

Abstract
In seeking to understand mental health and disease, it is fundamental to identify the biological substrates that draw together the experiences and physiological processes that underlie observed psychological changes. Mitochondria are subcellular organelles best known for their central role in energetics, producing adenosine triphosphate (ATP) to power most cellular processes. Converging lines of evidence indicate that mitochondria play a key role in the biological embedding of adversity. Preclinical research documents effects of stress exposure on mitochondrial structure and function and recent human research suggests alterations constituting recalibrations, both adaptive and non-adaptive. Current research suggests dynamic relationships among stress exposure, neuroendocrine signaling, inflammation, and mitochondrial function. These complex relationships are implicated in disease risk, and their elucidation may provide for prevention and treatment of stress- and trauma-related disorders. We review and evaluate the evidence for mitochondrial dysfunction as a consequence of stress exposure and a contributing factor to psychiatric disease.
Keywords: mitochondria, psychosocial stress, early life adversity, allostatic load
INTRODUCTION
Human experience is marked by a broad range of stressors, adversities, and traumas. These vary by severity, timing, chronicity, and type of exposure. Sources of stress differ over time, according to geographical region, economic status, and a myriad of other variables. As investigators seek to improve understanding of key determinants of early experience, the sequelae of early life adversity are brought into focus––from the scope of broadly-defined population health, down to the molecular mechanisms underlying the complex interactions of genes and environment. A unifying and critical question underlies the work of the developmental psychologist, the epidemiologist, and the molecular neuroscientist: why should early experiences shape one’s development and health outcomes throughout life? (Lewis 1997, Hartman et al. 2017). In the pursuit of understanding risk, resilience, and opportunities for therapeutic interventions, these lines of investigation convene on the fundamental principle that humans are adaptive––acutely, over the course of a lifetime, and evolutionarily. The human body employs a suite of environmentally responsive biobehavioral processes that promotes survival, and in an evolutionary context, increases the likelihood of survival across generations.
However, adaptation is a compromise. Responses that are advantageous in certain circumstances, can result in maladaptive consequences in other settings. Convincing evidence across various fields of inquiry has reliably demonstrated over the past two decades that adversity in childhood is associated with increased risk for both psychopathology and chronic health problems throughout the lifespan (Nanni et al. 2012, Havranek et al. 2015, Nemeroff, 2016, Jakubowski et al. 2018). The psychological and biophysical sequelae of stress exposure originate in evolutionary mechanisms designed to enable an individual to respond to an environmental threat. While acutely these may be adaptive, in excess, these processes produce broad systemic physiologic and psychological disruption.
The mitochondrion, well-known for its role in cellular energy production, represents a critical nexus of biological, psychological, and social factors that underlie the mechanisms and consequences of the stress response. Psychosocial factors impact biological processes through physiological systems that are highly integrated with mitochondrial functioning. Mitochondria are subcellular organelles with broad functions in energetics, cell-signaling, and hormone production. Mitochondrial structure and function are exquisitely responsive to the environment and serve as both a target and mediator of the stress response (Picard & McEwen 2018a, Picard & McEwen 2018b). As a highly energy-dependent organ, the brain is densely populated by mitochondria (Juster et al. 2016). Thus, there is a profound impact of mitochondrial dysfunction on psychological processes, with increasing evidence demonstrating associations of stress-related mitochondrial dysfunction and psychopathology (Tyrka et al. 2016, Picard et al. 2018b, Trumpff et al. 2019a). Animal models involving experimentally-induced defects in mitochondria and stress paradigms both produce pathophysiological states that suggest common mechanistic origins underlying these processes. Such processes are found in evidence from studies of the CNS, neuroendocrine, and immune systems, and may implicate mitochondrial dysfunction in stress-related disease.
In this selective review, we discuss the role of mitochondria in stress responding and stress-related psychiatric disorders. We begin by introducing mitochondria and the role they may play in the biological embedding of adverse experiences. We then discuss recent evidence demonstrating the role of mitochondrial within the stress response, with particular focus on the interaction of mitochondria with established endocrine, inflammatory, and epigenetic pathways that underlie stress physiology. These findings are integrated with recent studies suggesting a role for mitochondrial dysfunction in stress-related psychiatric disorders. Finally, we reflect on the limitations of current evidence and look toward future directions to better understand the unique contribution of mitochondria to psychological function and potential for treatment interventions. The literature covered here is not exhaustive but highlights key findings that characterize the current state of the field with respect to this highly dynamic organelle.
THE ORIGIN AND PHYSIOLOGICAL FUNCTIONS OF MITOCHONDRIA
Endosymbiosis, from the Greek for “within,” “together,” and “living,” is the process by which one cell comes to live within another. Mitochondria originated as a bacterial cell, engulfed by an ancestor of the modern mammalian cell about 1.5 billion years ago (Sagan 1967, Archibald 2015). With the exception of red blood cells, every cell of the human body contains hundreds to thousands of these life-sustaining organelles. Mitochondria maintain characteristics of their bacterial origin, including their own genome––the maternally-inherited, circular, double-stranded, mitochondrial DNA (mtDNA; McFarland et al. 2010, Kasahara & Kato 2018). Mitochondria play a critical role in a broad range of functions within the cell and throughout the body, from energetics, epigenetics, and inflammation, to hormone synthesis and metabolism (Shaughnessy et al. 2014, Juster et al. 2016, Picard et al. 2018a). They respond dynamically to metabolites and neuroendocrine factors, driving inflammatory processes, and regulating cell-division and cell-survival. Mitochondria sense, integrate, and signal information about their environments (Picard & McEwen 2018a).
Justifying the appellation “the powerhouse of the cell,” mitochondria are the cell’s primary site of energy flow. The genes of mtDNA encode for protein complexes that serve as the machinery for supplying cellular energy: the electron transport chain (ETC) and oxidative phosphorylation (Shaughnessy et al. 2014). Through a series of chemical reactions and conversions, the ETC transforms metabolites derived from food and oxygen into an electrochemical gradient (Wallace 2005). This gradient fuels a number of processes within mitochondria, including the synthesis of adenosine triphosphate (ATP), which serves as the energy currency used throughout the cell to power many essential processes of life, from DNA synthesis to neurotransmission and muscle contraction. The electrochemical gradient further provides for a number of additional critical functions of the mitochondria, including the transport of ions, proteins, and other molecules as well as the synthesis of steroid hormones (Hoffmann & Spengler 2018, Picard et al. 2018a).
The brain is a highly energy-dependent organ and is densely populated with mitochondria (Magistretti & Allaman 2015). Mitochondria play an important role in brain function (Srivastava et al. 2018). This is evident in inherited mitochondrial disease, a heterogeneous group of disorders caused by mutations in mtDNA and nuclear genes that encode for structural mitochondrial proteins or proteins involved in mitochondrial function. Many of these disorders are characterized by atrophy in cortical, brainstem, and cerebellar brain regions (Gorman et al. 2016) as well as a variety of neurological symptoms such as changes in vision, deafness, difficulty swallowing, diminished muscle tone, lack of coordination, neuropathy, and seizures (McFarland et al. 2010). Mitochondrial diseases are also associated with affective changes, with more than half of individuals with mitochondrial disease expressing comorbid psychiatric disorders (Kasahara & Kato 2018). Further, mitochondrial dysfunction is found in primary developmental and neuropsychiatric diseases such as autism (Goh et al. 2014), ADHD (Hwang et al. 2017), Alzheimer’s disease, and Parkinson’s disease (Yan et al. 2013, Flannery & Trushina 2019). Additionally, mitochondria play an important role in neural plasticity (Steib et al. 2014). Taken together, mitochondria are integral to brain function, providing intriguing potential psychiatric implications for mitochondrial dysfunction in the context of stress exposure.
ALLOSTASIS UNDER THE MICROSCOPE: MITOCHONDRIAL ALLOSTATIC LOAD
Defining Stress
The term “stress” can be used to describe a number of distinct concepts. Stress may refer to an event, a stressor, or stress exposure. It may describe one’s psychological state or the perception of stress, which is subjective. Stress may refer to a characterized set of physiological responses commonly understood to be activated in the setting of environmental challenges. In this chapter, we use clarifying terms to disambiguate these distinct concepts.
Stressful experiences or exposures encompass a wide variety of environmental demands on the individual. A stress exposure may have the potential for positive outcomes marked by reward, such as a job interview or an exam. In such situations, the stress-related physiological response is activated and deactivated over the time course of a discrete stressor. Acute stress may also occur in the setting of a profoundly negative experience, such as the death of a parent or spouse. In the wake of such events, personal resources and external support may allow the individual to prevail and continue to thrive. In so-called “toxic stress,” the exposure may be severe or chronic, and the individual lacks either internal or external resources needed to promote resilience, resulting in the absence of control over the environment. In such circumstances, the physiological and behavioral recalibrations may serve to protect some functions while disadvantaging others. Untethered, maladaptive processes serve to promote physiological dysregulation and pathology (Shonkoff et al. 2009). Chronic, prolonged exposure to toxic stress imparts both disorders of mental and physical health (Wegman & Stetler 2009, Rich-Edwards et al. 2010, Widom et al. 2015, Basu et al. 2017, Jakubowski et al. 2018).
Allostasis
Allostasis is a complex collection of physiological functions, which allows an organism to adapt and maintain stability in a dynamic environment (Sterling & Eyer 1988, McEwen & Stellar 1993). These biobehavioral responses are coordinated by the central nervous system (CNS) and extend throughout the body to optimize functioning according to varying environmental and internal demands. Multiple interwoven systems, from the CNS to the endocrine and immune systems, interact to produce dynamic, physiological changes. In excess, these processes are overextended, leading to maladaptive, pathological functioning (Danese & McEwen 2012, Danese & Lewis 2017).
