

Abstract
Due to its late diagnosis and dismal prognosis, pancreatic ductal adenocarcinoma (PDAC) is one of the most devastating solid malignancies, with only 9% of patients surviving after being diagnosed. A multidrug chemotherapeutic regimen FOL-F-IRIN-OX (combination of 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) offers survival benefits superior to that of gemcitabine single agent, but the treatment-related side effects are also severe. To overcome this therapeutic barrier, we developed polymeric micelles bearing active formats of irinotecan and oxaliplatin, SN38 and 1,2-diaminocyclohexane-platinum (II), DACHPt. Crosslinked micelles were prepared using amphiphilic PEG-b-poly(L-glutamic acid)/SN38 conjugates and subsequently loaded with DACHPt. The dual drug-loaded micelles exhibited improved colloidal stability, prolonged drug release and remarkable cytotoxicity in human pancreatic cancer cell lines and KrasG12D; Trp52R172H/+; Pdx-1 Cre murine tumor organoids models. In vivo, (SN38 + DACHPt)-loaded micelles displayed superior antitumor and antimetastatic activities without impairing safety. Our results suggest that nanomedicine mimicking irinotecan and oxaliplatin as parts of FOLFIRINOX regimen may further improve the feasibility of this multidrug treatment for patients with advanced pancreatic cancer.
Keywords: Pancreatic cancer, combination therapy, FOLFIRINOX, polymeric micelles, irinotecan, SN38, oxaliplatin, DACHPt
Graphic abstract
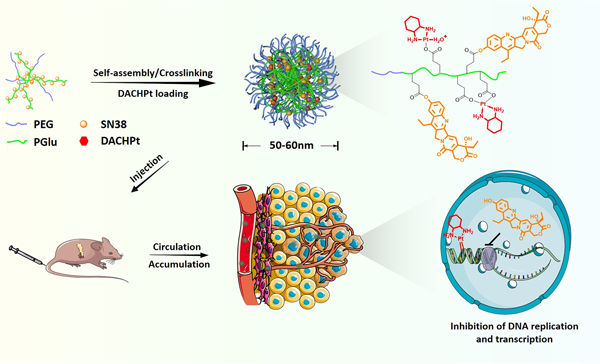
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) represents the most common type of pancreatic malignancies that account for about 3% of all cancers in the US. Despite its relatively rare occurrence, it is the fourth leading cause of death due to cancer with the lowest 5-year relative survival rate of 9% [1]. According to the American Cancer Society, an estimated 47,050 deaths and 57, 600 new pancreatic cancer cases are expected to be diagnosed in US population in 2020. The incidence of PDAC is expected to rise further in the future, and projections indicate that PDAC may surpass colorectal and breast cancer to rank as the second most common cause of cancer-related deaths by 2030 [2, 3]. Such a grim prognosis is mainly caused by the lack of distinctive symptoms in most PDAC patients, the absence of sensitive and non-invasive biomarkers for early diagnosis, and an aggressive nature and early metastatic spread of the disease. Surgical resection followed by adjuvant treatment is the only potentially curative option for PDAC. Approximately 85% of patients are diagnosed at an advanced stage of PDAC at the time of clinical presentation where the tumor is unresectable because of the local invasion beyond the pancreas or metastasis to distant organs [4]. Conventional chemotherapy remains the gold standard care for advanced PDAC. In the last two decades, gemcitabine has been a therapeutic backbone as a single agent or a part of multi-drug regimens mostly due to its acceptable toxicity profile and increased response rates [5, 6]. Development of resistance to gemcitabine-based regimens and frequent relapse led to the testing of alternative therapeutic regimens for PDAC patients. Triple regimen combining 5-Fluorouracil, Leucovorin (folinic acid), Irinotecan, and Oxaliplatin (FOL-F-IRIN-OX) emerged as the only combination regimen with statistically and clinically significant survival improvement of 11.1 months compared to gemcitabine (6.8 months, p < 0.001) and has a profound impact on the management of PDAC [7, 8]. However, positive responses to this therapy were also accompanied by severe treatment-related side effects. Thus, FOLFIRINOX is now reserved as a front-line therapy only for patients with good performance status. In some cases, modified FOLFIRINOX with reduced drug doses was also implemented in the clinic in the adjuvant setting for patients with resected PDAC (NCT01526135). Patient performance status is one of the factors in cancer care that plays an important predictive role in shaping prognosis and in determining the best treatment for a patient with cancer. Therefore, management of the toxicity of this multidrug regimen while sustaining its therapeutic activity may further improve its feasibility and expand the patient population that will benefit from this treatment.
Irinotecan, one of the active ingredients of FOLFIRINOX, is a camptothecin prodrug that is converted by carboxylesterases into its active metabolite 7 - ethyl - 10 - hydroxycamptothecin (SN38). This conversion is often inefficient and leads to large interpatient variability in drug exposure. SN38 inhibits topoisomerase 1 and eventually leads to inhibition of both DNA replication and transcription, which in turn causes cell death. SN38 is significantly more potent compared with irinotecan itself but its clinical utility is limited due to its very low solubility in water and toxicity issues. Oxaliplatin, a third-generation platinum derivative, acts by the formation of platinum-DNA adducts resulting in cell cycle arrest, inhibition of DNA replication and induction of apoptosis. The presence of diaminocyclohexane (DACH) ligand in oxaliplatin activates different cellular damage recognition mechanisms as compared with cisplatin and carboplatin, which has been attributed to the differences in cytotoxicity of various platinum complexes. Although better tolerated than cisplatin, oxaliplatin displays a characteristic profile of adverse events and peripheral neuropathy is probably the main dose-limiting toxicity caused by this drug.
Encapsulation of chemotherapeutic agents into delivery carriers of nanoscale sizes has been widely explored to mitigate their unfavorable toxicity profiles and improve treatment outcomes, especially for multidrug regimens. The unique characteristics of nanocarriers such as their dimensions, high loading capacity, and favorable pharmacokinetics, make them suitable for delivering chemotherapeutic drugs to the target tumor tissue [9]. One of such carriers, liposomal formulation of irinotecan, has been shown to improve the pharmacokinetics and tumor biodistribution of both irinotecan and its active metabolite SN38 [10]. A phase 3 multicenter clinical trial (NAPOLI-1) has demonstrated a survival advantage for the patients receiving liposomal irinotecan in combination with 5 - fluorouracil and leucovorin compared to the control arms (5 - fluorouracil plus leucovorin or liposomal irinotecan monotherapy) with a manageable safety profile [11, 12]. No substantial changes in quality-of-life measures compared to baseline were reported in any treatment group. As a result, liposomal irinotecan (Onivyde) received regulatory approval as a part of combination regimen for the treatment of patients with advanced metastatic PDAC who progressed following gemcitabine treatment. Currently, clinical trials were initiated to assess safety and preliminary efficacy of the combination of liposomal irinotecan with 5-fluorouracil, leucovorin and oxaliplatin as a preoperative regimen in resectable pancreatic cancer (NCT03528785, NCT03483038). Etirinotecan pegol (NKTR-102), a PEGylated form of irinotecan, has been engineered to provide the extended-release of irinotecan and is currently in advanced clinical development for metastatic breast cancer (NCT02915744) [13]. Micellar formulation of SN38 (NK012) is another nanomedicine under clinical evaluation [14, 15]. Polymeric micelles were prepared by self-assembly of amphiphilic block copolymers consisted of PEG and SN38-conjugated polyglutamate segments. It was found that NK012 can gradually release SN38 in a nonenzymatic manner and exerts remarkably more potent antitumor and reduced toxic effects against various human tumor xenografts compared with irinotecan. Phase I clinical trials demonstrated that NK012 produced a prolonged and enhanced accumulation of NK012 in tumor sites and was well tolerated at doses up to 28 mg/m2. This formulation is undergoing evaluation in Japan in patients with unresectable metastatic colorectal cancer who had previously received oxaliplatin-based chemotherapy [15]. Polymeric micelle platform was also utilized to develop nanosized formulations of platinum-based anticancer drugs [16]. Particularly, micellar formulation of 1, 2 - diaminocyclohexane-platinum (II) (DACHPt), the parent complex of oxaliplatin, was developed by the polymer-metal complex formation between DACHPt and the carboxylate moieties of PEG-b-poly(L-glutamic acid) copolymer. As shown in several preclinical models, DACHPt-loaded micelles were able to achieve substantial tumor deposition and demonstrated enhanced anticancer activity compared to oxaliplatin without evident toxicities. DACHPt-loaded micelles (NC-4016) are currently in early clinical trials in patients with advanced solid tumors or lymphoma.
Herein, we have built on this clinically translatable strategy and explored polymeric micelles as a dual-drug delivery platform for both SN38 and DACHPt to determine whether delivery of the active formats of irinotecan and oxaliplatin in one carrier particle may provide for better tailored combinatorial regimen in PDAC treatment. To this end, polymeric micelles were prepared using amphiphilic PEG-b-poly(L-glutamic acid)/SN38 conjugates and subsequently loaded with DACHPt. Intramicellar cross-linking was further utilized for structural reinforcement of the micellar carrier. Our results showed that dual-drug micellar formulation co-loaded at synergistic drugs ratio exhibits remarkable cytotoxicity in human pancreatic cancer cell lines and murine tumor organoids models. The in vitro studies were further corroborated with significantly reduced tumor burden and metastasis and better tolerability compared to clinically used free drug combination in an orthotopic model of pancreatic cancer indicating that nanomedicine mimicking irinotecan and oxaliplatin as parts of FOLFIRINOX regimen may further improve the feasibility of this multidrug treatment for patients with pancreatic cancer.
2. Materials and methods
2.1. Materials
The poly(ethylene glycol)-b-poly(L-glutamic acid) diblock copolymer, PEG114-PGlu150 (ĐM = 1.01) was purchased from Alamanda Polymers, Inc. (Madison, AL, USA). PEG114-PGlu80 copolymer (ĐM = 1.07) was synthesized according to previously described procedure [17]. 7 - ethyl - 10 - hydroxylcamptothecin (SN38) for the conjugation (> 98%) and biological study (> 99.5%) were supplied by Acros Organics (Thermo Fisher, NJ, USA) and Selleckchem (Houston, TX, USA), respectively. Dichloro-(1, 2- diaminocyclohexane)-platinum (II) (DACHPt), MTT reagent (3- (4, 5- Dimethylthiazol- 2- yl)- 2, 5- diphenyltetrazolium bromide), 1, 2- ethylenediamine (ED), 4- dimethylaminopyridine (DMAP) were obtained from Sigma-Aldrich (St Louis, MO, USA). 1- (3- dimethylaminopropyl)- 3- ethylcarbodiimide hydrochloride (EDC) was purchased from Alfa Aesar (Haverhill, MA, USA). Fetal bovine serum (FBS), trypsin, Dulbecco’s Modified Eagle’s Medium (DMEM) were purchased from Invitrogen Inc. (Carlsbad, CA, USA). Medium M3 Base was obtained from Incell Corp. (San Antonio, TX, USA). All reagent grade chemicals purchased from Sigma-Aldrich were used without any purification.
2.2. Preparation and characterization of drug-loaded micelles
Synthesis of PEG-P(Glu-Glu/SN38) conjugates.
SN38 was conjugated to PEG-PGlu copolymers via coupling reaction between the carboxyl group of PGlu and hydroxyl group at 10-position of SN38 in the presence of EDC and DMAP. The molar ratio of the glutamic acid residues of PEG-PGlu to SN38 was varied to obtain copolymers with different drug content. Briefly, PEG-PGlu was first converted into acid form by dissolving in water with addition of 1M HCl and formed suspension was freeze-dried. PEG114-PGlu150 (100 mg, 3.57 µmol of Glu units) and EDC (1.5 eq. with respect to SN38) were then suspended in anhydrous DMF (10 mL) and stirred for 10 min followed by addition of the required amount of SN38 and DMAP (1.5 eq. with respect to SN38). After stirring for 24 h at r.t. the reaction mixture was diluted by DI water (10 mL) and obtained turbid light-yellow mixture was placed into a dialysis bag (MWCO of 3.5kDa) and dialyzed against DI water for 48 h. The recovered suspension was centrifuged (2000 g, 5 min), filtered (0.45 µm) and clear solution of PEG-P(Glu-Glu/SN38) conjugate was freeze-dried. Conjugation and purity were confirmed by 1H NMR spectra recorded in TFA-d1 at 25 °C using a Bruker 500 MHz spectrometer and ultra-performance liquid chromatography (UPLC). The SN38 content in resulting PEG-P(Glu-Glu/SN38) copolymers was determined by alkaline digestion and UPLC analysis under isocratic conditions using Waters ACQUITY H-Plus UPLC system connected with a photodiode array (PDA) detector set at 380 nm. As stationary phase an ACQUITY UPLC® BEH C18 1.7µm column was used (2.1 × 50 mm), and a mobile phase of acetonitrile/water mixture (25/75, v/v, with 0.1% formic acid added) was applied at a flow rate of 0.5 mL/min.
Synthesis of cross-linked micelles.
