

Abstract
Nearly 200 distinct chemical modifications of RNAs have been discovered to date. Their analysis via direct methods has been possible in abundant RNA species, such as ribosomal, transfer or viral RNA, since several decades. However, their analysis in less abundant RNAs species, especially cellular messenger RNAs, was rendered possible only recently with the advent of high throughput sequencing techniques. Given the growing biomedical interest of the proteins that write, erase and read RNA modifications, ingenious new methods to enrich and identify RNA modifications at base resolution have been implemented, and more efforts are underway to render them more quantitative. Here, we review several crucial modification-specific (bio)chemical approaches and discuss their advantages and shortcomings for exploring the epitranscriptome.
Keywords: epitranscriptome, RNA modifications, methylation, acetylation, method, sequencing
Introduction
The chemical diversity of RNA polymers is vastly expanded by more than 200 chemical modifications that can occur at the phosphate, ribose and nucleobases in living organisms [1]. These chemical tags fine-tune RNA–DNA, RNA–RNA and RNA–protein interactions, eventually affecting gene expression networks and cellular functions. Most of RNA modifications are incorporated during or after transcription and are often called post-transcriptional modifications. Borrowing from the language of epigenetics, and from the Ancient Greek ε’πί (epí, ‘on top of’), RNA modifications are also called epitranscriptomic, to highlight that they carry another layer of information on top of the RNA sequence itself. In this review, we highlight methods developed to dissect this additional layer of information for a few base modifications with high biological impact.
Transfer RNAs (tRNAs) are the most chemically diverse RNA molecules. tRNAs have the advantage of being small (<100 nt), abundant in cells and carrying evolutionary conserved RNA modifications [2]. As a result, the study of tRNA modifications has driven forward technological advances in the last 6 decades. The state of the art for tRNA modification detection and quantification entails tRNA affinity purification with sequence specific probes, digestion with specific RNases and tandem mass spectrometry analysis [3]. Despite their abundance, full-length tRNAs remain difficult to sequence using RNA-seq techniques. The main culprit is the presence of specific tRNA modifications that create a roadblock for many commonly used reverse transcriptases (RTs) during the step of complementary DNA (cDNA) synthesis [4]. The two main strategies for overcoming this hurdle has been to either remove select tRNA modifications with appropriate eraser enzymes before RNA-seq library preparation [5, 6], or to use RTs, such as thermostable group II intron reverse transcriptases (TGIRTs) [7] that have the dual advantage of bypassing RNA modification roadblocks and of leaving a characteristic nucleotide misincorporation signature at RNA modification sites [8, 9].
Specific messenger RNAs (mRNAs) are significantly less abundant than specific tRNAs, making affinity purification techniques coupled to tandem mass spectrometry unviable for detecting specific mRNA modifications at single nucleotide resolution level. Instead, modifications on specific mRNAs and other less abundant RNAs are most often inferred through either direct or indirect (sequencing-by-synthesis [SBS]) sequencing methods (Figure 1). Indeed, many RNA modifications are now detectable by a direct sequencing method from Oxford Nanopore Technology (Figure 1), which decodes changes to an electrical current as nucleic acids are passed through a protein nanopore to provide the specific RNA sequence [10]. While still in development, this technology is very promising for identifying RNA modifications and their combinations in intact RNA molecules, but due to space limitations, we will not review this method here, and refer the reader to an excellent review by Novoa et al. [10] as a starting point. In SBS methods, the RNA is first reverse-transcribed into cDNA with RTs prior to library amplification, and this step is crucial for detection of RNA modifications at the single nucleotide resolution level. RNA modifications that do not affect Watson–Crick base pairing are RT silent, i.e. they do not change the output sequence, while RNA modifications that change Watson–Crick base pairing can induce either a nucleotide misincorporation or an RT block at the RNA modification site during cDNA synthesis as mentioned above. In this case, the RNA modification site can be inferred from the mismatch to the reference sequence, or from read pile up/drop-off (Figure 1). RT silent modifications are most often enriched with specific antibodies prior to RNA-seq. While effective in identifying approximate RNA modification locations, such methods cannot precisely identify which sites in the RNA-seq peaks are modified, nor can they quantify the modification stoichiometry for each site. Remarkably however, RT silent RNA modifications can be ‘unsilenced’ through specific antibody crosslinking, chemical, editing or digestion methods to allow their identification at single-nucleotide resolution level (Figure 1).
Figure 1.
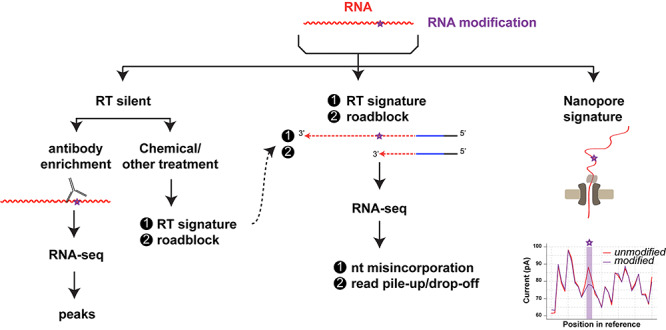
A given RNA modification may be detected by direct sequencing, such as the Oxford Nanopore Technology, or by SBS sequencing methods that involve an obligate RT into cDNA step. When the RNA modification does not change base pairing during RT (silent), the modification is enriched through specific antibodies, or ‘unsilenced’ through specific treatments. In the latter case, and when the RNA modification naturally changes the base pairing (RT signature) or creates a roadblock, it can inferred though mismatch to the reference or read pile-up or drop-off.
