

Abstract
A unified synthetic strategy to access tertiary four-membered carbo/heterocyclic boronic esters is reported. Use of a Cu(I) catalyst in combination with a modified dppbz ligand enables regioselective hydroboration of various trisubstituted benzylidenecyclobutanes and carbo/heterocyclic analogs. The reaction conditions are mild, and the method tolerates a wide range of medicinally relevant heteroarenes. The protocol can be conveniently conducted on gram-scale, and the tertiary boronic ester products undergo facile diversification into valuable targets. Reaction kinetics and computational studies indicate that the migratory insertion step is turnover-limiting and accelerated by electron-withdrawing groups on the dppbz ligand. Energy decomposition analysis (EDA) calculations reveal that electron-deficient P-aryl groups on the dppbz ligand enhance the T-shaped π/π interactions with the substrate and stabilize the migratory insertion transition state.
Keywords: Copper catalysis, hydroborations, benzylidenecyclobutanes, 4-membered rings, tertiary boronic esters, modified dppbz ligands, heterocycle
Graphical Abstract

Four-membered carbo/heterocycles are prized motifs in medicinal chemistry.[1] The unique properties and advantages of cyclobutanes, azetidines, oxetanes and other types of four-membered rings have recently been highlighted in numerous publications. For example, larger, saturated heterocycles tend to be more lipophilic and are cleared more rapidly relative to their four-membered ring counterparts.[2] In addition, four-membered rings can increase water solubility, reduce pKa and be used as bioisosteres.[3] For these reasons, numerous four-membered-ring-containing compounds have been developed to treat various medical indications, including PF-03654746,[4] linsitinib,[5] baricitinib,[6] and GNE-317[7] (Figure 1A). Despite the importance of four-membered ring motifs, preparative methods to access these structures remain relatively few in number, particular in comparison to the available methodology to synthesize five- and six-membered ring motifs. Specifically, appending a tertiary-substituted four-membered ring onto a target molecule of interest remains challenging.
Figure 1.

Overview of Proposed Approach to Preparation of Tertiary Boronic Esters.
Owing to their synthetic versatility, carbo/heterocyclic tertiary boronic esters have attracted significant attention in academia and industry (Figure 1B). For example, Hall and Pfizer co-workers recently demonstrated asymmetric copper-catalyzed 1,4-hydroboration of conjugated cyclobutenones to afford enantioenriched tertiary cyclobutylboronates.[8] In addition, decarboxylative and deoxygenative radical borylation of cyclobutyl precursors has been independently demonstrated by the Aggarwal, Baran, and Studer groups.[9] Deborylative alkylation developed by Morken and co-workers has also been used to access tertiary cyclobutylboronic esters from bis- or tris(boronate) precursors.[10] More recently, Aggarwal and Studer groups independently published elegant methods to generate both tertiary cyclobutyl and azetidinyl boronic esters from highly strained alkyl lithium intermediates through a 1,2-metallate rearrangement.[11,12] Additionally, the Brown group showed one example of a nickel-catalyzed arylboration of a benzylideneazetidine substrate to afford a tertiary azetidinylboronic ester product.[13] Although these existing methods are impressive and highly valuable, they also have limitations in terms of requiring harsh reaction conditions, employing air/moisture sensitive reagents, relying on precursors that are difficult to prepare, and exhibiting narrow substrate scope (e.g., heterocycle incompatibility). Moreover, to the best of our knowledge, a synthetic method to access tertiary boronic esters with a four-membered ring containing oxygen, sulfone or gem-difluoro groups has not been described.
We thus sought to develop a mild, heterocycle-tolerant, and unified method to afford various types of tertiary four-membered cyclic boronic esters. In this context, the Shi group and our laboratories have independently demonstrated copper–boryl catalytic systems with 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl (BINAP) as the ligand that can functionalize benzylidenecyclopropanes to afford tertiary cyclopropylboronic esters (Figure 1C).[14, 15] The reaction conditions are mild, and a wide variety of heterocycles are tolerated. Based on these precedents, we questioned whether copper–boryl catalysis could grant access to four-membered-ring variants. At the outset, however, we recognized a major challenge in the form of the strain energy difference between three- and four-membered ring systems. In the previous cyclopropane system, strain energy was used as a major driving force to achieve borylative addition to sterically hindered trisubstituted alkenes. On the other hand, the estimated strain energy difference between benzylidenecyclobutane and cyclobutane itself (2.7 kcal/mol) is significantly smaller than the difference between benzylidenecyclopropane and cyclopropane (13.5 kcal/mol).[16] Therefore, functionalizations of benzylidenecyclobutane are expected to suffer from a much lower thermodynamic driving force due to the decreased ring strain energy release. Indeed, the Shi group reported that benzylidenecyclobutane did not react well under their reaction conditions for aminoboration of benzylidenecyclopropanes.[14a]
With these considerations in mind, we reasoned that the low reactivity of benzylidenecyclobutanes could be overcome through the use of a tailored ancillary ligand on the Cu catalyst, which would fine-tune its steric and electronic properties and enhance non-covalent interactions with the substrate.[17] To this end, we conducted two-dimensional screening with diverse phosphine ligands and five representative benzylidene carbo/heterocycles: cyclobutane (1a), azetidine (1b), oxetane (1c), thietane 1,1-dioxide (1d), 3,3-difluorocyclobutane (1e) (Table 1). It is worth mentioning that these substrates were synthesized through simple Wittig reactions from inexpensive and commercially available starting materials. Under the previously published hydroboration reaction conditions, the simplest ligand, triphenylphosphine (PPh3), gave only trace amounts of products (entry 1).[14b] Notably, tri(pentafluorophenyl)phosphine ((C6F5)3P) and BINAP, which were successfully used for our previous cyclopropane system, were ineffective in the cyclobutane system, although BINAP gave moderate yields for more electronically activated 1b and 1c (entries 2–3).[14b] N-Heterocyclic carbene (NHC ligands) were briefly considered, but they did not significantly improve yields compared to BINAP in model studies (see Supporting Information). After further screening with bis-phosphine ligands (entries 4–6), we found that 1,2-bis(diphenylphosphino)benzene ligand (dppbz) gave improved results for all substrates. Inspired by recent reports describing the use of modified dppbz ligands in related transformations,[18] we envisioned that modification of the electronic property of dppbz ligands without changing the 1,2-bis(phosphino)benzene backbone could enhance the product yields. Indeed, para-fluorine substituted dppbz ligand (4-F-dppbz, entry 7) increased the yields of desired products for all substrates. More strongly electron-withdrawing 4-CF3-dppbz ligand further increased the product yield with the cyclobutane substrate but decreased the yields with substrates 1d and 1e (entry 8). Additional fluorine substitution on the meta-position (entry 9) led to diminished yields. Notably, introduction of an electron-donating group (entry 10) or a sterically bulky tert-butyl group in the meta-position (entry 11) significantly decreased the yield of the reaction. Across these optimization experiments, we did not observe any evidence of undesired side reactions, such as ring-opening; typically only unreacted starting materials along with the desired products could be detected.
