

Abstract
The causative agent of severe acute respiratory syndrome (SARS) is the SARS-associated coronavirus, SARS-CoV. The viral nucleocapsid (N) protein plays an essential role in viral RNA packaging. In this study, recombinant SARS-CoV N protein was shown to be dimeric by analytical ultracentrifugation, size exclusion chromatography coupled with light scattering, and chemical cross-linking. Dimeric N proteins self-associate into tetramers and higher molecular weight oligomers at high concentrations. The dimerization domain of N was mapped through studies of the oligomeric states of several truncated mutants. Although mutants consisting of residues 1–210 and 1–284 fold as monomers, constructs consisting of residues 211–422 and 285–422 efficiently form dimers. When in excess, the truncated construct 285–422 inhibits the homodimerization of full-length N protein by forming a heterodimer with the full-length N protein. These results suggest that the N protein oligomerization involves the C-terminal residues 285–422, and this region is a good target for mutagenic studies to disrupt N protein self-association and virion assembly.
Coronaviruses are responsible for ∼30% of human upper respiratory infections each year. In November 2002, a new coronavirus, known as the severe acute respiratory syndrome (SARS)1 -associated coronavirus, SARS-CoV, emerged in China and caused more than 8000 cases of SARS worldwide. Approximately 10% of these cases were fatal. Similar to other coronaviruses, SARS-CoV is an enveloped, single-stranded (ss) RNA virus. The SARS-CoV genome contains ∼29,700 nucleotides (1, 2), encoding the RNA-dependent RNA polymerase and four structural proteins: spike (S), envelope (E), membrane (M), and nucleocapsid (N). In addition, intergenic regions encode several open reading frames for nonstructural proteins of unknown function (1, 2). The S protein is a surface glycoprotein that mediates viral entry by binding to the cellular receptor angiotensin-converting enzyme 2 (ACE2) (3) and inducing membrane fusion. The receptor-binding domain has been mapped to amino acids 318–510 (4, 5), and structures of the heptad repeat region of S protein (6, 7) indicate that it is a class I membrane fusion protein. The M protein of coronavirus is the most abundant protein component of the envelope. This protein plays a predominant role in the formation and release of the virion envelope. When co-expressed with the E protein, virus-like particles with sizes and shapes similar to those of virions are assembled (8, 9). Recently, virus-like particles of SARS-CoV were obtained by recombinant expression of S, E, and M proteins in insect cells (10). Inside the envelope, the N protein associates with the genomic RNA to form a long, flexible, helical ribonucleo-protein. The N protein is typically 350–450 amino acids in length, highly basic, and serine-phosphorylated, but the extent and physiological relevance of phosphorylation is unclear (11, 12). In addition to its structural role, several additional functions are postulated for the N protein including viral RNA synthesis, transcription, translation, and virus budding (13, 14, 15).
Central to the process of virion assembly is the specific packaging of viral RNA into the virion. The mouse hepatitis virus N protein was first shown in 1986 to have RNA binding activity by NorthWestern assay (16). Based on sequence analysis, Parker and Masters (17) suggest that the N protein consists of three highly conserved domains separated by variable spacer regions. Although lacking sequence homology to previously described RNA-binding motifs, the central domain of N protein has been shown to be the RNA-binding region by several laboratories (18, 19, 20, 21). Recently, the structure of the N-terminal region consisting of residues 49–178 of SARS-CoV N protein has been determined by NMR (22). Although the structure reveals a fold similar to the U1A RNA-binding protein (22), whether this N-terminal domain indeed binds RNA remains to be determined.
Although the RNA binding property of the N protein has been extensively studied, the self-association activity of the N protein is rather poorly characterized. The results in this report show that the recombinant full-length N protein is dimeric with a propensity to form tetramers and higher molecular weight oligomers. The dimerization region is mapped to the C-terminal 138 residues, and the homodimer formation of the N homodimer is inhibited by excess truncated mutant containing the dimerization domain.
MATERIALS AND METHODS
Protein Expression and Purification—DNA encoding full-length SARS-CoV (strain GD01) N protein (amino acids 1–422) was cloned into plasmid pET21a (Novagen). The PCR primers contained the native initiating methionine codon and a stop codon immediately following the last residue of N protein. The correct DNA sequence was confirmed on both strands of the plasmid. BL21 Star Escherichia coli cells (Invitrogen) transformed with the expression plasmids were grown to the log phase, induced with 1 mm isopropyl-1-thio-β-d-galactopyranoside, and harvested by centrifugation 4 h after induction. The cell pellets were rinsed with phosphate-buffered saline (pH 7.4) and stored at -80 °C until protein purification. Thawed pellets were suspended in lysis buffer (25 mm HEPES at pH 8.0, 100 mm NaCl, and 1 mm EDTA) and lysed by French press. The cell lysate was centrifuged at 75,000 × g for 30 min at 4 °C. The supernatant was loaded onto a cation exchange column (Amersham Biosciences; SP Sepharose Fast Flow). The column was washed with 5 column volumes of lysis buffer followed by 2 column volumes of 25 mm HEPES at pH 8.0, 300 mm NaCl, and 1 mm EDTA. The bound N protein was eluted with 25 mm HEPES at pH 8.0, 1 m NaCl, and 1 mm EDTA. Fractions containing N protein were pooled and dialyzed against 25 mm HEPES at pH 8.0, 170 mm NaCl, and 1 mm EDTA, loaded onto a second cation exchange column (Amersham Biosciences; Source 15S), and eluted with a gradient of NaCl. Further purification was achieved by size exclusion chromatography in lysis buffer (Amersham Biosciences; Superdex 200).
