

Abstract
Glycosaminoglycans (GAGs) constitute a considerable fraction of the glycoconjugates found on cellular membranes and in the extracellular matrix of virtually all mammalian tissues. The essential role of GAG-protein interactions in the regulation of physiological processes has been recognized for decades. However, the underlying molecular basis of these interactions has only emerged since 1990s. The binding specificity of GAGs is encoded in their primary structures, but ultimately depends on how their functional groups are presented to a protein in the three-dimensional space. This review focuses on the application of NMR spectroscopy on the characterization of the GAG-protein interactions. Examples of interpretation of the complex mechanism and characterization of structural motifs involved in the GAG-protein interactions are given. Selected families of GAG-binding proteins investigated using NMR are also described.
Keywords: glycosaminoglycans, proteins, interaction, NMR, conformation
Introduction
Glycosaminoglycans (GAGs) are linear acidic heteropolysaccharides that exist in all mammals and are formed by repeating disaccharide units composed of N-acetyl-hexosamine and hexuronic or hexose (Table 1; Vasconcelos and Pomin, 2017). GAGs can have different sulfation patterns with different charge densities and heterogeneous monosaccharide compositions (Uhl et al., 2020). In addition to HA, GAGs are synthesized from the Golgi apparatus in the form of proteoglycans (Sasarman et al., 2016). According to the disaccharide composition and sulfation pattern, GAGs can be divided into several groups, including heparin/heparan sulfate (HS), chondroitin sulfate (CS)/dermatan sulfate (DS), keratan sulfate (KS) and hyaluronic acid (HA) (Pomin and Mulloy, 2018). Heparin/HS is composed of repeating disaccharide units of glucosamine (GlcNAc) and glucuronic acid (GlcA) or iduronic acid (IdoA). The initial substrate is [→4)-β-D-GlcA-(1→4)- α-D-GlcNAc-(1→] n. GlcNAc can be substituted by sulfate groups at the amide, 3 or/and 6 hydroxyl groups, and the persulfation can be written as GlcNS3S6S. GlcA can be converted into IdoA by C5 epimerase, and both can be modified by 2-O-sulfation (written as IdoA2S or GlcA2S). CS consists of repeating disaccharide units of glucuronic acid (GlcA) and galactosamine (GalNAc). The initial substrate is [→4)-β-D-GlcA-(1→3)- β-D-GalNAc-(1→] n. CS can undergo sulfation modification similar to heparin except for N-sulfation. However, due to the difference in glycosidic linkage, 3-O-sulfation in heparin becomes 4-O-sulfation. DS is obtained by converting GlcA in CS by C5-epimerase into IdoA. KS consists of repeating disaccharide units of Gal and GlcNAc, both of which can be 6-O-sulfated (Pomin, 2015). HA is the only GAG that is not modified by sulfation and is not synthesized as proteoglycans. It is composed of repeating disaccharide units of GlcA and GlcNAc. According to the monosaccharide composition and sulfation pattern, GAG disaccharides can have 408 possible compositions (Soares et al., 2017).
TABLE 1.
Structures and tissue distribution of glycosaminoglycans.
| Glycosaminoglycans | Degree of sulfation per disaccharide unit | Molecular weight range | Tissue distribution | |
| Heparin/Heparan sulfate (HS) | 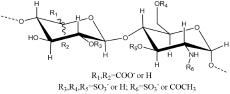 |
Heparin about 1.8∼2.4 | Heparin about 3∼30 kDa | Heparin in liver, lungs and skin; |
| HS about 0.8∼1.8 | HS about 10∼100 kDa | HS was widely distributed on the cell surface. | ||
| Chondroitin sulfate (CS) | 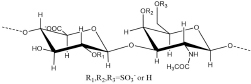 |
0.1–1.3 | 5∼50 kDa | cartilage, tendon, aorta, ligament |
| Dermatan sulfate (DS) |  |
< 1 | 15∼40 kDa | skin, blood vessels, heart valves |
| Hyaluronic acid (HA) |  |
0 | 4∼12000 kDa | synovial fluid, vitreous humour, ECM of loose connective tissue |
| Keratan sulfate (KS) | 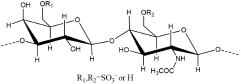 |
< 1 | 5∼25 kDa | KS I in cornea; KS II in cartilage aggregated; KS III in brain tissue |
As an important component of the extracellular matrix (ECM), GAGs play important roles in the construction of biological systems and the transduction of biological signals (Theocharis et al., 2016). Signal transduction occurs mainly through the interaction between GAGs and proteins, and these interactions are critical to the biological activity of these proteins. GAGs participate in a variety of physiological processes, including binding, activating and fixing a variety of protein ligands, such as growth factors, cytokines, chemokines, lipoproteins, proteases and their inhibitors, and other ECM components (Dyer et al., 2017; Rider and Mulloy, 2017; Crijns et al., 2020). GAGs are also associated with many pathological processes, including degenerative neurological diseases (Alzheimer’s disease), cardiovascular diseases (thrombosis and atherosclerosis) and cancer (Vigetti et al., 2016; Huynh et al., 2019; Morla, 2019). In the invasion of viruses, GAGs also play roles that cannot be ignored (such as in herpes simplex virus and COVID-19) (Liu et al., 2020). The interaction between GAGs and proteins occurs mainly through electrostatic forces. This puts forward requirements for amino acid sequences in proteins and meets some rules, such as the XBBXBX and XBBBXXBX heparin-binding sequences proposed by Cardin, where B is a basic amino acid and X is any amino acid (Cardin and Weintraub, 1989). However, long-term research has found that the interaction between GAGs and proteins is not simply determined by the primary structure sequence. A large number of studies have proven that hydrogen bonds and van der Waals forces sometimes even play roles far exceeding electrostatic forces in the interaction; a proper tertiary structure of the protein is also required (Rudd et al., 2017). This poses more serious and complex problems for studying the interactions between GAGs and proteins.
The interactions between GAGs and proteins are closely related to many factors, including saccharide unit composition, degree of sulfation, sulfation pattern, chain length, monosaccharide ring conformation and glycosidic linkage. The research methods used to characterize the interaction between GAGs and proteins mainly include gel electrophoresis (GE) (Nogueira et al., 2019), affinity chromatography (AC) (Sandoval et al., 2020), surface plasmon resonance (SPR) (Przybylski et al., 2020), biological layer interferometry (BLI) (Xiao et al., 2016), isothermal titration (ITC) (Zsila et al., 2018), microarray methods (Pomin and Wang, 2018b), crystal diffraction methods (X-ray) (Dahms et al., 2015), mass spectrometry (MS) (Yang and Chi, 2017), and nuclear magnetic resonance spectroscopy (NMR) (Kato and Peters, 2017). NMR is an insensitive technique compared with other analytical method for the study of interactions between biomolecules. The amount of sample needs to be in milligrams with high purity. In the study of proteins, NMR can characterize a protein with a molecular weight around 20 KD very well. However, proteins need to be isotope labeled by 15N and/or 13C when the molecular weight increases and can be studied up to 100 KD. The cross peaks will become broadening and overlapped severely for larger proteins. Even with the above limitations, NMR is still an irreplaceable technique in the characterization of the biomolecule interactions at the atomic level especially in the case of glycosamionoglycans. Both X-ray diffraction and NMR can provide more precise tertiary structure information, and they do not require sample derivatization and will not cause structural damage to the sample during the experiment. Due to the accuracy and refinement of the data, both types of data can be used for model construction. However, X-ray diffraction studies a crystal in solid state and provide only few conformations of the interaction. While, NMR studies a solution under physiological condition and records dynamic conformations during the whole interaction period. Glycosaminoglycans are very hard to obtain a crystal due to their high flexibilities and exchangeable conformations. The solution NMR can not only show the natural state of the complex, but also detect the change of the complex conformation on the ns-ms time scale (Pomin and Wang, 2018a). Compared with the immobilization study of crystal diffraction, solution NMR can also be used for the dynamic study of interactions under physiological conditions.
Nuclear magnetic resonance is widely used to study the conformation of GAGs alone or in complex with proteins (Pomin, 2014), but the information usually obtained indicates that there are multiple GAGs or complex structures in solution. According to NMR data, GAGs present different folds configurations in solution according to their type and environment (Mulloy, 2006), such as the controversial 3-folds and 4-folds coexisting left-handed helix of HA (Gargiulo et al., 2010), which will directly affect the distribution of acidic groups in space. Generally speaking, the conformational changes of GAGs are mainly caused by two factors, one is the ring conformation of monosaccharides, and the second is the flexibility of the glycosidic linkages (Skidmore et al., 2009). The conformation of the IdoA residue in heparin, HS and DS is different from that of the other three monosaccharides (GlcNAc, GalNAc, and GlcA). IdoA exist in the conformational equilibrium, with two chairs (1C4 and 4C1) and one shew-boat (2S0), instead of the fixed conformation 4C1 adopted in GalNAc, GlcNAc, or GlcA (Pomin, 2014). This gives these three different types of GAGs more flexible and various protein binding activities. This balance is affected by chain length, the degree of sulfation of adjacent monosaccharides, and its own 2-O sulfation (Haasnoot et al., 2020). When interacting with proteins, the conformational balance of IdoA will be tilted, such as binding to fibroblast growth factor-2 (FGF2), fibroblast growth factor-2 receptor (FGF2R), and eosinophil cationic protein (Ecp) (Hricovíni et al., 2002; García-Mayoral et al., 2013). In free state, when the conformational balance ratio is closer to the required binding state, the binding affinity is stronger (Hricovíni et al., 2002). Conversely, when the required conformation of the bound state cannot be achieved, the activity may be completely lost. But even if the protein has a clear tendency to a certain conformation of IdoA, there will generally be a conformational balance. The binding of AT III to heparin requires an absolute 2S0 conformation, but according to the NMR structure information, there is negligible 1C4 conformation in the whole binding process (Guerrini et al., 2006). Even though IdoA brings more variable binding conformational selectivity, recent studies have shown that GlcA has a better effect on the overall conformation of GAGs (Whitmore et al., 2020). In order to adapt to the ECM environment, the angle of the glycosidic linkages is allowed to change to a certain extent. The angle of the glycosidic linkages is affected by temperature, and the increase in temperature will result in a transition to the higher energy state (Hughes et al., 2017). When interacting with proteins, the glycosidic linkages can adopt proper orientations to meet the structural requirements during binding to proteins, and even cause the kinking of the GAGs polymer chain, thereby further enhancing the binding affinity (Hricovini, 2004). Compared with the obvious conformational equilibrium of IdoA, sometimes GAGs have α/β isomeric equilibrium at the reducing end (Silipo et al., 2008) and rapid intramolecular hydrogen bond exchange (Almond et al., 1998). Due to the flexibility of GAGs, there may be multiple interaction modes at the same binding domain in the GAG-protein interaction process (Tjong et al., 2007). In the interaction between GAGs and proteins, the structure of the proteins is normally changed or stabilized. The weak interaction between GAGs and proteins undergoes on the ns-ms time scale, so the conformation of the protein in the system will change over time. Due to the structural heterogeneity and conformational flexibility of GAGs or the dynamic changes of the complex, it is also very difficult to construct a model of complexes in solution (Almond, 2018).
Solution NMR can provide information about conformational changes and kinetic data during interactions between proteins and GAGs (Pomin and Wang, 2018a). NMR can also reveal the effects of different temperatures, pH values, salt concentrations, and ligand concentrations on the binding activity. There are three main goals in using NMR to study GAG-protein interactions: the first is to detect the amino acids involved in binding from the perspective of proteins, the second is to analyze the saccharide and its groups involved in binding from the perspective of GAGs, and the third is to observe the conformational changes and kinetic information during binding from the perspective of the interaction. To achieve these three goals, three technologies, chemical shift perturbation (CSP), saturation transfer difference (STD), and exchange-transferred nuclear Overhauser effect (trNOE), are initially used (Vignovich and Pomin, 2020), while other technologies, such as saturation transfer double difference (STDD) (Ledwitch et al., 2016), paramagnetic relaxation enhancement (PRE) (Orton et al., 2016), pseudocontact shifts (PCS) (Srb et al., 2019), and exchange-transferred rotating-frame Overhauser effect (ROE), have been developed to compensate for the shortcomings of the former. The latest pulse sequences have been developed to provide a more detailed and accurate description of the binding process, such as the gradient spectroscopic observation of water ligands (waterLOGSY) (Huang and Leung, 2019) and heteronuclear in-phase single quantum coherence experiment (HISQC) (Sepuru et al., 2018a). In addition, solid-state NMR has also been applied to study interactions involving ligands with low solubility (Malmos et al., 2016; Stewart et al., 2016). These techniques are based on four types of data: nuclear Overhauser effect (NOE), scalar coupling (J), residual dipole coupling (RDC) and chemical shift anisotropy (CSA). The purpose of this paper is to introduce some important findings of the application of NMR to the study of the interactions between GAGs and proteins (Table 2) and the review is classified according to the type of GAGs.
TABLE 2.
