

Abstract
Streptococcus pneumoniae is the leading cause of hospital community-acquired pneumonia. Patients with pneumococcal pneumonia may develop complicated parapneumonic effusions or empyema that can lead to pleural organization and subsequent fibrosis. The pathogenesis of pleural organization and scarification involves complex interactions between the components of the immune system, coagulation, and fibrinolysis. EPCR (endothelial protein C receptor) is a critical component of the protein C anticoagulant pathway. The present study was performed to evaluate the role of EPCR in the pathogenesis of S. pneumoniae infection–induced pleural thickening and fibrosis. Our studies show that the pleural mesothelium expresses EPCR. Intrapleural instillation of S. pneumoniae impairs lung compliance and lung volume in wild-type and EPCR-overexpressing mice but not in EPCR-deficient mice. Intrapleural S. pneumoniae infection induces pleural thickening in wild-type mice. Pleural thickening is more pronounced in EPCR-overexpressing mice, whereas it is reduced in EPCR-deficient mice. Markers of mesomesenchymal transition are increased in the visceral pleura of S. pneumoniae–infected wild-type and EPCR-overexpressing mice but not in EPCR-deficient mice. The lungs of wild-type and EPCR-overexpressing mice administered intrapleural S. pneumoniae showed increased infiltration of macrophages and neutrophils, which was significantly reduced in EPCR-deficient mice. An analysis of bacterial burden in the pleural lavage, the lungs, and blood revealed a significantly lower bacterial burden in EPCR-deficient mice compared with wild-type and EPCR-overexpressing mice. Overall, our data provide strong evidence that EPCR deficiency protects against S. pneumoniae infection–induced impairment of lung function and pleural remodeling.
Keywords: endothelial protein C receptor, mesothelium, pleural fibrosis, Streptococcus pneumoniae
Pleural infection is a common and increasing clinical problem that results in significant morbidity and mortality (1). It occurs most frequently in association with pneumonia (2). Streptococcus pneumoniae is the leading cause of bacterial related parapneumonic pleural space infections that result from community-acquired pneumonia (3). About 4.5 million people are affected by community-acquired pneumonia yearly in the United States alone, and it is the ninth leading cause of death when combined with influenza (4). About 20–40% of patients with pneumonia develop pleural effusions, and 10% of them develop complicated parapneumonic effusion or empyema (2). Complicated parapneumonic effusions may develop into a multiloculated fibrinopurulent collection, often leading to the pleural organization with subsequent pleural thickening and scarring (5). If advanced, pleural scarification can lead to restrictive lung disease and morbidity. The current understanding of the pathogenesis of pleural organization remains incomplete. However, complex interactions between resident and inflammatory cells, profibrotic mediators, coagulation, and fibrinolytic pathways are believed to be integral to pleural remodeling and fibrosis (6). Recent studies showed that proteases generated in coagulation and fibrinolytic pathways are capable of inducing mesomesenchymal transition (MesoMT) of pleural mesothelial cells, which contributes to pleural scarification (7, 8).
EPCR (endothelial protein C receptor) is a type I transmembrane receptor that augments protein C activation by thrombin-thrombomodulin complexes and promotes anticoagulation (9). EPCR was also shown to play a key role in mediating APC (activated protein C) and FVIIa (factor VIIa)-induced PAR1 (protease-activated receptor 1)-dependent cell signaling that elicits antiinflammatory and vascular barrier protective effects (10–13). In addition to APC and FVIIa, EPCR can also bind other ligands, including Mac-1 on neutrophils indirectly through its association with PR3 (proteinase-3) (14) or directly with Mac-1 on monocytes (15), TCR (T-Cell receptor) present on a subset of γδ T cells (16), and pfEMP1 (Plasmodium falciparum erythrocyte membrane protein 1) on red blood cells that are infected by the malarial parasite P. falciparum (17). The above observations raise the possibility that EPCR mediates novel signaling pathways and could affect various cellular processes that are yet to be identified (13).
EPCR expression was originally reported to be limited to the endothelium of large and microvascular blood vessels, liver sinusoidal endothelium, and spleen (18, 19). However, recent studies have shown that EPCR is also expressed on the surface of many different cell types, such as vascular smooth muscle cells, cardiomyocytes, neutrophils, monocytes, and hematopoietic stem cells (20). At present, there is no direct evidence that pleural mesothelial cells express EPCR. However, primary rabbit pleural mesothelial cells (PMC) isolated from the rabbit pleural effusions or normal rabbit lung tissue were shown to support thrombin-mediated activation of protein C (21), suggesting that pleural mesothelial cells are likely to express EPCR.
This study was designed to investigate the role of EPCR in the pathogenesis of S. pneumoniae–induced pleural infection and its impact on pleural organization and fibrosis. We found that EPCR deficiency confers a protective effect against S. pneumoniae–induced pleural fibrosis and lung impairment. Furthermore, our in vitro and in vivo data suggests that mesothelial EPCR promotes internalization of S. pneumoniae and thus shields it from antibiotics, improving its chances of survival and dissemination of the pathogen. Overall, our findings indicate that EPCR contributes to the pathogenesis of pleural remodeling after an S. pneumoniae–induced pleural injury.
Methods
Details and expanded methodology are included in the data supplement.
Primary Human PMC and Pleural Tissue Samples
Isolation, characterization, and culture of human PMC (HPMCs) from patients with congestive heart failure or postcoronary artery bypass pleural effusions were described previously (7, 22). Deidentified human lung tissues were obtained from the National Disease Research Interchange from surgical biopsies or autopsy specimens from patients with histologically near-normal pleural tissues, who died of causes otherwise unrelated to any pleural pathologic process. The Institutional Review Board at the University of Texas Health Science Center at Tyler approved the collection of pleural effusions to isolate HPMCs. The mesothelial origin of these cells was confirmed by the detection of the mesothelial marker, calretinin.
Mice
All animal procedures were approved by the Institutional Animal Use and Care Committee at the University of Texas Health Science Center at Tyler. The generation of EPCR-deficient mice (Procr−/−) and Tie2-mediated EPCR-overexpressing mice (Tie2-EPCR) was described previously (23, 24). Wild-type (WT) (C57BL/6J) mice were obtained from Jackson Laboratories or bred in-house. Mice aged 8–13 weeks, both male and female, were used for our studies. There were no discernible phenotypic differences between male and female mice except for weight (males, 25.02 ± 0.29 g; females, 20.02 ± 0.14 g). There were no significant differences among the genotypes in body weights.
Pleural Fibrosis Model
Pleural fibrosis was induced by intrapleural inoculation of S. pneumoniae (strain D39, capsular serotype 2, obtained from National Collection of Type Cultures) as described earlier with a few minor modifications (8, 25, 26). Briefly, mice were anesthetized with isoflurane gas, and S. pneumoniae were inoculated into the right pleural space by injecting 150 μl bacterial suspension (1.8 × 108–2.0 × 108 cfu/mouse) between the sixth and ninth rib using a 1-cc syringe with a 25-gauge, 5/8-inch needle. The control group received saline under the same conditions. Antibiotic treatment (ampicillin, 100 mg/kg) was initiated 4 hours after infection and was administered daily (single dose) by subcutaneous injection for 4 days. After the bacteria inoculation, mice were monitored periodically for signs of dehydration, weight loss, activity, posture, response, and behavior. Mice appearing dehydrated (detected by alterations of skin turgor) were subcutaneously injected with 200–500 μl warm 0.9% saline, as needed. Moribund animals were killed. After the administration of S. pneumoniae, mice were housed on a heating pad to maintain an ambient temperature of approximately 30°C.
Data Analysis
Statistical significance among the groups was analyzed by one-way ANOVA followed by Bonferroni multiple comparison test. When comparing two groups, the Mann-Whitney test was used to determine statistical significance. The values of each parameter within a group were expressed as the mean ± SEM. As we did not observe any discernible differences between male and female mice in their response to pleural S. pneumoniae infection, we combined the data from males and females for robust analysis.
Results
EPCR Expression in Pleural Mesothelium
To determine if EPCR is expressed on the healthy pleural mesothelium, normal human lung tissue sections were stained with EPCR-specific polyclonal antibodies. Robust expression of EPCR was seen on the normal pleural mesothelium covering the visceral surface of the lung. On the visceral pleura, EPCR expression was localized to the single layer of mesothelial cells (Figure 1A). As expected, robust EPCR expression was seen on the endothelium of blood vessels. EPCR expression in other lung cell types was very faint (Figure 1A). Next, we evaluated the expression of EPCR by Western blotting in primary HPMCs isolated from the pleural fluids collected from patients with congestive heart failure or after a coronary bypass surgery. EPCR was readily detectable in HPMCs isolated from both patients (Figure 1B). We also examined the expression of EPCR in various human pleural mesothelial cell lines (MeT-5A, MS-1, and M9K) and lung epithelial cell lines (A549 and Beas2B). All expressed EPCR in varying amounts (Figure 1C).
Figure 1.

The mesothelium expresses EPCR (endothelial protein C receptor). (A) Normal human lung tissue sections were stained with goat control IgG or goat antihuman EPCR antibodies. Scale bars, 100 µm. The mesothelium is indicated by black arrows. (B) Primary human mesothelial cells, isolated from patients with congestive heart failure or after coronary bypass pleural effusions, were grown to confluency. Cell lysates were prepared and subjected to immunoblot analysis and probed with an antihuman EPCR monoclonal antibody (JRK 1489). Lysates of immortalized, noncancerous, human mesothelial cell lines, MeT-5A and human mesothelial cells (HMC), were used as additional cell sources for mesothelial cell origin. (C) Lysates of different cell lines derived from the lungs were probed for EPCR expression by immunoblot analysis. Human umbilical vein endothelial cell (HUVEC) lysate was used as a positive control. (D) Lung tissue sections from wild-type mice were immunostained with goat control IgG or goat antimurine EPCR antibodies (top panel) or rabbit control IgG or rabbit antimurine tissue factor antibodies (bottom panel). EPCR staining and TF staining is indicated by black arrows. Scale bars, 20 µm. A549 = adenocarcinomic human alveolar basal epithelial cells; Beas2B = human bronchial epithelial cell line; BV = blood vessel; Hu = human; Meso. = mesothelial; MeT-5A = human pleural mesothelial cells; M9K and MS-1 = human mesothelion cell line; TF = tissue factor.
