

Abstract
Backgrounds & Aims:
Alcohol-related liver disease (ALD) is a major cause of chronic liver disease worldwide with limited therapeutic options. Interleukin-1 Receptor Associated Kinase 4 (IRAK4), the master kinase of TLRs/IL-1R-mediated signaling activation, is considered a novel therapeutic target in inflammatory diseases, but has not been investigated in the context of ALD.
Methods:
IRAK4 phosphorylation and IRAK1 protein were analyzed in liver from alcohol-related hepatitis patients and healthy controls. IRAK4 kinase activity-inactive knock-in (IRAK4 KI) mice and bone marrow chimeric mice were exposed to chronic ethanol-induced liver injury. IL-1β-induced IRAK4-mediated signaling and acute phase response were investigated in cultured hepatocytes. IRAK1/4 inhibitor was used to test the therapeutic potential for ethanol-induced liver injury in mice.
Results:
Increased IRAK4 phosphorylation and reduced IRAK1 protein were found in livers of alcoholic hepatitis patients. In the chronic ethanol-induced liver injury mouse model, hepatic inflammation and hepatocellular damage were attenuated in IRAK4 KI mice. IRAK4 kinase activity promotes expression of acute phase proteins in response to ethanol exposure, including C-reactive protein and serum amyloid A1(SAA1). SAA1 and IL-1β synergistically exacerbate ethanol-induced cell death ex vivo. Pharmacological blockage of IRAK4 kinase abrogated ethanol-induced liver injury, inflammation, steatosis, as well as acute phase gene expression and protein production in mice.
Conclusions:
Our data elucidate the critical role of IRAK4 kinase activity in the pathogenesis of ethanol-induced liver injury in mice and provide pre-clinical validation for use of an IRAK1/4 inhibitor as a new potential therapeutic strategy for the treatment of ALD.
Lay summary:
Herein, we have identified the role of IRAK4 kinase activity in the development of ethanol-induced liver injury in mice. Hepatocyte-specific IRAK4 is associated with acute-phase protein release in response to Interleukin-1β. Acute phase response and proinflammatory cytokine/chemokines synergistically exacerbate ethanol-induced hepatocyte cell death ex vivo. Pharmacological inhibition of IRAK4 kinase activity effectively attenuates ethanol-induced liver injury in mice.
Keywords: Alcoholic liver disease, IRAK4, inflammation, acute phase protein, hepatocyte
Graphical Abstract
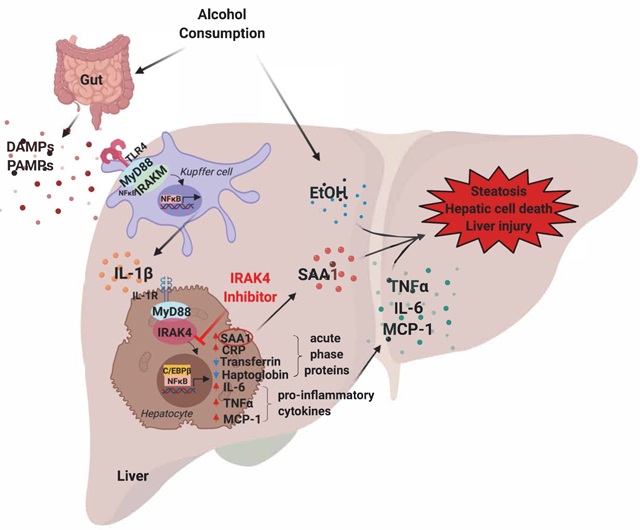
Introduction
Worldwide, alcohol abuse results in approximately 3.3 million deaths annually and alcohol-related liver disease (ALD) is a major cause of preventable mortality[1, 2]. ALD comprises a clinical-histologic spectrum including fatty liver, alcohol-related hepatitis (AH), fibrosis and cirrhosis. Patients with AH often show criteria of systemic inflammatory response syndrome (SIRS) and progress to multiple organ failure (MOF) and death; many AH patients fail to respond to medical treatment[3, 4].
Alcohol consumption disrupts the balance of gut microflora and increases intestinal permeability, resulting in increased translocation of bacterial products, such as lipopolysaccharide (LPS), into the circulation[5]. LPS recognition by Toll-like receptor 4 (TLR4) on hepatic resident macrophages (Kupffer cells) contributes to the activation of the inflammasome and release of pro-inflammatory cytokines, such as tumor necrosis factor α (TNFα), interleukin-1β (IL-1β) and interleukin-6 (IL-6), which in turn impact on hepatocyte function[6-8]. When pathogenic and inflammatory signals are sensed by hepatocytes, it triggers changes to the serum concentrations of hepatocyte secreted proteins, collectively referred to as acute phase proteins, including C-reactive protein (CRP) and Serum Amyloid A1 (SAA1)[9]. Acute phase proteins play a pivotal role in modulating the host’s overall immune response. Previous studies have shown that heavy drinkers have increased serum CRP concentrations compared to moderate drinkers[10]. Serum levels of CRP and SAA1 are increased in patients with alcoholic hepatitis, cirrhosis and hepatocellular carcinoma, and serum concentrations of CRP correlate with short-term 90-day mortality in these patients[3, 11]. These findings suggest that acute phase response is associated with the pathogenesis of ALD.
