

Abstract
Aims
Right ventricular dysfunction may arise because of pulmonary arterial hypertension (PAH). Development of new diagnostic methods able to identify PAH and allow for earlier treatment initiation, before the development of vascular remodelling and manifest right heart failure (HF), could potentially improve prognosis. Proteoglycans and inflammatory proteins are involved in vascular remodelling. We aimed to investigate their potential as biomarkers to differentiate PAH in a dyspnoeic population.
Methods and results
Plasma from 152 patients with PAH (n = 48), chronic thrombo‐embolic pulmonary hypertension (n = 20), pulmonary hypertension due to HF with reduced (n = 36) or preserved (n = 33) ejection fraction, and HF without pulmonary hypertension (n = 15) and 20 healthy controls were analysed with proximity extension assays. Haemodynamics were assessed in the patients with right heart catheterization. Plasma prolargin levels in PAH were lower compared with all the other studied disease groups (P < 0.001) but higher than the controls' levels (P = 0.003). Receiver operating characteristic curve of prolargin as a PAH‐differentiating marker in a pooled population, encompassing all the other studied disease groups, had a sensitivity of 74% and a specificity of 83.3% (area under the curve = 0.84, P < 0.001). Prolargin correlated with the mean right atrial pressure (r s = 0.65, P < 0.001), N‐terminal pro‐brain natriuretic peptide (r s = 0.64, P < 0.001), cardiac index (r s = −0.31, P = 0.029), stroke volume index (r s = −0.41, P = 0.004), right ventricular stroke work index (r s = −0.31, P = 0.032), six‐minute walking distance (r s = −0.41, P = 0.005), and mixed venous blood oxygen saturation (r s = −0.42, P = 0.003).
Conclusions
Plasma prolargin levels differentiate PAH patients from controls and the other investigated dyspnoea groups including HF. Its potential in PAH differentiation may be enhanced by inclusion in a multi‐marker panel. Larger studies are needed to evaluate its discriminative ability of PAH in relation to other dyspnoea aetiologies and its potential role in PAH risk stratification and pathobiology.
Keywords: Differentiation, Plasma biomarkers, Prolargin, Proteoglycans, Pulmonary hypertension
Introduction
Right ventricular dysfunction may arise because of several mechanisms, including pulmonary arterial hypertension (PAH). In PAH, excessive vasoconstriction and vascular remodelling may ensue, increasing pulmonary vascular resistance (PVR), ultimately leading to right ventricular dysfunction, which is a strong predictor of adverse outcomes. 1 , 2 Despite new vasodilator therapies, PAH is associated with high morbidity and mortality, where development of right heart failure (HF) is the principal cause of death. 2 PAH has a poor prognosis where idiopathic PAH has been shown to exhibit a median survival of 2.8 years from diagnosis if left untreated and only 1 year if related to rheumatological disease. 3 , 4 Progression of non‐specific symptoms, such as dyspnoea and fatigue, renders a delay from onset of symptoms to diagnosis with an average of ≥2 years. 5 This underscores the need for new diagnostic tools for earlier PAH diagnosis. Accordingly, the development of a screening method that may differentiate patients with PAH from those with other causes of unclear dyspnoea, as well as differentiate between different causes of pulmonary hypertension (PH), would be of great value to enable earlier diagnosis. This could potentially also enable earlier treatment initiation of PAH patients and improve their survival. Although several biomarkers have been evaluated, no one specifically differentiating PAH or pulmonary vascular remodelling has yet been demonstrated to be useful clinically. 4 , 6
Extracellular matrix (ECM) is a dynamic scaffold with a fundamental role in regulating vascular function. Collagen accumulation in the vascular wall in PAH leads to vessel fibrosis and increased stiffness. 7 , 8 In experimental PAH, ECM remodelling occurs early in PAH development, which precedes increases in mean pulmonary artery pressure (MPAP) and PVR. 9 Unadapted remodelling of ECM may also contribute to vessel stiffness. 10 Among ECM proteins, matrix metalloproteinases have specifically been suggested to play a role in vascular remodelling and PH development. 9
In a recent study, we found that plasma levels of matrix metalloproteinase (MMP)‐2, MMP‐7, MMP‐9, MMP‐12, and tissue inhibitor of metalloproteinase‐4 were elevated in PAH compared with healthy controls. 11 Plasma levels of MMP‐7 were furthermore lower in PAH compared with chronic thrombo‐embolic pulmonary hypertension (CTEPH), as well as HF with preserved or reduced ejection fraction (EF), with or without PH. Future studies investigating the relationship between MMP‐7 and PAH pathogenesis, pulmonary vascular remodelling, and congestion, as well as its clinical usefulness in differentiation and earlier diagnosis of PAH, were urged for. 11
Similarly, proteoglycans have a great influence on ECM development and signalling 12 , 13 and are known to play an important part in ECM remodelling in heart disease. 14 For instance, decorin interacts with transforming growth factor‐β, limiting pulmonary fibrosis, whereas glypican‐1 is involved in flow‐induced endothelial nitric oxide synthase activation. 15 , 16 Maladaptive immune response and inflammatory dysregulation have additionally been proposed to play a role in endothelial dysfunction and vascular remodelling. In PAH, a pro‐inflammatory phenotype of endothelial cells has been identified with elevated levels of E‐selectin, intercellular adhesion molecule‐1 (ICAM‐1), and vascular cell adhesion molecule, as well as characterized by excessive cytokine release. 10 Intriguingly, the ECM functions as a reservoir for factors that influences cell proliferation, differentiation, and migration, including growth factors and cytokines, which furthermore may influence the inflammatory response. 17 Accordingly, circulating inflammatory mediators are associated with a worse clinical outcome in PAH. 10
The aim of the present study was to investigate the plasma levels of proteoglycans and inflammatory proteins in different dyspnoea groups and their potential value as biomarkers in the differentiation of PH of various causes, with a focus on PAH, in relation to haemodynamics. We hypothesized that some proteoglycans and related proteins may potentially be used to differentiate the different subtypes of PH from each other, as proteoglycans may be involved in the pathology of pulmonary vascular remodelling in PAH.
Methods
Study population
The study population consisted of 152 patients ≥18 years under investigation of dyspnoea with baseline right heart catheterization (RHC) at diagnosis, enrolled in the Lund Cardio Pulmonary Register (LCPR) between September 2011 and March 2017. Patients with PAH (n = 48), CTEPH (n = 20), PH due to HF with preserved EF (HFpEF‐PH, n = 33), PH due to HF with reduced EF (HFrEF‐PH, n = 36), and HFrEF or HFpEF without PH (HF‐NON‐PH, n = 15, whereof HFrEF, n = 8 and HFpEF, n = 7), as well as 20 healthy controls, the last without RHC haemodynamics, were included. Patients lacking all haemodynamic data were excluded.
Patients with PH, exhibiting an MPAP ≥ 25 mmHg at rest at RHC, had been categorized into precapillary or postcapillary PH, exhibiting pulmonary arterial wedge pressure (PAWP) ≤15 or >15 mmHg, respectively. 4 HFpEF was defined as EF ≥ 50% and HFrEF as EF < 50%. To classify HF and identify intracardiac shunts, echocardiography and magnetic resonance imaging were used. Pulmonary scintigraphy was performed to identify CTEPH, and additional spirometry with diffusion capacity and/or high‐resolution computed tomography was performed to exclude Group 3 PH due to hypoxia and/or lung disease. For subgroup analysis of PAH, IPAH and familial PAH (FPAH) were considered as one entity (IPAH/FPAH, n = 23) and PAH due to connective tissue disease (CTD‐PAH, n = 25) as another.
All participants had given their informed written consent. The study was conducted in accordance with the Declaration of Helsinki and Istanbul and was approved by the local ethics committee in Lund (Dnr 2010/114, 2010/248, 2010/442, 2011/368, and 2015/270).
Plasma sampling and protein analyses
Venous blood samples were collected from the patients during RHC from the venous introducer, inserted in the right internal jugular vein. Venous blood samples were additionally collected from the healthy controls. All blood samples were centrifuged, and plasma was extracted and stored at −80°C in the LCPR cohort of the Region Skåne biobank.