DNA encoding different N protein fragments were cloned into a pET-like vector (pETTEV281), which contains an N-terminal His6 tag and the cleavage site of tobacco etch virus (TEV) protease. Cells were grown similarly to the full-length N protein, harvested, and broken by French press in lysis buffer (25 mm Tris at pH 7.2 and 150 mm NaCl). After centrifugation (75,000 × g, 30 min), the supernatant was loaded onto a cobalt column (Clontech) pre-equilibrated in lysis buffer. After washing with lysis buffer followed by lysis buffer plus 5 mm imidazole, the protein was eluted with lysis buffer plus 200 mm imidazole. Fractions containing the target protein were pooled and dialyzed into TEV protease cleavage buffer (25 mm Tris at pH 7.2, 25 mm NaCl, and 1 mm EDTA). TEV protease was added to 5% (w/w), and the reaction mixture was incubated at 30 °C for 7–10 h. SDS-PAGE was used to monitor the completeness of the cleavage. Cation exchange (Amersham Biosciences; Source 15S) or size exclusion chromatography (Amersham Biosciences; Superdex 200) was used to further purify the proteins.
Nucleic Acid Binding Assays—ssDNA oligonucleotides were synthesized by Integrated DNA Technologies, and yeast tRNA was obtained commercially (Roche Applied Science). N protein (10 μm final concentration) and oligonucleotides were mixed at increasing molar ratios of nucleic acid to protein from 1:10 to 10:1 in 25 mm HEPES at pH 8.0, 100 mm KOAc, and 1.7 mm Mg(OAc)2 and incubated at 30 °C for 30 min. The protein/nucleic acid mixtures were electrophoresed on 0.8% agarose gels in Tris acetate buffer (100 mm Tris at pH 8.0, 1.25 mm, NaOAc, and 1 mm EDTA) in the presence of ethidium bromide (0.5 μg/ml final concentration). Nucleic acid was directly visualized on a short wavelength UV transilluminator. The gels were then fixed in 40% (v/v) methanol with 10% (v/v) acetic acid, dried under vacuum, and stained with Coomassie Brilliant Blue R-250 to visualize protein bands.
Size Exclusion Chromatography Light Scattering (SEC-LS)—The molecular masses of full-length N proteins were determined using SEC-LS in the HHMI Biopolymer Facility and the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University by Ewa Folta-Stogniew. Briefly, a sample containing ∼300 μg of N protein was filtered through a 0.22-μm Durapore membrane (Millipore) and applied to a Superose 6 HR 10/30 column (Amersham Biosciences) coupled with an in-line Dawn EOS laser light-scattering apparatus (Wyatt Technology Corp.), refractometer (Wyatt Technology Corp.), and UV detector (Waters Corp.). The weight average molecular mass of the elution peak was calculated using ASTRA software as described (23).
Analytical Ultracentrifugation—Sedimentation velocity and equilibrium experiments were conducted using a Beckman Optimal XL-I ultracentrifuge with an An60 Ti four-hole rotor and the interference optical detection system. The protein samples were dialyzed to osmotic equilibrium against the buffer (25 mm HEPES at pH 8.0, 200 mm NaCl, and 1 mm EDTA), and solvent density was determined using an Anton-Paar DMA 5000 density meter. In the sedimentation velocity experiments, samples at 1 and 25 μm were spun at 42,000 rpm, 20 °C, and 150 scans were collected at 2-min intervals. Continuous sedimentation coefficient distributions were calculated using the c(s) module from the software package Sedfit (version 8.9). Analyses of the sedimentation velocity profiles were also performed by direct boundary modeling from solutions of the Lamm equation using the non-interaction discrete species module of Sedfit as described (24, 25). Sedimentation equilibrium studies were performed at three different protein concentrations (5, 34, and 65 μm) at 7000 rpm, 20 °C. The partial-specific volume of the protein was calculated using the program Sednterp. Time-independent and radial independent corrections were determined and subtracted from the final scans after the samples had reached equilibrium for the initial rotor speed only (26). Corrected data were modeled with either the program Winnonlin (version 1.06) for non-linear least squares analysis of equilibrium data or the program Sedphat (version 1.9) (26).
Cross-linking Experiments—Protein (5 μm in 20 mm HEPES at pH 8.0 with 100 mm NaCl) was chemically cross-linked with bis(sulfosuccinimidyl) suberate (BS3) (Pierce) (0–4 mm) at room temperature for 30 min and quenched by adding Tris (pH 6.8) to a 20 mm final concentration. Reaction mixtures were then analyzed by SDS-PAGE.
