

ABSTRACT
The structural integrity and functional stability of organelles are prerequisites for the viability and responsiveness of cells. Dysfunction of multiple organelles is critically involved in the pathogenesis and progression of various diseases, such as chronic obstructive pulmonary disease, cardiovascular diseases, infection, and neurodegenerative diseases. In fact, those organelles synchronously present with evident structural derangement and aberrant function under exposure to different stimuli, which might accelerate the corruption of cells. Therefore, the quality control of multiple organelles is of great importance in maintaining the survival and function of cells and could be a potential therapeutic target for human diseases. Organelle-specific autophagy is one of the major subtypes of autophagy, selectively targeting different organelles for quality control. This type of autophagy includes mitophagy, pexophagy, reticulophagy (endoplasmic reticulum), ribophagy, lysophagy, and nucleophagy. These kinds of organelle-specific autophagy are reported to be beneficial for inflammatory disorders by eliminating damaged organelles and maintaining homeostasis. In this review, we summarized the recent findings and mechanisms covering different kinds of organelle-specific autophagy, as well as their involvement in various diseases, aiming to arouse concern about the significance of the quality control of multiple organelles in the treatment of inflammatory diseases.
Abbreviations: ABCD3: ATP binding cassette subfamily D member 3; AD: Alzheimer disease; ALS: amyotrophic lateral sclerosis; AMBRA1: autophagy and beclin 1 regulator 1; AMPK: AMP-activated protein kinase; ARIH1: ariadne RBR E3 ubiquitin protein ligase 1; ATF: activating transcription factor; ATG: autophagy related; ATM: ATM serine/threonine kinase; BCL2: BCL2 apoptosis regulator; BCL2L11/BIM: BCL2 like 11; BCL2L13: BCL2 like 13; BECN1: beclin 1; BNIP3: BCL2 interacting protein 3; BNIP3L/NIX: BCL2 interacting protein 3 like; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CANX: calnexin; CAT: catalase; CCPG1: cell cycle progression 1; CHDH: choline dehydrogenase; COPD: chronic obstructive pulmonary disease; CSE: cigarette smoke exposure; CTSD: cathepsin D; DDIT3/CHOP: DNA-damage inducible transcript 3; DISC1: DISC1 scaffold protein; DNM1L/DRP1: dynamin 1 like; EIF2AK3/PERK: eukaryotic translation initiation factor 2 alpha kinase 3; EIF2S1/eIF2α: eukaryotic translation initiation factor 2 alpha kinase 3; EMD: emerin; EPAS1/HIF-2α: endothelial PAS domain protein 1; ER: endoplasmic reticulum; ERAD: ER-associated degradation; ERN1/IRE1α: endoplasmic reticulum to nucleus signaling 1; FBXO27: F-box protein 27; FKBP8: FKBP prolyl isomerase 8; FTD: frontotemporal dementia; FUNDC1: FUN14 domain containing 1; G3BP1: G3BP stress granule assembly factor 1; GBA: glucocerebrosidase beta; HIF1A/HIF1: hypoxia inducible factor 1 subunit alpha; IMM: inner mitochondrial membrane; LCLAT1/ALCAT1: lysocardiolipin acyltransferase 1; LGALS3/Gal3: galectin 3; LIR: LC3-interacting region; LMNA: lamin A/C; LMNB1: lamin B1; LPS: lipopolysaccharide; MAPK8/JNK: mitogen-activated protein kinase 8; MAMs: mitochondria-associated membranes; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MFN1: mitofusin 1; MOD: multiple organelles dysfunction; MTPAP: mitochondrial poly(A) polymerase; MUL1: mitochondrial E3 ubiquitin protein ligase 1; NBR1: NBR1 autophagy cargo receptor; NLRP3: NLR family pyrin domain containing 3; NUFIP1: nuclear FMR1 interacting protein 1; OMM: outer mitochondrial membrane; OPTN: optineurin; PD: Parkinson disease; PARL: presenilin associated rhomboid like; PEX3: peroxisomal biogenesis factor 3; PGAM5: PGAM family member 5; PHB2: prohibitin 2; PINK1: PTEN induced putative kinase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; RB1CC1/FIP200: RB1 inducible coiled-coil 1; RETREG1/FAM134B: reticulophagy regulator 1; RHOT1/MIRO1: ras homolog family member T1; RIPK3/RIP3: receptor interacting serine/threonine kinase 3; ROS: reactive oxygen species; RTN3: reticulon 3; SEC62: SEC62 homolog, preprotein translocation factor; SESN2: sestrin2; SIAH1: siah E3 ubiquitin protein ligase 1; SNCA: synuclein alpha; SNCAIP: synuclein alpha interacting protein; SQSTM1/p62: sequestosome 1; STING1: stimulator of interferon response cGAMP interactor 1; TAX1BP1: Tax1 binding protein 1; TBK1: TANK binding kinase 1; TFEB: transcription factor EB; TICAM1/TRIF: toll-like receptor adaptor molecule 1; TIMM23: translocase of inner mitochondrial membrane 23; TNKS: tankyrase; TOMM: translocase of the outer mitochondrial membrane; TRIM: tripartite motif containing; UCP2: uncoupling protein 2; ULK1: unc-51 like autophagy activating kinase; UPR: unfolded protein response; USP10: ubiquitin specific peptidase 10; VCP/p97: valosin containing protein; VDAC: voltage dependent anion channels; XIAP: X-linked inhibitor of apoptosis; ZNHIT3: zinc finger HIT-type containing 3.
KEYWORDS: Lysophagy, mitophagy, nucleophagy, pexophagy, reticulophagy, ribophagy
Introduction
Macroautophagy/autophagy is an evolutionarily conserved degradation system in which intracellular contents such as proteins, organelles, and lipids are degraded in a lysosome-dependent manner [1]. This tight regulatory process acts as an essential self-protective mechanism and is perturbed in many pathological conditions after activation by disparate stimuli, including deprivation of nutrients or energy, hypoxia, and even cell differentiation signals [1,2]. Upon stimulation, the autophagic machinery is recruited to the site of the phagophore and participates in the formation of autophagosomes, which subsequently fuse with lysosomes (in mammals) for cargo degradation. These degradation products can be further recycled to the cytoplasm for cellular replenishment [3,4]. The initiation of autophagic machinery is of great importance for cell survival and function because it removes damaged organelles and aberrant protein depositions, as well as supplying extra energy, as reported in cancer, neurodegenerative disorders, and inflammatory and metabolic diseases [5]. Taking sepsis as an example, mitophagy is extensively activated to remove damaged mitochondria and arrest further organ injury under septic exposure, while impaired mitophagy results in overactivation of NLRP3 (NLR family pyrin domain containing 3) inflammasomes and an increase in the mortality of septic animals [6]. Therefore, selective autophagy is essential for cell fate and function underlain by the quality control of organelles.
Selective autophagy is one of the major forms of autophagy, targeting the degradation of specific substrates, especially dysfunctional or superfluous organelles [7]. Organelle-specific autophagy is an essential mechanism for cellular homeostasis by removing damaged or redundant organelles in response to diverse stresses, such as energy deprivation, oxidative stress, and hypoxemia [8]. To date, multiple types of organelle-specific autophagy have been reported with regard to different organelles, including mitophagy, pexophagy, reticulophagy (endoplasmic reticulum), ribophagy, lysophagy, and nucleophagy (Figure 1). In addition to the involvement of common ATG (autophagy related) proteins and similar terminal phases of autophagosome-lysosome fusion, organelle-specific autophagic receptors are also needed to ensure the recognition of specific cargos for degradation (Table 1). These receptors are responsible for binding with autophagosomes via the LIR (LC3-interacting region) after the initiation of the autophagic process, depletion of which leads to aberrant formation of organelles and further results in disruption of cellular function and viability [9]. Thus, the failure of organelle-specific autophagy reportedly contributes to a wide range of diseases, varying from inflammatory disease to tumor invasion, which indicates that quality control of multiple organelles is of great importance for cell fate as well as the development of various diseases (Table 2). It is noteworthy that multiple-organelle dysfunction (MOD) might be a basic yet robust contributor to the disorder and even death of cells under an abnormal response. For instance, hypoxia, a common etiology of various severe conditions, initiates ER stress and functional changes in mitochondria concurrently, accompanied by activation of a variety of organelle-specific autophagies [10]. Additionally, the connection among different organelles is critically involved in cellular homeostasis. It has been confirmed that the ER and mitochondria present close physical contacts under exposure to hypoxia, which further contributes to the recruitment of multiple autophagy-associated proteins, such as ATG5 and ATG14 [11,12]. Therefore, the dysfunction of multiple organelle-specific autophagies might be a latent indicator for the destruction of cells and deterioration of critical functions, and quality control of multiple organelles will be a potential therapeutic target for maintaining cell viability and improving outcomes of patients. In this review, we highlighted the detailed mechanisms concerning cargo recognition in characterized organelle-specific autophagy pathways in mammals. In addition, we summarized the latest findings with regards to the correlation between defective organelle-specific autophagy and the pathogenesis of inflammatory diseases.
Figure 1.