Table 1.
Ligand optimization with representative substrates
 | ||||||
|---|---|---|---|---|---|---|
| entry | ligand |  |
 |
 |
 |
 |
| 1 | PPh3 | 2 | 4 | 3 | 6 | < 1 |
| 2 | (C6F5)3P | < 1 | < 1 | < 1 | 10 | < 1 |
| 3 | BINAP | 2 | 66 | 61 | 4 | 5 |
| 4 | dppe | 36 | 45 | 79 | 33 | 4 |
| 5 | dppp | 10 | 50 | 85 | 26 | 12 |
| 6 | dppbz | 49 | 88 | 91 | 34 | 29 |
| 7 | 4-F-dppbz | 62 | 95 | 97 | 53 | 74 |
| 8 | 4-CF3-dppbz | 74 | 99 | 99 | 29 | 33 |
| 9 | 3,4,5-F-dppbz | 15 | 72 | 67 | 6 | 32 |
| 10 | 4-OMe-dppbz | 17 | 39 | 13 | 45 | 18 |
| 11 | 3,5-t-Bu-dppbz | < 1 | 9 | 5 | 41 | 52 |
 | ||||||
Reaction conditions: 1 (0.2 mmol), CuCl (10 mol%), ligand (15 mol%), B2(pin)2 (0.3 mmol), NaOt-Bu (0.2 mmol), MeOH (0.2 mmol) in THF (0.5 mL) at room temperature.
Yields were determined by 1H NMR with the use of 1,3,5-trimethoxybenzene as an internal standard.
We explored the arene substituent scope using 4-F-dppbz as the ligand (Scheme 1). Interestingly, we observed a clear trend that electron-deficient aryl groups gave higher yields of the desired products than electron-rich aryl groups. Substrates containing electron-withdrawing trifluoromethyl or fluoride groups gave the desired products in good to excellent isolated yields (2f, 2g, 2l and 2m). On the other hand, more electron-donating substituents resulted in diminished reactivity (2h–2j); in particular, the highly electron-rich -NMe2 substrate was unreactive (2k). For low-yielding cases, unreacted starting material was observed as the major component of the crude reaction mixture. The cyano group was well-tolerated (2n), and an electron-donating -OMe group at the meta-position also delivered moderate yield (2o). In certain low-yielded cases, the 4-CF3-dppbz ligand provided moderately higher yields (2h, 2j and 2o; yields with 4-CF3-dppbz as ligand in parentheses).
Scheme 1.
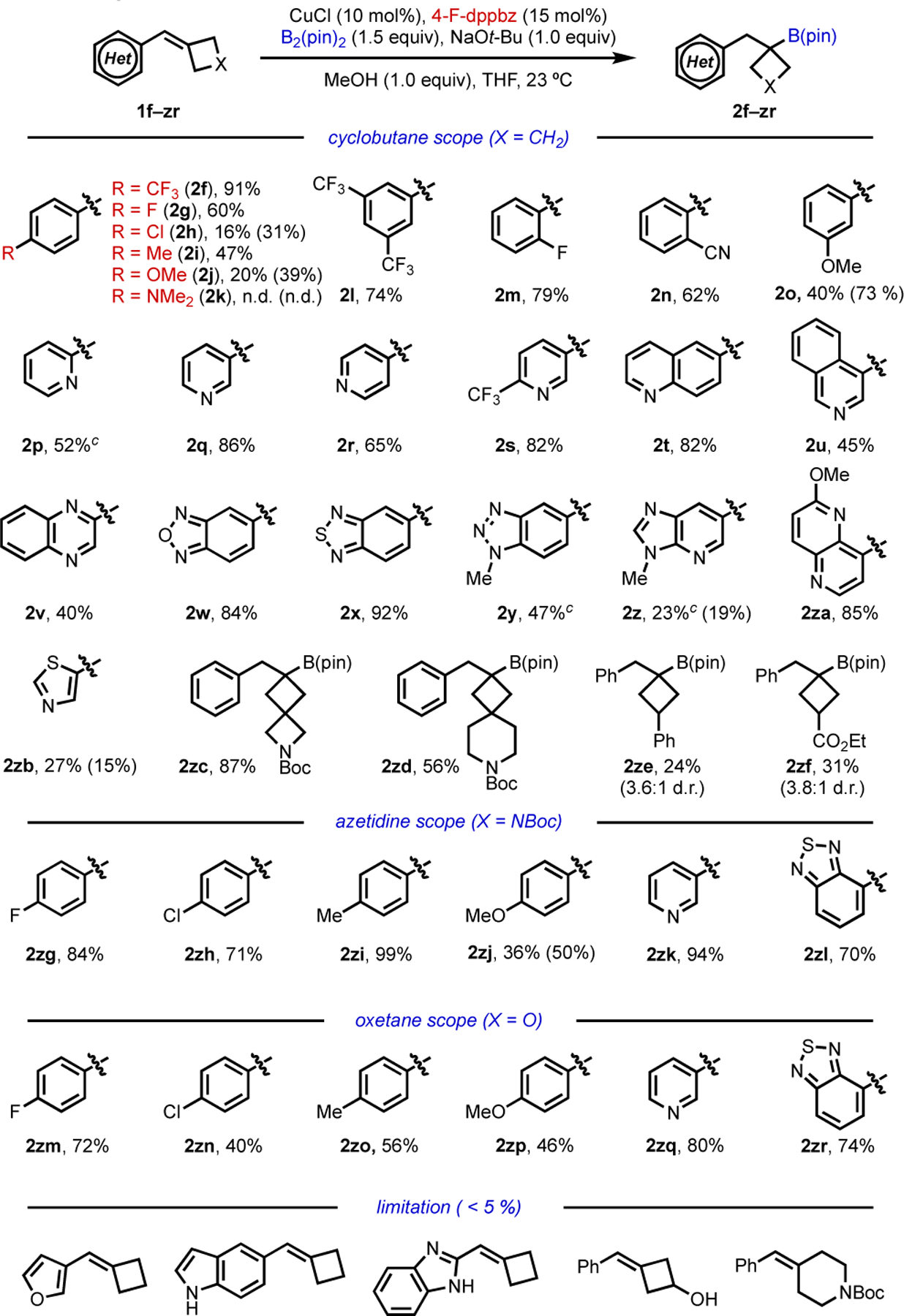
Hydroboration scope with substituted arenes and heterocycles.
