

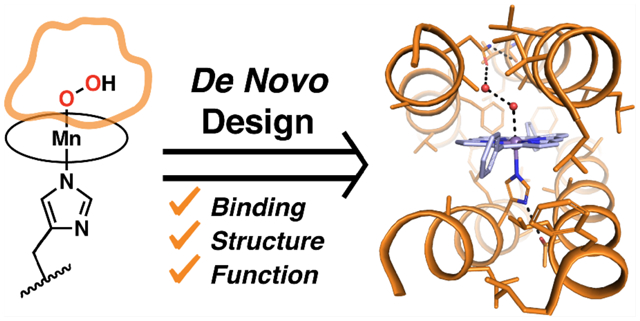
Abstract
De novo protein design offers the opportunity to test our understanding of how metalloproteins perform difficult transformations. Attaining high-resolution structural information is critical to understanding how such designs function. There have been many successes in the design of porphyrin-binding proteins; however, crystallographic characterization has been elusive, limiting what can be learned from such studies as well as the extension to new functions. Moreover, formation of highly oxidizing high-valent intermediates poses design challenges that have not been previously implemented: (1) purposeful design of substrate/oxidant access to the binding site and (2) limiting deleterious oxidation of the protein scaffold. Here we report the first crystallographically characterized porphyrin-binding protein that was programmed to not only bind a synthetic Mn–porphyrin but also maintain binding site access to form high-valent oxidation states. We explicitly designed a binding site with accessibility to dioxygen units in the open coordination site of the Mn center. In solution, the protein is capable of accessing a high-valent Mn(V)–oxo species which can transfer an O atom to a thioether substrate. The crystallographic structure is within 0.6 Å of the design and indeed contained an aquo ligand with a second water molecule stabilized by hydrogen bonding to a Gln side chain in the active site, offering a structural explanation for the observed reactivity.
Graphical Abstract
INTRODUCTION
Hemes are one of the most widely used and versatile cofactors employed in biology.1–3 Nature has evolved a variety of heme-binding protein scaffolds to perform a broad range of functions, such as ligand sensing and transport, electron transfer, protection from reactive oxygen species, and substrate oxidation. Free heme, without the aid of a protein scaffold, is prone to aggregation and has drastically reduced functionality. On the other hand, synthetic chemists have developed a massive library of metalloporphyrins to recapitulate the reactions performed in nature (albeit often less selectively and at slower rates) but also to perform reactions not found in nature, such as cyclopropanation and amination.2,4,5 While directed evolution has produced engineered heme proteins with impressive rates and selectivities for abiological reactions,6,7 de novo protein design offers the opportunity to test our understanding of protein structure–function relationships.8–12 A complete understanding of how proteins tune the function of heme cofactors combined with novel reactivity of synthetic cofactors would allow for the design of functional metalloproteins that can expand upon the scope of natural proteins.10
The de novo design of metalloproteins has seen many successes,8–10,13 but the ability to design cofactor-binding proteins with sub-Angstrom accuracy, without the need for evolution or experimental optimization of the scaffold, has only recently been achieved.14 De novo heme-binding helical bundles have been designed from first-principles, including maquettes10,15,16 and binary-patterned artificial superfamilies;17 however, these have not been successfully structurally characterized at high resolution. Although they have shown the ability to tune redox potentials of heme cofactors and can perform a limited range of peroxidase chemistry,15,17–20 maximizing the utility of these designed heme-binding proteins through further design refinements has been limited by the inability to acquire high-resolution structural information—providing function without structure. In an effort to address this issue of structural nonuniqueness, our group employed a strategy used by natural cofactor-binding proteins: a four-helix bundle was computationally designed to bind a photoreducible Zn porphyrin ((CF3)4PZn) (Figures 1and 2) adjacent to a well-packed distal core to ensure a well-packed, structurally unique protein. This led to a high-resolution NMR structure of PS1 showing sub-Angstrom accuracy compared to the design. However, this work was conducted using Zn(II) porphyrin, selected for photophysical applications, and therefore containing a redox-inactive metal ion.14
Figure 1.
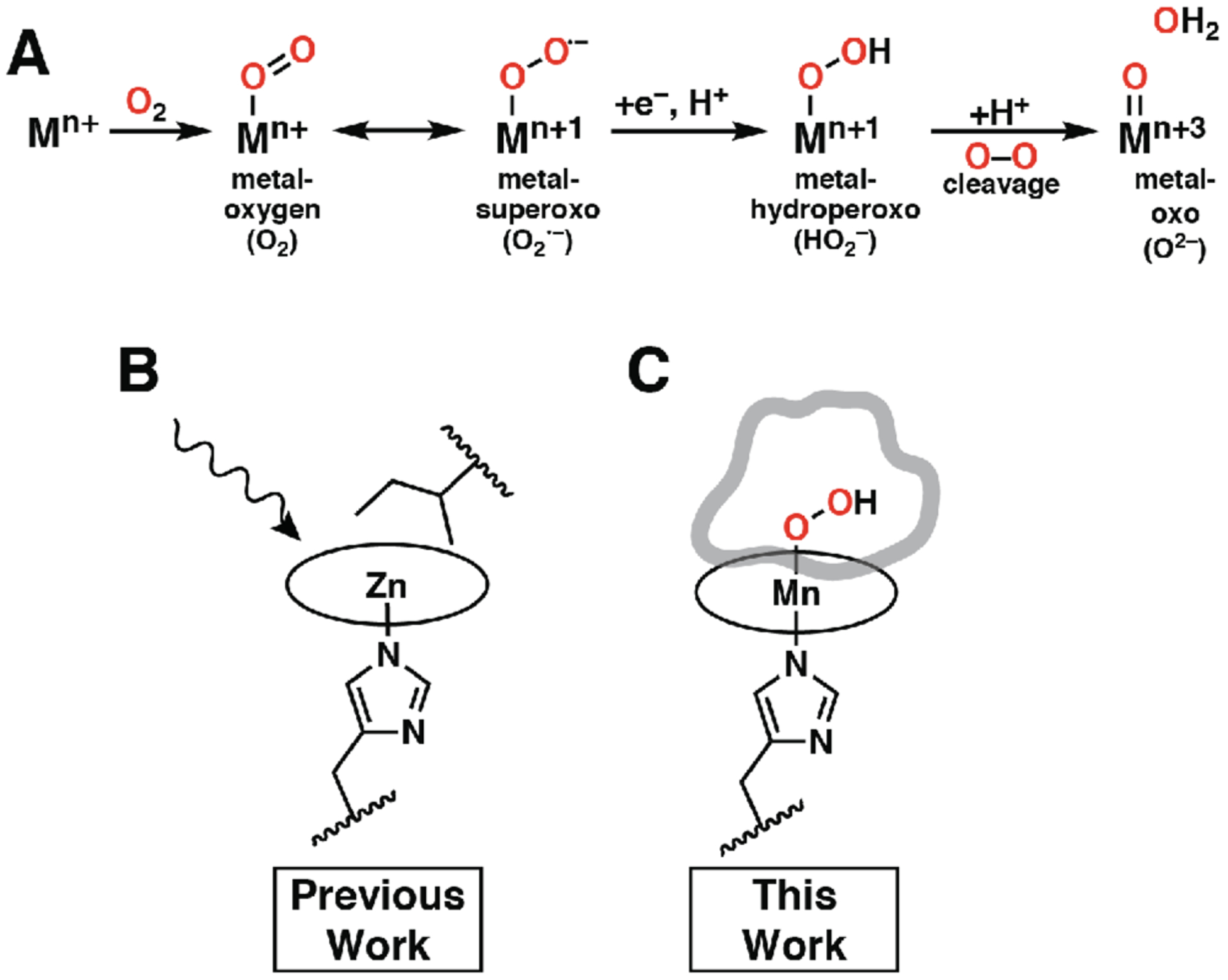
(A) Common metal–oxygen intermediates utilized in heme proteins. Illustrations of designed function used for (B) PS1 and (C) MPP1. PS1 was designed to undergo photophysical function; therefore, no binding site access was required, and an Ile residue was placed above the distal face of the porphyrin to maximize packing with the cofactor. The binding site of MPP1 was designed with adequate space for two O atoms above the Mn center for accessibility by using a dioxygen unit in the design. Circles represent porphyrin macrocycles.
