

Abstract
A diverse array of post-transcriptional modifications is found in RNA molecules from all domains of life. While the locations of RNA modifications are well characterized in abundant noncoding RNAs, modified sites in less abundant mRNAs are just beginning to be discovered. Recent work has revealed hundreds of previously unknown and dynamically regulated pseudouridines (Ψ) in mRNAs from diverse organisms. This unit describes Pseudo-seq, an efficient, high-resolution method for identification of Ψs genome-wide. This unit includes methods for isolation of RNA from S. cerevisiae, preparation of Pseudo-seq libraries from RNA samples, and identification of sites of pseudouridylation from the sequencing data. Pseudo-seq is applicable to any organism or cell type, facilitating rapid identification of novel pseudouridylation events.
Keywords: RNA modification, pseudouridine, next-generation sequencing
INTRODUCTION
RNA molecules contain a chemically diverse array of posttranscriptional modifications that modulate their structure, stability, and activity. Pseudouridine, the C5-glycoside isomer of uridine, is the most abundant post-transcriptional modification found within cellular RNAs owing to its presence at multiple positions in both rRNAs and tRNAs (Ge and Yu, 2013) (Fig. 4.25.1A). Ψ formation maintains the Watson-Crick base pairing potential of uridine, but allows formation of an extra hydrogen bond through the newly freed N1-H, which has a stabilizing effect on RNA secondary structure (Charette and Gray, 2000; Hudson et al., 2013). Recent work has revealed dynamic regulation of pseudouridylation in response to cellular stressors, suggesting regulatory potential for pseudouridylation(Wu et al., 2011; Carlile et al., 2014; Courtes et al., 2014; Lovejoy et al., 2014; Schwartz et al., 2014) Although the molecular and cellular functions of endogenous mRNA pseudouridylation are not yet known, artificially targeted pseudouridylation of stop codons promotes non-canonical base-pairing of Ψ in the ribosomal decoding center leading to efficient nonsense suppression (Karijolich and Yu, 2011; Fernández et al., 2013). Thus, regulated mRNA pseudouridylation has the potential to profoundly affect gene expression by altering the genetic code. A method for rapid identification of Ψs genome-wide would help to further elucidate the regulatory potential and function of these RNA modifications.
Figure 4.25.1.
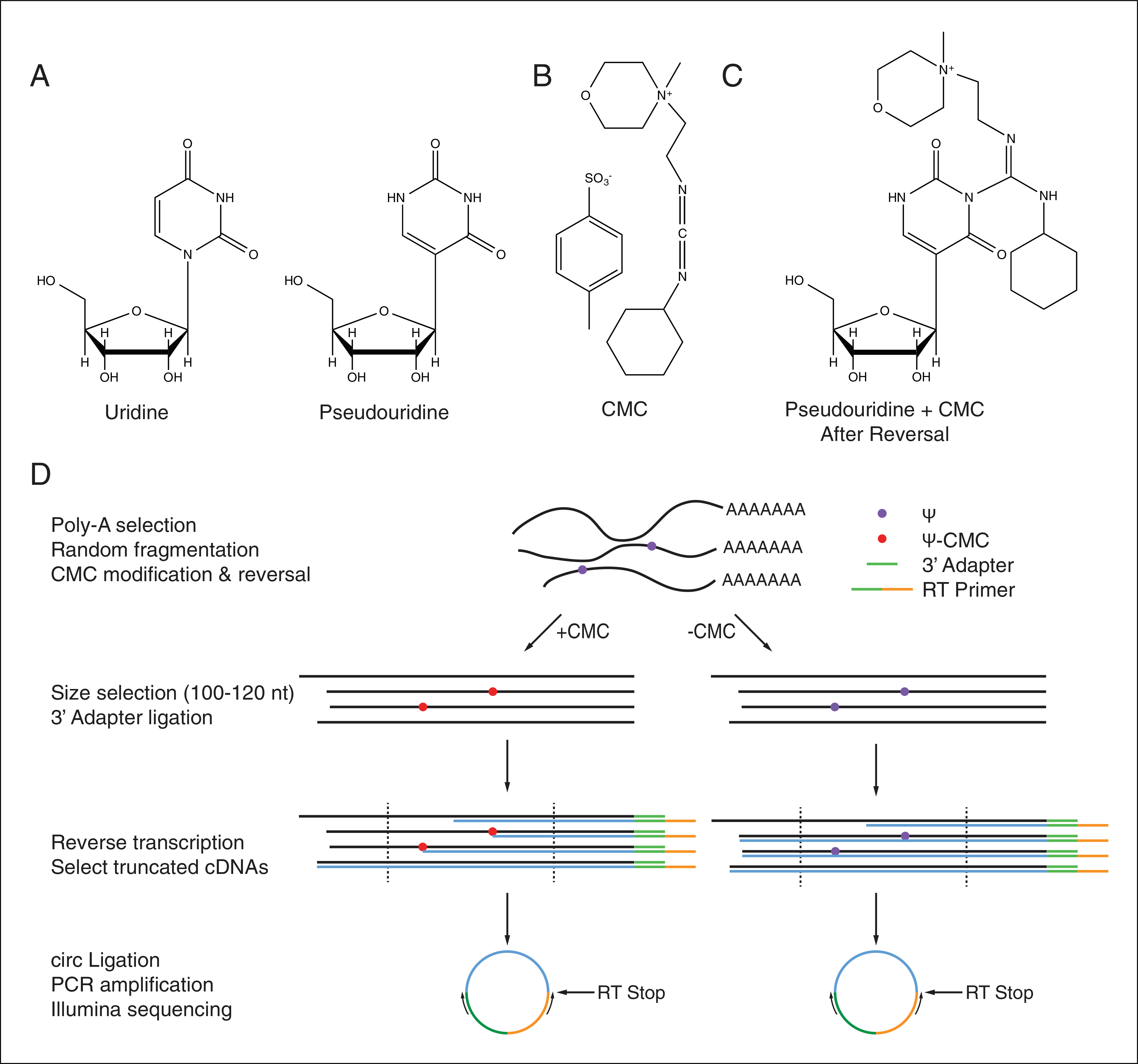
(A) The chemical structures of uridine and pseudouridine. (B) The structure of CMC. (C) The structure of Ψ-CMC adducts after alkaline reversal. (D) A schematic illustrating the steps in Pseudo-seq library preparation. CMC-dependent RT stops indicate sites of pseudouridylation. See Basic Protocols 1 and 2 for details.
This unit describes Pseudo-seq, a method for identifying sites of pseudouridylation genome-wide based on the Illumina sequencing platform (Carlile et al., 2014). Pseudo-seq exploits the fact that in vitro treatment of RNA with the carbodiimide N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMC) (Fig. 4.25.1B), can be used to specifically modify Ψ residues (Gilham and Ho, 1971). CMC forms covalent adducts with the N1 position of guanidine, the N3 position of uridine, and the N1 and N3 positions of pseudouridine; however, only the Ψ-N3-CMC adduct is stable under alkaline conditions, allowing for specific labeling of Ψ residues (Fig. 4.25.1C). These bulky Ψ-CMC adducts cause strong stops to reverse transcriptase (RT) one nucleotide 3′ of Ψ-CMC adducts, allowing their identification using primer extension (Bakin and Ofengand, 1993). Pseudo-seq adapts this assay to the Illumina platform, allowing for efficient genome-wide Ψ identification (Fig. 4.25.1D).
This unit provides detailed protocols that have been optimized for performing Pseudo-seq on S. cerevisiae. Basic Protocol 1 describes the isolation of total or poly(A)+ RNA from exponentially growing cultures of S. cerevisiae. Basic Protocol 2 describes preparation of Pseudo-seq libraries from the RNA obtained in Basic Protocol 1. This consists of a conventional Illumina library preparation protocol, which has been modified to include CMC modification and reversal steps, as well as changes in the size selection steps designed to allow enrichment of abortive RT products. Basic Protocol 3 provides an outline of the steps required for computational analysis of Pseudo-Seq data, including identification of novel pseudouridylation events, as well as genetic assignment of sites to known pseudouridylation factors. The protocols presented in this unit have been optimized for budding yeast. However, with simple modifications they can be applied to any growth condition, model organism, or cell type, provided a sufficient quantity of RNA can be obtained.
SAMPLE PREPARATION AND RNA ISOLATION
In this protocol, cultures of S. cerevisiae are grown to mid-log phase and are harvested by centrifugation. High-quality total RNA is then isolated from cell pellets by extraction with hot acid phenol, which is sufficient for the identification of Ψ in highly abundant transcripts such as rRNAs, and tRNAs (unit 13.12). Poly(A)+ RNAs are then enriched by selection with oligo(dT) cellulose, which allows for detection of Ψ residues in polyadenylated transcripts of lower abundance (Sambrook and Russell, 2001).
The growth conditions, culture size, and media used for propagation of budding yeast can be changed without significantly affecting these protocols, provided a sufficient quantity of RNA is recovered. Protocols for isolation of high-quality total RNA from other biological systems can be found elsewhere, and should be compatible with Pseudo-seq. If detection of Ψ in nonpolyadenylated transcripts of lower abundance is desired, a method of enrichment other than poly(A) selection may be employed. Performing Pseudo-seq on multiple biological replicates is essential for the ability to filter out false positives, especially in transcripts of lower abundance. The number of replicates required is flexible, provided changes in computational parameters are made. More details on the extent of biological replication required can be found in the Commentary.
