

Abstract
A new tetradentate, monoanionic, mixed N/O donor ligand (BNPAPh2O−) with second coordination sphere H-bonding groups has been synthesized for stabilization of terminal FeIII(OH)(X) complexes. The complex [FeII(BNPAPh2O)(OTf)] (1) reacts with O2 to give a mononuclear terminal FeIII(OH) complex, [FeIII(OH)(BNPAPh2O)(OTf)] (2), both of which were characterized by X-ray diffraction, electrospray ionization mass spectrometry, UV–vis, 1H and 19F nuclear magnetic resonance, 57Fe Mössbauer, and electron paramagnetic resonance spectroscopies. Treatment of 2 with carbon radicals (Ar3C·) gives Ar3COH and the FeII complex 1, in direct analogy with the elusive radical “rebound” process proposed for nonheme iron enzymes.
Dioxygen activation at nonheme iron centers is of fundamental importance for a range of enzymatic and synthetic catalysts.1–3 An archetypal example is seen in the alpha-ketoglutarate (α-KG) dependent nonheme iron enzyme taurine dioxygenase (TauD). The mechanism of TauD involves the binding of O2 to FeII, followed by electron transfer from the FeII/α-KG unit to cleave the O–O bond and give a ferryl (FeIV(O)) intermediate. The FeIV(O) species then cleaves the substrate C–H bond to give an FeIII(OH) complex and substrate radical, which recombines via an “oxygen rebound” mechanism to give the final hydroxylated product and the reduced FeII active site (Scheme 1).4–8 A related class of nonheme Fe enzymes are the α-KG-dependent halogenases, which exhibit an interesting mechanistic divergence at the ferric hydroxide stage, in which a coordinated halide, or other pseudohalide X (e.g., X = Cl−, Br−, N3−, NO2−),9–13 can preferentially recombine with the substrate radical in lieu of the hydroxide group. Recent efforts to model the latter processes have been described.14–18 The factors that may bias an FeIII(OH)(X) center toward rebound of X− versus OH− are still not well understood. A similar conundrum arises in the proposed mechanism for isopenicillin N synthase (IPNS), in which a nascent carbon radical recombines with a ligated sulfur center instead of the OH− ligand.19–21
Scheme 1.
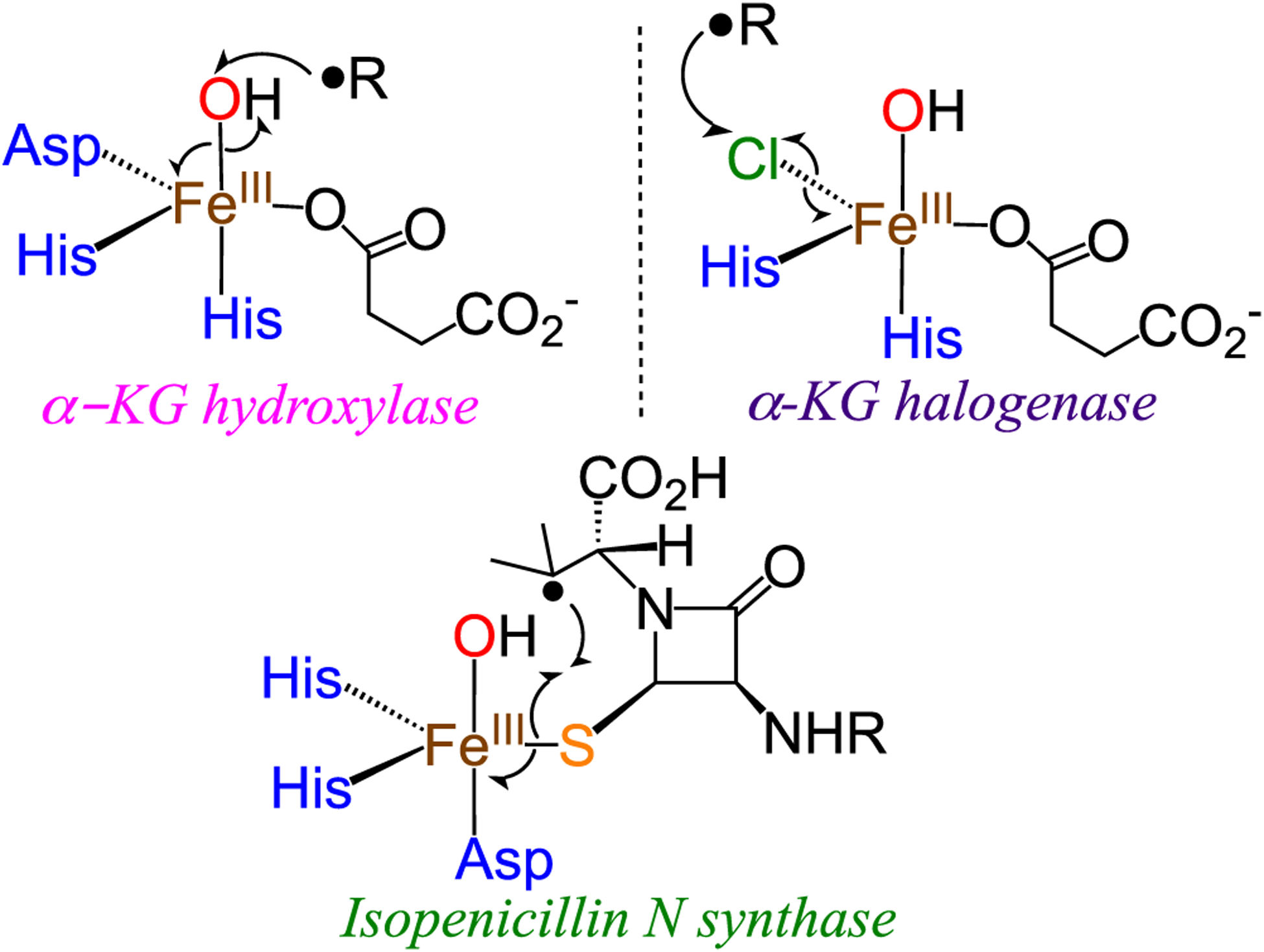
Selective Rebound Step in Different Nonheme Iron Enzymes
Our research group has made significant efforts toward constructing inorganic analogs of sulfur-ligated thiol dioxygenases,22–26 a subclass of nonheme Fe dioxygenases. Part of these efforts has involved discerning the general structural requirements for synthesizing mononuclear FeII complexes that facilitate O2 activation. In the course of this work, we have also become interested in examining the factors that contribute to the control of sulfur versus OH rebound in IPNS, as well as more generally, the factors that influence FeIII(OH) reactivity in hydroxylases and halogenases. We recently described the observation of C–O bond formation with a porphyrinoid, formally FeIV(OH) complex,27 and a nonheme FeIII(OMe) complex,28 in reactions with carbon radicals. However, reactivity between an isolated nonheme FeIII(OH) complex and a carbon radical, in direct analogy with the enzymatic processes described in Scheme 1, has not been achieved in any synthetic system to date. Such studies are hampered by the inherent difficulty of synthesizing well-defined, mononuclear FeIII(OH) complexes, which arises in part from their tendency to convert to O(H)-bridged, diferric species.29,30 Only a relatively small number of mononuclear, nonheme, terminal FeIII(OH) complexes have been structurally characterized.31–39
Herein we describe a new tetradentate ligand that was designed to fulfill three criteria: (1) stabilization of an FeIII(OH) unit, (2) O2 activation at an FeII center, and (3) inclusion of an open site cis to the OH group. This ligand led to the isolation of an FeIII(OH) complex that was obtained via activation of O2. We can react this complex with exogenous carbon radicals to give the hydroxylated product and the reduced ferrous form, providing a direct analog of the radical “rebound” step in nonheme iron oxygenases.