The plasma samples were analysed with proximity extension assay (PEA) and read out by quantitative PCR. In short, antibody pairs with complementing DNA oligonucleotide tails bind the target protein in the proximity of each other, creating a DNA sequence that is elongated by DNA polymerase and read out by quantitative PCR. 18 Plasma protein levels were analysed using the Proseek Multiplex Cardiovascular II, Cardiovascular III, and Oncology II 96‐plex immunoassay panels (Olink Proteomics, Uppsala, Sweden) and were reported in normalized protein expression values. Normalized protein expression is an arbitrary unit (AU) on a log2‐scale. The present study investigated a selection of proteins from the aforementioned Olink panels. Twelve proteoglycans and associated proteins were analysed, namely: decorin, glypican‐1, syndecan‐1, perlecan (also known as heparan sulfate proteoglycan 2), prolargin (also known as proline–arginine‐rich end leucine‐rich repeat protein), collagen alpha‐1(I) chain, integrin α‐V, integrin β‐2, integrin β‐5, melusin (also integrin beta‐1‐binding protein 2), vascular endothelial cadherin (VE‐cadherin, also known as cadherin‐5), and matrix extracellular phosphoglycoprotein (MEPE). In addition, 65 inflammatory plasma proteins were analysed including ALCAM, alpha‐taxilin, annexin A1, azurocidin, C–C motif chemokine 3 (CCL3), CCL15, CCL16, CCL17, CCL22, CCL24, CD4, CD48, CD70, CD93, CD160, CD163, CD207, carcinoembryonic antigen‐related cell adhesion molecule 8 (CEACAM8), chitotriosidase‐1, chitinase‐3‐like protein 1, C–X–C motif chemokine 1 (CXCL1), CXCL13, CXCL16, CXCL17, E‐selectin, Fc receptor‐like B (FcRLB), growth/differentiation factor 15 (GDF‐15), intercellular adhesion molecule‐2, ICOS ligand, interferon gamma receptor 1 (IFN‐gamma‐R1), low affinity immunoglobulin gamma Fc region receptor II‐b (IgG Fc receptor II‐b), interleukin (IL)‐1 receptor‐like 2 (IL‐1RL2), IL‐1 receptor antagonist protein (IL‐1RN), IL‐1 receptor type 1 (IL‐1RT1), IL‐1RT2, IL‐2 receptor subunit alpha (IL‐2‐RA), IL‐4 receptor subunit alpha (IL‐4R‐alpha), IL‐6, IL‐6R‐alpha, pro‐interleukin‐16 (pro‐IL‐16), IL‐17D, IL‐17 receptor A (IL‐17RA), IL‐18, IL‐18‐binding protein (IL‐18BP), IL‐27, junctional adhesion molecule A (JAM‐A), kidney injury molecule 1 (KIM‐1), T‐lymphocyte surface antigen Ly‐9 (Ly‐9), lymphotactin, macrophage receptor MARCO (MARCO), monocyte chemotactic protein 1 (MCP‐1), MHC class I polypeptide‐related sequence A and MHC class I polypeptide‐related sequence B (MIC‐A/B), myeloperoxidase, platelet endothelial cell adhesion molecule (PECAM‐1), peptidoglycan recognition protein 1 (Pglyrp1), polymeric immunoglobulin receptor (PIgR), progranulin, P‐selectin, P‐selectin glycoprotein ligand 1 (PSGL‐1), pulmonary surfactant‐associated protein D (PSP‐D), pentraxin‐related protein PTX3 (PTX3), SLAM family member 5 (SLAMF5), SLAMF7, toll‐like receptor 3 (TLR3), and trem‐like transcript 2 protein (TLT‐2). For consistency, N‐terminal pro‐brain natriuretic peptide (NT‐proBNP) was also analysed with PEA.
Right heart catheterization
RHC measurements were performed with Swan–Ganz catheters (Baxter Healthcare Corp., Santa Ana, CA), by experienced cardiologists, as part of clinical diagnosis. MPAP, PAWP, mean right atrial pressure (MRAP), and mean arterial pressure (MAP) were registered during RHC. Heart rate (HR) was measured by electrocardiogram. Thermodilution was used to measure cardiac output (CO). Mixed venous blood oxygen saturation (SvO2) was measured during RHC; six‐minute walking distance (6MWD) values at the time of RHC were collected from medical records.
Cardiac index (CI), stroke volume (SV), stroke volume index (SVI), left ventricular stroke work index (LVSWI), right ventricular stroke work index (RVSWI), PVR, and transpulmonary pressure gradient (TPG) were calculated by the following formulae: Body surface area (BSA) = (weight0.425 × height0.725 × 0.007184), CI = CO∕BSA, SV = CO∕HR, SVI = SV∕BSA, LVSWI = (MAP − PAWP) × SVI, RVSWI = (MPAP − MRAP) × SVI, TPG = MPAP − PAWP, and PVR = TPG∕CO.
Statistics
Statistical analyses were performed using GraphPad Prism Version 8.0.2 for Windows, GraphPad Software, San Diego, CA, www.graphpad.com. Data distribution was analysed using histograms. Proteins' levels were analysed with Kruskal–Wallis tests, which were then analysed by Benjamini and Hochberg false discovery rate (FDR) test (Q = 5%). 19 , 20 Significant Kruskal–Wallis tests, after FDR analysis, were followed by uncorrected Dunn's multiple comparison tests for all groups, which in turn were analysed with FDR (Q = 5%). The purpose of the FDR analyses was to limit false‐positive results. The threshold for statistical significance was set to P < 0.0021 after FDR analysis. For other statistical tests, a P‐value <0.05 was considered significant. All presented P‐values are raw. Mann–Whitney U test was used for subgroup analysis of PAH. Receiver operating characteristic curve was used to investigate prolargin as a predictor for PAH among the other dyspnoea exhibiting groups, that is, CTEPH, HFpEF‐PH, HFrEF‐PH, and HF‐NON‐PH. Youden's index was used to determine the ideal cut‐off. Correlation analyses, using Spearman's coefficient, were performed between prolargin and MRAP, MPAP, CI, SVI, RVSWI, PVR, as well as 6MWD, SvO2, and NT‐proBNP. Additional correlation analyses were performed between age and prolargin levels in controls, PAH, CTEPH, HFpEF‐PH, and HFrEF‐PH HF‐NON‐PH. The analysis of the 65 inflammatory proteins was corrected with a separate Benjamini and Hochberg FDR analysis (Q = 5%) with a threshold for the ANOVAs set to P < 0.033. For the following multiple comparisons, the threshold was set to P < 0.016.
Results
Population characteristics
The baseline characteristics of the study population have previously been described 11 and are presented in Table 1 , with the addition of medications.
Table 1.
Population characteristics
| Control | PAH | CTEPH | HFpEF‐PH | HFrEF‐PH | HF‐NON‐PH | |
|---|---|---|---|---|---|---|
| Patients n (% female) | 20 (50) | 48 (83) | 20 (65) | 33 (64) | 36 (19) | 15 (53) |
| Age (years) | 41 (26.8–50.5) | 71.5 (64–76) | 75 (70.8–77.8) | 75 (68.5–83) | 54 (47.3–59.5) | 60 (46–76) |
| BSA (m2) | 1.9 (1.8–2) | 1.7 (1.6–2) | 1.8 (1.8–2) | 1.9 (1.7–2.1) | 2 (1.9–2.1) | 2 (1.7–2.1) |
| Haemodynamics | ||||||
| MAP (mmHg) | 96 (89.4–104) | 98.5 (94–110.3) | 98 (91.5–104.5) | 79.5 (75.3–88.8) | 89 (80–96) | |
| MPAP (mmHg) | 43 (37–54.8) | 42 (35–54.3) | 34 (28.5–46) | 34.5 (29–40.8) | 20 (17–22) | |
| PAWP (mmHg) | 8 (6–11) | 9.5 (7–13) | 18 (16–22.5) | 25 (19–28) a | 15 (9–18) | |
| MRAP (mmHg) | 7 (4–11) | 5.5 (3.3–8) | 10 (6.5–14) | 14.5 (9–17) | 6 (2–16) | |
| HR (b.p.m.) | 77.5 (70–94.3) | 75 (69.5–88) | 70 (61.5–82.5) | 71 (68.3–86) | 72 (60–84) | |
| CO (L/min) | 3.8 (3.0–5.1) | 4 (3.5–4.7) | 4.5 (3.7–5.7) | 3.2 (2.8–4.0) | 3.3 (3.0–4.4) | |
| CI (L/min/m2) | 2.2 (1.8–2.8) | 2.3 (1.9–2.5) | 2.4 (2.1–2.8) | 1.6 (1.4–1.9) | 1.9 (1.6–2.2) | |
| SV (mL/beat) | 51.2 (40.8–56.3) | 56.3 (45.8–65.1) | 61.7 (48.8–83.7) | 45.1 (36.0–54.5) | 54.8 (44.8–58.8) | |
| SVI (mL/beat/m2) | 28.7 (22.6–35) | 30.5 (26.3–32.5) | 33.8 (28.1–42.3) | 22.5 (18.2–27.2) | 29 (25.2–31.9) | |
| PVR (WU) | 9.5 (6.2–11.8) | 9.3 (5.9–10.8) | 3.6 (2.4–4.9) | 3.0 (2.3–3.7) a | 1.5 (1.0–2.0) | |
| LWSWI (mmHg × mL/m2) | 2488 (2045–3213) | 2508 (2330–3187) | 2664 (2189–3308) | 1152 (957–1636) a | 2168 (1650–2716) | |
| RVSWI (mmHg × mL/m2) | 990.5 (807.2–1246) | 1111 (844.5–1298) | 831.5 (670.7–1140) | 439.6 (305.8–649.3) | 382.4 (195.5–494.5) | |
| NT‐proBNP (AU) | 3.1 (2.1–3.8) | 2.6 (1.0–4.2) | 2.9 (2.4–3.3) | 4.9 (4.1–5.4) | 3.2 (1.3–4.4) | |
| SvO2 (%) | 59.3 (51.1–66.2) | 62.5 (54.9–67.9) | 64.1 (57.8–66.8) | 50.3 (46.5–55.2) | 61.2 (58.5–69.2) | |
| Creatinine (μmol/L) | 90 (70.8–113.5) | 88 (73–122.5) | 99 (79–117) | 121 (90–145) | 93 (80.5–123) | |
| Medications n (%) | ||||||
| Beta‐blockers | 16 (34) b | 9 (50) c | 25 (75.8) | 35 (97.2) | 11 (73.3) | |
| ACE inhibitors | 10 (21.3) b | 2 (11.1) c | 12 (36.4) | 19 (52.8) | 3 (20) | |
| ARBs | 4 (8.5) b | 7 (38.9) c | 10 (30.3) | 14 (38.9) | 5 (33.3) | |
| MRAs | 11 (23.4) b | 3 (16.7) c | 9 (27.3) | 21 (58.3) | 7 (46.7) | |
ACE, angiotensin‐converting enzyme; ARBs, angiotensin II receptor blockers; AU, arbitrary units; BSA, body surface area; CI, cardiac index; CO, cardiac output; CTEPH, chronic thrombo‐embolic pulmonary hypertension; HF‐NON‐PH, heart failure without pulmonary hypertension; HFpEF‐PH, pulmonary hypertension due to heart failure with preserved ejection fraction; HFrEF‐PH, pulmonary hypertension due to heart failure with reduced ejection fraction; HR, heart rate; LVSWI, left ventricular stroke work index; MAP, mean arterial pressure; MPAP, mean pulmonary artery pressure; MRAP, mean right atrial pressure; MRAs, mineralocorticoid receptor antagonists; PAH, pulmonary arterial hypertension; PAWP, pulmonary arterial wedge pressure; PVR, pulmonary vascular resistance; RVSWI, right ventricular stroke work index; SV, stroke volume; SV, stroke volume index; WU, Wood units.
