

Abstract
The hexadehydro-Diels-Alder (HDDA) reaction is the thermal cyclization of an alkyne and a 1,3-diyne to generate a benzyne intermediate. This is then rapidly trapped, in situ, by a variety of species to yield highly functionalized benzenoid products. In contrast to nearly all other methods of aryne generation, no other reagents are required to produce a HDDA benzyne. The versatile and customizable nature of the process has attracted much attention due not only to its synthetic potential, but also because of the fundamental mechanistic insights the studies often afford. The authors have attempted to provide here a comprehensive compilation of publications appearing by mid-2020 that describe experimental results of HDDA reactions.
Graphical Abstract
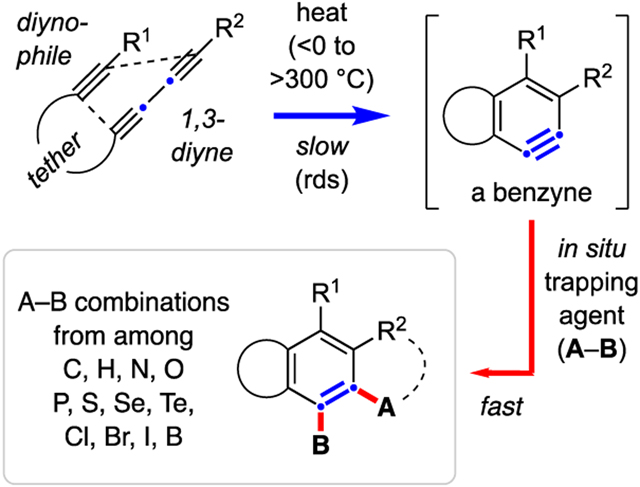
1. INTRODUCTION
1.1. Background: Dehydro-Diels-Alder Reactions (Figure 1)
Figure 1.
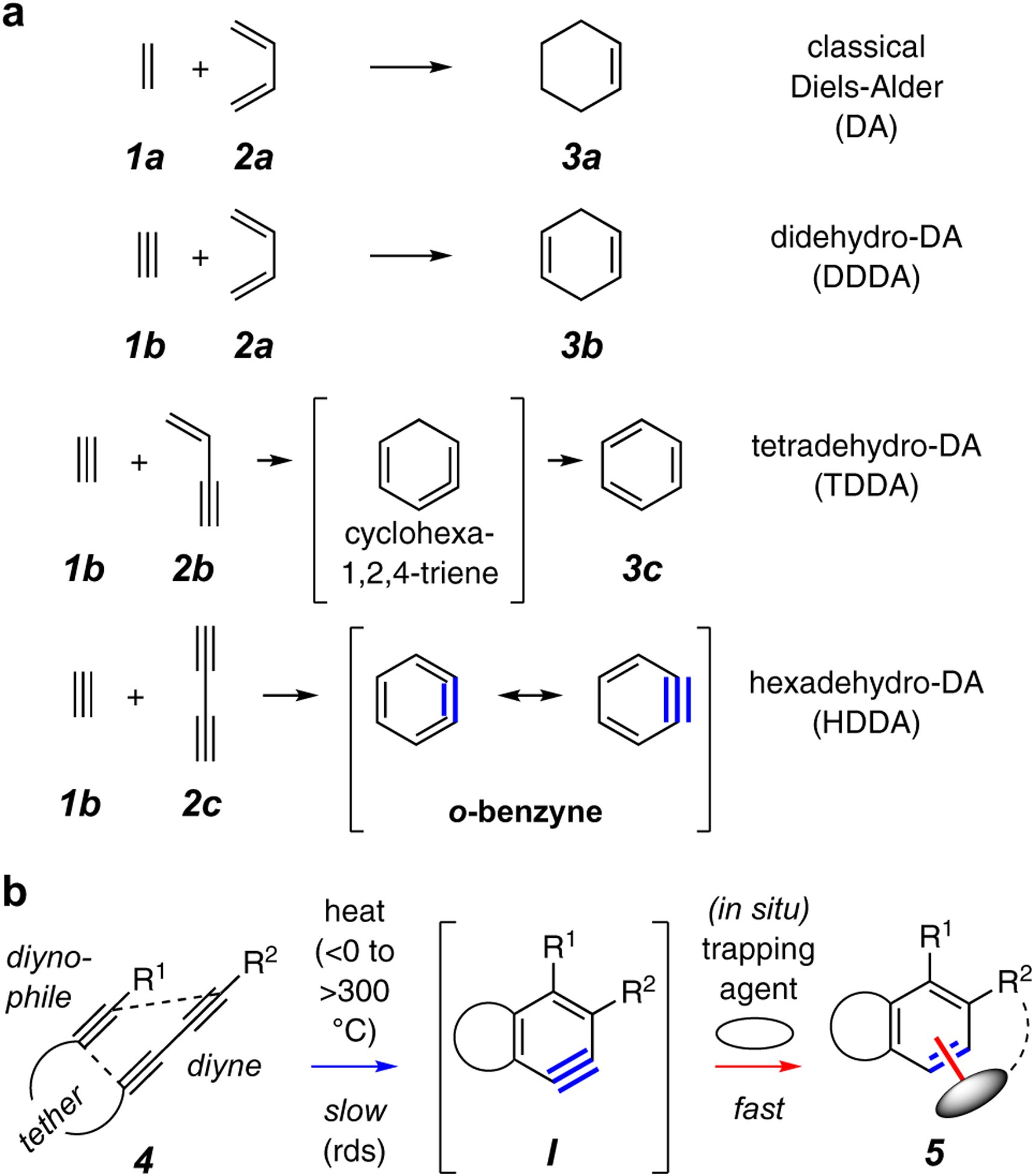
(a) The family of prototypical classical and various dehydro-Diels-Alder reactions of increasing oxidation states. (b) The generic intramolecular hexadehydro-Diels-Alder (HDDA) cyclization and benzyne-trapping reaction.
The Diels-Alder (DA) cycloaddition is one of the most widely known – and powerful – C–C bond forming reactions in organic chemistry.1 The reaction produces six-membered ring structures in an efficient, customizable, controllable, and predictable fashion, all grounded on a sound mechanistic foundation. The simplest DA reaction is the thermal [4+2] cycloaddition between ethylene (1a, the “dienophile”) and 1,3-butadiene (2a, the “diene”) to give cyclohexene (3a), as shown in Figure 1a. Variations involving progressively more highly oxidized substrates include the didehydro- and tetradehydro-Diels-Alder reactions.2,3,4 Of course, these result in more oxidized products, namely cyclohexadiene [3b, from ethyne (1b) and butadiene] and benzene [3c, from ethyne and 1,3-butenyne (2b)] through the intermediate strained allene 1,2,4-cyclohexatriene. The highest oxidized case – the hexadehydro variant – is exemplified by the reaction of ethyne with 1,3-butadiyne (2c) to generate 1,2-dehydrobenzene (or o-benzyne). This represents a special situation because benzyne is highly reactive and cannot unimolecularly reorganize to a stable, isolable entity. However, because benzynes (and arynes more broadly) represent one of the most versatile and useful of all reactive intermediates in organic chemistry, this variant offers special opportunity.
A generic version of this triyne-to-benzyne cycloisomerization, now involving a tether linking the 1,3-diyne to a distal diynophile and rendering the reaction intramolecular in nature, is shown in Figure 1b. The resulting benzyne intermediate, I, is produced in the presence of suitable trapping agents (oval) that can be either part of substrate 4 (e.g., bonded to R2) or altogether separate from 4. These processes lead to structurally complex, highly substituted and/or fused, benzenoid products 5 via intra- (cf. dashed arc in 5) or intermolecular processes, respectively. The overall transformation involves the initial rate-limiting cycloisomerization of the triyne (blue arrow) followed by rapid in situ trapping (red arrow). As demonstrated by the now many examples of published results, the highly customizable nature of both the hexadehydro-Diels-Alder (HDDA) substrate and the trapping reagent(s) provides considerable versatility for the creation of structurally sophisticated benzenoid products. Reviews of the early developments in HDDA chemistry have appeared.5,6,7
1.2. Earliest Examples of Hexadehydro-Diels-Alder Reaction (Figure 2)
Figure 2.
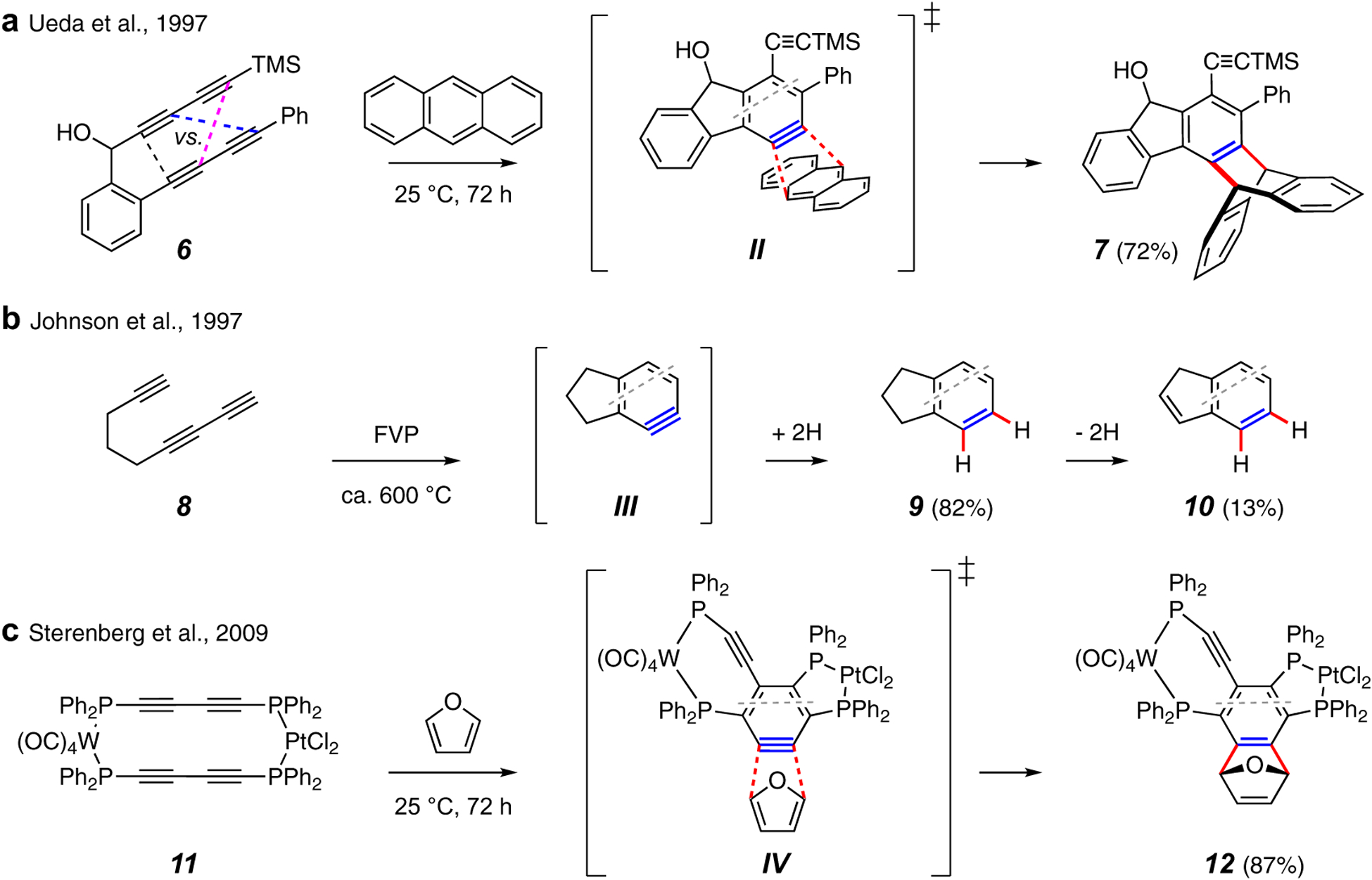
Earliest reports of cyclization of a triyne to a benzyne intermediate. (a)formattingasusedbyChemRev Ueda and coworkers (1997): trapping of the benzyne II with anthracene gives the triptycene 7. (b) Johnson and coworkers (1997): high temperature (short time) cycloisomerization of 8 to 10 via III and 9. (c) Sterenberg and Tsui (2009): intra-annular cyclization of dinuclear complex 11 in the presence of furan gives the adduct 12 via IV.