The new tetradentate ligand BNPAPh2OH, and FeII complex FeII(BNPAPh2O)(OTf) (1), were prepared as shown in Scheme 2. The neopentylamine substituents were included as potential stabilizing H-bond donors,40 a strategy that has been used by others to stabilize metal/oxygen species.32,33,35,41–44 In addition, an anionic alkoxide donor was installed to facilitate O2 activation. Metalation with FeII followed by crystallization gave yellow-green crystals of 1 suitable for X-ray diffraction (XRD). The structure of 1 (Figure 1) reveals a 5-coordinate (τ = 0.67)45 mononuclear iron(II) complex, with a triflate ligand occupying the fifth site. The neopentyl amine groups are oriented toward the triflate, forming two hydrogen bonds with N1(H)–O2 = 3.026(2) Å and N5(H)–O2 3.116(2) Å.
Scheme 2.
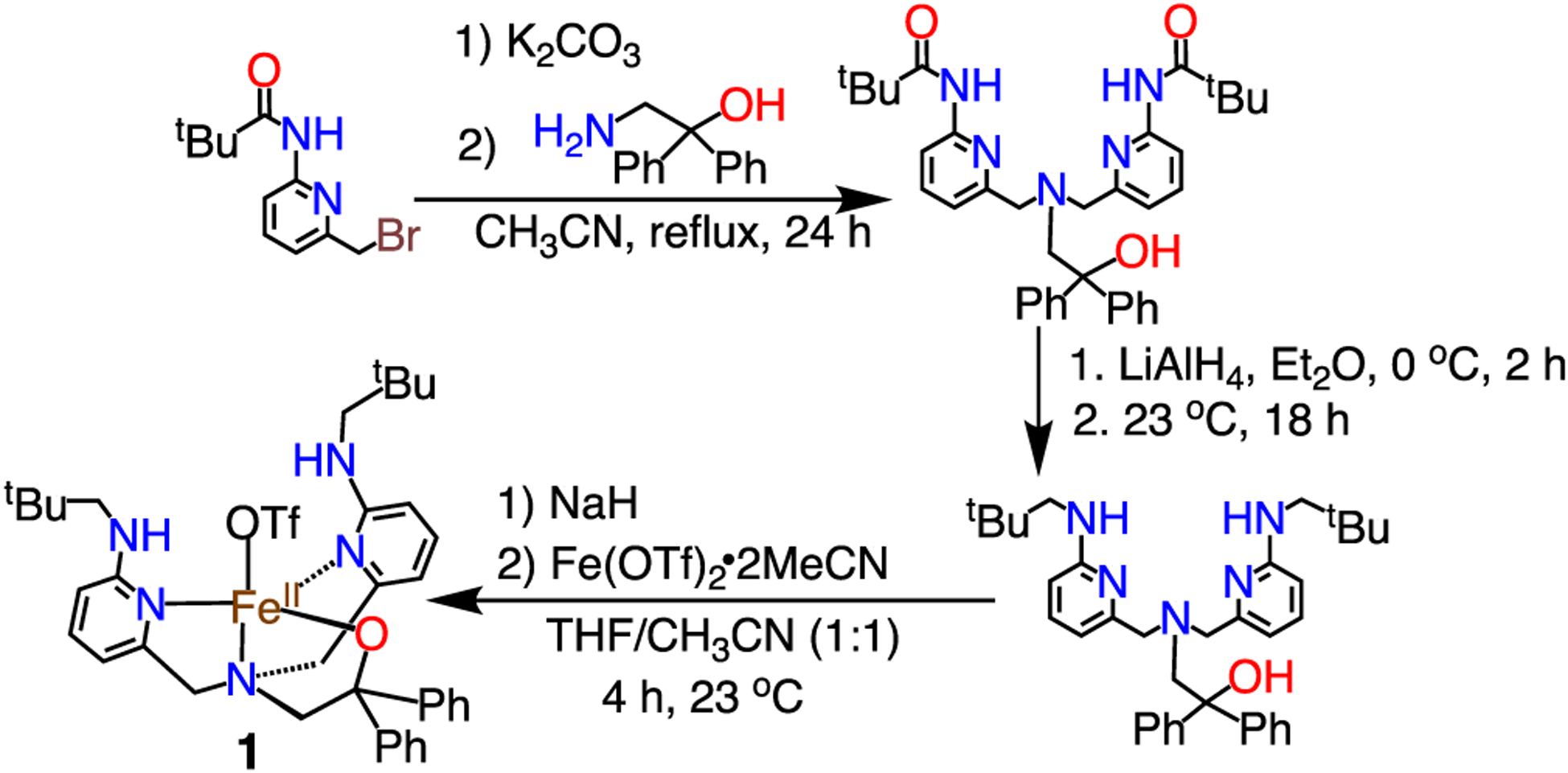
Synthesis of BNPAPh2OH and Complex 1
Figure 1.
