

Abstract
DNA methylation is one of the epigenetic changes, which plays a major role in regulating gene expression and, thus, many biological processes and diseases. There are several methods for determining the methylation of DNA samples. However, selecting the most appropriate method for answering biological questions appears to be a challenging task. The primary methods in DNA methylation focused on identifying the state of methylation of the examined genes and determining the total amount of 5-methyl cytosine. The study of DNA methylation at a large scale of genomic levels became possible following the use of microarray hybridization technology. The new generation of sequencing platforms now allows the preparation of genomic maps of DNA methylation at the single-open level. This review includes the majority of methods available to date, introducing the most widely used methods, the bisulfite treatment, biological identification, and chemical cutting along with their advantages and disadvantages. The techniques are then scrutinized according to their robustness, high throughput capabilities, and cost.
1. Introduction
Epigenetics is a broad concept used to describe various reversible genomic changes [1]. Epigenetics, in the literal sense of “Beyond Genetics,” refers to inherited changes in gene expression, which result from changes in chromosomes without altering the DNA sequence. Epigenetic mechanisms of gene regulation include DNA methylation, covalent changes in histones, and noncoding and messenger RNAs [2, 3]. DNA methylation, the covalent changes in cytosine, is one of the most widely studied changes in the field of epigenetics and provided a molecular mechanism through which the expression of the gene can be regulated [4, 5]. This process is capable of preserving and transmitting epigenetic information through the replication of DNA and cell division [6]. DNA methylation is associated with a wide range of biological processes, including deactivation of chromosome X, genomic imprinting, stem cell differentiation, gene expression control, and chromosomal stability [7]. These findings indicate that DNA methylation is one of the most important modifications which play an essential role in regulating the growth of the cells and their proliferation.
Under the catalysis of the MTase methyltransferase enzyme, the fifth carbon in the 5′-CG-3′ dinucleotide can selectively be replaced by a methyl group to form 5-methyl cytosine. About 60-90% of 5′-CG-3 dinucleotides are methylated in the eukaryotes, and the nonmethylated 5′-CG-3 dinucleotides often accumulate in the promoter of structural genes and the starting point for their transcription to form CpG islands [8]. The methylation of non-CpG regions in CHH and CHG (H = A, C, T) is seen in the embryonic stem cells and in the plants [9]. The DNA methylation pattern is hereditary and mutable [10]. The normal expression of the parental allele results in the correct expression of the gene, while abnormal DNA methylation in the parental allele typically causes different cancers, genetic diseases, and aging disorders [8, 11]. Therefore, performing research on DNA methylation plays a critical role in medicine, biological sciences, and biochemistry.
Given its importance in early development and aging, DNA methylation is a crucial epigenetic modification to profile. The detection of DNA methylation patterns is a rapidly advancing area of research, which promises the possibility of the methylation profiling to distinguish various tumor and cancer types, and possibly their response to chemotherapeutic agents. Detection of aberrancies of DNA methylation appears to be one of the most significant tests in early cancer diagnosis. These important findings regarding DNA methylation would not have been possible without the advancement of various profiling approaches. The accelerated development of array and sequencing technologies has significantly improved DNA methylation profiling, providing an unprecedentedly comprehensive view of the DNA methylation landscape. The present study will serve as an introduction to methods for DNA methylation analysis. This review provides an overview of the major profiling approaches, with a focus on the recent and promising genome-wide methodologies.
2. Techniques Based on Bisulfite Treatment
Initial studies on DNA methylation focused on identifying the position of methylation of the examined genes and determining the total content of 5-methyl cytosine [12]. Bisulfite treatment is one of the widely used and effective methods for categorizing 5-methyl cytosine and nonmethylated bases [8]. Exposing the genomic DNA to sodium bisulfite induces nonmethylated cytosine (C) deamination and converts it to uracil (U), while the methylated cytosine remains intact. Uracil is finally converted to thymine (T) following the polymerase chain reaction (PCR) [13]. As a result, the gene methylation information is transmitted to the sequence information. The gene features, such as melting point (temperature) and specific identification interactions, change due to the sequence changes (Figure 1). The differences between these features form the basis of the DNA methylation analysis (Figure 2).
Figure 1.
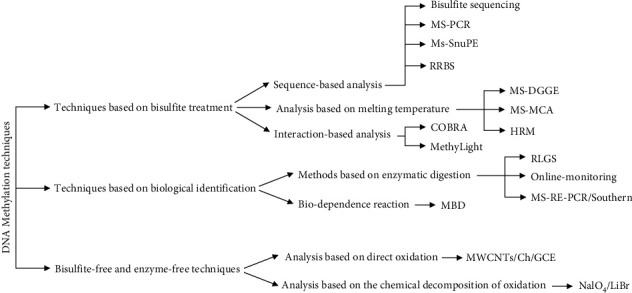
Schematic diagram of DNA methylation methods.
Figure 2.