aReaction conditions: 1 (0.2 mmol), CuCl (10 mol%), 4-F-dppbz (15 mol%), B2(pin)2 (0.3 mmol), NaOt-Bu (0.2 mmol), MeOH (0.2 mmol) in THF (0.5 mL) at room temperature. Percentages refer to the isolated yields of products. bThe values in parentheses correspond to isolated yields with 4-CF3-dppbz as ligand in place of 4-F-dppbz; d.r.=diasteromeric ratio. cThe final product was oxidized to the corresponding alcohol for ease of isolation.
Many heterocycles commonly employed in drug discovery were well tolerated under the reaction conditions. Ortho-, meta-, and para-substituted pyridines, the most common nitrogen-containing heterocycle, gave the products in good to excellent yields (2p–2s). We further explored more complex heterocycles, namely quinoline (2t), isoquinoline (2u), quinoxaline (2v), benzoxadiazole (2w), benzothiadiazole (2x), benzotriazole (2y), imidazolopyridine (2z), 1,5-naphthyridine (2za), and thiazole (2zb), which gave corresponding products in synthetically viable to excellent yields. In addition, benzylidene spirocyclic compounds also performed well under the reaction condition (2zc and 2zd). Substrates containing substituted cyclobutanes also furnished the desired products, albeit in low yields with moderate d.r. (2ze and 2zf).
In general, azetidine and oxetane substrates provided higher yields than cyclobutane substrates, presumably because the alkenes are more electronically activated due to the inductive effect of the heteroatom. Both azetidine and oxetane substrates containing para-fluoro, chloro, methyl, and methoxy substituents gave moderate to excellent yields (2zg–2zj and 2zm–2zp), and heterocycles were compatible as well (2zi, 2zj and 2zo, 2zr). The method is not without its limitations; notably, substrates containing an electron-rich furan ring or NH-containing azaheterocycles or free alcohol proved to be unreactive (<5% 1H NMR yield). Similarly, benzylidenepiperidine substrate could not be functionalized under these conditions.
We next turned our attention to the non-aryl containing substrates (Scheme 2). Notably, simple alkylidene azetidine substrate was unreactive under the reaction condition (2zs). On the other hand, electronically activated cyclobutylidene by ester group gave the desired product in excellent yield (2zt).[14c] Alkenylboronic ester and alkenylsilane substrates which have used in regioselective copper-catalyzed hydroboration were also evaluated, and moderate yield was obtained in alkenylboronic ester case while alkenylsilane was not reactive (2zu and 2zv).[14d, 14e]
Scheme 2.

Hydroboration scope with non-aryl containing substrates.
aReaction conditions: 1 (0.2 mmol), CuCl (10 mol%), 4-F-dppbz (15 mol%), B2(pin)2 (0.3 mmol), NaOt-Bu (0.2 mmol), MeOH (0.2 mmol) in THF (0.5 mL) at room temperature. Percentages refer to the isolated yields of products.
To highlight the utility of the tertiary boronic ester products, we prepared product 2b on gram scale and demonstrated several diversification reactions (Scheme 3). Oxidation to the corresponding tertiary alcohol 3 occurred in 93% yield.[19] A Zweifel–Aggarwal vinylation delivered desired product 4, and a Matteson homologation furnished primary boronic ester 5 in modest yield.[20, 21] Conversion to the tertiary trifluoroborate salt 6 occurred in 95% yield.[22] In addition, using trifluoroborate 6 as a tertiary radical precursor, Minisci[23] and Giese reactions[24] were performed to give the corresponding C–C coupled products 7 and 8 in synthetically useful yields.
Scheme 3.

Selected derivatization reaction with tertiary boronic ester 2b.
The crucial importance of the modified dppbz ligands and the pronounced reactivity trends based on substrate electronic properties prompted us to examine the mechanism of this transformation through kinetic and computational studies. First, in order to establish the turnover-limiting step, we measured the initial rates of four electronically different benzylidenecyclobutane substrates (1a, 1f, 1i and 1j) by 1H NMR (Figure 2A). Mirroring the yield trends noted above, substrate 1f containing an electron-withdrawing group reacted with faster initial rate than electron-neutral 1a or electron-rich 1i and 1j. This trend indicates that negative charge builds up at the benzylic carbon during the turnover-limiting step in the catalytic cycle, consistent with this step being migratory insertion since it is the only step in the catalytic cycle that would be expected to be accelerated by an electron-withdrawing group on the arene. We visualized this rate data using a Hammett plot and observed a positive ρ-value (Figure 2B). The data deviated from linearity in the case of substrate 1f, suggesting that the turnover-limiting step may have changed in the case of 1f, where migratory insertion is presumably fast. We next studied the effect of ligand modifications on the initial rate of 1a by comparing plain dppbz and modified dppbz ligands. 4-CF3 and 4-F-dppbz ligands clearly showed faster initial rates than plain dppbz ligand (see Figure S3 in the SI). Additionally, we attempted to examine ligand effects by preparing and characterizing the Cl-bridged Cu–ligand complexes with each of these three ligands. However, we did not observe clear trends from comparison of the X-ray crystal structures and 31P NMR shifts (see Table S11 in the SI).