TLR/IL-1R signaling plays a critical role in ALD[12], nonalcoholic steatohepatitis[13], liver fibrosis[14], and immune-mediated liver injury[15]. Upon ligand recognition, the majority of TLRs and IL-1R recruit myeloid differentiation primary response protein 88 (MyD88), followed by recruitment of IL-1R-associated kinase 4 (IRAK4) and other IRAKs to form the Myddosomes[16]. Clustering of IRAK4 molecules activates kinase activity, leading to phosphorylation, ubiquitination, and degradation of IRAK1. Subsequently, this activates downstream mitogen-activated protein kinase (MAPK) and activates nuclear factor κB (NFκB)[17-19]. Accumulating evidence illustrates that the kinase activity of IRAK4 is critical for the pathogenesis of many inflammatory diseases, whereas kinase-inactive (K213M, K214M) IRAK4 fails to mediate these responses[22]. IRAK4 kinase activity exacerbates modified LDL-mediated NFκB activation, while IRAK4 kinase activity-inactive knock-in (IRAK4 KI) mice have attenuated development of atherosclerosis[23]. IRAK4 kinase activity also contributes to LPS-induced TLR4-mediated septic shock and murine lupus[24]. However, the role of IRAK4 kinase activity during the development of ethanol-induced liver injury has not been investigated.
Preclinical research and clinical trials targeting IRAK4 kinase activity have been conducted in inflammatory diseases. A potent IRAK4 small-molecule inhibitor BMS-986126 attenuates murine lupus in mouse models[25]; small molecule inhibitors ND-2158 and ND-2110 suppress LPS-induced TNF production, alleviate collagen-induced arthritis, block gout formation, as well as ameliorate the activated B cell-like subtype of diffuse large B cell lymphoma in mouse models[26]. Recently, a potent IRAK1/4 inhibitor R191 was reported to suppress NFκB and Protein kinase B/Akt signaling, as well as show efficacy in Waldenström’s macroglobulinemia cell lines and in vivo[27]. These studies indicate that IRAK4 kinase inhibition is emerging as a promising drug target for treating inflammation-related diseases.
In this study, we aimed to understand the role of IRAK4 kinase activity during the pathogenesis of ethanol-induced liver injury in mice. IRAK4 KI mice and littermate control wild-type (WT) mice were subjected to chronic ethanol feeding to induce liver injury[21]. Compared to WT mice, IRAK4 KI mice were protected from ethanol-induced liver injury and inflammation, as well as hepatic steatosis. Mechanistically, IRAK4 kinase activity is required for IL-1β-mediated activation of the acute phase response hepatocytes. In addition, pharmacological inhibition of IRAK4 kinase activity using a potent IRAK1/4 inhibitor ameliorated ethanol-induced liver injury, inflammation, and steatosis in both chronic Lieber-DeCarli and Gao-binge models of ethanol-induced liver injury[28]. Our novel observations suggest that IRAK4 kinase activity contributes to ethanol-induced liver injury, indicating that functional inhibition of IRAK4 could be a potential therapeutic strategy for ALD patients.
Methods and Materials
Patient Samples
Liver tissue samples and paraffin embedded sections were provided by the National Institute on Alcohol Abuse and Alcoholism-supported Clinical Resource for Alcoholic Hepatitis Investigations at Johns Hopkins University (R24AA025017). The use of these tissue samples was approved by Institutional Review Board at Cleveland Clinic Foundation. Detailed information can be found in Supplementary information.
Mouse models
IRAK4 kinase-activity inactive knock-in mice (C57BL/6 background) have been previously described[22]. All procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Detailed methods can be found in Supplementary information.
Data analysis and statistics
Data are expressed as mean ± standard error of the mean (SEM), n = 4 - 6 Pair-fed, n= 6 - 8 EtOH-fed. Comparisons between groups were analyzed by Student’s t test or Two-way ANOVA analysis. Following comparisons were conducted by Bonferroni testing. GraphPad Prism 7 was used for data analysis and representation. p < 0.05 was considered significant.
Results
IRAK4 kinase activity is required for ethanol-induced liver injury
TLR/IL-1R signaling is implicated in the progression of ethanol-induced liver injury. Phosphorylation of IRAK4 was increased in livers of patients with AH undergoing liver transplant compared to liver explants from healthy controls and lower levels of IRAK1 protein were found in alcoholic hepatitis patients than healthy controls. This observation may be due to the ubiquitous degradation of IRAK1 upon TLR/IL-1R signaling pathway activation (Fig. 1A-B)[19]. Consistent with observations made in AH patients, IRAK4 was phosphorylated and IRAK1 protein level was decreased in livers of mice in response to chronic ethanol exposure (Fig. 1C). Based on this, we hypothesized that IRAK4 kinase activity contributes to ethanol-induced liver injury. To test this hypothesis, we subjected female IRAK4 KI mice and their littermate control WT mice to chronic ethanol feeding to model steatosis and mild inflammation. Body weight, ethanol consumption, induction of Cytochrome P450 2E1 (CYP2E1), or blood ethanol concentration were not affected by genotype (Table 1). Compared to pair-fed control mice, chronic ethanol-fed wild-type (WT) mice had increased alanine aminotransferase (ALT) and serum aspartate aminotransferase (AST) (Fig. 1D), indicating ethanol-induced hepatocellular damage. Furthermore, ethanol-fed mice also displayed increased liver steatosis and liver triglyceride content (Fig. 1E-F). IRAK4 KI mice, however, had reduced ethanol-induced liver injury, serum AST and AST levels (Fig. 1D), and attenuated hepatic steatosis compared to WT mice without affecting the expression levels fatty acid metabolism pathway critical enzymes (Fig. 1E-F, Suppl. Fig. 1C). These data supported the critical role of IRAK4 kinase activity in the pathogenesis of ethanol-induced liver injury.
Figure 1. IRAK4 kinase activity is required for ethanol-induced liver injury.

(A) Paraffin-embedded liver HC and AH sections were stained with anti-p-IRAK4 antibody. (B) Western blot analysis of homogenates of livers HC and AH using indicated antibodies. WT and IRAK4 KI mice were allowed free access to EtOH or pair-fed control diets (n = 4-6). (C) Western blot analysis of liver lysates from pair-fed and EtOH-fed WT mice using indicated antibodies. (D) AST and ALT activity was determined in plasma, and hepatic triglyceride content was measured in whole-liver homogenates. (E) Paraffin-embedded liver sections were stained with hematoxylin and eosin (H&E). (F) Frozen liver sections were subjected to Oil-red O staining. Values represent means ± SEM, n=4-6 per group. *, p < 0.05 by Student’s t test or Two-way ANOVA test. Abbreviations: IRAK4 KI, IRAK4 kinase-activity inactive knockin; EtOH, ethanol; TG, triglyceride; AH, alcoholic hepatitis; HC, healthy control.
