

ABSTRACT
Macroautophagic/autophagic degradation of nuclear components (or nuclear autophagy) is a poorly understood area in autophagy research. We previously reported the nuclear lamina protein LMNB1 (lamin B1) as a nuclear autophagy substrate in primary human cells, stimulating the investigation of nuclear autophagy in the mammalian system. We recently reported the sirtuin protein SIRT1 as a new selective substrate of nuclear autophagy in senescence and aging. Upon senescence of primary human cells, SIRT1 degradation is mediated by a direct nuclear SIRT1-LC3 interaction, followed by nucleus-to-cytoplasm shuttling of SIRT1 and autophagosome-lysosome degradation. In vivo, SIRT1 is downregulated by lysosomes in hematopoietic and immune organs upon natural aging in mice and in aged human T cells. Our study identified another substrate of nuclear autophagy and suggests a new strategy to promote SIRT1-mediated health benefits by suppressing its autophagic degradation.
Abbreviations: HSPC: hematopoietic stem and progenitor cells; NAD+: nicotinamide adenine dinucleotide; SASP: senescence-associated secretory phenotype
KEYWORDS: Aging, nuclear autophagy, senescence, SIRT1, sirtuin
Sirtuins are a conserved family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases and ADP-ribosyltransferases. SIRT1, located in the nucleus and cytoplasm, functions through deacetylation of its substrates that are regulators of metabolism and aging. Moderate overexpression or pharmaceutical activation of SIRT1 is usually associated with improved healthspan and/or lifespan in several organisms; therefore, strategies to stimulate SIRT1 catalytic activity are being pursued for aging intervention.
We began our study by investigating SIRT1 protein homeostasis in cellular senescence [1]. Senescence is a stable form of cell cycle arrest induced by telomere shortening or by cellular stress, and can be used as an in vitro tool to investigate the biological mechanisms of aging. Accumulation of senescent cells is observed in aged tissues, and clearance of senescent cells delays age-related pathologies, suggesting a causal relation between senescence and organismal aging. We observed downregulation of SIRT1 protein in senescent primary human cells triggered by multiple means, including replication exhaustion, oncogene expression, and DNA damage. The mRNA levels of SIRT1 do not decrease in these conditions. Addition of the lysosome inhibitor Lys05 restores SIRT1 protein in senescent cells, whereas addition of the proteasome inhibitor MG132 fails to do so, indicating that loss of SIRT1 protein in senescence is mediated by lysosomal degradation.
We subsequently discovered that macroautophagy (hereafter “autophagy”) mediates SIRT1 degradation in senescence. First, inhibition of autophagy by ATG7 knockdown prevents SIRT1 downregulation in senescent cells. Second, using imaging experiments, we found that while SIRT1 in proliferating primary human fibroblasts is localized in the nucleus, SIRT1 undergoes nucleus-to-cytoplasm shuttling and is targeted by phagophores (autophagosome precursors) and degraded by lysosomes in senescence. Third, SIRT1 directly interacts with the Atg8-family protein LC3B in the nucleus. Disruption of this interaction, using SIRT1 mutants that do not bind to LC3B, suppresses SIRT1 downregulation and its nucleus-to-cytoplasm transport in senescence. Whereas senescent cells actively degrade SIRT1, starved or quiescent cells do not show loss of SIRT1. Hence, SIRT1 is a selective substrate of nuclear autophagy upon cellular senescence (illustrated in Figure 1).
Figure 1.
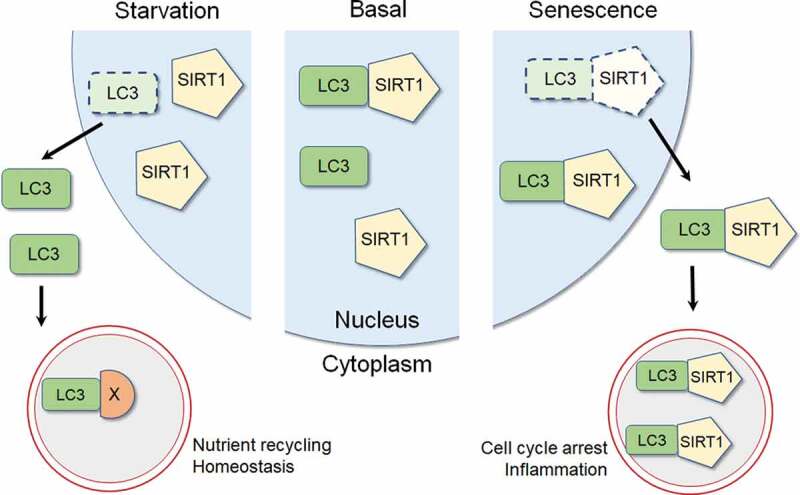
Schematic model for SIRT1 and autophagy. In the basal state, LC3 and SIRT1 interact in the nucleus. Upon starvation, SIRT1 remains in the nucleus while LC3 translocates from the nucleus to the cytoplasm to stimulate cytoplasmic autophagy that degrades cellular constituents (labeled as X), facilitating nutrient recycling and homeostasis. Upon senescence, LC3-SIRT1 interaction is enhanced, followed by nucleus-to-cytoplasm translocation of the LC3-SIRT1 complex, leading to cytoplasmic degradation of SIRT1 by autophagy. Degradation of SIRT1 in senescence is associated with induction of cell cycle arrest and pro-inflammatory responses
Next, we examined SIRT1 protein levels in natural aging. We found that SIRT1 protein remains largely the same in several tissues, comparing old mice and young mice. This observation is consistent with the reported health benefits of sirtuin-activating compounds investigated in old mice. We observed loss of SIRT1 protein in testes, spleen, thymus, hematopoietic stem and progenitor cells (HSPC) from old mice, and CD8+ CD28− T cells from aged human donors. The mRNA levels of Sirt1/SIRT1 in these tissues/cells remain unchanged. Inhibition of lysosomal degradation by Lys05 restores SIRT1 protein level. Hence, SIRT1 protein is selectively degraded in several tissues during aging, including the hematopoietic and immune organs/cells.