GAG binding proteins.
| Type of protein | Name of protein | Type of GAG | participating binding residues and Secondary structure | Affinity (Kd) | References |
| Chemokine | CCL5 | Heparin | 40S loop (R44KNR47), α helix (K55, K56) | 18 μM | Wang et al., 2011 |
| CS | 40S loop(R44KNR47), N loop (R17, L19, I15) | 0.25 μM | Deshauer et al., 2015 | ||
| CXCL1 | Heparin/HS | N terminus(R8), N-loop (H19, K21),40S turn (K45, R48), β3-strand (R49), C-helix (K60, K61, K65) | 50 μM | Sepuru and Rajarathnam, 2016; Sepuru et al., 2018b; Sepuru and Rajarathnam, 2019 | |
| CS/DS | N terminus(R8), N-loop (H19, K21), 40S turn (R48), | 4 μM | Sepuru and Rajarathnam, 2019 | ||
| CXCL2 | Heparin | N-loop (R17, K21), 40S turn (K45), C-helix (K61, K65, K69) | 25 μM | Sepuru et al., 2018b | |
| CXCL5 | Heparin/HS | N-loop (H23, K25), 40S turn (K49), β3 strand (K52), C-helix (Lys64, Lys65, Lys69, Lys76) | 30 μM | Sepuru et al., 2016; Sepuru and Rajarathnam, 2019 | |
| CS/DS | N-loop (H23, K25), 40S turn (K49), β3 strand (K52) | 3 μM | Sepuru and Rajarathnam, 2019 | ||
| CXCL7 | Heparin | N-loop (H15, K17), β3-strand(R44), C-helix (R54, K57, K61) | — | Brown et al., 2017 | |
| CXCL8 | Heparin | N-loop (K15, H18, K20, K23), C -helix (R60, K64, R68), β3-strand (R47), 50S loop (K54) | μM | Joseph et al., 2015 | |
| CXCL11 | Heparin | C- helix (K57SKQAR62) | — | Severin et al., 2010 | |
| CXCL12 | Heparin | C-helix (R12, A40), 20S loop (K24), 40S loop (N46) | μM | Laguri et al., 2011 | |
| CXCL13 | HS | C-helix (K60, R64, R67, H68), C-loop (K84, R85, R86) | 19 nM | Monneau et al., 2017 | |
| CXCL14 | Heparin | 10S loop(I12), β2-strand (I36, T37), 40S loop(K54), C -helix (R72), | — | Penk et al., 2019 | |
| CS/DS | 10S loop(I12), 40S loop(K54), C -helix (R72) | — | Penk et al., 2019 | ||
| Growth factor | FGF1 | Heparin | β1–β2 loop (N18), β8–β9 loop (N92), β10–β11 loop (K113), β11 strand (K118), β11–β12 loop (Q127, K128) | nM | Ogura et al., 1999 |
| FGF2 | Heparin | β1 strand (K27), β1–β2 loop (N28), β8–β9 loop (N102), β10–β11 loop (R121), β11 strand (K126), β11–β12 loop (Q135, K136) | nM | Faham et al., 1996 | |
| FGF7 | Heparin | β3 strand (R18), 40s loop(N92), β10(N114), 110s loop(Q115), 120s loop (V120, K124, Q129, K130, T131) | — | Ye et al., 2001 | |
| Serpin | AT III | Heparin | N-terminal end (K11, R13), A helix (R46, R47), D helix (K114, K125, R129, R132, K133, K136) | 20 nM | Olson et al., 2002 |
| Type II cytokines | IL-10 | Heparin/CS/DS | D helix (K99, R102, R104, R106), 110S loop (R107, K117, K119) | 0.41 mM | Künze et al., 2016 |
| IFNγ | Heparin | C-terminal end (D1:K125TGKRKR131, D2:R137GRR140) | 1.63 nM | Saesen et al., 2013 | |
| Roundabout 1 | HS | 80s loop (K81), 130s loop (V133, H134, G135, R136, K137), βA strand (I167, R169) | — | Gao et al., 2016 | |
| Cytokine | Pleiotrophin | CS | C-terminal TSR domain β-sheet (K60, K61, K69, K91, K92, K84, K86, K107) | 90 μM | Ryan et al., 2016 |
| Link protein | CD44 | HA | β1 strand (K38), 40s loop (R41, Y42), 70s loop (R78, Y79), 90s loop (N100, N101), 150s loop (R150), β9 strand (R154), 160s loop(R162) | μM | Banerji et al., 2007 |
| TSG-6 | HA | 10s loop (K11, Y12), 40s loop (H45, C47), β3 strand (A49), β3 strand (Y59), 60s loop (V62, K63), 80s loop (Y78, R81) | μM | Higman et al., 2014 | |
| Viral pathogen | viral CCL2 | Heparin | 10s loop(R18), 40s loop (K45, R46, R48) | 113 mM | Zhao and LiWang, 2010 |
| Defensins | Human β-defensin 2 | Heparin/DS | 20s loop(R22RYK25), β3 strand (K39), 40s loop(K40) | 5 mM | Seo et al., 2010 |
| RNase A | Eosinophil cationic protein | Heparin | α1 helix (R7, Q14, H15), β1 strand (Q40), loop4(H64), β6 strand(H128) | 15 μM | García-Mayoral et al., 2013 |
Heparin/Heparan Sulfate
Heparin is the most negatively charged polymer found in nature, and it is also the most studied in the GAG family (Conrad, 1997). One way to distinguish between heparin and HS is based on whether the mature body is still connected to the core protein. HS will be secreted out of the cell in the form of glycoproteins, most of which are fixed on the cell membrane to mediate many intercellular signaling pathways. Heparin is cleaved by β-endoglucuronidase and is combined with alkaline protease in the form of oligosaccharide chains to be stored in secretory granules (Oduah et al., 2016). The binding of heparin to protein mostly relies on its own high electronegativity and the positively charged domains in the protein. Hydrogen bonds and van der Waals forces also play important roles in the binding process. Moreover, the binding of heparin and protein is sometimes ion-dependent. For example, the binding of Langerin and heparin is mainly Ca2 + -dependent, although there are additional non-Ca2 + -dependent binding sites (Muñoz-García et al., 2015; Hanske et al., 2017; José García-Jiménez et al., 2019). HS can be divided into a high-sulfation domain (NS domain) and a low-sulfation domain (NA domain). Heparin essentially contains all possible sulfation modification structures of the NS domain due to the degree of high sulfation. Most of the biological functions of HS are concentrated in the NS domain, although the NA domain is more flexible and more suitable for bending. Due to the early large-scale clinical application of heparin, it was relatively easy to obtain. Early research mainly used heparin as a substitute for HS to carry out functional and structural studies. In approximately the past thirty years, the study of the interaction between heparin and various proteins has become a hot spot, and the gradual maturity of chemical enzyme synthesis has given this field new vitality. Heparin can induce the oligomerization or heteromerization of proteins, which can prevent proteins from being hydrolyzed by protein-degrading enzymes and increase or decrease the possibility of their binding to receptors.
Antithrombin III (AT III) is an absolutely conserved serine protease with two different glycosylation forms (α, β), consisting of three β-sheets (A-C) and nine α-helices (A-I) (Rezaie and Giri, 2020). Heparin is a cofactor of the antithrombin-mediated coagulation cascade, and the interaction between them directly affects the activities of factors IXa, Xa and IIa (Gray et al., 2012). Choay, J used chemical enzymatic synthesis of various heparin-related oligosaccharides to determine that the minimum specific sequence required for binding to AT III was the pentasaccharide A1GA2∗IA3 (Figure 1), which is also the only specific recognition sequence for heparin and protein binding found thus far (Thunberg et al., 1982; Choay et al., 1983). Although the specific pentasaccharide can meet the requirement of binding to AT III, it can only inhibit the activity of Xa. Inhibiting thrombin activity requires a heparin chain containing more than 16 saccharides, which can form a ternary complex with antithrombin and thrombin (Lane et al., 1984). The interaction between heparin and AT III was described as a three-state, two-step kinetic process (Figure 2; Olson et al., 1981), which assumed that AT III was in a balance of ‘native unactivated,’ ‘intermediate-activated’ and ‘fully activated’ states under physiological conditions (Roth et al., 2015). First, A1GA2∗ was driven by K125 and K114 to combine with the C- terminus of helix D in “native unactivated” AT III, and the reducing end faced the N-terminus (Desai et al., 1998). Then, accompanied by conformational changes in AT III (helix D extension, reactive center loop exposure, and closure of sheet A) and heparin (IdoA from equilibrium conformation between 1C4 and 2S0 to complete 2S0), each unit in the pentasaccharide was further combined with AT III (van Boeckel et al., 1994). The combined complex can interact with the target protease or enzymatically decompose, and heparin is dissociated accordingly. In the electrostatic binding of heparin and AT III, several sulfate groups of heparin-specific pentasaccharide (N-SO3 for A2∗ and A3, 6-O-SO3 for A1, and 3-O-SO3 for A2∗) and carboxyl groups were irreplaceable (Olson et al., 2002).
FIGURE 1.

Structures of AT III binding pentasaccharides (red circle) and their extended octasccharides. Sulfate groups in black dashed circles in the pentasaccharide, A1GA2*IA3, are essential for the binding to AT III. Bule squares showed the important amino acids in AT III contributing to the binding to heparin.
FIGURE 2.

Process of heparin binding to AT III. The binding of heparin with AT III is a reversible process. This process involves native unactivated (AT III, PDB code 1E05), intermediate-activated (AT III*, PDB code 1NQ9) and fully activated (AT III**, 1E03) states. During the binding process, IdoA transforms from conformational equilibrium to a complete 2S0 conformation (Jimenez-Barbero and Peters, 2003). The models of the three states are derived from X-ray. The reactive center loop (RCL) (red), sheet A (green), and helix D (gray blue) and the helix extension (dark blue) are highlighted in each state.
Further research using NMR focused on the specific role of each monosaccharide in the binding of heparin to AT III and the effect of extended pentasaccharide on the binding. The ratio of the 2S0 conformation in IdoA in the A1GA2∗IA3 sequence was 20% higher than that in the general heparin sequence (Ferro et al., 1987). In the three different chemically synthesized heparin pentasaccharides, the pentasaccharide had anticoagulant activity only when IdoA was in 2S0 (Das et al., 2001). Therefore, the proportion of 2S0 of IdoA in the heparin pentasaccharide sequence was one of the factors affecting the binding rate, which was affected by the degree of sulfation of glucosamine on both sides and its own 2-O-SO3 (Haasnoot et al., 2020). Although the absence of 2-O-SO3 in IdoA had no significant effect on the binding conformation, it resulted in a decrease in the proportion of free state 2S0 and a two-fold decrease in affinity (Stancanelli et al., 2018). At the same time, the flexibility of IdoA provided unlimited possibilities for the binding of heparin to protein. A recent study used IdoA2S instead of GlcA in the AT III binding sequence (Elli et al., 2020). The results showed that IdoA2S, which replaced GlcA, was in a pure 1C4 conformation when bound, and the affinity was tripled, which provided a basis for the application of bovine heparin. The unique structure of bovine heparin also provided unique ideas for the study of the specific mechanism of anticoagulation between heparin and AT III (Naggi et al., 2016). The 3-O-SO3 and 6-O-SO3 also had significant effects on the conformational balance of IdoA (Muñoz-García et al., 2012; Guerrini et al., 2013). The contribution of A2∗’s 3-O-SO3 to binding was in not only the conformation of heparin but also the formation of ‘intermediate-activated’ AT III (Lindahl et al., 1980; Casu et al., 1981). Octa-7 (Figure 1), an octasaccharide with extended reducing end, showed that adding an extra 3-O-SO3 to the A3 would increase the ratio of 2S0 in I by approximately 15%. The additional 3-O-SO3 formed new ionic bonds with R46 and R47. The extended disaccharide also had a certain contribution to the binding (by interacting with E113 and R24), and the binding force of otcasccharide and AT III was 40% higher than that of the specific pentasaccharide sequence and AT III. In the binding state, I and extended nonreducing end IdoA2S was completely in 2S0. In a similar structure (OCTA-1), due to the lack of 3-O-SO3 in the reducing end of A3, the extended IdoA2S was completely in 1C4 when bound, resulting in a substantial decrease in affinity (Guerrini et al., 2013). When extended reducing end IdoA2S’s 2-O-SO3 was removed (OCTA-2), the affinity increased slightly (Guerrini et al., 2008). In addition, there was little interaction between the reducing end extended disaccharide and AT III. In the other two octasaccharides with GlcA or IdoA as the extended nonreducing end (OCTA-3, OCTA-4), there was a significant polarization of affinity. The affinity of octasaccharide with GlcA as the nonreducing end was one order of magnitude higher than that with IdoA, which was in pure 2S0. In recent years, the appearance of low-molecular-weight heparin has become a research hotspot due to its unique fragments produced by cleavage or hydrolysis on anticoagulation. In Guerrini’s study, the affinity of two octasaccharides (OCTA-5, OCTA-6) containing specific pentasaccharide sequences derived from enoxaparin in binding with AT III decreased by 60-fold compared with the hexasaccharide with a complete pentasaccharide sequence. Because of the special pentasaccharide unit, the binding of the reducing end became weaker (Guerrini et al., 2010). The interaction difference of the octasaccharides with AT III showed that the substitution of different groups on heparin not only affected the binding strength with AT III but also changed the conformation during binding.