Given IRAK4 is a pivotal kinase downstream of the major innate immune TLR/IL-1R signaling pathway, we next checked whether inactivation of IRAK4 kinase activity could decrease hepatic inflammatory response after chronic-ethanol feeding. Compared to pair-fed mice, chronic ethanol-fed WT mice had increased mRNA expression of pro-inflammatory cytokine/chemokines in the liver, namely TNFα, IL-1β, IL-6 and monocyte chemoattractant protein 1 (MCP-1), but this induction was ameliorated in the ethanol-fed IRAK4 KI mice (Fig. 2A). Liver resident macrophages, Kupffer cells, identified by F4/80 immunohistochemistry staining, were increased in response to ethanol exposure, but this induction was not observed in IRAK4 KI mice (Fig. 2B). This was consistent with F4/80 mRNA expression from the whole liver mRNA extracts (Fig. 2A). Ly6C+ monocytes are recruited to the liver in response to ethanol exposure[30]. Indeed, we found that foci of Ly6C+ monocytes were increased in the livers of chronic ethanol-fed WT mice in comparison to pair-fed control mice. In contrast, the Ly6C+ foci were reduced in the liver sections of IRAK4 KI mice as compared to that of WT control mice (Fig. 2C). In addition, by in situ TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay, we found that chronic ethanol exposure-induced hepatic cell death was increased in WT, but not in IRAK4 KI mice (Fig. 2D). These observations suggested that IRAK4 kinase activity promotes the downstream signaling events that drive hepatic inflammatory response and cell death contributing to ethanol-induced liver injury.
Figure 2. IRAK4 kinase activity promotes ethanol-induced liver inflammation and hepatic cell death.

(A) Total mRNAs from livers of WT and IRAK4 KI mice were subjected to reverse-transcription polymerase chain reaction analysis (RT-qPCR). Paraffin-embedded liver sections were stained with (B) F4/80 and (C) Ly6C and (D) in situ TUNEL staining. Images were quantified by ImageJ software. Values represent means ± SEM, n=4-6 per group. *, p < 0.05 by Two-way ANOVA test.
IRAK4 kinase activity correlates to ethanol-induced acute phase response
TLR/IL-1R signaling activation is known to modulate acute phase gene expression in hepatocytes [31, 32]. Increased CRP and SAA1 are found in the serum from patients with AH, cirrhosis and hepatocellular carcinoma, and serum concentrations of CRP correlate with short-term 90-day mortality in these patients[3, 11]. Therefore, we examined whether ethanol-induced expression of acute phase reactants was dependent on IRAK4 kinase activity. Indeed, chronic ethanol-fed WT mice had increased mRNA expression of C-reactive protein (CRP) and Serum Amyloid A1(SAA1), and decreased mRNA expression of the negative acute phase reactants, Transferrin and Haptoglobin. However, the impact of ethanol exposure on acute gene expression was normalized in the IRAK4 KI mice (Fig. 3A). Consistent with the mRNA expression signatures in liver, the plasma concentration of CRP and SAA1 were increased in WT mice after ethanol feeding, but this induction was prevented in the ethanol-fed IRAK4 KI mice (Fig. 3B). C/EBPb is a key transcription factors for regulation of acute phase gene expression in hepatocytes [33-35]. We observed that ethanol feeding resulted in the induction of C/EBPb, which was greatly reduced in IRAK4 KI mice (Fig. 3C). Our previous study found that IRAKM is required for ethanol-induced inflammasome activation and IL-1β production in Kupffer cells via a mechanism that is independent of IRAK4 kinase activity[21]. Consistent with our previous data, the accumulation of cleaved IL-1β protein in response to ethanol feeding was equivalent between WT and IRAK4 KI mice (Fig. 3C). Taken together, these data suggested that IRAK4 kinase activity is required for ethanol-induced regulation of acute phase genes, which may contribute to liver injury in response to chronic ethanol exposure.
Figure 3. IRAK4 kinase activity is involved in ethanol-induced acute phase response.
