

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a new strain of a Coronaviridae virus that presents 79% genetic similarity to the severe acute respiratory syndrome coronavirus, has been recently recognized as the cause of a global pandemic by the World Health Organization, implying a major threat to world public health. SARS-CoV-2 infects host human cells by binding through the viral spike proteins to the ACE-2 (angiotensin-converting enzyme 2) receptor, fuses with the cell membrane, enters, and starts its replication process to multiply its viral load. Coronavirus disease (COVID-19) was initially considered a respiratory infection that could cause pneumonia. However, in severe cases, it extends beyond the respiratory system and becomes a multiorgan disease. This transition from localized respiratory infection to multiorgan disease is due to two main complications of COVID-19. On the one hand, it is due to the so-called cytokine storm: an uncontrolled inflammatory reaction of the immune system in which defensive molecules become aggressive for the body itself. On the other hand, it is due to the formation of a large number of thrombi that can cause myocardial infarction, stroke, and pulmonary embolism. The pulmonary endothelium actively participates in these two processes, becoming the last barrier before the virus spreads throughout the body. In this review, we examine the role of the pulmonary endothelium in response to COVID-19, the existence of potential biomarkers, and the development of novel therapies to restore vascular homeostasis and to protect and/or treat coagulation, thrombosis patients. In addition, we review the thrombotic complications recently observed in patients with COVID-19 and its potential threatening sequelae.
Keywords: endothelial cell, COVID-19, coagulation/thrombosis, pulmonary embolism
Role of the Endothelium in Patients with COVID-19
The endothelium is a dynamic organ responsible for essential body functions, and it consists of a single layer of endothelial cells (ECs) lining the entire vascular system (1). It regulates systemic blood flow and coagulation, initiates and amplifies the inflammatory response, and maintains vascular tone, structure, and homeostasis (1, 2). Homeostatic vessel function is crucial and is mediated by the balanced production of various hormones, neurotransmitters, and vasoactive factors (3). Distress to this balance contributes to vascular injury, dysfunction, and the pathogenesis of diverse vascular diseases (4) (Figure 1).
Figure 1.
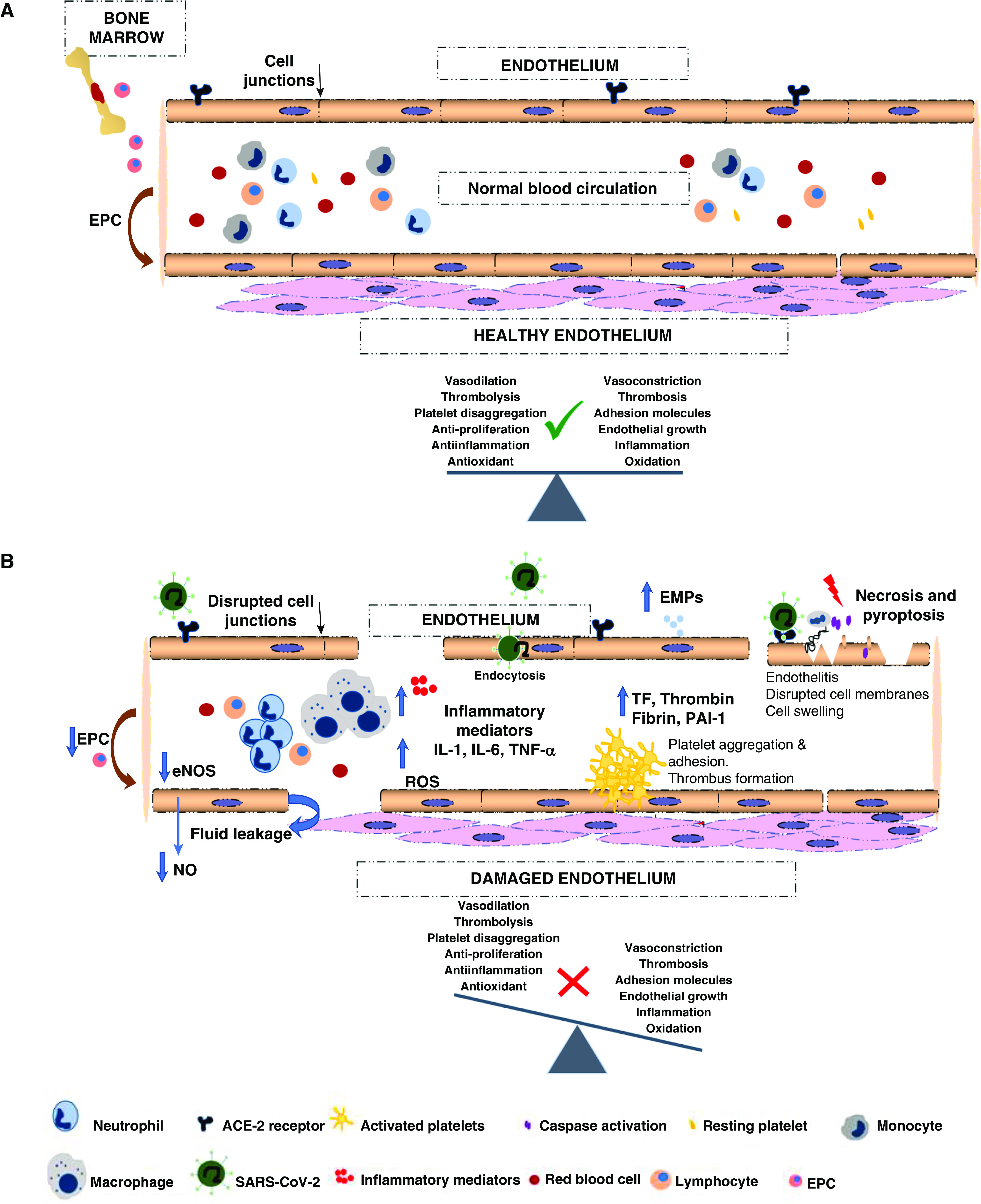
Potential mechanisms through which the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has effects on the endothelium. (A) Healthy endothelium: maintenance of a perfect dynamic balance among different factors that modulate systemic blood flow, coagulation, inflammatory responses, proliferation, and the oxidative state. (B) Damaged endothelium: the entrance of SARS-CoV-2 into endothelial cells triggers endothelial dysfunction associated with apoptosis and pyroptosis. Damage is produced by dysregulation of the balance between injury and repair capacity, leading to the release of inflammatory mediators that cause a state of abnormal systemic thromboinflammation, with an increase of oxidative and proliferative factors. Red arrows indicate EPC mobilization from the bone marrow to peripheral circulation. The black arrow points to the disrupted cell junctions. Blue arrows indicate increase in the decrease levels of inflammatory mediators and thrombotic factors. The red lightning bolt indicates injury, cell death (necrosis and pyroptosis). The green check mark indicates vascular homeostasis, and the red cross indicates damaged endothelium. EMP = endothelial microparticle; eNOS = endothelial NO synthase; EPC = endothelial progenitor cells; NO = nitric oxide; ROS = reactive oxygen species; TF = tissue factor.