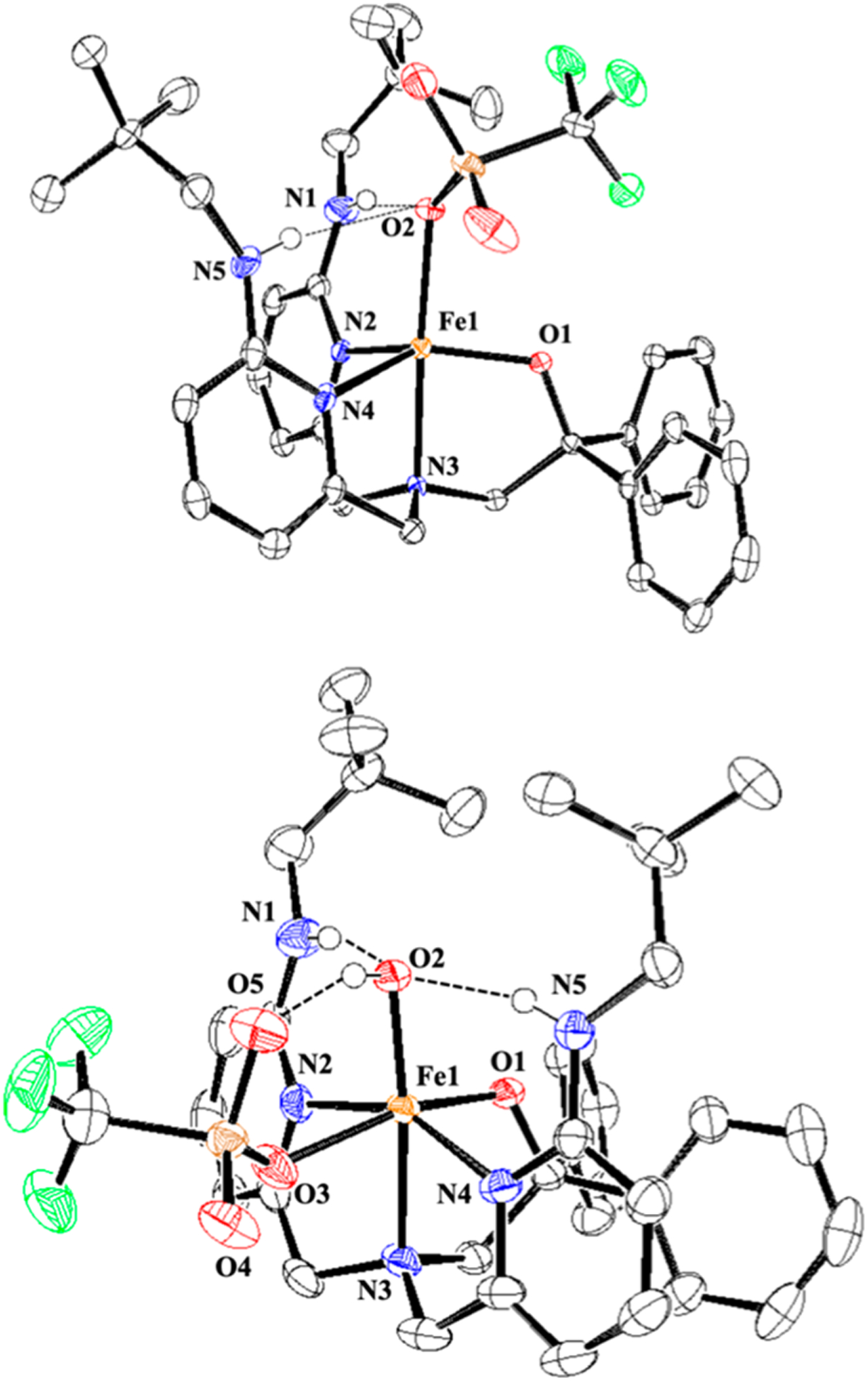
Displacement ellipsoid plots (50% probability level) for 1 and 2 at 110(2) K. Hydrogen atoms (except for N–H and O–H) have been omitted for clarity.
Complex 1 is pale yellow in CH3CN, with UV–vis maxima at 323 nm (ε = 9550 M−1 cm−1) and 420 nm (ε = 990 M−1 cm−1). The 1H nuclear magnetic resonance (NMR) spectrum of 1 in CD3CN reveals paramagnetic peaks from +100 to −20 ppm (Figure 2), indicative of a high-spin (hs) FeII complex. The 19F NMR spectrum in CD3CN shows a sharp peak at −78 ppm, whereas a broad peak at −74 ppm is observed in tetrahydrofuran (THF)-d8 (Figure S25), indicative of free and bound triflate ligand, respectively. These data suggest the CD3CN displaces the OTf− ligand and the H-bonded site is relatively labile, which would allow for O2 binding. Mössbauer spectroscopy on solid 1 at 80 K shows a sharp quadrupole doublet with δ = 1.03 and |ΔEQ| = 2.42 mm s−1, also typical of a hs-FeII center (Figure S15).
Figure 2.
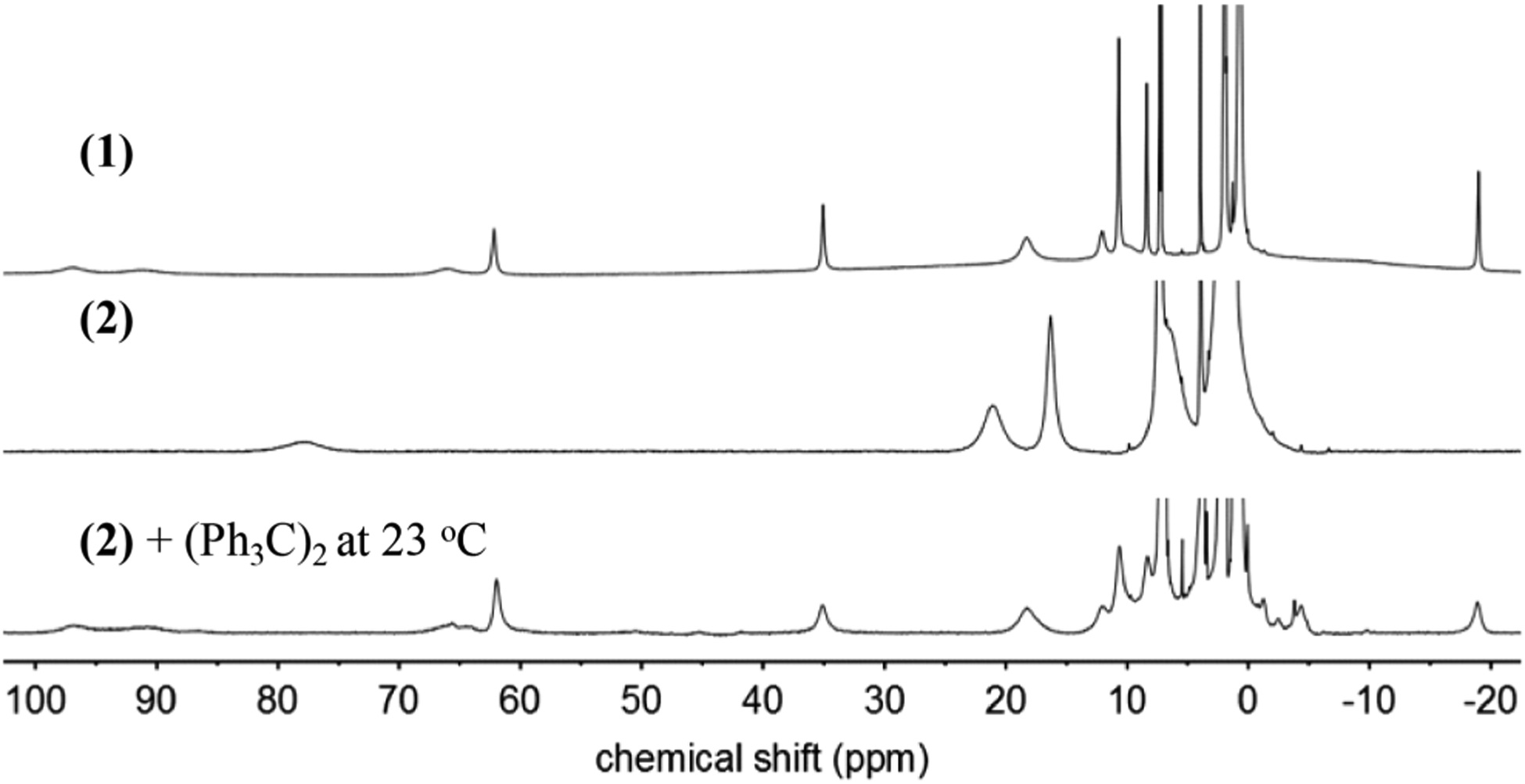
1H NMR spectra (CD3CN) of 1 (top), 2 (middle), and reaction of 2 with excess (Ph3C)2 (bottom) after removal of THF and dissolution in CD3CN.