PEG-P(Glu-Glu/SN38) micelles (SN38/m) were prepared using dialysis method: acid form of the conjugate was dissolved in DMF at concentration of 10 mg/mL followed by drop-wise addition of equal volume of water upon stirring. The resulting dispersion was then dialyzed against water for 24 h. The obtained micelles were further crosslinked using ED in the presence of EDC ([COOH]/[EDC] = 5, [ED]/[EDC] = 0.5) at r.t. overnight [17]. Byproducts of the crosslinking reaction were removed by exhaustive dialysis of the reaction mixtures against double distilled water for 48h. The resulting SN38/m dispersion was stored at 4 °C for further use. Cy5-labeled SN38/m (0.5 wt% Cy5) were synthesized by EDC coupling (at a molar ratio of [EDC]/[Cy5 amine] = 10) in the aqueous phase and purified by ultrafiltration using Centricon Plus-20 centrifugal filter units (MWCO 30kDa, Millipore, Billerica, MA, USA).
DACHPt loading.
DACHPt was first converted into DACHPt aqueous complex using previously reported procedure [18]. The aqueous dispersion of SN38/m micelles (1 mg/mL) was mixed with obtained aqueous solution of DACHPt at pH 8.0 at different molar ratio of DACHPt to the Glu units of micelles (0.25 or 0.15) followed by incubation at r.t. for 24h. Unbound DACHPt was removed by ultrafiltration using Centricon Plus-20 centrifugal filter units (MWCO 30kDa, Millipore, Billerica, MA, USA). Pt content in dual drug-loaded micelles (further denoted as (SN38 +DACHPt)/m) was measured by Pt (Pt194 / Pt195) assay using inductively coupled plasma mass spectrometry (ICP-MS, NexION 300Q, PerkinElmer, Waltham, MA, USA) calibrated with Pt (0.8 to 500 ng/mL). Effective hydrodynamic diameters (Deff), polydispersity indices (PDI) and ζ-potential of the micelles were determined by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK).
2.3. Drug release studies
Drug release profiles were examined in phosphate buffer saline (PBS, pH 7.4), acetate buffered saline (ABS, pH 5.5, 0.14 M NaCl), as well as in PBS in the presence of 10% FBS by dialysis method using a membrane with 3.5 kDa cutoff. The concentrations of SN38 and Pt (II) released were determined by UPLC and ICP-MS, respectively, and expressed as a percentage of the total SN38 or Pt (II) available vs. time.
The unbound fraction of SN38 and oxaliplatin in human whole blood (fu) was determined by an erythrocyte vs. plasma or buffer partitioning method as described before [19–21]. Plasma was separated from erythrocytes by centrifugation of whole human blood (Innovative Research, MI, USA) at 2500 g for 10 min. Plasma or PBS was then added to the tubes containing erythrocytes to a final concentration of 40% (Hematocrit) (n = 3) and vortexed to get a suspension. Irinotecan (IRIN, 20 μg) / oxaliplatin (OX, 10 μg) solution in DMSO or dual drug-loaded micelles (on the basis of 10 μg SN38 and OX, respectively) were added to the erythrocyte/plasma (or erythrocyte/PBS) dispersions and incubated at 37 °C for 2 h and 6 h, respectively. 10 μL aliquots were taken for CB or CB⁎ measurement of Pt content and 100 μL aliquots were taken for those measurement of IRIN or SN38. The samples were centrifuged at 2500 g for 10 min. Another 10 μL aliquots from the supernatant was used for Cp or Cb measurement of Pt while another 100 μL aliquots from the supernatant were taken for those measurement of IRIN or SN38. 10 μL aliquots for Pt measurements were digested by 90 μL mixture of conc. HNO3 and 30% H2O2 (4/1, v/v) using iridium as internal standard for 24 h and further diluted to 5 mL by DI water for ICP-MS measurement using gold as instrumental internal standard. Acetonitrile (900 μL) was added to all the 100 μL aliquots for IRIN or SN38 measurement, followed by sonicating and vortexing each sample thoroughly. Samples were then centrifuged (5000 g, 10 min), supernatants were collected, and solvent was evaporated to dryness under mild nitrogen flow at 50 °C. Acetonitrile (50 μL) was again added to all the tubes, followed by sonicating and vortexing, and IRIN or SN38 content was determined by UPLC. Spiking solution (irinotecan or SN38 in DMSO) was added to erythrocyte-plasma, plasma, erythrocyte-buffer, buffer, and acetonitrile solution, and processed in the same way as the samples. The concentration of drug associated with erythrocytes in the erythrocyte - plasma samples (CE) and erythrocyte - buffer samples (CE⁎) were determined using following equations:
where CB and CB⁎ were the concentration of drug in the erythrocyte-plasma and erythrocyte-buffer suspensions and Cp and Cb were the concentration of drug in the plasma and buffer.
The partition coefficients for erythrocyte-plasma (Pp) and erythrocyte-buffer (Pb), and fu (%) were determined by:
respectively.
Average fu ± SD (n = 3) were compared by One-Way ANOVA with Bonferroni’s Multiple Comparison Test.
2.4. Cell lines
T3M4, PANC-1 and CD18/HPAF human pancreatic carcinoma (PC) cell lines were obtained from the American Type Culture Collection (ATCC). Cells were cultured and maintained in DMEM supplemented with 10% (v / v) fetal bovine serum (FBS) in the presence of penicillin and streptomycin (100 U/mL and 0.1 mg/mL, respectively) at 37 °C in a humidified atmosphere containing 5% CO2. hTERT-HPNE immortalized normal human pancreatic ductal cell line was kindly provided by Dr. Michel Ouellette (UNMC, Omaha, NE) and was maintained in medium containing one volume of medium M3, three volumes of glucose-free DMEM with additional 2 mM L-glutamine and 1.5 g/L sodium bicarbonate, 5% FBS, 5.5 mM glucose, and 10 ng/ml EGF. Cells were cultured less than one month before initiating treatment studies. Cell lines were routinely inspected for their change in morphology, growth curve analysis, and presence of mycoplasma contamination; karyotyping analysis was performed in the cell lines to confirm their identify.
2.5. Cell viability assay
Cells were seeded in 96-well plates (2 x 103 cells/well) and were allowed to grow as a monolayer with 20–40% confluency for 24 h prior before drug treatment. Cells were exposed to various doses of free oxaliplatin or irinotecan alone, free drug combination, SN38/m, SN38/m plus free oxaliplatin, and (SN38 + DACHPt)/m for 48 or 72 h at 37 °C, followed by washing with PBS, and maintaining in DMEM with 10% FBS for additional 24 h. Cytotoxicity of all treatment groups was assessed by a standard MTT assay and the IC50 values were calculated using GraphPad Prism software. CompuSyn software was used to calculate the combination index (CI) for different drug treatments based on Chou and Talalay method as previously described [22]. The best-fit CI value at IC50 was used to show and compare the synergistic effects of different drug combinations and for different cell lines. The combination was considered as synergistic when values of CI were less than 1, while CI = 1 and CI > 1 values represented additive and antagonistic effects of drug combinations, respectively.
2.6. Wound healing assay
Wound healing assay in PC cells under various treatment conditions was performed according to previously described protocol with slight modifications to understand the treatment efficacy in metastatic potential [23]. PANC-1 (2 x 106 cells/well) or CD18/HPAF (2 x 105 cells/well) cells were seeded in a 24 well plate in triplicates supplemented with 10% FBS in DMEM. After overnight incubation, an artificial wound was created using a 200μl sterile pipette tip. The culture plates were washed with PBS to remove the damaged and detached cells. Cells were then treated with 25 ng/mL with respect to free OX if available or corresponding free IRIN or SN38/m for 48 hours. Images were taken at 0, 24 and 48 h at 10X magnification using Accuscope microscope assisted with Moticam 580 digital camera. The wound closure was calculated by measuring the wounds area using ImageJ software. The mean area between the edges at predetermined area of the control and treatment above-indicated time points were calculated and the arbitrary units were converted into percentage as follows: % of wound healing = (mean area of measurement at 0 h - mean area of measurement at certain time point) / mean area of measurement at 0 h multiplied by 100.
2.7. Colony formation assay
Colony formation assays were performed to understand the therapeutic impact in in vitro tumorigenic potential as previously described [24]. Briefly, PANC-1 (2 x 103 cells/well) or CD18/HPAF (1 x 103 cells/well) cells were plated in a six-well plate in triplicates. After 24 hours of incubation with complete media, cells were treated with free drugs or drug-loaded micelles at concentrations of 4, 10 and 25 ng/mL with respect to free OX. After 72 h the medium was discarded, cells were washed with PBS, and incubated in complete media for another 10–12 days at 37°C (media were refreshed every four days). The cells were washed with PBS, fixed with 100% methanol, stained with 0.4% crystal violet in methanol and colonies were counted using ImageJ software. The number of the colonies were converted into percentage as follows: % of colony formation = # of colonies in treated cells/ # of colonies in control multiplied by 100.
2.8. Cell uptake studies
Cells (200,000 cells/well) were seeded in 12-well plates, allowed to attach overnight and then were exposed to Cy5-labeled drug-loaded micelles at concentrations of 40 µM based on SN38 equivalents in DMEM with 10% FBS for 2 h at 37 °C Cell were then washed with cold PBS, trypsinized, centrifuged (1000 rpm, 3 min), washed again and resuspended in PBS (pH 7.4) for flow cytometry analysis. In select experiments, cells were pretreated for 30 min with inhibitors of endocytosis (10 µg/mL chlorpromazine, or 1 µg/mL filipin, or 2 mg/mL amiloride hydrochloride) then the same inhibitors were also present during subsequent incubation with the micelles for 1h. The mean fluorescence intensity and % gated cells were analyzed using Becton Dickinson FACStarPlus flow cytometer and FACSDiva software (Version 8.0, Becton Dickinson, San Jose, CA). At least 10,000 events were acquired in linear mode, gated to exclude debris and dead cells and visualized in logarithmic mode.
2.9. Ex vivo PC 3D-tumor organoids
The murine PC tumor organoids were developed and established from KrasG12D/+; Trp52R172H/+; Pdx-1 Cre- (KPC) autochthonous mouse model of 50 weeks of age [25]. A part of the tissue was formalin fixed for pathological confirmation and remaining tumor tissues were chopped into small fragments (~1 mm3) followed by enzymatic digestion with 0.012% (w/v) collagenase XI (Sigma, St Louis, MO) and 0.012% (w/v) dispase (GIBCO, Waltham, MA) in DMEM media containing 1% FBS. The minced tumor fragments were washed thrice in cell culture grade PBS followed by suspension in ice-cold Matrigel (BD Biosciences, San Jose, CA). The tissue matrigel suspension was subsequently transferred to the 48 well plate and maintained at 37 °C for 15 min to allow Matrigel to solidify followed by addition of organoid media (AdDMEM/F12 medium supplemented with 1X HEPES (Invitrogen, Carlsbad, CA), 1X penicillin/streptomycin (Invitrogen, Carlsbad, CA), 1X Glutamax (Invitrogen, Carlsbad, CA), 1X B27 (Invitrogen, Carlsbad, CA), 1 mM N-acetyl-Lcysteine (Sigma, St Louis, MO), 1 mg/mL recombinant RSPO1 (0.1 mg/mL Noggin recombinant protein (Peprotech, Rocky Hill, NJ), 50 ng/mL epidermal growth factor (EGF, Peprotech, Rocky Hill, NJ), 10nM Gastrin (Sigma, St Louis, MO), 10 mM Nicotinamide (Sigma, St Louis, MO), and 0.5 mM A83–01 (Tocris, Minneapolis, MN)) as described previously. The tumor organoids were treated with free drugs or drug-loaded micelles at 4 ng/mL of OX basis for 3 days. The morphological and structural variations of tumor organoids in each cohort were carefully monitored and the area of each tumor organoid was measured using EVOS FL Auto Cell Imaging System at 20X at day 0 and day 3.
2.10. In vivo antitumor activity and safety studies
All procedures described here were approved by and performed in accordance with the guidelines of University of Nebraska Medical Center Institutional Animal Care and Use Committee (IACUC). Pancreatic orthotopic implantation procedure of PC cells was performed as follows: CD18/HPAFLuc cells were trypsinized, washed and reconstituted in sterile PBS. Male/female athymic nude mice (6 – 8 weeks old) were anesthetized with 0.35 mL of intraperitoneal injection of a 4:1 mixture of ketamine (100 mg/mL) and xylazine (20 mg/mL) diluted 10 times in sterile water. The surgical site was sterilized and incised in the peritoneum at the right mid-abdomen region below the sternum by scissors. A total of 0.2 x 106 CD18/HPAFLuc cells in 20 µl sterile PBS was injected into the head of pancreas without causing injury and torsion. The abdomen was closed using a 2-layer suture with 5–0 chromic self-absorbable catgut and soft staple. The skin staples were removed 10 days after surgery and the Luc-expressing tumors were imaged by IVIS to randomly assign animals into treatment groups (n = 11): (1) 5% dextrose as control, (2) free drug combination (8 mg/kg IRIN and 4 mg/kg OX), (3) SN38/m (4 mg/kg SN38), (4) SN38/m plus free OX (4 mg/kg SN38 and 4 mg/kg OX), and (5) (SN38 + DACHPt)/m (4 mg/kg SN38 and 4 mg/kg on OX basis). A total of 4 treatments were administrated through tail vein injection once every 4 days (q4d x 4). Mice were monitored and weighed every 3 days. Orthotopic tumor growth and total tumor burden were monitored by palpation and whole body IVIS imaging (every 4 days during treatment and every 8 days afterwards). All the mice were sacrificed at day 47 after tumor cell inoculation. Detailed necropsy was performed to evaluate tumor burden and metastasis status in each group. Orthotopic tumors from each group were collected for immunohistochemistry (IHC) analysis. Blood was collected immediately after sacrificing mice by cardiac puncture into heparinized tubes and assayed for blood cell count using Vetscan HM5 (Abaxis, Union City, CA) and liver and kidney function markers (VetScan Comprehensive Diagnostic Profile, Vetscan VS2 Chemistry Analyzer, Abaxis, Union City, CA) according to manufacturer’s recommendations.