To date, in coronavirus disease (COVID-19), the origin of endothelial dysfunction is not known. Increasing evidence suggests the presence of a direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection of ECs, inducing endothelial damage (5). The endothelium of arteries and veins appears to be highly sensitive to SARS-CoV-2 infection. Both arterial and venous ECs and arterial smooth muscle cells express the ACE-2 (angiotensin-converting enzyme 2) receptor (6), being direct targets of SARS-CoV-2 and placing the whole vascular system at risk of injury. However, endothelial dysfunction could also occur secondarily to the activation of inflammatory and/or coagulation and complement cascades. It is probable that both mechanisms take place and prompt a vast alteration in the patient’s normal endothelial function. In this review, we aim to describe recent data related to both endothelial dysfunction mechanisms.
The physical evidence of SARS-CoV-2 infection arises from observational studies in postmortem samples. Varga and colleagues observed the presence of an immune response within the endothelium (endotheliitis) and congested small blood vessels (5). Transmission EM images of the kidney endothelium from patients with COVID-19 revealed the presence of intracellular SARS-CoV-2 viral structures in ECs (7). Pesaresi and colleagues, also analyzing EM images, demonstrated the presence of SARS-CoV-2 particles contained in vesicles in the extracellular compartment close to lung smooth muscle cells in lung, renal, and heart tissues (7).
Vascular walls of different organs of patients with COVID-19 showed EC destruction, disruption of intercellular junctions, cell swelling, and a loss of contact with the basal membrane (5). Different mechanisms, such as apoptosis, necrosis, or pyroptosis, could be the cause of all these processes. First, apoptosis is defined as a programmed cell death that includes several cellular transformations, such as cell shrinkage, chromatin condensation, and DNA fragmentation (8). During apoptosis, the cellular membrane remains intact and several apoptotic bodies are exposed on the cellular surface, prompting rapid phagocytosis to prevent the release of intracellular content and surrounding tissue damage or inflammation (4). Second, EC necrosis causes the loss of cell membrane integrity and uncontrolled leakage of intracellular contents into the extracellular space (8). This triggers an inflammatory response that attracts phagocytes to the damaged site and causes tissue injury and healing inhibition (4). Finally, pyroptosis shares similarities with and diverges from both apoptosis and necrosis. In pyroptosis, cell lysis and inflammation occur, like during cell necrosis, and membrane blebbing and caspase are activated in a manner similar to activation during apoptosis (9). Although all of these processes have been proposed to possibly occur in patients with COVID-19, the vast EC injury observed seems to be closer to pyroptosis or necrosis than to apoptosis (10).
A deeper understanding of the cellular processes behind these endothelial abnormalities by the use of specific assays that enable distinguishing between programmed cell death processes might shed light on the precise mechanisms that underlie the pathological changes occurring in the vascular wall of patients with COVID-19 as well as on their potentially lethal consequences.
Potential Biomarkers in COVID-19
Circulating biomarkers have emerged as promising noninvasive surrogates that may provide insights into endothelial functional status (11). Endothelial extracellular vesicles (EVs) and endothelial progenitor cells (EPCs) are examples of such circulating biomarkers.
Endothelial EVs are particles naturally released from ECs contained by a lipid bilayer and carry coding and noncoding RNA, proteins, DNA fragments, and lipids, which facilitate cell–cell communication (12, 13). An increased number of endothelial EVs has been reported in a variety of vascularly related disorders (14, 15), and their amounts inversely correlated with endothelial function (11). A recent study reported that EVs isolated from the plasma of patients with COVID-19 carried high concentration of proinflammatory cytokines, proteases, and peptidases, which correlated with disease severity (12). This suggests that EVs could be important players in COVID-19 pathophysiology and disease progression.
Bone marrow–derived circulating EPCs are progenitor cells mobilized into the circulation in response to vascular injury (11). They are involved in the maintenance of the endothelium and in the restoration of its normal function (16). A decreasing number of EPCs has been established as an independent prognostic risk factor associated with endothelial dysfunction and high cardiovascular risk (17, 18). In patients suffering from severe sepsis, increased levels of circulating ECs, both circulating mature ECs (CECs) and EPCs, have been found, which is consistent with vascular injury (19).
In addition, recent evidence has shown that CECs were increased in patients with COVID-19 compared with healthy individuals, and their numbers returned to basal levels after recovery (20).
These early results encourage further investigation into the role of endothelial EVs, CECs, and EPCs as biomarkers of endothelial damage in patients with COVID-19 and into monitoring their number and function as markers of the disease state or the response to therapy.