Complex 1 was reacted with excess, dry O2 gas in CH3CN at 23 °C, causing an immediate color change from pale yellow to dark orange, with new UV–vis features appearing at 315 nm (ε = 9000 M−1 cm−1), 365 nm (ε = 2400 M−1 cm−1), and 440 nm (ε = 950 M−1 cm−1). The new dark orange species is air-stable, and the CH3CN can be removed in vacuo to give an orange solid, which was dissolved in toluene/THF and layered with pentane to afford orange crystals in 24 h that were suitable for XRD.
Analysis by XRD revealed a six-coordinate FeIII(OH) complex, FeIII(BNPAPh2O)(OH)(OTf) (2) (Figure 1). A terminal hydroxide ligand occupies the hydrogen bonding site, and the axial triflate ligand of 1 has shifted to an equatorial position cis to the OH− ligand in 2. The hydroxide ligand forms two intramolecular hydrogen bonds with the neopentyl amine substituents, with N1(H)–O2 = 2.831(3) and N5(H)–O2 = 2.857(2) Å. There is a third H-bonding interaction between the OH− and the OTf− ligands, with O2(H)–O5 = 2.832(1) Å. The Fe–O(H) bond length is 1.880(1)Å, in the range of other H-bonded and non-H-bonded FeIII(OH) complexes.31–35 However, a proper comparison with a non-H-bonded analog of 2 is currently not available. The Fe–Npy and Fe–Namine distances are shorter by 0.1–0.2 Å when compared to the FeII analog 1, and the Fe–O1 distance is also contracted by 0.05 Å.
Complex 2 shows paramagnetic peaks in the 1H NMR spectrum from 150 to 1 ppm (Figure 2). The 19F NMR spectrum for 2 in CD3CN shows a sharp peak at −74.6 ppm, which corresponds to free OTf−, and indicates the OTf− ligand is likely displaced by solvent. In contrast, the same spectrum in the more weakly coordinating THF-d8 shows no peaks from +200 to −200 ppm, consistent with OTf− remaining coordinated (Figure S26). The EPR spectrum (20 K) reveals signals at g = 9.01, 4.23, which is consistent with an S = 5/2 ferric ion (Figure S14). Analysis of a crystalline sample of 2 by Mössbauer spectroscopy at 80 K reveals a broad quadrupole doublet which can be fit with δ = 0.47 mm s−1 and |ΔEQ| = 0.97 mm s−1 (Figure S16). The broadened spectrum comes from 2 likely being in an intermediate spin relaxation regime.46,47 The Mössbauer spectrum of 2 at 5 K shows a 6 line hyperfine splitting pattern which is expected for a high-spin FeIII species (Figure S17).
Density functional theory (DFT) was employed to obtain optimized structures for 1 and 2 at the BP86/6–311G*/6–31G* (for C and H atoms) level of theory. These optimized structures matched well with the structures from XRD for 1 and 2, and were then used for the calculation of Mössbauer spectroscopic parameters. The calculations gave δ = 1.05 and |ΔEQ| = 2.61 mm s−1 for 1, and δ = 0.47 and |ΔEQ| = 1.42 mm s−1 for 2. These parameters are in reasonable agreement with experiment for the solid-state samples.
Production of 2 by addition of excess O2 in acetonitrile followed by electrospray ionization mass spectrometry (ESI-MS) reveals the major peak at m/z = 636.85 (positive ion mode), corresponding to [2–OTf]+. In comparison, the use of isotopically labeled 18O2 leads to the major peak shifted by +2 units to m/z = 638.85. Addition of excess H218O to 1 in CH3CN, followed by exposure to excess, natural abundance O2, does not lead to any shift in the major peak at m/z 636.85. These 18O-labeling experiments show that the hydroxide ligand must originate from dioxygen.
A suggested mechanism is given in Scheme S1. Initial binding of O2 gives an FeIII(superoxo) species that then leads to a peroxo-bridged dimer, followed by homolytic O–O cleavage to give an FeIV(O) species. The latter species then abstracts hydrogen from an exogenous source (e.g., solvent) to give 2. Precedent for this mechanism is described for both heme and nonheme Fe and Mn complexes.33,48–50 Carrying out the reaction of 1 with O2 in the presence of excess 9,10-dihydroanthracene, a potential H atom donor, leads to production of anthracene in ~90% yield (Scheme 3), consistent with H atom abstraction by the putative ferryl intermediate. Attempts to observe intermediates in the reaction between 1 and O2 were carried out at lower temperature. However, at temperatures as low as −90 °C, only the formation of 2 was observed as seen at 23 °C, and at lower temperatures (e.g., −105 °C) no reaction occurs.
Scheme 3.