Materials
YPAD (see recipe)
S. cerevisiae
Deionized, distilled H2O
Liquid nitrogen
Acid phenol
AES buffer (see recipe)
Ice
Chloroform
25:24:1 acid phenol:chloroform:isoamyl alcohol
Sodium acetate, pH 5.3
Isopropanol
70% Ethanol
TES buffer (see recipe)
Oligo(dT) cellulose beads (NEB, cat. no. S1408)
TES + NaCl Buffer (see recipe)
5 M NaCl
1 M Tris·Cl, pH7.6
0.5 M EDTA, pH 8.0
20% (w/v) SDS
Shaking incubator
2-liter baffled flasks
Benchtop and refrigerated centrifuges
50- and 15-ml conical tubes
Pipets
Adjustable temperature water bath
Vortex mixer
Oak Ridge tubes (Thermo Scientific, cat. no. 3115-0030)
Rotating rack
Cellulose Acetate Syringe Filters (0.45 μm)
2-ml microcentrifuge tubes
200-μl PCR tubes
Additional reagents for quantification of cells and nucleic acids by spectrophotometry (appendix 3) and isopropanol precipitation of the poly(A)+ RNA with GlycoBlue (Support Protocol 1)
Yeast growth
-
1
Inoculate 10 ml of YPAD with a single yeast colony, and grow overnight with shaking at 30°C to a target OD600 ~ 7.0.
-
2
Dilute the overnight culture to OD600 = 0.05 in 750 ml prewarmed YPAD, and grow in a 2-liter baffled flask at 30°C with shaking at 200 rpm to OD600 = 1.0.
-
3
Harvest the cells by centrifuging for 5 min at ~16,000 × g, 4°C, and decant the supernatant.
-
4Resuspend the cell pellet in 25 ml ice-cold deionized distilled H2O, transfer into a clean 50-ml conical tube, centrifuge the cells for 5 min at ~3400 × g, 4°C, in a benchtop centrifuge, decant the supernatant, and then remove the residual wash with a pipet.The cell pellets should be approximately 5 ml.
-
5
Snap-freeze the cell pellets in liquid N2 in an open Dewar flask and store up to 6 months at −80°C, or proceed directly to RNA extraction.
RNA extraction
-
6
Resuspend the cell pellet in 5 ml acid phenol, and add 5 ml AES buffer.
-
7Incubate for 30 min in a 65°C water bath, and vortex every minute for ~10 sec.Frequent vigorous vortexing is necessary to maximize RNA recovery.
-
8
Place the tubes on ice for 10 min.
-
9
Add 5 ml chloroform, and centrifuge for 5 min at ~3400 × g, room temperature, in a benchtop centrifuge.
-
10
Transfer the aqueous phase into a clean 15-ml conical tube, and add 5 ml acid phenol:chloroform:isoamyl alcohol, and centrifuge as in step 9.
-
11Repeat step 10 until no protein is visible at the interface between the aqueous and organic phases.Usually two to three extractions are sufficient. There may be an SDS precipitate at the interface, which can be easily confused with protein.
-
12
Perform a final extraction with chloroform by transferring the aqueous phase into a clean 15-ml conical tube, adding 5 ml chloroform, and centrifuging as in step 9.
-
13
Transfer the aqueous phase into a clean Oak Ridge tube, add 1/9 vol 3 M sodium acetate, pH 5.3, and 1 vol isopropanol, pellet the RNA by centrifuging for 30 min at ~14,000 × g, 4°C, and decant the supernatant.
-
14
Wash twice, each time with 10 ml 70% ethanol, centrifuging for 10 min at ~14,000 × g, 4°C, after each wash. Air-dry the RNA pellet for ~3 min.
-
15Resuspend RNA in ~2 ml H2O if proceeding directly to library preparation, or in ~2 ml TES buffer if performing poly(A) selection.Expect a yield of ≥10 μg of RNA per ml of culture at OD600 = 1.0. A final concentration of ≥1 μg/μl is desired.
Enrichment of polyadenylated RNAs with oligo(dT) cellulose beads
Budding yeast transcripts have relatively short poly(A) tails compared to other organisms (Subtelny et al., 2014). We have found that poly(A) selection using oligo(dT) cellulose and relatively long incubations gives much greater recovery of yeast mRNA than rapid, magnetic bead based capture methods (e.g., Dynabeads). Dynabeads capture may be used for organisms with longer poly(A) tails.
-
6Transfer 1.5 ml of oligo(dT) beads (50% slurry) into a 15-ml conical tube.This volume of slurry is sufficient for one sample. However, beads can be prepared in batch for all samples (steps 16 to 19).
-
7
Pellet the beads by centrifuging for <1 min at 3000 × g, room temperature, in a benchtop centrifuge, and gently remove the supernatant by decanting.
-
8Wash the beads three times, each time with 3 ml H2O, centrifuging and removing supernatants as in step 17.The washes in steps 18 and 19 should be performed with 2 slurry volumes of wash buffer (H2O or TES + NaCl).
-
9Wash the beads twice, each time with 3 ml TES + NaCl Buffer, centrifuging and removing supernatants as in step 17.If preparing beads in batch, distribute equal volumes of beads (~750 μl bead bed volume) in wash buffer to individual 15-ml conical tubes for each sample before the final spin. Handling times for this protocol can be significantly reduced by using 15-ml tubes with attached “flip-top” caps.
-
10
Bring 7.5 to 10 mg of total RNA to a volume of 4.5 ml in TES Buffer in a 15-ml conical tube.
-
11
Denature the RNA by incubating for 15 min in a 65°C water bath, and then placing the tubes on ice.
-
12
Bring the concentration of NaCl to 0.55 M by adding 563 μl of 5 M NaCl to the RNA samples. Then transfer RNA to the oligo(dT) cellulose bead pellet, vortex briefly, and incubate on a rotating rack for 15 min at room temperature.
-
13Pellet the beads by centrifuging for <1 min at 3000 × g, room temperature, in a benchtop centrifuge. Transfer the supernatant into the original 15-ml conical tube, incubate in a 65°C water bath for 10 min, and place on ice for 2 min.This step re-denatures the unbound RNA in the supernatant to enhance poly(A) RNA binding to beads in the subsequent incubation step.
-
14Return the RNA to the beads, vortex briefly, and incubate on a rotating rack for 15 min at room temperature.This second incubation with oligo(dT) cellulose increases recovery of poly(A) RNA.
-
15
Pellet the beads, and discard the supernatant as in step 17.
-
16
Wash the beads three times, each time with 5 ml TES + NaCl buffer. For each wash incubate on a rotating rack for 2 min at room temperature. Pellet the beads as in step 17, and discard the supernatant.
-
17Add 2 ml ice-cold H2O to the bead pellet. Vortex briefly, pellet the beads, and discard the supernatant as in step 17.It is important to perform this step quickly to avoid loss of bound RNA. It is advisable to do no more than 4 samples at once.
-
18
Add 2 ml 55°C H2O to the bead pellet, and incubate in a 55°C water bath for 5 min. Pellet the beads as in step 17, and transfer the supernatant to a clean 15 ml conical tube.
-
19Repeat step 28 once more, and pool the eluates.This poly(A) selected RNA will be used in a second round of poly(A) selection. To skip this second round of selection proceed directly to step 39.
-
20
Wash the used beads once with 5 ml H2O, and once with 5 ml TES + NaCl Buffer. Pellet the beads and discard the supernatant after each wash as in step 17.
-
21
Bring the RNA eluates to a total volume of 5 ml in TES buffer by adding 50 μl 1 M Tris·Cl, pH 7.6, 10 μl 0.5 M EDTA, pH 8.0, 25 μl 20% SDS, and H2O to 5 ml (~900 μl).
-
22
Incubate the RNA for 10 min in a 65°C water bath, and then place on ice.
-
23
Bring the concentration of NaCl to 0.55 M by adding 626 μl of 5 M NaCl to RNA. Then transfer to the oligo(dT) cellulose bead pellet, vortex briefly, and incubate on a rotating rack for 15 min at room temperature.
-
24
Pellet the beads as in step 23. Transfer the supernatant to the original 15-ml conical tube, incubate in a 65°C water bath for 5 min, and place on ice for 2 min.
-
25
Return the RNA to the beads, vortex briefly, and incubate on a rotating rack for 15 min at room temperature.
-
26
Wash the beads three times with TES + NaCl buffer as in step 26.
-
27
Wash the beads once with ice-cold water as in step 27.
-
28
Elute poly(A)+ RNA twice with 1.8 ml 55°C H2O as in steps 28 to 29, pooling the eluates.
-
29Pass the eluates through a cellulose acetate filter with a syringe, and distribute the filtrate across four 2-ml microcentrifuge tubes.This step removes any residual beads from the eluates.
-
30
Isopropanol precipitate the poly(A)+ RNA with GlycoBlue as described in Support Protocol 1.
-
31Resuspend each pellet in 6 μl H2O, and combine the RNA into one 200-μl PCR tube (24 μl total per sample).The yield should be approximately 2 μg of poly(A)+ RNA.
ISOPROPANOL PRECIPITATION OF NUCLEIC ACIDS
This protocol describes precipitation of nucleic acids from samples of small volumes.
Materials
Sample: PolyA+ RNA (Basic Protocol 1); RNA fragments (step 4, Basic Protocol 3); gel-purified fragments obtained (from Support Protocol 3)
3 M sodium acetate, pH 5.3
Isopropanol
GlycoBlue (Invitrogen, cat. no. AM9516)
70% (v/v) ethanol
Microcentrifuge
- To each sample add 1/9 vol 3 M sodium acetate, pH 5.3, 1 vol isopropanol, and 2 μl GlycoBlue.Omit GlycoBlue if the concentration of nucleic acids is high, or if GlycoBlue added in previous steps carries over.
Precipitate at −20°C for 30 min to overnight, or for 10 min at −80°C.
Centrifuge for 30 min at max speed, 4°C, in a microcentrifuge, and discard the supernatant.