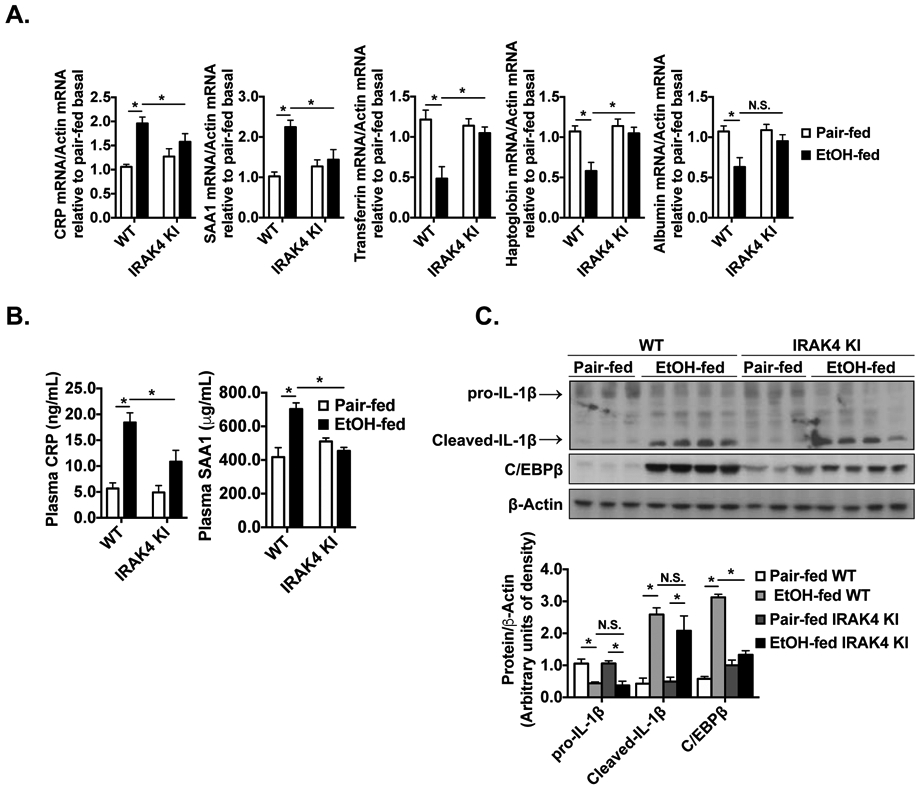
(A) Total mRNAs from livers of WT and IRAK4 KI mice were subjected to RT-qPCR. (B) CRP and SAA1 concentrations were measured in plasma from WT and IRAK4 KI mice by enzyme-linked immunosorbent assay (ELISA). (C) Tissue lysates from whole liver of WT and IRAK4 KI mice were analyzed by western blot using indicated antibodies. Images were quantified by ImageJ software. Values represent means ± SEM, n=4-6 per group. *, p < 0.05 by Two-way ANOVA test.
Inhibition of IRAK4 kinase activity on hepatocytes attenuates the TLR/IL-1R signaling pathway and acute phase protein production
We then investigated the cellular mechanism by which IRAK4 kinase activity promotes ethanol-induced acute phase gene induction in hepatocytes, contributing to ethanol-induced liver injury. Notably, ethanol exposure induces the activation of the NLRP3 inflammasome, leading to IL-1β cleavage and secretion by Kupffer cells[21, 36]. We previously reported that IRAK4 kinase activity is not required for low-dose LPS (100 pg/ml, pathophysiological concentration in AH patients) induced NFκB activation in macrophages (Suppl. Fig. 4)[21]. Our data showed that ethanol-induced IL-1β production was not different between WT and IRAK4 KI mice (Fig. 3C). IL-1β is known to act on hepatocytes to induce acute phase protein synthesis in hepatocytes[37]. Therefore, we hypothesized that IRAK4 kinase activity may function in non-myeloid cells, such as hepatocytes, to promote ethanol-induced IL-1β-mediated induction of acute phase proteins and other inflammatory genes. To test this hypothesis, we generated bone marrow chimeric mice: IRAK4 KI → WT (IRAK4 kinase-inactive in myeloid cells) and WT → IRAK4 KI (IRAK4 kinase-inactive in non-myeloid cells). Ethanol exposure-induced ALT, AST and hepatic steatosis were reduced in IRAK4 KI in non-myeloid chimera mice compared to IRAK4 KI in myeloid chimera mice (Fig 4. A-C). Furthermore, IRAK4 KI non-myeloid chimera mice had less proinflammatory mRNA expression and acute phase response following ethanol-feeding compared to IRAK4 KI in myeloid chimera mice (Fig. 4D-F). These data indicate that IRAK4 kinase activity in non-myeloid cells, likely hepatocytes, contributes to ethanol-induced liver injury and acute phase response.
Figure 4. Bone marrow chimeric IRAK4 KI recipient mice have decreased ethanol-induced liver injury compared to WT recipients.

(A) Paraffin-embedded liver sections were stained with H&E. (B) AST and ALT activity was determined in plasma. (C) Hepatic triglyceride content was measured in whole-liver homogenates. (D-E) Total mRNAs from livers of WT and IRAK4 KI chimeric recipient mice were subjected to RT-qPCR analysis. (F) CRP and SAA1 concentrations were measured in plasma of WT and IRAK4 KI chimeric mice by ELISA. Values represent means ± SEM, n=4-6 per group. *, p < 0.05 by Two-way ANOVA test.
To further study the mechanistic role of IRAK4 kinase activity in hepatocytes, primary hepatocytes were isolated from livers of WT and IRAK4 KI mice, and treated with IL-1β. Interestingly, IL-1β-induced IL-1R downstream signaling activation was ablated in IRAK4 KI cells, as IRAK1 degradation, induction of c-Jun N-terminal kinases (JNK) and IκBα phosphorylation were diminished in IRAK4 KI cells compared to WT cells (Fig. 5A). IL-1β-induced mRNA levels of IL-6, TNFα and MCP-1 were decreased in IRAK4 KI hepatocytes (Fig. 5B). Furthermore, IL-1β-induced C/EBPβ expression was also reduced in IRAK4 KI hepatocytes compared to WT cells (Fig. 5A). Consistently, CRP and SAA1 production in response to IL-1β were decreased in the culture media of hepatocytes from IRAK4 KI mice (Fig. 5C). Using a potent IRAK1/4 inhibitor[38], we found that IRAK1/4 inhibitor treatment ameliorated IL-1β-mediated signaling activation in primary hepatocytes from C57BL/6 mice (Fig. 5E), similar results were also observed in a hepatic cell line AML12 cells (Suppl. Fig. 2). To better understand how mechanistically IRAK4 kinase activity links to acute phase response, C/EBPβ was knocked-down in AML12 cells (Suppl. Fig. 3). The mRNA expression levels of acute phase proteins and proinflammatory genes were induced by IL-1β, but the induction was ameliorated by C/EBPβ knockdown or inhibition of IRAK4 kinase activity (Fig. 5F-H). These results suggest that IRAK4 kinase activity probably promoted the expression of the acute phase and inflammatory genes in hepatocytes via the induction of C/EBPβ. Furthermore, since it has been reported that SAA1 stimulation leads to stellate cell death[39], we investigated the potential effect of SAA1 on hepatocytes with and without the presence of ethanol. Ethanol or SAA1 alone did induce relative mild cell toxicity, while ethanol and SAA1 synergistically accelerated hepatocyte cell death (Fig. 5I). Consistently, ethanol/SAA1 combined stimulation led to activation of Caspase 3 (Fig. 5J), implicating the potential role of hepatocyte-derived acute phase protein SAA1 induced by IL-1-IRAK4 signaling in exacerbating liver injury of alcohol-fed mice. In addition to acute phase proteins, IRAK4 kinase activity and C/EBPβ were also required for IL-1-induced inflammatory gene expression in hepatocytes, including IL-6, TNF and MCP-1 (Fig. 5G), which probably also contributed to alcohol-induced liver injury.