Endothelial Injury: Thromboinflammation
Endothelial dysfunction and a subsequent dysregulation of coagulation and inflammatory responses represent an initial and essential step in the vascular clinical manifestations of patients with COVID-19. Complement activation, IFN induction (21), and generation of a proinflammatory feedback loop induced by SARS-CoV-2 infection are suggested to be important in the generation of EC damage. As always in nature, a tight equilibrium is the key, and like in other fatal respiratory viruses–associated diseases, COVID-19 very often results in a hyperinflammatory response with immune dysregulation and excessive infiltration of inflammatory cells into the lung tissue. This promotes severe lung injury and increases the risk of vascular hyperpermeability, multiorgan failure, and death (22, 23).
Persistent inflammation impairs biological anticoagulant activities, altering the hemostatic equilibrium to favor an increase of platelet reactivity, EC dysfunction, and thrombus formation (24). Inflammatory cytokines such as IL-1, IL-6, and TNF-α are important mediators involved in coagulation activation by upregulating prothrombotic factors and inhibiting fibrinolytic activity (24–26). Thrombotic and inflammatory mechanisms are largely interrelated and dependent on each other, and dysregulation can develop into a fatal vicious cycle (27). The mechanisms that combine both pathways are TF (tissue factor)–mediated thrombin generation, dysregulation of anticoagulant production (28), and reduction of fibrinolysis (26, 29). In natural conditions, when the levels of thrombin are increased in the coagulation process, thrombin interacts with thrombomodulin, a membrane protein present on the surface of ECs that promotes anticoagulation by protein C activation. In addition, anticoagulant proteins, such as antithrombin or TFPI (TF pathway inhibitor), are often impaired during inflammation, contributing further to the propagation of coagulation (30).
Another key enzyme involved in coagulation is plasmin, which is generated from plasminogen. It can degrade fibrin clots. During inflammatory events, TNF-α and IL-1β proinflammatory cytokines induce the production of PAI-1, the main plasminogen activator, in vascular ECs. Furthermore, platelet α granules also contain large amounts of PAI-1, which release upon activation (31). Future research aiming to identify risk factors to mitigate processes such as hyperinflammation and thrombosis are required to improve the survival of patients with COVID-19.
Thrombotic Complications in Patients with COVID-19
Thrombotic complications have been consistently observed in patients with severe COVID-19 (32, 33). The loss of even a small number of ECs due to SARS-CoV-2 infection could lead to disruption of the endothelial barrier, vascular leakage, and exposure of TF-expressing cells. This leads to an abnormal activation of the coagulation system that provokes small-vessel vasculitis and microthrombosis (34). Histological analysis in patients with COVID-19 has shown alveolar capillary microthrombi rates 9 times higher than those of patients with influenza A pneumonia (35).
A significant increase in blood levels of D-dimer, generated when cross-linked fibrin is broken down, and fibrin and fibrinogen degradation products was observed among patients with COVID-19 compared with healthy control subjects (P < 0.001) (36). In a retrospective study, Guan and colleagues reported that 46.4% of patients with COVID-19 presented with high D-dimer concentrations (37). Fogarty and colleagues also showed that 67% of their cohort (n = 88) had significantly higher D-dimer levels (32) (Table 1). It is been shown that higher levels of blood D-dimer is an indication of thrombosis, and its levels correlate with the probability of having pulmonary embolism (PE) (21). Patients with severe COVID-19 showed higher values of D-dimer and fibrinogen degradation products than those with milder manifestations (P < 0.05) (33). However, recent observers noted one patient with a 10-fold increase D-dimer levels on admission who did not have increased IL-6 (38). This suggests that in some patients, D-dimer levels might not go hand-in-hand with systemic inflammation, indicating the possibility of a primary thrombotic disease.
Table 1.
Relevant Parameters Related to COVID-19 Development and Severity
| Study | Ref. | Platelets |
CRP | D-Dimer |
Other Parameters | Incidence of TC | Population (Nonsevere/Severe) | ||
|---|---|---|---|---|---|---|---|---|---|
| Nonsevere | Severe | Nonsevere | Severe | ||||||
| Chen et al. (2020) | 42 | ↓ | — | ↑ | ↑ | — | ↓ PT; ↑ Serum ferritin | — | 99 |
| Cui et al. (2020) | 50 | ≈ | — | — | ↑ | ↑↑ | ↑ APTT; ↓ Lymphs | 25% VTE | 81 (61/20) |
| Fogarty et al. (2020) | 32 | ≈ | — | ↑↑ | ↑ | ↑↑ | ↑ FIB; ↑ PT | — | 83 (50/33) |
| Guan et al. (2020) | 37 | ↓ | ↓↓ | ↑ | ↑ | ↑↑ | ↓ Lymphs | — | 1,099 (926/173) |
| Han et al. (2020) | 54 | — | — | — | ↑ | ↑↑ | ↓ AT; ↑ FDP; ↑ FIB | — | 94 (49/45) |
| Helms et al. (2020) | 49 | ≈ | — | — | ↑ | — | ↑ FIB; ↑↑ FVII (50/57); ↑↑ vWF | 18% TEC; 16.7% PE; 2% DVT; 1.6% CIA; 0.7% LI; 0.7% MI; 2.7% | 150 (0/150) |
| Huang et al. (2020) | 43 | ↓ | ↓↓ | — | ↑ | ↑↑ | ↑ PT | — | 41 (28/13) |
| Klok et al. (2020) | 47 | — | — | — | — | — | — | 27% VTE; 21.9% PE | 184 (0/184) |
| Llitjos et al. (2020) | 48 | — | ≈ | ↑↑ | — | ↑↑ | — | 69% VTE; 23% PE | 26 (0/26) |
| Ranucci et al. (2020) | 57 | ≈ | — | — | ↑ | — | ↑ APTT; ↑ FIB | — | 16 (0/16) |
| Wichmann et al. (2020) | 52 | — | — | ↑ | — | ↑ | — | 33% PE | 12 (deceased) |
| Wu et al. (2020) | 58 | ↓↓ | ↓ | ↑↑ | ↑ | ↑ | ↑ APTT; ↑ PT; ↑ Serum ferritin | — | 201 (148/53) |
| Zhang et al. (2020) | 64 | — | — | ↑↑ | ↑ | ↑↑ | ↓ Lymphs | — | 140 (58/82) |
| Zhou et al. (2020) | 55 | ↓ | — | ↑ | — | — | ↓ Lymphs | — | 191 (137/54) |
Definition of abbreviations: ↑ = increase; ↑↑ = high increase; ↓ = decrease; ↓↓ = high decrease; APTT = activated partial thromboplastin time; AT = antithrombin; CIA = cerebral ischemic attack; COVID-19 = coronavirus disease; CRP = C-reactive protein; DVT = deep vein thrombosis; FDP = fibrin degradation products; FIB = fibrinogen; HC = hemorragic complications; LI = limb ischemia; lymphs = lymphocytes; MI = mesenteric ischemia; PE = pulmonary embolism; PT = prothrombin time; Ref. = reference; TC = thrombotic complications; TEC = thromboembolic complications; VTE = venous thromboembolism.