Variables are presented as median (inter‐quartile range).
Indicates n = 35 due to one patient being unable to go through all tests.
Indicates n − 1.
Indicates n − 2.
Plasma prolargin differentiates pulmonary arterial hypertension from other pulmonary hypertension aetiologies
Plasma prolargin levels were higher in PAH compared with controls (P = 0.003), but lower compared with the other disease groups (P < 0.001) (Figure 1A ). Receiver operating characteristic curve analysis of prolargin levels in PAH vs. CTEPH, HFpEF‐PH, HFrEF‐PH, and HF‐NON‐PH pooled together resulted in an area under the curve of 0.84 (95% confidence interval, 0.77–0.91) (P < 0.001), with a sensitivity of 74% and a specificity of 83.3% with a cut‐off value of 6.625 AU (Figure 1B ).
FIGURE 1.
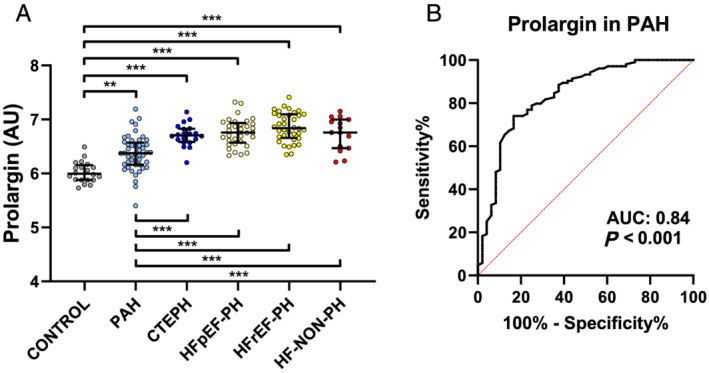
Plasma levels of prolargin in pulmonary hypertension subgroups and heart failure. (A) Pulmonary arterial hypertension (PAH) is differentiated from all other groups by prolargin. Plasma prolargin levels were higher than controls (P = 0.003) and lower than all other disease groups (P < 0.001). (B) Receiver operating characteristic curve of prolargin in PAH vs. chronic thrombo‐embolic pulmonary hypertension (CTEPH), pulmonary hypertension due to heart failure with preserved ejection fraction (HFpEF‐PH), pulmonary hypertension due to heart failure with reduced ejection fraction (HFrEF‐PH), and heart failure without pulmonary hypertension (HF‐NON‐PH) pooled together. AU, arbitrary units; AUC, area under the curve. **P < 0.01; ***P < 0.001. In (A), Kruskal–Wallis test was performed to analyse the differences between the groups. Statistical significance was considered P < 0.0021, after correction for multiple comparisons with Benjamini and Hochberg false discovery rate (Q = 5%).
Prolargin correlated positively with MRAP (r s = 0.65, P < 0.001) and NT‐proBNP (r s = 0.64, P < 0.001) but negatively with CI (r s = −0.31, P = 0.029), SVI (r s = −0.41, P = 0.004), RVSWI (r s = −0.31, P = 0.032), 6MWD (r s = −0.41, P = 0.05), and SvO2 (r s = −0.42, P = 0.003). Prolargin did not correlate with MPAP (P = 0.11) or PVR (P = 0.18) (Figure 2 ). Subgroup analysis of prolargin levels in IPAH/FPAH vs. CTD‐PAH revealed no difference between the groups (P = 0.13). Plasma levels of all ECM‐related proteins are found in Table 2 and Supporting Information, Figure S1 and the P‐values in Supporting Information, Table S1 .
FIGURE 2.
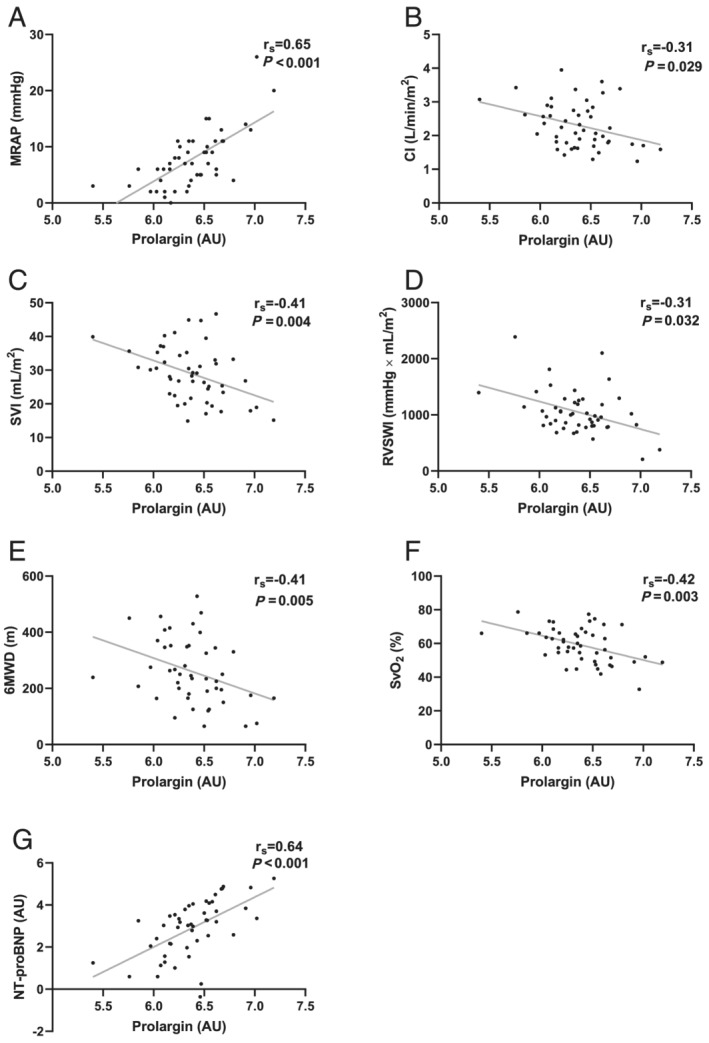
Correlations of prolargin with haemodynamic parameters. Prolargin correlated (A, G) positively with mean right atrial pressure (MRAP) and N‐terminal pro‐brain natriuretic peptide (NT‐proBNP) and (B–F) negatively with cardiac index (CI), stroke volume index (SVI), right ventricular stroke work index (RVSWI), six‐minute walking distance (6MWD), and mixed venous oxygen saturation (SvO2). AU, arbitrary units.
Table 2.
Proteins' levels and comparisons of all groups
| Proteins (AU) | Control | PAH | CTEPH | HFpEF‐PH | HFrEF‐PH | HF‐NON‐PH |
|---|---|---|---|---|---|---|
| n = 20 | n = 48 | n = 20 | n = 33 | n = 36 | n = 15 | |
| Proteoglycans | ||||||
| Prolargin | 6.00 (5.88–6.16) | 6.38 (6.16–6.57) a | 6.71 (6.59–6.83) a , b | 6.76 (6.57–6.94) a , b | 6.84 (6.66–7.10) a , b | 6.76 (6.47–7) a , b |
| Decorin | 4.34 (4.2–4.53) | 4.69 (4.43–4.98) a | 4.89 (4.7–5.07) a | 5.06 (4.77–5.31) a , b | 4.99 (4.8–5.22) a , b | 4.93 (4.79–5.58) a , b |
| Syndecan‐1 | 5.81 (5.41–6.22) f | 6.45 (6.16–6.91) a | 6.28 (6.00–6.63) a | 6.42 (6.15–7.00) a | 7.02 (6.51–7.82) a – d , f | 6.3 (5.88–6.56) e |
| Perlecan | 6.04 (5.75–6.24) | 6.52 (6.16–6.92) a | 6.56 (6.34–6.78) a | 6.79 (6.48–7.00) a | 6.85 (6.42–7.22) a , b | 6.57 (6.22–6.83) a |
| Glypican‐1 | 3.91 (3.62–4.05) f | 3.62 (3.25–3.88) a | 3.65 (3.39–4.07) | 3.94 (3.72–4.05) b | 3.84 (3.61–4.19) b , f | 3.82 (3.63–4.13) |
| Other adhesion proteins | ||||||
| MEPE | 2.86 (2.64–3.23) | 2.25 (1.81–2.63) a | 2.42 (2.12–2.86) a | 2.31 (2.11–2.60) a | 2.95 (2.56–3.13) b – d | 2.62 (2.32–2.70) b |
| VE‐cadherin | 3.08 (2.75–3.34) | 2.94 (2.52–3.36) | 2.97 (2.72–3.15) | 2.99 (2.91–3.44) | 3.52 (3.21–3.7) a – d | 3.14 (3.00–3.50) |
| Collagen alpha‐1(I)chain | 2.18 (1.75–2.57) | 1.81 (1.48–2.37) | 1.64 (1.24–2.08) a | 1.94 (1.6–2.34) | 2.17 (1.89–2.54) b , c | 1.66 (1.48–2.14) e |
| Melusin | 4.97 (4.16–6.04) | 4.46 (3.36–5.93) | 4.03 (3.35–4.62) | 4.51 (3.58–5.80) | 3.88 (2.73–4.99) a | 4.67 (3.75–5.68) |
| Integrin α‐V | 2.75 (2.60–2.88) f | 2.6 (2.44–2.78) | 2.72 (2.50–2.84) | 2.75 (2.51–2.93) | 2.62 (2.45–2.89) f | 2.66 (2.41–2.81) |
| Integrin β‐2 | 4.34 (4.11–4.45) | 4.20 (3.68–4.56) | 4.12 (3.85–4.45) | 4.23 (3.92–4.62) | 3.93 (3.52–4.43) | 4.01 (3.87–4.21) |
| Integrin β‐5 | 7.72 (7.05–7.88) f | 7.59 (7.26–7.74) | 7.53 (7.25–7.65) | 7.67 (7.45–7.94) | 7.52 (7.23–7.87) f | 7.54 (7.41–7.94) |
CTEPH, chronic thrombo‐embolic pulmonary hypertension; HF‐NON‐PH, heart failure without pulmonary hypertension; HFpEF‐PH, pulmonary hypertension due to heart failure with preserved ejection fraction; HFrEF‐PH, pulmonary hypertension due to heart failure with reduced ejection fraction; MEPE, matrix extracellular phosphoglycoprotein; PAH, pulmonary arterial hypertension; VE‐cadherin, vascular endothelial cadherin.
