

Abstract
Chondroblastoma is currently classified as a benign neoplasm; however, chondroblastoma and chondroblastoma-like osteosarcoma have morphologic overlap, raising the possibility that some tumors diagnosed as chondroblastoma-like osteosarcoma might actually represent malignant chondroblastoma. The H3F3B K36M point mutation, which has not been reported in osteosarcoma, is identified in 95% of chondroblastomas and is reliably detectable by immunohistochemistry (IHC). We reviewed 11 tumors diagnosed as atypical chondroblastoma, malignant chondroblastoma, or chondroblastoma-like osteosarcoma (median follow-up: 8.8 years; range: 4 months–26.4 years). Seven chondroblastomas with cytologic atypia and permeative growth were designated “malignant chondroblastoma”; six were H3K36M-positive by IHC. Relative to conventional chondroblastoma, malignant chondroblastoma occurred in older individuals (median: 52 years; range: 29–57 years) and arose at unusual sites. Three of four tumors with long-term follow-up recurred, and one patient died of widespread metastases. One was found to have chromosomal copy number alter4ations and a SETD2 mutation in addition to H3F3B K36M. The four remaining tumors were classified as chondroblastoma-like osteosarcoma. Chondroblastoma-like osteosarcoma occurred in younger patients (median: 21 years; range: 19–40 years) than malignant chondroblastoma. In contrast to malignant chondroblastoma, all had regions of malignant cells forming bone. Two of three patients with long-term follow-up developed recurrences, and two died of disease, one with widespread metastases. No mutations in H3F3A/H3F3B were detected by Sanger sequencing. While malignant chondroblastoma and chondroblastoma-like osteosarcoma show significant morphologic overlap, they have distinct clinical presentations and genetic findings. When considering this challenging differential diagnosis, IHC using histone H3 mutation-specific antibodies is a critical diagnostic adjunct.
Introduction
Chondroblastoma most commonly arises in the epiphyses of long bones in adolescents and young adults, with a median age at presentation of 15–20 years and a 15% local recurrence rate [1]. Rarely, patients with histologically conventional chondroblastoma develop lung metastases, often years after the initial presentation; these metastases generally have an indolent course [2]. The existence of atypical or malignant chondroblastoma has been controversial, and chondroblastoma has been re-classified from a tumor of intermediate biologic potential in the 4th edition of the World Health Organization classification to a benign neoplasm in the 5th edition [3, 4]. Further obfuscating the issue, chondroblastoma-like tumors with atypical or malignant histologic features and unusual radiologic findings have historically been diagnosed as chondroblastoma-like osteosarcoma.
The H3 histone family member 3B (H3F3B) K36M point mutation has been identified in 95% of chondroblastomas [2], and H3F3A and H3F3B G34W substitutions (H3G34W) occur in 85–90% of giant cell tumors of bone [5, 6]. Mutation-specific H3K36M and H3G34W antibodies can be used to detect these point mutations with >90% sensitivity and nearly 100% specificity [5, 7].
The genetic alterations in chondroblastoma-like osteosarcoma have not been systematically characterized, and the morphologic overlap between chondroblastoma and chondroblastoma-like osteosarcoma raises the question of whether some tumors diagnosed as chondroblastoma-like osteosarcoma represent malignant chondroblastoma. Here, we characterize the clinicopathologic features of 11 cases previously diagnosed as chondroblastoma-like osteosarcoma and atypical or malignant chondroblastoma.
Methods
Case selection
The archives of Brigham and Women’s Hospital, Massachusetts General Hospital, and Stanford Hospital were searched for cases containing “chondroblastoma-like osteosarcoma” in the diagnostic top line or report text, as well as cases containing “chondroblastoma” along with terms “atypical,” “malignant” or “metastatic.” Two cases were published previously [8, 9]. An additional case was provided by JDR. This study was approved by the institutional review boards of the authors’ institutions.
Sixteen candidate cases were found; hematoxylin and eosin-stained slides and immunohistochemistry (IHC) were re-reviewed concurrently by four co-authors (JLH, GPN, YPH and DJP). Based on this review, five cases were excluded: one, diagnosed as “atypical spindle cell neoplasm with features of chondroblastoma,” was found on review to have insufficient features for more definitive classification; one, diagnosed as “high-grade radiation-associated osteosarcoma with chondroblastoma-like features,” was excluded because of prior radiation therapy; one was excluded because it was determined on review to be a conventional giant cell tumor of bone, confirmed by H3G34W IHC; and two were excluded because they were reclassified as malignant giant cell tumors of bone, confirmed by H3G34W IHC. The remaining 11 cases were re-classified as “malignant chondroblastoma” or chondroblastoma-like osteosarcoma. Chondroblastomas with permeative growth and/or a degree of cytologic atypia beyond that of conventional chondroblastoma were designated “malignant chondroblastoma”; permeative growth has been previously proposed as a criterion for malignancy in chondroblastoma [10]. Tumors containing bone production by malignant cells were classified as chondroblastoma-like osteosarcoma; in borderline cases, consensus opinion was used to distinguish bone formation from the eosinophilic chondroid matrix of chondroblastoma. Resection or curettage specimens were available for review in all cases; primary tumors were not available in cases 1 and 9. Immunohistochemistry and molecular studies were used to support diagnoses when available.
Imaging studies were reviewed by a musculoskeletal radiologist (MAB). Radiographs were available for eight patients, CT for six patients, and MRI for four patients. A whole-body PET/CT was available for one patient.
Immunohistochemistry
IHC was performed on 4 μm formalin-fixed, paraffin-embedded (FFPE) tissue sections. Following pressure cooker antigen retrieval (Target Retrieval Solution [pH 6.1 citrate buffer]; Dako, Carpinteria, CA), rabbit monoclonal antibodies directed against histone H3.3 with mutations G34W (clone RM263; 1:2000 dilution; RevMAb Biosciences USA, South San Francisco, CA) and K36M (clone RM193; 1:4000 dilution; RevMAb Biosciences USA) were used along with the EnVision Plus detection system (Dako). An algorithmic approach was used, such that H3K36M IHC was performed first, and negative tumors were further tested with H3G34W IHC. IHC for BAP1 (clone C4; 1:30 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) was performed on one tumor following the same antigen retrieval methods.
H3F3A and H3F3B sequencing
H3F3A and H3F3B mutational hotspot sequencing was attempted for the five tumors with negative H3K36M and H3G34W IHC, using methods described previously [6]. In brief, DNA was isolated from 4 μm to 10 μm FFPE tissue sections using standard protocols (QIAamp DNA FFPE Tissue Kit, Qiagen, Valencia, CA). Genomic DNA primers covered mutational hotspots in codons 27, 34, and 36, with amplicon sizes of 128 bp (H3F3A) and 148 bp (H3F3B). PCR products were purified using ExoSAP-IT PCR Product Cleanup Reagent (Applied Biosystems, Foster City, CA), followed by Sanger sequencing.
Results
Integrated case review with clinicopathologic features
Upon re-review, 7 of 11 tumors were classified as malignant chondroblastoma and 4 of 11 were classified as chondroblastoma-like osteosarcoma (Table 1).
Table 1.
Clinicopathologic characteristics of seven malignant chondroblastomas and four chondroblastoma-like osteosarcomas in this cohort.
