

ABSTRACT
Lysosomes are digestive organelles in cells containing many hydrolases, and also serve as a signaling hub to integrate intracellular and extracellular inputs; therefore, the integrity of lysosomes is critical for cellular homeostasis. Many agents and conditions can damage lysosomal membranes, which lead to leakage of lysosomal acidic contents into the cytosol thus becoming harmful for cells. Accordingly, cells have developed several defense systems to cope with damaged lysosomes, but underlying mechanisms of each system and their cross-talks are unclear. In our recent study, we found that a master transcription factor regulating autophagy and lysosomal biogenesis, TFEB (transcription factor EB) is activated during lysosomal damage, and this activation depends on an autophagy-independent function of lipidated LC3, which localizes on lysosomes. We further showed that this regulatory mechanism is essential to prevent the progression of the crystal nephropathy that accompanies lysosomal damage.
KEYWORDS: Autophagy, LC3, lysosome, TFEB, TRPML1
Numerous endogenous and exogenous factors, including silica, crystals, pathogens and drugs induce lysosomal membrane permeabilization and/or lysosomal rupture. Because leaking lysosomes are harmful, cells have developed several defense systems, collectively called endolysosomal damage responses, which include sequestration of damaged lysosomes by a specific type of selective macroautophagy (hereafter autophagy), termed lysophagy, endosomal sorting complexes required for transport (ESCRT)-mediated lysosomal repair, and lysosomal biogenesis induced by TFEB. Autophagy is an evolutionarily conserved cytoplasmic degradation system in which newly generated phagophores, precursors to autophagosomes, randomly or selectively sequester cytoplasmic materials, followed by subsequent fusion of autophagosomes with lysosomes for degradation. Lysophagy selectively sequesters lysosomes with relatively large ruptures, whereas ESCRT repairs those with small areas of damage. TFEB is a master transcription factor regulating autophagy and lysosomal function. TFEB activity is regulated by phosphorylation by MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1). In nutrient-rich conditions, TFEB is phosphorylated on lysosomes by MTORC1, and the phosphorylated TFEB remains in the cytoplasm. Upon starvation, TFEB is dephosphorylated and translocates from the cytosol to the nucleus, where it upregulates genes involved autophagy and lysosomal biogenesis. However, roles of TEFB and its regulation during the lysosome damage response are poorly understood. In addition, although it is plausible that each pathway in the endolysosomal damage response must be coordinated to maintain lysosomal homeostasis, the exact cross-talk among these pathways remains elusive.
In our recent study [1], we found that TFEB is activated by lysosomal damage induced by a lysosomotropic agent, L-leucyl-L-leucine methyl ester (LLOMe), in mammalian cells. Importantly, knockdown of TFEB function by siRNA impairs the clearance of damaged lysosomes labeled by a damaged lysosome marker, LGALS3 (galectin 3), indicating TFEB activation is essential for lysosomal homeostasis after the lysosomal damage. We found that TFEB activation occurs in WT and some autophagy-deficient cells (e.g., rb1cc1/fip200, or atg9 KO). Surprisingly we found that TFEB activation is impaired in a subset of autophagy-deficient cells lacking ATG3, ATG5, ATG7 and ATG16L1. These genes belong to the ATG conjugation systems mediating the lipidation of LC3/Atg8, and lipidated LC3 localizes on autophagosomes upon induction of autophagy. Cells lacking LC3/Atg8 homologs (LC3A, LC3B, LC3C, GABARAP, GABARAPL1 and GABARAPL2) also show impaired TFEB activation during lysosomal damage, suggesting that the lipidation of LC3 rather than function of autophagy is critical for TFEB activation. Interestingly, starvation-induced TFEB activation does not require the function of the ATG conjugation systems and the presence of LC3/Atg8, indicating this lipidated LC3-mediated TFEB activation occurs specifically during lysosomal damage.