Platelets: A lower concentration of platelets has been reported by several authors. CRP: Elevated CRP levels have been found in patients with COVID-19. D-dimer: D-dimer is a predictive coagulation marker for adverse COVID-19 evolution. Other parameters: Different parameters related to coagulation have been studied. Incidence of TC: Significant TC are shown that might play a major role in COVID-19 development.
Thrombocytopenia has also been reported in patients with COVID-19, although abnormalities in prothrombin time, partial thromboplastin time, and platelet counts might not be present (39). Giannis and colleagues reported that 36.2% of patients with COVID-19 showed thrombocytopenia that increases with disease severity (40). Lippi and colleagues stated that a significantly lower platelet count was reported in the most severe COVID-19 cases (41). Besides this, Chen and colleagues and Huang and colleagues found thrombocytopenia in patients COVID-19 (42, 43) (Table 1). The reasons behind thrombocytopenia in patients with COVID-19 might be 1) bone marrow failure and reduced platelet production, 2) increased platelet damage due to viral antigens attached to platelet surface, and/or 3) platelet aggregation and formation of thrombi that lower the number of circulating platelets (44). Further studies are needed to determine the role of the platelets in the thrombotic complications in patients with COVID-19.
Pulmonary Thrombosis in Patients with COVID-19
Distinct coagulopathies have been described to be related to COVID-19, such as venous thromboembolism (VTE) and disseminated intravascular coagulation (DIC) (45, 46).
VTE
Deep vein thrombosis (DVT) is the formation of a blood clot inside the deep veins, usually in the leg. If the thrombotic embolism moves toward the lungs, it can develop into PE. VTE is the combination of both procedures. Recent data suggest that around 30–70% of patients with COVID-19 who are admitted to the ICU develop blood clots in the deep veins of the legs or lungs (47, 48). Helms and colleagues found that 18% of patients with COVID-19 present thromboembolic complications, including PE (16.7%) and DVT (2%), among others (49). Klok and colleagues reported a 27% rate of VTE in patients with COVID-19, although their study did not have a systematic assessment (47). Cui and colleagues showed a 25% incidence rate of VTE using a systematic assessment in patients with COVID-19 but without thromboprophylaxis (50). This study reports that D-dimer levels of 1,500 ng/ml predict with high sensitivity and specificity which patients will develop VTE. More recently, Llitjos and colleagues, with a systematic assessment of VTE using complete duplex-doppler ultrasound in patients with severe COVID-19, reported a 69% incidence rate of VTE and a 23% incidence rate of PE (48) (Table 1). Besides this, it has been found that VTE also happened in a percentage of patients treated with therapeutic anticoagulation from admission, highlighting the thrombogenicity of COVID-19 (48).
The incidence of coagulopathies during COVID-19 seems to be high and also rare compared with the incidence during other coronaviruses infections. Consistent with the hypothesis that coagulation activation may play a role in COVID-19 pathogenesis, postmortem studies have highlighted diffuse alveolar damage, edema, and marked pathological changes specifically involving the lung microvasculature, including disseminated microthrombi and significant hemorrhagic necrosis (51). Wichmann and colleagues reported that out of the total of autopsies of patients with COVID-19 performed (n = 12), 33% of the autopsies showed massive PE with or without underlying DVT, despite the absence of a history of VTE, indicating the possibility of an in situ pulmonary thrombosis (52) (Table 1). Male sex, older age (>60 yr), a history of smoking, dyspnea as an initial symptom, and the presence of preexisting comorbidities such as hypertension, type 2 diabetes mellitus, obesity, chronic obstructive pulmonary disease, and coronary heart disease, among others, might contribute to the development of VTE. All of these involve significant endothelial dysfunction.
DIC
DIC is observed by the presence of blood clots throughout the body that could block small blood vessels. Interestingly, 71.4% of nonsurvivors of COVID-19 versus 0.6% of survivors fulfilled the clinical criteria for a prothrombotic form of DIC, with a median time from admission to DIC manifestation of 4 days (33). The American Society of Hematology has recently named this new prothrombotic form of DIC “COVID-19–associated coagulopathy” (53). This new coagulopathy pattern is characterized by high levels of D-dimer and fibrinogen. These patients also present a rise in inflammation blood markers such as CRP (C-reactive protein ). CRP has been shown to facilitate monocyte–EC interactions (54) and to promote PAI-1 and TF activation. It has been reported that there are high levels of CRP in patients with COVID-19 (55). However, unlike classical DIC, thrombocytopenia in COVID-19–associated coagulopathy is mild (platelet count of ∼100 × 109/L) and often has minimal activated partial thromboplastin time (APTT) and/or prothrombin time (56). In alignment with these facts, Ranucci and colleagues reported higher APTT and fibrinogen in patients with COVID-19 (57). Moreover, elevated APTT and prothrombin time were found by Wu and colleagues (58) (Table 1).