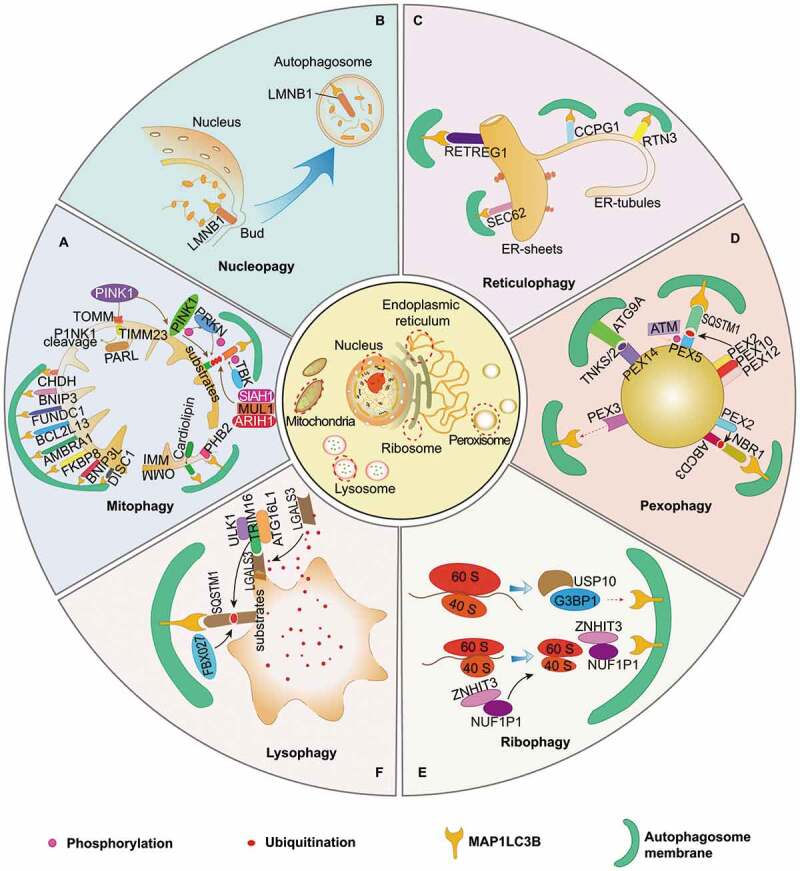
Quality control of multiple organelles by organelle-specific autophagy. (A) Mitophagy is of great importance in maintaining functional homeostasis of mitochondria, which is initiated by PINK1-PRKN-dependent and independent pathways. It requires TOMM and TIMM23 for the import and subsequent cleavage of PINK1 in functional mitochondria, while its damage and dysfunction are followed with PINK1 accumulation on the OMM. VDAC, RHOT1, MFN1/2 proteins act as phosphoubiquitin substrates for PINK1- and PRKN-dependent mitophagy. Outer mitochondrial membrane (OMM) proteins, including BNIP3L, FUNDC1, BNIP3, AMBRA1, BCL2LI3, FKBP8, CHDH, and DISC1, can detect damaged mitochondria and mediate mitophagy by interacting with MAP1LC3B protein directly. In the pathogenesis of mitochondrial dysfunction, IMM proteins, such as PHB2 and cardiolipin, are responsible for the initiation of mitophagy after translocation from IMM to OMM. Additionally, several post-transcriptional modification mechanisms are involved in regulating mitophagy: SIAH1, MUL1 and ARIH1 function as E3 ubiquitin ligases that target OMM proteins, and TBK1 can enhance mitophagy by phosphorylating autophagic receptors. (B) Nucleophagy is programmed for selective removal of nuclear components through the process of autophagy. LMNB1 and chromatin can be degraded via autophagic machinery under senescence exposure. (C) Four receptors reportedly contribute to sequestration of isolated cargos of ER into autophagosomes, including RETREG1, SEC62, CCPG1, and RTN3, which are essential for initiation of reticulophagy after binding with MAP1LC3B. Both RETREG1 and SEC62 are primarily responsible for the turnover of ER sheets, while CCGP1 and RTN3 for the ER-tubules. (D) Pexophagy is deemed great potential in quality control of peroxisome and might be induced by following mechanisms: overexpression of PEX3, activation of ATG9A-TNKS/TNKS2-PEX14 complex, phosphorylation and mono-ubiquitination of PEX5 via ATM and the PEX2-PEX10-PEX12 complex, which recognized by SQSTM1, as well as ABCD3-dependent NBR1-MAP1LC3B pathway in overexpression of PEX2. (E) NUFIP1 serves as the major receptor for ribophagy machinery via specific binding to MAP1LC3B in the assist of ZNHIT3. USP10 and G3BP1 are found to be the mammalian homologs of ribophagy receptors in yeast, suggesting their potential roles in mammal ribophagy. (F) Lysophagy is indispensable for quality control of lysosomes. LGALS3 can sense damaged lysosomes and further recruit TRIM16 to initiate autophagic machinery by ubiquitinating autophagy associated molecules, including ULK1 and ATG16L1. In addition, FBXO27 can serve as a ubiquitinating glycoprotein, which regulate the recruitment of autophagic machinery in SQSTM1-MAP1LC3B pathway
Table 1.
Brief description of receptors for organelle-specific autophagy
| Types | Locations | Receptors | Function | Refs |
|---|---|---|---|---|
| Mitophagy | Outer mitochondrial membrane | VDAC | Mediating degradation of mitochondria in PINK1- and PRKN-dependent signaling pathway | [29,30] |
| RHOT1 | ||||
| MFN1/2 | ||||
| BNIP3L | Mediating ubiquitin-independent degradation of mitochondria by directly interacting with MAP1LC3B | [40,45,49] | ||
| FUNDC1 | [39,44,46] | |||
| BNIP3 | [41] | |||
| AMBRA1 | [50,51] | |||
| BCL2LI3 | [52] | |||
| FKBP8 | [53] | |||
| CHDH | [54] | |||
| DISC1 | [55] | |||
| Inner mitochondrial membrane | PHB2 | [56] | ||
| Cardiolipin | [57] | |||
| Reticulophagy | ER-sheets | RETREG1 | Mediating the degradation of ER-sheets by interacting with LC3 | [79] |
| SEC62 | [86] | |||
| ER-tubules | CCPG1 | Involved in degradation of ER-tubules by binding with MAP1LC3B | [87] | |
| RTN3 | [85] | |||
| Nucleophagy | - | - | - | - |
| Lysophagy | - | - | - | - |
| Pexophagy | Peroxisomal membrane proteins | PEX5 | Mediating the degradation of peroxisomes via different receptors | [113,114] |
| PEX14 | [109–111] | |||
| ABCD3 | [116] | |||
| Ribophagy | 60S ribosomal subunits | NUFIP1 | Involved in degradation of ribosomes by interacting with MAP1LC3B | [129] |
Table 2.
The protective effects of organelle-specific autophagy on various inflammatory diseases
| Diseases | Multiple-organelle dysfunction | Quality control of multiple organelles by organelle-specific autophagy | Effects of organelle-specific autophagy | Refs |
|---|---|---|---|---|
| COPD | Dysfunction of mitochondria and severe disorganization of endoplasmic reticulum, Golgi, and lysosomes of lung fibroblasts | Induction of mitophagy | Improving survival of bronchial epithelial cells by removing damaged mitochondria and reducing production of ROS | [146] |
| Restraining excessive inflammatory response in small airway epithelial cells by limiting inflammasome activation | ||||
| Protecting against cellular senescence under CSE exposure | [147,149] | |||
| Cardiovascular diseases | Dysfunction of mitochondria and uncontrolled ER stress | Induction of mitophagy | Maintaining homeostasis of cardiomyocytes | [150] |
| Improving cardiomyopathy by limiting lethal inflammatory response | [151] | |||
| Restraining cardiomyocyte loss and promoting tissue remodeling and fibrosis | [152] | |||
| Protecting from hypertrophic cardiomyopathy by alleviating oxidative stress and insulin resistance | [153] | |||
| Exerting cytoprotective effects under exposure to atherosclerosis | [154] | |||
| Infectious diseases | Dysfunction of mitochondria and ER stress | Induction of mitophagy | Downregulating pro-inflammatory signals caused by S. aureus pneumonia | [157] |
| Induction of reticulophagy | Limiting the replication of various viruses, including Zika, Dengue, and Ebola | [159,160] | ||
| Maintaining cellular homeostasis under exposure to Gram-positive bacteria | [82] | |||
| Induction of lysophagy | Inhibiting the invasion of intracellular M. tuberculosis | [98,99] | ||
| Induction of nucleophagy | Maintaining integrity of epidermal barrier | [163] | ||
| Sepsis | Damaged mitochondria, persistent ER stress, and aggregation of damaged peroxisomes | Induction of mitophagy | Protecting against cell death by eliminating damaged mitochondria | [166] |
| Suppressing persistent NLRP3 inflammasome activation | [6] | |||
| Induction of pexophagy | Restoring redox balance | [169] | ||
| Neurodegenerative diseases | Dysfunction of mitochondria, impairment of RETREG1-mediated reticulophagy, aberrant response of peroxisomes, leakage of lysosomes, and failure of nucleophagy by mutation of LMNA | Induction of mitophagy | Maintaining viability of neurons by eliminating damaged mitochondria | [170,180] |
| Improving familial and sporadic Parkinson diseases by decreasing accumulation of SNCA in the substantia nigra | [177,178] | |||
| Restricting local inflammatory response | [179] | |||
| Induction of reticulophagy | Essential for the survival of sensory and autonomic neurons | [79] | ||
| Induction of pexophagy | Restoring function of peroxisomes and reducing ROS production | [113] | ||
| Induction of nucleophagy | Delaying cell senescence and alleviating degenerative disorders | [195–197] |
Organelle-specific autophagy
Mitophagy
Mitochondria are crucial for multiple intracellular processes as the main source of ATP and play a major role in initiating programmed cell death [13]. Damaged mitochondria, however, markedly disrupt cellular metabolic homeostasis, which further results in excessive generation of reactive oxygen species (ROS) and cell death. Thus, maintaining homeostatic quantity and quality control of mitochondria are prerequisite for the treatment of various diseases. Several mechanisms are reportedly involved in achieving functional homeostasis with structural integrity of mitochondria [14]. In addition to the mitochondrial ubiquitin-proteasome system, which specifically targets unfolded proteins, mitophagy has a major role in the quality control of mitochondria and is a possible therapeutic target for alleviating cell death and tissue injury [14,15]
Mitophagy refers to a protective effect in which damaged mitochondria are selectively degraded by lysosomes via the autophagic processes in response to diverse stimuli, including hypoxia, ROS, and respiratory chain inhibitors [16–20]. It can be initiated via both PINK1 (PTEN induced putative kinase 1)-PRKN (parkin RBR E3 ubiquitin protein ligase)-dependent and -independent pathways [21]. Of note, PINK1-PRKN-dependent mitophagy is the most thoroughly studied pathway in mammalian cells, as PINK1 is a Ser/Thr kinase, while PRKN is an E3-ubiquitin ligase [22,23]. Functional mitochondria translocate PINK1 into the inner mitochondrial membrane (IMM) with the help of the TOMM (translocase of the outer mitochondrial membrane) and TIMM23 (translocase of inner mitochondrial membrane 23) proteins, followed by the cleavage of PINK1 by PARL (presenilin associated rhomboid like). Nevertheless, damaged mitochondria with impaired membrane potential fail to import PINK1 to the IMM but give rise to the accumulation and stabilization of PINK1 on the outer mitochondrial membrane (OMM) under disparate lethal conditions [24–26]. Then, PINK1 activates several substrates, such as ubiquitin, and mediates the recruitment and activation of PRKN from the cytoplasm to the outer membrane of mitochondria [27,28]. In turn, PRKN amplifies the upstream signal of PINK1 activity, which links phosphoubiquitin chains to various mitochondrial surface-associated proteins, including VDAC (voltage dependent anion channels), RHOT1/MIRO1 (ras homolog family member T1), MFN1 (mitofusin 1), and MFN2 [21,29,30]. Phosphorylated MFN2 might conversely recruit PRKN, indicating a possible feed-forward loop of PRKN recruitment [30]. Autophagic cargo receptor proteins such as SQSTM1/p62 (sequestosome 1), OPTN (optineurin), CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), TAX1BP1 (Tax1 binding protein 1), and NBR1 (NBR1 autophagy cargo receptor) recognize and bind to the phosphoubiquitinated OMM proteins through their ubiquitin-binding domain and subsequently interact with MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) via LIR to start autophagosome formation [29,31]. Intriguingly, these receptors can be the target of activated TBK1 (TANK binding kinase 1), which enhances mitophagy by phosphorylating autophagic receptors, especially OPTN [32,33].
Another PINK1-dependent mechanism has been demonstrated to be responsible for initiating mitophagy, which involves CALCOCO2 and OPTN but does not need PRKN. These two receptors can not only recruit ULK1 (unc-51 like autophagy activating kinase) to ease mitophagy initiation but also directly bind to MAP1LC3B, resulting in the degradation of damaged mitochondria within autolysosomes [31,34]. In addition, CALCOCO2 can interact with MTPAP (mitochondrial poly[A] polymerase) via its N-terminal SKIP carboxyl homology domain to form a complex that contributes to the ubiquitin-independent recognition of damaged mitochondria by autophagy [35]. SNCAIP (synuclein alpha interacting protein [synphilin]) is a synuclein α-interacting protein that forms a complex with PINK1, and it further recruits SIAH1 (siah E3 ubiquitin protein ligase 1) to ubiquitinate mitochondrial proteins in the absence of PRKN [36]. Along with SIAH1, which serves as a ubiquitin E3 ligase, both MUL1 (mitochondrial E3 ubiquitin protein ligase 1) and ARIH1 (ariadne RBR E3 ubiquitin protein ligase 1) localize with depolarized mitochondria, ubiquitinating OMM proteins that subsequently recruit autophagosomes for engulfment and degradation of damaged mitochondria [37,38]. Moreover, ARIH1 mediates mitophagy in a PINK1-dependent manner, while MUL1 acts independently of PINK1 activation [38].