| # | Consensus diagnosis | Original diagnosis | Primary site | Age (y) | Sex | Size (cm) | Mit/ 2 mm2 | Infiltrative borders | Necrosis | IHC status | Surgery | Margin Status | Additional treatment | Follow-up | Recurrence | Metastasis | Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Malignant CB | CB | Left third rib | 49 | M | NA | 15 | N | N | H3K36M: POS | RES | NEG | None | 26 y 5 m | Y | Y (Fig. 2) | DD |
| 2 | Malignant CB | CB-OS | Right talus | 48 | M | 4 | 1 | Y | N | H3K36M: NEG H3G34W: NEG | RES (AMP) | NEG | Chemotherapy | 7 y 2 m | N | N | DOC |
| 3 | Malignant CB | CB vs. CB-OS | Left anterior 4th rib | 29 | M | 4 | 3 | Y | N | H3K36M: POS | RES | NEG | 2 cycles of chemotherapy | 4 m | N | N | NED |
| 4 | Malignant CB | CB vs. Chondrosarcoma | Left anterior 4th rib | 55 | M | 6.4 | 2 | Y | N | H3K36M: POS | RES | NEG | None | 9 m | N | N | NED |
| 5 | Malignant CB | CB | Right acromion | 37 | M | 8.6 | 1; 3 (R) | NA | N | H3K36M: POS | CUR & CRYO | - | None | 5 y 5 m | Y | N | NED |
| 6 | Malignant CB | CB-OS | Right scapula | 65 | M | 13.5 | 2 | Y | Y (focal) | H3K36M: POS | RES | NEG | None | 1 y | N | N | NED |
| 7 | Malignant CB | CB | Right radial head | 57 | F | 3.5 | 3; 3 (R) | Y | N | H3K36M: POS | CUR & CRYO | - | None | 9 y 11 m | Y | N | NED |
| 8 | CB-OS | CB-OS | Right 9th rib | 19 | M | 6.5 | 3 | Y | Y | H3K36M: NEG H3G34W: NEG | RES | NEG | Chemotherapy | 1 y 9 m | N | N | NED |
| 9 | CB-OS | CB | Left scapula | 40 | F | NA | 2; 5 (R) | NA | Y | H3K36M: NEG H3G34W: NEG | RES | Unknown | Unknown | 21 y | N | Y (lung, skull, brain) | DD |
| 10 | CB-OS | Atypical osteochondral neoplasm; LG OS (R) | Proximal humerus | 23 | M | 10.9 (R) | 1; 2 (R) | N | Y (at fracture site) | H3K36M: NEG H3G34W: NEG | CUR | - | None | 16 y 4 m | Y | N | NED |
| 11 | CB-OS | CB-OS | C7 vertebra | 19 | F | 5.1 | 4 | Y | N | H3K36M: NEG H3G34W: NEG | RES | POS | Radiation | 1 y 5 m | Y | N | DD |
indicates case number, Mit mitoses, CB chondroblastoma, CB-OS chondroblastoma-like osteosarcoma, LG low-grade, OS osteosarcoma, R recurrence, NA not available, Y yes, N no, RES oncologic resection, AMP amputation, CUR curettage, CRYO cryotherapy, DD deceased from disease, DOC deceased from other causes, NED no evidence of disease.
Imaging findings
Imaging was available for review for six patients with malignant chondroblastoma and two patients with chondroblastoma-like osteosarcoma. Both neoplasms had similar imaging findings. Malignant chondroblastomas manifested as expansile, lytic lesions on radiographs and CT with thin sclerotic margins, with or without areas of cortical break, and scattered internal trabeculations (Fig. 1). On MRI, lesions were hypointense on T1- and mixed intermediate to hyper- and hypointense signal on fluid-sensitive sequences and demonstrated contrast enhancement. FDG-PET/CT showed increased FDG uptake. All cases involved epiphyses or epiphysis equivalents. Chondroblastoma-like osteosarcoma exhibited lytic lesions with cortical break and scattered thin internal trabeculations (Fig. 1). Both cases involved epiphyses. Tumor recurrence demonstrated lytic expansile lesions with cortical destruction and associated soft tissue mass. In both tumor types, the imaging appearances of tumor recurrences were similar to the primary tumors.
Fig. 1. Imaging findings in malignant chondroblastoma and chondroblastoma-like osteosarcoma.
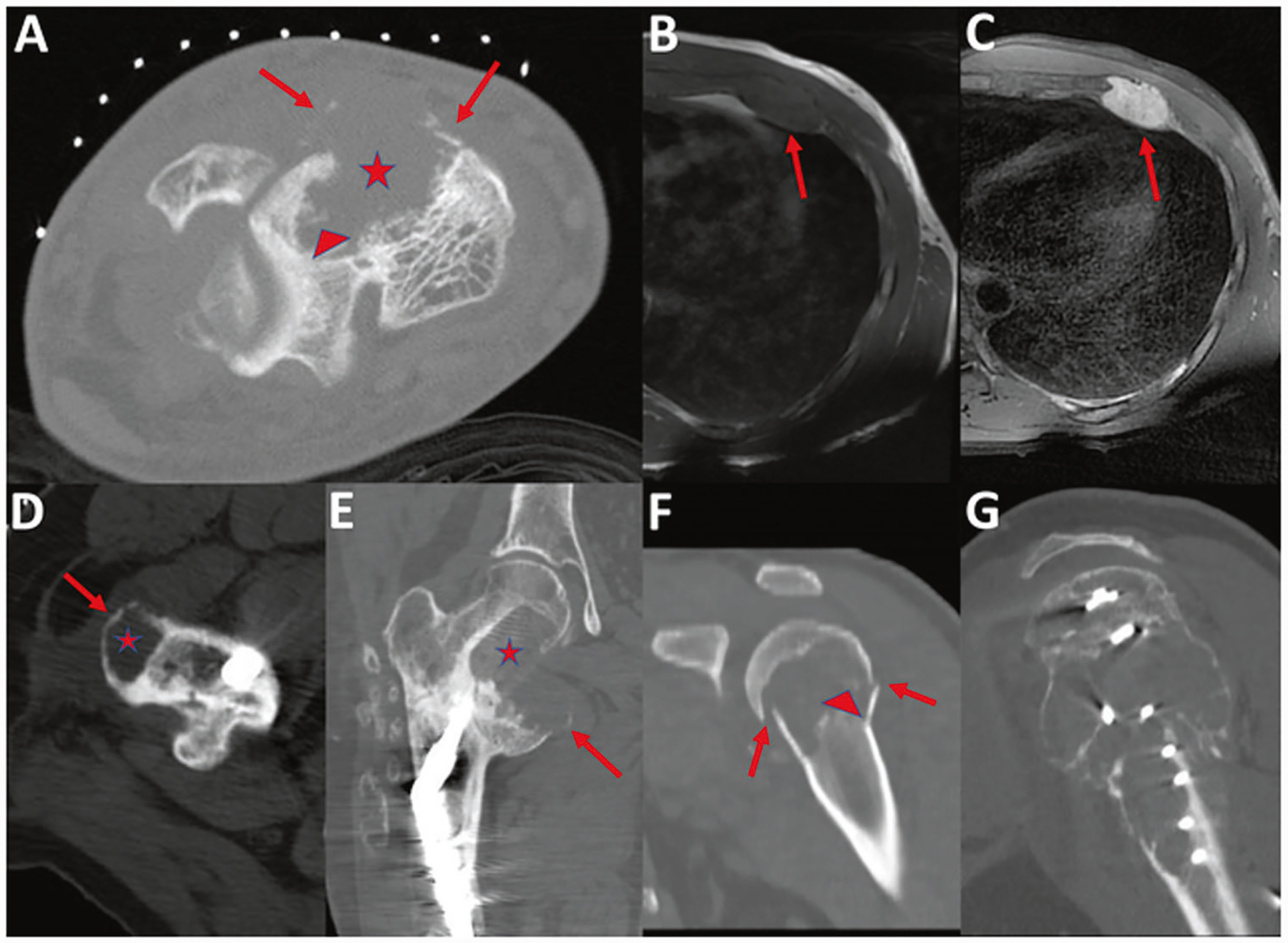
a Malignant chondroblastoma (case 2). Axial CT of the ankle performed as part of a CT-guided biopsy demonstrates an expansile lytic lesion in the talus (asterisk) with cortical destruction and areas of thin peripheral calcifications (arrows) and internal trabeculations (arrowhead). b–c Malignant chondroblastoma (case 3). Axial T1-weighted (b) and fat suppressed T2-weighted (c) MR images demonstrate T1 hypointense and T2 hyperintense signal of the lesion of the left anterior 4th rib (arrows). d–e Metastatic malignant chondroblastoma (case 1). Axial CT (d) demonstrates a lytic lesion (asterisk) in the greater trochanter, with thin sclerotic margins (arrow), consistent with tumor metastasis. Coronal reformatted CT (e) demonstrates tumor recurrence of the medial femoral neck with expansile lytic lesion extending into the lesser trochanter (asterisk) and thin sclerotic margin (arrow). This image represents the third recurrence at this metastatic site. Metastatic malignant chondroblastoma exhibits similar findings to those of primary tumors. f–g Chondroblastoma-like osteosarcoma (case 10). Coronal reformatted CT (f) demonstrates a lytic lesion with cortical break and pathologic fracture (arrow) of the proximal humeral metaphysis and epiphysis. Faint internal areas of trabeculation are present (arrowhead). Sagittal reformatted CT (g) performed 13 years later demonstrates tumor recurrence as a large lytic expansile lesion with internal trabeculations.
Malignant chondroblastoma
The median age at presentation of the seven patients with malignant chondroblastoma (Table 1, cases 1–7) was 52 years (range: 29–57 years). Six tumors occurred in males and one occurred in a female. Body sites for primary tumors were ribs (3 tumors), scapula (2), talus (1), and radius (1). The median tumor size was 6.4 cm (range: 3.5–13.5 cm). Five tumors were initially treated with en bloc resections, including a below-the-knee amputation for the talar tumor; the remaining two were treated with curettage and cryoablation. All five resections had negative surgical margins. The original diagnoses were chondroblastoma (6 tumors), chondroblastoma-like osteosarcoma (3), and chondrosarcoma (1) (three received multiple diagnoses, either at different institutions or at the time of tumor recurrence).
