

Abstract
Skeletal muscle is ideally suited and highly desirable as a target for therapeutic gene delivery because of its abundance, high vascularization, and high levels of protein expression. However, efficient gene delivery to skeletal muscle remains a current challenge. Besides the major obstacle of cell-specific targeting, efficient intracellular trafficking, or the cytosolic transport of DNA to the nucleus, must be demonstrated. To overcome the challenge of cell-specific targeting, herein we develop a generation 5-polyamidoamine dendrimer (G5-PAMAM) functionalized with a skeletal muscle-targeted peptide, ASSLNIA (G5-SMTP). Specifically, to demonstrate the feasibility of our approach, we prepared a complex of our G5-SMTP dendrimer with a plasmid encoding firefly luciferase and investigated its delivery to skeletal muscle cells. Luciferase assays indicated a threefold increase in transfection efficiency of C2C12 murine skeletal muscle cells using G5-SMTP when compared with nontargeting nanocarriers using unmodified G5. To further improve the transfection yield, we employed a cationic dynein light chain 8 protein (DLC8)-binding peptide (DBP) containing an internal sequence known to bind to the DLC8 of the dynein motor protein complex. Complexation of DBP with our targeting nanocarrier, that is, G5-SMTP, and our luciferase plasmid cargo resulted in a functional nanocarrier that showed an additional sixfold increase in transfection efficiency compared with G5-SMTP transfection alone. To our knowledge, this is the first successful use of two different functional nanocarrier components that enable targeted skeletal muscle cell recognition and increased efficiency of intracellular trafficking to synergistically enhance gene delivery to skeletal muscle cells. This strategy of targeting and trafficking can also be universally applied to any cell/tissue type for which a recognition domain exists.
Keywords: skeletal muscle targeting, PAMAM dendrimer, plasmid transfection, cytoplasmic transport, dynein light chain targeting
Graphical Abstract
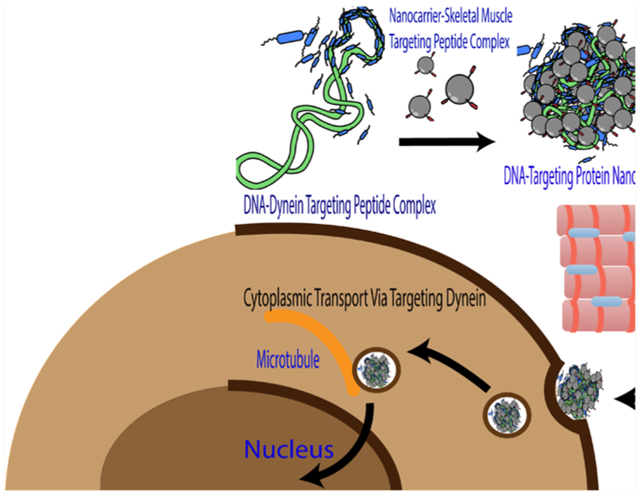
INTRODUCTION
Skeletal muscle has long been thought a suitable target for gene therapy for the amelioration of both muscle- and nonmuscle-related diseases.1,2 As one of the most abundant tissues in the body, skeletal muscle offers the obvious advantage of being easily accessible for virtually any type of gene delivery approach. Another advantage is that skeletal muscle tissue is multinucleated, offering multiple sites for continuous transgene expression of the delivered nucleic acid, leading to skeletal muscle tissue being described as a “protein factory”. For these reasons, skeletal muscle holds immense potential as a site for gene therapy applications against a large variety of diseases.
While promising, gene delivery to skeletal muscle must overcome several barriers to achieve high transfection efficiency.3,4 The most obvious extracellular barrier to gene delivery is cell-specific targeting. Most of the approaches for skeletal muscle-specific gene delivery have focused on direct transfer of nucleic acids by means of injection or electroporation.5 Direct injection has shown some feasibility, but transfection efficiency has been relatively low. Also, systemic gene therapy, for instance correction of a dysfunctional dystrophin gene, would be impossible to achieve with such a localized means of gene delivery. Electroporation has fared much better as a method of gene transfer,5,6 but it is also very localized, and tissue damage has been reported with this method.5 A third method of gene delivery applied to skeletal muscle cells has been the use of the adeno-associated virus (AAV).7 Already one of the most efficient vectors for gene delivery, AAV has also been modified to include the skeletal muscle-targeting ASSLNIA peptide on the surface of its capsid.8 Incorporation of this peptide into the AAV capsid has been shown to increase tropism of the virus to skeletal muscle as well as enhance transfection efficiency. However, the use of AAV for gene transfer is hampered by limitations on DNA packaging and potential vector immunogenicity.9 A higher transfection efficiency is crucial for gene delivery to be utilized as an actual therapeutic platform.
In addition to cell-specific targeting, various intracellular barriers to gene delivery must be overcome.10 Once targeted to the cell of interest, the DNA must traverse the plasma membrane, escape the endolysosomal compartment, migrate through the cytoplasm, and translocate across the nuclear membrane. Various polymeric gene delivery platforms have been reported, with functionalities that include endolysosomal escape and nuclear targeting.11–13 However, little attention has been paid to the cytoplasmic transport that takes place between escaping the endolysosome and arriving at the nucleus. It is known that viruses such as AAV can exploit the dynein motor protein complex, the molecular machinery responsible for transporting cellular cargo on microtubules, during cytoplasmic transport.14 Using this as inspiration, some groups have developed gene delivery platforms with targeting capabilities specific for the dynein motor protein complex and have observed enhanced transfection efficiency in vitro.15 However, there are no reports published on improving transfection efficiency by combining cellular targeting and intracellular trafficking.
Polymeric gene delivery has been extensively studied as an alternative means to conventional nonviral and viral delivery.16 Unlike the established methods previously discussed, polymeric gene delivery offers the advantages of reduced toxicity, low immunogenicity, and an increased capacity for DNA packaging. In addition, functional moieties can be integrated into polymers to confer the ability to deliver nucleic acids to specific tissue types17 as well as to enhance intracellular trafficking to the nucleus.18 Among the most characterized and utilized polymers for gene delivery are different generations of polyamidoamine (PAMAM) dendrimers.19 PAMAM dendrimers are hyperbranched molecules consisting of an ethylene diamine core and repeating monomers of ethylene diamine and methyl acrylate. With each successive addition of monomers, layers of branch points (generations) are formed,20 with the outermost generation containing terminal primary amine groups. It is through these amines, which are protonated at physiological pH,21 that PAMAM dendrimers interact electro-statically with the negatively charged phosphate backbone of DNA to form the transfection polyplex.22 PAMAM dendrimers with high positive charge can interact with the cell membrane as well as aid in endolysosomal escape for effective transport and release of cargo into the cytoplasm. In addition, the terminal amine groups can be functionalized with targeting moieties to increase tropism of the polyplex to specific cells of interest.17 For example, Bae et al. functionalized PAMAM dendrimers with peptides specific for mesenchymal stem cells,23 and Daftarian et al. developed a dendrimer functionalized with an MHC class II-targeted peptide for delivery of DNA vaccines to antigen-presenting cells.24 In another example, Liu et al. used PAMAM dendrimers complexed with E-selectin as a platform to deliver bone marrow stem cells for wound-healing purposes.25 Recently a new class of dendrimers, fluorinated dendrimers, has been shown to achieve very high gene transfection efficiency in several cell lines at very low N/P ratios.26–28
In this manuscript, we enhanced gene delivery within skeletal muscle cells using dendrimer/DNA polyplexes while concurrently promoting cell-specific interactions and cytoplasmic transport of the DNA cargo using targeting peptides. For this, we developed a generation 5 PAMAM dendrimer functionalized with the ASSLNIA skeletal muscle targeting peptide (G5-SMTP) for improved cell-type selectivity as well as a dynein-binding peptide (DBP) for enhanced transfection efficiency of our cargo DNA, an expression plasmid (pLuc) for luciferase reporter. We show that G5-SMTP exhibits increased affinity for skeletal muscle cells compared with unmodified G5. Furthermore, inclusion of the DBP further enhances G5-SMTP-mediated gene delivery to skeletal muscle cells. These methods in combination lead to enhanced transfection efficiency over standard polymeric gene delivery.
