. [DOI] [PubMed] [Google Scholar]
- (164).Müller CE; Wanka L; Jewell K; Schreiner PR Enantioselective Kinetic Resolution of Trans-Cycloalkane-1,2-Diols. Angew. Chem. Int. Ed 2008, 47, 6180–6183. [DOI] [PubMed] [Google Scholar]
- (165).Müller CE; Schreiner PR Organocatalytic Enantioselective Acyl Transfer onto Racemic as Well as meso Alcohols, Amines, and Thiols. Angew. Chem. Int. Ed 2011, 50, 6012–6042. [DOI] [PubMed] [Google Scholar]
- (166).Müller CE; Zell D; Hrdina R; Wende RC; Wanka L; Schuler SMM; Schreiner PR Lipophilic Oligopeptides for Chemo- and Enantioselective Acyl Transfer Reactions onto Alcohols. J. Org. Chem 2013, 78, 8465–8484. [DOI] [PubMed] [Google Scholar]
- (167).Wanka L; Cabrele C; Vanejews M; Schreiner PR γ-Aminoadamantanecarboxylic Acids Through Direct C–H Bond Amidations. Eur. J. Org. Chem 2007, 2007, 1474–1490. [Google Scholar]
- (168).Procházková E; Kolmer A; Ilgen J; Schwab M; Kaltschnee L; Fredersdorf M; Schmidts V; Wende RC; Schreiner PR; Thiele CM Uncovering Key Structural Features of an Enantioselective Peptide-Catalyzed Acylation Utilizing Advanced NMR Techniques. Angew. Chem. Int. Ed 2016, 55, 15754–15759. [DOI] [PubMed] [Google Scholar]
- (169).Shinisha CB; Sunoj RB On the Origins of Kinetic Resolution of Cyclohexane-1,2-Diols Through Stereoselective Acylation by Chiral Tetrapeptides. Org. Lett 2009, 11, 3242–3245. [DOI] [PubMed] [Google Scholar]
- (170).Müller CE; Zell D; Schreiner PR One-Pot Desymmetrization of Meso-1,2-Hydrocarbon Diols Through Acylation and Oxidation. Chem. Eur. J 2009, 15, 9647–9650. [DOI] [PubMed] [Google Scholar]
- (171).Müller CE; Hrdina R; Wende RC; Schreiner PR A Multicatalyst System for the One-Pot Desymmetrization/Oxidation of Meso-1,2-Alkane Diols. Chem. Eur. J 2011, 17, 6309–6314. [DOI] [PubMed] [Google Scholar]
- (172).Hrdina R; Müller CE; Schreiner PR Kinetic Resolution of Trans-Cycloalkane-1,2-Diols via Steglich Esterification. Chem. Commun 2010, 46, 2689–2690. [DOI] [PubMed] [Google Scholar]
- (173).Hofmann C; Schuler SMM; Wende RC; Schreiner PR En Route to Multicatalysis: Kinetic Resolution of Trans-Cycloalkane-1,2-Diols via Oxidative Esterification. Chem. Commun 2014, 50, 1221–1223. [DOI] [PubMed] [Google Scholar]
- (174).Hofmann C; Schümann JM; Schreiner PR Alcohol Cross-Coupling for the Kinetic Resolution of Diols via Oxidative Esterification. J. Org. Chem 2015, 80, 1972–1978. [DOI] [PubMed] [Google Scholar]
- (175).Hrdina R; Müller CE; Wende RC; Wanka L; Schreiner PR Enantiomerically Enriched Trans-Diols from Alkenes in One Pot: a Multicatalyst Approach. Chem. Commun 2012, 48, 2498–2500. [DOI] [PubMed] [Google Scholar]
- (176).Alachraf MW; Wende RC; Schuler SMM; Schreiner PR; Schrader W Functionality, Effectiveness, and Mechanistic Evaluation of a Multicatalyst-Promoted Reaction Sequence by Electrospray Ionization Mass Spectrometry. Chem. Eur. J 2015, 21, 16203–16208. [DOI] [PubMed] [Google Scholar]
- (177).Chen P; Qu J Backbone Modification of B-Hairpin-Forming Tetrapeptides in Asymmetric Acyl Transfer Reactions. J. Org. Chem 2011, 76, 2994–3004. [DOI] [PubMed] [Google Scholar]
- (178).Wiberg KB; Rablen PR Why Does Thioformamide Have a Larger Rotational Barrier Than Formamide? J. Am. Chem. Soc 1995, 117, 2201–2209. [Google Scholar]
- (179).Wiberg KB; Rush DJ Solvent Effects on the Thioamide Rotational Barrier: An Experimental and Theoretical Study. J. Am. Chem. Soc 2001, 123, 2038–2046. [DOI] [PubMed] [Google Scholar]
- (180).Lee HJ; Choi YS; Lee KB; Park J; Yoon CJ Hydrogen Bonding Abilities of Thioamide. J. Phys. Chem. A 2002, 106, 7010–7017. [Google Scholar]
- (181).la Cour TF Stereochemistry of Peptides Containing a Thioacyl Group. Int. J. Pept. Protein Res 1987, 30, 564–571. [DOI] [PubMed] [Google Scholar]
- (182).Artis DR; Lipton MA Conformations of Thioamide-Containing Dipeptides: a Computational Study. J. Am. Chem. Soc 1998, 120, 12200–12206. [Google Scholar]
- (183).Tran TT; Treutlein H; Burgess AW Conformational Analysis of Thiopeptides: (Φ,Ψ) Maps of Thio-Substituted Dipeptides. J. Comp. Chem 2001, 22, 1026–1037. [Google Scholar]
- (184).Gilli P; Pretto L; Bertolasi V; Gilli G Predicting Hydrogen-Bond Strengths from Acid–Base Molecular Properties. The pKa Slide Rule: Toward the Solution of a Long-Lasting Problem. Acc. Chem. Res 2009, 42, 33–44. [DOI] [PubMed] [Google Scholar]
- (185).Vasbinder MM; Jarvo ER; Miller SJ Incorporation of Peptide Isosteres Into Enantioselective Peptide-Based Catalysts as Mechanistic Probes. Angew. Chem. Int. Ed 2001, 40, 2824–2827. [DOI] [PubMed] [Google Scholar]
- (186).Dakin HD; West R A General Reaction of Amino Acids. J. Biol. Chem 1928, 78, 91–104. [Google Scholar]
- (187).Dakin HD; West R A General Reaction of Amino Acids. II. J. Biol. Chem 1928, 78, 745–756. [Google Scholar]
- (188).Vechia LD; de Souza ROMA; de Mariz e Miranda LS The Dakin-West Reaction: Past, Present and Future. Tetrahedron 2018, 74, 4359–4371. [Google Scholar]
- (189).Wende RC; Seitz A; Niedek D; Schuler SMM; Hofmann C; Becker J; Schreiner PR The Enantioselective Dakin-West Reaction. Angew. Chem. Int. Ed 2016, 128 (8), 2769–2773. [DOI] [PubMed] [Google Scholar]
- (190).Wagner JP; Schreiner PR London Dispersion in Molecular Chemistry—Reconsidering Steric Effects. Angew. Chem. Int. Ed 2015, 54, 12274–12296. [DOI] [PubMed] [Google Scholar]
- (191).Höfle G; Steglich W 4-Dialkylaminopyridines as Acylation Catalysts. III. Acylation of Sterically Hindered Alcohols. Synthesis 1972, 619–621. [Google Scholar]
- (192).Vedejs E; Chen X Kinetic Resolution of Secondary Alcohols. Enantioselective Acylation Mediated by a Chiral (Dimethylamino) Pyridine Derivative. J. Am. Chem. Soc 1996, 118, 1809–1810. [Google Scholar]
- (193).Spivey AC; Maddaford A; Fekner T; Redgrave AJ; Frampton CS Synthesis of C2-Symmetric Analogues of 4-(Pyrrolidino)Pyridine: New Chiral Nucleophilic Catalysts. J. Chem. Soc., Perkin Trans 1 2000, 3460–3468. [Google Scholar]
- (194).Wurz RP Chiral Dialkylaminopyridine Catalysts in Asymmetric Synthesis. Chem. Rev 2007, 107, 5570–5595. [DOI] [PubMed] [Google Scholar]
- (195).Mandai H; Irie S; Mitsudo K; Suga S Studies on the Synthesis of DMAP Derivatives by Diastereoselective Ugi Reactions. Molecules 2011, 16, 8815–8832. [Google Scholar]
- (196).Mandai H; Fujiwara T; Noda K; Fujii K; Mitsudo K; Korenaga T; Suga S Enantioselective Steglich Rearrangement of Oxindole Derivatives by Easily Accessible Chiral N,N-4-(Dimethylamino)Pyridine Derivatives. Org. Lett 2015, 17, 4436–4439. [DOI] [PubMed] [Google Scholar]
- (197).Schedel H; Kan K; Ueda Y; Mishiro K; Yoshida K; Furuta T; Kawabata T Asymmetric Desymmetrization of meso-Diols by C2-Symmetric Chiral 4-Pyrrolidinopyridines. Beilstein J. Org. Chem 2012, 8, 1778–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Kawabata T; Muramatsu W; Nishio T; Shibata T; Schedel H A Catalytic One-Step Process for the Chemo- and Regioselective Acylation of Monosaccharides. J. Am. Chem. Soc 2007, 129, 12890–12895 [DOI] [PubMed] [Google Scholar]
- (199).Cozett RE; Venter GA; Gokada MR; Hunter R Catalytic Enantioselective Acyl Transfer: The Case for 4-PPY with a C-3 Carboxamide Peptide Auxiliary Based on Synthesis and Modelling Studies. Org. Biomol. Chem 2016, 14, 10914–10925. [DOI] [PubMed] [Google Scholar]
- (200).Jacobsen EN Comprehensive Asymmetric Catalysis I–III; Pfaltz A, Yamamoto H, Eds.; Springer–Verlag: Berlin, 1999. [Google Scholar]
- (201).Dalko PI; Moisan L Enantioselective Organocatalysis. Angew. Chem. Int. Ed 2001, 40, 3726–3748. [DOI] [PubMed] [Google Scholar]
- (202).Tanaka K; Mori A; Inoue S The Cyclic Dipeptide Cyclo[(S)-Phenylalanyl-(S)-Histidyl] as a Catalyst for Asymmetric Addition of Hydrogen-Cyanide to Aldehydes. J. Org. Chem 1990, 55, 181–185. [Google Scholar]
- (203).Danda H; Nishikawa H; Otaka K Enantioselective Autoinduction in the Asymmetric Hydrocyanation of 3-Phenoxybenzaldehyde Catalyzed by Cyclo [(R)-Phenylalanyl-(R)-Histidyl]. J. Org. Chem 1991, 56, 6740–6741. [Google Scholar]
- (204).Kogut EF; Thoen JC; Lipton MA Examination and Enhancement of Enantioselective Autoinduction in Cyanohydrin Formation by Cyclo[(R)-His-(R)-Phe]. J. Org. Chem 1998, 63, 4604–4610. [Google Scholar]
- (205).Shvo Y; Gal M; Becker Y; Elgavi A Asymmetric Hydrocyanation of Aldehydes with Cyclo-Dipeptides: A New Mechanistic Approach. Tetrahedron: Asymmetry 1996, 7, 911–924. [Google Scholar]
- (206).Schoenebeck F; Houk KN Theoretical Study of the Catalysis of Cyanohydrin Formation by the Cyclic Dipeptide Catalyst Cyclo[(S)-His-(S)-Phe]. J. Org. Chem 2009, 74, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Akagawa K; Kudo K Asymmetric Induction by Helical Poly(Amino Acid)s in Cyanosilylation of Aldehydes. Tetrahedron Lett 2012, 53, 5981–5983. [Google Scholar]
