

Abstract
Ruthenium complexes bearing protic diimine ligands are cytotoxic to certain cancer cells upon irradiation with blue light. Previously reported complexes of the type [(N,N)2Ru(6,6’-dhbp)]Cl2 with 6,6’-dhbp = 6,6’-dihydroxybipyridine and N,N = 2,2’-bipyridine (bipy) (1A), 1,10-phenanthroline (phen) (2A), and 2,3-dihydro-[1,4]dioxino[2,3-f][1,10]phenanthroline (dop) (3A) show EC50 values as low as 4 μM (for 3A) vs. breast cancer cells upon blue light irradiation (Inorg. Chem. 2017, 56, 7519). Herein, subscript A denotes the acidic form of the complex bearing OH groups, and B denotes the basic form bearing O− groups. This photocytotoxicity was originally attributed to photodissociation, but recent results suggest that singlet oxygen formation is a more plausible cause of photocytotoxicity. In particular, bulky methoxy substituents enhance photodissociation but these complexes are non-toxic (Dalton Trans. 2018, 47, 15685). Cellular studies are presented herein that show the formation of reactive oxygen species (ROS) and apoptosis indicators upon treatment of cells with complex 3A and blue light. Singlet oxygen sensor green (SOSG) shows the formation of 1O2 in cell culture for cells treated with 3A and blue light. At physiological pH, complexes 1A-3A are deprotonated to form 1B-3B in situ. Quantum yields for 1O2 (ɸΔ ) are 0.87 and 0.48 for 2B and 3B respectively, and these are an order of magnitude higher than the quantum yields for 2A and 3A. The values for ɸΔ show an increase with 6,6’-dhbp derived substituents as follows: OMe < OH < O−. TD-DFT studies show that the presence of a low lying triplet metal-centered (3MC) state favors photodissociation and disfavors 1O2 formation for 2A and 3A (OH groups). However, upon deprotonation (O− groups), the 3MLCT state is accessible and can readily lead to 1O2 formation, but the dissociative 3MC state is energetically inaccessible. The changes to the energy of the 3MLCT state upon deprotonation have been confirmed by steady state luminescence experiments on 1A-3A and their basic analogs, 1B-3B. This energy landscape favors 1O2 formation for 2B and 3B and leads to enhanced toxicity for these complexes under physiological conditions. The ability to convert readily from OH to O− groups allowed us to investigate an electronic change that is not accompanied by steric changes in this fundamental study.
Graphical Abstract
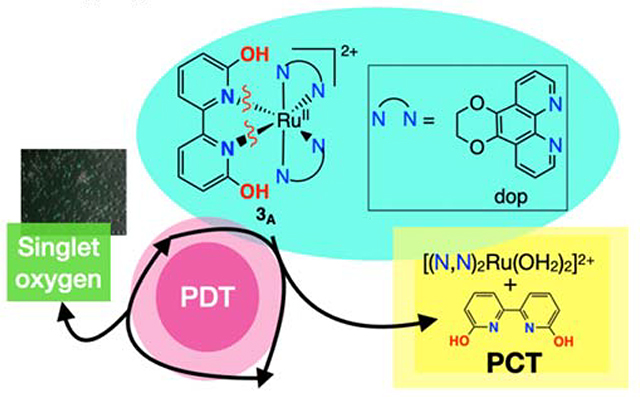
TOC Synopsis: Which pathway dominates for protic Ru compounds: singlet oxygen formation or photodissociation? Hypothesis: the protonation state of the OH groups determines the dominant pathway.
INTRODUCTION
Platinum (Pt)-based drugs are arguably among the most successful anticancer drugs. However, the side effects of treatment are severe and even life-threatening. Pt-based anticancer agents such as cisplatin act on all rapidly dividing cells, which includes healthy cells. There is an urgent need for therapies with greater selectivity for tumors over normal tissue. Tumor-specific therapeutics could take advantage of prodrugs that are activated in the unique environment of the cancer cell or by focused light to minimize off-target effects. One example is photodynamic therapy (PDT), an FDA-approved technique that uses a photosensitizer (PS), light, and oxygen to generate reactive oxygen species (ROS), importantly cytotoxic singlet oxygen (1O2).1–2 Photochemotherapy (PCT), while not FDA-approved, is an emerging light-based alternative to PDT that involves light-induced photoreactions that generate cytotoxic species (Scheme 1).3 PCT using metal complexes typically relies on light-triggered ligand dissociation to generate a ligand deficient metal center and the liberated ligand, both of which could be cytotoxic.4–6 PDT is generally thought to be more effective because the process is inherently catalytic in the PS (Schemes 1 and 3).7 Therefore, a very low concentration of a PS can generate a significant quantity of cytotoxic 1O2. In contrast, PCT is stoichiometric, and one metal complex typically generates one equivalent of aquated metal complex and one equivalent of free ligand (Scheme 1). While requiring higher dosing levels of metal complex, the PCT mechanism (in theory) does not require oxygen and thus could have added utility for treating hypoxic tumors. In practice, however, very few PCT agents maintain activity in hypoxia,8 and continued efforts are underway to address this shortcoming.
Scheme 1.

Ru complexes herein can utilize both PCT and PDT pathways. This paper focuses on the factors that influence which pathway is taken.
Scheme 3.

Photodissociation products vs. singlet oxygen. Which species is responsible for the observed toxicity? What role does charge play in determining the pathway taken?
Protic Ru(II) complexes as PCT agents have proven advantageous in our preliminary work9–12 because they are deprotonated at physiological pH, which results in improved cellular uptake due to an overall neutral charge and a more lipophilic metal complex.13 This paper focuses on the factors that influence which pathway (PDT versus PCT) is dominant in protic Ru(II) complexes. By understanding these factors, we can create better light-responsive agents that may generate 1O2 in normoxia but capitalize on PCT mechanisms in hypoxia.