Figure 2.
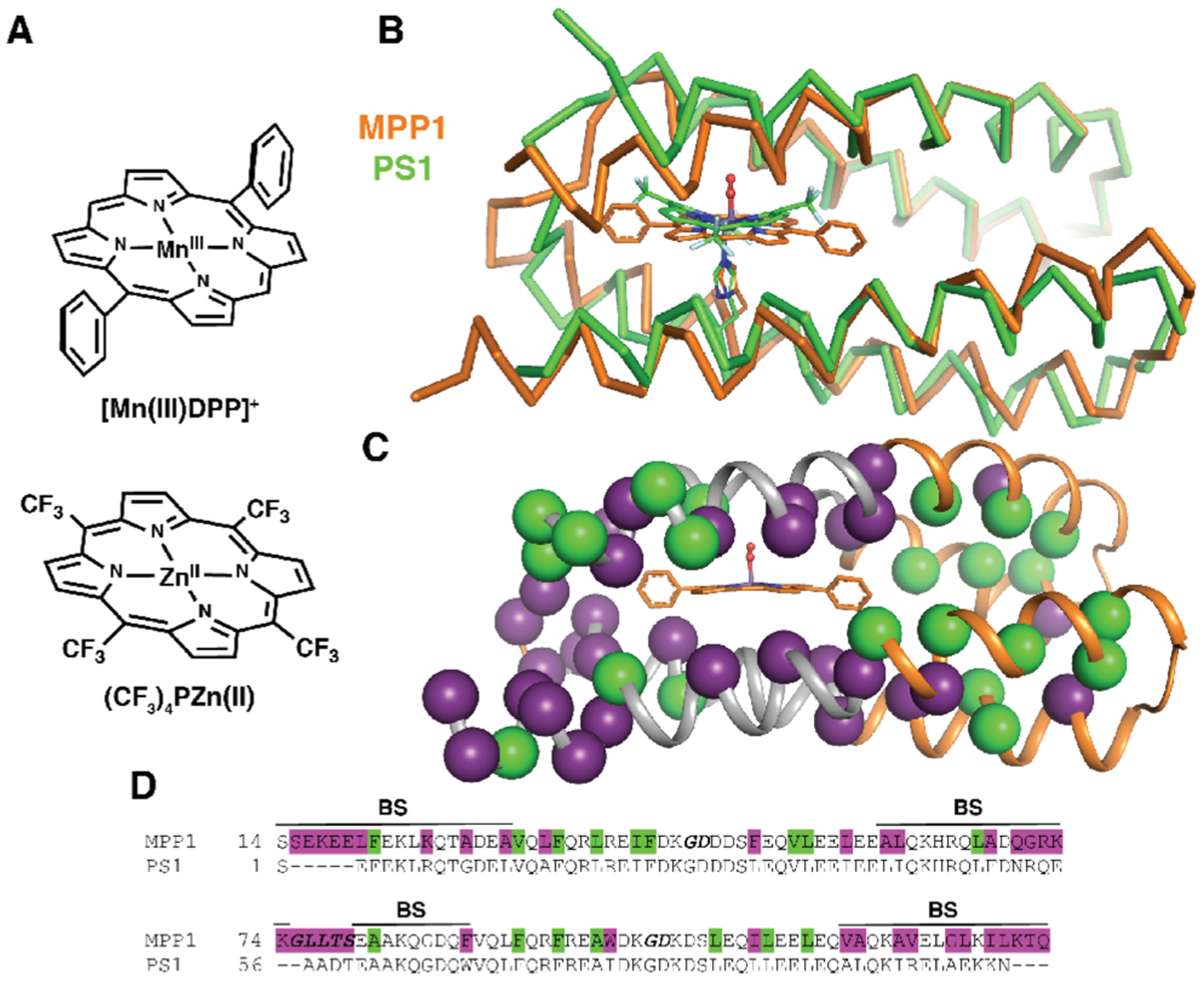
Comparison of MPP1 design to previously designed PS1. (A) ChemDraw representations of cofactors used for design, [Mn(III)DPP]+ (this work) and (CF3)4PZn(II) (previous work). (B) A ribbon representation of the MPP1 design overlaid with the NMR structure of PS1 illustrating the differences in backbone length and position. (C) A cartoon representation of the MPP1 design showing the changes in sequence relative to PS1 (purple spheres) to accommodate the new cofactor and residues that were allowed to change but did not (green spheres). (D) Sequence alignment of MPP1 and PS1 colored as in (C) with the binding site region designated as BS and loop regions bolded and italicized.
Here we explore and extend the principles used in the design of PS1 to create a non-natural manganese diphenylporphyrin-binding protein that was crystallographically characterized, providing us with information that is lacking in solution NMR: the exact positioning of the metal ion relative to the protein and the location of aqua ligands and associated water molecules within the binding site.
A common feature in many heme enzymes and synthetic model complexes is the binding of a dioxygen species (O2 or H2O2) which is then heterolytically cleaved to make a high-valent metal–oxo species with the release of water.2–4 Thus, the ability to accommodate two O atom units (e.g., one ligated and one distal water molecule) is critical to facilitating oxidative chemistry. Additionally, it is essential to be able to access high-valent intermediates without deleterious effects to the protein or cofactor. We achieve these enabling features for the first time in a de novo designed protein. Herein we report the computational design of the first crystallographically characterized de novo metalloporphyrin-binding protein.
Expanding on the design strategy for PS1 (dubbed Enfold),14 which simultaneously packed the binding site and folded core residues, we computationally designed a protein for a new abiological cofactor, MnDPP (Mn–diphenylporphyrin, Figure 2A). This cofactor is larger and contains a redox active metal (Mn) providing a good test of the breadth of our approach. The ability to access multiple redox states additionally expands the scope of possible reactivity with computationally designed proteins. Akin to native heme proteins, Mn–aryl porphyrins are capable of performing a variety of oxidative transformations, including epoxidation, sulfoxidation, and alcohol oxidation, via high-valent Mn-intermediates (i.e., Mn(IV) or Mn(V)).2 Formation of these strongly oxidizing intermediates presents additional design challenges that have not been previously implemented with precision in the de novo design of porphyrin proteins: (1) to purposefully design access to the metal center for oxidants and/or substrates and (2) to limit the ability of these powerful metal-based oxidants to deleteriously react with the protein scaffold itself. In the design of PS1, emphasis was placed on a well-folded core thereby restricting access to the cofactor.14 Natural oxidases and peroxidases often maintain access channels to the cofactor with water molecules during the resting state, which are then displaced during catalysis.3,21 We implement this concept in our design by using a dioxygen unit in the open coordination site of the Mn center. We have shown that this strategy provides access to the binding site for solvent, oxidants, and substrate and prevents detrimental damage to the binding site.
RESULTS
Design of MPP1.
The cofactor targeted in the design was a diphenyl porphyrin, MnDPP, with a dioxygen unit bound to the metal center (Figure 2). The inclusion of the dioxygen assures sufficient space for oxidant and substrate access to the cofactor. As compared to our earlier design of PS1, which binds to (CF3)4PZn, MnDPP represents a more extended cofactor with a flatter porphyrin ring. Previous proteins designed to bind diphenyl metalloporphyrins failed to result in sequences whose properties were amenable to high-resolution structure determination.22 Thus, the design and high-resolution structure determination of a MnDPP-binding protein represented a significant challenge. Moreover, the MnDPP cofactor is compatible with future work featuring complex cofactors that are bridged with diphenylporphyrins.23,24
The design of MPP1 (Manganese Porphyrin-binding Protein 1) began with the parameterized backbone of PS1,14 which includes an axial His ligand and a second-shell H-bonded Thr on an adjacent helix. The MnDPP was placed with its phenyl rings and the metal ion oriented along the central axis of the bundle (Figure 2). While PS1 was used as a starting template, the bundle needed to be extended to accommodate the larger diphenylporphyrin cofactor. Thus, an idealized helical bundle was fit to the structure of PS1, and the helices were elongated to match the length of the cofactor (plus an additional region intended to form a well-packed hydrophobic core). The dioxygen unit (formally OOH−) from an oxy-P450 structure25 (PDB 2A1M) was placed in the open coordination site on the Mn center (~1.8 Å Mn–O distance, trans to the His ligand, Figure 2B–C).