Figure 5. Hepatocyte-specific IRAK4 is associated with acute-phase protein release in response to IL-1β.


(A) Western blot analysis of IL-1β-treated whole cell lysates from cultured WT and IRAK4 KI primary hepatocytes, and (D) WT primary hepatocytes with or without IRAK4 inhibitor treatment. CRP and SAA1 concentrations were measured in culture media from WT and IRAK4 KI primary hepatocytes (C) or IRAK4 inhibitor treated WT primary hepatocytes (E) by ELISA. (B) Total mRNAs from WT and IRAK4 KI primary hepatocyte treated with or without IL-1β for 12 hours were subjected to RT-qPCR analysis. (F-G) Total mRNAs from AML12 cells with or without C/EBPβ knockdown with indicate treatments for 12 hours were subjected to RT-qPCR analysis. (H) CRP and SAA1 concentrations were measured by ELISA in Culture media from (F). (I) WT Primary hepatocytes were treated with EtOH (150 mM) for 20 hours, mouse recombinant SAA1 (5 μg/ml) or DMSO were then added for 8 hours. Cytotoxicity were assessed by MTS assay. (J) Western blot analysis of whole cell lysates from cultured primary hepatocytes from (I). Images were quantified by ImageJ software. The experiments were repeated for three times with similar results. Data represent mean ± SEM, *: p < 0.05. by Two- way ANOVA or Student’s t-test. IRAK4 Inh., IRAK1/4 inhibitor
Pharmacological inhibition of IRAK4 kinase activity attenuates ethanol-induced liver injury in mice
Given our observations that IRAK4 KI mice were protected from ethanol-induced liver injury (Fig. 1-3), and IRAK4 kinase-inactivation prevented IL-1β response in hepatocytes (Fig. 4-5), we hypothesized that blockade of IRAK4 kinase activity could be an effective therapeutic strategy. Therefore, we tested the efficacy of an IRAK1/4 inhibitor[27] as a potential treatment for chronic ethanol exposure, and as a preventative during Gao-binge ethanol feeding (Fig. 6A-B). The IRAK1/4 inhibitor treatment resulted in significant decreases in plasma ALT and AST levels in response to ethanol consumption compared to saline vehicle in both models (Fig. 6E). In addition, IRAK1/4 inhibitor treatment attenuated ethanol-induced lipid accumulation in hepatocytes in both models, suggesting that pharmacological inhibition of IRAK4 kinase activity is capable of attenuating ethanol-induced liver injury and steatosis (Fig. 6C-F).
Figure 6. Pharmacological inhibition of IRAK4 attenuates ethanol-induced liver injury.

C57BL/6J mice were allowed free access to ethanol or pair-fed control; during last week of feeding or certain time points, IRAK4/1 inhibitor were orally administered 10 mg/kg body weight twice/day (A, B). (C, D) Paraffin-embedded liver sections were stained with H&E. (E) AST and ALT activity was determined in plasma. (F) Hepatic triglyceride content was measured in whole-liver homogenates. Values represent means ± SEM, n=4-6 per group. *, p < 0.05 by Two-way ANOVA test.
mRNA expression of inflammatory markers was up-regulated in WT mice after either chronic or acute ethanol feeding, but not in IRAK1/4 inhibitor treated mice (Fig. 7A). Notably, in the chronic feeding model, the numbers of F4/80+ and Ly6C+ cells were increased in WT mice after ethanol feeding, but was decreased in IRAK1/4 inhibitor treated mice (Fig. 7B-C). Neutrophil infiltration is a hall mark of alcoholic hepatitis and has been shown to correlate with severity of alcoholic hepatitis[40]. Additionally, acute ethanol binge results in immune cell infiltration the in Gao-binge mouse model, including myeloperoxidase(MPO) positive neutrophils[41]. We found that MPO+ cells were accumulated in control mice after alcohol binge, but this accumulation was diminished with IRAK1/4 inhibitor treatment (Fig. 7D). Consistent with data from IRAK4 KI mice, the mRNA expression of CRP and SAA1 were reduced in the IRAK1/4 inhibitor treated group, whereas the negative regulators were upregulated (Fig. 8A). IRAK1/4 inhibitor treatment also reduced chronic ethanol feeding-induced upregulation of C/EBPβ, which is responsible for acute phase gene transcription, without affecting IL-1β cleavage (Fig. 8B). Furthermore, the serum concentration and hepatic production of CRP and SAA1 were attenuated in the IRAK1/4 inhibitor treated mice (Fig. 8C). Clearly, IRAK1/4 inhibitor treatment ameliorated ethanol-induced liver inflammation, immune cell recruitment and hepatocellular damage following chronic (mild) or Gao-binge (severe) ethanol feeding in mice.
Figure 7. Ethanol-induced hepatic inflammation is reduced with IRAK1/4 inhibitor treatment.