RESULTS
Expression and Purification of Recombinant SARS-CoV N Protein—The N protein of SARS-CoV is a highly basic protein of 422 residues, where 14.2 mol % are lysines and arginines as opposed to 8.5 mol % glutamates and aspartates. To study the biochemical properties of the SARS-CoV N protein, full-length N protein without additional tags was expressed in E. coli cells (Fig. 1A ). The protein was purified from the soluble fraction of cell lysate by cation exchange chromatography, taking advantage of its basic nature (isoelectric point = 10.9). The absorbance ratio at 260 and 280 nm is 0.47, indicating that the purified protein is free of nucleic acids. The molecular mass of the N protein was determined by matrix assisted laser desorption/ionization mass spectroscopy to be 45,877 Da, which is 148 Da less than the theoretical molecular mass (46,025 Da). The difference in the mass could be caused by posttranslational removal of the N-terminal methionine (mass difference of 131 Da), as is often seen in many prokaryotic and eukaryotic proteins (27, 28). The mass spectroscopy result suggests that the recombinant SARS-CoV N protein is not phosphorylated. Purified N protein elutes as a sharp peak with an apparent molecular mass of 250 kDa in the gel filtration chromatogram, suggesting the formation of higher molecular weight oligomers (Fig. 1B). However, sedimentation velocity experiments (see below) suggest that the N protein is asymmetric, and its average hydrodynamic radius might be much larger than that of a globular protein of equivalent mass. As a result, the retention time of N protein on a gel filtration column will be unusually short, and the apparent 250 kDa mass of the N protein oligomer is likely an overestimate.
Fig. 1.
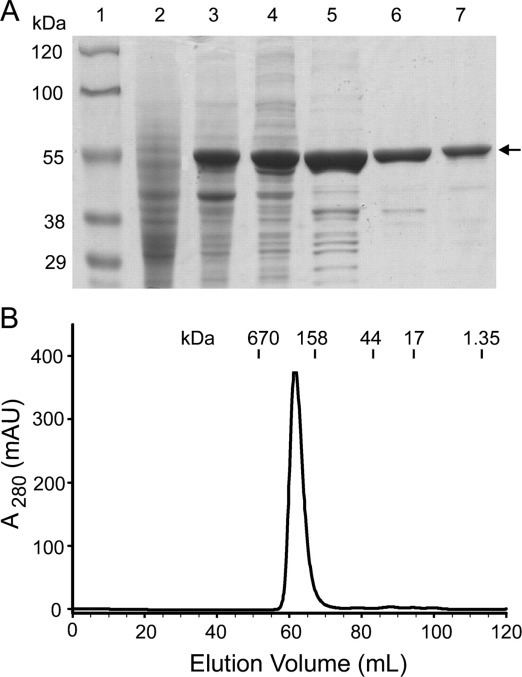
Expression and purification of recombinant SARS-CoV N protein.A, SDS-PAGE. Lanes are as follows: lane 1, protein marker; lane 2, lysate from uninduced cells; lane 3, lysate from cells induced by 1 mm isopropyl-1-thio-β-d-galactopyranoside for 4 h; lane 4, supernatant of the induced lysate centrifuged at 75,000 × g for 30 min; lane 5, elution from the first cation exchange column (SP-Sepharose); lane 6, elution from the second cation exchange column (Source 15S); lane 7, elution from size exclusion column (Superdex 200). The arrow points to the band corresponding to the N protein. B, fast protein liquid chromatography gel filtration chromatogram of purified N protein. The column (Superdex 200 16/60) was calibrated with a mixture of standard proteins with molecular masses as shown. AU, absorbance units.
Recombinant N Protein Binds to Nonspecific DNA and RNA—The binding of SARS-CoV N protein to nucleic acids was analyzed by native gel shift assays. Nonspecific RNA and DNA binding by native CoV N protein was reported previously (16, 18, 29, 30). Thus, oligonucleotide binding was used as a test for proper folding of the recombinant protein. Since a specific RNA sequence has not been identified as the ligand for the SARS-CoV N protein, nonspecific nucleic acids were used, including bakers' yeast tRNA and a 54-base ssDNA (Fig. 2 ). In these assays, the SARS-CoV N protein was kept at a constant concentration for all reactions, and only the amount of nucleic acid was changed. The reaction mixtures were subjected to agarose gel assays similar to those established for alphavirus in vitro assembly experiments (31). Briefly, the reaction mixtures were loaded onto a 0.8% agarose gel in the presence of ethidium bromide. Nucleic acids were visualized by UV irradiation, and protein bands were identified by staining the gel with Coomassie Blue. The formation of the N protein/nucleic acid complex is detected by co-migration of protein and nucleotide. In the absence of nucleotide binding, the N protein migrates in the opposite direction of complex due to the native gel conditions and its positive charge. As the amount of nucleic acid increases, N protein co-migrates into the gel, indicating the formation of an N protein/nucleic acid complex. A dramatic shift in the mobility of the nucleic acid in the protein/nucleic acid mixtures in comparison with free nucleic acid is observed. At a 1:1 molar ratio, the N protein/nucleic acid complex forms a discrete band, and at nucleic acids:protein molar ratios higher than 1:1, the complex migrates as a smear in the native agarose gel, indicating the presence of heterogeneity in the complex. Finally, the effect of ionic strength on the binding of nucleic acids was analyzed by adding KOAc to reaction mixtures. Complex formation is not impaired by up to 500 mm KOAc (data not shown), suggesting strong association of N protein with both ssDNA and tRNA.
Fig. 2.