In addition to the PINK1- and PRKN-dependent pathway, the PINK1-PRKN- independent mechanism also accounts for the induction and completion of the mitophagy process, especially in cells without PRKN expression. These OMM-localized receptors, including BNIP3L/NIX (BCL2 interacting protein 3 like), FUNDC1 (FUN14 domain containing 1), and BNIP3 (BCL2 interacting protein 3), own the unique ability of interacting with processed MAP1LC3B at phagophores via a common LIR motif in their cytosolic N-terminal domains [39–42]. BNIP3L is responsible for the selective mitochondrial clearance within the differentiation of reticulocytes, while both BNIP3 and FUNDC1 are transcriptional targets of HIF1A/HIF1 (hypoxia inducible factor 1 subunit alpha) [39,43,44]. In fact, the connection between those receptors and MAP1LC3B is tightly regulated by phosphorylation [45,46]. FUNDC1 is reportedly dephosphorylated by PGAM5 (PGAM family member 5) and phosphorylated by ULK1 correspondingly [46]. In addition, BNIP3 and BNIP3L synergistically function by forming homodimers and act as regulatory targets of PRKN, hinting the crosstalk between PINK1- and PRKN-dependent and -independent pathways [47–49]. Interestingly, AMBRA1 (autophagy and beclin 1 regulator 1) can initiate mitophagy in both PRKN-dependent and PRKN-independent manners [50,51]. A pool of AMBRA1 proteins localize in the OMM and possess LIR domains to enable the MAP1LC3B-LIR connection.
Likewise, both BCL2L13 (BCL2 like 13) and FKBP8 (FKBP prolyl isomerase 8) were recently found to be mitophagy receptors located on the OMM and can mediate mitophagy in PRKN-independent manners [52,53]. In addition, CHDH (choline dehydrogenase) accumulates in the OMM and interacts with SQSTM1 to recruit MAP1LC3B and autophagosomes in response to impaired mitochondria [54]. DISC1 (DISC1 scaffold protein) is a novel mitophagy receptor that contains an LIR domain, which is genetically associated with psychiatric disorders and Alzheimer disease (AD) [55].
Unlike OMM-localized mitophagy receptors, IMM-localized protein PHB2 (prohibitin 2) is a newly discovered receptor for regulating mitophagy that mainly depends on PRKN activation, and it is involved in the process of removing paternal mitochondria in Caenorhabditis elegans embryos [56]. Similarly, cardiolipin, a membrane lipid that specifically appears in the IMM, may function as an MAP1LC3B receptor in mammalian cortical neurons [57]. Translocation of cardiolipin from the IMM to the OMM is induced by lethal components that severely disrupt the integrity of mitochondria [57]. Furthermore, OMM-localized cardiolipin is reported to interact with MAP1LC3B directly [57].
The synergistic effects between mitophagy and other protective pathways for quality control of mitochondria are important for the functional homeostasis of mitochondria, as in mitophagy’s interplay with mitochondria-derived vesicles and mitochondrial dynamics in maintaining cellular homeostasis [58–61]. Circumscribed mitochondrial impairment induces the formation of mitochondria-derived vesicles, which further mediate selective sequestration and degradation of damaged portions of mitochondria, but without influencing the entire organelle [61]. Meanwhile, induction of mitophagy might be associated with downregulation of mitochondrial fusion and simultaneous upregulation of mitochondrial fission [47]. For example, DNM1L/DRP1 (dynamin 1 like), a typical mitochondrial fission-related factor, enables fission of depolarized mitochondria under regulation of PINK1 as well as the mitophagy receptors FUNDC1 and BCL2L13, indicating that initiation of mitophagy is accompanied by mitochondrial fission [48,59,62]. Another mechanism, involving DNM1L-independent mitochondrial clearance, is responsible for mitochondrial fission [63]. AMP-activated protein kinase (AMPK), as an example, is documented as an essential contributor to mitochondrial fission and can induce mitophagy by phosphorylating ULK1 [64]. Additionally, mitochondria-associated membranes (MAMs) have great capacity to modulate mitophagy-related mitochondrial fission [62]. Overall, mitophagy plays a vital protective role in the quality control of mitochondria not only by itself but also through synergistic effects with mitochondrial dynamics via the potential interactions between factors of mitochondrial dynamics and autophagic receptors.
Reticulophagy
The ER is an intracellular network structure that is responsible for the biosynthesis of lipids and proteins and serves as a calcium ion pool. It provides the vital place for modifying and folding both nascent integral membrane and secretory proteins, whose homeostasis is maintained under tight surveillance within the ER lumen. ER-localized sensors can detect the alteration of the ER environment and subsequently initiate several mechanisms for quality control of the ER. The most well-studied mechanism includes the unfolded protein response (UPR) and ER-associated degradation (ERAD) pathways [65]. ERAD refers to the translocation of unfolded proteins to the cytoplasm, which is followed by ubiquitin-proteasome system-dependent degradation [66]. The UPR acts to restore defective proteostasis upon detection of ER stress, which is reportedly a major response to accumulated unfolded proteins in the ER lumen. Indeed, the UPR promotes the capacity of the ER in dealing with unfolded proteins and attenuates protein synthesis by upregulating transcription of redox enzymes, chaperones, foldases, and lipid-synthesizing enzymes, thereby helping the cell to accommodate ER stress [67,68]. ER stress reveals perturbations in ER functions, which further initiate UPR and ER stress associated autophagy to reestablish the ER equilibrium, or else, unresolved ER stress is bound to cause cell death by inducing apoptosis [69,70]. Indeed, the crosstalk between UPR and autophagy or apoptosis determines the cell fate and is governed by multiple signaling pathways. The UPR is quickly initiated the moment accumulated unfolded proteins are detected by three major sensors, including EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3), ERN1/IRE1α (endoplasmic reticulum to nucleus signaling 1), and ATF6 (activating transcription factor 6). These activated pathways are capable of both promoting efficient autophagy and apoptotic pathways under expose to different types of ER stress. For example, activation of EIF2AK3 suppresses general protein translation by EIF2S1/eIF2α (eukaryotic translation initiation factor 2 subunit alpha), which promotes dedicated translational activity of ATF4. ATF4 is identified with potent induction of autophagy by regulating expression of autophagy associated genes, such as Atg5, Atg7, and Atg10 [71]. However, ATF4 turns pro-apoptotic by degrading XIAP (X-linked inhibitor of apoptosis) and stimulating DDIT3/CHOP (DNA-damage inducible transcript 3) pathway in the setting of persistent ER stress [72]. It has been reported that DDIT3 is mainly responsible for ER stress-associated apoptosis by promoting activation of BH3-only protein BCL2L11/BIM (BCL2 like 11) and suppressing activity of anti-apoptotic protein BCL2 (BCL2 apoptosis regulator) [73,74]. Furthermore, other signaling pathways are found with critical roles in regulating ER stress-associated autophagy and apoptosis. Stimulation of ERN1 induces dissociation of a BECN1-BCL2 complex and autophagy via augmenting activation of MAPK8/JNK (mitogen-activated protein kinase 8) which results in cellular apoptosis with persistent activation [75,76]. In addition, induction of autophagy is also capable of mediating degradation of ERN1 in OPTN-dependent way, while it presents with extensive aggregation and further drives deterioration of Crohn disease–like ileitis in atg16l1 deficient mice [77]. Therefore, efficient activation of both the UPR and autophagy machinery is beneficial for orchestrating protective ER stress, but cellular apoptosis is inevitable when confronted with uncontrolled ER stress. Indeed, inhibiting UPR pathways or blocking autophagic machinery is reported to jeopardize cellular viability and induce apoptosis [78]. However, the specific regulatory pathway from UPR to reticulophagy remains unclarified even though activation of the UPR is reportedly capable of inducing formation of ER-associated autophagosomes [79].
Reticulophagy represents a process in which subdomains of ER cisternae filled with faulty proteins and lipids are selectively sequestered into double-membrane phagophores, which eventually fuse with lysosomes for degradation [79,80]. It seems that reticulophagy coexists with one of the downstream signaling pathways of ER stress, and they share synergetic effects to restrain ER volume and counterbalance ER expansion [81–83]. In addition, reticulophagy is regarded as a back-up system when the ERAD pathway is insufficient for the degradation of ER proteins [84,85].
Reticulophagy receptors in mammals play key roles in mediating sequestration of isolated cargos of ER into autophagosomes, and some of them have been uncovered in recent years: RETREG1/FAM134B (reticulophagy regulator 1), SEC62 (SEC62 homolog, preprotein translocation factor), RTN3 (reticulon 3), and CCPG1 (cell cycle progression 1) [86–88]. Indeed, those receptors share common features that are all ER-resident proteins and comprise at least one LIR motif, allowing their binding to autophagosomes. Correspondingly, each reticulophagy pathway is responsible for a unique biological function because disparate cargo receptors mediate the autophagic degradation of distinct subdomains derived from ER at different stages of ER stress [80,86–89].
RETREG1 is the first discovered receptor for mediating reticulophagy, and it is involved in the pathogenesis of hereditary sensory and autonomic neuropathy type II [9]. Downregulation of RETREG1 literally leads to ER swelling, whereas its overexpression results in fragmentation and degradation of the ER membrane [80]. Similar to RETREG1, RTN3 protein contains an RHD that facilitates reticulophagy by promoting membrane curvature and ER scission. The difference is that RETREG1 primarily mediates remodeling and scission of ER sheets, while RTN3 gives rise to the fragmentation of ER tubules [80,86]. Once the stress is resolved, SEC62 presents with programmed initiation to further promote the so-called recovery reticulophagy, which aims to eliminate excessive ER membranes generated during the acute UPR phase [87]. The latest-discovered receptor, CCPG1, harbors two RB1CC1/FIP200 (RB1 inducible coiled-coil 1)-interacting regions, and it was implicated in the correlation between UPR and reticulophagy under basal conditions [88].
Nucleophagy
Healthy nuclear organization and function are the prerequisites for cellular viability, as the nucleus has a vital role in the maintenance and expression of the genome. Thus, selective degradation of portions of the nucleus has been observed in both yeast and mammals, suggesting an evolutionarily conserved mechanism to maintain nuclear integrity [90,91].
Nucleophagy is programmed for selective removal of nuclear components through the process of autophagy. Several types of nucleophagy have been characterized in S. cerevisiae, such as piecemeal microautophagy of the nucleus and late nucleophagy [91,92]. Piecemeal microautophagy of the nucleus selectively degrades nonessential nuclear contents by pinching off portions of the nuclear envelope [93,94]. During this process, two vital proteins, Vac8 and Nvj1, are required to form a junction between the nucleus and the vacuole [93,95]. The late nucleophagy is reportedly activated in yeast under prolonged exposure to nitrogen starvation, which is independent of Nvj1 as well as Vac8 [91].
ATG39, the recently discovered nucleophagy receptor in yeast, is a resident protein in the perinuclear ER and mediates the formation of vesicles containing intranuclear compartments [89]. However, no homologs of Atg39 have been demonstrated in mammals. Researchers have revealed that part of the nucleus can be degraded by autophagy in mammalian cells from nuclear envelopathies and emerinopathies caused by mutations in the genes encoding LMNA (lamin A/C) and EMD (emerin), respectively [90]. Upon oncogene-induced senescence in human cells, degradation of LMNB1 (lamin B1) and chromatin is mediated by the autophagy machinery via direct interaction with MAP1LC3B, suggesting a protective mechanism against tumorigenesis [92]. Moreover, a chronic stall in canonical autophagy under several neurodegenerative conditions might lead to the activation of alternative clearance pathways that require nucleophagy-based LMNB1 degradation [96].