Next, we assessed the expression of EPCR of normal mouse pleural tissue. Immunohistochemical analysis showed robust expression of EPCR on the mesothelium, like that observed in humans. Immunostaining of tissue factor (TF), a key procoagulant cofactor that triggers the activation of the coagulation cascade, showed that unperturbed mesothelial cells do not express detectable levels of TF. TF expression was restricted to the submesothelial layer and epithelial cells (Figure 1D).
EPCR Deficiency Protects against Impairment in Pulmonary Function in the S. pneumoniae–induced Pleural Empyema Model
S. pneumoniae–induced injury and organization were well standardized and characterized by our earlier studies (8, 25, 26). To initiate the investigation of the role of EPCR in pleural fibrosis, we used WT, EPCR-deficient, and Tie2-EPCR mice. Healthy mice across all genotypes were randomly assorted into two groups, the control group or the S. pneumoniae group. Control group mice were injected with saline (150 μl) into the pleural cavity, whereas S. pneumoniae group mice received S. pneumoniae in log-phase growth (1.8 × 108–2.0 × 108 cfu/mouse in 150 μl saline). Four hours after the infection, both groups of mice were treated with the first dose of ampicillin (100 mg/kg body weight), after which ampicillin was given once daily for 4 days. Mice were observed daily, and mouse sickness scores (MSS) were calculated based on the scoring criteria described in Table E1 in the data supplement. Scores were evaluated by two independent observers, blinded to the information on treatments and genotypes, to avoid biased conclusions. At 24 hours after infection, all mice inoculated with S. pneumoniae appeared very sick across all three genotypes with MSS scores exceeding the saline counterparts (Figure 2A). Weight loss was maximum across the genotypes at 48 hours after infection (Figure 2B). Infected mice regained most of the lost weight by Day 7. Minor differences in MSS and weight loss among the three genotypes were not statistically significant.
Figure 2.
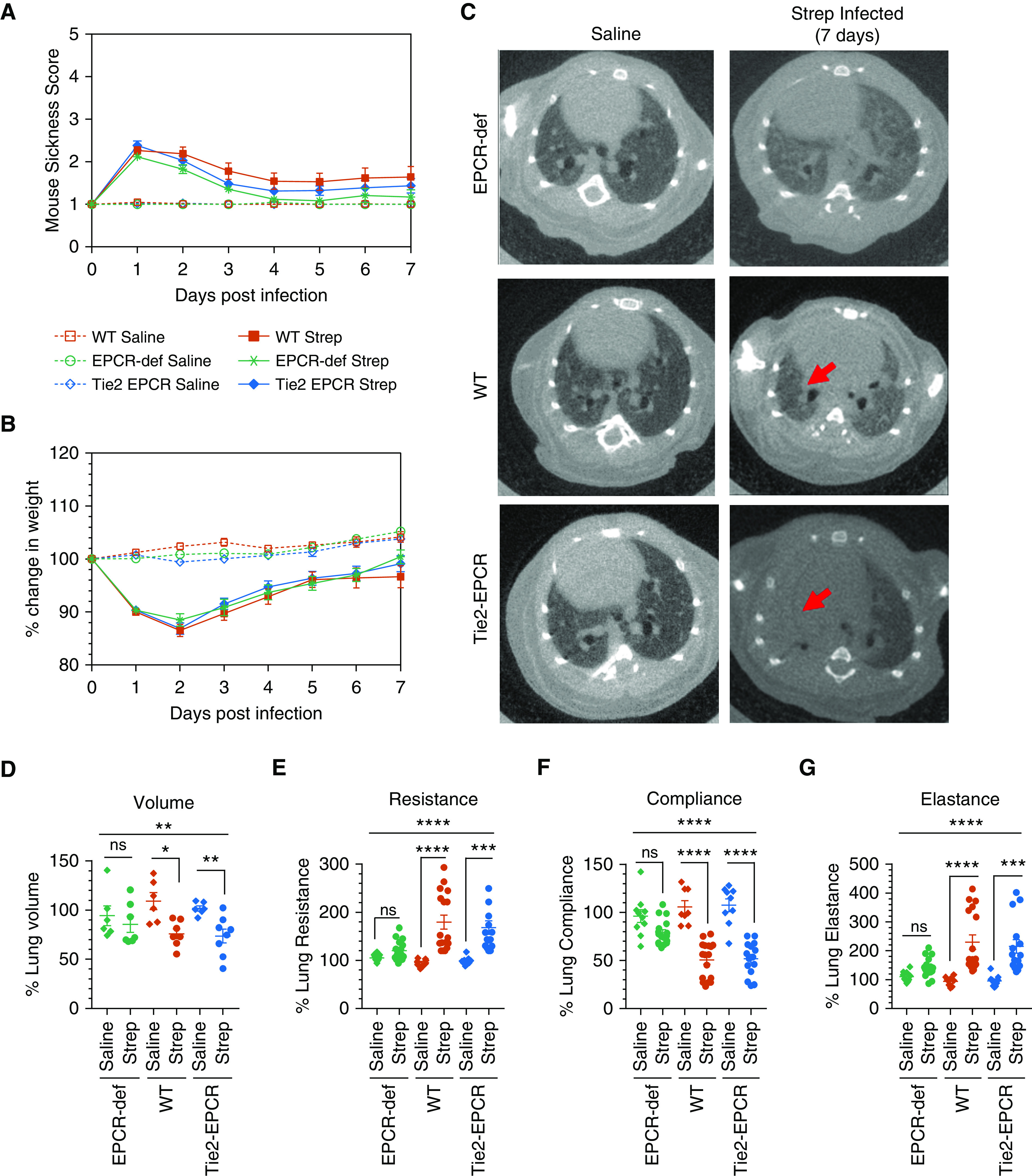
EPCR deficiency attenuates the impairment of pulmonary function in mice infected with Streptococcus pneumoniae. WT, Tie2-EPCR, and EPCR-deficient mice were inoculated with S. pneumoniae intrapleural. As a control, another group of mice was injected with intrapleural saline. Antibiotic treatment was initiated 4 hours after infection and given once daily for 4 days. The disease was allowed to progress for 7 days. Mice were monitored daily, and at the end of the seventh day after the infection, they were subjected to pulmonary functions testing and computed tomography imaging. (A) Mouse sickness score was determined daily for 7 days by monitoring fur, eyes, physical activity, posture, response, respiration, and weight loss, as described in Table E1 (saline, n = 10–13 mice per group; S. pneumoniae infection, n = 18–23 mice per group). (B) Percentage change in weight of saline-administered or S. pneumoniae–infected mice for 7 days. (C) Computed tomography imaging of the thoracic cavity of mice at Day 7 after infection. The red arrows indicate areas of pleural density +/− parenchymal atelectasis or lung infiltrates. (D) Lung volumes were calculated from three-dimensional renditions collected at full inspiration (renditions from n = 6–8 mice per group for both saline and S. pneumoniae–infected groups were analyzed). (E–G) Anesthetized mice were intubated with a 20-gauge intravenous cannula into the trachea, and other lung parameters, including resistance (E), compliance (F), and elastance (G), were obtained by the “snapshot perturbation method” using the flexiVent system (saline, n = 8–10 mice per group; S. pneumoniae infection, n = 15–17 mice per group). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. EPCR-def = EPCR-deficient; ns = no statistically significant difference; Strep = Streptococcus; WT = wild type.
After 7 days, both saline-administered and S. pneumoniae–infected mice were anesthetized and subjected to chest computed tomography scan imaging. The lungs of mice injected with saline appeared normal, whereas the lungs of S. pneumoniae–infected mice exhibited pleural effusions (Figure 2C). Parenchymal lung consolidation was generally more pronounced in the WT and Tie2-EPCR mice compared with EPCR-deficient mice (Figure 2C). Determination of lung volumes from the three-dimensional renditions of the computed tomography images showed a significant decrease in the lung volume in S. pneumoniae–infected WT (P < 0.05) and Tie2-EPCR mice (P < 0.01) compared with the lung volume of saline-injected mice of the corresponding genotype. By contrast, we found no significant decrease in the lung volumes of EPCR-deficient mice infected with S. pneumoniae compared with the saline control mice (P = 0.51; Figure 2D). These data suggest that EPCR plays an important role in the progression of pleural fibrosis initiated by S. pneumoniae infection.
Pulmonary function testing showed evidence of lung restriction and decreased compliance in S. pneumoniae–infected WT and Tie2-EPCR but not EPCR-deficient mice. Both WT and Tie2-EPCR mice infected with S. pneumoniae exhibited significantly reduced lung compliance (P < 0.0001) over their saline counterparts. There was no significant difference in the lung compliance of S. pneumoniae–infected and saline-administered EPCR-deficient mice (Figure 2F). S. pneumoniae–infected EPCR-deficient mice did not show any significant differences in lung resistance and elastance compared with their saline counterparts. S. pneumoniae infection significantly increased lung resistance and elastance in WT and Tie2-EPCR mice compared with their saline-treated control mice (Figures 2E and 2G).
EPCR-Overexpressing Mice Exhibit Increased Pleural Thickening in S. pneumoniae–induced Pleuritis
Gross examination of the pleural cavities of mice inoculated with S. pneumoniae showed gross pleural erythema and purulence on Day 7 after infection in WT and Tie2-EPCR mice. Histology of the lungs of mice infected with S. pneumoniae showed increased pleural thickening compared with the lungs of control mice treated with saline. Although pleural thickening was clearly evident in all infected WT mice, it was strikingly increased in Tie2-EPCR mice infected with S. pneumoniae (Figure 3A). Pleural thickening in S. pneumoniae–infected EPCR-deficient mice was minimal (pleural thickness: S. pneumoniae–infected mice, 16.15 ± 2.62 μm; saline control, 5.07 ± 0.234 μm) (Figures 3A and 3B). In EPCR-deficient infected mice, the visceral mesothelium was still single layered (Figure 3A). In contrast to EPCR-deficient mice, lungs of infected WT mice showed significantly increased pleural thickening (pleural thickness in S. pneumoniae–infected WT mice, 77.77 ± 6.908 μm; saline-administered mice, 4.81 ± 0.2 μm; P < 0.0001). The most dramatic increase in pleural thickness after the infection was observed in Tie2-EPCR mice (pleural thickness in S. pneumoniae–infected mice, 168.8 ± 17.74 μm; saline-administered mice, 5.59 ± 0.221 μm). The pleural thickening in WT and Tie2-EPCR mice was associated with extensive infiltrating inflammatory cells, including neutrophils (Figure 3A, insets). These results clearly suggest that EPCR expression influences pleural thickening after pleural infection.