Ruthenium anticancer agents are far less developed than their platinum counterparts, with no Ru-based coordination complexes FDA-approved to date (apart from the use of the 106Ru isotope in radiotherapy). However, Ru-based metallotherapeutics have much to offer, specifically in the area of light-based therapeutics. The pseudo octahedral geometries of Ru(II) and Ru(III) coordination complexes accommodate six ligands, and well-established synthetic methodology for variation of these very modular architectures makes it possible to rapidly examine a diverse structural landscape for building in and fine-tuning desired properties. As with other drug-like molecules, their three-dimensional structures, redox potentials, and relative lipophilicity/hydrophilicity indices can play a role in cellular uptake, localization, and mechanism of action.14
To date only two Ru(III) complexes (NAMI-A and NKP-1339/IT-139/BOLD-100), which are not light-activated, have advanced to clinical trials (Chart 1).15–18 NAMI-A, an antimetastatic agent, has since been abandoned after a Phase 2 study did not yield the desired efficacy (albeit in what may have been an inappropriate clinical trial design for an antimetastatic agent).18 IT-139 (formerly NKP-1339 but currently BOLD-100), was proposed to exhibit anticancer activity through modulation of ER stress, completed a Phase 1 study,17 and a Phase 2 study (Clinicaltrials.gov identifier NCT04421820) using BOLD-100 (formerly IT-139 and NKP-1339) in combination with FOLFOX for various advanced solid tumors is currently recruiting patients.
Chart 1.

Ruthenium complexes that entered clinical trials.
The only Ru(II) complex to advance to human clinical trials is TLD1433, and this light-triggered compound is being used as a PDT agent for treating non-muscle invasive bladder cancer (NMIBC).19–20 It has an extraordinarily high 1O2 quantum yield (near unit efficiency) and as a consequence is extremely phototoxic toward cancer cells.21 TLD1433 performed well in a Phase 1b clinical trial (Clinicaltrials.gov identifier NCT03053635) and is currently in a much larger Phase 2 clinical trial (Clinicaltrials.gov identifier NCT03945162. Together, the two Ru(III) complexes NAMI-A and BOLD-100 and the one Ru(II) complex TLD1433 that have advanced to clinical trials are opening the door to many translational investigations with Ru-based complexes as anticancer therapeutics, particularly in the field of light-responsive prodrugs.20
While Ru(II) complexes for PCT have not advanced to clinical trials, these agents are of special interest for their potential to act under hypoxic conditions. Glazer et al. first reported Ru(II) complexes that readily photodissociate strain-inducing ligands to bind DNA and cause cytotoxicity,22–24 and they and others have since investigated a variety of related systems.8, 25–27 The premise is that steric bulk near the metal center facilitates photodissociation of a ligand with visible light. This mechanism is not limited to the bis-heteroleptic diimine systems and has also been applied to Ru(II) tris-heteroleptic complexes derived from a combination of tri-, bi-, and monodentate ligands as well.5–6, 28–34 Turro has shown that some of these tris-heteroleptic complexes can both photodissociate ligands and generate 1O2, exploiting both PCT and PDT mechanisms in a single complex in a dual-action approach,4–6 and Glazer and McFarland have demonstrated similar dual-action capacity for bis-heteroleptic diimine complexes.22, 25
The work presented herein was inspired by some of these earlier PCT examples, especially the dual action systems, but with a focus on examining the role that charged ligands may play in determining the partitioning of excited state reactivity between the PCT and PDT mechanisms. McFarland, Sadler, and others have shown that changing the charge of the metal complex (e.g. with cyclometalated C,N bound ligands) can greatly alter the photocytotoxicity,14, 35–41 but these examples are presumed to rely on 1O2 as the photocytotoxic agent. The present work investigates Ru(II) bis-heteroleptic diimine complexes with ligands that bear ionizable groups that are positioned to induce strain in the inner coordination sphere of the complex. This arrangement presents a unique opportunity to directly influence access to the dissociative triplet metal-centered (3MC) state in systems that may otherwise generate 1O2 from triplet metal-to-ligand charge transfer (3MLCT) states.
Our prior publications focused on a series of compounds of the type [(N,N)2Ru(6,6’-dhbp)]2+ with 6,6’-dhbp = 6,6’-dihydroxybipyridine and N,N = 2,2’-bipyridine (bipy) (1A), 1,10-phenanthroline (phen) (2A), or 2,3-dihydro-[1,4]dioxino[2,3-f][1,10]phenanthroline (dop) (3A) (Scheme 2a).9–10 Herein, subscript A denotes the acidic and dicationic form of the metal complex bearing OH groups and subscript B denotes the basic and neutral form bearing O− groups. The 6,6’-dhbp ligand is protic, allowing the properties of the metal complexes to be controlled by deprotonation events. The OH groups are deprotonated at physiological pH, and the uptake of the resulting neutral Ru complexes is improved significantly.13 Of complexes 1A-3A (which form predominantly 1B-3B at physiological pH), the most photocytotoxic complex was 3, which displayed an EC50 of ~4 μM upon irradiation with blue light and a photocytotoxicity index (PI = EC50_dark/EC50_light) as high as 120 in breast cancer cells.10 This photocytotoxicity was initially attributed to photodissociation of the 6,6’-dhbp ligand, but in fact the evidence now supports a different mechanism described herein.
Scheme 2.

a) Deprotonation of XA = [(N,N)2Ru(6,6’-dhbp)]2+ complexes under physiological conditions changes the properties. X = 1, 2, or 3 and A denotes the acidic form bearing OH groups and B denotes the basic form bearing O− groups. b) Aprotic complexes 4 and 5. 4 (N,N = phen) and 5 (N,N = dop)
The 6,6’-dhbp ligand provides steric bulk near the metal center that helps promote photodissociation of this ligand (ɸPD is ~10−3 for 1A-3A, Table 1). Interestingly, deprotonation reduced the quantum yields for photodissociation by 1–2 orders of magnitude (Table 1) for 2 and 3. Thus, ɸPD values are 10−4 to 10−5 for 2B and 3B under physiological pH conditions. Deprotonation is mostly an electronic change without any steric component. In contrast, the complexes [(N,N)2Ru(6,6’-dmbp)]2+ with 6,6’-dmbp = 6,6’-dimethoxybipyridine and N,N = phen (4) or dop (5) were designed to be aprotic and sterically bulkier versions of 2 and 3 (Scheme 2b).11 The change from OH to OMe groups resulted in increased (by a factor of 3–12) quantum yields for photodissociation (Table 1).11 However, the complexes 4 and 5 were not phototoxic to breast cancer cells, which was attributed to a combination of poor uptake (as reflected in log(Do/w) values in Table 1) for these dicationic compounds (cf. 2 and 3) and low cytotoxicity for the products of photodissociation.11
Table 1.