This review is primarily focused on the psychological sequelae and psychiatric disorders associated with stress and trauma. However, it should be appreciated that due to the integrated and systemic nature of stress physiology, the pathology associated with adversity involves multiple organ systems and a variety of medical conditions. For example, allostatic processes are evident in the recalibration of cardiovascular functioning in order to meet the demands of a stressful environment. Sympathetic activation and exposure to glucocorticoids during an acute stressor provides for the brief, well-circumscribed cardiovascular activation with an associated elevation in blood pressure, which is useful for coping with or fleeing from a stressor. With chronic stress and associated dysregulated glucocorticoid exposure over time, individuals face increased risk of disease through arterial stiffening, coronary artery calcification, or aneurysm (Havranek et al. 2015). The concept of allostasis refers to stress-induced adaptations across a wide range of physiological systems. Pathology represents the end product of adaptive processes that have become dysfunctional in the organism’s attempt to adapt to environmental adversity.
Health behaviors represent a major category of risk and resilience factors. They integrate multiple domains of allostasis and represent behavioral responses to external and internal cues. Unhealthy behaviors, such as overeating and substance abuse, can serve to acutely alter negative affective states accompanying a stressor. While these behaviors may initially serve to manage and reduce the apprehension, anxiety, and tension associated with stress exposure, over time, they often exacerbate physiological perturbations and further increase the risk of systemic disease.
Mitochondrial Allostatic Load
Accumulating evidence supports a key role for mitochondria in the physiological response to both acute and chronic stress exposure, and, ultimately, the biological embedding of adversity in health and psychological functioning. In the setting of acute stress exposure, mitochondria respond dynamically to cues from stress-associated neuroendocrine, metabolic, and inflammatory pathways. With chronic exposure to stress, mitochondria demonstrate both structural and functional dysregulation. As illustrated in Figure 1, mitochondrial allostatic load (MAL), describes the cumulative adaptive and maladaptive effects of chronic stress exposure on mitochondrial structure and function and the resulting pathological changes across a wide range of bodily systems (Picard et al. 2014a, Picard & McEwen 2018a, Picard & McEwen 2018b). Distinct from traditional biomarkers of allostatic load, such as lipids and glucose, mitochondria are living organelles, and thus both shed light on dynamic responses to stressors and pose challenges to sensitive and specific measurement of their responses (Picard & McEwen 2018a). Below we describe the growing understanding of the role of mitochondria in physiological stress, from their interaction with primary stress mediators like glucocorticoids, secondary processes including metabolic and inflammatory mechanisms, and tertiary endpoints with a focus on psychiatric disease.
Figure 1:
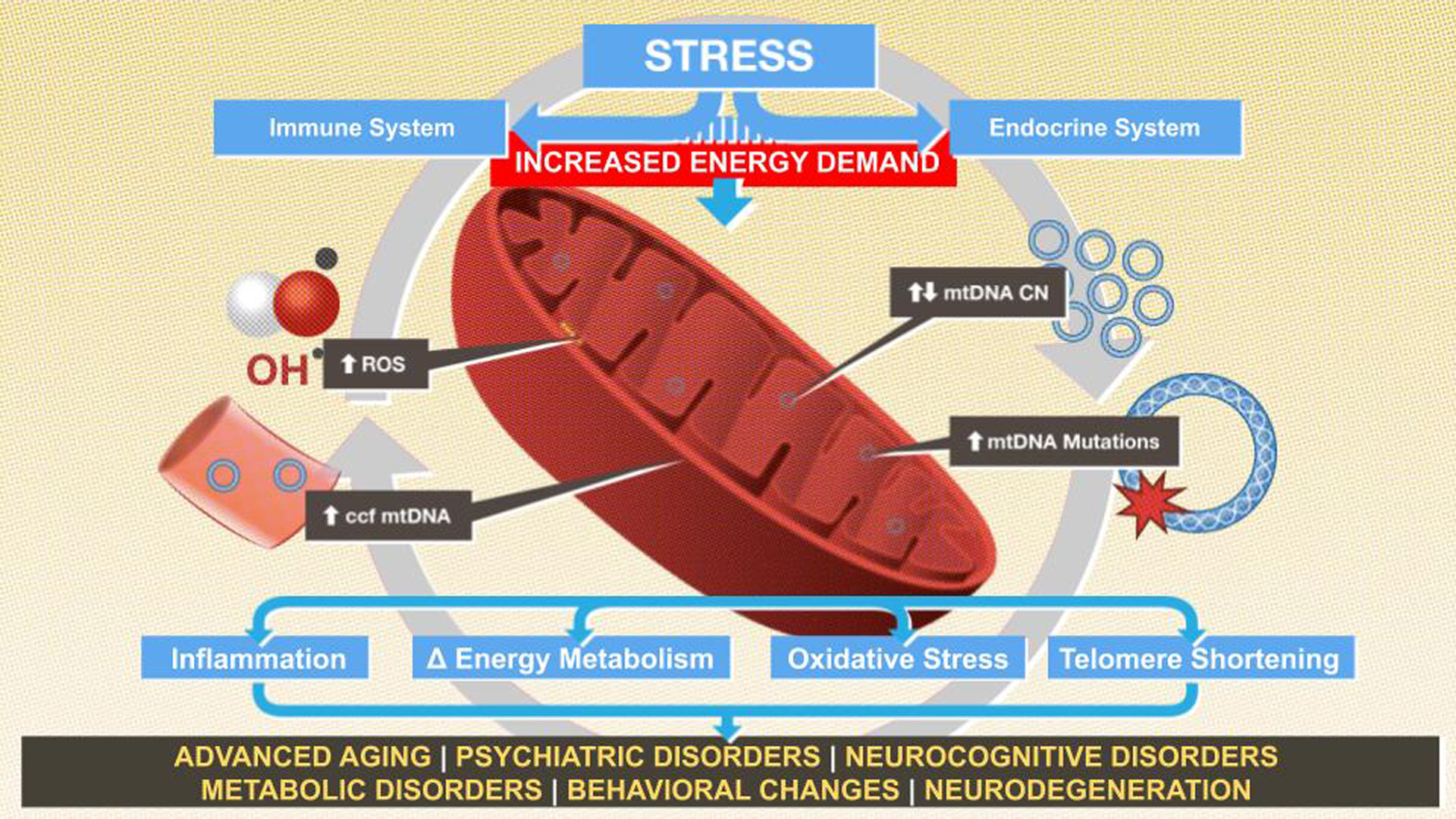
Conceptual framework for the interplay of stress exposure and mitochondrial alterations via the inflammatory and endocrine systems, which increases the risk of pathological states in vulnerable populations.
STRESS EXPOSURE: ESTABLISHED PATHWAYS & IMPLICATIONS FOR MITOCHONDRIA STRUCTURE & FUNCTION
Stress-Associated Endocrine Pathways
The endocrine pathways underlying the acute stress response are well-documented and serve as a foundation for the current understanding of stress physiology. In a threatening experience, the sympathetic-adrenal-medullary (SAM) axis is activated within seconds and catecholamines, such as adrenaline, are released from the adrenal gland. Within minutes, the hypothalamic-pituitary-adrenal (HPA) axis is stimulated, beginning with the release of corticotropin-releasing factor (CRF). CRF acts on the pituitary gland, precipitating the release of adrenocorticotropic hormone (ACTH). Systemically, ACTH stimulates the production of the glucocorticoid hormone, cortisol (Sapolsky et al. 2000). Glucocorticoids serve a number of important functions in the stress response, including negative feedback to the HPA axis, providing timely suppression of ongoing stress-related processes. The circulation of glucocorticoids also provides for the mobilization and availability of energy substrates in the form of glucose and lipids, which are critical substrates for mitochondrial function (Picard et al. 2014a, Picard & McEwen 2018a, Hoffman & Spengler 2018).
The negative feedback mechanism that provides for a well-circumscribed response to stress is altered with chronic or repeated stress, resulting in HPA axis dysregulation (Berens et al. 2017). Continued stress exposure drives hyperactivation of this system, leading to elevated levels of circulating cortisol. However, there is evidence that sustained HPA axis hyperactivation eventually results in under-responsive, hypoactive functioning (Nemeroff 2016, Bunea et al. 2017). Differential patterns of dysregulation appear to be mediated in part by the type of stressor, reported age at the time of stressor, and chronicity (Khoury et al. 2019).
Stress-Associated Inflammation
The stress response also engages the immune system in a complex manner, alternately activating and suppressing distinct immune functions (Segerstrom & Miller 2004, Steptoe et al. 2007, Rohleder 2014). The interaction of stress exposure and immune response is postulated to be a mechanism providing optimal inflammatory defense against injury and infection in adverse conditions (Marsland et al. 2017, Godoy et al. 2018). Following an acute stressor, catecholamines trigger the release of immune signaling molecules called cytokines. Cytokines are important mediators of the local and systemic inflammatory response to pathogens. They also potentiate the stress response, activating the release of cortisol, which conversely inhibits cytokine production, thus down-regulating stress-associated inflammation (Marsland et al. 2017, Godoy et al. 2018).
Individuals with chronic stress exposure in childhood demonstrate significant and graded elevations in inflammatory proteins as adults, an association not explained by mediators such as adult stressors or unhealthy behaviors (Danese et al. 2007, Takizawa et al. 2015, Baumeister et al. 2016). Adults with early life stress (ELS) have a greater inflammatory response to acute social stressors compared to those without a history of adversity (Carpenter et al. 2010). Chronic stress may lead to diminished glucocorticoid sensitivity to cytokine production (Meyer & Wirtz 2018). Taken together, this literature indicates that chronic psychosocial stress leads to elevated risk of inflammation and disease (Meyer & Wirtz 2018).