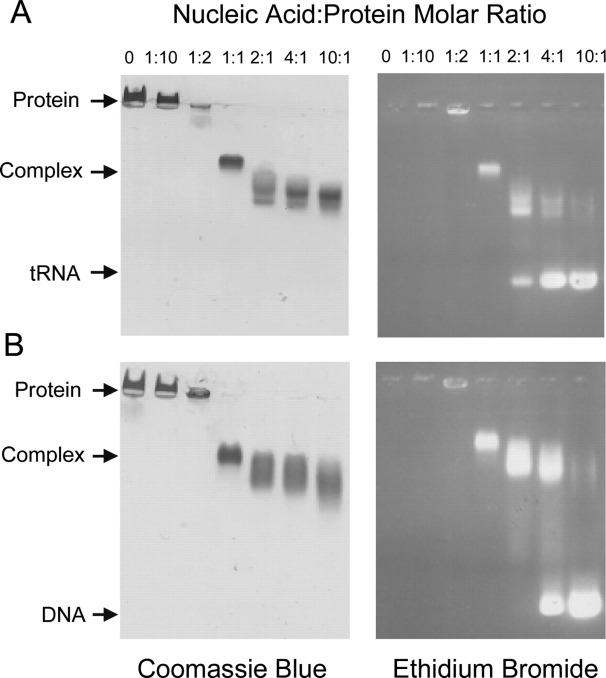
Nonspecific binding of SARS-CoV N protein with (A) yeast tRNA and (B) 54-mer ssDNA. N proteins were kept at 10 μm final concentration, and oligonucleotides were added at increasing nucleic acid:protein molar ratios. The reaction mixtures were electrophoresed on native 0.8% agarose gels. The first lanes of each gel contain protein only. The gels on the left are stained with Coomassie Blue for detection of protein, and the gels on the right are stained with ethidium bromide for detection of nucleic acid.
Self-association of the Recombinant N Protein—To accurately determine the oligomeric state of the SARS-CoV N protein, we carried out size exclusion chromatography coupled with continuous measurement of laser light-scattering and refractive index of the eluted sample (SEC-LS). In this approach, size exclusion chromatography only serves as a fractionation step. The molecular mass determination is independent of the elution position from the sizing column and depends only on the light-scattering and refractive index. Based on analyses of 14 protein standards, the molecular mass of proteins in solution can be determined by the SEC-LS technique with an accuracy of ± 5% (23). Recombinant N protein eluted from Superose 6 as a single peak (Fig. 3A ). The molecular mass of the eluted protein at each point of the chromatogram ranges from 75 to 115 kDa, indicating that the sample contains a mixture of molecules with different molar masses (Fig. 3A). The mass average of the molecular mass is 92.7 kDa, which is in excellent agreement with that of a dimer (theoretic value 92.0 kDa).
Fig. 3.
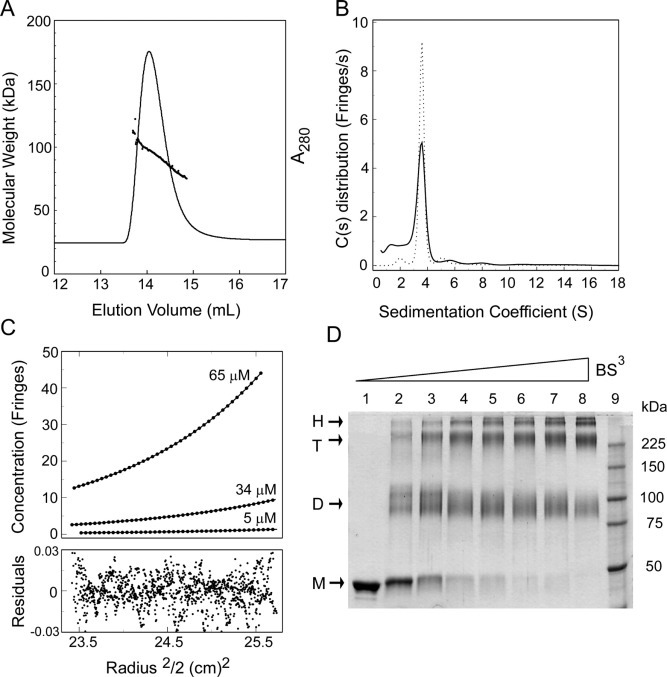
Oligomerization of the recombinant N protein.A, estimation of the native molecular mass by SEC-LS. The solid line corresponds to UV trace of the protein eluting from Superose 6 column, and the dotted line represents the average molecular weight calculated by ASTRA software at 5-μl intervals. B, sedimentation coefficient distributions calculated for N protein from sedimentation velocity studies. Shown are plots of the distribution of molecules with a given s value against the sedimentation coefficient (s) for proteins at 1 (solid line) and 25 μm (dotted line). These distributions are described by a corrected weight average sedimentation coefficient S and a calculated frictional ratio (f/fo) of 2.0. C, the effect of protein concentration on the equilibrium distribution of N protein from sedimentation equilibrium studies. Shown are plots of protein concentration in fringes against radius2/2 cm2 at three concentrations. Only 1 in 30 data points are shown, although all were used in the analysis. The lines are calculated for an ideal distribution that includes a dimer/tetramer equilibrium. The calculated mass for the dimer is 92.4 kDa, and the Kd is 2 mm. The residuals from the fit are shown in the lower panel. D, SARS-CoV N protein was cross-linked with various concentrations of BS3 (0–2 mm). The cross-linked products were analyzed by SDS-PAGE in an 8% gel. The arrows point to bands corresponding to monomer (M), dimer (D), tetramer (T), and higher order structures (H).