Wash the pellets with 750 μl ice-cold 70% ethanol, centrifuge for 10 min at max speed, 4°C, in a microcentrifuge, and discard the supernatant.
Air-dry the nucleic acid pellets for 2 min, and resuspend in an appropriate buffer.
REGENERATION OF OLIGO(dT) CELLULOSE BEADS
Oligo(dT) cellulose beads can be reused multiple times to save on reagent costs. First residual RNA is degraded by incubation of beads at alkaline pH, followed by extensive washing to neutralize the pH, and storage in TES + NaCl buffer.
Additional Materials (also see Basic Protocol 1)
Used oligo(dT) cellulose beads
0.1 N NaOH
Deionized distilled H2O
TES + NaCl buffer (see recipe)
- Resuspend used oligo(dT) Cellulose Beads in ≥ 2 bead bed vols 0.1 N NaOH, and incubate on a rotating rack for 1 hour at room temperature.Beads can be regenerated in batch by pooling beads from multiple samples.
Pellet the beads by centrifuging for <1 min at 3000 × g, room temperature, in a benchtop centrifuge, and gently remove the supernatant by decanting.
Resuspend the beads in 10 ml deionized distilled H2O, vortex briefly, pellet the beads, and discard the supernatant as in step 2. Repeat until the supernatant reaches a pH of 7.0.
Resuspend the beads in 1 bead bed volume of TES + NaCl buffer, and store up to 6 months at 4°C.
PSEUDO-SEQ LIBRARY PREPARATION
In this protocol a library suitable for sequencing on an Illumina HiSeq is prepared from total or poly(A)+ RNA isolated using Basic Protocol 1. It assumes that the final libraries will be submitted to a facility that will perform sample quality control and Illumina sequencing. Sample RNA is first fragmented uniformly by treatment with divalent cations. After fragmentation, Ψ residues are specifically derivatized by treatment of RNA with CMC, and subsequent alkaline reversal to remove CMC from G and U residues. In parallel RNA fragments are mock treated. Fragments of the desired size range are gel purified, and subjected to 3′ adapter ligation, providing a uniform primer binding site for RT. cDNA synthesis is carried out, followed by gel purification of truncated cDNAs. Intramolecular ligation is then used to circularize the purified cDNAs, which are subsequently amplified by PCR. These PCR products are gel purified, and are ready for Illumina Sequencing.
These protocols describe a conventional Illumina library preparation protocol that has been modified to facilitate the identification of Ψs. Other library preparation protocols should be compatible with Pseudo-seq with a few adaptations, provided the 3′ end of the cDNAs can be mapped from the sequencing data. These adaptations include in vitro CMC derivatization and reversal of RNA, size selection of a narrow range of RNA fragment sizes, and size selection of truncated rather than full-length cDNAs. It is important that CMC modification and reversal steps are carried out after RNA fragmentation to maximize the accessibility of Ψ residues to CMC.
Materials
Total or Poly(A)+ RNA obtained from Basic Protocol 1
Deionized distilled H2O
Ice
100 mM ZnCl2
40 mM EDTA, pH 8.0
GlycoBlue
BEU buffer (see recipe)
CMC (Sigma, cat. no. C106402)
0.5 M CMC in BEU buffer (make fresh immediately before CMC treatment)
3 M sodium acetate, pH 5.3
Ethanol
Sodium carbonate buffer (see recipe)
10 mM Tris·Cl, pH 8.0
RNasin Plus (Promega, cat. no. N2615)
T4 Polynucleotide Kinase (PNK; NEB, cat. no. M0201) containing: 10× PNK buffer
Calf intestinal alkaline phosphatase (CIP; NEB, cat. no. M0290)
2× RNA loading dye (see recipe)
10-bp DNA ladder (Invitrogen, cat. no. 10821-015)
0.5× TBE
SYBR Gold (Invitrogen, cat. no. S-11494)
3′ adapter: /5Phos/TGGAATTCTCGGGTGCCAAGG/3ddC/
100 μM adenylated 3′ adapter: AppTGGAATTCTCGGGTGCCAAGG/3ddC/
(see unit 26.4)
T4 RNA ligase (NEB, cat. no. M0204) containing:
T4 RNA ligase buffer
PEG 8000
100 μM Gel-purified RT Primer: /5Phos/GATCGTCGGACTGTAGAACTCTG AACCTGTCGGTGGTCGCCGTATCATT/iSp18/CACTCA/iSp18/GCCTT
GGCACCCGAGAATTCCA
10× RT buffer without Mg2+ (see recipe)
10 mM dNTPs
100 mM MgCl2
AMV RT (Promega, cat. no. M5108)
1 N NaOH
1 N HCl
CircLigase ssDNA ligase (Epicentre, cat. no. CL4115K)
1 mM ATP
50 mM MnCl2
Phusion High-Fidelity (HF) DNA polymerase (NEB, cat. no. M0530L) containing:
HF buffer
Forward PCR primer: AATGATACGGCGACCACCGA
Barcoded reverse PCR primers (XXXXXX Indicates unique barcodes):
CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA
6× DNA loading dye (see recipe)
Thermal cycler
200-μl PCR tubes
Thermomixer (Eppendorf)
1.5-ml microcentrifuge tubes
Microcentrifuge
Gel electrophoresis equipment
Rocker platform
UV-transilluminator
Additional reagents for the adenylation of oligonucleotides (unit 26.4), extracting RNA from the gel slices (Support Protocol 3), precipitating with GlycoBlue (Support Protocol 1), and denaturing and nondentauring polyacrylamide gel electrophoresis (PAGE) (unit 2.12).
RNA fragmentation
The fragmentation conditions can vary depending upon the type and source of RNA. Those presented here have been optimized for total or poly(A)+ RNA from S. cerevisiae. Use of RNA from other biological systems may require optimization of fragmentation temperature, reagent, and time.
-
1
To ~2 μg of poly(A)+ RNA or ~25 μg of total RNA in PCR tubes add H2O to a final volume of 54 μl, and then add 6 μl of 100 mM ZnCl2.
-
2
Incubate the fragmentation reactions at 94°C in a thermal cycler for 55 sec for poly(A)+ RNA or 5 min for total RNA.
-
3
Quench fragmentation reaction by placing on ice, and quickly adding 60 μl of 40 mM EDTA.
-
4
Precipitate the fragmentation reaction as described in Support Protocol 1. Omit GlycoBlue if poly(A)+ RNA is used, and add GlycoBlue if total RNA is used.
-
5
Resuspend the RNA fragments in 30 μl H2O.
CMC modification and reversal
Ψ residues can be specifically derivatized by treatment of RNA with CMC, which modifies Ψs, Us, and Gs. Reversal of CMC modification of Us and Gs occurs at alkaline pH, allowing for the specific labeling of Ψ residues. At this point in the protocol the RNA from an individual sample is split into CMC-treated and mock-treated samples, and all subsequent steps should be carried out on both samples.
-
6Make a fresh stock of 0.5 M CMC in BEU buffer.A concentration of 0.5 M corresponds to 212 mg/ml. A total of 100 μl is needed per sample.
-
7Split the RNA sample into two clean 200-μl PCR tubes for CMC-treatment (+CMC) and mock treatment (−CMC). For the +CMC sample, transfer 18 μl of RNA, and add 2 μl of H2O. For the −CMC sample transfer 12 μl of RNA, and add 8 μl of H2O.More RNA is added to the CMC-treated sample than the mock-treated sample to account for poorer recovery of CMC-derivatized RNA by ethanol precipitation.
-
8Add 2.9 μl of 40 mM EDTA (5 mM final) to RNA fragments. Incubate for 3 min at 80°C in a thermal cycler and then quickly place on ice.This denatures the RNA fragments increasing the extent of CMC derivatization.
-
9Add 100 μl of 0.5 M CMC in BEU buffer to the +CMC sample (0.4 M CMC final), and 100 μl of BEU buffer to the −CMC sample.The concentration of CMC can be varied. Lower concentrations of CMC may be used to map closely spaced Ψ residues.
-
10
Incubate the RNA for 45 min at 40°C with shaking at 1000 rpm in a Thermomixer.
-
11Transfer the +CMC and −CMC samples into clean 1.5-ml microcentrifuge tubes. To each tube add 2 μl GlycoBlue, 50 μl of 3 M sodium acetate (pH 5.3), and 1.0 ml ethanol.Use of these precipitation conditions is important since isopropanol precipitation leads to poor recovery of RNA after CMC treatment.
-
12
Precipitate at −20°C for 30 min to overnight.
-
13Centrifuge for 30 min at max speed, 4°C, in a microcentrifuge, and discard the supernatant.The +CMC RNA pellets will be more diffuse than the −CMC RNA pellets.
-
14
Wash the pellets twice, each time with 500 μl ice-cold 70% ethanol. Centrifuge for 10 min at max speed, 4°C, in a microcentrifuge, and discard the supernatant after each wash.
-
15
Air-dry the pellets for 2 min.
-
16
Resuspend both +CMC and −CMC samples in 30 μl sodium carbonate buffer.
-
17Incubate the RNA for 2 hr at 50°C with shaking at 1000 rpm in a Thermomixer.This step reverses CMC modification of U and G residues.
-
18
To each sample add 2 μl GlycoBlue, 1/9 vol 3 M sodium acetate, pH 5.3, and 2.5 vol ethanol.
-
19
Precipitate RNA as in steps 11 to 14.
-
20
Resuspend the pellets in 8 μl of 10 mM Tris·Cl, pH 8.0.