(A) Total mRNAs from livers of Saline and IRAK1/4 Inh. treated mice were subjected to RT-qPCR analysis. Paraffin-embedded liver sections were stained with (B) F4/80, (C) Ly6C and (D) MPO from chronic or acute-on-chronic ethanol feeding model. Images were quantified by ImageJ software. Values represent means ± SEM, n=4-8 per group. *, p < 0.05 by Two-way ANOVA test.
Figure 8. Pharmacological inhibition of IRAK4 reduces ethanol-induced acute phase response.

(A) Total mRNAs from livers of Saline and IRAK1/4 Inh. treated mice were subjected to RT-qPCR analysis. (B) Whole liver homogenates were analyzed by western blot using indicated antibodies. (C) Plasma cytokine concentrations were measured by ELISA. Images were quantified by ImageJ software. Values represent means ± SEM, n=4-8 per group. *, p < 0.05 by Two-way ANOVA test. (D) Schematic model.
Discussion
Alcohol-associated liver disease abuse causes a global burden on public health with limited definitive therapeutic methods available[42]. Here, we report that IRAK4 kinase activity contributes to the pathogenesis of ethanol-induced liver injury in mice. Increased IRAK4 phosphorylation and IRAK1 degradation were found in livers of AH patients and ethanol-fed mice. Accordingly, we made use of the IRAK4 KI mice and subjected the mice to chronic ethanol feeding. Ethanol-induced liver injury, hepatic inflammation and hepatocytes death were greatly reduced in IRAK4 KI mice compared with littermate WT controls. Interestingly, hepatic steatosis was also decreased in IRAK4 KI mice, without affecting the expression levels of critical enzymes in fatty acid metabolic pathways, including Cpt1, Acc1, and Fas. These data suggest that the reduction in hepatic inflammation and cell death in IRAK4 KI mice prevented excessive lipid accumulation (Suppl. Fig. 1C). Importantly, we found that ethanol-induced expression of acute phase proteins, CRP and SAA1, was attenuated in the IRAK4 KI mice. Furthermore, we demonstrated that IRAK4 kinase activity in hepatocytes is required for the acute-phase gene expression and protein production, associated with the up-regulation of C/EBPβ. Based on these mechanistic insights, a newly developed IRAK1/4 inhibitor was used to evaluate the therapeutic potential of targeting IRAK4 kinase activity in ethanol-induced liver injury in mice by pre-clinical chronic and Gao-binge ethanol-feeding models. Pharmacological inhibition of IRAK4 kinase activity in vivo abrogated ethanol-induced steatosis, hepatic inflammation, acute phase protein production, as well as hepatocellular injury.
Alcohol intake increases gut permeability and leads to accumulation of low levels of TLR ligands, such as LPS, in the circulation. Activation of Toll-like receptors (TLRs) in hepatic macrophages and injury to hepatocytes contribute to the pathogenesis of ALD. We previously reported that the MyD88-myeloid deficiency prevented the development of hepatic steatosis and inflammation. Mice deficient in IRAKM were also protected from chronic ethanol-induced liver injury in mice. Importantly, in response to low-dose LPS (100 pg/ml, pathophysiological concentration in AH patients), the IRAKM Myddosome was preferentially formed independently of IRAK4-kinase activity in macrophages[21]. Based on these previous findings, it is unlikely that IRAK4 kinase activity in myeloid cells would have a major impact on ethanol-induced liver injury (Suppl. Fig. 4). Therefore, we predicted that the protective role of IRAK4 kinase inactivation is likely due to loss of IRAK4-kinase activity in parenchymal cells, such as hepatocytes. Consistent with this prediction, ethanol-induced liver injury was reduced in IRAK4 KI in myeloid chimera mice compared to IRAK4 KI in non-myeloid chimera mice. These data indicated that TLR-IL-1R-mediated IRAK4-kinase-dependent signaling in hepatocytes makes a significant contribution to the pathogenesis of ethanol-induced liver injury.
Hepatocytes, as the predominant parenchymal cells in the liver, not only play a key role in ethanol metabolism, but also participate in host defense and protein synthesis during the pathogenesis of ALD. While hepatocytes metabolize ethanol, excessive ethanol exposure in hepatocytes results in an increase of oxidative damage to lipids, proteins and DNA, leading to hepatocytes apoptosis or necrosis[43]. Hepatocytes react to danger signals produced by dead hepatocytes as well as cytokines/chemokines produced by Kupffer cells, such as IL-1β. In response to these signals, hepatocytes synthesize and secrete acute phase proteins via activation of the critical transcription factor C/EBPβ, contributing to the clearance of danger signals and host defense. In our experiments, while ethanol stimulation alone induced mild cytotoxicity in primary hepatocytes, co-treatment of SAA1 significantly increased cell death and Caspase 3 cleavage, indicating the acute phase response may further exacerbate ethanol-induced hepatic cell death in an autocrine manner. Recently, deletion of IL-1RI in hepatocytes was shown to protects mice from acute liver failure, reduces hepatic and systemic inflammatory cytokine and chemokine levels, as well as attenuates neutrophil infiltration into the liver in response to injury[44]. These data suggested that IL-1β plays a critical in hepatic inflammation via interaction with hepatocytes. In the current study, we found that IL-1β-induced downstream signaling, acute phase protein production and proinflammatory genes were decreased in IRAK4 KI hepatocytes and IRAK4 inhibitor-treated cells. Therefore, targeting hepatocyte IRAK4 kinase activity might be a useful therapeutic strategy for the treatment of ALD.