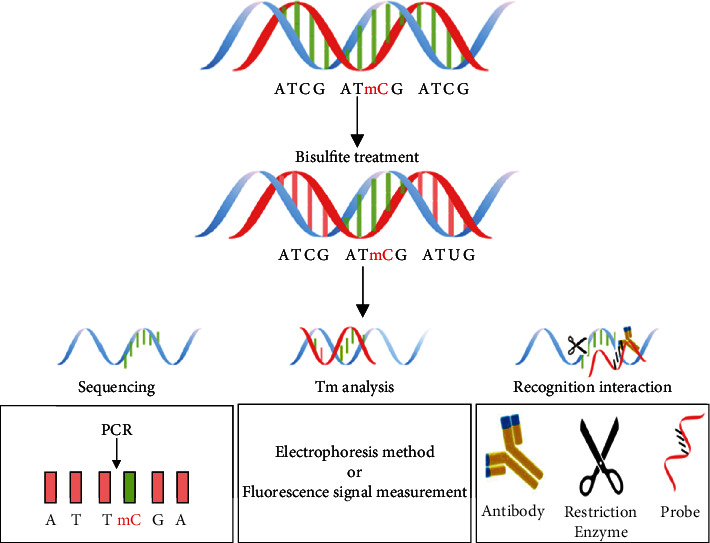
The main method for 5-methyl cytosine analysis after treatment with bisulfite.
2.1. Sequence-Based Analysis
The gene sequencing is a simple idea to differentiate 5-methyl cytosine from other bases. In 1992, Frommer proposed the bisulfite sequencing method to determine 5-methyl cytosine in the single strands. First, the DNA is treated with sodium bisulfite and multiplied by PCR. By determining the sequence of the resulting product, the 5-methyl cytosine position is detected. This technique is suitable for determining the multiple DNA methylation, while the need for cloning and sequencing processes makes the preparation process boring and long-lasting [14]. In 1996, Herman et al. developed an advanced sequencing-based technique called methylation-specific PCR (MS-PCR) [15]. In this method different from the previous bisulfite sequencing method, two specific promoters are predesigned according to the methylated and nonmethylated sequences. Following the treatment with sodium bisulfite, the target sequence is proliferated by PCR with two specific promoters. If the target sequence is methylated, there will be multiplied 5-methyl cytosine in the PCR products. This technique avoids the complex sequencing process. However, due to the need for the sequence primer design and the necessity for recognizing the position of methylation, this technique can be only used for qualitative determination [16]. A technique called methylation-sensitive single-nucleotide primer extension (Ms-SnuPE) was developed by Gonzalgo and Liang in 1997 to overcome the limitations of MS-PCR [17]. In this method, after treatment with bisulfite, the DNA is amplified by PCR and isolated by electrophoresis. The resulting products are then used as templates for the development of the primer [18, 19]. By electrophoretic separation and analyzing the expanded end product radiation, the C/T ratio is easily obtained and the relative ratio of methylated and nonmethylated sequences is achieved [20]. This method is not only suitable for the identification of the methylation position of each gene site but also able to determine the level of methylation. However, the tedious nature of primer design and the radioactive contamination will inevitably remain as constraints [9, 21]. By studying further in sequencing methods, the techniques based on new generation sequencing were developed and used as powerful analysis methods. In 2005, Meissner et al. proposed the reduced representation bisulfite-sequencing method (RRBS) [22], in which, the Msp I restriction enzyme is used for digesting the genome to enrich the CpG sites. Then, the sequencing process is done to obtain the methylation information of every single base. RRBS, which integrates the Msp1 restriction enzyme digestion, the bisulfite conversion, and the new generation of sequencing to analyze the methylation patterns of certain components, is more cost-effective than the WGBS since the focus of these methods is on enriching the regions rich in CpG near the identification sequence of the restriction enzyme [23–25].
The bisulfite sequencing of the whole WGBS genome (MethylC-seq; BS-seq) theoretically covers all the cytosine information [26]. In this method, the genomic DNA is purified and divided into fragments. The ends of the fragmented DNAs are restored; the adenine bases are added to the end of the 3′ (tail-A) of the DNA fragments, and the methylated adapters are attached to the DNA fragments [27]. The DNA fragments are selected in terms of size before sodium sulfite treatment and PCR amplification, and the resulting library is sequenced [28]. It should be noted that a large number of PCR cycles and inappropriate selection of DNA polymerase insensitive to uracil may lead to overrepresentation of methylated DNA data [29]. Starting with an adequate amount of genomic DNA can prevent the loss of information from the studied areas and excessive reproduction [13]. The main advantage of WGBS is its ability to assess the state of methylation almost from every CpG site, including the low-density CpG regions such as the “genetic deserts” between the genes, the incomplete methylated domains, and the final regulatory elements [29]. It can also determine the absolute level of DNA methylation and reveal the sequence methylation context.
2.2. Analysis Based on Melting Temperature
Bisulfite sequencing provides reliable support for extensive mapping of the genome; however, multiple operations and lower credible results due to complicated bases and incomplete bisulfite conversion are challenging [30]. Therefore, the idea of identification without sequencing and analysis for the unique melting temperature of the 5-methyl cytosine sequence drew the attention. In 1999, Aggerhoim developed a methylation-specific denaturing gradient gel electrophoresis (MS-DGGE) to determine the level of 5-methyl cytosine [31]. The DNA sequences treated with bisulfite are treated with DGGE, in which the solubility of the denaturing substance increases with the melting temperature from top to down [31]. The DNA strands are converted to dendrite and stopped in different areas of the gel due to the unique temperature of methylated and nonmethylated DNA strands. Depending on the isolation result, the 5-methyl cytosine level is measured [9]. This technique is cost-effective and can provide a comprehensive analysis of DNA methylation [32, 33]. However, the separation efficiency should be improved. In 2001, the methylation-specific melting-curve analysis (MS-MCA) was designed to determine the position of 5-methyl cytosine [34].