Variables are presented in arbitrary units (AU) as median (inter‐quartile range). Kruskal–Wallis tests were performed to analyse the differences between the groups. Statistical significance was considered P < 0.0021, after correction for multiple comparisons with Benjamini and Hochberg false discovery rate (Q = 5%).
Significant difference compared with controls.
Significant difference compared with PAH.
Significant difference compared with CTEPH.
Significant difference compared with HFpEF‐PH.
Significant difference compared with HFrEF‐PH.
n − 1.
Other plasma proteins
Plasma IL‐17D displayed higher levels in PAH compared to controls (P = 0.001), and lower than the other PH groups (P < 0.006), but not different compared with HF‐NON‐PH (P = 0.25) (Figure 3A and Table 3 ). Plasma levels of all inflammatory proteins are shown in Table 3 . Plasma decorin levels were higher in all disease groups compared with controls (P < 0.002). The decorin levels in PAH were lower than in the other disease groups (P < 0.01), except for CTEPH (P = 0.048) (Figure 3B ). Syndecan‐1 plasma levels were higher in HFrEF‐PH compared with all other PH groups, HF‐NON‐PH, and controls (P < 0.002) (Figure 3C ).
FIGURE 3.
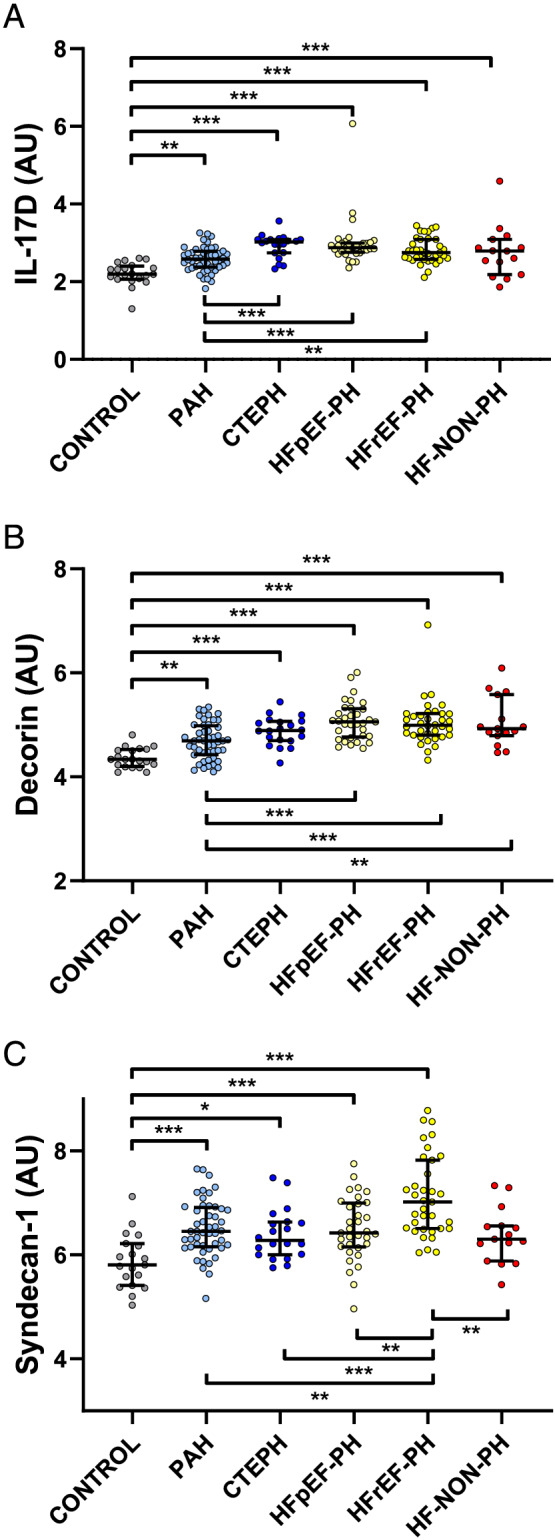
Plasma levels of interleukin‐17D (IL‐17D), decorin, and syndecan‐1 in pulmonary hypertension (PH) subgroups. (A) IL‐17D differentiates PAH from other PH. (B) Decorin differentiates PAH from heart failure with or without PH (P < 0.01). (C) Syndecan‐1 levels were higher in heart failure with reduced ejection fraction (P < 0.002) than all other groups. Other abbreviations as in Figure 1 . **P < 0.01; ***P < 0.001. Kruskal–Wallis tests were performed to analyse the differences between the groups. Statistical significance was considered P < 0.0021, after correction for multiple comparisons with Benjamini and Hochberg false discovery rate (Q = 5%).
Table 3.
Inflammatory proteins' levels and comparisons of all groups
| Plasma protein (AU) | Controls | PAH | CTEPH | HFpEF‐PH | HFrEF‐PH | HF‐NON‐PH |
|---|---|---|---|---|---|---|
| n = 20 | n = 48 | n = 20 | n = 33 | n = 36 | n = 15 | |
| ALCAM | 4.2 (4.1–4.4) | 4.3 (3.9–4.5) | 4.3 (4.2–4.6) | 4.5 (4.3–4.7) | 4.5 (4.2–4.8) | 4.5 (4.2–4.6) |
| Alpha‐taxilin | 6.4 (4.9–7) f | 5.5 (4.8–6.7) | 5.7 (5.3–6.2) | 6.2 (5.5–7.1) | 5.8 (4.6–6.5) f | 6.7 (5.3–7) |
| Annexin A1 | 1.4 (1.2–1.8) f | 1.8 (1.5–2.2) a | 1.9 (1.5–2.3) a | 1.7 (1.5–2) a | 1.7 (1.4–2) f | 1.7 (1.5–2) |
| Azurocidin | 1.6 (1.4–1.9) | 1.9 (1.4–2.5) a | 1.9 (1.7–2.5) a | 1.7 (1.5–2.1) | 1.9 (1.5–2.6) a | 1.8 (1.6–2.4) |
| CCL3 | 1.4 (1.2–1.7) | 2.6 (2.2–3) a | 2.6 (2.3–3) a | 2.8 (2.4–3.5) a | 2.5 (2.3–3.2) a | 2.4 (2.3–2.9) a |
| CCL15 | 6.1 (5.9–6.6) | 6.7 (6.4–7.1) a | 6.9 (6.4–7.2) a | 7 (6.6–7.7) a , b | 7.1 (6.7–7.7) a , b | 6.6 (6.5–7.1) a |
| CCL16 | 5.3 (4.9–5.6) | 5.6 (5.3–6) | 5.7 (5.4–5.9) | 5.9 (5.4–6.3) a | 6 (5.5–6.5) a , b | 5.9 (5.7–6.2) a |
| CCL17 | 7.1 (6.1–7.5) | 6.9 (6.3–7.6) | 7.5 (6.9–8) | 7.6 (6.3–8.6) | 6.9 (6.2–7.8) | 7.1 (6.2–8.5) |
| CCL22 | 1.9 (1.7–2.5) | 2 (1.5–2.6) | 1.7 (1.3–2) | 1.8 (1.5–2) | 1.9 (1.4–2.8) | 1.6 (1.4–1.9) |
| CCL24 | 4.9 (4.4–5.4) | 4.9 (4.1–5.2) | 5 (4.2–5.4) | 5.4 (5–5.8) b | 4.8 (4.3–5.8) | 4.6 (3.7–5.2) d |
| CD4 | 3.8 (3.6–4) | 4.2 (4–4.6) a | 4.4 (4.2–4.8) a | 4.5 (4.2–4.9) a | 4.6 (4.3–4.9) a , b | 4.3 (4.1–4.7) a |
| CD48 | 6.1 (5.9–6.3) f | 6.2 (5.9–6.5) | 6.3 (6.1–6.6) | 6.3 (6–6.6) | 6.2 (6–6.5) f | 6.3 (5.9–6.6) |
| CD70 | 3 (2.8–3.3) f | 3.6 (3.2–4) a | 3.4 (3.2–4.2) a | 3.7 (3.4–4) a | 3.2 (2.9–3.6) b , d , f | 3.3 (3–3.7) |
| CD93 | 9 (8.9–9.1) | 9 (8.5–9.4) | 9 (8.7–9.3) | 9.4 (9.1–9.7) a – c | 9.5 (9.2–9.8) a – c | 9 (8.8–9.4) e |
| CD160 | 4.3 (4.1–4.6) f | 4.6 (4.3–5) | 4.7 (4.3–5.2) | 4.9 (4.5–5.5) a | 4.8 (4.5–5.3) a , f | 4.7 (4.4–5.1) |
| CD163 | 6.8 (6.5–7.1) | 7 (6.5–7.4) | 7 (6.5–7.3) | 7 (6.8–7.3) | 7 (6.7–7.6) | 7.3 (6.6–7.9) |
| CD207 | 2.1 (2–2.4) f | 2.2 (2–2.5) | 2.2 (1.9–2.7) | 2.3 (2.1–2.9) | 2.4 (2.1–2.6) f | 2.1 (2–2.3) |
| CEACAM8 | 3.2 (3–3.7) | 4.1 (3.5–4.5) a | 4.3 (4–4.6) a | 4.2 (3.6–4.7) a | 4.3 (3.8–4.8) a | 3.7 (3.5–4.9) a |
| Chitotriosidase‐1 | 1.1 (0.64–2) | 2.4 (1.6–2.7) a | 2.4 (1.8–3) a | 2.8 (2.1–3.8) a | 1.9 (1.5–2.9) a , d | 2.1 (1.5–2.4) |
| Chitinase‐3‐like protein 1 | 4.3 (4–4.6) | 5.7 (5.3–6.2) a | 5.6 (5.1–6.2) a | 6 (5.6–7.2) a | 5.3 (4.6–6.4) a , d | 5.3 (4.9–6) a |
| CXCL1 | 8.3 (7.4–9.3) | 8.2 (7.5–8.9) | 8.5 (7.6–9.3) | 8.7 (7.8–9.1) | 8.2 (7.1–8.6) | 8.7 (8.1–8.9) |
| CXCL13 | 7.9 (7.6–8.3) f | 9 (8.6–9.7) a | 8.7 (8.4–9.1) a | 8.8 (8.5–9.8) a | 8.7 (8.4–9.2) a , f | 8.7 (8.4–9) a |
| CXCL16 | 5.7 (5.5–5.8) | 5.7 (5.5–6) | 5.7 (5.5–5.9) | 5.9 (5.7–6.1) a | 5.9 (5.8–6.2) a | 5.8 (5.6–6) |
| CXCL17 | 3.7 (3.2–4.5) f | 5.4 (5–5.6) a | 5.5 (5.1–6) a | 5.5 (5.1–5.7) a | 5.4 (5–5.9) a , f | 4.7 (4.2–5.4) c – e |
| E‐selectin | 1.7 (1.3–1.8) | 2.2 (1.6–2.5) a | 1.7 (1.4–2.4) | 1.7 (1.2–2.4) b | 1.7 (1.4–2.1) b | 1.6 (1.1–1.9) b |
| FcRLB | 1.1 (0.88–1.4) f | 1.6 (1.2–2) a | 1.7 (1.4–2) a | 2 (1.5–2.4) a | 2 (1.5–2.4) a , f | 1.9 (1.5–2.4) a |
| GDF‐15 | 3.2 (2.9–3.5) | 5.1 (4.6–5.8) a | 4.9 (4.4–5.3) a | 5.1 (4.6–5.6) a | 5.4 (4.5–5.9) a | 5 (4.4–5.3) a |
| ICAM‐2 | 4.1 (4–4.5) | 4.4 (4–4.8) | 4.3 (4–4.5) | 4.4 (4–4.8) | 4.6 (4.2–5) | 4.4 (4–4.7) |