Using a custom RosettaScript, the interior residues were allowed to vary through iterative cycles of flexible backbone sequence design. Due to the high expression levels and stability of PS1, most solvent-exposed residues were unchanged (Figure 2C–D). To minimize potential oxidation of the protein, Trp and Tyr residues were disallowed at positions within 12 Å of the cofactor, and Met and Cys residues were disallowed throughout during design. PS1 showed the importance of adding Gly and Ala residues to create a more polar binding site and avoid hydrophobic collapse in the absence of the cofactor,14 as can be an issue in heme-binding maquettes.26,27 Therefore, three Gly residues were constrained in close proximity to the cofactor to ensure sufficient flexibility in the binding region for apo-MPP1. Additionally, the final MPP1 sequence had seven Ala residues in the binding site. Together, these small residues were intended to enable easy access to the Mn center and minimize the number of oxidizable bonds in close proximity to the cofactor. Models were assessed for low-energy and good packing scores by analyzing packing score (pstat) and total energy score from Rosetta outputs. The initial design of the helical sequences was conducted without connecting loops because this approach provided the most flexibility to allow the protein to optimize its fit with the cofactor.
To create single-chain proteins, loops were designed using MASTER.28 In MASTER an input structural element (query) is compared to a library of three-dimensional protein structures using the backbone root-mean-square deviation (RMSD) as a similarity score. Library structural fragments that match the query to a given RMSD threshold are enumerated, and the designability of a query is quantified by the total number of structural matches found in the library. Short segments of the two helices (six residues) were queried against a database of helical proteins from the PDB searching for a particular loop length to connect them. This allows for the use of loops that are well represented in natural proteins and are good matches for the structural context of the designed backbone. Loop length and sequence are important parameters to consider in the design of highly stable parameterized helical back-bones;29,30 therefore, loops were clustered by RMSD, and clusters with highly represented loops were chosen. As a result, helices A and B (loopAB) and C and D (loopCD) were connected with short, highly designable (as evidenced by the tight clustering) nested Schellman plus N-capping loop motifs29–33 (Figure 3A) to ensure a stable folded core. MASTER proved particularly useful in choosing loopBC. To accommodate the larger cofactor and, as a result, longer interhelical distance between helices B and C, a longer loop was required. Overall, these loops were less well-represented in the PDB than loopsAB/CD, but a large cluster was found nonetheless (see the Supporting Information, Figure 3B, and Table S1). After the loops were built, additional rounds of flexible backbone sequence design were performed to ensure complementary packing of the interior of the bundle with the new loops. At this stage, three additional designs (MPP2–4) were prepared with different lengths for loopBC, including the original loop backbone from PS1 (MPP4) (Figure S1).
Figure 3.
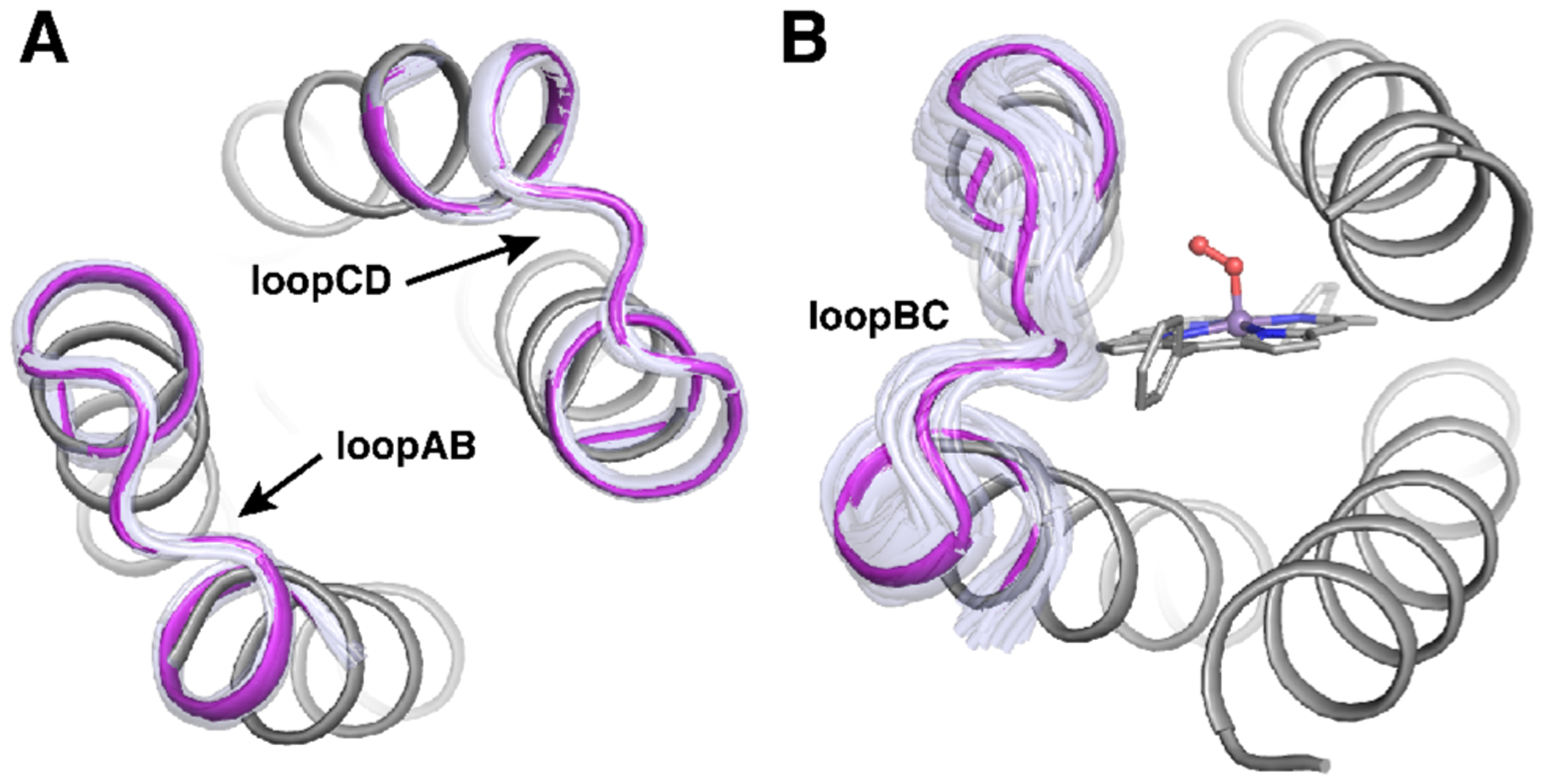
Design model of MPP1 showing representative clusters of loops. Clustered loops from MASTER for loopAB and loopCD (A) and loopBC (B). Loop clusters are shown in light purple and the chosen loop in magenta.
Despite coming from the same parameterized backbone, 65% of the interior residues in MPP1 changed relative to PS1, with the majority of changes occurring in the binding site (Figure 2C). Moreover, the sequence of MPP1 has no similarity with any natural protein (blast E value <0.05 against nonredundant protein database).
As a test of the structural stability of apo-MPP1, ab initio folding predictions were run on the designed sequence.34 As expected, the binding region appears more flexible relative to the conformationally unique folded core (Figure S2). As discussed above, the addition of Gly and Ala residues to the binding region, along with polar His and Thr residues, maintains an appropriately dynamic binding site. The lower RMSD clustering of the results for the folded core region, relative to the binding site, suggest a level of preorganization of the distal core residues.
Solution Characterization.
MPP1–MPP4 all express well in E. coli (20–30 mg/L) and were purified to homogeneity (Figure S3). Spectral titrations indicated that they each bound Mn(III)DPP with similar shifts in the absorption spectrum strongly suggesting that they accommodate the cofactor in a similar binding environment (Figure S4). As a preliminary test of unique solution structure, 1H NMR spectra were collected for holo-MPP1–4 (Figure S5). MPP1–2 appeared to be well structured in solution while MPP3–4 showed evidence of conformational averaging, similar to previously designed maquettes. MPP1 crystallized readily (see below), while MPP2 did not form crystals in preliminary crystallization trials. We therefore focused our efforts on MPP1.
