

Abstract
Platelets have been shown to play an important immunomodulatory role in the pathogenesis of various diseases through their interactions with other immune and nonimmune cells. Sepsis is a major cause of death in the United States, and many of the mechanisms driving sepsis pathology are still unresolved. Monocytes have recently received increasing attention in sepsis pathogenesis, and multiple studies have associated increased levels of platelet–monocyte aggregates observed early in sepsis with clinical outcomes in sepsis patients. These findings suggest platelet–monocyte aggregates may be an important prognostic indicator. However, the mechanisms leading to platelet interaction and aggregation with monocytes, and the effects of aggregation during sepsis are still poorly defined. There are few studies that have really investigated functions of platelets and monocytes together, despite a large body of research showing separate functions of platelets and monocytes in inflammation and immune responses during sepsis. The goal of this review is to provide insights into what we do know about mechanisms and biological meanings of platelet–monocyte interactions, as well as some of the technical challenges and limitations involved in studying this important potential mechanism in sepsis pathogenesis. Improving our understanding of platelet and monocyte biology in sepsis may result in identification of novel targets that can be used to positively affect outcomes in sepsis.
Keywords: Cell interactions, infection, inflammation, platelet satellitism, sepsis pathogenesis
INTRODUCTION
Platelet function is traditionally defined by effects on hemostasis and thrombosis. In recent years, the concept of platelet function has widened, with discovery of evidence supporting roles for platelets as tiny but versatile (and mighty) players in a range of biological activities, including immune responses (1–5). Platelet α-granules are released after platelet activation, and contain molecules with immunoregulatory functions, such as platelet-factor (platelet factor 4) and pro-platelet basic protein. These platelet-derived factors play a rather limited role in thrombosis, but can play a considerable role in regulating host immune responses, including recruitment and activation of innate immune cells, and platelets were shown to be involved in the regulation of the activation, trafficking, and differentiation of T cells through this chemokine and cytokine release (6–10). In addition to indirect communication through secreted factors, platelets have been reported to aggregate with other immune cells, primarily monocytes followed by neutrophils and to a lesser extent with lymphocytes, to form heterogeneous complexes (11, 12) Direct platelet–monocyte interaction to form heterogeneous cell complexes was discovered decades ago (13, 14), although our understanding of this phenomenon in disease processes and outcome is limited. There has been no official consensus on naming the interaction between platelets and monocytes, and it is referred to as a “platelet–monocyte complex” in some studies (15–17), and as platelet satellitism in other studies (18–23). Many review articles include discussion of the interaction between platelets and monocytes as a part of the wider interaction between platelets and immune cells, but few articles focus attention on platelet–monocyte aggregates as functional units.
Platelet–monocyte aggregates have been associated with various diseases, especially in cardiovascular disease. However, our understanding of their roles in this disease is limited. What we do know is that platelet–monocyte aggregates in circulation are a sensitive marker of platelet activation (24). A higher number of platelet–monocyte aggregates were detected in acute myocardial infarction patients compared with healthy controls and further elevated in patients with complications (16). Those studies seem to support the notion that platelet–monocyte aggregates play a proinflammatory role in cardiovascular diseases. However, an inverse correlation was found between inflammation in ulcerative colitis and circulating platelet–monocyte aggregates, which raised the possibility that platelet–monocyte complexes play an anti-inflammatory and protective role in that disease process (25).
In the case of sepsis, platelet–monocyte aggregates were reported to be elevated at very early time points (4 h) in a murine sepsis model (26), and have been shown to be associated with higher risk of mortality in older (age>=65) septic patients (27). Moreover, platelet aggregation with monocytes was shown to polarize monocytes toward M1 phenotype in a murine sepsis model, which suggests that platelet–monocyte aggregates play pro-inflammatory roles in sepsis (28). Therefore, the exact role of platelet–monocyte aggregates is likely to be disease-specific and more studies are needed to uncover important underlying mechanisms related to pathogenesis and outcome.
In this review, we focus on our current understanding of direct platelet–monocyte interactions to form aggregates in multiple disease processes, and then highlight implications of platelet–monocyte aggregates in sepsis. We hope to provide some insights on this understudied research area, and to inspire future work that may be important in determining novel sepsis therapeutics based on specific platelet–monocyte interactions.
MECHANISM OF PLATELET–MONOCYTE AGGREGATION
Multiple studies have tried to address the mechanism and meaning of platelet–monocyte aggregates in a variety of experimental and pathological contexts. As mentioned above, platelets play a role in attracting monocytes through the release of chemokines and cytokines that induce inflammation. Platelets can also release other inflammatory mediators, such as high mobility group box 1 protein (HMGB1) (29–31), which can activate monocytes, or potentially modulate monocyte immune phenotype and function (6, 8, 30, 32, 33).
Direct contact between platelets and various immune cells, including myeloid cells and lymphocytes, also occurs, and it is now realized that there is a reciprocal interaction affecting not only the immune cells, but also the platelets themselves. The phenomenon of bidirectional cell-to-cell contact between multiple platelets and a single leukocyte (predominantly neutrophils and monocytes) occurs in a variety of conditions including blood diseases (22, 34), infectious diseases (18), trauma (35–37), idiopathic pulmonary fibrosis (38), thromboembolic disease (39), diabetes (40), cardiovascular disease (15, 41), sickle cell disease (42, 43), and autoimmune disease (44). In many cases this type of interaction was termed platelet satellitism (20, 21, 23), and despite being a well-recognized phenomenon the mechanism and function behind platelet satellitism has not been well described. One in vitro study suggested that platelet activation is the major initiator of platelet–monocyte aggregates, as opposed to being a function of monocyte activation (45). Upon activation, platelets rapidly increase surface expression of cluster of differentiation (CD)62p (also known as P-selectin), which then is recognized by its cognate receptor, P-selectin glycoprotein ligand-1 (PSGL-1) on the surface of monocytes. This seems to be the initiating signal in platelet–monocyte aggregation and is further consolidated by potentiation of other downstream signaling and receptor interactions, including CD40L (CD154)-Macrophage-1 antigen, glycoprotein VI -CD147, or via interaction of intercellular adhesion molecule 1 with fibrinogen attached to the activated platelet (8, 46–48). Fibrinogen has also been reported to act as a bridge between platelets and monocytes (46), with some platelet autoantibodies shown to also act in this way to mediate platelet binding to monocytes (49). A summary of studies of platelet–monocyte aggregates formation was provided in Table 1
Table 1.
Summary of studies of molecules associated with platelet-monocyte aggregate formation
| Species | Ex vivo/in vivo | PMA detection method | Involved molecules | Main findings |
|---|---|---|---|---|
| Human | Ex vivo | Light microscopy and flowcytometry | GPIIb-IIIa on platelets | GPIIb-IIIa on platelets had a supportive role in the formation of PMA only in the presence of weak activation of platelets (50) |
| Human | Ex vivo | Flowcytometry | GPIIb-IIIa on platelets | GPIIb-IIIa antagonist inhibited the formation of PMA and monocyte tissue factor expression after platelets activation (51) |
| Mouse | Ex vivo | Flowcytometry | ICAM-1 on monocytes | ICAM-1 on monocytes bound to the fibrinogen attached to the activated platelets as to facilitate the formation of PMA (46) |
| Human and mouse | Ex vivo and in vivo | Flowcytometry | Monocyte EMMPRIN (CD147/basigin) and platelet GPVI | EMMPRIN-GPVI engagement facilitated formation of PMA in vivo and ex vivo (47) |
| Human | Ex vivo | Flowcytometry | Platelet P-selectin (CD62P) and monocyte PSGL-1 | CD62P-PSGL-1 engagement was responsible for formation of PMA (13) |
| Human | Ex vivo | Flowcytometry | Platelet P-selectin (CD62P) | Platelet activation (increased expression of CD62P) was, not monocyte activation, led to increased formation of PMA (45) |
| Human | Ex vivo | Flowcytometry | Platelet P-selectin (CD62P) and monocyte PSGL-1 | P-selectin-PSGL-1 engagement induced monocyte activation via upregulation of IL1β and TNFα by NF-κB translocation into nucleus (60) |
| Human | Ex vivo | Flowcytometry | Platelet P-selectin (CD62P) and monocyte PSGL-1 | P-selectin-PSGL-1 engagement induced monocyte upregulation of IL8 and tissue factor by phosphorylation of Lyn, which could be inhibited by IL10 (65) |
| Human | Ex vivo | Flowcytometry | Platelet P-selectin (CD62P) and Phosphatidylserine | P-selectin-mediated binding and Phosphatidylserine recognition on activated platelets induced monocyte secretion of IL8 and IL10 (53) |
Interestingly, nonactivated quiescent platelets have also shown capacity to bind to monocytes in some physiologic and pathophysiological conditions. Baseline aggregation of platelets with monocytes in whole blood was measured at up to 45% of all monocytes in one study (13), although these results vary widely across different studies. The mechanism of quiescent platelet association with monocytes has not been fully uncovered, but is thought to be quite different from activated platelet binding. Evidence from monoclonal blocking antibody studies suggests that GPIIb/IIIa (integrin αIIbβ3) expression on platelets is potentially involved in the formation of platelet–monocyte aggregates between quiescent platelets and monocytes, as blocking GPIIb/IIIa partially decreased the percentage of platelet–monocyte aggregates (13, 50, 51). Additionally, regardless of the status of platelet activation, the formation of platelet–monocyte aggregates is calcium (Ca2+)-dependent, and can be reversed by depletion of CaCl2 in cell culture media, or conversely promoted by supplementation of CaCl2 (52).