After treatment with bisulfite, the DNA strands are labeled with a fluorescence color. Depending on the measurement of the fluorescence intensity during melting, the melting curve of the DNA strands is obtained and their melting point is determined by optical cycle processing [32]. Since the melting point has a direct relationship with the CG value in the DNA sequence, the melting temperature of the nonmethylated DNA strand is lower than the methylated DNA strand due to the conversion of CG to UG during the bisulfite treatment [35]. Therefore, the DNA methylated and nonmethylated strands are separated from each other. However, it is difficult to accurately determine the state of methylation of each of the bases by using this technique. Rodríguez López et al. proposed the high-resolution melting (HRM) method to overcome this limitation in 2010 for the direct detection of 5-methyl cytosine [36]. This approach is different from MS-MCA. In HRM, saturated fluorescent color is used to label the DNA sample. A clear change in the fluorescence signal was achieved during the melting due to the capture of the target base position by the colors [37]. With high resolution, the methylation status of each of the bases was precisely determined [38].
2.3. Interaction-Based Analysis
Despite the analysis of melting temperatures, some certain interactions can be considered as alternatives for economic and efficient identifying the target sequence. Restriction enzymes and oligonucleotide probes are widely used in these interactions, which are the core of this approach [39, 40]. The combined bisulfite-restriction analysis (COBRA) was introduced to determine the status of 5-methyl cytosine [8]. After treatment with bisulfite and PCR, the BstU I restriction enzyme is added to the DNA sample, which detects and cuts the methylated 5′-CGCG-3′ sequence [8]. The reaction products are then analyzed by electrophoresis to determine the status of 5-methyl cytosine [41]. This technique can easily provide accurate results. However, its scope of use is limited due to the dependence on the restriction enzyme sequence [42]. At the beginning of the 21st century, enzymatic digestion technologies and oligo-probe protein hybridization were combined for the analysis of 5-methyl cytosine [43]. The MethyLight is the typical method. A specific oligo probe, Tagus, is designed and labeled with fluorophore and extinguisher for the 5′ and 3′ ends, respectively [44]. After bisulfite treatment, the DNA sample is incubated with the probe and analyzed using the real-time PCR [45]. The duplex strands are cut in 5-methyl cytosine by the restriction enzymes, and the fluorophore of the 5′ end is released. The 5-methyl cytosine level is measured from the produced fluorescence signal [46]. However, the use of restriction enzyme and the real-time PCR has made this method uneconomic and difficult.
3. Techniques Based on Biological Identification
Although the treatment with bisulfite is widely used to detect 5-methyl cytosine from other cytosines, it involves some issues such as incomplete conversion, false-positive results, and difficult and time-consuming use [44, 47]. In comparison, the biological identification methods, such as enzymatic digestion or biological reactions, can quickly detect the methylation with high specificity under mild reaction conditions [48]. Therefore, the 5-methyl-cytosine analysis methods based on biological identification have turned into the new center of attention (Figure 3).
Figure 3.
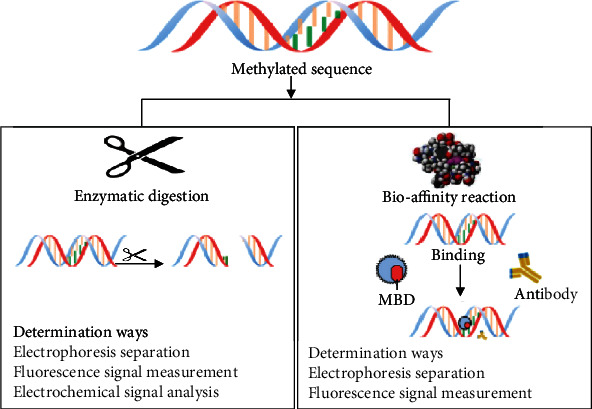
The main biological detection approach for 5-methyl cytosine and the method to determine identified targets.
3.1. Methods Based on Enzymatic Digestion
The methods based on enzymatic cutting use of different cutting (cleavage) characteristics of isoschizomers and nonisoschizomers. A pair of isoschizomers identify an identical sequence and have the same cutting point but show different sensitivity to the state of DNA methylation [13]. Methylation-sensitive restriction enzymes (MREs), including BstU I, Hpa II, Not I, and SmaI, only cut the nonmethylated target regions, and the methylated DNA stays intact [49]. The MRE cuttings are associated with the sequencing technologies to predict the DNA methylation levels at the genome level. In the MRE cutting sequence along with sequencing (MRE-seq), the MRE cuts the nonmethylated CpG regions in the genomic DNA and the resulting DNA fragments are selected and sequenced based on size [50]. The sequencing results reveal the location of nonmethylated CpG regions within the enzyme identification sites [51]. The MRE-seq provides the possibility of relative estimation of DNA methylation levels but has a relatively small coverage at the genome level since the identification sites containing CpG are limited [52, 53].