Stress-Associated Telomere Shortening
A large body of literature also documents that stress exposure alters telomeres, the DNA-protein complexes that cap the ends of chromosomes and confer chromosomal stability. DNA segments are routinely removed from the ends of chromosomes with each cell replication cycle and telomeres provide a buffer so that critical DNA segments are protected. Telomere shortening beyond a threshold leads to the arrest of the cell cycle, cellular senescence, and possibly DNA damage and cell death. Telomere shortening is associated with aging and with medical and psychiatric disease (Epel & Prather 2018). Importantly, telomere attrition is predictive of age-associated diseases, such as heart disease and diabetes. Furthermore, shorter telomeres are found in psychiatric disorders, including major depressive disorder (MDD) and anxiety disorders (Wei et al. 2016, Ridout et al. 2018). Intriguingly, telomeres are responsive to cumulative stress. Specifically, exposure to glucocorticoids, inflammation, and oxidative stress are associated with telomere attrition, providing a possible mechanism linking stress, aging, and disease (Ridout et al. 2016). Over the past decade, a growing body of evidence has demonstrated a robust inverse relationship between telomere length and early adversity (Hanssen et al. 2017, Li et al. 2017, Ridout et al. 2018, Epel & Prather 2018).
Mitochondria at the Intersection of Stress Pathways
The “fight or flight” response to a threat requires energy in the form of ATP to fuel survival behaviors. Energy is also required for the physiological processes that allow the body to adapt and respond to stress challenge. The stress response increases the availability of energy substrates, such as glucose, which is used as fuel by the brain (Magistretti & Allaman 2015). Mitochondria provide the energy that fuels key enzymatic reactions, transcription and translation of genetic material to provide various substrates, release and reuptake of neurotransmitters, the synthesis of hormones, sympathetic activation, behavioral adaptations, and structural changes to tissues and organs (Picard et al. 2018a). Further, steroid hormones such as glucocorticoids are both synthesized and metabolized by mitochondria (Picard et al. 2018a, Hoffmann & Spengler 2018). In order to meet these high energy demands, mitochondrial activity is activated by the presence of neuroendocrine and metabolic stress mediators, including glucocorticoids and circulating glucose (Du et al. 2009, Psarra et al. 2011, Picard et al. 2014a, Smith et al. 2018).
In response to acute stress exposure, neuroendocrine pathways stimulate mechanisms and behaviors to modulate utilization of energy stores, such as modifications in eating behaviors (shifting preference toward calorically dense macronutrients) and rapid mobilization of free fatty acids from central fat stores. Mitochondria are sensitive to these changes in metabolic, endocrine stressors, and stress-mediators. With high levels of glucocorticoid exposure, mitochondria have diminished calcium buffering capacity, an important function of mitochondria in maintaining the internal milieu of the cell. Rapid calcium flux or calcium overload in the cell promotes sensitization to cell death (Du et al. 2009, Picard et al. 2014a, Srivastava et al. 2018). In acute stress, circulating levels of important substrates, such as glucose or lipids, increase in order to provide the energy needed to respond to a stressor. In the context of hyperglycemia, mitochondria “band together,” undergoing fusion in order to promote survival. In conditions of severe or prolonged exposure to stress, these substrates are chronically elevated, and mitochondria become fragmented, increasing the risk of cell death (Shutt & McBride 2012, Hoffmann & Spengler 2018, Meyer et al. 2018). Prolonged fragmentation is associated with further oxidative stress and damage to mtDNA (Liesa & Shirihai 2013). Importantly, these changes impair the bioenergetic functions of mitochondria and are amplified over time (Picard & McEwen 2018a).
Consistent with their symbiotic origins, mitochondria communicate with other parts of the cell, providing signals about their functional status (Chandel 2015). Importantly, the mitochondrial response to stress is communicated both locally within the cell and systemically throughout the body. Exposure to stress-mediators precipitates mitochondrial release of signaling molecules, which are collectively referred to as mitokines. Mitokines serve as signals that indicate mitochondrial fitness, which is of particular importance in the context of environmental stressors. Mitokines include various mitochondrial metabolites, calcium, and reactive oxygen species (ROS; Chandel 2015, Shaughnessy et al. 2014). ROS are generated as a by-product of the energy-producing processes within mitochondria––at low levels, ROS support a number of key functions in the cell. When elevated, ROS overwhelm the cell’s antioxidant capacity and promote oxidative stress, causing cell death and damage to tissue (Meyer et al. 2018). As with other mitokines, ROS can drive local and systemic pathological processes involving oxidative stress, inflammation, metabolism, gene expression, and cell senescence (Picard & McEwen 2018a). These signals are particularly important in the nucleus, where the genome is largely under mitochondrial regulation (Picard et al. 2014b). These mechanisms allow mitochondria to influence broad physiological processes throughout the body (Picard & McEwen 2018b).
Another important mitokine is circulating cell-free mitochondrial DNA (ccf-mtDNA), which is present in low levels in healthy individuals, abundant in inflammatory disease, and significantly increased in critically ill hospital patients (Nakahira et al. 2013). ccf-mtDNA is generated by mitochondria in the event of stress, cell damage, or death, releasing mtDNA into the bloodstream, which then circulates freely throughout the body. The immune system recognizes ccf-mtDNA as foreign, stemming from its evolutionary origin as bacteria, which consequently induces systemic inflammation (Picard et al. 2014a, Zhang et al. 2010). While acute exposure to psychosocial stress has been associated with decreases in ROS, prolonged exposure is associated with mitochondrially-derived oxidative stress, which further activates a number of pro-inflammatory processes, including cytokine release and proinflammatory gene expression (Meyer & Wirtz 2018). Moreover, cytokines appear to stimulate mitochondrial production of glucocorticoids, in the immune-mediated propagation of the stress response (Meyer & Wirtz 2018).
Stressors in the environment are translated into biological and physiological changes through adaptation on the cellular level. Importantly, while mtDNA does not contain telomeres, telomere length on nuclear chromosomes and mitochondrial dysfunction have been linked, particularly in relation to ELS (Tyrka et al. 2016, Cai et al. 2015a). Increasing evidence suggests complex co-regulation of telomere length and mitochondrial function, with mitochondrially-derived oxidative stress driving telomere attrition and shorter telomeres leading to mitochondrial dysfunction and cell death (Picard & McEwen 2018a).
STRESS-INDUCED MITOCHONDRIAL DYSFUNCTION, AGING, & DISEASE RISK
Mitochondria & Aging:
Over the past two decades, there has been tremendous interest in the relationship between stress, molecular mechanisms of aging, and age- and stress-related conditions, including psychiatric disorders, cardiovascular disease, obesity, diabetes, and cancer (Lagouge & Larsson 2013, Epel & Prather 2018). Shortened telomere length, a marker of accelerated cellular aging, has been associated with age-related disease, stress exposure (particularly ELS), and psychiatric disorders, including MDD and anxiety (Epel & Prather 2018, Ridout et al. 2018, Tyrka et al. 2016, Cai et al. 2015a). Mitochondria have been increasingly recognized for their importance in both the stress response and the aging process, with current research focusing on the role of mitochondria in accelerated aging and the development of age-related disease (Han et al. 2019). Aging itself is associated with perturbations in mitochondrial structure and function, including impaired replication, alterations in mtDNA copy number (mtDNAcn), increased ROS production, mtDNA mutations, and organelle damage with resulting release of ccf-mtDNA (Lagouge & Larsson 2013, Bratic & Larsson 2013, Picard et al. 2014a). In the setting of chronic stress exposure, these changes may accumulate, accelerating aging and increasing an individual’s risk of developing age and stress-related metabolic conditions such as cardiovascular disease and diabetes (Ridout et al. 2016, Picard et al. 2014a). For example, increases in ccf-mtDNA levels have been shown to not only predict mortality in patients admitted to medical intensive care units but have also been associated with up to an eight-fold increased risk of death within 28 days of admission (Nakahira et al. 2013). Furthermore, considering that age-related conditions such as diabetes, cancer, and cardiovascular disease are accompanied by alterations in homeostatic mechanisms governing energy usage and metabolism, it is consistent that mtDNA mutations and mitochondrial dysfunction have been documented to contribute to the onset of these diseases (Wallace 2005).
The relationship between psychiatric conditions and cellular aging is important to consider as mental illness has negative associations with physical health, including increased risk of premature death and earlier-onset of age-related chronic medical conditions (Viron & Stern 2010). Further, coping with a psychiatric condition can be an inherent source of chronic stress, which may predispose patients to poorer health outcomes. For example, individuals with psychiatric conditions are more likely to engage in adverse health behaviors, including alcohol and substance use and poor dietary choices (Kilian et al. 2006), furthering metabolic risk. In the following sections, we will outline the role that mitochondria play in these pathologic processes and how they may mediate the relationship between stress and poor health outcomes.
Stress & the Brain
In the brain, cortico-limbic networks are designed to detect and adapt to environmental threats through coordination of the stress response. A perceived threat selectively activates brain regions that contribute to a coordinated biobehavioral response. The prefrontal cortex provides the overarching executive function of the brain––controlling cognition, learning, memory, and impulse control (Tottenham & Galván 2016). In the context of stress exposure, prefrontal inhibitory control over the amygdala is released, triggering a cascade of autonomic and neuroendocrine mediated mechanisms via the sympathetic nervous system and the hypothalamic-pituitary-adrenal (HPA) axis, respectively (Godoy et al. 2018, Bunea et al. 2017). The amygdala applies emotional valence, drives fear conditioning, and has downstream output to the sympathetic-adrenal-medullary (SAM) and neuroendocrine regulatory systems (Berens et al. 2017). The hippocampus provides for memory, cognition, and negative feedback to the HPA axis (Juster et al. 2016, McEwen 2007).