3′ end healing
-
6To the RNA samples add 0.5 μl of RNasin Plus, 1.25 μl 10× PNK buffer, 1.25 μl T4 PNK, and 1 μl CIP.RNA fragmentation with Zn2+ leaves a 2′,3′ cyclic phosphate, which must be healed, and removed to convert the RNA fragments to suitable substrates for adapter ligation.
-
7
Incubate for 1 hr at 37°C.
-
8
Add 12.5 μl of 2× RNA loading dye.
Size selection of RNA fragments
-
6
Prepare an 8% TBE-urea-polyacrylamide mini-gel (see unit 2.12), and pre-run for 20 min at 200 V.
-
7Prepare 10-bp ladder by mixing 0.5 μl of 10-bp DNA ladder, 9.5 μl H2O, and 10 μl of 2× RNA loading dye.The volumes above are for one gel lane. Prepare a sufficient quantity for loading on both sides of all samples. The ladder will be used to guide excision of RNA fragments from the gel.
-
8
Incubate the RNA samples for 2 min at 95°C, and place on ice until loading.
-
9Load the gel, and run for 50 min at 200 V.The bromophenol blue dye front will run off of the gel.
-
10
Disassemble the gel, and stain in 15 ml of 1:10,000 SYBR Gold in 0.5× TBE for 5 min at room temperature with gentle rocking.
-
11
Visualize the gel on a UV-transilluminator and photograph.
-
12Excise the gel-slices corresponding to several size ranges for each sample using a clean razor blade, and transfer each gel slice to a clean 1.5-ml microcentrifuge tube.For example, 80 to 100 nt, 100 to 120 nt, and 120 to 140 nt fragment ranges work well (Fig. 4.25.2).Proceed with a single range of fragment sizes, keeping the others as backup. While Pseudo-seq libraries can be made from a wide range of RNA fragment sizes, including shorter fragments (e.g., 60 to 70 nt), it is important that the size range used is relatively narrow (10 to 20 nt). This allows for robust separation of truncated from full-length cDNAs.
-
13
Extract RNA from the gel slice as described in Support Protocol 3.
-
14
Resuspend the RNA fragments in 5.5 μl H2O.
Figure 4.25.2.
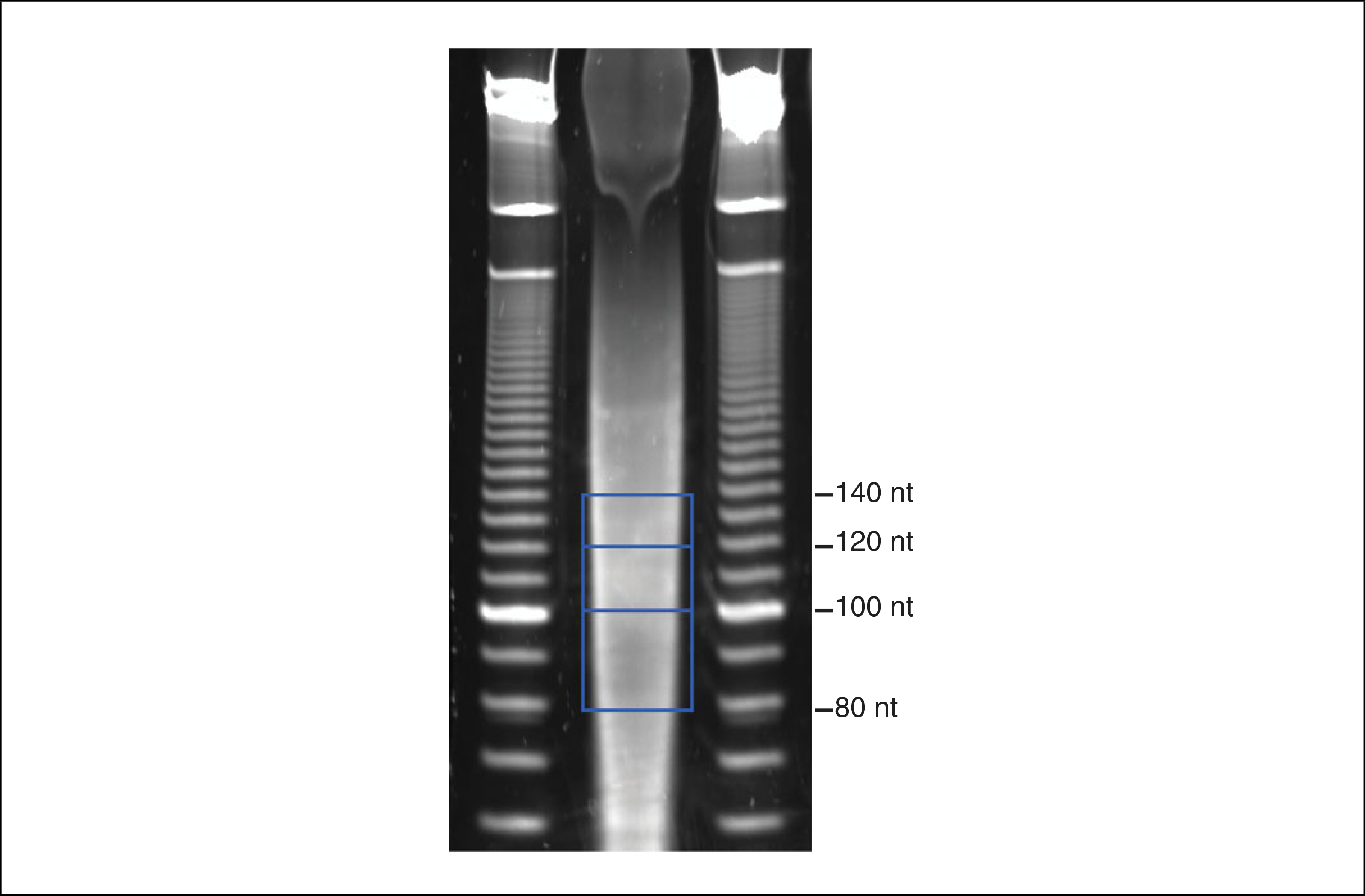
Purification of RNA fragments example gel. Regions excised from the gel are bounded by blue boxes, representing size ranges of 80 to 100 nt, 100 to 120 nt, and 120 to 140 nt.
3′ adapter ligation
This protocol uses adenylated adapters, which are available commercially, but are quite expensive. This expense can be avoided by in house adenylation of adapters with ImpA, which can be obtained by a relatively straightforward synthesis that can be performed using common equipment. Both steps are described in unit 26.4, Alternate Protocol.
-
6To the gel-purified RNA fragments add 0.5 μl adenylated 3′ adapter, 1.2 μl 10× T4 RNA ligase buffer, 3 μl PEG 8000, 1 μl RNasin Plus, and 1 μl T4 RNA Ligase.Do not add ATP to this reaction. Omitting ATP ensures that only the adenylated adapters can be ligated on to the 3′ ends of RNA fragments.
-
7
Incubate for 2.5 hr at 22°C.
-
8
Bring up to 270 μl in H2O, and precipitate with GlycoBlue as in Support Protocol 1.
-
9Resuspend the RNA pellets in 7 μl water.Ligation efficiency should be 70% to 90%, which can be assessed by running 0.8 μl of the precipitated ligation reaction on an 8% TBE-Urea-Polyacrylamide mini-gel.
Reverse transcription
-
6Transfer 6.2 μl of the resuspended ligation reactions into clean 200-μl PCR tubes and add 1 μl gel-purified RT primer (25 M stock), and 0.8 μl 10× RT buffer without Mg2+. In parallel, prepare a no RNA reaction with 6.2 μl H2O.It is very important that the RT primer be gel-purified to ensure that it is a uniform length, allowing robust separation of truncated from full-length cDNAs. Gel-purification should be performed in house, as gel-purified primers obtained commercially can be heterogeneous.
-
7
Anneal the RT primer to the RNA fragments. In a thermal cycler incubate at 65°C for 4 min, followed by 55°C for 2 min, 45°C for 2 min, and 42°C for 2 min. Centrifuge quickly to collect any condensation, and then place the samples on ice.
-
8
Prepare an extension master mix (per sample) as follows:
0.6 μl of 10× RT buffer without Mg2+
1.4 μl of 10 mM dNTPs
0.7 μl of 100 mM MgCl2
1.3 μl H2O
1.0 μl RNasin Plus
1.0 μl AMV RT.
-
9
Add 6 μl extension mix to the annealing reactions.
-
10
Incubate for 1 hr at 42°C in a thermal cycler.
-
11Add 1.5 μl of 1 N NaOH, and incubate for 15 min at 98°C in a thermal cycler.This step removes the RNA fragments.
-
12
Neutralize by adding 1.5 μl of 1 N HCl.
-
13
Add 17 μl of 2× RNA loading dye
Size selection of cDNA
-
6
Prepare an 8% TBE-urea-polyacrylamide mini-gel (see unit 2.12), and pre-run for 20 min at 200 V.
-
7Prepare 10-bp ladder by mixing 0.5 μl of 10-bp DNA Ladder, 9.5 μl H2O, and 10 μl 2× RNA loading dye.The volumes above are for one gel lane. Prepare a sufficient quantity for the number of samples used.
-
8
Incubate cDNA samples for 2 min at 95°C, and place on ice until loading.
-
9Load the gel, and run for 65 min at 200 V.Split RT samples across 2 lanes, and load one half of the no RNA RT sample in one well. Run the RT primer as close to the bottom of the gel as possible to maximize separation of truncated from full-length cDNAs. The xylene cyanol runs at ~75 nt, and the RT primer runs at 85 nt.
-
10
Disassemble the gel, and stain in 15 ml of 1:10,000 SYBR Gold in 0.5× TBE for 5 min at room temperature with gentle rocking.
-
11
Visualize the gel on a UV-transilluminator and photograph.