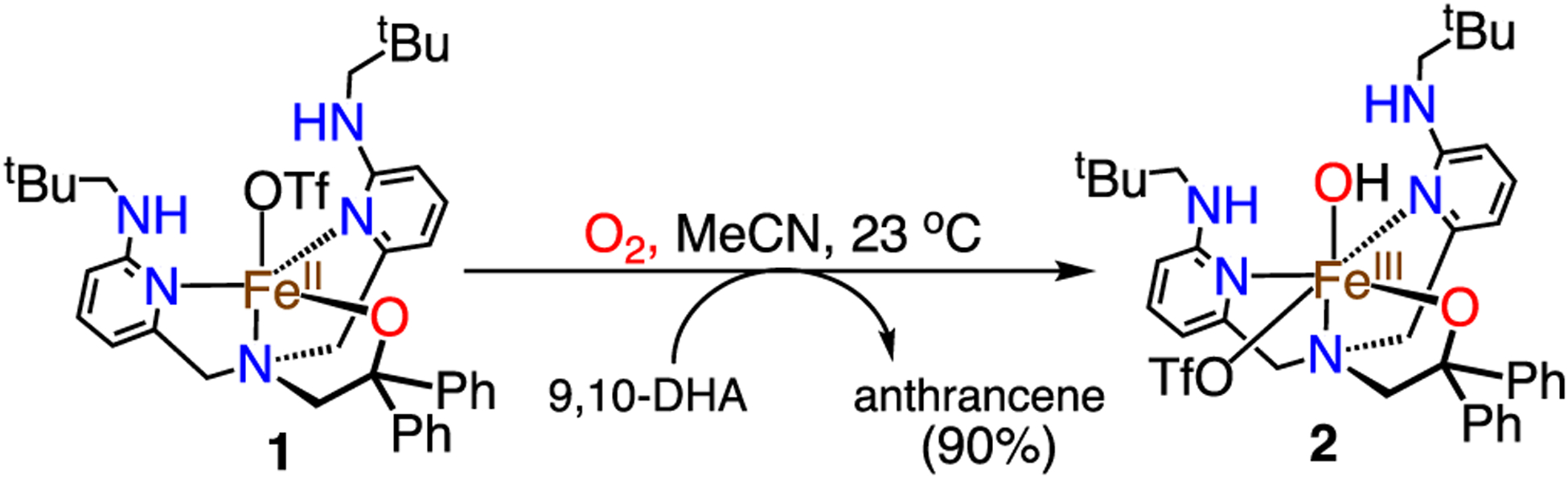
Activation of Dioxygen by 1
The similarity of complex 2 to the enzymatic species shown in Scheme 1 motivated us to examine its reactivity with carbon-based radicals. Addition of Ph3C· to 2 in THF at 23 °C causes a color change from dark orange to yellow over 4 h. The 1H NMR spectrum shows complete loss of 2, and the appearance of peaks for 1 (Figure 2). A new peak at δ 5.50 ppm was also formed, arising from the alcohol (Ph3COH) in 84% (±3) yield (Scheme 4).
Scheme 4.
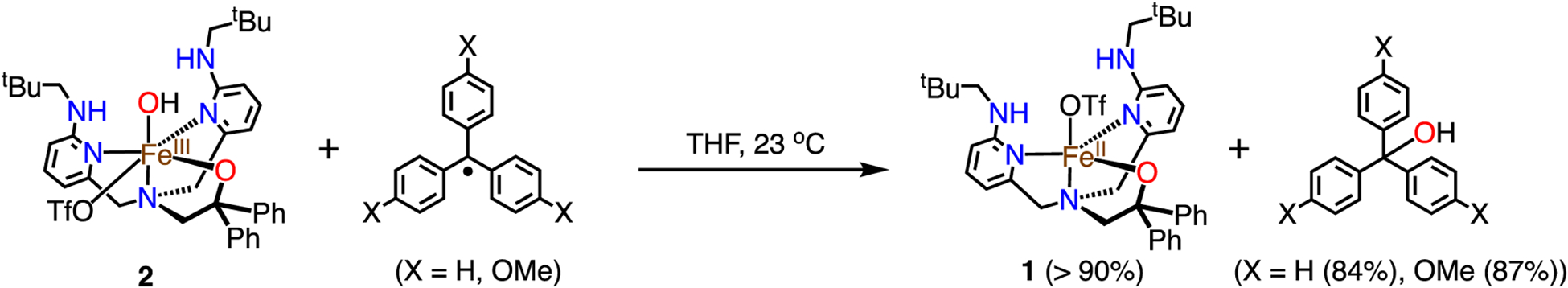
Reaction between 2 and Triarylmethyl Radicals
Changing to the more electron-rich (p-OMe-C6H4)3C· (1 equiv) causes faster conversion (~45 min) of 2 to 1 as seen by 1H NMR spectroscopy. Quantification of (p-OMe-C6H4)3COH gives a yield of 87% (±2) (Scheme 4), consistent with 1:1 stoichiometry between 2 and the radical.
The formation of the products (p-X-C6H4)3COH (X = H, OMe) was also confirmed by ESI-MS analysis (Figures S22 and 23). Incorporation of 18OH into 2 by exchange with H218O was quantitative, and reaction of (p-X-C6H4)3C· (X = H, OMe) with 18O-labeled 2, led to a two unit shift in m/z by ESI-MS analysis (Figures S28 and 29, 18O incorporation, 99%)). These results indicate that there is direct transfer of the OH ligand to the carbon radicals.
The reaction with the p-OMe derivative was monitored by Mössbauer spectroscopy (Figure 3). The spectrum for 57Fe-labeled complex 1 in 2-MeTHF(80 K) gives δ = 1.14 mm s−1 and |ΔEQ| = 2.24 mm s−1 (Figure 3a). The spectrum for complex 2 (Figure 3b) is broad, with δ = 0.47 mm s−1 and |ΔEQ| = 1.24 mm s−1 (see Figure S18 for fitting details). Reaction between 2 and (p-OMe-C6H4)3C· gives a quadrupole doublet matching 1 (δ = 1.14 mm s−1; |ΔEQ| = 2.23 mm s−1) (Figure 3c), which accounts for 90% of the total Fe.
Figure 3.