Figure 3.
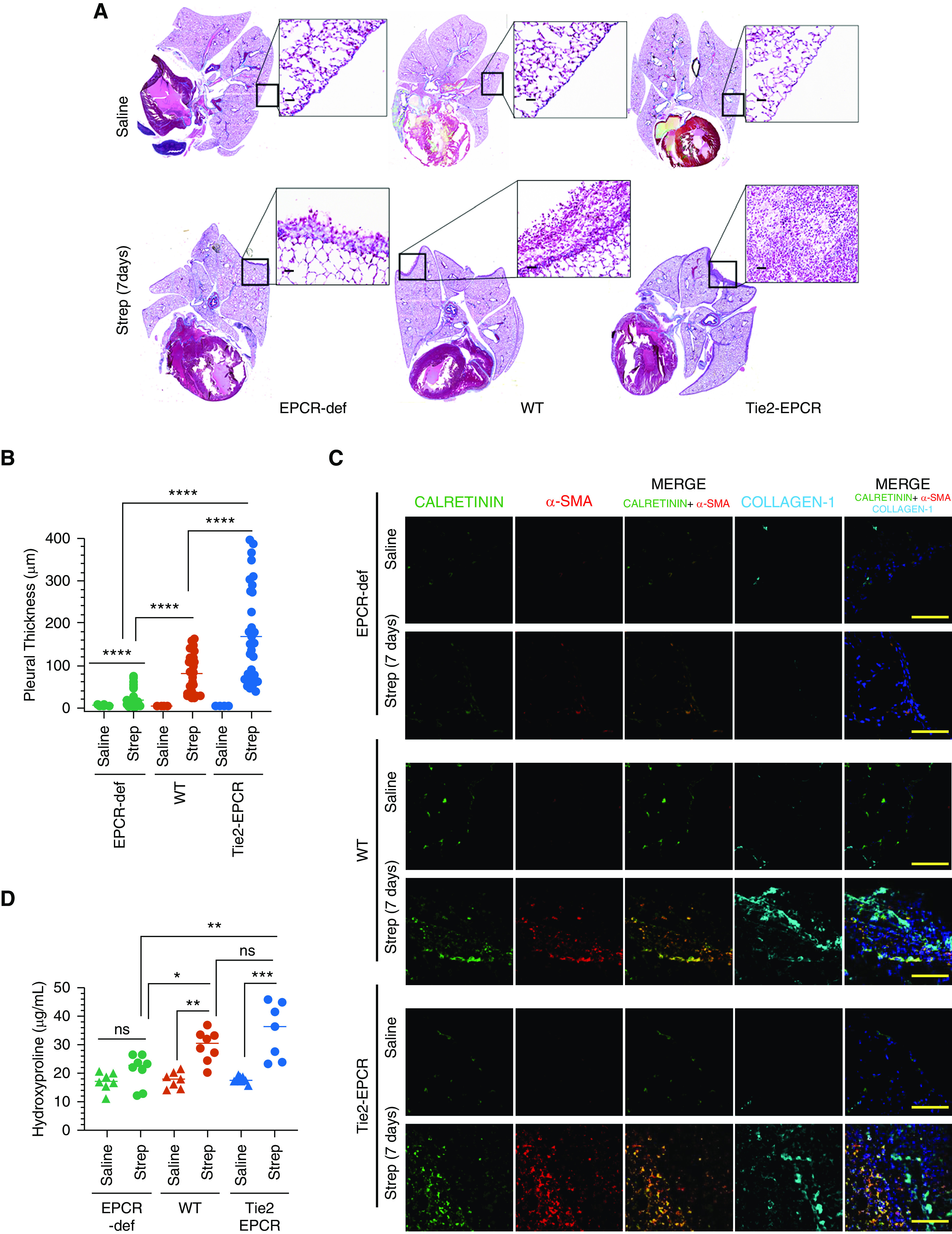
EPCR promotes pleural thickening induced by S. pneumoniae infection. WT, EPCR-overexpressing Tie2-EPCR, or EPCR-def mice were administered with saline or S. pneumoniae intrapleural. The lungs were harvested 7 days after infection. (A) Masons-Trichrome staining of lung tissue sections to assess collagen deposition. (B) Pleural thickening, as measured microscopically at random places covering the entire lungs using Nikon digital sight DS-Fi1 camera (Nikon Instruments) and NIS elements BR 3.2 (Nikon Instruments) software (number of fields examined per group, 34–44). (C) Immunofluorescence staining of lung tissue sections from saline-administered or S. pneumoniae–infected mice for calretinin (as a mesothelial marker), α-smooth muscle actin (α-SMA), and collagen-1. The images of Calretinin were merged with the images of α-SMA (middle column) or α-SMA+collagen (last column on the right). Scale bars, 100 µm. (D) Hydroxyproline content in the lung tissues of saline and S. pneumoniae–infected mice (7 d). Scale bars, 20 µm. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Picrosirius red staining of the lungs of mice infected with S. pneumoniae suggested increased collagen deposition in the thickened pleura of both WT and Tie2-EPCR mice (Figure E1), which was minimal or absent in EPCR-deficient mice. Collagen deposition in the pleura was also significantly higher in Tie2-EPCR mice compared with WT mice. Consistent with the above data, confocal image analysis of the infected lungs of WT and Tie2-EPCR mice revealed robust expression of α-smooth muscle actin (α-SMA) (red), which was colocalized with the mesothelial marker, calretinin (green), in the injured pleura (Figure 3C). On the contrary, S. pneumoniae–infected lungs of EPCR-deficient mice showed little α-SMA expression (Figure 3C). No α-SMA was observed in the lungs of saline-treated mice of all three genotypes (Figure 3C). In addition to α-SMA, an extensive collagen-1 deposition was also seen in the visceral pleura in both S. pneumoniae–infected WT and Tie2-EPCR mice lungs. No collagen-1 deposition was seen in the pleura of EPCR-deficient mice infected intrapleural with S. pneumoniae (Figure 3D). Measurement of hydroxyproline content, which accurately reflects the amount of collagen, in the peripheral lung tissue showed no significant increase in hydroxyproline content in S. pneumoniae–infected EPCR-deficient mice over sham-infected EPCR-deficient mice. In contrast, hydroxyproline content was significantly higher in the lung tissues of S. pneumoniae–infected WT and Tie2-EPCR mice compared with respective control mice (Figure 3D).
EPCR Deficiency Significantly Reduces Infiltration of Neutrophils and Macrophages in Pleural Fibrosis
In response to injury, such as bacterial infection, PMC releases several proinflammatory cytokines, which results in an influx of a large number of inflammatory cells (27). These inflammatory cells play a major role in the pathogenesis of pleural fibrosis (27). There was extensive infiltration of neutrophils into the thickened pleura of S. pneumoniae–infected WT and Tie2-EPCR mice but not in EPCR-deficient mice (Figure 4A). The extent of neutrophil infiltration into the fibrotic area of the lungs in infected Tie2-EPCR mice (137.2 ± 12.96 cells/field) was significantly greater than WT mice (89.05 ± 8.86 cells/field; P < 0.001) (Figure 4A). Neutrophil infiltration into the lungs of S. pneumoniae–infected EPCR-deficient mice was minimal (26.13 ± 7.38). Similar to neutrophil infiltration, macrophage infiltration was also significantly higher in pleural fibrotic areas of infected WT (222.6 ± 19.8 cells/field) and Tie2-EPCR mice (296.3 ± 23.52 cells/field) compared with EPCR-deficient mice (85.8 ± 10 cells/field) (P < 0.001) (Figure 4B).
Figure 4.
