

Abstract
Spurred by newly developed drug delivery systems (DDSs), side effects of cancer chemotherapy could be reduced by using multifunctional nanoplatforms. However, the facile synthesis of effective DDSs remains a challenge. Here, a six-arginine-tailed anti-epidermal growth factor receptor (EGFR) affibody was employed to easily synthesize the highly reactive oxygen species (hROS)- and trypsin-responsive 11-mercaptoundecanoic acid-modified gold nanoclusters (MUA-Au NCs) for tumor-targeted drug delivery. The polyarginine moiety of affibody sealed methotrexate (MTX)-loaded MUA-Au NCs through charge effect, as well as leaving the rest targeting fragment of the affibody to specifically bind tumor overexpressed EGFR. As the shell of MUA-Au NCs-MTX-Affibody (MAMA), polyarginine chains of affibody could be digested by trypsin, helping to release MTX from MAMA. The released MTX accelerated destroying MUA-Au NCs through inducing the generation of hROS. Specifically targeting EGFR-overexpressed tumors, quickly delivering a sufficient amount of drug to the tumor, subsequently increasing the local MTX and hROS levels, and safely eliminating the biocompatible structure from kidney, endowed MAMA greater treatment effectiveness and lower side effect than chemotherapy, especially in pancreatic cancer due to its high trypsin level. This simply fabricated DDS may find applications in high effective cancer therapy, especially for tumors with high trypsin activity.
Keywords: Cancer therapy, Drug delivery, Gold nanocluster, Reactive oxygen species, Trypsin
1. Introduction
Nanotechnology-based drug delivery systems (DDSs) hold enormous potential to efficiently encapsulate chemotherapeutic drugs and achieve targeting drug release to tumor tissues with limited cytotoxicity to normal cells [1]. An ideal DDSs design needs several essential elements including biocompatibility with negligible toxicity, stable drug encapsulation, and specific targeting [2]. Gold nanostructures have been extensively studied as DDSs in cancer therapy, due to their good biocompatibility and easy in surface modification [3]. Shells and coatings have been usually used for encapsulating the loaded drug into the gold nanostructure [4]. Antibodies and affibodies of protein overexpressed on the cancer cells have been added to the surface of the DDSs shells and coatings for specifically targeting tumor biomarkers [5,6]. However, complicated functionalization of DDSs requires multiple reactions with repeated purifications, resulting in drug leaking and low yields of the final product. It remains a demanding challenge to simply fabricate DDSs without sophisticated modifications.
Meanwhile, compared to conventional nanocarriers in clinical applications or under clinical trials, DDSs are superior in controllability which reduces adverse off-target effects [7]. Since proliferation and progression of cancer are closely related to reactive oxygen species (ROS), various responsive groups of ROS have been exploited to synthesize DDSs for controlled drug release in tumors [8]. However, due to insufficient intracellular levels of endogenous ROS usually result in relatively low therapeutic efficiency, inducing the generation of high dosage ROS in tumors targeted by ROS-responsive DDSs is a promising strategy to potentiate the therapeutic outcome in cancer therapy. Besides, highly ROS (hROS) as strong oxides including hydroxyl radical (•OH), peroxynitrite (ONOO−), and hypochlorite (ClO−) can oxidize nucleic acids, proteins, and lipids, damaging the living cells and activating the programmed cell death [9]. Therefore, developing a new DDS which can control release drug into targeted tumors, as well as increase the in situ levels of hROS in tumors are highly desired.
The other method to minimize the potential side effects of nanocarriers is providing the DDS with high affinity and selectivity to bind the aim targets. Epidermal growth factor receptor (EGFR) has been reported as an overexpressed biomarker in many cancers, especially certain gastrointestinal carcinomas [10,11]. Antibodies for EGFR have been used as targeting moieties for high-contrast tumor imaging and as potential drugs for cancer treatment because of their superior affinities with target and increased accumulation on the tumor surface [12,13]. However, the antibodies have low clearance rates and low tumor penetration capability due to their large size, resulting in the low signal-to-noise ratio for tumor imaging and unsatisfactory effectiveness for cancer treatment [14]. Affibody has been developed as a class of engineered protein that retains the desirable high affinity and specificity of the antibody, but with small size and chemical robustness for medical application [15,16]. The specific selectivities, short circulation, high renal clearance, biocompatibility, and ease of synthesis make affibody suitable for use in DDS.
Herein, we report a novel strategy to easily synthesize a highly biocompatible tumor-targeting DDS that exhibits the desired properties of hROS and trypsin dual responsive drug-controlled release, as well as inducing hROS generation to accelerate drug release and cell damage in tumors for enhanced cancer therapy. Methotrexate (MTX) is selected as the loaded chemotherapeutic drug, which has been used clinically in high doses as a folate antagonism to block the folate pathway and stimulate ROS production for decreasing metastasis and inducing apoptosis of cancer [17]. As shown in Scheme 1, hROS-responsive 11-mercaptoundecanoic acid-modified gold nanoclusters (MUA-Au NCs) are one-pot synthesized, then cancer-targeting ligand, six-arginine-tailed anti- EGFR affibodies are prepared. MTX is loaded into MUA-Au NCs to form MUA-Au NCs-MTX (MAM). Finally, positively charged polyarginine tails of affibodies easily self-assemble onto the negatively charged surface of MAM, forming a shell to seal in the loaded drug, whereas EGFR targeting fragments of affibodies remains on the surface of MUA-Au NCs-MTX-Affibody (MAMA) for tumor targeting. Upon active targeting to EGFR overexpressed tumor, endogenous upregulated trypsin in cancer cells can digest polyarginine chains of affibodies to release MTX. In situ released MTX further induces hROS production, achieving subsequent amplification of high-dosage MTX release by destroying Au NCs structure, which is favorable for both chemotherapy and high oxidative stress therapy of cancer.
Scheme 1.
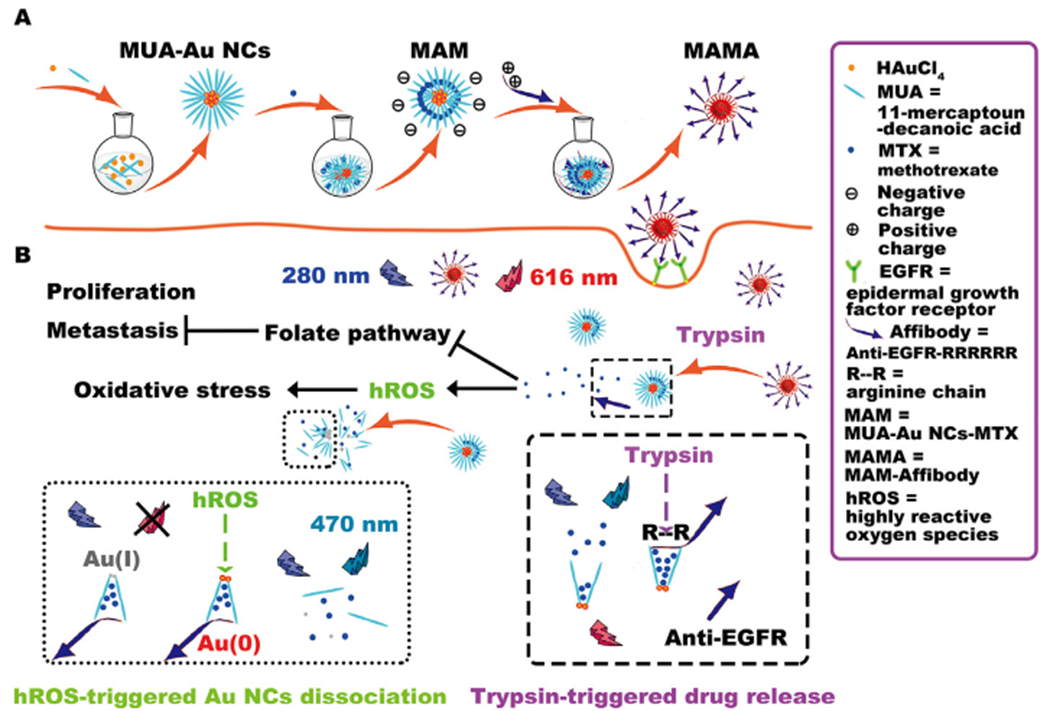
Schematic illustration of the synthesis of MAMA, as well as drug release and fluorescence change strategies for monitoring MTX induced hROS generation using MAMA.
2. Material and methods
2.1. Materials
Human cancer cell lines HepG2, BxPC3, and normal cell line hTERT-HPNE were obtained from Institute of Biochemistry and Cell Biology (SIBS, CAS, Shanghai, China) and grown in Dulbecco’s Modified Eagle medium (DMEM) and RPMI 1640 media with 10% FBS subsequently maintained in a 37 °C with 5% CO2 incubator, respectively. Female Nude mice (18–20 g, 6 weeks old, purchased from Charles River Laboratories) were selected for establishing the xenograft mice models. 5 × 107 HepG2 or BxPC3 cells in 100 μL of PBS were separately inoculated subcutaneously in each mouse at the shoulder to create two different groups of mice models. All animal studies were conducted under the protocol approved by the Stanford University Institutional Animal Care and Use Committee. Cell culture media, fetal bovine serum (FBS), phosphate-buffered saline (PBS), penicillin/streptomycin, mouse serum, and acid-soluble collagen solution were bought from Gibco (Grand Island, NY, US). Trypsin activity colorimetric assay kit was obtained from BioVision Inc. (Milpitas, CA, US). Other chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, MO, US).