Clinical follow-up was available for all seven patients (median: 3.2 years; range: 4 months–26.4 years). Three tumors recurred (Table 1); when considering patients with >1 year of follow-up, 3 of 4 (75%) recurred at intervals of 3.8, 5.4, and 6.7 years. Both tumors treated with curettage recurred. One of five (20%) tumors treated with resection recurred, despite negative margins. Two patients received adjuvant chemotherapy; neither experienced recurrence or metastasis. One patient with a rib primary (case 1) who developed recurrence also developed femur, hip soft tissue, scalp, chin, and cerebellar metastases over a 26-year period (Fig. 2). This patient expired 27.5 years after the initial presentation.
Fig. 2. Timeline for widely metastatic malignant chondroblastoma (case 1).
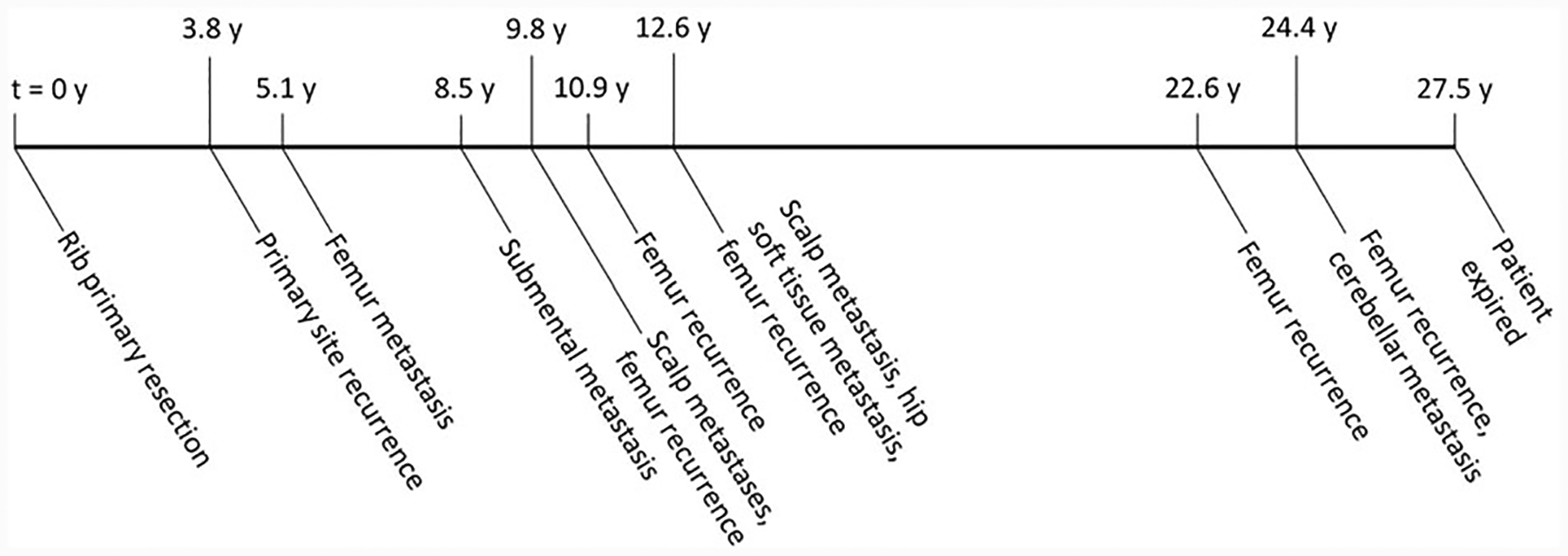
The long axis represents time, with time at initial presentation designated “t = 0;” events are described below the axis, and the time elapsed since initial presentation is shown above the axis for each event. This patient presented with a rib mass at 49 years of age and developed recurrence and metastases over a long time period, dying 27.5 years after initial presentation with a likely cerebellar metastasis (not biopsied). The tumor in this case had lymphovascular invasion as well as a mitotic rate of up to 15 mitoses per 2 mm2, a much higher mitotic rate than the other malignant chondroblastomas in this series. The course of disease in this patient calls into question the notion that chondroblastoma is a uniformly benign neoplasm.
There were three histologic patterns in malignant chondroblastoma: four tumors closely resembled conventional chondroblastoma but with infiltrative borders and/or increased cytologic atypia (Figs. 3–4); two tumors contained sheets of neoplastic chondroblasts with a paucity of osteoclastic giant cells (Fig. 5); and one tumor had tenosynovial giant cell tumor-like cytology, but with marked cytologic atypia (Fig. 6). Five tumors had infiltrative borders; in one tumor, it was not possible to assess invasion on the curettage specimen. The primary tumor in case 1 was reported to be circumscribed but to exhibit lymphovascular invasion. One had focal necrosis. Six tumors had mitotic rates ranging from 1–3 mitoses per 2 mm2, and the remaining tumor, which metastasized (case 1; Figs. 2–3), had up to 15 mitoses per 2 mm2. Lymphovascular invasion was identified in two tumors, including the one that recurred and metastasized. In contrast to chondroblastoma-like osteosarcoma, no tumors exhibited bone production by cytologically malignant cells.
Fig. 3. Histologic features of malignant chondroblastoma giving rise to widespread metastases.
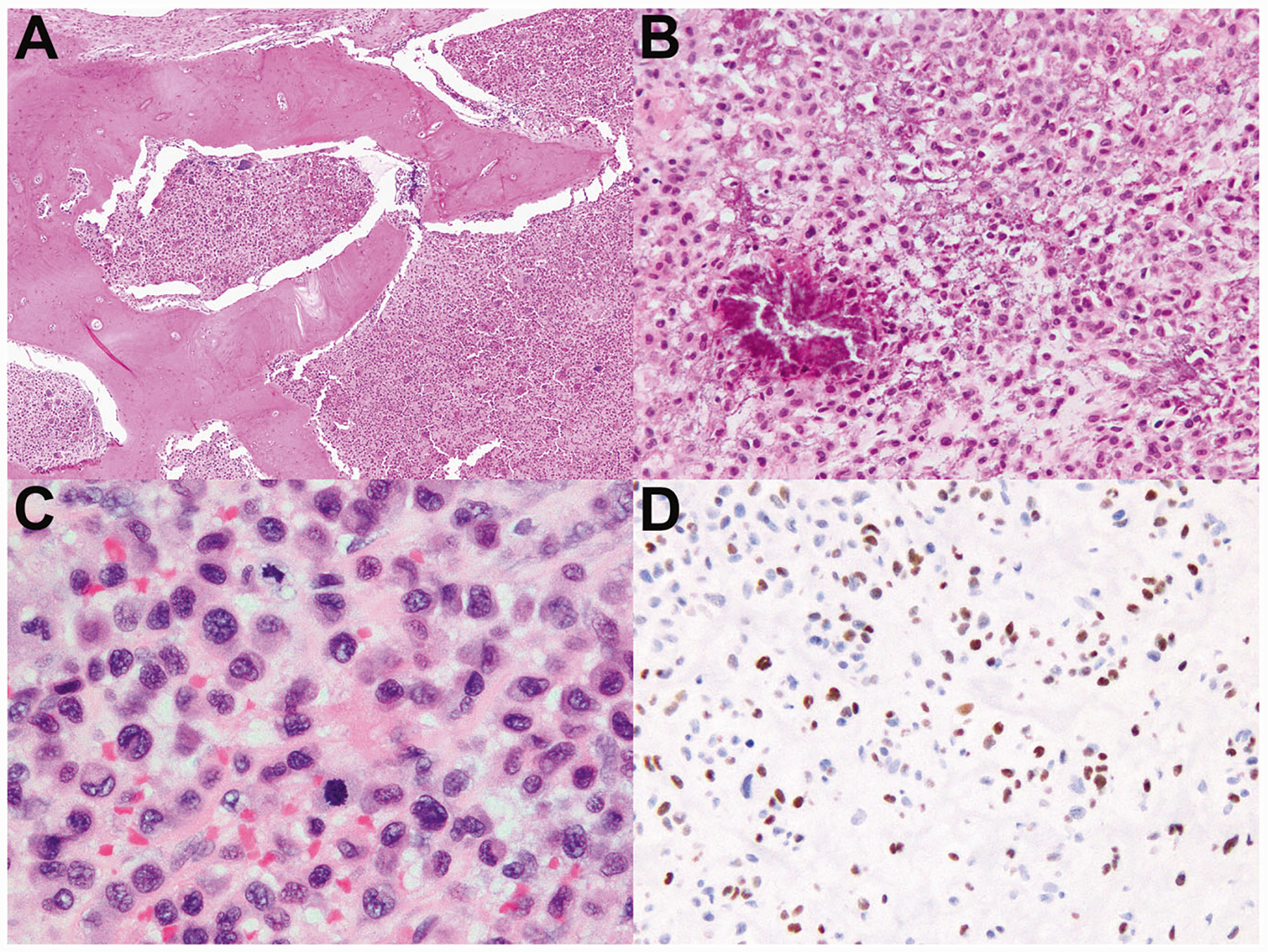
a–d Are images of case 1, which gave rise to widespread metastases eventually leading to patient demise (Fig. 2). a This metastatic focus of malignant chondroblastoma exhibits permeative growth through bony trabeculae. b So-called “chicken-wire” calcifications are present multifocally in this tumor, as is common in conventional chondroblastoma. c This tumor exhibits frequent mitoses and greater cytologic atypia than conventional chondroblastoma. d Immunohistochemistry demonstrates nuclear positivity for H3K36M, which is essentially 100% specific for chondroblastoma.