Growing evidence shows that patients with AH have a high incidence of death due to the development of Multiple organ failure (MOF). The presence of systemic inflammatory response syndrome (SIRS) is a major predictor of MOF and death in AH, identifying the main inflammatory mediators that cause the systemic inflammatory response and targeting on systemic inflammation may provide a novel therapeutic approach to these patients[3, 45]. Elevated levels of IL-1β, which is mainly produced by macrophages via TLR signaling, amplifies the inflammatory cascade and contributes to the pathogenesis of ALD[20, 46], suggesting targeting of TLR/IL-1R downstream signaling pathway may be a promising therapeutic strategy. In support of this, in vivo intervention with recombinant IL-1R antagonist, IL-1Ra, markedly reduced serum ALT, steatosis, fibrosis markers, and inflammation in the liver of mice[36]. Clinical trials are currently underway to test the efficacy of inhibition IL-1 signaling in ALD. IL-1R antagonists (Anakinra) in the treatment of alcoholic hepatitis has completed (ClinicalTrials.gov Identifier: NCT01809132), and another clinical trial using an IL-1β monoclonal antibody inhibitor (Canakinumab) is ongoing (ClinicalTrials.gov Identifier: NCT03775109). While IL-1Ra shows therapeutic effects in treating ALD, inhibition of the master kinase IRAK4 might be even more efficacious to block TLR/IL-1R dual signaling pathway-dependent pathogenesis during ALD. On the other hand, prognostic value of acute phase proteins has been evaluated in patients with alcoholic hepatitis and cirrhosis. CRP levels were found to be a true prognostic marker in patients with cirrhosis, particularly powerful in the event of advanced liver failure[47]. Furthermore, a recent study also shows that serum levels of transferrin predicts mortality in patients with severe alcoholic hepatitis[48]. These studies suggest that the levels of acute phase reactants may become useful prognostic biomarkers for patients with alcohol-related liver disease, and serum acute phase reactants may also be exploited as effective companion biomarkers for IRAK4 inhibitor therapy for ALD in the future.
In summary, our study illustrates that IRAK4 kinase activity in hepatocytes promotes ethanol-induced liver steatosis, hepatocellular death, inflammation, as well as liver injury. Administration of IRAK1/4 inhibitor attenuates ethanol-induced liver injury. These findings underline the significance of TLR/IL-1R signaling as a potential therapeutic target in ALD, and pre-clinical efficacy of IRAK1/4 inhibition as a new therapeutic strategy for the treatment of ALD.
Supplementary Material
Highlights:
IRAK4 kinase activity is required for ethanol-induced liver injury in mice.
Hepatocyte-specific IRAK4 is associated with acute-phase protein release in response to Interleukin-1β.
Acute phase response and proinflammatory cytokine/chemokines synergistically exacerbate ethanol-induced cell death ex vivo.
Pharmacological inhibition of IRAK4 kinase activity attenuates ethanol-induced liver injury in mice.
Targeting IRAK4 kinase activity might be a potential therapeutic strategy for the treatment of Alcohol-associated Liver Disease.
Acknowledgments:
We would like to thank the financial sources from the National Institutes of Health (1R01AA023722, to X.L. and L.E.N; 2PO1HL029582-26A1 and PO1CA062220-16A1 to X.L; P50AA024333, to L.E.N., K99AA026648 to K.L.P).
Footnotes
Conflict of Interest Statement: Vanessa Taylor is an employee of Rigel Pharmaceuticals. The other authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement:
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
References
- [1].WHO. Global status report on alcohol and health. 2014. [Google Scholar]
- [2].Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Michelena J, Altamirano J, Abraldes JG, Affo S, Morales-Ibanez O, Sancho-Bru P, et al. Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology. 2015;62:762–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mathurin P, O'grady J, Carithers RL, Phillips M, Louvet A, Mendenhall CL, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut. 2011;60:255–60. [DOI] [PubMed] [Google Scholar]
- [5].Stärkel P, Schnabl B. Bidirectional communication between liver and gut during alcoholic liver disease. Seminars in liver disease: Thieme Medical Publishers; 2016. p. 331–9. [DOI] [PubMed] [Google Scholar]
- [6].Uesugi T, Froh M, Arteel GE, Bradford BAU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–8. [DOI] [PubMed] [Google Scholar]
- [7].Cohen JI, Chen X, Nagy LE. Redox signaling and the innate immune system in alcoholic liver disease. Antioxid Redox Signal. 2011;15:523–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Petrasek J, Csak T, Szabo G. Toll-like receptors in liver disease. Advances in clinical chemistry: Elsevier; 2013. p. 155–201. [DOI] [PubMed] [Google Scholar]
- [9].Dinarello CA. Interleukin-1 and the pathogenesis of the acute-phase response. N Engl J Med. 1984;311:1413–8. [DOI] [PubMed] [Google Scholar]
- [10].Imhof A, Froehlich M, Brenner H, Boeing H, Pepys MB, Koenig W. Effect of alcohol consumption on systemic markers of inflammation. The Lancet. 2001;357:763–7. [DOI] [PubMed] [Google Scholar]
- [11].She S, Xiang Y, Yang M, Ding XC, Liu XY, Ma LN, et al. C-reactive protein is a biomarker of AFP-negative HBV-related hepatocellular carcinoma. International Journal of Oncology. 2015;47:543–54. [DOI] [PubMed] [Google Scholar]
- [12].Imaeda AB. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. Journal of Clinical Investigation. 2009;119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32. [DOI] [PubMed] [Google Scholar]
- [15].Dolganiuc A Dendritic cells in hepatitis C infection: can they (help) win the battle? Journal of Gastroenterology. 2011. [DOI] [PubMed] [Google Scholar]
- [16].Lin S-C, Lo Y-C, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Qin J, Yao J, Cui G, Xiao H, Kim TW, Fraczek J, et al. TLR8-mediated NF-kappaB and JNK activation are TAK1-independent and MEKK3-dependent. J Biol Chem. 2006;281:21013–21. [DOI] [PubMed] [Google Scholar]
- [18].Li X IRAK4 in TLR/IL-1R signaling: Possible clinical applications. European journal of immunology. 2008;38:614–8. [DOI] [PubMed] [Google Scholar]