2.2. Synthesis of MUA-Au NCs, MAM, and MAMA
4 mg 11-MUA was added into 10 mL of HAuCl4 solution (10 mmol/L), and then NaOH solution (100 μL, 1 mol/L) was added dropwise into the solution [18]. The MUA-Au NCs solution could be obtained and purified through high-speed centrifugation after the mixture was left to react for 24 h at room temperature. 10 mL MUA-Au NCs (0.2 mg/mL) was added 250 μmol of MTX with stirring for 4 h to get MAM. The obtained solution was dialyzed in membrane tubing with a molecular weight cutoff of 3 kDa against ultrapure water to remove free molecules and ions and then stored at 4 °C. Six-arginine tailed anti-EGFR affibody (VDNKFNKEMWAAWEEIRNLPNLNGWQMITAFASLVDDPSQSANLLAEAKKLNDAQAPK) was synthesized, purified by HPLC with a 2998 photodiode array detector (Waters, Milford, MA, USA), and characterized by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) (SimulTOF 200 Combo, Marlborough, MA, US). 20 mg affibody was dissolved in 10 mL prepared MAM solution with stirring for 4 h at room temperature to get MAMA. The obtained solution was dialyzed in membrane tubing with a molecular weight cutoff of 12 kDa against ultrapure water to remove free molecules and ions and then stored at 4 °C.
2.3. Characterization of MUA-Au NCs, MAM, and MAMA
The fluorescence spectra and UV–Vis absorption spectra of MA, MAM, and MAMA were recorded by using a TECAN Infinite® M1000 PRO microplate reader (Mānnedorf, Switzerland). Transmission electron microscopy (TEM) JEM-2100 (JEOL Ltd., Japan) was used for MUA-Au NCs, MAM, and MAMA characterization. The size distribution and zeta potentials of MA, MAM, and MAMA were determined by DLS at room temperature using a Zetasizer Nano-ZS90 (Malvern Instruments, Worcestershire, UK), and the Fourier Transform Infrared (FT-IR) spectra of MUA-Au NCs with and without affibody were recorded using a Bruker Tensor 27 FT-IR spectrometer equipped with a Digi Tech TM detector (Karlsruhe, Germany).
2.4. ROS- and enzymes-responsive fluorescent change and drug release
The •OH was generated by mixing Fenton’s Reagent with H2O2. HClO and H2O2 stock solution were used to supply the ClO− and H2O2 [19]. The ROS- and enzymes-responsive fluorescent change was investigated by incubating MAMA with H2O2, ClO−, •OH, proteinase K, and trypsin with stirring for 20 min at room temperature, respectively. Different concentrations of •OH and different activities of trypsin were used for further drug-releasing tests of MAMA. Briefly, 190 μL MAMA was mixed with 10 μL of •OH with different concentrations, and trypsin with different activities for 12 h, respectively. The fluorescent of MTX from MAMA was measured and calculated to corresponding drug concentration.
2.5. Western blot analysis and cell uptake of MAMA
Proteins were abstracted from HepG2, BxPC3, and hTERT-HPNE cells using lysis buffer to quantify EGFR expression of these cells. After the protein concentrations were determined, equivalent 40 μg of proteins were boiled in PBS with a 5 × loading buffer. The samples were separated by 15% SDS-PAGE gel electrophoresis and transferred to PVDF membranes (Millipore, Bedford, MA, USA). The membrane was blocked in 5% nonfat milk and incubated with the primary antibody of EGFR and the appropriate horseradish peroxidase-conjugated secondary antibodies (Sangon, Shanghai, China). Protein bands were visualized by the enhanced chemiluminescence (ECL) reagent (Sangon, Shanghai, China). To study the integrin targeting property of MAMA, HepG2 and hTERT-HPNE cells were incubated in a serum-free medium containing MAMA for 20 min and then rinsed with PBS and replaced with fresh cell medium, respectively. To further validate that the EGFR targeting property of MAMA, pre-blocking experiments were designed with HepG2 cells incubated with 2 mg/mL anti-EGFR antibody for 20 min before incubating with MAMA. The cells were then fixed using 4% phosphate-buffered paraformaldehyde and stained with 4′6-diamidino-2-phenylindole (DAPI) for 10 min. The cells were imaged by a CLSM (Zeiss LSM880, Jena, Germany). The excitation wavelength of 616 nm was recorded as MAMA under UV excitation with 454 nm emission of DAPI as a reference for nucleus staining.
2.6. hROS induced by MTX and cytotoxicity of MAMA
To understand the hROS-responsive fluorescent quenching of MAMA in cells, HepG2 and BxPC3 cells were seeded at a concentration of 5 × 104 cells/plate on 35 mm confocal plates for overnight growth. 100 μL MAMA was applied to each cell culture plate with 100 μL medium for a 12 h incubation. The cells were imaged by a CLSM at 2 h, 7 h, and 12 h incubation with MAMA. Trypsin inhibitor TLCK was used to co-incubated with MAMA for 12 h. The FI of MAMA at 616 nm was measured at different time points. Different concentrations of MTX were added into cells to test the hROS induced by MTX. The hROS was measured from cells by aminophenyl fluorescein at 515 nm emission after 2 h, 7 h, and 12 h incubation with MTX. Folate was used as MTX competitor to co-incubated with MTX and cells for 12 h. The FI change of MAMA after 12 h incubation with HepG2 and BxPC3 cells were imaged by three-dimensional airyscan, and the FI change of MAMA from cell member to nuclei was measured by line-scan. The cytotoxicity of MUA-Au NCs, MAMA, MAM, and free MTX incubated with cells were studied by Cell Counting Kit-8 (CCK-8) assay (Dojindo, Japan) using HepG2, BxPC3, and hTERT-HPNE cells as former reported, respectively [20]. Cells were evaluated after 2, 12 and 24 h incubation 190 μL medium with 10 μL either MAM or MAMA and 0.24 μmol free MTX, respectively. The FI at 450 nm was measured using the TECAN Infinite® M1000 PRO microplate reader after another 1 h CCK-8 incubation.
2.7. Synthesis of MN and 68Ga-radiolabeling
To a solution of MTX (10 mg, 22 μmol) in DMSO (1 mL) was added EDC (4.3 mg, 28 μmol) and NHS (3.2 mg, 28 μmol). After 1 h, ethylenediamine (2.3 mg, 38 μmol) and DIPEA (77 μL, 440 μmol) in DMSO (1 mL) was added at 0 °C and maintain for 1 h. Then the reaction was allowed to warm up to room temperature and stirred overnight. Distilled water (1 mL) was added to quench the reaction and the product was purified by HPLC on a reverse-phase C4 column eluted with acetonitrile/water (containing 0.1% trifluoroacetic acid). MTX-NH2 mass spectrometry (MALDI-TOF MS) m/z: [M +] calculated for C22H29N10O4 + : 497.24, found: m/z 497.35. To a solution of MTX-NH2 (10 mg, 20 μmol) in DMSO (2 mL) was added NODAGA-NHS ether (22 mg, 30 μmol) and DIPEA (70 μL, 400 μmol). The reaction mixture was stirred overnight at room temperature. Distilled water (1 mL) was added to quench the reaction and the product was purified by HPLC on a reverse-phase C4 column eluted with acetonitrile/water (containing 0.1% trifluoroacetic acid). MN (MALDI-TOF MS) m/z: [M+] calculated for C37H52N13O11+ : 854.39, found: m/z 854.61. 68Ga labeling was performed as reported earlier [21]. A 68Ge/68Ga-generator (Isotope, Moscow, Russia) was eluted with 0.1 M aqueous HCl. MTX-NODAGA was pre-loaded to MUA-Au NCs to form MAMNA and MAMN with and without affibody sealing, respectively. MAMNA, MAMN, and MN were diluted in 0.1 M sodium acetate buffer (pH 4.5) and mixed with pH adjusted 68Ga for 20 min. 68Ga-MN was purified by HPLC with a radioactivity detector, 68Ga-MAMNA and 68Ga-MAMN were purified by PD-10 column with PBS as the mobile phase.
2.8. In vivo PET/CT imaging and biodistribution studies
100 μCi radioactive doses of 68Ga-MAMNA, 68Ga-MAMN, 68Ga-MN, and 68Ga-MAMNA with anti-EGFR antibody in 100 μL PBS were separately injected into four groups of mice models with HepG2 tumors and another four groups of mice models with BxPC3 tumors via the tail vein 1 h before PET/CT imaging using Inveon μPET/CT (Siemens, Munich, Germany), respectively (n = 3, each group). PET/CT data were reconstructed by software Inveon Research Workplace (IRW 4.2). The region of interested PET signal was quantified and presented as a percentage of the injected dose per gram (% ID/g). Major organs were collected and wet-weighed at 2 h post-injection. The radioactivity uptake by the tissues was measured by gamma-counter (Perkin-Elmer, Waltham, MA, US) and presented as % ID/g (mean ± SD).
2.9. Cell apoptosis and necrosis of tumors
Tumors were removed from six groups of HepG2 tumor-bearing mice and another six groups of BxPC3 tumor-bearing mice after a onetime injection of MAMA, MAM, and the same amount of free MTX for 2 and 4 h, respectively (n = 3, each group). The extracted tumors were put into cold culture media and minced into 2–4 mm fragments, then incubated with dissociation solutions for 30 min at 37 °C [22]. The fragments were filtered through a 70 μm nylon mesh cell strainer (BD Biosciences, San Diego, CA, USA). The released cells were centrifuged at 1200 rpm for 5 min. MTX positive cells were collected by sorter Carmen (BD Influx, Heidelberg, Germany). Then, the collected MTX positive cells were stained with Annexin V-FITC and PI according to the protocol. The cells were incubated in the dark at room temperature for 15 min. Finally, the percentage of apoptotic cells with positive or negative MUA-Au NCs signal under UV excitation at 2 and 4 h post-injection of MAMA, MAM, and free MTX were calculated by Falstaff (BD Aria II, Heidelberg, Germany). The necrotic rates of cells at 4 h postinjection of MAMA, MAM, and free MTX were assessed by FACScan flow cytometry.