- (208).Berkessel A Asymmetric Epoxidation of Olefins with Hydrogen Peroxide—Catalysis by an Aspartate-Containing Tripeptide. Angew. Chem. Int. Ed 2008, 47, 3677–3679. [DOI] [PubMed] [Google Scholar]
- (209).Hoveyda AH Catalyst Discovery Through Combinatorial Chemistry. Chem. Biol 1998, 5, R187–R191. [DOI] [PubMed] [Google Scholar]
- (210).Shimizu KD; Snapper ML; Hoveyda AH High-Throughput Strategies for the Discovery of Catalysts. Chem. Eur. J 1998, 4, 1885–1889. [Google Scholar]
- (211).Nitta H; Yu D; Kudo M; Mori A; Inoue S Peptide-Titanium Complex as Catalyst for Asymmetric Addition of Hydrogen Cyanide to Aldehyde. J. Am. Chem. Soc 1992, 114, 7969–7975. [Google Scholar]
- (212).Krueger CA; Kuntz KW; Dzierba CD; Wirschun WG; Gleason JD; Snapper ML; Hoveyda AH Ti-Catalyzed Enantioselective Addition of Cyanide to Imines. A Practical Synthesis of Optically Pure α-Amino Acids. J. Am. Chem. Soc 1999, 121, 4284–4285. [Google Scholar]
- (213).Porter JR; Traverse JF; Hoveyda AH; Snapper ML Enantioselective Synthesis of Arylamines Through Zr-Catalyzed Addition of Dialkylzincs to Imines. Reaction Development by Screening of Parallel Libraries. J. Am. Chem. Soc 2001, 123, 984–985. [DOI] [PubMed] [Google Scholar]
- (214).Deng H; Isler MP; Snapper ML; Hoveyda AH Aluminum-Catalyzed Asymmetric Addition of TMSCN to Aromatic and Aliphatic Ketones Promoted by an Easily Accessible and Recyclable Peptide Ligand. Angew. Chem. Int. Ed 2002, 41, 1009–1012. [DOI] [PubMed] [Google Scholar]
- (215).Degrado SJ; Mizutani H; Hoveyda AH Modular Peptide-Based Phosphine Ligands in Asymmetric Catalysis: Efficient and Enantioselective Cu-Catalyzed Conjugate Additions to Five-, Six-, and Seven-Membered Cyclic Enones. J. Am. Chem. Soc 2001, 123, 755–756. [DOI] [PubMed] [Google Scholar]
- (216).Brown MK; Degrado SJ; Hoveyda AH Highly Enantioselective Cu-Catalyzed Conjugate Additions of Dialkylzinc Reagents to Unsaturated Furanones and Pyranones: Preparation of Air-Stable and Catalytically Active Cu-Peptide Complexes. Angew. Chem. Int. Ed 2005, 44, 5306–5310. [DOI] [PubMed] [Google Scholar]
- (217).Kang SY; Park YS Discovery of Dipeptide-Derived Catalysts for the Enantioselective Addition of Dimethylzinc to Aldehydes. Eur. J. Org. Chem 2012, 2012, 1703–1706. [Google Scholar]
- (218).Reetz MT Lipases as Practical Biocatalysts. Curr. Opin. Chem. Biol 2002, 6, 145–150. [DOI] [PubMed] [Google Scholar]
- (219).Ghanem A Trends in Lipase-Catalyzed Asymmetric Access to Enantiomerically Pure/Enriched Compounds. Tetrahedron 2007, 63, 1721–1754. [Google Scholar]
- (220).Winkler FK; D’Arcy A; Hunziker W Structure of Human Pancreatic Lipase. Nature 1990, 343, 771–774. [DOI] [PubMed] [Google Scholar]
- (221).Brady L; Brzozowski AM; Derewenda ZS; Dodson E; Dodson G; Tolley S; Turkenburg JP; Christiansen L; Huge-Jensen B; Norskov L A Serine Protease Triad Forms the Catalytic Centre of a Triacylglycerol Lipase. Nature 1990, 343, 767–770. [DOI] [PubMed] [Google Scholar]
- (222).Akagawa K; Kudo K Solvolysis of Formylphenyl Esters by a Bifunctional Peptide Catalyst. Chem. Lett 2016, 45, 300–302. [Google Scholar]
- (223).Haque TS; Little JC; Gellman SH Stereochemical Requirements for β-Hairpin Formation: Model Studies with Four-Residue Peptides and Depsipeptides. J. Am. Chem. Soc 1996, 118, 6975–6985. [Google Scholar]
- (224).Fisk JS; Mosey RA; Tepe JJ The Diverse Chemistry of Oxazol-5-(4H)-Ones. Chem. Soc. Rev 2007, 36, 1432–1440. [DOI] [PubMed] [Google Scholar]
- (225).Gottwald K; Seebach D Ring Opening with Kinetic Resolution of Azlactones by Ti-TADDOLates. Tetrahedron 1999, 55, 723–738. [Google Scholar]
- (226).Berkessel A; Mukherjee S; Cleemann F; M ller TN; Lex J Second-Generation Organocatalysts for the Highly Enantioselective Dynamic Kinetic Resolution of Azlactones. Chem. Commun 2005, 1898–1900. [DOI] [PubMed] [Google Scholar]
- (227).Lee JW; Ryu TH; Oh JS; Bae HY; Bin Jang H; Song CE Self-Association-Free Dimeric Cinchona Alkaloid Organocatalysts: Unprecedented Catalytic Activity, Enantioselectivity and Catalyst Recyclability in Dynamic Kinetic Resolution of Racemic Azlactones. Chem. Commun 2009, 7224–7226. [DOI] [PubMed] [Google Scholar]
- (228).Brown SA; Parker MC; Turner NJ Dynamic Kinetic Resolution: Synthesis of Optically Active α-Amino Acid Derivatives. Tetrahedron: Asymmetry 2000, 11, 1687–1690. [Google Scholar]
- (229).Xie L; Hua W; Chan A; Leung YC Asymmetric Alcoholysis of 2-Phenyl-5(4H)-Oxazolones by the Catalytic Mixture of Cyclo[(S)-His-(S)-Phe] with Chiral Auxiliaries. Tetrahedron: Asymmetry 1999, 10, 4715–4728. [Google Scholar]
- (230).Metrano AJ; Miller SJ Peptide-Catalyzed Conversion of Racemic Oxazol-5(4H)-ones into Enantiomerically Enriched α-Amino Acid Derivatives. J. Org. Chem 2014, 79, 1542–1554. [DOI] [PubMed] [Google Scholar]
- (231).Richter F; Blomberg R; Khare SD; Kiss G; Kuzin AP; Smith AJT; Gallaher J; Pianowski Z; Helgeson RC; Grjasnow A; et al. Computational Design of Catalytic Dyads and Oxyanion Holes for Ester Hydrolysis. J. Am. Chem. Soc 2012, 134, 16197–16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Meninno S; Lattanzi A Organocatalytic Asymmetric Reactions of Epoxides: Recent Progress. Chem. Eur. J 2016, 22, 3632–3642. [DOI] [PubMed] [Google Scholar]
- (233).Chimni SS; Kumar V; Bala N Design of Peptidyl Thiourea Derivatives as Organocatalysts for the Asymmetric Ring Opening of Meso-Stilbene Oxides. Asian J. Org. Chem 2014, 3, 700–705. [Google Scholar]
- (234).Dalko PI; Moisan L In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed 2004, 43, 5138–5175. [DOI] [PubMed] [Google Scholar]
- (235).Revell JD; Wennemers H Peptidic Catalysts Developed by Combinatorial Screening Methods. Curr. Opin. Chem. Biol 2007, 11, 269–278. [DOI] [PubMed] [Google Scholar]
- (236).Raghothama SR; Awasthi SK; Balaram P B-Hairpin Nucleation by Pro-Gly β-Turns. Comparison of d-Pro-Gly and l-Pro-Gly Sequences in an Apolar Octapeptide. J. Chem. Soc., Perkin Trans 2 1998, 137–144. [Google Scholar]
- (237).Yamashita Y; Yasukawa T; Yoo W-J; Kitanosono T; Kobayashi S Catalytic Enantioselective Aldol Reactions. Chem. Soc. Rev 2018, 47, 4388–4480. [DOI] [PubMed] [Google Scholar]
- (238).Trost BM; Brindle CS The Direct Catalytic Asymmetric Aldol Reaction. Chem. Soc. Rev 2010, 39, 1600–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Bahmanyar S; Houk KN; Martin HJ; List B Quantum Mechanical Predictions of the Stereoselectivities of Proline-Catalyzed Asymmetric Intermolecular Aldol Reactions. J. Am. Chem. Soc 2003, 125, 2475–2479. [DOI] [PubMed] [Google Scholar]
- (240).Seebach D; Beck AK; Badine DM; Limbach M; Eschenmoser A; Treasurywala AM; Hobi R; Prikoszovich W; Linder B Are Oxazolidinones Really Unproductive, Parasitic Species in Proline Catalysis? Thoughts and Experiments Pointing to an Alternative View. Helv. Chim. Acta 2007, 90, 425–471. [Google Scholar]
- (241).Sharma AK; Sunoj RB Enamine Versus Oxazolidinone: What Controls Stereoselectivity in Proline-Catalyzed Asymmetric Aldol Reactions? Angew. Chem. Int. Ed 2010, 49, 6373–6377. [DOI] [PubMed] [Google Scholar]
- (242).Blackmond DG; Moran A; Hughes M; Armstrong A Unusual Reversal of Enantioselectivity in the Proline-Mediated α-Amination of Aldehydes Induced by Tertiary Amine Additives. J. Am. Chem. Soc 2010, 132, 7598–7599. [DOI] [PubMed] [Google Scholar]
- (243).Burés J; Armstrong A; Blackmond DG Kinetic Correlation Between Aldehyde/Enamine Stereoisomers in Reactions Between Aldehydes with α-Stereocenters and Chiral Pyrrolidine-Based Catalysts. Chem. Sci 2012, 3, 1273–1275. [Google Scholar]
- (244).Horecker BL; Tsolas O; Lai C-Y in The Enzymes, 1975; Vol. 7, p 213–258. [Google Scholar]
- (245).Metrano AJ; Miller SJ Peptide-Based Catalysts Reach the Outer Sphere through Remote Desymmetrization and Atroposelectivity. Acc. Chem. Res 2019, 52, 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Wu F-C; Da C-S; Du Z-X; Guo Q-P; Li W-P; Yi L; Jia Y-N; Ma X N-Primary-Amine-Terminal β-Turn Tetrapeptides as Organocatalysts for Highly Enantioselective Aldol Reaction. J. Org. Chem 2009, 74, 4812–4818. [DOI] [PubMed] [Google Scholar]
- (247).Bayat S; Tejo BA; Abdulmalek E; Salleh AB; Normi YM; Rahman MB A. Rational Design of Mimetic Peptides Based on Aldo-Ketoreductase Enzyme as Asymmetric Organocatalysts in Aldol Reactions. RSC Adv 2014, 4, 38859–38868. [Google Scholar]
- (248).D’Elia V; Zwicknagl H; Reiser O Short α/β-Peptides as Catalysts for Intra- and Intermolecular Aldol Reactions. J. Org. Chem 2008, 73, 3262–3265. [DOI] [PubMed] [Google Scholar]
- (249).De Pol S; Zorn C; Klein CD; Zerbe O; Reiser O Surprisingly Stable Helical Conformations in α/β-Peptides by Incorporation of cis-β-Aminocyclopropane Carboxylic Acids. Angew. Chem. Int. Ed 2004, 43, 511–514. [DOI] [PubMed] [Google Scholar]
- (250).Burgess K; Li S; Rebenspies J Chiral 1,3-Cyclobutane Amino Acids: Syntheses and Extended Conformations. Tetrahedron Lett 1997, 38, 1681–1684. [Google Scholar]