Currently, the administration of low-molecular-weight heparin for all hospitalized patients with COVID-19 in the absence of active bleeding is recommended as an effective treatment to prevent the risk of DIC and death. Tang and colleagues reported lower mortality rates and a better prognosis in heparin-treated patients with COVID-19 than in nontreated patients with COVID-19 (59). Preliminary data from Mycroft-West and colleagues suggested that heparin might present further antiviral properties, protecting the mucosa of the respiratory tract and consequently preventing SARS-CoV-2 infection (60). However, randomized studies to evaluate heparin-administration efficacy are required.
Intussusceptive Angiogenesis
Vessel growth has been also observed to be higher in the lungs of patients with COVID-19 than in patients who died of influenza A pneumonia or in healthy control subjects (35). Intussusceptive angiogenesis or nonsprouting angiogenesis is a dynamic and still quite enigmatic process in which an existing capillary wall called the intussusceptive pillar invaginates into the lumen and splits a single vessel in two (61). Intussusceptive angiogenesis occurs in normal development and under pathological conditions such as lung fibrosis, inflammation, or cancer (61). Recently, Ackermann and colleagues reported that the level of intussusceptive angiogenesis might be an important factor in COVID-19 pathogenesis (35). It is possible that the presence of DIC throughout the body in some patients with COVID-19 could modify the dynamics of the microvascular networks and induce intussusceptive angiogenesis. Further studies in patients with COVID-19 assessing the levels of growth and angiogenic factors such as VEGF (vascular endothelial growth factor), FGF-2 (fibroblast growth factor-2), HGF (hepatocyte growth factor), angiopoietin-1, and ephrin-B and its receptors, among others, will be of great interest to assess the magnitude of vascular injury after SARS-CoV-2 infection.
Post–Pulmonary Thrombosis Syndrome after COVID-19: The Imminent Sequelae
Another important aspect to take into account is post–pulmonary thrombosis syndrome (27). This syndrome consists of persistent thrombotic lesions and involves long-term functional limitations, including those for patients with a chronic thromboembolic disease without pulmonary hypertension (62) and those for patients with chronic thromboembolic pulmonary hypertension. It is reported that 2–4% of patients with acute PE within 2 years after the embolic event will develop chronic thromboembolic pulmonary hypertension, but this is mostly underdiagnosed (63). It is currently unknown which potential sequelae, in the lungs and at other organ levels, may occur over the medium to long term in survivors of SARS-CoV-2 (27), and determining this will require the development of future studies.
In previous epidemics of other coronaviruses, respiratory deterioration as measured by imaging and lung-function techniques was observed to persist up to 1 year after the apparent clinical recovery of the patients (64). Therefore, it is likely to be similar in survivors of COVID-19, and it is likely that significant pulmonary vascular alterations may appear in the short to medium term after overcoming the acute infection. All of these possible consequences will be seen in the coming months or years.
Preventive Therapies of Endothelial Damage in COVID-19
There are several potential strategies that could be used to prevent SARS-CoV-2 infection. In this review, two different approaches that are being considered for COVID-19 treatment are going to be described.
Targeting the ACE-2 receptor could directly impede SARS-CoV-2 infection by ADAM17 levels increment. ADAM17 is a metalloproteinase involved in the shedding of different membrane-anchored cytokines, cell adhesion molecules, and enzymes such as ACE-2 (65). The increase of ADAM17 levels might accelerate ACE-2 shedding and reduce the chances of SARS-CoV-2 infectivity. Interestingly, higher shedding of ACE-2 has been observed in women than in men, which could potentially explain why men seem to be a more susceptible population (66). However, downregulation of ACE-2 might present unfavorable effects. ACE-2 is an essential enzyme in the renin–angiotensin system and is key in regulating systemic blood pressure and fluid and electrolyte balance balance. Studies with animal models have shown that pharmacological activation of ACE-2 was able to lower systemic blood pressure, attenuate platelet attachment to vessels, increase antiinflammatory mechanisms, and reduce the probabilities of thrombus formation (67). Future studies are essential to accurately assess the benefits and damages of the presence and/or activity or absence and/or inhibition of ACE-2 for SARS-CoV-2 infection and thrombogenic capabilities as well as to design novel therapies for restoring vascular homeostasis to treat and protect patients with COVID-19.
Nitric oxide (NO) is also being explored as an experimental treatment for patients with COVID-19. NO, generated from l-arginine by NOS (NO synthase), is an important vasoactive factor that regulates vasodilatation, inflammation, thrombosis, and the immune response and inhibits important events that contribute to the development of vascular-remodeling diseases (56). As extensive EC injury has been reported in COVID-19, it would be of interest to study the iNOS (inducible NOS) levels and NO production after SARS-CoV-2 infection. Interestingly, the involvement of NO in apoptosis induction has been reported in different cell systems and after NS1 dengue virus infection. When the NO precursor arginine and the eNOS (endothelial NOS) cofactor tetrahydrobiopterin are not available, eNOS fails to produce NO and promote the formation of reactive oxygen species, causing endothelial dysfunction (56).
It has been demonstrated that an imbalance between vasodilators, such as NO and prostacyclin, and vasoconstrictors, such as ET-1 (endothelin-1) and thromboxane, results in the disruption of basal pulmonary vascular tone, vascular injury repair, and growth (68). Accordingly, it is possible that patients currently being treated with specific drugs aimed at protecting endothelial well-being, such as vasodilators, angiotensin II antagonist drugs, endothelin receptor antagonists, and antiproliferative or antithrombotic treatments, may confer a protective benefit in SARS-CoV-2 infection or in the development of severe complications (Figure 2).
Figure 2.