2.10. Therapeutic effects and histology analysis
When the tumors reached around 50 mm3, the mice carried each kind of tumors were randomly assigned into four groups (eight mice per group): saline only, MTX only, MAM, and MAMA. The total concentration of MTX administered, whether the free drug or bound in the carriers, was kept consistent at 8 mg/kg. All groups were treated every two days for ten iterations. The tumor volumes of all the groups were monitored every four days with a digital caliper, and the tumor volume was calculated following formula: width2 × length/2 [23]. The body weight of mice was measured using balance every four days. The BxPC3 tumor-bearing mice were sacrificed after the last treatment for 4 h. Tumors and the major clearance organs of MAMA including livers and kidneys were collected, fixed in 4% formalin overnight, and then embedded in optimal cutting temperature compound [24]. The frozen tissues were sectioned by Leica CM1850 Cryostat (Buffalo Grove, IL, US), and the slices were stained by H&E, then visualized using Nano-Zoomer S60 (Hamamatsu, Japan). CLSM was used for observing the MAMA distribution and MTX released conditions from histological sections.
2.11. Statistical analysis
All data were representative results from at least three independent experiments and expressed as means ± SD. Statistical analyses were performed using the t-test, *p < .05, **p < .01, ***p < .001 and ****p < .0001 were considered statistically significant.
3. Results and discussion
MAMA was prepared in three steps and the characterization of the product coming from each step as shown in Fig. 1. The average diameter of MUA-Au NCs, MAM, and MAMA was 1.1 nm, 3.4 nm, and 9 nm, respectively (Fig. 1A). The molecular weight of synthesized affibody was 7519.1 g/mol with 99.9% purity after high-performance liquid chromatography (HPLC) (Fig. S1A, B). The pronounced IR absorption bands of affibody occurred at 1650 cm−1 (ν-CO-NH-), 1590 cm−1 () 1210 cm−1 (νCn) and 1100 cm−1 (νCn) (Fig. S1C). Changing of size and IR absorption band confirmed the successful affibody modification. The surface charge decreased from ‒17 mV of MUA-Au NCs to − 32 mV after modified by the affibody (Fig. S1D). The polyarginine moiety of affibody with strong ~ + 5 charge at pH 7 attaches to MAM by charge effect, leaving the mild negatively-charged anti-EGFR fragment of affibody outside of MAMA to form a stable structure with negative charges on the outermost surface (Fig. S1E, F). The size changes of MAMA when it was incubated with different ROS and trypsin in solution were evaluated using dynamic light scattering (DLS), respectively (Fig. 1B, C). When incubated with high levels of different ROS, the size of MAMA was almost unchanged due to the integrity of the structure being maintained. These results suggest stable binding between the negatively charged carboxylic acids and positively charged polyarginine of the affibody. After incubating with different enzymes, proteinase K and trypsin significantly decreased the size of MAMA through cleaving affibody and digesting the polyarginine chain of affibody, respectively. It was shown that the enzymes were the. main factors for the size decreasing of MAMA.
Fig. 1.
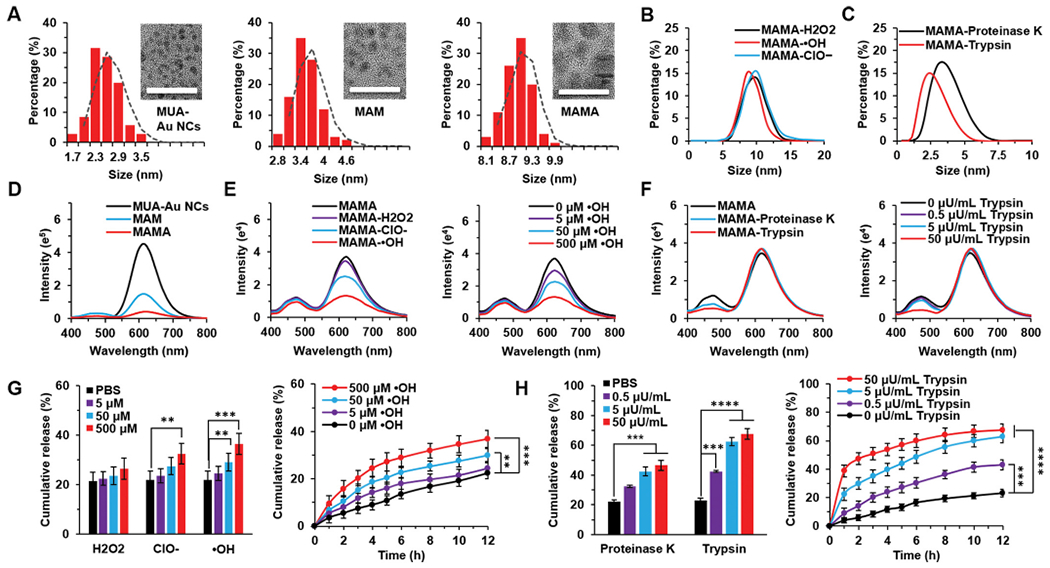
Size, fluorescent intensity and drug release of MAMA under ROS and enzyme incubation. (A) Size characterization and transmission electron microscopy (TEM) images of MUA-Au NCs, MAM, and MAMA. Scale bar = 20 nm. (B) The dynamic light scattering (DLS)-measured size distribution of MAMA incubated with ROS. (C) The DLS-measured size distribution of MAMA incubated with enzymes. (D) Fluorescence spectra of MAMA. (E) Fluorescence spectra of MAMA incubated with H2O2, ClO−, and •OH, respectively. (F) Fluorescence spectra of MAMA incubated with proteinase K and trypsin. (G) The drug release of MAMA incubated with H2O2, ClO−, and •OH, respectively. (H) The drug release of MAMA incubated with proteinase K and trypsin. All result represents the mean ± standard deviation (SD) (**P < .01, ***P < .001, and ****P < .0001).
The fluorescence spectra of MUA-Au NCs showed the maximal fluorescence emission wavelength at 616 nm under UV 254 nm excitation. MAM displayed an additional blue emission peak at 470 nm under UV light due to carrying MTX which is weakly fluorescent (Fig. 1D). After loading MTX, the fluorescence intensity (FI) of MAM and MAMA turned weak. It indicated that the fluorescence quantum yield of gold nanoclusters was largely impacted by charge transfer from the surface ligands to the metal core via S—Au bonds [25]. A change in ligand structure or electron-donating ability would impact emission. And also, the FI of MUA-Au NCs decreased when MTX attached to the surface of Au NCs [26]. Significant changes in 616 nm with minimal changes in 470 nm FI of MAMA were found when incubated with the hROS ClO− and •OH, but weak ROS H2O2 barely affected the FI of MAMA (Fig. 1E). It was because of that hROS could quench the fluorescent signal of MAMA through changing the valence of Au NCs from Au (0) to Au (I), the decomposition of Au NCs resulted in the loss of fluorescence [27]. The FI quenching of Au NCs was positively correlated with the concentration increasing of •OH, which indicated that MAMA had a potential for sensitively detecting hROS. Since there was no significant change in 470 nm FI of MAMA after incubation with either ClO− or •OH, hROS was not the main factor for the drug release of MAMA. After incubated with enzymes, only 470 nm FI of MAMA was significantly decreased due to MTX release without disrupting the MUA-Au NCs structure, especially when incubated with trypsin (Fig. 1F). Compare with proteinase K which broadly cleaved the carboxylic group of aliphatic and aromatic amino acids, trypsin could selectively cleave peptide chains at the carboxyl side of arginine and digest the arginine shell of MAMA [28,29]. The FI decreasing of MTX was positively correlated with the activities increasing of trypsin, which suggested that MTX release was mainly triggered by high trypsin. As a serine protease family member, trypsin level has been reported higher in pancreatic cancer and also has a potential role in cancer invasion [30,31]. The various fluorescence patterns could provide a trackable signal of the position of MAMA as well as monitoring drug release and hROS change in vitro. Meanwhile, the trypsin-responsive drug release of MAMA is promising for pancreatic cancer treatment.
To evaluate the potential of the arginine chain as the shell of MAMA, MTX loading concentration and drug-releasing efficiency of MAM and MAMA were tested in solution. After confirming the relationship between the FI of MTX at 470 nm and different concentrations of MTX, MTX at different concentrations was mixed with the same amount of MUA-Au NCs to calculate the maximum drug loading capacity of MUA-Au NCs (Fig. S2A). The maximum drug loading of 0.2 mg MUA-Au NCs was achieved by using 2.38 μmol MTX after 4 h mixture at pH 7.4, 37 °C (Fig. S2B and S2C). When the drug loading efficiency was as high as 95.2%, the concentration of MTX loaded into MUA-Au NCs was 1.08 mg/mL, which was comparable to concentrations of MTX used clinically [32]. Consider the charge of MAM and six-arginine tails would be affected by pH, drug-releasing efficiency of MAM with or without six-arginine tails of affibody as a shell was evaluated under physiological conditions (pH 7.4) and acidic tumor microenvironment (pH 6.6). Due to the open system of MAM, the cumulative release of MTX was 66.0% and 66.7% after 12 h incubation in phosphate-buffered saline (PBS) at pH 7.4 and pH 6.6, 37 °C, respectively. When 2 mg/mL six-arginine-tailed affibodies were applied as the capping shell of MAM to form MAMA, the cumulative MTX release decreased to 24.65% and 20.38% after 12 h incubation in PBS at pH 7.4 and pH 6.6, 37 °C, respectively (Fig. S2D). After calculation, 20–25 affibodies were sufficient to seal 1 MAM which carries 10–15 MTX, forming 1 MAMA unit [33]. Because the theoretical isoelectric point of the six arginine chain is around 13, it is easy for the six arginine chain to self-assemble on the negatively charged surface when the environmental pH is lower than 13. Since that there was little FI quenching of MUA-Au NCs at 616 nm when trypsin helped to release 80% of MTX from MAMA through digesting the arginine shell, the high drug loading capacity of MAMA mainly relied on the van der Waals and hydrophobic interactions between MTX and MUA chains (Fig. S3) [26]. The excellent drug loading capacity of MAMA and the successful sealing function of a six-arginine-tailed affibody suggests the great potential of MAMA as a drug carrier in both physiological and cancerous environment.