- (251).Aguilera J; Moglioni AG; Moltrasio GY; Ortuño RM Stereodivergent Synthesis of the First Bis(Cyclobutane) γ-Dipeptides and Mixed γ-Oligomers. Tetrahedron: Asymmetry 2008, 19, 302–308. [Google Scholar]
- (252).Aguilera J; Cobos JA; Gutiérrez-Abad R; Acosta C; Nolis P; Illa O; Ortuño RM The Role of the Chiral cis-1,3-Disubstituted 2,2-Dimethylcyclobutane Motif in the Conformational Bias of Several Types of γ-Peptides. Eur. J. Org. Chem 2013, 3494–3503. [Google Scholar]
- (253).Illa O; Porcar-Tost O; Robledillo C; Elvira C; Nolis P; Reiser O; Branchadell V; Ortuño RM Stereoselectivity of Proline/Cyclobutane Amino Acid-Containing Peptide Organocatalysts for Asymmetric Aldol Additions: A Rationale. J. Org. Chem 2018, 83, 350–363. [DOI] [PubMed] [Google Scholar]
- (254).Milbeo P; Maurent K; Moulat L; Lebrun A; Didierjean C N-Pyrrolidine-Based α/β-Peptides Incorporating ABOC, a Constrained Bicyclic β-Amino Acid, for Asymmetric Aldol Reaction Catalysis. Tetrahedron 2016, 72, 1706–1715. [Google Scholar]
- (255).Songis O; Didierjean C; Laurent C; Martinez J; Calmès M Asymmetric Diels–Alder Cycloaddition of 1-Aminocyclohexadiene to Chiral Acrylate: Synthesis of Enantiopure Bridgehead-Aminobicyclo[2.2.2]Octane-2-Carboxylic Acid Derivatives. Eur. J. Org. Chem 2007, 3166–3172. [Google Scholar]
- (256).André C; Legrand B; Deng C; Didierjean C; Pickaert G; Martinez J; Averlant-Petit MC; Amblard M; Calmes M (S)-ABOC: A Rigid Bicyclic β-Amino Acid as Turn Inducer. Org. Lett 2012, 14, 960–963. [DOI] [PubMed] [Google Scholar]
- (257).Krattiger P; Kovasy R; Revell JD; Ivan S; Wennemers H Increased Structural Complexity Leads to Higher Activity: Peptides as Efficient and Versatile Catalysts for Asymmetric Aldol Reactions. Org. Lett 2005, 7, 1101–1103. [DOI] [PubMed] [Google Scholar]
- (258).Ötvös SB; Mándity IM; Fülöp F Asymmetric Aldol Reaction in a Continuous-Flow Reactor Catalyzed by a Highly Reusable Heterogeneous Peptide. J. Catal 2012, 295, 179–185. [Google Scholar]
- (259).Aprile C; Giacalone F; Gruttadauria M; Marculescu AM; Noto R; Revell JD; Wennemers H New Ionic Liquid-Modified Silica Gels as Recyclable Materials for l-Proline- or H-Pro-Pro-Asp-NH2-Catalyzed Aldol Reaction. Green Chem 2007, 9, 1328–1334. [Google Scholar]
- (260).Zhao XS; Bao XY; Guo W; Lee FY Immobilizing Catalysts on Porous Materials. Mater. Today 2006, 9, 32–39. [Google Scholar]
- (261).Jebors S; Enjalbal C; Amblard M; Mehdi A; Subra G; Martinez J Bioorganic Hybrid OMS by Straightforward Grafting of Trialkoxysilyl Peptides. J. Mat. Chem. B 2013, 1, 2921–2925. [DOI] [PubMed] [Google Scholar]
- (262).Yan J; Wang L Asymmetric Aldol Reactions Catalyzed by Efficient and Recyclable Silica-Supported Proline-Based Peptides. Chirality 2009, 21, 413–420. [DOI] [PubMed] [Google Scholar]
- (263).Bonnefoy J; Legrand A; Quadrelli EA; Canivet J; Farrusseng D Enantiopure Peptide-Functionalized Metal-Organic Frameworks. J. Am. Chem. Soc 2015, 137, 9409–9416. [DOI] [PubMed] [Google Scholar]
- (264).Gruttadauria M; Salvo AMP; Giacalone F; Agrigento P; Noto R Enhanced Activity and Stereoselectivity of Polystyrene-Supported Proline-Based Organic Catalysts for Direct Asymmetric Aldol Reaction in Water. Eur. J. Org. Chem 2009, 5437–5444. [Google Scholar]
- (265).Lei M; Shi L; Li G; Chen S; Fang W; Ge Z; Cheng T; Li R Dipeptide-Catalyzed Direct Asymmetric Aldol Reactions in the Presence of Water. Tetrahedron 2007, 63, 7892–7898. [Google Scholar]
- (266).Hu X-M; Zhang D-X; Zhang S-Y; Wang P-A Highly Modular Dipeptide-Like Organocatalysts for Direct Asymmetric Aldol Reactions in Brine. RSC Adv 2015, 5, 39557–39564. [Google Scholar]
- (267).Córdova A; Notz W; Barbas CF III Direct Organocatalytic Aldol Reactions in Buffered Aqueous Media. Chem. Commun 2002, 3024–3025. [DOI] [PubMed] [Google Scholar]
- (268).Hayashi Y; Aratake S; Okano T; Takahashi J; Sumiya T; Shoji M Combined Proline–Surfactant Organocatalyst for the Highly Diastereo- and Enantioselective Aqueous Direct Cross-Aldol Reaction of Aldehydes. Angew. Chem. Int. Ed 2006, 45, 5527–5529. [DOI] [PubMed] [Google Scholar]
- (269).Maya V; Raj M; Singh VK Highly Enantioselective Organocatalytic Direct Aldol Reaction in an Aqueous Medium. Org. Lett 2007, 9, 2593–2595. [DOI] [PubMed] [Google Scholar]
- (270).Bayat S; Tejo BA; Salleh AB; Abdmalek E; Normi YM Various Polar Tripeptides as Asymmetric Organocatalyst in Direct Aldol Reactions in Aqueous Media. Chirality 2013, 25, 726–734. [DOI] [PubMed] [Google Scholar]
- (271).Andreu C; del Olmo M; Asensio G Effect of Addition of Lewis/Bronsted Acids in the Asymmetric Aldol Condensation Catalyzed by Trifluoroacetate Salts of Proline-Based Dipeptides. Tetrahedron 2012, 68, 7966–7972. [Google Scholar]
- (272).Chandrasekhar S; Johny K; Reddy CR Proline–Threonine Dipeptide as an Organocatalyst for the Direct Asymmetric Aldol Reaction. Tetrahedron: Asymmetry 2009, 20, 1742–1745. [Google Scholar]
- (273).Al-Momani LA Asymmetric Direct Aldol Reaction Using Modified Hydroxyproline (Hyp) Derivatives. Jordan J. Chem 2018, 13, 179–189. [Google Scholar]
- (274).Vlasserou I; Sfetsa M; Gerokonstantis D-T; Kokotos CG; Moutevelis-Minakakis P Combining Prolinamides with 2-Pyrrolidinone: Novel Organocatalysts for the Asymmetric Aldol Reaction. Tetrahedron 2018, 74, 2338–2349. [Google Scholar]
- (275).Breña-Valle LJ; Sánchez RC; Cruz-Almanza R Diastereoselective Alkylation of 1-Benzyl-(5S)-Substituted 2-Pyrrolidinones. Tetrahedron: Asymmetry 1996, 7, 1019–1026. [Google Scholar]
- (276).Chen F; Huang S; Zhang H; Liu F; Peng Y Proline-Based Dipeptides with Two Amide Units as Organocatalyst for the Asymmetric Aldol Reaction of Cyclohexanone with Aldehydes. Tetrahedron 2008, 64, 9585–9591. [Google Scholar]
- (277).De Nisco M; Pedatella S; Ullah H; Zaidi JH; Naviglio D; Özdamar Ö; Caputo R Proline–β3-Amino-Ester Dipeptides as Efficient Catalysts for Enantioselective Direct Aldol Reaction in Aqueous Medium. J. Org. Chem 2009, 74, 9562–9565. [DOI] [PubMed] [Google Scholar]
- (278).Triandafillidi I; Bisticha A; Voutyritsa E; Galiatsatou G; Kokotos CG tert-Butyl Ester or Benzylamide of the Dipeptide Pro-Gly as Organocatalysts for the Asymmetric Aldol Reaction. Tetrahedron 2015, 71, 932–940. [Google Scholar]
- (279).Bisticha A; Triandafillidi I; Kokotos CG tert-Butyl Esters of Peptides as Organocatalysts for the Asymmetric Aldol Reaction. Tetrahedron: Asymmetry 2015, 26, 102–108. [Google Scholar]
- (280).Psarra A; Kokotos CG; Moutevelis-Minakakis P tert-Butyl Esters of Tripeptides Based on Pro-Phe as Organocatalysts for the Asymmetric Aldol Reaction in Aqueous or Organic Medium. Tetrahedron 2014, 70, 608–615. [Google Scholar]
- (281).Tsandi E; Kokotos CG; Kousidou S; Ragoussis V; Kokotos G Sulfonamides of Homoproline and Dipeptides as Organocatalysts for Michael and Aldol Reactions. Tetrahedron 2009, 65, 1444–1449. [Google Scholar]
- (282).Huang W; Tian H; Xu H; Zheng L; Liu Q; Zhang S L-Valine Dipeptide Organocatalysts with Two Amide Units for the Direct Asymmetric Aldol Reaction in Brine. Catal. Lett 2011, 141, 872–876. [Google Scholar]
- (283).Wang B; Chen G.-h.; Liu L.-y.; Chang W-X; Li J A Novel Proline-Valinol Thioamide Small Organic Molecule for a Highly Enantioselective Direct Aldol Reaction. Adv. Synth. Catal 2009, 351, 2441–2448. [Google Scholar]
- (284).Rodríguez B; Bruckmann A; Bolm C A Highly Efficient Asymmetric Organocatalytic Aldol Reaction in a Ball Mill. Chem. Eur. J 2007, 13, 4710–4722. [DOI] [PubMed] [Google Scholar]
- (285).Hernández JG; Juaristi E Asymmetric Aldol Reaction Organocatalyzed by (S)-Proline-Containing Dipeptides: Improved Stereoinduction under Solvent-Free Conditions. J. Org. Chem 2011, 76, 1464–1467. [DOI] [PubMed] [Google Scholar]
- (286).Hernández JG; Juaristi E Efficient Ball-Mill Procedure in the ‘Green’ Asymmetric Aldol Reaction Organocatalyzed by (S)-Proline-Containing Dipeptides in the Presence of Water. Tetrahedron 2011, 67, 6953–6959. [Google Scholar]
- (287).Machuca E; Rojas Y; Juaristi E Synthesis and Evaluation of (S)-Proline-Containing α,β-Dipeptides as Organocatalysts in Solvent-Free Asymmetric Aldol Reactions under Ball-Milling Conditions. Asian J. Org. Chem 2015, 4, 46–53. [Google Scholar]
- (288).Machuca E; Juaristi E Organocatalytic Activity of α,α-Dipeptide Derivatives of (S)-Proline in the Asymmetric Aldol Reaction in Absence of Solvent. Evidence for Non-Covalent π-π Interactions in the Transition State. Tetrahedron Lett 2015, 56, 1144–1148. [Google Scholar]
- (289).Lehn J-M Perspectives in Chemistry—Steps Towards Complex Matter. Angew. Chem. Int. Ed 2013, 52, 2836–2850. [DOI] [PubMed] [Google Scholar]
- (290).Tena-Solsona M; Nanda J; Diaz-Oltra S; Chotera A; Ashkenasy G; Escuder B Emergent Catalytic Behavior of Self-Assembled Low Molecular Weight Peptide-Based Aggregates and Hydrogels. Chem. Eur. J 2016, 22, 6687–6694. [DOI] [PubMed] [Google Scholar]