EXPERIMENTAL SECTION
Materials.
Ethidium bromide, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), ethylenediaminetetraacetic acid (EDTA), Dulbecco’s phosphate-buffered saline without calcium and magnesium (PBS), and G5 PAMAM dendrimer were purchased from Sigma-Aldrich (St. Louis, MO). G5-SMTP was synthesized and purified by 21st Century Biochemicals (Marlboro, MA). VivoTag-S 680XL was obtained from PerkinElmer (Waltham, MA). Plasmid pLuc was obtained from Promega (Madison, WI). His-tagged DLC-8 was purchased from Abcam (Cambridge, MA). DBP (CHHHKKKKETQTKKKHHHC) was synthesized and purified by Biomatik (Wilmington, DE). Ni-NTA agarose beads were obtained from Qiagen (Valencia, CA). GelRed was purchased from VWR (Radnor, PA). C2C12 cell culture was obtained from American Type Culture Collection (ATCC) (Manassas, VA).
Dendrimer–Plasmid DNA and Dendrimer–Plasmid DNA–DBP Polyplex Formulation.
Polyplex preparation was performed according to the charge ratio (±), which is the ratio of positively charged moieties on a dendrimer polycation (+) to negatively charged plasmid DNA phosphate groups (−). For dendrimer–plasmid DNA polyplex formation, 50 μL of G5 (0.035–1.4 g/L) or G5-skeletal muscle targeted peptide (G5-SMTP) (0.0375–1.5 g/L) and 50 μL of pLuc (0.1 g/L) in HEPES buffer (20 mM HEPES, pH 7.4) were mixed together and incubated at room temperature for at least 15 min prior to use. G5-SMTP–plasmid DNA–DBP polyplexes were formed by mixing equal volumes of DBP, pLuc, and dendrimer solutions. Briefly, one volume of DBP (0.1 or 0.2 μg/μL) and one volume of pLuc (0.1 g/L) were mixed in HEPES buffer and incubated at room temperature for 15 min. After incubation, one volume of G5-SMTP solution (0.0375–1.5 g/L) was added to the DBP–pLuc mixture and incubated at room temperature for 15 min.
Gel Retention Assay.
Gel electrophoresis was performed on 0.7% (w/v) agarose gels containing 2.5 μL of GelRed and prepared in 50 mM Trisacetate buffer containing 2 mM EDTA. Polyplexes were prepared by adding 10 μL of varying concentrations of G5 (0.035–1.4 g/L) or G5-SMTP (0.0375–1.5 g/L) solution to 10 μL of pLuc solution (0.1 g/L). Samples were incubated at room temperature for at least 15 min and then analyzed by agarose gel electrophoresis.
Ethidium Bromide Exclusion Assay.
Ethidium bromide solution (500 mg/L) was added to pLuc solution (0.1 g/L) at a 4:1 base-pair/ethidium bromide molar ratio, and the resultant solution was incubated at room temperature for 1h. Samples were then prepared by adding equal volumes of G5 (0.035–1.4 g/L) or G5-SMTP (0.0375–1.5 g/L) solution to pLuc intercalated with ethidium bromide. The samples were incubated at room temperature for at least 15 min prior to analysis. Measurements were made by reading the fluorescence intensity at an excitation of 510 nm and emission of 595 nm using a Varian Cary Eclipse fluorescence spectrophotometer.
Size and Zeta Potential Characterization.
Dendrimer polyplexes were prepared by adding varying concentrations of G5 (0.035–1.4 g/L) or G5-SMTP (0.0375–1.5 g/L) in 50 μL aliquots to 50 μL of pLuc solution (0.1 g/L). The samples were incubated at room temperature for at least 15 min prior to analysis. After incubation, each sample was diluted in 900 μL of deionized water and analyzed for size and zeta potential using a Malvern Zetasizer. DBP–DNA polyplexes were prepared by adding 50 μL of DBP solution (0.1 or 0.2 g/L) to 50 μL of pLuc solution (0.1 g/L), followed by incubation at room temperature for 15 min. After incubation, 50 μL of G5-SMTP (0.0375–1.5 g/L) or G5 (0.035–1.4 g/L) solution was added to DBP–pLuc polyplex solution and incubated for at least 15 min prior to analysis. Each sample was then diluted in 850 μL of deionized water and analyzed in the same manner as the G5-SMTP–DNA polyplexes. Scanning electron microscopy (SEM) images were obtained using DBP–DNA–G5-SMTP polyplexes prepared at a charge ratio of 1.
Fluorescent Labeling of G5 and G5-SMTP.
Both G5 and G5-SMTP were labeled with the near-infrared fluorescent label VivoTag-S 680XL according to the manufacturer’s instructions. This fluorophore contains a N-hydroxy succinimidyl ester group which was reacted with an amine group on G5/G5-SMTP dendrimers to form a stable amide linkage. VivoTag-S680 XL was conjugated to the dendrimers at a mole ratio of 5:1. A solution of VivoTag-S 680XL in anhydrous dimethyl sulfoxide was added dropwise to the G5/G5-SMTP solution under continuous stirring and was allowed to react for 2 h at room temperature as per manufacturer’s instructions. After completion of the reaction, unconjugated VivoTag-S 680XL was removed by dialysis using PBS buffer. The average number of dyes conjugated to G5/G5-SMTP was calculated and found to be 2.2 for G5 PAMAM and 1.8 for G5 PAMAM–SMTP.
In Vitro Cell Contact Assays.
C2C12 cells were seeded at a density of 7.5 × 104 cells per well on 24-well plates and incubated in Dulbecco’s modified Eagle’s medium (DMEM) [supplemented with 10% fetal bovine serum (FBS)] at 37 °C, 5% CO2 until they reached approximately 80–90% confluence. The cells were then washed twice with PBS and replenished with 0.5 mL of DMEM. Polyplexes were prepared by adding 10 μL of G5-SMTP (0.0375–1.5 g/L) or G5 (0.035–1.4 g/L) labeled with VivoTag 680XL to 10 μL of pLuc (0.1 g/L) and were incubated at room temperature for at least 15 min. Polyplex samples were then added directly to the cells, and the cells were incubated for 15 min. After the incubation, cells were washed three times with PBS and trypsinized to detach them from the plates. The cells were then collected in polystyrene round-bottom tubes, centrifuged, and washed with PBS. After washing, cells were fixed overnight at 4 °C with 4% paraformaldehyde in PBS. After overnight fixation, the cells were centrifuged to remove the paraformaldehyde and washed with PBS before resuspension in 0.5 mL of PBS for analysis. A total of 10 000 cells were analyzed by flow cytometry.
Confocal Microscopy of Cell Binding.
C2C12 cells were seeded at a density of 2.5 × 105 cells per well on 6-well plates containing sterile glass coverslips in each well. Cells were incubated as previously described for roughly 24 h, washed twice with PBS, and replenished with 2 mL of DMEM. Polyplexes were prepared by adding 10 μL of VivoTag 680XL-labeled G5-SMTP (0.0375–1.5 g/L) or VivoTag 680XL-labeled G5 (0.035–1.4 g/L) with similar fluorescence intensity into 10 μL of pLuc (0.1 g/L) at a charge ratio of 10 and were incubated at room temperature for at least 15 min. Polyplex samples were then added directly to the cells, and the cells were incubated for 1 h. After incubation, cells were washed three times with PBS and fixed overnight at 4 °C with 4% paraformaldehyde in PBS. After overnight fixation, cells were incubated at 37 °C, 5% CO2 with 2 mL of DAPI (1 μg/mL) for 15 min. The fixed cells were then washed twice with PBS to remove excess DAPI. After washing, the glass coverslips were removed from their wells, mounted on glass microscope slides, and sealed with nail polish. Cells were then imaged using confocal microscopy.
In Vitro Binding Pulldown Assay.