2.11. Histological and immunohistochemical analysis
For histopathological analysis, tissue specimens were instantly fixed in 10% buffered formalin for 72 h and stored in 75% ethanol. The tissues were embedded in paraffin and 5 µm sections were cut and stained with hematoxylin and eosin as well as probed with requisite antibodies as described previously [26]. Briefly, tissue slides were baked overnight at 65 °C. Next day, tissues were deparaffinized, hydrated and antigens were recovered by boiling in citrate buffer (pH 6, 0.05% Tween 20). Tissue sections were blocked with 2.5 % horse serum and incubated with indicated primary antibody (Ki67, cleaved caspase-3, TUNEL, etc.) overnight at 4 °C. Tissues were washed and incubated with HRP-conjugated or fluorescence-labeled secondary antibody. Subsequently, tissues slides were washed and developed using VECTASTAIN Elite ABC Universal PLUS kit (Vector Laboratories, Burlingame, CA) for colorimetric detection and counterstained with hematoxylin or DAPI. Tissues were dehydrated, dried, and mounted with Cytoseal 60 (Thermo Scientific, Waltham, MA) and quantified and evaluated by Definiens Tissue Studio 64 [Dual] 4.1 with corresponding modules (Definiens, Inc., Cambridge, MA), ZEN 3.0 blue edition (Oberkochen, Germany) or ImageJ.
2.12. Data Analysis
Statistical comparisons were carried out using Students’ t-test or One-Way ANOVA with diverse multiple comparison test. P values less than 0.05 were considered significant. Analysis of variance with diverse multiple comparison test was performed using GraphPad Prism 6 (GraphPad Software, Inc., San Diego, CA).
3. Results and Discussion
3.1. Synthesis and characterization of dual drug-loaded micelles
A micellar carrier design is based on diblock copolymer, PEG-PGlu, in which part of the Glu units are modified with SN38 moieties. The incorporation of hydrophobic SN38 into block copolymers facilitates their self-assembly into micellar aggregates. The remaining PGlu segments can serve as a reservoir for the incorporation of DACHPt through coordination interactions with COOH functionalities and PEG chains afford stability of the drug-loaded micelles in aqueous dispersion (Figure 1). The association behavior of hydrophobically modified copolymers depends on polymer composition and the hydrophobic SN38 content. For these reasons we used PEG-PGlu copolymers with a constant PEG chain length (114 repeating units) and differed in the lengths of PGlu blocks (80, 150, and 240 repeating units) as precursors for the synthesis of polymer-SN38 conjugates and feed ratios of SN38/Glu units were varied (7–40 mol % relative to total Glu units) to obtain PEG-P(Glu-Glu/SN38) conjugates with different degrees of SN38 grafting (Table S1). A series of PEG-P(Glu-Glu/SN38) conjugates has been synthesized according to previously reported procedure [27] and their chemical structures were confirmed by 1H NMR (Figure S1). All synthesized polymer-drug conjugates were able to self-assemble into polymeric micelles upon direct dissolution in PBS. However, the formed aggregates were quite large in size (> 200 nm) with a broad size distribution. Therefore, PEG-P(Glu-Glu/SN38) conjugates were converted into acid form, dissolved in DMF and micelles were prepared by dialysis in water. DLS measurements revealed formation of uniform (unimodal, relatively narrow particle size distribution, PDI <0.2) micelles with diameters in the range of 45 – 85 nm at pH 6, except in the case of the conjugates based on PEG114-PGlu240 copolymers (~ 120 nm) (Table 1).
Figure 1.
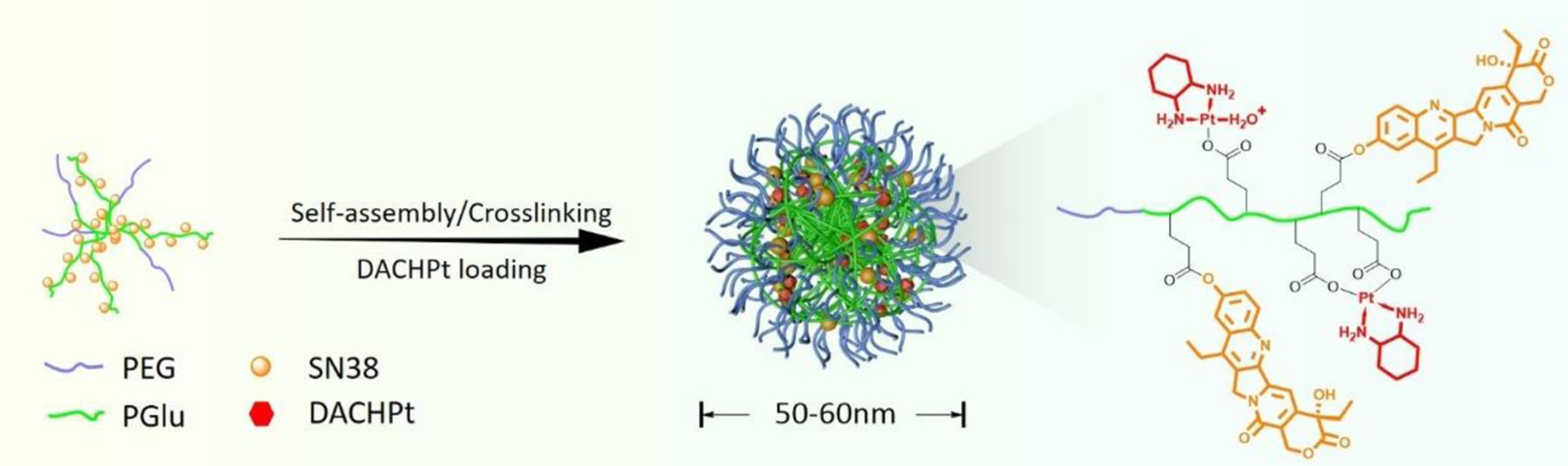
Schematic illustration of dual drug-loaded micelle preparation.
Table 1.
Characteristics of polymer-SN38 conjugates and corresponding micelles.
| Copolymer | SN38 (%mol) |
Deff (nm)b | PDI | SN38 content (%w/w)c | |
|---|---|---|---|---|---|
| Feed | Yield a | ||||
| PEG114-PGlu80 | 12.5 | 7.9 | 71 ± 7 | 0.13± 0.05 | 14 |
| 25 | 10.6 | 77 ± 4 | 0.08 ± 0.01 | 18 | |
| PEG114-PGlu150 | 6.7 | 2.1 | 60 ± 7 | 0.20 ± 0.06 | 5 |
| 13.3 | 5.6 | 45 ± 2 | 0.15 ± 0.03 | 12 ± 2 | |
| 26.7 | 6.1 | 73 ± 5 | 0.16 ± 0.01 | 13 | |
| 40 | 7.2 | 83 ± 2 | 0.15± 0.02 | 15 | |
| PEG114-PGlu240 | 8.3 | 4.2 | 114 ± 5 | 0.16 ± 0.03 | 10 |
| 16.7 | 5.5 | 127 ± 4 | 0.08 ± 0.02 | 13 | |
SN38 content in polymer conjugates was determined by alkaline digestion and UPLC analysis.
Effective diameter (Deff) and polydispersity index (PDI) of SN38/m were determined by DLS in water, pH 6, 25°C. Copolymer concentration 1mg/mL.
SN-38 content in the micelles is expressed as the mass of incorporated drug per mass of drug-loaded micelles.
It is well recognized that the size of the nanocarriers is one of the design parameters that is critical for achieving favorable biodistribution and tumor accumulation. In this respect, it was recently demonstrated by Kataoka’s group that polymeric micelles smaller than 50 nm would be preferable to achieve efficient penetration and accumulation in pancreatic tumors that are characterized by hypovascularity and thick fibrosis in tumor microenvironments. Thus, for the following studies we selected micelles with the smallest size of 45 nm (PDI 0.15) based on PEG114-PGlu150 copolymer with estimated SN38 content of ca. 12 wt% and low level of free drug (about 0.2% w/w). As it was reported, such multimolecular assemblies are stabilized by hydrophobic and π-π stacking interactions between SN38 moieties [27]. We also expected that association behavior of SN38 conjugated copolymers will be affected by the presence of polyanion PGlu chains bearing a substantial number of unmodified carboxylic groups that become ionized as pH increases. Indeed, SN38-conjugated micelles were prepared at pH 6 when PGlu segments are only partially ionized. When pH values of the micelle dispersion were gradually increased to pH 7, the sharp increase in size and appearance of large aggregates with broad size distribution were detected by DLS suggesting formation of heterogeneous particle populations (Figure S2). This may be explained by destabilization of multimolecular micelles due to ionization of the additional carboxylic groups and increased electrostatic repulsion of PGlu chains within the micelle corona. Overall, these data suggest that self-assembly of PEG-P(Glu-Glu/SN38) conjugates is driven by fine interplay between electrostatic and hydrophobic interactions. As the dispersion stability of the polymeric micelles is a crucial factor for their systemic application, we utilized crosslinking strategy to reinforce the stability of the proposed micellar carrier. To this end, preformed SN38-loaded micelles were used as templates for the covalent crosslinking of PGlu chains within the volume of the micelles as we previously described [17]. The targeted extent of crosslinking (20%) was controlled by the molar ratio of crosslinker (ethylenediamine) to carboxylic acid groups of the Glu residues. Importantly, after completion of the crosslinking reaction and purification, the size of crosslinked micelles (further designated as SN38/m) in dispersion was similar to that of the precursor micelles (ca. 45 nm, pH 6) confirming that the formation of crosslinks was limited to intramicellar reactions. In contrast to their non-crosslinked counterparts, SN38/m remained their integrity in a wide range of the pH values (Figure S2). An increase of pH from 6 to 8 is accompanied by an increase of the particles size with no substantial changes in size detected above pH 8 where the ionization of the PGA chains is essentially complete. The observed pH-induced dimensional changes (nanogel-like behavior) of SN38/m are consistent with polyelectrolyte behavior of cross-linked PGlu chains. Notably, these changes were reversible, and the size distribution of SN38/m remained narrow (PDI ≤ 0.13).
An ionic network of PGlu chains in SN38/m was further used for DACHPt loading. DACHPt was first converted to aqueous complex and then incubated with an aqueous SN38/m dispersion for 24 h at pH 8 when SN38/m are fully swollen, and the bulk of the carboxylate groups is accessible to DACHPt binding through coordination interactions. By varying the feed concentrations of DACHPt we prepared two dual-drug loaded (SN38 +DACHPt)/m formulations with 6% and 12% DACHPt loading that correspond to 2:1 and 1:1 weight ratio (SN38:DACHPt) of drugs in the micelles, respectively. As expected, the net negative charge of SN38/m decreased upon DACHPt loading consistent with progressive neutralization of PGlu segments (Table 2). The particle size of (SN38 +DACHPt)/m was slightly larger (51–55 nm) than that of SN38/m. It is likely that formed DACHPt-PGlu adducts, which occupies larger volume than the carboxylic groups of PGlu chains, may prevent the collapse of the PGlu network within the micelles upon the decrease of pH values from pH 8 (loading) to pH 6 (purification). An increase in the molar ratio of DACHPt to carboxylate group in the micelles also led to a decrease in pH-dependent swelling ability of (SN38 +DACHPt)/m compared to that of the SN38/m precursor. For (SN38 +DACHPt)/m with higher degree of DACHPt loading (1:1) the swelling of the micelles was practically completely suppressed as their overall size at pH 7.4 remained nearly the same as at pH 6 (Table 2). All prepared SN38/m and (SN38 +DACHPt)/m formulations remained stable in dispersion displaying no aggregation or precipitation with loading capacity preserved for as long as three months.
Table 2.
Physicochemical characteristics of drug-loaded micelles.a
| Sample | DI water, pH 6.0 |
PBS, pH 7.4 |
|||||
|---|---|---|---|---|---|---|---|
| Deff (nm) | PDI | ζ-potential (mV) | LC (%, w/w) c |
Deff (nm) | PDI | ||
| SN38 | DACHPt | ||||||
| SN38/m | 45 ± 3 | 0.13 ± 0.03 | −32 ± 5 | 12 ± 2 | - | 78 ± 6 | 0.18 ± 0.02 |
| SN38 + DACHPt)/m (2:1) b | 51 ± 3 | 0.12 ± 0.01 | −17 ± 6 | 11 | 6 | 66 ± 5 | 0.15 ± 0.05 |
| SN38 + DACHPt)/m (1:1) b | 55 ± 5 | 0.15 ± 0.02 | −7 ± 4 | 12 ± 2 | 12 ± 2 | 58 ± 2 | 0.16 ± 0.04 |
Size (Deff), PDI, and ζ-potential were determined by DLS. All measurements were carried out at 1 mg/mL polymer concentration and 25°C. Data presented as mean ± SD, n=3.