Another way platelets and monocytes can interact is via monocyte phagocytosis of the platelet. This is known to occur in dengue fever (53) and Hodgkin lymphoma (22). In those cases, it is thought that monocytes recognize activated platelets through exposed phosphatidylserine (PS) on the platelet surface, similar to classic recognition of apoptotic cells by monocytes and macrophages. Monocytes are therefore able to phagocytose the platelets via PS receptor and scavenger receptor interaction (Fig. 1) (22, 53). Some major unanswered questions include whether aggregation of platelets with monocytes is a first step in phagocytosis, if the phagocytosis itself is targeted or if phagocytosis plays a specific role in modulating immune responses (54).
Fig. 1. Monocyte/macrophage phagocytosis of platelets.
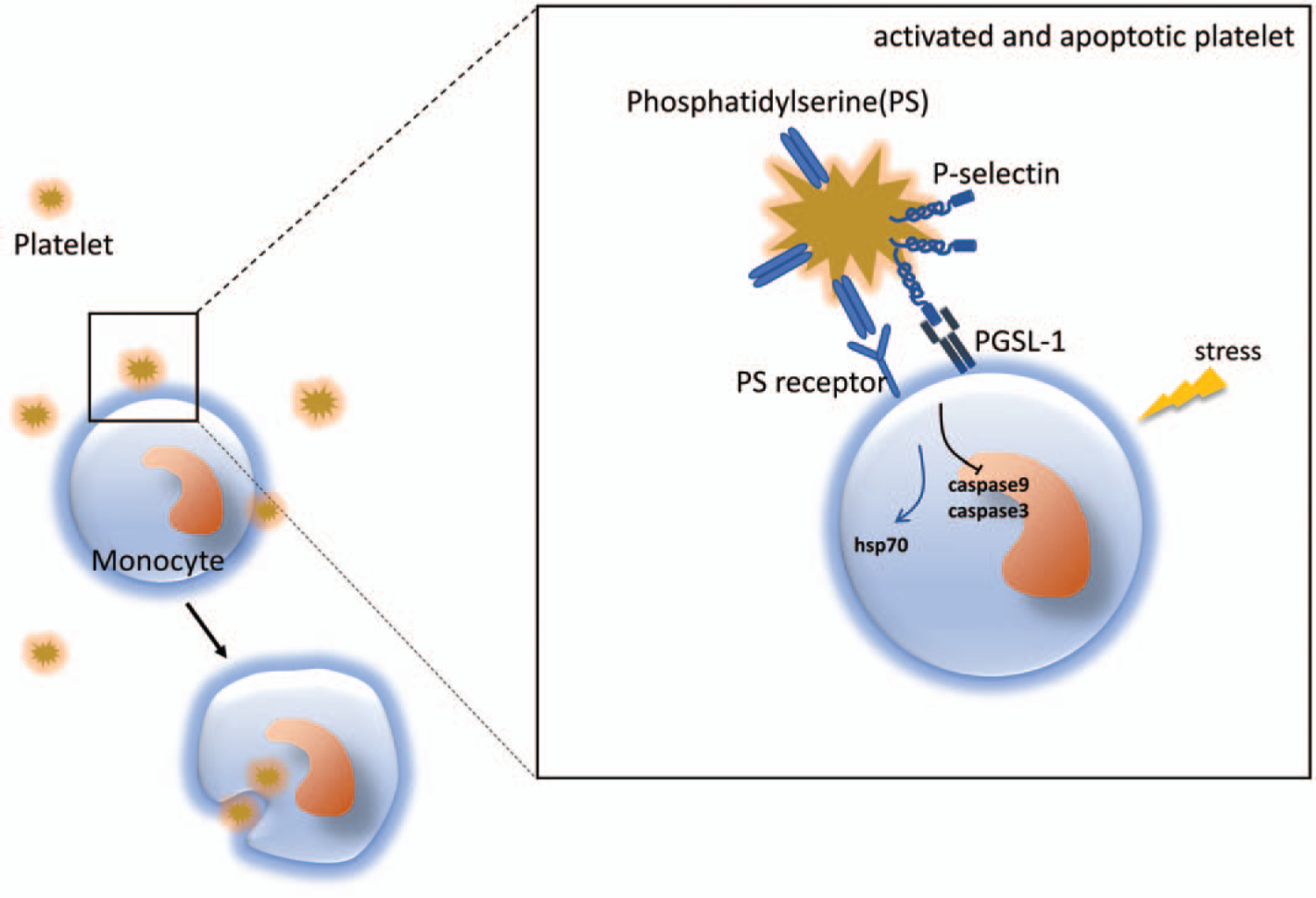
Activated platelets expose phosphatidylserine (PS) on their membrane and aggregate with monocytes via P-selectin-PGSL-1 interaction. The engagement of PS and its receptor triggers the process of phagocytosis, which promotes monocyte survival by regulation of apoptotic pathways. Phagocytosis of platelets may also participate in regulation other biological processes, such as metabolism.
PLATELET AGGREGATION WITH DIFFERENT SUBTYPES OF MONOCYTES
Monocytes in peripheral blood can be divided into three major groups in mice based on their surface expression of Ly6c: Ly6chi, Ly6cint, and Ly6clow (55). In humans the subgroups are defined by monocyte expression of CD14 and CD16: classical monocyte (CD14++CD16‒); intermediate monocyte (CD14++CD16+); non-classical monocyte (CD14+CD16++) (56). Platelets have been shown to aggregate with different subtypes of monocytes at different percentages in volunteers without cardiovascular disease history (CD14++CD16‒, 8.1±3.4%; CD14++CD16+, 21.2±14%; CD14+CD16‒, 18±12.6%; CD14+CD16+, 22.3±14.3%) (57), or similarly in a study of assessing platelet–monocyte aggregates in patients with acute myocardial infarction, they also found different percentages in healthy adult volunteers (CD14++CD16‒, 3.23±0.16%; CD14++CD16+, 10.29±1.37%; CD14+CD16++, 3.23±0.32%) (16). There were also differences in platelet–monocyte aggregation between healthy control volunteers (n=23) and patients who had suffered acute myocardial infarction (n=31) (16). The authors defined the platelet–monocyte aggregates using known cell markers (CD16 and CD14 for monocyte, CD41a for platelet) by flow cytometry, with double-positive surface marking of CD14 and CD41a defined as platelet–monocyte aggregates). The data showed that in acute myocardial infarction patients there was an marked increase of platelet–monocyte aggregates compared with healthy volunteers (11.13±1.61% vs. 4.35±0.23%, P=0.00004), and there were more platelet–monocyte aggregates formed with intermediate monocytes (CD14++CD16‒, 7.90±1.32%; CD14++CD16+, 18.41±1.99%; CD14+CD16++, 6.76±0.84%), in contrast to more platelet–monocyte aggregates of classical monocytes in healthy volunteers (16). Another study measured platelet–monocyte aggregates in both healthy volunteers (n=73) and patients with cardiovascular diseases (CVD) (n=345) by flow cytometry. In this study, platelet–monocyte aggregates were defined as double positive for CD61 and CD14 (41). The data from this study showed that platelet–monocyte aggregates were significantly elevated in patients with CVD (21.8% vs. 9.4%, P < 0.001), particularly in patients with peripheral artery disease (approximately 30%), compared with healthy controls. Therefore, they concluded that platelet–monocyte aggregates could be used as a robust marker of platelet activation and monocyte inflammatory response. Interestingly, they also reported that classical monocytes were less capable of aggregating with platelets compared with non-classical monocytes in healthy controls, which was contradictory to Loguinova et al. (16). The disparity was difficult to explain and was possibly due to different sample collection, processing or measurement techniques, all of which can affect platelet activation and aggregation. Notably, though these two studies both detect platelet–monocyte aggregates by flow cytometry using surface markers, they were not able to distinguish definitively between platelets attached to the cell surface of monocytes or monocytes with engulfed platelets. Further studies are needed, potentially using enhanced techniques to unravel the whole process in more detail. However, the results of these two studies do suggest that platelets can preferentially aggregate with different subtypes of monocytes. This may be of great significance in pathogenesis, diagnosis, and treatment of platelet-associated diseases.
Platelet-derived extracellular vesicles (EV) may also be involved in regulating the association of platelets with specific monocyte subtypes. Indeed, one study suggested that platelet-derived EVs preferentially associated with classical and intermediate monocytes (58). However, the mechanism underlying attraction to one subtype over another is not well understood. There is a known higher level of expression of PSGL-1 on the surface of murine Ly6chi monocytes, and human classical monocytes (CD14++CD16‒), at baseline in healthy individuals. Alterations in this expression may therefore influence aggregation of platelets and monocytes in the healthy versus disease states (59).