Since the beginning of the 21st century, enzyme-related techniques have been widely used in DNA methylation studies. The routine method is the restriction-landmark genomic scanning (RLGS) approach [54]. DNA samples are digested by the Not I restriction enzyme, leading to the identification of methylated sites. The sequences are then labeled with 32P-dCTP and 32P-dGTP and digested with the methylation-insensitivity restriction enzyme (EcoR V) [55]. After initial separation, the product is again treated with another methylation-insensitivity restriction enzyme (Hinf I) with a high frequency by performing one-dimensional electrophoresis and subjected to two-dimensional electrophoresis [56]. After two stages of screening, the methylation status of several CpG sites is examined. However, the use of the technique is complex and the result is uncertain [57]. An advanced enzymatic digestion technique, called methylation-sensitive restriction endonuclease-PCR/southern (MS-RE-PCR), was developed [58].
The DNA samples are treated with Hpa II and Msp I restriction enzymes. Both the Hpa II and Msp I enzymes can specifically detect the 5′-CCGG-3 sequences [59, 60]. The Hpa II and Msp I are unable to cut the nonmethylated C in the 5′-CCGG-3, while the Msp I can cut the methylated C in the 5′-CmCGG-3′. The digested DNA samples are analyzed with Hpa II and Msp I enzymes using the PCR or southern blot, and the status of DNA methylation is obtained in several methylation sites. This is a simple and cost-effective method, but not suited for complex gene samples [61]. In 2007, the methylation-sensitive restriction endonuclease was used to monitor the online activity of MTase and examine the methylation status [62]. In this method, a molecular beam radius labeled with a pair of fluorophore and extinguisher is used as the oligo probe. The probe first shows a weak fluorescence signal in its pinhead. Under the Dam MTase catalysis, the 5-methyl cytosine fragments are produced in the probe. The addition of Dpn I endonuclease then cuts the strand specifically at the methylated site and increases the fluorophore signal due to the separation of the fluorophore and the extinguisher [63]. Therefore, the MTase activity and the DNA methylation status are controlled by a clear fluorescence signal. Nevertheless, the procedure and the cost of fluorescent labeling are the weaknesses of this technique.
3.2. Biodependence Reaction
Some biomolecules such as MBD, anti-5-methyl-cytosine IgG1 antibodies, and Zinc Finger Proteins (ZF) show the inherent ability of the methylation site-specificity and can be directly used to decompose the DNA methylation [64]. The results of the MBD-based approach, which is based on the ability of MBD proteins to specifically bind to the methylated DNA sequences, can be represented using the microarray (MBD-chip) or sequencing technologies (MBDCap-seq/MethylCap-seq) [13]. Concurrently, an MBD-based approach to screen and identify the 5-methyl cytosine in the genome was introduced. The functional area of the polypeptide exposed to the MBD can combine with 5-methyl cytosine fragments and can combine directly with the 5-methyl cytosine surface [13]. Therefore, in combination with column chromatography, the 5-methyl cytosine is directly separated from other strands.
4. Bisulfite-Free and Enzyme-Free Techniques
Although the bisulfite conversion methods and biological approaches have shown specificity to characterize the position of DNA methylation, there are still issues of reaction efficiency, costs of analysis, and operational complexity in the analysis of 5-methyl cytosine. An enzyme- and bisulfite-free method for DNA methylation should overcome these constraints and have many advantages such as speed, convenience, and low cost (Figure 4).
Figure 4.
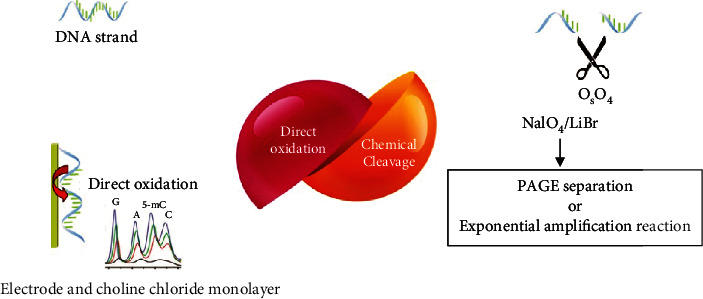
Two methods of analysis without bisulfite and enzyme. One measures the signals produced in the oxidation reaction, and the other measures the broken target pieces by performing chemical reactions.
4.1. Analysis Based on Direct Oxidation
Thymine and 5-methyl cytosine are oxidized by osmium tetroxide in dual C5-C6 bonds. Therefore, there is a potential proper way to use direct electrochemical oxidation of pyrimidine for the analysis of DNA methylation [65]. In 2010, an electrochemical method was proposed for the general analysis of DNA methylation [66]. The choline chloride monolayer-supported multiwalled carbon nanotubes (MWCNTs) (MWCNTs/Ch/GCE) were made [67]. The MWCNTs provide significant electrocatalytic activities for DNA bases. All of the bases of purine, guanine, pyrimidine, thymine, adenine, cytosine, and 5-methyl cytosine are detected by their unique oxidation signals [68]. In the following years, an advanced method was designed to improve the capacity of determining the mixing bases using overoxidized polypyrrole guided by MWCNTs/GCE (PPyox/MWCNTs/GCE) [69]. This technique has an extraordinary specificity and precision and can be used to rapidly determine the mixing bases without using the enzyme, probe, or bisulfite.