Our in vivo aging results using mice and human samples suggest a new avenue in targeting SIRT1 for health benefits. While several natural and synthetic sirtuin-activating compounds have been developed to stimulate SIRT1 catalytic activity, we reason that these compounds may not exert their full effects in hematopoietic and immune systems in aged individuals, due to the substantial loss of SIRT1 in these tissues. Immunosenescence is an important aspect of aging, because aging is associated with declines of both innate and adaptive arms of the immune system, accompanied by chronic, sterile inflammation in the absence of infection. These alterations of the immune system reduce the capacity for immuno-surveillance of infection and cancer. Meanwhile, chronic inflammation underlying aging promotes most, if not all, age-associated diseases. Previous studies reported that genetic deletion of SIRT1 in immune cells or HSPC in young mice phenocopies several aspects of the age-associated alterations of the immune and hematopoietic systems, suggesting that loss of SIRT1 is causal to the deteriorations of the two systems in aging. Therefore, our study revealed a new mechanism for immunosenescence, and suggests the stabilization of SIRT1 protein as a new strategy to promote healthy, productive aging.
We note that the behavior of SIRT1 in senescence highly resembles that of LMNB1, the first characterized mammalian nuclear autophagy substrate. Both proteins are specifically degraded in senescence and not upon starvation or quiescence, and are shuttled to the cytoplasm by interacting with LC3B. LC3B interactions with SIRT1 and LMNB1 are increased in senescence and not upon starvation or quiescence, correlating with senescence-specific degradation of the two substrates. The mechanisms and signaling pathways for nuclear autophagy in senescence are unclear. We speculate that senescence-specific phosphorylation/dephosphorylation of LMNB1 and SIRT1 may confer the increased affinity to Atg8-family proteins. This represents a major future direction for exploration.
What is the biological significance of nuclear autophagy in senescence? Our current knowledge is that nuclear autophagy promotes two important features of senescence: the cell cycle arrest program and the pro-inflammatory program, both are implicated in tumor suppression. Autophagy-mediated downregulation of LMNB1 reinforces cell cycle arrest when cells encounter activated oncogenes or DNA damage. Blocking LMNB1 autophagic degradation by disrupting LMNB1-LC3B interaction results in prolonged cell cycle progression and increased growth of RAS-induced colonies. Autophagic degradation of SIRT1 may stimulate the pro-inflammatory responses of senescence. Senescent cells secret a large array of pro-inflammatory cytokines, chemokines, growth factors, and proteases, collectively termed as the senescence-associated secretory phenotype (SASP), which promotes immune-mediated clearance of senescent cells, acting as a tumor-suppressive mechanism. SIRT1 was reported to negatively regulate the expression of several senescence-associated inflammatory factors, including IL6, IL8, and IL1B/IL-1β. Hence, downregulation of SIRT1 by nuclear autophagy relieves the inhibition of SASP imposed by SIRT1, therefore stimulating immuno-surveillance of pre-malignancy. While acute induction of senescence restrains tumorigenesis, senescence and nuclear autophagy during natural aging may impair tissue renewal and promote chronic inflammation and age-associated diseases. For example, SIRT1 loss could facilitate the decline of the immune system during aging. This notion is consistent with the “antagonistic pleiotropy hypothesis”, in which a phenotypic trait is beneficial to the fitness in young organisms, but is detrimental to the organisms’ fitness later in life. Hence, our study suggests that selectively targeting the degradation of nuclear autophagy substrates in aging could be a new approach to suppress aging and aging-associated diseases.
Funding Statement
L.W. is supported by NIH [grant K99AG065500]. C.X. is supported by a Glenn/AFAR Scholarship for Research in the Biology of Aging from American Federation of Aging Research (AFAR). T.J. is supported by TOPPFORSK [grant number 249884] of the Research Council of Norway. S.L.B. is supported by NIH [grant P01AG031862]. Z.D. is supported by NIH [grant R00AG053406 and R35GM137889], and Glenn Foundation for Medical Research and AFAR [Grant for Junior Faculty].
Disclosure statement
No potential conflicts of interest were disclosed.
Reference
- [1].Xu C, Wang L, Fozouni P, et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol. 2020;22(10):1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]