Fig. 4. Malignant chondroblastoma resembling conventional chondroblastoma but with increased atypia and infiltrative borders.
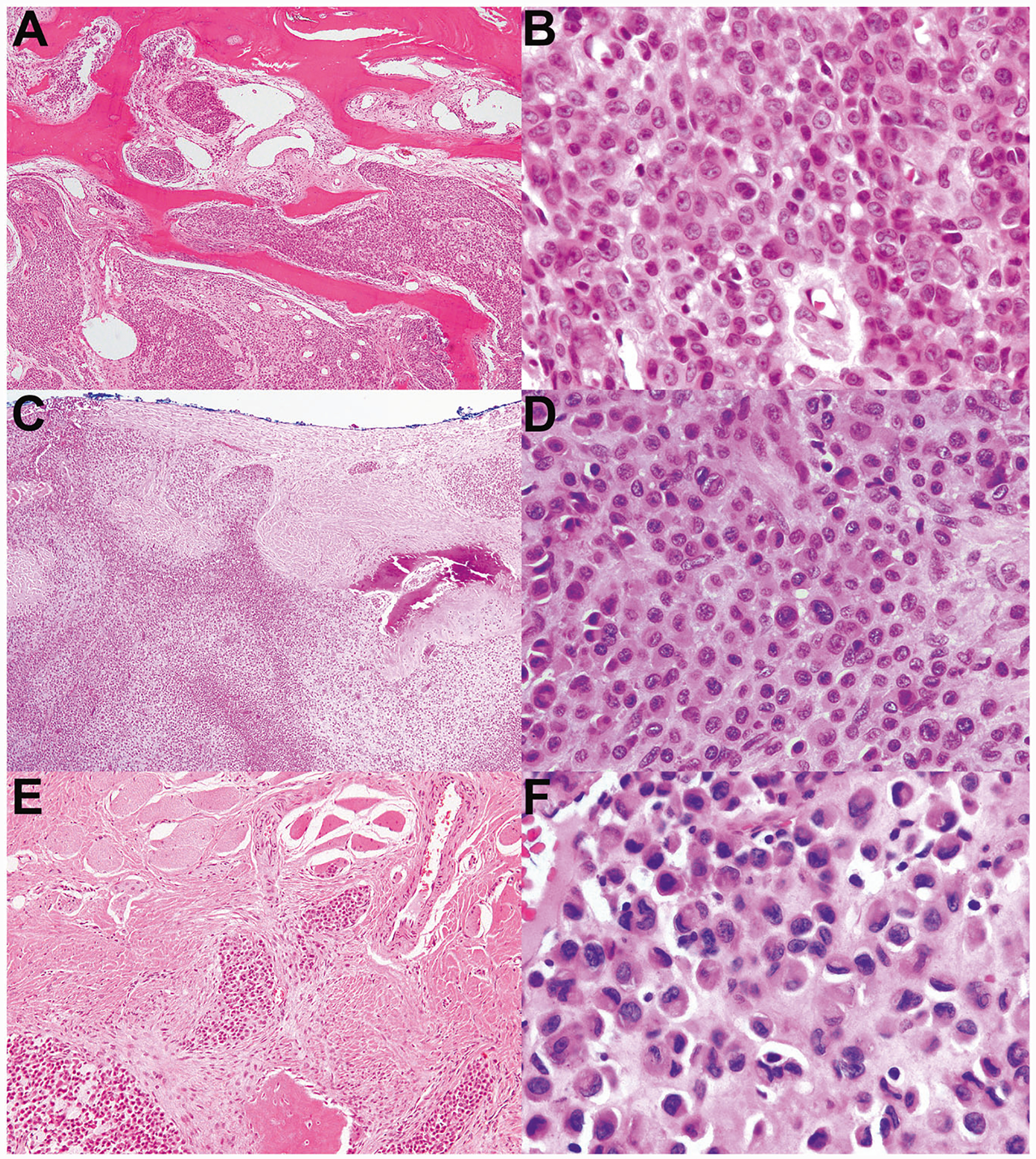
a–f Correspond to cases 2–4, respectively. These three malignant chondroblastomas contained eosinophilic chondroid matrix, numerous admixed osteoclastic giant cells, and neoplastic chondroblasts with eccentric folded nuclei and eosinophilic cytoplasm, all characteristic of conventional chondroblastoma. However, all three exhibited significant cytologic atypia and infiltrative borders, both of which would be unusual for conventional chondroblastoma. a–b This tumor occurred in the talus and had an infiltrative growth pattern. b The degree of cytologic atypia shown here is beyond that of conventional chondroblastoma. c This tumor exhibits a permeative growth pattern; here, it is infiltrating through the cortex of the rib and extending into adjacent soft tissue. d There is significant cytologic atypia, including nuclear pleomorphism and scattered large, hyperchromatic nuclei. e This tumor disrupts the cortex of the rib and extends into adjacent skeletal muscle. f Compared with other malignant chondroblastomas in this series, this tumor had relatively mild nuclear atypia; however, it was found to be markedly infiltrative upon consensus review and was therefore classified as malignant.
Fig. 5. Giant-cell-poor malignant chondroblastoma.
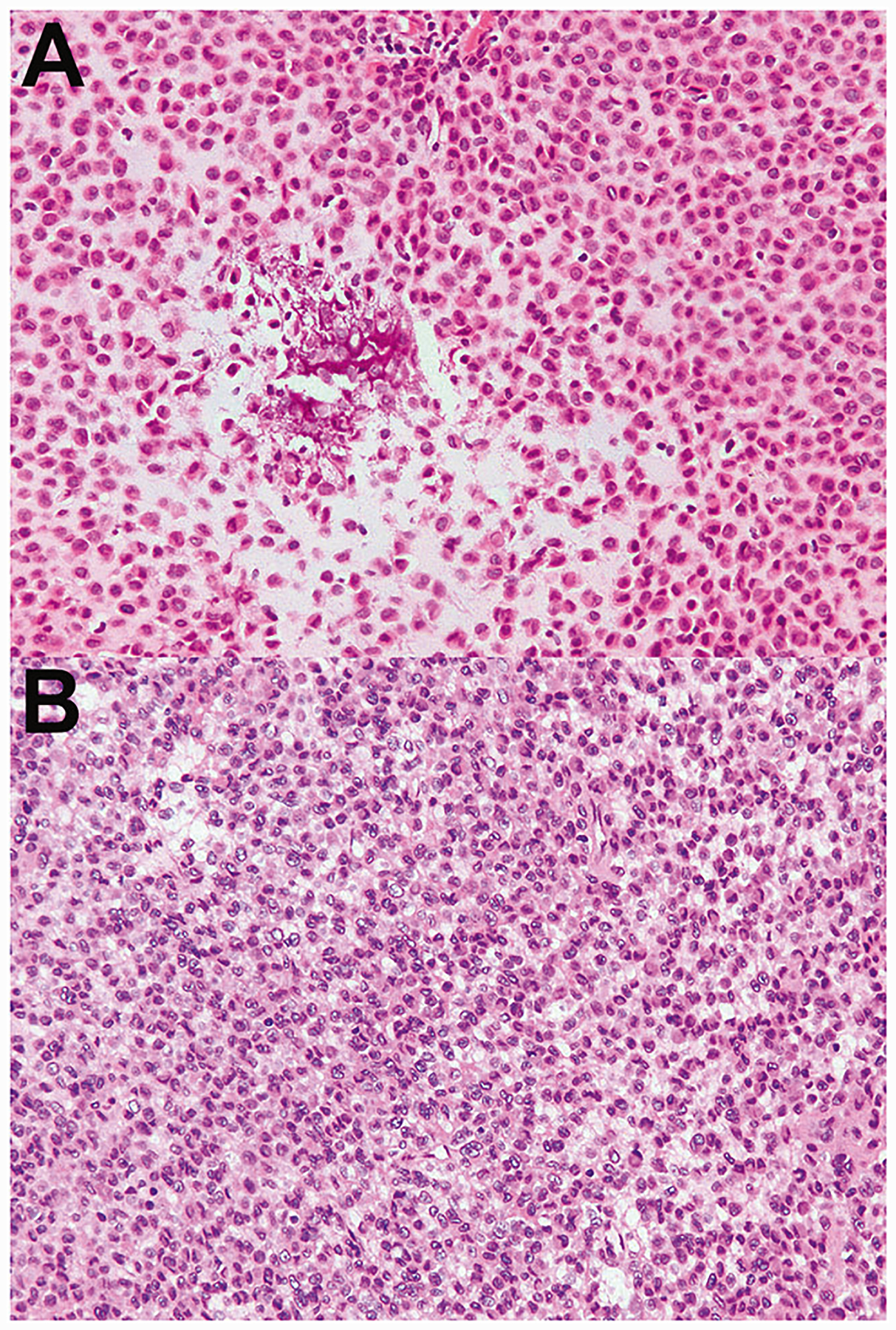
a–b Correspond to cases 5–6, respectively. Two malignant chondroblastomas occurred in the scapula and contained sheets of chondroblasts, with a paucity of giant cells. a Occasional areas of “chicken-wire” calcification are seen; note the lack of admixed giant cells. This tumor recurred at an interval of 5.4 years. b There are sheets of neoplastic chondroblasts and only rare giant cells. This tumor was found to contain lymphovascular invasion.