- (291).Soares BM; Aguilar AM; Silva ER; Coutinho-Neto MD; Hamley IW; Reza M; Ruokolainen J; Alves WA Chiral Organocatalysts Based on Lipopeptide Micelles for Aldol Reactions in Water. Phys. Chem. Chem. Phys 2017, 19, 1181–1189. [DOI] [PubMed] [Google Scholar]
- (292).Pelin JNBD; Gerbelli BB; Soares BM; Aguilar AM; Alves WA Amyloidogenic Model Peptides as Catalysts for Stereoselective Aldol Reactions. Catal. Sci. Technol 2019, 9, 4304–4313. [Google Scholar]
- (293).Du Z-X; Zhang L-Y; Fan X-Y; Wu F-C; Da C-S Highly Enantioselective Biomimetic Intramolecular Dehydration: Kinetic Resolution of β-Hydroxy Ketones Catalyzed by β-Turn Tetrapeptides. Tetrahedron Lett 2013, 54, 2828–2832. [Google Scholar]
- (294).Szőllősi G; Csampai A; Somlai C; Fekete M; Bartok M Unusual Enantioselectivities in Heterogeneous Organocatalyzed Reactions: Reversal of Direction Using Proline Di- Versus Tri-Peptides in the Aldol Addition. J. Mol. Catal. A: Chem 2014, 382, 86–92. [Google Scholar]
- (295).Akagawa K; Sakamoto S; Kudo K Resin-Supported Acid- and Base-Catalyzed One-Pot Sequential Reaction Including an Enantioselective Step. Tetrahedron Lett 2007, 48, 985–987. [Google Scholar]
- (296).Akagawa K; Takigawa S; Mano E; Kudo K Sequential Oxidation/Asymmetric Aldol Reaction of Primary Alcohols by Resin-Supported Catalysts. Tetrahedron Lett 2011, 52, 770–773. [Google Scholar]
- (297).Ryland BL; Stahl SS Practical Aerobic Oxidations of Alcohols and Amines with Homogeneous Copper/Tempo and Related Catalyst Systems. Angew. Chem. Int. Ed 2014, 53, 8824–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Akagawa K; Higuchi J; Yoshikawa I; Kudo K Kinetic Resolution of Ansa Cyclophanes by Peptide-Catalyzed Aldol/Retro-Aldol Reactions. Eur. J. Org. Chem 2018, 5278–5281. [Google Scholar]
- (299).Gibson SE; Knight JD [2.2]Paracyclophane Derivatives in Asymmetric Catalysis. Org. Biomol. Chem 2003, 1, 1256–1269. [DOI] [PubMed] [Google Scholar]
- (300).Hassan Z; Spuling E; Knoll DM; Lahann J; Bräse S Planar Chiral [2.2]Paracyclophanes: From Synthetic Curiosity to Applications in Asymmetric Synthesis and Materials. Chem. Soc. Rev 2018, 47, 6947–6963. [DOI] [PubMed] [Google Scholar]
- (301).Kotha S; Shirbhate ME; Waghule GT Selected Synthetic Strategies to Cyclophanes. Beilstein J. Org. Chem 2015, 11, 1274–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Kanger T; Kriis K; Laars M; Kailas T; Müürisepp A-M; Pehk T; Lopp M Bimorpholine-Mediated Enantioselective Intramolecular and Intermolecular Aldol Condensation. J. Org. Chem 2007, 72, 5168–5173. [DOI] [PubMed] [Google Scholar]
- (303).Kon K; Kohari Y; Murata M Unnatural Tripeptide as Highly Enantioselective Organocatalyst for Asymmetric Aldol Reaction of Isatins. Tetrahedron Lett 2019, 60, 415–418. [Google Scholar]
- (304).Angelici G; Falgiani A; Luppi G; Kaptein B; Broxterman QB; Tomasini C Atom Economic and Highly Syn-Selective Prolinamide-Catalyzed Cross-Aldol Addition of Hydroxyacetone to Aromatic Aldehydes. Synth. Commun 2008, 38, 1137–1146. [Google Scholar]
- (305).Messerer M; Wennemers H Reversing the Enantioselectivity of a Peptidic Catalyst by Changing the Solvent. Synlett 2011, 499–502. [Google Scholar]
- (306).Krattiger P; Kovàsy R; Revell JD; Wennemers H Using Catalyst–Substrate Coimmobilization for the Discovery of Catalysts for Asymmetric Aldol Reactions in Split-and-Mix Libraries. QSAR Comb. Sci 2005, 24, 1158–1163. [Google Scholar]
- (307).Clemente FR; Houk KN Computational Evidence for the Enamine Mechanism of Intramolecular Aldol Reactions Catalyzed by Proline. Angew. Chem. Int. Ed 2004, 43, 5766–5768. [DOI] [PubMed] [Google Scholar]
- (308).List B; Hoang L; Martin HJ New Mechanistic Studies on the Proline-Catalyzed Aldol Reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 5839–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Wiesner M; Revell JD; Wennemers H Tripeptides as Efficient Asymmetric Catalysts for 1,4-Addition Reactions of Aldehydes to Nitroolefins–a Rational Approach. Angew. Chem. Int. Ed 2008, 47, 1871–1874. [DOI] [PubMed] [Google Scholar]
- (310).Wiesner M; Revell JD; Tonazzi S; Wennemers H Peptide Catalyzed Asymmetric Conjugate Addition Reactions of Aldehydes to Nitroethylene—A Convenient Entry Into γ2-Amino Acids. J. Am. Chem. Soc 2008, 130, 5610–5611. [DOI] [PubMed] [Google Scholar]
- (311).Wiesner M; Neuburger M; Wennemers H Tripeptides of the Type H-D-Pro-Pro-Xaa-NH2 as Catalysts for Asymmetric 1,4-Addition Reactions: Structural Requirements for High Catalytic Efficiency. Chem. Eur. J 2009, 15, 10103–10109. [DOI] [PubMed] [Google Scholar]
- (312).Wiesner M; Upert G; Angelici G; Wennemers H Enamine Catalysis with Low Catalyst Loadings - High Efficiency via Kinetic Studies. J. Am. Chem. Soc 2010, 132, 6–7. [DOI] [PubMed] [Google Scholar]
- (313).Bächle F; Duschmalé J; Ebner C; Pfaltz A; Wennemers H Organocatalytic Asymmetric Conjugate Addition of Aldehydes to Nitroolefins: Identification of Catalytic Intermediates and the Stereoselectivity-Determining Step by ESI-MS. Angew. Chem. Int. Ed 2013, 52, 12619–12623. [DOI] [PubMed] [Google Scholar]
- (314).Arakawa Y; Wiesner M; Wennemers H Efficient Recovery and Reuse of an Immobilized Peptidic Organocatalyst. Adv. Synth. Catal 2011, 353, 1201–1206. [Google Scholar]
- (315).Arakawa Y; Wennemers H Enamine Catalysis in Flow with an Immobilized Peptidic Catalyst. ChemSusChem 2012, 6, 242–245. [DOI] [PubMed] [Google Scholar]
- (316).Duschmalé J; Kohrt S; Wennemers H Peptide Catalysis in Aqueous Emulsions. Chem. Commun 2014, 50, 8109–8112. [DOI] [PubMed] [Google Scholar]
- (317).Duschmalé J; Wennemers H Adapting to Substrate Challenges: Peptides as Catalysts for Conjugate Addition Reactions of Aldehydes to α,β-Disubstituted Nitroolefins. Chem. Eur. J 2012, 18, 1111–1120. [DOI] [PubMed] [Google Scholar]
- (318).Kastl R; Wennemers H Peptide-Catalyzed Stereoselective Conjugate Addition Reactions Generating All-Carbon Quaternary Stereogenic Centers. Angew. Chem. Int. Ed 2013, 52, 7228–7232. [DOI] [PubMed] [Google Scholar]
- (319).Wennemers H; Schnitzer T, A Stereoselective Tripeptide Catalyst for Conjugate Addition Reactions of Acetophenones to Dicyanoolefins. Synlett 2017, 28, 1282–1286. [Google Scholar]
- (320).Grünenfelder CE; Kisunzu JK; Wennemers H Peptide-Catalyzed Stereoselective Conjugate Addition Reactions of Aldehydes to Maleimide. Angew. Chem. Int. Ed 2016, 128, 8713–8716. [DOI] [PubMed] [Google Scholar]
- (321).Grünenfelder CE; Kisunzu JK; Trapp N; Kastl R; Wennemers H Crystal Structures of Peptidic Catalysts of the H-d-Pro-Pro-Xaa Type. Peptide Science 2017, 108, e22912, 1–7. [DOI] [PubMed] [Google Scholar]
- (322).Bartlett GJ; Choudhary A; Raines RT; Woolfson D n→π* Interactions in Proteins. Nat. Chem. Biol 2010, 6, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Fischer G Chemical Aspects of Peptide Bond Isomerisation. Chem. Soc. Rev 2000, 29, 119–127. [Google Scholar]
- (324).Dugave C; Demange L Cis-Trans Isomerization of Organic Molecules and Biomolecules: Implications and Applications. Chem. Rev 2003, 103, 2475–2532. [DOI] [PubMed] [Google Scholar]
- (325).Schnitzer T; Wennemers H Influence of the Trans/Cis Conformer Ratio on the Stereoselectivity of Peptidic Catalysts. J. Am. Chem. Soc 2017, 139, 15356–15362. [DOI] [PubMed] [Google Scholar]
- (326).Schnitzer T; Wennemers H Effect of γ-Substituted Proline Derivatives on the Performance of the Peptidic Catalyst H-dPro-Pro-Glu-NH2. Synthesis 2018, 50, 4377–4382. [Google Scholar]
- (327).Siebler C; Maryasin B; Kuemin M; Erdmann RS; Rigling C; Grünenfelder C; Ochsenfeld C; Wennemers H Importance of Dipole Moments and Ambient Polarity for the Conformation of Xaa–Pro Moieties – A Combined Experimental and Theoretical Study. Chem. Sci 2015, 6, 6725–6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Rigling C; Kisunzu JK; Duschmalé J; Häussinger D; Wiesner M; Ebert M-O; Wennemers H Conformational Properties of a Peptidic Catalyst: Insights from NMR Spectroscopic Studies. J. Am. Chem. Soc 2018, 140, 10829–10838. [DOI] [PubMed] [Google Scholar]
- (329).Montalbetti CAGN; Falque V Amide Bond Formation and Peptide Coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar]
- (330).Znabet A; Ruijter E; de Kanter FJJ; Köhler V; Helliwell M; Turner NJ; Orru RVA Highly Stereoselective Synthesis of Substituted Prolyl Peptides Using a Combination of Biocatalytic Desymmetrization and Multicomponent Reactions. Angew. Chem. Int. Ed 2010, 49, 5289–5292. [DOI] [PubMed] [Google Scholar]
- (331).de la Torre AF; Rivera DG; Ferreira MAB; Corrêa AG; Paixão MW Multicomponent Combinatorial Development and Conformational Analysis of Prolyl Peptide–Peptoid Hybrid Catalysts: Application in the Direct Asymmetric Michael Addition. J. Org. Chem 2013, 78, 10221–10232. [DOI] [PubMed] [Google Scholar]