Heparin plays a key role in the regional aggregation and oligomerization of fibroblast growth factor (FGF), protecting it from denaturation and degradation and inducing its binding to the receptor (FGFR) (Korsensky and Ron, 2016). FGF is a growth factor family with 23 members, and its structure is highly related (12 β strands form the classic β-trefoil structure) (Li et al., 2016). The receptor proteins of FGF include four categories (FGFR1-4), which are composed of three immunoglobulin (Ig)-like domains, which can be subdivided into seven categories according to the difference in Ig3 (Cheng et al., 2017). FGFR Ig2 is a key site for the binding of FGF and FGFR mediated by heparin (Kan et al., 1993). In the study of the effect of FGF and heparin, acidic fibroblast growth factor (aFGF, FGF1) and basic fibroblast growth factor (bFGF, FGF2) were the most classic models (Schlessinger et al., 2000). Studies have shown that the binding of heparin to FGF does not change the FGF conformation, and the binding domain is mainly located at the β1-2 and β10-11 strands (Canales-Mayordomo et al., 2006). Although there is clear evidence in the study of Crystallography, in the free state, 116-120 (131-136) of FGF1 (FGF2) constitute βXI structure (Zhu et al., 1991). However, Moy’s NMR study on the structure of FGF2 in solution showed that there was no evidence to prove the existence of βXI (Moy et al., 1995). It is speculated that this is the structural change caused by the combination with HSPG, and this change is very important for the combination. This was confirmed in the subsequent NMR structural study of FGF1, Ogura pointed out that in the binding state, the 116-120 sequence has an obvious tendency of β-chain structure (Ogura et al., 1999). In addition, K125 in FGF2 and K118 in FGF1 had high affinity in binding with heparin. Therefore, the β11 chain was considered to be the key structure for the binding of FGF to heparin. In the combination of FGF2 and heparin, 2-O-SO3 and N-SO3 were necessary (Yu et al., 2014), and additional 6-O-SO3 was required for FGF1 (Guerrini et al., 2002). However, in the study using 48 kinds of heparin disaccharides to bind FGF1, 3-O-SO3 provided a stronger binding ability, and further C6 sulfation seemed to have a negative effect on the binding (Hu et al., 2012). In the study of the binding of heparin to FGF, 1C4 might have been the more favorable conformation (Canales et al., 2005; Guglieri et al., 2008). Interestingly, a recent study showed that specific AT-binding sequences can bind to FGFR2 Ig2 as a high-affinity complex, and IdoA remained in a high proportion of 2S0 (Nieto et al., 2011). Some experiments have shown that the combination of FGF and heparin seem to require a certain regular sequence of monosaccharide units or a special sulfation pattern (Ojeda et al., 2002). The mirror image of the carbohydrate structure also caused a significant reduction or loss of activity (Muñoz-García et al., 2013). For FGF1, only a single 6-sulfated tetrasaccharide was needed to induce its dimerization (Hricovíni et al., 2002). However, for FGF2 to be fully activated, heparin fragments of approximately decasaccharide might be required (Moy et al., 1997), although there was also evidence that tetrasaccharides could induce FGF2 dimerization (Guglieri et al., 2008). Heparin can induce FGF dimerization, but whether it is a critical step is controversial. Some NMR data showed that heparin, which formed a high-affinity complex with FGF, did not induce the dimerization of FGF but still had high activity (Canales et al., 2006).
In the study of the FGF-FGFR-heparin binding model (Figure 3), the crystal study gave two hypotheses: a 2:2:1 trans-binding model and a 2:2:2 cis-binding model (Pellegrini, 2001). NMR research in recent years has explained the formation process of the 2:2:2 model. Nieto used FGF1 and FGFR2 Ig2 and two heparin oligosaccharides to study the mechanism (Nieto et al., 2013). In the activity experiment, FGF1 and FGF2 had different requirements for heparin. In deheparinized cells, FGF2 activity was completely lost. However, after pretreatment of the cells with heparin, the activity recovered. FGF1 requires the presence of an additional heparin-like stabilizer myo-inositol hexasulfate (MIHS). It is speculated that the role of heparin in FGF1 was not limited to mediating the binding of FGF and FGFR. There was a second binding site in the FGF-FGFR complex, which was a clear cis-dimer binding model mark. Subsequent speculation suggested that the signaling pathway should be regarded as follows: FGFR dimerization was initially induced by GAGs, and then FGF and the ternary complex formed a higher-order aggregate and activated the subsequent enzyme cascade. Schieborr investigated the interactions among FGF1/FGF2, FGFR4 Ig2, and three different heparin polysaccharides (Saxena et al., 2010). The experimental results showed that the hexasaccharide could meet all the binding site requirements for inducing FGF dimerization, but the stability of the resulting complex was extremely poor. STD experiments showed that the combination of octasaccharide and FGF2 had a positive synergistic effect, but due to the lack of heparin structure data, the exact mechanism needs further experimental verification. Heparin was proven to have an extremely low dimerization ability for inducing FGFR4 Ig2, which was clear proof of the trans-dimer model in the description by Pomin (2016). However, the NMR data suggested there was a secondary binding site in the FGF-FGF Ig2 complex, which was again a clear cis-dimer binding model. Schieborr proposed that hexasaccharides and octasaccharide could mediate FGF2 signaling pathways under different mechanisms, and the positive synergistic effect of octasaccharide was due to the different residues involved in the binding. However, while there should theoretically be an FGF/FGFR/heparin 4:2:2 complex in the pathway, there were no data to support its existence. The existence of the FGF/FGFR/heparin 2:2:1 model was clearly supported by Brown’s ITC data, but no NMR evidence was obtained (Brown et al., 2013).
FIGURE 3.

Model of FGF-FGFR-heparin complex obtained by X-ray. FGF1-FGFR2-heparin decasaccharide (A) (PDB code 1E0O) and its amplified figure (B), FGF2-FGFR1-heparin decasaccharide (C) (PDB code 1FQ9) and its amplified figure (D). In the carton models, the heparin binding domains are shown in red. In the amplified figures, different kinds of heparin binding domains are shown in different colors according to the amino acid residues.
CXCL12 has six different splicing variants (CXCL12α-φ) in humans and is the only CXC chemokine with differential gene splicing (Janssens et al., 2017b). The complex of CXCL12 and the receptor CXCR4 mediates many physiological functions, including physiological processes such as hematopoiesis, embryonic development, vascular repair, and inflammation (Murphy and Heusinkveld, 2018). CD26, a leukocyte-activating antigen, can be cleaved CXCL12 between the N-terminal P2 and V3 residues (Janssens et al., 2017a). The cleaved product has a reduced affinity for CXCR4 and cannot activate it any more. Research on the binding domain of CXCL12 and heparin/HS can be traced back to 1999. The K24HLK27 base sequence in the β1-strand of the β-sheet, conforming to the BBXB rule, was verified in a mutation experiment (Amara et al., 1999). Sadir believed that R41 and R43 in the β2 strand were additional binding sites, in addition to K1 at the N-terminus as a potential binding site (Sadir et al., 2001). The binding between heparin/HS and K1 in CXCL12 was believed to protect CXCL12 from being cleaved by CD26 (Sadir et al., 2004). Murphy first used X-ray crystallography to study the interaction between CXCL12 and heparin/HS and proposed two binding domains in CXCL12: one at the interface of the dimer and the other in the N-loop region and the N-terminal helix similar to the binding domain in CXCL8 (Murphy et al., 2007). Using 13C-labeled octasaccharides in the NMR experiment, Laguri determined that the heparin-binding sequence was related to the GlcN-3, GlcA-4, and GlcN-5 units of the octasaccharides (Laguri et al., 2011). N-sulfation and 6-O-sulfation are essential for binding. The nonreducing end monosaccharide and reducing end disaccharide of the octasaccharide formed additional contact with the N-terminus of CXCL12 (R8 and R12 are the most prominent), and a consistent molecular binding model was constructed. However, Ziarek proposed a controversial molecular model (Ziarek et al., 2013). He believed that heparin and two CXCL12 molecules should drive the formation of the polymer in an almost orthogonal conformation, instead of the previously proposed interface of two CXCL12 molecules (composed of a β1 strand and the N-terminus). The data indicated that the binding site in CXCL12 should be on the six-strand of the β-sheet, while the N-terminus was not involved. The main residues involved in binding included K20, K24, K27, K41, K43 and R47, while A8 and A12 provided additional binding. It was proposed that the reason why heparin protected CXCL12 from CD26 cleavage was not the preemptive combination but the coverage of K1 caused by dimerization. Panitz’s study proved that the interaction affinity between heparin and CXCL12 was much higher than that of other GAGs, and the degree of sulfation was not the only factor influencing the binding (Panitz et al., 2016). The binding sites in CXCL12 with other GAGs were similar to heparin, with the exception of a second binding site for CS compared to heparin (R20, A21, N30, K64).
Type II cytokines have six secondary structure elements (A-F) to form an α-helical structure, of which A, C, D, and F adopt the classic four-helix topology, while B and E exist as the connecting structure (Pestka et al., 2004). Interleukin-10 (IL-10), interferon-γ (IFNγ) and interleukin-26 (IL-26) are the three proteins in this family that exist in the form of dimers. Although IL-10 and IFN-γ had the same protein folding mode, their binding with heparin split into two completely different manners. STD data indicated that when IL-10 bound to heparin, the degree of sulfation rather than the site had a greater impact on the binding (Künze et al., 2014), although the effect of 6-O-SO3 on affinity was 2-3 times greater than the effects of N-SO3 and 2-O-SO3. Data showed that there was a hydrogen bond or strong van der Waals force between IL-10 and the methyl group in the N-acetyl residue of the saccharides. As the heparin chain length increases, the affinity increases. When the chain length reached eight sugars, the affinity suddenly increased. It was calculated using STD data that when IL-10 bound to a heparin oligosaccharide with more than eight sugars, the Hill coefficient was approximately 2. This indicated that heparin and each monomer of the IL-10 dimer were bound, and the binding was synergistically positive. It was speculated that the binding site in IL-10 was located at the C-terminus of the D helix and the basic amino acid cluster L101RLRLRRCHRF111 of the adjacent DE loop. This heparin-binding domain existed in both monomers, which also supported the positive synergistic combination of octasaccharide and IL-10. NOE data showed that the conformation of a tetrasaccharide in the binding center did not change much. Further PCS data confirmed that the binding domain of IL-10 with heparin was in the 101-111 basic amino acid cluster (Gehrcke and Pisabarro, 2015). This domain is absolutely conserved in IL-10 from various sources, and it is also located in the binding domain of IL-10R2 and IL-10. The reason why GAG had an inhibitory effect on IL-10 might be due to the low-affinity IL-10R2 competing with heparin for binding.
Unlike IL-10, the binding domain of IFN-γ with heparin was located at the C-terminus. IFN-γ had four clusters of enriched basic amino acids, but only two C-terminal domains, K125-R131 (D1) and R137-R140 (D2), interacted with heparin (Vanhaverbeke et al., 2004). NOE data showed that the interaction between the protein and heparin had no effect on the conformation of the protein, and only the electrostatic force contributed to the binding without any other interaction force. The increase in sugar chain length increased not only the affinity between heparin and IFNγ but also the bending degree of the whole sugar chain. The binding of IFNγ to heparin protected the D1 domain from protease hydrolysis, and D1 acts as the main binding domain to heparin. ITC experiments have shown that D2 is not necessary for the binding of IFNγ to heparin, but removing D2 will increase the binding of IFNγ to heparin (Döbeli et al., 1988). Further studies have shown that the combination of D1 with heparin was mainly a thermodynamic process, while the combination of D2 with heparin was a kinetic process (Saesen et al., 2013). The main function of D2 was to strengthen the binding of IFNγ with heparin. The binding of the C-terminus of IFNγ to heparin is a two-step process. First, D1 bound to heparin, and the binding site was oriented. Then, D2 combined with heparin to strengthen the binding. The binding of IFNγ to its receptor includes two domains, one of which is the C-terminus. Therefore, HSPG on the cell surface competed with the IFNγ receptor for binding; and the addition of exogenous heparin could also reduce the IFNγ concentration on the cell surface. The inhibitory effect of heparin on the activity of certain proteins might be due to its competition with the protein receptor for binding, which led to the decreased or even disappearance of the binding affinity between the receptor and the protein. IL-10 inhibits the activity of IFNγ, so its mechanism might be more complicated. Studying the interaction between GAGs and proteins of a specific sequence may help to develop a more thorough understanding of the mechanism.
Chondroitin Sulfate
According to the type of uronic acid and sulfation, common CS can be divided into five categories: nonsulfated chondroitin sulfate (CS-O), 4-O-sulfated chondroitin sulfate (CS-A), 6-O-sulfated chondroitin sulfate (CS-C), 2, 4-O-disulfated chondroitin sulfate (CS-D), and 4,6-O-disulfated chondroitin sulfate (CS-E) (Yang et al., 2020). CS-B (DS) has all of the sulfation modification types of the above five types of CS, but its uronic acid is epimerized into IdoA. Oversulfated chondroitin sulfate (OSCS) was sulfated at all sites that could be sulfated, and it was one of the culprits that triggered the “heparin crisis” in 2008 (Zhu et al., 2019). There is a special kind of 3-O-sulfated chondroitin sulfate (CS-K) in marine organisms that has a high affinity for growth factors (Palhares et al., 2019).