Fig. 6. Malignant chondroblastoma with tenosynovial giant cell tumor-like neoplastic cells.
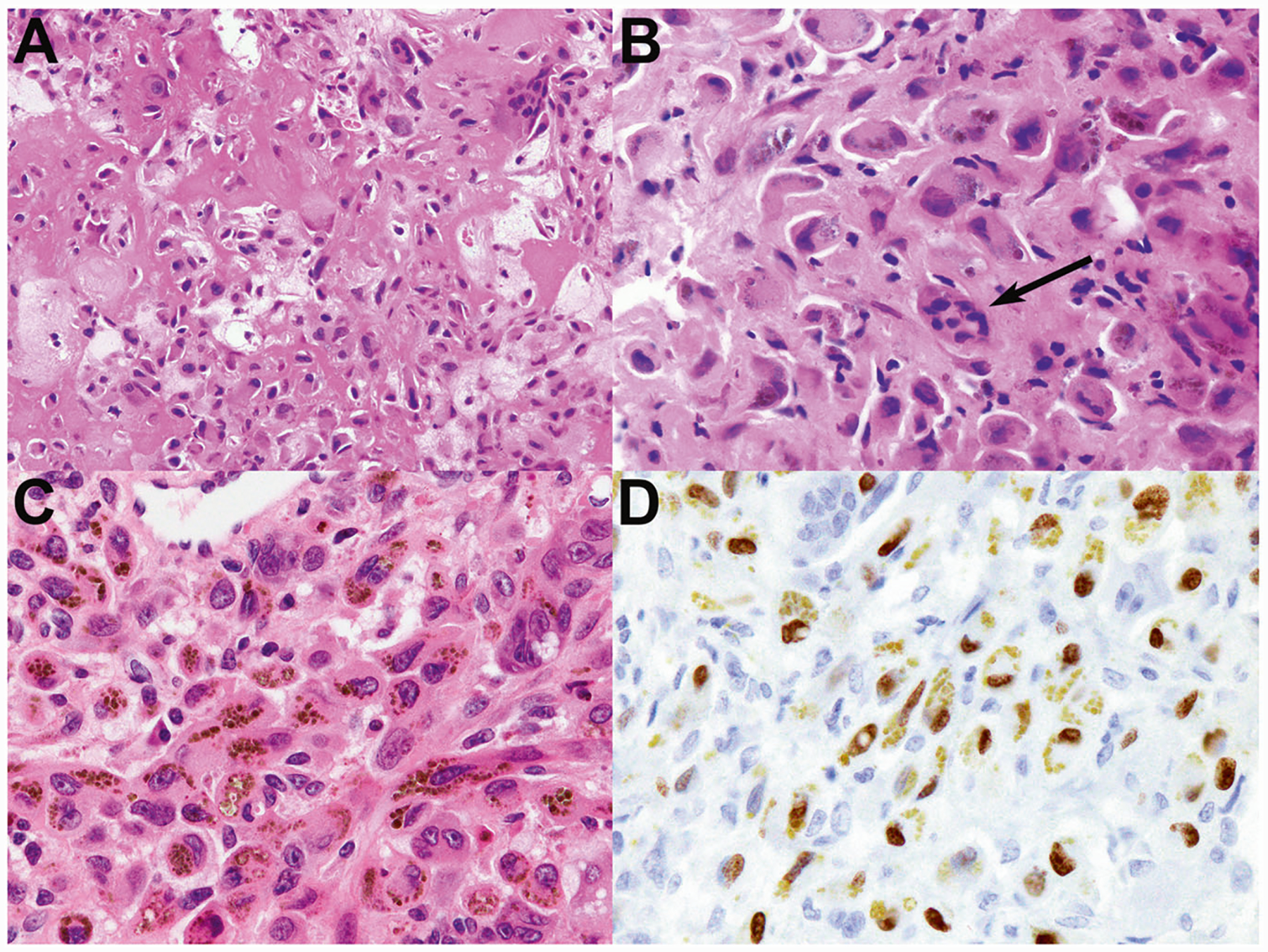
a This malignant chondroblastoma had regions of eosinophilic chondroid matrix that resembled bone formation, highlighting the difficulty in distinguishing these features. b There is focally marked nuclear pleomorphism; note the size of some neoplastic nuclei relative to those of a nearby osteoclastic giant cell (arrow). c Other regions of this tumor had hemosiderin-laden neoplastic cells with pale cytoplasm and pleomorphic nuclei with vesicular chromatin, resembling the neoplastic cells of tenosynovial giant cell tumor. There were scattered admixed osteoclastic giant cells. This tumor recurred 9.9 years after initial resection. d Immunohistochemistry demonstrates dark brown nuclear positivity for H3K36M, in contrast to the golden-brown cytoplasmic hemosiderin.
IHC was positive for H3K36M in 6 of 7 cases (Figs. 1d and 6d). Case 2 was H3K36M- and H3G34W-negative and failed DNA isolation for Sanger sequencing. Based on morphology, including the lack of bone production by cytologically malignant cells, this tumor was classified as malignant chondroblastoma.
Next-generation sequencing was performed for clinical diagnostic workup of two tumors (cases 3 and 4). The only identified mutation in case 3 was H3F3B K36M. Case 4 had the H3F3B K36M missense mutation (35% allele fraction) and additionally was found to harbor SETD2 K1937* nonsense mutation (26% allele fraction) and copy number gains and losses in chromosomes 9 and 10.
Chondroblastoma-like osteosarcoma
Four tumors were classified as chondroblastoma-like osteosarcoma (Table 1, cases 8–11). Two occurred in males and two occurred in females, with a median age at presentation of 21 years (range: 19–40 years). Primary tumors were located in the humerus (1 tumor), rib (1), scapula (1), and C7 vertebra (1). Only case 10 occurred in a long bone (the humerus). The median tumor size was 6.5 cm (range: 5.1–10.9 cm). Three tumors were initially treated with en bloc resections, and the fourth was treated with curettage. Margins status was known for two of three resections; one had negative margins and the other had positive margins. Original diagnoses were chondroblastoma-like osteosarcoma (2 tumors), low-grade osteosarcoma (1), and chondroblastoma (1).
Follow-up was available for all patients (median duration: 9.0 years; range: 1.4 years–26 years). Two tumors recurred and one metastasized; the only tumor that did not recur or metastasize was resected with negative margins in a patient who received adjuvant chemotherapy. The proximal humerus tumor recurred 14.5 years after initial resection. Two patients died of disease: one patient with a C7 vertebral tumor expired 17 months after initial diagnosis due to locally destructive growth. The second patient who died of disease had a scapular primary tumor and developed widespread metastases including metastases to the lungs (interval after initial diagnosis: 4, 8, and 13 years), distal humerus (23 years), skull (24 years), and brain (25 years).