- (332).Scatena GS; de la Torre AF; Cass QB; Rivera DG; Paixão MW Multicomponent Approach to Silica-Grafted Peptide Catalysts: a 3D Continuous-Flow Organocatalytic System with On-Line Monitoring of Conversion and Stereoselectivity. ChemCatChem 2014, 6, 3208–3214. [Google Scholar]
- (333).Cortes-Clerget M; Gager O; Monteil M; Pirat J-L; Migianu-Griffoni E; Deschamp J; Lecouvey M Novel Easily Recyclable Bifunctional Phosphonic Acid Carrying Tripeptides for the Stereoselective Michael Addition of Aldehydes with Nitroalkenes. Adv. Synth. Catal 2015, 358, 34–40. [Google Scholar]
- (334).Tsogoeva S; Jagtap S Dual Catalyst Control in the Chiral Diamine-Dipeptide-Catalyzed Asymmetric Michael Addition. Synlett 2004, 2624–2626. [Google Scholar]
- (335).Freund M; Schenker S; Tsogoeva SB Enantioselective Nitro-Michael Reactions Catalyzed by Short Peptides on Water. Org. Biomol. Chem 2009, 7, 4279–4284. [DOI] [PubMed] [Google Scholar]
- (336).Durini M; Sahr FA; Kuhn M; Civera M; Gennari C; Piarulli U Bifunctional 2,5-Diketopiperazines as Efficient Organocatalysts for the Enantioselective Conjugate Addition of Aldehydes to Nitroolefins. Eur. J. Org. Chem 2011, 2011, 5599–5607. [Google Scholar]
- (337).Akagawa K; Yamashita T; Sakamoto S; Kudo K Friedel–Crafts-Type Alkylation in Aqueous Media Using Resin-Supported Peptide Catalyst Having Polyleucine. Tetrahedron Lett 2009, 50, 5602–5604. [Google Scholar]
- (338).Akagawa K; Suzuki R; Kudo K Effect of the Helical Tether of a Resin-Supported Peptide Catalyst for Friedel–Crafts-Type Alkylation in Water. Adv. Synth. Catal 2012, 354, 1280–1286. [Google Scholar]
- (339).Akagawa K; Umezawa R; Kudo K Asymmetric One-Pot Sequential Friedel–Crafts-Type Alkylation and α-Oxyamination Catalyzed by a Peptide and an Enzyme. Beilstein J. Org. Chem 2012, 8, 1333–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (340).Akagawa K; Kudo K Construction of an All-Carbon Quaternary Stereocenter by the Peptide-Catalyzed Asymmetric Michael Addition of Nitromethane to β-Disubstituted α,β-Unsaturated Aldehydes. Angew. Chem. Int. Ed 2012, 51, 12786–12789. [DOI] [PubMed] [Google Scholar]
- (341).Akagawa K; Nishi N; Sen J; Kudo K Peptide-Catalyzed Consecutive 1,6- and 1,4-Additions of Thiols to α,β,γ,δ-Unsaturated Aldehydes. Org. Biomol. Chem 2014, 12, 3581–3585. [DOI] [PubMed] [Google Scholar]
- (342).Akagawa K; Sugiyama M; Kudo K Asymmetric Michael Addition of Boronic Acids to a γ - Hydroxy- α,β -Unsaturated Aldehyde Catalyzed by Resin-Supported Peptide. Org. Biomol. Chem 2012, 10, 4839–6. [DOI] [PubMed] [Google Scholar]
- (343).Copeland GT; Miller SJ Selection of Enantioselective Acyl Transfer Catalysts from a Pooled Peptide Library Through a Fluorescence-Based Activity Assay: An Approach to Kinetic Resolution of Secondary Alcohols of Broad Structural Scope. J. Am. Chem. Soc 2001, 123, 6496–6502. [DOI] [PubMed] [Google Scholar]
- (344).Krattiger P; McCarthy C; Pfaltz A; Wennemers H Catalyst–Substrate Coimmobilization: A Strategy for Catalysts Discovery in Split-and-Mix Libraries. Angew. Chem. Int. Ed 2003, 42, 1722–1724. [DOI] [PubMed] [Google Scholar]
- (345).Akagawa K; Sakai N; Kudo K Histidine-Containing Peptide Catalysts Developed by a Facile Library Screening Method. Angew. Chem. Int. Ed 2015, 54, 1822–1826. [DOI] [PubMed] [Google Scholar]
- (346).Akagawa K; Satou J; Kudo K Exploration of Structural Frameworks for Reactive and Enantioselective Peptide Catalysts by Library Screenings. J. Org. Chem 2016, 81, 9396–9401. [DOI] [PubMed] [Google Scholar]
- (347).Lautens M; Klute W; Tam W Transition Metal-Mediated Cycloaddition Reactions. Chem. Rev 1996, 96, 49–92. [DOI] [PubMed] [Google Scholar]
- (348).Lebel H; Marcoux J-F; Molinaro C; Charette AB Stereoselective Cyclopropanation Reactions. Chem. Rev 2003, 103, 977–1050. [DOI] [PubMed] [Google Scholar]
- (349).Ebner C; Carreira EM Cyclopropanation Strategies in Recent Total Syntheses. Chem. Rev 2017, 117, 11651–11679. [DOI] [PubMed] [Google Scholar]
- (350).Wu W; Lin Z; Jiang H Recent Advances in the Synthesis of Cyclopropanes. Org. Biomol. Chem 2018, 16, 7315–7329. [DOI] [PubMed] [Google Scholar]
- (351).Simmons HE; Smith RD A New Synthesis of Cyclopropanes from Olefins. J. Am. Chem. Soc 1958, 80, 5323–5324. [Google Scholar]
- (352).Charette AB; Beauchemin A Simmons-Smith Cyclopropanation Reaction. In Organic Reactions; Wiley: Hoboken, NJ, USA, 2004; Vol. 188, pp 1–415. [Google Scholar]
- (353).Furukawa J; Kawabata N; Nishimura J A Stereospecific Synthesis of Cyclopropane Derivatives from Olefins. Tetrahedron Lett 1968, 9, 3495–3498. [Google Scholar]
- (354).Sawada S; Takehana K; Inouye Y Partial Asymmetric Synthesis in the Simmons-Smith Reaction. II. J. Org. Chem 1968, 33, 1767–1770. [Google Scholar]
- (355).Yang Z; Lorenz JC; Shi Y Exploring New Reactive Species for Cyclopropanation. Tetrahedron Lett 1998, 39, 8621–8624. [Google Scholar]
- (356).Long J; Yuan Y; Shi Y Asymmetric Simmons-Smith Cyclopropanation of Unfunctionalized Olefins. J. Am. Chem. Soc 2003, 125, 13632–13633. [DOI] [PubMed] [Google Scholar]
- (357).Long J; Du H; Li K; Shi Y Catalytic Asymmetric Simmons–Smith Cyclopropanation of Unfunctionalized Olefins. Tetrahedron Lett 2005, 46, 2737–2740. [Google Scholar]
- (358).Long J; Xu L; Du H; Li K; Shi Y Mechanistic Studies on a Dipeptide-Promoted Asymmetric Cyclopropanation of Unfunctionalized Olefins. Org. Lett 2009, 11, 5226–5229. [DOI] [PubMed] [Google Scholar]
- (359).Charette AB; Beauchemin A; Francoeur S Acyloxymethylzinc Reagents: Preparation, Reactivity, and Solid-State Structure of This Novel Class of Cyclopropanating Reagents. J. Am. Chem. Soc 2001, 123, 8139–8140. [DOI] [PubMed] [Google Scholar]
- (360).Nakamura M; Hirai A; Nakamura E Reaction Pathways of the Simmons–Smith Reaction. J. Am. Chem. Soc 2003, 125, 2341–2350. [DOI] [PubMed] [Google Scholar]
- (361).Fournier J-F; Charette AB Improved Protocol for the Diastereoselective Cyclopropanation of Alkenes Using Geminal Dizinc Carbenoids: A Study on the Effect of Zinc Iodide. Eur. J. Org. Chem 2004, 1401–1404. [Google Scholar]
- (362).Kunz RK; MacMillan DWC Enantioselective Organocatalytic Cyclopropanations. the Identification of a New Class of Iminium Catalyst Based Upon Directed Electrostatic Activation. J. Am. Chem. Soc 2005, 127, 3240–3241. [DOI] [PubMed] [Google Scholar]
- (363).Lakhdar S; Appel R; Mayr H How Does Electrostatic Activation Control Iminium-Catalyzed Cyclopropanations? Angew. Chem. Int. Ed 2009, 48, 5034–5037. [DOI] [PubMed] [Google Scholar]
- (364).Hartikka A; Arvidsson PI Tetrazolic Acid Functionalized Dihydroindol: Rational Design of a Highly Selective Cyclopropanation Organocatalyst. J. Org. Chem 2007, 72, 5874–5877. [DOI] [PubMed] [Google Scholar]
- (365).Li W; Li X; Ye T; Wu W; Liang X; Ye J Asymmetric Organocatalytic Michael/a-Alkylation Reaction of α,β-Unsaturated Aldehyde with Chloroacetophenone. Tetrahedron Lett 2011, 52, 2715–2718. [Google Scholar]
- (366).Akagawa K; Takigawa S; Nagamine IS; Umezawa R; Kudo K Peptide-Catalyzed Diastereo- and Enantioselective Cyclopropanation of Aromatic α,β-Unsaturated Aldehydes. Org. Lett 2013, 15, 4964–4967. [DOI] [PubMed] [Google Scholar]
- (367).Wei Y; Shi M Recent Advances in Organocatalytic Asymmetric Morita–Baylis–Hillman/Aza-Morita–Baylis–Hillman Reactions. Chem. Rev 2013, 113, 6659–6690. [DOI] [PubMed] [Google Scholar]
- (368).Scherer A; Gladysz JA A Promising New Catalyst Family for Enantioselective Cycloadditions Involving Allenes and Imines: Chiral Phosphines with Transition Metal–CH2–P: Linkages. Tetrahedron Lett 2006, 47, 6335–6337. [Google Scholar]
- (369).Jean L; Marinetti A Phosphine-Catalyzed Enantioselective [3+2] Annulations of 2,3-Butadienoates with Imines. Tetrahedron Lett 2006, 47, 2141–2145. [Google Scholar]
- (370).Fang Y-Q; Jacobsen EN Cooperative, Highly Enantioselective Phosphinothiourea Catalysis of Imine–Allene [3 + 2] Cycloadditions. J. Am. Chem. Soc 2008, 130, 5660–5661. [DOI] [PubMed] [Google Scholar]
- (371).Xu Z; Lu X A Novel [3+2] Cycloaddition Approach to Nitrogen Heterocycles via Phosphine-Catalyzed Reactions of 2,3-Butadienoates or 2-Butynoates and Dimethyl Acetylenedicarboxylate with Imines: A Convenient Synthesis of Pentabromopseudilin. J. Org. Chem 1998, 63, 5031–5041. [Google Scholar]
- (372).Evans CA; Miller SJ Amine-Catalyzed Coupling of Allenic Esters to α,β-Unsaturated Carbonyls. J. Am. Chem. Soc 2003, 125, 12394–12395. [DOI] [PubMed] [Google Scholar]
- (373).Cowen BJ; Saunders LB; Miller SJ Pyridylalanine (Pal)-Peptide Catalyzed Enantioselective Allenoate Additions to N-Acyl Imines. J. Am. Chem. Soc 2009, 131, 6105–6107. [DOI] [PubMed] [Google Scholar]
- (374).Saunders LB; Cowen BJ; Miller SJ Pyridylalanine (Pal)-Peptide Catalyzed Enantioselective Allenoate Additions to N-Acyl Imines Proceed via an Atypical “Aza-Morita–Baylis–Hillman” Mechanism. Org. Lett 2010, 12, 4800–4803. [DOI] [PubMed] [Google Scholar]