To determine whether the polydispersity of the N protein measured by SEC-LS resulted from co-elution of dimers with higher molecular weight oligomers, both velocity and equilibrium sedimentation analytical ultracentrifugation were performed. Sedimentation velocity measures the rate at which molecules move in response to centrifugal force and provides information about the mass and shape of the molecule. The sedimentation coefficient distributions of N protein samples at 1 and 25 μm show one major peak, centered at a sedimentation coefficient of 3.9 ± 0.3 S, which corresponds to the N protein dimer (Fig. 3B). Based on the fact that the dimer peak remains as the major peak at 1 μm concentration, the dissociation constant of the dimer/monomer is likely to be less than 1 μm. In addition, there are several small peaks in Fig. 3B, suggesting that the sample is heterogeneous. Because the sample is more than 95% pure, as suggested by mass spectroscopy, the presence of molecules with multiple sedimentation coefficient values is possibly caused by reversible self-association of the N protein and not by impurities. However, the dimer/oligomer equilibriums may not be fast on the velocity time scale, which could lead to significant changes in species distributions over the course of an experiment. It is therefore difficult and less accurate to derive the molecular mass and binding constants (Kd values) from sedimentation velocity data.
Therefore, to further characterize the self-association of the N protein, equilibrium sedimentation experiments were carried out. At equilibrium, the balance of sedimentation force and diffusion of the sample produces a gradient in protein concentration across the centrifuge cell. The concentration distribution at equilibrium is independent of the shape of the molecule and depends only on the molecular mass. This technique is particularly valuable for studying self-association of proteins because the overall mass distribution reflects the equilibrium of the association process. Centrifugation was carried out for 48 h at three protein concentrations (5, 34, and 65 μm), and the data were then simultaneously analyzed (Fig. 3C). Fitting a single species model to these data showed significant asymmetry in the residuals (data not shown), suggesting that the sample is a mixture of different species. A satisfactory fit was obtained when a reversible dimer/tetramer association model was used (Fig. 3C). The calculated mass is 92.4 ± 0.5 kDa for the dimer, which is consistent with the SEC-LS measurements. The dissociation constant for the tetramer/dimer formation is estimated at 2 mm.
Because both SEC-LS and ultracentrifugation measurements suggest that the dimeric N protein is in equilibrium with higher molecular weight species, we carried out chemical cross-linking experiments to covalently stabilize different oligomers. Purified N protein was incubated with the cross-linking reagent BS3 at a series of BS3 concentrations from 0.05 to 2 mm. The reaction products were analyzed by SDS-PAGE (Fig. 3D). In the presence of the cross-linker, three protein bands are evident, migrating at apparent molecular weights corresponding to one, two, and four N protein polypeptide chains. As the concentration of cross-linking reagent is increased, bands corresponding to oligomers larger than a tetramer also appear. The absence of a trimeric band suggests that dimeric, not monomeric, N protein is the building block of the oligomers. To test whether the oligomeric formation is dependent of nucleic acids, the N protein was incubated with RNase and DNase prior to cross-linking. Identical results were obtained with and without RNase/DNase treatment (not shown), indicating that the N protein forms a dimer in the absence of nucleic acids.
Mapping the Dimerization Domain—To determine the primary sequence requirements for N protein homo-interactions, several constructs encoding truncated N protein were overexpressed and purified (Fig. 4A ). An N-terminal His6 tag was engineered in all constructs to facilitate purification and was removed by TEV protease digestion before analysis. The first two constructs, NF1 and NF2 (NF stands for N protein fragment), residues 1–210 and 211–422, respectively, split the full-length protein into two halves. NF3 and NF4, residues 1–284 and 285–422, respectively, were designed to further narrow down the dimerization domain. The oligomerization state of each construct was first probed by chemical cross-linking (Fig. 4B). NF1, which contains the N-terminal 210 residues of the N protein, remained monomeric even in the presence of 4 mm BS3. The C-terminal half of the protein (NF2) cross-linked to a dimer, suggesting that the dimerization domain is located in the C-terminal region. Next, smaller fragments of the N protein were subcloned to pinpoint the dimerization domain. Among them, only NF3 (residues 1–284) and NF4 (residues 285–422) were stably expressed in bacteria and remained soluble during purification. Cross-linking experiments showed that NF3 is monomeric and that NF4 forms dimers (Fig. 4B). Unlike the full-length N protein, NF2 and NF4 do not form oligomers larger than dimers in cross-linking experiments. The concentration of cross-linking reagent was increased to 4 mm, which is twice that used for the full-length N. However, no obvious band corresponding to oligomers larger than dimers was observed.
Fig. 4.
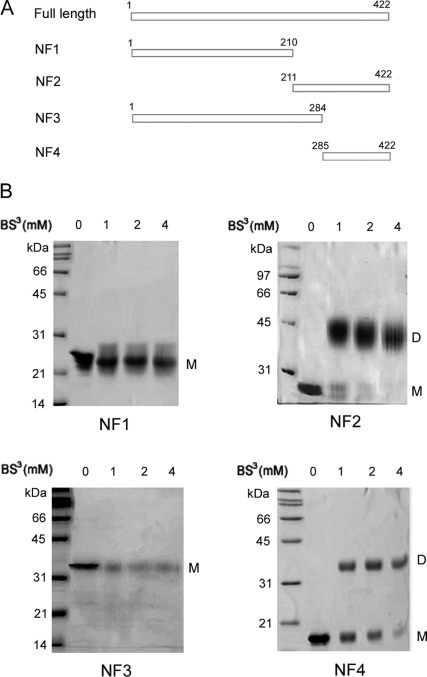
Mapping the regions involved in dimerization.A, schematic diagram of different N protein constructs. B, cross-linking of N protein fragments NF1–4. Purified untagged proteins were treated with BS3 at 0, 1, 2, and 4 mm concentration. The reaction mixtures were fractionated by SDS-PAGE (10% for NF2, 12% for NF1, NF3, and NF4) and stained with Coomassie Blue. Bands corresponding to monomer and dimer are labeled as M and D, respectively.