The chondroblastoma-like osteosarcomas were found on consensus review to contain regions of cytologically malignant cells producing bone, distinguishing them from malignant chondroblastoma. All four tumors exhibited more striking cytologic atypia than the malignant chondroblastomas. Assigning a histologic grade to these unusual osteosarcomas is difficult and defies conventional criteria; however, the lack of aggressive behavior and lack of significant nuclear pleomorphism seem to support classification as low-to-intermediate grade rather than high-grade sarcomas. Across all tumors, mitoses ranged from 1–5 per 2 mm2, all three assessable primary tumors had infiltrative borders, and 3 of 4 tumors had at least focal necrosis. There was a wide range of morphologic patterns, including multiple patterns in the same tumor (Figs. 7–8). Case 8 had regions showing morphologic overlap with chondroblastoma, including eosinophilic chondroid matrix and neoplastic cells with brightly eosinophilic cytoplasm and eccentric nuclei; however, there was also marked nuclear atypia, lymphovascular invasion, and bone production (Fig. 7a, b). Case 9 had regions resembling chondroblastoma but also had bone production by malignant-appearing cells, and it contained areas with epithelioid to spindled neoplastic cells with basophilic cytoplasm and vesicular nuclei resembling giant cell tumor of bone (Fig. 7c, d). Case 10 had areas with eosinophilic chondroid matrix and focal bone production; in other regions, there were sheets of mononuclear tumor cells with amphophilic cytoplasm, eccentric nuclei, and cytoplasmic hemosiderin, mimicking tenosynovial giant cell tumor (Fig. 8a, b). Case 11 had regions of malignant cells producing bone, while other areas bore a striking resemblance to chondroblastoma (Fig. 8c, d).
Fig. 7. Chondroblastoma-like osteosarcoma, cases 8–9.
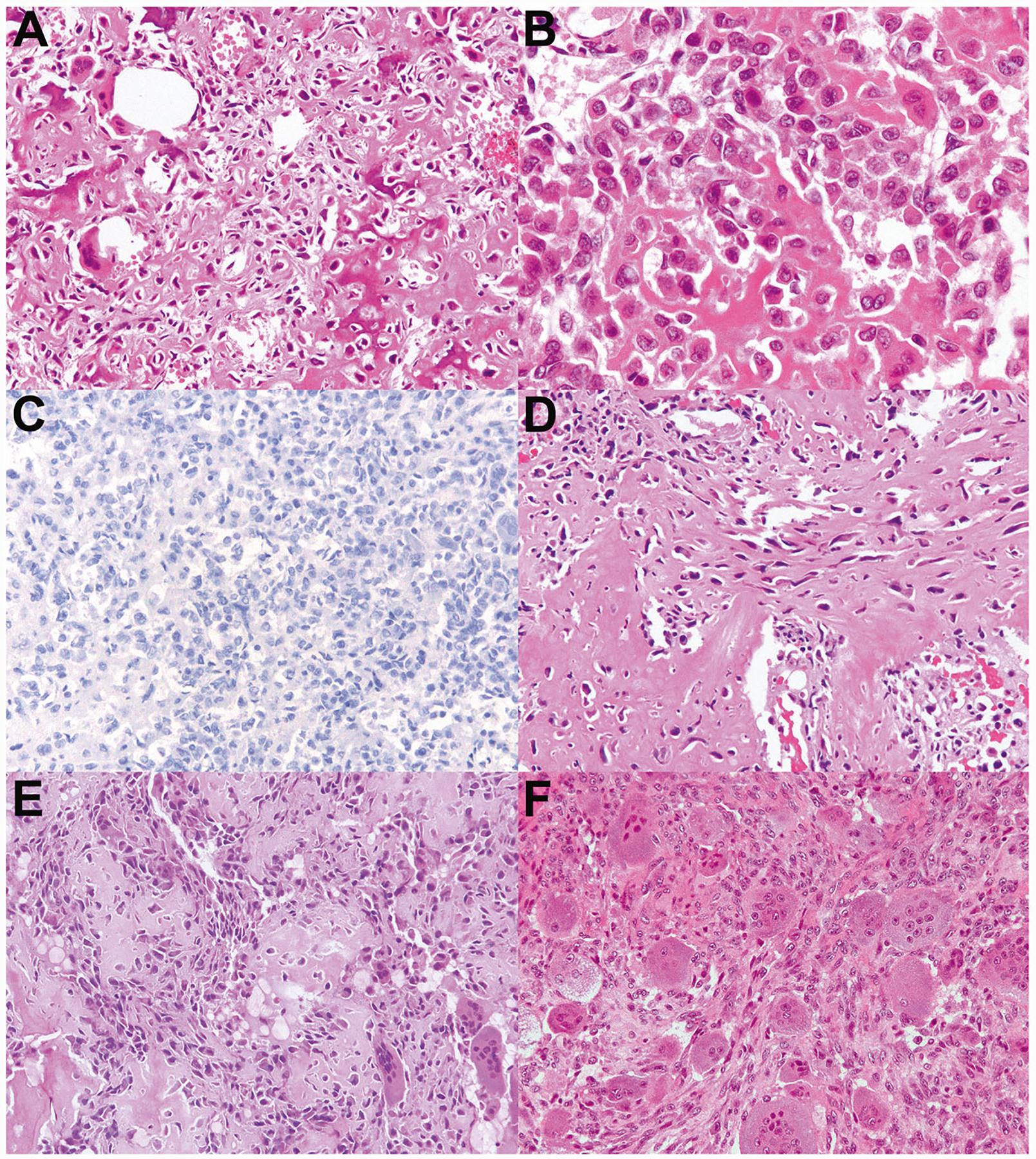
a–c Case 8 contains regions of hyperchromatic, focally spindled neoplastic cells forming bone, consistent with the diagnosis of osteosarcoma. However, in other areas (b) the tumor bears a striking resemblance to chondroblastoma. c H3K36M immunohistochemistry is negative, and Sanger sequencing confirmed wild-type H3F3A and H3F3B sequences. This tumor, which occurred in the rib of a 19-year-old male, was also found to have lymphovascular invasion. d–f Case 9 occurred in the scapula of a 40-year-old woman who developed widespread metastases over the next 24 years and ultimately died of disease. d Similar to case 8, this tumor contains regions of bone formation by malignant cells with hyperchromatic nuclei and focally spindled cytomorphology; (e) it also contains regions that bear a resemblance to chondroblastoma: eosinophilic neoplastic cells with eccentric nuclei and scattered osteoclast-like giant cells. f This tumor also had regions of mononuclear to spindled neoplastic cells with basophilic cytoplasm, resembling giant cell tumor of bone.
Fig. 8. Chondroblastoma-like osteosarcoma, cases 10–11.
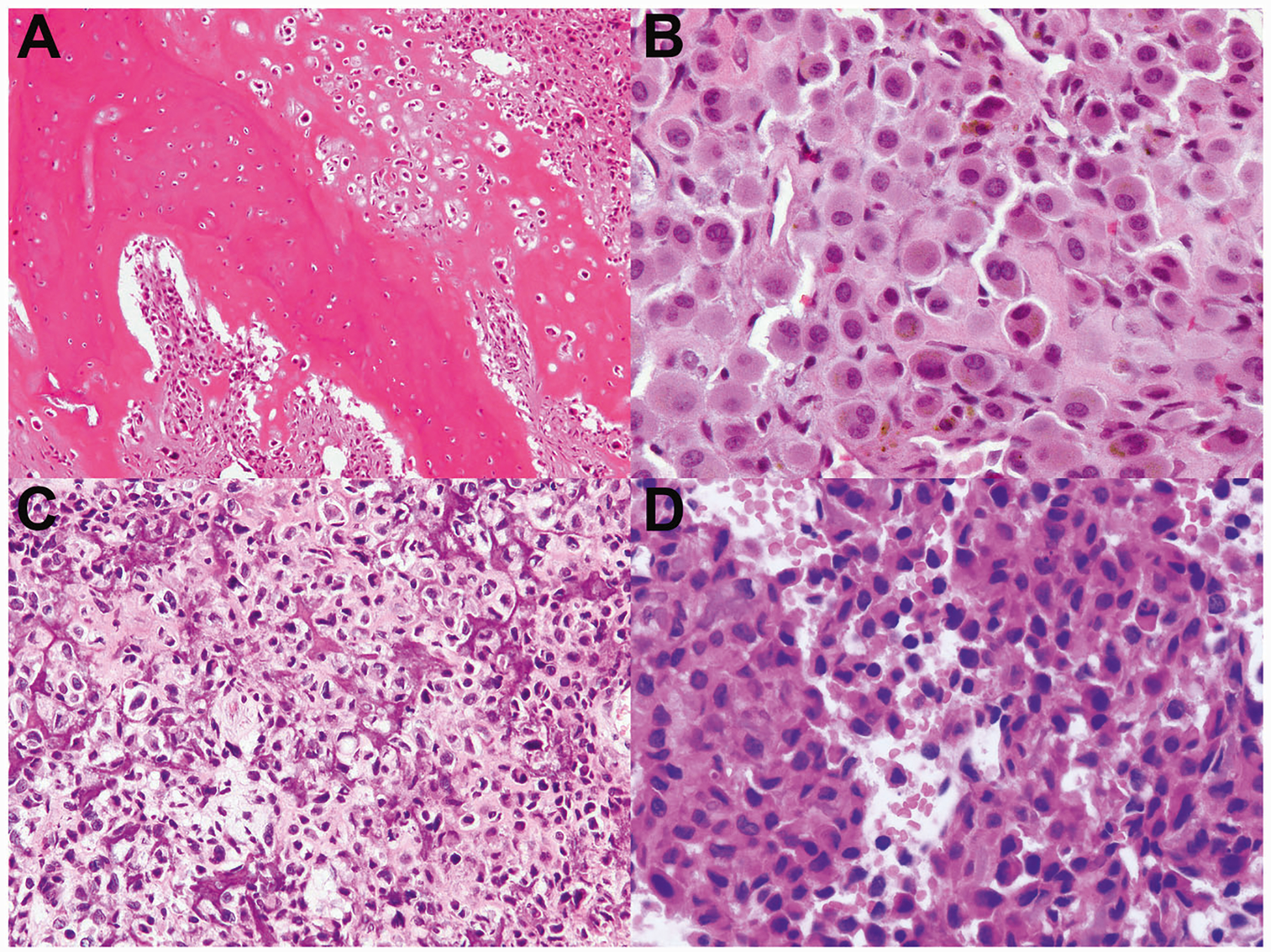
a–b Case 10 had areas of eosinophilic chondroid matrix with chondroblastoma-like neoplastic cells, bone formation (a), and regions of tenosynovial giant cell tumor-like neoplastic cells containing cytoplasmic hemosiderin (b). This tumor arose in the humerus and recurred after an interval of 14.5 years. c–d Case 11 occurred in the C7 vertebral bone; the patient died of disease due to uncontrolled locally aggressive growth 17 months after presentation. c This tumor had nuclear atypia, and regions of bone production, resembling conventional osteosarcoma. d Other regions bore a striking resemblance to chondroblastoma.