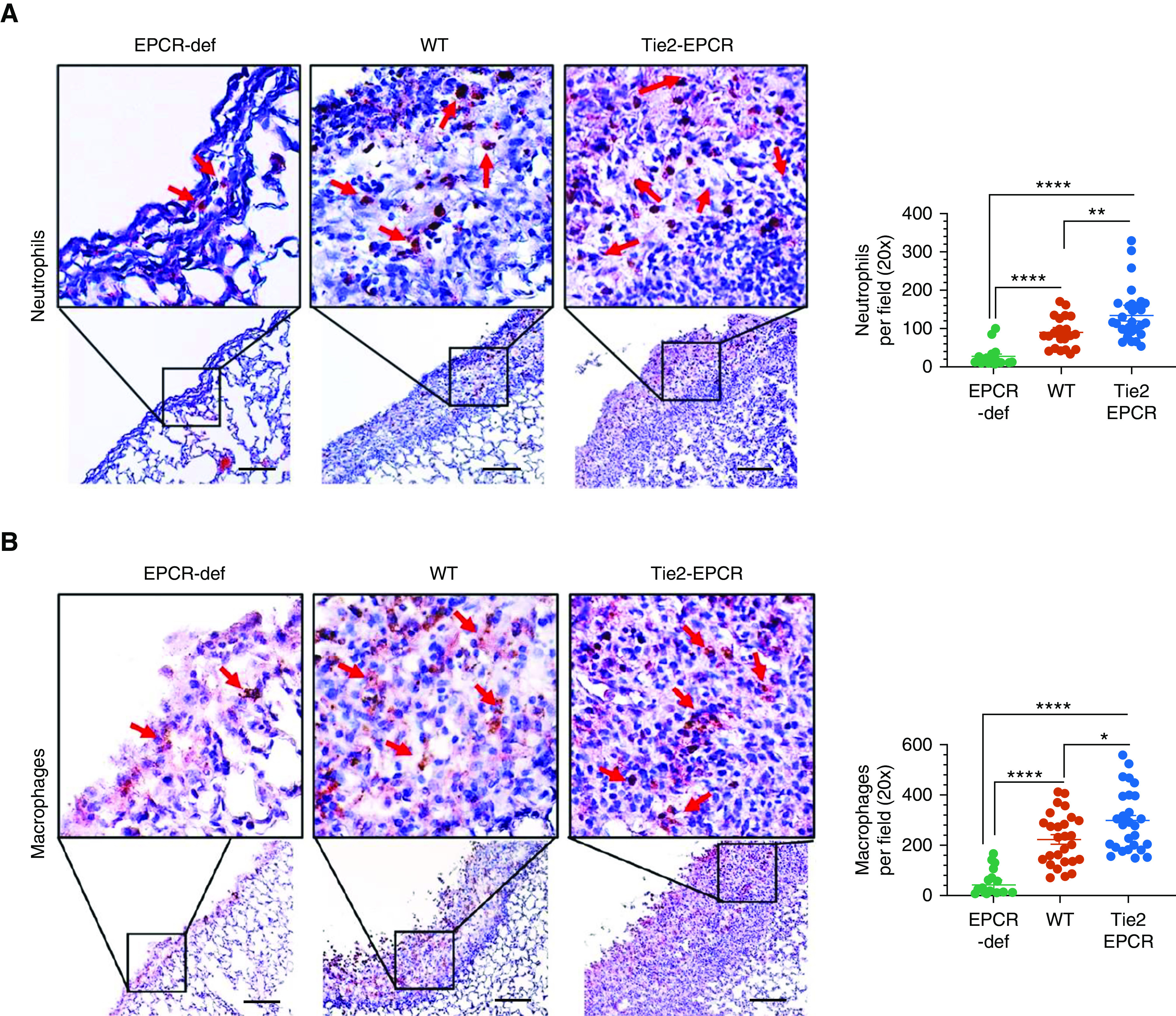
Increased infiltration of neutrophils and macrophages into the fibrotic pleura in EPCR-expressing mice after S. pneumoniae pleural infection. WT, Tie2-EPCR, or EPCR-def mice were administered with saline or S. pneumoniae intrapleural. After 7 days, lungs and pleural lavages were harvested from these mice. The lung tissue sections were stained with antimouse Ly6G to identify neutrophils (n = 15–28 fields per group) (A) or with anti-F4/80 to mark macrophages (n = 19–25 fields per group) (B). The number of neutrophils and macrophages in multiple fields was counted and averaged. These data were shown in the scatter dot plot graph, the right side of the images. Red arrows point to neutrophils or macrophages. Scale bars, 100 µm. *P < 0.05, **P < 0.01, and ****P < 0.0001.
Next, we analyzed the presence of immune cells in the pleural lavages of saline-administered and S. pneumoniae–infected mice. In all three genotypes (i.e., WT, Tie2-EPCR, and EPCR-deficient mice), S. pneumoniae infection markedly elevated the levels of inflammatory cells in the pleural cavity (Figure E2). Flow cytometric analysis of pleural lavages showed that the number of neutrophils, macrophages (both M1 and M2), and γδ-T cells were significantly higher in the pleural lavages isolated from S. pneumoniae–infected mice across all the genotypes compared with their saline counterparts. However, we found no significant differences in the number of various immune cell types, except macrophages, in the pleural lavages of infected mice expressing EPCR (WT and Tie2-EPCR mice) and mice lacking EPCR (EPCR-deficient mice). In the case of macrophages, both WT and Tie-2 EPCR mice exhibited a significantly higher number of macrophages in the pleural lavage compared with EPCR-deficient mice (Figure E2). The discrepancy in the pleural inflammatory cell recruitment data between immunohistochemistry and flow cytometry could be due to differences in the methodologies and the regions analyzed. In immunohistochemistry, the evaluation of inflammatory cell recruitment was restricted to cell counts in the fibrotic areas of the pleura. An analysis of inflammatory cell recruitment by flow cytometry was performed using cells isolated from pleural lavages, which may reflect the contribution of cells present in small pleural effusions or those mobilized from the pleural surface. Their percentages were normalized to CD45-positive cells, which identifies most hematopoietic cells.
We next analyzed profibrotic (TGF-β) and proinflammatory cytokines (IL-1β, IL-6, IFNγ, and TNF-α) in the pleural lavages collected from saline-administered and S. pneumoniae–infected mice 7 days after infection (Figure E3). No statistically significant differences were found in the levels of cytokines tested among S. pneumoniae–infected WT, Tie2-EPCR, and EPCR-deficient mice. Although not reaching statistical significance, the levels of TGF-β were elevated in S. pneumoniae–infected EPCR-expressing WT and Tie2-EPCR mice compared with infected EPCR-deficient mice (Figure E3D). Both WT and Tie2-EPCR mice exhibited increased IL-1β levels (P = 0.084 and P = 0.05, respectively) compared with the EPCR-deficient mice (Figure E3E). Because coagulation plays a key role in the pathogenesis of pleural organization, we next measured thrombin-antithrombin (TAT) levels in the pleural lavages as an index of thrombin generation. No significant differences were found in the TAT levels in the pleural lavages of WT and Tie2-EPCR compared with that of EPCR-deficient mice (Figure E4).
EPCR-Deficient Mice Exhibit Enhanced Coagulation and Fibrin Deposition during the Early Stage of S. pneumoniae Infection
To obtain a better understanding of the role of EPCR in the development of pleural fibrosis in S. pneumoniae infection, we evaluated various parameters that contribute to the pathogenesis of pleural fibrosis early after pleural infection (i.e., 48 hours after S. pneumoniae instillation). Levels of IL-6, TNF-α, and IFN-γ were increased in all genotypes compared with their noninfected counterparts. More importantly, levels of these cytokines were significantly higher in the pleural lavages of S. pneumoniae–infected WT and Tie2-EPCR mice compared with S. pneumoniae–infected EPCR-deficient mice (Figures 5A–5C). However, elevated IL-1β levels in the infected mice were not statistically significant among the three genotypes (Figure 5D). Determination of TGF-β levels in the pleural lavage revealed that S. pneumoniae infection significantly increased TGF-β levels in WT and Tie2-EPCR mice but not in EPCR-deficient mice (Figure 5E).
Figure 5.
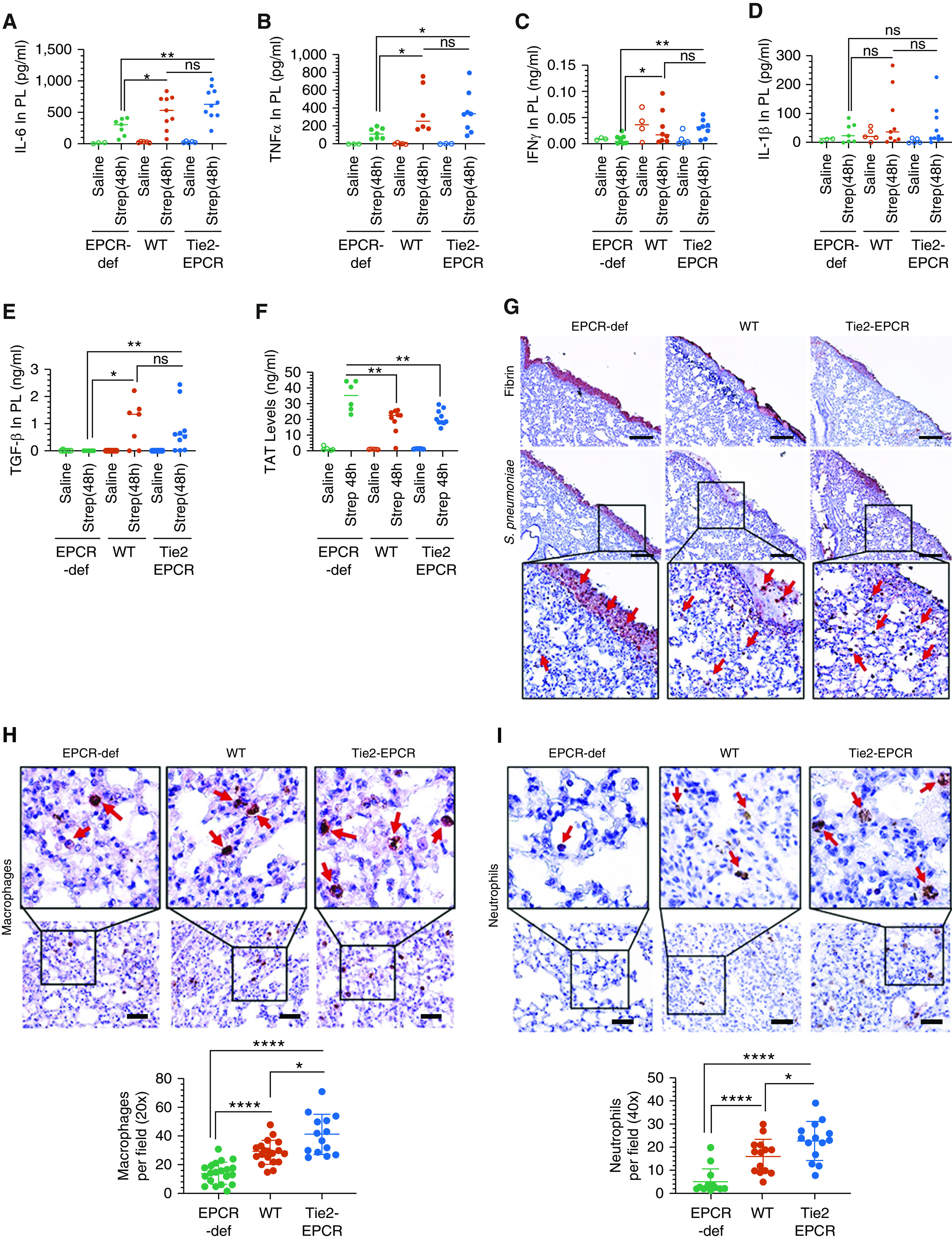
EPCR-def mice exhibit reduced inflammation but enhanced coagulation and pleural fibrin deposition. Saline or S. pneumoniae was administered into the pleural cavity of WT, Tie2-EPCR, or EPCR-def mice. After 48 hours, mice were killed, and pleural lavages and tissues were collected. (A–E) Pleural lavages (PL) were analyzed for the presence of proinflammatory cytokines IL-6 (A), TNF-α (B), IFN-γ (C), IL-1β (D), and TGF-β (E). (F) Thrombin-antithrombin complex levels were also estimated in the pleural lavages (saline, n = 3–5 mice per group; S. pneumoniae infection, n = 7–10 mice per group). (G–I) Lung tissue sections were stained by immunohistochemistry for fibrin (G, top panel) and S. pneumoniae (type 2 serotype) (G, bottom panel, insets were enlarged digitally), macrophages (F4/80 antigen) (H), and neutrophils (Ly6G antigen) (I). Macrophages and neutrophils number was counted from 14 to 20 fields from tissue sections originated from three to five mice/group. Red arrows point to (G) S. pneumoniae, (H) macrophages, or (I) neutrophils. Scale bars, 100 µm. *P < 0.05, **P < 0.01, and ****P < 0.0001.