| ICOS ligand | 3.4 (3.2–3.5) f | 3.3 (3.2–3.5) | 3.3 (3.2–3.5) | 3.4 (3.2–3.6) | 3.3 (3.2–3.5) f | 3.3 (3.2–3.6) |
| IFN‐gamma‐R1 | 3.4 (3.3–3.5) f | 3.5 (3.2–3.7) | 3.6 (3.3–3.7) | 3.9 (3.6–4.1) a , b | 3.8 (3.5–4.1) a , b , f | 3.6 (3.4–3.9) |
| IgG Fc receptor II‐b | 1.7 (1.4–2) | 1.6 (1–2) | 2.1 (1.4–2.3) | 1.7 (1.4–2.2) | 1.6 (0.63–2.1) | 1.6 (0.98–2.3) |
| IL‐1RL2 | 4.2 (3.8–4.6) | 4.4 (4.1–4.6) | 4.5 (4.4–5) | 4.6 (4.3–4.7) | 4.4 (4.2–4.7) | 4.4 (4.3–4.8) |
| IL‐1RN | 3.3 (3–3.9) | 4.2 (3.9–5) a | 4.6 (3.9–5.2) a | 4.2 (4–4.7) a | 4.2 (3.8–4.7) a | 4.2 (3.4–5) a |
| IL‐1RT1 | 6 (5.9–6.1) | 6.3 (5.9–6.5) | 6.1 (5.9–6.2) | 6.2 (5.9–6.5) | 6.4 (6.2–6.7) a – c | 6.2 (5.9–6.7) |
| IL‐1RT2 | 4.4 (4.2–4.6) | 4.2 (3.9–4.4) a | 4.2 (4–4.4) | 4.5 (4.2–4.8) b , c | 4.6 (4.3–4.9) b , c | 4.4 (4.3–4.6) |
| IL‐2‐RA | 3.3 (3.2–3.5) | 3.7 (3.1–4.2) | 3.4 (3.2–4) | 3.8 (3.5–4.4) a , c | 3.7 (3.3–4.2) | 3.5 (3.1–3.9) |
| IL‐4R‐alpha | 2.2 (2.1–2.5) | 2.9 (2.5–3.4) a | 3 (2.7–3.2) a | 3.1 (2.8–3.6) a | 3.2 (2.9–3.4) a | 3 (2.8–3.4) a |
| IL‐6 | 1.7 (1.2–2.2) f | 3.4 (2.6–4.5) a | 2.7 (2–3.7) a | 3.5 (2.6–4.3) a | 3.7 (3–4.4) a , f | 3.3 (2.2–3.9) a |
| IL‐6R‐alpha | 10 (10–11) | 10 (10–11) | 10 (10–11) | 10 (10–11) | 10 (9.9–11) | 11 (10–11) |
| pro‐IL‐16 | 5.1 (4.4–5.2) | 5.1 (4.8–5.7) a | 5.6 (5.3–5.7) a | 5.6 (5.4–6) a , b | 5.3 (5–5.8) a | 5.4 (5.1–5.7) a |
| IL‐17D | 2.2 (2.1–2.4) | 2.6 (2.4–2.8) a | 3 (2.7–3.1) a , b | 2.9 (2.8–3) a , b | 2.8 (2.6–3.1) a , b | 2.8 (2.2–3.1) a |
| IL‐17RA | 3.6 (3.2–3.8) | 3.6 (3–3.9) | 3.6 (3.4–3.9) | 3.7 (3.3–4) | 3.7 (3.2–4.1) | 3.7 (3.3–4) |
| IL‐18 | 8.2 (7.7–8.4) | 8.4 (8–8.9) | 8.4 (8.1–8.9) | 8.5 (8.1–8.8) | 8.5 (8.1–8.7) | 8.7 (8.1–8.9) |
| IL‐18BP | 5.5 (5.4–5.7) | 5.9 (5.3–6.3) | 5.7 (5.5–6) | 6.2 (5.7–6.4) a | 5.7 (5.4–6.2) | 5.9 (5.5–6.2) |
| IL‐27 | 3.7 (3.6–3.9) | 4.3 (4–4.6) a | 4.5 (4.1–5) a | 4.5 (4.2–4.9) a | 4.6 (4.3–5) a , b | 4.5 (4–4.8) a |
| JAM‐A | 6.1 (5.3–6.5) | 5.7 (4.8–6.6) | 5.8 (5.4–6.2) | 6.2 (5.8–6.8) | 5.9 (5.5–6.4) | 6.4 (5.7–7.1) |
| KIM‐1 | 7.7 (7–8.1) | 9 (8.2–9.6) a | 9.6 (9.1–10) a | 9.6 (9.3–11) a , b | 8.9 (8.1–9.8) a , d | 9.5 (9.4–9.9) a |
| Ly‐9 | 4.7 (4.6–4.9) f | 4.9 (4.7–5.1) | 4.9 (4.7–5.2) | 4.9 (4.7–5.3) | 4.9 (4.7–5.1) f | 4.9 (4.6–5) |
| Lymphotactin | 4.6 (4.4–4.9) | 5.1 (4.7–5.7) | 5.7 (5.5–5.9) a , b | 5.9 (5.3–6.3) a , b | 5.8 (5.3–6.3) a , b | 5.3 (5.2–5.6) a |
| MARCO | 5.9 (5.8–6) | 6 (5.8–6.2) | 6.2 (6–6.4) a , b | 6.3 (6.1–6.5) a , b | 6.1 (5.9–6.2) d | 6.3 (6.1–6.4) a , b |
| MCP‐1 | 2.2 (2–2.4) | 2.8 (2.4–3) a | 2.9 (2.5–3.1) a | 2.8 (2.5–3.2) a | 2.6 (2.4–2.8) a | 2.6 (2.4–2.7) a |
| MIC‐A/B | 4 (3.4–4.2) f | 4.1 (3.6–4.5) | 3.9 (3.6–4.4) | 4.1 (3.8–4.6) | 4.3 (4–4.8) f | 3.8 (3.6–4.8) |
| Myeloperoxidase | 3.5 (3.4–3.7) | 3.6 (3.2–3.8) | 3.6 (3.4–4) | 3.6 (3.4–3.9) | 3.6 (3.5–3.9) | 3.6 (3.2–3.9) |
| PECAM‐1 | 5.4 (4.9–5.7) | 5.2 (4.5–5.8) | 5.1 (4.9–5.6) | 5.4 (5.1–5.8) | 5.2 (4.9–5.5) | 5.6 (5.1–5.8) |
| Pglyrp1 | 6.5 (6.2–6.7) | 6.8 (6.5–7.2) a | 7.1 (6.8–7.3) a | 7 (6.8–7.5) a | 6.8 (6.5–7.4) a | 6.9 (6.4–7.3) a |
| PIgR | 5.6 (5.5–5.7) | 5.8 (5.7–5.9) a | 5.9 (5.8–6) a | 5.8 (5.7–5.9) a | 5.9 (5.8–5.9) a | 5.8 (5.7–5.9) a |
| Progranulin | 3.2 (3–3.3) | 3.3 (3–3.6) | 3.2 (3.1–3.6) | 3.4 (3.2–3.6) | 3.6 (3.2–3.8) a , b | 3.4 (3.2–3.9) |
| P‐selectin | 9.3 (9–10) | 9.3 (8.7–9.8) | 9.5 (9–10) | 9.4 (8.8–9.9) | 9.2 (8.8–9.6) | 9.5 (9–10) |
| PSGL‐1 | 4.3 (4.2–4.5) | 4.3 (4.1–4.5) | 4.4 (4.3–4.6) | 4.3 (4.2–4.6) | 4.4 (4.2–4.4) | 4.3 (4.2–4.6) |
| PSP‐D | 1.6 (1.4–2.4) | 2.8 (2–3.6) a | 2.6 (2.1–3.2) a | 2.4 (1.8–2.9) | 2.9 (2.3–3.3) a | 2.1 (1.3–2.4) b , c , e |
| PTX3 | 2.7 (2.2–2.8) | 3.3 (3–3.6) a | 3.2 (2.8–3.6) a | 3 (2.6–3.4) | 3.2 (2.7–3.5) a | 3 (2.7–3.6) |
| SLAMF5 | 5.3 (5.1–5.7) | 5.1 (4.7–5.4) | 5.3 (5.1–5.7) | 5.4 (5.2–5.8) b | 5.2 (4.9–5.6) | 5.4 (5.1–5.7) b |
| SLAMF7 | 1.3 (1.1–2) | 2.1 (1.6–2.5) a | 2.2 (1.8–2.6) a | 2.2 (1.8–2.6) a | 2.1 (1.9–2.5) a | 2.2 (2.1–2.5) a |
| TLR3 | 5.1 (4.7–5.4) f | 4.7 (4.3–5.1) | 5.2 (4.2–5.6) | 4.8 (4.4–5.4) | 5.3 (4.7–5.6) b , f | 5.2 (4.9–5.8) b |
| TLT‐2 | 4 (3.8–4.3) | 3.8 (3.2–4.3) | 3.8 (3.5–4.2) | 4 (3.7–4.4) | 3.8 (3.5–4.1) | 3.9 (3.6–4.4) |
CTEPH, chronic thrombo‐embolic pulmonary hypertension; HF‐NON‐PH, heart failure without pulmonary hypertension; HFpEF‐PH, pulmonary hypertension due to heart failure with preserved ejection fraction; HFrEF‐PH, pulmonary hypertension due to heart failure with reduced ejection fraction; PAH, pulmonary arterial hypertension.
Variables are presented in arbitrary units (AU) as median (inter‐quartile range). Proteins' abbreviations are found in methods. Kruskal–Wallis tests were performed to analyse the differences between the groups. Statistical significance was considered P < 0.016, after correction for multiple comparisons with Benjamini and Hochberg false discovery rate (Q = 5%).
Significant difference compared with controls.
Significant difference compared with PAH.
Significant difference compared with CTEPH.
Significant difference compared with HFpEF‐PH.
Significant difference compared with HFrEF‐PH.
Indicates n − 1.
Discussion