4.2. Analysis Based on the Chemical Decomposition of Oxidation
Recently, several new chemical methods based on chemical oxidation decomposition have been proposed to separate 5-methyl cytosine [70]. Inspired by the capability to separate 5-methyl cytosine from cytosine by OsO4, Yamada et al. [71] developed a method for detecting 5-methyl cytosine through light-sensitive oxidation using the 2-methyl-1,4-naphthoquinone-chromophore. Since then, an advanced method has been reported by using NaIO4/LiBr [72]. The C5-C6 dual bond of 5-methyl cytosine in the DNA is selectively oxidized by NaIO4/LiBr and then broken down using the hot piperidine method [73]. The DNAs chemically decomposed are analyzed by electrophoresis of the polyacrylamide gel (PAGE), and the status of DNA methylation is easily achieved [74]. The chemical cut-off method shows high efficiency for detecting the methylation sites. However, its sequence nonselectivity and undesirable sensitivity prevent its application in the genetic methylation analysis.
5. Conclusion
Many techniques have been developed to determine the status of DNA methylation over the past two decades. These diverse methods have three stages, providing the possibility of selecting and analyzing the methylated DNA from nonmethylated DNA in the target genome. In the first stage, the methylated and nonmethylated fragments can be distinguished through three main groups from the pretreatment methods, including the conversion of bisulfite, digestion with methylation-sensitive restriction enzymes, and dependence of the methylated DNA. The bisulfite treatment, extensively used in various platforms, relies on the conversion of nonmethylated cytosine to uracil and the nonalteration of the methylated cytosine. In this method, the methylated and nonmethylated fragments are identified by differences in the sequence. The restriction enzymes, sensitive and insensitive to methylation, including Hpa II and Msp I, are used to measure the methylation of genes. The Hpa II is stopped in the presence of 5-methyl cytosine in the four-nucleotide CCGG sequence, while the Msp I is not affected by DNA methylation. Therefore, the analysis of cut-off DNA products can be used for the detection of CpG methylation in specific genomic locations. The physical separation can be done by using antibodies and proteins binding to methylated cytosine.
The second stage is the replication of treated DNA with PCR. The last step is DNA methylation using various techniques like MS-PCR, COBRA, bisulfite-free sequencing, or microarray analysis. These techniques have been developed to determine the status of DNA methylation in general, specific genes, or genomic-wide surfaces. Initially, the analyses focused on the approaches of special locations. But now, the analyses can be performed on a large genomic scale using the new powerful technologies, including the DNA methylation microarrays and broad genomic free-bisulfite sequencing. However, recent advances have been made in the DNA methylation analysis but some barriers limit the application of these methods. (i) Significant errors during PCR amplification, DNA extraction, and the process of bisulfite conversion led to false results. (ii) Both PCR and bisulfite tests are time-consuming and boring. (iii) High-throughput technologies are costly and very complicated to run in standard laboratories. The appropriate approach in DNA methylation analysis depends on the study objectives. No DNA methylation analysis method will be appropriate for all applications. By knowing the type of information presented in each method, the researchers will be able to choose the most appropriate method based on their research needs.
Acknowledgments
This study was supported by the Islamic Azad University, Drug Applied Research Center, Tabriz University of Medical Sciences, and Zabol University.
Abbreviations
- MS-PCR:
Methylation-specific PCR
- Ms-SnuPE:
Methylation-sensitive single-nucleotide primer extension
- RRBS:
Reduced representation bisulfite-sequencing
- MS-DGGE:
Methylation-specific denaturing gradient gel electrophoresis
- MS-MCA:
Methylation-specific melting-curve analysis
- COBRA:
Combined bisulfite-restriction analysis
- MREs:
Methylation-sensitive restriction enzymes
- RLGS:
Restriction-landmark genomic scanning.
Data Availability
The datasets used in this study are available from the corresponding author on reasonable request.
Additional Points
Transparency Document. The transparency document associated with this article can be found in the online version.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- 1.Kader F., Ghai M. DNA methylation and application in forensic sciences. Forensic science international. 2015;249:255–265. doi: 10.1016/j.forsciint.2015.01.037. [DOI] [PubMed] [Google Scholar]
- 2.Syedmoradi L., Esmaeili F., Norton M. L. Towards DNA methylation detection using biosensors. The Analyst. 2016;141(21):5922–5943. doi: 10.1039/C6AN01649A. [DOI] [PubMed] [Google Scholar]