Early adversity can have a significant impact on these neural networks, as they grow and mature during childhood, adolescence, and adulthood. In addition to the psychological, social, and emotional responses to the traumatic experience, the effects of extreme and chronic stress on cortico-limbic brain circuitry may result in enduring difficulties with emotion regulation, fear learning, and executive function. Extreme or chronic early stress has been linked to abnormalities in cognitive functions regulated by the prefrontal cortex, including attention, impulsivity, and executive functioning (Danese & McEwen 2012). Individuals with a history of significant adversity often have difficulty with emotion regulation, including hypervigilance, anxiety, anger, and compromised behavioral control (Tottenham & Galván 2016).
There has been substantial evidence in support of volumetric brain changes secondary to stress exposure. An example of this is shrinkage of the prefrontal grey matter appreciated in children and adults with a history of ELS (McEwen & Morrison 2013). Adults with ELS also demonstrate consistent reduced hippocampal volume (Lupien et al. 2018). Both increases and decreases in amygdala volume have been appreciated; however, the direction of these changes is likely moderated by the type of exposure as well as its timing (Berens et al. 2017). Several potential mechanisms for these changes have been proposed, in particular, the “neurotoxicity” hypothesis, whereby mediators, such as glucocorticoids, disrupt growth, maturation, and survival of neurons (Lupien et al. 2018). Multiple other stress-mediators have been identified as potentially neurotoxic in development, including cytokines, excitatory amino acids, endogenous opioids, brain-derived neurotrophic factor (BDNF) (Zimmerman et al. 2019, Berens et al. 2017), telomere shortening, and mitochondrial dysfunction (Epel & Prather 2018).
Mitochondria & the Brain: Stress-Induced Alterations
The mitochondrion has been identified as a potential mechanism by which activation of the stress-response system induces changes in brain morphology, development, and capabilities. With increased energy demands from the stress-response system, mitochondria produce increased ROS, which overwhelm the antioxidant capacity of the cells and can result in mtDNA mutations (Lagouge & Larsson 2013). The resulting oxidative stress has been postulated to play a role in the pathogenesis of neuropsychiatric disorders, particularly depression and dementia. In such disease processes, it is suggested that the accumulation of ROS and mtDNA mutations, among other abnormalities, causes impaired cell functioning and replication, leading to apoptosis and neuronal atrophy (Irie et al. 2003, Forlenza & Miller 2006, Maes et al. 2009, Irie et al. 2001, Yan et al. 2013). This proposed mechanism has been supported by animal models wherein stress exposure, either by corticosterone or immobilization, generated impaired mitochondrial functioning within the cortex, hypothalamus, and hippocampus (Madrgial et al. 2001, Gong et al. 2011) along with depressive- and anxiety-like behaviors (Yang et al. 2016). Furthermore, alterations to mitochondrial proteins and functioning via the oxidative phosphorylation pathway within hippocampal synapses have been appreciated in rodent models of stress-induced depression (Xie et al. 2018). These findings indicate that mitochondria may play a critical role in neuronal integrity and synaptic transmission, thus, contributing to the development of stress-related disease.
Primary Mitochondrial Disease, the Brain, & Behavior
Stress is not the only pathway by which the brain is influenced by mitochondrial aberrations. For example, individuals with primary mitochondrial disorders have appreciable hyperintensities within the basal ganglia, cerebellum, and brainstem along with atrophy in the cerebellum and cerebrum and white matter leukoencephalopathy (Bricout et al. 2014, Friedman et al. 2010). Furthermore, animal models reveal that mtDNA mutations influence brain development, leading to appreciable malformations in the hippocampus and cerebrum (Ross et al. 2013) and that mitochondrial dysfunction is associated with neurodegeneration in the amygdala and hippocampus (Romero-Granados et al. 2011). Notably, structural changes to these brain regions are involved in the stress response and are appreciated in neuropsychiatric conditions, including anxiety, depression (Epel & Prather 2018), PTSD (Gurvits et al. 1996), Parkinson’s disease, and Alzheimer’s disease (Weintraub et al. 2012).
Alterations to mtDNA and mitochondrial function have also been associated with behavioral and psychiatric symptoms. In humans, mitochondrial disorders are often associated with psychiatric disorders, including schizophrenia, bipolar disorder, and depression (Shao et al. 2008, Rosebush et al. 2017). This link has been further supported by data from animal models. For example, rodents with mtDNA mutations have been shown to develop alterations in circadian rhythms, consistent with the manic phase of bipolar disorder, which were exacerbated by antidepressants and alleviated by lithium treatment (Kasahara et al. 2006). Additionally, manipulation of mitochondrial functioning in rodents has been shown to generate alterations in anxious social behaviors (Hollis et al. 2015). Specifically, mitochondrial dysfunction resulted in altered brain metabolism and produced anxiety-like behaviors with detrimental effects on social standing, whereas augmented mitochondrial functioning resulted in improved social standing in high-anxiety rodents (Hollis et al. 2015). In another rodent study, impaired mitochondrial activity interfered with learning acquisition and memory consolidation during maze tasks designed to test hippocampal and amygdala functioning (Romero-Granados et al. 2011).
The Role of Mitochondria in Neuropsychiatric Conditions
Mitochondrial impairments and alterations have been appreciated in individuals diagnosed with primary neuropsychiatric conditions. Positron emission tomography (PET) scans are a useful tool for monitoring cellular metabolism and have been employed in these populations to index mitochondrial performance. PET imaging has revealed reduced metabolic rates in the brains of patients with schizophrenia (Mitelman et al. 2018), MDD (Su et al. 2014), and bipolar disorder, particularly in the depressive phase (Shao et al. 2008). Furthermore, deficits in the components of the mitochondrial ETC have been associated with the pathogenesis of neuropsychiatric diseases, including Alzheimer’s and Parkinson’s diseases, MDD, schizophrenia, and bipolar disorder, with the strongest evidence in neurodegenerative disorders (Holper et al. 2019).
Mitochondrial involvement in neuropsychiatric disease states can also be evaluated using peripheral molecular biomarkers, namely mtDNAcn, a marker of mitochondrial biogenesis, and ccf-mtDNA, a potential indicator of mitochondrial stress. There is evidence of increased mtDNAcn and mtDNA deletions within the neurons of patients with Parkinson’s disease (Bury et al. 2017) as well as increased mtDNA deletions and oxidative damage in the brains of those with Alzheimer’s disease (Corral-Debrinski el al. 1994, Wang et al. 2005). Additional research has also appreciated a similar relationship between elevated mtDNAcn in peripheral blood cells and autism spectrum disorders (Chen et al. 2015, Yoo et al. 2016) and has suggested that mitochondrial dysfunction may underlie a subtype of autism (Goh et al. 2014).
Our group examined leukocyte mtDNAcn in 290 healthy unmedicated adults with and without a history of ELS, including childhood maltreatment and parental loss, as well as with and without a lifetime history of depressive, anxiety, and substance-use disorders. We observed that both ELS and lifetime psychopathology were independently associated with elevations of mtDNAcn and shorter leukocyte telomere length (Tyrka et al. 2016). Furthermore, in the largest study to date, Cai and colleagues studied 11,670 Chinese women with and without recurrent MDD, and found that higher salivary mtDNAcn and shorter telomere length were associated with MDD (Cai et al. 2015a). These authors also appreciated higher mtDNAcn in depressed patients versus controls in two additional smaller samples (Cai et al. 2015a). Moreover, other investigations have reported higher leukocyte mtDNA levels in adults with MDD (Tsujii et al. 2019) and suicide (Otsuka et al. 2017). However, some studies have found no difference in salivary or leukocyte mtDNAcn (Verhoeven et al. 2018, He et al. 2014, Tymofiyeva et al. 2018) or a decrease of mtDNAcn in older adults with MDD (Kim et al. 2011). Another recent study found no difference in mtDNAcn between a small number of depressed patients and healthy controls, however mtDNAcn was negatively correlated with severity of depression, and blood cells from depressed patients had impairments in repair and degradation of mtDNA in response to oxidative stress (Czarny et al. 2019). These biomarkers have also been examined in other psychiatric disorders. There is evidence for an association between higher mtDNAcn and lifetime anxiety disorders as well as substance use disorders (Tyrka et al. 2016), mixed depression, anxiety and stress-related disorders (Wang et al. 2017), and anxiety symptoms in adolescents (Tymofiyeva et al. 2018). However, one study found lower mtDNAcn in the granulocytes of male combat veterans with PTSD compared to those without PTSD (Bersani et al. 2016). Findings are mixed for bipolar disorder, with evidence of decreased mtDNAcn (Wang et al. 2017, Tsujii et al. 2019, Yamaki et al. 2018), increased mtDNAcn (Fries et al. 2017), or no significant difference in comparison with controls (de Sousa et al. 2014, Yamaki et al. 2018).