N6-Methyladenosine (m6A)
Adenosine can be methylated on the exocyclic amino group (-NH2) at the sixth position of the purine ring to form N6-methyladenosine (m6A). It is one of the most prevalent RNA modifications, especially in mRNA, where it is primarily written by METTL3/14 complex [11], and plays crucial roles in mRNA metabolism and associated cellular processes [12–14]. m6A requires sophisticated methods for its site specific detection and quantitation, because it does not disrupt Watson–Crick base pairing and is silent during reverse transcription [15]. m6A antibody-based enrichment and next-generation sequencing approaches (MeRIP-seq or m6A-seq) were pioneered by Meyer et al. [16], and Dominissini et al. [17] in 2012, and have proved to be successful for transcriptome-wide m6A mapping in many biological settings (reviewed in [18]). Another technique, termed site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET) was developed by the Pan group and is useful for validating the precise location of the m6A residue and its modification ratio one site at a time [19]. In addition, several metabolic tagging techniques using alkyne or allyl analogs of the methyl group donor S-Adenosyl-Methionine (SAM) that induce RT block [15] or misincorporation signature [20] have been reported. For example, the latter method uses Se-allyl-L-selenohomocysteine as SAM analog for in-cell experiments [20]. Once the allyl-handle is metabolically incorporated as N6-allyladenosine at the position of m6A by methyltransferases, through iodine induced cyclization, mismatch signatures at m6A sites can be generated. The MAZTER-seq method takes advantage of the fact that the endoribonuclease mazF cuts immediately upstream of an ACA sequence but not (m6A)CA [21], to detect and quantify the m6A sites occurring within the (m6A)CA motif. MAZTER-seq was successfully applied to profile and quantify m6A at single-nucleotide resolution at 16–25% of expressed sites in mRNA [22]. In another elegant method called DART-seq [23], Meyer fused the m6A-binding domain YTH from the YTHDF2 m6A reader to the deaminase domain (BE1) of APOBEC1 to induce a C to U editing in the invariable C residue of the m6A writer consensus domain (Gm6AC →Gm6AU) to enable antibody-free single nucleotide resolution m6A mapping and quantitation. In [23], the cells were transiently transfected with an APOBEC1-YTH construct limiting its use to cell lines or model organisms engineered to express APOBEC1-YTH, however Gm6AC →Gm6AU editing post-RNA extraction with recombinant APOBEC1-YTH should be feasible and render m6A mapping from low input samples—such as patient samples—finally possible.
5-Methylcytosine (m5C)
Similar to DNA, RNA can be methylated on the carbon 5 of the pyrimidine ring of cytosines to form 5-methylcytosine or m5C (Figure 2). m5C is abundant in tRNA and rRNA, as well as in other RNA species, including mRNA where it may be the second most abundant base modification after m6A [24]. Although RNA m5C modification has been less studied than m6A, it is clearly a very important modification carried out by eight different m5C RNA methyltransferases (NSUN1-7, and TRMT1/DNMT2) in humans, most of which have important biological roles relevant to disease and cancer [25, 26]. m5C is RT silent, but similarly to DNA, bisulfite treatment coupled to RNA-seq has been used to map m5C at single nucleotide resolution [27–30]. Indeed, due to the electron rich C5-C6 double bond in the m5C pyrimidine ring, bisulfite treatment converts C but not m5C to U (Figure 2). Thus, upon bisulfite treatment, reverse transcription-polymerase chain reaction (RT-PCR) and sequencing, C gives rise to T, while m5C stays C (Figure 2). However, bisulfite reaction suffers from RNA degradation and incomplete C to U conversion, especially in double stranded or highly structured regions (i.e. G-C clamps) [31], limiting m5C mapping to abundant RNAs. Other methods have been developed to improve the sensitivity of m5C detection. For example, in the Aza-IP method developed by Khoddami and Cairns [32], cells are metabolically labeled with the 5-azacytidine (5-azaC) C analog, which randomly gets incorporated into RNA instead of C. When this occurs at a target m5C RNA site, a covalent bond (C–S) forms between the C6 position of the base and the sulfur atom of a cysteine residue in the catalytic pocket of specific RNA methyltransferases [32]. An alternative, UV-crosslink-free version of iCLIP named miCLIP (methylation-individual nucleotide resolution crosslinking and immmunoprecipitation) was developed by the Frye lab to identify NSUN2 m5C RNA methyltransferase targets [33]. In miCLIP, NSUN2 is engineered to not release NSUN2 RNA targets due to irreversible covalent bond formation between the NSUN2-C271A mutant and the RNA catalytic intermediate. Thus, NSUN2 target RNAs could be enriched and identified through IP of the protein–RNA complex and RNA-seq of the recovered target RNAs [33]. Osmium reaction-based tagging of m5C followed by liquid chromatography coupled to mass spectrometry (LC–MS) has also been reported [34, 35]. It takes advantage of the C5-methyl group that increases electron density along the C5=C6 double bond in m5C and forms a stable m5C–Os–ligand ternary complex (ligand=bipyridine). However, it suffers from structural problems and high reactivity toward m5U (T) [34, 35].
Figure 2.