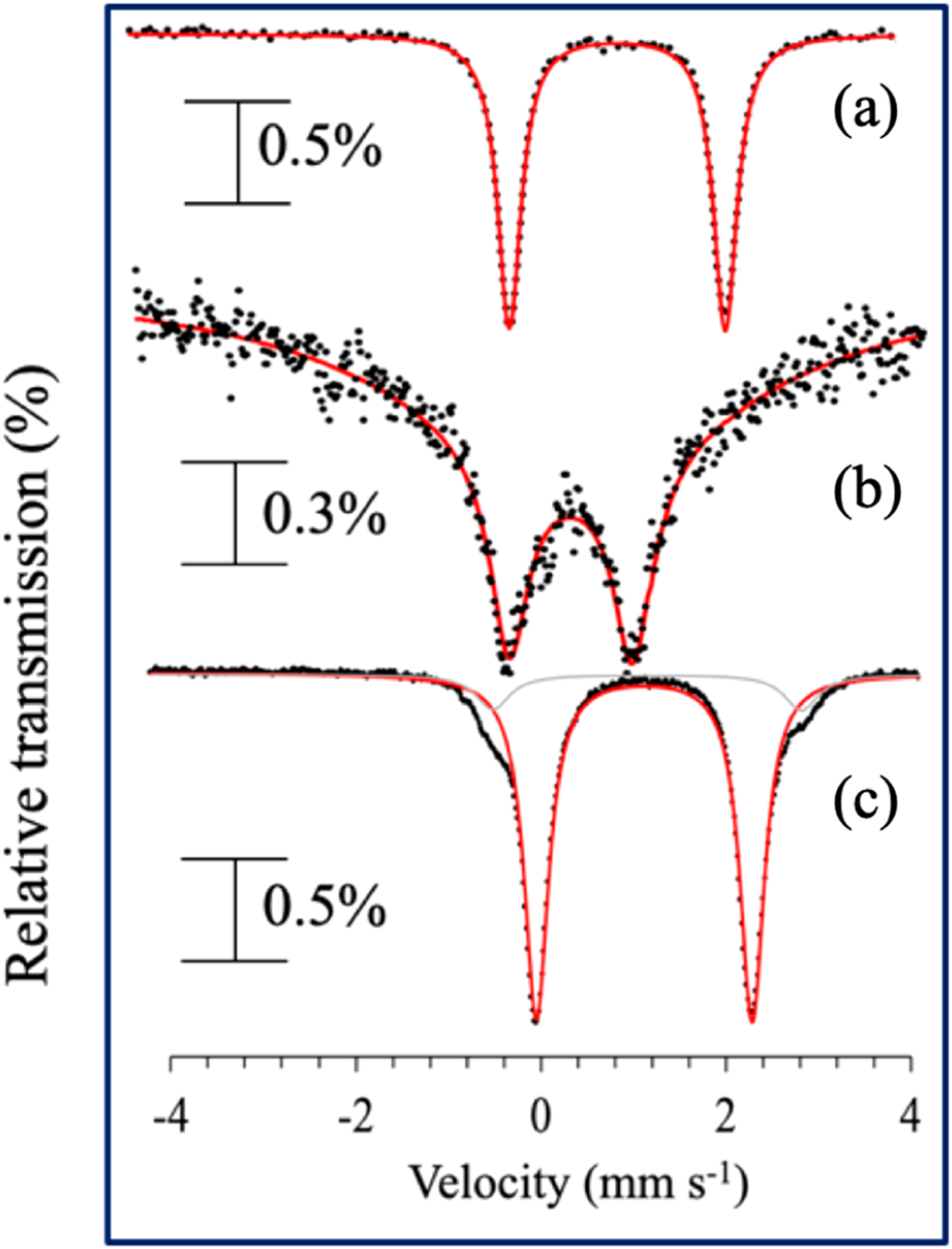
Zero field 57Fe Mössbauer spectra (80 K) of (a) 1, (b) 2 in 2-MeTHF, and (c) reaction of 2 with (p-OMe-C6H4)3C· (at 23 °C) in 2-MeTHF. Black circles = experimental data; red line = best fit.
Electrochemical analysis of 2 shows a reversible wave in the cyclic voltammogram with E1/2 = −0.69 V (ΔEpp = 170 mV) vs Fc+/Fc in CH3CN. This reduction potential is in line with other FeIII(OH) complexes (Table S8). The redox potentials for (p-X-C6H4)3C+/(p-X-C6H4)3C· are −0.11 V (X = H) and −0.58 V (X = OMe), in the same solvent,51 indicating that outer-sphere electron-transfer (ET) from (p-X-C6H4)3C· to 2 should be endergonic by 13.4 kcal mol−1 for X = H, and 2.5 kcal mol−1 for X = OMe. Thus, ET to 2 is thermodynamically unfavorable for both carbon radicals, suggesting that a concerted process may be operative (Scheme S2).
In conclusion, the incorporation of an anionic donor combined with H-bond groups in the second coordination sphere of a new tetradentate ligand provided access to an FeII complex that activates O2 and yields a stable, terminal FeIII(OH) complex. Reaction of the latter complex with trityl radical derivatives led to the first direct observation52 of a model “rebound” reaction to give the hydroxylated carbon product and the one-electron-reduced FeII complex. It may be somewhat surprising that smooth transfer of OH• from Fe to the radical occurs, because of the significant steric encumbrance and tight H-bond network in 2. Determining what inherent factors (e.g., H-bonding, sterics, redox potential, donor atoms) influence O2 activation and Fe(OH)/radical reactivity will be the focus of future studies.
In addition, the new BNPAPh2O− ligand leaves a potential labile equatorial site on the metal to build Fe(OH)(X) complexes. Efforts to replace the equatorial OTf− in 2 with other anionic donors are underway.
Supplementary Material
ACKNOWLEDGMENTS
The NIH (GM119374 to D.P.G.) is gratefully acknowledged for financial support. Computer time was provided by the Maryland Advanced Research Computing Center (MARCC). We also thank Prof. Guy N. L. Jameson for valuable discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b03329.