In the interaction with chemokines, the main function of GAG was to locally aggregate chemokines to increase their binding to G-coupled protein receptors and to form a concentration gradient required for the migration of leukocytes, among which HS was dominant (Rajarathnam et al., 2018). However, CS also played an important role in the interaction with certain chemokines, such as the chemokine CCL5 (regulated upon activation of normal T cell expressed and secreted factor, RANTES). CS plays an important role in a variety of biological pathways mediated by CCL5, such as inducing T cell apoptosis and monocyte blockade. Deshauer studied the interaction between two CS hexasaccharides and CCL5 and used TEMPO to label CS for PRE experiments to study the binding sites in depth (Deshauer et al., 2015). In the titration of CCL5 with CS444 (GlcA-GalNAc4S- GlcA-GalNAc4S- GlcA-GalNAc4S), there were obvious chemical shift changes in the 40S loop, the N-terminus and the N loop (Figure 4). At a ratio of 1:1, the chemical shift had no significant change. When CS644 (GlcA-GalNAc6S- GlcA-GalNAc4S- GlcA-GalNAc4S) is used for titration, there are only small chemical shift disturbances at these three binding sites. However, when the ratio of CS644:CCL5 was more than 1:1, R17 and L19 in the N loop showed obvious chemical shift disturbances. In the PRE experiment, CS444 data showed that its reducing end was close to the 40S loop BBXB sequences. However, CS644 had additional chemical shift changes at Y3, A16, and R21, indicating that CS644 was also close to the 20S loop, N-loop and N-terminus, which suggested that the combination of CS644 and CCL5 was more heterogeneous. It can be seen that the type of GAG, the degree of sulfation and the ring conformation had a huge influence on the binding conformation between GAG and protein, which was also reflected in Pichert’s CXCL8 and CS hexasaccharide interaction study (Pichert et al., 2012).
FIGURE 4.

Complex of CCL5 dimer and CS466. In the carton models, the chondroitin sulfate binding domains are shown in red. In the amplified figures, different kinds of chondroitin sulfate binding domains are shown in different colors according to the amino acid residues.
Midkine (MK) and pleiotropic protein (PTN) form the MK/PTN cytokine family, which is a heparin-binding nerve growth factor. They are highly similar in structure and share more than 50% of the amino acid sequence (Herradon et al., 2019). They consist of two TSR domains with a hinge connection. Each domain consists of three antiparallel β-strands to form β-sheets. The C-terminal domain (CTD) of PTN is the main CS-binding domain, which has an affinity far greater than that of the N-terminal domain (NTD) (Ryan et al., 2016). CTD has two basic residue clusters (cluster 1: K69, K91, K92 and cluster 2: K84, K86, K107). The electrostatic potential diagram showed that the two sides of the β-sheet can be coplanar. According to the PRE data, CS-A preferred cluster 2, while CS-E preferred cluster 1. The data showed that K54 in NTD was close to the paramagnetic center, but NTD had only a few residues with side chains and HN atom transfer perturbation. The hydrophobic hinge can arrange two lysines (K60 and K61) near CTD cluster 1 to participate in the binding of CS. Although there was no clear reason to prove the effect of the C-terminus on the binding of CS to PTN, the affinity of CS-A, but not CS-E, to the C-terminal truncated PTN was greatly reduced. CS-E had a greater affinity than CS-A, which might be the reason why the PTN/MK family was associated with many tumorous inflammations (Weckbach et al., 2018). Unlike PTN, according to STD data, CS-E can simultaneously bind to the two domains of the midkine (Solera et al., 2016).
Tumor necrosis factor-stimulated gene-6 (TSG-6) is a classic HA-binding protein that shows different binding modes with CS compared to HA (Park et al., 2016). The combination of CS and Link- TSG-6 had at least two binding sites, and 4-O-sulfation was preferred. The slow exchange site was similar to the HA-binding site, but there were still some differences due to the sulfation pattern of CS. STD data indicated that there was a second group of rapid exchange binding sites, which were close to the heparin-binding site according to the model based on PRE data. The change in the relaxation rate ratio R2/R1 indicated that the initial combination of CS and Link- TSG-6 can induce dimerization. The dimerization interface and the CS binding site were located on opposite sides, so CS plays a neutralizing role rather than functioning as a bridge in inducing dimerization.
Dermatan Sulfate
Although DS was similar in structure to CS, the existence of IdoA gave it unparalleled structural flexibility. For example, in combination with hepatocyte growth factor/scattering factor (HGF/SF), the presence or absence of IdoA was the key to the combination of GAG with HGF/SF (Deakin et al., 2009). The binding mode of DS and NK1 (HGF/SF heparin-binding domain) was similar to that of heparin, although the affinity was slightly lower. The binding was concentrated in the N domain. Although crystallographic data proved that the K1 domain was involved in binding, this binding was based on the premise of dimerization. However, the NMR data showed that in solution, the low-molecular-weight GAGs would not induce its dimerization.
Sepuru used medium-length GAG to study the interaction with CXCL1 or CXCL5 in the presence of monomers and dimers through CSP experiments (Sepuru and Rajarathnam, 2019). The two binding sites in CXCL1 with HS were on the opposite sides of the protein, the α-domain (H19, K21, K45, K60, K61, K65) and the β-domain (R8, K29, R48, K49). The results showed that CXCL1 and HS were combined in a ratio of 1:2, and ITC experiments verified this result. The binding sites of CXCL1 with CS and DS are located in the γ-domain (R8, H19, K21, K45, K49). The binding domain of CXCL5 with GAG was similar to that of CXCL1, but there was no obvious specificity for GAG species. Neither CXCL1 nor CXCL5 bound to GAG involved helices, which was different from the previous proposal that helices are an important binding site for the interaction of chemokines that activate CXCR2 with GAG. In the HADDOCK model, the interaction between DS and CXCL1 involved two sulfate groups, two carboxyl groups and two N-acetyl groups, and the interaction model with CXCL5 involved two sulfate groups, one N-acetyl and one hydroxyl group. The molecular docking models of CS and DS with different structures were quite different. They involved different residue-binding groups and positions. This was consistent with the differences in the interaction morphology of GAG with different structures proposed previously. This was also reflected in the combination of CXCL14 and DS (Penk et al., 2019). The binding of DS and heparin with CXCL14 occurred in the C-terminal helix, part of the N-terminus and the transition between the second and third β-sheets (Y44-Q47). However, the maximum perturbation in the combination of DS and CXCL14 was associated with R72, while I36 and T37 were more affected in terms of heparin. DS and CS also had significant differences in N-terminal disturbances. The interaction between DS and protein was also dependent on chain length and sulfation pattern. In the study of the interaction between tau protein and DS, tau was favored for 6-O-sulfation (Zhao et al., 2017). Disulfated DS had a higher affinity than monosulfated DS, although the affinity of both was less than that of heparin.
Decorin binding protein B (DBPB) bound to DS in a different binding mode than DBPA, mainly through the linker between helices 1 and 2, the C-terminal tail, and the alkaline patch (Feng and Wang, 2015). In the PRE experiment, there were no clear data indicating that the C-terminal tail was involved in binding. It was speculated that this was because the binding occurs at the nonreducing end of DS, while the TEMPO label was at the reducing end of DS. The mutation data showed that the three sites all had a promoting effect on binding, and the C-terminus played a key role in binding. The most obvious difference between DBPB and DBPA was only the C-terminal disulfide bond, which again emphasized the influence of protein structure on binding. Due to the lack of disulfide bonds, the C-terminus could exist in multiple conformations when combined with DS, which was also thermodynamically favorable. Although the BXBB sequence in DBPA remained highly dynamic in DBPB, it did not contribute much to the binding due to the exposure of the C-terminus and the position of the linker in DBPB.
Hyaluronic Acid
Hyaluronic acid has a different synthesis site (plasma membrane) and a different synthesis form (non-glycoprotein) compared to other GAGs. HA will not undergo further modification; thus, the interaction between it and the protein seems to be structurally specific. The hydrogen bonds and intramolecular hydrogen bonds with water molecules gave it a complex β-sheet structure (Taweechat et al., 2020). In the double helix structure of HA, every two monosccharide flip 180°. HA, as a structural scaffold, widely exists in the epithelial tissue, connective tissue and nerve tissue of vertebrates and regulates the physical and chemical processes of tissue hydration and penetration. The interaction between HA and HA-binding protein (hyaluroadhesin) mediates various physiological activities, such as cell signal transduction, wound repair, tissue regeneration, leukocyte rolling adhesion and inflammation (Fallacara et al., 2018). Most HA-binding proteins belong to the link protein superfamily. Some other proteins (such as receptor for hyaluronan-mediated motility, RHAMM) and peptides (thymosin α1, Tα1) bound to HA are independent of the link module (Naor, 2016).
The 14 human link proteins can be divided into three categories (A, B, C) according to their structural composition (Kohda et al., 1996). TSG-6 was the most typical type A Link protein, and its HA-binding domain (HABD) was the only Link module (Figure 5; Day and Milner, 2019). The link module was composed of 100 amino acids and structured by two β-sheets and two α-helices, which were stabilized by two extremely conserved disulfide bonds. The two β-sheets were composed of four and two β-strands. Type B Link protein used CD44 as a template. It extended the β-sheet at the C- and N-termini on the basis of type A (adding four β strands), and the HABD of type B was redefined (Senbanjo and Chellaiah, 2017). The type C link protein was composed of two links in series, both of which participate in binding with HA. This subcategory included aggrecan, versican and HAPLN1-4, but detailed research on its structure is lacking. The binding of HA and protein had very strict requirements on the tertiary structure of the protein. This was most obvious in the type C Link protein, which did not interact with GAGs other than HA. In one study, three link modules were connected in series, but the binding activity with HA was completely lost (Cai et al., 2004).
FIGURE 5.

HA binding domains (HABD) of TSG-6 [(A) PDB code 1O7B; (B) PDB code 2PF5] and CD44 [(C) PDB code 1POZ; (D) PDB code 1UUH]. In the models, the TSG-6 or CD44 residues participate in binging are shown in red. The HABD of TSG-6 was the only Link module. The link module was structured by two β-sheets and two α-helices. The two β-sheets were composed of four and two β-strands. CD44 extended the β-sheet at the C- and N-termini on the basis of TSG6 (adding four β strands), and the HABD of CD44 was redefined. Unlike the NMR model (C), due to the low charge density caused by the conformational balance, the crystal (D) does not have a secondary structure in residues 62-73.
Kahmann proposed that the binding of Link-TSG-6 and HA was concentrated in the β4/β5 loop. The association was accompanied by the rearrangement of C47 and C68 disulfide bonds (Kahmann et al., 2000). In the previously proposed B(X)7B rule motif (R5EARSGKYK13), R5 and K13 had no obvious evidence of involvement in binding, but K11 was the main binding residue. In Blundell’s subsequent research, it was shown that the folding of the link module remains unchanged during the combination (Blundell et al., 2003). The largest structural change was found in β4/β5. K11 also changed its orientation and became more oriented. For Y59 and Y58, the benzene rings did not rotate due to ring stacking. Due to the derived polarity of the binding, the two ends of the binding were located at K11 and R81. Higman proposed that in the free state, the β4/β5 loop of TSG-6 was highly dynamic. In this state, there was a conformation that exposes aromatic residues and captured HA by stacking interactions and then rearranged structural elements, such as the β4/β5 loop (Higman et al., 2007). There were two structural elements that were obviously solidified, one of which was G10 located at the corner of α1/β1, and the other was K54 of β3/β4. K54 was far from the HA-binding site but played an important role in the binding of heparin to TSG-6. Its solidification explained the problem that HA and heparin could not bind to TSG-6 at the same time, although they have different binding sites.
In the 2014 study, HA and hybrid HA of different lengths were used to study the interaction with Link-TSG-6 (Higman et al., 2014). Although the heptasaccharide with the reducing end of GlcA (HA7AA) had a complete binding structure, the entropy was unfavorable. Therefore, the octasaccharide with the reducing end of GlcNAc (HA8AN) was defined as the minimum unit required for binding. HSQC data clearly showed that HA8NA and HA7AA had two binding modes, with the reducing end GlcA bound to K63/H45 as the dominant one. The affinity of HA8NA was twice that of HA8AN, while the affinity of the two heptasaccharides had no such difference. The reason for the difference in specific affinity is unknown. In the binding model of HA8AN and TSG-6, H45 and K63 appear to be new binding residues. They bound to the reducing terminal disaccharide of the octasaccharide to make the binding tighter. The binding of HA and Link-TSG-6 was mainly through ionic interactions, ring-stacking interactions, hydrogen bonding, van der Waals forces and hydrophobic repulsion. Since the binding occurred on two interfaces, this imposed an inevitable requirement for the distortion of the two glycosidic bonds between the fifth and seventh residues. For heptasaccharides, the significant reduction in the affinity of hexasaccharides might be due to the lack of multiple groups of binding, resulting in instability of the distortion of glycosidic bonds. The CS part of hybrid HA will also be distorted during binding, but due to the lack of structural elements and the lack of hydrogen bonds during binding, the affinity was far lower than that of HA. However, due to the existence of binding, this provided a certain explanation for the chondroprotective function of TSG-6. CS, Heparin and HA had different binding modes with TSG-6, giving TSG-6 complex biological functions.