The first reports of cyclization of a multiyne to product(s) indicative of a benzyne intermediate came coincidentally and independently from the research groups of Ueda8 (Figure 2a) and Johnson9 (Figure 2b) in 1997. Tetrayne 6 was observed to cyclize at ambient temperature in the presence of added anthracene to produce the adduct 7 by way of a transition structure (TS) similar to that shown as II. A second minor isomer (not shown) arising from closure of the tetrayne substrate in the complementary sense (see dashed bonds in 6) was also isolated. This early example, proceeding even at ambient temperature, demonstrated the enhanced reactivity of tetraynes relative to analogous triynes as well as the issue of regioselectivity in the direction of ring closure in unsymmetrical tetraynes. In Johnson’s studies the minimalistically tethered triyne 8 was subjected to flash vacuum pyrolysis conditions, giving rise, via III, to a mixture of indane (9), indene (10), and “some soot” – a more or less balanced equation.9 A dozen years later, the mixed bimetallic tetrayne complex 11 (Figure 2c) was observed by researchers in the Sterenberg group to give rise to a doubly fused benzyne, which was captured by furan to form 12 via TS IV.10 Over a decade, Ueda and coworkers proceeded to study the cyclizations of a number of substrates related to 611,12,13,14,15,16,17,18 in efforts primarily motivated by their interest in using radical character in their o-benzyne intermediates to react with DNA12,18 in analogy to enediyne-derived 1,4-diradicals19 (i.e., p-benzyne). That said, it is now recognized that o-benzyne derivatives react overwhelmingly by polar processes wherein the strained alkyne behaves as a soft (i.e., polarizable) electrophile rather than via single-electron processes involving radical intermediates.
It was not until 2012 that the substantially greater generality of this cycloisomerization transformation became recognized through a report of work done in the Hoye laboratories.20 It was also in this publication that the term “hexadehydro-Diels-Alder” (HDDA) was coined. The HDDA reaction offers certain advantages over classical methods for generation and trapping of arynes. First, because the initial cyclization event requires only heating, no additional reagents are required. This is in contrast to nearly all other methods of forming arynes, including the most commonly used Kobayashi method.21 The ability to access this class of reactive intermediate in a pristine environment (i.e., reagent- and byproduct-free) has led to the discovery of some new types of trapping reactions as well as the uncovering of certain mechanistic details that provide new fundamental understanding of aryne reactions. At a strategic level, the HDDA reaction constitutes a de novo construction of the aromatic ring from an acyclic substrate, which is entirely complementary to classical benzyne generation. The latter always starts with a preformed aromatic ring; accordingly, in traditional benzyne chemistry creation and trapping constitutes a net substitution reaction at two adjacent carbons of the benzenoid precursor.
1.3. Organization of this Review
Here are some organizational and formatting details to help orient readers. Beyond those shown in Figure 2, examples of specific reactions are found in Sections 4 and 5. Those in subsections 4.1–4.13 are categorized according to the types of the two new bonds that are formed at each of the benzyne carbon atoms as a result of the trapping reaction. The HDDA substrates (tethered triynes or tetraynes) and the final, isolated reaction products are designated by Arabic numbers. Intermediates or transition structures (TSs) that are helpful in rationalizing the overall outcome of each reaction are shown within brackets and are distinguished by Roman numerals. Some of these species are well supported by experimental and/or computational experiments, but others are offered by the researchers as their most reasonable rationale to account for the transformation.
We have attempted to include (or at least comment on) at least one reaction from each of the published reports in the last decade in which experimental outcomes are described. Any omission or oversight in achieving that goal is unintentional. Many publications provide multiple examples, in some cases dozens, of the same type of transformation or of trapping reactions that fall into more than one class. The reactions selected for inclusion here are meant to be exemplary of all of the major types of reaction pathways, but by no means do they represent a full compilation of all known HDDA transformations. A Reaxys search of, minimally, a 1,3-diyne mapped to a benzenoid product shows >600 unique reactions.
We typically have elected not to show the reaction solvent unless it explicitly participates in the trapping reaction (e.g., as a member of a 3-component reaction). Some solvents are essentially inert, but others (e.g., benzene or toluene) will, slowly, trap the benzyne if the other trapping processes are relatively slow. The rate of any trapping is a function of both i) the inherent reactivity of the trapping agent and ii) the concentration (or effective molarity) of that agent. The number of equivalents of an external trapping agent is also not shown, unless only a slight excess was used, implying that an agent of that type is particularly effective at capturing the benzyne. In most instances, trapping agents are trivially available, are used in excess, and the effect of using less has not been explicitly evaluated. The indicated yields are those of isolated material, often for single runs of relatively small-scale experiments; large error bars should be presumed.
2. DIYNE-TO-DIYNOPHILE SUBSTRATE TETHERS (Figure 3)
Figure 3.
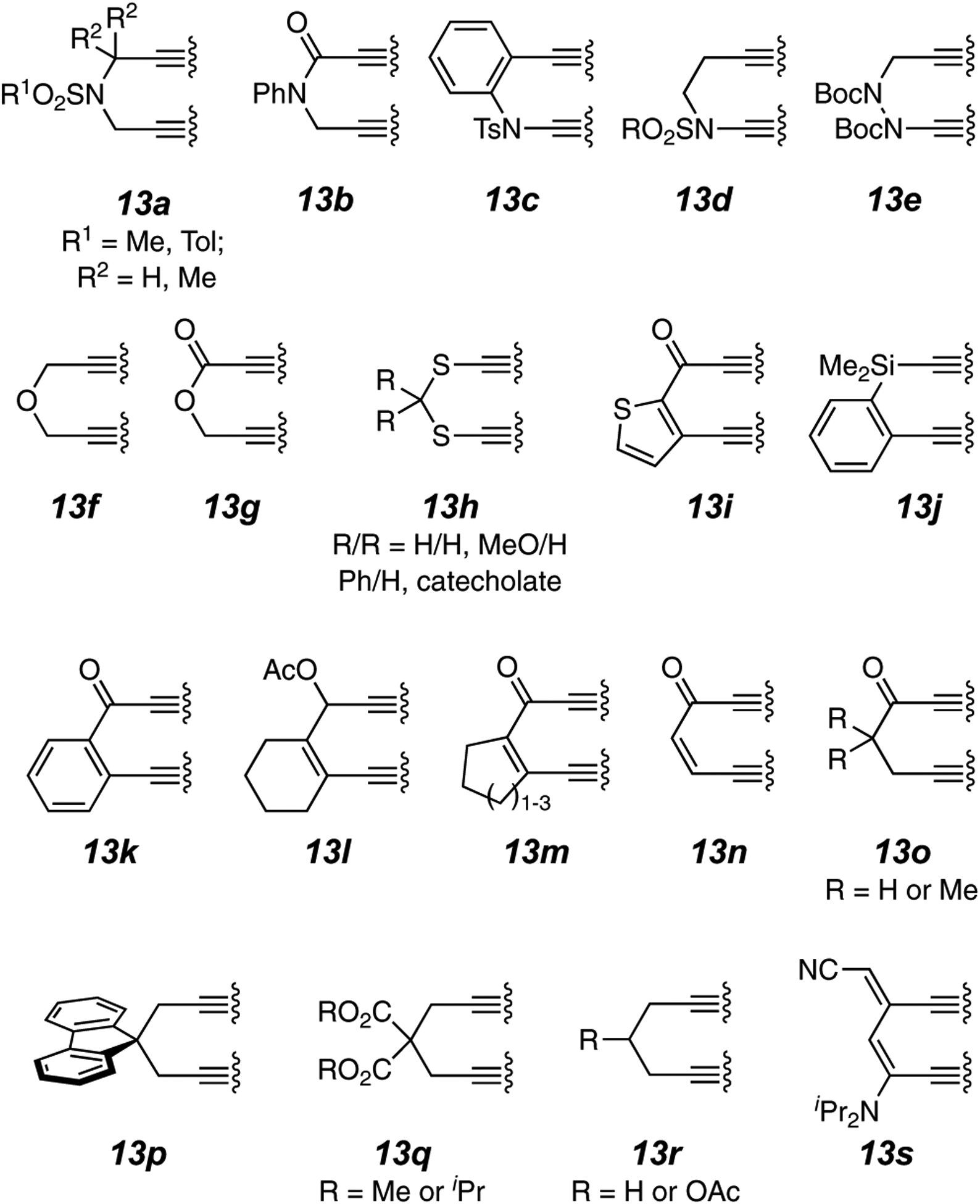
The majority of the three-atom tethers present in reported HDDA polyyne substrates.
The tether or linker connects the diyne and diynophile portions of the HDDA substrate. Almost exclusively, tethers are composed of three atoms, which allows for optimum distancing and orientation of the two reacting moieties to enable the cyclization event. The linker atoms are most commonly second-row elements and can be either sp3- or sp2-like in geometry. Essentially all of the 3-atom tethers used to date in an HDDA substrate are portrayed in Figure 3. Of these, the most common are of types 13a, 13k, and 13q. This choice is based, in part, on the ease of synthetic access to the triyne or tetrayne substrates. It is not uncommon for the substrates to be prepared by a sequence involving three to seven chemical reactions.
The nature of the tether influences the ease of the initial, rate-determining thermal cycloisomerization event and, therefore, the temperature and time used for the HDDA reaction.22 A few substrates cyclize at ambient temperature (or even below), many react conveniently in the 70–140 °C range, and a few require even higher temperature. Other than these differences in the rate of cyclization to the corresponding benzyne, there are relatively few instances in which a given trapping reaction would not be expected to be compatible with a given tethering moiety. It also can be anticipated that additional designer tether substructures can be invented to give access to products having specific fused rings of interest in the setting of, e.g., studies of target-directed synthesis of natural products.23,24,25 The nature of the substituent at the remote terminus of the diyne or diynophile is also known to significantly influence the rate of the HDDA cyclization.26 Because of these multiple rate-influencing factors, it is difficult to rank the relative rates of cyclization, especially so given that in only a few studies have investigators reported the half-life of the initial, rate-limiting HDDA cyclization. More commonly, the reaction temperature, time, and isolated yield of product are given. However, even if such reactions have been carefully monitored by, e.g., tlc, there is considerable variance in when the observer chooses to stop the reaction. Did that happen after, say, 4, 10, or 20 half-lives? The short answer is that neither the researcher nor the reader can judge without more careful analysis. We would encourage the practice of determining and reporting the half-life of cyclization of a given substrate when possible. Not surprisingly, mechanistic aspects of these cycloisomerizations have also been examined computationally.27,28,29,30,31,32,33
3. BENZYNE TRAPPING AGENTS
The trapping agent is the species that (rapidly) captures the reactive benzyne intermediate after the initial (rate-limiting) cyclization event. A wide variety of both inter- and intramolecular traps have been demonstrated. In many cases, the final product arises only after a series of intermediate events. Additionally, several reports have described the development of multicomponent reactions34 wherein more than one trapping agent has been engaged to produce structurally more complex products. One consideration for those using or designing new trapping reaction strategies is that the reagent itself cannot undergo reaction with the substrate polyyne faster than its rate of conversion to the HDDA-generated benzyne.35 Fortunately, such incompatibility is not a serious limitation, and that allows for substantial versatility in trapping strategy.
4. TRAPPING REACTIONS LEADING TO PRODUCTS HAVING NEW TYPES OF ADJACENT σ-BONDS AT THE BENZYNE CARBONS
4.1. Introduction of C–C and C–C bonds
4.1.1. Intramolecular Trap (Figure 4)
Figure 4.