X-ray crystallographic data for 1 (CIF)
X-ray crystallographic data for 2 (CIF)
Synthesis, 1H NMR, 19F NMR, ESI-MS, CV, 57Fe Mössbauer data, UV–vis, EPR data, supporting schemes S1–S3, supporting tables S1–S8 and supporting figures S1–S30 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Sahu S; Goldberg DP Activation of Dioxygen by Iron and Manganese Complexes: A Heme and Nonheme Perspective. J. Am. Chem. Soc 2016, 138, 11410–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kovaleva EG; Lipscomb JD Versatility of biological nonheme Fe(II) centers in oxygen activation reactions. Nat. Chem. Biol 2008, 4, 186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hong S; Lee Y-M; Ray K; Nam W Dioxygen activation chemistry by synthetic mononuclear nonheme iron, copper and chromium complexes. Coord. Chem. Rev 2017, 334, 25–42. [Google Scholar]
- (4).Mitchell AJ; Dunham NP; Martinie RJ; Bergman JA; Pollock CJ; Hu K; Allen BD; Chang W.-c.; Silakov A; Bollinger JM Jr.; Krebs C; Boal AK Visualizing the Reaction Cycle in an Iron(II)- and 2-(Oxo)-glutarate-Dependent Hydroxylase. J. Am. Chem. Soc 2017, 139, 13830–13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bollinger JM Jr.; Chang W.-c.; Matthews ML; Martinie RJ; Boal AK; Krebs C Chapter 3. Mechanisms of 2-Oxoglutarate-Dependent Oxygenases: The Hydroxylation Paradigm and Beyond. In 2-Oxoglutarate-Dependent Oxygenases; Schofield CJ, Hausinger RP, Eds.; The Royal Society of Chemistry, 2015; pp 95–122. [Google Scholar]
- (6).Martinez S; Hausinger RP Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-Dependent Oxygenases. J. Biol. Chem 2015, 290, 20702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Timmins A; de Visser SP A Comparative Review on the Catalytic Mechanism of Nonheme Iron Hydroxylases and Halogenases. Catalysts 2018, 8, 314. [Google Scholar]
- (8).Wojcik A; Radon M; Borowski T Mechanism of O2 Activation by alpha-Ketoglutarate Dependent Oxygenases Revisited. A Quantum Chemical Study. J. Phys. Chem. A 2016, 120, 1261–74. [DOI] [PubMed] [Google Scholar]
- (9).Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Jr. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 17723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Matthews ML; Chang WC; Layne AP; Miles LA; Krebs C; Bollinger JM Jr. Direct nitration and azidation of aliphatic carbons by an iron-dependent halogenase. Nat. Chem. Biol 2014, 10, 209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Timmins A; Quesne MG; Borowski T; de Visser SP Group Transfer to an Aliphatic Bond: A Biomimetic Study Inspired by Nonheme Iron Halogenases. ACS Catal. 2018, 8, 8685–8698. [Google Scholar]
- (12).Pratter SM; Ivkovic J; Birner-Gruenberger R; Breinbauer R; Zangger K; Straganz GD More than just a Halogenase: Modification of Fatty Acyl Moieties by a Trifunctional Metal Enzyme. ChemBioChem 2014, 15, 567–574 [DOI] [PubMed] [Google Scholar]
- (13).Mitchell AJ; Dunham NP; Bergman JA; Wang B; Zhu Q; Chang W -c.; Liu, X.; Boal, A. K. Structure-Guided Reprogramming of a Hydroxylase To Halogenate Its Small Molecule Substrate. Biochemistry 2017, 56, 441–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Puri M; Biswas AN; Fan R; Guo Y; Que L Jr. Modeling Non-Heme Iron Halogenases: High-Spin Oxoiron(IV)–Halide Complexes That Halogenate C–H Bonds. J. Am. Chem. Soc 2016, 138, 2484–2487. [DOI] [PubMed] [Google Scholar]
- (15).Planas O; Clémancey M; Latour J-M; Company A; Costas M Structural modeling of iron halogenases: synthesis and reactivity of halide-iron(iv)-oxo compounds. Chem. Commun 2014, 50, 10887–10890. [DOI] [PubMed] [Google Scholar]
- (16).Rana S; Biswas JP; Sen A; Clémancey M; Blondin G; Latour J-M; Rajaraman G; Maiti D Selective C–H halogenation over hydroxylation by non-heme iron(iv)-oxo. Chem. Sci 2018, 9, 7843–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rana S; Bag S; Patra T; Maiti D Catalytic Electrophilic Halogenations and Haloalkoxylations by Non-Heme Iron Halides. Adv. Synth. Catal 2014, 356, 2453–2458. [Google Scholar]
- (18).Chatterjee S; Paine TK Hydroxylation versus Halogenation of Aliphatic C–H Bonds by a Dioxygen-Derived Iron–Oxygen Oxidant: Functional Mimicking of Iron Halogenases. Angew. Chem., Int. Ed 2016, 55, 7717–7722. [DOI] [PubMed] [Google Scholar]
- (19).Tamanaha E; Zhang B; Guo Y; Chang WC; Barr EW; Xing G; St. Clair J; Ye S; Neese F; Bollinger JM Jr.; Krebs C Spectroscopic Evidence for the Two C-H-Cleaving Intermediates of Aspergillus nidulans Isopenicillin N Synthase. J. Am. Chem. Soc 2016, 138, 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ge W; Clifton IJ; Stok JE; Adlington RM; Baldwin JE; Rutledge PJ Isopenicillin N Synthase Mediates Thiolate Oxidation to Sulfenate in a Depsipeptide Substrate Analogue: Implications for Oxygen Binding and a Link to Nitrile Hydratase? J. Am. Chem. Soc 2008, 130, 10096–10102. [DOI] [PubMed] [Google Scholar]
- (21).Kawatsu T; Lundberg M; Morokuma K Protein Free Energy Corrections in ONIOM QM:MM Modeling: A Case Study for Isopenicillin N Synthase (IPNS). J. Chem. Theory Comput 2011, 7, 390–401. [DOI] [PubMed] [Google Scholar]
- (22).Gordon JB; Vilbert AC; Siegler MA; Lancaster KM; Moënne-Loccoz P; Goldberg DP A Nonheme Thiolate-Ligated Cobalt Superoxo Complex: Synthesis and Spectroscopic Characterization, Computational Studies, and Hydrogen Atom Abstraction Reactivity. J. Am. Chem. Soc 2019, 141, 3641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Badiei YM; Siegler MA; Goldberg DP O2 activation by bis(imino)pyridine iron(II)-thiolate complexes. J. Am. Chem. Soc 2011, 133, 1274–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gordon JB; McGale JP; Prendergast JR; Shirani-Sarmazeh Z; Siegler MA; Jameson GNL; Goldberg DP Structures, Spectroscopic Properties, and Dioxygen Reactivity of 5- and 6-Coordinate Nonheme Iron(II) Complexes: A Combined Enzyme/Model Study of Thiol Dioxygenases. J. Am. Chem. Soc 2018, 140, 14807–14822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).McQuilken AC; Jiang Y; Siegler MA; Goldberg DP Addition of Dioxygen to an N4S(thiolate) Iron(II) Cysteine Dioxygenase Model Gives a Structurally Characterized Sulfinato–Iron(II) Complex. J. Am. Chem. Soc 2012, 134, 8758–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jiang Y; Widger LR; Kasper