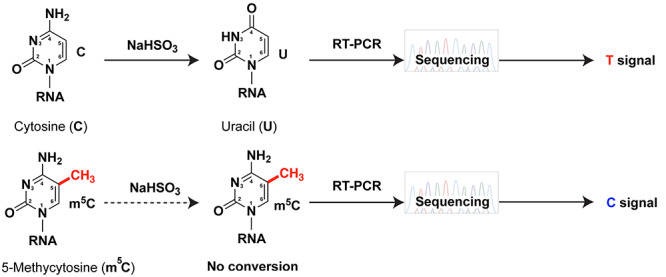
Chemistry behind bisulfite sequencing. Bisulfite deaminates cytosine (C) to uracil (U), giving rise to a T signal during sequencing. m5C has an electron rich C5-C6 bond and is resistant to deamination, exhibiting a C signal.
Pseudouridine (Ψ)
Pseudouridine or Ψ is a highly abundant RNA modification catalyzed by a large number of Ψ-synthases (PUS, 13 different enzymes in humans), which break the nitrogen-carbon glycosidic bond between the ribose and U, rotate the base by 180° and form a carbon–carbon glycosidic bond instead [36–39]. This isomerization allows for an additional hydrogen bond donor in Ψ compared to U contributing to better thermodynamic (structural) stability and base-stacking [36–39]. Ψ detection is problematic for two reasons; firstly, it is an RT silent modification, and secondly, it is also a mass spectrometry silent modification due to Ψ having the same mass as U. [39]. However Ψ can be ‘unsilenced’ by chemical labeling with carbodiimides or carbodiimide derivatives that enable its detection by mismatch signatures during sequencing [39–43] or by using several tag-free or tag-on mass spectrometry based approaches [44–46] (Figure 3). Interestingly, a recent report also indicated base-skipping/deletion at/near the position of Ψ during bisulfite sequencing [27].
Figure 3.
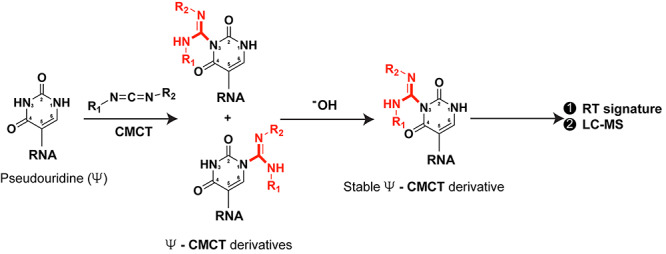
Labeling of Pseudouridine (Ψ) by carbodiimide CMCT (N-cyclohexyl-N′-β-(4-methylmorpholinium) ethylcarbodiimide p-tosylate) reagent. CMCT forms bulky derivatives which upon alkaline hydrolysis give rise to a single derivative exhibiting distinct RT signature. Using this chemistry, Ψ can be identified by sequencing and LC–MS. (R1 = cyclohexyl group, R2 = ethyl-4-methylmorpholinium group).
N4-acetylcytosine (ac4C)
Cytosines can undergo acetylation on the exocyclic amino group (–NH2) at the fourth position of the pyrimidine ring to form ac4C in a reaction requiring ATP, Acetyl-CoA, the NAT10 acetyltransferase [47], and in specific cases the U13 snoRNA or the THUMPD1 adaptor protein [48]. Ac4C stabilizes G–C interactions and fine-tunes mRNA translation efficiency and stability [49]. Ac4C is also RT silent, and recently, two different approaches attempted to reveal the ac4C epitranscriptome in human cell lines. The ac4C antibody-based acRIP-seq approach identified numerous ac4C sites in mRNA [49]. The other approach exploited the fact that sodium borohydride (NaBH4) or sodium cyanoborohydride (NaCNBH3) reduces ac4C to give rise to T upon RT-PCR, whereas unmodified C remains C [50] (Figure 4). The sodium cyanoborohydride treatment coupled to RNA-seq mapped ac4C in ribosomal RNA and a few tRNAs in human cells, but only mapped ac4C to a few mRNAs—only when both NAT10 and THUMPD1 were overexpressed [51]. It was suggested that the ac4C antibody suffers from non-specificity, however the sites of ac4C identified by acRIP-seq disappeared in HeLa-NAT10-KO cells [49]. Thus, it is likely that the discrepancies between the two approaches come from yet to be identified differences, including differences in cell lines growth conditions and/or ratio of modification. This discrepancy could be solved by coupling the acRIP-seq to sodium cyanoborohydride treatment, which could concomitantly enrich the ac4C modified RNAs and directly test the specificity of the antibody.
Figure 4.
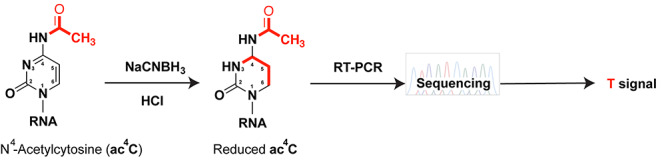
Sodium cyanoborohydride (NaCNBH3) mediated reduction of ac4C. In acidic media, the hydride reduces ac4C to N4-acetyltetrahydrocytidine, which generates a T signal instead of a C signal during sequencing.