As a different approach to oligomer size assessment, the native molecular masses of all four truncated constructs were determined by SEC-LS. As shown in Table I , the molecular masses of NF1 and NF3 match well with the monomer masses predicted by amino acid composition. NF2 is predicted to have a molecular mass of 23.4 kDa, and the mass determined by SEC-LS is 49.0 kDa. NF4 is also confirmed to be a dimer with a mass of 31.8 kDa (theoretical dimer, 30.8 kDa). In addition, analytical ultracentrifugation results were consistent with that of the chemical cross-linking and SEC-LS analysis (data not shown). Taken together, our results demonstrate that the C-terminal 138 residues are necessary and sufficient for N protein dimerization.
Table I.
Oligomeric states of SARS-CoV N protein fragments
| Construct | Residue range | Theoretical MMa of a monomer | MMa determined by SEC-LS | Oligomeric stateb |
|---|---|---|---|---|
| kDa | kDa | |||
| NF1 | 1–210 | 22.6 | 23.6 | Monomer |
| NF2 | 211–422 | 23.4 | 49.0 | Dimer |
| NF3 | 1–284 | 30.6 | 35.0 | Monomer |
| NF4 | 285–422 | 15.4 | 31.8 | Dimer |
MM, molecular mass
As determined by chemical cross-linking and analytical ultracentrifugation
Inhibition of N Protein Dimerization—Having identified the dimerization domain of the N protein, we analyzed whether this domain inhibits the homotypic interactions of the full-length N protein. If the interaction mediated by the C-terminal 138 residues is as strong as that of the full-length protein, excess NF4 should drive the heterodimer formation of the full-length N and NF4 proteins and thereby inhibit the formation of the full-length N/N homodimer. Purified full-length N protein was mixed with NF4 at a molar ratio of 1:10 to promote heterodimer formation. Based on the analytical ultracentrifugation results, the N protein remains as a dimer at 1 μm concentration; thus, 4 m urea was used to facilitate dissociation of the N/N and NF4/NF4 homodimers. The reaction mixture was then dialyzed to remove urea, and size exclusion chromatography was used to separate different species in the mixture (Fig. 5A ). Two peaks, at 12.35 (peak A) and 13.61 ml (peak B), were observed in the gel filtration profile. Comparison of these peaks with the profiles of N homodimer and NF4 homodimer indicates that peak B overlaps perfectly with that of the NF4 homodimer (not shown), whereas there is a 1-ml shift between peak A and that of full-length N homodimer (Fig. 5A, dotted line). The slower migration rate of peak A suggests that it contains a species smaller than an N/N homodimer. The protein composition of elution fractions was analyzed by SDS-PAGE followed by silver staining (Fig. 5B). Peak A contains both N and NF4, whereas peak B only contains NF4. To determine whether the N and NF4 found in peak A formed a heterodimer, the elution fractions were treated with 1 mm BS3 at room temperature for 30 min and analyzed by SDS-PAGE. Purified N and NF4 were also cross-linked and loaded on the same gel for comparison. Although the cross-linking experiments reveal an NF4 homodimer band in peak B, the peak A fraction contains a prominent band migrating between the expected positions of the N/N and NF4/NF4 homodimers (Fig. 5B). Since there is no cross-linked band corresponding to N/N homodimer or NF4/NF4 homodimer in peak A, we conclude that peak A contains mainly N/NF4 heterodimer. A weak band at the similar position is also observed in the non-cross-linked peak A sample. This band is likely to be an SDS-resistant N/NF4 heterodimer. It is also possible that a contaminant protein with a similar molecular weight co-elutes with the N/NF4 heterodimer during purification.
Fig. 5.
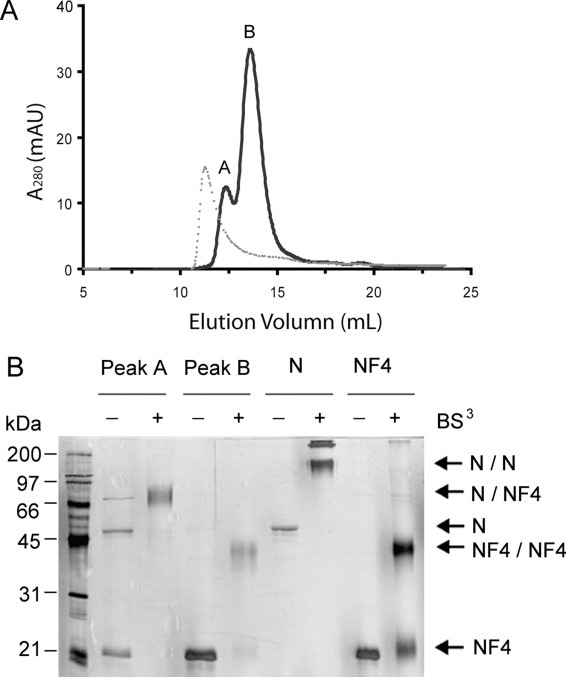
Inhibition of the full-length N protein dimerization.A, gel filtration profile (Superdex 200 16/30) of the mixture of full-length N and NF4 at 1:10 molar ratio (solid line). Peak A has an elution volume of 12.35 ml, and peak B elutes at 13.61 ml. The profile of the full-length N protein alone on the same column reveals a single elution peak at 11.32 ml (dotted line). AU, absorbance units. B, protein composition and cross-linking of the gel filtration elution fractions of N/NF4 mixture, visualized by silver staining after SDS-PAGE. Fractions of peaks A and B in panel A were treated with 1 mm BS3. Samples containing only full-length N protein or NF4 were also cross-linked and loaded on the same gel. N/N, full-length N protein homodimer; N/NF4, heterodimer of the full-length N and NF4; NF4/NF4, homodimer of NF4.