Pleural S. pneumoniae infection increased pleural lavage TAT levels at 48 hours in all three genotypes, indicating early activation of coagulation in infected mice (Figure 5F). However, TAT levels in infected EPCR-deficient mice (34.76 ± 3.82 ng/ml) were significantly higher compared with TAT levels in infected WT (19.48 ± 2.57 ng/ml) or Tie2-EPCR mice (21.27 ± 1.48 ng/ml) (Figure 5F). Consistent with the increased activation of coagulation, immunohistochemical analysis of fibrin showed increased fibrin deposition on the visceral pleural mesothelium in EPCR-deficient mice compared with EPCR-expressing WT and Tie2-EPCR mice (Figure 5G, top panel). Immunohistochemical analysis of lung tissue sections with S. pneumoniae serotype 2–specific polyclonal antibodies against bacterial capsular proteins revealed that most of the bacteria were trapped in the fibrin clot on the outside of the lung on the mesothelium in EPCR-deficient mice with very few bacteria in the lung parenchyma (Figure 5G, bottom panel, red arrows). By contrast, a large number of bacteria were seen in the lung parenchyma of WT and Tie2-EPCR mice (Figure 5G, bottom panel, red arrows).
We next stained the lung tissue sections of S. pneumoniae–infected mice (48 h) with specific markers against macrophages (F4/80) and neutrophils (Ly6-G). Correlating with higher proinflammatory cytokine levels, EPCR-expressing WT and Tie2-EPCR mice exhibited significantly higher macrophage infiltration into the lungs (28.7 ± 2.0 and 41 ± 3.74, respectively) compared with EPCR-deficient mice (14.3 ± 1.7) (Figure 5H). Similarly, WT and Tie2-EPCR mice also exhibited significantly higher neutrophils (16.3 ± 2 and 22.8 ± 2.2, respectively) in the lungs compared with EPCR-deficient mice (5.0 ± 1.5) (Figure 5I). Both macrophage and neutrophil counts were significantly higher in Tie2-EPCR mice compared with WT mice (P < 0.05).
The decrement of cells and cytokines observed in the pleural lavages of the mice with EPCR deficiency likely reflects the relative preservation of the pleural architecture versus the extensive inflammation and organization that were observed in WT and EPCR-overexpressing mice, recapitulating trends observed in patients and animal models of empyema (28).
EPCR-Expressing Mouse Lung Tissues Exhibit Increased Bacterial Burden
In our earlier studies, we established that the antibiotic treatment renders the pleural cavity sterile after 3 days of antibiotic treatment in WT mice infected with S. pneumoniae intrapleural (8). To investigate the possibility that EPCR regulates the survival and dissemination of bacteria after pleural infection, we next assessed the bacterial burden in WT, Tie2-EPCR, and EPCR-deficient mice at 48 hours after infection. By that time, the infected mice received two doses of antibiotics, at 4 hours and 24 hours after infection. S. pneumoniae could be cultured from the pleural lavages and lung lysates of all three genotypes (Figures 6A and 6B). However, the number of bacteria present in WT and Tie2-EPCR was markedly higher compared with EPCR-deficient mice. Furthermore, S. pneumoniae was also readily detectable in blood, spleen, liver, and kidneys of WT and Tie2-EPCR mice (Figures 6C–6F). In contrast, these organs contained very few bacteria in EPCR-deficient mice infected with S. pneumoniae.
Figure 6.

EPCR expression increases bacterial burden and dissemination in S. pneumonia infection. WT, Tie2-EPCR, or EPCR-def mice were administered with S. pneumoniae intrapleural. After 48 hours, after two doses of antibiotics at 4 hours and 24 hours after infection, mice were killed, and various organs were harvested. Bacterial burden was assessed by culturing appropriately diluted tissue homogenates on blood agar plates. Bacterial burden in (A) pleural lavages, (B) lungs, (C) blood, (D) spleen, (E) liver, and (F) kidney. Compiled data of three independent experiments are presented (n = 7–9 per group). (G) Lung tissue sections of S. pneumoniae–infected mice were stained for tissue gram staining. Images were captured using the Lionheart FX automated microscope (BioTek, VT). S. pneumoniae bacteria appear as blackish-blue dots (shown by blue arrows). (H) Staining of lung tissue sections with antipneumococcal antibody against serotype 2 capsular protein. Images were photographed using Olympus BX41 light microscope. Red staining represents S. pneumoniae bacteria (shown by red arrows). Scale bars, 100 µm (top panel) and 20 µm (bottom panel). **P < 0.01 and ***P < 0.001. cfu = colony forming unit.
To further investigate the possibility that EPCR expression influences the bacterial burden, we next assessed S. pneumoniae–infected lung sections of WT, Tie2-EPCR, and EPCR-deficient mice collected at Day 7 after infection with tissue gram staining. These analyses revealed the presence of bacteria in the thickened fibrotic sections of both WT and Tie2-EPCR mice (Figure 6G, blue arrows). Although we could also detect bacteria in EPCR-deficient mice, their numbers were markedly reduced versus EPCR-expressing mice. Next, we immunostained the sections with S. pneumoniae serotype 2–specific polyclonal antibodies against the capsular proteins of the bacteria. We similarly observed a large bacterial burden in the injured pleura of both WT and Tie2-ECPR mice, whereas EPCR-deficient mice exhibited few bacteria (Figure 6H, red arrows, Figure E5).
EPCR Promotes the Internalization of S. pneumoniae in Mesothelial Cells
Many studies showed the internalization and dissemination of S. pneumoniae across tissue barriers (29, 30). It is therefore possible that EPCR could aid the internalization of S. pneumoniae, which allows bacteria to evade killing by antibiotics. To investigate this possibility, we evaluated the internalization of S. pneumoniae and EPCR dependency in PMC (MeT-5A). MeT-5A cells were stably transfected with EPCR-specific shRNA to establish the MeT-5A (-EPCR) cell line or transfected with an empty vector control MeT-5A (VC) (Figure 7A). MeT-5A, MeT-5A (VC), and MeT-5A (-EPCR) cells were exposed to S. pneumoniae for 2 hours (1 cell:50 bacteria) to allow adherence and internalization of the bacteria. Bacteria that were not internalized were killed by antibiotic treatment for 1 hour, and then the extent of internalization was assessed. As shown in Figure 7B, bacterial internalization was markedly lower in MeT-5A (-EPCR) cells (17.22 ± 3.69 cfu/well) compared with MeT-5A cells (75.38 ± 7.65 cfu/well) and MeT-5A (VC) cells (60.0 ± 8.56 cfu/well). In the absence of cells, bacteria treated with the same amount of antibiotics did not yield any colonies, indicating that the antibiotics added were sufficient to kill all bacteria that are not internalized.
Figure 7.