RECIPROCAL EFFECTS OF PLATELET–MONOCYTE AGGREGATES
Platelet effects on monocyte activation and function are reported to be dependent on platelet activation status and even though resting platelets can interact with monocytes, they have little impact on monocyte function (60). One explanation may be due to the differences in P-selectin expression level on activated and inactivated platelets, but much of the biology is as yet unclear.
Interaction of human-activated platelets with monocytes can induce production of chemokine and cytokines, including Tumor Necrosis Factor α (TNFα), Interleukin 1β, chemokine (C-X-C motif) ligand 8 (Interleukin 8) (CXCL8 IL8), and chemokine (C-C motif) ligand 2 (monocyte chemoattractant protein-1) (61, 62). Coculture of human monocytes with activated autologous platelets also induced upregulation of monocyte CD16 and Cyclooxygenase-2 expression indicating increased monocyte proinflammatory capacity (63). Additionally, purified P-selectin from human platelets primed autologous monocytes for cytokine secretion, including TNFα, Interleukin 1β, IL6, CXCL8 (IL8), IL12, and CCL4 (MIP-1β) (64, 65), as well as expression of tissue factor (65, 66). These data highlight the potential importance of P-selectin expression on platelets and P-selectin-PSGL-1 signaling in regulating monocyte inflammatory responses. Downstream signaling pathways within monocytes are also activated, including protein tyrosine kinases and mitogen-activated protein kinases and Src family kinase Lyn (65, 67, 68). However, one study showed that inhibition of human platelet–monocyte aggregate formation via PSGL-1 or P-selectin was not sufficient to attenuate platelet-mediated monocyte activation, suggesting other pathways and receptor–ligand interactions may be equally important in producing a full inflammatory response (69). Interestingly, other studies found that activated platelets do not always activate monocytes to induce production of proinflammatory cytokines, and in some situations human platelet–monocyte interactions can actually dampen inflammation by increasing monocyte expression of IL10, and reducing expression of TNFα (70, 71). Taken together these conflicting findings show just how much more we have to learn about platelet–monocyte interactions and the complexity of these interactions. It is also likely that the outcome of platelet–monocyte interactions is influenced by the surrounding inflammatory milieu, and so will likely be pathology and tissue specific.
In addition to the modulation of the immunophenotype of monocytes, platelets can also exert other effects. Human platelet phagocytosis by monocytes has been shown to play a significant role in the regulation of monocyte apoptosis (72). A large percentage of monocytes cultured in serum-deprived media will be induced to undergo apoptosis, but addition of activated or nonactivated platelets, and uptake of platelets by phagocytosis, can significantly attenuate monocyte apoptosis in a number-dependent manner via inhibition of caspase9 and caspase3 pathway and induction of heat shock protein 70 pathway (Fig. 1) (72). Human-activated platelets have also been shown to increase monocyte cell surface expression of β1 and β2 integrins, which can increase adherence to endothelial cells and facilitate monocyte transmigration to the site of inflammation (73). Human platelets can also influence monocyte differentiation in the circulation, and convert monocytes into immunoregulatory cells under certain circumstances (49). One example of this is in tuberculosis, where platelets can induce monocytes to differentiate into epithelioid-like multinucleated giant foam cells that have immunosuppressive capacity (74). Microarray data from this study indicated platelet-influenced gene signature alteration of monocytes during their differentiation into these giant foam cells (74).
Monocytes also have a great impact on platelets via the podoplanin–CLEC2 axis, though there are very limited data available. Podoplanin, a small membrane glycoprotein, is relatively well conserved across species and widely expressed by various normal cell types, including but not limited to lymphatic endothelial cells, kidney podocytes, fibroblastic reticular cells, as well as monocytes and was reported to play important roles in inflammation and cancer (75–78). C-type lectin receptor 2 (CLEC-2) was reported to be the cognate receptor of podoplanin and highly expressed by immune cells and platelets. However, the podolanin–CLEC2 interaction has only been extensively investigated in platelets, with CLEC2 recently identified as a novel platelet activation receptor. Engagement of CLEC2 on platelets leads to platelet aggregation (79), which plays a critical role in maintaining the normal physiological functions of lymphatic vessels (80, 81). Platelet interactions through podoplanin–CLEC2 have also been shown to be important in various physiological and pathophysiological conditions, including but not limited to hemostasis, thrombosis, brain development, and immune responses (79, 82). Readers are referred to reference (79) for a review of the functions of CLEC2. A recent study also reported that the podoplanin–CLEC2 axis is implicated in a murine sepsis model (intraperitoneal lipopolysaccharide and cecal ligation and puncture). Activation of this pathway was shown to inhibit the progression of inflammation by reducing the release of proinflammatory cytokines and chemokines in serum as part of the cytokine storm associated with poor outcomes in sepsis (83). Since podoplanin is expressed by monocytes and CLEC2 is expressed by platelets, it is reasonable to speculate that these surface proteins may also be involved in platelet–monocyte aggregate formation, although this has not been definitively shown to date.
Another perspective is to view the platelet–monocyte aggregate as a functional unit since platelets and monocytes have mutual effects on each other’s phenotype when forming aggregates. Therefore, the aggregates may have distinct phenotype than platelets or monocytes alone. Indeed, human platelets, monocytes, and platelet–monocyte aggregates have been shown to have sophisticated interactions with endothelial cells in cardiovascular diseases (84). Specifically, human platelet–monocyte aggregates have been shown to be more capable of tethering and rolling on endothelial cells (85, 86). Moreover, human platelet–monocyte aggregates also showed increased capability of transmigration compared with monocytes without platelets attached (87). It is not yet clear whether there are similar functions for platelet–monocyte aggregates in sepsis, but it is possible that inflammatory responses in epithelium may be regulated by platelet–monocyte aggregates.
OVERVIEW OF PLATELETS IN SEPSIS
Platelets are known to be activated in septic patients (88, 89) through various mechanisms including, but not limited to, the excessive formation of thrombin (90) (which is a potent activator of platelets (91)), extensive exposure of collagen with upregulation of expression of von Willebrand factor and tissue factor on activated endothelial cells (92), as well as C1q binding to its receptor on platelets (93). The activation of platelets results in the release of many bio-active molecules encapsulated within granules (alpha-granules, dense granules, and lysosomes) of platelets, including coagulation regulators, chemokine, growth factors, and adhesion molecules. These have broad interactions with both the coagulation and immune systems, and are involved in sepsis-induced coagulation disorders and inflammatory dysfunction during sepsis. In addition, platelets have been shown to constitutively express functional toll like receptor (TLR4) (94, 95), which can be activated by pathogen-associated molecular patterns and damage-associated molecular patterns. Engagement of TLR4 activates platelets in a non-canonical manner, and promotes pro-thrombotic, procoagulant responses, and immune responses (95). Therefore, platelet-TLR4 activation bridges the thrombosis and immune responses during sepsis (96). The role of platelets as sentinel innate immune cells and the activation of innate immune signaling pathways leading to enhanced thrombosis is known as immunothrombosis (97). Interestingly, NACHT, LRR, and PYD domains-containing protein 3 inflammasome was also activated in platelets and contributed to excessive IL-1β release and multi-organ injuries in a rat sepsis model (cecal ligation and puncture) (98). Therefore, activated platelets may participate and shape the pathophysiology of sepsis in a direct manner. On the other hand, activated platelets themselves have been shown to directly bind to neutrophils. This interaction activates and shapes the immunophenotype of neutrophils, producing more proinflammatory cells with higher capacities for adhesion, phagocytosis, and generation of reactive oxygen species (99). Moreover, activated platelets were shown to guide neutrophil transmigration via supporting neutrophil adhesion to endothelial cells through sequential action of P-selectin and the beta 2-integrin CD11b/CD18 (100). Apart from immune cells, activated platelets can also directly interact with endothelial cells, which are active participants in inflammation and play a fundamental regulatory role in the progression of sepsis (101). It was reported that activated human platelets could engage with human umbilical vein endothelial cells via CD40L (CD154)–CD40 interactions, and subsequently induce expression of adhesion molecules and proinflammatory chemokines (CXCL8 (IL8) and chemokine (C-C motif) ligand 2 (monocyte chemoattractant protein-1)) in coculture condition in vitro (101). Importantly, activated platelets, as mentioned above, which play important roles in inflammatory responses and activation of endothelial cells, were also reported to be a major player in the development of multi-organ dysfunction in sepsis (102). One example of this is in sepsis-induced acute lung injury (ALI) (103), where platelets and leukocytes accumulate inappropriately and platelets directly bind to neutrophils via P-selectin-PSGL interaction to stimulate neutrophils to release granules. The granule contents are implicated in the degradation of surfactant proteins, apoptosis of epithelial cells, and induction of coagulation disorders during sepsis (104). Another mechanism of platelet influence on ALI is via promotion of excessive formation of neutrophil extracellular traps (NETs), which subsequently cause secondary tissue damage and inflammation (105, 106). Platelet-derived granzyme B has also been implicated in tissue damage through induction of cell apoptosis in spleen and lung. This was reported to be dependent on cell-to-cell direct contact and could be attenuated by platelet GPIIb/IIIa receptor inhibitor (107). Platelets also get involved in other organ injuries induced by sepsis, such as acute kidney injury and sepsis-associated cardiopathy (reviewed in (102, 108)). Additionally, platelets were also reported to be able to engulf bacteria by themselves, such as Staphylococcus aureus, and thus mitigate the clearance of the pathogens by phagocytes and serve as a nidus for prolonged infection (109).