- [19].Cui W, Xiao N, Xiao H, Zhou H, Yu M, Gu J, et al. β-TrCP-mediated IRAK1 degradation releases TAK1-TRAF6 from the membrane to the cytosol for TAK1-dependent NFκB activation. Molecular and cellular biology. 2012:MCB. 00722–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou H, Yu M, Roychowdhury S, Sanz-Garcia C, Pollard KA, McMullen MR, et al. Myeloid-MyD88 Contributes to Ethanol-Induced Liver Injury in Mice Linking Hepatocellular Death to Inflammation. Alcoholism: Clinical and Experimental Research. 2017;41:719–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhou H, Yu M, Zhao J, Martin BN, Roychowdhury S, McMullen MR, et al. IRAKM-Mincle axis links cell death to inflammation: Pathophysiological implications for chronic alcoholic liver disease. Hepatology. 2016;64:1978–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, et al. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J Exp Med. 2007;204:1025–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim TW, Febbraio M, Robinet P, Dugar B, Greene D, Cerny A, et al. The critical role of IL-1 receptor-associated kinase 4-mediated NF-kappaB activation in modified low-density lipoprotein-induced inflammatory gene expression and atherosclerosis. J Immunol. 2011;186:2871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Murphy M, Pattabiraman G, Manavalan TT, Medvedev AE. Deficiency in IRAK4 activity attenuates manifestations of murine Lupus. Eur J Immunol. 2017;47:880–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dudhgaonkar S, Ranade S, Nagar J, Subramani S, Prasad DS, Karunanithi P, et al. Selective IRAK4 inhibition attenuates disease in murine lupus models and demonstrates steroid sparing activity. The Journal of Immunology. 2017;198:1308–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kelly PN, Romero DL, Yang Y, Shaffer AL 3rd, Chaudhary D, Robinson S, et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med. 2015;212:2189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ni H, Shirazi F, Baladandayuthapani V, Lin H, Kuiatse I, Wang H, et al. Targeting Myddosome Signaling in Waldenström's Macroglobulinemia with the Interleukin-1 Receptor-Associated Kinase 1/4 Inhibitor R191 Clinical Cancer Research. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8:627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Khanova E, Wu R, Wang W, Yan R, Chen YB, French SW, et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology. 2018;67:1737–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Craig J McClaina DBH, Zhenyuan Songa, Ion Deaciucc, Shirish Barvea. Monocyte activation in alcoholic liver disease. Alcohol. 2002;27:53–61. [DOI] [PubMed] [Google Scholar]
- [31].Gabay C, Kushner I. Acute-Phase Proteins and Other Systemic Responses to Inflammation. New England Journal of Medicine. 1999;340:448–54. [DOI] [PubMed] [Google Scholar]
- [32].Crispe IN. Hepatocytes as immunological agents. The Journal of Immunology. 2016;196:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, et al. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. The EMBO journal. 1990;9:1897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, et al. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proceedings of the National Academy of Sciences. 1993;90:10193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Giltiay NV, Karakashian AA, Alimov AP, Ligthle S, Nikolova-Karakashian MN. Ceramide-and ERK-dependent pathway for the activation of CCAAT/enhancer binding protein by interleukin-1β in hepatocytes Journal of lipid research. 2005;46:2497–505. [DOI] [PubMed] [Google Scholar]
- [36].Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. The Journal of clinical investigation. 2012;122:3476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Baumann H, Gauldie J. Regulation of hepatic acute phase plasma protein genes by hepatocyte stimulating factors and other mediators of inflammation. Molecular biology & medicine. 1990;7:147–59. [PubMed] [Google Scholar]
- [38].Markovtsov VV, Lamagna C, Chan M, Yi S, Young C, Frances R, et al. Potential role for R191, potent and selective IRAK4 kinase inhibitor, in treatment of hematologic malignancies. AACR; 2016. [Google Scholar]
- [39].Siegmund SV, Schlosser M, Schildberg FA, Seki E, De Minicis S, Uchinami H, et al. Serum amyloid A induces inflammation, proliferation and cell death in activated hepatic stellate cells. PLoS One. 2016;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jaeschke H Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol. 2002;27:23–7. [DOI] [PubMed] [Google Scholar]
- [41].Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58:1814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Leggio L, Lee MR. Treatment of Alcohol Use Disorder in Patients with Alcoholic Liver Disease. Am J Med. 2017;130:124–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Castilla R, Gonzalez R, Fouad D, Fraga E, Muntane J. Dual effect of ethanol on cell death in primary culture of human and rat hepatocytes. Alcohol and Alcoholism. 2004;39:290–6. [DOI] [PubMed] [Google Scholar]
- [44].Gehrke N, Hövelmeyer N, Waisman A, Straub BK, Weinmann-Menke J, Wörns MA, et al. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. Journal of hepatology. 2018;68:986–95. [DOI] [PubMed] [Google Scholar]
- [45].Sujan R, Cruz-Lemini M, Altamirano J, Simonetto D, Maiwall R, Axley P, et al. A Validated Score Predicts Acute Kidney Injury and Survival in Patients with Alcoholic Hepatitis: A Multicentric International Prospective Cohort Study. Liver Transplantation. 2018. [DOI] [PubMed] [Google Scholar]
- [46].Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nature reviews Gastroenterology & hepatology. 2015;12:387. [DOI] [PubMed] [Google Scholar]
- [47].Di Martino V, Coutris C, Cervoni JP, Dritsas S, Weil D, Richou C, et al. Prognostic value of C-reactive protein levels in patients with cirrhosis. Liver Transplantation. 2015;21:753–60. [DOI] [PubMed] [Google Scholar]
- [48].Atkinson SR, Hamesch K, Spivak I, Guldiken N, Cabezas J, Argemi J, et al. Serum Transferrin Is an Independent Predictor of Mortality in Severe Alcoholic Hepatitis. American Journal of Gastroenterology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.