2′-O-methylation (Nm)
2′-O-methylation, i.e. methylation of the 2′-OH group in ribose, is one of the most common modifications observed in almost every class of RNAs, including mRNAs and piRNAs [52, 53]. Please note that the whole nucleotide is referred to as Nm, where N is any base. 2′-O-methylation disrupts hydrogen bonds due to the increase in lipophilic surface and thus has significant effects including stabilization of helical structures, protection of RNA from various endonucleases, or fine-tuning of RNA-protein interactions [52]. Due to the presence of 2′-OMe group, Nm is resistant to alkaline hydrolysis (Figure 5A) or periodate oxidation (Figure 5B). These properties have been coupled to RNA-seq to map Nm sites in transcriptome-wide scale [54–57]. However, given that Nm mapping relies on read pile-up or drop-off at the Nm site, instead of misincorporation, the confidence in the identified sites is weakened, while the rate of Nm modification is difficult to estimate. For example, Nm-seq identified 7412 sites in non-ribosomal RNAs species from HEK293 cells, with a consensus sequence of 10 nt present in 33% of all sites [57]. However, close inspection of this consensus sequence revealed that it is nearly identical to the 3′ adaptor used in the Nm-seq library, suggesting that a third of identified Nm sites may be due to a commonly observed mispriming artifact [58]. In spite of this, the rest of the reads may represent bona fide Nm sites, highlighting the need for more robust methods for systematic detection and validation of Nm sites. Indeed, internal Nm modifications may have important roles in gene expression regulation, as suggested by a recent study showing that similar to rRNA, Fibrillarin can be guided by snoRNAs U32A and U51 to modify a specific residue within the protein-coding region of peroxidasin to enhance the stability, but inhibit the translation of the mRNA into protein [59].
Figure 5.
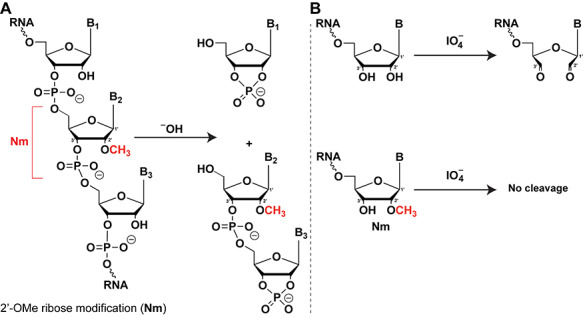
(A) Detection of Nm (2′-O-Me) by alkaline hydrolysis. Due to the presence of 2′-OH, RNA gets hydrolyzed in basic conditions, while 2′-OMe containing nucleotide is resistant to cleavage. (B) Detection of Nm (2′-O-Me) by periodate based oxidation. Periodate is capable of oxidizing 1,2-diol (2′,3′-OH groups), but not Nm due to the presence of 2′-OMe group.
7-methylguanosine (m7G)
Guanines can be methylated at the N7-position of the purine ring (m7G) inducing a positive charge. The most widely known function of m7G modification is related to it being an obligate feature of eukaryotic mRNA caps [60]. However, methylation of m7G by METTL1 was recently mapped in internal sites in other RNA species [61–65] by exploiting an observation in the 1970’s that sodium borohydride efficiently reduces m7G in basic conditions, and the reduced base is prone to forming an abasic site in acidic conditions [66, 67] (Figure 6). Since the abasic site consists of an equilibrating mixture of the ring-closed acetal (major) and the ring-opened aldehyde (minor), it can be covalently bound with hydrazine probes (e.g. biotin-hydrazine) for enrichment and detection purposes [61–65]. Moreover, given that m7G sites can be directly traced by the abasic site signatures generated during cDNA synthesis and sequencing [61–64], it is paramount to inspect that the sequence of the RNAs enriched by this method contain these abasic site signatures, in order to distinguish background RNAs from RNAs with bona fide m7G sites.
Figure 6.
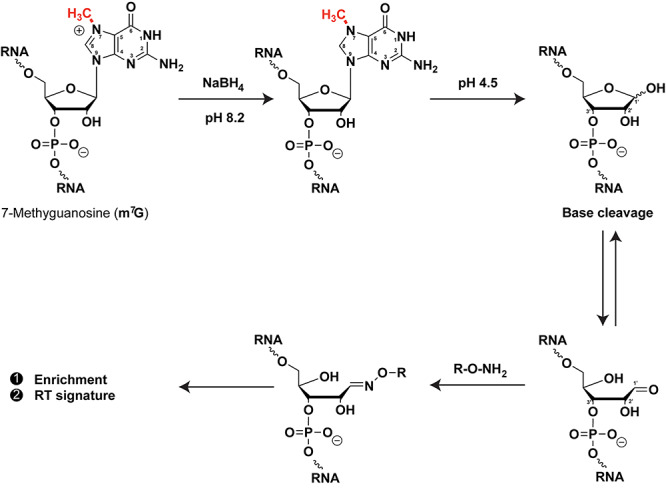
Sodium borohydride (NaBH4) mediated detection of m7G. NaBH4 reduces m7G and the reduced base is prone to cleavage forming an abasic site in acidic media. Abasic sites can be covalently linked with hydrazine probes (e.g. biotin-hydrazine) for enrichment and detection purposes. R = linker containing biotin moiety.