GD; Siegler MA; Goldberg DP Iron(II)-Thiolate S-Oxygenation by O2: Synthetic Models of Cysteine Dioxygenase. J. Am. Chem. Soc 2010, 132, 12214–12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zaragoza JPT; Yosca TH; Siegler MA; Moënne-Loccoz P; Green MT; Goldberg DP Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc 2017, 139, 13640–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pangia TM; Davies CG; Prendergast JR; Gordon JB; Siegler MA; Jameson GNL; Goldberg DP Observation of Radical Rebound in a Mononuclear Nonheme Iron Model Complex. J. Am. Chem. Soc 2018, 140, 4191–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Brines LM; Coggins MK; Poon PCY; Toledo S; Kaminsky W; Kirk ML; Kovacs JA Water-Soluble Fe(II)–H2O Complex with a Weak O–H Bond Transfers a Hydrogen Atom via an Observable Monomeric Fe(III)–OH. J. Am. Chem. Soc 2015, 137, 2253–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ching W-M; Zhou A; Klein JEMN; Fan R; Knizia G; Cramer CJ; Guo Y; Que L Jr. Characterization of the Fleeting Hydroxoiron(III) Complex of the Pentadentate TMC-py Ligand. Inorg. Chem 2017, 56, 11129–11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ogo S; Wada S; Watanabe Y; Iwase M; Wada A; Harata M; Jitsukawa K; Masuda H; Einaga H Synthesis, Structure, and Spectroscopic Properties of [FeIII(tnpa)(OH)(PhCOO)]ClO4: A Model Complex for an Active Form of Soybean Lipoxygenase-1. Angew. Chem., Int. Ed 1998, 37, 2102–2104. [DOI] [PubMed] [Google Scholar]
- (32).Ogo S; Yamahara R; Roach M; Suenobu T; Aki M; Ogura T; Kitagawa T; Masuda H; Fukuzumi S; Watanabe Y Structural and Spectroscopic Features of a cis (Hydroxo)-FeIII-(Carboxylato) Configuration as an Active Site Model for Lipoxygenases. Inorg. Chem 2002, 41, 5513–5520. [DOI] [PubMed] [Google Scholar]
- (33).MacBeth CE; Golombek AP; Young VG; Yang C; Kuczera K; Hendrich MP; Borovik AS Activation by Nonheme Iron Complexes: A Monomeric Fe(III)-Oxo Complex Derived From O2. Science 2000, 289, 938. [DOI] [PubMed] [Google Scholar]
- (34).Çelenligil-Çetin R; Paraskevopoulou P; Dinda R; Staples RJ; Sinn E; Rath NP; Stavropoulos P Synthesis, Characterization, and Reactivity of Iron Trisamidoamine Complexes That Undergo Both Metal- and Ligand-Centered Oxidative Transformations. Inorg. Chem 2008, 47, 1165–1172. [DOI] [PubMed] [Google Scholar]
- (35).Soo HS; Komor AC; Iavarone AT; Chang CJ A Hydrogen-Bond Facilitated Cycle for Oxygen Reduction by an Acid- and Base-Compatible Iron Platform. Inorg. Chem 2009, 48, 10024–10035. [DOI] [PubMed] [Google Scholar]
- (36).Cook SA; Ziller JW; Borovik AS Iron(II) Complexes Supported by Sulfonamido Tripodal Ligands: Endogenous versus Exogenous Substrate Oxidation. Inorg. Chem 2014, 53, 11029–11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kleinlein C; Bendelsmith AJ; Zheng S-L; Betley TA C–H Activation from Iron(II)-Nitroxido Complexes. Angew. Chem., Int. Ed 2017, 56, 12197–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chambers MB; Groysman S; Villagrán D; Nocera DG Iron in a Trigonal Tris(alkoxide) Ligand Environment. Inorg. Chem 2013, 52, 3159–3169. [DOI] [PubMed] [Google Scholar]
- (39).Mukherjee J; Lucas RL; Zart MK; Powell DR; Day VW; Borovik AS Synthesis, Structure, and Physical Properties for a Series of Monomeric Iron(III) Hydroxo Complexes with Varying Hydrogen-Bond Networks. Inorg. Chem 2008, 47, 5780–5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Berreau LM; Allred RA; Makowska-Grzyska MM; Arif AM Synthesis and structure of a nitrogen/sulfur-ligated zinc hydroxide complex. Chem. Commun 2000, 1423–1424. [Google Scholar]
- (41).Ford CL; Park YJ; Matson EM; Gordon Z; Fout AR A bioinspired iron catalyst for nitrate and perchlorate reduction. Science 2016, 354, 741–43. [DOI] [PubMed] [Google Scholar]
- (42).Gordon Z; Drummond MJ; Matson EM; Bogart JA; Schelter EJ; Lord RL; Fout AR Tuning the Fe(II/III) Redox Potential in Nonheme Fe(II)–Hydroxo Complexes through Primary and Secondary Coordination Sphere Modifications. Inorg. Chem 2017, 56, 4852–4863. [DOI] [PubMed] [Google Scholar]
- (43).Dahl EW; Dong HT; Szymczak NK Phenylamino derivatives of tris(2-pyridylmethyl)amine: hydrogen-bonded perox-odicopper complexes. Chem. Commun 2018, 54, 892–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Dahl EW; Kiernicki JJ; Zeller M; Szymczak NK Hydrogen Bonds Dictate O2 Capture and Release within a Zinc Tripod. J. Am. Chem. Soc 2018, 140, 10075–10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Addison AW; Rao TN; Reedijk J; van Rijn J; Verschoor GC Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc., Dalton Trans 1984, 1349–1356. [Google Scholar]
- (46).Mørup S Magnetic Relaxation Phenomena. In Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications; Gütlich P, Bill E, Trautwein AX, Eds.; Springer: Berlin, Heidelberg, 2011; pp 201–234. [Google Scholar]
- (47).Gupta R; Lacy DC; Bominaar EL; Borovik AS; Hendrich MP Electron Paramagnetic Resonance and Mössbauer Spectroscopy and Density Functional Theory Analysis of a High-Spin FeIV–Oxo Complex. J. Am. Chem. Soc 2012, 134, 9775–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wijeratne GB; Corzine B; Day VW; Jackson TA Saturation Kinetics in Phenolic O–H Bond Oxidation by a Mononuclear Mn(III)–OH Complex Derived from Dioxygen. Inorg. Chem 2014, 53, 7622–7634. [DOI] [PubMed] [Google Scholar]
- (49).Lee Y-M; Hong S; Morimoto Y; Shin W; Fukuzumi S; Nam W Dioxygen Activation by a Non-Heme Iron(II) Complex: Formation of an Iron(IV)–Oxo Complex via C–H Activation by a Putative Iron(III)–Superoxo Species. J. Am. Chem. Soc 2010, 132, 10668–10670. [DOI] [PubMed] [Google Scholar]
- (50).Kim SO; Sastri CV; Seo MS; Kim J; Nam W Dioxygen Activation and Catalytic Aerobic Oxidation by a Mononuclear Nonheme Iron(II) Complex. J. Am. Chem. Soc 2005, 127, 4178–4179. [DOI] [PubMed] [Google Scholar]
- (51).Volz H; Lotsch W Polarographische reduktion von triarylmethylkationen. Tetrahedron Lett. 1969, 10, 2275–2278. [Google Scholar]
- (52).While this paper was under review, a report of a nonheme FeIII(OH) complex reacting with (p-H-C6H4)3C· to give FeII and alcohol was described Drummond MJ; Ford CL; Gray DL; Popescu CV; Fout AR J. Am. Chem. Soc 2019, 141, 6639–6650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.