- 3.Alizadeh N., Memar M. Y., Moaddab S. R., Kafil H. S. Aptamer-assisted novel technologies for detecting bacterial pathogens. Biomedicine & Pharmacotherapy. 2017;93:737–745. doi: 10.1016/j.biopha.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Teschendorff A. E., Relton C. L. Statistical and integrative system-level analysis of DNA methylation data. Nature Reviews Genetics. 2018;19(3):129–147. doi: 10.1038/nrg.2017.86. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki M. M., Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature Reviews Genetics. 2008;9(6):465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 6.Schübeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- 7.Nazor K. L., Altun G., Lynch C., et al. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10(5):620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L., Xu Y.-Z., Xiao X.-F., et al. Development of techniques for DNA-methylation analysis. TrAC Trends in Analytical Chemistry. 2015;72:114–122. doi: 10.1016/j.trac.2015.03.025. [DOI] [Google Scholar]
- 9.Kurdyukov S., Bullock M. DNA methylation analysis: choosing the right method. Biology. 2016;5(1):p. 3. doi: 10.3390/biology5010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhattacharjee R., Moriam S., Nguyen N.-T., Shiddiky M. J. A. A bisulfite treatment and PCR-free global DNA methylation detection method using electrochemical enzymatic signal engagement. Biosensors and Bioelectronics. 2019;126:102–107. doi: 10.1016/j.bios.2018.10.020. [DOI] [PubMed] [Google Scholar]
- 11.Soozangar N., Sadeghi M. R., Jeddi F., Somi M. H., Shirmohamadi M., Samadi N. Comparison of genome-wide analysis techniques to DNA methylation analysis in human cancer. Journal of cellular physiology. 2018;233(5):3968–3981. doi: 10.1002/jcp.26176. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y., Wu F., Tan L., et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Molecular Cell. 2011;42(4):451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yong W.-S., Hsu F.-M., Chen P.-Y. Profiling genome-wide DNA methylation. Epigenetics & chromatin. 2016;9(1):p. 26. doi: 10.1186/s13072-016-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urich M. A., Nery J. R., Lister R., Schmitz R. J., Ecker J. R. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nature protocols. 2015;10(3):475–483. doi: 10.1038/nprot.2014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herman J. G., Graff J. R., Myöhänen S., Nelkin B. D., Baylin S. B. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(18):9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Candiloro I. L. M., Mikeska T., Dobrovic A. Assessing combined methylation–sensitive high resolution melting and pyrosequencing for the analysis of heterogeneous DNA methylation. Epigenetics. 2014;6(4):500–507. doi: 10.4161/epi.6.4.14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalgo M. L., Liang G. Methylation-sensitive single-nucleotide primer extension (Ms-SNuPE) for quantitative measurement of DNA methylation. Nature protocols. 2007;2(8):1931–1936. doi: 10.1038/nprot.2007.271. [DOI] [PubMed] [Google Scholar]
- 18.Boyd V. L. Compositions, methods, and kits for analyzing dna methylation. 2010. Google Patents.
- 19.Kafil H. S., Mobarez A. M. Spread of enterococcal surface protein in antibiotic resistant Entero-coccus faecium and Enterococcus faecalis isolates from urinary tract infections. The Open Microbiology Journal. 2015;9(1):14–17. doi: 10.2174/1874285801509010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jost J., Saluz H. DNA methylation: molecular biology and biological significance. Basel: Birkhäuser; 2013. [DOI] [Google Scholar]
- 21.Laird P. W. Principles and challenges of genome-wide DNA methylation analysis. Nature Reviews Genetics. 2010;11(3):191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 22.Meissner A., Gnirke A., Bell G. W., Ramsahoye B., Lander E. S., Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic acids research. 2005;33(18):5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyle P., Clement K., Gu H., et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome biology. 2012;13(10):p. R92. doi: 10.1186/gb-2012-13-10-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garrett-Bakelman F. E., Sheridan C. K., Kacmarczyk T. J., et al. Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. Journal of Visualized Experiments. 2015;96, article e52246 doi: 10.3791/52246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo H., Zhu P., Wu X., Li X., Wen L., Tang F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Research. 2013;23(12):2126–2135. doi: 10.1101/gr.161679.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonora G., Rubbi L., Morselli M., et al. DNA methylation estimation using methylation-sensitive restriction enzyme bisulfite sequencing (MREBS) PLoS One. 2019;14(4) doi: 10.1371/journal.pone.0214368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-Garcerà M., Gigli E., Sanchez-Quinto F., et al. Fragmentation of contaminant and endogenous DNA in ancient samples determined by shotgun sequencing; prospects for human palaeogenomics. PLoS One. 2011;6(8) doi: 10.1371/journal.pone.0024161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heyn H., Vidal E., Ferreira H. J., et al. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome biology. 2016;17(1):p. 11. doi: 10.1186/s13059-016-0879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olova N., Krueger F., Andrews S., et al. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome biology. 2018;19(1):p. 33. doi: 10.1186/s13059-018-1408-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rauluseviciute I., Drabløs F., Rye M. B. DNA methylation data by sequencing: experimental approaches and recommendations for tools and pipelines for data analysis. Clinical Epigenetics. 2019;11(1) doi: 10.1186/s13148-019-0795-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yokoyama S., Kitamoto S., Yamada N., et al. The application of methylation specific electrophoresis (MSE) to DNA methylation analysis of the 5′CpG island of mucin in cancer cells. BMC cancer. 2012;12(1):p. 67. doi: 10.1186/1471-2407-12-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernández H. G., Tse M. Y., Pang S. C., Arboleda H., Forero D. A. Optimizing methodologies for PCR-based DNA methylation analysis. BioTechniques. 2013;55(4):181–197. doi: 10.2144/000114087. [DOI] [PubMed] [Google Scholar]