Platelet effects may not all be detrimental, and platelets are known to participate in the defense response against pathogens via facilitating leukocytes recruitment and infiltration as well as promoting their capability of pathogen clearance (31, 110–113). For example, platelets promotion of the formation of neutrophil NETs through interaction with neutrophils is vital for trapping and clearing invading pathogens (106, 111, 114). Moreover, a previous study from our lab group has shown that platelet-derived HMGB1 played an essential role in clearance of bacteria in murine polymicrobial sepsis model (cecal ligation and puncture) via promotion of recruitment of neutrophils through platelet chemokines (platelet factor 4 and regulated on activation, normal T cell expressed and secreted), as well as via increased production of reactive oxygen species (31). For a more complete review of the roles platelets in defense against pathogens, the reader is referred to reviews in (4, 110, 113).
In summary, platelets actively are involved in the pathophysiologic process of sepsis at multiple levels, and are at the crossroads of the clotting cascade, immune response, and endothelial cell activation (2, 108, 115). On one hand, platelets contribute to dysregulated inflammatory responses and endorgan injuries. On the other hand, platelets also actively participate in defense responses to clear invading pathogens and help restore host homeostasis. Like many other events in sepsis, mechanism intended to act locally can lead to damage and dysfunction when the process becomes more disseminated (113).
PLATELET–MONOCYTE AGGREGATES IN SEPSIS PATHOGENESIS
A recent study showed that circulating platelet–monocyte aggregates measured by flow cytometry were significantly elevated as early as 4 h (approximately sepsis 90% vs. sham 60%) and subsequently peaked at 24 h (approximately sepsis 95% vs. sham 55%), with levels persisting until 48 h in a model of murine peritoneal polymicrobial sepsis model (26). The number of platelets attached per monocyte was also significantly increased within 24 h after sepsis, which was identified by the significantly elevated mean fluorescence intensity of CD41 on monocytes by almost 20-fold (26). Similarly, results from another study showed that coculture of human platelets and monocytes in the presence of LPS led to significant increases of platelet–monocyte aggregation at 24 h (28). Interestingly, platelet–platelet aggregation, platelets–monocyte aggregation, or platelet internalization (phagocytosis) by monocytes all were shown to facilitate polarization of CD14+ monocytes toward the proinflammatory M1 phenotype in the presence of LPS. These effects were dependent on cell–cell contact, where glycoprotein Ib (GPIb)–CD11b interactions were proven to play critical roles in the engagement of platelet and monocytes (28). Notably, adding platelets 24 h or 48 h after initiation of monocyte differentiation was not able to shift the balance toward M1 phenotype after LPS stimulation, which highlights the importance of early contact of platelets and monocytes (or formation of platelet–monocyte aggregates) in the outcome of immune and inflammatory responses (28).
In clinical settings of sepsis, platelet–monocyte aggregates significantly increased in septic patients compared with age and gender matched non-septic SIRS patients (11.70±2.94% vs. 5.64±1.53%) (116). Specifically, classical monocytes (CD14++CD16‒) were mainly involved in platelet association to form platelet–monocyte aggregates, while intermediate (CD14++CD16+) and non-classical monocytes (CD14+CD16++) remained unchanged. Importantly, elevated platelet–monocyte aggregates in circulation were associated increased mortality in septic patients (116). Similarly, increased platelet–monocyte aggregates were associated with higher risk of mortality in older septic patients (age>=65) and may be useful as a predictive factor for outcomes in this older population. However, in younger septic patients (age<65), no association was found between platelet–monocyte aggregates and mortality, suggesting differences in pathophysiology of sepsis in aging that deserve additional scrutiny (27). Additionally, it has been observed that the level of platelet–monocyte aggregates varies between gram-positive and gram-negative bacterial sepsis, with higher platelet–monocyte aggregate formation in patients infected with gram-positive bacteria evidenced by higher median fluorescence intensity signal of platelet marker CD61 (15.6 [13.7–17.1] vs. 8.5[8.1–9.5]) (17). The mechanism behind this observation was not clear but the authors speculated that the differences may relate to toxins released from gram-positive bacteria (17).
Collectively, though many studies focus on the functions of platelets in sepsis, there are fewer that investigate the role of platelet–monocyte aggregation in sepsis (Table 2). Based on current literature, we have proposed a paradigm of platelet–monocyte aggregation and its biological effects in sepsis (Fig. 2). More specific and definitive investigations are needed to provide better understanding of the biological mechanisms, importance, and effects of platelet–monocyte aggregates formed during sepsis.
Table 2.
Summary of studies investigating platelet-monocyte aggregate formation in sepsis
| Species | Experimental model | PMA detection method | PMA change | Involved molecules | Implications |
|---|---|---|---|---|---|
| Mouse | CLP (in vivo) blood Samples | Flow cytometry | ↑ | Not studied | PMA was used as an early marker of platelet activation (26) |
| Human | Coculture platelets and monocyte in presence of LPS (ex vivo) | Flow cytometry | ↑ | Glycoprotein Ib (GPIb)–CD11b axis | Platelets induced monocytes toward type 1 macrophage (M1) phenotype (28) |
| Human | Observational prospective study (in vivo). Blood Samples | Flow cytometry | ↑ | Not studied | Increased PMA was associated with higher mortality. Percentage of PMA varied in monocyte subsets (116) |
| Human | Prospective cohort study (in vivo) blood samples | Flow cytometry | ↑ | Not studied | Elevated PMA was associated with higher mortality in older patients (>=65) but not in younger patients (<65) (27) |
| Human | Prospective cohort study (in vivo) blood samples | Flow cytometry | ↑ | Not studied | PMA was higher in gram-positive bacteria caused sepsis than gram-negative (17) |
Fig. 2. Platelet–monocyte interactions.
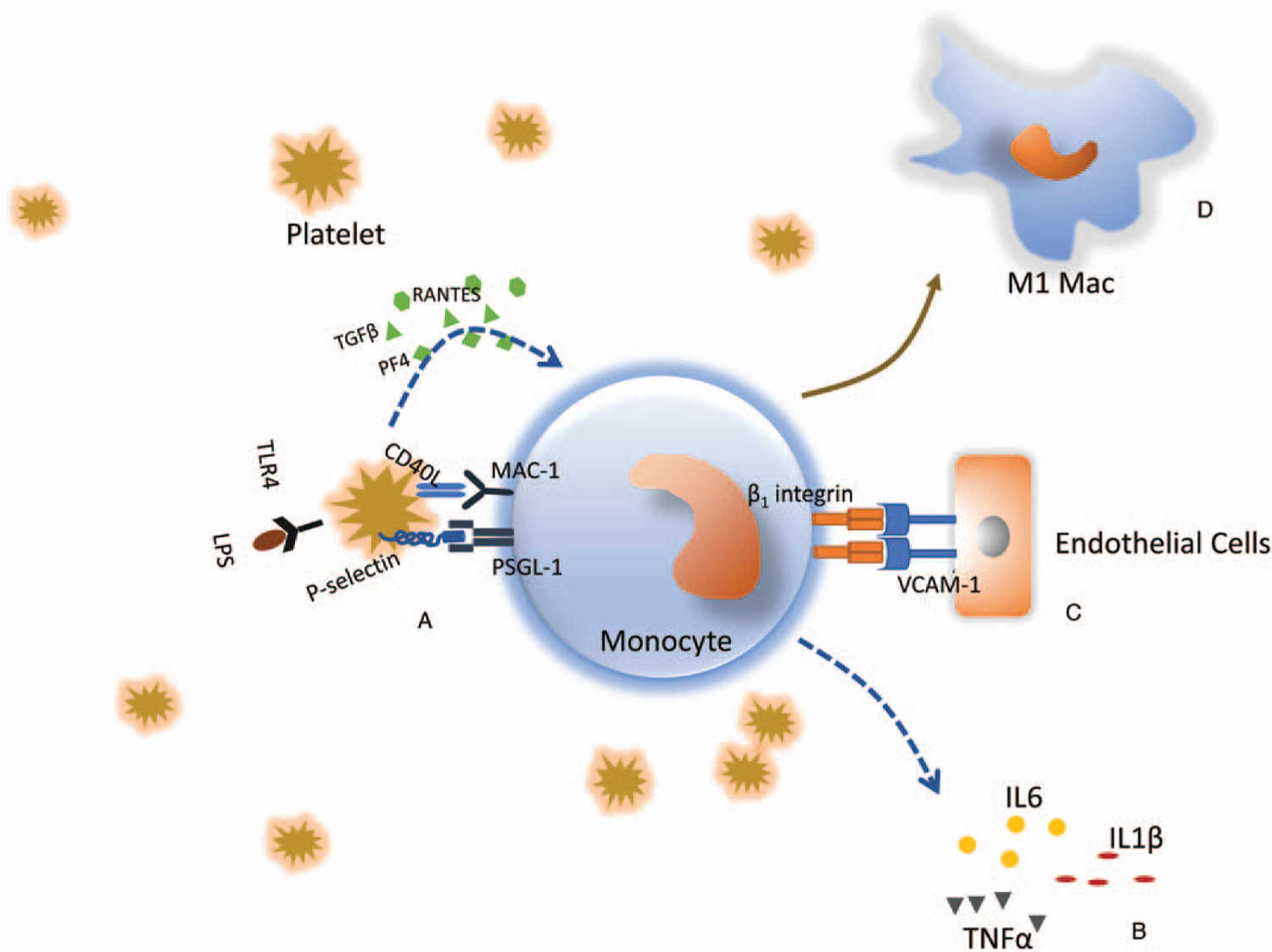
A, Activated platelets upregulate surface expression of P-selectin, which binds to receptor P-selectin glycoprotein ligand-1 (PSGL-1). This initiates aggregation of platelets and monocytes. Aggregation is further consolidated by potentiation of other ligand–receptor interactions, including CD40L (CD154) with CD11b/CD18 (MAC-1). Simultaneously, activated platelets release chemokines and cytokines (e.g., PF4, RANTES, TGFβ), which bind to cognate receptors on monocytes, initiating intracellular signal transduction and alteration of monocyte function. B, Monocytes are activated after aggregation with platelets, and release a variety of proinflammatory cytokines (e.g., IL6, IL1β, TNFα), which contribute to the dysregulated inflammatory response after sepsis. C, Aggregation of platelets and monocytes induces expression of adherent molecules on monocyte cell surface and promotes monocyte–endothelial interactions and potentiates monocyte transmigration to the infection site. D, Platelets–monocyte aggregation induces M1 polarization of monocytes.