DISCUSSION
In this study, the SARS-CoV N protein in E. coli was expressed and purified to homogeneity. Native gel shift experiments show that N protein binds to yeast tRNA and ssDNA. The nonspecific RNA and DNA binding observed here is consistent with what is shown for native coronavirus N proteins (16, 18, 29, 30), suggesting that the recombinant N protein obtained in this study is properly folded. The N protein is postulated to interact with a specific RNA sequence, the packaging signal, to initiate efficient encapsulation of genomic RNA in the virions. However, both nonspecific (16, 18, 29, 30) and sequence-specific (21, 30, 32, 33) RNA binding is observed with coronavirus N proteins. In addition, Makino and colleagues (34) have recently shown that the M protein, not the N protein, interacts selectively with RNAs containing the packaging signal. To understand how the viral genomic RNA is recognized and packaged into the virion, the protein-RNA interactions must be studied in a system in which the packaging signal is well characterized. Unfortunately, the SARS-CoV packaging signal has not been identified yet. Thus, we carried out the in vitro RNA/DNA binding experiments simply to demonstrate that the recombinant N protein was folded into its native structure.
Self-association of mouse hepatitis virus N proteins is known to occur in the virion nucleocapsid, and the N/N interaction is resistant to RNase A treatment (35). The oligomerization states of coronavirus N proteins are reported to be dimeric (36), trimeric (16, 30), and higher (37). In this report, three independent methods, analytical ultracentrifugation, SEC-LS, and chemical cross-linking, were used to study the self-association of the SARS-CoV N protein. These methods consistently show that recombinant SARS-CoV N protein forms dimers in the absence of nucleic acids. In addition, the dimeric N proteins self-associate into tetramers with an estimated Kd of 2 mm. These results suggest an explanation for the controversy in the literature regarding the oligomeric states of coronavirus N proteins, in that both dimers and tetramers can form in the absence of nucleic acid.
Recently, two different regions were identified by two-hybrid assays to be important for N protein oligomerization: a serine/arginine-rich motif between residues 184–196 (37) and the C-terminal 209 residues (36). To clarify the discrepancy, truncated constructs of SARS-CoV N protein were purified, and their oligomeric states were determined by chemical cross-linking and SEC-LS. Two N-terminal fragments, residues 1–210 and residues 1–284, both yield monomeric proteins that do not associate into oligomers. Two C-terminal constructs, NF2 (residues 211–422) and NF4 (residues 285–422), form stable dimers in solution. These results indicate that the C-terminal 138 residues are necessary and sufficient to mediate the dimer formation of the SARS-CoV N protein. In contrast to the full-length N protein, no higher order oligomers were observed in NF2 and NF4 cross-linking experiments, suggesting that the dimer-dimer association of the N protein requires the presence of the N-terminal domain.
We showed that excess NF4, consisting of residues 285–422, prevents full-length N/N homodimerization in vitro. It would be of interest to test in vivo whether simultaneous expression of NF4 will inhibit assembly of the helical core. We believe that the dimer formation of the N protein, mediated by the C-terminal 138 amino acids, is likely to be the first step in the formation of the nucleocapsid core. Association of N protein dimers is likely to drive further assembly of the core. RNA binding may enhance dimer-dimer interactions by neutralizing the high positive charge of the N proteins and thereby trigger the formation of tetramers and higher order structures. Mutations in the N protein dimerization region would be expected to influence the helical core formation and/or stability. Therefore, the C-terminal 138 residues are a good target region to design mutations to disrupt SARS-CoV virion assembly.
Acknowledgments
We thank Connie Bonham for mass spectroscopy, Ewa Folta-Stogniew for conducting the SEC-LS analysis, Etti Harms for the gift of pETTEV281 vector, and Purdue Cancer Center forDNA sequencing facilities. We also thank Richard Kuhn and members of the Kuhn laboratory for many helpful discussions.
Footnotes
The abbreviations used are: SARS, severe acute respiratory syndrome; CoV, coronavirus; SARS-CoV, SARS-associated coronavirus; S, spike; E, envelope; M, membrane; N, nucleocapsid; ss, single-stranded; SEC-LS, size exclusion chromatography coupled with in-line measurement of laser light scatter and refractive index; BS3, bis(sulfosuccinimidyl) suberate; TEV, tobacco etch virus.