Dual-drug loaded micelles were prepared at different DACHPt loading. Drug content in the micelles expressed as (SN38 : DACHPt) weight ratios.
Loading capacity (LC) is expressed as mass of incorporated drug per mass of drug-loaded micelles (w/w). SN38 and DACHPt content were determined by UPLC and ICP-MS, respectively.
3.2. Drug release studies
The drug release profiles from (SN38 + DACHPt)/m (1:1 w/w drug ratio) were evaluated by equilibrium dialysis under pH conditions mimicking bloodstream (PBS, pH 7.4) and late endosomal compartments (ABS, pH 5.5), respectively. As shown in Figure 2A, (SN38 + DACHPt)/m displayed relatively slow release of SN38 reaching approximately 26% after 24 h incubation at pH 7.4 at 37 °C. Noteworthy, the release rate of SN38 from single SN38/m was significantly faster with nearly 60% loaded SN38 released within 24 h (insert in Figure 2A). Indeed, at pH 7.4 SN38/m are more swollen compared to (SN38 + DACHPt)/m (Table 2) with ester bonds readily accessible for hydrolysis which leads to an increased rate of the SN38 release. The rate of the SN38 release from (SN38 + DACHPt)/m was further decreased under acidic conditions which is consistent with the slower rate of the hydrolytic breakdown of phenyl ester bond in weak acidic environment [28]. Release of Pt species from (SN38 + DACHPt)/m at pH 7.4 was faster (about 41%) than that of SN38 during the same time period (Figure 2B) and also was slowdown at pH 5.5. This observation may be ascribed to a differences in the structure of the core of (SN38 + DACHPt)/m: since the release of conjugated SN38 is suppressed at the acidic conditions, the core of (SN38 + DACHPt)/m may be more dense, which in turn may hamper the in and out diffusion of water, chloride ions and Pt species. As expected, the rates of release of both drugs were accelerated in the presence of serum albumin in the medium (10% FBS, PBS). This effect was more pronounced for SN38 in dual-drug formulation apparently due to combined hydrolysis and enzymatically mediated mechanisms of the drug release (Figure 2A).
Figure 2.
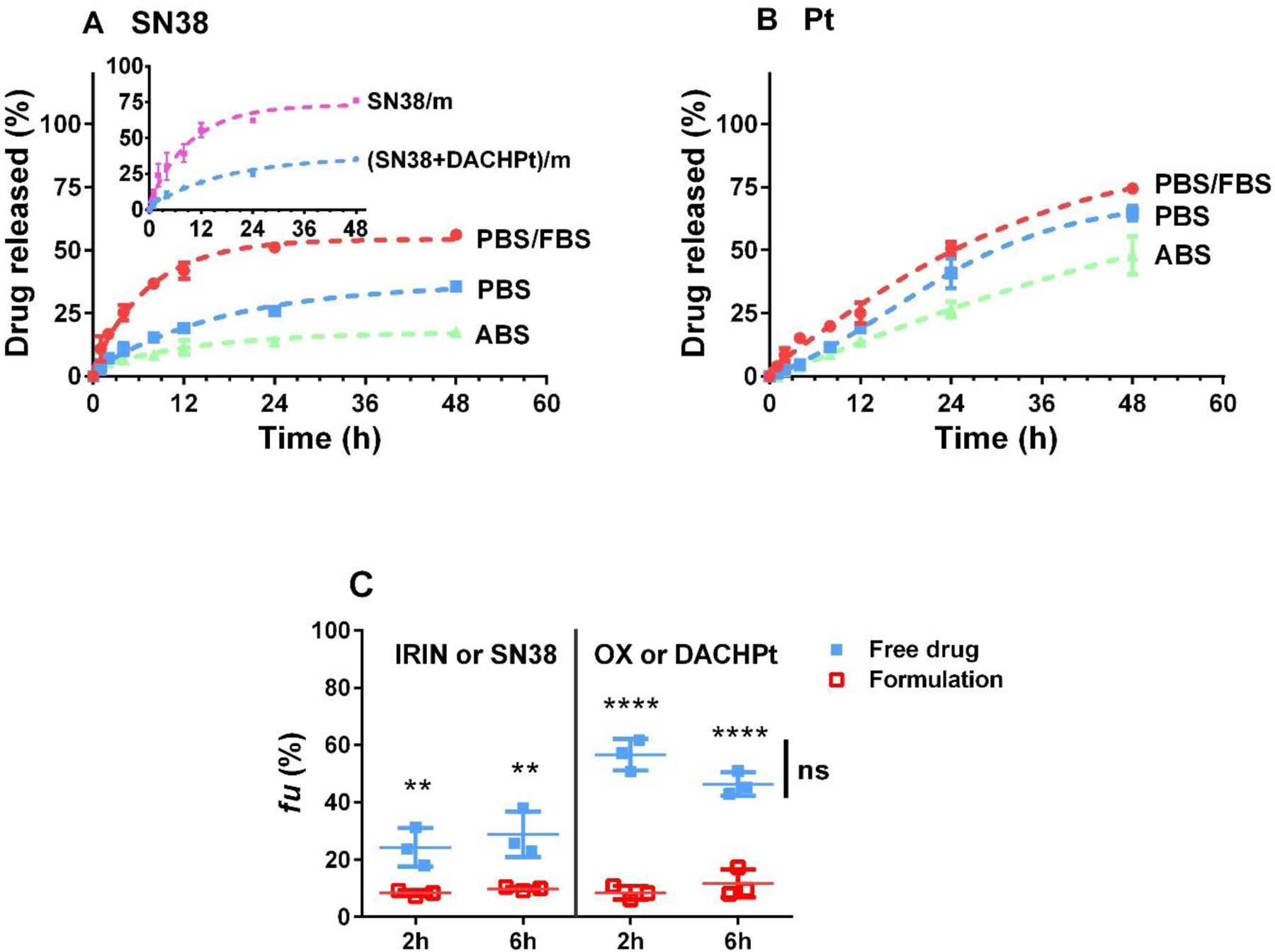
Drug release profiles of (A) SN38 and (B) Pt from (SN38 + DACHPt)/m (1 :1 w/w) in PBS (pH 7.4), ABS (pH 5.5) and PBS in the presence of 10% FBS at 37 °C. Data are expressed as the mean ± SD (n = 3). Insert in (A) shows a comparison of SN38 release profiles from SN38/m and (SN38 + DACHPt)/m in PBS. (C) Average percentage of unbound fraction (fu) of each drug in human whole blood post incubation with (SN38 + DACHPt)/m or combination of free drugs (IRIN + OX) for 2 and 6 hours, at 37 °C. Data are presented as the mean (fu) ± SD (n = 3) and compared by one-way ANOVA with Bonferroni’s multiple comparison test. (**p < 0.01, ****p < 0.0001, ns - not significant).
To further assess the possible in vivo stability and drug release from (SN38 + DACHPt)/m, we utilize another in vitro test that has been designed to evaluate the stability of polymeric micelle formulations and predict their pharmacokinetic behavior in vivo [19, 21]. According to this erythrocyte vs. buffer or plasma partitioning method, (SN38 + DACHPt)/m or a mixture of free drugs (IRIN + OX) were incubated for either 2 h or 6 h with to the erythrocyte/plasma (or erythrocyte/PBS) dispersions to mimic the dilution of the formulations in the blood. The unbound fraction (fu) of each drug was determined by UPLC (SN38 or IRIN) or ICP-MS (Pt). A decrease in the fu value for drugs co-loaded into micelles vs. free drugs indicates that a higher proportion of drug remains within the polymer micelles (Figure 2C). For example, after 2 h of incubation time, the (SN38 + DACHPt)/m formulation exhibited significantly lower unbound drug fractions compared to free drug combination (8.3 ± 0.8% for SN38 vs. 24.3 ± 5.5% for IRIN and 8.4 ± 2.0% for DACHPt vs. 56.6 ± 4.5% for OX, p < 0.01). Notably, the fu values for both drugs in (SN38 + DACHPt)/m remained practically the same after 6 h exposure to human blood, implying its good stability. Moreover, similar fu values for both drugs (10–12%) at different time points also suggest that after encapsulation in one carrier, the two drugs can be released in synchronized fashion, which may be beneficial for achieving synergistic anticancer activity of the (SN38 + DACHPt)/m formulation.
3.3. Cytotoxicity of (SN38 + DACHPt)/m against PC cell lines and 3D tumor organoids
Previous studies demonstrated that SN38 and OX reciprocally affect the cellular response to DNA damage, which results in synergistic activity of these drugs [29]. To further understand whether co-formulation into micellar carrier can retain or even improve the synergistic activity of these drug combination, we tested cytotoxicity of (SN38 + DACHPt)/m in a panel of PC cell lines by MTT assay. We first screened (SN38 + DACHPt)/m formulations with different drug ratios and compared them with single SN38/m, free OX or their combination in T3M4 cells after exposure to micelles/drugs (Table S1). We found that SN38/m were more effective in killing T3M4 cells as the IC50 value for SN38/m was order of magnitude lower than that for free IRIN or OX. Notably, all SN38 micellar formulations displayed higher cytotoxicity compared to that of free IRIN plus OX combination applied to the cells at the clinically used drug weight ratio (IRIN:OX = 2:1). This agrees with known higher potency of SN38 relative to IRIN [30]. When comparing dual drug formulations, (SN38 + DACHPt)/m or SN38/m plus free OX, and single SN38/m, the IC50 values (expressed in SN38 equivalents) were not significantly different from that of SN38/m (Table S1) which can be explained by relatively low sensitivity of T3M4 cells to OX. Among dual drug formulations, (SN38 + DACHPt)/m combination with 2:1 (SN38:DACHPt) drug weight ratio was the least effective. As (SN38 + DACHPt)/m (1:1) displayed smaller size at physiological conditions (Table 2) and therefore may be more suitable for accessing tumors and accumulating there, we selected (SN38 + DACHPt)/m (1:1) to confirm their activity in PC cells with differential sensitivity to the drugs in the next set of experiments. As summarized in Table 3, the combination of SN38/m plus free OX was more cytotoxic than the SN38/m formulation alone in all three PC cell lines studied.
Table 3.
IC50 values for drug-loaded micelles and free drugs against PC cell lines.a
| Sample | Drug ratio, w/w | IC50 (nM) |
||
|---|---|---|---|---|
| PANC-1 | CD18/HPAF | T3M4 | ||
| 48h |
||||
| IRIN | - | 943 ± 47 | 168 ± 14 | 435 ± 68 |
| OX | - | 236 ± 23 | 413 ± 3 | 2070 ± 162 |
| IRIN + OX | 2 | 90 ± 5 CI =1.23 ± 0.02) |
39 ± 4 (CI = 0.72 ± 0.03) |
121 ± 14 (CI = 0.57 ± 0.11) |
| SN38/m | - | 30 ± 4 | 8.7 ± 1.0 | 6.0 ± 2.2 |
| SN38/m + OX | 1 | 7.5 ± 0.1 (CI = 0.27 ± 0.03) |
1.3 ± 0.1 (CI = 0.15 ± 0.01) |
2.6 ± 0.7 (CI = 0.42 ± 0.05) |
| (SN38 + DACHPt)/m | 1 | 19.0 ± 2.4 (CI = 0.68 ± 0.01) |
8.5 ± 1.3 (CI = 1.04 ± 0.03) |
6.5 ± 1.7 (CI = 1.22 ± 0.25) |
| 72h |
||||
| IRIN | - | 263 ± 24 | 83 ± 8.5 | 935 ± 148 |
| OX | - | 184 ± 14 | 309 ± 28 | 3511 ± 392 |
| IRIN + OX | 2 | 62 ± 6 (CI = 1.38 ± 0.10) |
20 ± 1 (CI = 0.69 ± 0.00) |
261 ± 44 (CI = 0.64 ± 0.18) |
| SN38/m | - | 23 ± 1 | 5.3 ± 0.1 | 4.0 ± 1.2 |
| SN38/m + OX | 1 | 5.5 ± 1.2 (CI = 0.26 ± 0.06) |
0.82 ± 0.09 (CI = 0.17 ± 0.00) |
1.9 ± 0.7 (CI = 0.46 ± 0.03) |
| (SN38 + DACHPt)/m | 1 | 4.6 ± 1.6 (CI = 0.22 ± 0.08) |
0.67 ± 0.01 (CI = 0.14 ± 0.02) |
1.0 ± 0.4 (CI = 0.30 ± 0.11) |
Cells were seeded at the density of 3,000 cell/well and exposed to various drug formulations for 48h or 72h followed by culturing in fresh completed media for 24h. IC50 values for drug combinations were calculated with respect to OX equivalents. Data are presented as mean ± SD (n = 3). CI - combination index, calculated at IC50.