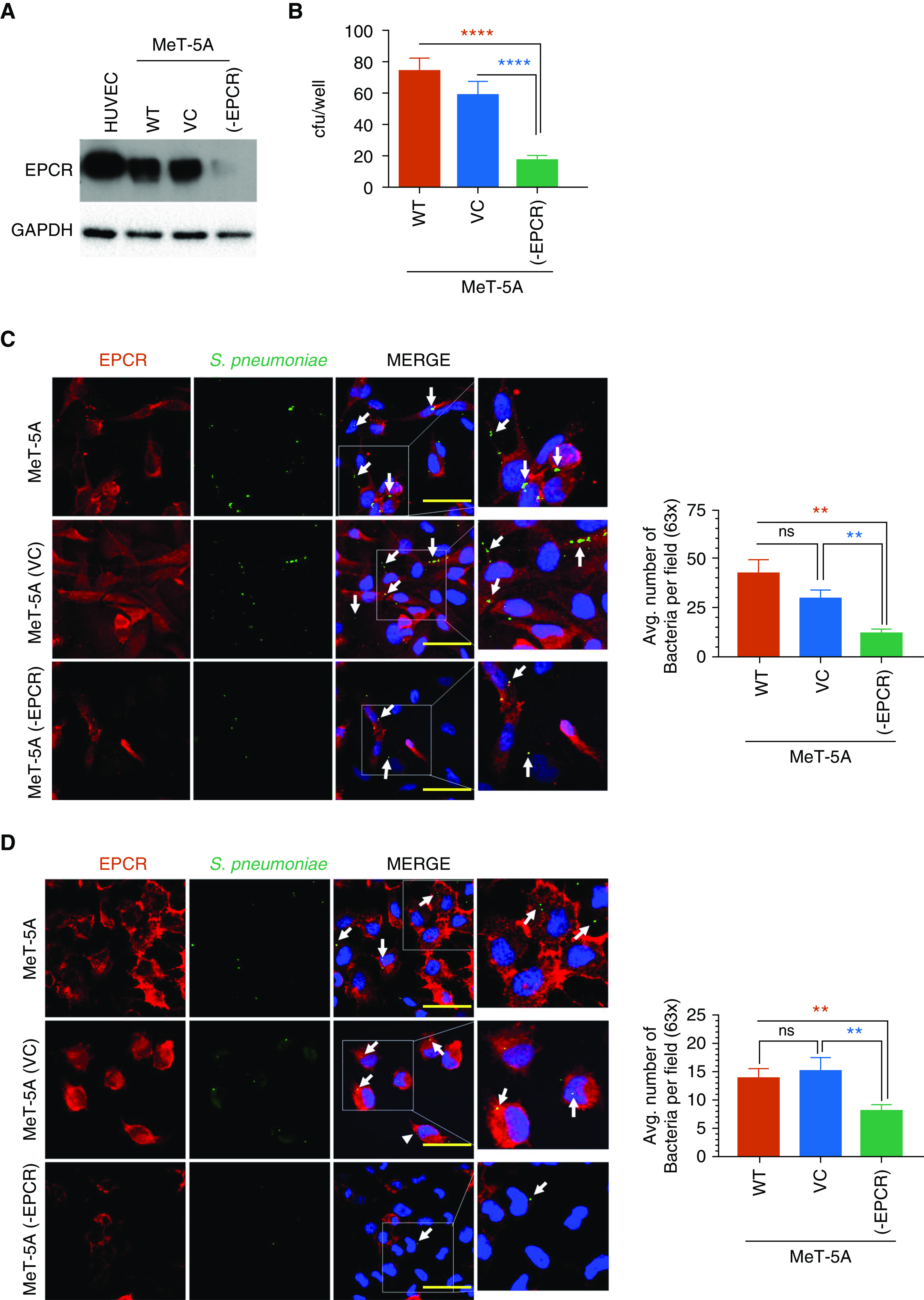
EPCR aids in the internalization of S. pneumoniae and protects it from antibiotics. (A) Immunoblot analysis of EPCR expression in MeT-5A cells and MeT-5A cells stably transfected with vector control (MeT-5A [VC]) or EPCR shRNA (MeT-5A [-EPCR]). Cell lysates of HUVEC were used as a control for EPCR expression. (B) Antibiotic protection assay. MeT-5A, MeT-5A (VC), and MeT-5A (-EPCR) cells were grown to confluency and exposed to S. pneumoniae in a log-phase (1 × 107 cfu/ml). After 2 hours incubation at 37°C, the supernatant medium containing bacteria was removed, cells were washed three times, and then an antibiotic solution containing ampicillin (10 μg/ml) and gentamicin (200 μg/ml) was added to the cells to kill bacteria. After 1 hour, the monolayers were washed and lysed with 1 ml of 0.025% ice-cold Triton X-100. Cell homogenates were plated on blood agar plates to culture bacteria. The next day, the number of bacteria colonies formed were counted. (C and D) S. pneumoniae was fluorescently labeled with 5(6)-Carboxyfluorescein N-succinimidyl ester. Labeled bacteria (1 × 107 cfu/ml) were added to MeT-5A, MeT-5A (VC) and MeT-5A (-EPCR) cells grown on glass coverslips. After 2 hours, nonadherent bacteria were removed, and cells were washed with ice-cold PBS. In some wells, the adherent, noninternalized bacteria were quenched by incubating the cells with 0.5% Trypan blue for 10 minutes. The cells were then fixed and stained for EPCR using Mab JRK 1500 antihuman EPCR antibody, followed by AF594-labeled secondary antibodies. (C and D) Representative confocal images of cells with adherent and internalized bacteria (before quenching) (C) or internalized bacteria (after quenching). White arrows point bacteria. (D). The number of bacteria was counted from multiple fields, and the quantified data was shown in right side panels. Scale bars, 20 µm. **P < 0.01 and ****P < 0.0001. Avg. = average.
Next, to visualize EPCR-dependent adherence and internalization, bacteria were fluorescently labeled with 5(6)-Carboxyfluorescein N-succinimidyl ester (FAM-NSE) and added to MeT-5A, MeT-5A (VC), or MeT-5A (-EPCR) cells (1 cell:50 bacteria). After 2 hours, nonadherent bacteria were removed, and the cells were imaged without (Figure 7C) or with (Figure 7D) quenching the fluorescence of adherent bacteria. As shown in Figures 7C and 7D, we observed a greater number of bacteria in MeT-5A and MeT-5A (VC) cells compared with MeT-5A (-EPCR) (white arrows) These results further support the concept that EPCR promotes the internalization of S. pneumoniae.
S. pneumoniae was shown to be internalized in epithelial and endothelial cells via dynamin-dependent or dynamin-independent pathways (31, 32). Our earlier studies showed that EPCR-mediated internalization of its ligands was mediated via a dynamin- and caveolar-dependent pathway (33). Therefore, we investigated whether the internalization of S. pneumoniae in mesothelial cells is mediated via a dynamin- and caveolar-dependent pathway. Pretreatment of MeT-5A cells with β-methyl cyclodextrin, which disrupts caveolae or dynasore, an inhibitor of dynamin GTPase activity, attenuated the shielding of the bacteria from antibiotics (Figure E6), suggesting that S. pneumoniae was internalized via a caveolar- and dynamin-dependent pathway.
Discussion
A close relationship exists between infection and inflammation and the activation of coagulation (34). The efficacy to eradicate the invading pathogen is dependent on the inflammatory responses of the host and the amplitude of the coagulation system (35). EPCR has been implicated as a key receptor in the complex interaction between coagulation and inflammation (36, 37). The data presented herein show that EPCR enhances pleural organization and fibrosis and restricts pulmonary function in S. pneumoniae–infected mice. EPCR deficiency diminishes S. pneumoniae growth and dissemination, attenuates inflammation, and protects against pleural fibrosis. Our data also show that EPCR facilitates the internalization of S. pneumoniae in PMC, and the EPCR-mediated internalization shields the bacteria from antibiotics.
The extent of pleural fibrosis in S. pneumoniae–infected mice appeared to correlate with EPCR expression levels. EPCR-deficient mice exhibited minimal thickening of pleura, whereas pleural fibrosis is extensive in EPCR-overexpressing Tie2-EPCR mice. WT mice expressing normal levels of EPCR showed an intermediate level of pleural fibrosis. The extent of pleural fibrosis did not correlate with significant differences in pulmonary function testing in WT and Tie2-EPCR mice. This finding may be attributable to heterogeneity of the pleural thickening, a modest impact of heterogeneously increased pleural thickness in EPCR-overexpressing Tie2-EPCR mice on pulmonary function, the possibility that there is a threshold of pleural thickness that does not result in pleurodesis but causes lung restriction, or a combination of all these factors.
In general, EPCR plays a protective role against bacterial infection, as it supports cytoprotective and antiinflammatory signaling induced by activated protein C (38–41). However, our current data suggest that EPCR may be detrimental in S. pneumoniae infection. Our data are consistent with earlier findings that showed EPCR is detrimental in certain infections (42, 43). EPCR was shown to impair antibacterial responses in a mouse model against pneumococcal pneumonia and sepsis caused by S. pneumoniae (42). EPCR-overexpressing mice showed increased bacterial outgrowth in the lungs, blood, liver, spleen, and kidneys, and EPCR deficiency significantly reduced bacterial burden (42). Overexpression of EPCR was also shown to be detrimental in pneumonia-derived sepsis caused by Burkholderia pseudomallei (Melioidosis) (43). EPCR-overexpressing Tie2-EPCR mice enhanced the survival of the bacteria in the lungs as well as increased the dissemination of the bacteria to distant organs. However, in the above study, EPCR-deficient mice did not show a reduction in bacterial dissemination and pulmonary damage (43). Differences between our findings and the above study are likely due to specific responses associated with different bacteria and potential differences between the injury in the lung parenchyma versus the pleural space.
At present, the mechanisms by which EPCR promotes bacterial growth and dissemination and pleural fibrosis remain to be better understood. It is generally believed that activation of coagulation is beneficial in infections, as it limits pathogen dissemination and supports pathogen killing (44). Local fibrin deposition was shown to play a critical role in host defense against bacterial infection (44–46). Earlier studies indicate that fibrin can provide protective function during peritoneal bacterial infections by promoting the clearance of bacteria by modulating host inflammatory response through leukocyte engagement (47) or by physically trapping bacteria, thereby directly limiting the dissemination (48, 49). The latter possibility appears to be true in the current study, as the downregulation of the protein C anticoagulant pathway in EPCR-deficient mice led to extensive fibrin deposition on the visceral pleural mesothelium at the early phase of infection. Most of the bacteria appeared to be trapped in the fibrin in EPCR-deficient mice. In WT and Tie2-EPCR mice, in which fibrin deposition was much lower in the pleura, bacteria were readily detectable in the lung parenchyma, including the subpleural, peripheral lung.
Pathogenic bacteria use multiple strategies to promote their entry and survival in the host. S. pneumoniae was shown to bind specific cellular receptors in endothelial cells and epithelial cells, such as PAFR (platelet-activated factor receptor), PIGR (polymeric immunoglobulin receptor), and PECAM-1 (platelet endothelial cell adhesion molecule-1) (50–53). S. pneumoniae hijacks the internalization and recycling pathways of these receptors to gain entry into host cells, survive, and penetrate into tissues (53). Our current studies show, for the first time, that mesothelial cell EPCR facilitates S. pneumoniae internalization, as knockdown of EPCR reduced internalization of the bacteria. Our data indicate that the internalization of the bacteria via the EPCR-dependent mechanism promotes bacterial survival even in the presence of a lethal dose of antibiotics. This is in agreement with earlier studies in which S. pneumoniae was shown to resist the lethal dose of antibiotics after becoming internalized into brain microvascular endothelial cells (30). Internalization also helps the pathogen to escape immune surveillance as well as clearance (53). This could contribute to increased bacterial burden and dissemination in EPCR-expressing WT and Tie2-EPCR mice compared with EPCR-deficient mice in our model system, in which mice were treated with antibiotics after the installation of S. pneumoniae in the pleural cavity.