For the binding pulldown assay, 65 μg of His-tagged DLC8 and 6 μg of DBP were mixed in 100 μL of lysis buffer and allowed to incubate for 1 h at room temperature. In a separate tube, 100 μL of Ni-NTA agarose beads were added and centrifuged to remove the supernatant. The beads were then resuspended in 200 μL of lysis buffer and centrifuged to remove the supernatant. The His-tagged DLC8 and DBP reaction mixture was then added to the Ni-NTA agarose beads, mixed, and allowed to incubate for 1 h at room temperature. After the reaction, the solution was centrifuged at 2000 rpm for 5 min, and the supernatant was transferred to a separate tube and analyzed as the “supernatant”. The resultant Ni-NTA agarose bead pellet was resuspended in 100 μL of wash buffer and centrifuged at 2000 rpm for 5 min. The supernatant was transferred to a separate tube and analyzed as “wash 1”. This step was repeated once more to obtain “wash 2”. After the Ni-NTA agarose beads were washed twice, they were resuspended in 100 μL of elution buffer and centrifuged at 2000 rpm for 5 min. The supernatant was transferred to a separate tube and analyzed as “elution 1”. This step was repeated once more to obtain “elution 2”. Samples were prepared for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis by mixing 15 μL of each supernatant with 15 μL of Laemmli sample buffer containing 5% β-mercaptoethanol. Samples were then boiled at 90 °C for 10 min. A volume of 15 μL of each sample was then loaded onto a 16% Tris–tricine gel and run at 30 V for 1 h, followed by 90 V for approximately 3–4 h.
In Vitro Transfection Assays.
C2C12 cells were seeded at a density of 7.5 × 104 cells per well on 24-well plates and incubated in DMEM (supplemented with 10% FBS) at 37 °C, 5% CO2 until they reached approximately 80–90% confluence. The cells were then washed twice with PBS and replenished with 0.5 mL DMEM prior to transfection. Two different studies were performed to evaluate the transfection efficiency of pLuc DNA, as quantified using a Promega Luciferase Assay kit according to the manufacturer’s instructions. In the first, the performance of polyplex samples (G5-plasmid, and G5-SMTP-plasmid) was compared. In the second, the transfection efficiency of G5-SMTP-plasmid and G5-SMTP–DBP–plasmid was compared. In both studies, 1 μg of pLuc DNA was employed as the plasmid DNA, and polyplexes were added directly to the cells. The cells were then incubated for 24 h. After incubation, the cells were washed twice with PBS and lysed by adding 0.4 mL of 1× Promega reporter lysis buffer to each well. One freeze–thaw cycle was performed by freezing the cells at −80 °C and then thawing to complete the lysis. The resultant cell lysates were scraped from the 24-well plates and analyzed for luciferase expression. Data were normalized to the total protein concentration obtained from a BCA assay.
DNA Vaccination in Mice Using G5-SMTP Polyplexes.
We evaluated the ability of the platform to deliver a model DNA vaccine in vivo. We employed a DNA vaccine comprising a pcDNA3.1 plasmid encoding the gene for chicken ovalbumin (pcOVA), a well-established antigen model in immunological studies. For our study, we employed 8-week-old female BALB/c mice. We evaluated three groups (five animals per group), PBS control, pcOVA DNA control, and G5-SMTP–pCOVA DNA group. Animals were vaccinated at day 0, and booster was given on day 14. Blood samples were collected at days 0, 7, and 21. The blood samples collected were centrifuged to obtain the serum from which the antibodies were analyzed. The serum was serially diluted and added to microtiter plates coated with the chicken ovalbumin antigen. After incubation for 1 h, wash steps were performed using PBS buffer. An alkaline phosphatase-conjugated anti-mouse IgG secondary antibody was then added and quickly followed by the imaging substrate p-nitrophenyl phosphate, and the absorbance at 405 nm was observed. Antibody titer was defined as the lowest dilution that yields an absorbance reading above the calculated limit of detection (mean of the blank + 3 standard deviations).
RESULTS AND DISCUSSION
Here, we describe the design of a generation 5 PAMAM dendrimer functionalized with the ASSLNIA skeletal muscle targeting peptide (G5-SMTP) to improve delivery of DNA to skeletal muscle cells. In addition, we developed a cationic DBP with an internal sequence known to interact with dynein light chain 8 (DLC8) of the dynein motor protein complex. DBP functioned to improve intracellular trafficking for further enhancement of skeletal muscle transfection efficiency using our targeted dendrimer/DNA polyplex. The mechanism of polymeric gene delivery, especially in reference to dendrimer-based transfection, has been extensively discussed in the literature.29–32 Briefly, PAMAM dendrimers have been shown to go through the clathrin-dependent endocytosis pathway and are able to traverse cell monolayers via paracellular and transcellular pathways. The studies performed in this manuscript include biophysical characterization of G5-SMTP–plasmid DNA polyplexes, evaluation of the ability of these polyplexes to target skeletal muscle cells, comparison of transfection efficiency with and without DBP, and pilot in vivo transfection studies as described below.
Formation of Dendrimer–Plasmid DNA Polyplexes.
Electrostatic interactions between the negatively charged phosphate groups of the luciferase-producing pLuc DNA and positively charged amine groups of G5 PAMAM dendrimers drive DNA condensation in much the same way as histones condense genomic DNA. As the charge ratio is increased by the addition of more dendrimer, plasmid DNA is further condensed, resulting in the formation of polyplexes (Figure 1). Binding of DNA to dendrimer has been shown in the literature to be mainly due to ionic interactions,33,34 which is what we expect with our polyplexes. Further, to show that the binding of plasmid is internal and not just a surface interaction, we studied condensation using UV–visible spectroscopy and a nuclease protection assay (Figures S3 and S4). Both studies confirmed a stable binding of plasmid DNA to dendrimer. Because G5-SMTP comprises a G5 PAMAM dendrimer functionalized with ASSLNIA peptide, we sought to determine whether the presence of the peptide interfered with polyplex formation. Therefore, we evaluated the ability of G5-SMTP to condense plasmid DNA by preparing polyplexes at varying charge ratios (Figure 2).
Figure 1.

A) Schematic showing G5 PAMAM dendrimer modified with ASSLNIA peptide (shown in red). The inset box provides the primary structure of the peptide. (B) Illustration of polyplex formation, with histone-like aggregation of the G5-SMTP around the much larger plasmid DNA.
Figure 2.