The in vitro MTX release rate was further evaluated for MAMA upon treating with ROS and enzymes. hROS ClO− and •OH were observed to mediate much faster MTX release compared with weak ROS H2O2, which also agreed with their better fluorescence quenching to MAMA at 616 nm (Fig. 1E, G). We ascribe this selective drug release to the valence change-based decomposition of Au NCs [34]. The in vitro MTX release profile under •OH was further quantitatively evaluated. Compare with MAMA at 0 μM •OH, the drug release was minimal at 5 μM •OH, a comparable upper level of •OH in mouse serum (Fig. S4A). At an elevated level of •OH at 50 μM, which was similar to the hROS level in cancer cells, there was a ~ 8.0% more cumulative MTX release than MAMA at 0 μM •OH upon incubation for 12 h. Whereas at 500 μM •OH, 37.0% total MTX release was observed upon treating MAMA for 12 h. Drug release analysis with •OH as the trigger in solution suggested that normal level of hROS (~1 μM) would barely help on drug-releasing of MAMA, and only a small amount of MTX could be released from hROS-responsive MAMA in cancer cells, but higher hROS level would release more MTX. Since there was no significant increase of MTX release after 2 h incubated MAMA with 500 μM •OH at pH 7.4, 37 °C, hROS-responsive drug release of MAMA was a time-dependent process. Enzyme-responsive drug release of MAMA was also monitored in vitro (Fig. 1H). For the control group, the cumulative release of MTX remained at 23.0% after 12 h incubation in PBS at pH 7.4, 37 °C. Whereas the MTX release of MAMA increased readily after incubated MAMA with proteinase K and trypsin, respectively, at 5 nU/mL and 50 nU/mL, which also agreed with the decrease of MAMA FI at 470.
nm after enzyme incubation (Fig. 1F). The cumulative release of MTX of MAMA incubated with trypsin was higher than that incubated with the same activity proteinase K after 12 h, which indicated that trypsin could promote a much faster drug release by selectively cleave arginine chain and digest shell of MAMA. After quantitatively evaluated the in vitro MTX releasing profile of MAMA incubated with trypsin, 63.0% total MTX release was observed upon treating MAMA at 5 μU/mL trypsin for 12 h, which was almost the same amount as MTX release of open system MAM after 12 h incubation in PBS at pH 7.4, 37 °C. It suggested that 5 μU/mL trypsin was enough to digest polyarginine shell upon treating MAMA for 12 h. Drug release analysis with trypsin as the trigger in solution showed that low activity of trypsin (~0.5 μU/mL) in serum would barely help on fast drug-releasing of MAMA, but 30.0% and 47.0% MTX would be released from MAMA at trypsin activity of pancreatic cancer cells (~5 μU/mL) and pancreas (~50 μU/mL) upon 2 h incubation at pH 7.4, 37 °C, respectively (Fig. S4B). It showed that the trypsin-responsive drug release of MAMA was an activity-dependent process. Hence, hROS and enzymes could mediate the decomposing of Au NCs and the polyarginine shell digestion of MAMA to release MTX, and trypsin was the main trigger to quickly release the drug.
To investigate the EGFR targeting property of MAMA based on the decoration of the anti-EGFR fragment of affibody, cellular uptake of MAMA towards HepG2, BxPC3, and hTERT-HPNE was compared. Before commencing cell studies, we investigated the EGFR expression level of three cell lines (Fig. 2A). The expression of EGFR in HepG2 was 12.0-fold higher than in hTERT-HPNE, with BxPC3 levels falling in between. The cellular uptake of MAMA was monitored in hTERT-HPNE cells with lower expressed EGFR and HepG2 cells with overexpressed EGFR which were separately incubated with MAMA for 30 min. We also performed pre-blocking experiments based on confocal laser scanning microscopy (CLSM) imaging and quantitative FI analysis of MAMA at 616 nm (Fig. 2B, C). HepG2 cells were pretreated with excess free anti-EGFR antibody before incubation with MAMA for pre-blocking study. The FI of HepG2 cells was stronger than that of hTERT-HPNE cells, or HepG2 cells pre-blocking by the free anti-EGFR antibody. Thus, the anti-EGFR fragment of affibody on the surface of MAMA could facilitate a high affinity to EGFR overexpressed cancer cells. After quantification, the intensity of the fluorescent signal of MAMA at 616 nm was positively correlated with the EGFR expression level of cells. These results suggested the superiority of EGFR targeting property of MAMA, which indicated that MAMA could provide a great selection to deliver the drug specifically to EGFR expressed cells and also explored a potential method for conveniently evaluating EGFR expression level of cells directly by its FI.
Fig. 2.
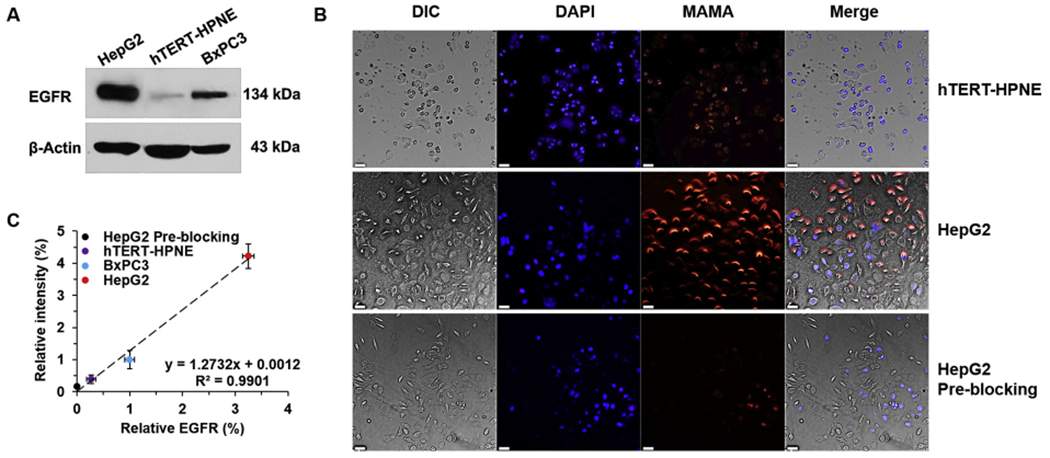
EGFR targeting and imaging properties of MAMA in cells. (A) EGFR western-blot analysis of cells. (B) Confocal images of cells incubated with MAMA for 30 min. Bars = 50 μm. (C) The quantitative relationship between relative EGFR protein level and relative intensity of MAMA in cells. All result represents the mean ± standard deviation (SD).
Besides, 29.1% and 37.4% cumulative drug release of MAMA was found when separately.
incubated MAMA with either cell culture medium or mouse blood for 12 h, respectively (Fig. S5A). No significant drug release of MAMA was observed upon treating MAMA with cell culture media and PBS for 12 h, which supported the notion that MAMA could stably deliver MTX to cells. The FI changing of MAMA was then examined in HepG2 and BxPC3 cells under UV excitation (Fig. 3A). CLSM imaging revealed that the FI of MAMA at 616 nm exhibited a time-dependent quenching in both HepG2 and BxPC3, which gradually decreased as the incubation time increased. Upon incubation for 2 h, the red fluorescence signal from MUA-Au NCs of MAMA in HepG2 cells was ~1.5-fold higher than that in BxPC3 cells (Fig. 3B). Previous data showed that EGFR expression level of HepG2 cells was ~4.2-fold higher that of BxPC3 cells (Fig. 2A), after calculated by the equation between FI of MAMA at 616 nm and expression level of EGFR (Fig. 2C), theoretically, ~5.3-fold higher signal from MUA-Au NCs of MAMA should get from HepG2 cells compared with BxPC3 cells. The lower red fluorescence signal of MAMA in the real test was caused by 26.1% higher hROS in HepG2 cells after 2 h incubation with MAMA (Fig. S5B). Even hROS in BxPC3 cells was not as high as that in HepG2 cells, the red fluorescence signal of MAMA still sharply decreased in BxPC3 cells upon incubation for 7 h. After 12 h incubation, the blue signal from MTX of MAMA clearly showed up in BxPC3 cells, and red fluorescent spots of MAMA existed in HepG2 cells. The change of fluorescence signal from red to blue could be eliminated by adding Tosyl-L-lysyl-chloromethane hydrochloride (TLCK) as the inhibitor of trypsin to BxPC3 cells before incubation with MAMA, but in contrast, the FI change in HepG2 cells was negligible after using inhibitor. As shown previously, trypsin could help to release MTX from MAMA without changing the FI of MUA-Au NCs (Fig. 1F), and hROS was the main factor to quench the FI of MAMA at 616 nm (Fig. 1E). Since neither TLCK nor affibody could affect the FI from MUA-Au NCs of MAMA (Fig. S5C), it suggested that MTX released from MAMA could help to quench the FI from MUA-Au NCs in BxPC3 cells. Interestingly, after incubated with the same amount of MAMA for 12 h, the intracellular hROS in BxPC3 was 13.2% higher than that in HepG2 (Fig. S5B). Compare with that hROS level of BxPC3 cells was lower than that of HepG2 cells upon incubation with MAMA for 2 h, the variables after 7 h incubation with MAMA were more MTX release due to 3.0-fold higher trypsin activity in BxPC3 cells than that in HepG2 cells (Fig. S5D). To better understand the relationship between MTX release and hROS increase in cells without influenced by high trypsin activity, the hROS state of HepG2 cells was evaluated upon treating with different concentrations of MTX (Fig. 3C). After 2 h, 5 μM MTX helped to increase ~ 10.0% hROS in HepG2 cells compared with low concentration MTX treated groups. ~8.0% and ~ 16.0% relative hROS was increased in HepG2 cells after 7 h incubation with 1 and 5 μM MTX, respectively. Upon incubation with 0.5, 1, and 5 μM MTX for 12 h, ~8.0%, ~37.0%, and ~ 57.0% hROS increase in HepG2 cells were found, respectively, compared to the cells without MTX, suggesting higher MTX induced faster hROS increase in HepG2. Notably, pretreated cells with MTX competitor, folate, could attenuate the hROS increased by MTX in all these groups. These results showed that MAMA could significantly enhance the intracellular hROS level by releasing as much as MTX to cells.