MPP1 tightly binds Mn(III)DPP (Kd = 122 ± 19 nM) despite the cofactor’s poor aqueous solubility. The cofactor was added from a DMSO stock solution, and binding occurs within the mixing time, resulting in a red shift in the Mn(III)DPP split Soret absorption manifold and Q-band transitions (Figure 4A–B). The absorbance at 463 nm is consistent with binding of an axial His ligand.35 In addition, the measured extinction coefficient (ε463 = 65 000 M−1 cm−1) of the Soret band suggests that the Mn(III) center is six-coordinate in solution, consistent with the design of His and water as axial ligands under these conditions.36,37 Although MnDPP is achiral, binding to the active site is expected to result in induced chirality if it is held rigidly within the cofactor binding site. Indeed, the visible CD spectrum of holo-MPP1 shows a clear induced Cotton effect associated with the Soret band transitions centered at 391 and 463 nm, matching the electronic absorption spectrum (Figure 4). It should be noted that despite starting from a parameterized PS1 helical bundle, MPP1 does not bind tightly to (CF3)4PZn (Figure S6).
Figure 4.
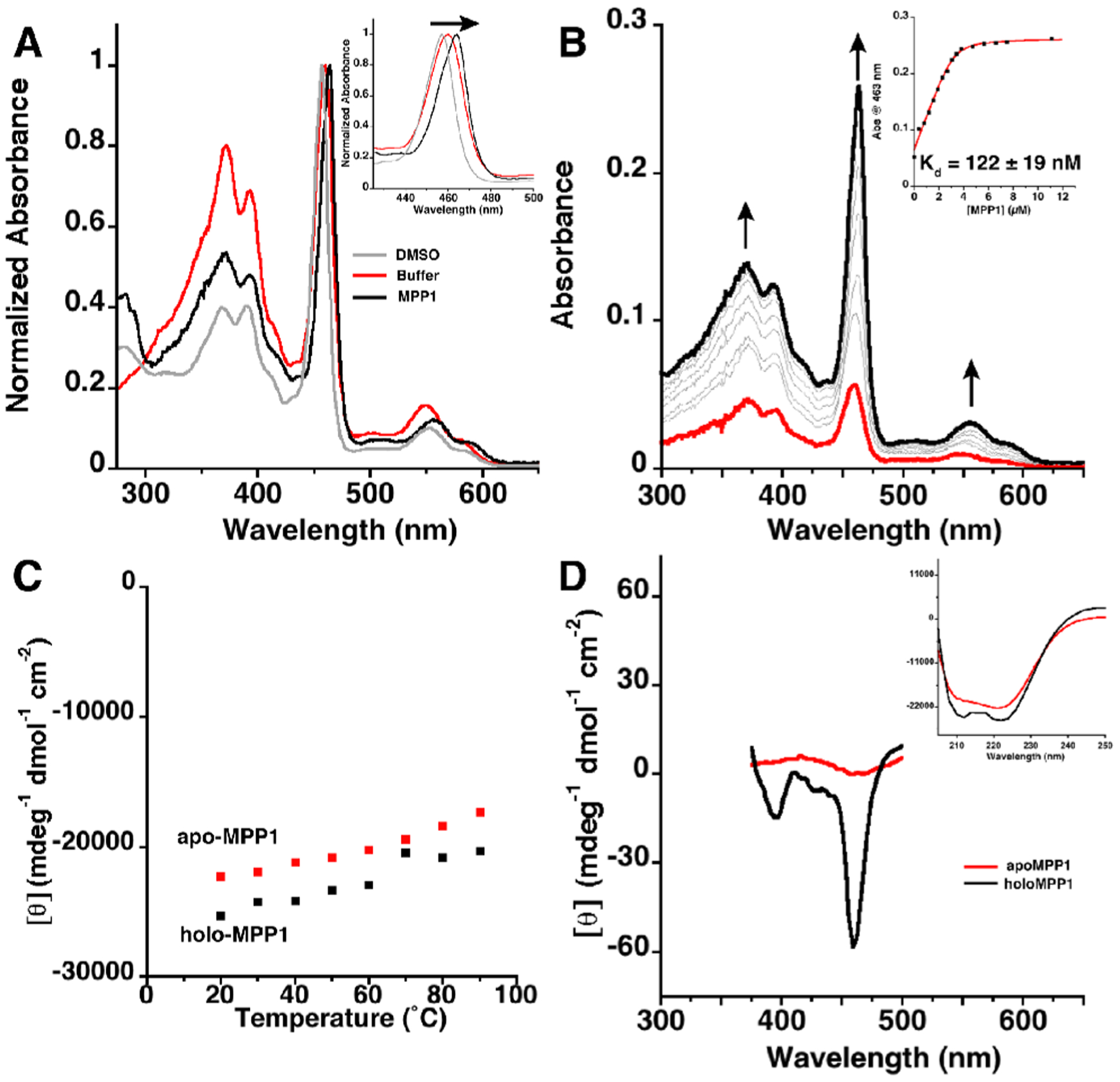
(A) Electronic absorption spectra showing the red shift for the Mn(III)DPP Soret band in DMSO, buffer, and MPP1. Inset: Zoom in of Soret bands. (B) Electronic absorption spectra showing the titration of apo-MPP1 into a solution of Mn(III)DPP to determine Kd. Inset: Plot of absorption at 463 nm versus MPP1 concentration and fit (in red). Kd error is from fit. (C) Temperature-dependent CD plot showing the thermal stability of apo- and holo-MPP1 (ellipticity measured at 222 nm). (D) CD spectra in UV (inset) and visible regions of apo- and holo-MPP1 showing the induced Cotton effect. Buffer conditions were 50 mM MES, 150 mM NaCl pH 7.5.
MPP1 was designed to be exceptionally well-folded, leading to extreme thermal stability. This allows for ease of study as well as the possibility of doing catalytic reactions that require high temperatures. As designed, apo- and holo-MPP1 are thermostable, with Tm > 90 °C (Figure 4C). This level of thermostability is on par with (or better than) thermophilic proteins from extremophiles. In fact, the induced CD of the Soret bands is still evident at 90 °C (Figure S7). Furthermore, the protein prevents aggregation of the Mn-cofactor which readily occurs in the absence of the protein.
To determine the effect of the protein environment on the Mn(III/II) reduction potential, spectrophotometric dithionite titration experiments were performed (see the Supporting Information).38 The E1/2(Mn III/II) for holo-MPP1 was found to be −107 (±5) mV vs SHE (pH 7, Figure S8).15,20,39 Because redox-coupled protonation/deprotonation play critical roles in catalysis, it was important to assess changes in the protonation state accompanying changes in the oxidation state of the Mn ion. Therefore, we confirmed and extended these results using an independent, spectroelectrochemical method that employs a true reference electrode to measure changes in Mn(III/II) reduction potential as a function of pH (see the Supporting Information, Figure 5, Figure S8, and Table S2). A midpoint potential of −145 mV vs SHE was observed at pH 7, in agreement with the value measured by spectrophotometric solution titration. The observed midpoint potential is constant from pH 8 to 7. At pH 6, it became less negative by 30 mV, indicative of partial protonation of a group that is thermodynamically coupled to the Mn(II/III) ion (Figure S9 and Table S2). At lower pH, one might expect full protonation in the reduced state, and, therefore, a full 60 mV/pH would be observed, as expected for a 1e–/1H+ process. Unfortunately, measurements at lower pH were complicated by possible protonation of the His ligand.
Figure 5.