CHALLENGES AND LIMITATIONS OF CURRENT EXPERIMENTAL METHODS TO STUDY PLATELET–MONOCYTE AGGREGATES
Platelets can be activated extremely easily in vitro. Even during blood extraction, a small amount of agitation is enough to activate platelets and cause significant artifacts in the assessment of platelet–monocyte aggregation. This presents a significant technical challenge when investigating mechanisms of platelet–monocyte association in specific pathologic conditions. Moreover, the number of monocytes in mouse peripheral blood is very limited, and so most of the studies investigating mechanisms of platelet–monocyte interactions use human blood as the source of monocytes and platelets for in vitro coculture experiments. While experiments using human samples can be illuminating and provide high relevance to human disease, mechanistic studies are often only possible in animal model systems.
Flow cytometry is the most commonly used method to detect platelet–monocyte aggregates in blood. However, data show that flow cytometry results varied greatly with different sample processing (e.g., anticoagulant used, blood procurement method) (11, 117, 118). Besides, given that monocyte may aggregate with varying number of platelets/or platelet-derived vesicles, the flowcytometry method may be not sensitive enough to differentiate those varying sizes of platelet–monocyte aggregates. Moreover, in most of the cases, flow cytometry is not always available right after sample preparation and delay of detection of platelet–monocyte aggregates is very likely to increase artifacts (117). Recently, a new technology referred to as image flow was used to study platelet–monocyte interactions. Image flow combines the microscopy with flow cytometry and makes the process in the flow cytometer visible and all cells with fluorescence signal can be inspected individually (119). This new technology extends our tools of exploring the mechanism and effects of platelet–monocyte aggregates. However, the inherent defects of this new technology are similar to conventional flow cytometry and the results may vary greatly due to different sample collection and processing, which could obscure in depth data interpretation. Therefore, standardizing sample collection and stringently following processing workflow protocols is probably needed to help make data between investigators comparable.
Notably, many studies discussed in this review employed in vitro coculture techniques to investigate how platelets affect the functions of monocytes, where platelets were activated by various known agonists and then cocultured with autologous monocytes. This approach limits the ability to define the importance of disease conditions on platelet interaction with monocytes. This inevitably raises the question of pathological relevance of in vitro coculture studies in recapitulating “real-world” interactions between platelets and monocytes.
On the gene level, to our knowledge, very few data have been collected to analyze monocyte or platelet gene expression profiles and their alteration after their aggregation. Recently, single-cell sequencing has shown increasing power in the investigation of cell biology on gene expression level (120). The ability of single-cell sequencing to resolve gene changes to a high level and differentiate between cell subtypes should reveal signatures of monocytes after interacting with platelets compared with those without platelet interaction. Moreover, gene comparison data would potentially reveal new mechanisms and functions associated with interaction, and define new effects of platelet–monocyte interaction. However, single-cell sequencing will not be able to distinguish platelet–monocyte aggregates from platelets phagocytosed by monocytes. Also, the low abundance of mRNA in platelets means that the application of single-cell sequencing in exploring the transcriptome of platelets remains to be validated, although bulk RNA sequencing of platelets has been successfully performed.
Phagocytosis of platelets by monocytes has been shown to occur in many cases. However, the mechanism and cellular signaling of this biological phenomenon is not well described. One of the critical reasons is the lack of robust and efficient investigating strategies. What happens to platelets after phagocytosis is still uncertain. One study proposed a very intriguing signaling pathway while investigating the interaction between platelets and macrophages, which implicated amyloid precursor protein, a platelet constituent protein. Amyloid cleavage into beta-amyloid peptide by macrophages following platelet phagocytosis is thought to activate macrophages and increase expression of inflammatory mediators (121). However, this pathway will require further confirmation.
FUTURE PERSPECTIVES
Despite some progress in the field the mechanism of regulation of platelet–monocyte aggregates remains poorly understood and little is known about the molecular regulation network behind it. A better understanding of the regulation network could be the basis of novel drug discovery and intervention approaches. One area of interest is in regulation of platelet activation, as this plays an essential role in the formation of platelet–monocyte aggregates. There are already a multitude of drugs (e.g., aspirin and clopidogrel) that interfere with platelet activation that could potentially regulate platelet–monocyte aggregation. Another interesting area of study is the location of platelet–monocyte aggregates. Almost all published literature report detection of aggregates only in the blood/circulation using flow cytometry. It is possible that platelet–monocyte aggregates form in the tissue, but more studies are needed to validate this speculation. Moreover, aggregation of varying numbers of platelets and/or platelet-derived vesicles may cause a varying size of the aggregates. But little is known about the biological indications of the size of platelets–monocyte aggregates. Additionally, there are questions about where platelet–monocyte aggregates go after they are formed, how long they stay in circulation, and how they are resolved in sepsis. Therefore, more studies definitely seem appropriate potentially making use of the new techniques.
To date, data relating to platelet–monocyte interactions in sepsis are quite limited. Given that platelet–monocytes were reported to participate in pathophysiology of other diseases associated with dysregulated inflammation (e.g., cardiovascular disease and trauma, as mentioned in this review), it is reasonable to speculate that all those findings regarding the mechanisms of platelet–monocyte aggregation and possible roles they play in those conditions are likely to be shared by sepsis. However, this hypothesis does need to be definitively tested and other factors potentially affecting the formation of platelet–monocyte aggregates in sepsis, such as the role of gender and age, need to be defined.
Interestingly, antiplatelet drugs (aspirin and clopidogrel) have been shown to play a very prominent role in the regulation of inflammation and immune cells (neutrophils and monocytes) activation (122, 123). Pretreatment of mice subjected endotoxemia with clopidogrel was shown to suppress pro-inflammatory genes expression. In clinical settings, aspirin and P2Y12 inhibitors (clopidogrel) have been observed to have a significant anti-inflammatory role and decrease the level of C-reactive protein, P-selectin, and platelet–leukocyte aggregates in peripheral blood of patients, which therefore has been proposed as a potential target for the treatment of sepsis (124, 125). The point here is that antiplatelet drugs may potentially affect the formation of platelet–monocyte aggregates, which is likely to be a mechanism of how antiplatelet drugs function in modulating the pathophysiology of sepsis. Therefore, future studies may focus on how antiplatelet drugs affect platelet–monocyte aggregation.
SUMMARY AND CONCLUDING REMARKS
Sepsis is still a worldwide leading cause of death, characterized as early excessive inflammatory response followed by complex regulation of immune dysregulation. Great effort has been put into studying the roles of platelets, monocytes, and their interactions in the pathogenesis of sepsis. However, the mechanism of platelet–monocyte interaction in sepsis is poorly understood. It is commonly accepted that activated platelets bind to monocytes predominantly via P-selectin and subsequently trigger downstream signaling pathways to alter monocyte phenotype. Meanwhile, many other surface molecules are at least partially involved in this interaction and further consolidate it. Nevertheless, the exact roles of these aggregates in sepsis are still not fully uncovered, in part, because of the limitations of existing techniques for investigation. There is potential for new data to have a significant impact in our knowledge of platelet–monocyte interactions, and to lead to novel therapeutics.
ACKNOWLEDGMENTS
The authors sincerely acknowledge Dr Zhongjie Yi and Dr Ping Sun for their constructive suggestions and discussions in composing this manuscript.
Funding Support:
NIH R01GM102146 and R01HL141080 to MJS; R35-GM127027 to TRB; R35GM119526 and R01HL141080 to MDN.