- (375).Mbofana CT; Miller SJ Diastereo- and Enantioselective Addition of Anilide-Functionalized Allenoates to N-Acylimines Catalyzed by a Pyridylalanine-Based Peptide. J. Am. Chem. Soc 2014, 136, 3285–3292. [DOI] [PubMed] [Google Scholar]
- (376).Aroyan C; Dermenci A; Miller SJ The Rauhut–Currier Reaction: A History and its Synthetic Application. Tetrahedron 2009, 65, 4069–4084. [Google Scholar]
- (377).Chandra Bharadwaj K Intramolecular Morita–Baylis–Hillman and Rauhut–Currier Reactions. A Catalytic and Atom Economic Route for Carbocycles and Heterocycles. RSC Adv 2015, 5, 75923–75946. [Google Scholar]
- (378).Aroyan CE; Miller SJ Enantioselective Rauhut–Currier Reactions Promoted by Protected Cysteine. J. Am. Chem. Soc 2007, 129, 256–257. [DOI] [PubMed] [Google Scholar]
- (379).Aroyan CE; Dermenci A; Miller SJ Development of a Cysteine-Catalyzed Enantioselective Rauhut–Currier Reaction. J. Org. Chem 2010, 75, 5784–5796. [DOI] [PubMed] [Google Scholar]
- (380).Osuna S; Dermenci A; Miller SJ; Houk KN The Roles of Counterion and Water in a Stereoselective Cysteine-Catalyzed Rauhut–Currier Reaction: A Challenge for Computational Chemistry. Chem. Eur. J 2013, 19, 14245–14253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Dermenci A; Selig PS; Domaoal RA; Spasov KA; Anderson KS; Miller SJ Quasi-Biomimetic Ring Contraction Promoted by a Cysteine-Based Nucleophile: Total Synthesis of Sch-642305, Some Analogs and Their Putative Anti-HIV Activities. Chem. Sci 2011, 2, 1568–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Chu M; Mierzwa R; Xu L; He L; Terracciano J; Patel M; Gullo V; Black T; Zhao W; Chan TM; et al. Isolation and Structure Elucidation of Sch-642305, a Novel Bacterial DNA Primase Inhibitor Produced by Penicillium Verrucosum. J. Nat. Prod 2003, 66, 1527–1530. [DOI] [PubMed] [Google Scholar]
- (383).Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3296. [DOI] [PubMed] [Google Scholar]
- (384).Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed 2015, 128, 58–106. [DOI] [PubMed] [Google Scholar]
- (385).Otto H-H; Schirmeister T Cysteine Proteases and Their Inhibitors. Chem. Rev 1997, 97, 133–172. [DOI] [PubMed] [Google Scholar]
- (386).Stubbe J; van Der Donk WA Protein Radicals in Enzyme Catalysis. Chem. Rev 1998, 98, 705–762. [DOI] [PubMed] [Google Scholar]
- (387).Licht S; Gerfen GJ; Stubbe J Thiyl Radicals in Ribonucleotide Reductases. Science 1996, 271, 477–481. [DOI] [PubMed] [Google Scholar]
- (388).Yoon TP; Jacobsen EN Privileged Chiral Catalysts. Science 2003, 299, 1691–1693. [DOI] [PubMed] [Google Scholar]
- (389).Subramanian H; Moorthy R; Sibi MP Thiyl Radicals: From Simple Radical Additions to Asymmetric Catalysis. Angew. Chem. Int. Ed 2014, 53, 13660–13662. [DOI] [PubMed] [Google Scholar]
- (390).Dénès F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- (391).Roberts BP Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem. Soc. Rev 1999, 28, 25–35. [Google Scholar]
- (392).Haque MB; Roberts BP Enantioselective Radical-Chain Hydrosilylation of Prochiral Alkenes Using Optically Active Thiol Catalysts. Tetrahedron Lett 1996, 37, 9123–9126. [Google Scholar]
- (393).Haque MB; Roberts BP; Tocher DA Enantioselective Radical-Chain Hydrosilylation of Alkenes Using Homochiral Thiols as Polarity-Reversal Catalysts. J. Chem. Soc., Perkin Trans 1 1998, 2881–2890. [Google Scholar]
- (394).Hashimoto T; Kawamata Y; Maruoka K An Organic Thiyl Radical Catalyst for Enantioselective Cyclization. Nat Chem 2014, 6, 702–705. [DOI] [PubMed] [Google Scholar]
- (395).Ryss JM; Turek AK; Miller SJ Disulfide-Bridged Peptides That Mediate Enantioselective Cycloadditions Through Thiyl Radical Catalysis. Org. Lett 2018, 20, 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Faber K Non-Sequential Processes for the Transformation of a Racemate Into a Single Stereoisomeric Product: Proposal for Stereochemical Classification. Chem. Eur. J 2001, 7, 5004–5010. [DOI] [PubMed] [Google Scholar]
- (397).Alexeeva M; Enright A; Dawson MJ; Mahmoudian M; Turner NJ Deracemization of α-Methylbenzylamine Using an Enzyme Obtained by in Vitro Evolution. Angew. Chem. Int. Ed 2002, 41, 3177–3180. [DOI] [PubMed] [Google Scholar]
- (398).Lackner AD; Samant AV; Toste FD Single-Operation Deracemization of 3H-Indolines and Tetrahydroquinolines Enabled by Phase Separation. J. Am. Chem. Soc 2013, 135, 14090–14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (399).Ji Y; Shi L; Chen M-W; Feng G-S; Zhou Y-G Concise Redox Deracemization of Secondary and Tertiary Amines with a Tetrahydroisoquinoline Core via a Nonenzymatic Process. J. Am. Chem. Soc 2015, 137, 10496–10499. [DOI] [PubMed] [Google Scholar]
- (400).Shin NY; Ryss JM; Zhang X; Miller SJ; Knowles RR Light-Driven Deracemization Enabled by Excited-State Electron Transfer. Science 2019, 366, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (402).Hölzl-Hobmeier A; Bauer A; Silva AV; Huber SM; Bannwarth C; Bach T Catalytic Deracemization of Chiral Allenes by Sensitized Excitation with Visible Light. Nature 2018, 564, 240–243. [DOI] [PubMed] [Google Scholar]
- (403).Mok PLH; Roberts BP Enantioselective α-Hydrogen-Atom Abstraction from an Ester by an Optically Active Amine-Boryl Radical. J. Chem. Soc., Chem. Commun 1991, 150–152. [Google Scholar]
- (404).Du J; Skubi KL; Schultz DM; Yoon TP A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Lu Z; Shen M; Yoon TP [3+2] Cycloadditions of Aryl Cyclopropyl Ketones by Visible Light Photocatalysis. J. Am. Chem. Soc 2011, 133, 1162–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Hurtley AE; Cismesia MA; Ischay MA; Yoon TP Visible Light Photocatalysis of Radical Anion Hetero-Diels-Alder Cycloadditions. Tetrahedron 2011, 67, 4442–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Hoveyda AH; Hird AW; Kacprzynski MA Small Peptides as Ligands for Catalytic Asymmetric Alkylations of Olefins. Rational Design of Catalysts or of Searches That Lead to Them? Chem. Commun 2004, 1779–1785. [DOI] [PubMed] [Google Scholar]
- (408).Yoon TP Photochemical Stereocontrol Using Tandem Photoredox-Chiral Lewis Acid Catalysis. Acc. Chem. Res 2016, 49, 2307–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).France S; Weatherwax A; Lectka T Recent Developments in Catalytic, Asymmetric α-Halogenation: A New Frontier in Asymmetric Catalysis. Eur. J. Org. Chem 2005, 475–479. [Google Scholar]
- (410).Ueda M; Kano T; Maruoka K Organocatalyzed Direct Asymmetric α-Halogenation of Carbonyl Compounds. Org. Biomol. Chem 2009, 7, 2005–2012. [DOI] [PubMed] [Google Scholar]
- (411).Denmark SE; Kuester WE; Burk MT Catalytic, Asymmetric Halofunctionalization of Alkenes: A Critical Perspective. Angew. Chem. Int. Ed 2012, 51, 10938–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Tan CK; Yeung Y-Y Recent Advances in Stereoselective Bromofunctionalization of Alkenes Using N-Bromoamide Reagents. Chem. Commun 2013, 49, 7985–7996. [DOI] [PubMed] [Google Scholar]
- (413).Landry ML; Burns NZ Catalytic Enantioselective Dihalogenation in Total Synthesis. Acc. Chem. Res 2018, 51, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Gathergood N; Zhuang W; Jørgensen KA Catalytic Enantioselective Friedel–Crafts Reactions of Aromatic Compounds with Glyoxylate: A Simple Procedure for the Synthesis of Optically Active Aromatic Mandelic Acid Esters. J. Am. Chem. Soc 2000, 122, 12517–12522. [Google Scholar]
- (415).Paras NA; MacMillan DW New Strategies in Organic Catalysis: The First Enantioselective Organocatalytic Friedel-Crafts Alkylation. J. Am. Chem. Soc 2001, 123, 4370–4371. [DOI] [PubMed] [Google Scholar]
- (416).Jia Y-X; Zhong J; Zhu S-F; Zhang C-M; Zhou Q-L Chiral Brønsted Acid Catalyzed Enantioselective Friedel-Crafts Reaction of Indoles and Alpha-Aryl Enamides: Construction of Quaternary Carbon Atoms. Angew. Chem. Int. Ed 2007, 46, 5565–5567. [DOI] [PubMed] [Google Scholar]
- (417).Bringmann G; Gulder T; Gulder TAM; Breuning M Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products. Chem. Rev 2011, 111, 563–639. [DOI] [PubMed] [Google Scholar]
- (418).LaPlante SR; D Fader L; Fandrick KR; Fandrick DR; Hucke O; Kemper R; Miller SPF; Edwards PJ Assessing Atropisomer Axial Chirality in Drug Discovery and Development. J. Med. Chem 2011, 54, 7005–7022. [DOI] [PubMed] [Google Scholar]
- (419).Tang W; Zhang X New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev 2003, 103, 3029–3070. [DOI] [PubMed] [Google Scholar]
- (420).Bringmann G; Price Mortimer AJ; Keller PA; Gresser MJ; Garner J; Breuning M Atroposelective Synthesis of Axially Chiral Biaryl Compounds. Angew. Chem. Int. Ed 2005, 44, 5384–5427. [DOI] [PubMed] [Google Scholar]
- (421).Bringmann G; Menche D Stereoselective Total Synthesis of Axially Chiral Natural Products via Biaryl Lactones. Acc. Chem. Res 2001, 34, 615–624. [DOI] [PubMed] [Google Scholar]