-
12Excise the gel-slices corresponding to truncated cDNAs (Fig. 4.25.3) using a clean razor blade, and transfer each gel slice to a clean 1.5-ml microcentrifuge tube.Excised cDNAs should contain at least 25 nt added to the primer. Shorter products contain non-specific stops. Additionally, avoid the 105 nt non-specific product present in the no RT control, and avoid the full-length cDNA band.
-
13
Extract the cDNAs from the gel slices as described in Support Protocol 3.
-
14
Resuspend in 15 μl of 10 mM Tris·Cl, pH 8.0.
Figure 4.25.3.
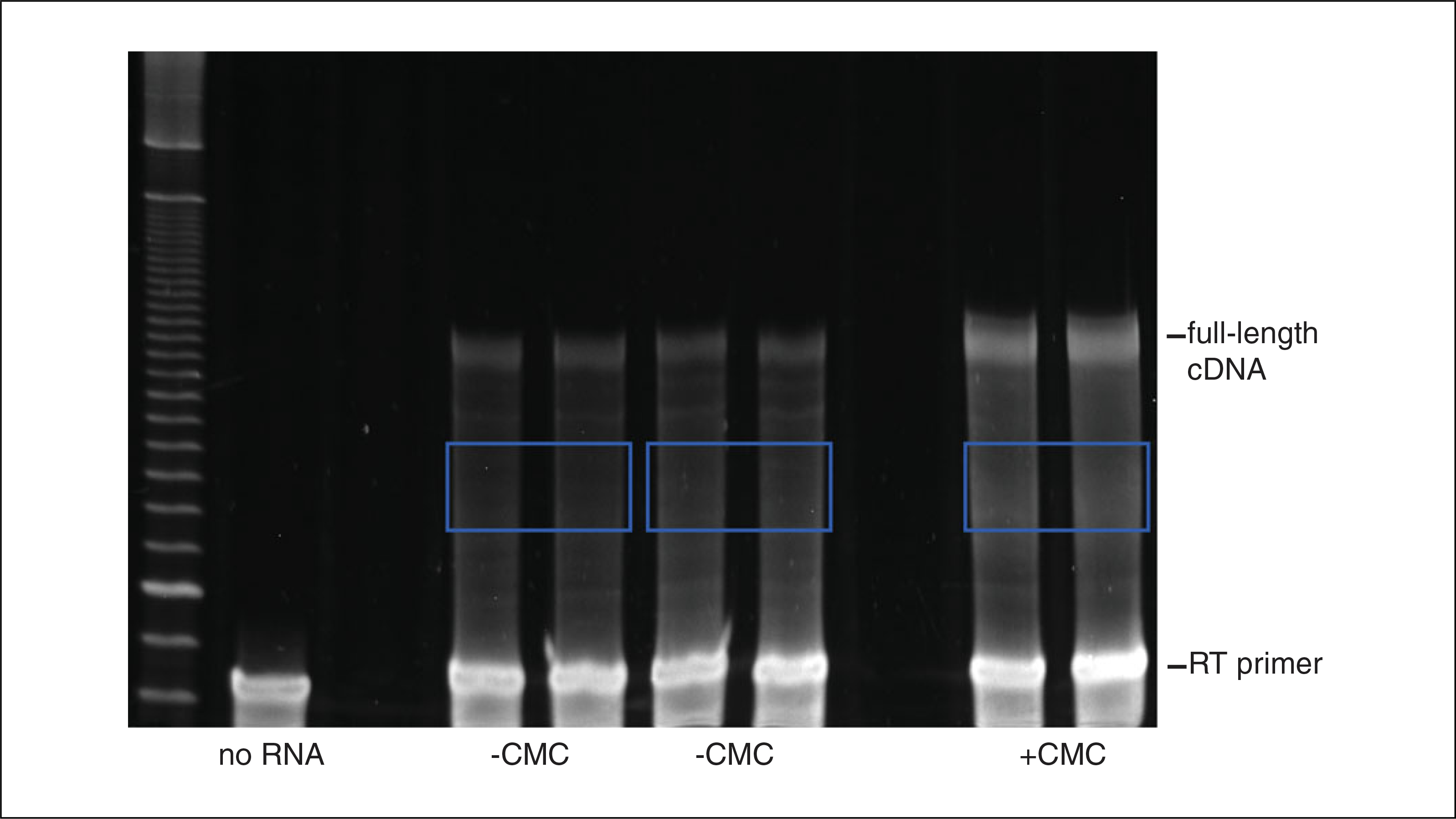
Purification of truncated cDNAs example gel. Regions excised from the gel (two −CMC samples, and one +CMC sample) are bounded by blue boxes.
cDNA circularization
-
6
Prepare circularization master mix (per sample) as follows:
2 μl of 10× CircLigase buffer
1 μl of 1 mM ATP
1 μl of 50 mM MnCl2.
-
7
Add 4 μl circularization master mix to each cDNA sample and mix well.
-
8
Add 1 μl of 0.5× CircLigase in 1× CircLigase buffer to each sample, and mix well.
-
9Incubate for 60 min at 60°C in a thermal cycler.Longer incubations may increase ligation efficiency.
-
10Incubate for 10 min at 80°C in a thermal cycler to heat inactivate.After heat inactivation, reactions can be stored at −20°C.
PCR amplification
-
6
Prepare a PCR master mix for each library, enough for 4.5 reactions. Per library:
15 μl HF buffer
1.5 μl 10 mM dNTPs
3.78 μl 10 μM forward PCR primer
3.78 μl 10 μM barcoded reverse PCR primers
52.6 μl H2O
0.75 μl Phusion.Use a different barcoded reverse primer for each sample that will be sequenced on the same HiSeq lane. -
7
Add 4.5 μl circularized cDNA sample to PCR master mix, mix well, and pipet 16.7 μl into each of four separate 200-μl PCR tubes.
-
8
Perform PCR amplification by placing all tubes into a thermal cycler and starting a program with the specified conditions. Remove tubes successively after the extension phase of cycles 12, 14, 16, and 18, placing tubes on ice.
1 cycle: 30 sec 98°C (initial denaturation)
18 cycles: 10 sec 98°C (denaturation)
20 sec 60°C (annealing)
40 sec 72°C (extension).
-
9
Add 3.4 μl of 6× DNA loading dye to the reactions.
-
10
Prepare 10-bp ladder by mixing 1 μl of 10-bp ladder, 15.7 μl H2O, and 3.3 μl 6× DNA loading dye.
-
11
Prepare an 8% TBE- Polyacrylamide mini-gel (see unit 2.12).
-
12
Load the sample and run for 40 min at 200 V.
-
13
Disassemble the gel, and stain in 15 ml of 1:10,000 SYBR Gold in 0.5× TBE for 5 min at room temperature with gentle rocking.
-
14
Visualize the gel on a UV-transilluminator and photograph.
-
15Excise the insert containing PCR bands (Fig. 4.25.4) using a clean razor blade, and transfer each gel slice to a clean 1.5-ml microcentrifuge tube.Avoid the 105-bp no insert band, and do not cut bands from lanes that are saturated, or have high-molecular-weight products. The size of the PCR product will depend on the range of RNA fragment sizes used for library preparation.
-
16
Extract the PCR products from the gel slices as described in Support Protocol 3.
-
17Resuspend in 10 μl of 10 mM Tris·Cl, pH 8.0.The gel-purified PCR product is suitable for Illumina sequencing. If more material is needed scale up the PCR reaction to 50 μl.
Figure 4.25.4.
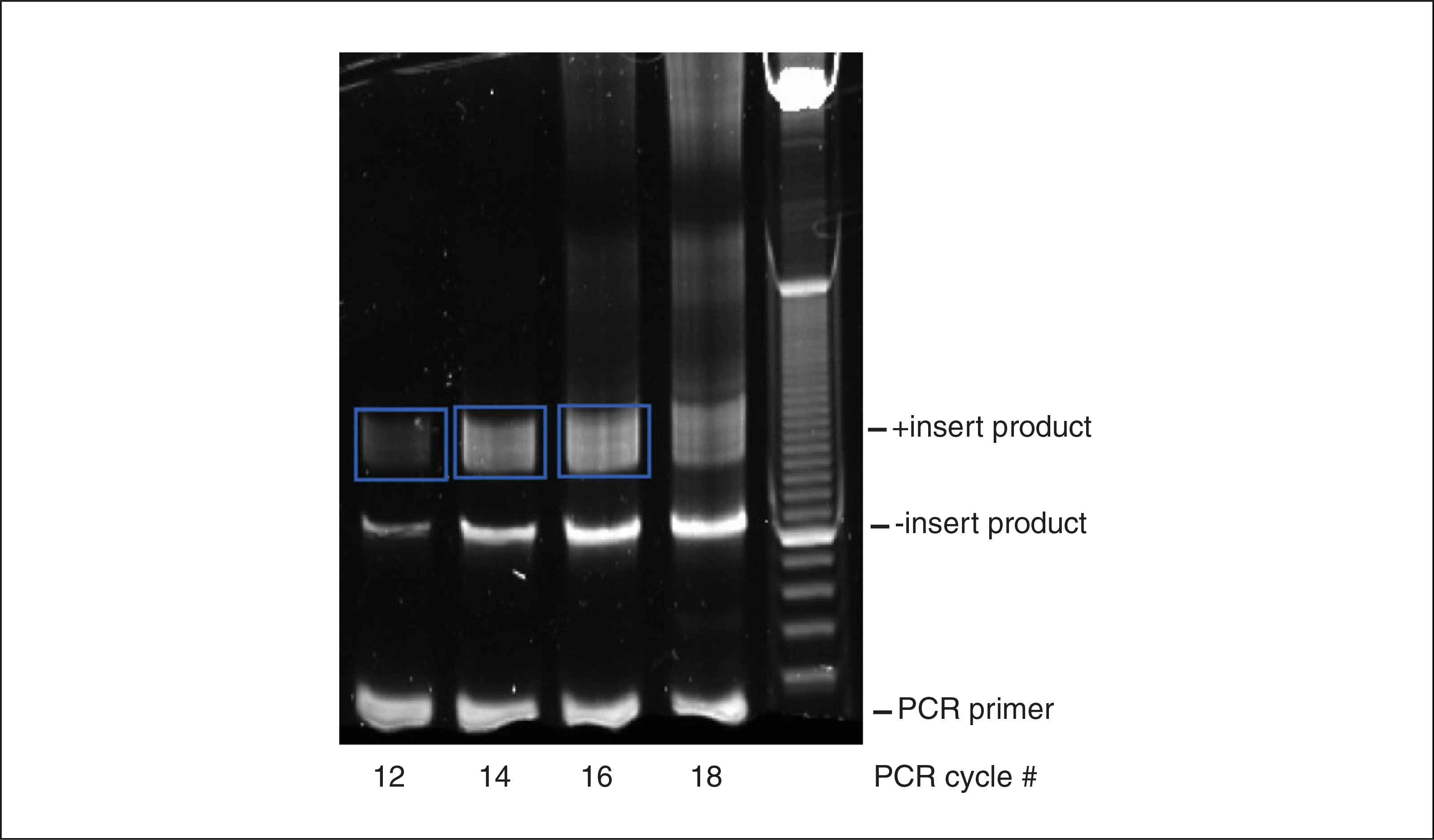
Purification of PCR products example gel. Regions excised from the gel are bounded by blue boxes. The lane for 18 cycles is too saturated.