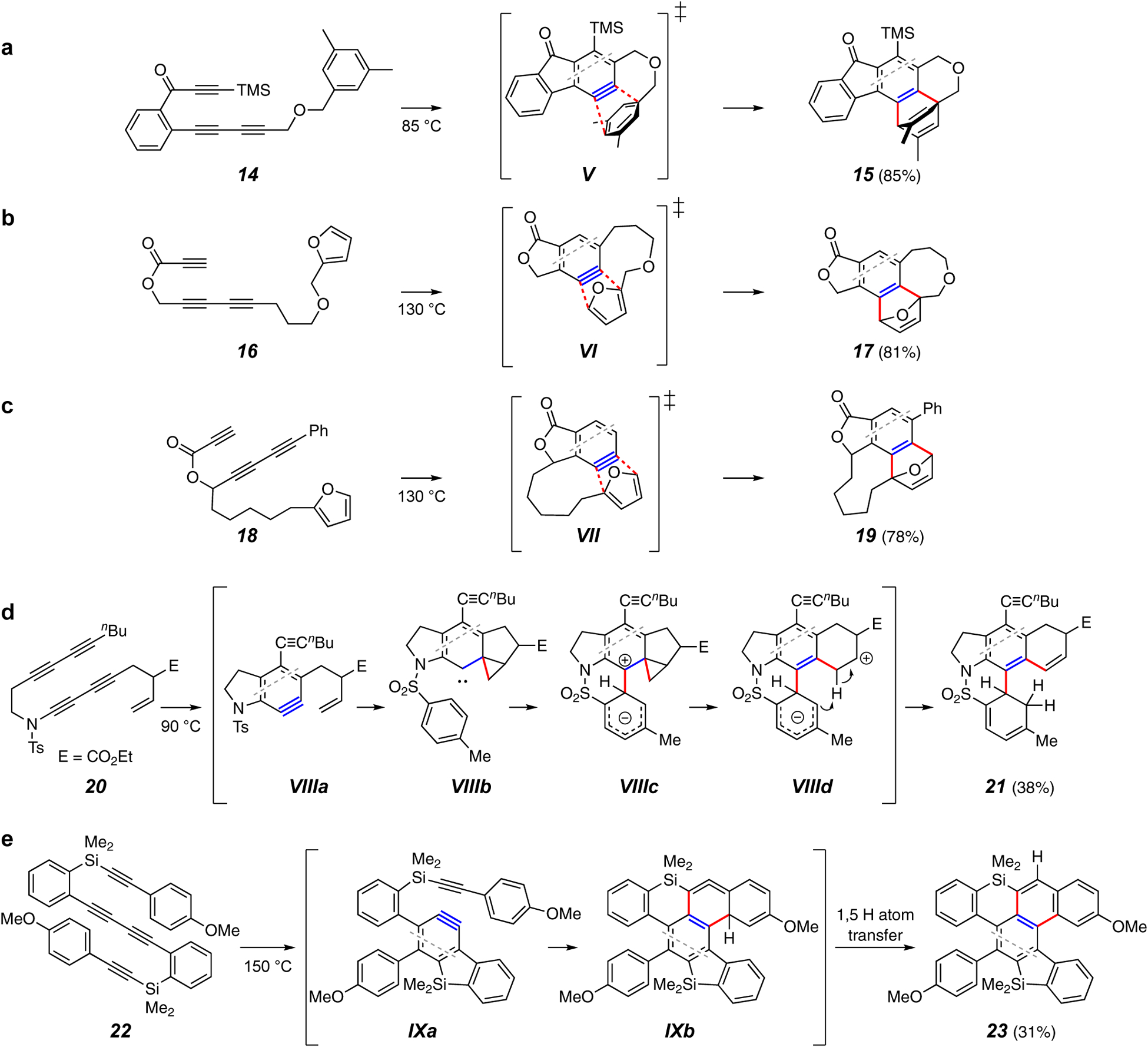
Intramolecular traps forming two C–C bonds.
Reactions involving trapping agents that lead to the formation of two new carbon-carbon bonds are among the more common HDDA transformations. Representative variants of this process, all of an intramolecular nature, are displayed in Figure 4. Among the initial array of HDDA reactions20 was an intramolecular trapping of the pendant aromatic moiety present in substrate 14 by a DA reaction of the benzyne with that arene. This yielded adduct 15 via V (Figure 4a). It is worth noting that although DA cycloadditions of higher acenes are quite common, arynes comprise one of the very few dienophiles capable of dearomatizing simple benzenoid arenes.36,37 A similar trapping was observed with the furan-containing substrates 16 and 18 via VI and VII, respectively (Figures 4b and 4c).38, This work demonstrated that a variety of ring sizes, including medium-sized cycles (cf. 17 and 19), could be formed by intramolecular trapping and that the trapping agent could be appended to an atom within the diyne-to-diynophile tether (cf. 18).
An interesting intramolecular formation of two C–C bonds by successive elementary mechanistic events was reported by Lee and coworkers, who showed that the toluenesulfonyl group in substrates such as 20 was not innocent, as is evident from the structure of the unusual product 21 (Figure 4d).39 This is explained through a complex sequence of events in which the benzyne VIIIa is proposed to first be captured by the remote, pendant alkene to yield the carbene VIIIb. This is sufficiently nucleophilic to engage the electron deficient toluenesulfonamide ring through an ipso attack. Fragmentation and proton transfer produces the dearomatized product 21 (cf. VIIIc and VIIId). This remarkable process constitutes capture of the benzyne by two separate intramolecular moieties and highlights the level of mechanistic complexity that can be observed in some types of trapping reactions.
Another example of sequential formation of two C–C bonds to the benzyne can be found in a recent report from Shibata et al.40 The conversion of benzyne IXa (from 22, Figure 4e) to product 23 proceeds via the strained allene IXb by way of a tetradehydro-DA (TDDA) reaction. This result also demonstrates the viability of an unusual silicon-containing tether in substrate 22. Finally, see Figure 24a in the Addendum for an interesting intramolecular trapping of a naphthyne, itself generated by an HDDA reaction in which a classical benzyne functioned as the diynophile.
Figure 24.
Reactions identified during manuscript review or published since initial submission.
4.1.2. Intermolecular Trap (Figure 5)
Figure 5.
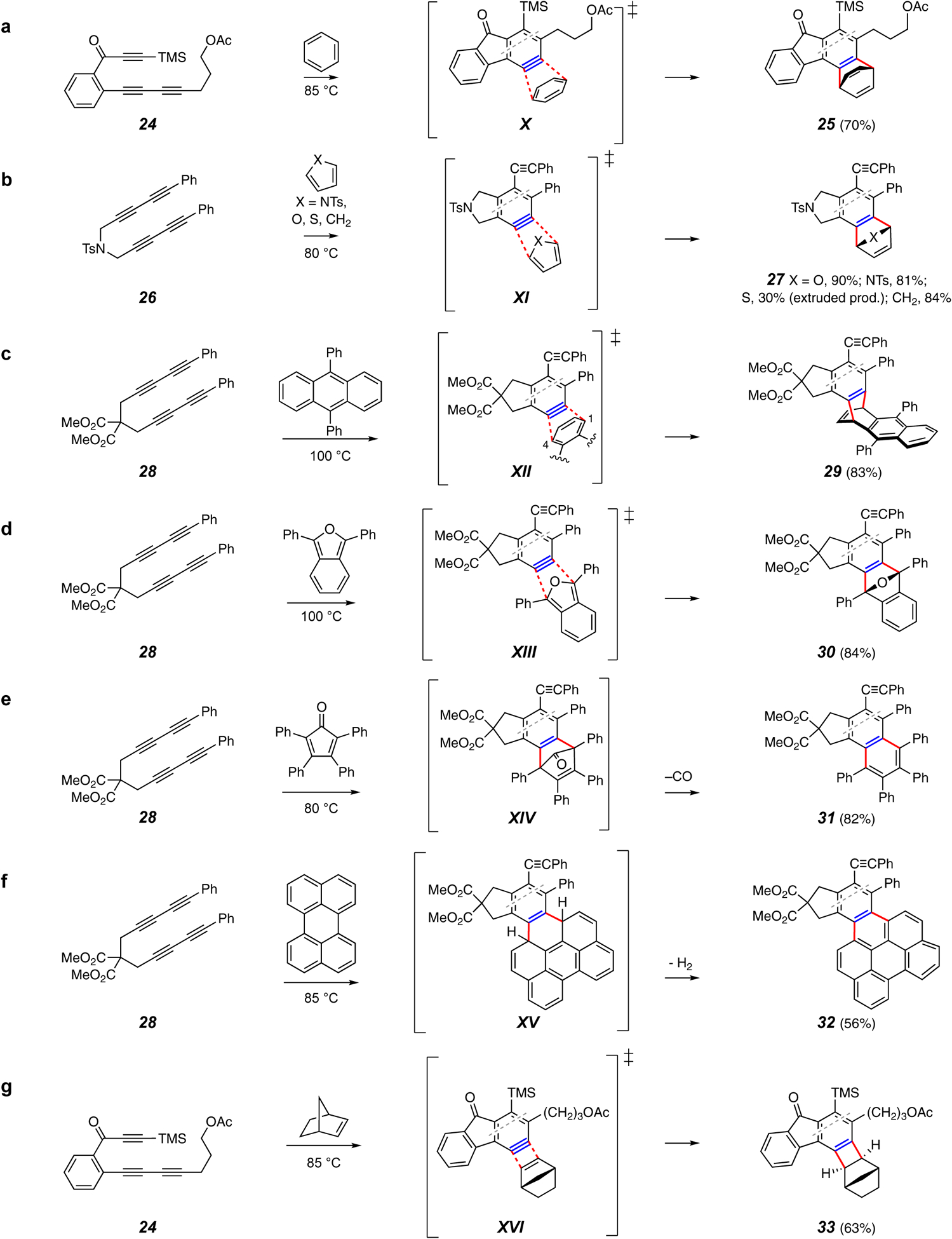
Intermolecular traps forming two C–C bonds.
There are numerous examples of processes involving intermolecular reactions with agents that produce two new C–C bonds (Figure 5). A number of these reactions are closely analogous to those shown above in Figure 4. For example intermolecular DA reactions with benzene;20 5-membered cyclic dienes (furans,41 pyrroles,41 thiophenes, and cyclopentadiene),42 anthracenes,43 and isobenzofurans44 are quite common (cf. X–XIII, Figure 5a–d). Furans are particularly reactive and efficient traps and are frequently used as probe molecules when examining new types of HDDA substrates. See Figure 24b in the Addendum for an example in which an imidazole was used as the trapping agent, giving, following oxidation, a fused isoindole-1,3-dione product.