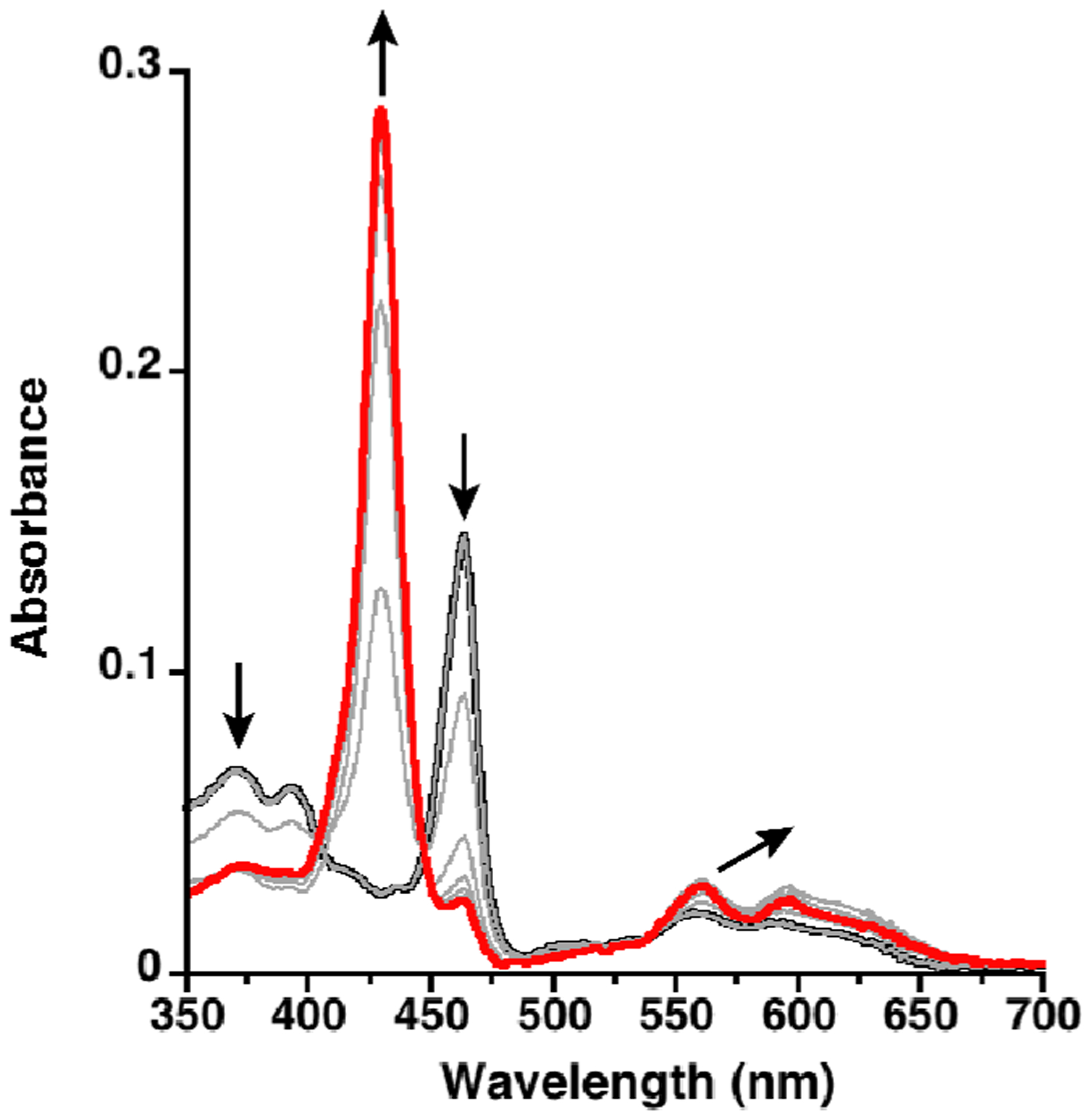
Spectroelectrochemical titration of Mn(III)MPP1 (black) to Mn(II)MPP1 (red) in the presence of redox mediator indigo-5,5′-disulfonate (IDIS). The potential was decreased from 10 to −390 mV vs SHE in 50 mV increments, holding each potential until no further spectral changes were observed. The spectra of IDIS under the same conditions have been subtracted for clarity.
The cofactor can be reduced from Mn(III) to Mn(II) upon addition of 1 equiv of dithionite to a degassed solution, causing a blue shift in the Soret. The Mn(II) species is stable in the absence of dioxygen; however, after oxygenation the spectrum quantitatively evolves to that for the Mn(III) species indicating that (1) the binding site is accessible to dioxygen and (2) the intermediate species does not deleteriously affect the binding environment (Figure S10).
Formation of Mn(V) Species and Sulfoxidation Reaction.
To design functional metalloproteins, there must be access to the cofactor for the oxidant and substrate to reach the metal center, but the protein scaffold must also support a high-valent species without deleterious effects. Mn–porphyrin catalysts often go through a Mn(V)–oxo intermediate as the competent oxidant via a two-electron oxidation from Mn(III) to Mn(V).4 These species have been well studied in the literature, with diagnostic absorption spectra.40–42 To test MPP1’s ability to support formation of these highly oxidizing species, the Mn(III) holo-protein was oxidized in solution with sodium periodate (NaIO4), a two-electron oxidant and O atom transfer agent, in both MES and phosphate buffer conditions (Figures S11–F12). Upon addition, the Soret transition centered at 463 nm associated with the Mn(III) species decreases with a concomitant evolution of a band at 431 nm that matches the literature for similar species assigned as a Mn(V)–oxo species (Figure 6).40–42 This reaction proceeds with approximately 50% conversion based on the absorbance of the Mn(III) species. At room temperature, the Mn(V) species is unstable and slowly converts back to the starting Mn(III) spectrum with a half-life of ~12 min (Figure S11). However, addition of thioanisole to the preformed Mn(V) species leads to a rapid decrease in the 431 nm band and return of the Mn(III) starting spectrum. Analysis of the reaction mixture by mass spectroscopy verified the formation of methyl phenyl sulfoxide with no evidence for further oxidation to phenyl methyl sulfone (Figure 6C and Figures S13–S18). This sulfoxidation reaction is consistent with the assignment of a Mn(V)–oxo species forming upon the addition of periodate to the Mn(III) holo-protein.43,44 It should be noted that in the absence of the protein, the cofactor does not form this species and, instead, slowly decomposes in the presence of NaIO4 (Figure S19), strongly suggesting that the protein environment helps stabilize this species.
Figure 6.
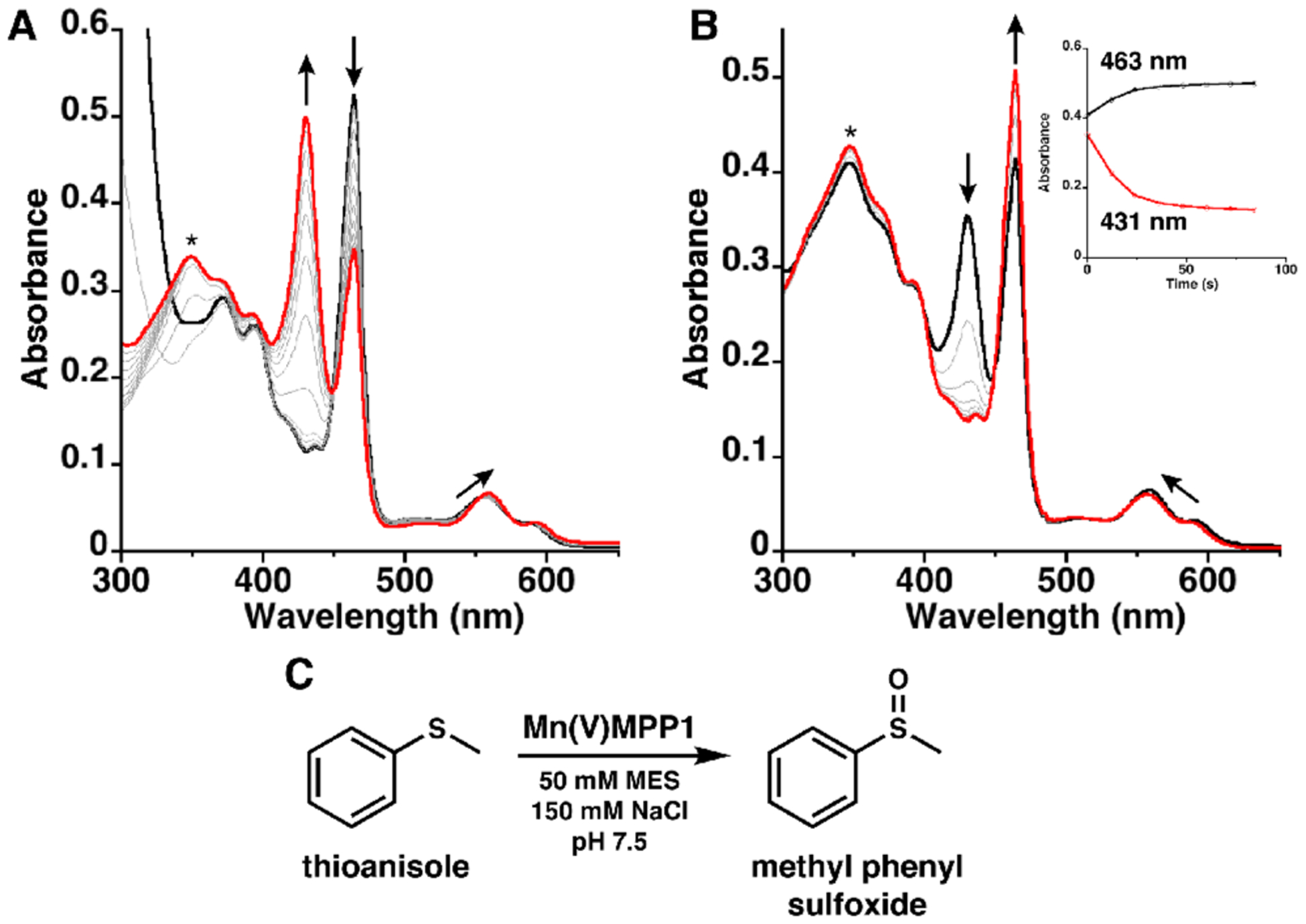
(A) Electronic absorption spectra showing the reaction of Mn(III)MPP1 (black) with excess NaIO4 to form a Mn(V)–oxo species monitored with 60 s scans. (B) Reaction of the Mn(V)–oxo species (red) with 10 equiv of thioanisole showing the loss of the Mn(V)–oxo bands and conversion back to the Mn(III) spectrum (black) monitored with 12 s scans. Inset: Time trace monitoring absorbances at 463 and 431 nm. (C) Scheme showing sulfoxidation of thioanisole. (*) indicates the absorbance from NaIO4 (see the Supporting Information).