A limitation of the current study is it is difficult to ascertain the relative contribution of mesothelial EPCR versus EPCR present on vascular endothelial cells and other cell types in S. pneumoniae–induced pleural fibrosis. Besides endothelial and mesothelial cells, EPCR is expressed in many other cell types (13). EPCR-deficient mice used in the current study lack EPCR in all cell types. Therefore, it is not feasible to dissect the contribution of mesothelial EPCR and EPCR present in other cell types in the pathogenesis of pleural fibrosis. Overexpression of EPCR in Tie2-EPCR is primarily limited to endothelial cells on large vessels and capillaries in the lung (24). Earlier studies showed no increased EPCR expression in Mac-1, CD19, CD4, and CD8 positive leukocytes in Tie2-EPCR mice (24). Consistent with these data, we found no increased expression of EPCR in PMC in Tie2-EPCR mice (Figure E7). This could explain why we found a similar bacterial burden and dissemination in both WT and Tie2-EPCR mice. However, it raises the question of why pleural thickening is more pronounced in Tie2-EPCR mice. The pleural mesothelium is covered by a fine network of capillaries. Both bacterial and viral infections are known to result increased angiogenesis in vivo, a physiologic response to facilitate the wound healing process (54, 55). The thickened pleura of S. pneumoniae–infected lungs of both WT and Tie2-EPCR mice exhibited neoangiogenesis, and these newly formed blood vessels stained positive for EPCR (Figure E8). The reduced neoangiogenesis in EPCR-deficient mice could be due to less severe injury of these mice in response to S. pneumoniae infection, or EPCR may be playing a role in neoangiogenesis. The origin of these newly formed pleural vessels is unknown, but earlier studies indicated the potential contribution of mesothelial and mesothelial-derived cells in neovascularization (56–59). It is possible that enhanced EPCR expression in existing capillaries on the pleural mesothelium or newly formed capillaries in Tie2-EPCR mice could support the progression of pleural fibrosis in these mice.
EPCR has shown to bind directly to Mac1 expressed on monocytes to promote their adherence to the endothelium (15). Soluble EPCR was shown to bind to PR3 (proteinase-3) on activated neutrophils and affect cell–cell adhesion and neutrophil-signaling events, partially assisted by Mac1 (14). Pleural thickening in the WT and EPCR-overexpressing mice was associated with significantly increased infiltration of immune cells, such as neutrophils and macrophages. Infiltration of neutrophils and macrophages into the lungs was significantly higher in Tie2-EPCR mice compared with WT mice. These interactions may be responsible partly for the release of cytokine mediators, which promote pleural remodeling and fibrosis. Although we found a significant increase in IL-6, TNF-α, IFN-γ, and TGF-β levels in the pleural lavage in WT and Tie2-EPCR at 48 hours after the infection, differences between them are not statistically significant. It is possible that cytokine levels between the infected WT and Tie2-EPCR might be significantly different at the very early stages of infection. Here, we wish to point out that differences seen in cytokine levels between EPCR-deficient mice and EPCR-expressing WT and Tie2-EPCR at Day 2 of the infection were not evident at Day 7 of the infection.
Our work and that of others showed that PMC contribute to the pathogenesis of pleural fibrosis by undergoing MesoMT, by which PMC assume characteristics of myofibroblasts by expressing increased levels of α-SMA and collagen I (60–63). Our earlier studies showed that coagulation proteases thrombin and factor Xa induce MesoMT (7, 25). The enhanced pleural fibrosis seen in Tie2-EPCR mice could also result at least in part from increased APC generation in these mice and APC-induced MesoMT. Earlier studies showed significantly higher circulating APC levels in Tie2-EPCR mice compared with WT after LPS administration (24). Consistent with these data, we found elevated levels of APC in the pleural lavage of S. pneumoniae–infected Tie2-EPCR mice (Table E2). Furthermore, our preliminary studies also show that APC induces MesoMT markers such as α-SMA and collagen-1 in HPMCs (Figure E9).
Conclusions
The role of EPCR in pleural organization and fibrosis after S. pneumoniae empyema has not, to our knowledge, been previously explored. In the present study, we show that EPCR promotes pleural fibrosis and significantly restricts pulmonary function. EPCR deficiency protected against pleural fibrosis. Our data indicate that EPCR may contribute to the progression of pleural fibrosis by multiple mechanisms, such as downregulation of the activation of coagulation that results in reduced fibrin deposition on the visceral pleura; internalization of the pathogen by PMC, which could promote bacteria survival and dissemination; and through APC-driven MesoMT. Further studies are required to determine the relative importance of the above mechanisms and the contribution of mesothelial EPCR and EPCR expressed on endothelial cells or other cell types to the progression of pleural fibrosis initiated by S. pneumoniae infection.
Supplementary Material
Footnotes
Supported by National Institutes of Health grants R01-HL-107483 and R01-HL124055 (L.V.M.R), R01-HL-130402 (S.I.), and R01-HL-130133 and R01-HL-142853 to (T.A.T.); and The Dr. and Mrs. James Vaughn Professorship in Biomedical Research endowment funds (L.V.M.R).
Author Contributions: S.K. performed most of the experiments described in the manuscript, analyzed data, and wrote the first draft of the manuscript. J.M. assisted S.K. in performing the experiments. W.K. and S.M. performed flow cytometry and analyzed the flow cytometry data. C.T.E. provided breeding pairs of EPCR (endothelial protein C receptor) deficient and Tie2-EPCR mice and EPCR antibodies. S.K., T.A.T., S.I., L.V.M.R., and U.R.P. contributed to the design of the research and analysis of data. L.V.M.R. and U.R.P. supervised the research and wrote the manuscript. All authors reviewed the manuscript and approved the final version of the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0328OC on February 18, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rosenstengel A. Pleural infection-current diagnosis and management. J Thorac Dis. 2012;4:186–193. doi: 10.3978/j.issn.2072-1439.2012.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Light RW. Parapneumonic effusions and empyema. Proc Am Thorac Soc. 2006;3:75–80. doi: 10.1513/pats.200510-113JH. [DOI] [PubMed] [Google Scholar]
- 3.Ferreira-Coimbra J, Sarda C, Rello J. Burden of community-acquired pneumonia and unmet clinical needs. Adv Ther. 2020;37:1302–1318. doi: 10.1007/s12325-020-01248-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2010. Natl Vital Stat Rep. 2013;61:1–117. [PubMed] [Google Scholar]
- 5.Davies HE, Davies RJ, Davies CW BTS Pleural Disease Guideline Group. Management of pleural infection in adults: British thoracic society pleural disease guideline 2010. Thorax. 2010;65:ii41–ii53. doi: 10.1136/thx.2010.137000. [DOI] [PubMed] [Google Scholar]
- 6.Mutsaers SE, Prele CM, Brody AR, Idell S. Pathogenesis of pleural fibrosis. Respirology. 2004;9:428–440. doi: 10.1111/j.1440-1843.2004.00633.x. [DOI] [PubMed] [Google Scholar]
- 7.Owens S, Jeffers A, Boren J, Tsukasaki Y, Koenig K, Ikebe M, et al. Mesomesenchymal transition of pleural mesothelial cells is PI3K and NF-κB dependent. Am J Physiol Lung Cell Mol Physiol. 2015;308:L1265–L1273. doi: 10.1152/ajplung.00396.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucker TA, Jeffers A, Boren J, Quaid B, Owens S, Koenig KB, et al. Organizing empyema induced in mice by Streptococcus pneumoniae: effects of plasminogen activator inhibitor-1 deficiency. Clin Transl Med. 2016;5:17. doi: 10.1186/s40169-016-0097-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci USA. 1996;93:10212–10216. doi: 10.1073/pnas.93.19.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 11.Rezaie AR. The occupancy of endothelial protein C receptor by its ligand modulates the par-1 dependent signaling specificity of coagulation proteases. IUBMB Life. 2011;63:390–396. doi: 10.1002/iub.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pendurthi UR, Rao LVM. Endothelial cell protein C receptor-dependent signaling. Curr Opin Hematol. 2018;25:219–226. doi: 10.1097/MOH.0000000000000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohan Rao LV, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood. 2014;124:1553–1562. doi: 10.1182/blood-2014-05-578328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurosawa S, Esmon CT, Stearns-Kurosawa DJ. The soluble endothelial protein C receptor binds to activated neutrophils: involvement of proteinase-3 and CD11b/CD18. J Immunol. 2000;165:4697–4703. doi: 10.4049/jimmunol.165.8.4697. [DOI] [PubMed] [Google Scholar]
- 15.Fink K, Busch HJ, Bourgeois N, Schwarz M, Wolf D, Zirlik A, et al. Mac-1 directly binds to the endothelial protein C-receptor: a link between the protein C anticoagulant pathway and inflammation? PLoS One. 2013;8:e53103. doi: 10.1371/journal.pone.0053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willcox CR, Pitard V, Netzer S, Couzi L, Salim M, Silberzahn T, et al. Cytomegalovirus and tumor stress surveillance by binding of a human γδ T cell antigen receptor to endothelial protein C receptor. Nat Immunol. 2012;13:872–879. doi: 10.1038/ni.2394. [DOI] [PubMed] [Google Scholar]
- 17.Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498:502–505. doi: 10.1038/nature12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laszik Z, Mitro A, Taylor FB, Jr, Ferrell G, Esmon CT. Human protein C receptor is present primarily on endothelium of large blood vessels: implications for the control of the protein C pathway. Circulation. 1997;96:3633–3640. doi: 10.1161/01.cir.96.10.3633. [DOI] [PubMed] [Google Scholar]
- 19.Ye X, Fukudome K, Tsuneyoshi N, Satoh T, Tokunaga O, Sugawara K, et al. The endothelial cell protein C receptor (EPCR) functions as a primary receptor for protein C activation on endothelial cells in arteries, veins, and capillaries. Biochem Biophys Res Commun. 1999;259:671–677. doi: 10.1006/bbrc.1999.0846. [DOI] [PubMed] [Google Scholar]