Other processes are unique to intermolecular trappings. For example, HDDA-generated benzynes can be trapped with tetraphenylcyclopentadienone (28 to 31 via CO extrusion from the DA adduct XIV, Figure 5e)45 or perylenes (28 to 32 via H2 extrusion from the DA adduct XV, Figure 5f46). Highly conjugated polycyclic products such as those formed here are of interest for potential application in organic electronic devices.45 Strained alkenes are known to add in a net [2+2] fashion to benzynes, consistent with what is observed for the HDDA product 33 from 24 (cf. XVI, Figure 5g).20
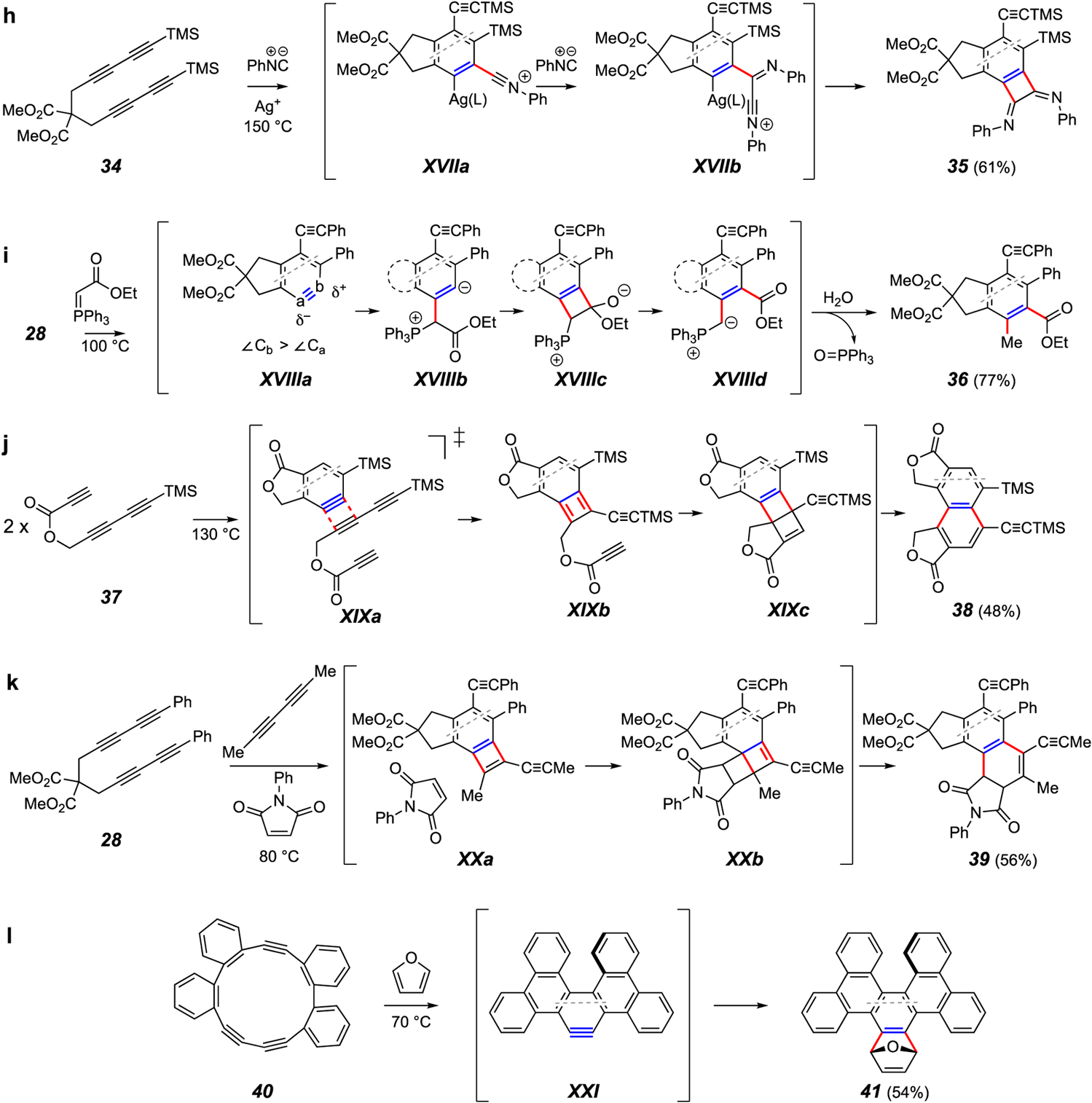
Recently, researchers in the Lee lab reported a procedure for coupling two equivalents of an isonitrile with an HDDA aryne in the presence of a silver catalyst (e.g., 34 to 35; Figure 5h).47 This was rationalized as involving an initial nucleophilic attack on the aryne by the isonitrile to form XVIIa, after which a second molecule attacks the now-electrophilic carbon of that adduct. The intermediate XVIIb then cyclizes to form the interesting benzocyclobutene-1,2-diimine 35. This process was suitable for a variety of aryl (although not alkyl) isonitriles.
Hu and coworkers coupled the HDDA benzyne with stabilized Wittig ylides to introduce adjacent methyl and ethoxycarbonyl groups onto the aryne (e.g., 28 to 36; Figure 5i).48 Following nucleophilic attack onto the benzyne XVIIIa by the ylide carbon atom, the intermediate XVIIIb was envisioned to cyclize to the tetrahedral intermediate XVIIIc. Opening of the strained ring to the new, stabilized ylide XVIIId and capture by water would then account for formation of 3 I note that the reviewers did not have A notable aspect of this transformation is its regioselectivity. Normally, nucleophiles would be expected to preferentially attack a fused benzyne such as XVIIIa at carbon atom “b” because of the distortion of the aryne ring. That is, DFT computations show that the internal bond angle at atom “b” is significantly larger than that at atom “a”. In accord with the distortion analysis of unsymmetrical aryne reactivity,31,49,50,51 the atom with larger internal angle is more electrophilic (greater p-character in its in-plane π-bond). In the case of addition of the ylide to XVIIIa, the large size of that nucleophile makes steric factors override the inherent electronic bias. Namely, the phenyl ring is effectively bulkier than the methylene carbon pinched back into the five-membered ring, steering the attack to atom “a”.
In the absence of any other trapping agents in the reaction environment, HDDA polyyne substrates will often cannibalize themselves, producing intractable oligomeric product mixtures. However, in certain instances, the nature of the substrate polyyne is such that its reaction with its progenitor benzyne is selective, leading to dimeric products. For example (Figure 5j), the ester triyne 37 gave rise to the unsymmetrical dimer 38 in a process envisioned to proceed through a net [2+2] cycloaddition (cf. XIXa) to give the benzocyclobutadiene XIXb, which is then trapped intramolecularly by a pendant alkyne, giving rise to the penultimate Dewar naphthalene XIXc.52 As an extension of this process, it was found that the added external diyne 2,4-hexadiyne (Figure 5k) could effectively trap the benzyne generated from 28 to produce the transient benzocyclobutadiene XXa.52 When a reactive DA dienophile such as N-phenylmaleimide was also present, the final three-component adduct 39 was formed via opening of the strained DA adduct XXb.
An interesting intra-annular cyclization of the cyclic triyne 40 gave the dibenzopicene derivative 41 (Figure 5l).53 Use of furan as the trapping agent for benzyne XXI represents another example of how popular it is to employ that very efficient process to benchmark reactions that produce novel HDDA benzynes.
4.2. Introduction of C–C and C–O bonds (Figure 6)
Figure 6.
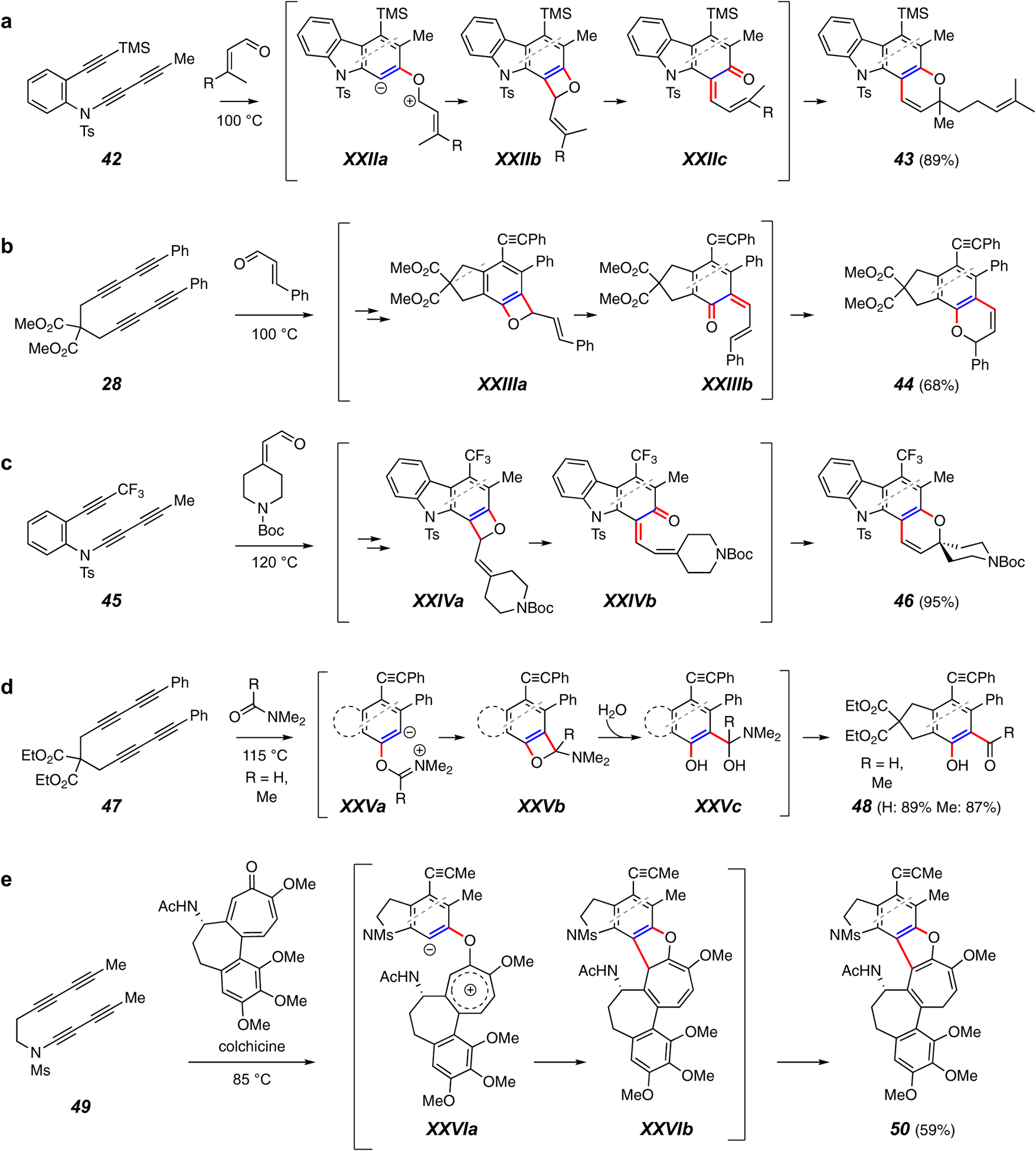
Trapping reactions forming a C–C and a C–O bond. (a-c) Reactions with enals begins with benzoxetene formation prior to electrocyclic opening and reclosure to produce benzopyrans. (d) Nucleophilic amides capture benzynes to product o-hydroxy aldehydes and ketones such as 48. (e) The cycloheptatrienone in colchicine captures an HDDA-produced benzyne to give the benzofuran derivative 50.
Trapping reactions that form both a C–C and C–O bond are rarer than the C–C and C–C counterparts discussed above. The most common motif in this category is the overall [4+2] trapping of the benzyne by an enal54 to produce a benzopyran derivative such as 43,24 44,55 or 4656 (Figures 6a–c). The process begins with a net [2+2] cyclization of the aldehyde carbonyl group with the HDDA benzyne to produce a benzoxetene such as XXIIb, XXIIIa, or XIVa. It is likely that these benzoxetenes are not formed directly by a concerted [2+2] cycloaddition but by a stepwise process, proceeding through a zwitterionic intermediate, shown explicitly as XXIIa for the reaction of 42 with, for example, citral enroute to the benzopyran 43. Under the thermal reaction conditions, the strained oxetene ring undergoes a 4π-electrocyclic ring opening to the dienone XXIIc, which, by a 6π-electrocyclic ring closure, generates 43. Notably, this compound served as the penultimate intermediate in a chemical synthesis of the carbazole-based natural product mahanimbine.24 This demonstrates a complementary synthesis strategy for the preparation of complex molecules – namely, a de novo construction of the benzenoid core. The reaction of 45 to 46 shows trapping by an exocyclic enal, which produces a spirocyclic product (Figure 6c).56
In a related type of reaction, salicylaldehydes and salicylketones can be produced by trapping of the benzyne with tertiary amides (e.g., 47 to 48, Figure 6d). Initial zwitterion XXVa closes to XXVb, which is envisioned to be opened through attack by adventitious water to give the tetrahedral intermediate XXVc.57 Note that this is another example in which the steric bulk of the aryl group ortho to the benzyne diverts the in-plane attack by the nucleophile, here the amide carbonyl oxygen, preferably to the carbon distal to the aryl (cf. Figure 5i).