Structural Characterization.
Holo-MPP1 is the first crystallographically characterized de novo designed porphyrin-binding protein, a significant step forward in understanding how to build binding sites to create functional proteins. The structure was solved to 1.75 Å resolution to reveal that the backbone matches the design exceptionally well (0.6 Å all backbone RMSD, Figure 7). LoopBC is well-structured; however, it is displaced relative to the design, likely because of an N-terminal 6-His tag and TEV cleavage sites. This tag, which was not included in the design, interacts with the loopBC in the crystal structure (Figure 7A and Figure S20). The protein crystallized with an additional MnDPP unit at a symmetry interface on the surface bound to E58 (see the Supporting Information, Figure S21). However, in aqueous solution, the protein is monomeric and binds a single equivalent of cofactor (Figure 4B and Figure S3). Thus, the additional intersubunit cofactor must only assemble under the crystallization conditions.
Figure 7.
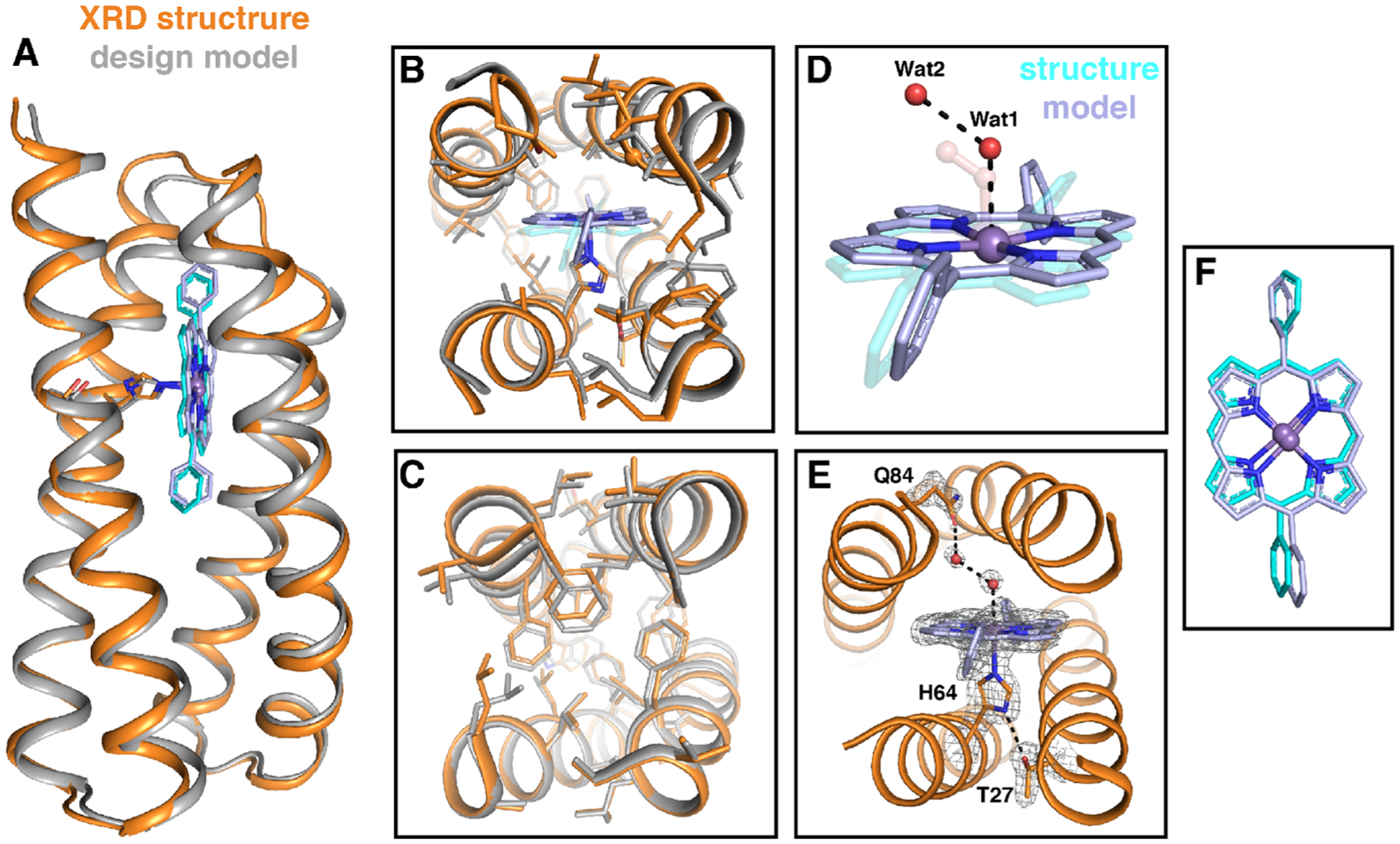
Structural comparison of the designed model of MPP1 (gray) and the crystal structure (PDB: 7JRQ, orange). (A) Cartoon representation showing an extremely good backbone match between the design and structure (0.6 Å all backbone RMSD). Comparisons of the binding site region (B) and folded core region (C). Comparison of the placement of Wat1 and Wat2 (red spheres) relative to the dioxygen unit in the design (transparent) (D) and extended H-bonding network from the binding site to the surface via a water network (E). The positions of T27, H64, MnDPP, Wat1, Wat2, and Q84 in (E) are indicated by the F0–Fc omit map (gray mesh, contoured at 3σ). (F) Comparison of observed cofactor placement (gray) relative to the model (cyan). Glycine residues are shown as Cα spheres.
The His-bound cofactor sits precisely where it was placed in the design, predicting the Mn position within 0.5 Å, less than the length of a C–H bond. The only difference is a ~ 55° rotation in the torsion angle of one of the macrocycle’s meso-phenyl groups (Figure 7). In addition to the extraordinary similarity between the structure and design, the structure confirmed Mn–His coordination and revealed two water molecules (Wat1 and Wat2) within the binding site (Figure 7). Wat1 is a weakly coordinating sixth ligand to the Mn center (Wat1–Mn distance 2.62(1) Å), which agrees with the solution absorption spectrum of MPP1 and matches the design. The Wat1-Mn distance is similar to the Mn–O(water) distance found in a Mn-porphyrinoid-substituted myoglobin.45 Wat2 forms an H-bonding network that includes Wat1 and Gln84. As expected, the Wat1–Mn and Wat1–Wat2 distances are longer than the analogous distances in the design because they are not part of a covalent dioxygen unit, and rather a H-bonded network. However, the two water molecules do sit along the same vector as the O–O bond in the design with an identical Mn–O–O angle of 124° (Figure 7D). As in natural proteins, these water molecules within the binding site may act as a placeholder to maintain an access channel. This is the first structural evidence of binding site access for a designed porphyrin-binding protein, a major advance toward the development of functional metalloproteins from scratch, and, we believe, a result of purposefully designed access using a dioxygen unit.