Lysophagy
Lysosomes act as a “digestive apparatus” for cellular metabolism and are filled with diverse hydrolytic enzymes that degrade unwanted intracellular components via the autophagy machinery. Rupture of the lysosome leads to leakage of contents, which is detrimental to the cell and even results in cell death. Therefore, clearance of damaged and dysfunctional lysosomes is important for maintaining cellular homeostasis. Of note, lysosomes play a crucial role in the terminal step of autophagy, while researchers also report that lysosomes themselves can be targeted for autophagy under the impairment state [97]. The mechanism protects cells from the adverse effects of lysosomal damage, termed lysophagy [97,98].
Upon acute insults per se, the ruptured lysosomes are sequestered by autophagosomes after recruitment of autophagic machinery, where the lysosomes recover from low pH and have their degradation capacity restored [97]. The recovery of acidity and proteolysis activity for damaged lysosomes is presumably due to fusion with other intact lysosomes through the autophagy pathway. Concomitantly, β-galactosides are released from damaged lysosomes, which are subsequently sensed by LGALS3/GAL3 (galectin 3), a key lysophagy marker [97]. Furthermore, the atypical TRIM (tripartite motif containing) family E3 ligase TRIM16 is recruited to the binding site of LGALS3, which is synergistically localized at damaged lysosomes and initiates the autophagic response by ubiquitinating autophagy-associated molecules, including ULK1 and ATG16L1. Thus, the TRIM16-LGALS3 complex may serve as a platform for the formation of lysophagy-related autophagosomes, which effectively bring the autophagy machinery in proximity to damaged lysosomes [99,100]. Accordingly, many studies have consistently indicated that damaged lysosomes are degraded in ubiquitin-SQSTM1-MAP1LC3B-dependent patterns, implicating a similarity toward other types of organelle-selective autophagy [97,98]. Like the autophagic receptor SQSTM1, which targets K63-linked ubiquitin chains, it is assumed that ubiquitin-directed AAA-ATPase VCP/p97 (valosin containing protein) triggers the removal of a subset of damaged lysosomes that are modified with K48-linked ubiquitin chains via lysophagy. Interfering with VCP can lead to the accumulation of K48-linked conjugates, and VCP is important for lysophagy under the persistent exposure of damaged lysosomes [101]. In addition, FBXO27 (F-box protein 27), a recently reported glycoprotein-specific F-box protein, is a part of the SCF complex and was identified as a ubiquitinating glycoprotein in regulating the recruitment of autophagic machinery to damaged lysosomes [102]. Lysophagy-independent mechanisms for the quality control of lysosomes have been uncovered, such as the endosomal sorting complex required for transport machinery, which is rapidly recruited to injured endolysosomes for augmenting the repair of injured organelles [103].
Pexophagy
The peroxisome is a small yet highly dynamic organelle that plays a central role in the metabolism of lipids and the reduction of ROS. The homeostasis of peroxisomes is critical for cellular integrity, which can be achieved via a tightly regulated interplay between peroxisome biogenesis and degradation [104,105]. Of note, pexophagy is deemed to have great potential in quality control of peroxisomes as the selective degradation of peroxisomes by autophagy machinery [106].
However, no pexophagy-specific receptor has been found in mammalian cells, and mammal pexophagy primarily relies on ubiquitination of peroxisomal proteins and further interaction with autophagic receptors, including SQSTM1 and NBR1 [107,108]. It has been indicated that overexpression of NBR1 and SQSTM1 induces clustering and degradation of peroxisomes, though SQSTM1 is dispensable for pexophagy, while NBR1 overexpression presents with excessive pexophagy activation. Additionally, SQSTM1-NBR1 binding enhances the efficiency of NBR1-mediated pexophagy [107].
Previously, researchers have shown that overexpression of PEX3 (peroxisomal biogenesis factor 3) leads to ubiquitination and clustering of peroxisomes, and it subsequently induces pexophagy. They further confirmed that NBR1-mediated pexophagy was not blocked in PEX3 mutant cells, indicating that PEX3 plays a dispensable role in pexophagy induction [109]. Under starvation, PEX14 is implicated in pexophagy by directly interacting with MAP1LC3B [110]. Intriguingly, PEX14 does not contain an LIR for MAP1LC3B binding, suggesting that NBR1 or SQSTM1 induces a conformational change of PEX14, which binds to MAP1LC3B through exposed transmembrane domains [111]. In a recent article, it was revealed that PEX14 interacted with the poly(ADP-ribose) polymerase family members TNKS (tankyrase) and TNKS2 to form an ATG9A-TNKS/TNKS2-PEX14 complex that is involved in a noncanonical pexophagy pathway. Both TNKS and TNKS2 act as bridges that connect PEX14 and ATG9A to enhance pexophagy, and the ATG9A-TNKS/TNKS2-PEX14 complex might be a potential pexophagy receptor [112].
PEX5, another peroxisome-localized protein, serves as an import receptor to facilitate translocation of peroxisomal proteins into autophagosomes [113]. The role of PEX5 in pexophagy has been studied by several researchers using the ROS stimulation model [114,115]. Phosphorylation and subsequent mono-ubiquitination of PEX5 result in pexophagy induction in a SQSTM1-dependent manner [114]. Notably, ATM (ATM serine/threonine kinase) is critically involved in this process by phosphorylating PEX5 at S141. Phosphorylation of PEX5 then is mono-ubiquitinated at K209 by the peroxisomal E3 ubiquitin ligase complex that is composed of PEX2, PEX10, and PEX12, which are recognized by SQSTM1 and are subsequently recruited to the autophagic response [114,116]. It has been shown that overexpression of PEX2 induces NBR1-dependent pexophagy, and peroxisomal membrane proteins such as ABCD3 (ATP binding cassette subfamily D member 3) can be the targets of PEX2 during amino acid starvation [117].
The peroxisomal matrix protein CAT (catalase) has been demonstrated to play a role in pexophagy induced by serum starvation. Blockade of CAT can lead to the accumulation of ROS in peroxisomes, in turn resulting in pexophagy [118]. Other stimuli also reportedly trigger pexophagy, such as EPAS1/HIF-2α (endothelial PAS domain protein 1), whereas the mechanism and actual receptors that are required for this process remain elusive [119,120].
Ribophagy
Ribosomes are the main site for protein translation and are responsible for the synthesis of almost half of cellular proteins [121]. The processes of protein translation and biogenesis of ribosomes require high energy consumption, which is under rigorous surveillance. Thus, the degradation of ribosomes and simultaneous downregulation of protein synthesis seem critical for cellular survival, especially upon nutrient starvation [121,122]. The quality control process of ribosomes is attributed to a selective form of autophagy termed ribophagy, in which ribosomes are selectively engulfed into autophagosomes and subsequently degraded by lysosomes [123]. It was reported that the ubiquitin protease Ubp3 and its cofactor Bre5 are involved in this process in yeast, which mainly targets the 60S subunit without influencing 40S subunits [123]. In addition, both Cdc48 and Doa1/Ufd3, which are binding chaperones for E3 ligase Rsp5 and Ubp3-Bre5, respectively, are evident in Ubp3-Bre5-dependent ribophagy [124,125]. Recently, USP10 (ubiquitin specific peptidase 10) and G3BP1 (G3BP stress granule assembly factor 1) were found to be the mammalian homologs of Ubp3 and Bre5, respectively, indicating the evolutionary conservation of ribophagy [126].
Accumulating evidence has indicated that ribosomes are engulfed by autophagosomes in mammalian cells, but their degradation proceeds with specific kinetics, which is different from other intracellular proteins and organelles [127,128]. Mammalian ribophagy has been further confirmed in the development of neurodegeneration, suggesting the critical role of ribophagy in the function and viability of cells [129]. NUFIP1 (nuclear FMR1 interacting protein 1) reportedly serves as the receptor for the selective degradation of ribosomes in mammalian cells via specific binding to MAP1LC3B through the LIR motif within the NUFIP1 sequence [130]. Notably, the interaction between NUFIP1 and its binding partner ZNHIT3 (zinc finger HIT-type containing 3) is indispensable during starvation-induced ribophagy [130]. The exact target of NUFIP1 is roughly located at the 60S ribosomal subunit, while the actual ligand for this process needs further investigation [130].
Crosstalk between multiple organelles underlying selective autophagy
The crosstalk between various organelles is of great importance in maintaining cellular homeostasis, and it is also noteworthy as a physical response to different stimuli. Taking the ER as an example, it is a major store for intracellular calcium, which is responsible for impaired homeostasis of both mitochondria and lysosomes under aberrant release from the ER, hinting that calcium might be an essential signal molecule for the communication between the ER and other organelles [131]. These connections occur with the initiation and completion of organelle-specific autophagy processes. It has been documented that the induction of mitophagy is involved in lysosomal biogenesis by enhancing the expression of TFEB (transcription factor EB), which further increases the expression of lysosomal proteins, such as GBA (glucocerebrosidase beta) and CTSD (cathepsin D) [132]. In turn, upregulated expression of TFEB improves the clearance of damaged mitochondria by inducing mitophagy, indicating that the interplay between multiple organelles is a protective layer in the quality control of multiple organelles [132]. The intracellular contacts between various organelles are necessary for quality control of other organelles by promoting organelle-specific autophagy, especially for ER-mitochondria contact sites, known as MAMs, which is most studied as a physiological response to various stimuli, including hypoxia, nutrient deprivation and ER stress, and it shows a close connection under stress exposure [12]. Multiple autophagy-associated proteins are recruited to this ER-mitochondria contact site for the quality control of both organelles through autophagic processes, including BECN1, ATG14, and ATG5 [13]. Researchers have demonstrated that both PINK1 and BECN1 are re-localized at MAMs following mitophagic stimuli, which in turn promote the formation of MAMs and autophagosome, thereby potentiating mitophagy [133]. Mitophagy receptor BNIP3L localizes to both the ER and mitochondria, which are reported to regulate the crosstalk between the two organelles in regulating apoptosis [134,135]. Likewise, another mitophagy receptor FUNDC1 is demonstrated to accumulate at MAMs, which can initiate mitochondrial fission prior to mitophagy under hypoxia. FUNDC1 promotes mitophagic activity via indirect binding to ER-resident protein CANX (calnexin). As mitophagy proceeds, the association between FUNDC1 and CANX is attenuated, FUNDC1 can also interact with DNM1L and recruits it to the MAMs where mitochondrial fission occurs. As shown in FUNDC1-depleted cells, translocation of DNM1L is remarkably abated with substantially elongated mitochondria during hypoxia. Thus, in response to hypoxia, FUNDC1 functions synergistically with DNM1L, which integrates both mitophagy and mitochondrial fission at MAMs [62,136]. Of note, depletion of MAMs tether-inducing protein PACS2 obviously impairs mitophagy in vascular smooth muscle cells, which ultimately leads to apoptosis and cell death [137]. Additionally, tether-inducing protein MFN2 that serves as a substrate of PINK1, abundantly localizes in MAMs and is demonstrated to play a role in regulating ER stress [138–140]. Therefore, the functional yet structural well-being of MAMs are prerequisites for the induction of mitophagy in diverse stimuli and are crucial for the crosstalk between ER and mitochondria. In addition, these contacts play critical roles in efficient pexophagy as well, as their disruption can result in severe dysfunction of pexophagy [141].