Fig. 3.
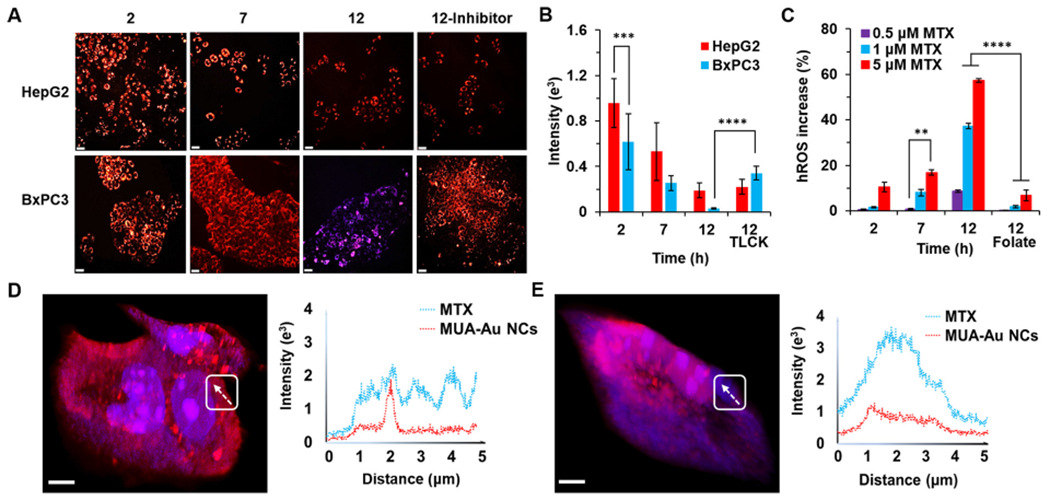
The FI changing of MAMA in cells. (A) CLSM images of MAMA under UV excitation after incubation with HepG2 and BxPC3 cells for 2, 7, and 12 h with or without trypsin inhibitor (TLCK). Scale bar = 50 μm. (B) The FI of MAMA at 616 nm in A. (C) Relative hROS increase in BxPC3 cells induced by different concentrations of MTX after 2, 7, and 12 h with or without folate incubation. Three-dimensional confocal imaging of MAMA and the line-scan profile in HepG2 cells (D) and BxPC3 cells (E) were shown after 12 h incubation. Bar = 5 μm. All result represents the mean ± standard deviation (SD) (**P < .01, ***p < .001, and ****P < .0001).
Such an MTX release and hROS increase in cells were further visualized with three-dimensional airyscan imaging (Fig. 3D, E). Under UV excitation, the red fluorescent emission of MAMA turned weaker in BxPC3 cells than that in HepG2 cells after 12 h incubation, which suggested that more structure of MUA-Au NCs was decomposed by higher hROS in BxPC3 cells. Blue signal was originated from the emission of MTX itself. Compared with HepG2 cells, the FI of MTX in BxPC3 cells was stronger and more diffuse which would be consistent with more drug release and subsequent diffusion of the drug throughout the cells. The fluorescent changes of MAMA including red signal decreasing and blue signal increasing were in good agreement with the hROS increase and MTX release in cells. To further probe the intracellular fate of MAMA, FI changes of MAMA from cell membrane to the cytoplasm were investigated. Airyscan imaging and the line-scan profiles also revealed that there were several peaks with low FI of MTX mixed with the same FI of MUA-Au NCs in HepG2 cells, indicating that MTX was trapped into MAMA without being fully released even after high hROS helped to decompose MUA-Au NCs structure. Whereas in BxPC3 cells, a strong and wide FI peak of MTX and low FI of MUA-Au NCs were showed when MAMA moved from membrane to the center of cells, revealing a highly efficient drug release of MAMA due to high trypsin activity in BxPC3 cells, which subsequently helped to elevate hROS level, finally decomposing MUA-Au NCs structure. Herein, the large amount of MTX release from MAMA would guarantee the increase of hROS level in cells, resulting in complete drug release from MAMA accompany with fluorescent quenching of MUA-Au NCs. All these cell study results showed the synthesized MAMA was the same as the designed structure, with expected fluorescent changes during hROS- and trypsin-responsive drug release in cells, as well as hROS induced by MTX release. The data suggested that MAMA would be a promising drug delivery and controlled-release system for EGFR-overexpressed cancers, especially for EGFR-overexpressed cells with high trypsin activityr.
Finally, to further investigate whether the more MTX release from MAMA and subsequent hROS increase could provide more cytotoxicity, cell viability assays were performed (Fig. S6A). Firstly, the biocompatibility of MAMA was evaluated. Almost no loss of viability was found in hTERT-HPNE cells treated with 0.2 mg/mL MAMA with no MTX loaded into the system for 24 h, which demonstrated the biocompatible backbone of MAMA. Then MAMA was incubated with hTERT-HPNE, HepG2, and BxPC3 cells, respectively, for 2, 12, and 24 h. There was no significant loss of viability in hTERT-HPNE cells treated with MAMA, compared to those treated with free MTX for 24 h. However, a significant decrease in cell survival was observed in both HepG2 and BxPC3 cells treated with MAMA, compared to those treated with the same concentration of free MTX for 24 h. The results indicate that MAMA helps to deliver more MTX to the targeted cells, subsequently increasing the cytotoxicity to specific cells. Moreover, the sharp decrease of cell viability in BxPC3 after 12 h incubation with MAMA shows the fast drug release of MAMA in the cells with high trypsin activity. To better understand the higher cytotoxicity was mainly caused by more MTX release due to higher trypsin activity in BxPC3 cells, or the hROS increase induced by MTX release, BxPC3 cells were pretreated by glutathione (GSH) to attenuate the oxidative stress of hROS increasing. It showed that hROS contributed ~66.0% relative cell viability decrease in BxPC3 cells, suggesting higher cytotoxicity in BxPC3 cells attributed to hROS increase, which was induced by more MTX release from MAMA (Fig. S6B). Strong hROS not only could help to release more MTX from MAMA but also could mediate cell apoptosis, combining with MTX caused inhibition of cell proliferation by blocking the folate pathway, MAMA could decrease cell viability by increasing oxidative stress and attenuating the metastasis. The specific targeting and effective MTX-releasing properties of MAMA were highly promising for the treatment of EGFR-overexpressed cells with high trypsin activity.
The decoration of affibody endowed the potential enrichment effect of MAMA in EGFR-overexpressed cells. To investigate whether MAMA could efficiently deliver the drug to the tumor in vivo, MTX was modified to incorporate gallium-68 (68Ga), a radioisotope that could label the conjugated MTX later for positron emission tomography (PET) imaging to mimic the drug delivery of MAMA (Fig. S7). 1,4,7-triaza-cyclononane,1-gluteric acid-4,7-acetic acid (NODAGA) as a macrocyclic chelator had a strong affinity to 68Ga [35]. MTX was modified with NODAGA to form MTX-NODAGA (MN) which was loaded into MUA-Au NCs for 4 h to form MUA-Au NCs-MTX-NODAGA (MAMN), and then the whole structure was encapsulated by affibody to form MUA-Au NCs-MTX-NODAGA-Affibody (MAMNA). The masses of MTX and MN were identified by MALDI-TOF to confirm the structures (Fig. S8A, B). Since that the stability of MAMNA and MAMA was similar in mouse blood, as well as MAMNA and MAMN showed no significant difference (~2%) in drug loading efficiency compared with MAMA and MAM after 6 h incubation, respectively, which indicated that MN could well represent MTX to.
mimic the drug delivery of MAMA in vivo (Fig. S9A, B). After radiolabeled by 68Ga, the purified 68Ga-MAMNA, 68Ga-MAMN, and 68Ga-MN were employed to image the drug delivery and biodistribution of MAMA, MAM, and MTX, respectively, in vivo. To this aim, HepG2 and BxPC3 tumor-bearing mice were intravenously (i.v.) injected with different tracers, respectively. The different levels of 68Ga-MN accumulated in tumors were clearly illustrated through visual evaluation of the PET images with computed tomography (CT) imaging as the reference of body position (Fig. 4A). 68Ga-MAMNA delivered 12.0- and 5.6-times higher amounts of 68Ga-MN to HepG2 and BxPC3 tumors, respectively, than did 68Ga-MAMN at 1 h post-injection. Compared to 68Ga-MN, 68Ga-MAMNA significantly delineated the subcutaneous HepG2 and BxPC3 tumors with 10- and 2.8-times the 68Ga-MN uptake, respectively, at 1 h post-injection, indicating the stable drug loading and efficient delivery functions of 68Ga-MAMNA (Fig. 4B). Excess free anti-EGFR antibody blocked both HepG2 and BxPC3 tumor uptake of 68Ga-MAMNA, which further supported EGFR-specific targeting of 68Ga-MAMNA. Biodistribution of the tracers in mice organs was determined at 2 h post-injection, and high activities of tracers were observed in the renal cortex and bladder of all groups of mice (Fig. 4C). Tumor/muscle ratios were 14.33 ± 0.30 and 9.33 ± 0.36 of 68Ga-MAMNA, 1.00 ± 0.24 and 1.67 ± 0.25 of 68Ga-MAMN, as well as 1.48 ± 0.28,2.41 ± 0.21 of 68Ga-MN in HepG2 and BxPC3 tumor-bearing mice, respectively, at 2 h post-injection (n = 3). Compared to 68Ga-MN, 68Ga-MAMNA uptake was 6.0–6.2-fold lower in the heart and 7.7–7.8-fold lower in the liver at 2 h post-injection, suggesting that 68Ga-MAMNA reduced the free 68Ga-MN in vivo, thus decreasing the side effects of MTX on the heart and liver [36]. Improved targeting, efficient drug delivery, and decreased untargeted accumulation are all benefits to the proposed MAMA that may allow lower amounts of MTX to be administered while receiving high treatment effectiveness and low side effect.