DISCUSSION
The long history of de novo designed porphyrin-binding proteins has taught us much, but the ability to gain structural insight is invaluable. As the first crystallographically characterized de novo designed porphyrin-binding protein, MPP1 illustrates the importance in determining the protein structure to further our understanding of the design features necessary to produce functional proteins from scratch. This work expands on the Enfold14 strategy to include purposefully designed substrate access by including a two O atom unit within the binding site. Additionally, it demonstrates the generality of this approach―all parts of the protein are important: a well-folded distal core, shape complementary binding site, and appropriate space reserved for external access. When designed in unison, the resulting protein structure was highly predictable without the need for evolution or experimental optimization. All four sequences designed bound the cofactor with high affinity, although two showed evidence of conformational averaging in solution. Of the two that showed an NMR spectrum consistent with a unique structure, one was crystallized and structurally interrogated.
The structure showed the presence of two water molecules in an H-bonding network that sit above the Mn proximal to the designed location of the dioxygen unit. This finding suggests that MPP1, as designed, is capable of accommodating two O atom units during the activation of dioxygen. Moreover, the designed metalloprotein can access a Mn(V)–oxo species and perform O atom transfer to thioanisole. With the structure of MPP1 in hand, we can redesign the binding site to include an active site customized for substrate. This work is a substantial step forward in protein design and paves the way for functional metalloproteins designed from scratch.
METHODS
Protein Design.
Full methods and scripts regarding the design of MPP1 are given in the Supporting Information. Briefly, the parameterized backbone of PS1 was redesigned to bind Mn(III)DPP with a dioxygen unit bound in the open coordination site via a customized Rosetta script for flexible backbone sequence design. The flexible backbone protocol was the following: distance and angle constraints between Mn and His were loaded, the model was repacked, the backbone and then side chains were relaxed, and three trials of a Monte Carlo flexible backbone subprotocol were performed and filtered for models with nativelike packing (pstat ≥0.48). Models were analyzed for packing and total energy. The designs of MPP2–4 proceeded in the same fashion.
Protein Expression and Purification.
Full details are given in the Supporting Information. Briefly, the gene coding for the protein sequence of MPP1 was ordered from IDT, which was cloned into the IPTG-inducible pET-21a plasmid (cloning site NdeI-BamHI). The sequence also coded for an N-terminal 6xHis-tag followed by a TEV protease cleavage sequence, followed by the designed sequence.
Porphyrin-Binding Titration to Determine Kd.
Mn(III)DPP(Cl) was purchased from Frontier Scientific and used as received. Mn(III)DPP(Cl) (3.4 μM) was solubilized in a 1 mL solution of 50 mM MES, 150 mM NaCl, pH 7.5 buffer from a DMSO stock solution. One microliter of a 308 μM stock of apo-MPP1 was titrated into the 1 mL solution containing the porphyrin, and an electronic absorption spectrum was measured until >2.5 equiv of protein was added. Absorbance changes at 463 nm, due to His–Mn coordination-induced spectral shifts of the porphyrin, were fit to a single-site, protein–ligand-binding model.
Preparation of Holo-MPP.
First, 1.2 equiv (60 μL) of a 10 mM DMSO solution of Mn(III)DPP(Cl) was added to 1 mL of a 500 μM apo-MPP1 solution in 50 mM MES, 150 mM NaCl, pH 7.5 and allowed to sit for 5 min at room temperature. The protein/cofactor solution was centrifuged at 14 000g in an Amicon Ultra –0.5 mL centrifuge filter (10 kDa MW cutoff) for 3 min, 3 times, replacing the buffer to 0.5 mL after each spin. Finally, the solution was spun for 2 min at 12 000g in an Amicon ultrafree-MC GV filter. The holo-MPP1 was stored at 4 °C for short-term storage or −20 °C for long-term storage. Binding tests of (CF3)4PZn were carried out with the same method.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Yibing Wu for recording and analyzing NMR data, Dr. Nick Polizzi for helpful discussions about Rosetta protocols, and Dr. Jack Nicoludis for helpful discussions about XRD data processing. We also thank A. S. Borovik and Dr. Kelsey Miller for allowing us to use their synchrotron beamtime. X-ray diffraction experiments were performed at the Stanford Synchrotron Radiation Light Source on beamline 12-2. This work was primarily supported by NIGMS F32 GM130029, NSF grants 1709506 and CHE-1709497, and AFOSR FA9550-19-1-0331.
ABBREVIATIONS
- DPP
diphenylporphyrin
- MPP1–4
manganese porphyrin-binding protein
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10136.
Additional methods for protein design, spectroelectro-chemistry, Mn(V) formation and reactivity, and crystallography, Figures S1–S21, and Tables S1–S3 (PDF)
Accession Codes
Atomic coordinates for holo-MPP1 have been deposited in the Protein Data Bank with accession number 7JRQ.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c10136
The authors declare no competing financial interest.
Contributor Information
Samuel I. Mann, Department of Pharmaceutical Chemistry and the Cardiovascular Research Institute, University of California at San Francisco, San Francisco, California 94158-9001, United States;.
Animesh Nayak, Department of Chemistry, Duke University, Durham, North Carolina 27708, United States.
George T. Gassner, Department of Chemistry and Biochemistry, San Francisco State University, San Francisco, California 94132, United States
Michael J. Therien, Department of Chemistry, Duke University, Durham, North Carolina 27708, United States;.
William F. DeGrado, Department of Pharmaceutical Chemistry and the Cardiovascular Research Institute, University of California at San Francisco, San Francisco, California 94158-9001, United States;.
