

Description
A 3-year-old boy born to consanguineously married couple (third cousins) presented with features of regression of attained developmental milestones. There was normal development till age 14 months when the child seemingly lost ability to sit without and stand with support. At 3 years of age, the child needed support for sitting, was unable to hold objects and communicated only with signs. There was similar history in son and daughter of maternal uncle with daughter succumbing to her symptoms at 4 years of age. On examination, all four limbs’ muscle tone was increased with exaggerated deep tendon reflexes. Electroencephalogram was normal. Patient was referred for MRI of head.
MRI of brain was performed without intravenous contrast on a 3 T machine (Signa HDxt 3.0T, GE Healthcare, Milwaukee, Wisconsin, USA). MRI showed severe symmetrical bilateral cerebellar atrophy (figure 1) with parenchymal hyperintensity, suggestive of gliosis. Claval hypertrophy (nucleus gracilis hypertrophy) was noted with severe optic chiasma atrophy (figure 1). ‘Drooping’ of posterior half of corpus callosum was seen. Susceptibility-weighted imaging showed symmetrical hypointensities of bilateral globus pallidi and substantia nigra (figure 2), suggestive of mineral deposition which was abnormal for age. MRI findings were suggestive of phospholipase-associated neurodegeneration (PLAN) or infantile neuroaxonal dystrophy (INAD). Another differential was glycosylation type 1A disorder which also shows cerebellar atrophy but with no mineral deposition or claval hypertrophy.1 Whole exome sequencing with confirmatory sanger sequencing was performed which revealed homozygous missense c.1756G>A p.Gly586Arg mutation of PLA2G6 gene. This mutation has already been described in literature2 with RefSeq transcript NM_006208.3 (ENPP1):c.1756G>A (p.Gly586Arg). Child was started on supportive and physical therapy and family was counselled regarding natural history of disease and probability of next child being afflicted with same.
Figure 1.
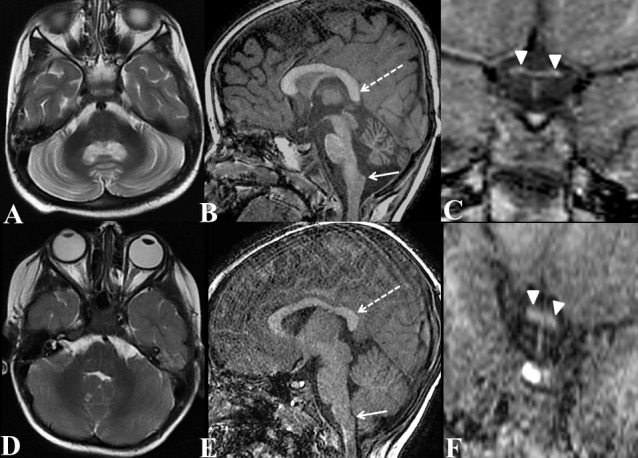
Axial T2W (A) of brain, sagittal T1W of brain (B) and coronal T1W image of optic chiasma (C) of our patient with homozygous p.Gly586Arg variant in PLA2G6; compared with axial T2W (D) of brain, sagittal T1W of brain (E) and coronal T1W image of optic chiasma (D) of a normal 3-year-old boy (age and gender matched normal subject). Axial T2W image (A) shows diffuse symmetrical atrophy with gliosis (hyperintensities) of bilateral cerebellar hemispheres (compare with normal cerebellum of normal child of same age in D). In addition, sagittal T1W image of brain (B) shows claval (nucleus gracilis) hypertrophy (arrow) with drooping of splenium of corpus callosum (broken arrow), (compare with normal claval and splenium of corpus callosum of normal child of same age in E). Coronal T1W image of optic chiasma (C) shows diffuse optic chiasma atrophy (compare with normal optic chiasma of normal child of same age in F).
Figure 2.
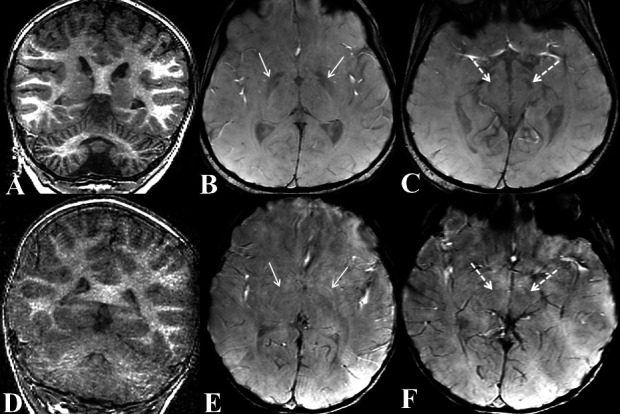
Coronal T1W (A) of cerebellar hemispheres and axial susceptibility-weighted imaging (SWI) images at the level of basal ganglia (B) and midbrain (C) of our patient; compared with Coronal T1W (A) of cerebellar hemispheres and axial SWI images at the level of basal ganglia (B) and midbrain (C) of a normal 3-year-old boy (age and gender matched normal subject). Coronal T1W image (A) shows diffuse symmetrical atrophy of bilateral cerebellar hemispheres (compare with normal cerebellum of normal child of same age in D). In addition, SWI shows hypointensities in symmetrical manner involving bilateral globus pallidi (arrows in B) and substantia nigra (broken arrows in C); suggesting mineral deposition (compare with normal SWI appearance of globus pallidi and substantia nigra in a normal child of same age in E and F, respectively).