At the same time, (SN38 + DACHPt)/m presented a trend toward lower cytotoxicity compared to the combination of SN38/m plus free OX against these cell lines at 48h, which is likely can be attributed to slower release of active Pt species from the dual drug-loaded micelles compared to free OX. Release of SN38 from (SN38 + DACHPt)/m is also slower compared to that from SN38/m and thereby can be an additional reason for the differential cytotoxicity observed. However, similar inhibitory activities and strong drug synergy (CI values less than 0.25) were observed for both (SN38 + DACHPt)/m and (SN38/m + OX) when the treatment time was extended to 72h (Table 3, Figure S3). In comparison to PC cells, normal, immortalized pancreatic duct cells (hTERT-HPNE) were substantially less sensitive to the free drugs or their micellar formulations with several orders of magnitude higher IC50 values (Table 4). Notably, OX in free form or in combination with SN38/m was more toxic to normal pancreatic cells than IRIN or SN38/m alone. In contrast, (SN38 + DACHPt)/m exhibited less toxicity relative to free OX suggesting that therapeutic approach with (SN38 + DACHPt)/m may hold a benefit of reduced treatment-associated toxicity to normal tissues.
Table 4.
IC50 values for drug-loaded micelles and free drugs against normal hTERT-HPNE pancreatic cells.a
| Sample | Drug ratio, w/w | IC50 (mM) |
|
|---|---|---|---|
| 48h | 72h | ||
| IRIN | - | >500 | >500 |
| OX | - | 0.7 ± 0.2 | 2.4 ± 0.6 |
| IRIN + OX | 2 | 0.5 ± 0.3 | 3.5 ± 1.4 |
| SN38/m | - | >10 | >10 |
| SN38/m + OX | 1 | 1.4 ± 0.5 | 3.1± 1.3 |
| (SN38 + DACHPt)/m | 1 | >10 | 9.5 ± 2.7 |
Cells were seeded at the density of 3,000 cell/well and exposed to various drug formulations for 48h or 72h followed by culturing in fresh completed media for 24h. IC50 values for drug combinations were calculated with respect to OX equivalents. Data are presented as mean ± SD (n = 3).
To get insight into mechanism of the interactions of single and double drug-loaded micelles with cancer cells, Cy5-labeled SN38/m and (SN38 + DACHPt)/m were incubated with CD18/HPAF cells, followed by analysis using flow cytometry. SN38/m showed a fast uptake as already after 30 min practically all cells were gated (Figure 3A). It is also clear that the uptake continues after that with an increase of the mean fluorescence over time (Figure 3B). In comparison, the time course of the uptake of (SN38 + DACHPt)/m appears somewhat slower as about 55 % gated cells were reached in 2h (Figure 3A). Similar trend was observed in normal hTERT-HPNE pancreatic cells (Figure S4). Laser scanning confocal microscopy images inferred that the fluorescence appears to be primarily distributed as dots inside the cells and is indicative of the vesicular uptake of both SN38/m and (SN38 + DACHPt)/m. Consistent with the difference in cellular uptake detected by flow cytometry, the mean fluorescence value per cell was higher for SN38/m than that for (SN38 + DACHPt)/m. We also observed that cells treated with SN38/m displayed more intense fluorescence signal along the cell membrane compared to the signal by the (SN38 + DACHPt)/m, suggesting that physicochemical properties of SN38/m may facilitate their adhesion to the cellular surface and the subsequent uptake. (Figure 3C). Interestingly, we did not observe an alternation in the cellular uptake of the micelles in the serum-free medium suggesting that the adsorbed proteins as a corona on the micelle surfaces are not likely to contribute to the differences in micelle-uptake levels (Figure 3D). The flow cytometry analysis further demonstrated that the cellular uptake of the both micelles was blocked at 4°C, which suggests energy-dependent endocytosis pathways for internalization (Figure 3D). To further elucidate the specific internalization pathways of these micelles, the uptake of Cy5-labeled micelles was examined under conditions blocking different endocytosis pathways using pretreatment with chlorpromazine (inhibitor of clathrin-mediated endocytosis), filipin (inhibitor of caveolae-mediated endocytosis), or amiloride hydrochloride (inhibitor of macropinocytosis) for 30 min at subtoxic concentrations. In each case, the flow cytometry analysis was performed 60 min after exposure of the CD18/HPAF cells to the micelles with or without the corresponding inhibitors. As it shown in Figure 3D, the uptake of both SN38/m and (SN38 + DACHPt)/m was significantly inhibited by all three inhibitors, however, to a different extent. The greatest inhibition of micelles entry was observed with filipin and amiloride hydrochloride suggesting that the most prominent pathways for the micelle’s uptake were the caveolae-mediated endocytosis and macropinocytosis. Although chlorpromazine inhibited micelles uptake to some extent, its effect was much less pronounced than that of the filipin or amiloride hydrochloride. It should be noted that for all three inhibitors, the observed inhibition effects for SN38/m and (SN38 + DACHPt)/m did not vary significantly from each other. Taken together, our inhibition studies suggest that the internalization of drug-loaded micelles is mediated by more than one cellular uptake mechanisms that appear to be independent on material composition. An enhanced in vitro cell internalization rate for SN38 when compared with (SN38 + DACHPt)/m is probably due to the larger size and greater negative ζ - potential [31]. We cannot exclude the possibility that difference in elasticity of drug-loaded micelles can be another influencing factor with respect to important aspects of cellular uptake and intracellular distribution. Indeed, it was recently demonstrated that soft nanolipogels, an alginate encapsulating liposomes, exhibited significantly greater uptake into neoplastic and non-neoplastic cells relative to their elastic counterparts [32]. Moreover, as is seen for most types of nanomedicines, particle uptake is varying with cell type. Further comprehensive studies will be necessary to elucidate the effects of the architecture and mechanical properties of the drug loaded micelles on their interactions with the cells, their fate and the resulting cellular responses.
Figure 3.
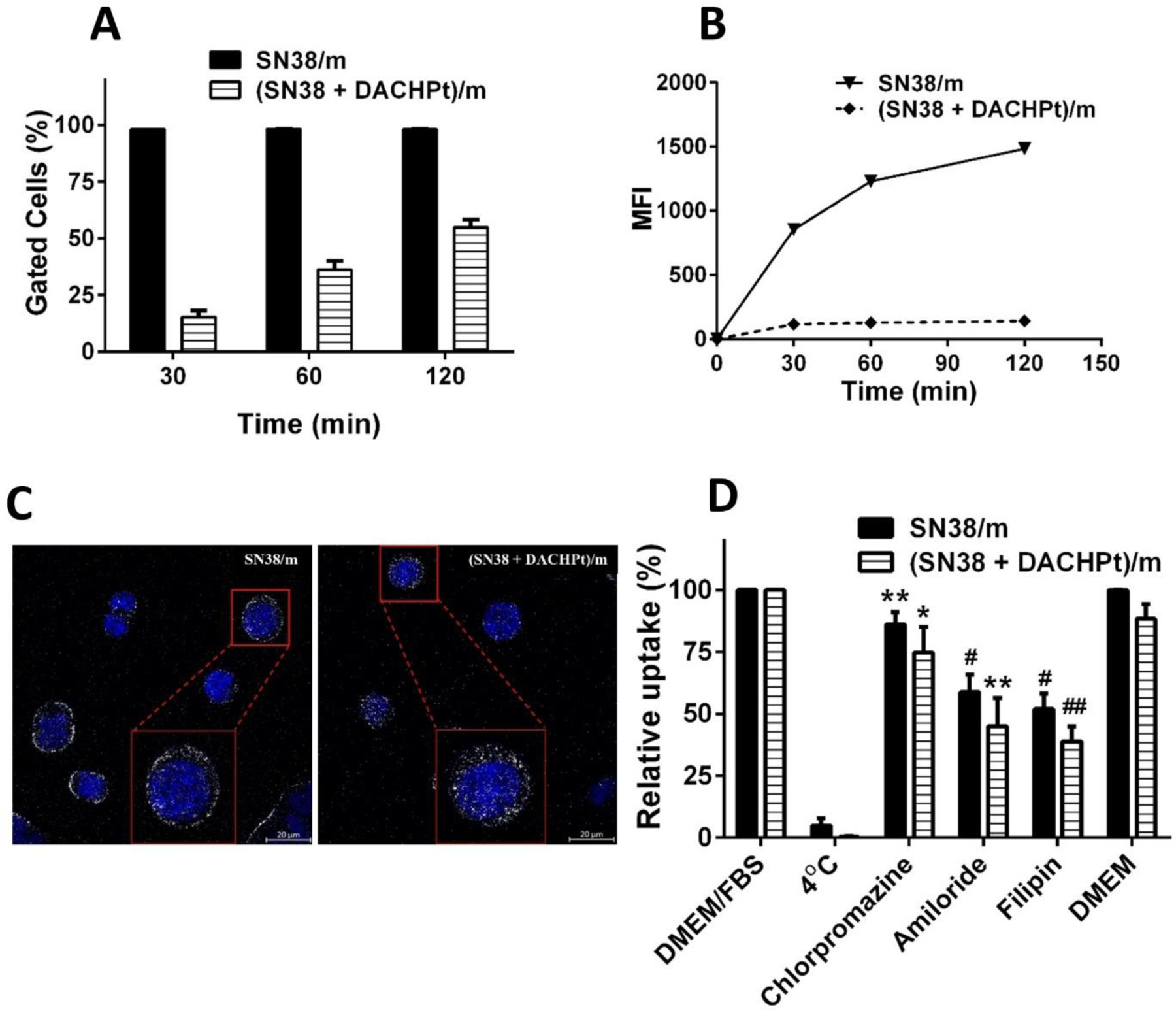
Cellular uptake of Cy5-labeled SN38/m and (SN38 + DACHPt)/m in CD18/HPAF cells as a function of incubation time at 37 °C as investigated by flow cytometry and expressed in (A) % gated cells and (B) mean fluorescence intensity (MFI). (C) Representative confocal images of the uptake at 60 min post-treatment with micelles and magnified images of the boxed areas (red). White, Cy5; blue, DAPI. Scale bars represent 20 µm. (D) Effect of endocytosis inhibitors on the uptake of drug-loaded micelles. Cells were pretreated with corresponding inhibitors or at 4°C for 30 minutes followed by adding Cy5-labeled SN38/m or (SN38 + DACHPt)/m at concentration of 40 µM based on SN38 equivalents and incubated for another 60 min. The relative cell uptake (%) represents the uptake of micelles by cells treated with endocytic inhibitors normalized by the uptake by nontreated ones. Data presented as mean ± SD (n = 3). Statistical comparisons between the control group (no inhibitor) and the corresponding inhibitor group were determined by Student t-test (*p < 0.05, **p < 0.01, #p < 0.001, ##p < 0.0001).
To further validate the results of MTT assays, the growth inhibitory effect of micellar formulations against PANC-1 and CD18/HPAF cells was evaluated using a clonogenic assay. Both cells displayed concentration-dependent decrease of the number of colonies after a 48h treatment with IRIN plus OX combination (Figure 4A and 4B). Consistent with MTT assay results, all SN38 formulations exhibited the most potent cytotoxicity and significantly (p <0.01) reduced the colony-forming potential of PC cells compared with drug free combination at concentrations as low as 10 nM based on OX equivalents.
Figure 4.
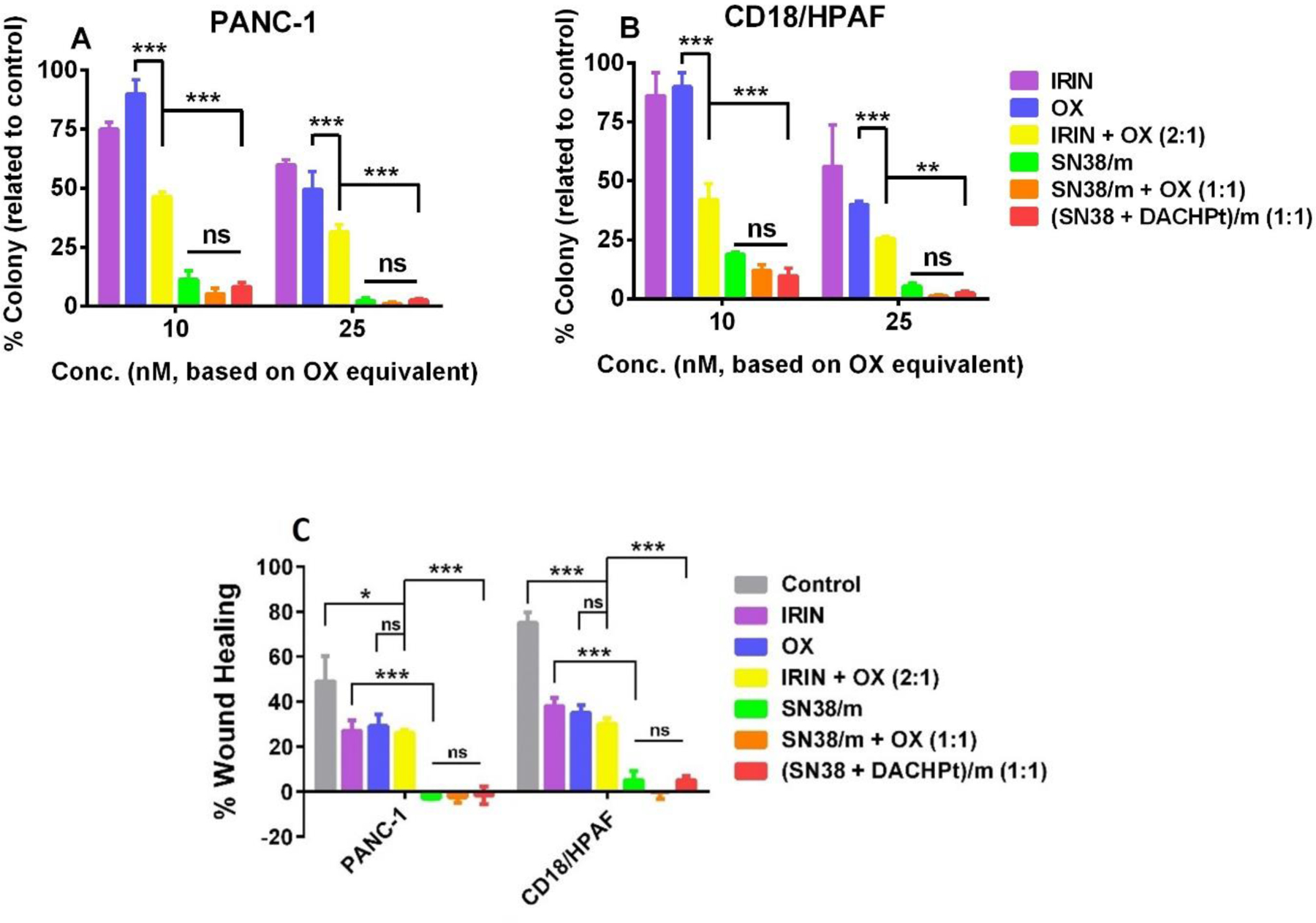
Drug-loaded micelle formulations suppress colony-formation ability and migration of PC cells. (A, B) Colony formation assay was performed in PANC-1 and CD18/HPAF cells that were treated with free drugs and drug-loaded micelle formulations for 48 h at indicated concentrations based on OX equivalents. (C) Inhibition of the migration of PC cells as determined by wound healing assay. Cells were treated with various drug formats for 24 h at concentration of 60 nM based on OX equivalents. Data presented as mean ± SD (n = 3). Statistical significance was determined using one-way ANOVA with Bonferroni’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ns - not significant).