Fig. 4.
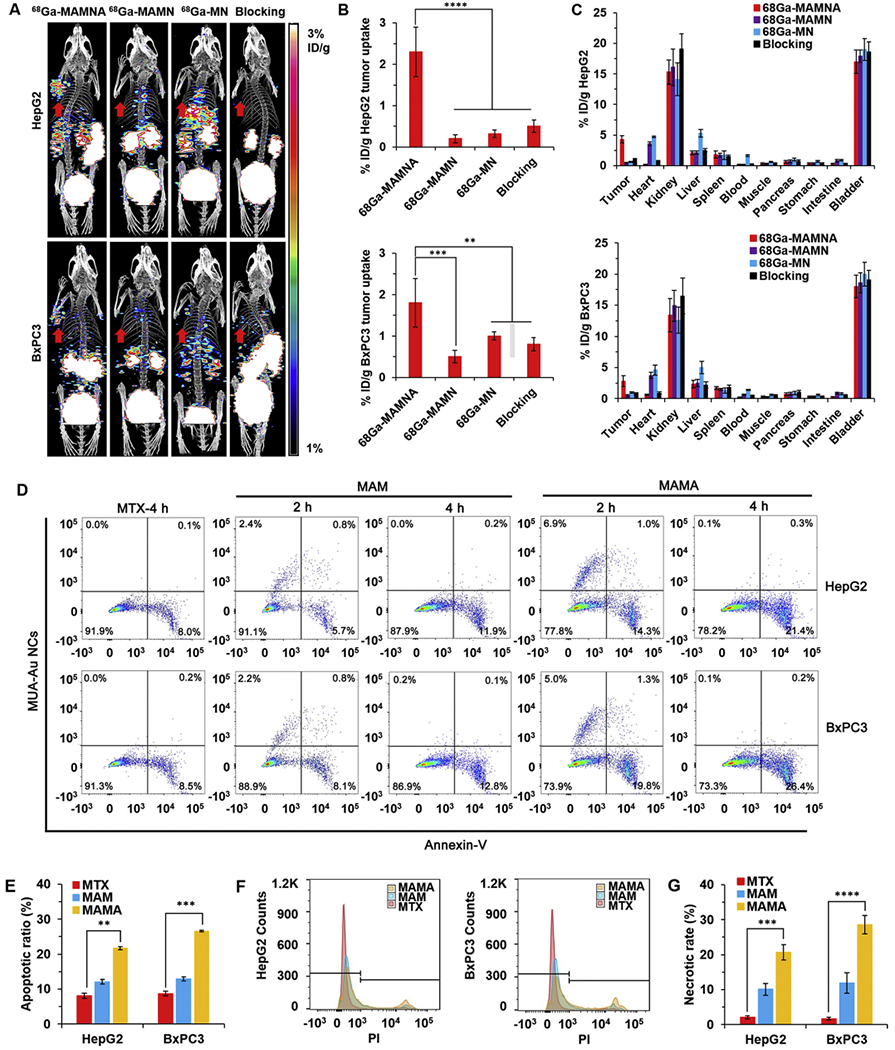
Fast drug delivery of MAMNA and MTX caused apoptosis of tumor in vivo. (A) In vivo positron emission tomography/computed tomography (PET/CT) imaging of HepG2 and BxPC3 tumor-bearing mice (n = 3) at 1 h post-injection of 68Ga-MAMNA, 68Ga-MAMN, 68Ga-MN and 68Ga-MAMNA with EGFR antibody as blocking control. Red arrows showed the tumor region of mice. (B) HepG2 and BxPC3 tumor uptake of tracers. (C) The biodistribution of radiolabelled probes after 2 h postinjection in HepG2 and BxPC3 tumor-bearing mice. (D) Cell apoptosis of HepG2 and BxPC3 tumors extracted from mice after 2 and 4 h different treatments were analyzed by flow cytometer with Annexin V staining. (E) Statistical analysis for the percentage of apoptotic cells in D. (F) Determination of cell necrotic rates upon different treatments for 4 h detected by propodium iodide (PI) based on flow cytometry analysis (FACS). (G) Statistical analysis for the percentage of necrotic cells in F. All result represents the mean ± standard deviation (SD) (**P < .01, ***p < .001, and ****p < .0001). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To further investigate whether hROS induced by MTX released from MAMA could occur in vivo, HepG2 and BxPC3 tumor-bearing mice were intravenously injected with MAMA, MAM, and the same amount of free MTX for 2 and 4 h, respectively, followed by the apoptotic analysis of cells extracted from mice tumors (Fig. 4D). All cells digested from tumors which had MTX positive signal under UV excitation were sorted and collected for the apoptosis analysis. The cells with positive MUA-Au NCs signal in MAMA treated group was more than that in MAM treated group at 2 h post-injection in HepG2 and BxPC3 tumors, which indicated that more MAMA was accumulated in tumors than MAM. The cells with the positive signal of MUA-Au NCs from MAMA in HepG2 tumor (6.9%) was higher than that in BxPC3 tumors (5.0%) at 2 h postinjection, verified EGFR targeting capability of MAMA in vivo. There was no fluorescence of MUA-Au NCs from MAMA or MAM in HepG2 and BxPC3 tumors at 4 h post-injection, which suggested that hROS of HepG2 and BxPC3 tumors were increased to the level which could completely decompose the MUA-Au NCs structure at 4 h post-injection. Fluorescein-annexin V (V-FITC) staining and MUA-Au NCs fluorescence assay further demonstrated that hROS increase was associated with cell apoptosis in vivo. The apoptotic ratio caused by MAMA, 21.7% and 26.6% in HepG2 and BxPC3 tumors, were significantly higher than that of free MTX (8.1% and 8.7%), and MAM (12.1% and 12.9%) at 4 h postinjection, respectively (Fig. 4E). Compare with HepG2 tumors, MAMA uptake of fewer cells from BxPC3 tumors obtained a higher apoptotic ratio at 2 h post-injection, which were in good agreement with the cytotoxicity results that increasing of hROS was induced by MTX quick release from MAMA due to high trypsin activity in BxPC3 cells (Fig. S5B, D). The antitumor effects were further confirmed by propodium iodide (PI) staining followed by flow cytometry analysis (FACS). The cell necrotic rates of HepG2 and BxPC3 tumors in the MAMA treated group got 9.9- and 16.8-folds higher than that in the free MTX treated group at 4 h post-injection, respectively (Fig. 4F). Compared to the free MTX treatment, the high dose of MTX releasing from MAMA at tumor site provided higher toxicity to the tumor, which was confirmed by getting the higher cell apoptotic and necrotic rates from the tumor treated by MAMA in vivo. The highest cell apoptotic ratio and the cell necrotic rate were achieved by MAMA treated BxPC3 tumor, suggesting its most effective inhibition of EGFR-overexpressed pancreatic cancer.
Encouraged by the superior anticancer effectiveness of MAMA in vitro and in vivo, animal experiments were carried out to evaluate the long-term therapeutic effectiveness by the dual-responsive EGFR-targeting of MTX release from MAMA and subsequent induced hROS increase. In vivo anticancer effectiveness of MAMA was evaluated by intravenous injection into HepG2 and BxPC3 tumor-bearing mice. After the tumors had developed to about 50 mm3, the mice carrying HepG2 and BxPC3 tumors, respectively, were divided into three groups: MAMA-, MAM-, and free MTX-treated (n = 8 per group). Given that the biological half-life of MTX is about 8–12 h, all groups were treated every two days for ten iterations [37]. Moreover, because of the maximum dose of MTX used in clinical treatment is 30 mg/week, thus the highest MTX concentration in.
various samples which could be administrated via the tail vein to the tumor-bearing is 8 mg/kg per dose [38]. The relative tumor volumes and relative body weight in each group were monitored during the treating process (Fig. 5A, B). The groups of saline and MTX did not show obvious inhibition of tumor growth, besides, MAM displayed moderate inhibition, whereas the group of MAMA exhibited the most significant inhibition in HepG2 and BxPC3 tumor-bearing mice. The images of excised tumors from all mice after treatment showed that the tumor size from the MAMA treated group was the smallest (Fig. 5C). Furthermore, the inhibition ratios were determined for each group in HepG2 and BxPC3 tumor-bearing mice after three weeks of treatment, respectively, 18.8% and 33.3% for MTX, 43.8% and 58.3% for MAM, and 68.8% and 83.3% for MAMA. The body weight changes were calculated for each group in HepG2 and BxPC3 tumor-bearing mice after three weeks of treatment, respectively, 20.0% and 26.1% loss for MTX, 10.0% and 17.4% loss for MAM, 5.0% and 4.3% loss for MAMA. Compare with free MTX treated groups, the HepG2 and BxPC3 tumor volumes decreased by 61.5% and 75%, respectively, simultaneously with a 23.8% and 22.7% relative increase of body weight after three weeks of MAMA treatment in the corresponding two groups of mice. Taken together, it indicates that MAMA has greater antitumor effectiveness and lower side effect than either using MAM as the DDS or the same concentration of free MTX in vivo. MAMA significantly inhibited BxPC3 tumor growth compared to HepG2 models, verifying that MAMA is a good candidate to treat EGFR-overexpressed pancreatic cancer.
Fig. 5.