PLAN is a rare disease with prevalence of approximately 1:1 000 000.3 Based on age of onset, PLAN is subdivided into INAD, atypical neuroaxonal dystrophy, dystonia–Parkinsonism and autosomal recessive early-onset parkinsonism (known as PARK14).4 Our case showed typical MRI features. Mineral deposition in globus pallidi and substantia nigra may not be seen in early stages and become evident/progress later on subsequent imaging.2 Some studies have suggested claval hypertrophy as early marker for PLAN.5 On histopathology, claval nuclei show characteristic ‘spheroid bodies’.5 However, claval hypertrophy is not pathognomonic for PLAN and can be seen in other causes of pontocerebellar atrophy and need not necessarily represent nucleus gracilis hypertrophy due to spheroid globules.4 Symmetric cerebellar atrophy has been seen in all documented cases.6 Up to 90% of patients show optic nerves and/or chiasma atrophy.7 PLA2G6 gene is involved in mitochondrial and cell membrane homeostasis and spheroid bodies represent degenerated mitochondrial membranes.6 PLAN is usually inherited in autosomal recessive manner due to biallelic deleterious variants in PLA2G6. We present a case with classical neuroradiological findings and a missense variant, which has been described previously.4 6 PLAN is progressive and currently no treatment exists with current treatments relying on supportive and physical therapy.
Patient’s perspective.
I was very much concerned about by child’s disease and was anxious to know the cause of his illness and its treatment. After MRI, our doctors ordered a test hoping to finally understand the cause of illness. Subsequently, genetic testing proved the anomaly of the disease. While we are devastated about the non-curable nature of the disease, we are now prepared with the knowledge of upcoming possibilities with respected to our child’s disease. We are also getting testing of our other relatives done. We have also been counselled about chances of next child having the same disease.
Learning points.
Phospholipase-associated neurodegeneration/infantile neuroaxonal dystrophy is an extremely rare autosomal recessive disorder caused due to biallelic loss-of-function variants in PLA2G6.
Patients typically present with halt of neurodevelopment and regression of attained milestones.
MRI shows typical constellation of findings such as claval hypertrophy, drooping of posterior half of corpus callosum, optic nerve atrophy and early mineralisation of globus pallidi and substantia nigra. These can guide in performing relevant genetic analysis for confirmation.
Footnotes
Contributors: SS wrote the manuscript and compiled the references and was involved in obtaining patient consent. SCM was involved in researching recent and relevant literature for the topic. AI helped in preparing the images and also adding the relevant figure captions. HL was involved in editing the final draft, ensuring the relvancy of data presented and references. These have participated sufficiently in this submission to take public responsibility for its contents. The manuscript has been approved by all authors. There is no conflict of interest.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Feraco P, Mirabelli-Badenier M, Severino M, et al. The shrunken, bright cerebellum: a characteristic MRI finding in congenital disorders of glycosylation type 1A. AJNR Am J Neuroradiol 2012;33:2062–7. 10.3174/ajnr.A3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Illingworth MA, Meyer E, Chong WK, et al. Pla2G6-Associated neurodegeneration (plan): further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab 2014;112:183–9. 10.1016/j.ymgme.2014.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregory A, Kurian MA, Maher ER. PLA2G6-associated neurodegeneration. : Pagon RA, Adam MP, Bird TD, . Gene Reviews™. Seattle (WA: University of Washington, Seattle, 1993-2013. [PubMed] [Google Scholar]
- 4.Paisan-Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 2009;65:19–23. 10.1002/ana.21415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farina L, Nardocci N, Bruzzone MG, et al. Infantile neuroaxonal dystrophy: neuroradiological studies in 11 patients. Neuroradiology 1999;41:376–80. 10.1007/s002340050768 [DOI] [PubMed] [Google Scholar]
- 6.Kurian MA, Morgan NV, MacPherson L, et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (plan). Neurology 2008;70:1623–9. 10.1212/01.wnl.0000310986.48286.8e [DOI] [PubMed] [Google Scholar]
- 7.Maawali A, Yoon G, Halliday W. Hypertrophy of the clava, a new MRI sign in patients with PLA2G6 mutations. : Poster presentation. American Society of Human Genetics Meeting, 2011. [Google Scholar]