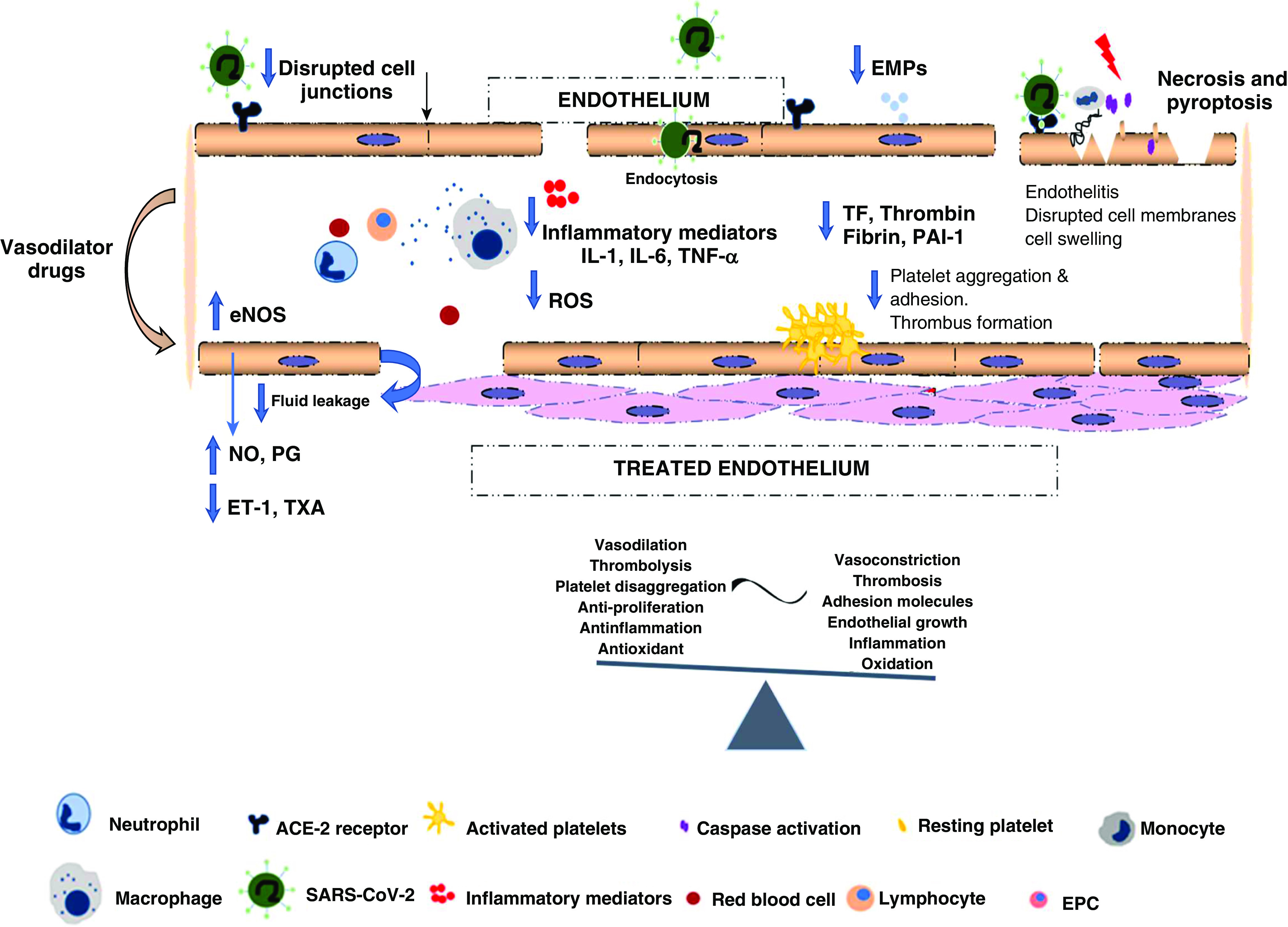
Treated endothelium: vasodilator drugs used nowadays may produce a protective effect on the endothelium that can mitigate the development to severe stages of coronavirus disease (COVID-19). Red arrows indicate EPC mobilization from the bone marrow to peripheral circulation. The black arrow points to the disrupted cell junctions. Blue arrows indicate increase in the decrease levels of inflammatory mediators and thrombotic factors. The red lightning bolt indicates injury, cell death (necrosis and pyroptosis). The green check mark indicates vascular homeostasis, and the red cross indicates damaged endothelium. ET-1 = endothelin-1; PG = prostacyclins; TXA = thromboxane.
Recent studies have used inhaled NO in patients with COVID-19 and severe hypoxemia, indicating that inhaled-NO therapy may have a preventive and/or rescue role because of its potent vasodilator effect on pulmonary circulation (69–71).
There are still too many open questions about the mechanisms underlying the pathophysiology of SARS-CoV-2 infection, the thromboinflammatory complications, and the potential and magnitude of post–SARS-CoV-2 sequelae. As always, time will tell, and all the present and future studies will be essential for setting strong foundations to enable establishing novel strategies to prevent COVID-19, treat patients with COVID-19, and prepare for other fatal infections.