Previously, lysosomal calcium efflux through the lysosomal calcium channel MCOLN1/TRPML1 has been shown to activate TFEB through the dephosphorylation of TFEB by PPP3/calcineurin. Thus, we wondered if the calcium leaked from damaged lysosomes triggers lipidated LC3-dependent TFEB activation. Indeed, we found that an MCOLN1 agonist, ML-SA1 treatment, activates TFEB in WT cells, but not in cells lacking the ATG conjugation systems or all LC3/Atg8 homologs, suggesting that calcium efflux from lysosomes is sufficient to trigger TFEB activation in an LC3 lipidation-dependent manner. However, knockdown of PPP3CB and/or PPP3CA, components of PPP3/calcineurin do not largely abolish TFEB activation induced by lysosomal damage, indicating that other phosphatases are involved. In contrast, phosphorylation of the Ser211 residue on TFEB, which is directly phosphorylated by MTORC1, is decreased in WT cells upon lysosomal damage but not in ATG conjugation system-deficient cells, suggesting LC3 lipidation is essential to prevent MTORC1-dependent phosphorylation of TFEB. Interestingly, phosphorylation of another MTORC1 substrate, RPS6KB/S6K is not altered upon lysosomal damage or ML-SA1 treatment, implying that MTORC1 substrate-specific recruitment on lysosomes might be affected by lipidation of LC3.
We sought to determine why ATG conjugation system-deficient mutants show impaired TFEB activation by ML-SA1. Remarkably, calcium imaging using Fura-2 reveals ATG conjugation-deficient cells show reduced intracellular calcium increase upon ML-SA1 treatment, indicating that MCOLN1 capacity is impaired in the absence of LC3 lipidation. To address how MCOLN1 capacity is affected in the absence of LC3 lipidation, we examined the localization of LC3 during lysosomal damage. Intriguingly, lipidated LC3 localizes on lysosomes in addition to autophagosomes upon lysosomal damage. Lysosomal LC3 is also observed in autophagy-deficient cells (e.g., rb1cc1, or atg9 KO), but not ATG conjugation-deficient cells, again confirming it is an autophagy-independent process. Similar LC3 recruitment on lysosomes is observed with ML-SA1 treatment without obvious lysosomal damage marked by LGALS3, suggesting that lysosomal calcium efflux suffices to induce LC3 lipidation on lysosomes. Moreover, we found that all LC3 paralogs interact with MCOLN1, and presumably the interaction is essential for efficient calcium efflux from lysosomes. LC3- and MCOLN1-mediated TFEB activation might be especially important after ESCRT roughly repairs damaged lysosomes. Indeed, TFEB is activated even in ATG conjugation system-deficient cells when ESCRT function is impaired.
Crystal nephropathy is one example of lysosomal damage-induced tissue injury. We thus addressed if LC3 lipidation-dependent TFEB activation has critical roles in this pathological condition. Oxalate nephropathy is a representative of crystal nephropathy caused by calcium oxalate (CaOx) crystals. We confirmed that administration of CaOx leads to the deposition of CaOx crystals and induces LGALS3-positive lysosomal damage and TFEB nuclear localization in proximal tubular epithelial cells of mouse kidney. Similar to our in vitro experiments, this TFEB activation is diminished in proximal tubule-specific atg5 knockout mouse kidney, suggesting the presence of LC3 lipidation-dependent TFEB activation. We further found kidney injury induced by CaOx crystals is exacerbated in proximal tubule-specific tfeb knockout mice compared to control. Moreover, human crystal nephropathy specimens show clear lysosomal damage and reduced TFEB expression compared to controls, highlighting the correlation between TFEB expression and progression of crystal nephropathy.