Proteoglycans are of critical importance for ECM interaction, 21 which in addition to a dysregulated inflammatory response constitute factors that may affect the pathobiology of PAH including endothelial dysfunction and vascular remodelling. 9 , 10 We, therefore, investigated whether plasma proteoglycans and inflammatory proteins differentiated PAH from other causes of PH and dyspnoea. The present study indicates that prolargin potentially could be used in a future multi‐marker panel, to in a dyspnoea population differentiate PAH patients from CTEPH, HFrEF, and HFpEF with or without PH, as well as controls. These findings are of interest a such plasma measurements could be used to enable earlier PAH diagnosis and treatment initiation, before the onset of right‐sided HF, with the aim to improve outcome and prognosis in PAH.
Prolargin is a small leucine‐rich repeat proteoglycan expressed in the ECM of collagen‐rich tissues such as skin, sclera, tendon, lung, and heart. 22 Prolargin mediates cell adhesion to the ECM by binding cell surface glycosaminoglycans 23 and is an inhibitor of the complement system. 24 Inflammation and dysregulated immunity are important in pulmonary endothelial dysfunction and vascular remodelling in PAH and PH. 10 In addition, inflammation and HF are closely interconnected and reinforce each other, thereby creating a vicious circle. 25 The complement system has been implicated in the development of PAH, with increased C3d deposition in the lungs of IPAH patients and a hypoxia mouse model. In contrast, C3 deficiency attenuated PAH in a mouse model of chronic hypoxia‐induced PAH. 26 The present study demonstrated increased levels of prolargin in PH, as well as HF compared with controls, an increase that may be in response to inflammatory processes. Prolargin inhibits the formation of the membrane attack complex by decreasing C9 polymerization, as well as inhibits the alternative pathway by interacting with C3. 24 Accordingly, prolargin is suggested to down‐regulate and limit the pathological activation in inflammatory diseases such as rheumatoid arthritis. 24 Thus, inflammation is a plausible driver for the increase of prolargin. Whether the lesser increase of prolargin in PAH relative to the other disease groups mean that the inflammation is of a lower grade or involve other pathways in PAH remains uncertain.
Prolargin levels may rise in response to increased complement and/or inflammation, acting as a negative regulator of complement activation. The greater levels of prolargin in HF with or without PH, as well as CTEPH, compared with PAH, may thus indicate greater inflammatory activity in these groups leading to higher corresponding levels of prolargin. The complement system is dysregulated in HF with an overactivated alternative complement pathway 27 and increased levels of membrane attack complexes. 28 On the other hand, the lower levels of prolargin in PAH vs. the other disease groups may represent aberrant signalling in PAH with less complement inhibition, thus maintaining and increasing the inflammatory response.
The present findings suggest that prolargin potentially could be a useful addition to current diagnostic tools, for differentiation of PAH, with good sensitivity and specificity, from the other causes of dyspnoea, such as CTEPH, HFpEF‐PH, HFrEF‐PH, and HF‐NON‐PH. However, the intermediate plasma prolargin levels between healthy controls and the other disease groups might not be ideal and may limit its potential as a sole discriminator of PAH. To further evaluate the usefulness of prolargin, future ‘deep phenotyping’, combining patterns of several proteins may be a viable route to increase the combined accuracy in PAH differentiation. 6 Our correlation analyses indicate that increased prolargin levels are associated with worsening right heart function. Prolargin correlated positively with MRAP and NT‐proBNP and negatively with 6MWD and SvO2. In the 2015 European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines risk assessment tool, high levels of NT‐proBNP, low 6MWD, high MRAP, and low SvO2 are separate parameters indicating an increased risk of mortality in PAH. 4 Coupled with the negative correlation with SVI and RVSWI and no correlation with PVR, the last, a parameter that, however, is not included in the ESC/ERS guidelines risk score, it may suggest that prolargin levels partially reflect decompensation, as the haemodynamic parameters included in the ESC/ERS guidelines risk score also in part reflect the state of right‐sided dysfunction. 2 , 4 Prolargin may therefore possibly be useful in the context of PAH risk assessment as a marker of disease progression and may potentially offer new insights in the inflammatory processes in PAH development. As we found that prolargin levels were higher in the studied HF groups compared with PAH, and correlated with haemodynamics related to decompensation, we hypothesized that the increase in prolargin may reflect PAWP. However, the levels of prolargin were not consistent in the precapillary groups; that is, there was a difference between PAH and CTEPH, and no difference was observed between CTEPH and the postcapillary groups. This suggests that PAWP, related to volume overload and pulmonary congestion, 29 does not account for the increase in prolargin.