Figure 2.

Hammett analysis of hydroboration of substituted benzylidenecyclobutanes.
Computational Studies.
To investigate the reaction mechanisms and the origin of ligand and substrate effects on reactivity, we performed density functional theory (DFT) calculations at the M06/SDD-6–311+G(d,p)/SMD(THF)//M06L/LANL2DZ-6–31G(d) level of theory. We computed the reaction energy profiles based on the proposed reaction mechanisms shown in Scheme 4 (see Scheme S1–S6 in the SI). The productive catalytic cycle involves the σ-bond metathesis of LCuOMe (IM1) with B2(pin)2 to form copper(I)–boryl species IM2, which undergoes migratory insertion of 1a to form benzylic Cu(I) intermediate IM3. Protodecupration of IM3 yields the boronic ester and regenerates the copper(I)–alkoxy complex IM1.
Scheme 4.

Proposed reaction mechanism of hydroboration of benzylidenecyclobutanes.
We first investigated the origins of product selectivity in the hydroboration of benzylidenecyclobutanes. Unlike the previously reported hydroboration of benzylidenecylcopropanes,[14b] the above experiments reveal that benzylidenecyclobutanes do not readily undergo β-carbon elimination (TS2, Scheme 4) from the benzylic Cu(I) intermediate IM3 to form the putative ring-opened alkenyl boronate products (9), despite the similar ring strain energies of cyclopropane and cyclobutane rings (27.5 and 26.3 kcal/mol, respectively). Our calculations on the reaction energy profiles of the hydroboration of benzylidenecyclobutane 1a with 4-CF3-dppbz-supported Cu catalyst suggest the β-carbon elimination (TS2a) is disfavored by 1.3 and 11.3 kcal/mol in terms of activation free energy and activation enthalpies, respectively, compared to the competing protodecupration pathway (TS3a). Although both TS3a and TS2a adopt a tetrahedral geometry around the copper center, the β-carbon elimination transition state (TS2a) is more sensitive to steric effects from the ligand due to a greater substrate binding angle (∠CCuO = 74.4° in TS3a compared to ∠CCuC = 88.2° in TS2a). Therefore, steric repulsions between the substrate and the ligand destabilize the protodecupration transition state (Figure 3). (For the full reaction energy profiles and a more detailed comparison of benzylidenecyclobutanes (BCBs) to benzylidenecyclopropanes (BCPs), see SI).
Figure 3.
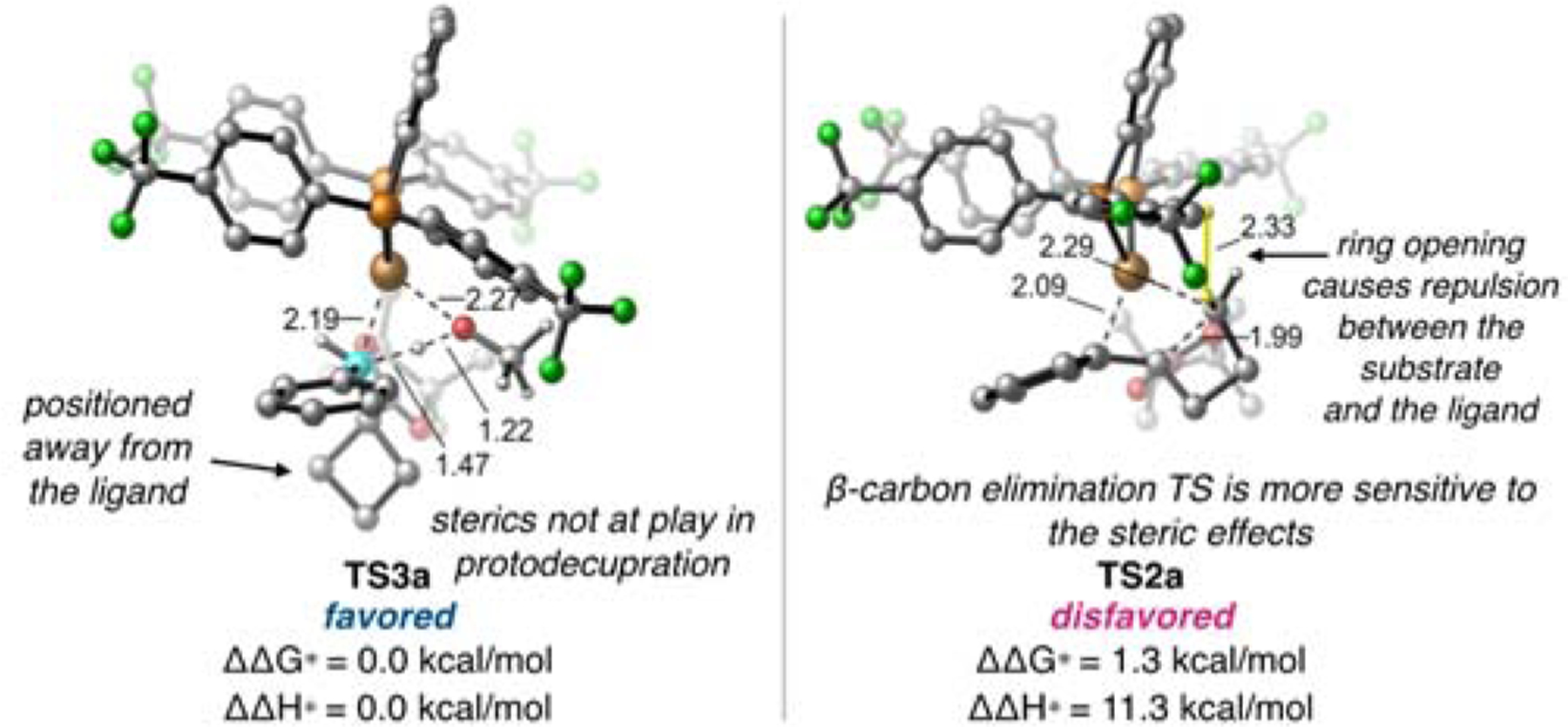
Protodecupration (left) and β-carbon elimination (right) transition states with 4-CF3-dppbz ligand.