Collectively, we discovered that during the lysosomal damage response LC3 is recruited on lysosomes in addition to phagophores, and the lysosomal LC3 has an essential role to activate TFEB through an autophagy-independent function. Upon lysosomal damage, low level calcium efflux from lysosomes induces LC3 lipidation and its localization on lysosomes (Figure 1). Lysosomal lipidated LC3 then interacts with MCOLN1, and we assume the interaction is critical for larger amounts of calcium efflux leading to TFEB activation, partly through preventing MTORC1-mediated TFEB phosphorylation (Figure 1). Although actual targets remain elusive, TFEB might contribute to clearance of damaged lysosomes through activating lysophagy and lysosomal biogenesis. Why LC3 lipidation is required for TFEB activation during lysosomal damage but not during starvation is of particular interest. How LC3 is recruited on lysosomes and affects MCOLN1 also need to be clarified in future studies.
Figure 1.
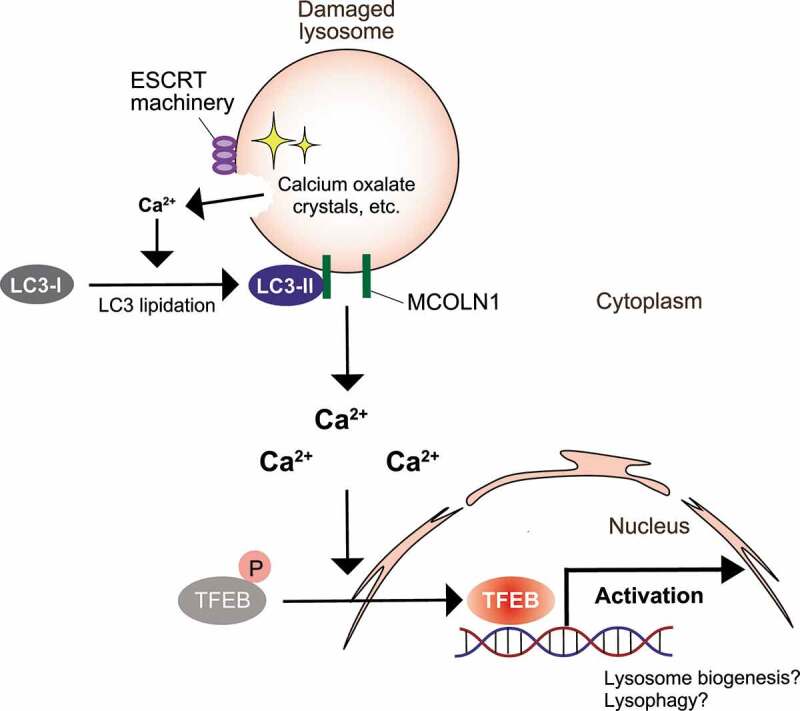
Model of ATG conjugation system-dependent TFEB activation during the lysosomal damage response. Upon lysosomal damage, small amounts of calcium efflux from lysosomes induce LC3 lipidation and recruit it on lysosomes. Lipidated LC3 on lysosomes then interacts with a calcium channel, MCOLN1, further facilitating large amounts of calcium efflux essential for TFEB activation. This system is particularly important after ESCRT roughly repairs the ruptured lysosomes. Activated TFEB is essential for lysosomal homeostasis after lysosomal damage, presumably through activation of lysophagy and/or lysosomal biogenesis, but the exact downstream targets need to be clarified
Funding Statement
SN is supported by AMED-PRIME (17gm6110003h0001), JSPS KAKENHI, Senri Life Science Foundation, Takeda Science Foundation, Nakajima Foundation, MSD Life Science Foundation, Astellas Foundation for Research on Metabolic Disorders, and Mochida Memorial Foundation for Medical and Pharmaceutical Research. TY is supported by JST CREST (JPMJCR17H6), AMED (grant numbers JP17gm5010001 and JP17gm0610005) and an HFSP Research grant.
Disclosure statement
S.N. and T.Y. have applied for a patent for assays related to this work. T.Y. is founder for AutoPhagyGO.
Reference
- [1].Nakamura S, Shigeyama S, Minami S, et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat Cell Biol. 2020;22(1252–1263). DOI: 10.1038/s41556-020-00583-9 [DOI] [PubMed] [Google Scholar]