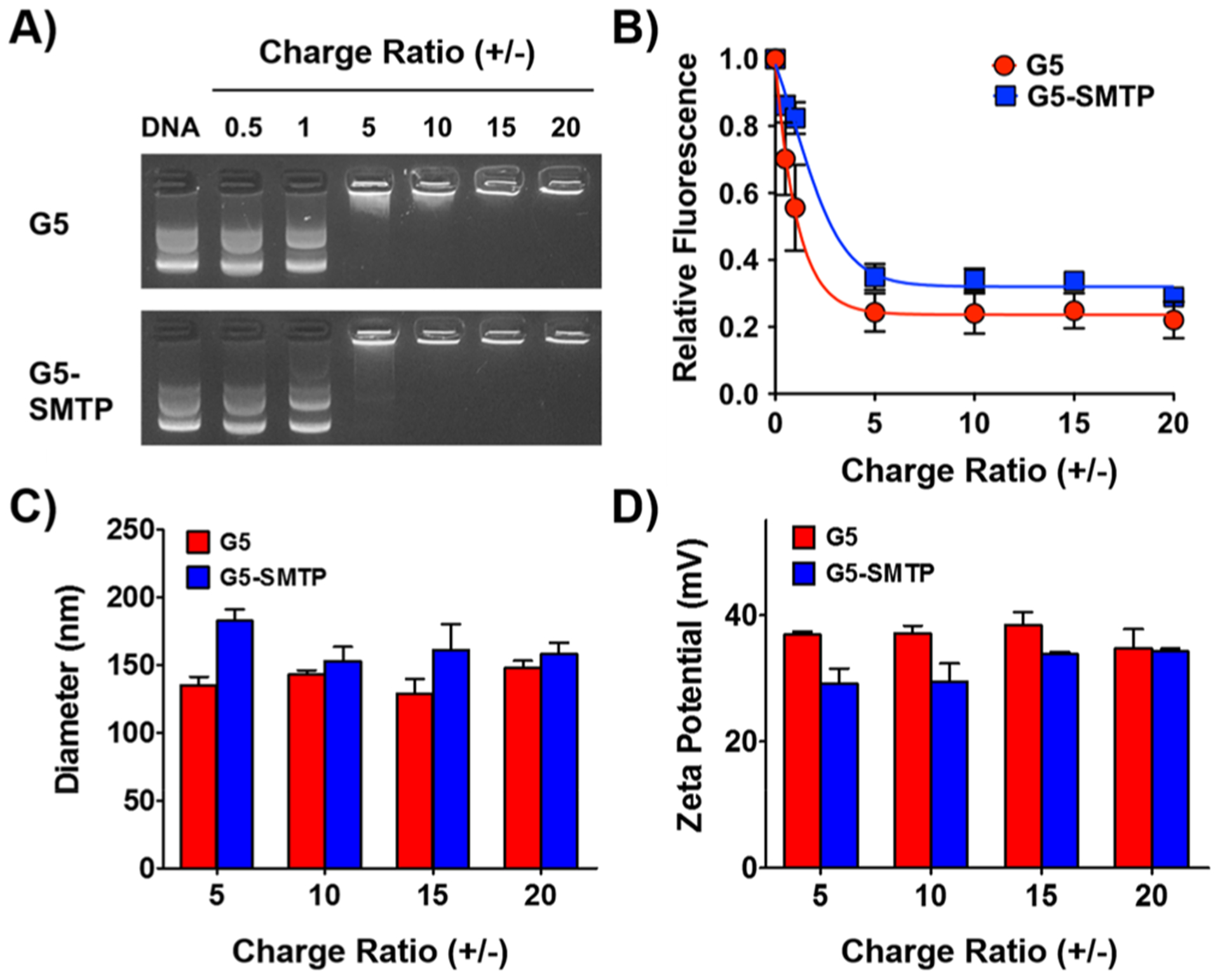
Analysis and characterization of G5-SMTP polyplex formation. The ability of G5-SMTP to form polyplexes with pLuc was determined by (A) gel retention assay and (B) ethidium bromide exclusion assay. Characterization of (C) size and (D) surface charge were determined by dynamic light scattering (DLS) and measuring the zeta potential, respectively. Experiments were performed in triplicate, with data representing the means and standard error of the mean.
We first investigated G5-SMTP–pLuc polyplex formation by means of a gel retention assay (Figure 2A). On an agarose gel, free DNA will migrate toward the cathode when subjected to an electrical current. However, we expect that the DNA that is condensed by the dendrimer will exceed the pore size of the gel and will be retained in the loading well. G5-SMTP condensed the plasmid DNA in a very similar manner to G5, with full condensation of DNA taking place at a charge ratio of 5 and higher. The ethidium bromide exclusion assay corroborated this observation, as evidenced by the decrease in fluorescence from the released ethidium bromide up to a charge ratio of 5 (Figure 2B). Taken together, our gel retention and ethidium bromide exclusion experiments show that the presence of ASSLNIA does not interfere with ability of G5-SMTP to form polyplexes with plasmid DNA.
Diameter and Zeta Potential of Dendrimer–Plasmid DNA Polyplexes.
Uniform nanoparticle size is an important factor to consider in gene delivery. Nanoparticles within the size range of 50–1000 nm are taken up by cells through endocytosis.35,36 With this in mind, we evaluated the diameters of the G5-SMTP–pLuc polyplexes using DLS (Figure 2C). Because complete polyplex formation was observed at charge ratios of 5 and above in our gel retention and ethidium bromide analyses, we performed DLS analysis using polyplexes at charge ratios of 5 and higher. Both G5 and G5 SMTP–pLuc polyplexes showed an average diameter of 150 nm (Figure 2C). The particle diameters overall were under 200 nm, which is within the range for efficient endocytosis. Furthermore, these results indicate that the presence of the ASSLNIA peptide in G5-SMTP does not result in significant size variation of the resulting particles.
Another consideration for efficient gene delivery is nanoparticle surface charge.35 It has been reported that nanoparticles containing an overall positive charge exhibit more efficient cell attachment and internalization.37 Therefore, we evaluated our G5-SMTP polyplexes for zeta potential as a measure of net surface charge. A relatively uniform and positive zeta potential was observed across all charge ratios for polyplexes comprising G5/G5-SMTP and pLuc (Figure 2D). Overall, our size and zeta potential characterizations together show that G5-SMTP forms polyplexes with plasmid DNA that are uniform and within the size necessary for cellular delivery.
G5-SMTP Polyplexes Exhibit Increased Affinity to Skeletal Muscle Cells.
Because ASSLNIA has been shown to bind skeletal muscle cells,38 we hypothesized that the presence of the peptide provides G5-SMTP with a greater-than-background affinity for skeletal muscle cells. To test this hypothesis, we performed a cell contact assay, where a C2C12 cell population was briefly incubated with fluorescently labeled polyplexes. We analyzed these cells using flow cytometry for fluorescence intensity because of a positive association between fluorescence intensity and the amount of cell-bound dendrimer (Figure 3A). At charge ratios between 5 and 20, we observed an overall trend of increasing fluorescence intensity for cells incubated with G5-SMTP–pLuc compared with G5-pLuc. These data indicate that there is a greater amount of G5-SMTP–pLuc bound to cells compared with G5-pLuc. These observations are further corroborated by our confocal studies performed using G5-SMTP–pLuc and G5-pLuc polyplexes prepared at charge ratio of 10 (Figure 3B). Taken together, our cell contact assay and confocal studies reveal that G5-SMTP has a higher affinity for skeletal muscle cells relative to the unmodified G5.
Figure 3.

In vitro cell contact assays. C2C12 cells were incubated for 15 min with pLuc polyplexes prepared using either G5 or G5-SMTP labeled with VivoTag 680XL. After incubation, cells were collected and analyzed for VivoTag 680XL fluorescence by flow cytometry. (A) Graphical representation of flow cytometry data obtained for charge ratios ranging from 5 to 20. Data represent the average of at least two experiments, with the error bars representing standard error. (B) Confocal studies of polyplex cell binding were performed by incubating C2C12 cells for 1 h with G5 or G5-SMTP polyplexes at a charge ratio of 10 that were labeled with VivoTag 680XL. After incubation, cells were washed and fixed with 4% paraformaldehyde prior to imaging with confocal microscopy. Bars:100 μ.
In Vitro Transfection Using G5-SMTP Polyplexes.
Next, we sought to determine whether this translated to increased transfection efficiency in vitro (Figure 4). We transfected C2C12 cells with polyplexes composed of pLuc and either G5-SMTP or G5 at varying charge ratios. We assayed for luciferase expression at 24 h post-transfection. No discernible effect on transfection efficiency in C2C12 cells was observed for polyplexes at charge ratios below 20. However, G5-SMTP–pLuc polyplexes at a charge ratio of 20 exhibited over a threefold increase in transfection compared to unmodified G5-pLuc polyplexes at the same ratio. These data indicate that G5-SMTP can mediate a charge ratio-dependent increase in transfection efficiency because of its affinity for skeletal muscle cells.
Figure 4.