Metastases are the key hallmarks of PDAC, and majority of patients diagnosed with this disease have already progressed to advanced stages with complications involving distant metastasis at the time of diagnosis. Migration of cancer cells is the prerequisite of local invasion and distant metastasis and, thus, controlling cancer cell migration is an effective strategy for treating the disease. To assess whether (SN38 + DACHPt)/m and other drug combinations can prevent migration of PC cells, we performed wound healing assay in PANC-1 and CD18/HPAF cells (Figure 4C). A scratch wound was created in the confluent monolayers of PC cells followed by treatment with free drugs and drug-loaded micelle formulations for 24 h. The assessment of wound healing assay reveals that the treatment of PC cells with free drugs or their combination at concentrations well below their IC50 values resulted in approximately 2-fold decrease in the collective migration of the cells compared to untreated controls. In contrast, cell migration was practically completely abolished after cells exposure to any of SN38 formulations. Taken together, the results obtained from cell viability and migration studies suggest that SN38 plays predominant role in observed superior cytotoxic activity of single SN38/m micelle compared to clinically used combination of free IRIN and OX. Furthermore, SN38/m enter cancer cells via endocytosis, where SN38 can be liberated in the endocytic vesicles. Such vesicles may serve as reservoirs of the drug available to the cell over a period of time. The potency of SN38/m was further increased when combined with free OX or used as dual drug-loaded (SN38 + DACHPt)/m formulation. Nevertheless, we did not find statistically significant differences between (SN38 + DACHPt)/m and SN38/m combination under static in vitro conditions in 2D cell cultures.
While traditional cell lines provide easy models of PDAC, these cells are growing in two-dimensional culture and lack the essential tumor microenvironment that leads to structural and functional organization of the tumor tissue. Tumor-derived 3D organoids recapitulate the characteristics of the primary tumors in terms of cellular composition, genomic and mutational landscape and tissue function and were shown to represent a useful model system to identify molecular pathways that correlated with disease progression as well as to predict drug treatment outcomes [33–35]. In this context, we have generated pancreatic tumor organoids from autochthonous KrasG12D; Trp52R172H/+; Pdx-1 Cre PDAC mouse tumors to model tumor responses to the treatments with drug-loaded micelle in ex vivo assay.
The mouse pancreatic tumor organoids were continuously exposed to free drugs or drug-loaded micelle formulations for three days and assessed for growth inhibitory effects. Organoids treatment with free IRIN plus OX combination or single SN38/m micelles led to only marginal decrease in organoids size compared to untreated controls (Figure 5). Notably, a statistically significant reduction of growth of mouse pancreatic tumor organoids was observed upon treatment with (SN38 + DACHPt)/m formulation as compared to SN38/m (p < 0.05) or free drug combination (p < 0.01). Moreover, histological analysis showed more necrotic and apoptotic cells in (SN38 + DACHPt)/m-treated tumor organoids compared to other treatment groups (Figure S5). This ex vivo experiments support our argument that co-loading of SN38 and DACHPt into the micelles can further improve the therapeutic outcomes of this multi-drug regimen in PDAC.
Figure 5.
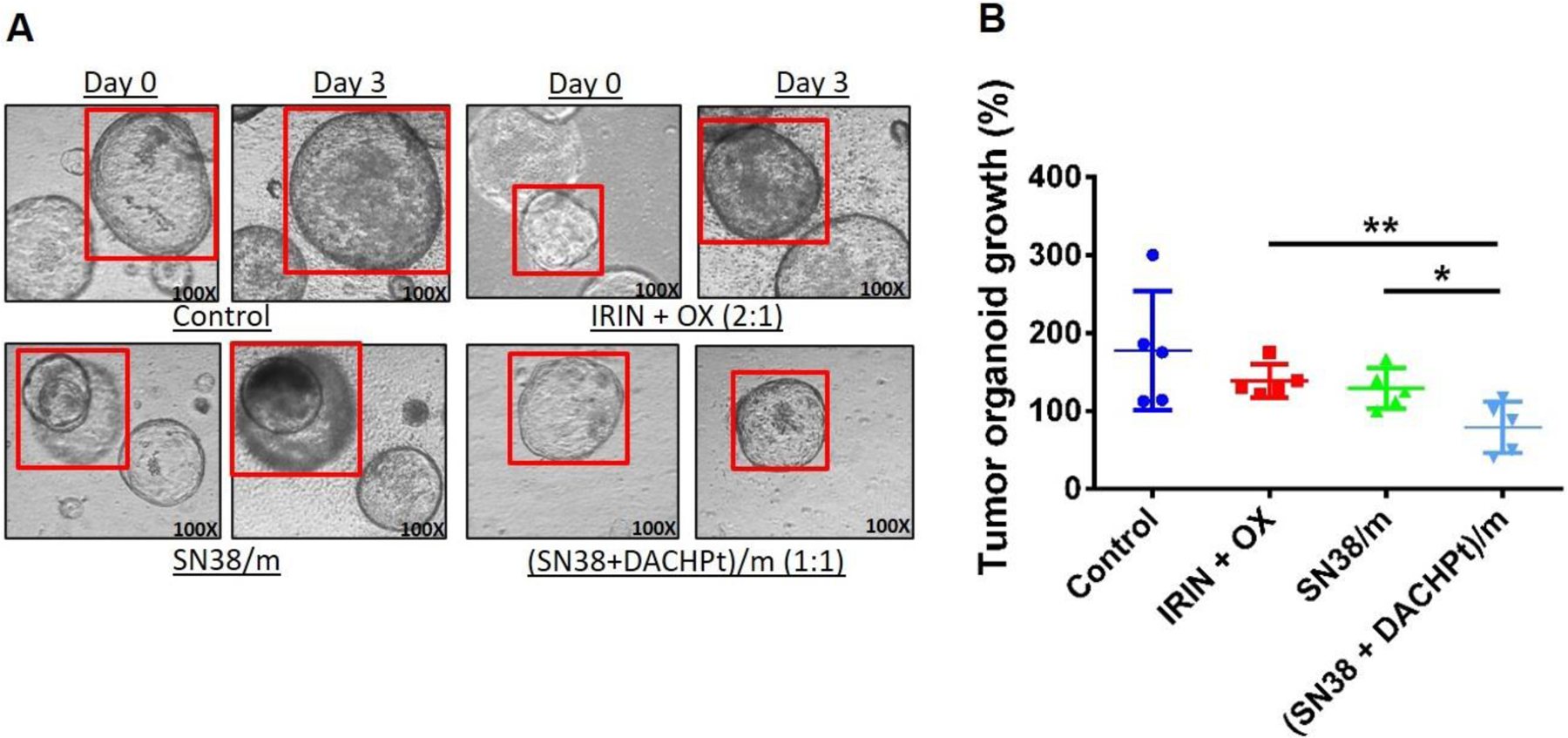
(SN38 + DACHPt)/m treatment inhibit growth of mouse pancreatic tumor organoids. (A) Representative light microscope images of tumor organoids treated with IRIN + OX (2:1) combination, SN38/m, or (SN38 + DACHPt)/m on Day 0 and Day 3. (B) The effect of various treatments on the growth of tumor organoids on Day 3. The tumor organoids were treated with free drugs or drug-loaded micelles for 3 days at concentration of 10 nM based on OX equivalents. Data are expressed as mean ± SD (n = 5), *p < 0.05, **p < 0.01.
3.4. Antitumor activity in orthotopic PC murine model
The antitumor effect of (SN38 + DACHPt)/m was evaluated in mice bearing orthotopic CD18/HPAF luciferase-tagged human PC xenografts. Tumor burden was monitored by whole body bioluminescent imaging (BLI) and treatments were initiated on day 10 post tumor cell implantation. The mice (n = 11) were injected intravenously 4 times every 4 days with vehicle (5% dextrose), the free IRIN + OX combination (8 mg/kg and 4 mg/kg, respectively, dose ratio is mimicking FOLFIRINOX regimen), SN38/m, SN38/m plus free OX, and (SN38 + DACHPt)/m at equivalent doses of 4 mg/kg with respect to OX or SN38 (Figure 6A). The treatment with combination of IRIN + OX led to small but significant (p < 0.05) delay of the tumor growth compared to untreated animals (Figure 6B). However, it also resulted in noticeable decrease in body weight during the time of treatment (Figure 6C). On day 12, the IRIN + OX group had about 8% body weight loss decrease in body weight compared to control. This result was not surprising as the severe toxicity of this multidrug regimen has been observed in clinic. The growth of tumors was further slowed down by single drug SN38/m micelles, however there was no significant difference between SN38/m and (IRIN + OX) groups. In contrast, both SN/38 plus free OX and (SN38 + DACHPt)/m regimens were found to effectively suppress growth of the tumors. Of great interest, however, was the difference in activity of the (SN38 + DACHPt)/m vs mixture of single drug-loaded SN38/m + free OX at the same dose ratio: inhibition of tumor growth was significantly enhanced in animals treated with (SN38 + DACHPt)/m compared to those treated with the SN38/m plus free OX combination (p < 0.05). Of note, SN38/m plus free OX treatment displayed comparable or even superior effect in in vitro settings compared to the (SN38 + DACHPt)/m (Table 3), which appeared to be a result of OX immediate availability to the tumor cells compared to delayed Pt release from (SN38 + DACHPt)/m. However, OX has a short elimination half-life (10 – 25 min) in vivo [36, 37]. while incorporation of DACHPt into the micelles has been shown to extend the blood circulation of the platinum drug after bolus injection [16]. It is likely, that the improved therapeutic efficacy of (SN38 + DACHPt)/m observed in vivo is associated with the altered pharmacokinetics and biodistribution of the micelles compare to free drug, which led to differential accumulation of platinum drugs in the tumor. The Pt content in blood, tumor and vital organs of the treated animals was studied 24 h after the last injection (day 14) and data summarized in Figure 6D. About 12-fold higher Pt content in plasma samples and significantly reduced association with blood cells were detected in animals treated with (SN38 + DACHPt)/m compared to treatments that included free OX. These data are in the good agreement with the results of our in vitro hematologic stability study (Figure 2C). It seems that the DACHPt retention in micelles and higher plasma exposure may facilitate enhanced Pt tumor accumulation leading to improved overall efficacy of the treatment. Unfortunately, tumors were practically nonexistent in animals treated with (SN38 + DACHPt)/m and no tumor Pt content data from this group could be gained. Future comprehensive pharmacokinetics and biodistribution studies will be needed to confirm this possibility and assess the level and functional consequences of accumulation of both drugs at the tumor site.
Figure 6.