N1-methyladenosine (m1A)
N1-methyladenosine is a major modification primarily observed in conserved sites in tRNA and rRNA [68]. The m1A nucleoside is positively charged and able of disrupting Watson–Crick base pairing. Consequently, m1A creates RT roadblock and/or misincorporation signatures in ratios that are specific of the adjoining nucleotides and used reverse transcriptases [69–71]. These properties have been exploited to map m1A at single nucleotide resolution in RNA-seq experiments performed with or without pre-enrichment with an m1A antibody [5–9, 69–76]. The stoichiometry of m1A modification in tRNA and rRNA is high, leading to excellent concordance of sites detected by mass spectrometry and RNA-seq [69–71], however the extent of m1A in cytoplasmic mRNA has been debated [75], going from a dozen, to hundreds or thousands of sites identified [69–71, 74, 76]. While most of the cited publications agree that the m1A antibody enriches sequences mapping to the 5′ ends of mRNAs, they diverge in the number of ‘called’ m1A sites. This may be due to a combination of issues, including m1A antibody specificity, accuracy of read mapping, and the ratio of misincorporation by RTs within the m1A-antibody enriched sequences [75]. At least two publications have reported that the ratio of misincorporation is not linearly correlated to the ratio of m1A modifications when using either TGIRT [69] or an RT specifically evolved from the human immunodeficiency virus RT catalytic domain (p66) to increase m1A readthrough and misincorporation rate [71]. This problem may be the primary reason why, apart from a few overlapping called m1A sites among datasets, there is variability in the numbers and identity of ‘called’ m1A sites even within the same cell line. The case of m1A exemplifies the difficulty of accurately identifying low stoichiometry RNA modification sites in RNA even in the advantageous situation of a modification that is not RT silent. Going forward, as previously proposed [18], evolution of new RTs with improved readthrough and higher misincorporation rate as in the elegant study from the He and Dickinson labs [71], should solve many of the issues related to the bioinformatic analysis of misincorporation signatures for detecting RNA modifications with low stoichiometry [77].
Remarks
Numerous (bio)chemical methods have been reported to date to identify/quantify a growing spectrum of RNA modifications. Here we described several methods targeting crucial modifications such as N6-methyladenosine (m6A), 5-methylcytosine (m5C), pseudouridine (Ψ), N4-acetylcytosine (ac4C), 7-methylguanosine (m7G), N1-methyladenosine (m1A) etc.
Antibody-based techniques require high input of RNA and often show background/non-specificity during enrichment. Thus, multiple controls and/or orthogonal approaches should be used to correlate/verify the results whenever possible. One should keep in mind this drawback before using any modification-specific antibody techniques.
LC–MS based methods are very sensitive for detecting modification. But impurity present in RNA samples or degraded RNA might give rise to ‘false positive’ results. Moreover, for low abundance RNAs (e.g. mRNA or miRNA), mass spectrometry dependent methods can be difficult to perform.
Reproducibility of results using available methods might vary from person to person (or lab to lab) depending on multiple crucial factors such as pH, reagent concentration, reaction/incubation time, solubility, salt concentration, temperature and RT used. RNA modifications are sensitive to environmental effects, e.g. heat shock, oxidative/reductive stress. Therefore, handing cells/tissues with proper care and proper experimental settings are extremely important to obtain reproducible results. Notably, epitranscriptomic profile or RNA modification levels may largely be context-specific and thus vary from one cell line to another cell line and from one organism to another.
Many of the techniques described here have been crucial for deciphering the roles of RNA modifiers in cellular processes involved in normal development and disease. However, many are laborious and often suffer from non-specificity, RNA degradation, background and structural problems, or inappropriate bioinformatic analysis. Moreover, RNA-friendly chemical reactions might require proper optimization to gain better target selectivity and yield. Thus, more extensive research, and more sensitive and accurate methods are still required to explore in depth the epitranscriptome and the panoply of proteins that write, read and erase them. This is paramount to elucidating their roles in disease and cancer, with the ultimate goal of targeting them for therapeutic benefit.
Summary points
Detection at single nucleotide resolution level is paramount to uncover the molecular mechanism of action of RNA modifications.
RNA modifications in rare RNAs are inferred through either direct or indirect sequencing methods.
RNA modifications that affect base pairing can induce reverse transcription (RT) roadblock or nucleotide misincorporation, thus enabling detection at single nucleotide resolution.
RNA modifications that do not affect base pairing are called RT silent, but can be ‘unsilenced’ through specific chemical or biochemical treatments to induce RT roadblock or nucleotide misincorporation.
Turja Kanti Debnath is a postdoctoral fellow in the Xhemalce lab, where he is working on developing methods to detect RNA modifications. His interest in RNA modifications and epigenetics started during his PhD in Chemical Biology in the Okamoto lab at the University of Tokyo in Japan.
Blerta Xhemalçe, PhD is an associate professor of Molecular Biosciences at the University of Texas at Austin and a member of the LiveStrong Cancer Institutes. Her lab aims to unravel molecular mechanisms of gene expression regulation involving RNA interference, RNA modifications and epigenetics in the context of cancer.
Contributor Information
Turja K Debnath, Department of Molecular Biosciences, University of Texas at Austin, 2500 Speedway, 78712 Austin TX, USA.
Blerta Xhemalçe, Department of Molecular Biosciences, University of Texas at Austin, 2500 Speedway, 78712 Austin TX, USA.
Funding
BX is supported by the United States National Institutes of Health Grant R01 GM127802.