MOD appears to be a noteworthy issue with regards to altered connections among multiple organelles. For instance, mitochondria and ER can simultaneously develop severe dysfunction in lung cells, as evidenced by the overproduction of ROS, hyperpolarization of mitochondrial potential together with protracted ER stress under exposure to perfluoroalkyl acids [142]. Both mitophagy and reticulophagy can be excessively activated in the quality control of these exposed organelles, implying that strategies that combine upregulation of both selective autophagies might be a potential remedy for cell death [142]. In addition, reticulophagy and mitophagy were found to be activated at various intervals during the course of nonalcoholic fatty liver disease, hinting a sequential dysfunction of both organelles [143]. Therefore, MOD consists of two or more organelles suffering from dysfunction at the same time or in sequence under various stimuli, which provides a novel insight into the therapeutic significance of the quality control of multiple organelles.
Organelle-specific autophagy and inflammatory diseases
Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease (COPD) is one of the most prevalent pulmonary diseases worldwide among elderly people, characterized by air flow limitation due to exposure to chronic inflammation, but the precise underlying mechanisms remain elusive. Numerous studies have shown that cigarette smoke exposure (CSE) is the main reason for the pathogenesis and progression of COPD, which contributes to the excessive inflammatory response in airways, alveoli, and microvasculature [144]. Therefore, controlling the inflammatory response is of great importance in improving the outcomes of COPD patients. In addition, a tremendous oxidative burden has been identified as the major contributor to the abnormal response as well as intractable inflammation under CSE exposure, which is caused by dysfunction of mitochondria [145]. The ER, Golgi, and lysosomes of lung fibroblasts also have extensive disorganization in COPD patients, along with unresolved ER stress and massive apoptosis [146]. Thus, MOD seems to be a key contributor to deterioration and a potential marker for early warning of COPD conditions, which provides novel insights for exploring effective therapeutic targets for COPD that underly the quality control of multiple organelles.
Indeed, autophagy is a potential therapeutic target in alleviating COPD mediated by the inflammatory response, and the functional status of autophagy is a prerequisite for the treatment of COPD due to its great capacity in eliminating aberrant proteins and the quality control of multiple organelles. Taking mitophagy as an example, many studies have indicated that CSE leads to the accumulation of damaged mitochondria by impairing mitophagy in a deteriorative state [147–150]. The induction of mitophagy shows noteworthy protective effects on human bronchial epithelial cells by removing damaged mitochondria and reducing the production of ROS [147]. Likely, mitophagy downregulates the excessive inflammatory response and acts as a protective mechanism for COPD patients. Disturbed activation of mitophagy, either by inhibiting the PINK or the PRKN signaling pathway, results in elevated ROS generation and inflammasome activation in small airway epithelial cells from COPD patients [147]. Consistently, CSE is confirmed as a main contributor to augmenting cellular senescence during the progression of COPD by inducing mitochondrial dysfunction and mitophagy impairment, along with accumulation of damaged mitochondrial DNA [148]. It has been demonstrated that PRKN is the rate-limiting factor for PINK1- and PRKN-dependent mitophagy, and its deletion significantly accelerates cellular senescence in response to CSE, together with deteriorated pathological changes in lung tissues [150]. Thus, reversing dysfunctional mitophagy should be handled as a high priority for the treatment of COPD.
Other researchers have reported enhanced expression of PINK1, the necroptosis regulator RIPK3/RIP3 (receptor interacting serine/threonine kinase 3), and the fission regulator DNM1L in lung epithelial cells from COPD patients, indicating extensive activation of both mitophagy and necroptosis under persistent CSE stimulation [149]. The relationship between mitophagy and necroptosis is important for the progression and outcome of COPD patients. In other models, such as inflammatory bowel diseases, however, activation of mitophagy by ATG16L reveals protective effects via inhibiting RIPK3-dependent necroptosis [151]. Meanwhile, TAX1BP1, a selective autophagy receptor, is also demonstrated to play a pro-survival role in sepsis by suppressing necroptosis induced by TICAM1/TRIF (toll-like receptor adaptor molecule 1) [152]. Therefore, the crosstalk between organelle-sepcific autophgy and necroptosis might be divergent under exposure with disparate stimuli and disease models. In addition, the role of organelle-specific autophagy might be different in regulating necroptosis due to its diverse types, which should be taken into consideration for further researches.
Cardiovascular disease
In recent years, it has become increasingly clear that the functional stability of multiple organelles is of great significance for the function and survival of cardiomyocytes. Taking mitochondria as an example, efficient mitophagy is required for maintaining the homeostasis of cardiomyocytes, while its failure contributes to severe cardiomyopathy [153]. Activation of mitophagy can protect cardiomyocytes from lethal inflammation by promoting an anti–inflammatory response, as uncontrolled inflammation is one of the major factors for the pathogenesis of cardiomyopathy. Therefore, mitophagy might be a potential therapeutic target for cardiovascular disease because of its protective effect in restraining cardiomyocyte loss and promoting tissue remodeling and fibrosis [154,155]. In accordance with the above view, it has been revealed that activation of mitophagy is capable of alleviating oxidative stress, insulin resistance, and mitochondrial dysfunction by downregulating LCLAT1/ALCAT1 (lysocardiolipin acyltransferase 1), a lysocardiolipin acyltransferase that is critically involved in the pathogenesis of hypertrophic cardiomyopathy [156]. In contrast, deficiency of mitophagy caused by ablation of PINK1 or PRKN significantly exacerbates the cytotoxic activity of human vascular smooth muscle cells in response to oxidized low-density lipoproteins, while overexpression of PINK1 or PRKN shows cytoprotective effects in the setting of atherosclerosis [157]. Dysfunction of other organelles, e.g. the ER, also accounts for the aberrant performance of cardiomyocytes, as well as being one of the important factors for cardiovascular disease. It was reported that both impaired mitochondrial function and ER stress were associated with the pathogenesis and progression of cardiomyopathy caused by Trypanosoma cruzi infection, accompanied by aberrant lipid metabolism and accumulation of lipid droplets in cardiomyocytes [158]. Administration of 2-aminopurine, an inhibitor of ER stress, showed improvement in cardiac pathology, indicating that maintaining the functional homeostasis of both mitochondria and the ER might be a priority for the viability and response of cardiomyocytes [158]. However, the significance of reticulophagy and other kinds of organelle-specific autophagy in cardiovascular diseases remains largely unclear.
Infection and sepsis
A growing body of evidence suggests that autophagy is involved in eliminating invading pathogens and maintaining the balance of the inflammatory response after induction by diverse pathogens or their associated products, such as viral DNA or lipopolysaccharide (LPS). Deficient autophagy indeed contributes to uncontrolled infection and excessive inflammation, which are both major causes for the development of sepsis [159]. In fact, autophagy is considered a potential therapeutic target for sepsis because of its great capacity to eliminate aberrant intracellular proteins and its role in the quality control of multiple organelles. For instance, mitophagy is extensively activated in the alveolar region of S. aureus-induced pneumonia in a mouse model, and it acts as a pro-survival factor by limiting pro-inflammatory signals caused by S. aureus pneumonia [160]. As ER lumen is always exploited by various bacteria and viruses for their replication and assemble, hinting reticulophagy could serve as an intracellular defensive mechanism by eliminating pathogens within the ER lumen, while its deficiency resulted in the aggregation of bacteria and viruses at the ER [161,162]. RETREG1-mediated reticulophagy reportedly limits the replication of various viruses, including Zika, Dengue and Ebola, which are abated by interfering with the activation of reticulophagy [162,163]. Mechanistically, researchers have found that NS2B3, a virus protease complex, strongly suppresses the reticulophagy pathway by enhancing the cleavage of RETREG1 within the RHD domain [162]. Likewise, RTN3, another recently characterized receptor for reticulophagy, has great ability to reduce virus replication [164,165]. In addition, the STING1 (stimulator of interferon response cGAMP interactor 1)-dependent cell-autonomous response is confirmed to resolve ER stress and maintain cellular viability in Gram-positive bacterial challenge by promoting reticulophagy, indicating a key role of reticulophagy in resolving infection [83].
Multiple studies have demonstrated that other kinds of organelle-specific autophagy, such as lysophagy and nucleophagy, are involved in eliminating invading pathogens and resolving the inflammatory response. In this context, lysophagy presents with obvious activation after stimulation with M. tuberculosis, which limits the invasion of intracellular M. tuberculosis in LGALS3- and TRIM16-dependent ways [99,100]. Induction of nucleophagy specifically removes nuclei in the terminal phase of keratinocyte differentiation, while its failure results in parakeratosis [166]. Therefore, accumulating evidence suggests that dysfunction of multiple organelles might be a major reason for the pathogenesis and progression of various infectious diseases, which further sheds light on the therapeutic potential of the quality control of multiple organelles with regard to cellular viability and functional stability (Figure 2).
Figure 2.