Comparison of acidity, log(Do/w) (previously reported)10–11, 13, quantum yields for photodissociation (ΦPD) (previously reported)9–11, and quantum yields for singlet oxygen (ΦΔ) (reported herein) upon irradiation at 450 nm for complexes 1–5.
| compound | Structure | pKa avg | log(Do/w)a at pH 7.4 | ΦPD at pH 5.0b | ΦPD at pH 7.5c | ΦΔ for XAd | ΦΔ for XBd |
|---|---|---|---|---|---|---|---|
| 1A | [(bipy)2Ru(6,6’-dhbp)]2+ | 6.3 | 1.4(1) | 0.0058(5) | 0.0012(1) | 0.041(2) | 0.18(2) |
| 2A | [(phen)2Ru(6,6’-dhbp)]2+ | 6.0(1) | 1.6(1) | 0.0020(2) | 0.000036(1) | 0.048(2) | 0.87(9) |
| 3A | [(dop)2Ru(6,6’-dhbp)]2+ | 5.9(1) | 1.8(1) | 0.001(1) | 0.00022(3) | 0.048(2) | 0.48(5) |
| 4 | [(phen)2Ru(6,6’-dmbp)]2+ | N/A | −1.3(2) | 0.024(6) | N/A | 0.01(1)e | N/A |
| 5 | [(dop)2Ru(6,6’-dmbp)]2+ | N/A | −1.1(1) | 0.0030(2) | N/A | 0.01(1)e | N/A |
Log(Do/w) is the octanol-water partition coefficient measured as described in our prior publication.13 Do/w is defined as the total concentration of all ruthenium complex (in various protonation states) in octanol divided by the total concentration of all ruthenium complex in water for the equilibrated biphasic mixture.
In aqueous solution at pH 5.0, mostly the XA form is present if using a protic ligand (in 1-3).
In aqueous solution at pH 7.5, mostly the XB form is present if using a protic ligand (in 1-3).
Isolated XA or XB (in 1-3) was used in CD3OD. The aprotic complexes 4 and 5 are included in the XA column because they carry the same charge as 1A-3A.
Photodissociation also occured during these experiments to quantify singlet oxygen, thereby reducing the accuracy of this measurement. Incident photons could have led to either product.
Complexes 1-5 generate [(N,N)2Ru(H2O)2]2+ upon photodissociation (N,N = bipy, phen or dop) (Scheme 3), yet the complexes with the lowest ɸPD values (2B, 3B) displayed the greatest photocytotoxicity. Bonnet recently presented evidence that for Glazer’s compound, [(bipy)2Ru(6,6’-Me2-bipy)]2+ (G, Chart 2), the methylated bipy ligand rather than the metal is the source of toxicity.22, 42 Bonnet stated that [(bipy)2Ru(OH2)2]2+ (from photodissociation of G) is nontoxic by synthesizing another molecule that is expected to also form this photoproduct in cells. The photoproduct is the same one that we obtain from photodissociation of 1A (Scheme 3). We have previously conducted experiments to probe whether the aquated metal complex or the free ligand are toxic (Scheme 3). We independently synthesized the products from the photodissociation of 1-3, namely [(N,N)2Ru(OH2)2]SO4 (N,N = bipy, phen, dop), and these were nontoxic (EC50 >100 μM) under our experimental conditions.10 However, Glazer and co-workers performed extensive biological characterization on G, and demonstrated direct DNA damage in live cells through metalation of the nucleic acids at levels comparable to cisplatin, and induction of the DNA damage response as measured by immunoblotting for reporters such as H2AX.33, 43 They also reported that G produces a phenotype in bacteria that is virtually identical to cisplatin. Thus, the source of the biological activity of these photoejecting Ru(II) complexes is a subject of some controversy.
Chart 2.

Glazer’s compound (G) which photodissociates to generate [(bipy)2Ru(OH2)2]2+ which binds DNA.
With regard to the free ligand being the source of cytotoxicity, some pyridone derivatives44–46 do exhibit drug-like properties. However, 6,6’-dhbp, as the free ligand, is nontoxic (EC50 >100 μM) against several breast cell lines under the assay conditions employed.10 Nevertheless, we acknowledge that there are numerous issues in attempting to quantify the cytotoxicity of free ligands, knowing that they (i) may aggregate / precipitate in the buffers and media used for making stock solutions, (ii) likely do not enter cells in the same manner as the intact metal complex, (iii) may not localize to the same part of the cell as the intact metal complex, and (iv) are expected to avidly engage with metal ions both in the buffers and media as well as the cell, among other things. With these considerations in mind, the data presented herein (Table 1) suggests that the photodissociation products, both the aquated metal complexes and their free ligands, are of low toxicity with the caveat that the considerations described above were not explored.
Herein, we discuss several of these complexes in terms of an alternative mechanism for photocytotoxicity that involves 1O2 generation. The protic complexes in particular allow us to investigate the role of charge, as it relates to both the ligand and metal complex, in determining which pathway dominates: photodissociation versus 1O2 formation.
RESULTS AND DISCUSSION
Cellular Studies Measuring Reactive Oxygen Species (ROS), 1O2, and Apoptosis.
Cellular studies focused on complex 3 because it had the best photocytotoxicity index (PI) toward breast cancer cell lines in our previous study. MCF7 cells were dosed with complex 3A (which forms 3B in situ) at a concentration of 5 μM (which is near the EC50 value), and the percentage of cells showing intracellular ROS activity and late apoptosis were measured for the dark condition (solid bars) and with a sublethal dose of 450 nm blue light (striped bars) (Figure 1). This study indicates that complex 3A, when activated by blue light, induces the formation of ROS and triggers apoptosis. Notably, no ROS were induced with 3A in the dark (i.e., not significantly different than the negative control), but there was a moderately increased level of apoptosis, which suggests a different mechanism for the low level of dark cytotoxicity observed in our past work. ROS production for 3A (in contrast to the positive control) was clearly light-triggered, which was also demonstrated with MDA-MB-231 cells using the same type of assay (see Figures S1 and S2 in the Supporting Information).
Figure 1.

Left: Apoptosis indicators in MCF7 as measured by Annexin WPK Right: ROS indicators in MCF7. +ROS is the pyocyanin positive control as an ROS inducer. For both plots, solid bars Indicate incubation with no drug, positive control, or 3A for 48 h in the dark. Striped bars indicate incubation followed by irradiation with blue tight.