The HABD in CD44 was mainly located in the link module, C-terminal extension and α1-helix. Two N-linked glycosylation sites (N25 and N100) were also located in the HABD (Takeda et al., 2003). Teriete pointed out that octasaccharide might be the smallest unit that satisfies all binding requirements (Teriete et al., 2004). All binding sites were located on the same plane, but due to the scattered distribution, there might be two incompatible binding modes. One used N100/N101 to R150/R154, similar to the combination of TSG-6 and HA. The other used K38/R162 as the terminal binding, and the binding was farther away from the charged area. The data showed that the binding is accompanied by a structural rearrangement. Takeda proposed that the parallel sheets of β8 and β0 involved rearrangement, which might be related to the special structure of β8 (Takeda et al., 2006). More thorough structural changes were located at the C-terminal extensions of α3 and β9, and their structure changed from a regular to a randomized structure after the combination. This result was in conflict with crystal studies, which showed that binding did not involve changes in C-terminal extension (Banerji et al., 2007). But unlike other studies, the protein used by Banerji is of mouse origin. And in the model established in this study, the complex is in two conformational equilibrium (type A and B, Figure 6). The difference between the two conformations is the orientation of R45 (human CD44 R41). Ogino also proposed that CD44 was in the balance of two conformations in the unbound or bound state (Ogino et al., 2010). In the unbound state, it had a regular structure and low HA affinity, which was conducive to cell rolling. In the combined state, it was mainly a random structure with high HA affinity, which was conducive to cell adhesion. The balance of these two states was conducive to the physiological activity of CD44-mediated cell rolling.
FIGURE 6.

The HA-binding site in mouse CD44. [(A) PDB code 2JCQ; (C) PDB code 2JCR] The ribbon diagram of mouse CD44 (type A and B complex). (B,D) Surface representation of the HA binding site in the type A and B crystal complex.
In terms of RHAMM, two amino acid clusters were mainly involved in binding with HA: the first was the proposed BX7B structure (K531-K541), and the second was K553-K562 (Ziebell and Prestwich, 2004). Studies have shown that the second binding site plays a major role in binding. Studies on Tα1 indicated that the binding is mainly related to its terminal L16KEKK20 (Mandaliti et al., 2017). The combination of HA and these two substances occurred mainly through electrostatic forces, which was different from the role of HA with TSG-6 and CD44. The combination of HA and CD44 was mainly through hydrogen bonding and van der Waals forces, while the combination with TSG-6 was mainly through electrostatic forces and aromatic accumulation.
Kertan Sulfate
Kertan sulfate is the only GAG without any acidic uronic acid residue, and its interaction with proteins mainly depends on structural characteristics and sulfation modification. KS is mainly distributed in the cornea and cartilage tissue and is divided into three categories (I-III) according to the distribution and connection with glycoproteins (Caterson and Melrose, 2018). KS plays an important role in brain development, neurodevelopment and regeneration, implantation and fertilization and maintains the balance of tissue hydration properties (Ota et al., 2018; Melrose, 2019; Miller et al., 2020). KS has many protein partners, including tyrosine protein kinases, inflammatory cytokines, growth factors, chemokines, cytoskeletal cells, and lectins. Only a few studies of the interaction between KS and protein have been investigated using NMR (Huckerby, 2002).
Galectin 3 (Gal-3) seems to be one of KS’s most tacit partners, and its distribution is extremely close to that of KS. The interaction between full-length Gal-3 and KS has been studied using HSQC; the disturbance was found to be in the β1, β3, β4, β5, β6, and β10 strands, and the β10 strand was the most important strand. The binding domain can be on the S- and F-faces in Gal-3. When the N-terminal tail of Gal-3 was truncated, KS interaction on the S-face became more obvious. The presence of other negatively charged regions did not affect the binding between KS and the Gal-3 S-face according to MD data. In the binding state, the conformations of the F-face and the N-terminal tail were changed. The binding was mainly concentrated on the left side of the S-face, which facilitated its combination with other proteins or heteropolymerization with other galectins. However, the pulse field gradient NMR data showed that KS did not induce oligomerization of Gal-3. Desulfated KS had far less affinity than KS, and the chemical shift disturbances on the F-face and N-terminal tail were greatly reduced.
Conclusion
Glycosaminoglycans, as common glycoproteins in biological systems, are involved in many physiological and pathological processes. The study of their structure and interaction with proteins has received extensive attention, but the study of molecular perspectives is only the tip of the iceberg. This not only is due to the delay of carbohydrate research but is also related to the limitations of technology. The information produced by NMR is incomparable to all other technologies. For example, it can provide information about the binding affinity constant, on/off chemical exchange rate, binding site and atomic information, but high-precision research is more demanding for technology. In particular, regarding the special existence of GAG, its highly complex structure not only endows it with rich biological functions but also brings incomparable difficulties for research. The study of the interactions between GAG and proteins using NMR is based on complete structural characterizations of GAG and/or proteins, which face huge obstacles. Biosynthesis carriers of GAG are difficult to find, while chemical and enzymatic syntheses are limited to a few scientists. This in turn makes it difficult to obtain isotope-labeled GAG. Because the binding of GAG and protein has obvious multibinding characteristics, it will cause oligomerization and even precipitation. The application of NMR technology is mainly limited by several factors, including the length of the oligosaccharides, the molecular weight of the proteins, and the concentration range and stability of the complex. However, with the renewal and iteration of technology, the rise of high magnetic flux nuclear magnetic spectrometry and enzymatic chemical synthesis has injected a steady stream of vitality into interaction research. The study of the interaction between GAG and proteins is helpful for understanding various physiological and pathological mechanisms and has a huge impetus for drug development.
Author Contributions
CB and LJ participated in preparation, creation, initial draft writing and review of this article. Both authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Almond A. (2018). Multiscale modeling of glycosaminoglycan structure and dynamics: current methods and challenges. Curr. Opin. Struct. Biol. 50 58–64. 10.1016/j.sbi.2017.11.008 [DOI] [PubMed] [Google Scholar]
- Almond A., Brass A., Sheehan J. K. (1998). Dynamic exchange between stabilized conformations predicted for hyaluronan tetrasaccharides: comparison of molecular dynamics simulations with available NMR data. Glycobiology 8 973–980. 10.1093/glycob/8.10.973 [DOI] [PubMed] [Google Scholar]
- Amara A., Lorthioir O., Valenzuela A., Magerus A., Thelen M., Montes M., et al. (1999). Stromal cell-derived factor-1alpha associates with heparan sulfates through the first beta-strand of the chemokine. J. BiolChem. 274 23916–23925. 10.1074/jbc.274.34.23916 [DOI] [PubMed] [Google Scholar]
- Banerji S., Wright A. J., Noble M., Mahoney D. J., Campbell I. D., Day A. J., et al. (2007). Structures of the CD44–hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat. Struct. Mol. Biol. 14 234–239. 10.1038/nsmb1201 [DOI] [PubMed] [Google Scholar]
- Blundell C. D., Mahoney D. J., Almond A., DeAngelis P. L., Kahmann J. D., Teriete P., et al. (2003). The link module from ovulation- and inflammation-associated protein TSG-6 changes conformation on hyaluronan binding. J. Biol. Chem. 278 49261–49270. 10.1074/jbc.M309623200 [DOI] [PubMed] [Google Scholar]
- Brown A., Robinson C. J., Gallagher J. T., Blundell T. L. (2013). Cooperative heparin-mediated oligomerization of fibroblast growth factor-1 (FGF1) precedes recruitment of FGFR2 to ternary complexes. Biophys. J. 104 1720–1730. 10.1016/j.bpj.2013.02.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A., Sepuru K., Rajarathnam K. (2017). Structural basis of native CXCL7 monomer binding to CXCR2 receptor N-domain and glycosaminoglycan heparin. Int. J. Mol. Sci. 18:508. 10.3390/ijms18030508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S., Dufner-Beattie J. L., Prestwich G. D. (2004). A selective protein sensor for heparin detection. Analytical Biochem. 326 33–41. 10.1016/j.ab.2003.11.017 [DOI] [PubMed] [Google Scholar]
- Canales A., Angulo J., Ojeda R., Bruix M., Fayos R., Lozano R., et al. (2005). Conformational flexibility of a synthetic glycosylaminoglycan bound to a fibroblast growth factor. FGF-1 recognizes both the 1C4 and 2SO conformations of a bioactive heparin-like hexasaccharide. J. Am. Chem. Soc. 127 5778–5779. 10.1021/ja043363y [DOI] [PubMed] [Google Scholar]
- Canales A., Lozano R., López Méndez B., Angulo J., Ojeda R., Nieto P. M., et al. (2006). Solution NMR structure of a human FGF-1 monomer, activated by a hexasaccharide heparin-analogue. FEBS J. 273 4716–4727. 10.1111/j.1742-4658.2006.05474.x [DOI] [PubMed] [Google Scholar]
- Canales-Mayordomo A., Fayos R., Angulo J., Ojeda R., Martín-Pastor M., Nieto P. M., et al. (2006). Backbone dynamics of a biologically active human FGF-1 monomer, complexed to a hexasaccharide heparin-analogue, by 15N NMR relaxation methods. J. Biomol. NMR 35 225–239. 10.1007/s10858-006-9024-y [DOI] [PubMed] [Google Scholar]
- Cardin A. D., Weintraub H. J. (1989). Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9 21–32. 10.1161/01.atv.9.1.21 [DOI] [PubMed] [Google Scholar]
- Casu B., Oreste P., Torri G., Zoppetti G., Choay J., Lormeau J. C., et al. (1981). The structure of heparin oligosaccharide fragments with high anti- (factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem. J. 197 599–609. 10.1042/bj1970599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterson B., Melrose J. (2018). Keratan sulfate, a complex glycosaminoglycan with unique functional capability. Glycobiology 28 182–206. 10.1093/glycob/cwy003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W., Wang M., Tian X., Zhang X. (2017). An overview of the binding models of FGFR tyrosine kinases in complex with small molecule inhibitors. Eur. J. Med. Chem. 126 476–490. 10.1016/j.ejmech.2016.11.052 [DOI] [PubMed] [Google Scholar]
- Choay J., Petitou M., Lormeau J. C., Sinaÿ P., Casu B., Gatti G. (1983). Structure-activity relationship in heparin: a synthetic pentasaccharide with high affinity for antithrombin III and eliciting high anti-factor Xa activity. Biochem. Biophys. Res. Commun. 116 492–499. 10.1016/0006-291X(83)90550-8 [DOI] [PubMed] [Google Scholar]
- Conrad H. E. (1997). Heparin-Binding Proteins. Illinois: Academic Press. [Google Scholar]
- Crijns H., Vanheule V., Proost P. (2020). Targeting chemokine—glycosaminoglycan interactions to inhibit inflammation. Front. Immunol. 11:483. 10.3389/fimmu.2020.00483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahms S. O., Mayer M. C., Roeser D., Multhaup G., Than M. E. (2015). Interaction of the amyloid precursor protein-like protein 1 (APLP1) E2 domain with heparan sulfate involves two distinct binding modes. Acta Crystallographica Sec. D Biol. Crystallography 71 494–504. 10.1107/S1399004714027114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S. K., Mallet J. M., Esnault J., Driguez P. A., Duchaussoy P., Sizun P., et al. (2001). Synthese konformativ fixierter kohlenhydrate: eine skew-bootkonformation der L-Iduronsäure in Heparin bestimmt dessen antithrombotische Aktivität. Angewandte Chemie 113 1723–1726. 10.1002/1521-3757(20010504)113:93.0.CO;2-N [DOI] [Google Scholar]
- Day A. J., Milner C. M. (2019). TSG-6: a multifunctional protein with anti-inflammatory and tissue-protective properties. Matrix Biol. 78 60–83. 10.1016/j.matbio.2018.01.011 [DOI] [PubMed] [Google Scholar]
- Deakin J. A., Blaum B. S., Gallagher J. T., Uhrín D., Lyon M. (2009). The binding properties of minimal oligosaccharides reveal a common heparan sulfate/dermatan sulfate-binding site in hepatocyte growth factor/scatter factor that can accommodate a wide variety of sulfation patterns. J. Biol. Chem. 284 6311–6321. 10.1074/jbc.M807671200 [DOI] [PubMed] [Google Scholar]