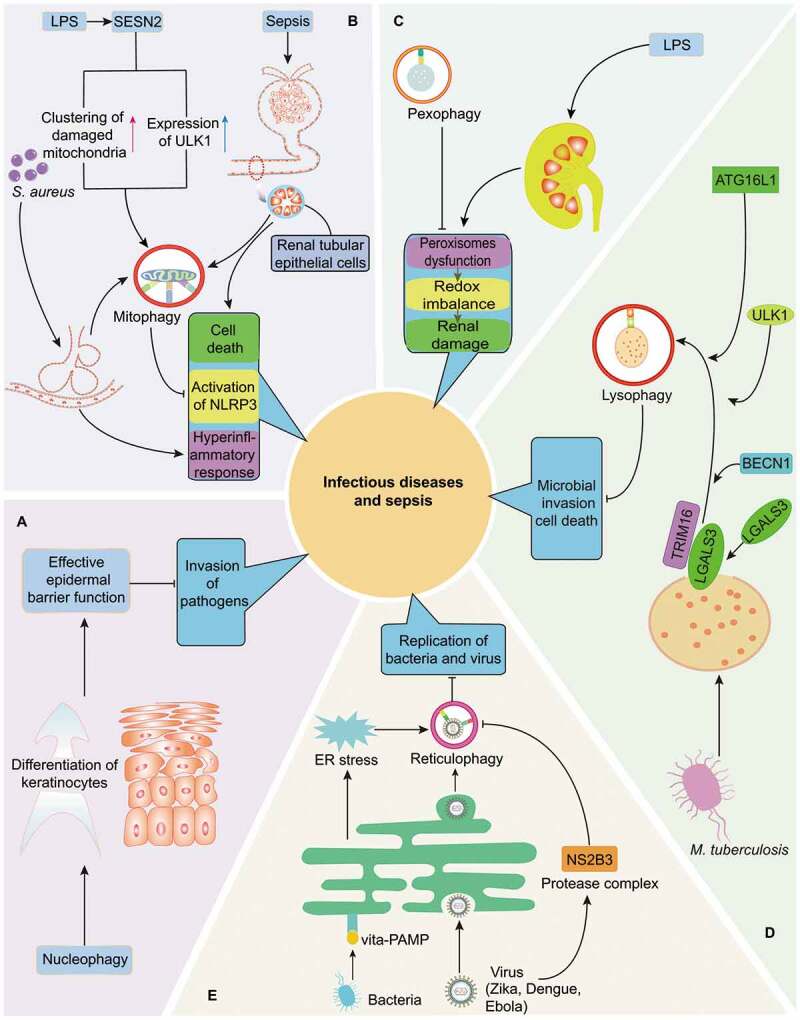
Quality control of multiple organelles through organelle-specific autophagy in infection and sepsis. (A) Nucleophagy is critically involved in preventing the invasion of pathogens by maintaining the integrity of epidermal barrier. (B) As initiated by LPS induced SESN2 upregulation, S. aureus-induced pneumonia as well as sepsis-related renal dysfunction, mitophagy is essential for the balance of inflammatory response and survival of cells in infection or septic challenge by limiting persistent NLRP3 inflammasome activation and eliminating damaged mitochondria respectively. (C) Induction of pexophagy attenuates LPS-mediated renal damage by restoring dysfunction of peroxisomes and redox imbalance. (D) Induction of lysophagy reportedly limits the invasion of intracellular M. tuberculosis in LGALS3- and TRIM16-dependent pathways, accompanied with recruitment of core autophagy proteins, including BECN1, ULK1 and ATG16L1. (E) Reticulophagy can promote the elimination of bacteria and virus via following mechanisms: resolving ER stress under exposure of Gram-positive bacteria and directly limiting replication of Zika, Dengue, and Ebola viruses. However, reticulophagy can be suppressed by NS2B3, a virus protease complex that may cleave RETREG1 within RHD domain
Sepsis is defined as a dysregulated host response to infection, accompanied by multiple-organ dysfunction and high mortality, and is the leading cause of poor outcome in intensive care units [167]. It is well-known that sepsis initiates a complex immune response with the concomitant occurrence of pro-inflammatory and anti–inflammatory responses but presenting with disturbed homeostasis. Most septic patients rapidly switch into a more protracted immunosuppressive phase right after an initial hyperinflammation, which appears to be the main cause for deleterious outcomes [168]. Therefore, restoring the balance of the host inflammatory response and maintaining cell fate are of great significance in the treatment of sepsis. Mitophagy is activated in renal tubular epithelial cells of septic patients, and it protects against cell death by eliminating damaged mitochondria [169]. Mitophagy has been reported to involve multiple protective mechanisms to improve the survival and outcome in septic settings. SESN2 (sestrin 2), known as a stress-inducible protein, is beneficial for septic models by inhibiting the activation of the NLR family, including the NLRP3 inflammasome and the production of pro-inflammatory cytokines [6]. It has been indicated that SESN2 induces mitophagy priming mainly in the following two ways: on one hand, it facilitates perinuclear clustering of damaged mitochondria by mediating SQSTM1 aggregation and its recruitment to Lys63-linked ubiquitin on the mitochondrial surface; on the other hand, it promotes the expression of ULK1 to recruit autophagic machinery for mitophagy activation [6]. Thus, mitophagy is a promising target for improving the outcome of septic cases, as its deficiency via knockout of either pink1 or prkn results in increased susceptibility to polymicrobial sepsis [170].
Other kinds of organelle-specific autophagy, however, are rarely reported to protect cells during septic challenge. The quality control of ER shows great benefits for septic animal post-burn surgery, as persistent ER stress is responsible for the abnormal response of immune cells as well as poor outcomes [171]. However, there is no evidence of the role of reticulophagy in the development of sepsis associated with ER quality control. In addition, pexophagy is reported to be a default response to endotoxic stimulation, as its ablation leads to aggregation of damaged peroxisomes, redox imbalance, and exacerbated renal damage in LPS-induced acute kidney injury model, implicating its involvement in the pathogenesis of septic complications [172].
Neurodegenerative disease
It has been globally accepted that neuroinflammation is a common etiology of the pathogenesis of neurodegenerative diseases [173,174]. Multiple measurements that aim to control chronic inflammation and maintain the viability of neuronal cells are considered effective treatments for patients with neurodegenerative disease. The functional status of multiple organelles appears to be a prerequisite for the response and viability of neuronal cells. The intact function of mitochondria is indispensable for the development and survival of neurons, while disruption of mitophagy may contribute to neurodegeneration by inducing aggregation of dysfunctional mitochondria [175]. In recent reports, impairment of mitophagy was noted in the AD model, along with evident dysfunction of mitochondria, which was positively correlated with the aggregation of pathogenic proteins [176,177]. Likely, failure of mitophagy has a close relationship to the pathogenesis of AD, as a similar status of mitophagy was revealed in neurons from both sporadic AD patients and familial AD patients [178,179]. Furthermore, deficiency of mitophagy caused by mutation of PRKN and PINK1 is associated with the development of familial and sporadic Parkinson disease (PD) due to accumulation of SNCA (synuclein alpha) in the substantia nigra, which is rescued by PRKN overexpression [180,181]. Additionally, PINK1 deficiency was reported to augment the local inflammatory response owing to the accumulation of massive neuroinflammation in brain tissue [182]. Therefore, the protective effects of mitophagy in limiting PD progression depend on PINK1 and PRKN, which clear damaged mitochondria [173,183].
However, whether the reticulophagy process is associated with the pathogenesis of neurodegenerative diseases suffer from a lack of direct evidence. Mutation of RETREG1 has been confirmed to contribute to the pathogenesis of hereditary sensory and autonomic neuropathy type II, but the specific mechanisms need further investigation [9,184]. Researchers have demonstrated that impairment of RETREG1-mediated reticulophagy jeopardizes the survival of sensory and autonomic neurons [80]. In addition to RETREG1, RTN3 has been shown to be involved in the development of neurodegenerative diseases, especially in AD [185,186]. Dysfunction of peroxisomes seems to be related to the progression of various neurodegenerative disorders, including AD and PD [187]. The aberrant response of peroxisomes results in excessive ROS production, which in turn activates the ATM kinase to promote the phosphorylation and subsequent autoubiquitination of PEX5 for the initiation of pexophagy [114]. Thus, pexophagy might be beneficial in neurodegenerative disorders as an essential mechanism for the quality control of peroxisomes.
Of note, dysfunction of lysosomes contributes to deteriorative neurodegeneration, as mutations in genes that are involved in the lysosomal damage response and lysophagy, such as VCP, SQSTM1, OPTN or TBK1, reportedly account for the pathogenesis of neurodegenerative diseases [188–190]. The mutations of VCP, SQSTM1 and TBK1 can be detected in both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), while OPTN mutation is identified only in ALS as well as ALS-FTD, a subtype of ALS, which presents itself with FTD symptoms [189,191–195]. Additionally, endocytosed amyloid-β, a major etiological factor for the development of AD, was confirmed to promote activation of the NLRP3 inflammasome by inducing leakage of lysosomes, suggesting that abnormal homeostasis of lysosomes might be detrimental for the balance of the local inflammatory response [174]. Although there is a lack of direct evidence regarding the protective role of lysophagy in neurodegenerative processes, the quality control of lysosomes is beneficial for the modulation of neuronal cells in neurodegenerative diseases. Moreover, mutation of LMNB1, which undergoes essential binding with MAP1LC3B for nucleophagy, is documented as an etiology for human degenerative disorder, adult-onset demyelinating leukodystrophy [92,196,197]. Activation of LMNB1 markedly delays cell senescence and alleviated degenerative disorders, implying that nucleophagy triggered by LMNB1 might exert a protective impact on neurodegenerative diseases [198–200].
Taken together, the above data demonstrate that dysfunction of multiple organelles is critically involved in the development of various neurodegenerative diseases with compromised viability and uncontrolled local inflammation. Therefore, quality control of multiple organelles could be a novel interventional strategy for neurodegenerative disorders, which could be a much more efficient and accurate approach than current ones, based on the surge of research on organelle-specific autophagy [201].
Conclusions and clinical perspectives
Organelle-specific autophagy indeed shows great benefits in the defense against various diseases through the quality control of specific organelles, thereby maintaining the survival and function of cells. However, there is not always only a single organelle that suffers from structural derangement and functional failure under exposure to the same stimuli. Taking sepsis as an example, dysfunction of mitochondria is noted in dendritic cells at 24 h after construction of a septic animal model, which presents with evident derangement of ER and intractable ER stress [171,202]. Other conditions, such as hypoxia, can cause significant inhibition of glucose oxidation, which result in an imbalance of mitochondrial calcium flux and dysfunction of mitochondria caused by refractory ER stress [203]. Interestingly, the interplay between multiple organelles is also critically involved in the pathogenesis of various diseases. It has been confirmed that calcium flow from the ER to mitochondria is essential for the functional maintenance of mitochondria, which is closely dependent on the status of the ER and UCP2 (uncoupling protein 2), an important mediator of the transportation of calcium [203]. Disrupted delivery of calcium, either by persistent ER stress or suppression of UCP2 expression, leads to marked dysfunction of mitochondria [11,203]. In fact, the expression of UCP2 has been identified as a specific marker for the severity of septic patients and a promising therapeutic target for sepsis thanks to its great capacity in regulating NLRP3 inflammasome [204,205]. Therefore, MOD might be a valuable indicator for the progression and severity of different diseases because of its critical involvement in cell fate and function. Quality control of multiple organelles deserves further investigation in terms of specific molecular mechanisms and associated clinical significance, as it might be a potential therapeutic target for inflammatory diseases.