Variability in these findings could be due to differences in sample characteristics, such as severity and chronicity of the psychiatric disorder, the presence of other medical conditions, and medication use. Another potential source of this variability is the cell or tissue source of the mtDNA, as some studies have examined blood (which has a mixture of cell types) and others have sampled saliva (which contains a mixture of blood and oral cells). However, the largest study to date found that the cellular composition of saliva differed only slightly for cases and controls and did not explain the differences in mtDNA (Cai et al. 2015b). It is important to note that whole blood samples, and possibly saliva, contain mtDNA comprised of intracellular as well as ccf-mtDNA and, thus, studies of mtDNAcn in whole blood cannot distinguish between these effects. Additionally, recent work documents that ccf-mtDNA has a robust increase in response to psychosocial stress in healthy adults (Trumpff et al. 2019a, Hummel et al. 2018) and there is evidence that this may be due to stress-induced glucocorticoid signaling (Trumpff et al. 2019a). A recent study examined both ccf-mtDNA and intracellular mtDNA from peripheral blood mononuclear cells (PBMCs) and found that in comparison to healthy controls, depressed patients had elevations in ccf-mtDNA but not PBMC mtDNAcn (Lindqvist et al. 2018). In another study, ccf-mtDNA was also elevated in individuals following a suicide attempt when compared to non-suicidal controls (Lindqvist et al. 2016). Furthermore, ccf mtDNA was positively correlated with cortisol in the dexamethasone suppression test, linking impaired HPA axis function to cellular or mitochondrial damage (Lindqvist et al. 2016).
Stress & Neuropsychopathology: Mitochondrial Involvement
Considering that stress and trauma are major risk factors for psychopathology, that stress exposure activates systems known to be pathogenic for a variety of disorders, and that mitochondria play a prominent role in coordinating these responses, mitochondria may provide the biological link between psychosocial stress and psychiatric outcomes. This concept is supported by the preclinical work discussed above demonstrating that both stress-induced mitochondrial abnormalities and experimentally-induced mitochondrial dysfunction produce behavioral changes that resemble psychiatric symptoms. As previously discussed, recent studies have begun to examine mitochondrial indices in humans with psychiatric conditions; only two of those studies have assessed the role of stress exposure (Cai et al. 2015a, Tyrka et al. 2016).
In our group’s study of healthy, un-medicated adults, childhood adversity, including moderate-severe maltreatment on the Childhood Trauma Questionnaire (CTQ; Bernstein et al. 1994) and/or childhood parental loss (death or desertion identified via interview), was linked to elevated mtDNAcn, even among those with no lifetime psychiatric disorder (Tyrka et al. 2016). This effect was not accounted for by measures of subclinical psychiatric symptoms, recent perceived stress, or resilience. The effect for those with both early stress and lifetime psychiatric disorders was greater than for those with early stress alone, but not surprisingly, the burden of early adversity was also greater in the former group. Conversely, lifetime depressive, anxiety, and substance use disorders were also associated with elevations in mtDNAcn, even in the absence of early adversity, and this was not explained by recent perceived stress or symptom burden. Thus, early adversity and lifetime psychiatric conditions were each uniquely linked to mtDNAcn, possibly due to shared neuroendocrine, inflammatory, or oxidative stress mechanisms.
Our group also recently reported similar findings in a sample of 256 maltreated and comparison preschool-aged children (Ridout et al. 2019). Maltreatment, as well as other measures of adversity, were significant positive predictors of salivary mtDNAcn. Furthermore, elevations in mtDNAcn were also associated with the presence of internalizing behaviors, consistent with symptoms of anxiety and depression.
In the large study of Chinese women with and without recurrent MDD introduced above, Cai and colleagues reported that higher mtDNAcn and shorter telomere length were seen with both a measure of 16 lifetime traumatic events as well as an abbreviated measure of childhood sexual abuse, and that the maltreatment effects were stronger with increasingly severe abuse (Cai et al. 2015a). However, the association of these stress measures and the molecular measures were conditional on the presence of MDD. Furthermore, in contrast to the study by our group (Tyrka et al. 2016), there was no significant effect of the measured stress exposures among non-MDD controls. Conversely, MDD had associations with higher mtDNAcn and shorter telomere length that were independent of the stress measures.
These authors also examined an animal stress model and found that four weeks of stress exposure resulted in increased mtDNAcn and reduced telomere length in blood and saliva (Cai et al. 2015a). The increases in mtDNAcn at four weeks were associated with impaired mitochondrial function in the liver, and at least partially resolved after four more weeks without stress exposure. Further experiments revealed that administration of the glucocorticoid corticosterone was sufficient to induce changes to mtDNAcn and telomere length after four weeks, suggesting a mechanism for these molecular signatures via the HPA-axis (Cai et al. 2015a). The authors hypothesized that the extent and persistence of molecular changes due to adversity may be influenced by individual genetic or additional unmeasured environmental factors and those with depressive manifestations may have larger or longer-lasting effects. In a related study, a greater mtDNA mutation load was seen with depressed participants compared to controls, with at least one of these mutations associated with the amount of mtDNA (Cai et al. 2015b). Furthermore, the authors used animal models to demonstrate that this increase in mutation burden can be generated from stress exposure, thus drawing together changes in mitochondrial genomic sequence and copy number with the stress response (Cai et al. 2015b).
Most work assessing mitochondria in humans has examined mtDNA, which is easily and reliably isolated and measured from frozen tissue. Alterations in mtDNAcn are thought to reflect mitochondrial biogenesis and increases may occur in response to impairments in mitochondrial function (Giordano et al. 2014). However, mtDNAcn is an indirect measure, and variability in associations with psychiatric conditions may reflect a variety of underlying processes affecting mtDNAcn. Picard and colleagues (Picard et al. 2016) developed the Mitochondrial Health Index (MHI), which integrates measures of both mitochondrial mass (how many mitochondria or their volume) and mitochondrial enzymatic activity (reflecting mitochondrial function) to represent the overall functional capacity of mitochondria. In a sample of 91 mothers, approximately half of whom were experiencing chronic stress in the form of caring for a child with autism, both stress exposure and mood were related to MHI, with lower MHI in individuals with more perceived stress, and higher MHI associated with positive mood. Interestingly, there was no significant relationship between the individual components of the MHI and caregiving status, indicating the MHI may be a valuable composite measure of mitochondrial functional capacity.
It is also possible to directly measure mitochondrial functional capacity, but reliable measurement of mitochondrial activity requires live, intact cells, thus curbing its methodological feasibility for certain studies (van der Windt et al. 2016). Although there are concerns regarding mitochondrial viability from frozen tissue, some investigations have analyzed mitochondrial functioning using cryogenically preserved cells. In a study of 30 women with variable levels of childhood maltreatment as measured by the CTQ, Boeck et al. (2016) found that maltreatment was linked to greater ROS production, oxidative stress, and mitochondrial oxygen consumption in a dose-dependent fashion, and ROS and mitochondrial activity were associated with the release of inflammatory cytokines.
The literature described above highlights how chronic or severe psychosocial stressors can alter biological processes that accelerate cellular aging and potentially increase disease risk through mitochondrial mechanisms. It is also important to acknowledge that age-related, neurological, and psychiatric illnesses can be an inherent source of stress, thus, may impact mitochondrial function, independent of other sources of adversity. Moreover, mitochondrial dysfunction occurs through inborn genetic errors and disease states that are unrelated to psychosocial stressors. Meaningful early progress has begun to characterize the role of mitochondria in adversity, the aging process, and the pathophysiology of stress- and age-related disease processes. There is increasing awareness of the function of mitochondria in disease pathogenesis, but there is still more progress to be made. In the remaining section we describe the limitations of current evidence and provide suggestions for future directions.
CURRENT LIMITATIONS & FUTURE DIRECTIONS
Accumulating evidence from animal models and recent human studies has identified mitochondria as a critical intersection point for psychosocial factors and stress physiology. Despite improved understanding of the wide-ranging roles for this organelle in regulating key stress-related processes, many questions remain. The mechanisms that underlie these processes are highly complex, due in part to their dynamic integration with neuroendocrine, inflammatory, and epigenetic systems––all of which are influenced to varying degrees by genetic, environmental, and developmental factors. As illustrated in Figure 1, the relationship between stress, mitochondria, and psychiatric disorders is non-linear, with bidirectional interaction among multiple interwoven physiological systems. Very few human studies have been conducted thus far, and the results are mixed, highlighting the limitations to our current understanding and encouraging careful consideration by investigators moving forward.
While animal models have provided critical insight into the mitochondrial response to acute and chronic stress exposure, and can identify behaviors similar to those seen with psychiatric conditions, human studies are necessary to understand the complex cognitive and affective phenotypes observed in stress-associated psychiatric disorders. Clinical studies have assessed mitochondrial function in response to stressors of varying nature, chronicity, and reported age of stress exposure. As with any human study, there are complexities associated with measuring and disentangling sources of variance. Whereas animal models allow for strict control of genetic and environmental factors as well as experimental designs that isolate causal factors and directly measure outcomes, human studies inherently present the opportunity for interference by a vast number of potential confounders––some of which may serve as critical moderators or mediators of disease risk. Participants have individual genetic predispositions, unique personal experiences, and varying degrees of resilience and coping. They have distinct patterns of health-influencing behaviors––some of which may be clandestine, potential recent stressors, substance histories, and underlying medical problems that may meaningfully contribute to mitochondrial functioning. Medications, such as antidepressants, appear to alter energy metabolism and may affect mitochondrial processes (Adzic, 2016). This broad variability makes it difficult to precisely measure the relationship of stress exposure and mitochondrial function in human samples. Intensive measurement, or control of a broad range of exposures, behaviors, and health conditions is costly and generally only possible in smaller studies, which may have limited generalizability. However, when these factors go unmeasured, error is increased and effect sizes are diminished.