EXTRACTION OF NUCLEIC ACIDS FROM POLYACRYLAMIDE GELS
This protocol describes the elution of nucleic acids, either RNA or DNA, from polyacrylamide gel slices.
Materials
Gel slices
DNA elution buffer (see recipe)
RNA elution buffer (see recipe)
1.5-ml microcentrifuge tubes
Rocker platform
Spin-X columns (Corning, cat. no. 8162)
Microcentrifuge
Additional reagents and equipment for precipitating the filtered, eluted RNA with GlycoBlue (Support Protocol 1)
Place the gel slice in a clean 1.5-ml microcentrifuge tube
Add 400 μl of the appropriate elution buffer: DNA elution buffer for DNA or RNA elution buffer for RNA.
Elute overnight at room temperature for DNA, or 4°C for RNA with gentle rocking.
- Transfer the supernatant into a Spin-X column, and centrifuge for 3 min min at max speed, room temperature, in a microcentrifuge.This step removes residual polyacrylamide gel fragments from the samples.
Precipitate the filtered, eluted RNA with GlycoBlue as described in Support Protocol 1.
COMPUTATIONAL ANALYSIS OF PSEUDO-SEQ DATA
This protocol describes the identification of sites of pseudouridylation from Illumina sequencing of the Pseudo-seq libraries prepared as described in Basic Protocol 2. Analysis of Pseudo-seq data uses a combination of publically available computational tools, and custom scripts that must be prepared by the user. Therefore, familiarity with analysis of next-generation sequencing data, and Python or another comparable scripting language is essential. This protocol provides usage instructions for publically available tools, and a detailed outline of the steps carried out by custom scripts, which should be sufficient to successfully complete Pseudo-seq data analysis.
The first steps in analysis involve parsing the Illumina sequencing reads, and their subsequent mapping to the target genome. If sequencing libraries have been multiplexed using barcoded PCR primers, the sequencing reads must first be de-multiplexed, that is reads should be separated by barcode. Reads should then be trimmed of the 3′ adapter sequence. Processed reads can then be mapped to the genome. These steps can be combined into an automated pipeline using custom bash scripts.
Post-mapping analyses aimed at Ψ identification are performed with custom scripts prepared by the user. Briefly, the +CMC and −CMC libraries are scaled to the same size, and then a Pseudo-seq peak value is calculated for each U residue in the transcriptome. Those sites with peak values exceeding a specified cutoff in a certain fraction of replicate libraries are identified as Ψs.
Materials
Illumina FASTQ files: The sequence files generated from an Illumina sequencing run (there should be a FASTQ file for each library submitted for sequencing)
S. cerevisiae Bowtie Index: Bowtie indices for common organisms are available, as are instructions for building bowtie index for a genome, which can be found in the Bowtie2 documentation
S. cerevisiae Splice Junctions: A TopHat readable file of splice junctions (a description of these files can be found in the TopHat documentation)
S. cerevisiae Transcript Annotations: Feature annotations in GFF format can be obtained from http://yeastgenome.org, and UTR boundaries can be found from published work (e.g., Xu et al., 2009; Arribere and Gilbert, 2013).
Python: A powerful programming language that can be used for analysis of Pseudo-Seq data (installation instructions can be found at http://python.org; other programming languages may be substituted for Python)
Cutadapt: A package for trimming adapter sequences from next-generation sequencing data (documentation can be found at https://code.google.com/p/cutadapt/)
Bowtie2: A package for aligning sequencing reads to a reference genome (documentation can be found at http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
TopHat: A package for mapping RNA-seq reads to splice-junctions; note that TopHat is dependent on Bowtie and SAMtools (documentation can be found at http://ccb.jhu.edu/software/tophat/index.shtml)
SAMtools: A set of utilities for the manipulation of genome alignments in SAM format (documentation can be found at http://samtools.sourceforge.net/)
Parsing and mapping of reads
-
1De-multiplex the FASTQ files. Match the read’s index (barcode) sequence to one of the indices used for barcoding, allowing up to one mismatch. Reads assigned to a given index sequence should be written to a new FASTQ file for that index.Each read in a FASTQ file is defined by four lines of information. The first line begins with @, and contains the name of the read. Included in this line is the read’s index, which is located between the # and / characters. The next three lines contain the sequence, an alternate read name, and Phred quality scores, respectively.
@D5FF8JN1:7:1101:1461:2111#AGGTTT/1
TTTGCTCGAATATATTAGCATGGAATAATAGAATTGGAATTCTCGGGTGC
+D5FF8JN1:7:1101:1461:2111#AGGTTT/1
abbeeeeegggggiiiiiihihighhhihhiigiihicfhgchceghhhiThe index sequences are the reverse complements of barcodes in the Barcoded Reverse PCR Primers. Reads that cannot be assigned to an index used should be stored in a file for unmapped barcodes. This step is performed by a custom script, which uses simple string comparisons for assigning indices. -
2
Compress the de-multiplexed FASTQ files. In a UNIX shell enter the command:
gzip sequence_file.fastq
Alternately, if all FASTQ files are in the same directory enter the command:
gzip *.fastqWhile this step is not necessary, compressing FASTQ files saves considerable storage space, and subsequent computational steps are compatible with compressed data. Compressed FASTQ files will have the extension fastq.gz. -
3
Trim adapter sequences, and filter reads by length using Cutadapt. For each compressed FASTQ file, enter the following command in a UNIX shell:
cutadapt -a ADAPTER_SEQUENCE -- overlap 3 --minimum- length 18 -o trimmed_reads.fastq.gz input.fastq.gz
ADAPTER_SEQUENCE is the sequence of the adapter to trim. --overlap 3 specifies that at least 3 bases of overlap between a read and an adapter sequence are required for trimming. -- minimum-length discards reads shorter than 18 nucleotides. input.fastq.gz and trimmed_reads.fastq.gz are the input and output file names respectively.A minimum length of 18 works well for S. cerevisiae, however this parameter can be changed for other systems. -
4
Map the trimmed reads to the target genome and annotated splice junctions using TopHat. For each compressed FASTQ file of trimmed reads enter the following command in a UNIX shell:
tophat2 --no-novel-juncs --no-novel-indels --raw-juncs splice_junctions bowtie_index trimmed_reads.fastq.gz
--no-novel-juncs and --no-novel-indels specify that TopHat should not search for novel splice junctions or indels, respectively. splice_junctions is the file containing splice junction annotations, bowtie_index is the genome’s bowtie index, and trimmed_reads.fastq.gz is the file containing the trimmed reads generated in step 3.
TopHat will create several output files. The relevant file for subsequent analysis is called accepted_hits.bam. This file should be renamed with an informative, unique file name.
-
5
Use SAMtools to read the accepted_hits.bam file for each library, and use a custom script to assign the mapped read 5′ ends, which correspond to the cDNA 3′ ends, to their corresponding genomic positions. The genome can then be split into transcribed units.
-
6
Determine the total number of mappable reads in the library, the total number of reads mapping to rRNA, and the total number of reads mapping to coding sequences using a custom script. These values will be used to scale libraries in step 7.
Computational identification of sites of pseudouridylation
These steps are performed using custom scripts. Steps 7 to 12 should be performed for each +CMC and −CMC library pair. A more detailed discussion of how to choose the parameters involved is included in the Commentary section below.
-
7
For each +CMC and −CMC library pair, scale the size of the −CMC to the size of the +CMC library. Scale by multiplying the reads at each position in the −CMC library to the ratio of reads in the +CMC library to reads in the−CMC library.
For identification of sites in ncRNAs, the ratio should be calculated based on the total mapped reads in the library, or the reads mapping to rRNA. For sites in mRNAs, scale based on the ratio of reads mapping to coding sequences.Scaling is necessary to ensure that differences in library size do not affect Pseudo-seq peak values, described in step 11. A larger +CMC library will artificially inflate peak values, and the converse is true for a larger −CMC library. -
8Filter transcripts by read coverage. Remove transcripts from further analysis with fewer than rc reads per nt on average, where rc is an empirically determined read coverage threshold.This step may be skipped with a sufficient number of biological replicates, in this case use an rc value of 0. Empirical determination of rc is described in the Commentary section below.