References
- 1. Cantara WA, Crain PF, Rozenski J, et al. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res 2011;39(Database issue):D195–D201. doi: 10.1093/nar/gkq28Epub 2010 Nov 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev 2010;24(17):1832–1860. doi: 10.101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross RL, Cao X, Limbach PA. Mapping post-transcriptional modifications onto transfer ribonucleic acid sequences by liquid chromatography tandem mass spectrometry. Biomolecules 2017;7(1):21. doi: 10.3390/biom7010021. [DOI] [Google Scholar]
- 4. Wilusz JE. Removing roadblocks to deep sequencing of modified RNAs. Nat Methods 2015;12(9):821–822. doi: 10.1038/nmeth.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng G, Qin Y, Clark WC, et al. Efficient and quantitative high-throughput tRNA sequencing. Nat Methods 2015;12(9):835–837. doi: 10.1038/nmeth.3478. Epub 2015 Jul 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cozen AE, Quartley E, Holmes AD, et al. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods 2015;12(9):879–884. doi: 10.1038/nmeth.3508. Epub 2015 Aug 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mohr S, Ghanem E, Smith W, et al. Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA 2013;19(7):958–970. doi: 10.1261/rna.039743.113Epub 2013 May 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katibah GE, Qin Y, Sidote DJ, et al. Broad and adaptable RNA structure recognition by the human interferon-induced tetratricopeptide repeat protein IFIT5. Proc Natl Acad Sci U S A 2014;111(33):12025–12030. doi: 10.1073/pnas.1412842111Epub 2014 Aug 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reinsborough CW, Ipas H, Abell NS, et al. BCDIN3D regulates tRNAHis 3′ fragment processing. PLoS Genet 2019;15(7):e1008273. doi: 10.1371/journal.pgeneCollection 2019 Jul. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Novoa EM, Mason CE, Mattick JS. Charting the unknown epitranscriptome. Nat Rev Mol Cell Biol 2017;18(6):339–340. doi: 10.1038/nrm.2017.49Epub May 10. [DOI] [PubMed] [Google Scholar]
- 11. Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 2014;10(2):93–95. doi: 10.1038/nchembio.432Epub 2013 Dec 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA modifications in gene expression regulation. Cell 2017;169(7):1187–1200. doi: 10.016/j.cell.2017.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang H, Weng H, Chen J. The biogenesis and precise control of RNA m(6)a methylation. Trends Genet 2020;36(1):44–52. doi: 10.1016/j.tig.2019.10.011Epub Dec 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother 2019;112:108613. doi: 10.1016/j.biopha.2019.108613Epub 2019 Feb 19. [DOI] [PubMed] [Google Scholar]
- 15. Hartstock K, Nilges BS, Ovcharenko A, et al. Enzymatic or in vivo installation of Propargyl groups in combination with click chemistry for the enrichment and detection of methyltransferase target sites in RNA. Angew Chem Int Ed Engl 2018;57(21):6342–6346. doi: 10.1002/anie.201800188Epub 2018 Mar 22. [DOI] [PubMed] [Google Scholar]
- 16. Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 2012;149(7):1635–1646. doi: 10.016/j.cell.2012.05.003Epub May 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012;485(7397):201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 18. Shelton SB, Reinsborough C, Xhemalce B. Who watches the watchmen: roles of RNA modifications in the RNA interference pathway. PLoS Genet 2016;12(7):e1006139. doi: 10.1371/journal.pgeneCollection 2016 Jul. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu N, Parisien M, Dai Q, et al. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 2013;19(12):1848–1856. doi: 10.261/rna.041178.113Epub 2013 Oct 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shu X, Cao J, Cheng M, et al. A metabolic labeling method detects m(6)a transcriptome-wide at single base resolution. Nat Chem Biol 2020;16(8):887–895. doi: 10.1038/s41589-020-0526-9Epub 2020 Apr 27. [DOI] [PubMed] [Google Scholar]
- 21. Imanishi M, Tsuji S, Suda A, et al. Detection of N(6)-methyladenosine based on the methyl-sensitivity of MazF RNA endonuclease. Chem Commun (Camb) 2017;53(96):12930–12933. doi: 10.1039/c7cc07699a. [DOI] [PubMed] [Google Scholar]
- 22. Garcia-Campos MA, Edelheit S, Toth U, et al. Deciphering the "m(6)a code" via antibody-independent quantitative profiling. Cell 2019;178(3):731–47.e16. doi: 10.1016/j.cell.2019.06.013Epub Jun 27. [DOI] [PubMed] [Google Scholar]
- 23. Meyer KD. DART-seq: an antibody-free method for global m(6)a detection. Nat Methods 2019;16(12):1275–1280. doi: 10.038/s41592-019-0570-0Epub 2019 Sep 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perry RP, Kelley DE, Friderici K, et al. The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the is 5´ terminus. Cell 1975;4(4):387–394. doi: 10.1016/0092-8674(75)90159-2. [DOI] [PubMed] [Google Scholar]
- 25. Motorin Y, Lyko F, Helm M. 5-methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res 2010;38(5):1415–1430. doi: 10.093/nar/gkp117Epub 2009 Dec 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bohnsack KE, Höbartner C, Bohnsack MT. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: mechanisms, cellular functions, and links to disease. Genes (Basel) 2019;10(2):102. doi: 10.3390/genes10020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khoddami V, Yerra A, Mosbruger TL, et al. Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc Natl Acad Sci U S A 2019;116(14):6784–6789. doi: 10.1073/pnas.1817334116Epub 2019 Mar 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Squires JE, Patel HR, Nousch M, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 2012;40(11):5023–5033. doi: 10.1093/nar/gks144Epub 2012 Feb 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schaefer M. RNA 5-Methylcytosine analysis by Bisulfite sequencing. Methods Enzymol 2015;560:297–329. doi: 10.1016/bs.mie.2015.03.007Epub May 13. [DOI] [PubMed] [Google Scholar]