- (422).Yu C; Huang H; Li X; Zhang Y; Wang W Dynamic Kinetic Resolution of Biaryl Lactones via a Chiral Bifunctional Amine Thiourea-Catalyzed Highly Atropo-Enantioselective Transesterification. J. Am. Chem. Soc 2016, 138, 6956–6959. [DOI] [PubMed] [Google Scholar]
- (423).Kakiuchi F; Le Gendre P; Yamada A; Ohtaki H; Murai S Atropselective Alkylation of Biaryl Compounds by Means of Transition Metal-Catalyzed C–H/Olefin Coupling. Tetrahedron: Asymmetry 2000, 11, 2647–2651. [Google Scholar]
- (424).Gustafson JL; Lim D; Miller SJ Dynamic Kinetic Resolution of Biaryl Atropisomers via Peptide-Catalyzed Asymmetric Bromination. Science 2010, 328, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (425).Denmark SE; Burk MT Lewis Base Catalysis of Bromo- and Iodolactonization, and Cycloetherification. Proc. Natl. Acad. Sci. USA 2010, 107, 20655–20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Kamrath MZ; Garand E; Jordan PA; Leavitt CM; Wolk AB; Van Stipdonk MJ; Miller SJ; Johnson MA Vibrational Characterization of Simple Peptides Using Cryogenic Infrared Photodissociation of H 2-Tagged, Mass-Selected Ions. J. Am. Chem. Soc 2011, 133, 6440–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Garand E; Kamrath MZ; Jordan PA; Wolk AB; Leavitt CM; McCoy AB; Miller SJ; Johnson MA Determination of Noncovalent Docking by Infrared Spectroscopy of Cold Gas-Phase Complexes. Science 2012, 335, 694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Gustafson JL; Lim D; Barrett KT; Miller SJ Synthesis of Atropisomerically Defined, Highly Substituted Biaryl Scaffolds Through Catalytic Enantioselective Bromination and Regioselective Cross-Coupling. Angew. Chem. Int. Ed 2011, 50, 5125–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Barrett KT; Miller SJ Enantioselective Synthesis of Atropisomeric Benzamides Through Peptide-Catalyzed Bromination. J. Am. Chem. Soc 2013, 135, 2963–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Clayden J; Moran WJ; Edwards PJ; LaPlante SR The Challenge of Atropisomerism in Drug Discovery. Angew. Chem. Int. Ed 2009, 48, 6398–6401. [DOI] [PubMed] [Google Scholar]
- (431).LaPlante SR; Edwards PJ; Fader LD; Jakalian A; Hucke O Revealing Atropisomer Axial Chirality in Drug Discovery. ChemMedChem 2011, 6, 505–513. [DOI] [PubMed] [Google Scholar]
- (432).Cuyegkeng MA; Mannschreck A Liquid Chromatography on Triacetycellulose, 14 Chromatographic Separation of Enantiomers and Barriers to Enantiomerization of Axially Chiral Aromatic Carboxamides. Chem. Ber 1987, 120, 803–809. [Google Scholar]
- (433).Bragg RA; Clayden J; Morris GA; Pink JH Stereodynamics of Bond Rotation in Tertiary Aromatic Amides. Chem. Eur. J 2002, 8, 1279–1289. [DOI] [PubMed] [Google Scholar]
- (434).Barrett KT; Miller SJ Regioselective Derivatizations of a Tribrominated Atropisomeric Benzamide Scaffold. Org. Lett 2015, 17, 580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Barrett KT; Metrano AJ; Rablen PR; Miller SJ Spontaneous Transfer of Chirality in an Atropisomerically Enriched Two-Axis System. Nature 2014, 508, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Hirsch DR; Metrano AJ; Stone EA; Storch G; Miller SJ; Murelli RP Troponoid Atropisomerism: Studies on the Configurational Stability of Tropone-Amide Chiral Axes. Org. Lett 2019, 21, 2412–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (437).Meck C; D’Erasmo MP; Hirsch DR; Murelli RP The Biology and Synthesis of α-Hydroxytropolones. Med. Chem. Commun 2014, 5, 842–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Murelli RP; D’Erasmo MP; Hirsch DR; Meck C; Masaoka T; Wilson JA; Zhang B; Pal RK; Gallicchio E; Beutler JA; et al. Synthetic A-Hydroxytropolones as Inhibitors of HIV Reverse Transcriptase Ribonuclease H Activity. Med. Chem. Commun 2016, 7, 1783–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Zhang B; D’Erasmo MP; Murelli RP; Gallicchio E Free Energy-Based Virtual Screening and Optimization of RNase H Inhibitors of HIV-1 Reverse Transcriptase. ACS Omega 2016, 1, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (440).Diener ME; Metrano AJ; Kusano S; Miller SJ Enantioselective Synthesis of 3-Arylquinazolin-4(3H)-Ones via Peptide-Catalyzed Atroposelective Bromination. J. Am. Chem. Soc 2015, 137, 12369–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).Khan I; Zaib S; Batool S; Abbas N; Ashraf Z; Iqbal J; Saeed A Quinazolines and Quinazolinones as Ubiquitous Structural Fragments in Medicinal Chemistry: An Update on the Development of Synthetic Methods and Pharmacological Diversification. Bioorg. Med. Chem 2016, 24, 2361–2381. [DOI] [PubMed] [Google Scholar]
- (442).Metrano AJ; Abascal NC; Mercado BQ; Paulson EK; Miller SJ Structural Studies of β-Turn-Containing Peptide Catalysts for Atroposelective Quinazolinone Bromination. Chem. Commun 2016, 52, 4816–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Zhou AQ; O’Hern CS; Regan L Revisiting the Ramachandran Plot from a New Angle. Protein Sci 2011, 20, 1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (444).Yan XC; Metrano AJ; Robertson MJ; Abascal NC; Tirado-Rives J; Miller SJ; Jorgensen WL Molecular Dynamics Simulations of a Conformationally Mobile Peptide-Based Catalyst for Atroposelective Bromination. ACS Catal 2018, 8, 9968–9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Crawford JM; Stone EA; Metrano AJ; Miller SJ; Sigman MS Parameterization and Analysis of Peptide-Based Catalysts for the Atroposelective Bromination of 3-Arylquinazolin-4(3H)-Ones. J. Am. Chem. Soc 2018, 140, 868–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Laforteza BN; Chan KSL; Yu J-Q Enantioselective Ortho-C–H Cross-Coupling of Diarylmethylamines with Organoborons. Angew. Chem. Int. Ed 2015, 127, 11295–11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (447).Su B; Zhou T-G; Xu P-L; Shi Z-J; Hartwig JF Enantioselective Borylation of Aromatic C–H Bonds with Chiral Dinitrogen Ligands. Angew. Chem. Int. Ed 2017, 129, 7311–7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Hurtley AE; Stone EA; Metrano AJ; Miller SJ Desymmetrization of Diarylmethylamido Bis(Phenols) Through Peptide-Catalyzed Bromination: Enantiodivergence as a Consequence of a 2 Amu Alteration at an Achiral Residue Within the Catalyst. J. Org. Chem 2017, 82, 11326–11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Schmidt F; Stemmler RT; Rudolph J; Bolm C Catalytic Asymmetric Approaches Towards Enantiomerically Enriched Diarylmethanols and Diarylmethylamines. Chem. Soc. Rev 2006, 34, 454–470. [DOI] [PubMed] [Google Scholar]
- (450).Payne JT; Butkovich PH; Gu Y; Kunze KN; Park HJ; Wang D-S; Lewis JC Enantioselective Desymmetrization of Methylenedianilines via Enzyme-Catalyzed Remote Halogenation. J. Am. Chem. Soc 2018, 140, 546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Zhang W; Liu N; Schienebeck CM; Decloux K; Zheng S; Werness JB; Tang W Catalytic Enantioselective Halolactonization of Enynes and Alkenes. Chem. Eur. J 2012, 18, 7296–7305. [DOI] [PubMed] [Google Scholar]
- (452).Whitehead DC; Yousefi R; Jaganathan A; Borhan B An Organocatalytic Asymmetric Chlorolactonization. J. Am. Chem. Soc 2010, 132, 3298–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Jaganathan A; Garzan A; Whitehead DC; Staples RJ; Borhan B A Catalytic Asymmetric Chlorocyclization of Unsaturated Amides. Angew. Chem. Int. Ed 2011, 50, 2593–2596. [DOI] [PubMed] [Google Scholar]
- (454).Whitehead DC; Fhaner M; Borhan B A Peptide Bromoiodinane Approach for Asymmetric Bromolactonization. Tetrahedron Lett 2011, 52, 2288–2291. [Google Scholar]
- (455).Braddock DC; Cansell G; Hermitage SA Ortho-Substituted Iodobenzenes as Novel Organocatalysts for Bromination of Alkenes. Chem. Commun 2006, 44, 2483–2483. [DOI] [PubMed] [Google Scholar]
- (456).Steinreiber J; Ward TR Artificial Metalloenzymes as Selective Catalysts in Aqueous Media. Coord. Chem. Rev 2008, 252, 751–766. [Google Scholar]
- (457).Rosati F; Roelfes G Artificial Metalloenzymes. ChemCatChem 2010, 2, 916–927. [Google Scholar]
- (458).Ward TR Artificial Metalloenzymes Based on the Biotin-Avidin Technology: Enantioselective Catalysis and Beyond. Acc. Chem. Res 2011, 44, 47–57. [DOI] [PubMed] [Google Scholar]
- (459).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (460).Luchaco-Cullis CA; Mizutani H; Murphy KE; Hoveyda AH Modular Pyridinyl Peptide Ligands in Asymmetric Catalysis: Enantioselective Synthesis of Quaternary Carbon Atoms Through Copper-Catalyzed Allylic Substitutions. Angew. Chem 2001, 113, 1504–1508. [DOI] [PubMed] [Google Scholar]
- (461).Gilbertson SR; Chen G; McLoughlin M Versatile Building Block for the Synthesis of Phosphine-Containing Peptides: The Sulfide of Diphenylphosphinoserine. J. Am. Chem. Soc 1994, 116, 4481–4482. [Google Scholar]