Simultaneously, the connection between different types of organelle-specific autophagy appears to be a noteworthy issue for cellular function and homeostasis, as well as the development of many inflammatory diseases. Of note, some molecules that are responsible for one kind of organelle-specific autophagy also play pivotal roles in the quality control of another organelle. For example, USP30 has been confirmed as an integral protein of the mitochondrial membrane that mediates mitophagy via the PINK-dependent signaling pathway, while it has recently been reported to be critical in inducing pexophagy in a PINK-independent manner, hinting at the essential connection between different types of organelle-specific autophagy and the synergetic quality control of multiple organelles. It is our belief that a deep understanding of autophagy might promote a surge in research on the quality control of intracellular components and arouse broad concern about intracellular self-protective mechanisms in inflammatory disorders. Organelle-specific autophagy, taking a step further, takes control of multiple organelles in more efficient and accurate ways. Therefore, it is reasonable to seek the quality control of multiple organelles by synchronously modulating the activity of different types of organelle-specific autophagy, in turn guiding the development of selective autophagy-based therapies for human diseases.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81730057, 81842025, 81801935), the National Key Research and Development Program of China (No. 2017YFC1103302), and the Key Project of Military Medical Innovation Program of Chinese PLA (No. 18CXZ026).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Mizushima N, Komatsu M.. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [2].Anding AL, Baehrecke EH.. Autophagy in cell life and cell death. Curr Top Dev Biol. 2015;114:67–91. [DOI] [PubMed] [Google Scholar]
- [3].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kim MJ, Bae SH, Ryu JC, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 2016;12:1272–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jin M, Liu X, Klionsky DJ. SnapShot: selective autophagy. Cell. 2013;152:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017;41:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kurth I, Pamminger T, Hennings JC, et al. Mutations in FAM134B, encoding a newly identified golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009;41:1179–1181. [DOI] [PubMed] [Google Scholar]
- [10].Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and selective autophagy in cancer development and therapy. Front Cell Dev Biol. 2018;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Simmen T, Herrera-Cruz MS. Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr Opin Cell Biol. 2018;53:61–69. [DOI] [PubMed] [Google Scholar]
- [12].Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. [DOI] [PubMed] [Google Scholar]
- [13].Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. [DOI] [PubMed] [Google Scholar]
- [14].Romanello V, Sandri M. mitochondrial quality control and muscle mass maintenance. Front Physiol. 2015;6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Von Stockum S, Nardin A, Schrepfer E, et al. Mitochondrial dynamics and mitophagy in parkinson’s disease: A fly point of view. Neurobiol Dis. 2016;90:58–67. [DOI] [PubMed] [Google Scholar]
- [16].Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang J, Ney PA. NIX induces mitochondrial autophagy in reticulocytes. Autophagy. 2008;4:354–356. [DOI] [PubMed] [Google Scholar]
- [19].Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Frank M, Duvezin-Caubet S, Koob S, et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta. 2012;1823:2297–2310. [DOI] [PubMed] [Google Scholar]
- [21].Ryter SW, Bhatia D, Choi ME. Autophagy: a lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid Redox Signal. 2019;30:138–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Durcan TM, Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015;29:989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu L, Sakakibara K, Chen Q, et al. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24:787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Aerts L, Craessaerts K, De Strooper B, et al. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem. 2015;290:2798–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010;8:e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates parkin e3 ligase activity by phosphorylating serine 65. Open Biol. 2012;2:120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. [DOI] [PubMed] [Google Scholar]
- [28].Kane LA, Lazarou M, Fogel AI, et al. PINK1 phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. [DOI] [PubMed] [Google Scholar]
- [30].Chen Y, Dorn GW 2nd.. PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Richter B, Sliter DA, Herhaus L et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Nation Acad Sci USA. 2016; 113:4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Nation Acad Sci USA. 2016; 113:E3349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in parkinson’s disease. Neuron. 2015;85:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Furuya N, Kakuta S, Sumiyoshi K, et al. NDP52 interacts with mitochondrial RNA poly(A) polymerase to promote mitophagy. EMBO Rep. 2018;19:e46363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Szargel R, Shani V, Abd Elghani F, et al. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet. 2016;25:3476–3490. [DOI] [PubMed] [Google Scholar]
- [37].Yun J, Puri R, Yang H, et al. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife. 2014;3:e01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Villa E, Proics E, Rubio-Patino C, et al. Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep. 2017;20:2846–2859. [DOI] [PubMed] [Google Scholar]
- [39].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. [DOI] [PubMed] [Google Scholar]
- [40].Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hanna RA, Quinsay MN, Orogo AM, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Springer MZ, Macleod KF. In brief: mitophagy: mechanisms and role in human disease. J Pathol. 2016;240:253–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chourasia AH, Boland ML, Macleod KF. Mitophagy and cancer. Cancer Metab. 2015;3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wu W, Tian W, Hu Z, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rogov VV, Suzuki H, Marinkovic M, et al. Phosphorylation of the mitochondrial autophagy receptor nix enhances its interaction with LC3 proteins. Sci Rep. 2017;7:1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen G, Han Z, Feng D, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362–377. [DOI] [PubMed] [Google Scholar]
- [47].Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lee Y, Lee HY, Hanna RA, et al. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;301:H1924–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gao F, Chen D, Si J, et al. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet. 2015;24:2528–2538. [DOI] [PubMed] [Google Scholar]
- [50].Strappazzon F, Nazio F, Corrado M, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015;22:419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Van Humbeeck C, Cornelissen T, Hofkens H, et al. Parkin interacts with ambra1 to induce mitophagy. J Neurosci. 2011;31:10249–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bhujabal Z, Birgisdottir AB, Sjottem E, et al. FKBP8 recruits LC3A to mediate parkin-independent mitophagy. EMBO Rep. 2017;18:947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Park S, Choi SG, Yoo SM, et al. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy. 2014;10:1906–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang ZT, Lu MH, Zhang Y, et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell. 2018;18:e12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wei Y, Chiang WC, Sumpter R Jr., et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gong G, Song M, Csordas G, et al. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pryde KR, Smith HL, Chau KY, et al. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol. 2016;213:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].McLelland GL, Lee SA, McBride HM, et al. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016;214:275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Soubannier V, McLelland GL, Zunino R, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–141. [DOI] [PubMed] [Google Scholar]
- [62].Wu W, Li W, Chen H, et al. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy. 2016;12:1675–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Burman JL, Pickles S, Wang C, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol. 2017;216:3231–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Toyama EQ, Herzig S, Courchet J, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ruggiano A, Foresti O, Carvalho P. Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol. 2014;204:869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Smith M, Wilkinson S. ER homeostasis and autophagy. Essays Biochem. 2017;61:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lee JS, Mendez R, Heng HH, et al. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am J Transl Res. 2012;4:102–113. [PMC free article] [PubMed] [Google Scholar]
- [69].Urra H, Dufey E, Lisbona F, et al. When ER stress reaches a dead end. Biochim Biophys Acta. 2013;1833:3507–3517. [DOI] [PubMed] [Google Scholar]
- [70].Cai Y, Arikkath J, Yang L, et al. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy. 2016;12:225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].B’Chir W, Maurin AC, Carraro V, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hiramatsu N, Messah C, Han J, et al. Translational and posttranslational regulation of XIAP by eIF2alpha and ATF4 promotes ER stress-induced cell death during the unfolded protein response. Mol Biol Cell. 2014;25:1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. [DOI] [PubMed] [Google Scholar]
- [74].McCullough KD, Martindale JL, Klotz LO, et al. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wu HM, Wang J, Zhang B, et al. CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in staphylococcus aureus-stimulated macrophage. Life Sci. 2016;161:51–59. [DOI] [PubMed] [Google Scholar]
- [76].Oh SH, Lim SC. Endoplasmic reticulum stress-mediated autophagy/apoptosis induced by capsaicin (8-methyl-N-vanillyl-6-nonenamide) and dihydrocapsaicin is regulated by the extent of c-jun NH2-terminal kinase/extracellular signal-regulated kinase activation in WI38 lung epithelial fibroblast cells. J Pharmacol Exp Ther. 2009;329:112–122. [DOI] [PubMed] [Google Scholar]
- [77].Tschurtschenthaler M, Adolph TE, Ashcroft JW, et al. Defective ATG16L1-mediated removal of IRE1alpha drives Crohn’s disease-like ileitis. J Exp Med. 2017;214:401–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Song S, Tan J, Miao Y, et al. Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress. J Cell Physiol. 2017;232:2977–2984. [DOI] [PubMed] [Google Scholar]
- [79].Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. 2007;3:285–287. [DOI] [PubMed] [Google Scholar]
- [80].Khaminets A, Heinrich T, Mari M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. [DOI] [PubMed] [Google Scholar]
- [81].Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Schuck S, Gallagher CM, Walter P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci. 2014;127:4078–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Moretti J, Roy S, Bozec D, et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. 2017;171:809–23.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Houck SA, Ren HY, Madden VJ, et al. Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol Cell. 2014;54:166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hidvegi T, Ewing M, Hale P, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. [DOI] [PubMed] [Google Scholar]
- [86].Grumati P, Morozzi G, Holper S, et al. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife. 2017;6:e25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Fumagalli F, Noack J, Bergmann TJ, et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol. 2016;18:1173–1184. [DOI] [PubMed] [Google Scholar]
- [88].Smith MD, Harley ME, Kemp AJ, et al. ccpg1 is a non-canonical autophagy cargo receptor essential for er-phagy and pancreatic ER proteostasis. Dev Cell. 2018;44:217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mochida K, Oikawa Y, Kimura Y, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522:359–362. [DOI] [PubMed] [Google Scholar]
- [90].Park YE, Hayashi YK, Bonne G, et al. Autophagic degradation of nuclear components in mammalian cells. Autophagy. 2009;5:795–804. [DOI] [PubMed] [Google Scholar]
- [91].Mijaljica D, Prescott M, Devenish RJ. A late form of nucleophagy in saccharomyces cerevisiae. PloS One. 2012;7:e40013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Dou Z, Xu C, Donahue G, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kvam E, Goldfarb DS. Nucleus-vacuole junctions and piecemeal microautophagy of the nucleus in scerevisiae. Autophagy. 2007;3:85–92. [DOI] [PubMed] [Google Scholar]
- [94].Kraft C, Reggiori F, Peter M. Selective types of autophagy in yeast. Biochim Biophys Acta. 2009;1793:1404–1412. [DOI] [PubMed] [Google Scholar]
- [95].Krick R, Muehe Y, Prick T, et al. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell. 2008;19:4492–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Baron O, Boudi A, Dias C, et al. Stall in canonical autophagy-lysosome pathways prompts nucleophagy-based nuclear breakdown in neurodegeneration. Curr Biol. 2017;27:3626–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32:2336–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hung YH, Chen LM, Yang JY, et al. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat Commun. 2013;4:2111. [DOI] [PubMed] [Google Scholar]
- [99].Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and trim16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Fraiberg M, Elazar Z. A TRIM16-galactin3 complex mediates autophagy of damaged endomembranes. Dev Cell. 2016;39:1–2. [DOI] [PubMed] [Google Scholar]
- [101].Papadopoulos C, Kirchner P, Bug M, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017;36:135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Yoshida Y, Yasuda S, Fujita T et al. Ubiquitination of exposed glycoproteins by SCF(FBXO27) directs damaged lysosomes for autophagy. Proc Nat Acad Sci USA. 2017; 114:8574–8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Skowyra ML, Schlesinger PH, Naismith TV, et al. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360:eaar5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Tripathi DN, Walker CL. The peroxisome as a cell signaling organelle. Curr Opin Cell Biol. 2016;39:109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Eberhart T, Kovacs WJ. Pexophagy in yeast and mammals: an update on mysteries. Histochem Cell Biol. 2018;150:473–488. [DOI] [PubMed] [Google Scholar]
- [106].Zientara-Rytter K, Subramani S. Autophagic degradation of peroxisomes in mammals. Biochem Soc Trans. 2016;44:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Deosaran E, Larsen KB, Hua R, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2013;126:939–952. [DOI] [PubMed] [Google Scholar]