REFERENCES
- (1).Reedy CJ; Gibney BR Heme Protein Assemblies. Chem. Rev 2004, 104 (2), 617–650. [DOI] [PubMed] [Google Scholar]
- (2).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118 (5), 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114 (7), 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Baglia RA; Zaragoza JPT; Goldberg DP Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes. Chem. Rev 2017, 117 (21), 13320–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lyaskovskyy V; Suarez AI; Lu H; Jiang H; Zhang XP; de Bruin B Mechanism of cobalt(II) porphyrin-catalyzed C-H amination with organic azides: radical nature and H-atom abstraction ability of the key cobalt(III)-nitrene intermediates. J. Am. Chem. Soc 2011, 133 (31), 12264–73. [DOI] [PubMed] [Google Scholar]
- (6).Chen K; Arnold FH Engineering new catalytic activities in enzymes. Nat. Catal 2020, 3 (3), 203–213. [Google Scholar]
- (7).Brandenberg OF; Fasan R; Arnold FH Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Korendovych IV; DeGrado WF De novo protein design, a retrospective. Q. Rev. Biophys 2020, 53, No. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pinter TBJ; Koebke KJ; Pecoraro VL Catalysis and Electron Transfer in De Novo Designed Helical Scaffolds. Angew. Chem., Int. Ed 2020, 59 (20), 7678–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Watkins DW; Armstrong CT; Anderson JL De novo protein components for oxidoreductase assembly and biological integration. Curr. Opin. Chem. Biol 2014, 19, 90–8. [DOI] [PubMed] [Google Scholar]
- (11).Huang PS; Boyken SE; Baker D The coming of age of de novo protein design. Nature 2016, 537 (7620), 320–7. [DOI] [PubMed] [Google Scholar]
- (12).Cheng RP Beyond de novo protein design–de novo design of non-natural folded oligomers. Curr. Opin. Struct. Biol 2004, 14 (4), 512–20. [DOI] [PubMed] [Google Scholar]
- (13).Lombardi A; Pirro F; Maglio O; Chino M; DeGrado WF De Novo Design of Four-Helix Bundle Metalloproteins: One Scaffold, Diverse Reactivities. Acc. Chem. Res 2019, 52 (5), 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Polizzi NF; Wu Y; Lemmin T; Maxwell AM; Zhang S-Q; Rawson J; Beratan DN; Therien MJ; DeGrado WF De novo design of a hyperstable non-natural protein–ligand complex with sub-Å accuracy. Nat. Chem 2017, 9 (12), 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Farid TA; Kodali G; Solomon LA; Lichtenstein BR; Sheehan MM; Fry BA; Bialas C; Ennist NM; Siedlecki JA; Zhao Z; Stetz MA; Valentine KG; Anderson JLR; Wand AJ; Discher BM; Moser CC; Dutton PL Elementary tetrahelical protein design for diverse oxidoreductase functions. Nat. Chem. Biol 2013, 9 (12), 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Koder RL; Anderson JL; Solomon LA; Reddy KS; Moser CC; Dutton PL Design and engineering of an O2 transport protein. Nature 2009, 458 (7236), 305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kamtekar S; Schiffer JM; Xiong H; Babik JM; Hecht MH Protein design by binary patterning of polar and nonpolar amino acids. Science 1993, 262 (5140), 1680–5. [DOI] [PubMed] [Google Scholar]
- (18).Moffet DA; Foley J; Hecht MH Midpoint reduction potentials and heme binding stoichiometries of de novo proteins from designed combinatorial libraries. Biophys. Chem 2003, 105 (2–3), 231–239. [DOI] [PubMed] [Google Scholar]
- (19).Moffet DA; Certain LK; Smith AJ; Kessel AJ; Beckwith KA; Hecht MH Peroxidase Activity in Heme Proteins Derived from a Designed Combinatorial Library. J. Am. Chem. Soc 2000, 122 (31), 7612–7613. [Google Scholar]
- (20).Shifman JM; Gibney BR; Sharp RE; Dutton PL Heme redox potential control in de novo designed four-alpha-helix bundle proteins. Biochemistry 2000, 39 (48), 14813–21. [DOI] [PubMed] [Google Scholar]
- (21).Sevrioukova IF; Poulos TL Understanding the mechanism of cytochrome P450 3A4: recent advances and remaining problems. Dalton Trans. 2013, 42 (9), 3116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cochran FV; Wu SP; Wang W; Nanda V; Saven JG; Therien MJ; DeGrado WF Computational De Novo Design and Characterization of a Four-Helix Bundle Protein that Selectively Binds a Nonbiological Cofactor. J. Am. Chem. Soc 2005, 127 (5), 1346–1347. [DOI] [PubMed] [Google Scholar]
- (23).Jiang T; Bai Y; Zhang P; Han Q; Mitzi DB; Therien MJ Electronic structure and photophysics of a supermolecular iron complex having a long MLCT-state lifetime and panchromatic absorption. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (34), 20430–20437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Polizzi NF; Eibling MJ; Perez-Aguilar JM; Rawson J; Lanci CJ; Fry HC; Beratan DN; Saven JG; Therien MJ Photoinduced Electron Transfer Elicits a Change in the Static Dielectric Constant of a de Novo Designed Protein. J. Am. Chem. Soc 2016, 138 (7), 2130–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nagano S; Poulos TL Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam - Implications for the dioxygen activation mechanism. J. Biol. Chem 2005, 280 (36), 31659–31663. [DOI] [PubMed] [Google Scholar]
- (26).Skalicky JJ; Gibney BR; Rabanal F; Bieber Urbauer RJ; Dutton PL; Wand AJ Solution Structure of a Designed Four-α-Helix Bundle Maquette Scaffold. J. Am. Chem. Soc 1999, 121 (21), 4941–4951. [Google Scholar]
- (27).Huang SS; Koder RL; Lewis M; Wand AJ; Dutton PL The HP-1 maquette: From an apoprotein structure to a structured hemoprotein designed to promote redox-coupled proton exchange. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (15), 5536–5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhou J; Grigoryan G Rapid search for tertiary fragments reveals protein sequence-structure relationships. Protein Sci. 2015, 24 (4), 508–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Engel DE; DeGrado WF Alpha-alpha linking motifs and interhelical orientations. Proteins: Struct., Funct., Genet 2005, 61 (2), 325–37. [DOI] [PubMed] [Google Scholar]
- (30).Lahr SJ; Engel DE; Stayrook SE; Maglio O; North B; Geremia S; Lombardi A; DeGrado WF Analysis and design of turns in alpha-helical hairpins. J. Mol. Biol 2005, 346 (5), 1441–54. [DOI] [PubMed] [Google Scholar]
- (31).Sagermann M; Martensson LG; Baase WA; Matthews BW A test of proposed rules for helix capping: implications for protein design. Protein Sci. 2002, 11 (3), 516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schellman C Protein Folding ;Elsevier: New York, 1980; pp 53–61. [Google Scholar]
- (33).Hovmoller S; Zhou T; Ohlson T Conformations of amino acids in proteins. Acta Crystallogr., Sect. D: Biol. Crystallogr 2002, 58 (5), 768–776. [DOI] [PubMed] [Google Scholar]
- (34).Bradley P; Misura KM; Baker D Toward high-resolution de novo structure prediction for small proteins. Science 2005, 309 (5742), 1868–71. [DOI] [PubMed] [Google Scholar]
- (35).Yonetani T; Asakura T Comparison of manganese porphyrin-containing cytrochrome c peroxidase, horseradish peroxidase, and myoglobin. J. Biol. Chem 1969, 244, 4580–4588. [PubMed] [Google Scholar]
- (36).Williamson MM; Hill CL Isolation and characterization of a five-coordinate manganese(III) porphyrin cation. Crystal and molecular structure of aquo(tetraphenylporphinato)manganese(III) triflate. Inorg. Inorg. Chem 1986, 25 (26), 4668–4671. [Google Scholar]
- (37).Hill CL; Williamson MM Structural and electronic properties of six-coordinate manganese(III) porphyrin cations. Crystal and molecular structure of bis(N,N-dimethylformamide)-(tetraphenylporphinato)manganese(III) perchlorate, [MnIII TPP-(DMF)2]+ClO4. Inorg. Chem 1985, 24 (18), 2836–2841. [Google Scholar]
- (38).Gassner GT The styrene monooxygenase system. Methods Enzymol. 2019, 620, 423–453. [DOI] [PubMed] [Google Scholar]
- (39).Lin R; Immoos CE; Farmer PJ Unusual voltammetry of manganese-substituted myoglobin in surfactant film: evidence for two redox pathways. JBIC, J. Biol. Inorg. Chem 2000, 5 (6), 738–47. [DOI] [PubMed] [Google Scholar]
- (40).Jin N; Groves JT Unusual Kinetic Stability of a Ground-State Singlet Oxomanganese(V) Porphyrin. Evidence for a Spin State Crossing Effect. J. Am. Chem. Soc 1999, 121 (12), 2923–2924. [Google Scholar]
- (41).Groves JT; Lee J; Marla SS Detection and Characterization of an Oxomanganese(V) Porphyrin Complex by Rapid-Mixing Stopped-Flow Spectrophotometry. J. Am. Chem. Soc 1997, 119 (27), 6269–6273. [Google Scholar]
- (42).Nam W; Kim I; Lim MH; Choi HJ; Lee JS; Jang HG Isolation of an oxomanganese(V) porphyrin intermediate in the reaction of a manganese(III) porphyrin complex and H2O2 in aqueous solution. Chem. - Eur. J 2002, 8 (9), 2067–2071. [DOI] [PubMed] [Google Scholar]
- (43).Prokop KA; Neu HM; de Visser SP; Goldberg DP A Manganese(V)–Oxo π-Cation Radical Complex: Influence of One-Electron Oxidation on Oxygen-Atom Transfer. J. Am. Chem. Soc 2011, 133 (40), 15874–15877. [DOI] [PubMed] [Google Scholar]
- (44).Mandimutsira BS; Ramdhanie B; Todd RC; Wang H; Zareba AA; Czernuszewicz RS; Goldberg DP A stable manganese(V)-oxo corrolazine complex. J. Am. Chem. Soc 2002, 124 (51), 15170–1. [DOI] [PubMed] [Google Scholar]
- (45).Oohora K; Kihira Y; Mizohata E; Inoue T; Hayashi T C(sp3)-H bond hydroxylation catalyzed by myoglobin reconstituted with manganese porphycene. J. Am. Chem. Soc 2013, 135 (46), 17282–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.