Next, we turned our attention to the origins of ligand and substrate effects on the barriers to alkene migratory insertion, since the experimental mechanistic studies (vide infra) suggested this is the turnover-limiting step of the catalytic cycle. Previous reports and our DFT calculations (see SI) indicated several stable dimeric and oligomeric copper(I)–alkoxide[25] or copper(I)–boryl species (e.g. i and ii, see Scheme 4) may be the off-cycle resting state(s) before the migratory insertion step. The computed energies required to convert these off-cycle complexes to the monomeric copper(I)–boryl species (IM2) do not correlate with the experimental reactivity, indicating the observed reactivity trend is not a result of dimer or oligomer dissociation. On the other hand, the calculated activation free energies of the migratory insertion transition states with respect to the monomeric copper(I)–boryl species agree well with the experimentally determined relative reactivities (Figure 4b and 4c), indicating the reactivity is mainly affected by the stability of the migratory insertion transition state. To gain more insights into factors that promote the catalyst/substrate interactions in this step, we sought to understand the differences in reactivity via the ligand–substrate interaction model analysis. Using an energy decomposition approach reported earlier,[17a, 17b] the computed activation energy for each reaction is dissected using the following equation:
where the distortion energy (ΔEdist) is the sum of the energies required to distort the LCuB(pin) complex and the substrate into their transition state geometries. The through-space interaction energy between the phosphine ligand and the benzylidenecyclobutane substrate (ΔEint-space) is calculated from the interaction energy of a hypothetical supramolecular complex of the ligand and the substrate at the transition state geometry in the absence of the CuB(pin) moiety. The rest of the catalyst/substrate interaction energy is defined as the through-bond interaction (ΔEint-bond) between the LCuB(pin) and the substrate in the transition state. Using the second-generation ALMO-EDA method implemented in Q-Chem 5.2, the through-space interaction energy (ΔEint-space) is further dissected according to the following equation:
where ΔEelstat, ΔEpauli, ΔEdisp, ΔEpol, and ΔEct correspond to several different types of non-covalent interactions, namely electrostatic, Pauli repulsions, dispersion interactions, intrafragment polarization, and interfragment charge transfer, respectively.
Figure 4.

Summary of ligand-substrate interactions and energy decomposition analysis.
The EDA calculations revealed the dominant factors that control the reactivity trends in reactions with different ligands (Figure 4b) and with different para-substituted benzylidenecyclobutanes (Figure 4c). The higher reactivity with the 4-CF3-dppbz ligand (entry 8, Table 1) compared to those with dppbz and 4-F-dppbz is primarily due to the more favorable electrostatic interactions between the ligand and the substrate (ΔEelstat, Figure 4b). Examination of the migratory insertion transition state geometries indicated an edge-to-face (T-shaped) interaction between one of the P–Ar groups on the ligand and the phenyl group on the substrate (Figure 4a). Therefore, an electron-withdrawing substituent on the “edge” arene (i.e., on the ligand) would enhance the T-shaped π/π interaction through more favorable electrostatic interactions.[26] On the other hand, when the electron-withdrawing substituent is installed on the “face” arene (i.e., on the benzylidenecyclobutane), the T-shaped π/π interaction becomes weaker, as evidenced by the slightly less favorable ΔEelstat term in the reaction with 1f (R’ = CF3) than that with 1a (R’ = H) and 1j (R’ = OCH3) (Figure 4c). Therefore, although the ligand effects on reactivity are controlled by the T-shaped π/π interaction between the ligand and the substrate, such interactions are not the dominant factor leading to the higher reactivity of electron-deficient benzylidenecyclobutanes (Figure 2). The EDA calculations show that the transition state with 1f (R = CF3) is stabilized by multiple factors, including smaller distortion energy (ΔEdist) and Pauli repulsion energy (ΔEpauli), as well as more favorable through-bond interaction energy (ΔEint-bond) (Figure 4c). These results indicate that the reaction with 1f (R = CF3) has an earlier transition state with diminished catalyst/substrate distortion and steric repulsions. Although the transition state is early, the bonding interaction (ΔEint-bond) between LCuB(pin) and the electron-deficient benzylidenecyclobutane (1f) is still the strongest among the three substrates because of the more electron-deficient π-cloud that promotes migratory insertion.[27]
In conclusion, we have demonstrated that copper catalyzed hydroboration of benzylidene four-membered rings to afford synthetically useful tertiary boronic ester products. The role of modified 4-F and 4-CF3-dppbz ligands in enhancing catalytic reactivity was elucidated through kinetic and computational studies. Specifically, DFT and EDA calculations revealed the T-shaped π/π interactions between the ligand and the substrate influence the reactivity with different modified dppbz ligands. On the other hand, the reactivity differences of substituted benzylidenecyclobutane substrates are mainly affected by the through-bond interactions between the catalyst and the substrates with varying electronic properties. A refined understanding of these types of non-covalent interactions in catalysis offers exciting opportunities in rationally designing ligands that incorporate requisite substrate recognition motifs to amplify reactivity.
Supplementary Material
ACKNOWLEDGMENT
This work was financially supported by the National Institutes of Health (5R35GM125052-02; R35GM128779), The Scripps Research Institute, and Pfizer, Inc. We gratefully acknowledge the Kwanjeong Educational Foundation (Graduate Fellowship to T.K.) Calculations were performed at the Center for Research Computing at the University of Pittsburgh, the Extreme Science and Engineering Discovery Environment (XSEDE), and the TACC Frontera supercomputers.
Footnotes
The authors declare no competing financial interest.