- 30. Khoddami V, Yerra A, Cairns BR. Experimental approaches for target profiling of RNA cytosine methyltransferases. Methods Enzymol 2015;560:273–296. doi: 10.1016/bs.mie.2015.03.008Epub May 28. [DOI] [PubMed] [Google Scholar]
- 31. Hussain S, Aleksic J, Blanco S, et al. Characterizing 5-methylcytosine in the mammalian epitranscriptome. Genome Biol 2013;14(11):215. doi: 10.1186/gb4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khoddami V, Cairns BR. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat Biotechnol 2013;31(5):458–464. doi: 10.1038/nbt.2566Epub 013 Apr 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hussain S, Sajini AA, Blanco S, et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep 2013;4(2):255–261. doi: 10.1016/j.celrep.2013.06.029Epub Jul 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Debnath TK, Okamoto A. Osmium tag for post-transcriptionally modified RNA. Chembiochem 2018;19(15):1653–1656. doi: 10.002/cbic.201800274Epub 2018 Jun 26. [DOI] [PubMed] [Google Scholar]
- 35. Tserovski L, Helm M. Diastereoselectivity of 5-Methyluridine Osmylation is inverted inside an RNA chain. Bioconjug Chem 2016;27(9):2188–2197. doi: 10.1021/acs.bioconjchem.6b00403Epub 2016 Sep 1. [DOI] [PubMed] [Google Scholar]
- 36. Hamma T, Ferré-D'Amaré AR. Pseudouridine synthases. Chem Biol 2006;13(11):1125–1135. doi: 10.016/j.chembiol.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 37. Charette M, Gray MW. Pseudouridine in RNA: what, where, how, and why. IUBMB Life 2000;49(5):341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- 38. Davis DR. Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res 1995;23(24):5020–5026. doi: 10.1093/nar/23.24.5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li X, Ma S, Yi C. Pseudouridine: the fifth RNA nucleotide with renewed interests. Curr Opin Chem Biol 2016;33:108–116. doi: 10.1016/j.cbpa.2016.06.014Epub Jun 24. [DOI] [PubMed] [Google Scholar]
- 40. Schwartz S, Bernstein DA, Mumbach MR, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 2014;159(1):148–162. doi: 10.1016/j.cell.2014.08.028Epub Sep 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carlile TM, Rojas-Duran MF, Zinshteyn B, et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014;515(7525):143–146. doi: 10.1038/nature13802Epub 2014 Sep 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lei Z, Yi C. A Radiolabeling-free, qPCR-based method for locus-specific Pseudouridine detection. Angew Chem Int Ed Engl 2017;56(47):14878–14882. doi: 10.1002/anie.201708276Epub 2017 Oct 19. [DOI] [PubMed] [Google Scholar]
- 43. Sun L, Xu Y, Bai S, et al. Transcriptome-wide analysis of pseudouridylation of mRNA and non-coding RNAs in Arabidopsis. J Exp Bot 2019;70(19):5089–5600. doi: 10.1093/jxb/erz273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taucher M, Ganisl B, Breuker K. Identification, localization, and relative quantitation of pseudouridine in RNA by tandem mass spectrometry of hydrolysis products. Int J Mass Spectrom 2011;304(2-3):91–97. doi: 10.1016/j.ijms.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mengel-Jørgensen J, Kirpekar F. Detection of pseudouridine and other modifications in tRNA by cyanoethylation and MALDI mass spectrometry. Nucleic Acids Res 2002;30(23):e135. doi: 10.1093/nar/gnf135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamaki Y, Nobe Y, Koike M, et al. Direct determination of Pseudouridine in RNA by mass spectrometry coupled with stable isotope Labeling. Anal Chem 2020;92(16):11349–11356. doi: 10.1021/acs.analchem.0c02122Epub 2020 Jul 28. [DOI] [PubMed] [Google Scholar]
- 47. Ito S, Horikawa S, Suzuki T, et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J Biol Chem 2014;289(52):35724–35730. doi: 10.1074/jbc.C114.602698Epub 2014 Nov 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sharma S, Langhendries JL, Watzinger P, et al. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res 2015;43(4):2242–2258. doi: 10.1093/nar/gkv075Epub 2015 Feb 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arango D, Sturgill D, Alhusaini N, et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 2018;175(7):1872–86.e24. doi: 10.016/j.cell.2018.10.030Epub Nov 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thomas JM, Briney CA, Nance KD, et al. A chemical signature for cytidine acetylation in RNA. J Am Chem Soc 2018;140(40):12667–12670. doi: 10.1021/jacs.8b06636Epub 2018 Sep 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sas-Chen A, Thomas JM, Matzov D, et al. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature 2020;583(7817):638–643. doi: 10.1038/s41586-020-2418-2Epub 2020 Jun 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dimitrova DG, Teysset L, Carré C. RNA 2'-O-methylation (nm) modification in human diseases. Genes (Basel) 2019;10(2):117. doi: 10.3390/genes10020117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ayadi L, Galvanin A, Pichot F, et al. RNA ribose methylation (2'-O-methylation): occurrence, biosynthesis and biological functions. Biochim Biophys Acta Gene Regul Mech 2019;1862(3):253–269. doi: 10.1016/j.bbagrm.2018.11.009Epub Dec 17. [DOI] [PubMed] [Google Scholar]