In the present study, we additionally analysed 65 inflammatory plasma proteins and found that most of these proteins were in general elevated compared with controls. However, this increase was in an unspecific fashion in relation to the various causes of dyspnoea. IL‐17D displayed a pattern similar to prolargin, with intermediate levels between the controls and the other PH groups. However, there was no difference between the levels of plasma IL‐17D in PAH and HF‐NON‐PH. IL‐17D is a member of the IL‐17 cytokine family, comprising six factors (IL‐17A–IL‐17F). 30 Although highly unexplored, it has been shown that IL‐17D stimulates the production of GM‐CSF, IL‐6, and IL‐8. 31 In the present study, we found a general increase in plasma IL‐6 levels across all disease groups including, but not specific to, PAH. Interestingly, excessive local secretion of IL‐6 was proposed among other cytokines to have a role in influencing the structural and functional changes in the pulmonary vasculature in PAH. 10 A Phase II study of the IL‐6 inhibitor tocilizumab in PAH patients did not demonstrate an overall change in PVR, but a potential modest effect was found in some patients with CTD‐PAH. The study was, however, underpowered. 32 , 33 A subsequent study found that serum IL‐6 levels were most abundant in CTD‐PAH and portopulmonary hypertension‐associated PAH compared with IPAH, and high levels of IL‐6 were observed in pulmonary arterial smooth muscle cells from PAH patients. 34 Hence, in line with the authors' conclusion, a more specified selection pre‐intervention with IL‐6 inhibitors could be achieved by enrolling patients with high IL‐6 levels. 34 In fact, a similar approach has previously been applied in a randomized controlled trial investigating the role of canakinumab, an IL‐1β inhibitor, which included the use of high‐sensitivity C‐reactive protein levels as an eligibility criterion for enrolment. 35 In the present study, the levels of the inflammatory proteins were studied at the diagnostic RHC, and thus, the relation between the studied dyspnoea groups may differ if measured at different disease stages. Taken together, we speculate that IL‐17D may, in part, contribute to the pathobiology of PAH by increasing IL‐6 production. However, this increase seems not specific to PAH as we observed a general increase in IL‐17D in all dyspnoea groups (Figure 3A ). Given the paucity of studies on IL‐17D, its role in PAH, PH, and HF remains to be elucidated.
Decorin is a proteoglycan synthesized by fibroblasts, stressed vascular endothelial cells, and smooth muscle cells. It has bifunctional properties acting as both a structural ECM component and a signalling molecule. Decorin interacts with receptor tyrosine kinases like epidermal growth factor receptor, insulin‐like growth factor 1 receptor, vascular endothelial growth factor receptor 2, and innate immunity receptors. 22 Decorin furthermore inhibits transforming growth factor‐β and connective tissue growth factor, counteracting the profibrotic effects and ECM accumulation. 15 , 36 In addition, decorin has antiproliferative, antitumorigenic, and proapoptotic functions via interaction with epidermal growth factor receptor signalling. 22 Circulating decorin levels have been shown to increase during inflammation in sepsis patients. 37 Additionally, decorin plays a role in down‐regulating levels of ICAM‐1 and syndecan‐1. 38 ICAM‐1 is central for leucocyte migration into tissues, 39 and increased levels of decorin may, therefore, counteract inflammation and remodelling. Although plasma decorin in the present study did not differentiate PAH patients from CTEPH, it differentiated PAH from HFpEF‐PH and HFrEF‐PH, as well as HF without PH. Decorin could, therefore, be a potential new differentiator between those causes of dyspnoea, as HFpEF‐PH and PAH, sometimes may be difficult to differentiate from each other. 40
Syndecan‐1 is part of the glycocalyx on endothelial cell surfaces and is required for endothelial cell remodelling in response to flow. 16 Syndecan‐1 is implicated in cardiac fibrosis and is associated with worse clinical outcomes in HFrEF. 41 This is in line with our results where syndecan‐1 levels were higher in HFrEF‐PH than in all the other disease groups. However, contrary to our results, Nijst et al. did not find any difference in syndecan‐1 levels between stable or decompensated HFrEF and healthy controls, which they speculated may be due to a wide distribution of syndecan‐1 in their healthy subjects. 42 We have recently found in another study that increased levels of syndecan‐1 in HF patients normalized towards the healthy controls' levels after heart transplantation. 43 The present study demonstrated an increase of syndecan‐1 across the PH groups with the highest levels in HFrEF‐PH. This suggests that the increased levels of syndecan‐1 may, in part, be attributable to pulmonary vasculature congestion or structural changes. In addition, this pattern indicates that plasma syndecan‐1 may be a future biomarker to identify HFrEF‐PH, which warrants future larger investigations.
The use of PEA allows for precise plasma measurements of different proteins with little cross‐reactivity.18 Biomarkers' levels were measured in patients naïve to PAH specific treatment and thus without the interference of PAH drugs. In the present study, we investigated circulating protein levels. Whether these truly reflect local levels in the tissues and/or remodelling in the pulmonary vasculature or elsewhere were not investigated but need further attention in the future. Even though our analyses were corrected with FDR to limit mass significance, false‐positive results may be present. However, as the aim of our study was to investigate the potential value of proteoglycans and inflammatory proteins as biomarkers, some uncertainty is allowed. The present study is limited by the relatively small patient population, and control group, the latter also somewhat younger than the disease groups. To address this issue, we investigated whether there was an association between prolargin and age in each group and compared the groups for consistency. However, no consistent pattern of correlation was found between prolargin and age in the groups studied. With a median age at diagnosis of 71.5 years, the PAH population was somewhat older than anticipated. It is however consistent with the Swedish Pulmonary Arterial Hypertension Register as well as the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension, which reported median ages of 67 and 71 years at diagnosis of IPAH, respectively. 44 , 45
Conclusion
In conclusion, the present study demonstrates that prolargin may be a new potential biomarker for differentiation of PAH. Its discriminating potential of PAH, with a sensitivity of 74% and a specificity of 83.3%, from the other dyspnoea related aetiologies CTEPH, HFpEF‐PH, HFrEF‐PH, and HF without PH, warrants further investigation and could be enhanced by prolargin being a part of a multi‐marker panel to allow for future ‘deep phenotyping’ of PAH patients. Prolargin may also influence and/or be a potential marker of impaired heart function, indicated by its associations with MRAP, NT‐proBNP, CI, and SVI. Future directions of research could be to investigate the change of prolargin levels in PAH and HF over time, as well as its potential in PAH risk assessment and pathobiology. Importantly, larger studies using matched controls are needed to evaluate the definite use of prolargin as a marker for PAH in a broader population, also involving diseases other than PH, which exhibit dyspnoea.
Conflict of interest
M.A. reports an unrestricted research grant from the Swedish Society of Pulmonary Hypertension. A.A. reports no conflicts of interest. H.B. reports personal lecture fees from Actelion Pharmaceuticals Sweden AB. H.B. has received unrestricted research grants from the Swedish Society of Pulmonary Hypertension on behalf of GlaxoSmithKline. G.R. reports personal lecture fees from Actelion Pharmaceuticals, Bayer Healthcare, GlaxoSmithKline, and Nordic Infucare outside the submitted work. G.R. has received unrestricted research grants from Actelion Pharmaceuticals and GlaxoSmithKline. G.R. is and has been primary investigator or co‐investigator in clinical PAH trials for GlaxoSmithKline, Actelion Pharmaceuticals, Pfizer, Bayer, and United Therapeutics and in clinical heart transplantation immunosuppression trials for Novartis.
Funding
This work was supported by unrestricted research grants from the Swedish Society of Pulmonary Hypertension, ‘Avtal om Läkarutbildning och Forskning’ (ALF), and Actelion Pharmaceuticals AB. The foundations had no role in the literature review selection, analysis, and interpretation of the data or publication of the manuscript.
Supporting information
Figure S1. Protein levels in disease groups and controls. PAH: Pulmonary arterial hypertension; CTEPH: Chronic thromboembolic pulmonary hypertension (PH); HFpEF‐PH: PH due to heart failure (HF) with preserved ejection fraction (EF); HFrEF‐PH: PH due to HF with reduced EF; HF‐NON‐PH: HF without PH; MEPE: Matrix extracellular phosphoglycoprotein; VE‐cadherin: Vascular endothelial cadherin; AU: Arbitrary units; Kruskal‐Wallis tests with multiple comparisons, significant difference after Benjamini and Hochberg FDR analysis (Q = 5%) shown as * p<0.021; ** p<0.01; *** p<0.001.
Table S1. P‐values of significant Kruskal‐Wallis and multiple comparisons
Acknowledgements
We acknowledge the support of the staff at The Hemodynamic Lab, The Section for Heart Failure and Valvular Disease, VO Heart and Lung Medicine, Skåne University Hospital, Lund, Sweden, and The Department of Clinical Sciences, Lund, Cardiology, Lund University, Lund, Sweden. Special thanks to Anneli Ahlqvist for support in assembling plasma samples and LCPR registration. In addition, we acknowledge the biobank services and retrieval of blood samples from LCPR performed at Lab medicine Skåne, University, and Regional Laboratories, Region Skåne, Sweden.
Arvidsson, M. , Ahmed, A. , Bouzina, H. , and Rådegran, G. (2021) Plasma proteoglycan prolargin in diagnosis and differentiation of pulmonary arterial hypertension. ESC Heart Failure, 8: 1230–1243. 10.1002/ehf2.13184.