Supporting Information
Detailed experimental and computational procedures, compound characterization, Cartesian coordinates of the calculated structures. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Liebman JF; Greenberg A A Survey of Strained Organic Molecules. Chem. Rev 1976, 76, 311–365. [Google Scholar]; (b) Marson CM New and Unusual Scaffolds in Medicinal Chemistry. Chem. Soc. Rev 2011, 40, 5514–5533. [DOI] [PubMed] [Google Scholar]; (c) Brandi A; Cicchi S; Cordero FM Novel Syntheses of Azetidines and Azetidinones. Chem. Rev 2008, 108, 3988–4035. [DOI] [PubMed] [Google Scholar]
- (2).(a) St. Jean DJ Jr.; Fotsch C Mitigating Heterocycle Metabolism in Drug Discovery. J. Med. Chem 2012, 55, 6002–6020. [DOI] [PubMed] [Google Scholar]; (b) Bull JA; Croft RA; Davis OA; Doran R; Morgan KF Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev 2016, 116, 12150–12233. [DOI] [PubMed] [Google Scholar]; (c) Izquierdo S; Rúa F; Sbai A; Parella T; Álvarez-Larena Á; Branchadell V; Ortuño RM (+)- and (−)-2-Aminocyclobutane-1-carboxylic Acids and Their Incorporation into Highly Rigid β-Peptides: Stereoselective Synthesis and a Structural Study. J. Org. Chem 2005, 70, 7963–7971. [DOI] [PubMed] [Google Scholar]
- (3).(a) Wuitschik G; Carreira EM; Wagner B; Fischer H; Parrilla I; Schuler F; Rogers-Evans M; Müller K Oxetanes in Drug Discovery: Structural and Synthetic Insights. J. Med. Chem 2010, 53, 3227–3246. [DOI] [PubMed] [Google Scholar]; (b) Meanwell NA Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- (4).Wager TT; Pettersen BA; Schmidt AW; Spracklin DK; Mente S; Butler TW; Howard H Jr.; Lettiere DJ; Rubitski DM; Wong DF; Nedza FM; Nelson FR; Rollema H; Raggon JW; Aubrecht J; Freeman JK; Marcek JM; Cianfrogna J; Cook KW; James LC; Chatman LA; Iredale PA; Banker MJ; Homiski MK; Munzner JB; Chandrasekaran RY Discovery of Two Clinical Histamine H3 Receptor Antagonists: trans-N-Ethyl-3-fluoro-3-[3-fluoro-4-(pyrrolidinylmethyl)- phenyl]cyclobutanecarboxamide (PF-03654746) and trans-3-Fluoro-3-[3-fluoro-4-(pyrrolidin-1-ylmethyl)phenyl]-N-(2-methylpropyl)cyclobutanecarboxamide (PF-03654764). J. Med. Chem 2011, 54, 7602–7620. [DOI] [PubMed] [Google Scholar]
- (5).Mulvihill MJ; Cooke A; Rosenfeld-Franklin M; Buck E; Foreman K; Landfair D; O’Connor M; Pirritt C; Sun Y; Yao Y; Arnold LD; Gibson NW; Ji Q-S Discovery of OSI-906: A Selective and Orally Efficacious Dual Inhibitor of the IGF-1 Receptor and Insulin Receptor. Future Med. Chem 2009, 1, 1153–1171. [DOI] [PubMed] [Google Scholar]
- (6).Smolen JS; Genovese MC; Takeuchi T; Hyslop DL; Macias WL; Rooney T; Chen L; Dickson CL; Camp JR; Cardillo TE; Ishii T; Winthrop KL J. Rheum 2019, 46, 7–18. [DOI] [PubMed] [Google Scholar]
- (7).Salphati L; Heffron TP; Alicke B; Nishimura M; Barck K; Carano RA; Cheong J; Edgar KA; Greve J; Kharbanda S; Koeppen H; Lau S; Lee LB; Pang J; Plise EG; Pokorny JL; Reslan HB; Sarkaria JN; Wallin JJ; Zhang X; Gould SE; Olivero AG; Philllips HS Targeting the PI3K Pathway in the Brain—Efficacy of a PI3K Inhibitor Optimized to Cross the Blood–Brain Barrier. Clin. Cancer Res 2012, 18, 6239–6248. [DOI] [PubMed] [Google Scholar]
- (8).Clement HA; Boghi M; McDonald RM; Bernier L; Coe JW; Farrell W; Helal CJ; Reese MR; Sach NW; Lee JC; Hall DG High-Throughput Ligand Screening Enables the Enantioselective Conjugate Borylation of Cyclobutenones to Access Synthetically Versatile Tertiary Cyclobutylboronates. Angew. Chem. Int. Ed 2019, 58, 18405–18409. [DOI] [PubMed] [Google Scholar]
- (9).(a) Fawcett A; Pradeilles J; Wang Y; Mutsuga T; Myers EL; Aggarwal VK Photoinduced decarboxylative borylation of carboxylic acids. Science 2017, 357, 283–286. [DOI] [PubMed] [Google Scholar]; (b) Baran PS; Li C; Wang J; Chatterjee AK; Kumar M; Yu S; Johnson KA; Qin T; Shang M Cu- and Ni-Catalyzed Decarboxylative Borylation Reactions. U.S. Patent WO 2018175173, September 27, 2018. [Google Scholar]; (c) Friese FW; Studer A Deoxygenative Borylation of Secondary and Tertiary Alcohols. Angew. Chem. Int. Ed 2019, 58, 9561–9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Hong K; Liu X; Morken JP Simple Access to Elusive α‐Boryl Carbanions and Their Alkylation: An Umpolung Construction for Organic Synthesis. J. Am. Chem. Soc 2014, 136, 10581–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu X; Ming W; Zhang Y; Friedrich A; Marder TB Copper-Catalyzed Triboration: Straightforward, Atom-Economical Synthesis of 1,1,1-Triborylalkanes from Terminal Alkynes and HBpin. Angew. Chem. Int. Ed 2019, 58, 18923–18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Fawcett A; Biberger T; Aggarwal VK Carbopalladation of C–C σ-bonds enabled by strained boronate complexes. Nat. Chem 2019, 11, 117–122. [DOI] [PubMed] [Google Scholar]; (b) Silvi M; Aggarwal VK Radical Addition to Strained σ‐Bonds Enables the Stereocontrolled