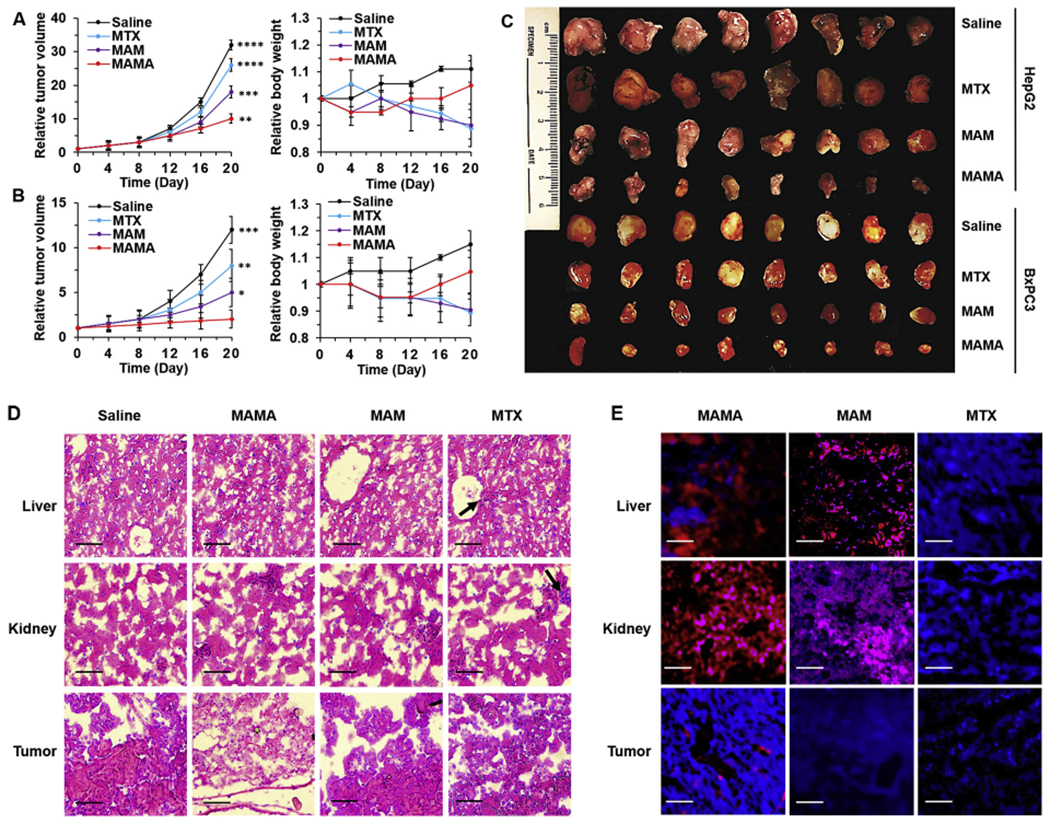
In vivo cancer inhibition and side effects evaluation. Relative tumor volume and body weight changes of (A) HepG2 and (B) BxPC3 tumor-bearing mice under different treatment for three weeks. (C) Photograph of tumors extracted from mice under different treatments for three weeks. Representative images of (D) Hematoxylin and eosin (H&E) staining and (E) Confocal laser scanning microscopy (CLSM) imaging of the liver, kidney, and tumor sections harvested from different treatment groups of BxPC3 tumor-bearing mice. Black arrows showed the toxic damages of tissues. Scale bar = 50 μm. All result represents the mean ± standard deviation (SD) (*P < .05, **P < .01, ***p < .001, and ****p < .0001).
Furthermore, the potential side effects of the major clearance organs in BxPC3 tumor-bearing mice were investigated after three weeks of MAMA treatment. The corresponding histological changes of organs were checked by hematoxylin and eosin (H&E) staining and CLSM imaging of the major MAMA accumulation and clearance organs including the liver, kidney, and tumor collected and sliced from BxPC3 tumor-bearing mice. Compared to normal tissues, MTX caused severe toxicity to the liver and kidney, which was indicated by cell atrophy, focal necrosis of tissue, congestion in central veins of the liver, as well as damage with dilatation in the subcapsular space of kidney tubular and glomerular structures (Fig. 5D). However, the tumor tissues were not significantly destroyed by free MTX or MAM, demonstrating the high hepatotoxicity, nephrotoxicity, and low tumor uptake of MTX due to their non-targeting drug delivery. MAMA markedly increased the necrosis of the BxPC3 tumor, simultaneously protecting the liver and kidney during three weeks of MAMA treatment. From CLSM images of the liver, kidney, and tumor sections harvested from different treatment groups of BxPC3 tumor-bearing mice, more MTX, represented by brighter fluorescent signal at 470 nm under UV excitation, was delivered to the tumor by MAMA than by MAM or no carrier after 4 h circulation in vivo (Fig. 5E). After statistical analysis for the FI of MUA-Au NCs from MAMA and MAM in the liver, kidney, and tumor, respectively, it was confirmed that MAMA and MAM were mainly eliminated by the kidney (Fig. S10). Strong FI of MUA-Au NCs and weak FI of MTX in the kidney of BxPC3 tumor-bearing mice indicated that the structures of MAMA and MAM were not completely decomposed during circulation in vivo for 4 h. It suggested that MAMA and MAM could be quickly excluded from the body by kidney and urine tract before MTX release, preventing side effects caused by free MTX. MAMA contributed 48.7% and 73.8% less MTX accumulation in the liver, 38.4% and 30.3% less MTX accumulation in kidney than MAM and free MTX, respectively. Meanwhile, MAMA delivered 3.5- and 7.5- fold MTX to the tumor compared to MAM and free MTX in BxPC3 tumor-bearing mice at 4 h post-injection. The animal experiment results supported the EGFR targeting, drug carrier sealing capabilities and trypsin-responsive drug release of the affibody, which could help MAMA efficiently deliver more MTX specifically to the EGFR-overexpressed tumors, reduce side effects compared to MAM or free MTX treatment, and get better effectiveness in the tumor with high trypsin activity. The MTX accumulation in the tumor could not only enhance the chemotherapeutic effects but also induce hROS increase, decomposing the MUA-Au NCs structure, quenching the fluorescent signal of MUA-Au NCS, helping completely release MTX from MAMA, finally resulting in proliferation inhibition and apoptosis of tumor cells. As anticipated, MAMA exhibits promising characteristics to be hROS and trypsin dual-responsive DDS for cancer treatment, especially for EGFR-overexpressed pancreatic cancer therapy without causing significant hepatotoxicity and nephrotoxicity in vivo.
4. Conclusions
In summary, a polyarginine-tailed anti-EGFR affibody has been successfully synthesized and has been incorporated into the MTX loaded MUA-Au NCs to form theranostic system MAMA by self-assembly based on charge effect between negatively charged -COOH terminal of MUA and positively charged polyarginine tails. MAMA has excellent biocompatibility, the adequate loading capacity of MTX, precise targeting to EGFR-overexpressed cells, and hROS and trypsin dual-responsive drug release in cells. The red fluorescence of MUA-Au NCs from MAMA can provide references for quickly evaluating the EGFR expression level of cells, and the blue fluorescence of MTX gives a visualized guidance for drug release from MAMA in cells. hROS can quench the fluorescence of MUA-Au NCs by decomposing MUA-Au NCs structure with little drug release, but trypsin working on digesting the polyarginine shell of MAMA mainly contributes to MTX release. High MTX accumulation in cells subsequently induces hROS increase, which helps to decompose the MUA-Au NCs structure and completely release MTX from MAMA. As a DDS, the adequate drug loading efficiency, specific targeting capability, highly efficient drug delivery, as well as hROS and trypsin dual-responsive drug release endow MAMA with excellent therapeutic effect on EGFR-overexpressed tumors through increasing the concentration of MTX accumulated in the cancer cells, subsequently inducing hROS increase in tumor local. MTX as a chemotherapeutic drug can decrease proliferation and metastasis of cancer cells through blocking the folate pathway, meanwhile, hROS increase efficiently inhibits cancer cells via apoptosis. Moreover, the polyarginine sealing of MAMA protects the major clearance organs away from the severe side effects of free MTX. Therefore, the simple capping design using polyarginine-tailed cancer-targeting peptides can serve as a generalized method for easily developing more multifunctional, powerful, and safe DDS like MAMA for cancer treatment, especially for better pancreatic cancer therapy in the future.
Supplementary Material
Acknowledgment
This work was supported, in part, by the National Natural Science Foundation of China (81201141) and the Clinical Capability Construction Project for Liaoning Provincial Hospitals (LNCCC-D50-2015 and LNCCC-C09-2015). All experiments involving animals were approved by the Administrative Panel on Laboratory Animal Care of Stanford University. We are very grateful to Michael Moseley (Stanford University, USA) for helping with the revised manuscript. We are also pleased to acknowledge the Stanford Small Animal Imaging Facility and Stanford Nano Shared Facilities for all the instrument support.
Footnotes
Declaration of Competing Interest
The authors declare no competing financial interest.