References
- 1. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: S13–S24. [DOI] [PubMed] [Google Scholar]
- 2. Konstam Marvin A, Kiernan Michael S, Bernstein D, Bozkurt B, Jacob M, Kapur Navin K, Kociol Robb D, Lewis Eldrin F, Mehra Mandeep R, Pagani Francis D, Raval Amish N, Ward C. Evaluation and management of right‐sided heart failure: a scientific statement from the American Heart Association. Circulation 2018; 137: e578–e622. [DOI] [PubMed] [Google Scholar]
- 3. D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT, Levy PS, Pietra GG, Reid LM, Reeves JT, Rich S, Vreim CE, Williams GW, Wu M. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991; 115: 343–349. [DOI] [PubMed] [Google Scholar]
- 4. Galiè N, Humbert M, Vachiery J‐L, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37: 67–119. [DOI] [PubMed] [Google Scholar]
- 5. Humbert M, Gerry Coghlan J, Khanna D. Early detection and management of pulmonary arterial hypertension. Eur Respir Rev 2012; 21: 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frost A, Badesch D, Gibbs JSR, Gopalan D, Khanna D, Manes A, Oudiz R, Satoh T, Torres F, Torbicki A. Diagnosis of pulmonary hypertension. Eur Respir J 2019; 53: 1801904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu J, Shi G‐P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim Biophys Acta 2014; 1842: 2106–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Botney MD, Liptay MJ, Kaiser LR, Cooper JD, Parks WC, Mecham RP. Active collagen synthesis by pulmonary arteries in human primary pulmonary hypertension. Am J Pathol 1993; 143: 121–129. [PMC free article] [PubMed] [Google Scholar]
- 9. Thenappan T, Chan SY, Weir EK. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2018; 315: H1322–H1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2019; 53: 1801887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arvidsson M, Ahmed A, Bouzina H, Rådegran G. Matrix metalloproteinase 7 in diagnosis and differentiation of pulmonary arterial hypertension. Pulm Circ 2019; 9: 2045894019895414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol 2014; 15: 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schaefer L. Proteoglycans, key regulators of cell–matrix dynamics. Matrix Biol 2014; 35: 1–2. [DOI] [PubMed] [Google Scholar]
- 14. Christensen G, Herum KM, Lunde IG. Sweet, yet underappreciated: proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol 2019; 75‐76: 286–299. [DOI] [PubMed] [Google Scholar]
- 15. Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF‐β‐mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol 2001; 280: L1327–L1334. [DOI] [PubMed] [Google Scholar]
- 16. Ebong EE, Lopez‐Quintero SV, Rizzo V, Spray DC, Tarbell JM. Shear‐induced endothelial NOS activation and remodeling via heparan sulfate, glypican‐1, and syndecan‐1. Integr Biol 2014; 6: 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaday GG, Lider O. Extracellular matrix moieties, cytokines, and enzymes: dynamic effects on immune cell behavior and inflammation. J Leukoc Biol 2000; 67: 149–159. [DOI] [PubMed] [Google Scholar]
- 18. Assarsson E, Lundberg M, Holmquist G, Björkesten J, Bucht Thorsen S, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, Andersson A‐C, Lindstedt P, Stenvang J, Gullberg M, Fredriksson S. Homogenous 96‐plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014; 9: e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 1995; 57: 289–300. [Google Scholar]
- 20. Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat 2001; 29: 1165–1188. [Google Scholar]
- 21. Takawale A, Sakamuri SS, Kassiri Z. Extracellular matrix communication and turnover in cardiac physiology and pathology. Compr Physiol 2015; 5: 687–719. [DOI] [PubMed] [Google Scholar]
- 22. Hultgardh‐Nilsson A, Boren J, Chakravarti S. The small leucine‐rich repeat proteoglycans in tissue repair and atherosclerosis. J Intern Med 2015; 278: 447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bengtsson E, Lindblom K, Tillgren V, Aspberg A. The leucine‐rich repeat protein PRELP binds fibroblast cell‐surface proteoglycans and enhances focal adhesion formation. Biochem J 2016; 473: 1153–1164. [DOI] [PubMed] [Google Scholar]
- 24. Happonen KE, Furst CM, Saxne T, Heinegard D, Blom AM. PRELP protein inhibits the formation of the complement membrane attack complex. J Biol Chem 2012; 287: 8092–8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Linthout S, Tschope C. Inflammation—cause or consequence of heart failure or both? Curr Heart Fail Rep 2017; 14: 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bauer EM, Zheng H, Comhair S, Erzurum S, Billiar TR, Bauer PM. Complement C3 deficiency attenuates chronic hypoxia‐induced pulmonary hypertension in mice. PLoS ONE 2011; 6: e28578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shahini N, Michelsen AE, Nilsson PH, Ekholt K, Gullestad L, Broch K, Dahl CP, Aukrust P, Ueland T, Mollnes TE, Yndestad A, Louwe MC. The alternative complement pathway is dysregulated in patients with chronic heart failure. Sci Rep 2017; 7: 42532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lappegård KT, Garred P, Jonasson L, Espevik T, Aukrust P, Yndestad A, Mollnes TE, Hovland A. A vital role for complement in heart disease. Mol Immunol 2014; 61: 126–134. [DOI] [PubMed] [Google Scholar]
- 29. Malfatto G, Blengino S, Perego GB, Branzi G, Villani A, Facchini M, Parati G. Transthoracic impedance accurately estimates pulmonary wedge pressure in patients with decompensated chronic heart failure. Congest Heart Fail 2012; 18: 25–31. [DOI] [PubMed] [Google Scholar]
- 30. Kolls JK, Lindén A. Interleukin‐17 family members and inflammation. Immunity 2004; 21: 467–476. [DOI] [PubMed] [Google Scholar]
- 31. Liu X, Sun S, Liu D. IL‐17D: a less studied cytokine of IL‐17 family. Int Arch Allergy Immunol 2020; 181: 618–623. [DOI] [PubMed] [Google Scholar]
- 32. Toshner M, Rothman AMK. IL‐6 in pulmonary hypertension: why novel is not always best. Eur Respir J 2020; 55: 2000314. [DOI] [PubMed] [Google Scholar]
- 33. Hernández‐Sánchez J, Harlow L, Church C, Gaine S, Knightbridge E, Bunclark K, Gor D, Bedding A, Morrell N, Corris P, Toshner M. Clinical trial protocol for TRANSFORM‐UK: a therapeutic open‐label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm Circ 2017; 8: 2045893217735820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simpson CE, Chen JY, Damico RL, Hassoun PM, Martin LJ, Yang J, Nies M, Griffiths M, Vaidya RD, Brandal S, Pauciulo MW, Lutz KA, Coleman AW, Austin ED, Ivy DD, Nichols WC, Everett AD. Cellular sources of interleukin‐6 and associations with clinical phenotypes and outcomes in pulmonary arterial hypertension. Eur Respir J 2020; 55: 1901761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida‐Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017; 377: 1119–1131. [DOI] [PubMed] [Google Scholar]
- 36. Vial C, Gutierrez J, Santander C, Cabrera D, Brandan E. Decorin interacts with connective tissue growth factor (CTGF)/CCN2 by LRR12 inhibiting its biological activity. J Biol Chem 2011; 286: 24242–24252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Merline R, Moreth K, Beckmann J, Nastase MV, Zeng‐Brouwers J, Tralhao JG, Lemarchand P, Pfeilschifter J, Schaefer RM, Iozzo RV, Schaefer L. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA‐21. Sci Signal 2011; 4: ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seidler DG, Mohamed NA, Bocian C, Stadtmann A, Hermann S, Schafers K, Schafers M, Iozzo RV, Zarbock A, Gotte M. The role for decorin in delayed‐type hypersensitivity. J Immunol 2011; 187: 6108–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lawson C, Wolf S. ICAM‐1 signaling in endothelial cells. Pharmacol Rep 2009; 61: 22–32. [DOI] [PubMed] [Google Scholar]
- 40. Rosenkranz S, Gibbs JSR, Wachter R, De Marco T, Vonk‐Noordegraaf A, Vachiéry J‐L. Left ventricular heart failure and pulmonary hypertension. Eur Heart J 2016; 37: 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tromp J, van der Pol A, Klip IT, de Boer RA, Jaarsma T, van Gilst WH, Voors AA, van Veldhuisen DJ, van der Meer P. Fibrosis marker syndecan‐1 and outcome in patients with heart failure with reduced and preserved ejection fraction. Circ Heart Fail 2014; 7: 457–462. [DOI] [PubMed] [Google Scholar]
- 42. Nijst P, Cops J, Martens P, Swennen Q, Dupont M, Tang WHW, Mullens W. Endovascular shedding markers in patients with heart failure with reduced ejection fraction: results from a single‐center exploratory study. Microcirculation 2018; 25: e12432. [DOI] [PubMed] [Google Scholar]
- 43. Ahmed A, Ahmed S, Arvidsson M, Bouzina H, Lundgren J, Rådegran G. Prolargin and matrix metalloproteinase‐2 in heart failure after heart transplantation and their association with haemodynamics. ESC heart failure 2020; 7: 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rådegran G, Kjellström B, Ekmehag B, Larsen F, Rundqvist B, Blomquist SB, Gustafsson C, Hesselstrand R, Karlsson M, Kornhall B, Nisell M, Persson L, Ryftenius H, Selin M, Ullman B, Wall K, Wikström G, Willehadson M, Jansson K. Characteristics and survival of adult Swedish PAH and CTEPH patients 2000–2014. Scand Cardiovasc J 2016; 50: 243–250. [DOI] [PubMed] [Google Scholar]
- 45. Hoeper MM, Huscher D, Ghofrani HA, Delcroix M, Distler O, Schweiger C, Grunig E, Staehler G, Rosenkranz S, Halank M, Held M, Grohé C, Lange TJ, Behr J, Klose H, Wilkens H, Filusch A, Germann M, Ewert R, Seyfarth H‐J, Olsson KM, Opitz CF, Gaine SP, Vizza CD, Vonk‐Noordegraaf A, Kaemmerer H, Gibbs JSR, Pittrow D. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol 2013; 168: 871–880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Protein levels in disease groups and controls. PAH: Pulmonary arterial hypertension; CTEPH: Chronic thromboembolic pulmonary hypertension (PH); HFpEF‐PH: PH due to heart failure (HF) with preserved ejection fraction (EF); HFrEF‐PH: PH due to HF with reduced EF; HF‐NON‐PH: HF without PH; MEPE: Matrix extracellular phosphoglycoprotein; VE‐cadherin: Vascular endothelial cadherin; AU: Arbitrary units; Kruskal‐Wallis tests with multiple comparisons, significant difference after Benjamini and Hochberg FDR analysis (Q = 5%) shown as * p<0.021; ** p<0.01; *** p<0.001.
Table S1. P‐values of significant Kruskal‐Wallis and multiple comparisons