In an unusual (and unexpected) mode of reaction, the benzyne produced from 49 (Figure 6e) is thought to engage the natural product colchicine at its ketonic oxygen to generate the tropylium zwitterion XXVIa.58 This collapses to the cycloheptatriene XXVIb, which isomerizes to adduct 50 by a final [1.5] hydrogen atom migration to account for product formation.
4.3. Introduction of C–C and C–N bonds (Figure 7)
Figure 7.
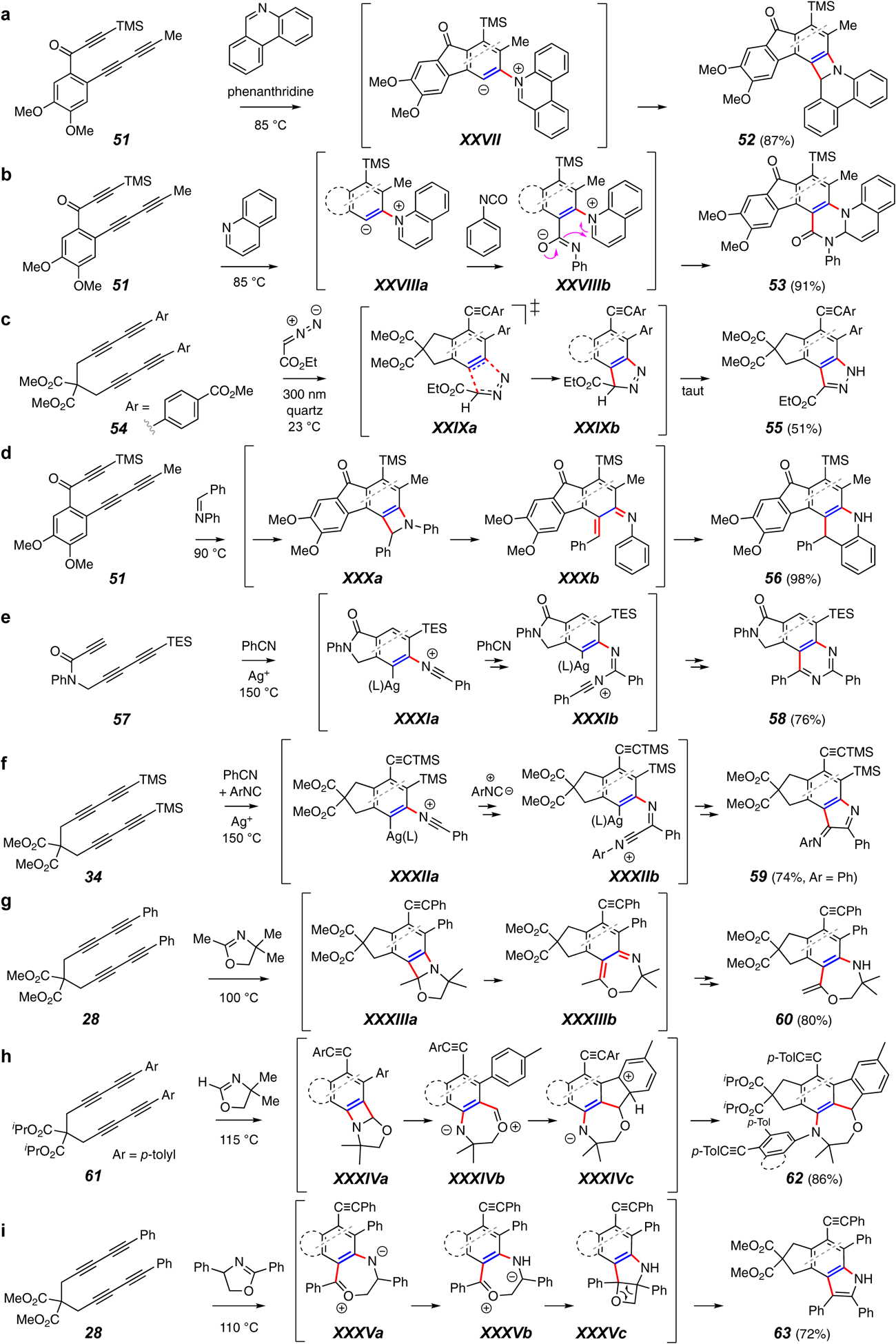
Trapping reactions forming a C–C and a C–N bond.
A variety of traps lead to the formation of products containing new C–C and C–N bonds (Figure 7). Cyclization of 51 in the presence of phenanthridine leads to the relatively rare benzazete derivative 52 (Figure 7a).59 The intermediate zwitterion XXVII is invoked. Such intermediates can be further exploited in three-component fashion, as demonstrated, for example, by the reaction of 51 in the presence of quinoline (or isoquinoline) and electrophiles such as phenylisocyanate (or reactive carbonyls, maleimides, dimethyl acetylene dicarboxylate, etc.) to give 53 (or analogous products, Figure 7b).59 This implies that the lifetime of the zwitterion XXVIIIa is sufficiently long that it can encounter the third component to produce the presumed penultimate precursor XXVIIIb.
HDDA benzynes are excellent 1,3-dipolarophiles, as exemplified by the reaction of 54 in the presence of ethyl diazoacetate (Figure 7c).60 The initially formed adduct XXIXb, formed via the cycloaddition portrayed in XXIXa, tautomerizes to the more stable benzopyrazole 55. This reaction is also notable because the cyclization of 54 (and related diarylated tetrayne substrates) can not only be induced thermally but, at much lower temperatures, by irradiation – a photo-HDDA reaction (discussed further in section 5.1 below).
Simple imines also trap the reactive benzynes. For example, the preparation of the acridine derivative 56 is shown in Figure 7d.61 In this case the intermediate azetidine ring in XXXa is sufficiently fragile to undergo electrocyclic opening to XXXb, analogous to the benzoxetene openings shown in Figures 6a–c. Electrocyclization and rearomatization leads to 56.
Lee group researchers have shown that added Ag(I) salts can promote the three-component reactions of nitriles with HDDA benzynes. For example, triyne 57 incorporated two molecules of benzonitrile to give the quinazoline derivative 58 (Figure 7e).62 Intermediate species like XXXIa and XXXIb were proposed to account for this mechanistically intriguing transformation. Even more interesting are reactions in which both a nitrile and an isonitrile are incorporated into a three-component product. For example, substrate 34 produced the unsymmetrical bis-imine 59 (Figure 7f).47 The complementary and constitutionally isomeric nature of products 59, 58, and 35 (cf. Figure 5h) is particularly notable.
Hu and coworkers have recently reported a thorough examination of the trapping of HDDA benzynes with oxazolines. Three variants of this chemistry, each producing a novel product architecture, are shown in Figure 7g–i.63 The nature of the substituent at the 2-position of the oxazoline is the key factor in determining the reaction pathway. The conversion of 28 to 60 (Figure 7g) shows that oxazolines bearing an alkyl substituent in their 2-position form azetidine and ring-opened o-quinomethide imines like XXXIIIa and XXXIIIb en route to product. In contrast, oxazolines unsubstituted at C2 add with reversed regiochemical orientation (cf. XXXIVa vs. XXXIIIa), and the resulting o-quinomethide XXXIVb is now sufficiently electrophilic to be captured by the pendent phenyl ring in Friedel-Crafts fashion to give the penultimate XXXIVc (Figure 7h). Finally, 2-arylated isoquinolines (Figure 7i) produce products that experience loss of formaldehyde (e.g., 63 from 28, suggested to proceed via XXXVa-c).
4.4. Introduction of C–C and C–H bonds
4.4.1. Intramolecular Trap (Figure 8)
Figure 8.
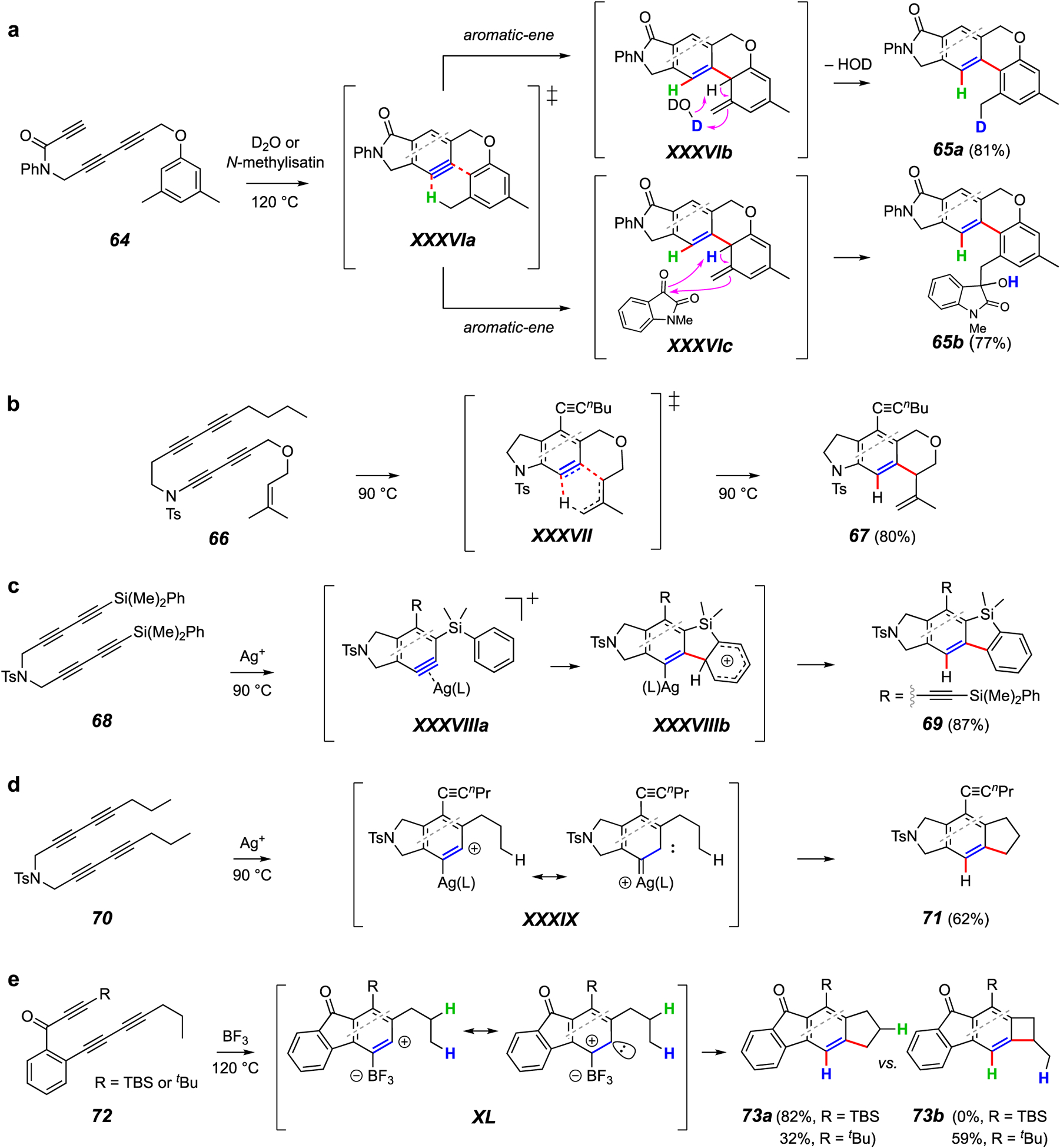
(Intramolecular) trapping reactions forming a C–C and a C–H bond.