Appendix A. Supplementary data
References
- [1].Senapati S, Mahanta AK, Kumar S, Maiti P, Controlled drug delivery vehicles for cancer treatment and their performance, Signal Transduction Targeted Ther 3 (2018) 7, 10.1038/s41392-017-0004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sahandi Zangabad P, Karimi M, Mehdizadeh F, Malekzad H, Ghasemi A, Bahrami S, Zare H, Moghoofei M, Hekmatmanesh A, Hamblin MR, Nanocaged platforms: modification, drug delivery and nanotoxicity. Opening synthetic cages to release the tiger, Nanoscale 9 (2017) 1356–1392, 10.1039/C6NR07315H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhao N, Pan Y, Cheng Z, Liu H, Gold nanoparticles for cancer theranostics: a brief update, J. Innovative Opt. Health Sci 9 (2016), 10.1142/S1793545816300044. [DOI] [Google Scholar]
- [4].Wen J, Yang K, Xu Y, Li H, Liu F, Sun S, Construction of a triple-stimuli-responsive system based on cerium oxide coated mesoporous silica nanoparticles, Sci. Rep 6 (2016) 38931, 10.1038/srep38931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yao VJ, D’Angelo S, Butler KS, Theron C, Smith TL, Marchiò S, Gelovani JG, Sidman RL, Dobroff AS, Brinker CJ, Bradbury ARM, Arap W, Pasqualini R, Ligand-targeted theranostic nanomedicines against cancer, J. Control. Release 240 (2016) 267–286, 10.1016/j.jconrel.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhao N, Qin Y, Liu H, Cheng Z, Tumor-targeting peptides: ligands for molecular imaging and therapy, Anti Cancer Agents Med. Chem 18 (2018) 74–86, 10.2174/1871520617666170419143459. [DOI] [PubMed] [Google Scholar]
- [7].Himri I, Guaadaoui A, Chapter 1 - Cell and organ drug targeting: Types of drug delivery systems and advanced targeting strategies, in: Grumezescu AM (Ed.), Nanostructures for the Engineering of Cells, Tissues and Organs, William Andrew Publishing, 2018, pp. 1–66. [Google Scholar]
- [8].Zhang W, Hu X, Shen Q, Xing D, Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy, Nat. Commun 10 (2019) 1704, 10.1038/s41467-019-09566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Redza-Dutordoir M, Averill-Bates DA, Activation of apoptosis signalling pathways by reactive oxygen species, Biochim. Biophys. Acta, Mol. Cell Res 1863 (2016) 2977–2992, 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- [10].Arienti C, Pignatta S, Tesei A, Epidermal growth factor receptor family and its role in gastric cancer, Front. Oncol 9 (2019) 1308, 10.3389/fonc.2019.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shih YH, Luo TY, Chiang PF, Yao CJ, Lin WJ, Peng CL, Shieh MJ, EGFR-targeted micelles containing near-infrared dye for enhanced photothermal therapy in colorectal cancer, J. Control. Release 258 (2017) 196–207, 10.1016/j.jconrel.2017.04.031. [DOI] [PubMed] [Google Scholar]
- [12].Papouchado B, Erickson LA, Rohlinger AL, Hobday TJ, Erlichman C, Ames MM, Lloyd RV, Epidermal growth factor receptor and activated epidermal growth factor receptor expression in gastrointestinal carcinoids and pancreatic endocrine carcinomas, Mod. Pathol 18 (2005) 1329–1335, 10.1038/modpathol.3800427. [DOI] [PubMed] [Google Scholar]
- [13].Kanwar SS, Nautiyal J, Majumdar APN, EGFR(S) inhibitors in the treatment of gastro-intestinal cancers: what’s new? Curr. Drug Targets 11 (2010) 682–698, 10.2174/138945010791170851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fu R, Carroll L, Yahioglu G, Aboagye EO, Miller PW, Antibody fragment and affibody immunoPET imaging agents: radiolabelling strategies and applications, ChemMedChem 13 (2018) 2466–2478, 10.1002/cmdc.201800624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Frejd FY, Kim KT, Affibody molecules as engineered protein drugs, Exp. Mol. Med 49 (2017) e306, 10.1038/emm.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ståhl S, Gräslund T, Eriksson Karlström A, Frejd FY, Nygren PÅ, Löfblom J, Affibody molecules in biotechnological and medical applications, Trends Biotechnol 35 (2017) 691–712, 10.1016/j.tibtech.2017.04.007. [DOI] [PubMed] [Google Scholar]
- [17].Hagner N, Joerger M, Cancer chemotherapy: targeting folic acid synthesis, Cancer Manag. Res 2 (2010) 293–301, 10.2147/CMR.S10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sun J, Zhang J, Jin Y, 11-Mercaptoundecanoic acid directed one-pot synthesis of water-soluble fluorescent gold nanoclusters and their use as probes for sensitive and selective detection of Cr3+ and Cr6+, J. Mater. Chem. C 1 (2013) 138–143, 10.1039/C2TC00021K. [DOI] [Google Scholar]
- [19].Li KB, Chen FZ, Zhang S, Shi W, Han DM, Cai C, Chen CX, A nile red-based near-infrared fluorescent probe for endogenous hydrogen polysulfides in living cells, Anal. Methods 9 (2017) 6443–6447, 10.1039/C7AY02376F. [DOI] [Google Scholar]
- [20].Pan YJ, Chen YY, Wang DR, Wei C, Guo J, Lu D-R, Chu CC, Wang CC, Redox/pH dual stimuli-responsive biodegradable nanohydrogels with varying responses to dithiothreitol and glutathione for controlled drug release, Biomaterials 33 (2012) 6570–6579, 10.1016/j.biomaterials.2012.05.062. [DOI] [PubMed] [Google Scholar]
- [21].Xu B, Li X, Yin J, Liang C, Liu L, Qiu Z, Yao L, Nie Y, Wang J, Wu K, Evaluation of 68Ga-labeled MG7 antibody: a targeted probe for PET/CT imaging of gastric cancer, Sci. Rep 5 (2015) 8626, 10.1038/srep08626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Petit V, Massonnet G, Maciorowski Z, Touhami J, Thuleau A, Némati F, Laval J, Château-Joubert S, Servely JL, Vallerand D, Fontaine JJ, Taylor N, Battini JL, Sitbon M, Decaudin D, Optimization of tumor xenograft dissociation for the profiling of cell surface markers and nutrient transporters, Lab. Investig 93 (2013) 611–621, 10.1038/labinvest.2013.44. [DOI] [PubMed] [Google Scholar]
- [23].Lin Z, Zhang Y, Cai H, Zhou F, Gao H, Deng L, Li R, A PD-L1-based cancer vaccine elicits antitumor immunity in a mouse melanoma model, Mol. Ther. Oncolytics 14 (2019) 222–232, 10.1016/j.omto.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Belvedere R, Bizzarro V, Forte G, Dal Piaz F, Parente L, Petrella A, Annexin A1 contributes to pancreatic cancer cell phenotype, behaviour and metastatic potential independently of Formyl Peptide receptor pathway, Sci. Rep 6 (2016) 29660, 10.1038/srep29660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wu Z, Jin R, On the ligand’s role in the fluorescence of gold nanoclusters, Nano Lett 10 (2010) 2568–2573, 10.1021/nl101225f. [DOI] [PubMed] [Google Scholar]
- [26].Chen Z, Qian S, Chen X, Gao W, Lin Y, Protein-templated gold nanoclusters as fluorescence probes for the detection of methotrexate, Analyst 137 (2012) 4356–4361, 10.1039/C2AN35786K. [DOI] [PubMed] [Google Scholar]
- [27].Xie Y, Xianyu Y, Wang N, Yan Z, Liu Y, Zhu K, Hatzakis NS, Jiang X, Functionalized gold nanoclusters identify highly reactive oxygen species in living organisms, Adv. Funct. Mater 28 (2018) 1702026, 10.1002/adfm.201702026. [DOI] [Google Scholar]
- [28].Olsen JV, Ong SE, Mann M, Trypsin cleaves exclusively C-terminal to arginine and lysine residues, Mol. Cell. Proteomics 3 (2004) 608–614, 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- [29].Mótyán JA, Tóth F, Tőzsér J, Research applications of proteolytic enzymes in molecular biology, Biomolecules 3 (2013) 923–942, 10.3390/biom.3040923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Soreide K, Janssen E, Körner H, Baak J, Trypsin in colorectal cancer: molecular biological mechanisms of proliferation, invasion, and metastasis, J. Pathol 209 (2006) 147–156, 10.1002/path.1999. [DOI] [PubMed] [Google Scholar]
- [31].Zhu J, Miao XR, Tao KM, Zhu H, Liu ZY, Yu DW, Chen QB, Qiu HB, Lu ZJ, Trypsin-protease activated receptor-2 signaling contributes to pancreatic cancer pain, Oncotarget 8 (2017) 61810–61823, 10.18632/oncotarget.18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sandlund JT, Laver JH, Methotrexate-exploring dosing and administration in ALCL, Nat. Rev. Clin. Oncol 6 (2009) 440–441, 10.1038/nrclinonc.2009.107. [DOI] [PubMed] [Google Scholar]
- [33].Hinterwirth H, Kappel S, Waitz T, Prohaska T, Lindner W, Lämmerhofer M, Quantifying thiol ligand density of self-assembled monolayers on gold nanoparticles by inductively coupled plasma–mass spectrometry, ACS Nano 7 (2013) 1129–1136, 10.1021/nn306024a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Luo Z, Yuan X, Yu Y, Zhang Q, Leong DT, Lee JY, Xie J, From aggregation-induced emission of Au(I)–thiolate complexes to ultrabright Au(0)@Au(I)–thiolate core–shell nanoclusters, J. Am. Chem. Soc 134 (2012) 16662–16670, 10.1021/ja306199p. [DOI] [PubMed] [Google Scholar]
- [35].Vatsa R, Shukla J, Kumar S, Chakraboarty S, Dash A, Singh G, Mittal BR, Effect of macro-cyclic bifunctional chelators DOTA and NODAGA on radiolabeling and in vivo biodistribution of Ga-68 cyclic RGD dimer, Cancer Biother. Radiopharm 34 (2019) 427–435, 10.1089/cbr.2019.2811. [DOI] [PubMed] [Google Scholar]
- [36].Abdel-Daim MM, Khalifa HA, Abushouk AI, Dkhil MA, Al-Quraishy SA, Diosmin attenuates methotrexate-induced hepatic, renal, and cardiac injury: a biochemical and histopathological study in mice, Oxidative Med. Cell. Longev 2017 (2017) 1–10, 10.1155/2017/3281670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tetef ML, Margolin KA, Doroshow JH, Akman S, Leong LA, Morgan RJ Jr., Raschko JW, Slatkin N, Somlo G, Longmate JA, Carroll MI, Newman EM, Pharmacokinetics and toxicity of high-dose intravenous methotrexate in the treatment of leptomeningeal carcinomatosis, Cancer Chemother. Pharmacol 46 (2000) 19–26, 10.1007/s002800000118. [DOI] [PubMed] [Google Scholar]
- [38].Bello AE, Perkins EL, Jay R, Efthimiou P, Recommendations for optimizing methotrexate treatment for patients with rheumatoid arthritis, Open Access Rheumatol 9 (2017) 67–79, 10.2147/OARRR.S131668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.