References
- 1.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 2.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 3.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babcock G.J., Esshaki D.J., Thomas W.D., Jr., Ambrosino D.M. J. Virol. 2004;78:4552–4560. doi: 10.1128/JVI.78.9.4552-4560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Supekar V.M., Bruckmann C., Ingallinella P., Bianchi E., Pessi A., Carfi A. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17958–17963. doi: 10.1073/pnas.0406128102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y., Lou Z., Liu Y., Pang H., Tien P., Gao G.F., Rao Z. J. Biol. Chem. 2004;279:49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vennema H., Godeke G.J., Rossen J.W., Voorhout W.F., Horzinek M.C., Opstelten D.J., Rottier P.J. EMBO J. 1996;15:2020–2028. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bos E.C., Luytjes W., van der Meulen H.V., Koerten H.K., Spaan W.J. Virology. 1996;218:52–60. doi: 10.1006/viro.1996.0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mortola E., Roy P. FEBS Lett. 2004;576:174–178. doi: 10.1016/j.febslet.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilbur S.M., Nelson G.W., Lai M.M., McMillan M., Stohlman S.A. Biochem. Biophys. Res. Commun. 1986;141:7–12. doi: 10.1016/S0006-291X(86)80326-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stohlman S.A., Lai M.M. J. Virol. 1979;32:672–675. doi: 10.1128/jvi.32.2.672-675.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai M.M., Cavanagh D. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He R., Leeson A., Andonov A., Li Y., Bastien N., Cao J., Osiowy C., Dobie F., Cutts T., Ballantine M., Li X. Biochem. Biophys. Res. Commun. 2003;311:870–876. doi: 10.1016/j.bbrc.2003.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tahara S.M., Dietlin T.A., Nelson G.W., Stohlman S.A., Manno D.J. Adv. Exp. Med. Biol. 1998;440:313–318. doi: 10.1007/978-1-4615-5331-1_41. [DOI] [PubMed] [Google Scholar]
- 16.Robbins S.G., Frana M.F., McGowan J.J., Boyle J.F., Holmes K.V. Virology. 1986;150:402–410. doi: 10.1016/0042-6822(86)90305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parker M.M., Masters P.S. Virology. 1990;179:463–468. doi: 10.1016/0042-6822(90)90316-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masters P.S. Arch. Virol. 1992;125:141–160. doi: 10.1007/BF01309634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson G.W., Stohlman S.A. J. Gen. Virol. 1993;74:1975–1979. doi: 10.1099/0022-1317-74-9-1975. [DOI] [PubMed] [Google Scholar]
- 20.Peng D., Koetzner C.A., McMahon T., Zhu Y., Masters P.S. J. Virol. 1995;69:5475–5484. doi: 10.1128/jvi.69.9.5475-5484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson G.W., Stohlman S.A., Tahara S.M. J. Gen. Virol. 2000;81:181–188. doi: 10.1099/0022-1317-81-1-181. [DOI] [PubMed] [Google Scholar]
- 22.Huang Q., Yu L., Petros A.M., Gunasekera A., Liu Z., Xu N., Hajduk P., Mack J., Fesik S.W., Olejniczak E.T. Biochemistry. 2004;43:6059–6063. doi: 10.1021/bi036155b. [DOI] [PubMed] [Google Scholar]
- 23.Folta-Stogniew E., Williams K.R. J. Biomol. Techniques. 1999;10:51–63. [PMC free article] [PubMed] [Google Scholar]
- 24.Schuck P. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuck P., Perugini M.A., Gonzales N.R., Howlett G.J., Schubert D. Biophys. J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R.A., Rouault T.A., Schuck P. Anal. Biochem. 2004;326:234–256. doi: 10.1016/j.ab.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 27.Flinta C., Persson B., Jornvall H., von Heijne G. Eur. J. Biochem. 1986;154:193–196. doi: 10.1111/j.1432-1033.1986.tb09378.x. [DOI] [PubMed] [Google Scholar]
- 28.Wolf B.P., Sumner L.W., Shields S.J., Nielsen K., Gray K.A., Russell D.H. Anal. Biochem. 1998;260:117–127. doi: 10.1006/abio.1998.2695. [DOI] [PubMed] [Google Scholar]
- 29.Baric R.S., Nelson G.W., Fleming J.O., Deans R.J., Keck J.G., Casteel N., Stohlman S.A. J. Virol. 1988;62:4280–4287. doi: 10.1128/jvi.62.11.4280-4287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cologna R., Spagnolo J.F., Hogue B.G. Virology. 2000;277:235–249. doi: 10.1006/viro.2000.0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tellinghuisen T.L., Hamburger A.E., Fisher B.R., Ostendorp R., Kuhn R.J. J. Virol. 1999;73:5309–5319. doi: 10.1128/jvi.73.7.5309-5319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molenkamp R., Spaan W.J. Virology. 1997;239:78–86. doi: 10.1006/viro.1997.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stohlman S.A., Baric R.S., Nelson G.N., Soe L.H., Welter L.M., Deans R.J. J. Virol. 1988;62:4288–4295. doi: 10.1128/jvi.62.11.4288-4295.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Narayanan K., Maeda A., Maeda J., Makino S. J. Virol. 2000;74:8127–8134. doi: 10.1128/jvi.74.17.8127-8134.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narayanan K., Kim K.H., Makino S. Virus Res. 2003;98:131–140. doi: 10.1016/j.virusres.2003.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Surjit M., Liu B., Kumar P., Chow V.T., Lal S.K. Biochem. Biophys. Res. Commun. 2004;317:1030–1036. doi: 10.1016/j.bbrc.2004.03.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He R., Dobie F., Ballantine M., Leeson A., Li Y., Bastien N., Cutts T., Andonov A., Cao J., Booth T.F., Plummer F.A., Tyler S., Baker L., Li X. Biochem. Biophys. Res. Commun. 2004;316:476–483. doi: 10.1016/j.bbrc.2004.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]