Measures of mitochondrial mass and function are limited by the lack of universal procedures for a number of laboratory processes, such as storing and collecting samples from these living, metabolically active organelles. The methodological choices made by investigators may substantially impact findings. While freezing samples does allow for analytes to be run en masse, it may also increase the likelihood of degradation of the sample, resulting in modified biological activity of the sample (Han et al. 2019). This is particularly an issue for studies of mitochondrial functional capacity, whereas mtDNA is more stable. Furthermore, there is a tremendous amount of variation in tissue sampling, including in vitro versus in vivo studies, cells from blood versus saliva, tissues from living versus post-mortem samples, and cells from human versus animal sources, among others. As with other examinations of peripheral markers of biological processes relevant to stress and risk for disease, the inclusion of whole blood or saliva containing mixed cell populations raises cellular heterogeneity as a possible confounder (Han et al. 2019).
Although research on the role of mitochondria in the biological response to adversity, trauma, and psychopathology is at a relatively early stage, most of the complexities discussed above apply to the preceding decades of research on the biological embedding of stress exposure and the molecular mechanisms and sequelae of psychiatric conditions. The state of the field has also opened the door to a wide variety of additional questions, including the role of biological sex, which influences mitochondrial function (Klinge 2017, Trumpff et al. 2019b). Other questions include: how reversible are these mitochondrial changes and what is the role of treatment, via psychotherapy, other behavioral approaches, or psychotropic medications, in helping to reverse these changes or stem their effects?
In this review, we have discussed emerging evidence indicating that chronic stress generates maladaptive alterations in mitochondria, which contribute to allostatic processes, ultimately promoting aging and disease. Together with the extensive body of literature on early adversity, these findings collectively highlight the critical value of early screening and intervention for childhood maltreatment. Preliminary assessments of the impact of early intervention reveal that trauma-informed treatments may help to reverse physiological effects of childhood adversity (Slopen et al. 2014). The research reviewed here provides a foundation for the opportunity to expand our understanding of risk and resilience and identify therapeutic interventions targeting these biological mechanisms. Further studies are needed that seek to elucidate how mitochondria contribute to neuronal circuits that regulate specific behaviors, vulnerability, and resilience to stress exposure. Continued efforts across relevant fields should be focused on translating this emerging area of research into clinical applications that improve the health outcomes of vulnerable patient populations.
REFERENCES
- 1.Adzic M, Brkic Z, Bulajic S, Mitic M, Radojcic MB. 2016. Antidepressant Action on Mitochondrial Dysfunction in Psychiatric Disorders. Drug Dev Res 77: 400–06 [DOI] [PubMed] [Google Scholar]
- 2.Archibald JM. 2015. Endosymbiosis and Eukaryotic Cell Evolution. Current biology : CB 25: R911–21 [DOI] [PubMed] [Google Scholar]
- 3.Basu A, McLaughlin KA, Misra S, Koenen KC. 2017. Childhood Maltreatment and Health Impact: The Examples of Cardiovascular Disease and Type 2 Diabetes Mellitus in Adults. Clin Psychol (New York) 24: 125–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumeister D, Akhtar R, Ciufolini S, Pariante CM, Mondelli V. 2016. Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-alpha. Molecular psychiatry 21: 642–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berens AE, Jensen SKG, Nelson CA 3rd. 2017. Biological embedding of childhood adversity: from physiological mechanisms to clinical implications. BMC Med 15: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein DP, Fink L, Handelsman L, Foote J, Lovejoy M, et al. 1994. Initial reliability and validity of a new retrospective measure of child abuse and neglect. Am J Psychiatry 151: 1132–6 [DOI] [PubMed] [Google Scholar]
- 7.Bersani FS, Morley C, Lindqvist D, Epel ES, Picard M, et al. 2016. Mitochondrial DNA copy number is reduced in male combat veterans with PTSD. Prog Neuropsychopharmacol Biol Psychiatry 64: 10–7 [DOI] [PubMed] [Google Scholar]
- 8.Boeck C, Koenig AM, Schury K, Geiger ML, Karabatsiakis A, et al. 2016. Inflammation in adult women with a history of child maltreatment: The involvement of mitochondrial alterations and oxidative stress. Mitochondrion 30: 197–207 [DOI] [PubMed] [Google Scholar]
- 9.Bratic A, Larsson NG. 2013. The role of mitochondria in aging. The Journal of clinical investigation 123: 951–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bricout M, Grevent D, Lebre AS, Rio M, Desguerre I, et al. 2014. Brain imaging in mitochondrial respiratory chain deficiency: combination of brain MRI features as a useful tool for genotype/phenotype correlations. Journal of medical genetics 51: 429–35 [DOI] [PubMed] [Google Scholar]
- 11.Bunea IM, Szentagotai-Tatar A, Miu AC. 2017. Early-life adversity and cortisol response to social stress: a meta-analysis. Translational psychiatry 7: 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bury AG, Pyle A, Elson JL, Greaves L, Morris CM, et al. 2017. Mitochondrial DNA changes in pedunculopontine cholinergic neurons in Parkinson disease. Annals of neurology 82: 1016–21 [DOI] [PubMed] [Google Scholar]
- 13.Cai N, Chang S, Li Y, Li Q, Hu J, et al. 2015a. Molecular signatures of major depression. Current biology CB 25: 1146–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai N, Li Y, Chang S, Liang J, Lin C, et al. 2015b. Genetic Control over mtDNA and Its Relationship to Major Depressive Disorder. Current biology : CB 25: 3170–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH. 2010. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 35: 2617–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandel NS. 2015. Evolution of Mitochondria as Signaling Organelles. Cell Metab 22: 204–6 [DOI] [PubMed] [Google Scholar]
- 17.Chen S, Li Z, He Y, Zhang F, Li H, et al. 2015. Elevated mitochondrial DNA copy number in peripheral blood cells is associated with childhood autism. BMC psychiatry 15: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, et al. 1994. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 23: 471–6 [DOI] [PubMed] [Google Scholar]
- 19.Czarny P, Wigner P, Strycharz J, Swiderska E, Synowiec E, et al. 2019. Mitochondrial DNA copy number, damage, repair and degradation in depressive disorder. The world journal of biological psychiatry : the official journal of the World Federation of Societies of Biological Psychiatry: 1–11 [DOI] [PubMed] [Google Scholar]
- 20.Danese A, McEwen BS. 2012. Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiology & behavior 106: 29–39 [DOI] [PubMed] [Google Scholar]
- 21.Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. 2007. Childhood maltreatment predicts adult inflammation in a life-course study. Proceedings of the National Academy of Sciences of the United States of America 104: 1319–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danese A, Lewis SJ. 2017. Psychoneuroimmunology of Early-Life Stress: The Hidden Wounds of Childhood Trauma? Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 42: 99–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Sousa RT, Uno M, Zanetti MV, Shinjo SM, Busatto GF, et al. 2014. Leukocyte mitochondrial DNA copy number in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 48: 32–5 [DOI] [PubMed] [Google Scholar]
- 24.Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, et al. 2009. Dynamic regulation of mitochondrial function by glucocorticoids. Proceedings of the National Academy of Sciences of the United States of America 106: 3543–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Epel ES, Prather AA. 2018. Stress, Telomeres, and Psychopathology: Toward a Deeper Understanding of a Triad of Early Aging. Annual review of clinical psychology 14: 371–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flannery PJ, Trushina E. 2019. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol Cell Neurosci 98: 109–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forlenza MJ, Miller GE. 2006. Increased serum levels of 8-hydroxy-2’-deoxyguanosine in clinical depression. Psychosomatic medicine 68: 1–7 [DOI] [PubMed] [Google Scholar]
- 28.Friedman SD, Shaw DW, Ishak G, Gropman AL, Saneto RP. 2010. The use of neuroimaging in the diagnosis of mitochondrial disease. Developmental disabilities research reviews 16: 129–35 [DOI] [PubMed] [Google Scholar]
- 29.Fries GR, Bauer IE, Scaini G, Wu MJ, Kazimi IF, et al. 2017. Accelerated epigenetic aging and mitochondrial DNA copy number in bipolar disorder. Translational psychiatry 7: 1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, et al. 2014. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain : a journal of neurology 137: 335–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Godoy LD, Rossignoli MT, Delfino-Pereira P, Garcia-Cairasco N, de Lima Umeoka EH. 2018. A Comprehensive Overview on Stress Neurobiology: Basic Concepts and Clinical Implications. Frontiers in behavioral neuroscience 12: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goh S, Dong Z, Zhang Y, DiMauro S, Peterson BS. 2014. Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder: evidence from brain imaging. JAMA psychiatry 71: 665–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong Y, Chai Y, Ding JH, Sun XL, Hu G. 2011. Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neuroscience letters 488: 76–80 [DOI] [PubMed] [Google Scholar]
- 34.Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, et al. 2016. Mitochondrial diseases. Nature reviews. Disease primers 2: 16080. [DOI] [PubMed] [Google Scholar]