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Stocker TJ, Ishikawa-Ankerhold H, Massberg S, Schulz C: Small but mighty: platelets as central effectors of host defense. Thromb Haemost 117(4):651–661, 2017. [DOI] [PubMed] [Google Scholar]
- 2.Thomas MR, Storey RF: The role of platelets in inflammation. Thromb Haemost 114(3):449–458, 2015. [DOI] [PubMed] [Google Scholar]
- 3.Vogel S, Thein SL: Platelets at the crossroads of thrombosis, inflammation and haemolysis. Br J Haematol 180(5):761–767, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeaman MR: Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol 12(6):426–437, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Garraud O, Hamzeh-Cognasse H, Pozzetto B, Cavaillon JM, Cognasse F: Bench-to-bedside review: platelets and active immune functions—new clues for immunopathology? Crit Care 17(4):236, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrell CN, Aggrey AA, Chapman LM, Modjeski KL: Emerging roles for platelets as immune and inflammatory cells. Blood 123(18):2759–2767, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rondina MT, Garraud O: Emerging evidence for platelets as immune and inflammatory effector cells. Front Immunol 5:653, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kral JB, Schrottmaier WC, Salzmann M, Assinger A: Platelet interaction with innate immune cells. Transfus Med Hemother 43(2):78–88, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam FW, Vijayan KV, Rumbaut RE: Platelets and their interactions with other immune cells. Compr Physiol 5(3):1265–1280, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Assinger A, Schrottmaier WC, Salzmann M, Rayes J: Platelets in sepsis: an update on experimental models and clinical data. Front Immunol 10:1687, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li N, Goodall AH, Hjemdahl P: A sensitive flow cytometric assay for circulating platelet-leucocyte aggregates. Br J Haematol 99(4):808–816, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Nagasawa A, Matsuno K, Tamura S, Hayasaka K, Shimizu C, Moriyama T: The basis examination of leukocyte-platelet aggregates with CD45 gating as a novel platelet activation marker. Int J Lab Hematol 35(5):534–541, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR: Dynamics of leukocyte-platelet adhesion in whole blood. Blood 78(7):1730–1737, 1991. [PubMed] [Google Scholar]
- 14.Chandler AB, Hand RA: Phagocytized platelets: a source of lipids in human thrombi and atherosclerotic plaques. Science 134(3483):946–947, 1961. [DOI] [PubMed] [Google Scholar]
- 15.Glezeva N, Gilmer JF, Watson CJ, Ledwidge M: A central role for monocyte-platelet interactions in heart failure. J Cardiovasc Pharmacol Ther 21(3):245–261, 2016. [DOI] [PubMed] [Google Scholar]
- 16.Loguinova M, Pinegina N, Kogan V, Vagida M, Arakelyan A, Shpektor A, Margolis L, Vasilieva E: Monocytes of different subsets in complexes with platelets in patients with myocardial infarction. Thromb Haemost 118(11):1969–1981, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tunjungputri RN, van de Heijden W, Urbanus RT, de Groot PG, van der Ven A, de Mast Q: Higher platelet reactivity and platelet-monocyte complex formation in gram-positive sepsis compared to gram-negative sepsis. Platelets 28(6):595–601, 2017. [DOI] [PubMed] [Google Scholar]
- 18.Vidranski V, Laskaj R, Sikiric D, Skerk V: Platelet satellitism in infectious disease? Biochem Med (Zagreb) 25(2):285–294, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skinnider LF, Musclow CE, Kahn W: Platelet satellitism—an ultrastructural study. Am J Hematol 4(2):179–185, 1978. [DOI] [PubMed] [Google Scholar]
- 20.Kjeldsberg CR, Swanson J: Platelet satellitism. Blood 43(6):831–836, 1974. [PubMed] [Google Scholar]
- 21.Chakrabarti I: Platelet satellitism: a rare, interesting, in vitro phenomenon. Indian J Hematol Blood Transfus 30(3):213–214, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bain BJ, Czako B: Monocyte adhesion with platelet satellitism and phagocytosis in Hodgkin lymphoma. Am J Hematol 93(12):1561, 2018. [DOI] [PubMed] [Google Scholar]
- 23.Kasprzycka E, Zak J, Ratomski K, Wysocka J, Hryniewicz K: [Platelet satellitism]. Wiad Lek 59(7–8):557–559, 2006. [PubMed] [Google Scholar]
- 24.Gerrits AJ, Frelinger AL 3rd, Michelson AD: Whole blood analysis of leukocyte-platelet aggregates. Curr Protoc Cytom 78:6, 2016. [DOI] [PubMed] [Google Scholar]
- 25.Zamora C, Canto E, Nieto JC, Garcia-Planella E, Gordillo J, Ortiz MA, Suarez-Calvet X, Perea L, Julia G, Juarez C, et al. : Inverse association between circulating monocyte-platelet complexes and inflammation in ulcerative colitis patients. Inflamm Bowel Dis 24(4):818–828, 2018. [DOI] [PubMed] [Google Scholar]
- 26.Vardon Bounes F, Memier V, Marcaud M, Jacquemin A, Hamzeh-Cognasse H, Garcia C, Series J, Sie P, Minville V, Gratacap MP, et al. : Platelet activation and prothrombotic properties in a mouse model of peritoneal sepsis. Sci Rep 8(1):13536, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rondina MT, Carlisle M, Fraughton T, Brown SM, Miller RR 3rd, Harris ES, Weyrich AS, Zimmerman GA, Supiano MA, Grissom CK: Platelet-monocyte aggregate formation and mortality risk in older patients with severe sepsis and septic shock. J Gerontol A Biol Sci Med Sci 70(2):225–231, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carestia A, Mena HA, Olexen CM, Ortiz Wilczynski JM, Negrotto S, Errasti AE, Gomez RM, Jenne CN, Carrera Silva EA, Schattner M: Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep 28(4):896.e5–908.e5, 2019. [DOI] [PubMed] [Google Scholar]
- 29.Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, Schaffer TE, Bohn E, Frick JS, Borst O, et al. : Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest 125(12):4638–4654, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogel S, Rath D, Borst O, Mack A, Loughran P, Lotze MT, Neal MD, Billiar TR, Gawaz M: Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem Biophys Res Commun 478(1): 143–148, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou H, Deng M, Liu Y, Yang C, Hoffman R, Zhou J, Loughran PA, Scott MJ, Neal MD, Billiar TR: Platelet HMGB1 is required for efficient bacterial clearance in intra-abdominal bacterial sepsis in mice. Blood Adv 2(6):638–648, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Semple JW, Italiano JE Jr, Freedman J: Platelets and the immune continuum. Nat Rev Immunol 11(4):264–274, 2011. [DOI] [PubMed] [Google Scholar]
- 33.Weyrich AS, Zimmerman GA: Platelets: signaling cells in the immune continuum. Trends Immunol 25(9):489–495, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Djaldetti M, Fishman P: Satellitism of platelets to monocytes in a patient with hypogammaglobulinaemia. Scand J Haematol 21(4):305–308, 1978. [DOI] [PubMed] [Google Scholar]
- 35.Kopcinovic LM, Pavic M: Platelet satellitism in a trauma patient. Biochem Med (Zagreb) 22(1):130–134, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zipperle J, Altenburger K, Ponschab M, Schlimp CJ, Spittler A, Bahrami S, Redl H, Schochl H: Potential role of platelet-leukocyte aggregation in trauma-induced coagulopathy: ex vivo findings. J Trauma Acute Care Surg 82(5): 921–926, 2017. [DOI] [PubMed] [Google Scholar]
- 37.Vulliamy P, Gillespie S, Armstrong PC, Allan HE, Warner TD, Brohi K: Histone H4 induces platelet ballooning and microparticle release during trauma hemorrhage. Proc Natl Acad Sci U S A 116(35):17444–17449, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fahim A, Crooks MG, Morice AH, Hart SP: Increased platelet binding to circulating monocytes in idiopathic pulmonary fibrosis. Lung 192(2):277–284, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI: Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 104(13):1533–1537, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Elalamy I, Chakroun T, Gerotziafas GT, Petropoulou A, Robert F, Karroum A, Elgrably F, Samama MM, Hatmi M: Circulating platelet-leukocyte aggregates: a marker of microvascular injury in diabetic patients. Thromb Res 121(6):843–848, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Allen N, Barrett TJ, Guo Y, Nardi M, Ramkhelawon B, Rockman CB, Hochman JS, Berger JS: Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis 282:11–18, 2019. [DOI] [PubMed] [Google Scholar]
- 42.Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T: Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol 24(2):81–88, 2002. [DOI] [PubMed] [Google Scholar]