Synthesis of Cyclobutyl Boronic Esters. J. Am. Chem. Soc 2019, 141, 9511–9515. [DOI] [PubMed] [Google Scholar]; (c) Wang D; Mück-Lichtenfeld C; Studer A Hydrogen Atom Transfer Induced Boron Retaining Coupling of Organoboronic Esters and Organolithium Reagents. J. Am. Chem. Soc 2019, 141, 14126–14130; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fawcett A; Murtaza A; Gregson CHU; Aggarwal VK Strain-Release-Driven Homologation of Boronic Esters: Application to the Modular Synthesis of Azetidines. J. Am. Chem. Soc 2019, 141, 4573–4578; [DOI] [PubMed] [Google Scholar]; (e) Bennett SH; Fawcett A; Denton EH; Biberger T; Fasano V; Winter N; Aggarwal VK Difunctionalization of C–C σ-Bonds Enabled by the Reaction of Bicyclo[1.1.0]butyl Boronate Complexes with Electrophiles: Reaction Development, Scope, and Stereochemical Origins. J. Am. Chem. Soc 2020, 142, 16766–16775; [DOI] [PubMed] [Google Scholar]; (f) Guo L; Noble A; Aggarwal VK α‐Selective Ring‐Opening Reactions of Bicyclo[1.1.0]butyl Boronic Ester with Nucleophiles. Angew. Chem. Int. Ed 2020, DOI: 10.1002/anie.202011739. [DOI] [PubMed] [Google Scholar]
- (12).For miscellaneous methodologies, see:; (a) Shu C; Noble A; Aggarwal VK Photoredox-Catalyzed Cyclobutane Synthesis by a Deboronative Radical Addition–Polar Cyclization Cascade. Angew. Chem. Int. Ed 2019, 58, 3870–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davenport R; Silvi M; Noble A; Hosni Z; Fey N; Aggarwal VK Visible Light-Driven Strain-Increase Ring Contraction Allows the Synthesis of Cyclobutyl Boronic Esters. Angew. Chem. Int. Ed 2020, 59, 6525–6528. [DOI] [PubMed] [Google Scholar]; (c) Wang X; Li L; Gong T; Xiao B; Lu X; Fu Y Vicinal Diboration of Alkyl Bromides via Tandem Catalysis. Org. Lett 2019, 21, 4298–4302. [DOI] [PubMed] [Google Scholar]; (d) Hollis WG Jr.; Lappenbusch WC; Everberg KA; Woleben CM The Use of Alkenylboronate Esters in [2+2] Enone-Olefin Photocycloadditions. Tetrahedron Lett. 1993, 34, 7517–7520. [Google Scholar]; (e) Conner ML; Brown MK Synthesis of 1,3-Substituted Cyclobutanes by Allenoate-Alkene [2 + 2] Cycloaddition. J. Org. Chem 2016, 81, 8050–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Giustra ZX; Yang X; Chen M; Bettinger HF; Liu S-Y Accessing 1,2-Substituted Cyclobutanes through 1,2-Azaborine Photoisomerization. Angew. Chem. Int. Ed 2019, 58, 18918–18922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen L; Lear AR; Gao P; Brown MK Nickel-Catalyzed Arylboration of Alkenylarenes: Synthesis of Boron- Substituted Quaternary Carbons and Regiodivergent Reactions. Angew. Chem. Int. Ed 2019, 58, 10956–10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Jiang H; Tang X; Shi M Copper-Catalyzed Regio- and Enantioselective Aminoboration of Alkylidenecyclopropanes: The Synthesis of Cyclopropane-Containing β-Aminoalkylboranes. Chem. Commun 2016, 52, 5273–5276. [DOI] [PubMed] [Google Scholar]; (b) Medina JM; Kang T; Erbay TG; Shao H; Gallego GM; Yang S; Tran-Dubé M; Richardson PF; Derosa J; Helsel RT; Patman RL; Wang F; Ashcroft CP; Braganza JF; McAlpine I; Liu P; Engle KM Cu-Catalyzed Hydroboration of Benzylidenecyclopropanes: Reaction Optimization, (Hetero)Aryl Scope, and Origins of Pathway Selectivity. ACS Catal. 2019, 9, 11130–11136; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mun S; Lee J; Yun J Copper-Catalyzed β-Boration of α,β-Unsaturated Carbonyl Compounds: Rate Acceleration by Alcohol Additives. Org. Lett 2006, 8, 4887–4889; [DOI] [PubMed] [Google Scholar]; (d) Lee Y; Jang H; Hoveyda AH Vicinal Diboronates in High Enantiomeric Purity through Tandem Site-Selective NHC–Cu-Catalyzed Boron–Copper Additions to Terminal Alkynes. J. Am. Chem. Soc 2009, 131, 18234–18235; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ito H; Kosaka Y; Nonoyama K; Sasaki Y; Sawamura M Synthesis of Optically Active Boron–Silicon Bifunctional Cyclopropane Derivatives through Enantioselective Copper(I)-Catalyzed Reaction of Allylic Carbonates with a Diboron Derivative. Angew, Chem. Int. Ed 2009, 47, 7424–7427. [DOI] [PubMed] [Google Scholar]
- (15).For a recent review of copper–boryl catalysis, see:; Hemming D, Fritzemeier R, Westcott SA, Santos WL, Steel PG, Copper-Boryl Mediated Organic Synthesis. Chem. Soc. Rev 2018, 47, 7477–7494. [DOI] [PubMed] [Google Scholar]
- (16).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Book: California, 2006; pp 110–112. [Google Scholar]