- [108].Kim PK, Hailey DW, Mullen RT et al. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Nat Acad Sci USA. 2008; 105:20567–20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yamashita S, Abe K, Tatemichi Y, et al. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy. 2014;10:1549–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hara-Kuge S, Fujiki Y. The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp Cell Res. 2008;314:3531–3541. [DOI] [PubMed] [Google Scholar]
- [111].Jiang L, Hara-Kuge S, Yamashita S, et al. Peroxin Pex14p is the key component for coordinated autophagic degradation of mammalian peroxisomes by direct binding to LC3-II. Genes Cells. 2015;20:36–49. [DOI] [PubMed] [Google Scholar]
- [112].Li X, Han H, Zhou MT, et al. Proteomic analysis of the human tankyrase protein interaction network reveals its role in pexophagy. Cell Rep. 2017;20:737–749. [DOI] [PubMed] [Google Scholar]
- [113].Ma C, Agrawal G, Subramani S. Peroxisome assembly: matrix and membrane protein biogenesis. J Cell Biol. 2011;193:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Zhang J, Tripathi DN, Jing J, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015;17:1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Nordgren M, Francisco T, Lismont C, et al. Export-deficient monoubiquitinated PEX5 triggers peroxisome removal in SV40 large T antigen-transformed mouse embryonic fibroblasts. Autophagy. 2015;11:1326–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].El Magraoui F, Baumer BE, Platta HW, et al. The RING-type ubiquitin ligases Pex2p, Pex10p and Pex12p form a heteromeric complex that displays enhanced activity in an ubiquitin conjugating enzyme-selective manner. FEBS J. 2012;279:2060–2070. [DOI] [PubMed] [Google Scholar]
- [117].Sargent G, van Zutphen T, Shatseva T, et al. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J Cell Biol. 2016;214:677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lee JN, Dutta RK, Maharjan Y, et al. Catalase inhibition induces pexophagy through ROS accumulation. Biochem Biophys Res Commun. 2018;501:696–702. [DOI] [PubMed] [Google Scholar]
- [119].Schonenberger MJ, Kovacs WJ. Hypoxia signaling pathways: modulators of oxygen-related organelles. Front Cell Dev Biol. 2015;3:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Schonenberger MJ, Krek W, Kovacs WJ. EPAS1/HIF-2alpha is a driver of mammalian pexophagy. Autophagy. 2015;11:967–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. [DOI] [PubMed] [Google Scholar]
- [122].Lafontaine DL. A ‘garbage can’ for ribosomes: how eukaryotes degrade their ribosomes. Trends Biochem Sci. 2010;35:267–277. [DOI] [PubMed] [Google Scholar]
- [123].Kraft C, Deplazes A, Sohrmann M, et al. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–610. [DOI] [PubMed] [Google Scholar]
- [124].Ossareh-Nazari B, Bonizec M, Cohen M, et al. Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep. 2010;11:548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kraft C, Peter M. Is the Rsp5 ubiquitin ligase involved in the regulation of ribophagy? Autophagy. 2008;4:838–840. [DOI] [PubMed] [Google Scholar]
- [126].Soncini C, Berdo I, Draetta G. Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene. 2001;20:3869–3879. [DOI] [PubMed] [Google Scholar]
- [127].Ashford TP, Porter KR. Cytoplasmic components in hepatic cell lysosomes. J Cell Biol. 1962;12:198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kristensen AR, Schandorff S, Hoyer-Hansen M, et al. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics. 2008;7:2419–2428. [DOI] [PubMed] [Google Scholar]
- [129].Baltanas FC, Casafont I, Weruaga E, et al. Nucleolar disruption and cajal body disassembly are nuclear hallmarks of DNA damage-induced neurodegeneration in purkinje cells. Brain Pathol. 2011;21:374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Wyant GA, Abu-Remaileh M, Frenkel EM, et al. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science. 2018;360:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Garrity AG, Wang W, Collier CM, et al. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife. 2016;5:e15887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ivankovic D, Chau KY, Schapira AH, et al. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem. 2016;136:388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Gelmetti V, De Rosa P, Torosantucci L, et al. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy. 2017;13:654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Tagaya M, Arasaki K. Regulation of mitochondrial dynamics and autophagy by the mitochondria-associated membrane. Adv Exp Med Biol. 2017;997:33–47. [DOI] [PubMed] [Google Scholar]
- [135].Diwan A, Matkovich SJ, Yuan Q, et al. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest. 2009;119:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Wu W, Lin C, Wu K, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016;35:1368–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Moulis M, Grousset E, Faccini J, et al. The multifunctional sorting protein pacs-2 controls mitophagosome formation in human vascular smooth muscle cells through mitochondria-ER contact sites. Cells. 2019;8:E638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. [DOI] [PubMed] [Google Scholar]
- [139].Dorn GW 2nd, Song M, Walsh K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J Mol Cell Cardiol. 2015;78:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Zorzano A, Hernandez-Alvarez MI, Sebastian D, et al. Mitofusin 2 as a driver that controls energy metabolism and insulin signaling. Antioxid Redox Signal. 2015;22:1020–1031. [DOI] [PubMed] [Google Scholar]
- [141].Liu X, Wen X, Klionsky DJ. ER-mitochondria contacts are required for pexophagy in Saccharomyces cerevisiae. Contact (Thousand Oaks). 2018;2:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Xin Y, Wan B, Yang Y, et al. Perfluoroalkyl acid exposure induces protective mitochondrial and endoplasmic reticulum autophagy in lung cells. Arch Toxicol. 2018;92:3131–3147. [DOI] [PubMed] [Google Scholar]
- [143].Pang L, Liu K, Liu D, et al. Differential effects of reticulophagy and mitophagy on nonalcoholic fatty liver disease. Cell Death Dis. 2018;9:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. gold executive summary. Am J Respir Crit Care Med. 2017;195:557–582. [DOI] [PubMed] [Google Scholar]
- [145].Jiang Y, Wang X, Hu D. Mitochondrial alterations during oxidative stress in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2017;12:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Weidner J, Jarenback L, Aberg I, et al. Endoplasmic reticulum, golgi, and lysosomes are disorganized in lung fibroblasts from chronic obstructive pulmonary disease patients. Physiol Rep. 2018;6:e13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Ito S, Araya J, Kurita Y, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11:547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Ahmad T, Sundar IK, Lerner CA, et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J. 2015;29:2912–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Mizumura K, Cloonan SM, Nakahira K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124:3987–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Araya J, Tsubouchi K, Sato N, et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy. 2019;15:510–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. 2017;214:3687–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Lim J, Park H, Heisler J, et al. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. eLife. 2019;8:e44452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Tong M, Saito T, Zhai P, et al. mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 2019;124:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. 2017;113:389–398. [DOI] [PubMed] [Google Scholar]
- [155].Travers JG, Kamal FA, Robbins J, et al. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118:1021–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Liu X, Ye B, Miller S, et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol Cell Biol. 2012;32:4493–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Swiader A, Nahapetyan H, Faccini J, et al. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget. 2016;7:28821–28835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Ayyappan JP, Lizardo K, Wang S, et al. Inhibition of ER stress by 2-aminopurine treatment modulates cardiomyopathy in a murine chronic chagas disease model. Biomol Ther. 2019;27:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016;166:288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Suliman HB, Kraft B, Bartz R, et al. Mitochondrial quality control in alveolar epithelial cells damaged by S. aureus pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. 2017;313:L699–l709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Junjhon J, Pennington JG, Edwards TJ, et al. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J Virol. 2014;88:4687–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Lennemann NJ, Coyne CB. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy. 2017;13:322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Chiramel AI, Dougherty JD, Nair V, et al. FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of ebola virus strains makona and mayinga. J Infect Dis. 2016;214:S319–s25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Aktepe TE, Liebscher S, Prier JE, et al. The host protein reticulon 3.1A is utilized by flaviviruses to facilitate membrane remodelling. Cell Rep. 2017;21:1639–1654. [DOI] [PubMed] [Google Scholar]
- [165].Wu MJ, Ke PY, Hsu JT, et al. Reticulon 3 interacts with NS4B of the hepatitis C virus and negatively regulates viral replication by disrupting NS4B self-interaction. Cell Microbiol. 2014;16:1603–1618. [DOI] [PubMed] [Google Scholar]
- [166].Akinduro O, Sully K, Patel A, et al. Constitutive autophagy and nucleophagy during epidermal differentiation. J Invest Dermatol. 2016;136:1460–1470. [DOI] [PubMed] [Google Scholar]
- [167].Cecconi M, Evans L, Levy M, et al. Sepsis and septic shock. Lancet. 2018;392:75–87. [DOI] [PubMed] [Google Scholar]
- [168].Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Takasu O, Gaut JP, Watanabe E, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Kang R, Zeng L, Xie Y, et al. A novel PINK1- and PARK2-dependent protective neuroimmune pathway in lethal sepsis. Autophagy. 2016;12:2374–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Zhu XM, Yao FH, Yao YM, et al. Endoplasmic reticulum stress and its regulator XBP-1 contributes to dendritic cell maturation and activation induced by high mobility group box-1 protein. Int J Biochem Cell Biol. 2012;44:1097–1105. [DOI] [PubMed] [Google Scholar]
- [172].Vasko R, Ratliff BB, Bohr S, et al. Endothelial peroxisomal dysfunction and impaired pexophagy promotes oxidative damage in lipopolysaccharide-induced acute kidney injury. Antioxid Redox Signal. 2013;19:211–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Sliter DA, Martinez J, Hao L, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Cheng A, Yang Y, Zhou Y, et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016;23:128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Kondadi AK, Wang S, Montagner S, et al. Loss of the m-AAA protease subunit AFG(3)L(2) causes mitochondrial transport defects and tau hyperphosphorylation. EMBO J. 2014;33:1011–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Hou Y, Ghosh P, Wan R, et al. Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1-42 on neural progenitor cell proliferation. Neurobiol Aging. 2014;35:975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Martin-Maestro P, Gargini R, A Sproul A, et al. Mitophagy failure in fibroblasts and ipsc-derived neurons of alzheimer’s disease-associated presenilin 1 mutation. Front Mol Neurosci. 2017;10:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Martin-Maestro P, Gargini R, Perry G, et al. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum Mol Genet. 2016;25:792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Lonskaya I, Hebron ML, Algarzae NK, et al. Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson’s disease. Neuroscience. 2013;232:90–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Winslow AR, Chen CW, Corrochano S, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190:1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Torres-Odio S, Key J, Hoepken -H-H, et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation. 2017;14:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Ashrafi G, Schlehe JS, LaVoie MJ, et al. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206:655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Murphy SM, Davidson GL, Brandner S, et al. Mutation in FAM134B causing severe hereditary sensory neuropathy. J Neurol Neurosurg Psychiatry. 2012;83:119–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Shi Q, Prior M, He W, et al. Reduced amyloid deposition in mice overexpressing RTN3 is adversely affected by preformed dystrophic neurites. J Neurosci. 2009;29:9163–9173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Small SA, Gandy S. Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Cipolla CM, Lodhi IJ. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol Metab. 2017;28:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Teyssou E, Takeda T, Lebon V, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125:511–522. [DOI] [PubMed] [Google Scholar]
- [189].Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. [DOI] [PubMed] [Google Scholar]
- [190].Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636. [DOI] [PubMed] [Google Scholar]
- [191].Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Fecto F, Siddique T. UBQLN2/P62 cellular recycling pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Muscle Nerve. 2012;45:157–162. [DOI] [PubMed] [Google Scholar]
- [193].Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Bucelli RC, Arhzaouy K, Pestronk A, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. 2015;85:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].van Beek N, Klionsky DJ, Reggiori F. Genetic aberrations in macroautophagy genes leading to diseases. Biochim Biophys Acta Mol Cell Res. 2018;1865:803–816. [DOI] [PubMed] [Google Scholar]
- [196].Salama R, Sadaie M, Hoare M, et al. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Dobrzynska A, Gonzalo S, Shanahan C, et al. The nuclear lamina in health and disease. Nucleus. 2016;7:233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Shimi T, Butin-Israeli V, Adam SA, et al. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011;25:2579–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Shah PP, Donahue G, Otte GL, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Lukasova E, Kovar Ik A, Bac Ikova A, et al. Loss of lamin B receptor is necessary to induce cellular senescence. Biochem J. 2017;474:281–300. [DOI] [PubMed] [Google Scholar]
- [201].Wu MY, Song JX, Wang SF, et al. Selective autophagy: the new player in the fight against neurodegenerative diseases? Brain Res Bull. 2018;137:79–90. [DOI] [PubMed] [Google Scholar]
- [202].Friedenberg SG, Strange HR, Guillaumin J, et al. Effect of disrupted mitochondria as a source of damage-associated molecular patterns on the production of tumor necrosis factor alpha by splenocytes from dogs. Am J Vet Res. 2016;77:604–612. [DOI] [PubMed] [Google Scholar]
- [203].Dromparis P, Paulin R, Sutendra G, et al. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res. 2013;113:126–136. [DOI] [PubMed] [Google Scholar]
- [204].Jiang ZM, Yang QH, Zhu CQ. UCP2 in early diagnosis and prognosis of sepsis. Eur Rev Med Pharmacol Sci. 2017;21:549–553. [PubMed] [Google Scholar]
- [205].Moon JS, Lee S, Park MA, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]