- 43.Vats R, Brzoska T, Bennewitz MF, Jimenez MA, Pradhan-Sundd T, Tutuncuoglu E, Jonassaint J, Gutierrez E, Watkins SC, Shiva S, et al. : Platelet extracellular vesicles drive inflammasome-IL-1beta-dependent lung injury in sickle cell disease. Am J Respir Crit Care Med 201(1):33–46, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ: Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol 115(2):451–459, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Singh MV, Davidson DC, Kiebala M, Maggirwar SB: Detection of circulating platelet-monocyte complexes in persons infected with human immunodeficiency virus type-1. J Virol Methods 181(2):170–176, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Armstrong PC, Kirkby NS, Chan MV, Finsterbusch M, Hogg N, Nourshargh S, Warner TD: Novel whole blood assay for phenotyping platelet reactivity in mice identifies ICAM-1 as a mediator of platelet-monocyte interaction. Blood 126(10):e11–e18, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schulz C, von Bruhl ML, Barocke V, Cullen P, Mayer K, Okrojek R, Steinhart A, Ahmad Z, Kremmer E, Nieswandt B, et al. : EMMPRIN (CD147/basigin) mediates platelet-monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J Thromb Haemost 9(5):1007–1019, 2011. [DOI] [PubMed] [Google Scholar]
- 48.Seizer P, Borst O, Langer HF, Bultmann A, Munch G, Herouy Y, Stellos K, Kramer B, Bigalke B, Buchele B, et al. : EMMPRIN (CD147) is a novel receptor for platelet GPVI and mediates platelet rolling via GPVI-EMMPRIN interaction. Thromb Haemost 101(4):682–686, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Inui M, Tazawa K, Kishi Y, Takai T: Platelets convert peripheral blood circulating monocytes to regulatory cells via immunoglobulin G and activating-type Fcgamma receptors. BMC Immunol 16:20, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanderson HM, Fox SC, Robbins RA, Losche W, Spangenberg P, Heptinstall S: Role of GPIIb-IIIa in platelet-monocyte and platelet-neutrophil conjugate formation in whole blood. Platelets 9(3–4):245–250, 1998. [DOI] [PubMed] [Google Scholar]
- 51.Steiner S, Seidinger D, Huber K, Kaun C, Minar E, Kopp CW: Effect of glycoprotein IIb/IIIa antagonist abciximab on monocyte-platelet aggregates and tissue factor expression. Arterioscler Thromb Vasc Biol 23(9):1697–1702, 2003. [DOI] [PubMed] [Google Scholar]
- 52.Shantsila E, Montoro-Garcia S, Lip GY: Monocytes circulate in constant reversible interaction with platelets in a [Ca2+]-dependent manner. Platelets 25(3):197–201, 2014. [DOI] [PubMed] [Google Scholar]
- 53.Hottz ED, Medeiros-de-Moraes IM, Vieira-de-Abreu A, de Assis EF, Vals-de-Souza R, Castro-Faria-Neto HC, Weyrich AS, Zimmerman GA, Bozza FA, Bozza PT: Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J Immunol 193(4):1864–1872, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freedman JE, Loscalzo J: Platelet–monocyte aggregates. Circulation 105(18):2130–2132, 2002. [DOI] [PubMed] [Google Scholar]
- 55.Mildner A, Marinkovic G, Jung S: Murine monocytes: origins, subsets, fates, and functions. Microbiol Spectr 4(5). [DOI] [PubMed] [Google Scholar]
- 56.Ziegler-Heitbrock L: The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol 81(3):584–592, 2007. [DOI] [PubMed] [Google Scholar]
- 57.Boudjeltia KZ, Brohee D, Piro P, Nuyens V, Ducobu J, Kherkofs M, Van Antwerpen P, Cauchie P, Remacle C, Vanhaeverbeek M: Monocyte-platelet complexes on CD14/CD16 monocyte subsets: relationship with ApoA-I levels. A preliminary study. Cardiovasc Pathol 17(5):285–288, 2008. [DOI] [PubMed] [Google Scholar]
- 58.Weiss R, Groger M, Rauscher S, Fendl B, Eichhorn T, Fischer MB, Spittler A, Weber V: Differential interaction of platelet-derived extracellular vesicles with leukocyte subsets in human whole blood. Sci Rep 8(1):6598, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, Huo Y, Xia L: P-selectin glycoprotein ligand-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation 117(25):3227–3237, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bournazos S, Rennie J, Hart SP, Fox KA, Dransfield I: Monocyte functional responsiveness after PSGL-1-mediated platelet adhesion is dependent on platelet activation status. Arterioscler Thromb Vasc Biol 28(8):1491–1498, 2008. [DOI] [PubMed] [Google Scholar]
- 61.Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, Prescott SM, Zimmerman GA: Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest 97(6):1525–1534, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dixon DA, Tolley ND, Bemis-Standoli K, Martinez ML, Weyrich AS, Morrow JD, Prescott SM, Zimmerman GA: Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Invest 116(10):2727–2738, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Passacquale G, Vamadevan P, Pereira L, Hamid C, Corrigall V, Ferro A: Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS One 6(10):e25595, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki J, Hamada E, Shodai T, Kamoshida G, Kudo S, Itoh S, Koike J, Nagata K, Irimura T, Tsuji T: Cytokine secretion from human monocytes potentiated by P-selectin-mediated cell adhesion. Int Arch Allergy Immunol 160(2):152–160, 2013. [DOI] [PubMed] [Google Scholar]
- 65.Christersson C, Johnell M, Siegbahn A: Tissue factor and IL8 production by P-selectin-dependent platelet-monocyte aggregates in whole blood involves phosphorylation of Lyn and is inhibited by IL10. J Thromb Haemost 6(6):986–994, 2008. [DOI] [PubMed] [Google Scholar]
- 66.Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie BC, Furie B: P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci U S A 91(19):8767–8771, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hidari KI, Weyrich AS, Zimmerman GA, McEver RP: Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J Biol Chem 272(45):28750–28756, 1997. [DOI] [PubMed] [Google Scholar]
- 68.Ba X, Chen C, Gao Y, Zeng X: Signaling function of PSGL-1 in neutrophil: tyrosine-phosphorylation-dependent and c-Abl-involved alteration in the F-actin-based cytoskeleton. J Cell Biochem 94(2):365–373, 2005. [DOI] [PubMed] [Google Scholar]
- 69.Stephen J, Emerson B, Fox KA, Dransfield I: The uncoupling of monocyte-platelet interactions from the induction of proinflammatory signaling in monocytes. J Immunol 191(11):5677–5683, 2013. [DOI] [PubMed] [Google Scholar]
- 70.Gudbrandsdottir S, Hasselbalch HC, Nielsen CH: Activated platelets enhance IL-10 secretion and reduce TNF-alpha secretion by monocytes. J Immunol 191(8):4059–4067, 2013. [DOI] [PubMed] [Google Scholar]
- 71.Takeda Y, Marumo M, Nara H, Feng ZG, Asao H, Wakabayashi I: Selective induction of anti-inflammatory monocyte-platelet aggregates in a model of pulsatile blood flow at low shear rates. Platelets 27(6):583–592, 2016. [DOI] [PubMed] [Google Scholar]
- 72.Lang D, Dohle F, Terstesse M, Bangen P, August C, Pauels HG, Heidenreich S: Down-regulation of monocyte apoptosis by phagocytosis of platelets: involvement of a caspase-9, caspase-3, and heat shock protein 70-dependent pathway. J Immunol 168(12):6152–6158, 2002. [DOI] [PubMed] [Google Scholar]
- 73.Martins PAdC, van Gils JM, Mol A, Hordijk PL, Zwaginga JJ: Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of β1 and β2 integrins. J Leukoc Biol 79(3):499–507, 2006. [DOI] [PubMed] [Google Scholar]
- 74.Feng Y, Dorhoi A, Mollenkopf HJ, Yin H, Dong Z, Mao L, Zhou J, Bi A, Weber S, Maertzdorf J, et al. : Platelets direct monocyte differentiation into epithelioid-like multinucleated giant foam cells with suppressive capacity upon mycobacterial stimulation. J Infect Dis 210(11):1700–1710, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Astarita JL, Acton SE, Turley SJ: Podoplanin: emerging functions in development, the immune system, and cancer. Front Immunol 3:283, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Quintanilla M, Montero-Montero L, Renart J, Martin-Villar E: Podoplanin in inflammation and cancer. Int J Mol Sci 20(3):707, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ugorski M, Dziegiel P, Suchanski J: Podoplanin—a small glycoprotein with many faces. Am J Cancer Res 6(2):370–386, 2016. [PMC free article] [PubMed] [Google Scholar]
- 78.Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, Khan M, Dominguez-Medina CC, Lax S, Carvalho-Gaspar M, et al. : Inflammation drives thrombosis after Salmonella infection via CLEC-2 on platelets. J Clin Invest 125(12):4429–4446, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suzuki-Inoue K, Inoue O, Ozaki Y: The novel platelet activation receptor CLEC-2. Platelets 22(5):380–384, 2011. [DOI] [PubMed] [Google Scholar]