Supplementary Material
Footnotes
Originally Published in Press as DOI: 10.1165/rcmb.2020-0359PS on November 12, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Smolders VF, Zodda E, Quax PHA, Carini M, Barberà JA, Thomson TM, et al. Metabolic alterations in cardiopulmonary vascular dysfunction. Front Mol Biosci. 2019;5:120. doi: 10.3389/fmolb.2018.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eelen G, de Zeeuw P, Treps L, Harjes U, Wong BW, Carmeliet P. Endothelial cell metabolism. Physiol Rev. 2018;98:3–58. doi: 10.1152/physrev.00001.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. 2010;4:302–312. doi: 10.2174/1874192401004010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winn RK, Harlan JM. The role of endothelial cell apoptosis in inflammatory and immune diseases. J Thromb Haemost. 2005;3:1815–1824. doi: 10.1111/j.1538-7836.2005.01378.x. [DOI] [PubMed] [Google Scholar]
- 5.Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280, e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pesaresi M, Pirani F, Tagliabracci A, Valsecchi M, Procopio AD, Busardò FP, et al. SARS-CoV-2 identification in lungs, heart and kidney specimens by transmission and scanning electron microscopy. Eur Rev Med Pharmacol Sci. 2020;24:5186–5188. doi: 10.26355/eurrev_202005_21217. [DOI] [PubMed] [Google Scholar]
- 8.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: a new frontier in cancer. Biomed Pharmacother. 2020;121:109595. doi: 10.1016/j.biopha.2019.109595. [DOI] [PubMed] [Google Scholar]
- 10.Yang M. SSRN. 2020. Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV infection [preprint] [accessed 2020 Jan 29; updated 2020 Feb 5]. Available from: [DOI] [Google Scholar]
- 11.García-Lucio J, Peinado VI, de Jover L, Del Pozo R, Blanco I, Bonjoch C, et al. Imbalance between endothelial damage and repair capacity in chronic obstructive pulmonary disease. PLoS One. 2018;13:e0195724. doi: 10.1371/journal.pone.0195724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnamachary B, Cook C, Spikes L, Chalise P, Dhillon NK. The potential role of extracellular vesicles in COVID-19 associated endothelial injury and pro-inflammation [preprint] medRxiv. 2020 doi: 10.1101/2020.08.27.20182808. [accessed 2020 Sep 1]. Available from: [DOI]
- 13.Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thulin Å, Christersson C, Alfredsson J, Siegbahn A. Circulating cell-derived microparticles as biomarkers in cardiovascular disease. Biomarkers Med. 2016;10:1009–1022. doi: 10.2217/bmm-2016-0035. [DOI] [PubMed] [Google Scholar]
- 15.Jansen F, Nickenig G, Werner N. Extracellular vesicles in cardiovascular disease: potential applications in diagnosis, prognosis, and epidemiology. Circ Res. 2017;120:1649–1657. doi: 10.1161/CIRCRESAHA.117.310752. [DOI] [PubMed] [Google Scholar]
- 16.Burger D, Touyz RM. Cellular biomarkers of endothelial health: microparticles, endothelial progenitor cells, and circulating endothelial cells. J Am Soc Hypertens. 2012;6:85–99. doi: 10.1016/j.jash.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Koutroumpi M, Dimopoulos S, Psarra K, Kyprianou T, Nanas S. Circulating endothelial and progenitor cells: evidence from acute and long-term exercise effects. World J Cardiol. 2012;4:312–326. doi: 10.4330/wjc.v4.i12.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt-Lucke C, Rössig L, Fichtlscherer S, Vasa M, Britten M, Kämper U, et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111:2981–2987. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- 19.Cribbs SK, Martin GS, Rojas M. Monitoring of endothelial dysfunction in critically ill patients: the role of endothelial progenitor cells. Curr Opin Crit Care. 2008;14:354–360. doi: 10.1097/MCC.0b013e3282fc216d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancuso P, Gidaro A, Gregato G, Raveane A, Cremonesi P, Quarna J, et al. Circulating endothelial progenitors are increased in COVID-19 patients and correlate with SARS-CoV-2 RNA in severe cases. J Thromb Haemost. 2020;18:2744–2750. doi: 10.1111/jth.15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kearon C, de Wit K, Parpia S, Schulman S, Afilalo M, Hirsch A, et al. PEGeD Study Investigators. Diagnosis of pulmonary embolism with d-dimer adjusted to clinical probability. N Engl J Med. 2019;381:2125–2134. doi: 10.1056/NEJMoa1909159. [DOI] [PubMed] [Google Scholar]
- 22.Ye Q, Wang B, Mao J. Cytokine storm in COVID-19 and treatment. J Infect. 2020;80:607–613. doi: 10.1016/j.jinf.2020.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. 2020;8:e46–e47. doi: 10.1016/S2213-2600(20)30216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417–430. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- 25.Lordan R, Tsoupras A, Zabetakis I. Platelet activation and prothrombotic mediators at the nexus of inflammation and atherosclerosis: potential role of antiplatelet agents. Blood Rev. doi: 10.1016/j.blre.2020.100694. [online ahead of print] 21 Apr 2020; DOI: 10.1016/j.blre.2020.100694. [DOI] [PubMed] [Google Scholar]
- 26.Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26–S34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- 27.d’Alessandro E, Becker C, Bergmeier W, Bode C, Bourne JH, Brown H, et al. Scientific Reviewer Committee. Thrombo-inflammation in cardiovascular disease: an expert consensus document from the third Maastricht consensus conference on thrombosis. Thromb Haemost. 2020;120:538–564. doi: 10.1055/s-0040-1708035. [DOI] [PubMed] [Google Scholar]
- 28.Levi M, Keller TT, van Gorp E, ten Cate H. Infection and inflammation and the coagulation system. Cardiovasc Res. 2003;60:26–39. doi: 10.1016/s0008-6363(02)00857-x. [DOI] [PubMed] [Google Scholar]
- 29.Pérez-Gómez F, Bover R. The new coagulation cascade and its possible influence on the delicate balance between thrombosis and hemorrhage [in Spanish] Rev Esp Cardiol. 2007;60:1217–1219. doi: 10.1157/13113924. [DOI] [PubMed] [Google Scholar]
- 30.Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margetic S. Inflammation and haemostasis. Biochem Med (Zagreb) 2012;22:49–62. [PMC free article] [PubMed] [Google Scholar]
- 32.Fogarty H, Townsend L, Ni Cheallaigh C, Bergin C, Martin-Loeches I, Browne P, et al. COVID19 coagulopathy in Caucasian patients. Br J Haematol. 2020;189:1044–1049. doi: 10.1111/bjh.16749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological findings and complications of COVID-19. Am J Hematol. 2020;95:834–847. doi: 10.1002/ajh.25829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song J-C, Wang G, Zhang W, Zhang Y, Li W-Q, Zhou Z. Chinese expert consensus on diagnosis and treatment of subacute combined degeneration. Chin J Neurol. 2020;53:269–273. [Google Scholar]