IHC for H3G34W and H3K36M was negative in all chondroblastoma-like osteosarcomas. Next-generation sequencing was performed for diagnostic workup of one tumor (case 9), which was found to harbor BAP1 rearrangement involving intron 9, a SETD2 truncation mutation in exon 14, and a LRP1B H1221N missense mutation; BAP1 protein expression was retained by IHC. All tumors underwent Sanger sequencing for this study. Cases 8, 9, and 10 were H3F3A and H3F3B wild-type. PCR amplification failed in case 11, likely due to DNA damage secondary to sample decalcification.
Discussion
The concept of atypical or malignant chondroblastoma has been controversial; reflecting this controversy, chondroblastoma had been classified as a tumor of intermediate biologic potential in the 4th edition of the World Health Organization classification [3], and it was reclassified as a benign neoplasm in the 5th edition [4]. While the rare phenomenon of indolent lung metastases has been well documented [11], there are few reports of “malignant” chondroblastoma [10, 12, 13], and to our knowledge there are no recently reported cases with molecular characterization. The uncertainty regarding atypical or malignant chondroblastoma is due in part to the differential diagnostic consideration of chondroblastoma-like osteosarcoma: tumors with chondroblastoma-like histology but with unusual cytologic atypia and/or infiltrative growth were more likely to have been classified as chondroblastoma-like osteosarcoma. Now that the molecular features of chondroblastoma have been characterized, we sought to determine whether at least some tumors diagnosed as chondroblastoma-like osteosarcoma might actually represent malignant chondroblastoma.
We found that 3 of 5 tumors originally diagnosed as chondroblastoma-like osteosarcoma harbored the H3K36M mutation characteristic of chondroblastoma. Two of these tumors closely resembled conventional chondroblastoma but showed permeative growth and a higher degree of cytologic atypia; the third had mild nuclear atypia but was markedly infiltrative, large (13.5 cm), and showed sheets of chondroblasts with a paucity of giant cells. All of these tumors were designated “malignant chondroblastoma” based on a combination of permeative growth and cytologic atypia. Permeation of cancellous bone/marrow, invasion into the haversian system, and infiltration of extra-osseous soft tissue are features of malignancy in most bone tumors. In the literature, one of the few reported metastatic chondroblastomas leading to patient demise exhibited permeative growth but had no nuclear atypia [10]; in our series, we found that most tumors with infiltrative growth also had nuclear atypia that was beyond the mild atypia of conventional chondroblastoma. Ultimately, we feel that permeative growth and cytologic atypia are both unusual enough features that either should raise the diagnostic consideration of malignant chondroblastoma. No malignant chondroblastoma in our series exhibited bone production by cytologically atypical cells. However, admittedly distinguishing bone fide bone production from the eosinophilic chondroid matrix of chondroblastoma can be challenging and subjective, although we did not have difficulty achieving group consensus for the cases in this series.
The imaging findings in malignant chondroblastoma and chondroblastoma-like osteosarcoma substantially overlapped. All tumors presented as expansile lytic lesions with scattered internal trabeculations. Cortical destruction and associated soft tissue mass were seen in both tumor types. Malignant chondroblastoma and chondroblastoma-like osteosarcoma involved the epiphysis or epiphysis equivalents, similar to conventional chondroblastoma. However, malignant chondroblastoma showed more aggressive features with cortical destruction and associated soft tissue mass, which are not seen in conventional chondroblastoma.
Malignant chondroblastoma and chondroblastoma-like osteosarcoma have a broader differential diagnosis: there was one chondroblastoma-like osteosarcoma in the series that bore a striking resemblance to malignant giant cell tumor of bone in areas but had bone production by malignant-appearing cells and in other areas resembled chondroblastoma; this tumor had negative H3G34W IHC and wild-type H3F3A and H3F3B sequences. There were also cases of chondroblastoma and chondroblastoma-like osteosarcoma with neoplastic cells reminiscent of tenosynovial giant cell tumor; interestingly, in the temporomandibular joint the reverse phenomenon occurs, with tenosynovial giant cell tumor mimicking chondroblastoma [14]. Lastly, one chondroblastoma-like osteosarcoma in our series showed focal cartilage production, raising the consideration of clear cell chondrosarcoma. We conclude that the morphologic features of malignant chondroblastoma are less distinctive than the features of its conventional counterpart, and that malignant chondroblastoma significantly overlaps with chondroblastoma-like osteosarcoma and other entities; thus, molecular testing (or immunohistochemical surrogates) is essential to resolve this differential diagnosis. It would be particularly difficult (if not impossible) to distinguish among these tumor types on limited needle biopsies on morphologic grounds, reinforcing the notion that ancillary testing is critically important for diagnosis.
In contrast to conventional chondroblastoma, which occurs in the long bone of adolescents and young adults, malignant chondroblastoma presented in older individuals with a distinct anatomic distribution (Table 1). The association between older age at presentation and unusual body sites has been observed previously [15], although such series have not shown these tumors to behave more aggressively than conventional chondroblastoma overall [16]. Three of 4 tumors in our study with >1 year of follow-up recurred, much higher than the 15% recurrence rate of conventional chondroblastoma [1], and one patient developed widespread metastases and died of disease. The tumor that gave rise to disseminated metastases had a much higher mitotic rate (15 mitoses per 2 mm2) than the others (1–3 mitoses per 2 mm2; Table 1), and there is a prior report of an unusually locally aggressive tumor that also had a high mitotic rate [12]. These findings suggest that, among tumors with malignant features, the mitotic rate might be a predictor of more aggressive behavior. However, one limitation of our study is the lack of long-term follow-up in three of seven cases; more tumors need to be studied to make this determination. Given that malignant chondroblastoma recurred in 3 of 4 patients with long-term follow-up, including both patients who were treated with curettage, oncologic surgical resection should be considered.
Chondroblastoma-like osteosarcoma was associated with a younger age at presentation than malignant chondroblastoma (median age: 21 years vs. 52 years). Two tumors recurred, one over 14 years after initial resection. Two patients died of disease, one due to local recurrence in the C7 vertebra and one due to widespread metastases over a 26-year period of time. The behavior of this small set of tumors contrasts with that of conventional osteosarcoma, which has a worse prognosis and faster disease progression when metastases are present. Overall, our findings suggest that chondroblastoma-like osteosarcoma is best considered a low-to-intermediate grade sarcoma with the potential for locally aggressive behavior and late metastases.
Chondroblastoma is characterized by the histone H3F3B K36M point mutation in 95% of tumors [17]. To our knowledge, there are no convincing reports of H3K36M in tumors other than chondroblastoma, including the over 1 million tumors in the Catalogue of Somatic Mutations in Cancer (COSMIC) database [18]. Thus, positive H3K36M IHC appears to be 100% specific for resolving the differential diagnosis of malignant chondroblastoma and chondroblastoma-like osteosarcoma.