- 80.Suzuki-Inoue K, Inoue O, Ding G, Nishimura S, Hokamura K, Eto K, Kashiwagi H, Tomiyama Y, Yatomi Y, Umemura K, et al. : Essential in vivo roles of the C-type lectin receptor CLEC-2: embryonic/neonatal lethality of CLEC-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC-2-deficient platelets. J Biol Chem 285(32):24494–24507, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bertozzi CC, Schmaier AA, Mericko P, Hess PR, Zou Z, Chen M, Chen CY, Xu B, Lu MM, Zhou D, et al. : Platelets regulate lymphatic vascular development through CLEC-2-SLP-76 signaling. Blood 116(4):661–670, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Navarro-Nunez L, Langan SA, Nash GB, Watson SP: The physiological and pathophysiological roles of platelet CLEC-2. Thromb Haemost 109(6):991–998, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, Grygielska B, Smith SW, Skordilis K, Watson SP: The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun 8(1):2239, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Gils JM, Zwaginga JJ, Hordijk PL: Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol 85(2):195–204, 2009. [DOI] [PubMed] [Google Scholar]
- 85.da Costa Martins P, van den Berk N, Ulfman LH, Koenderman L, Hordijk PL, Zwaginga JJ: Platelet-monocyte complexes support monocyte adhesion to endothelium by enhancing secondary tethering and cluster formation. Arterioscler Thromb Vasc Biol 24(1):193–199, 2004. [DOI] [PubMed] [Google Scholar]
- 86.Theilmeier G, Lenaerts T, Remacle C, Collen D, Vermylen J, Hoylaerts MF: Circulating activated platelets assist THP-1 monocytoid/endothelial cell interaction under shear stress. Blood 94(8):2725–2734, 1999. [PubMed] [Google Scholar]
- 87.van Gils JM, da Costa Martins PA, Mol A, Hordijk PL, Zwaginga JJ: Trans-endothelial migration drives dissociation of plateletmonocyte complexes. Thromb Haemost 100(2):271–279, 2008. [PubMed] [Google Scholar]
- 88.Claushuis TAM, Van Der Veen AIP, Horn J, Schultz MJ, Houtkooper RH, Van ‘t Veer C, Van Der Poll T: Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets; 2018;1–9, 2018. [DOI] [PubMed] [Google Scholar]
- 89.Gawaz M, Fateh-Moghadam S, Pilz G, Gurland HJ, Werdan K: Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur J Clin Invest 25(11):843–851, 1995. [DOI] [PubMed] [Google Scholar]
- 90.Schouten M, Wiersinga WJ, Levi M, van der Poll T: Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol 83(3):536–545, 2008. [DOI] [PubMed] [Google Scholar]
- 91.Coughlin SR: Thrombin signalling and protease-activated receptors. Nature 407(6801):258–264, 2000. [DOI] [PubMed] [Google Scholar]
- 92.Linden MD: Platelet physiology. Methods Mol Biol 992:13–30, 2013. [DOI] [PubMed] [Google Scholar]
- 93.Peerschke EI, Reid KB, Ghebrehiwet B: Platelet activation by C1q results in the induction of alpha IIb/beta 3 integrins (GPIIb-IIIa) and the expression of P-selectin and procoagulant activity. J Exp Med 178(2):579–587, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wijten P, van Holten T, Woo LL, Bleijerveld OB, Roest M, Heck AJ, Scholten A: High precision platelet releasate definition by quantitative reversed protein profiling—brief report. Arterioscler Thromb Vasc Biol 33(7):1635–1638, 2013. [DOI] [PubMed] [Google Scholar]
- 95.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P: Platelets express functional Toll-like receptor-4. Blood 106(7):2417–2423, 2005. [DOI] [PubMed] [Google Scholar]
- 96.Schattner M: Platelet TLR4 at the crossroads of thrombosis and the innate immune response. J Leukoc Biol 105(5):873–880, 2019. [DOI] [PubMed] [Google Scholar]
- 97.Vulliamy P, Kornblith LZ, Kutcher ME, Cohen MJ, Brohi K, Neal MD: Alterations in platelet behavior after major trauma: adaptive or maladaptive? Platelets; 2020;1–10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cornelius DC, Baik CH, Travis OK, White DL, Young CM, Austin Pierce W, Shields CA, Poudel B, Williams JM: NLRP3 inflammasome activation in platelets in response to sepsis. Physiol Rep 7(9):e14073, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peters MJ, Dixon G, Kotowicz KT, Hatch DJ, Heyderman RS, Klein NJ: Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. Br J Haematol 106(2):391–399, 1999. [DOI] [PubMed] [Google Scholar]
- 100.Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA: Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood 88(1):146–157, 1996. [PubMed] [Google Scholar]
- 101.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA: CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 391(6667):591–594, 1998. [DOI] [PubMed] [Google Scholar]
- 102.Greco E, Lupia E, Bosco O, Vizio B, Montrucchio G: Platelets and multi-organ failure in sepsis. Int J Mol Sci 18(10):2200, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Matthay MA, Ware LB, Zimmerman GA: The acute respiratory distress syndrome. J Clin Invest 122(8):2731–2740, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grommes J, Alard JE, Drechsler M, Wantha S, Morgelin M, Kuebler WM, Jacobs M, von Hundelshausen P, Markart P, Wygrecka M, et al. : Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med 185(6):628–636, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H, Wu D, Liu B, Li H, Li H, et al. : Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep 6:37252, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim SJ, Jenne CN: Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Semin Immunol 28(6):546–554, 2016. [DOI] [PubMed] [Google Scholar]
- 107.Sharron M, Hoptay CE, Wiles AA, Garvin LM, Geha M, Benton AS, Nagaraju K, Freishtat RJ: Platelets induce apoptosis during sepsis in a contact-dependent manner that is inhibited by GPIIb/IIIa blockade. PLoS One 7(7):e41549, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.de Stoppelaar SF, van ‘t Veer C, van der Poll T: The role of platelets in sepsis. Thromb Haemost 112(4):666–677, 2014. [DOI] [PubMed] [Google Scholar]
- 109.Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM: Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 99(11):4021–4029, 2002. [DOI] [PubMed] [Google Scholar]
- 110.Li Z, Yang F, Dunn S, Gross AK, Smyth SS: Platelets as immune mediators: their role in host defense responses and sepsis. Thromb Res 127(3):184–188, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lipinska-Gediga M: Platelets in sepsis—are there any new aspects? Anaesthesiol Intensive Ther 49(2):167–172, 2017. [DOI] [PubMed] [Google Scholar]
- 112.Nurden AT: The biology of the platelet with special reference to inflammation, wound healing and immunity. Front Biosci (Landmark Ed) 23:726–751, 2018. [DOI] [PubMed] [Google Scholar]
- 113.Rayes J, Bourne JH, Brill A, Watson SP: The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res Pract Thromb Haemost 4(1):23–35, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, et al. : Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13(4):463–469, 2007. [DOI] [PubMed] [Google Scholar]
- 115.Dewitte A, Lepreux S, Villeneuve J, Rigothier C, Combe C, Ouattara A, Ripoche J: Blood platelets and sepsis pathophysiology: a new therapeutic prospect in critical ill patients? Ann Intensive Care 7(1):115, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wu Q, Ren J, Hu D, Wu X, Li G, Wang G, Gu G, Chen J, Li R, Li Y, et al. : Monocyte subsets and monocyte-platelet aggregates: implications in predicting septic mortality among surgical critical illness patients. Biomarkers 21(6):509–516, 2016. [DOI] [PubMed] [Google Scholar]
- 117.Harding SA, Din JN, Sarma J, Jessop A, Weatherall M, Fox KA, Newby DE: Flow cytometric analysis of circulating platelet-monocyte aggregates in whole blood: methodological considerations. Thromb Haemost 98(2):451–456, 2007. [PubMed] [Google Scholar]
- 118.Ibeagha-Awemu EM, Ibeagha AE, Zhao X: The influence of different anti-coagulants and sample preparation methods on measurement of mCD14 on bovine monocytes and polymorphonuclear neutrophil leukocytes. BMC Res Notes 5:93, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hui H, Fuller KA, Erber WN, Linden MD: Imaging flow cytometry in the assessment of leukocyte-platelet aggregates. Methods 112:46–54, 2017. [DOI] [PubMed] [Google Scholar]
- 120.Papalexi E, Satija R: Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol 18(1):35–45, 2018. [DOI] [PubMed] [Google Scholar]
- 121.De Meyer GR, De Cleen DM, Cooper S, Knaapen MW, Jans DM, Martinet W, Herman AG, Bult H, Kockx MM: Platelet phagocytosis and processing of beta-amyloid precursor protein as a mechanism of macrophage activation in atherosclerosis. Circ Res 90(11):1197–1204, 2002. [DOI] [PubMed] [Google Scholar]
- 122.Thomas MR, Storey RF: Effect of P2Y12 inhibitors on inflammation and immunity. Thromb Haemost 114(3):490–497, 2015. [DOI] [PubMed] [Google Scholar]
- 123.Schrottmaier WC, Kral JB, Badrnya S, Assinger A: Aspirin and P2Y12 Inhibitors in platelet-mediated activation of neutrophils and monocytes. Thromb Haemost 114(3):478–489, 2015. [DOI] [PubMed] [Google Scholar]
- 124.Muhlestein JB: Effect of antiplatelet therapy on inflammatory markers in atherothrombotic patients. Thromb Haemost 103(1):71–82, 2010. [DOI] [PubMed] [Google Scholar]
- 125.Akinosoglou K, Alexopoulos D: Use of antiplatelet agents in sepsis: a glimpse into the future. Thromb Res 133(2):131–138, 2014. [DOI] [PubMed] [Google Scholar]