MDA-MB-231 cells were also tested for the presence of 1O2 upon treatment with 5 μM of 3A and blue light (Figure 2; via Singlet Oxygen Sensor Green (SOSG)). Singlet oxygen was detected in only ~2% of cells from the control samples (no metal complex and 3A in the dark), but in contrast, 1O2 was present in 33% of the cells for treated with 3A and irradiated. Since SOSG is specific for detecting 1O2 and not hydroxyl radicals or superoxide, these results suggest that 1O2 may be the ROS responsible for the observed photocytotoxicity with 3A.
Figure 2.

Left: SOSG luminescence is greatest for MDA-MB-231 cells treated with 3A (5 μM) and Irradiated with blue light. Right: Flow cytometry shows an increase in the number of cells with SO present with blue light and 3A.
Singlet Oxygen Quantum Yields.
The quantum yields for 1O2 formation () were determined for 1-5 using the direct method based on the 1O2 luminescence centered at 1268 nm (Table 1). The protic complexes were studied as the isolated acidic forms (1A-3A) bearing OH groups and the isolated basic forms (1B-3B). All measurements were performed in CD3OD as the preferred solvent given that water quenches the luminescence of 1O2, and acetonitrile has been shown to accelerate photodissociation.9 Values for ɸΔ were 0.041 for 1A and 0.048 for 2A and 3A, but the deprotonated analogs 1B, 2B, and 3B gave ɸΔ values of 0.18, 0.87 and 0.48, respectively. Quantum yields for 1O2 are enhanced 4.4- to 20-fold by deprotonation of the 6,6’-dhbp ligand, which generates the corresponding neutral Ru(II) complexes. Thus, for 3B (the form present at physiological pH), ɸΔ is three orders of magnitude greater than ɸPD. Likewise, for 2B, ɸΔ is four orders of magnitude greater than ɸPD. Considering that 1O2 generation is catalytic whereas photodissociation is stoichiometric, 1O2 is the most plausible source of photocytotoxicity for complexes such as 3, which was our most photocytotoxic complex (highest PI value).
The 1O2 quantum yields for 4 and 5 were calculated to be near 0.01 (Table 1), which shows that the change from OH (in 2A and 3A) to OMe groups reduces 1O2 production five-fold. Complexes 4 and 5 undergo a significant amount of photodissociation during the experiments designed to quantify 1O2 production, which results in greater error in ɸΔ for these complexes given that incident photons could have led to either product and also because the photoproducts themselves may then absorb incident photons. Nonetheless, it is clear that for a given amount of incident photons, the yield of 1O2 will be at least 87- to 48-fold higher for 2B and 3B (with O− groups) versus for 4 and 5 (with OMe groups).
Such effects based upon changing the charge of the ligand were demonstrated by McFarland et al. where complexes of the type [(bipy)2Ru(N,N)]2+ (ɸΔ = 0.8 to 0.9) had much higher 1O2 quantum yields than their cyclometallated analogs [(bipy)2Ru(N,C)]+ (ɸΔ = 0.07 to 0.08). The change from a neutral to an anionic ligand decreased the quantum yields for 1O2 formation by 10-fold, which is the opposite of what we observe (with an anionic ligand increasing ɸΔ).38 However, the key differences relative to this study were a lack of photodissociation as a competing pathway and the localization of the negative charge on a carbon atom, which is directly bound to the metal.47
Computational Studies of the Excited States Generated Upon Irradiation.
The observation that complexes bearing O− groups are less susceptible to photodissociation and have higher 1O2 quantum yields demonstrates that the OH (2A, 3A) vs. O− (2B, 3B) groups result in electronic effects that influence the energies of the excited states (vide infra) and thus the efficiency of 1O2 formation versus photodissociation. The energy of the 3MLCT state relative to the 3MC state is of particular importance in determining the predominant mechanism, whereby lower 3MLCT energies relative to the 3MC energies would be expected to favor 1O2 production over photodissociation. The 3MLCT energies of the protic and deprotonated forms of 1-3 were estimated by their phosphorescence (Figures S15–S19 and Table S4). The acidic forms (1A-3A) had luminescence maxima near 615 nm (ranging from 610–630 nm depending on the compound and the solvent, ~2.02 eV) whereas the basic forms (1B-3B) produced emission at substantially longer wavelengths. The doubly deprotonated neutral complexes yielded emission maxima near 710 nm (ranging from 696–734 nm, ~1.71 eV), with lower-intensity transitions clearly visible near 900 nm and tailing out past 1000 nm for 1B (Figure S15). With this is mind, time-dependent density functional theory (TD-DFT) studies were carried out on complexes 2 and 3 to probe the influence of protonation state on the relative energies of the lowest-lying 3MLCT states that were determined experimentally but also on the nonemissive 3MC states that are much more difficult to assess experimentally. These two complexes were selected for the study because they are the more photocytotoxic complexes and also show the largest enhancement in ɸΔ upon deprotonation.
We note that many experimental characteristics of 3MC excited states remain largely unknown in Ru polypyridyl complexes today. This includes the energy, whether it is bound or dissociative, and the existence of another potential on the way to non-radiative decay or ligand dissociation as noted by Mukata et al. and others.48 Over decades of Ru polypyridyl photophysical literature, transient absorption measurements do not reveal the nature of the 3MC state. More recently, time-resolved infrared vibrational spectroscopy (TR-IR) was able to detect the 3MC state through a detailed analysis of the temporal evolution of vibrational bands under different experimental conditions but, of course, not the energies.48,49 While transient absorption spectroscopy has provided experimental evidence of the ultrafast quenching of 3MLCT states by the 3MC state, these complexes are specially designed to have unusually stable 3MC states.50 This type of experiment does not yield information on the energy of the 3MC state relative to the 3MLCT, and DFT computations are relied upon herein to assert this difference. We use caution in interpreting DFT computations because they are better at predicting relative energy differences vs. absolute energies and furthermore DFT cannot probe every possible pathway involving 2, 3, and other biomolecules in cells. Nonetheless, the DFT computations are informative when considered with the above caveats and the results herein are qualitatively supported by luminescence, photodissociation, and singlet oxygen experiments.