Many reactions produce a C–C and a C–H bond. These again have been segregated into intra- and intermolecular categories (Figures 8 and 9, respectively). HDDA benzynes were used to establish the first high-yielding “aromatic ene” reactions.64 An example is given in Figure 8a. HDDA cyclization of 64, which bears a designed placement of a meta-alkylated aryl substituent, produces the enophilic benzyne, which abstracts a benzylic hydrogen atom (cf. XXXVIa) to yield the isotoluene derivative XXXVIb. Water-catalyzed proton shuttle, demonstrated by use of added D2O, rearomatizes this species to give the product 65a. Alternatively, under anhydrous conditions, an added enophile such as N-methylisatin captures the isotoluene in a bimolecular Alder-ene reaction as portrayed in XXXVIb to efficiently provide the adduct 65b.
Figure 9.
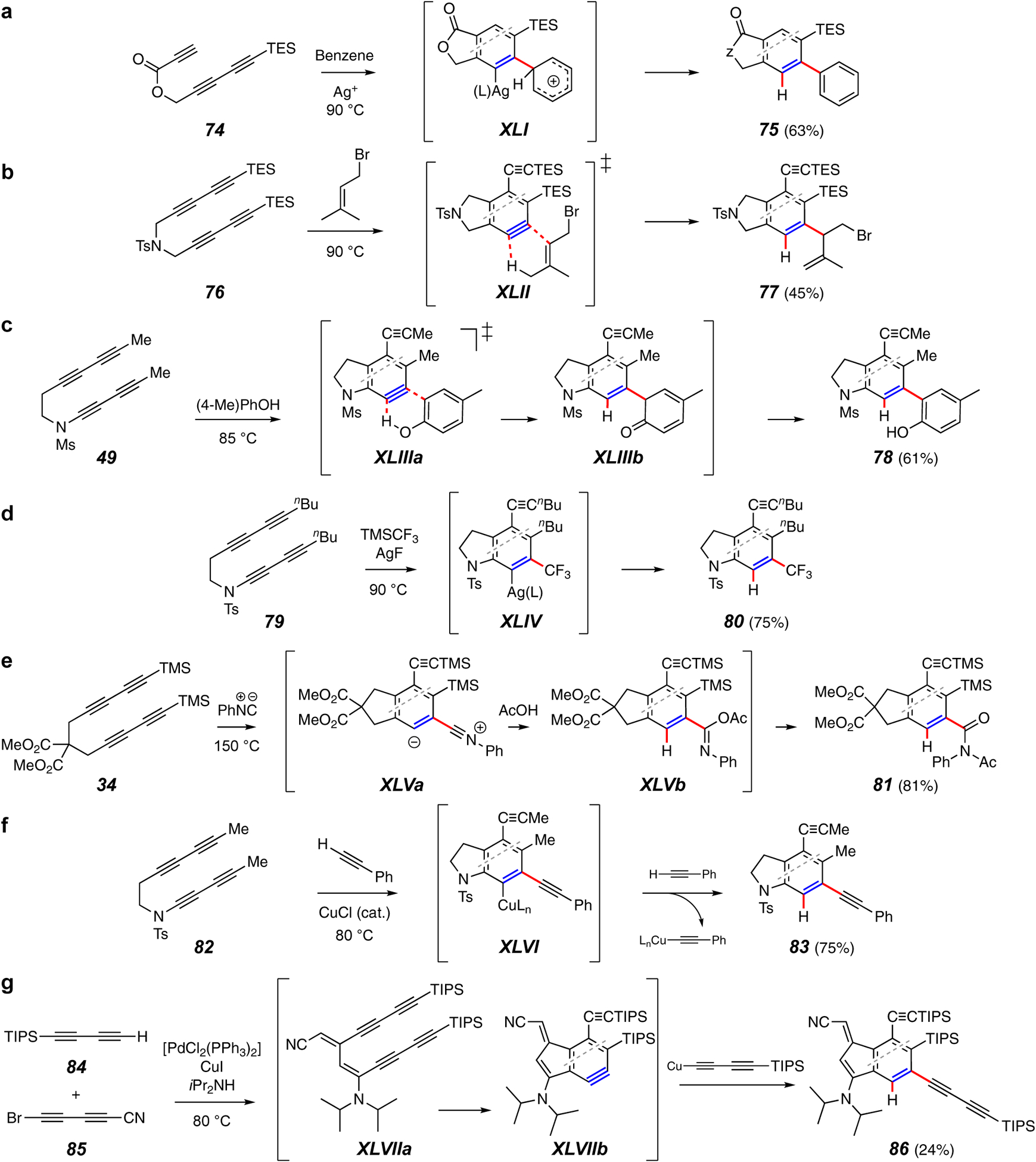
(Intermolecular) trapping reactions forming a C–C and a C–H bond.
Lee and coworkers have provided numerous examples of thermal cyclization of substrates bearing pendant alkenes. For example, 66 produced a benzyne that then underwent an intramolecular Alder-ene reaction (cf. XXXVII) to produce product 67.65 Lee et al. have also exploited silver ion additives66,67 to identify new modes of HDDA benzyne trapping reactions that lead to net C-H insertion (Figure 8c and 8d). In the case of tetrayne 68, it is envisioned that the pendant phenyl group is captured by an electrophilic carbon in the benzyne•Ag(I) complex XXXVIIIa to produce a phenonium ion XXXVIIIb en route to the product 69 (Figure 8c).68 Alternatively, electrophilic carbene character in the complex XXXIX is thought to enable insertion into a methyl C–H bond in the pendant n-propyl group to give 71 (Figure 8d).69,70
An analogous overall C–H insertion process, this time mediated by boron trifluoride as the additive, was used to convert, for example, 72 into the cyclopentano- and cyclobutanofluorenone derivatives 73a and 73b (Figure 8e).71 The different outcomes imposed by the TBS vs. t-Bu substituents in 72 are notable and suggest that differences in the extent of buttressing change the internal bond angles in XL in perhaps small but certainly important ways allowing for formation of the strained four-membered ring in product 73b. The carbene-like character suggested by structure XL is unprecedented for a boron-containing species, which further highlights the unique reactivity that can be uncovered by studies of HDDA reactions. For an additional example that demonstrates how subtle structural changes can be exploited to change the course of outcome in a similar carbenoid C–H insertion reaction, see Figure 24c in the Addendum.
4.4.2. Intermolecular Trap (Figure 9)
In the intermolecular mode of C–C and C–H bond-forming processes, some of the same reactivity patterns are observed. Bimolecular versions of the silver-catalyzed net electrophilic aromatic substitution (EAS) with an aromatic solvent molecule (74 to 75, Figure 9a)68 and the Alder ene reaction (76 to 77, Figure 9b)72,73 have both been demonstrated. Phenolic trapping agents capture HDDA benzynes at their ortho position (e.g., 49 to 78, Figure 9c).74 This process is viewed as a phenol-ene reaction (cf. XLIIIa to XLIIIb) and is notable because this mode of phenol trapping is not observed for classically generated benzynes, where the presence of basic reagents typically promotes oxygen attack and formation of diaryl ethers.
Net trifluoromethane addition to the benzyne can be accomplished by use of the Prakash reagent (CF3TMS) in the presence of an equivalent of silver fluoride in acetonitrile solvent (e.g., 79 to 80, Figure 9d).75 This conversion is presumed to involve capture of the benzyne by in situ-generated trifluoromethylsilver (AgCF3) to produce XLIV.
In another isonitrile-based multicomponent trapping scheme, substrate 34, when heated in the presence of both PhNC and acetic acid, gives the 1:1:1 adduct 81 (Figure 9e).76 The initial adduct XLVa is protonated and captured by acetate to produce the isoimide XLVb, which, finally, rearranges to the imide 81. Alcohols and sulfonamides were also shown to serve in the capacity of protic nucleophiles in analogous processes.
A Cu(I)-catalyzed process for the hydroalkynylation of HDDA benzynes has been developed (e.g., 82 to 83, Figure 9f).77 Initial Cu-acetylide addition to the benzyne is proposed to form the adduct XLVI, which then disproportionates with another molecule of alkyne to continue the cycle. In another Cu(I)-promoted reaction of a terminal alkyne, HDDA reactivity expressed itself in an unplanned way (Figure 9g).78 A mixture of substrates 84 and 85 produced the unusual HDDA substrate XLVIIa, which then cycloisomerized to XLVIIb and was captured by a third molecule of the terminal diyne 84.
4.5. Introduction of a C–C and a C–Br, C–S, C–Se, or C–Te bond (Figure 10)
Figure 10.
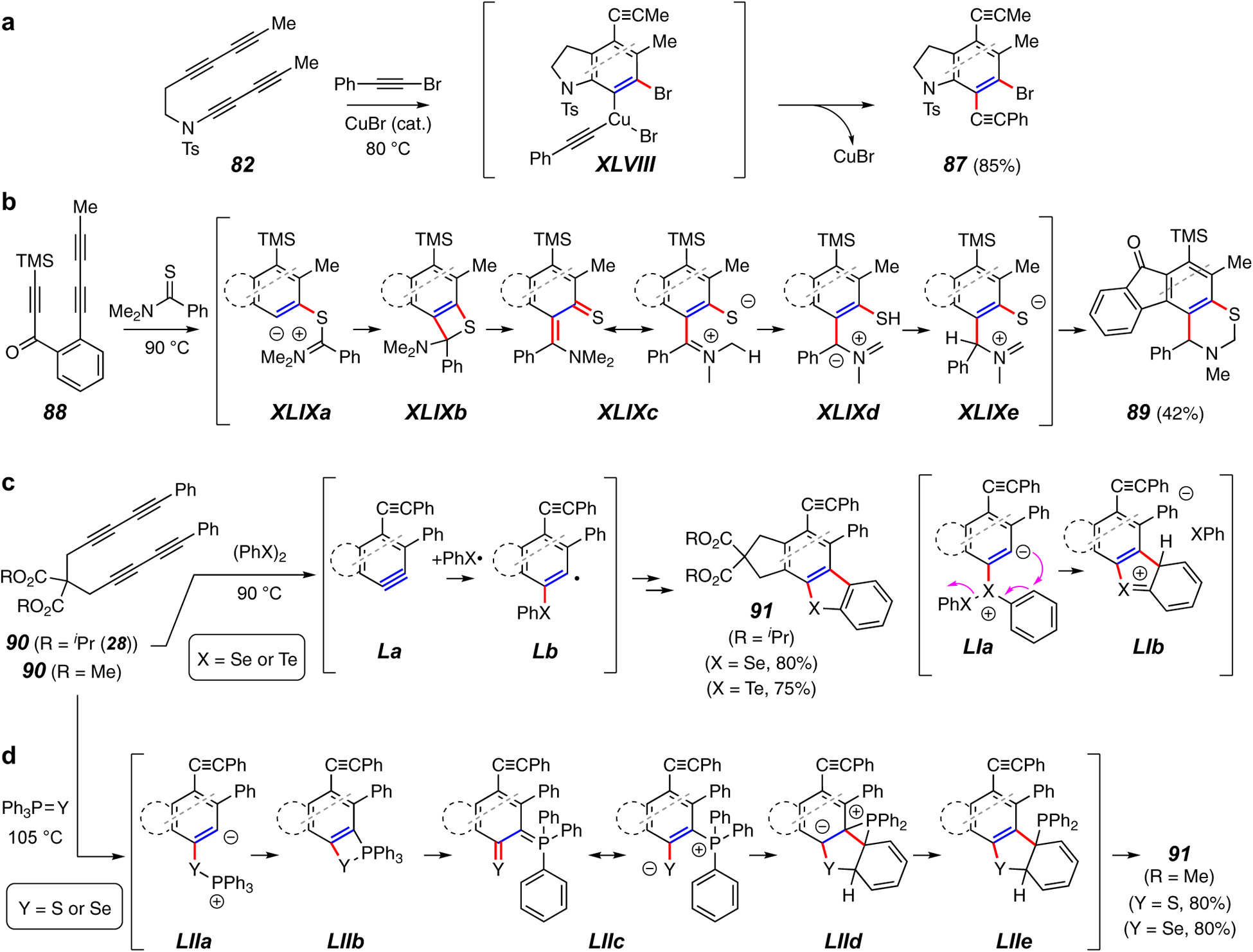
Trapping reactions forming a C–C and a C–Br, C–S, C–Se, or C–Te bond.