- 33.Jeong H. M., Lee S., Chae H., et al. Efficiency of methylated DNA immunoprecipitation bisulphite sequencing for whole-genome DNA methylation analysis. Epigenomics. 2016;8(8):1061–1077. doi: 10.2217/epi-2016-0038. [DOI] [PubMed] [Google Scholar]
- 34.Islam M. N., Yadav S., Haque M. H., et al. Optical biosensing strategies for DNA methylation analysis. Biosensors and Bioelectronics. 2017;92:668–678. doi: 10.1016/j.bios.2016.10.034. [DOI] [PubMed] [Google Scholar]
- 35.Smith E., Jones M. E., Drew P. A. Quantitation of DNA methylation by melt curve analysis. BMC Cancer. 2009;9(1):p. 123. doi: 10.1186/1471-2407-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodríguez López C. M., Guzmán Asenjo B., Lloyd A. J., Wilkinson M. J. Direct detection and quantification of methylation in nucleic acid sequences using high-resolution melting analysis. Analytical chemistry. 2010;82(21):9100–9108. doi: 10.1021/ac1024057. [DOI] [PubMed] [Google Scholar]
- 37.Słomka M., Sobalska-Kwapis M., Wachulec M., Bartosz G., Strapagiel D. High resolution melting (HRM) for high-throughput genotyping—limitations and caveats in practical case studies. International journal of molecular sciences. 2017;18(11):p. 2316. doi: 10.3390/ijms18112316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hussmann D., Hansen L. L. DNA Methylation Protocols. Springer; 2018. Methylation-sensitive high resolution melting (MS-HRM) pp. 551–571. [DOI] [PubMed] [Google Scholar]
- 39.Bhattacharjee R., Moriam S., Umer M., Nguyen N.-T., Shiddiky M. J. A. DNA methylation detection: recent developments in bisulfite free electrochemical and optical approaches. The Analyst. 2018;143(20):4802–4818. doi: 10.1039/C8AN01348A. [DOI] [PubMed] [Google Scholar]
- 40.Asgharzadeh M., Kafil H. S., Roudsary A. A., Hanifi G. R. Tuberculosis transmission in northwest of Iran: using MIRU-VNTR, ETR-VNTR and IS6110-RFLP methods. Infection, Genetics and Evolution. 2011;11(1):124–131. doi: 10.1016/j.meegid.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 41.Umer M., Herceg Z. Deciphering the epigenetic code: an overview of DNA methylation analysis methods. Antioxidants & redox signaling. 2013;18(15):1972–1986. doi: 10.1089/ars.2012.4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dhingra T., Mittal K., Sarma G. S. Analytical techniques for DNA methylation–an overview. Current Pharmaceutical Analysis. 2014;10(1):71–85. doi: 10.2174/157341291001140102111956. [DOI] [Google Scholar]
- 43.Rackham O. J. L., Dellaportas P., Petretto E., Bottolo L. WGBSSuite: simulating whole-genome bisulphite sequencing data and benchmarking differential DNA methylation analysis tools. Bioinformatics. 2015;31(14):2371–2373. doi: 10.1093/bioinformatics/btv114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laird P. W., Trinh B. N., Campan M., Weisenberger D. J. DNA methylation analysis by digital bisulfite genomic sequencing and digital MethyLight. 2016. Google Patents. [DOI] [PMC free article] [PubMed]
- 45.Campan M., Weisenberger D. J., Trinh B., Laird P. W. DNA Methylation Protocols. Springer; 2018. MethyLight and digital MethyLight. pp. 497–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olkhov-Mitsel E., Bapat B. Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers. Cancer medicine. 2012;1(2):237–260. doi: 10.1002/cam4.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gholizadeh P., Pormohammad A., Eslami H., Shokouhi B., Fakhrzadeh V., Kafil H. S. Oral pathogenesis of Aggregatibacter actinomycetemcomitans. Microbial Pathogenesis. 2017;113:303–311. doi: 10.1016/j.micpath.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 48.Zhang S., Huang J., Lu J., et al. A novel fluorescent biosensor based on dendritic DNA nanostructure in combination with ligase reaction for ultrasensitive detection of DNA methylation. Journal of Nanobiotechnology. 2019;17(1) doi: 10.1186/s12951-019-0552-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mayne B. T., Leemaqz S. Y., Buckberry S., et al. msgbsR : An R package for analysing methylation-sensitive restriction enzyme sequencing data. Scientific Reports. 2018;8(1):2190–2198. doi: 10.1038/s41598-018-19655-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stevens M., Cheng J. B., Li D., et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Research. 2013;23(9):1541–1553. doi: 10.1101/gr.152231.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirschner S. A., Hunewald O., Mériaux S. B., Brunnhoefer R., Muller C. P., Turner J. D. Focussing reduced representation CpG sequencing through judicious restriction enzyme choice. Genomics. 2016;107(4):109–119. doi: 10.1016/j.ygeno.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 52.Wischnitzki E., Burg K., Berenyi M., Sehr E. M. Plant Synthetic Promoters. Springer; 2016. Selecting hypomethylated genomic regions using MRE-Seq; pp. 83–102. [DOI] [PubMed] [Google Scholar]