- Desai U. R., Petitou M., Björk I., Olson S. T. (1998). Mechanism of heparin activation of antithrombin: evidence for an induced-fit model of allosteric activation involving two interaction subsites. Biochemistry 37 13033–13041. 10.1021/bi981426h [DOI] [PubMed] [Google Scholar]
- Deshauer C., Morgan A. M., Ryan E. O., Handel T. M., Prestegard J. H., Wang X. (2015). Interactions of the chemokine CCL5/RANTES with medium-sized chondroitin sulfate ligands. Structure 23 1066–1077. 10.1016/j.str.2015.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döbeli H., Gentz R., Jucker W., Garotta G., Hartmann D. W., Hochuli E. (1988). Role of the carboxy-terminal sequence on the biological activity of human immune interferon (IFN-γ). J. Biotechnol. 7 199–216. 10.1016/0168-1656(88)90052-1 [DOI] [Google Scholar]
- Dyer D. P., Migliorini E., Salanga C. L., Thakar D., Handel T. M., Richter R. P. (2017). Differential structural remodelling of heparan sulfate by chemokines: the role of chemokine oligomerization. Open Biol. 7:160286. 10.1098/rsob.160286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elli S., Stancanelli E., Wang Z., Petitou M., Liu J., Guerrini M. (2020). Degeneracy of the antithrombin binding sequence in heparin: 2-O-sulfated iduronic acid can replace the critical glucuronic acid. Chem. Eur. J. 26 11814–11818. 10.1002/chem.202001346 [DOI] [PubMed] [Google Scholar]
- Faham S., Hileman R. E., Fromm J. R., Linhardt R. J., Rees D. C. (1996). Heparin structure and interactions with basic fibroblast growth factor. Science 271 1116–1120. 10.1126/science.271.5252.1116 [DOI] [PubMed] [Google Scholar]
- Fallacara A., Baldini E., Manfredini S., Vertuani S. (2018). Hyaluronic acid in the third millennium. Polymers 10:701. 10.3390/polym10070701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W., Wang X. (2015). Structure of decorin binding protein B from borrelia burgdorferi and its interactions with glycosaminoglycans. Biochim. Biophys. Acta Proteins Proteomics 1854 1823–1832. 10.1016/j.bbapap.2015.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro D. R., Provasoli A., Ragazzi M., Torri G., Casu B., Gatti G., et al. (1987). Evidence for conformational equilibrium of the sulfated l-iduronate residue in heparin and in synthetic heparin mono- and oligosaccharides: NMR and force-field studies. Cheminform 108 6773–6778. 10.1002/chin.198710100 [DOI] [Google Scholar]
- Gao Q., Chen C., Zong C., Wang S., Ramiah A., Prabhakar P., et al. (2016). Structural aspects of heparan sulfate binding to robo1–Ig1–2. ACS Chem. Biol. 11 3106–3113. 10.1021/acschembio.6b00692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mayoral M. F., Canales Á, Díaz D., López-Prados J., Moussaoui M., de Paz J. L., et al. (2013). Insights into the glycosaminoglycan-mediated cytotoxic mechanism of eosinophil cationic protein revealed by NMR. ACS Chem. Biol. 8 144–151. 10.1021/cb300386v [DOI] [PubMed] [Google Scholar]
- Gargiulo V., Morando M. A., Silipo A., Nurisso A., Pérez S., Imberty A., et al. (2010). Insights on the conformational properties of hyaluronic acid by using NMR residual dipolar couplings and MD simulations. Glycobiology 20 1208–1216. 10.1093/glycob/cwq067 [DOI] [PubMed] [Google Scholar]
- Gehrcke J., Pisabarro M. T. (2015). Identification and characterization of a glycosaminoglycan binding site on interleukin-10 via molecular simulation methods. J. Mol. Graphics Modelling 62 97–104. 10.1016/j.jmgm.2015.09.003 [DOI] [PubMed] [Google Scholar]
- Gray E., Hogwood J., Mulloy B. (2012). The anticoagulant and antithrombotic mechanisms of heparin. Handb. Exp. Pharmacol. 207 43–61. 10.1007/978-3-642-23056-1_3 [DOI] [PubMed] [Google Scholar]
- Guerrini M., Agulles T., Bisio A., Hricovini M., Lay L., Naggi A., et al. (2002). Minimal heparin/heparan sulfate sequences for binding to fibroblast growth factor-1. Biochem. Biophys. Res. Commun. 292 222–230. 10.1006/bbrc.2002.6634 [DOI] [PubMed] [Google Scholar]
- Guerrini M., Elli S., Gaudesi D., Torri G., Casu B., Mourier P., et al. (2010). Effects on molecular conformation and anticoagulant activities of 1,6-anhydrosugars at the reducing terminal of antithrombin-binding octasaccharides isolated from low-molecular-weight heparin enoxaparin. J. Med. Chem. 53 8030–8040. 10.1021/jm100771s [DOI] [PubMed] [Google Scholar]
- Guerrini M., Elli S., Mourier P., Rudd T. R., Gaudesi D., Casu B., et al. (2013). An unusual antithrombin-binding heparin octasaccharide with an additional 3-O-sulfated glucosamine in the active pentasaccharide sequence. Biochem. J. 449 343–351. 10.1042/BJ20121309 [DOI] [PubMed] [Google Scholar]
- Guerrini M., Guglieri S., Beccati D., Torri G., Viskov C., Mourier P. (2006). Conformational transitions induced in heparin octasaccharides by binding with antithrombin III. Biochem. J. 399 191–198. 10.1042/BJ20060656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini M., Guglieri S., Casu B., Torri G., Mourier P., Boudier C., et al. (2008). Antithrombin-binding octasaccharides and role of extensions of the active pentasaccharide sequence in the specificity and strength of interaction. J. Biol. Chem. 283 26662–26675. 10.1074/jbc.M801102200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglieri S., Hricoviìni M., Raman R., Polito L., Torri G., Casu B., et al. (2008). Minimum FGF2 binding structural requirements of heparin and heparan sulfate oligosaccharides as determined by NMR spectroscopy. Biochemistry 47 13862–13869. 10.1021/bi801007p [DOI] [PubMed] [Google Scholar]
- Haasnoot C. A. G., de Gelder R., Kooijman H., Kellenbach E. R. (2020). The conformation of the idopyranose ring revisited: How subtle O-substituent induced changes can be deduced from vicinal 1H-NMR coupling constants. Carbohydrate Res. 496:108052. 10.1016/j.carres.2020.108052 [DOI] [PubMed] [Google Scholar]
- Hanske J., Wawrzinek R., Geissner A., Wamhoff E., Sellrie K., Schmidt H., et al. (2017). Calcium-independent activation of an allosteric network in langerin by heparin oligosaccharides. Chem.BioChem. 18 1183–1187. 10.1002/cbic.201700027 [DOI] [PubMed] [Google Scholar]
- Herradon G., Ramos-Alvarez M. P., Gramage E. (2019). Connecting metainflammation and neuroinflammation through the PTN-MK-RPTPβ/ζ axis: relevance in therapeutic development. Front. Pharmacol. 10:377. 10.3389/fphar.2019.00377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higman V. A., Blundell C. D., Mahoney D. J., Redfield C., Noble M. E. M., Day A. J. (2007). Plasticity of the TSG-6 HA-binding loop and mobility in the TSG-6-HA complex revealed by NMR and X-ray crystallography. J. Mol. Biol. 371 669–684. 10.1016/j.jmb.2007.05.073 [DOI] [PubMed] [Google Scholar]
- Higman V. A., Briggs D. C., Mahoney D. J., Blundell C. D., Sattelle B. M., Dyer D. P., et al. (2014). A refined model for the TSG-6 link module in complex with hyaluronan. J. Biol. Chem. 289 5619–5634. 10.1074/jbc.M113.542357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hricovini M. (2004). Structural aspects of carbohydrates and the relation with their biological properties. Curr. Med. Chem. 11 2565–2583. 10.2174/0929867043364414 [DOI] [PubMed] [Google Scholar]
- Hricovíni M., Guerrini M., Bisio A., Torri G., Naggi A., Casu B. (2002). Active conformations of glycosaminoglycans. NMR determination of the conformation of heparin sequences complexed with antithrombin and fibroblast growth factors in solution. Semin. Thrombosis Hemostasis 28 325–334. 10.1055/s-2002-34301 [DOI] [PubMed] [Google Scholar]
- Hu Y., Zhong Y., Chen Z., Chen C., Shi Z., Zulueta M. M. L., et al. (2012). Divergent synthesis of 48 heparan sulfate-based disaccharides and probing the specific sugar–fibroblast growth factor-1 interaction. J. Am. Chem. Soc. 134 20722–20727. 10.1021/ja3090065 [DOI] [PubMed] [Google Scholar]
- Huang R., Leung I. K. H. (2019). Protein-small molecule interactions by waterLOGSY. Methods Enzymol. 615 477–500. 10.1016/bs.mie.2018.08.020 [DOI] [PubMed] [Google Scholar]
- Huckerby T. (2002). The keratan sulphates: structural investigations using NMR spectroscopy. Prog. Nuclear Magn. Resonance Spect. 40 35–110. 10.1016/S0079-6565(01)00040-1 [DOI] [Google Scholar]
- Hughes A., Meneghetti M., Huang T., Hung S., Elli S., Guerrini M., et al. (2017). Investigating the relationship between temperature, conformation and calcium binding in heparin model oligosaccharides. Carbohydrate Res. 438 58–64. 10.1016/j.carres.2016.12.002 [DOI] [PubMed] [Google Scholar]
- Huynh M. B., Ouidja M. O., Chantepie S., Carpentier G., Maïza A., Zhang G., et al. (2019). Glycosaminoglycans from Alzheimer’s disease hippocampus have altered capacities to bind and regulate growth factors activities and to bind tau. PLoS One 14:e209573. 10.1371/journal.pone.0209573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens R., Mortier A., Boff D., Ruytinx P., Gouwy M., Vantilt B., et al. (2017a). Truncation of CXCL12 by CD26 reduces its CXC chemokine receptor 4- and atypical chemokine receptor 3-dependent activity on endothelial cells and lymphocytes. Biochem. Pharmacol. 132 92–101. 10.1016/j.bcp.2017.03.009 [DOI] [PubMed] [Google Scholar]
- Janssens R., Struyf S., Proost P. (2017b). The unique structural and functional features of CXCL12. Cell. Mol. Immunol. 15 299–311. 10.1038/cmi.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Barbero J., Peters T. (2003). NMR spectroscopy of glycoconjugates. Weinheim: Wiley-VCH. 74. [Google Scholar]
- José García-Jiménez M., Corzana F., De Paz J. L., Nieto P. M. (2019). Langerin-heparin interaction: analysis of the binding to the non-lectin site. Nat. Prod. Commun. 14 1934578X–1985159X. 10.1177/1934578X19851597 [DOI] [Google Scholar]
- Joseph P. R. B., Mosier P. D., Desai U. R., Rajarathnam K. (2015). Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: structural plasticity mediates differential binding interactions. Biochem. J. 472 121–133. 10.1042/BJ20150059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahmann J. D., O’Brien R., Werner J. M., Heinegard D., Ladbury J. E., Campbell I. D., et al. (2000). Localization and characterization of the hyaluronan-binding site on the link module from human TSG-6. Structure 8 763–774. 10.1016/s0969-2126(00)00163-5 [DOI] [PubMed] [Google Scholar]
- Kan M., Wang F., Xu J., Crabb J. W., Hou J., McKeehan W. L. (1993). An essential heparin-binding domain in the fibroblast growth factor receptor kinase. Science 259 1918–1921. 10.1126/science.8456318 [DOI] [PubMed] [Google Scholar]
- Kato K., Peters T. (2017). NMR in glycoscience and glycotechnology. London: Royal Society of Chemistry. [Google Scholar]
- Kohda D., Morton C. J., Parkar A. A., Hatanaka H., Inagaki F. M., Campbell I. D., et al. (1996). Solution structure of the link module: a hyaluronan-binding domain involved in extracellular matrix stability and cell migration. Cell 86 767–775. 10.1016/s0092-8674(00)80151-8 [DOI] [PubMed] [Google Scholar]
- Korsensky L., Ron D. (2016). Regulation of FGF signaling: recent insights from studying positive and negative modulators. Semin. Cell. Dev. Biol. 53 101–114. 10.1016/j.semcdb.2016.01.023 [DOI] [PubMed] [Google Scholar]
- Künze G., Gehrcke J., Pisabarro M. T., Huster D. (2014). NMR characterization of the binding properties and conformation of glycosaminoglycans interacting with interleukin-10. Glycobiology 24 1036–1049. 10.1093/glycob/cwu069 [DOI] [PubMed] [Google Scholar]
- Künze G., Köhling S., Vogel A., Rademann J., Huster D. (2016). Identification of the glycosaminoglycan binding site of interleukin-10 by NMR spectroscopy. J. Biol. Chem. 291 3100–3113. 10.1074/jbc.M115.681759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguri C., Sapay N., Simorre J., Brutscher B., Imberty A., Gans P., et al. (2011). 13C-labeled heparan sulfate analogue as a tool to study protein/heparan sulfate interactions by NMR Spectroscopy: application to the CXCL12α Chemokine. J. Am. Chem. Soc. 133 9642–9645. 10.1021/ja201753e [DOI] [PubMed] [Google Scholar]