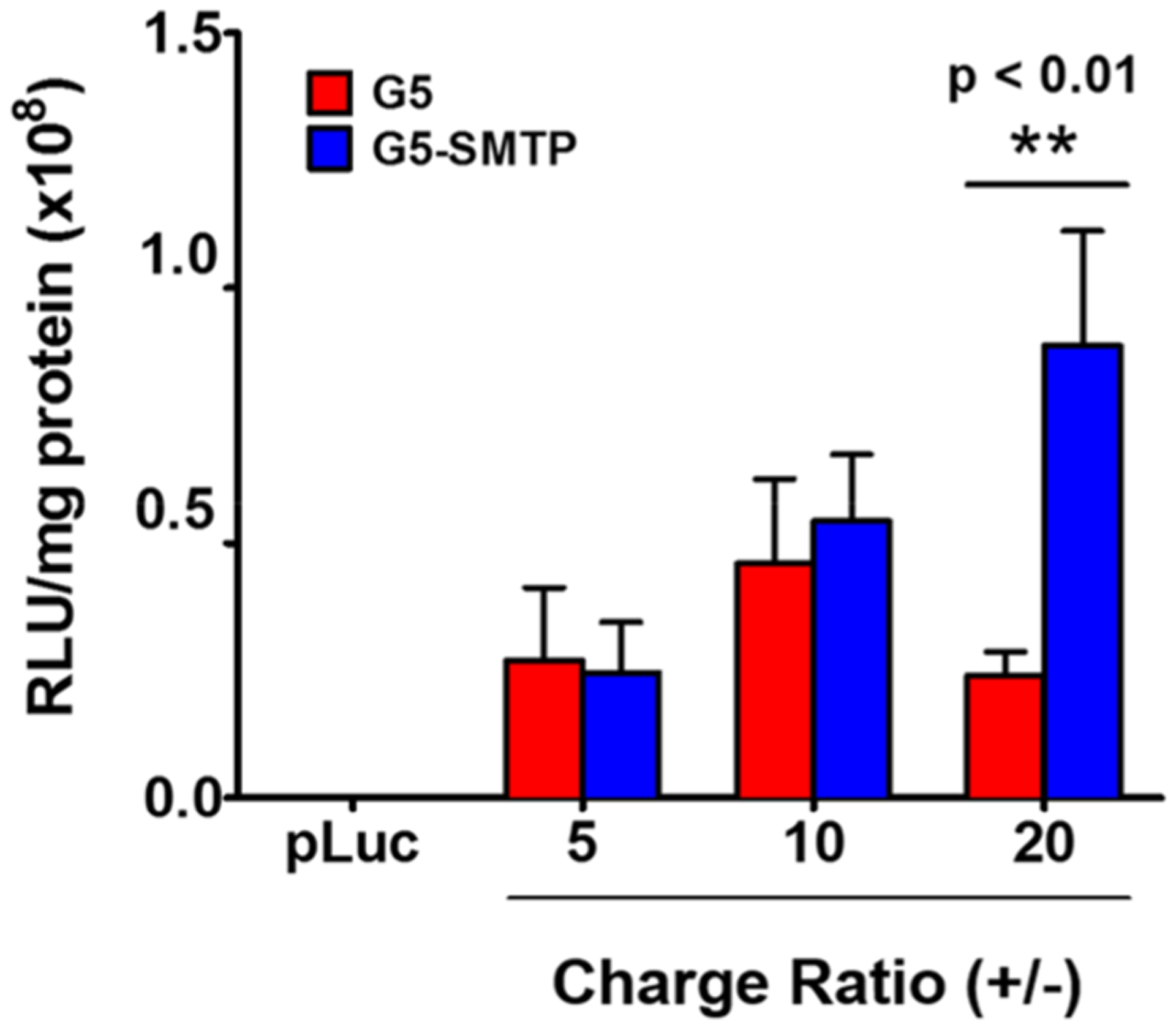
In vitro luciferase assays. C2C12 cells were transfected with G5 or G5-SMTP polyplexes prepared at varying charge ratios and analyzed for luciferase expression 24 h post-transfection. Experiments were performed in triplicate, with the data representing the mean and standard error of the mean.
Binding of DBP to DLC8.
DBP was designed with an internal binding sequence known to interact with the DLC8 component of the dynein motor protein complex (Figure 5A). Therefore, we set out to evaluate the ability of our peptide to interact with its intended target by a pull-down assay followed by SDS-PAGE analysis (Figure 5B). His-tagged DLC8 and DBP were mixed, and the resulting solution was then added to Ni-NTA resin to immobilize the His-tagged DLC8. If DBP interacted with its intended target, then it would be pulled down along with DLC8, and bands corresponding to both DLC8 and DBP would be visible on an SDS-PAGE gel following elution. If no interaction with DLC8 took place, then free DBP would remain in the supernatant and be washed away, leaving only a band corresponding to DLC8 in the elution. Following high stringency elution with imidazole, bands corresponding to DLC8 and DBP are both visible in the lanes labeled “elution 1” and “elution 2” in the SDS-PAGE gel image shown in Figure 5B. This combined with the fact that there were no DBP bands seen in the supernatant or wash steps indicate that the DBP peptide did interact with DLC8.
Figure 5.
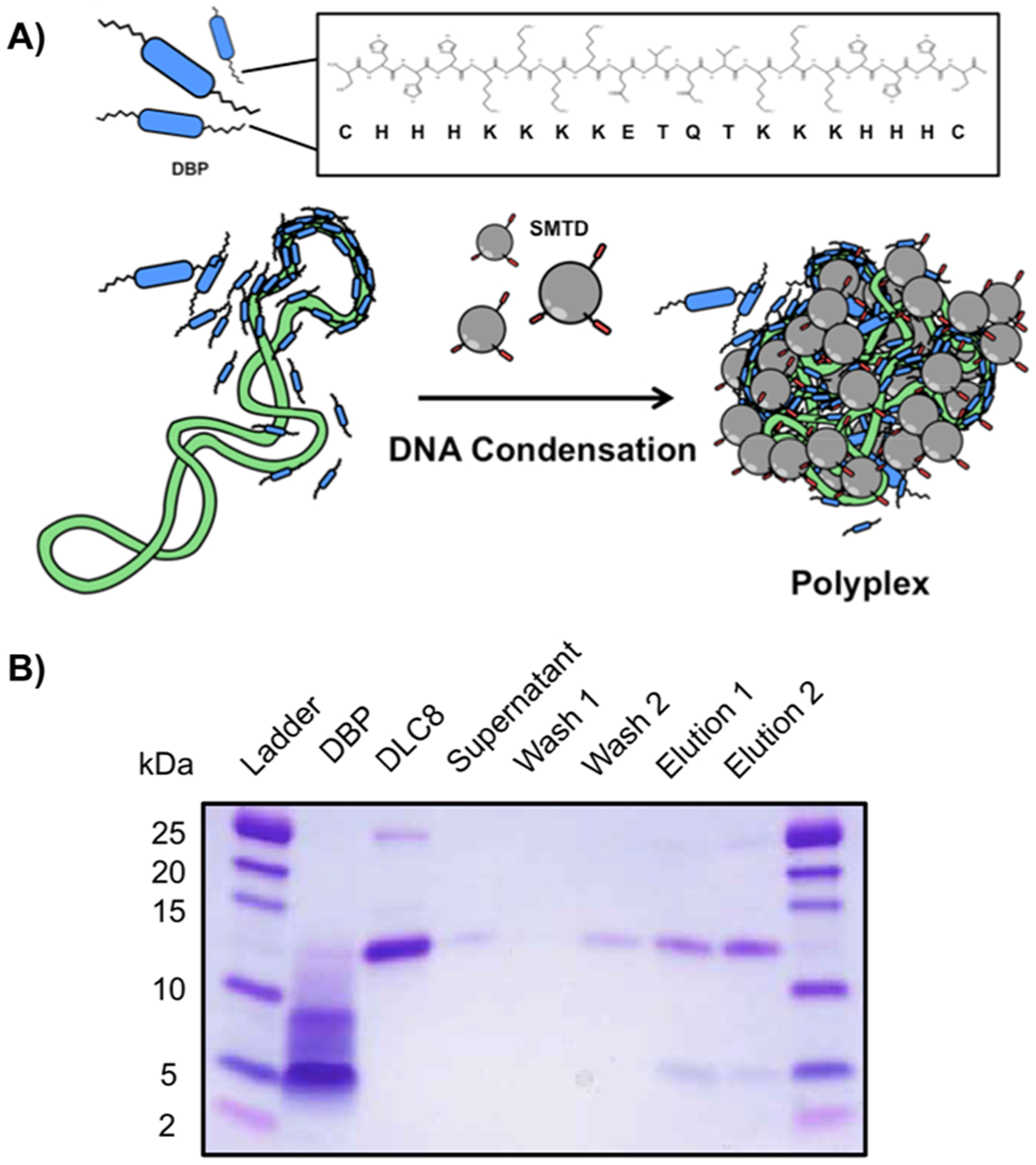
Association of DBP with DLC8. (A) Schematic illustrating the formation of the polyplexes with DBP, G5-SMTP, and plasmid DNA. DBP is in blue, the plasmid DNA is in green and the SMTP is in gray. The primary sequence of DBP is also shown in the inset. (B) SDS-PAGE analysis of the in vitro binding pull-down assay between DBP and DLC-8.