The OH bearing dicationic complexes 2A and 3A will be discussed first. Excitation with blue light at 450 nm provides 2.75 eV of energy, which populates the 1MLCT excited states for 2A and 3A. After a vertical excitation with blue light, the relaxed excited state formed will be the singlet MLCT state for 2A and 3A (Figure 3) at 2.3–2.4 eV above the ground state. These 1MLCT states undergo rapid intersystem crossing (ISC) to generate the 3MLCT state for 2A and 3A (ISC quantum yields are typically near unity for Ru(II) diimine complexes3). While the 3MLCT state could interact with 3O2 to generate 1O2, the presence of a 3MC excited state for both 2A and 3A (at 1.7 eV) that lies below the 3MLCT state (at 2.06–2.08 eV, cf. [Ru(bipy)3]2+ is at 2.1 eV above the ground state) indicates that the internal conversion to the 3MC state may be competitive. The presence of this low-lying 3MC state is not unheard of50–51 but is a marked departure from most examples in the literature, where 3MC states are typically higher in energy than 3MLCT states.3, 52–54 This 3MC state has substantial antibonding character between Ru(II) and 6,6’-dhbp (Figure 4 shows the orbital diagram for 3MC on 2A) and is expected to lead to photodissociation products. Taken together, these results suggest a mechanistic rationale for why the OH bearing dicationic complexes favor photodissociation over 1O2 formation.
Figure 3.

Free energy diagram for energetically accessible excited states of 2A (in blue) and 3A (in red) from PBE0-D3/BS1. The lowest energy, thermally accessible triplet excited state is 3MC, which leads to ligand loss and photodissoclation products. The 3MLCT state would lead to singlet oxygen, but this most likely quickly converts to 3MC for 2A and 3A before It comes in contact with oxygen.
Figure 4.

Orbital depiction of the 3MC excited state for 2A shows that this state Is antibonding between Ru and 6,6’-dhbp.
The lowest-lying singlet excited states for the neutral, deprotonated complexes, 2B and 3B appear substantially different in both character and energy (Figure 5) compared to 2A and 3A. For 2B and 3B, the lowest-lying singlet excited states are ligand-to-ligand charge transfer (1LLCT) at 1.3–1.4 eV above the ground state. The 1LLCT state involves charge transfer from the deprotonated 6,6’-dhbp ligand to either the phen (2B) or dop (3B) ligand. This change in state is likely due to the increased π electron density donated from an O− vs an OH group. These excited states are expected to undergo ISC very rapidly to the 3MLCT states. In contrast to 2A and 3A, for 2B and 3B the 3MC state is energetically uphill and inaccessible at 1.9 eV. Thus, ligand photodissociation does not occur readily for 2B and 3B (and this is reflected in the experimental values of ~10−4 to 10−5 for ɸPD at pH 7.5 in Table 1). The key reason for the difference in accessibility for 3MC is that the energy of the 3MLCT state is much lower for 2B and 3B at 1.21–1.26 eV (versus 2.06–2.08 eV for 2A and 3A), which was also confirmed experimentally from phosphorescence measurements.
Figure 5.

Free energy diagram for energetically accessible excited states of 2B (In blue) and 3B (in red) from PBE0-D3/BS1. The 3MLCT state is readily accessed and should lead to singlet oxygen formation. Furthermore, the 3MC (which leads to ligand loss) is not accessible and this explains the relative lack of photodissociation for 2B and 3B.
For 2B and 3B, the combination of raising the energy of 3MC and lowering the energy of the 3MLCT state makes the 3MC state inaccessible. Given that the only accessible excited state is 3MLCT for these basic forms, this state persists long enough to interact with 3O2 and to generate 1O2 in much higher quantum yields of 0.48 to 0.87 (Table 1). These results explain the difference in pathways that are available for the basic versus the acidic form of these 6,6’-dhbp complexes. Namely, the basic form can undergo 1O2 formation (with favorable ɸΔ) without forming photodissociation products due to an inaccessible 3MC state with antibonding character. In contrast, the acidic forms are able to readily access the 3MC state and photodissociate the 6,6’-dhbp ligand in a pathway that competes effectively with 1O2 formation. To further verify these interpretations, computations were repeated with different functional/basis set combinations with similar results (see Figures S8–S10 and Tables S2–S3).
The protonation state also has a measurable effect on the ground state absorption spectra for these complexes. For both complexes 2 and 3, higher pH (more basic) results in a red shift of about 50 nm (0.3 eV)10 and significant peak broadening (Figure 6, right). Absorption spectra for both the acidic and basic forms were simulated from TD-DFT single-point computations on the ground state geometries and are also shown (Figure 6, left). The trend in both λmax and spectral shape between the acidic and basic forms correlates nicely between the simulated spectra and experiment. For both 2 and 3 the simulated λmax red-shift by about 50 nm and the peak broadens significantly. Additional overlays between simulated and experimental spectra are provided in Figures S11–S14.
Figure 6.

Left: Simulated UV-VIS absorption spectra for complex 2 (top) and complex 3 (bottom) from TD-PBE0-D3// PBE0-D3/BS1 Right: Experimental UV-Vis absorption spectra for complex 2 (top) and complex 3 (bottom) as a function of pH as previously reported.10
CONCLUSIONS
Many studies have investigated how synthetic changes to ligands can influence the quantum yields for 1O2 and photodissociation. However, very few studies have looked at the influence of ligand protonation states55 on both the magnitude of these quantum yields and the competition between the pathways (Scheme 4). Deprotonation of complexes 1A-3A (bearing OH groups) to form 1B-3B (bearing O− groups) represents an electronic change and a change in complex charge (from 2+ to neutral) without any significant steric change. We observe that deprotonation both increases the quantum yield for 1O2 and decreases the quantum yield of photodissociation products. Furthermore, since 1O2 formation is catalytic (whereas photodissociation is stoichiometric) it can result in a greater amount of toxic species generated in cells. These results are confirmed by cellular studies, which show that complex 3 generates 1O2 in cells. Furthermore, the combined evidence shows that the products of photodissociation appear to be of low toxicity. This explains why 3B displays good photocytotoxicity indices despite exceedingly low quantum yields for photodissociation, and furthermore efforts to enhance quantum yields for photodissociation results in low PI values (and reduced ɸΔ values) for 4 and 5 (bearing OMe groups and a 2+ charge). The enhanced photocytotoxicity index of 3 in cells (vs. complexes 1 and 2) is related to enhanced cellular uptake for complex 3 due to the more lipophilic structure.10, 13 Our study herein did not reveal any significant differences in the quantum yields for 1O2 formation for 2 vs. 3. Both 2B and 3B are efficient at 1O2 formation in the deprotonated state, but those quantum yields decrease 10- to 20-fold upon protonation. Overall this study reveals that the substituent and the complex charge leads to an increase in ɸΔ values in this order: OMe < OH < O− that is in part determined by an apparent competition between photodissociation and singlet oxygen formation (Scheme 4).