- 35.Gurvits TV, Shenton ME, Hokama H, Ohta H, Lasko NB, et al. 1996. Magnetic resonance imaging study of hippocampal volume in chronic, combat-related posttraumatic stress disorder. Biological psychiatry 40: 1091–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han LKM, Verhoeven JE, Tyrka AR, Penninx B, Wolkowitz OM, et al. 2019. Accelerating research on biological aging and mental health: Current challenges and future directions. Psychoneuroendocrinology 106: 293–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanssen LM, Schutte NS, Malouff JM, Epel ES. 2017. The Relationship Between Childhood Psychosocial Stressor Level and Telomere Length: A Meta-Analysis. Health Psychol Res 5: 6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartman S, Li Z, Nettle D, Belsky J. 2017. External-environmental and internal-health early life predictors of adolescent development. Dev Psychopathol 29: 1839–49 [DOI] [PubMed] [Google Scholar]
- 39.Havranek EP, Mujahid MS, Barr DA, Blair IV, Cohen MS, et al. 2015. Social Determinants of Risk and Outcomes for Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 132: 873–98 [DOI] [PubMed] [Google Scholar]
- 40.He Y, Tang J, Li Z, Li H, Liao Y, et al. 2014. Leukocyte mitochondrial DNA copy number in blood is not associated with major depressive disorder in young adults. PLoS One 9: e96869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann A, Spengler D. 2018. The Mitochondrion as Potential Interface in Early-Life Stress Brain Programming. Frontiers in behavioral neuroscience 12: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollis F, van der Kooij MA, Zanoletti O, Lozano L, Canto C, Sandi C. 2015. Mitochondrial function in the brain links anxiety with social subordination. Proceedings of the National Academy of Sciences of the United States of America 112: 15486–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holper L, Ben-Shachar D, Mann JJ. 2019. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 44: 837–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hummel EM, Hessas E, Muller S, Beiter T, Fisch M, et al. 2018. Cell-free DNA release under psychosocial and physical stress conditions. Translational psychiatry 8: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hwang IW, Hong JH, Kwon BN, Kim HJ, Lee NR, et al. 2017. Association of mitochondrial DNA 10398 A/G polymorphism with attention deficit and hyperactivity disorder in Korean children. Gene 630: 8–12 [DOI] [PubMed] [Google Scholar]
- 46.Irie M, Asami S, Ikeda M, Kasai H. 2003. Depressive state relates to female oxidative DNA damage via neutrophil activation. Biochemical and biophysical research communications 311: 1014–8 [DOI] [PubMed] [Google Scholar]
- 47.Irie M, Asami S, Nagata S, Ikeda M, Miyata M, Kasai H. 2001. Psychosocial factors as a potential trigger of oxidative DNA damage in human leukocytes. Japanese journal of cancer research : Gann 92: 367–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jakubowski KP, Cundiff JM, Matthews KA. 2018. Cumulative childhood adversity and adult cardiometabolic disease: A meta-analysis. Health Psychol 37: 701–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juster RP, Russell JJ, Almeida D, Picard M. 2016. Allostatic load and comorbidities: A mitochondrial, epigenetic, and evolutionary perspective. Dev Psychopathol 28: 1117–46 [DOI] [PubMed] [Google Scholar]
- 50.Kasahara T, Kato T. 2018. What Can Mitochondrial DNA Analysis Tell Us About Mood Disorders? Biological psychiatry 83: 731–38 [DOI] [PubMed] [Google Scholar]
- 51.Kasahara T, Kubota M, Miyauchi T, Noda Y, Mouri A, et al. 2006. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Molecular psychiatry 11: 577–93, 23 [DOI] [PubMed] [Google Scholar]
- 52.Khoury JE, Bosquet Enlow M, Plamondon A, Lyons-Ruth K. 2019. The association between adversity and hair cortisol levels in humans: A meta-analysis. Psychoneuroendocrinology 103: 104–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kilian R, Becker T, Kruger K, Schmid S, Frasch K. 2006. Health behavior in psychiatric in-patients compared with a German general population sample. Acta Psychiatr Scand 114: 242–8 [DOI] [PubMed] [Google Scholar]
- 54.Kim MY, Lee JW, Kang HC, Kim E, Lee DC. 2011. Leukocyte mitochondrial DNA (mtDNA) content is associated with depression in old women. Arch Gerontol Geriatr 53: e218–21 [DOI] [PubMed] [Google Scholar]
- 55.Klinge CM. 2017. Estrogens regulate life and death in mitochondria. J Bioenerg Biomembr 49: 307–24 [DOI] [PubMed] [Google Scholar]
- 56.Lagouge M, Larsson NG. 2013. The role of mitochondrial DNA mutations and free radicals in disease and ageing. Journal of internal medicine 273: 529–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewis M 1997. Altering fate: why the past does not predict the future. New York: Guilford Press. Xii, 238 [Google Scholar]
- 58.Li Z, He Y, Wang D, Tang J, Chen X. 2017. Association between childhood trauma and accelerated telomere erosion in adulthood: A meta-analytic study. Journal of psychiatric research 93: 64–71 [DOI] [PubMed] [Google Scholar]
- 59.Liesa M, Shirihai OS. 2013. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17: 491–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lindqvist D, Fernstrom J, Grudet C, Ljunggren L, Traskman-Bendz L, et al. 2016. Increased plasma levels of circulating cell-free mitochondrial DNA in suicide attempters: associations with HPA-axis hyperactivity. Translational psychiatry 6: e971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lindqvist D, Wolkowitz OM, Picard M, Ohlsson L, Bersani FS, et al. 2018. Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 43: 1557–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lupien SJ, Juster RP, Raymond C, Marin MF. 2018. The effects of chronic stress on the human brain: From neurotoxicity, to vulnerability, to opportunity. Frontiers in neuroendocrinology 49: 91–105 [DOI] [PubMed] [Google Scholar]
- 63.Madrigal JL, Olivenza R, Moro MA, Lizasoain I, Lorenzo P, et al. 2001. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 24: 420–9 [DOI] [PubMed] [Google Scholar]
- 64.Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E. 2009. Increased 8-hydroxy-deoxyguanosine, a marker of oxidative damage to DNA, in major depression and myalgic encephalomyelitis / chronic fatigue syndrome. Neuro endocrinology letters 30: 715–22 [PubMed] [Google Scholar]
- 65.Magistretti PJ, Allaman I. 2015. A cellular perspective on brain energy metabolism and functional imaging. Neuron 86: 883–901 [DOI] [PubMed] [Google Scholar]
- 66.Marsland AL, Walsh C, Lockwood K, John-Henderson NA. 2017. The effects of acute psychological stress on circulating and stimulated inflammatory markers: A systematic review and meta-analysis. Brain, behavior, and immunity 64: 208–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McEwen BS. 2007. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiological reviews 87: 873–904 [DOI] [PubMed] [Google Scholar]
- 68.McEwen BS, Morrison JH. 2013. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron 79: 16–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McEwen BS, Stellar E. 1993. Stress and the individual. Mechanisms leading to disease. Archives of internal medicine 153: 2093–101 [PubMed] [Google Scholar]
- 70.McFarland R, Taylor RW, Turnbull DM. 2010. A neurological perspective on mitochondrial disease. Lancet Neurol 9: 829–40 [DOI] [PubMed] [Google Scholar]
- 71.Meyer A, Laverny G, Bernardi L, Charles AL, Alsaleh G, et al. 2018. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front Immunol 9: 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meyer T, Wirtz PH. 2018. Mechanisms of Mitochondrial Redox Signaling in Psychosocial Stress-Responsive Systems: New Insights into an Old Story. Antioxid Redox Signal 28: 760–72 [DOI] [PubMed] [Google Scholar]
- 73.Mitelman SA, Bralet MC, Mehmet Haznedar M, Hollander E, Shihabuddin L, et al. 2018. Positron emission tomography assessment of cerebral glucose metabolic rates in autism spectrum disorder and schizophrenia. Brain Imaging Behav 12: 532–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, et al. 2013. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS medicine 10: e1001577; discussion e77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nanni V, Uher R, Danese A. 2012. Childhood maltreatment predicts unfavorable course of illness and treatment outcome in depression: a meta-analysis. Am J Psychiatry 169: 141–51 [DOI] [PubMed] [Google Scholar]
- 76.Nemeroff CB. 2016. Paradise Lost: The Neurobiological and Clinical Consequences of Child Abuse and Neglect. Neuron 89: 892–909 [DOI] [PubMed] [Google Scholar]
- 77.Otsuka I, Izumi T, Boku S, Kimura A, Zhang Y, et al. 2017. Aberrant telomere length and mitochondrial DNA copy number in suicide completers. Scientific reports 7: 3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Picard M, Juster RP, McEwen BS. 2014a. Mitochondrial allostatic load puts the ‘gluc’ back in glucocorticoids. Nature reviews. Endocrinology 10: 303–10 [DOI] [PubMed] [Google Scholar]
- 79.Picard M, McEwen BS. 2018a. Psychological Stress and Mitochondria: A Conceptual Framework. Psychosomatic medicine 80: 126–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Picard M, McEwen BS. 2018b. Psychological Stress and Mitochondria: A Systematic Review. Psychosomatic medicine 80: 141–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Picard M, McEwen BS, Epel ES, Sandi C. 2018a. An energetic view of stress: Focus on mitochondria. Frontiers in neuroendocrinology 49: 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Picard M, Prather AA, Puterman E, Cuillerier A, Coccia M, et al. 2018b. A Mitochondrial Health Index Sensitive to Mood and Caregiving Stress. Biological psychiatry 84: 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Picard M, Wallace DC, Burelle Y. 2016. The rise of mitochondria in medicine. Mitochondrion 30: 105–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, et al. 2014b. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proceedings of the National Academy of Sciences of the United States of America 111: E4033–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Psarra AM, Sekeris CE. 2011. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochimica et biophysica acta 1813: 1814–21 [DOI] [PubMed] [Google Scholar]