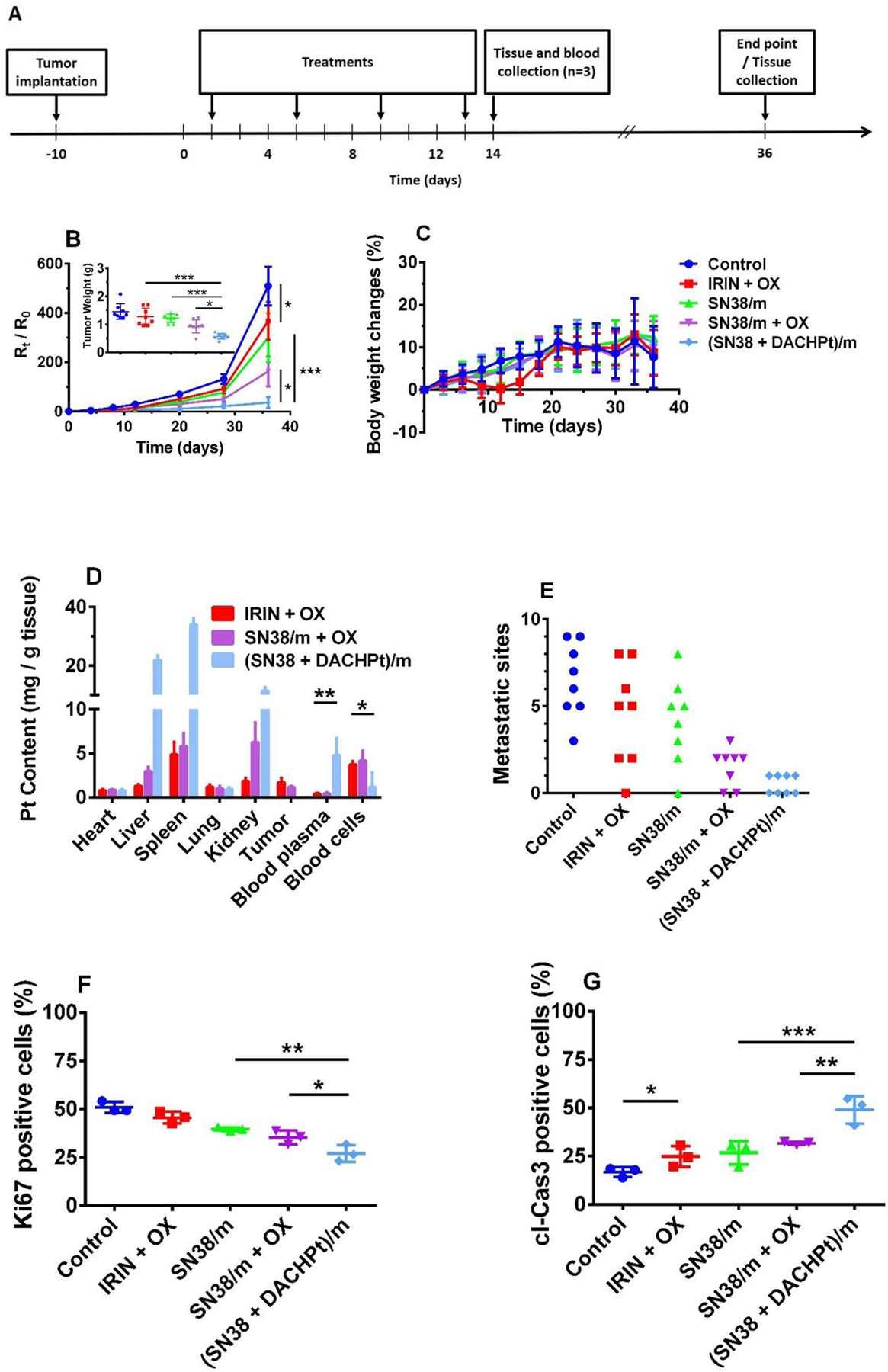
(SN38 + DACHPt)/m inhibit growth of orthotopic CD18/HPAF human pancreatic cancer xenografts. (A) Tumor inoculation and treatment schedule. (B) Changes in tumor burden as assessed by bioluminescence imaging (expressed in relative radiance units, Rt/R0, to the day when treatment was initiated for individual animal) following treatments with 5% dextrose (control), IRIN + OX combination (IRIN 8 mg/kg, OX 4 mg/kg), SN38/m (SN38 4 mg/kg), SN38/m plus free OX(SN38 4 mg/kg, OX 4 mg/kg), and (SN38 + DACHPt)/m (SN38 4 mg/kg, OX equiv. 4 mg/kg). Insert represents tumor weights at the experimental endpoint (day 36). Data are presented as mean ± SEM (n= 8). (C) Body weight changes throughout the experiment. (D) Blood and tissue distribution of Pt in different treatment groups as determined by ICP- MS. Mice were sacrificed at day 14 (n = 3). (E) The number of metastatic sites at distant organs in the various treatment groups. Each point corresponds to an individual animal. Quantification of (F) Ki67 positive and (G) caspase-3 positive cells in tumor tissue from mice from various groups (n = 3, based on the analysis of the whole tumor slice). Statistical significance was determined using one-way ANOVA with Turkey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001).
Necropsy analysis performed at the end of the observation period (day 36) further confirmed BLI data. Large and locally invasive tumors with metastatic spread into in distant organs like liver, spleen, kidney, stomach, diaphragm, intestine and lymph nodes were present in the control group (Figure S6). No substantial reduction in primary tumor burden was achieved by free IRIN + OX combination or single SN38/m (Figure 6B, insert) and all animals in these groups developed metastases at multiple sites (Figure 6E). It appears that SN38/m plus free OX combination provided a moderate reduction in tumor size compared to SN38/m and was more effective in suppression of metastatic spread. The treatment with (SN38 + DACHPt)/m led to the most potent inhibitory effect as evident by smaller size of the primary tumors and pronounced antimetastatic activity. Indeed, no sign of metastasis was detected in 50% of animals in this group and only one metastatic foci could be seen in each of other animals. To further assess the pathological significance of antitumor effect of different drug treatments, we performed immunohistochemical staining of the excised tumor sections for proliferation and apoptosis markers (Ki67 and cleaved caspase-3). As shown in Figure 6F, the number of proliferating cells was significantly lower in the tumors of animals in (SN38 + DACHPt)/m group as compared to animals treated with SN38/ m + OX combination (p < 0.05) or free IRIN+OX (p < 0.01) and was correlated with the observed reversed trend in number of apoptotic cell death in these tumor samples (Figure 6G, Figure S7). The results imply the co-loaded (SN38 + DACHPt)/m enhances the intrinsic cytotoxicity. This could explain the continued suppression of tumor growth seen in animals treated with (SN38 + DACHPt)/m even after 3 weeks post treatment, which in turn may lead to impairment of metastatic-related processes.
One of the defining hallmarks of PDAC is a pronounced desmoplastic microenvironment that plays a critical role in tumor initiation, progression and metastasis [38, 39]. It consists of extracellular matrix (ECM) components such as collagen and hyaluronan and non-neoplastic cells including cancer associated fibroblasts and immune cells. The excessive deposition of ECM components, which is produced and maintained by cancer-associated fibroblasts, generates high interstitial fluid pressures, leads to compression of blood vessels and acts as a barrier for effective delivery of chemotherapeutics. By Sirus Red staining of the tumor tissues, we observed high collagen density throughout the tumor lesion in the control group (Figure 7A) that was decreased by applied treatments. Among the formulations studied, the most substantial disruption and a significant decrease in collagen coverage (~ 4-fold compare to control group) was detected in (SN38 + DACHPt)/m-treated xenografts. These changes were paralleled with the decrease of the fibroblast activation marker, α-SMA as determined using immunofluorescence (Figure 7B). These results may suggest that α-SMA expressing fibroblast population is reduced as a result of cytotoxic action of drugs or there is a shift from an activated to a quiescent state of cancer-associated fibroblasts as a result of the treatments, which may diminish collagen accumulation in primary tumors, reduce tumor fibrosis and normalize ECM. We also cannot exclude the possibility that fibroblasts recruitment to the tumor tissue of animals treated with (SN38 + DACHPt)/m is reduced because of the delayed tumor growth. Using CD31staining of vascular endothelial cells, we observed a moderate but significant increase in the number of CD31-positive cells in response to treatments with SN38/m plus free OX and (SN38 + DACHPt)/m regimens compared to control implying that the density of intratumoral blood vessels appeared to be higher in these groups (Figure 7B). In addition, the density of the intact and closed vessels with lumen was also increased in these groups (Figure 7C), suggesting that ECM normalization may lead to vessel decompression. Several previous studies have demonstrated that besides their direct cytotoxic activity, some chemotherapeutics or their nanoformulations can also contribute into remodeling of tumor microenvironment. ECM-normalizing effect of nab-paclitaxel has been proposed as one of the contributing factors underlying its therapeutic activity in combination with gemcitabine [40]. An enhanced antitumor effect of lipid-based formulation of IRIN (Irinophore C) against glioblastoma murine models was attributed to its ability to normalize vasculature within tumor. It was proposed that sustain release of IRIN from the nanocarrier may mimic metronomic (anti-angiogenic) dosing schedule [41]. Though mechanism leading to observed remodeling of tumor microenvironment in response to (SN38 + DACHPt)/m remains unclear and will require additional studies, it may contribute to observed antitumor activity of this formulation. It may be beneficial in context of improving tumor delivery of 5-fluorouracil as a part of the combination regimen.
Figure 7.
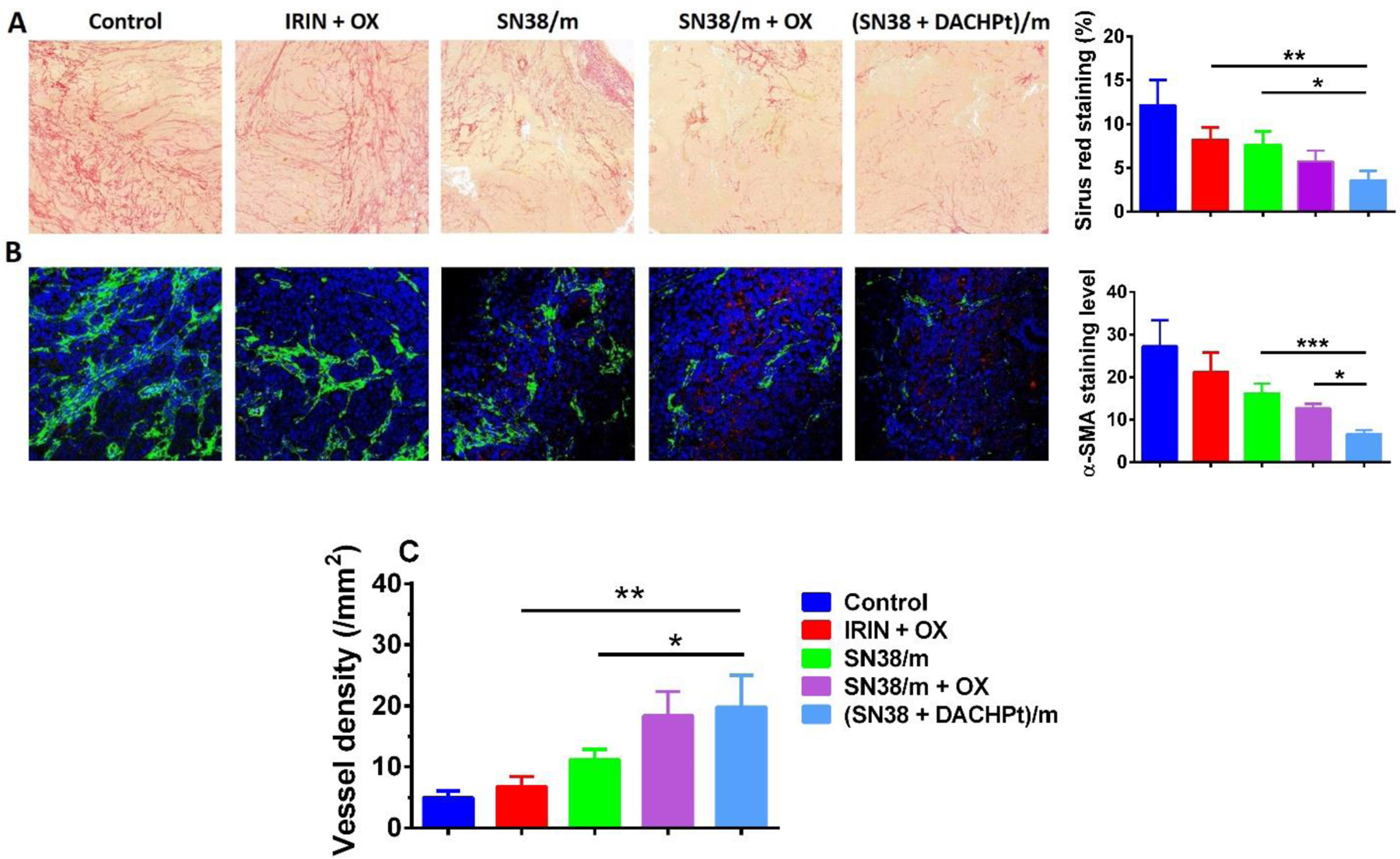
Morphological changes in CD18/HPAF after different treatments. (A) Sirus Red staining of the collagen and quantification of staining; (B) IHC staining of α-SMA (green) and blood vessel (CD31, red) with nuclear counterstain (blue), expression levels were quintified using ImageJ software (random fields for each tumor section, n = 8); (C) density of the intact and closed blood vessels with lumen > 5 mm were quantified using Definiens Tissue Studio software. *p < 0.05, **p<0.01, ***p<0.001.