Bromoalkynylation of the HDDA benzyne derived from 82 (Figure 10a) is a complement to the hydroalkynylation shown above in Figure 9f. The Cu(III) species XLVIII is proposed to account, following reductive elimination, for the production of products such as 87. Cross-coupling of the aryl bromide in these products demonstrated the potential for using this transformation to access products having two different types of adjacent C–C bonds (cf. Section 4.1.2).
Thioamides react with HDDA benzynes to produce benzothiazine derivatives (Figure 10b).79 Initial [2+2] cyclization of the HDDA benzyne from 88 with the thioamide generates adduct XLIXb via zwitterion XLIXa. Opening of the four-membered ring reveals the o-thioquinonemethide XLIXc. Proton transfer isomerizes this to the isomeric zwitterion XLIXe via XLIXd, which collapses to the thiazine ring in 89.
Hu and coworkers have reported some similarly complicated reaction pathways that produce dibenzo-thiophene, -selenophene, and -tellurophene derivatives (cf. 91, Figure 10c and 10d). In the first of these (Figure 10c)80 the benzyne from 90 is generated in the presence of diphenyl diselenide or diphenyl ditelluride, resulting in formation of 91 (X or Se or Te, respectively). It is proposed that the trapping involves addition of a PhX• radical to La to produce Lb, which then undergoes radical addition to the Ph group. An alternative consideration is the dipolar process implied by intermediates LIa and LIb. The same type of products were formed with a substantially different trapping agent and mechanism when the tetrayne 28 was heated in the presence of triphenylphosphine sulfide or selenide (Figure 10d).81 Upon HDDA cyclization of the initial substrate, the P=Y moiety undergoes a [2+2] cyclization to LIIb, likely via the zwitterion LIIa. This then ring opens to species LIIc having zwitterionic character. An unusual ring-closing event then occurs wherein a phosphine phenyl group is engaged by the sulfur or selenium atom to yield the polycyclic intermediate LIId. Aromatization to LIIe and loss of diphenylphosphine then accounts for formation of 91.
4.6. Introduction of C–O and C–Si bonds (Figure 11)
Figure 11.
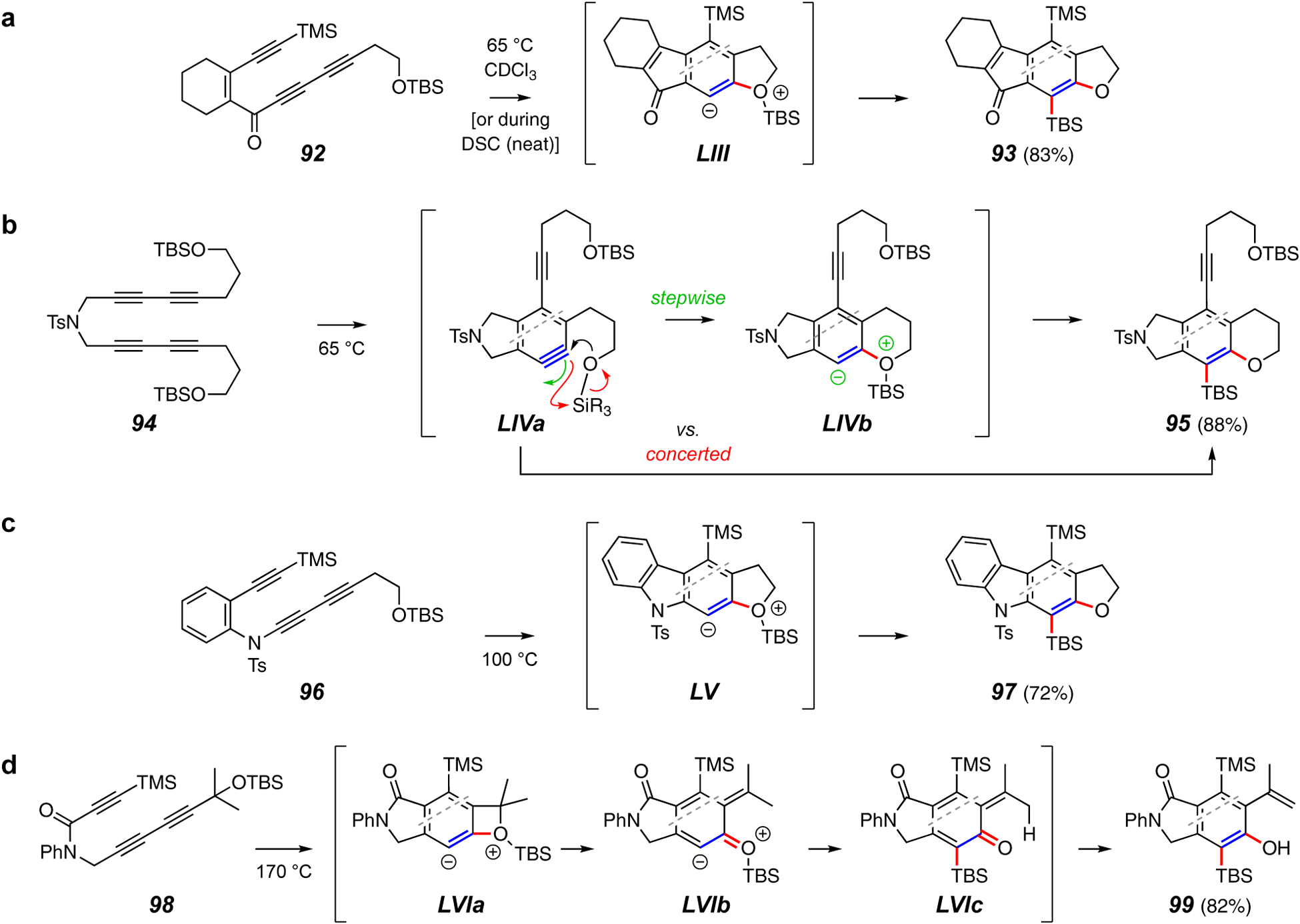
Trapping reactions forming a C–O and a C–Si bond.
Trapping processes that yield a C–O and a C–Si bond are not known for classically generated arynes. This is the type of reaction in the first (unplanned) HDDA cyclization observed in the Hoye laboratories that propelled investigations revealing the generality of these thermal cyclizations.20 A typical example is the conversion of 92 to 93 (Figure 11a). Intermediate zwitterion LIII, formed from trapping the initial benzyne by the oxygen atom of the pendant OTBS group, was envisioned to proceed via a retro-Brook-like rearrangement. Interestingly, this reaction proceeded not only in solution22 but also reasonably cleanly as a neat sample in a differential scanning calorimetry pan during DSC monitoring of the onset of exothermic reaction of substrate 92.82 This is noteworthy because it shows that intramolecular trapping can be faster than competitive reaction of the benzyne with an alkyne in an additional molecule of the triyne substrate, even at the highest possible substrate concentration.
Computational and experimental studies of related substrates such as 94 suggested that in addition to a stepwise process via the zwitterion LIVb, a concerted insertion of the benzyne π-bond into the O–Si bond in benzyne LIVa to provide 95 directly was energetically feasible (Figure 11b).83 This OTBS trapping strategy has often been employed when first probing the feasibility of a new class of HDDA substrate. For example, the carbazolyne derived from the diynamide 96 was converted to 97 via zwitterion LV or in the concerted alternative (Figure 11c).24
A more unusual and remarkably efficient transformation is shown in Figure 11d.84 Substrate 98, bearing a pendant siloxyethyl substituent, presumably undergoes analogous cyclization and intramolecular nucleophilic attack to yield the initial, now strained, zwitterionic intermediate LVIa. Ring-opening, perhaps electrocyclic, of the four-membered ring leads to the dearomatized LVIb, poised for rapid transfer of the silicon group to provide the o-quinonemethide LVIc. Intramolecular proton transfer gives the phenolic product 99.
4.7. Introduction of C–O and C–H bonds (Figure 12)
Figure 12.
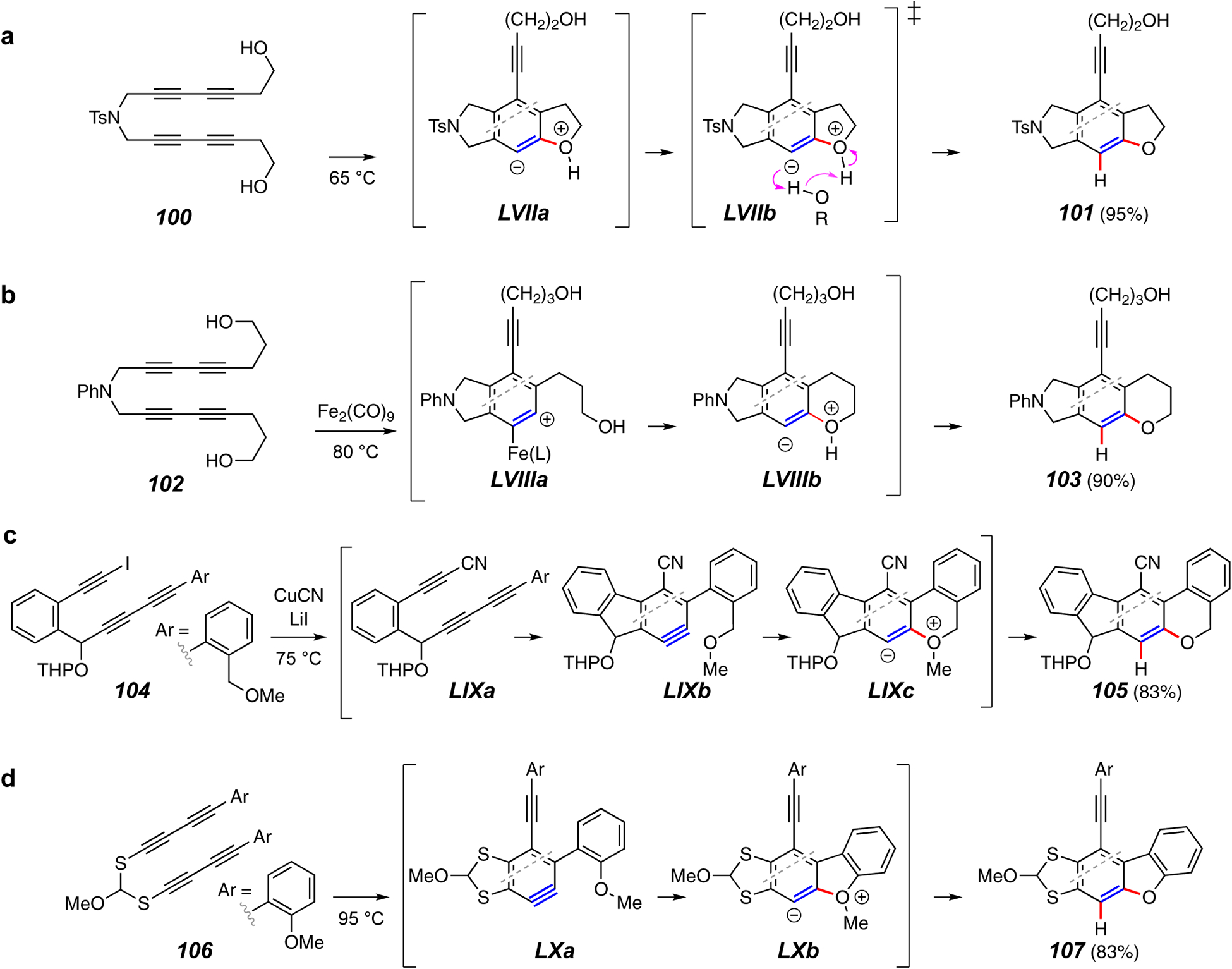
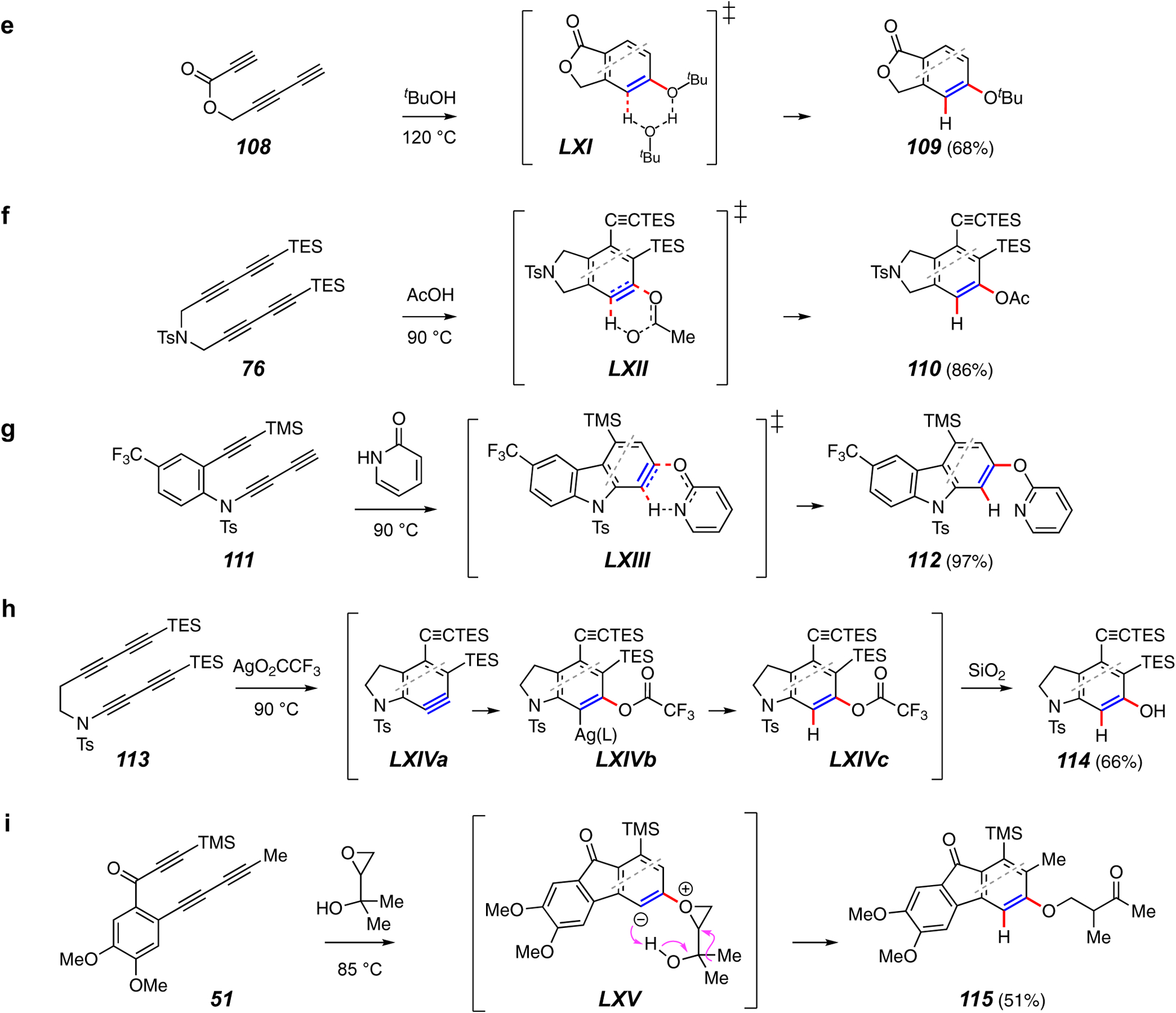