- 54. Birkedal U, Christensen-Dalsgaard M, Krogh N, et al. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew Chem Int Ed Engl 2015;54(2):451–455. doi: 10.1002/anie.201408362Epub 2014 Nov 21. [DOI] [PubMed] [Google Scholar]
- 55. Marchand V, Blanloeil-Oillo F, Helm M, et al. Illumina-based RiboMethSeq approach for mapping of 2'-O-me residues in RNA. Nucleic Acids Res 2016;44(16):e135. doi: 10.1093/nar/gkw547Epub 2016 Jun 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhu Y, Pirnie SP, Carmichael GG. High-throughput and site-specific identification of 2'-O-methylation sites using ribose oxidation sequencing (RibOxi-seq). RNA 2017;23(8):1303–1314. doi: 10.261/rna.061549.117Epub 2017 May 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dai Q, Moshitch-Moshkovitz S, Han D, et al. Nm-seq maps 2'-O-methylation sites in human mRNA with base precision. Nat Methods 2017;14(7):695–698. doi: 10.1038/nmeth.4294Epub 2017 May 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grozhik AV, Jaffrey SR. Distinguishing RNA modifications from noise in epitranscriptome maps. Nat Chem Biol 2018;14(3):215–225. doi: 10.1038/nchembio.2546. [DOI] [PubMed] [Google Scholar]
- 59. Elliott BA, Ho HT, Ranganathan SV, et al. Modification of messenger RNA by 2'-O-methylation regulates gene expression in vivo. Nat Commun 2019;10(1):3401. doi: 10.1038/s41467-019-11375-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cowling VH. Regulation of mRNA cap methylation. Biochem J 2009;425(2):295–302. doi: 10.1042/BJ20091352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang LS, Liu C, Ma H, et al. Transcriptome-wide mapping of internal N(7)-Methylguanosine Methylome in mammalian mRNA. Mol Cell 2019;74(6):1304–16.e8. doi: 10.016/j.molcel.2019.03.036Epub Apr 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Enroth C, Poulsen LD, Iversen S, et al. Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res 2019;47(20):e126. doi: 10.1093/nar/gkz736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin S, Liu Q, Jiang YZ, et al. Nucleotide resolution profiling of m(7)G tRNA modification by TRAC-Seq. Nat Protoc 2019;14(11):3220–3242. doi: 10.1038/s41596-019-0226-7Epub 2019 Oct 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lin S, Liu Q, Lelyveld VS, et al. Mettl1/Wdr4-mediated m(7)G tRNA Methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol Cell 2018;71(2):244–55.e5. doi: 10.1016/j.molcel.2018.06.001Epub Jul 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pandolfini L, Barbieri I, Bannister AJ, et al. METTL1 promotes let-7 MicroRNA processing via m7G methylation. Mol Cell 2019;74(6):1278–90.e9. doi: 10.016/j.molcel.2019.03.040Epub Apr 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wintermeyer W, Zachau HG. Tertiary structure interactions of 7-methylguanosine in yeast tRNA Phe as studied by borohydride reduction. FEBS Lett 1975;58(1):306–309. doi: 10.1016/0014-5793(75)80285-7. [DOI] [PubMed] [Google Scholar]
- 67. Behm-Ansmant I, Helm M, Motorin Y. Use of specific chemical reagents for detection of modified nucleotides in RNA. J Nucleic Acids 2011;2011:408053. doi: 10.4061/2011/408053Epub 2011 Apr 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Boccaletto P, Machnicka MA, Purta E, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res 2018;46(D1):D303–D307. doi: 10.1093/nar/gkx30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li X, Xiong X, Zhang M, et al. Base-resolution mapping reveals distinct m(1)a Methylome in nuclear- and mitochondrial-encoded transcripts. Mol Cell 2017;68(5):993–1005.e9. doi: 10.16/j.molcel.2017.10.019Epub Nov 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Safra M, Sas-Chen A, Nir R, et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017;551(7679):251–255. doi: 10.1038/nature24456Epub 2017 Oct 25. [DOI] [PubMed] [Google Scholar]
- 71. Zhou H, Rauch S, Dai Q, et al. Evolution of a reverse transcriptase to map N(1)-methyladenosine in human messenger RNA. Nat Methods 2019;16(12):1281–1288. doi: 10.038/s41592-019-0550-4Epub 2019 Sep 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hauenschild R, Tserovski L, Schmid K, et al. The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res 2015;43(20):9950–9964. doi: 10.1093/nar/gkv895Epub 2015 Sep 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tserovski L, Marchand V, Hauenschild R, et al. High-throughput sequencing for 1-methyladenosine (m(1)a) mapping in RNA. Methods 2016;107:110–121. doi: 10.1016/j.ymeth.2016.02.012Epub Feb 24. [DOI] [PubMed] [Google Scholar]
- 74. Li X, Xiong X, Wang K, et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol 2016;12(5):311–316. doi: 10.1038/nchembio.2040Epub 16 Feb 10. [DOI] [PubMed] [Google Scholar]
- 75. Schwartz S. M(1)a within cytoplasmic mRNAs at single nucleotide resolution: a reconciled transcriptome-wide map. RNA 2018;24(11):1427–1436. doi: 10.261/rna.067348.118Epub 2018 Aug 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016;530(7591):441–446. doi: 10.1038/nature16998Epub 2016 Feb 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sas-Chen A, Schwartz S. Misincorporation signatures for detecting modifications in mRNA: not as simple as it sounds. Methods 2019;156:53–59. doi: 10.1016/j.ymeth.2018.10.011Epub Oct 23. [DOI] [PubMed] [Google Scholar]