Scheme 4.

Changes to the OR group in the ligand within 1–5 controls the dominant pathway, PD = photodissociation.
TD-DFT studies suggest a rationale for why complexes 2A and 3A (bearing OH groups) favor photodissociation. The presence of a low lying 3MC state with substantial antibonding character between the metal and the 6,6’-dhbp ligand allows photodissociation to occur readily. Furthermore, any photodissociation that occurs will remove the possibility of subsequent 1O2 formation from that particular molecule because the aquated species is of low photocytotoxicity and is presumably a poor 1O2 generator. For 2A and 3A, the 3MLCT state (that can potentially lead to 1O2 formation) is on the pathway accessed upon light irradiation, but it likely does not persist for long enough to form significant amounts of 1O2. In contrast, complexes 2B and 3B (bearing O− groups) favor 1O2 formation due to a low lying 3MLCT state that can readily interact with 3O2. The presence of a much lower-lying 3MLCT state for 1B-3B was confirmed experimentally by luminescence experiments, with this state being substantially higher in energy for 1A-3A. For the deprotonated complexes, the 3MC state with antibonding character is high in energy and inaccessible, which greatly reduces access to the photodissociation pathway. While DFT results have uncertainties that are difficult to quantify without an experimental estimate of the 3MC energy levels, qualitatively the DFT results are consistent with our experimental data including photodissociation quantum yields which are much higher for 2A and 3A vs. 2B and 3B. Of course, at physiological pH complexes 2B and 3B are the dominant species in cellular media and inside the cells, even in the case of cancer cells displaying a Warburg phenotype.13, 56 Thus, the production of 1O2 by these complexes is consistent with the observed photocytotoxicity indices. It appears that any photodissociation does not in fact lead to photocytotoxicity. This suggests that the path forward towards enhancing photocytotoxicity in new dihydroxybipyridine derivatives would include synthetic efforts to reduce photodissociation and enhance 1O2 formation while improving cellular uptake.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge support from the NIH (R15-GM132803-01) to ETP and YK and the NSF (CHE 1800201) to CEW. Preliminary studies were also supported by the National Science Foundation EPSCoR Track 2 Grant (OIA-1539035) to ETP and CEW, the Research Grants Committee (RGC) at UA to ETP and YK, and the Alabama Commission on Higher Education Fellowship (to SP). We thank NSF MRI program (CHE 1726812, PI Cassady) and UA for the purchase of a MALDI TOF/TOF MS and Dr. Qiaoli Liang for MS experimental work. The computational work was completed with resources provided by the Alabama Supercomputer Authority, the Mississippi State University High Performance Computing Collaboratory, and the Mississippi Center for Supercomputing Research. SAM and CGC thank the National Cancer Institute (NCI) of the National Institutes of Health (NIH) (Award R01CA222227) for support in part of this work. The content in this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest Statement: SAM has a potential research conflict of interest due to a financial interest with Theralase Technologies, Inc. and PhotoDynamic, Inc. A management plan has been created to preserve objectivity in research in accordance with The University of Texas at Arlington (UTA) policy. The other authors declare no competing financial interest.
ASSOCIATED CONTENT
*Supporting Information
Experimental details for the synthesis of new compounds (1B, 2B, 3B); experimental details for cellular studies and assays for ROS, apoptosis and singlet oxygen; experimental details and results for the computational studies including the assignment of excited states; phosphorescence experiments; molecular coordinates list (PDF) and an XYZ molecular coordinates file.
REFERENCES
- 1.Dolmans DEJGJ; Fukumura D; Jain RK, Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [DOI] [PubMed] [Google Scholar]
- 2.Thota S; Rodrigues DA; Crans DC; Barreiro EJ, Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem 2018, 61, 5805–5821. [DOI] [PubMed] [Google Scholar]
- 3.White JK; Schmehl RH; Turro C, An overview of photosubstitution reactions of Ru(II) imine complexes and their application in photobiology and photodynamic therapy. Inorg. Chim. Acta 2017, 454, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knoll JD; Albani BA; Turro C, New Ru(II) Complexes for Dual Photoreactivity: Ligand Exchange and 1O2 Generation. Acc. Chem. Res 2015, 48, 2280–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huisman M; White JK; Lewalski VG; Podgorski I; Turro C; Kodanko JJ, Caging the uncageable: using metal complex release for photochemical control over irreversible inhibition. Chem. Commun 2016, 52, 12590–12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loftus LM; White JK; Albani BA; Kohler L; Kodanko JJ; Thummel RP; Dunbar KR; Turro C, New Ru II Complex for Dual Activity: Photoinduced Ligand Release and 1O2 Production. Chem. Eur. J 2016, 22, 3704–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Z; Cowan JA, Catalytic Metallodrugs: Substrate-Selective Metal Catalysts as Therapeutics. Chem. Eur. J 2017, 23, 14113–14127. [DOI] [PubMed] [Google Scholar]