Formation of Polyplexes with DBP, Plasmid DNA, and G5-SMTP.
Because DBP was designed, in part, as a polycation for the packaging of DNA (Figure 5A), we investigated whether the peptide was indeed capable of mediating polyplex formation with plasmid DNA in G5-SMTP dendrimers (Figure 6). DBP forms polyplexes with plasmid DNA at charge ratios of 1 and 2, with ratios above 2 leading to the formation of insoluble aggregates (data not shown). As a result, only polyplexes at these two ratios were evaluated. Polyplexes comprising pLuc and DBP at a charge ratio of 1 were complexed with G5-SMTP at charge ratio of 2, yielding a DBP/pLuc/G5-SMTP ratio of 1:1:2. For simplicity, we will maintain this ordering scheme and refer to this final construct as the delivery complex in the text. In a similar manner, we also prepared delivery complexes at ratios of 2:2:1 and 2:2:2, all of which exhibited average diameters under 350 nm (Figure 6A). We also evaluated control complexes prepared between DBP and DNA as well as G5-SMTP and DNA at similar ratios (Figure 6A). The DBP–DNA polyplexes exhibited the smallest variability in diameter compared with intact delivery complex and polyplexes without DBP. This is likely due to the shorter and linear nature of DBP, which may allow for further DNA condensation. Despite the more uniform range of diameters observed for DBP–DNA polyplexes, the zeta potential observed was quite negative at −30 mV (Figure 6b)—possibly because of significant regions of the plasmid DNA backbone that were still surface-exposed despite being largely condensed by the peptides. Conversely, delivery complexes exhibited zeta potentials of approximately +30 mV (Figure 6B). These data suggest that the combination of DBP and G5-SMTP are sufficient to neutralize the negative charge of the plasmid DNA. Figure 6C shows a SEM image of delivery complexes prepared at a charge ratio of 1 to demonstrate polyplex structures with sizes ~150–200 nm. SEM also indicated some aggregation, which was expected under the conditions necessary for SEM sample preparation.
Figure 6.
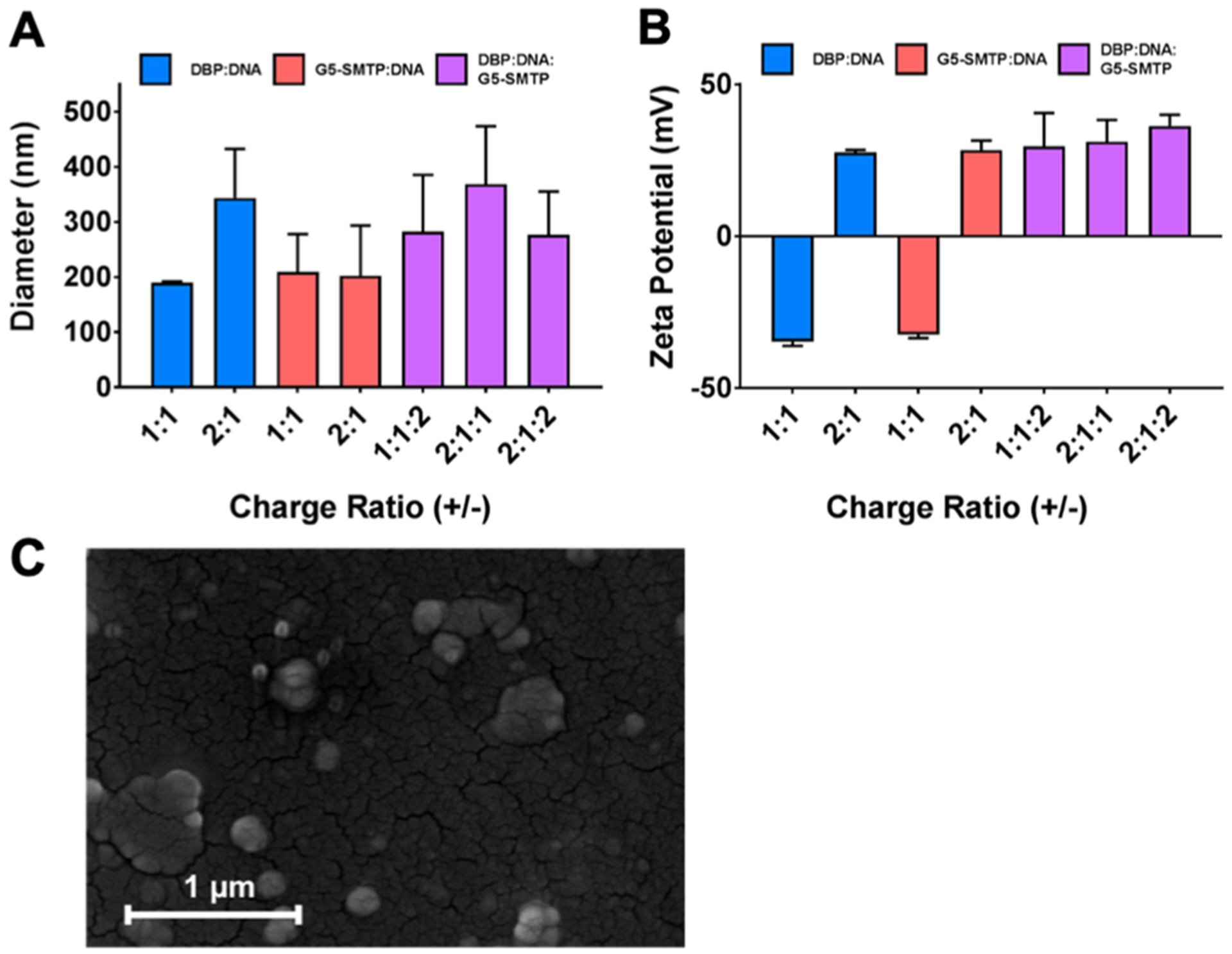
Characterization of DBP polyplexes. A series of polyplexes containing DBP, G5-SMTP, or a combination of both were prepared and characterized in terms of (A) size (B) and zeta potential. Data represent the average of at least two experiments, with the error bars representing standard error of the mean. (C) SEM image of DBP polyplexes prepared at a charge ratio of 1.
Transfection of DNA in C2C12 Cells Using the Delivery Complex.
We next sought to investigate the effect of DBP on the transfection efficiency of the prepared polyplexes in C2C12 cells (Figure 7). When DBP–DNA at charge ratio of 1 and G5-SMTP at charge ratio of 2 were combined in a single formulation, a significantly higher transfection efficiency was observed than DBP–DNA or G5-SMTP–DNA polyplexes (Figure 7A). An additional sixfold increase in transfection efficiency was observed for the delivery complex compared with the use of G5-SMTP–DNA. Interestingly, no discernible difference in transfection efficiency was observed when DNA was delivered with any combination of unmodified G5 or DBP (Figure 7B), indicating that DBP acts with G5-SMTP in a synergistic manner to enhance the delivery of plasmid DNA in C2C12 cells.
Figure 7.

In vitro luciferase assays with DBP polyplexes. C2C12 cells were transfected with DBP–DNA polyplexes prepared at varying charge ratios with either (A) G5-SMTP or (B) G5. Analysis of luciferase expression was performed 24 h post-transfection. Experiments were performed in triplicate, with the data representing the mean and standard error of the mean.
Determination of Humoral Response after DNA Vaccination.
To evaluate the performance of the platform in vivo, we determined the humoral response after vaccination with a chicken albumin DNA by measuring antibody titer using indirect enzyme-linked immunosorbent assay. Blood samples collected throughout the vaccination schedule were centrifuged to obtain the serum, from which the antibodies were analyzed. The serum was serially diluted and added to microtiter plates coated with chicken ovalbumin antigen. After incubation and washing steps, an alkaline phosphatase-conjugated anti-mouse IgG secondary antibody was added for detection of serum antibodies. The substrate, p-nitrophenyl phosphate, absorbance was measured at 405 nm. Analysis of blood serum collected on day 7 revealed that mice vaccinated with G5-SMTP–pcOVA polyplex exhibited a significantly higher antibody response compared with the pcOVA group at titers of 1:100, 1:200, 1:400, and 1:800 (Figure 8, day 7). After the booster, mice vaccinated with G5-SMTP–pcOVA continued to show significantly elevated antibody response at titers up to 1:800 compared with mice vaccinated with pcOVA (Figure 8, day 21). Collectively, our data show that G5-SMTP polyplexes at N/P ratios higher than 20 exhibited significantly higher transfection efficiency in skeletal muscle cell. Our in vivo data also confirmed the effectiveness of our strategy to transfect both differentiated and undifferentiated skeletal muscle cells. Additionally, this phenomenon translated into an increased humoral response in mice vaccinated with G5-SMTP–pcOVA polyplexes.