- 53.Khanna A., Czyz A., Syed F. EpiGnome™ Methyl-Seq Kit: a novel post-bisulfite conversion library prep method for methylation analysis. Nature Methods. 2013;10(10):iii–iiv. doi: 10.1038/nmeth.f.369. [DOI] [Google Scholar]
- 54.Freije W., Nusskern D. Materials and methods for assaying for methylation of CpG islands associated with genes in the evaluation of cancer. Google Patents. 2008 [Google Scholar]
- 55.Qin X., Xu J., Zhong Y. Multidisciplinary Management of Liver Metastases in Colorectal Cancer. Clinical and Translational Oncology. 2020;22(5):647–662. doi: 10.1007/s12094-019-02182-z. [DOI] [PubMed] [Google Scholar]
- 56.Erny G. L., Acunha T., Simó C., Cifuentes A., Alves A. Background correction in separation techniques hyphenated to high-resolution mass spectrometry - thorough correction with mass spectrometry scans recorded as profile spectra. Journal of Chromatography A. 2017;1492:98–105. doi: 10.1016/j.chroma.2017.02.052. [DOI] [PubMed] [Google Scholar]
- 57.Vidaki A., Ballard D., Aliferi A., Miller T. H., Barron L. P., Court D. S. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Science International: Genetics. 2017;28:225–236. doi: 10.1016/j.fsigen.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu W. Functional Nucleic Acids Detection in Food Safety. Springer; 2016. Genotoxicity detection at the molecular level in food safety assessment: conventional methods and developments; pp. 417–442. [DOI] [Google Scholar]
- 59.Petell C. J., Loiseau G., Gandy R., Pradhan S., Gowher H. A refined DNA methylation detection method using MspJI coupled quantitative PCR. Analytical Biochemistry. 2017;533:1–9. doi: 10.1016/j.ab.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fulneček J., Kovařík A. How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genetics. 2014;15(1):p. 2. doi: 10.1186/1471-2156-15-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cawthon R. M. A novel PCR method directly quantifies sequence features that block primer extension. bioRxiv. 2018 doi: 10.1101/325308. [DOI] [Google Scholar]
- 62.Thu K. L., Pikor L. A., Kennett J. Y., Alvarez C. E., Lam W. L. Methylation analysis by DNA immunoprecipitation. Journal of cellular physiology. 2010;222(3):522–531. doi: 10.1002/jcp.22009. [DOI] [PubMed] [Google Scholar]
- 63.Luo X., Li Y., Zheng J., Qi H., Liang Z., Ning X. The determination of DNA methyltransferase activity by quenching of tris (2, 2′-bipyridine) ruthenium electrogenerated chemiluminescence with ferrocene. Chemical Communications. 2015;51(46):9487–9490. doi: 10.1039/C5CC02817E. [DOI] [PubMed] [Google Scholar]
- 64.Hudson N. O., Buck-Koehntop B. A. Zinc finger readers of methylated DNA. Molecules. 2018;23(10):p. 2555. doi: 10.3390/molecules23102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanaka K., Tainaka K., Kamei T., Okamoto A. Direct labeling of 5-methylcytosine and its applications. Journal of the American Chemical Society. 2007;129(17):5612–5620. doi: 10.1021/ja068660c. [DOI] [PubMed] [Google Scholar]
- 66.Wang P., Mai Z., Dai Z., Zou X. Investigation of DNA methylation by direct electrocatalytic oxidation. Chemical Communications. 2010;46(41):7781–7783. doi: 10.1039/c0cc00983k. [DOI] [PubMed] [Google Scholar]
- 67.Wang P., Wu H., Dai Z., Zou X. Simultaneous detection of guanine, adenine, thymine and cytosine at choline monolayer supported multiwalled carbon nanotubes film. Biosensors and Bioelectronics. 2011;26(7):3339–3345. doi: 10.1016/j.bios.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 68.Zhou Y., Fang Y., Ramasamy R. P. Non-covalent functionalization of carbon nanotubes for electrochemical biosensor development. Sensors. 2019;19(2):p. 392. doi: 10.3390/s19020392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang P., Chen H., Tian J., Dai Z., Zou X. Electrochemical evaluation of DNA methylation level based on the stoichiometric relationship between purine and pyrimidine bases. Biosensors and Bioelectronics. 2013;45:34–39. doi: 10.1016/j.bios.2013.01.057. [DOI] [PubMed] [Google Scholar]
- 70.Ma S., Sun H., Li Y., Qi H., Zheng J. Discrimination between 5-hydroxymethylcytosine and 5-methylcytosine in DNA via selective electrogenerated chemiluminescence (ECL) labeling. Analytical chemistry. 2016;88(20):9934–9940. doi: 10.1021/acs.analchem.6b01265. [DOI] [PubMed] [Google Scholar]
- 71.Yamada H., Tanabe K., Nishimoto S.-i. Sensitive discrimination between cytosine and 5-methylcytosine in DNA by a modified invader method. Nucleic Acids Symposium Series. 2006;50(1):163–164. doi: 10.1093/nass/nrl081. [DOI] [PubMed] [Google Scholar]
- 72.Bareyt S., Carell T. Selective detection of 5-methylcytosine sites in DNA. Angewandte Chemie International Edition. 2008;47(1):181–184. doi: 10.1002/anie.200702159. [DOI] [PubMed] [Google Scholar]
- 73.Klungland A., Robertson A. B. Oxidized C5-methyl cytosine bases in DNA: 5-hydroxymethylcytosine; 5-formylcytosine; and 5-carboxycytosine. Free Radical Biology and Medicine. 2017;107:62–68. doi: 10.1016/j.freeradbiomed.2016.11.038. [DOI] [PubMed] [Google Scholar]
- 74.Ranade S. S., Chung C. B., Zon G., Boyd V. L. Preparation of genome-wide DNA fragment libraries using bisulfite in polyacrylamide gel electrophoresis slices with formamide denaturation and quality control for massively parallel sequencing by oligonucleotide ligation and detection. Analytical Biochemistry. 2009;390(2):126–135. doi: 10.1016/j.ab.2009.04.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used in this study are available from the corresponding author on reasonable request.