- (17).(a) Lu G; Liu RY; Yang Y; Fang C; Lambrecht DS; Buchwald SL; Liu P Ligand–Substrate Dispersion Facilitates the Copper-Catalyzed Hydroamination of Unactivated Olefins. J. Am. Chem. Soc 2017, 139, 16548–16555. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thomas AA; Speck K; Kevlishvili I; Lu Z; Liu P; Buchwald SL Mechanistically Guided Design of Ligands That Significantly Improve the Efficiency of CuH-Catalyzed Hydroamination Reactions. J. Am. Chem. Soc 2018, 140, 13976–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Saper NI, Ohgi A, Small DW, Semba K, Nakao Y, Hartwig JF Nickel-catalysed anti-Markovnikov hydroarylation of unactivated alkenes with unactivated arenes facilitated by non-covalent interactions. Nat. Chem 2020, 12, 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Neel AJ, Hilton MJ, Sigman MS Toste DF Exploiting non-covalent π interactions for catalyst design. Nature 2017, 543, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Hatakeyama T; Hashimoto T; Kondo Y; Fujiwara Y; Seike H; Takaya K; Tamada Y; Ono T; Nakamura M Iron-Catalyzed Suzuki–Miyaura Coupling of Alkyl Halides. J. Am. Chem. Soc 2010, 132, 10674–10676. [DOI] [PubMed] [Google Scholar]; (b) Ito S; Itoh T; Nakamura M Diastereoselective Carbometalation of Oxa- and Azabicyclic Alkenes under Iron Catalysis. Angew. Chem. Int. Ed 2011, 50, 454–457. [DOI] [PubMed] [Google Scholar]; (c) Miki Y; Hirano K; Satoh T; Miura M Copper-Catalyzed Intermolecular Regioselective Hydroamination of Styrenes with Polymethylhydrosiloxane and Hydroxylamines. Angew. Chem. Int. Ed 2013, 52, 10830–10834. [DOI] [PubMed] [Google Scholar]; (d) Nishikawa D; Hirano K; Miura M Copper-Catalyzed Regio- and Stereoselective Aminoboration of Alkenylboronates. Org. Lett 2016, 18, 4856–4859. [DOI] [PubMed] [Google Scholar]; (e) Nishikawa D; Sakae R; Miki Y; Hirano K; Miura M Copper-Catalyzed Regioselective Ring-Opening Hydroamination of Methylenecyclopropanes. J. Org. Chem 2016, 81, 12128–12138. [DOI] [PubMed] [Google Scholar]; (f) Iwamoto H; Kubota K; Ito H Highly Selective Markovnikov Hydroboration of Alkyl-Substituted Terminal Alkenes with a Phosphine-Copper(I) Catalyst. Chem. Commun 2016, 52, 5916–5919. [DOI] [PubMed] [Google Scholar]; (g) Kato K; Hirano K; Miura M Synthesis of β-Boryl-α-Aminosilanes by Copper-Catalyzed Aminoboration of Vinylsilanes. Angew. Chem. Int. Ed 2016, 55, 14400–14404. [DOI] [PubMed] [Google Scholar]; (h) Fujihara T; Sawada A; Yamaguchi T; Tani Y; Terao J; Tsuji Y Boraformylation and Silaformylation of Allenes. Angew. Chem. Int. Ed 2017, 56, 1539–1543. [DOI] [PubMed] [Google Scholar]; (i) Kato K; Hirano K; Miura M Copper/Bisphosphine Catalysts in the Internally Borylative Aminoboration of Unactivated Terminal Alkenes with Bis(pinacolato)diboron. J. Org. Chem 2017, 82, 10418–10424. [DOI] [PubMed] [Google Scholar]; (j) Takata T; Hirano K; Miura M Synthesis of α-Trifluoromethylamines by Cu-Catalyzed Regio- and Enantioselective Hydroamination of 1-Trifluoromethylalkenes. Org. Lett 2019, 21, 4284–4288. [DOI] [PubMed] [Google Scholar]; (k) Nishino S; Hirano K; Miura M Copper-Catalyzed Electrophilic Amination of gem-Diborylalkanes with Hydroxylamines Providing α-Aminoboronic Acid Derivatives. Org. Lett 2019, 21, 4759–4762. [DOI] [PubMed] [Google Scholar]
- (19).Farthing CN; Marsden SP Chiral Vinyl Dioxazaborocines in Synthesis: Asymmetric Cuprate Additions to β-boronyl Acrylates and Vinyl Sulfones. Tetrahedron Lett. 2000, 41, 4235–4238. [Google Scholar]
- (20).(a) Zweifel G; Arzoumanian H; Whitney CC A Convenient Stereoselective Synthesis of Substituted Alkenes via Hydroboration-Iodination of Alkynes. J. Am. Chem. Soc 1967, 89, 3652–3653. [Google Scholar]; (b) Sonawane RP; Jheengut V; Rabalakos C; Larouche-Gauthier R; Scott HK; Aggarwal VK Enantioselective Construction of Quaternary Stereogenic Centers from Tertiary Boronic Esters: Methodology and Applications. Angew. Chem. Int. Ed 2011, 50, 3760–3763. [DOI] [PubMed] [Google Scholar]
- (21).(a) Matteson DS α-Halo Boronic Esters: Intermediates for Stereodirected Synthesis. Chem. Rev 1989, 89, 1535–1551. [Google Scholar]; (b) Sadhu KM; Matteson DS (Chloromethyl)lithium: Efficient Generation and Capture by Boronic Esters and a Simple Preparation of Diisopropyl (Chloromethyl)boronate. Organometallics 1985, 4, 1687–1689. [Google Scholar]
- (22).Vedejs E; Chapman RW; Fields SC; Lin S; Schrimpf MR Conversion of Arylboronic Acids into Potassium Aryltrifluoroborates: Convenient Precursors of Arylboron Difluoride Lewis Acids. J. Org. Chem 1995, 60, 3020–3027. [Google Scholar]
- (23).Molander GA; Colombel V; Braz VA Direct Alkylation of Heteroaryls Using Potassium Alkyl- and Alkoxymethyltrifluoroborates. Org. Lett 2011, 13, 1852–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Greiser T; Weiss E Kristallstruktur des kupfer(I)-tert-butoxids, [(CH3)3COCu]4. Chem. Ber 1976, 109, 3142–3146. [Google Scholar]
- (26).Wheeler SE; Houk KN Origin of Substituent Effects in Edge-to-Face Aryl–Aryl Interactions. Mol. Phys 2009, 107, 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dang L; Zhao H; Marder T DFT Studies of Alkene Insertions into Cu–B Bonds in Copper(I) Boryl Complexes. Organometallics 2007, 26, 2824–2832. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.