- 8.Roque III J; Havrylyuk D; Barrett PC; Sainuddin T; McCain J; Colón K; Sparks WT; Bradner E; Monro S; Heidary D; Cameron CG; Glazer EC; McFarland SA, Strained, Photoejecting Ru(II) Complexes that are Cytotoxic Under Hypoxic Conditions. Photochem. Photobiol 2020, 96, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hufziger KT; Thowfeik FS; Charboneau DJ; Nieto I; Dougherty WG; Kassel WS; Dudley TJ; Merino EJ; Papish ET; Paul JJ, Ruthenium Dihydroxybipyridine Complexes are Tumor Activated Prodrugs due to Low pH and Blue Light Induced Ligand Release. J. Inorg. Biochem 2014, 130, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu F; Park S; Martinez K; Gray JL; Thowfeik FS; Lundeen JA; Kuhn AE; Charboneau DJ; Gerlach DL; Lockart MM; Law JA; Jernigan KL; Chambers N; Zeller M; Piro NA; Kassel WS; Schmehl RH; Paul JJ; Merino EJ; Kim Y; Papish ET, Ruthenium Complexes are pH-Activated Metallo Prodrugs (pHAMPs) with Light-Triggered Selective Toxicity Toward Cancer Cells. Inorg. Chem 2017, 56, 7519–7532. [DOI] [PubMed] [Google Scholar]
- 11.Qu F; Martinez K; Arcidiacono AM; Park S; Zeller M; Schmehl RH; Paul JJ; Kim Y; Papish ET, Sterically demanding methoxy and methyl groups in ruthenium complexes lead to enhanced quantum yields for blue light triggered photodissociation. Dalton Trans 2018, 47, 15685–15693. [DOI] [PubMed] [Google Scholar]
- 12.Papish ET; Paul JJ; Merino EJ, Ruthenium Complexes as Tumor Activated Metallo-Prodrugs. 2014, Patent Application filed with US Patent Office [Google Scholar]
- 13.Park S; Gray JL; Altman SD; Hairston AR; Beswick BT; Kim Y; Papish ET, Cellular uptake of protic ruthenium complexes is influenced by pH dependent passive diffusion and energy dependent efflux. J. Inorg. Biochem 2020, 203, 110922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaiddon C; Pfeffer M, The Fate of Cycloruthenated Compounds: From C-H Activation to Innovative Anticancer Therapy. Eur. J. Inorg. Chem 2017, 2017, 1–17. [Google Scholar]
- 15.Alessio E, Thirty Years of the Drug Candidate NAMI-A and the Myths in the Field of Ruthenium Anticancer Compounds: A Personal Perspective. Eur. J. Inorg. Chem 2017, 2017, 1549–1560. [Google Scholar]
- 16.Trondl R; Heffeter P; Kowol CR; Jakupec MA; Berger W; Keppler BK, NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci 2014, 5, 2925–2932. [Google Scholar]
- 17.Leijen S; Burgers SA; Baas P; Pluim D; Tibben M; van Werkhoven E; Alessio E; Sava G; Beijnen JH; Schellens JHM, Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest. New Drugs 2015, 33, 201–214. [DOI] [PubMed] [Google Scholar]
- 18.Burris HA; Bakewell S; Bendell JC; Infante J; Jones SF; Spigel DR; Weiss GJ; Ramanathan RK; Ogden A; Von Hoff D, Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: a first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monro S; Colón KL; Yin H; Roque J; Konda P; Gujar S; Thummel RP; Lilge L; Cameron CG; McFarland SA, Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev 2019, 119, 797–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McFarland SA; Mandel A; Dumoulin-White R; Gasser G, Metal-based photosensitizers for photodynamic therapy: the future of multimodal oncology? Curr. Opin. Chem. Biol 2020, 56, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi G; Monro S; Hennigar R; Colpitts J; Fong J; Kasimova K; Yin H; DeCoste R; Spencer C; Chamberlain L; Mandel A; Lilge L; McFarland SA, Ru(II) dyads derived from α-oligothiophenes: A new class of potent and versatile photosensitizers for PDT. Coord. Chem. Rev 2015, 282–283, 127–138. [Google Scholar]
- 22.Howerton BS; Heidary DK; Glazer EC, Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents. J. Am. Chem. Soc 2012, 134, 8324–8327. [DOI] [PubMed] [Google Scholar]
- 23.Hidayatullah AN; Wachter E; Heidary DK; Parkin S; Glazer EC, Photoactive Ru(II) Complexes With Dioxinophenanthroline Ligands Are Potent Cytotoxic Agents. Inorg. Chem 2014, 53, 10030–10032. [DOI] [PubMed] [Google Scholar]
- 24.Wachter E; Heidary DK; Howerton BS; Parkin S; Glazer EC, Light-activated ruthenium complexes photobind DNA and are cytotoxic in the photodynamic therapy window. Chem. Commun 2012, 48, 9649. [DOI] [PubMed] [Google Scholar]
- 25.Sainuddin T; Pinto M; Yin H; Hetu M; Colpitts J; McFarland SA, Strained ruthenium metal–organic dyads as photocisplatin agents with dual action. J. Inorg. Biochem 2016, 158, 45–54. [DOI] [PubMed] [Google Scholar]
- 26.Havrylyuk D; Heidary DK; Nease L; Parkin S; Glazer EC, Photochemical Properties and Structure–Activity Relationships of RuII Complexes with Pyridylbenzazole Ligands as Promising Anticancer Agents. Eur. J. Inorg. Chem 2017, 2017, 1687–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohler L; Nease L; Vo P; Garofolo J; Heidary DK; Thummel RP; Glazer EC, Photochemical and Photobiological Activity of Ru(II) Homoleptic and Heteroleptic Complexes Containing Methylated Bipyridyl-type Ligands. Inorg. Chem 2017, 56, 12214–12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li A; White JK; Arora K; Herroon MK; Martin PD; Schlegel HB; Podgorski I; Turro C; Kodanko JJ, Selective Release of Aromatic Heterocycles from Ruthenium Tris(2-pyridylmethyl)amine with Visible Light. Inorg. Chem 2016, 55, 10–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herroon MK; Sharma R; Rajagurubandara E; Turro C; Kodanko JJ; Podgorski I, Photoactivated inhibition of cathepsin K in a 3D tumor model. Biol. Chem 2016, 397, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arora K; White JK; Sharma R; Mazumder S; Martin PD; Schlegel HB; Turro C; Kodanko JJ, Effects of Methyl Substitution in Ruthenium Tris(2-pyridylmethyl)amine Photocaging Groups for Nitriles. Inorg. Chem 2016, 55, 6968–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tu Y-J; Mazumder S; Endicott JF; Turro C; Kodanko JJ; Schlegel HB, Selective Photodissociation of Acetonitrile Ligands in Ruthenium Polypyridyl Complexes Studied by Density Functional Theory. Inorg. Chem 2015, 54, 8003–8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Askes SHC; Bahreman A; Bonnet S, Activation of a Photodissociative Ruthenium Complex by Triplet-Triplet Annihilation Upconversion in Liposomes. Angew. Chem. Int. Ed 2013, 53, 1029–1033. [DOI] [PubMed] [Google Scholar]