Figure 8.
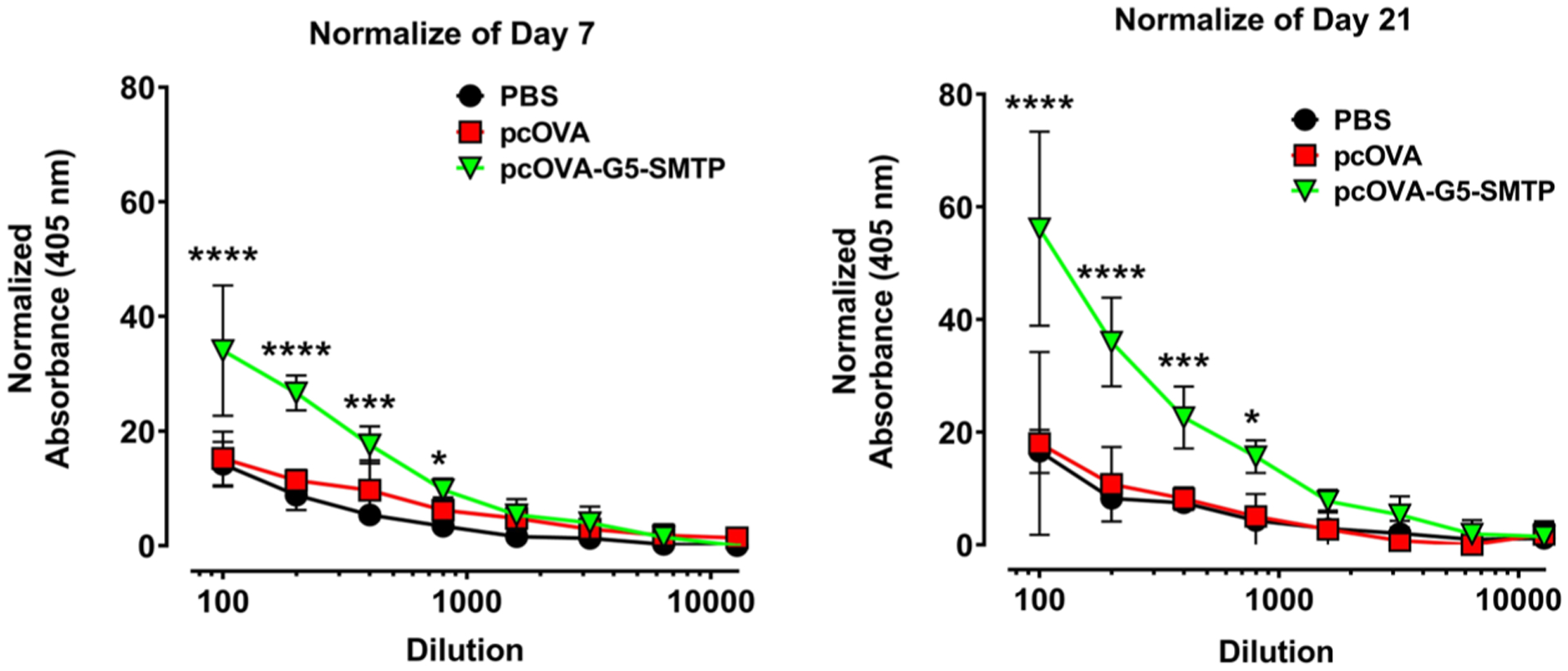
Determination of humoral response to polyplexes. The graph on the left shows humoral response measured using blood drawn on day 7 and the graph on the right shows the response measured using the blood drawn on day 21.
CONCLUSIONS
Our studies demonstrated that the ASSLNIA peptide-modified G5-SMTP exhibited enhanced affinity for skeletal muscle cells compared with unmodified G5 PAMAM dendrimer. This increase in affinity translated into a higher transfection efficiency in C2C12 cells in vitro and in vivo. To further enhance the transfection efficiency within skeletal muscle, DBP was combined with G5-SMTP to enhance intracellular transport by binding to the dynein motor complex. DBP was shown to interact with its target, DLC8, and allowed the formation of polyplexes with plasmid DNA. DBP was also shown to enhance the transfection efficiency of G5-SMTP-mediated gene delivery to skeletal muscle cells. As a result, the synergistic effects of a cell-specific platform like G5-SMTP, when combined with the enhanced transfection efficiency of DBP, represent a promising approach to gene delivery in skeletal muscle. Dendrimer-based gene delivery has shown promising results in preclinical studies; however, the clinical translation of dendrimers has not yet been achieved. The large cargo capacity of dendrimers combined with well-known and controllable chemical conjugations are advantageous properties. With more studies performed to moderate the toxicity of these polymers, their clinical applications are sure to expand. Our study demonstrated that targeted gene delivery using polymer-conjugated bioactive peptides would provide enhanced delivery efficiency while potentially reducing off-target effects and toxicity. The utility of this platform derives from its cargo capacity, allowing the delivery of many types of therapeutic nucleic acid directly to skeletal muscle. In the future, our platform can be modified further to incorporate newly developed functional moieties to expand the repertoire of intracellular trafficking strategies in skeletal muscle gene delivery.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank NIGMS (R01GM114321, R01GM127706) and the National Science Foundation (CHE-1506740) for the funding support. S.D. thanks the Miller School of Medicine of the University of Miami for the Lucille P. Markey Chair in Biochemistry and Molecular Biology.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.8b01313.