- (462).Landis CR; Clark TP Solid-Phase Synthesis of Chiral 3,4-Diazaphospholanes and Their Application to Catalytic Asymmetric Allylic Alkylation. Proc. Natl. Acad. Sci. USA 2004, 101, 5428–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Ma D; Cai Q Copper/Amino Acid Catalyzed Cross-Couplings of Aryl and Vinyl Halides with Nucleophiles. Acc. Chem. Res 2008, 41, 1450–1460. [DOI] [PubMed] [Google Scholar]
- (464).Shao Q; Wu K; Zhuang Z; Qian S; Yu J-Q From Pd(OAc)2 to Chiral Catalysts: the Discovery and Development of Bifunctional Mono-N-Protected Amino Acid Ligands for Diverse C-H Functionalization Reactions. Acc. Chem. Res 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Gilbertson SR; Collibee SE; Agarkov A Asymmetric Catalysis with Libraries of Palladium β-Turn Phosphine Complexes. J. Am. Chem. Soc 2000, 122, 6522–6523. [Google Scholar]
- (466).Christensen CA; Meldal M Solid-Phase Synthesis of a Peptide-Based P,S-Ligand System Designed for Generation of Combinatorial Catalyst Libraries. J. Comb. Chem 2007, 9, 79–85. [DOI] [PubMed] [Google Scholar]
- (467).Laungani AC; Breit B Supramolecular PhanePhos-Analogous Ligands Through Hydrogen-Bonding for Asymmetric Hydrogenation. Chem. Commun 2008, 844–846. [DOI] [PubMed] [Google Scholar]
- (468).Pye PJ; Rossen K; Reamer RA; Tsou NN; Volante RP; Reider PJ A New Planar Chiral Bisphosphine Ligand for Asymmetric Catalysis: Highly Enantioselective Hydrogenations Under Mild Conditions. J. Am. Chem. Soc 1997, 119, 6207–6208. [Google Scholar]
- (469).Laungani AC; Slattery JM; Krossing I; Breit B Supramolecular Bidentate Ligands by Metal-Directed in Situ Formation of Antiparallel β-Sheet Structures and Application in Asymmetric Catalysis. Chem. Eur. J 2008, 14, 4488–4502. [DOI] [PubMed] [Google Scholar]
- (470).Loughlin WA; Tyndall JDA; Glenn MP; Fairlie DP Beta-Strand Mimetics. Chem. Rev 2004, 104, 6085–6117. [DOI] [PubMed] [Google Scholar]
- (471).Kokan Z; Kirin SI The Application of “Backdoor Induction” in Bioinspired Asymmetric Catalysis. RSC Adv 2012, 2, 5729–5729. [Google Scholar]
- (472).Herrick RS; Jarret RM; Curran TP; Dragoli DR; Flaherty MB; Lindyberg SE; Slate RA; Thornton LC Ordered Conformations in Bis(Amino Acid) Derivatives of 1,1′-Ferrocenedicarboxylic Acid. Tetrahedron Lett 1996, 37, 5289–5292. [Google Scholar]
- (473).Kirin SI; Kraatz H-B; Metzler-Nolte N Systematizing Structural Motifs and Nomenclature in 1,N′-Disubstituted Ferrocene Peptides. Chem. Soc. Rev 2006, 35, 348–354. [DOI] [PubMed] [Google Scholar]
- (474).Kokan Z; Kirin SI “Backdoor Induction” of Chirality in Asymmetric Hydrogenation with Rhodium(I) Complexes of Amino Acid Substituted Triphenylphosphane Ligands. Eur. J. Org. Chem 2013, 8154–8161. [Google Scholar]
- (475).Kokan Z; Glasovac Z; Majerić Elenkov M; Gredičak M; Jerić I; Kirin SI “Backdoor Induction” of Chirality: Asymmetric Hydrogenation with Rhodium(I) Complexes of Triphenylphosphane-Substituted β-Turn Mimetics. Organometallics 2014, 33, 4005–4015. [Google Scholar]
- (476).Milo A; Neumann R An Achiral Manganese Salen Catalyst Encapsulated in a Peptidic Phosphonate Homochiral Solid for the Enantioselective Formation of Diols by Consecutive Epoxidation and Hydration Reactions. Chem. Commun 2011, 47, 2535–2537. [DOI] [PubMed] [Google Scholar]
- (477).Milo A; Neumann R Achiral Ruthenium Catalyst Encapsulated in Titanium Phosphonate Homochiral Peptide-Based Solids for Enantioselective Hydrogenation of Ketones to Secondary Alcohols. ACS Catal 2012, 2, 2531–2536. [Google Scholar]
- (478).Guisado-Barrios G; Muñoz BK; Kamer PCJ; Lastdrager B; van der Marel G; Overhand M; Vega-Vázquez M; Martin-Pastor M Cyclic Decapeptide Gramicidin S Derivatives Containing Phosphines: Novel Ligands for Asymmetric Catalysis. Dalton Trans 2013, 42, 1973–1978. [DOI] [PubMed] [Google Scholar]
- (479).Burck S; van Assema SGA; Lastdrager B; Slootweg JC; Ehlers AW; Otero JM; Dacunha-Marinho B; Llamas-Saiz AL; Overhand M; van Raaij MJ; et al. Bisphosphine-Functionalized Cyclic Decapeptides Based on the Natural Product Gramicidin S: a Potential Scaffold for Transition-Metal Coordination. Chem. Eur. J 2009, 15, 8134–8145. [DOI] [PubMed] [Google Scholar]
- (480).Cussó O; Giuliano MW; Ribas X; Miller SJ; Costas M A Bottom Up Approach Towards Artificial Oxygenases by Combining Iron Coordination Complexes and Peptides. Chem. Sci 2017, 8, 3660–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Chen MS; White MC Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. [DOI] [PubMed] [Google Scholar]
- (482).Gómez L; Garcia-Bosch I; Company A; Benet-Buchholz J; Polo A; Sala X; Ribas X; Costas M Stereospecific C–H Oxidation with H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew. Chem. Int. Ed 2009, 48, 5720–5723. [DOI] [PubMed] [Google Scholar]
- (483).Coquière D; Bos J; Beld J; Roelfes G Enantioselective Artificial Metalloenzymes Based on a Bovine Pancreatic Polypeptide Scaffold. Angew. Chem. Int. Ed 2009, 48, 5159–5162. [DOI] [PubMed] [Google Scholar]
- (484).Zheng L; Marcozzi A; Gerasimov JY; Herrmann A Conformationally Constrained Cyclic Peptides: Powerful Scaffolds for Asymmetric Catalysis. Angew. Chem. Int. Ed 2014, 53, 7599–7603. [DOI] [PubMed] [Google Scholar]
- (485).Arnesano F; Scintilla S; Natile G Interaction Between Platinum Complexes and a Methionine Motif Found in Copper Transport Proteins. Angew. Chem. Int. Ed 2007, 46, 9062–9064. [DOI] [PubMed] [Google Scholar]
- (486).Pellegrino S; Facchetti G; Contini A; Gelmi ML; Erba E; Gandolfi R; Rimoldi I Ctr-1 Mets7 Motif Inspiring New Peptide Ligands for Cu(I)-Catalyzed Asymmetric Henry Reactions Under Green Conditions. RSC Adv 2016, 6, 71529–71533. [Google Scholar]
- (487).Candeias NR; Gois PMP; Afonso CAM Rh(II)-Catalyzed Intramolecular C–H Insertion of Diazo Substrates in Water: Scope and Limitations. J. Org. Chem 2006, 71, 5489–5497. [DOI] [PubMed] [Google Scholar]
- (488).Popp BV; Ball ZT Structure-Selective Modification of Aromatic Side Chains with Dirhodium Metallopeptide Catalysts. J. Am. Chem. Soc 2010, 132, 6660–6662. [DOI] [PubMed] [Google Scholar]
- (489).Sambasivan R; Ball ZT Metallopeptides for Asymmetric Dirhodium Catalysis. J. Am. Chem. Soc 2010, 132, 9289–9291. [DOI] [PubMed] [Google Scholar]
- (490).Sambasivan R; Ball ZT Determination of Orientational Isomerism in Rhodium(II) Metallopeptides by Pyrene Fluorescence. Org. Biomol. Chem 2012, 10, 8203–8204. [DOI] [PubMed] [Google Scholar]
- (491).Sambasivan R; Ball ZT Screening Rhodium Metallopeptide Libraries “on Bead”: Asymmetric Cyclopropanation and a Solution to the Enantiomer Problem. Angew. Chem. Int. Ed 2012, 51, 8568–8572. [DOI] [PubMed] [Google Scholar]
- (492).Sambasivan R; Zheng W; Burya SJ; Popp BV; Turro C; Clementi C; Ball ZT A Tripodal Peptide Ligand for Asymmetric Rh(II) Catalysis Highlights Unique Features of on-Bead Catalyst Development. Chem. Sci 2014, 5, 1401–1407. [Google Scholar]
- (493).Cherney AH; Kadunce NT; Reisman SE Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C–C Bonds. Chem. Rev 2015, 115, 9587–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (494).Schäfer P; Sidera M; Palacin T; Fletcher SP Asymmetric Cross-Coupling of Alkyl, Alkenyl and (Hetero)Aryl Nucleophiles with Racemic Allyl Halides. Chem. Commun 2017, 53, 12499–12511. [DOI] [PubMed] [Google Scholar]
- (495).Swift EC; Jarvo ER Asymmetric Transition Metal-Catalyzed Cross-Coupling Reactions for the Construction of Tertiary Stereocenters. Tetrahedron 2013, 69, 5799–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (496).Kim B; Chinn AJ; Fandrick DR; Senanayake CH; Singer RA; Miller SJ Distal Stereocontrol Using Guanidinylated Peptides as Multifunctional Ligands: Desymmetrization of Diarylmethanes via Ullman Cross-Coupling. J. Am. Chem. Soc 2016, 138, 7939–7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (497).Chinn AJ; Kim B; Kwon Y; Miller SJ Enantioselective Intermolecular C–O Bond Formation in the Desymmetrization of Diarylmethines Employing a Guanidinylated Peptide-Based Catalyst. J. Am. Chem. Soc 2017, 139, 18107–18114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Kwon Y; Chinn AJ; Kim B; Miller SJ Divergent Control of Point and Axial Stereogenicity: Catalytic Enantioselective C–N Bond-Forming Cross-Coupling and Catalyst-Controlled Atroposelective Cyclodehydration. Angew. Chem. Int. Ed 2018, 57, 6251–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (499).Zahrt AF; Athavale SV; Denmark SE Quantitative Structure-Selectivity Relationships in Enantioselective Catalysis: Past, Present, and Future. Chem. Rev 2020, 120, 1620–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (500).Reid JP; Sigman MS Comparing Quantitative Prediction Methods for the Discovery of Small-Molecule Chiral Catalysts. Nat. Rev. Chem 2018, 2, 290–305. [Google Scholar]
- (501).Dobson CM Chemical Space and Biology. Nature 2004, 432, 824–828. [DOI] [PubMed] [Google Scholar]