There remains the question of whether malignant chondroblastoma has additional genetic alterations not present in conventional chondroblastoma. Two tumors in our study underwent next-generation sequencing during diagnostic workup, and both were found to have H3F3B K36M mutations. One had no additional mutations; the other had a nonsense SETD2 K1937* mutation and copy number gains and losses on chromosomes 9 and 10. In chondroblastoma, the H3K36M-mutant protein tightly binds histone methyltransferases SETD2 and MMSET, competitively inhibiting their binding to the co-expressed wild-type histone H3 protein, thus leading to a reduction in H3K36 methylation and aberrant H3K27 methylation [19, 20]. It is conceivable that the likely loss-of-function SETD2 K1937* mutation could lead to more pronounced aberrancies in histone H3 methylation. More generally, the presence of mutations and copy number alterations in addition to H3K36M supports the notion that some cases of malignant chondroblastoma may represent more biologically advanced tumors, although this hypothesis remains speculative at this time.
In contrast to chondroblastoma, the pathogenesis of chondroblastoma-like osteosarcoma has not been systematically studied; one-sequenced case in the literature was found to harbor a FN1-FGFR1 fusion and had elevated FGF23 gene-expression levels, raising the question of whether the tumor would be better classified as a phosphaturic mesenchymal tumor [21]. In general, osteosarcoma commonly harbors TP53 mutations and exhibits marked genomic instability [22, 23]. A recent study also uncovered frequent somatic copy number alterations in BRCA1 and associated proteins BAP1, PTEN, and PALB2 [24]. BAP1 somatic copy number variations were found in 38% of 31 sequenced osteosarcomas in one study [24], and BAP1 loss has been reported in osteosarcoma cell lines [25]. A case of chondroblastoma-like osteosarcoma in our series was found to have a rearrangement in BAP1. IHC demonstrated retained BAP1 expression, although there remains the possibility of a functionally impaired protein. This case was also found to have a SETD2 deletion; one might speculate that this alteration could lead to altered methylation patterns of histone H3 akin to those seen in chondroblastoma, and that this mutation might account for the morphologic overlap with chondroblastoma. More work is needed to determine whether histone H3 methylation abnormalities are a recurrent feature of chondroblastoma-like osteosarcoma.
In summary, malignant chondroblastoma appears to occur in a different clinical setting than conventional chondroblastoma, with an older age at presentation and a different body site distribution. One malignant chondroblastoma in this series gave rise to widespread metastases, with the patient ultimately dying of disease; this clinical behavior supports the notion that these tumors should be considered malignant, as does the presence of cytologic atypia and permeative growth, which are criteria for malignancy in other bone tumors. One malignant chondroblastoma was found to have mutations and copy number alterations in addition to the H3K36M mutation, suggesting that it might be more biologically advanced than conventional chondroblastoma. Lastly, there is substantial morphologic overlap between malignant chondroblastoma and chondroblastoma-like osteosarcoma, and 3 of 5 tumors in this study originally diagnosed as chondroblastoma-like osteosarcoma were reclassified as malignant chondroblastoma; while the production of bone by cytologically malignant cells can distinguish these tumor types, this high misclassification rate underscores the importance of testing for the presence of H3K36M when considering this differential diagnosis.
Acknowledgements
We would like to thank the following pathologists for referring consult cases and providing clinical follow-up: Dr Youssef Al Hmada (Jackson, MS), Prof Wojtek Biernat (Gdańsk, Poland), and Dr Teresita Zdunek (Chicago, IL).
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
This paper was presented in part at the 109th Annual Meeting of the United States and Canadian Academy of Pathology in Los Angeles, CA, February 29 – March 5, 2020.
References
- 1.Konishi E, Nakashima Y, Mano M, Tomita Y, Kubo T, Araki N, et al. Chondroblastoma of extra-craniofacial bones: clinicopathological analyses of 103 cases. Pathol Int. 2017;67:495–502. [DOI] [PubMed] [Google Scholar]
- 2.Lu C, Ramirez D, Hwang S, Jungbluth A, Frosina D, Ntiamoah P, et al. Histone H3K36M mutation and trimethylation patterns in chondroblastoma. Histopathology. 2019;74:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kilpatrick SE, Romeo S. Chondroblastoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of tumours of soft tissue and bone, 4th ed. Lyon, France: IARC Press; 2013. p. 240, 262–3. [Google Scholar]
- 4.Amary F, Bloem JL, Cleven AHG, Konishi E. Chondroblastoma. In: WHO Classification of Tumours Editorial Board. WHO classification of soft tissue and bone tumours, 5th ed. Lyon, France: IARC Press; 2020. [Google Scholar]
- 5.Amary F, Berisha F, Ye H, Gupta M, Gutteridge A, Baumhoer D, et al. H3F3A (Histone 3.3) G34W immunohistochemistry. Am J Surg Pathol. 2017;41:1059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaefer IM, Fletcher JA, Nielsen GP, Shih AR, Ferrone ML, Hornick JL, et al. Immunohistochemistry for histone H3G34W and H3K36M is highly specific for giant cell tumor of bone and chondroblastoma, respectively, in FNA and core needle biopsy. Cancer Cytopathol. 2018;126:552–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amary MF, Berisha F, Mozela R, Gibbons R, Guttridge A, O’Donnell P, et al. The H3F3 K36M mutant antibody is a sensitive and specific marker for the diagnosis of chondroblastoma. Histopathology. 2016;69:121–7. [DOI] [PubMed] [Google Scholar]
- 8.Mayo-Smith W, Rosenberg AE, Khurana JS, Kattapuram SV, Romero LH. Chondroblastoma of the rib. A case report and review of the literature. Clin Orthop Relat Res. 1990;251:230–4. [PubMed] [Google Scholar]
- 9.Al Hmada Y, Bernieh A, Morris RW, Lewin J, Allen T. Chondroblastoma-like Osteosarcoma. Arch Pathol Lab Med. 2020;144:15–17. [DOI] [PubMed] [Google Scholar]
- 10.Kyriakos M, Land VJ, Penning HL, Parker SG. Metastatic chondroblastoma. Report of a fatal case with a review of the literature on atypical, aggressive, and malignant chondroblastoma. Cancer. 1985;55:1770–89. [DOI] [PubMed] [Google Scholar]
- 11.Huvos AG, Higinbotham NL, Marcove RC, O’Leary P. Aggressive chondroblastoma. Review of the literature on aggressive behavior and metastases with a report of one new case. Clin Orthop Relat Res. 1977;126:266–72. [PubMed] [Google Scholar]
- 12.Hull MT, Gonzalez-Crussi F, DeRosa GP, Graul RS. Aggressive chondroblastoma. Report of a case with multiple bone and soft tissue involvement. Clin Orthop Relat Res. 1977;126:261–5. [PubMed] [Google Scholar]
- 13.Lin PP, Thenappan A, Deavers MT, Lewis VO, Yasko AW. Treatment and prognosis of chondroblastoma. Clin Orthop Relat Res. 2005;438:103–9. [DOI] [PubMed] [Google Scholar]
- 14.Oda Y, Izumi T, Harimaya K, Segawa Y, Ishihara S, Komune S, et al. Pigmented villonodular synovitis with chondroid metaplasia, resembling chondroblastoma of the bone: a report of three cases. Mod Pathol. 2007;20:545–51. [DOI] [PubMed] [Google Scholar]
- 15.Dahlin DC, Ivins JC. Benign chondroblastoma. A study of 125 cases. Cancer. 1972;30:401–13. [DOI] [PubMed] [Google Scholar]
- 16.John I, Inwards CY, Wenger DE, Williams DD, Fritchie KJ. Chondroblastomas presenting in adulthood: a study of 39 patients with emphasis on histological features and skeletal distribution. Histopathology. 2020;76:308–17. [DOI] [PubMed] [Google Scholar]
- 17.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang D, Gan H, Lee J-H, Han J, Wang Z, Riester SM, et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. 2016;352:1344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saba KH, Cornmark L, Rissler M, Fioretos T, Åström K, Haglund F, et al. Genetic profiling of a chondroblastoma-like osteosarcoma/malignant phosphaturic mesenchymal tumor of bone reveals a homozygous deletion of CDKN2A, intragenic deletion of DMD, and a targetable FN1-FGFR1 gene fusion. Genes Chromosom Cancer. 2019;58:731–6. [DOI] [PubMed] [Google Scholar]
- 22.Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci USA. 2014;111:E5564–E5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovac M, Blattmann C, Ribi S, Smida J, Mueller NS, Engert F, et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun. 2015;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engert F, Kovac M, Baumhoer D, Nathrath M, Fulda S. Osteosarcoma cells with genetic signatures of BRCAness are susceptible to the PARP inhibitor talazoparib alone or in combination with chemotherapeutics. Oncotarget. 2017;8:48794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]