Cytotoxicity study, in vitro transfection of muscle cells with EGFP, UV–vis analysis of polyplex formation, protection of plasmid DNA from degradation by serum nucleases, comparison of in vitro transfection using C2C12 and HeLa cells, and in vitro transfection of muscle cells with EGFP: comparison with Xfect (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lu QL; Bou-Gharios G; Partridge TA Non-viral gene delivery in skeletal muscle: a protein factory. Gene Ther. 2003, 10, 131–142. [DOI] [PubMed] [Google Scholar]
- (2).Marino M; Scuderi F; Provenzano C; Bartoccioni E Skeletal muscle cells: from local inflammatory response to active immunity. Gene Ther. 2011, 18, 109–116. [DOI] [PubMed] [Google Scholar]
- (3).Jones CH; Chen C-K; Ravikrishnan A; Rane S; Pfeifer BA Overcoming nonviral gene delivery barriers: perspective and future. Mol. Pharm 2013, 10, 4082–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Escoffre J-M; Teissié J; Rols M-P Gene Transfer: How Can the Biological Barriers Be Overcome? J. Membr. Biol 2010, 236, 61–74. [DOI] [PubMed] [Google Scholar]
- (5).Hartikka J; Sukhu L; Buchner C; Hazard D; Bozoukova V; Margalith M; Nishioka WK; Wheeler CJ; Manthorp M; Sawdey M Electroporation-facilitated delivery of plasmid DNA in skeletal muscle: plasmid dependence of muscle damage and effect of poloxamer 188. Mol. Ther 2001, 4, 407–415. [DOI] [PubMed] [Google Scholar]
- (6).Kamimura K; Suda T; Zhang G; Liu D Advances in Gene Delivery Systems. Pharm. Med 2011, 25, 293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Forbes SC; Bish LT; Ye F; Spinazzola J; Baligand C; Plant D; Vandenborne K; Barton ER; Sweeney HL; Walter GA Gene transfer of arginine kinase to skeletal muscle using adeno-associated virus. Gene Ther. 2014, 21, 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yu C-Y; Yuan Z; Cao Z; Wang B; Qiao C; Li J; Xiao X A muscle-targeting peptide displayed on AAV2 improves muscle tropism on systemic delivery. Gene Ther. 2009, 16, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Boisgerault F; Mingozzi F The Skeletal Muscle Environment and Its Role in Immunity and Tolerance to AAV Vector-Mediated Gene Transfer. Curr. Gene Ther 2015, 15, 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ruponen M; Honkakoski P; Ronkko S; Pelkonen J; Tammi M; Urtti A Extracellular and intracellular barriers in nonviral gene delivery. J. Controlled Release 2003, 93, 213–217. [DOI] [PubMed] [Google Scholar]
- (11).Wang Z; Chen C; Liu R; Fan A; Kong D; Zhao Y Two birds with one stone: dendrimer surface engineering enables tunable periphery hydrophobicity and rapid endosomal escape. Chem. Commun 2014, 50, 14025–14028. [DOI] [PubMed] [Google Scholar]
- (12).Weiss V; Argyo C; Torrano AA; Strobel C; Mackowiak SA; Schmidt A; Datz S; Gatzenmeier T; Hilger I; Bräuchle C; Bein T Dendronized mesoporous silica nanoparticles provide an internal endosomal escape mechanism for successful cytosolic drug release. Microporous Mesoporous Mater. 2016, 227, 242–251. [Google Scholar]
- (13).Thuy LT; Mallick S; Choi JS Polyamidoamine (PAMAM) dendrimers modified with short oligopeptides for early endosomal escape and enhanced gene delivery. Int. J. Pharm 2015, 492, 233–243. [DOI] [PubMed] [Google Scholar]
- (14).Castle MJ; Perlson E; Holzbaur EL; Wolfe JH Long-distance Axonal Transport of AAV9 Is Driven by Dynein and Kinesin-2 and Is Trafficked in a Highly Motile Rab7-positive Compartment. Mol. Ther 2014, 22, 554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tanaka K; Kanazawa T; Sugawara K; Horiuchi S; Takashima Y; Okada H A cytoplasm-sensitive peptide vector cross-linked with dynein light chain association sequence (DLCAS) enhances gene expression. Int. J. Pharm 2011, 419, 231–234. [DOI] [PubMed] [Google Scholar]
- (16).Jin L; Zeng X; Liu M; Deng Y; He N Current progress in gene delivery technology based on chemical methods and nanocarriers. Theranostics 2014, 4, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kong L; Alves CS; Hou W; Qiu J; Möhwald H; Tomás H; Shi X RGD peptide-modified dendrimer-entrapped gold nanoparticles enable highly efficient and specific gene delivery to stem cells. ACS Appl. Mater. Interfaces 2015, 7, 4833–4843. [DOI] [PubMed] [Google Scholar]
- (18).Ray M; Tang R; Jiang Z; Rotello VM Quantitative Tracking of Protein Trafficking to the Nucleus Using Cytosolic Protein Delivery by Nanoparticle-Stabilized Nanocapsules. Bioconjugate Chem. 2015, 26, 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yang J; Zhang Q; Chang H; Cheng Y Surface-Engineered Dendrimers in Gene Delivery. Chem. Rev 2015, 115, 5274–5300. [DOI] [PubMed] [Google Scholar]
- (20).Hu W; Hoyer J; Neundorf I; Govender P; Smith GS; Schatzschneider U Synthesis of CpM(CO)3-DAB and -PAMAM Dendrimer Conjugates and Preliminary Evaluation of Their Biological Activity. Eur. J. Inorg. Chem 2015, 2015, 1505–1510. [Google Scholar]
- (21).Freeman EC; Weiland LM; Meng WS Modeling the proton sponge hypothesis: examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J. Biomater. Sci., Polym. Ed 2013, 24, 398–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Qamhieh K; Nylander T; Black CF; Attard GS; Dias RS; Ainalem M-L Complexes formed between DNA and poly(amidoamine) dendrimers of different generations - modelling DNA wrapping and penetration. Phys. Chem. Chem. Phys 2014, 16, 13112–13122. [DOI] [PubMed] [Google Scholar]
- (23).Bae Y; Lee S; Green ES; Park JH; Ko KS; Han J; Choi JS Characterization of basic amino acids-conjugated PAMAM dendrimers as gene carriers for human adipose-derived mesenchymal stem cells. Int. J. Pharm 2016, 501, 75–86. [DOI] [PubMed] [Google Scholar]
- (24).Daftarian P; Kaifer AE; Li W; Blomberg BB; Frasca D; Roth F; Chowdhury R; Berg EA; Fishman JB; Al Sayegh HA; Blackwelder P; Inverardi L; Perez VL; Lemmon V; Serafini P Peptide-Conjugated PAMAM Dendrimer as a Universal DNA Vaccine Platform to Target Antigen-Presenting Cells. Cancer Res. 2011, 71, 7452–7462. [DOI] [PubMed] [Google Scholar]
- (25).Liu Z-J; Daftarian P; Kovalski L; Wang B; Tian RX; Castilla DM; Dikici E; Perez VL; Deo S; Daunert S; Velazquez OC Directing and Potentiating Stem Cell-Mediated Angiogenesis and Tissue Repair by Cell Surface E-Selectin Coating. PLoS One 2016, 11, No. e0154053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang M; Liu H; Li L; Cheng Y A fluorinated dendrimer achieves excellent gene transfection efficacy at extremely low nitrogen to phosphorus ratios. Nat. Commun 2014, 5, 3053. [DOI] [PubMed] [Google Scholar]
- (27).Wang H; Wang Y; Wang Y; Hu J; Li T; Liu H; Zhang Q; Cheng Y Self-Assembled Fluorodendrimers Combine the Features of Lipid and Polymeric Vectors in Gene Delivery. Angew. Chem., Int. Ed. Engl 2015, 54, 11647–11651. [DOI] [PubMed] [Google Scholar]
- (28).Cheng YY Fluorinated Polymers in Gene Delivery. Acta Polym. Sin 2017, 8, 1234–1245. [Google Scholar]
- (29).Shcharbin DG; Klajnert B; Bryszewska M Dendrimers in gene transfection. Biochemistry 2009, 74, 1070–1079. [DOI] [PubMed] [Google Scholar]
- (30).Guillot-Nieckowski M; Eisler S; Diederich F Dendritic vectors for gene transfection. New J. Chem 2007, 31, 1111–1127. [Google Scholar]
- (31).El-Sayed M; Rhodes CA; Ginski M; Ghandehari H Transport mechanism(s) of poly (amidoamine) dendrimers across Caco-2 cell monolayers. Int. J. Pharm 2003, 265, 151–157. [DOI] [PubMed] [Google Scholar]
- (32).Saovapakhiran A; D’Emanuele A; Attwood D; Penny J Surface modification of PAMAM dendrimers modulates the mechanism of cellular internalization. Bioconjugate Chem. 2009, 20, 693–701. [DOI] [PubMed] [Google Scholar]
- (33).Chen W; Turro NJ; Tomalia DA Using Ethidium Bromide To Probe the Interactions between DNA and Dendrimers†. Langmuir 2000, 16, 15–19. [Google Scholar]
- (34).Pandita D; Madaan K; Kumar S; Poonia N; Lather V Dendrimers in drug delivery and targeting: Drug-dendrimer interactions and toxicity issues. J. Pharm. BioAllied Sci 2014, 6, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Braun CS; Vetro JA; Tomalia DA; Koe GS; Koe JG; Russell Middaugh C Structure/function relationships of polyamidoamine/DNA dendrimers as gene delivery vehicles. J. Pharm. Sci 2005, 94, 423–436. [DOI] [PubMed] [Google Scholar]
- (36).Barar J Bioimpacts of nanoparticle size: why it matters? Bioimpacts 2017, 5, 113–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Verma A; Stellacci F Effect of Surface Properties on Nanoparticle-Cell Interactions. Small 2010, 6, 12–21. [DOI] [PubMed] [Google Scholar]
- (38).Ebner D; Bialek P; El-Kattan A; Ambler C; Tu M Strategies for Skeletal Muscle Targeting in Drug Discovery. Curr. Pharm. Des 2015, 21, 1327–1336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.