-
9
Locate all Us in the transcripts passing the read coverage cutoff specified in step 8.
-
10For each U, determine the number of reads whose 5′ ends map 1 nucleotide 3′ of the U (3′ position) for both the +CMC and −CMC libraries (r+ and r−, respectively). In addition, determine the number of reads whose 5′ ends map to a window of size ws centered at, but exclusive of the 3′ position for both the +CMC and −CMC libraries (wr+ and wr−, respectively). These read counts will be used to calculate the Pseudo-seq peak values in step 11.For budding yeast, a value of 150 works well for window size (ws).
-
11For each U calculate a Pseudo-seq peak value according to Equation 4.25.1 below, whose variables are described above in step 10:
-
12For each +CMC and −CMC library, pair flag U’s whose peak value is greater than p.These flagged U’s represent a combination of real sites of pseudouridylation, and false positives. Filtering for reproducibility in step 13 can be used to eliminate false positives.
-
13
Filter the U’s for those that are flagged in n or more of N +CMC and −CMC library replicate pairs. These positions are called as pseudouridines.
GENETIC ASSIGNMENT OF Ψs TO KNOWN PSEUDOURIDYLATION FACTORS
Predicted Ψs can be assigned to known pseudouridylation factors by performing Pseudo-seq on two biological replicates of yeast strains deleted for the pseudouridylation factor being tested.
Materials
Pseudo-seq libraries from yeast strains deleted for a pseudouridylation factor: Prepared as in Basic Protocol 2, analyzed as in Basic Protocol 3.
For each identified Ψ calculate the median Pseudo-seq peak height for all +CMC and −CMC library pairs.
For each identified Ψ calculate the median total reads (wr+ + wr−) in the window centered at that site for all +CMC and −CMC library pairs.
Call a Ψ as factor-dependent if the peak heights in both biological replicate Pseudo-seq libraries deleted for that factor are less than 25% of the median peak height from step 1, and at least one of the two replicate libraries has greater than 25% of the median total window reads from step 2.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see appendix 2.
AES buffer
50 mM sodium acetate, pH 5.3
10 mM EDTA, pH 8.0
1% (w/v) SDS
Store up to 1 year at room temperature
BEU buffer
7 M Urea
4 mM EDTA, pH 8.0
50 mM Bicine, pH 8.5
Filter sterilize using a 0.2-μm filter
Store up to 6 months at room temperature
Adjust the final pH to ~9.0 using NaOH and HCl
DNA elution buffer
300 mM NaCl
10 mM Tris·Cl, pH 8.0 (appendix 2)
Store up to 1 year at room temperature
DNA loading dye, 6×
30% (v/v) Glycerol
0.025% (w/v) bromphenol blue
0.025% (w/v) xylene cyanol FF
Store up to 1 year at room temperature
RNA elution buffer
300 mM sodium acetate, pH 5.3
1 mM EDTA, pH 8.0
100 U/ml RNasin Plus
Add the RNasin immediately before use
RNA loading dye, 2×
95% (v/v) formamide
5 mM EDTA, pH 8.0
0.025% (w/v) SDS
0.025% (w/v) bromphenol blue
0.025% (w/v) xylene cyanol FF
Divide into 0.5- to 1.0-ml aliquots
Store up to 6 months at −20°C
RT buffer w/o Mg2+, 10×
500 mM Tris·Cl, pH 8.6 (appendix 2)
600 mM NaCl
100 mM DTT
Store up to 6 months at −20°C
Sodium carbonate buffer
50 mM Na2CO3, pH 10.4
2 mM EDTA, pH 8.0
Prepare from stocks of 1 M Na2CO3, pH 10.4, and 0.5 M EDTA, pH 8.0, and adjust the final pH to 10.4 using sodium bicarbonate
Filter sterilize using 0.2-μm filter
Store up to at least 2 years at −20°C
TES buffer
10 mM Tris·Cl, pH 7.6 (appendix 2)
1 mM EDTA, pH 8.0
0.1% (w/v) SDS
Store up to 1 year at room temperature
TES + NaCl buffer
10 mM Tris·Cl, pH 7.6 (appendix 2)
1 mM EDTA, pH 8.0
0.1% (w/v) SDS
0.5 M NaCl
Store up to 1 year at room temperature
YPAD
1% (w/v) Bacto yeast extract
2% (w/v) Bacto peptone
2% (w/v) glucose
0.004% (w/v) adenine sulfate
Store up to 1 year at room temperature
COMMENTARY
Background Information
There is growing interest in both the transcriptome-wide identification of RNA modifications and the functional significance of these modifications. There are more than 100 different post-transcriptional RNA modifications known, the majority of which occur in tRNAs (Cantara et al., 2011). Until recently, only three were known to occur in mRNAs, m6A, m5C, and inosine. Next-generation sequencing techniques have been increasingly employed to allow genome-wide mapping of these modifications (Li et al., 2009; Meyer et al., 2012; Squires et al., 2012; Dominissini et al., 2013). We developed Pseudo-seq to allow the genome wide mapping of pseudouridine, the most abundant modified base in RNA.
Pseudouridine was first identified as a constituent of RNA more than fifty years ago by chromatographic analysis of nucleotides derived from bulk cellular RNA from budding yeast (Davis and Allen, 1957; Cohn, 1960). Early mapping of Ψ residues used a painstaking combination of procedures including RNase digestion, column chromatography, paper electrophoresis, and paper chromatography. These techniques were used to determine the sequences of the first tRNAs in the 1960s, which led to the identification of specific sites pseudouridylation in these molecules (Holley et al., 1965). Subsequent studies used a similar set of techniques to map Ψ positions in rRNA (Choi and Busch, 1978; Gupta and Randerath, 1979; Tanaka et al., 1980).
A breakthrough in the ability to map Ψ residues with single nucleotide resolution was introduced by Bakin and Ofengad in 1993 (Bakin and Ofengand, 1993). This method, described above, allowed the rapid identification of Ψ residues in a specific RNA of interest. While able to accurately identify sites of pseudouridylation, this primer extension assay is limited in both throughput, and in the set of transcripts that can be analyzed. Primer extension must be performed on a specific transcript of interest, which depending on length, may require primer extension from multiple sites. This assay is also highly dependent on transcript abundance, typically being limited to highly abundant transcripts such as rRNA, tRNAs, and snRNAs.
Pseudo-seq adapts Bakin and Ofengand’s primer extension assay to the Illumina sequencing platform. Ligation of a 3′ adapter provides a uniform primer binding site for reverse transcription, replacing gene specific primers. This allows synthesis of cDNAs for all RNA fragments present in a sample using a universal RT primer. Pseudo-seq also depends on two important modifications to next-generation sequencing library preparation. First, CMC treatment and reversal of RNA is added to allow specific labeling of Ψ residues. Second is the addition of a selection for truncated cDNAs, which should be enriched in CMC dependent stops at Ψ residues.
The ability of next-generation sequencing to provide transcriptome wide data provides Pseudo-seq with several advantages over the classic primer extension assay used for Ψ mapping. First is throughput. The primer extension assay is limited to one or a few genes of interest. However, Pseudo-seq can be used to identify Ψs in all transcripts, Second, next-generation sequencing provides sufficient coverage for Ψ identification in transcripts of a range of abundances. Primer extension is most easily carried out on highly abundant transcripts, severely limiting the fraction of the transcriptome accessible to this method. Finally, read counts generated by Pseudo-seq are more easily and reliably quantified than the intensity of radioactively labeled bands on gels. These advantages make Pseudo-seq an efficient discovery tool for new sites of modifications, allowing analysis of a large fraction of the transcriptome not accessible to previous methods.
Critical Parameters
The Pseudo-seq library preparation protocol is robust. However, as with any sequencing library protocol it is important that the libraries submitted for sequencing are of high quality. In particular the size distribution of the library should be in the expected range, and the library concentration should be high enough for successful Illumina sequencing. This protocol assumes that quality control will be performed by the facility performing the sequencing. There are also additional considerations specific to Pseudo-seq library preparation. Efficient CMC modification and reversal are important for the specific derivatization of Ψ residues. Thus the use of freshly prepared CMC during the modification step, and the use of properly stored Sodium Carbonate Buffer during reversal are important. It is also critical that truncated cDNAs be efficiently separated from full-length cDNAs. Size selection of a narrow range of RNA fragment sizes (10–20 nt) helps to minimize the extent of overlap between truncated and full-length cDNAs. During the cDNA size selection step it is important to carefully avoid the full-length cDNA band. Nonspecific cDNAs must also be avoided. These include the non-specific, no insert band, and products with fewer than 25 nt added to the RT primer.
The most important parameters to be considered during the planning stage of the experiment include sequencing depth, and the extent of biological replication. For budding yeast at least 6 million reads mapping to coding sequences for each library is ideal (≥ 12 million mappable reads per ± CMC library pair). This number is dependent on the transcriptome size, and should be scaled accordingly. Read coverage is also dependent upon the target transcripts, and can be scaled down considerably for highly abundant transcripts such as tRNAs, and rRNAs. The extent of biological replication is also critical, as the use of replicate data allows for false positives to be efficiently filtered from transcripts of lower abundance. For budding yeast, 14 biological replicates allows robust Ψ identification in transcripts of relatively low abundance. It should be noted that strains deleted for pseudouridylation factors can be used as biological replicates alongside wild-type replicates.