- 86.Rich-Edwards JW, Spiegelman D, Lividoti Hibert EN, Jun HJ, Todd TJ, et al. 2010. Abuse in childhood and adolescence as a predictor of type 2 diabetes in adult women. Am J Prev Med 39: 529–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ridout KK, Carpenter LL, Tyrka AR. 2016. The Cellular Sequelae of Early Stress: Focus on Aging and Mitochondria. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 41: 388–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ridout KK, Khan M, Ridout SJ. 2018. Adverse Childhood Experiences Run Deep: Toxic Early Life Stress, Telomeres, and Mitochondrial DNA Copy Number, the Biological Markers of Cumulative Stress. BioEssays : news and reviews in molecular, cellular and developmental biology 40: e1800077. [DOI] [PubMed] [Google Scholar]
- 89.Ridout KK, Parade SH, Kao HT, Magnan S, Seifer R, et al. 2019. Childhood maltreatment, behavioral adjustment, and molecular markers of cellular aging in preschool-aged children: A cohort study. Psychoneuroendocrinology 107: 261–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rohleder N 2014. Stimulation of systemic low-grade inflammation by psychosocial stress. Psychosomatic medicine 76: 181–9 [DOI] [PubMed] [Google Scholar]
- 91.Romero-Granados R, Fontan-Lozano A, Aguilar-Montilla FJ, Carrion AM. 2011. Postnatal proteasome inhibition induces neurodegeneration and cognitive deficiencies in adult mice: a new model of neurodevelopment syndrome. PLoS One 6: e28927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rosebush PI, Anglin RE, Rasmussen S, Mazurek MF. 2017. Mental illness in patients with inherited mitochondrial disorders. Schizophr Res 187: 33–37 [DOI] [PubMed] [Google Scholar]
- 93.Ross JM, Stewart JB, Hagstrom E, Brene S, Mourier A, et al. 2013. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501: 412–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sagan L 1967. On the origin of mitosing cells. J Theor Biol 14: 255–74 [DOI] [PubMed] [Google Scholar]
- 95.Sapolsky RM, Romero LM, Munck AU. 2000. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 21: 55–89 [DOI] [PubMed] [Google Scholar]
- 96.Segerstrom SC, Miller GE. 2004. Psychological stress and the human immune system: a meta-analytic study of 30 years of inquiry. Psychol Bull 130: 601–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shao L, Martin MV, Watson SJ, Schatzberg A, Akil H, et al. 2008. Mitochondrial involvement in psychiatric disorders. Annals of medicine 40: 281–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, et al. 2014. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect 122: 1271–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shonkoff JP, Boyce WT, McEwen BS. 2009. Neuroscience, molecular biology, and the childhood roots of health disparities: building a new framework for health promotion and disease prevention. JAMA 301: 2252–9 [DOI] [PubMed] [Google Scholar]
- 100.Shutt TE, McBride HM. 2013. Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochimica et biophysica acta 1833: 417–24 [DOI] [PubMed] [Google Scholar]
- 101.Slopen N, Koenen KC, Kubzansky LD. 2014. Cumulative adversity in childhood and emergent risk factors for long-term health. J Pediatr 164: 631–8 e1–2 [DOI] [PubMed] [Google Scholar]
- 102.Smith RL, Soeters MR, Wust RCI, Houtkooper RH. 2018. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr Rev 39: 489–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Srivastava R, Faust T, Ramos A, Ishizuka K, Sawa A. 2018. Dynamic Changes of the Mitochondria in Psychiatric Illnesses: New Mechanistic Insights From Human Neuronal Models. Biological psychiatry 83: 751–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Steib K, Schaffner I, Jagasia R, Ebert B, Lie DC. 2014. Mitochondria modify exercise-induced development of stem cell-derived neurons in the adult brain. The Journal of neuroscience : the official journal of the Society for Neuroscience 34: 6624–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Steptoe A, Hamer M, Chida Y. 2007. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain, behavior, and immunity 21: 901–12 [DOI] [PubMed] [Google Scholar]
- 106.Sterling P & Eyer J. 1988. Allostatsis: A new paradigm to explain arousal pathology. Handbook of life stress, cognition, and health. Chichester ; New York: Wiley. xxxiii, pp. 629–49 [Google Scholar]
- 107.Su L, Cai Y, Xu Y, Dutt A, Shi S, Bramon E. 2014. Cerebral metabolism in major depressive disorder: a voxel-based meta-analysis of positron emission tomography studies. BMC psychiatry 14: 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takizawa R, Danese A, Maughan B, Arseneault L. 2015. Bullying victimization in childhood predicts inflammation and obesity at mid-life: a five-decade birth cohort study. Psychological medicine 45: 2705–15 [DOI] [PubMed] [Google Scholar]
- 109.Tottenham N, Galvan A. 2016. Stress and the adolescent brain: Amygdala-prefrontal cortex circuitry and ventral striatum as developmental targets. Neurosci Biobehav Rev 70: 217–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Trumpff C, Marsland AL, Basualto-Alarcon C, Martin JL, Carroll JE, et al. 2019a. Acute psychological stress increases serum circulating cell-free mitochondrial DNA. Psychoneuroendocrinology 106: 268–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trumpff C, Marsland AL, Sloan RP, Kaufman BA, Picard M. 2019b. Predictors of ccf-mtDNA reactivity to acute psychological stress identified using machine learning classifiers: A proof-of-concept. Psychoneuroendocrinology 107: 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tsujii N, Otsuka I, Okazaki S, Yanagi M, Numata S, et al. 2019. Mitochondrial DNA Copy Number Raises the Potential of Left Frontopolar Hemodynamic Response as a Diagnostic Marker for Distinguishing Bipolar Disorder From Major Depressive Disorder. Front Psychiatry 10: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tymofiyeva O, Henje Blom E, Ho TC, Connolly CG, Lindqvist D, et al. 2018. High levels of mitochondrial DNA are associated with adolescent brain structural hypoconnectivity and increased anxiety but not depression. J Affect Disord 232: 283–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tyrka AR, Parade SH, Price LH, Kao HT, Porton B, et al. 2016. Alterations of Mitochondrial DNA Copy Number and Telomere Length With Early Adversity and Psychopathology. Biological psychiatry 79: 78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van der Windt GJ, Chang CH, Pearce EL. 2016. Measuring Bioenergetics in T Cells Using a Seahorse Extracellular Flux Analyzer. Curr Protoc Immunol 113: 3 16B 1–3 16B 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Verhoeven JE, Revesz D, Picard M, Epel EE, Wolkowitz OM, et al. 2018. Depression, telomeres and mitochondrial DNA: between- and within-person associations from a 10-year longitudinal study. Molecular psychiatry 23: 850–57 [DOI] [PubMed] [Google Scholar]
- 117.Viron MJ, Stern TA. 2010. The impact of serious mental illness on health and healthcare. Psychosomatics 51: 458–65 [DOI] [PubMed] [Google Scholar]
- 118.Wallace DC. 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual review of genetics 39: 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. 2005. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. Journal of neurochemistry 93: 953–62 [DOI] [PubMed] [Google Scholar]
- 120.Wang X, Sundquist K, Rastkhani H, Palmer K, Memon AA, Sundquist J. 2017. Association of mitochondrial DNA in peripheral blood with depression, anxiety and stress- and adjustment disorders in primary health care patients. Eur Neuropsychopharmacol 27: 751–58 [DOI] [PubMed] [Google Scholar]
- 121.Wegman HL, Stetler C. 2009. A meta-analytic review of the effects of childhood abuse on medical outcomes in adulthood. Psychosomatic medicine 71: 805–12 [DOI] [PubMed] [Google Scholar]
- 122.Wei YB, Martinsson L, Liu JJ, Forsell Y, Schalling M, et al. 2016. hTERT genetic variation in depression. J Affect Disord 189: 62–9 [DOI] [PubMed] [Google Scholar]
- 123.Weintraub D, Dietz N, Duda JE, Wolk DA, Doshi J, et al. 2012. Alzheimer’s disease pattern of brain atrophy predicts cognitive decline in Parkinson’s disease. Brain : a journal of neurology 135: 170–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Widom CS, Horan J, Brzustowicz L. 2015. Childhood maltreatment predicts allostatic load in adulthood. Child Abuse Negl 47: 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xie H, Huang H, Tang M, Wu Y, Huang R, et al. 2018. iTRAQ-Based Quantitative Proteomics Suggests Synaptic Mitochondrial Dysfunction in the Hippocampus of Rats Susceptible to Chronic Mild Stress. Neurochem Res 43: 2372–83 [DOI] [PubMed] [Google Scholar]
- 126.Yamaki N, Otsuka I, Numata S, Yanagi M, Mouri K, et al. 2018. Mitochondrial DNA copy number of peripheral blood in bipolar disorder: The present study and a meta-analysis. Psychiatry Res 269: 115–17 [DOI] [PubMed] [Google Scholar]
- 127.Yan MH, Wang X, Zhu X. 2013. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free radical biology & medicine 62: 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang N, Ren Z, Zheng J, Feng L, Li D, et al. 2016. 5-(4-hydroxy-3-dimethoxybenzylidene)-rhodanine (RD-1)-improved mitochondrial function prevents anxiety- and depressive-like states induced by chronic corticosterone injections in mice. Neuropharmacology 105: 587–93 [DOI] [PubMed] [Google Scholar]
- 129.Yoo HJ, Park M, Kim SA. 2017. Difference in mitochondrial DNA copy number in peripheral blood cells between probands with autism spectrum disorders and their unaffected siblings. The world journal of biological psychiatry : the official journal of the World Federation of Societies of Biological Psychiatry 18: 151–56 [DOI] [PubMed] [Google Scholar]
- 130.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, et al. 2010. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zimmermann KS, Richardson R, Baker KD. 2019. Maturational Changes in Prefrontal and Amygdala Circuits in Adolescence: Implications for Understanding Fear Inhibition during a Vulnerable Period of Development. Brain Sci 9 [DOI] [PMC free article] [PubMed] [Google Scholar]