- 20.Gleeson EM, O’Donnell JS, Preston RJ. The endothelial cell protein C receptor: cell surface conductor of cytoprotective coagulation factor signaling. Cell Mol Life Sci. 2012;69:717–726. doi: 10.1007/s00018-011-0825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iakhiaev A, Idell S. Activation and degradation of protein C by primary rabbit pleural mesothelial cells. Lung. 2006;184:81–88. doi: 10.1007/s00408-005-2566-z. [DOI] [PubMed] [Google Scholar]
- 22.Idell S, Zwieb C, Kumar A, Koenig KB, Johnson AR. Pathways of fibrin turnover of human pleural mesothelial cells in vitro. Am J Respir Cell Mol Biol. 1992;7:414–426. doi: 10.1165/ajrcmb/7.4.414. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Zheng X, Gu JM, Ferrell GL, Brady M, Esmon NL, et al. Extraembryonic expression of EPCR is essential for embryonic viability. Blood. 2005;106:2716–2722. doi: 10.1182/blood-2005-01-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Zheng X, Gu J, Hunter J, Ferrell GL, Lupu F, et al. Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost. 2005;3:1351–1359. doi: 10.1111/j.1538-7836.2005.01385.x. [DOI] [PubMed] [Google Scholar]
- 25.Jeffers A, Qin W, Owens S, Koenig KB, Komatsu S, Giles FJ, et al. Glycogen synthase kinase-3β inhibition with 9-ING-41 attenuates the progression of pulmonary fibrosis. Sci Rep. 2019;9:18925. doi: 10.1038/s41598-019-55176-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tucker T, Tsukasaki Y, Sakai T, Mitsuhashi S, Komatsu S, Jeffers A, et al. Myocardin is involved in mesothelial-mesenchymal transition of human pleural mesothelial cells. Am J Respir Cell Mol Biol. 2019;61:86–96. doi: 10.1165/rcmb.2018-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jantz MA, Antony VB. Pleural fibrosis. Clin Chest Med. 2006;27:181–191. doi: 10.1016/j.ccm.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Komissarov AA, Rahman N, Lee YCG, Florova G, Shetty S, Idell R, et al. Fibrin turnover and pleural organization: bench to bedside. Am J Physiol Lung Cell Mol Physiol. 2018;314:L757–L768. doi: 10.1152/ajplung.00501.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilkosz S, Edwards LA, Bielsa S, Hyams C, Taylor A, Davies RJ, et al. Characterization of a new mouse model of empyema and the mechanisms of pleural invasion by Streptococcus pneumoniae. Am J Respir Cell Mol Biol. 2012;46:180–187. doi: 10.1165/rcmb.2011-0182OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ring A, Weiser JN, Tuomanen EI. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J Clin Invest. 1998;102:347–360. doi: 10.1172/JCI2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manalee V, Survee SA, Bhutda S, Kamatha KG, Kim KG, Banerjee A. Streptococcus pneumoniae utilizes a novel dynamin independent pathway for entry and persistence in brain endothelium. Curr Res Microb Sci. 2020;1:62–68. doi: 10.1016/j.crmicr.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asmat TM, Agarwal V, Saleh M, Hammerschmidt S. Endocytosis of Streptococcus pneumoniae via the polymeric immunoglobulin receptor of epithelial cells relies on clathrin and caveolin dependent mechanisms. Int J Med Microbiol. 2014;304:1233–1246. doi: 10.1016/j.ijmm.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Nayak RC, Sen P, Ghosh S, Gopalakrishnan R, Esmon CT, Pendurthi UR, et al. Endothelial cell protein C receptor cellular localization and trafficking: potential functional implications. Blood. 2009;114:1974–1986. doi: 10.1182/blood-2009-03-208900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levi M, Keller TT, van Gorp E, ten Cate H. Infection and inflammation and the coagulation system. Cardiovasc Res. 2003;60:26–39. doi: 10.1016/s0008-6363(02)00857-x. [DOI] [PubMed] [Google Scholar]
- 35.van der Poll T, Herwald H. The coagulation system and its function in early immune defense. Thromb Haemost. 2014;112:640–648. doi: 10.1160/TH14-01-0053. [DOI] [PubMed] [Google Scholar]
- 36.Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83:536–545. doi: 10.1189/jlb.0607373. [DOI] [PubMed] [Google Scholar]
- 37.Esmon CT. Structure and functions of the endothelial cell protein C receptor. Crit Care Med. 2004;32(Suppl):S298–S301. doi: 10.1097/01.ccm.0000126128.64614.81. [DOI] [PubMed] [Google Scholar]
- 38.Taylor FB, Jr, Stearns-Kurosawa DJ, Kurosawa S, Ferrell G, Chang AC, Laszik Z, et al. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood. 2000;95:1680–1686. [PubMed] [Google Scholar]
- 39.Sinha RK, Wang Y, Zhao Z, Xu X, Burnier L, Gupta N, et al. PAR1 biased signaling is required for activated protein C in vivo benefits in sepsis and stroke. Blood. 2018;131:1163–1171. doi: 10.1182/blood-2017-10-810895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C: biased for translation. Blood. 2015;125:2898–2907. doi: 10.1182/blood-2015-02-355974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schouten M, de Boer JD, Kager LM, Roelofs JJ, Meijers JC, Esmon CT, et al. The endothelial protein C receptor impairs the antibacterial response in murine pneumococcal pneumonia and sepsis. Thromb Haemost. 2014;111:970–980. doi: 10.1160/TH13-10-0859. [DOI] [PubMed] [Google Scholar]
- 43.Kager LM, Schouten M, Wiersinga WJ, de Boer JD, Lattenist LC, Roelofs JJ, et al. Overexpression of the endothelial protein C receptor is detrimental during pneumonia-derived gram-negative sepsis (Melioidosis) PLoS Negl Trop Dis. 2013;7:e2306. doi: 10.1371/journal.pntd.0002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antoniak S. The coagulation system in host defense. Res Pract Thromb Haemost. 2018;2:549–557. doi: 10.1002/rth2.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun H. The interaction between pathogens and the host coagulation system. Physiology (Bethesda) 2006;21:281–288. doi: 10.1152/physiol.00059.2005. [DOI] [PubMed] [Google Scholar]
- 46.Sun H, Wang X, Degen JL, Ginsburg D. Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood. 2009;113:1358–1364. doi: 10.1182/blood-2008-07-170506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flick MJ, Du X, Witte DP, Jirousková M, Soloviev DA, Busuttil SJ, et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113:1596–1606. doi: 10.1172/JCI20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dunn DL, Simmons RL. Fibrin in peritonitis. III. The mechanism of bacterial trapping by polymerizing fibrin. Surgery. 1982;92:513–519. [PubMed] [Google Scholar]
- 49.Rotstein OD. Role of fibrin deposition in the pathogenesis of intraabdominal infection. Eur J Clin Microbiol Infect Dis. 1992;11:1064–1068. doi: 10.1007/BF01967800. [DOI] [PubMed] [Google Scholar]
- 50.Gradstedt H, Iovino F, Bijlsma JJ. Streptococcus pneumoniae invades endothelial host cells via multiple pathways and is killed in a lysosome dependent manner. PLoS One. 2013;8:e65626. doi: 10.1371/journal.pone.0065626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iovino F, Molema G, Bijlsma JJ. Platelet endothelial cell adhesion molecule-1, a putative receptor for the adhesion of Streptococcus pneumoniae to the vascular endothelium of the blood-brain barrier. Infect Immun. 2014;82:3555–3566. doi: 10.1128/IAI.00046-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iovino F, Molema G, Bijlsma JJ. Streptococcus pneumoniae Interacts with pIgR expressed by the brain microvascular endothelium but does not co-localize with PAF receptor. PLoS One. 2014;9:e97914. doi: 10.1371/journal.pone.0097914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weiser JN, Ferreira DM, Paton JC. Streptococcus pneumoniae: transmission, colonization and invasion. Nat Rev Microbiol. 2018;16:355–367. doi: 10.1038/s41579-018-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frantz S, Vincent KA, Feron O, Kelly RA. Innate immunity and angiogenesis. Circ Res. 2005;96:15–26. doi: 10.1161/01.RES.0000153188.68898.ac. [DOI] [PubMed] [Google Scholar]
- 55.Osherov N, Ben-Ami R. Modulation of host angiogenesis as a microbial survival strategy and therapeutic target. PLoS Pathog. 2016;12:e1005479. doi: 10.1371/journal.ppat.1005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elmadbouh I, Chen Y, Louedec L, Silberman S, Pouzet B, Meilhac O, et al. Mesothelial cell transplantation in the infarct scar induces neovascularization and improves heart function. Cardiovasc Res. 2005;68:307–317. doi: 10.1016/j.cardiores.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 57.Mutsaers SE, Wilkosz S. Structure and function of mesothelial cells. Cancer Treat Res. 2007;134:1–19. doi: 10.1007/978-0-387-48993-3_1. [DOI] [PubMed] [Google Scholar]
- 58.Shelton EL, Bader DM. Thymosin β4 mobilizes mesothelial cells for blood vessel repair. Ann N Y Acad Sci. 2012;1269:125–130. doi: 10.1111/j.1749-6632.2012.06713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colunga T, Hayworth M, Kreß S, Reynolds DM, Chen L, Nazor KL, et al. Human pluripotent stem cell-derived multipotent vascular progenitors of the mesothelium lineage have utility in tissue engineering and repair. Cell Rep. 2019;26:2566–2579, e10. doi: 10.1016/j.celrep.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Decologne N, Kolb M, Margetts PJ, Menetrier F, Artur Y, Garrido C, et al. TGF-beta1 induces progressive pleural scarring and subpleural fibrosis. J Immunol. 2007;179:6043–6051. doi: 10.4049/jimmunol.179.9.6043. [DOI] [PubMed] [Google Scholar]
- 61.Li Y, Wang J, Asahina K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc Natl Acad Sci USA. 2013;110:2324–2329. doi: 10.1073/pnas.1214136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nasreen N, Mohammed KA, Mubarak KK, Baz MA, Akindipe OA, Fernandez-Bussy S, et al. Pleural mesothelial cell transformation into myofibroblasts and haptotactic migration in response to TGF-beta1 in vitro. Am J Physiol Lung Cell Mol Physiol. 2009;297:L115–L124. doi: 10.1152/ajplung.90587.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tucker TA, Jeffers A, Alvarez A, Owens S, Koenig K, Quaid B, et al. Plasminogen activator inhibitor-1 deficiency augments visceral mesothelial organization, intrapleural coagulation, and lung restriction in mice with carbon black/bleomycin-induced pleural injury. Am J Respir Cell Mol Biol. 2014;50:316–327. doi: 10.1165/rcmb.2013-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.