- Lane D. A., Denton J., Flynn A. M., Thunberg L., Lindahl U. (1984). Anticoagulant activities of heparin oligosaccharides and their neutralization by platelet factor 4. Biochem. J. 218 725–732. 10.1042/bj2180725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledwitch K. V., Barnes R. W., Roberts A. G. (2016). Unravelling the complex drug–drug interactions of the cardiovascular drugs, verapamil and digoxin, with P-glycoprotein. Biosci. Rep. 36:e309. 10.1042/BSR20150317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wang C., Xiao J., McKeehan W. L., Wang F. (2016). Fibroblast growth factors, old kids on the new block. Semin. Cell Dev. Biol. 53 155–167. 10.1016/j.semcdb.2015.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl U., Backstrom G., Thunberg L., Leder I. G. (1980). Evidence for a 3-O-sulfated D-glucosamine residue in the antithrombin-binding sequence of heparin. Proc. Natl. Acad. Sci. U S A. 77 6551–6555. 10.1073/pnas.77.11.6551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Li J., Arnold K., Pawlinski R., Key N. S. (2020). Using heparin molecules to manage COVID-2019. Res. Prac. Thrombosis Haemostasis 4 518–523. 10.1002/rth2.12353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmos K. G., Bjerring M., Jessen C. M., Nielsen E. H. T., Poulsen E. T., Christiansen G., et al. (2016). How glycosaminoglycans promote fibrillation of salmon calcitonin. J. Biol. Chem. 291 16849–16862. 10.1074/jbc.M116.715466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandaliti W., Nepravishta R., Pica F., Vallebona P. S., Garaci E., Paci M. (2017). Thymosin α1 interacts with hyaluronic acid electrostatically by its terminal sequence LKEKK. Molecules 22:1843. 10.3390/molecules22111843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melrose J. (2019). Keratan sulfate (KS)-proteoglycans and neuronal regulation in health and disease: the importance of KS-glycodynamics and interactive capability with neuroregulatory ligands. J. Neurochem. 149 170–194. 10.1111/jnc.14652 [DOI] [PubMed] [Google Scholar]
- Miller M. C., Cai C., Wichapong K., Bhaduri S., Pohl N. L. B., Linhardt R. J., et al. (2020). Structural insight into the binding of human galectins to corneal keratan sulfate, its desulfated form and related saccharides. Sci. Rep. 10 1–18. 10.1038/s41598-020-72645-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monneau Y. R., Luo L., Sankaranarayanan N. V., Nagarajan B., Vivès R. R., Baleux F., et al. (2017). Solution structure of CXCL13 and heparan sulfate binding show that GAG binding site and cellular signalling rely on distinct domains. Open Biol. 7:170133. 10.1098/rsob.170133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morla S. (2019). Glycosaminoglycans and glycosaminoglycan mimetics in cancer and inflammation. Int. J. Mol. Sci. 20:1963. 10.3390/ijms20081963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy F. J., Safran M., Seddon A. P., Kitchen D., Bohlen P., Aviezer D., et al. (1997). Properly oriented heparin-decasaccharide-induced dimers are the biologically active form of basic fibroblast growth factor. Biochemistry 36 4782–4791. 10.1021/bi9625455 [DOI] [PubMed] [Google Scholar]
- Moy F. J., Seddon A. P., Campbell E. B., Böhlen P., Powers R. (1995). 1H, 15N, 13C and 13CO assignments and secondary structure determination of basic fibroblast growth factor using 3D heteronuclear NMR spectroscopy. J. Biomol. NMR 6 245–254. 10.1007/BF00197806 [DOI] [PubMed] [Google Scholar]
- Mulloy B. (2006). Progress in the structural biology of chondroitin sulfate. Adv. Pharmacol. 53:49. 10.1016/S1054-3589(05)53004-7 [DOI] [PubMed] [Google Scholar]
- Muñoz-García J. C., Chabrol E., Vivès R. R., Thomas A., de Paz J. L., Rojo J., et al. (2015). Langerin–heparin interaction: two binding sites for small and large ligands as revealed by a combination of nmr spectroscopy and cross-linking mapping experiments. J. Am. Chem. Soc. 137 4100–4110. 10.1021/ja511529x [DOI] [PubMed] [Google Scholar]
- Muñoz-García J. C., López-Prados J., Angulo J., Díaz-Contreras I., Reichardt N., de Paz J. L., et al. (2012). Effect of the substituents of the neighboring ring in the conformational equilibrium of iduronate in heparin-like trisaccharides. Chem. Eur. J. 18 16319–16331. 10.1002/chem.201202770 [DOI] [PubMed] [Google Scholar]
- Muñoz-García J. C., Solera C., Carrero P., de Paz J. L., Angulo J., Nieto P. M. (2013). 3D structure of a heparin mimetic analogue of a FGF-1 activator. A NMR and molecular modelling study. Organic Biomol. Chem. 11:8269. 10.1039/c3ob41789a [DOI] [PubMed] [Google Scholar]
- Murphy J. W., Cho Y., Sachpatzidis A., Fan C., Hodsdon M. E., Lolis E. (2007). Structural and functional basis of CXCL12 (stromal cell-derived Factor-1α) binding to heparin. J. Biol. Chem. 282 10018–10027. 10.1074/jbc.M608796200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P. M., Heusinkveld L. (2018). Multisystem multitasking by CXCL12 and its receptors CXCR4 and ACKR3. Cytokine 109 2–10. 10.1016/j.cyto.2017.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naggi A., Gardini C., Pedrinola G., Mauri L., Urso E., Alekseeva A., et al. (2016). Structural peculiarity and antithrombin binding region profile of mucosal bovine and porcine heparins. J. Pharmaceutical Biomed. Analysis 118 52–63. 10.1016/j.jpba.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Naor D. (2016). Editorial: interaction between hyaluronic acid and its receptors (CD44, RHAMM) regulates the activity of inflammation and cancer. Front. Immunol. 7:39. 10.3389/fimmu.2016.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto L., Canales Á, Fernández I. S., Santillana E., González-Corrochano R., Redondo-Horcajo M., et al. (2013). Heparin modulates the mitogenic activity of fibroblast growth factor by inducing dimerization of its receptor. A 3D view by using NMR. Chem. BioChem. 14 1732–1744. 10.1002/cbic.201300313 [DOI] [PubMed] [Google Scholar]
- Nieto L., Canales Á, Giménez-Gallego G., Nieto P. M., Jiménez-Barbero J. (2011). Conformational selection of the AGA∗IAM heparin pentasaccharide when bound to the fibroblast growth factor receptor. Chem. Eur. J. 17 11204–11209. 10.1002/chem.201101000 [DOI] [PubMed] [Google Scholar]
- Nogueira A. V., Rossi G. R., Iacomini M., Sassaki G. L., Trindade E. S., Cipriani T. R. (2019). Viscera of fishes as raw material for extraction of glycosaminoglycans of pharmacological interest. Int. J. Biol. Macromol. 121 239–248. 10.1016/j.ijbiomac.2018.09.156 [DOI] [PubMed] [Google Scholar]
- Oduah E., Linhardt R., Sharfstein S. (2016). Heparin: past, present, and future. Pharmaceuticals 9:38. 10.3390/ph9030038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino S., Nishida N., Umemoto R., Suzuki M., Takeda M., Terasawa H., et al. (2010). Two-state conformations in the hyaluronan-binding domain regulate CD44 adhesiveness under flow condition. Structure 18 649–656. 10.1016/j.str.2010.02.010 [DOI] [PubMed] [Google Scholar]
- Ogura K., Nagata K., Hatanaka H., Habuchi H., Kimata K., Tate S., et al. (1999). Solution structure of human acidic fibroblast growth factor and interaction with heparin-derived hexasaccharide. J. Biomol. NMR 13 11–24. 10.1023/A:1008330622467 [DOI] [PubMed] [Google Scholar]
- Ojeda R., Angulo J., Nieto P. M., Martín-Lomas M. (2002). The activation of fibroblast growth factors by heparin: synthesis and structural study of rationally modified heparin-like oligosaccharides. Can. J. Chem. 80 917–936. 10.1139/v02-023 [DOI] [Google Scholar]
- Olson S. T., Ingemar B. R., Bock S. C. (2002). Identification of critical molecular interactions mediating heparin activation of antithrombin: implications for the design of improved heparin anticoagulants. Trends Cardiovas. Med. 12 198–205. 10.1016/S1050-1738(02)00160-3 [DOI] [PubMed] [Google Scholar]
- Olson S. T., Srinivasan K. R., Björk I., Shore J. D. (1981). Binding of high affinity heparin to antithrombin III. Stopped flow kinetic studies of the binding interaction. J. Biol. Chem. 256 11073–11079. 10.1016/0165-022X(81)90075-0 [DOI] [PubMed] [Google Scholar]
- Orton H. W., Kuprov I., Loh C., Otting G. (2016). Using paramagnetism to slow down nuclear relaxation in protein NMR. J. Phys. Chem. Lett. 7 4815–4818. 10.1021/acs.jpclett.6b02417 [DOI] [PubMed] [Google Scholar]
- Ota F., Hirayama T., Kizuka Y., Yamaguchi Y., Fujinawa R., Nagata M., et al. (2018). High affinity sugar ligands of C-type lectin receptor langerin. Biochim. Biophys. Acta Gen. Subj. 1862 1592–1601. 10.1016/j.bbagen.2018.04.004 [DOI] [PubMed] [Google Scholar]
- Palhares L. C. G. F., Brito A. S., de Lima M. A., Nader H. B., London J. A., Barsukov I. L., et al. (2019). A further unique chondroitin sulfate from the shrimp litopenaeus vannamei with antithrombin activity that modulates acute inflammation. Carbohydrate Polymers 222:115031. 10.1016/j.carbpol.2019.115031 [DOI] [PubMed] [Google Scholar]
- Panitz N., Theisgen S., Samsonov S. A., Gehrcke J., Baumann L., Bellmann-Sickert K., et al. (2016). The structural investigation of glycosaminoglycan binding to CXCL12 displays distinct interaction sites. Glycobiology 26 1209–1221. 10.1093/glycob/cww059 [DOI] [PubMed] [Google Scholar]
- Park Y., Jowitt T. A., Day A. J., Prestegard J. H. (2016). Nuclear magnetic resonance insight into the multiple glycosaminoglycan binding modes of the link module from human TSG-6. Biochemistry 55 262–276. 10.1021/acs.biochem.5b01148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L. (2001). Role of heparan sulfate in fibroblast growth factor signalling: a structural view. Curr. Opinion Struct. Biol. 11 629–634. 10.1016/s0959-440x(00)00258-x [DOI] [PubMed] [Google Scholar]
- Penk A., Baumann L., Huster D., Samsonov S. A. (2019). NMR and molecular modeling reveal specificity of the interactions between CXCL14 and glycosaminoglycans. Glycobiology 29 715–725. 10.1093/glycob/cwz047 [DOI] [PubMed] [Google Scholar]
- Pestka S., Krause C. D., Sarkar D., Walter M. R., Shi Y., Fisher P. B. (2004). Interleukin-10 and related cytokines and receptors. Ann. Rev. Immunol. 22 929–979. 10.1146/annurev.immunol.22.012703.104622 [DOI] [PubMed] [Google Scholar]
- Pichert A., Samsonov S. A., Theisgen S., Thomas L., Baumann L., Schiller J., et al. (2012). Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology 22 134–145. 10.1093/glycob/cwr120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomin V., Mulloy B. (2018). Glycosaminoglycans and proteoglycans. Pharmaceuticals 11:27. 10.3390/ph11010027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomin V., Wang X. (2018a). Glycosaminoglycan-protein interactions by nuclear magnetic resonance (NMR) spectroscopy. Molecules 23:2314. 10.3390/molecules23092314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomin V. H. (2014). Solution NMR conformation of glycosaminoglycans. Prog. Biophys. Mol. Biol. 114 61–68. 10.1016/j.pbiomolbio.2014.01.001 [DOI] [PubMed] [Google Scholar]
- Pomin V. H. (2015). Keratan sulfate: an up-to-date review. Int. J. Biol. Macromol. 72 282–289. 10.1016/j.ijbiomac.2014.08.029 [DOI] [PubMed] [Google Scholar]
- Pomin V. H. (2016). Paradigms in the structural biology of the mitogenic ternary complex FGF: FGFR: heparin. Biochimie 127 214–226. 10.1016/j.biochi.2016.05.017 [DOI] [PubMed] [Google Scholar]
- Pomin V. H., Wang X. (2018b). Synthetic oligosaccharide libraries and microarray technology: a powerful combination for the success of current glycosaminoglycan interactomics. Chem. MedChem. 13 648–661. 10.1002/cmdc.201700620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybylski C., Gonnet F., Saesen E., Lortat-Jacob H., Daniel R. (2020). Surface plasmon resonance imaging coupled to on-chip mass spectrometry: a new tool to probe protein-GAG interactions. Analytical Bioanalytical Chem. 412 507–519. 10.1007/s00216-019-02267-2 [DOI] [PubMed] [Google Scholar]
- Rajarathnam K., Sepuru K. M., Joseph P. R. B., Sawant K. V., Brown A. J. (2018). Glycosaminoglycan interactions fine-tune chemokine-mediated neutrophil trafficking: structural insights and molecular mechanisms. J. Histochem. Cytochem. 66 229–239. 10.1369/0022155417739864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie A. R., Giri H. (2020). Antithrombin: an anticoagulant, anti-inflammatory and antibacterial serpin. J. Thrombosis Haemostasis 18 528–533. 10.1111/jth.14724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider C. C., Mulloy B. (2017). Heparin, heparan sulphate and the TGF-β cytokine superfamily. Molecules 22:713. 10.3390/molecules22050713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth R., Swanson R., Izaguirre G., Bock S. C., Gettins P. G. W., Olson S. T. (2015). Saturation mutagenesis of the antithrombin reactive center loop P14 residue supports a three-step mechanism of heparin allosteric activation involving intermediate and fully activated states. J. Biol. Chem. 290 28020–28036. 10.1074/jbc.M115.678839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd T. R., Preston M. D., Yates E. A. (2017). The nature of the conserved basic amino acid sequences found among 437 heparin binding proteins determined by network analysis. Mol. BioSyst. 13 852–865. 10.1039/C6MB00857G [DOI] [PubMed] [Google Scholar]
- Ryan E., Shen D., Wang X. (2016). Structural studies reveal an important role for the pleiotrophin C-terminus in mediating interactions with chondroitin sulfate. FEBS J. 283 1488–1503. 10.1111/febs.13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadir R., Baleux F., Grosdidier A., Imberty A., Lortat-Jacob H. (2001). Characterization of the stromal cell-derived factor-1α-heparin complex. J. Biol. Chem. 276 8288–8296. 10.1074/jbc.M008110200 [DOI] [PubMed] [Google Scholar]