- 35.Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casey K, Iteen A, Nicolini R, Auten J. COVID-19 pneumonia with hemoptysis: acute segmental pulmonary emboli associated with novel coronavirus infection. Am J Emerg Med. 2020;38:1544, e1–1544, e3. doi: 10.1016/j.ajem.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. China Medical Treatment Expert Group for COVID-19. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oudkerk M, Büller HR, Kuijpers D, van Es N, Oudkerk SF, McLoud T, et al. Diagnosis, prevention, and treatment of thromboembolic complications in COVID-19: report of the National Institute for Public Health of the Netherlands. Radiology. 2020;297:E216–E222. doi: 10.1148/radiol.2020201629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Connors JM, Levy JH. Thromboinflammation and the hypercoagulability of COVID-19. J Thromb Haemost. 2020;18:1559–1561. doi: 10.1111/jth.14849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giannis D, Ziogas IA, Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J Clin Virol. 2020;127:104362. doi: 10.1016/j.jcv.2020.104362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020;506:145–148. doi: 10.1016/j.cca.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu P, Zhou Q, Xu J. Mechanism of thrombocytopenia in COVID-19 patients. Ann Hematol. 2020;99:1205–1208. doi: 10.1007/s00277-020-04019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kollias A, Kyriakoulis KG, Dimakakos E, Poulakou G, Stergiou GS, Syrigos K, et al. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: emerging evidence and call for action. Br J Haematol. 2020;189:846–847. doi: 10.1111/bjh.16727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Makatsariya AD, Grigoreva KN, Mingalimov MA, Bitsadze VO, Khizroeva JK, Tretyakova MV, et al. Coronavirus disease (COVID-19) and disseminated intravascular coagulation syndrome. Obstet Gynecol Reprod. 2020;14:123–131. [Google Scholar]
- 47.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Llitjos JF, Leclerc M, Chochois C, Monsallier JM, Ramakers M, Auvray M, et al. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost. 2020;18:1743–1746. doi: 10.1111/jth.14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–1098. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020;18:1421–1424. doi: 10.1111/jth.14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deshpande C. Thromboembolic findings in COVID-19 autopsies: pulmonary thrombosis or embolism? Ann Intern Med. 2020;173:394–395. doi: 10.7326/M20-3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wichmann D, Sperhake J-P, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med. 2020;173:268–277. doi: 10.7326/M20-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135:2033–2040. doi: 10.1182/blood.2020006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han H, Yang L, Liu R, Liu F, Wu KL, Li J, et al. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. 2020;58:1116–1120. doi: 10.1515/cclm-2020-0188. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Y, Han T, Chen J, Hou C, Hua L, He S, et al. Clinical and autoimmune characteristics of severe and critical cases of COVID-19. Clin Transl Sci. 2020;13:1077–1086. doi: 10.1111/cts.12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837, 837a–837d. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ranucci M, Ballotta A, Di Dedda U, Bayshnikova E, Dei Poli M, Resta M, et al. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost. 2020;18:1747–1751. doi: 10.1111/jth.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180:934–943. doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang N, Bai H, Chen X, Gong J, Li D, Sun Z, et al. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18:1094–1099. doi: 10.1111/jth.14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mycroft-West CJ, Yates EA, Skidmore MA. Marine glycosaminoglycan-like carbohydrates as potential drug candidates for infectious disease. Biochem Soc Trans. 2018;46:919–929. doi: 10.1042/BST20170404. [DOI] [PubMed] [Google Scholar]
- 61.De Spiegelaere W, Casteleyn C, Van den Broeck W, Plendl J, Bahramsoltani M, Simoens P, et al. Intussusceptive angiogenesis: a biologically relevant form of angiogenesis. J Vasc Res. 2012;49:390–404. doi: 10.1159/000338278. [DOI] [PubMed] [Google Scholar]
- 62.Otero R, Oribe M, Ballaz A, Jimenez D, Uresandi F, Nauffal D, et al. Echocardiographic assessment of pulmonary arterial pressure in the follow-up of patients with pulmonary embolism. Thromb Res. 2011;127:303–308. doi: 10.1016/j.thromres.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 63.Martinez C, Wallenhorst C, Teal S, Cohen AT, Peacock AJ. Incidence and risk factors of chronic thromboembolic pulmonary hypertension following venous thromboembolism, a population-based cohort study in England. Pulm Circ. 2018;8:2045894018791358. doi: 10.1177/2045894018791358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang P, Li J, Liu H, Han N, Ju J, Kou Y, et al. Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: a 15-year follow-up from a prospective cohort study. Bone Res. 2020;8:8. doi: 10.1038/s41413-020-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palau V, Riera M, Soler MJ. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol Dial Transplant. 2020;35:1071–1072. doi: 10.1093/ndt/gfaa093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rizzo P, Vieceli Dalla Sega F, Fortini F, Marracino L, Rapezzi C, Ferrari R. COVID-19 in the heart and the lungs: could we “Notch” the inflammatory storm? Basic Res Cardiol. 2020;115:31. doi: 10.1007/s00395-020-0791-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fraga-Silva RA, Sorg BS, Wankhede M, Dedeugd C, Jun JY, Baker MB, et al. ACE2 activation promotes antithrombotic activity. Mol Med. 2010;16:210–215. doi: 10.2119/molmed.2009.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 69.Tavazzi G, Marco P, Mongodi S, Dammassa V, Romito G, Mojoli F. Inhaled nitric oxide in patients admitted to intensive care unit with COVID-19 pneumonia. Crit Care. 2020;24:508. doi: 10.1186/s13054-020-03222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferrari M, Santini A, Protti A, Andreis DT, Iapichino G, Castellani G, et al. Inhaled nitric oxide in mechanically ventilated patients with COVID-19. J Crit Care. 2020;60:159–160. doi: 10.1016/j.jcrc.2020.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parikh R, Wilson C, Weinberg J, Gavin D, Murphy J, Reardon CC. Inhaled nitric oxide treatment in spontaneously breathing COVID-19 patients. Ther Adv Respir Dis. 2020;14:1753466620933510. doi: 10.1177/1753466620933510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.