Except IRIN + OX group, mice treated with micelle-based formulations barely showed body weight loss during the treatment suggesting that they were well tolerated. Yet, analysis of blood samples collected after final dosing (day 14) showed a substantial decrease in WBCs count in animals treated with SN38/m plus free OX as well as in IRIN + OX group manifesting acute hematological toxicity (Table S2). In addition, (IRIN + OX)- treated animals demonstrated some degree of hepatotoxicity as elevated ALP levels were detected in the blood samples. Both of these toxicities, leucopenia and hepatotoxicity, are commonly associated with IRIN- and OX-based regimens. Importantly, albeit much higher platinum exposure of liver, spleen and kidney in the (SN38 + DACHPt)/m treatment group relative to free OX treatments (Figure 6D), the serum levels of markers of hepatic and renal toxicity remained within normal range. Furthermore, histological examination of H&E-stained tissue sections (liver, spleen, kidney, and lung) from animals sacrificed at the experimental endpoint (day 36) did not show any evidence of toxicity, providing additional proof that (SN38 + DACHPt)/m formulation was well-tolerated by the animals (Figure S8). Overall, our proof-of-concept studies demonstrated that polymeric micelles co-loaded with SN38 and DACHPt at synergistic drug ratio exerted an enhanced antitumor activity compared not only with the clinically used free drug combination but also with the mixture of the SN38-loaded micelles and free oxaliplatin administered at the same dose.
4. Conclusions.
In summary, we have engineered a drug carrier based on biodegradable polypeptide-based micelles formed by polymer-SN38 conjugates and loaded with DACHPt that was specifically designed to address the current limitations of intensive, multimodality FOLFIRINOX therapy of pancreatic cancer. These studies emphasized the importance of both drugs in the same polymeric carrier and further shown that therapy based on co-loaded micelles effectively suppresses tumor growth and prevents the development of metastasis in an orthotopic model of pancreatic cancer outperforming the delivery of clinically used free drug combination. The combination of improved therapeutic outcomes and reduced systemic toxicity by nanomedicines mimicking Irinotecan and Oxaliplatin regimen warranted its further evaluation in combination with 5-fluorouracil and need to be validated in other tumor models. It would be particularly interesting to establish the activity of the designed formulations in a genetically engineered mouse models that recapitulate the real setting of the disease and allows testing in immunooncological context. Research in these directions is currently underway in our laboratories.
Supplementary Material
Acknowledgements
TKB would like to thank Professor Henry Kopecek, to whom this special issue is dedicated to, for inspiring her as well as several generations of researchers in the field of biomedical engineering and therapeutics, for leading by example of passion and creativity and for outstanding contributions that helped define the field. This work was partly supported by a Cancer Nanotechnology Platform Partnership grant (U01 CA198910) of the National Cancer Institute Alliance for Nanotechnology in Cancer (TKB) and (CA183459) from the National Cancer Institute to SKB. We acknowledge the assistance of the Nanomaterials Core Facility of the Center for Biomedical Research Excellence (CoBRE), Nebraska Center for Nanomedicine supported by the Institutional Development Award from the National Institutes of General Medical Sciences (P30GM127200) and are grateful to the confocal microscopy core facility at University of Nebraska Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Pancreatic Cancer: Statistics, Cancer.net, https://www.cancer.net/cancer-types/pancreatic-cancer/statistics, (2020).
- [2].Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM, Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States, Cancer research, 74 (2014) 2913–2921. [DOI] [PubMed] [Google Scholar]
- [3].A.C. Society, Cancer Facts and Figures 2020 [Google Scholar]
- [4].Balaban EP, Mangu PB, Yee NS, Locally Advanced Unresectable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline Summary, Journal of oncology practice, 13 (2016) 265–269. [DOI] [PubMed] [Google Scholar]
- [5].Seufferlein T, Bachet J, Van Cutsem E, Rougier P, Group EGW, Pancreatic adenocarcinoma: ESMO–ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up, Annals of oncology, 23 (2012) vii33–vii40. [DOI] [PubMed] [Google Scholar]
- [6].Soni KS, Thomas D, Caffrey T, Mehla K, Lei F, O’Connell KA, Sagar S, Lele SM, Hollingsworth MA, Radhakrishnan P, A polymeric nanogel-based treatment regimen for enhanced efficacy and sequential administration of synergistic drug combination in pancreatic cancer, Journal of Pharmacology and Experimental Therapeutics, 370 (2019) 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vaccaro V, Sperduti I, Milella M, FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer, New England Journal of Medicine, 365 (2011) 768–769. [DOI] [PubMed] [Google Scholar]
- [8].Suker M, Beumer BR, Sadot E, Marthey L, Faris JE, Mellon EA, El-Rayes BF, Wang-Gillam A, Lacy J, Hosein PJ, Moorcraft SY, Conroy T, Hohla F, Allen P, Taieb J, Hong TS, Shridhar R, Chau I, van Eijck CH, Koerkamp BG, FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis, The Lancet Oncology, 17 (2016) 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shi J, Kantoff PW, Wooster R, Farokhzad OC, Cancer nanomedicine: progress, challenges and opportunities, Nature Reviews Cancer, 17 (2017) 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chiang N-J, Chao T-Y, Hsieh R-K, Wang C-H, Wang Y-W, Yeh CG, Chen L-T, A phase I dose-escalation study of PEP02 (irinotecan liposome injection) in combination with 5-fluorouracil and leucovorin in advanced solid tumors, BMC cancer, 16 (2016) 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang-Gillam A, Li C-P, Bodoky G, Dean A, Shan Y-S, Jameson G, Macarulla T, Lee K-H, Cunningham D, Blanc JF, Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial, The Lancet, 387 (2016) 545–557. [DOI] [PubMed] [Google Scholar]
- [12].Lei F, Xi X, Batra SK, Bronich TK, Combination therapies and drug delivery platforms in combating pancreatic cancer, Journal of Pharmacology and Experimental Therapeutics, 370 (2019) 682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tripathy D, Tolaney SM, Seidman AD, Anders CK, Ibrahim N, Rugo HS, Twelves C, Dieras V, Müller V, Tagliaferri M, ATTAIN: Phase III study of etirinotecan pegol versus treatment of physician’s choice in patients with metastatic breast cancer and brain metastases, Future Oncology, 15 (2019) 2211–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Burris HA, Infante JR, Greco FA, Thompson DS, Barton JH, Bendell JC, Nambu Y, Watanabe N, Jones SF, A phase I dose escalation study of NK012, an SN-38 incorporating macromolecular polymeric micelle, Cancer chemotherapy and pharmacology, 77 (2016) 1079–1086. [DOI] [PubMed] [Google Scholar]
- [15].Hamaguchi T, Tsuji A, Yamaguchi K, Takeda K, Uetake H, Esaki T, Amagai K, Sakai D, Baba H, Kimura M, A phase II study of NK012, a polymeric micelle formulation of SN-38, in unresectable, metastatic or recurrent colorectal cancer patients, Cancer chemotherapy and pharmacology, 82 (2018) 1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cabral H, Kataoka K, Progress of drug-loaded polymeric micelles into clinical studies, Journal of Controlled Release, 190 (2014) 465–476. [DOI] [PubMed] [Google Scholar]
- [17].Desale SS, Cohen SM, Zhao Y, Kabanov AV, Bronich TK, Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer, Journal of Controlled Release, 171 (2013) 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Oberoi HS, Nukolova NV, Zhao Y, Cohen SM, Kabanov AV, Bronich TK, Preparation and in vivo evaluation of dichloro (1, 2-diaminocyclohexane) platinum (II)-loaded core cross-linked polymer micelles, Chemotherapy research and practice, 2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aliabadi HM, Brocks DR, Mahdipoor P, Lavasanifar A, A novel use of an in vitro method to predict the in vivo stability of block copolymer based nano-containers, Journal of controlled release, 122 (2007) 63–70. [DOI] [PubMed] [Google Scholar]
- [20].Soni KS, Lei F, Desale SS, Marky LA, Cohen SM, Bronich TK, Tuning polypeptide-based micellar carrier for efficient combination therapy of ErbB2-positive breast cancer, Journal of controlled release: official journal of the Controlled Release Society, 264 (2017) 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schuhmacher J, Bühner K, Witt-Laido A, Determination of the Free Fraction and Relative Free Fraction of Drugs Strongly Bound to Plasma Proteins, Journal of Pharmaceutical Sciences, 89 (2000) 1008–1021. [DOI] [PubMed] [Google Scholar]
- [22].Han Y, He Z, Schulz A, Bronich TK, Jordan R, Luxenhofer R, Kabanov AV, Synergistic combinations of multiple chemotherapeutic agents in high capacity poly (2-oxazoline) micelles, Molecular pharmaceutics, 9 (2012) 2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Seshacharyulu P, Ponnusamy MP, Rachagani S, Lakshmanan I, Haridas D, Yan Y, Ganti AK, Batra SK, Targeting EGF-receptor (s)-STAT1 axis attenuates tumor growth and metastasis through downregulation of MUC4 mucin in human pancreatic cancer, Oncotarget, 6 (2015) 5164–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bafna S, Singh AP, Moniaux N, Eudy JD, Meza JL, Batra SK, MUC4, a multifunctional transmembrane glycoprotein, induces oncogenic transformation of NIH3T3 mouse fibroblast cells, Cancer research, 68 (2008) 9231–9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kaushik G, Ponnusamy MP, Batra SK, Concise Review: Current Status of Three-Dimensional Organoids as Preclinical Models, Stem Cells, 36 (2018) 1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Uz M, Kalaga M, Pothuraju R, Ju J, Junker WM, Batra SK, Mallapragada S, Rachagani S, Dual delivery nanoscale device for miR-345 and gemcitabine co-delivery to treat pancreatic cancer, Journal of Controlled Release, 294 (2019) 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S.-i., Hamaguchi T, Matsumura Y, Novel SN-38–incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor–secreting bulky tumors, Cancer research, 66 (2006) 10048–10056. [DOI] [PubMed] [Google Scholar]
- [28].Baddar FG, Abdel Wahhab S, Awad Boshra M., Guindy Nabila M., Wahba M, Malek BAA, Phenolic Esters. Part 1. Rate of Hydrolysis of Phenyl, p-Tolyi, and p-Chlorophenyl Hydrogen Succinate, Journal of the Chemical Society B: Physical Organic, (1970) 739–745. [Google Scholar]
- [29].Zeghari-Squalli N, Raymond E, Cvitkovic E, Goldwasser F, Cellular pharmacology of the combination of the DNA topoisomerase I inhibitor SN-38 and the diaminocyclohexane platinum derivative oxaliplatin, Clinical cancer research, 5 (1999) 1189–1196. [PubMed] [Google Scholar]
- [30].Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K, Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11, Cancer research, 51 (1991) 4187–4191. [PubMed] [Google Scholar]
- [31].Sahay G, Alakhova DY, Kabanov AV, Endocytosis of nanomedicines, Journal of controlled release, 145 (2010) 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guo P, Liu D, Subramanyam K, Wang B, Yang J, Huang J, Auguste DT, Moses MA, Nanoparticle elasticity directs tumor uptake, Nature communications, 9 (2018) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Boj SF, Hwang C-I, Baker LA, Chio IIC, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Organoid models of human and mouse ductal pancreatic cancer, Cell, 160 (2015) 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, van Boxtel R, Huch M, Rivera KD, Wilson JP, Feigin ME, Ohlund D, Handly-Santana A, Ardito-Abraham CM, Ludwig M, Elyada E, Alagesan B, Biffi G, Yordanov GN, Delcuze B, Creighton B, Wright K, Park Y, Morsink FH, Molenaar IQ, Borel Rinkes IH, Cuppen E, Hao Y, Jin Y, Nijman IJ, Iacobuzio-Donahue C, Leach SD, Pappin DJ, Hammell M, Klimstra DS, Basturk O, Hruban RH, Offerhaus GJ, Vries RG, Clevers H, Tuveson DA, Organoid models of human and mouse ductal pancreatic cancer, Cell, 160 (2015) 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Boj SF, Hwang CI, Baker LA, Engle DD, Tuveson DA, Clevers H, Model organoids provide new research opportunities for ductal pancreatic cancer, Mol Cell Oncol, 3 (2016) e1014757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ehrsson H, Wallin I, Yachnin J, Pharmacokinetics of oxaliplatin in humans, Medical oncology, 19 (2002) 261–265. [DOI] [PubMed] [Google Scholar]
- [37].Graham Martin A., Lockwood Graham F., Greenslade Dennis, Brienza Silvano, Bayssas Martine, Gamelin E, Clinical Pharmacokinetics of Oxaliplatin: A Critical Review, Clin Cancer Res, 6 (2000) 1205–1218. [PubMed] [Google Scholar]
- [38].Kota J, Hancock J, Kwon J, Korc M, Pancreatic cancer: Stroma and its current and emerging targeted therapies, Cancer Lett, 391 (2017) 38–49. [DOI] [PubMed] [Google Scholar]
- [39].Zhang B, Hu Y, Pang Z, Modulating the Tumor Microenvironment to Enhance Tumor Nanomedicine Delivery, Front Pharmacol, 8 (2017) 952–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas P, Megias D, Guerra C, Muñoz M, Quijano Y, Cubillo A, Rodriguez-Pascual J, Stromal disrupting effects of nab-paclitaxel in pancreatic cancer, British journal of cancer, 109 (2013) 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Verreault M, Wehbe M, Strutt D, Masin D, Anantha M, Walker D, Chu F, Backstrom I, Kalra J, Waterhouse D, Determination of an optimal dosing schedule for combining Irinophore C™ and temozolomide in an orthotopic model of glioblastoma, Journal of Controlled Release, 220 (2015) 348–357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.