The parameters used for Ψ calling during data analysis are also important. These include the read coverage cutoff (rc), the Pseudo-seq peak value cutoff (p), and the number of replicate ± CMC library pairs in which a position must be flagged (n). For Ψ identification in an experiment with 14 biological replicates, the following parameters work well: rc = 0.0, p = 1.0, and n = 10. However, if fewer biological replicates are used then more stringent cutoffs should be used. These cutoffs can be determined empirically by varying the parameters, and examining the read distributions around sites called as Ψs. Called pseudouridylation events should have a prominent peak of reads one nucleotide 3′ of the identified Ψ, which should be considerably more prominent than background reads in the vicinity of the called site. Examining the reads surrounding known Ψs in rRNAs, tRNAs or snRNAs should provide examples of what reliable Ψ peaks look like in highly expressed transcripts. Additionally, reads mapping to the rRNA can be randomly downsampled, simulating transcripts with low read coverage. Computational prediction of Ψs using downsampled rRNA can be used to assess computational parameters.
Troubleshooting
Successful library construction is highly dependent on the yield of CMC-derivatized RNA fragments. If there are low levels of RNA fragments in the desired size range, the fragmentation time can be increased or decreased, depending on whether the sample was under-or over-fragmented. There can be significant differences in fragmentation times between different types of RNA samples. The yield of RNA recovered by precipitation after the CMC modification and reversal steps can be low, due to inefficient precipitation of CMC modified RNA. If this is the case then the ratio of Sodium Acetate, pH 5.3, and Ethanol to RNA sample should be increased. Alternately, the CMC treatment time or concentration can be decreased. A low yield in 3′ adapter ligation could be due to the presence of unadenylated adaptor, or poor 3′ end healing of RNA fragments. These issues can be addressed by gel purification of the adapter, and increasing the incubation time for 3′ end healing, respectively. Some investigators suggest performing 3′-end healing after gel-purification of the desired fragment size to increase efficiency. In this case the reactions should be cleaned by phenol-chloroform extraction and the RNA recovered by ethanol precipitation before proceeding.
It is important to minimize PCR amplification bias in Pseudo-seq libraries. If PCR products are saturated, or have higher molecular weight bands, try reducing the number of PCR cycles used. To reduce the effects of PCR bias, 8 or more (as sequencing space allows) random nucleotides can be inserted into the RT primer. This allows identical reads, which likely arise during PCR amplification, to be collapsed.
If there are few reads at the expected positions for positive control Ψs it is possible that the CMC modification was inefficient. Use fresh CMC, or increase the CMC concentration used. If there are few reads at 5′ positive control Ψs in groups of closely spaced Ψs it is possible that the extent of CMC modification was too great (see below). Reduce the concentration of CMC used, or reduce the treatment time. Finally, if there are prominent CMC-dependent peaks that occur 1 nt 3′ of G residues, or negative control U residues it is likely that the CMC reversal step was inefficient. Make fresh sodium carbonate buffer, and ensure the pH is 10.4, or increase the incubation time during the reversal step.
Anticipated Results
The Pseudo-seq library preparation protocol is robust to a variety of factors, including RNA fragment size, CMC concentration, RT enzyme choice, and cDNA size range. Variation of these conditions does not significantly affect the ability to detect known Ψs within the rRNA.
Examining the reads across the rRNA locus using a genome browser, such as UCSC or IGV, can be used to quickly assess library quality. The +CMC library should have strong peaks of reads that occur 1 nt 3′ of known sites of pseudouridylation. The background reads in the +CMC library should be relatively uniform, and low compared to the positive control peaks. The −CMC library will have a somewhat uniform distribution of reads across the rRNA locus. Peaks present in both +CMC and −CMC libraries represent sites of CMC-independent RT stops. Similar patterns should be observed for other ncRNAs with known Ψs, such as tRNAs and snRNAs.
From experience, the use of the Ψ calling parameters described above on 14 biological replicate libraries prepared from S. cerevisiae should lead to the calling of approximately 200 potential sites of pseudouridylation. However, it is not possible to predict, a priori, the number of sites that will be identified from other growth conditions or biological systems, or from varying the Ψ calling parameters.
Time Considerations
If performing Pseudo-seq on S. cerevisiae, the steps from culture growth to sequencing library submission take 7 days. A typical experimental timeline is as follows: Day 1: culture growth and RNA extraction; day 2: Poly(A) selection; day 3: RNA fragmentation, and CMC modification and reversal; day 4: 3′-end healing and RNA fragment size selection; day 5: 3′ adapter ligation, reverse transcription, and cDNA size selection; day 6: cDNA circularization, PCR amplification, and PCR product gel purification; and day 7: sample submission to a sequencing facility. Provided the needed computational tools, and custom scripts are in place, data analysis should take 2 to 4 days.
Acknowledgements
We thank members of the Gilbert laboratory for helpful discussions regarding this protocol. This work was supported by grants to W.V.G. from National Institutes of Health (GM094303, GM081399), and the American Cancer Society—Robbie Sue Mudd Kidney Cancer Research Scholar Grant (RSG-13-396-01-RMC). T.M.C. was supported by the American Cancer Society New England Division (Ellison Foundation Postdoctoral Fellowship).
Literature Cited
- Arribere JA and Gilbert WV 2013. Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res. 23:977–987. doi: 10.1101/gr.150342.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakin A and Ofengand J 1993. Four newly located pseudouridylate residues in Escherichia coli 23 S ribosomal RNA are all at the peptidyl-transferase center: Analysis by the application of a new sequencing technique. Biochemistry 32:9754–9762. doi: 10.1021/bi00088a030. [DOI] [PubMed] [Google Scholar]
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FAP, Fabris D, and Agris PF 2011. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, and Gilbert WV 2014. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515:143–146. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charette M and Gray MW 2000. Pseudouridine in RNA: What, where, how, and why. IUBMB Life 49:341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- Choi YC and Busch H 1978. Modified nucleotides in T1 RNase oligonucleotides of 18 S ribosomal RNA of the Novikoff hepatoma. Biochemistry 17:2551–2560. doi: 10.1021/bi00606a015. [DOI] [PubMed] [Google Scholar]
- Cohn WE 1960. Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: Isolation, structure, and chemical characteristics. J. Biol. Chem. 235:1488–1498. [PubMed] [Google Scholar]
- Collart MA and Oliviero S 2001. Preparation of yeast RNA. Curr. Protoc. Mol. Biol 23:13.12.1–13.12.5. [DOI] [PubMed] [Google Scholar]
- Courtes FC, Gu C, Wong NSC, Dedon PC, Yap MGS, and Lee D-Y 2014. 28 S rRNA is inducibly pseudouridylated by the mTOR pathway translational control in CHO cell cultures. J. Biotechnol. 174:16–21. doi: 10.1016/j.jbiotec.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FF and Allen FW 1957. Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem 227:907–915. [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, and Rechavi G 2013. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Fernández IS, Ng CL, Kelley AC, Wu G, Yu Y-T, and Ramakrishnan V 2013. Unusual base pairing during the decoding of a stop codon by the ribosome. Nature 500:107–110. doi: 10.1038/nature12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J and Yu Y-T 2013. RNA pseudouridylation: New insights into an old modification. Trends Biochem. Sci. 38:210–218. doi: 10.1016/j.tibs.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilham PT and Ho N 1971. Reaction of pseudouridine and inosine with N-cyclohexyl-N’-β-(4-methylmorpholinium) ethylcarbodiimide. Biochemistry 10:3651–3657. doi: 10.1021/bi00796a003. [DOI] [PubMed] [Google Scholar]
- Gupta RC and Randerath K 1979. Rapid print-readout technique for sequencing of RNAs containing modified nucleotides. Nucleic Acids Res. 6:3443–3458. doi: 10.1093/nar/6.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley RW, Everett GA, Madison JT, and Zamir A 1965. Nucleotide sequences in theyeast alanince transfer ribonucleic acid. J. Biol. Chem 240:2122–2128. [PubMed] [Google Scholar]
- Hudson GA, Bloomingdale RJ, and Znosko BM 2013. Thermodynamic contribution and nearest-neighbor parameters of pseudouridine-adenosine base pairs in oligoribonucleotides. RNA 19:1474–1482. doi: 10.1261/rna.039610.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karijolich J and Yu Y-T 2011. Converting non-sense codons into sense codons by targeted pseudouridylation. Nature 474:395–398. doi: 10.1038/nature10165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon J-K, Aach J, Xie B, Leproust E, Zhang K, Gao Y, and Church GM 2009. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- Lovejoy AF, Riordan DP, and Brown PO 2014. Transcriptome-Wide Mapping of Pseudouridines: Pseudouridine Synthases Modify Specific mRNAs in S. cerevisiae. PloS One 9:e110799. doi: 10.1371/journal.pone.0110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR 2012. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Lagos-Quintana M, and Tuschl T 2005. Cloning of small RNA molecules. Curr. Protoc. Mol. Biol 72:26.4.1–26.4.18. [DOI] [PubMed] [Google Scholar]
- Sambrook J and Russell DW 2001. Molecular Cloning. CSHL Press. [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, Fink G, and Regev A 2014. Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell 159:148–162. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, and Preiss T 2012. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40:5023–5033. doi: 10.1093/nar/gks144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, and Bartel DP 2014. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 508:66–71. doi: 10.1038/nature13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Dyer TA, and Brownlee GG 1980. An improved direct RNA sequence method; its application to Vida faba 5.8 S ribosomal RNA. Nucleic Acids Res. 8:1259–1272. doi: 10.1093/nar/8.6.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Xiao M, Yang C, and Yu Y-T 2011. U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. EMBO J 30:79–89. doi: 10.1038/emboj.2010.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, and Steinmetz LM 2009. Bidirectional promoters generate pervasive transcription in yeast. Nature 457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]