- 33.Meijer MS; Bonnet S, Diastereoselective Synthesis and Two-Step Photocleavage of Ruthenium Polypyridyl Complexes Bearing a Bis(thioether) Ligand. Inorg. Chem 2019, 58, 11689–11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lameijer LN; van de Griend C; Hopkins SL; Volbeda A-G; Askes SHC; Siegler MA; Bonnet S, Photochemical Resolution of a Thermally Inert Cyclometalated Ru(phbpy)(N–N)(Sulfoxide)+ Complex. J. Am. Chem. Soc 2019, 141, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z; Sadler PJ, Organoiridium Complexes: Anticancer Agents and Catalysts. Acc. Chem. Res 2014, 47, 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng L; Chen Y; Huang H; Wang J; Zhao D; Ji L; Chao H, Cyclometalated Ruthenium(II) Anthraquinone Complexes Exhibit Strong Anticancer Activity in Hypoxic Tumor Cells. Chem. Eur. J 2015, 21, 15308–15319. [DOI] [PubMed] [Google Scholar]
- 37.Bautista HR; Díaz ROS; Shen LQ; Orvain C; Gaiddon C; Le Lagadec R; Ryabov AD, Impact of cyclometalated ruthenium(II) complexes on lactate dehydrogenase activity and cytotoxicity in gastric and colon cancer cells. J. Inorg. Biochem 2016, 163, 28–38. [DOI] [PubMed] [Google Scholar]
- 38.Sainuddin T; McCain J; Pinto M; Yin H; Gibson J; Hetu M; McFarland SA, Organometallic Ru(II) Photosensitizers Derived from π-Expansive Cyclometalating Ligands: Surprising Theranostic PDT Effects. Inorg. Chem 2016, 55, 83–95. [DOI] [PubMed] [Google Scholar]
- 39.Huang H; Zhang P; Chen H; Ji L; Chao H, Comparison Between Polypyridyl and Cyclometalated Ruthenium(II) Complexes: Anticancer Activities Against 2D and 3D Cancer Models. Chem. Eur. J 2015, 21, 715–725. [DOI] [PubMed] [Google Scholar]
- 40.Motley TC; Troian-Gautier L; Brennaman MK; Meyer GJ, Excited-State Decay Pathways of Tris(bidentate) Cyclometalated Ruthenium(II) Compounds. Inorg. Chem 2017, 56, 13579–13592. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh G; Colón KL; Fuller A; Sainuddin T; Bradner E; McCain J; Monro SMA; Yin H; Hetu MW; Cameron CG; McFarland SA, Cyclometalated Ruthenium(II) Complexes Derived from α-Oligothiophenes as Highly Selective Cytotoxic or Photocytotoxic Agents. Inorg. Chem 2018, 57, 7694–7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuello-Garibo J-A; Meijer MS; Bonnet S, To cage or to be caged? The cytotoxic species in ruthenium-based photoactivated chemotherapy is not always the metal. Chem. Commun 2017, 53, 6768–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun Y; Heidary DK; Zhang Z; Richards CI; Glazer EC, Bacterial Cytological Profiling Reveals the Mechanism of Action of Anticancer Metal Complexes. Mol. Pharm 2018, 15, 3404–3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Groutas WC; Stanga MA; Brubaker MJ; Huang TL; Moi MK; Carroll RT, Substituted 2-Pyrones, 2-Pyridones, and Other Congeners of Elasnin as Potential Agents for the Treatment of Chronic Obstructive Lung Diseases? J. Med. Chem 2002, 28, 1106–1109. [DOI] [PubMed] [Google Scholar]
- 45.Jacinto Demuner A; Moreira Valente VM; Almeida Barbosa LC; Rathi A; Donohoe TJ; Thompson AL, Synthesis and Phytotoxic Activity of New Pyridones Derived from 4-Hydroxy-6-Methylpyridin-2(1H)-one. Molecules 2009, 14, 4973–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dutta U; Deb A; Lupton DW; Maiti D, The regioselective iodination of quinolines, quinolones, pyridones, pyridines and uracil. Chem. Commun 2015, 51, 17744–17747. [DOI] [PubMed] [Google Scholar]
- 47.Albani BA; Peña B; Dunbar KR; Turro C, New cyclometallated Ru(II) complex for potential application in photochemotherapy? Photochem. Photobiol. Sci 2014, 13, 272–280. [DOI] [PubMed] [Google Scholar]
- 48.Mukuta T; Tanaka S. i.; Inagaki A; Koshihara S.-y.; Onda K, Direct Observation of the Triplet Metal-Centered State in [Ru(bpy)3]2+ Using Time-Resolved Infrared Spectroscopy. ChemistrySelect 2016, 1, 2802–2807. [Google Scholar]
- 49. Direct observation of the 3MC state has been rare, with reference 50 providing one example. Therein, [(6-methyl-2,2’-bipyridine)3Ru]2+ has a 3MC state of unusually low energy due to steric strain. Most other Ru polypyridyl complexes have 3MC states that are difficult to study by transient absorption spectroscopy and are more typically studied by computations. The difficulties lie in the typically short lifetimes of the 3MC states and the rates at which other states are populated, which results in a low steady state population of the 3MC state.
- 50.Sun Q; Mosquera-Vazquez S; Lawson Daku LM; Guénée L; Goodwin HA; Vauthey E; Hauser A, Experimental Evidence of Ultrafast Quenching of the 3MLCT Luminescence in Ruthenium(II) Tris-bipyridyl Complexes via a 3dd State. J. Am. Chem. Soc 2013, 135, 13660–13663. [DOI] [PubMed] [Google Scholar]
- 51.Loftus LM; Rack JJ; Turro C, Photoinduced ligand dissociation follows reverse energy gap law: nitrile photodissociation from low energy 3MLCT excited states. Chem. Commun 2020, 56, 4070–4073. [DOI] [PubMed] [Google Scholar]
- 52.Wagenknecht PS; Ford PC, Metal centered ligand field excited states: Their roles in the design and performance of transition metal based photochemical molecular devices. Coord. Chem. Rev 2011, 255, 591–616. [Google Scholar]
- 53.Juris A; Balzani V; Barigelletti F; Campagna S; Belser P; von Zelewsky A, Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev 1988, 84, 85–277. [Google Scholar]
- 54.Collin J-P; Sauvage J-P, Transition Metal-complexed Catenanes and Rotaxanes as Light-driven Molecular Machines Prototypes. Chem. Lett 2005, 34, 742–747. [Google Scholar]
- 55.Reichardt C; Sainuddin T; Wächtler M; Monro S; Kupfer S; Guthmuller J; Gräfe S; McFarland S; Dietzek B, Influence of Protonation State on the Excited State Dynamics of a Photobiologically Active Ru(II) Dyad. J. Phys. Chem. A 2016, 120, 6379–6388. [DOI] [PubMed] [Google Scholar]
- 56.Cardone RA; Casavola V; Reshkin SJ, The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.