- Sadir R., Imberty A., Baleux F., Lortat-Jacob H. (2004). Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 279 43854–43860. 10.1074/jbc.M405392200 [DOI] [PubMed] [Google Scholar]
- Saesen E., Sarrazin S., Laguri C., Sadir R., Maurin D., Thomas A., et al. (2013). Insights into the mechanism by which interferon-γ basic amino acid clusters mediate protein binding to heparan sulfate. J. Am. Chem. Soc. 135 9384–9390. 10.1021/ja4000867 [DOI] [PubMed] [Google Scholar]
- Sandoval D. R., Gomez Toledo A., Painter C. D., Tota E. M., Sheikh M. O., West A. M. V., et al. (2020). Proteomics-based screening of the endothelial heparan sulfate interactome reveals that C-type lectin 14a (CLEC14A) is a heparin-binding protein. J. Biol. Chem. 295 2804–2821. 10.1074/jbc.RA119.011639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasarman F., Maftei C., Campeau P. M., Brunel-Guitton C., Mitchell G. A., Allard P. (2016). Biosynthesis of glycosaminoglycans: associated disorders and biochemical tests. J. Inherited Metabolic Dis. 39 173–188. 10.1007/s10545-015-9903-z [DOI] [PubMed] [Google Scholar]
- Saxena K., Schieborr U., Anderka O., Duchardt-Ferner E., Elshorst B., Gande S. L., et al. (2010). Influence of heparin mimetics on assembly of the FGF⋅FGFR4 signaling complex. J. Biol. Chem. 285 26628–26640. 10.1074/jbc.M109.095109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J., Plotnikov A. N., Ibrahimi O. A., Eliseenkova A. V., Yeh B. K., Yayon A., et al. (2000). Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell. 6 743–750. 10.1016/S1097-2765(00)00073-3 [DOI] [PubMed] [Google Scholar]
- Senbanjo L. T., Chellaiah M. A. (2017). CD44: a multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell. Dev. Biol. 5:18. 10.3389/fcell.2017.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo E. S., Blaum B. S., Vargues T., De Cecco M., Deakin J. A., Lyon M., et al. (2010). Interaction of Human β-Defensin 2 (HBD2) with glycosaminoglycans. Biochemistry 49 10486–10495. 10.1021/bi1011749 [DOI] [PubMed] [Google Scholar]
- Sepuru K. M., Iwahara J., Rajarathnam K. (2018a). Direct detection of lysine side chain NH3+ in protein-heparin complexes using NMR spectroscopy. Analyst 143 635–638. 10.1039/c7an01406f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepuru K. M., Nagarajan B., Desai U. R., Rajarathnam K. (2016). Molecular basis of chemokine CXCL5-glycosaminoglycan interactions. J. Biol. Chem. 291 20539–20550. 10.1074/jbc.M116.745265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepuru K. M., Nagarajan B., Desai U. R., Rajarathnam K. (2018b). Structural basis, stoichiometry, and thermodynamics of binding of the chemokines KC and MIP2 to the glycosaminoglycan heparin. J. Biol. Chem. 293 17817–17828. 10.1074/jbc.RA118.004866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepuru K. M., Rajarathnam K. (2016). CXCL1/MGSA Is a novel glycosaminoglycan (GAG)-binding chemokine. J. Biol. Chem. 291 4247–4255. 10.1074/jbc.M115.697888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepuru K. M., Rajarathnam K. (2019). Structural basis of chemokine interactions with heparan sulfate, chondroitin sulfate, and dermatan sulfate. J. Biol. Chem. 294 15650–15661. 10.1074/jbc.RA119.009879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin I. C., Gaudry J., Johnson Z., Kungl A., Jansma A., Gesslbauer B., et al. (2010). Characterization of the chemokine CXCL11-heparin interaction suggests two different affinities for glycosaminoglycans. J. Biol. Chem. 285 17713–17724. 10.1074/jbc.M109.082552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silipo A., Zhang Z., Cañada F. J., Molinaro A., Linhardt R. J., Jiménez Barbero J. (2008). Conformational analysis of a dermatan sulfate-derived tetrasaccharide by nmr, molecular modeling, and residual dipolar couplings. Chembiochem 9 240–252. 10.1002/cbic.200700400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skidmore M. A., Guimond S. E., Rudd T. R., Fernig D. G., Turnbull J. E., Yates E. A. (2009). The activities of heparan sulfate and its analogue heparin are dictated by biosynthesis, sequence, and conformation. Connective Tissue Res. 49 140–144. 10.1080/03008200802148595 [DOI] [PubMed] [Google Scholar]
- Soares P. A. G., Queiroz I. N. L., Pomin V. H. (2017). NMR structural biology of sulfated glycans. J. Biomol. Struct. Dynamics 35 1069–1084. 10.1080/07391102.2016.1171165 [DOI] [PubMed] [Google Scholar]
- Solera C., Macchione G., Maza S., Kayser M. M., Corzana F., de Paz J. L., et al. (2016). Chondroitin sulfate tetrasaccharides: synthesis, three-dimensional structure and interaction with midkine. Chem. Eur. J. 22 2356–2369. 10.1002/chem.201504440 [DOI] [PubMed] [Google Scholar]
- Srb P., Svoboda M., Benda L., Lepšík M., Tarábek J., Šícha V., et al. (2019). Capturing a dynamically interacting inhibitor by paramagnetic NMR spectroscopy. Phys. Chem. Chem. Phys. 21 5661–5673. 10.1039/C9CP00416E [DOI] [PubMed] [Google Scholar]
- Stancanelli E., Elli S., Hsieh P., Liu J., Guerrini M. (2018). Recognition and conformational properties of an alternative antithrombin binding sequence obtained by chemoenzymatic synthesis. Chem. BioChem 19 1178–1188. 10.1002/cbic.201800095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart K. L., Hughes E., Yates E. A., Akien G. R., Huang T., Lima M. A., et al. (2016). Atomic details of the interactions of glycosaminoglycans with amyloid-β fibrils. J. Am. Chem. Soc. 138 8328–8331. 10.1021/jacs.6b02816 [DOI] [PubMed] [Google Scholar]
- Takeda M., Ogino S., Umemoto R., Sakakura M., Kajiwara M., Sugahara K. N., et al. (2006). Ligand-induced structural changes of the CD44 hyaluronan-binding domain revealed by NMR. J. Biol. Chem. 281 40089–40095. 10.1074/jbc.M608425200 [DOI] [PubMed] [Google Scholar]
- Takeda M., Terasawa H., Sakakura M., Yamaguchi Y., Kajiwara M., Kawashima H., et al. (2003). Hyaluronan recognition mode of CD44 revealed by cross-saturation and chemical shift perturbation experiments. J. Biol. Chem. 278 43550–43555. 10.1074/jbc.M308199200 [DOI] [PubMed] [Google Scholar]
- Taweechat P., Pandey R. B., Sompornpisut P. (2020). Conformation, flexibility and hydration of hyaluronic acid by molecular dynamics simulations. Carbohydrate Res. 493:108026. 10.1016/j.carres.2020.108026 [DOI] [PubMed] [Google Scholar]
- Teriete P., Banerji S., Noble M., Blundell C. D., Wright A. J., Pickford A. R., et al. (2004). Structure of the regulatory hyaluronan binding domain in the inflammatory leukocyte homing receptor CD44. Mol. Cell. 13 483–496. 10.1016/s1097-2765(04)00080-2 [DOI] [PubMed] [Google Scholar]
- Theocharis A. D., Skandalis S. S., Gialeli C., Karamanos N. K. (2016). Extracellular matrix structure. Adv. Drug Delivery Rev. 97 4–27. 10.1016/j.addr.2015.11.001 [DOI] [PubMed] [Google Scholar]
- Thunberg L., Bäckström G., Lindahl U. (1982). Further characterization of the antithrombin-binding sequence in heparin. Carbohydrate Res. 100 393–410. 10.1016/S0008-6215(00)81050-2 [DOI] [PubMed] [Google Scholar]
- Tjong S., Chen T., Huang W., Wu W. (2007). Structures of heparin-derived tetrasaccharide bound to cobra cardiotoxins: heparin binding at a single protein site with diverse side chain interactions†. Biochemistry 46 9941–9952. 10.1021/bi700995v [DOI] [PubMed] [Google Scholar]
- Uhl F. E., Zhang F., Pouliot R. A., Uriarte J. J., Rolandsson E. S., Han X., et al. (2020). Functional role of glycosaminoglycans in decellularized lung extracellular matrix. Acta Biomater 102 231–246. 10.1016/j.actbio.2019.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boeckel C. A., Grootenhuis P. D., Visser A. (1994). A mechanism for heparin-induced potentiation of antithrombin III. Nat. Struct. Biol. 1:423. 10.1038/nsb0794-423 [DOI] [PubMed] [Google Scholar]
- Vanhaverbeke C., Simorre J., Sadir R., Gans P., Lortat-Jacob H. (2004). NMR characterization of the interaction between the C-terminal domain of interferon-gamma and heparin-derived oligosaccharides. Biochem. J. 384 93–99. 10.1042/BJ20040757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos A., Pomin V. (2017). The sea as a rich source of structurally unique glycosaminoglycans and mimetics. Microorganisms 5:51. 10.3390/microorganisms5030051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigetti D., Passi A., Luca G. D., Caon I., Moretto P., Angelo M. L., et al. (2016). Extracellular matrix in atherosclerosis: hyaluronan and proteoglycans insights. Curr. Med. Chem. 23 2958–2971. 10.2174/0929867323666160607104602 [DOI] [PubMed] [Google Scholar]
- Vignovich W. P., Pomin V. H. (2020). Saturation transfer difference in characterization of glycosaminoglycan-protein interactions. SLAS Technol. 25 307–319. 10.1177/2472630320921130 [DOI] [PubMed] [Google Scholar]
- Wang X., Watson C., Sharp J. S., Handel T. M., Prestegard J. H. (2011). Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 19 1138–1148. 10.1016/j.str.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckbach L., Preissner K., Deindl E. (2018). The role of midkine in arteriogenesis, involving mechanosensing, endothelial cell proliferation, and vasodilation. Int. J. Mol. Sci. 19:2559. 10.3390/ijms19092559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore E. K., Martin D., Guvench O. (2020). Constructing 3-dimensional atomic-resolution models of nonsulfated glycosaminoglycans with arbitrary lengths using conformations from molecular dynamics. Int. J. Mol. Sci. 21:7699. 10.3390/ijms21207699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C., Lian W., Zhou L., Gao N., Xu L., Chen J., et al. (2016). Interactions between depolymerized fucosylated glycosaminoglycan and coagulation proteases or inhibitors. Thrombosis Res. 146 59–68. 10.1016/j.thromres.2016.08.027 [DOI] [PubMed] [Google Scholar]
- Yang J., Chi L. (2017). Characterization of structural motifs for interactions between glycosaminoglycans and proteins. Carbohydrate Res. 452 54–63. 10.1016/j.carres.2017.10.008 [DOI] [PubMed] [Google Scholar]
- Yang J., Shen M., Wen H., Luo Y., Huang R., Rong L., et al. (2020). Recent advance in delivery system and tissue engineering applications of chondroitin sulfate. Carbohydrate Polymers 230:115650. 10.1016/j.carbpol.2019.115650 [DOI] [PubMed] [Google Scholar]
- Ye S., Luo Y., Lu W., Jones R. B., Linhardt R. J., Capila I., et al. (2001). Structural basis for interaction of FGF-1, FGF-2, and FGF-7 with different heparan sulfate motifs. Biochemistry 40 14429–14439. 10.1021/bi011000u [DOI] [PubMed] [Google Scholar]
- Yu F., Roy S., Arevalo E., Schaeck J., Wang J., Holte K., et al. (2014). Characterization of heparin–protein interaction by saturation transfer difference (STD) NMR. Analytical Bioanalytical Chem. 406 3079–3089. 10.1007/s00216-014-7729-4 [DOI] [PubMed] [Google Scholar]
- Zhao B., LiWang P. J. (2010). Characterization of the interactions of vMIP-II, and a dimeric variant of vMIP-II, with glycosaminoglycans. Biochemistry 49 7012–7022. 10.1021/bi100549y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Huvent I., Lippens G., Eliezer D., Zhang A., Li Q., et al. (2017). Glycan determinants of heparin-tau interaction. Biophys. J. 112 921–932. 10.1016/j.bpj.2017.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Komiya H., Chirino A., Faham S., Fox G. M., Arakawa T., et al. (1991). Three-dimensional structures of acidic and basic fibroblast growth factors. Science 251 90–93. 10.1126/science.1702556 [DOI] [PubMed] [Google Scholar]
- Zhu Y., Zhang F., Linhardt R. J. (2019). “Heparin Contamination and Issues Related to Raw Materials and Controls,” in The Science and Regulations of Naturally Derived Complex Drugs, eds Sasisekharan R., Lee S. L., Rosenberg A., Walker L. A. (Cham: Springer International Publishing; ), 191–206. 10.1007/978-3-030-11751-1_11 [DOI] [Google Scholar]
- Ziarek J. J., Veldkamp C. T., Zhang F., Murray N. J., Kartz G. A., Liang X., et al. (2013). Heparin oligosaccharides inhibit chemokine (CXC Motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC Motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 288 737–746. 10.1074/jbc.M112.394064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell M. R., Prestwich G. D. (2004). Interactions of peptide mimics of hyaluronic acid with the receptor for hyaluronan mediated motility (RHAMM). J. Comp. Aided Mol. Design 18 597–614. 10.1007/s10822-004-5433-8 [DOI] [PubMed] [Google Scholar]
- Zsila F., Juhász T., Kohut G., Beke-Somfai T. (2018). Heparin and heparan sulfate binding of the antiparasitic drug imidocarb: circular dichroism spectroscopy, isothermal titration calorimetry, and computational studies. J. Phys. Chem. B 122 1781–1791. 10.1021/acs.jpcb.7b08876 [DOI] [PubMed] [Google Scholar]