Trapping reactions forming a C–O and a C–H bond.
Although there are many examples of traps that form a C–O and a C–H bond, many proceed by the same basic reaction pathway. As an example, consider the cyclization of tetrayne 100 to the benzofuran derivative 101 (Figure 12a).20 It is tempting to envision a mechanism that begins in parallel with that for the silyl ether addition (cf. Figure 11a) – namely, initial formation of the zwitterion LVIIa. Because unimolecular proton transfer within this species would face a stereoelectronically imposed barrier, a proton shuttle mechanism via a transition structure conveyed by LVIIb was thought likely.
A related, diironnonacarbonyl-promoted reaction is shown by the example of 102 to 103 (Figure 12b).85 A number of additional metal-based additives led to the same outcome. However, results from an important control experiment – that is, heating 102 at the same temperature in the absence of any additive – was not described. Because the same product, 103, would be expected to form at the same reaction temperature, the case for catalysis (cf. LVIIIa and LVIIIb) may not have been definitively established.
In 2008 Ueda and coworkers reported cyclization of the methyl ether-containing substrate 104, which produced the demethylated benzopyran derivative 105 (Figure 12c).86 This reaction is additionally notable because the starting material is a triyne that, upon conversion to the cyano analog LIXa (with CuCN), proceeded to undergo the HDDA isomerization to the benzyne LIXb. Nucleophilic attack by the methoxy oxygen gives the zwitterion LIXc, which proceeds through protonation and demethylation to 105.
The tetrayne substrate 106 contains o-methoxyphenyl substituents. Upon being heated, it is converted to the benzopyranyl product 107 via initial formation of zwitterion LXa (Figure 12d).87 Protonation by chloroform solvent (or adventitious water) and demethylation can account for product formation and the fates of the electrophilic methyl group – MeCl, MeCCl3, MeOH, and MeOMe – were detected as byproducts by direct 1H NMR monitoring of the reaction mixture. In this study, it was also shown that the sulfides in products from these types of bis-diynylsulfide precursors serve as a traceless tether. That is, they could be reductively cleaved (Ra-Ni) to establish an overall process that is the equivalent of an otherwise unknown bimolecular HDDA reaction.87
Bimolecular analogs of the unimolecular OH additions given in Figures 12a and 12b are also known (e.g., 108 to 109, Figure 12e).20 Later studies,88 both computational and experimental, support the idea that bimolecular alcohol addition is a process involving alcohol dimers, as suggested by the TS LXI. This view is related to the six-atom TSs that can be envisioned for additions of carboxylic acids89 and 2-pyridone to the benzyne (cf. TSs LXII and LXIII), which can account for the conversion of, e.g., 76 to 110 (Figure 12f90) and 111 to 112 (Figure 12g24).
In another Ag(I)-promoted reaction, researchers in the Lee group showed that silver trifluoroacetate promoted addition of trifluoroacetic acid to the benzyne LXIVa derived from 113 (Figure 12h).89 The labile aryl trifluoroacetate LXIVc, suggested to arise via LXIVb, was cleaved to the phenol 114 upon exposure to silica gel.
An example of a trapping reactions with a glycidol derivative is shown in Figure 12i.91 Trapping of the benzyne from 51 leads to products such as the ketone 115, from which it is clear that a carbon skeleton rearrangement has occurred. The zwitterion LXV, arising from attack by the epoxide oxygen, is poised to undergo a 1,2-methyl migration to account for the outcome. Several additional rearrangement or cleavage pathways were uncovered for glycidols with various substitution patterns.
4.8. Introduction of a C–N and a C–N or a C–S bond (Figure 13)
Figure 13.
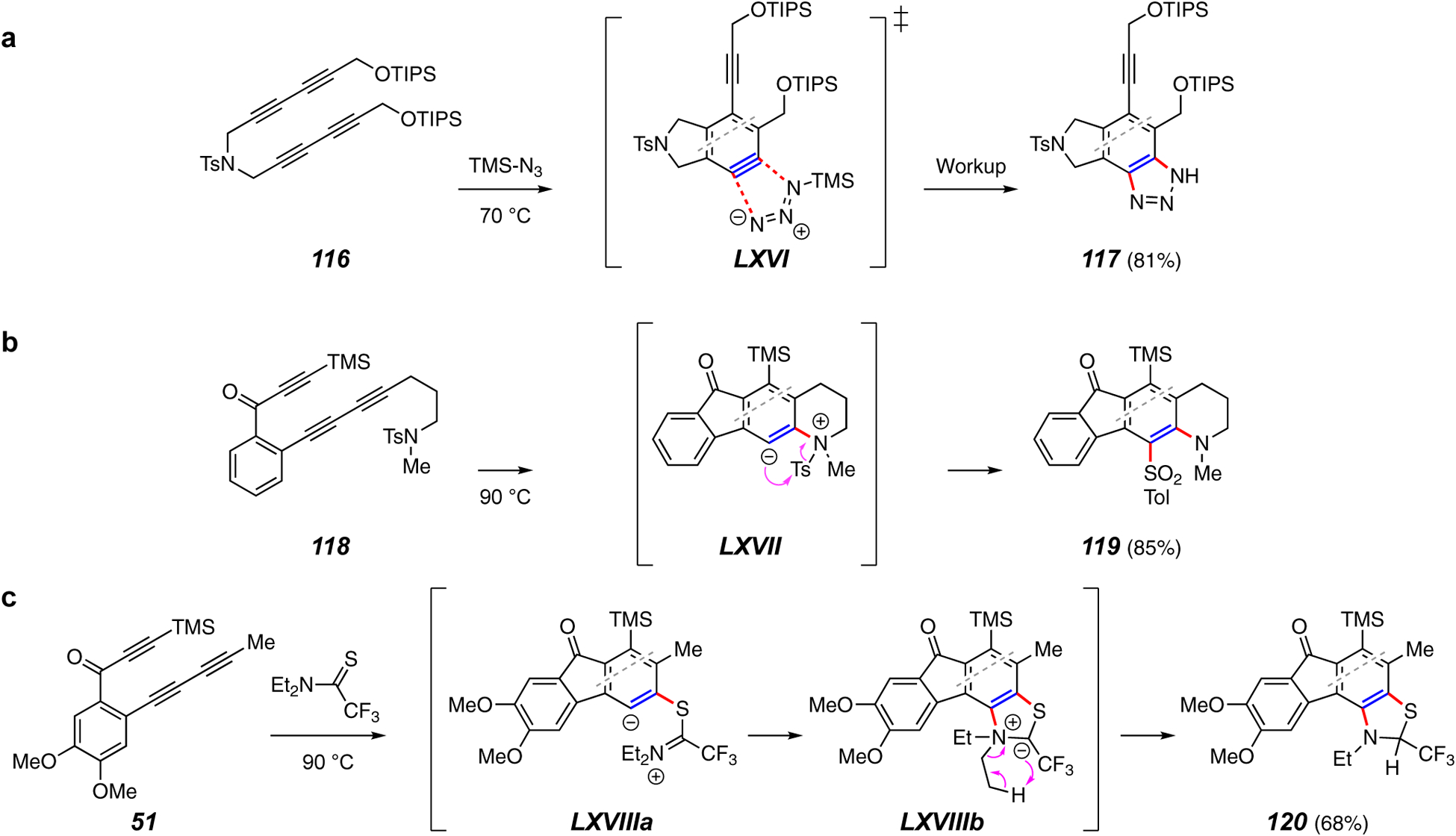
Trapping reactions forming two C–N bonds or a C–N and a C–S bond.
Trapping reactions that create both a C–N and a C–N or a C–N and a C–S bond are rare. The only method for the formation of two C–N bonds is the 1,3-dipolar cycloaddition reaction of the HDDA benzyne with an azide, a reaction well known92 for classically generated21 arynes. For example, use of trimethylsilyl azide gives the benzotriazole 117 when used to trap the benzyne from 116 (cf. LXVI, Figure 13a).41
Two methods for producing a C–N and a C–S bond have been described. The first involves intramolecular trapping by an aryl sulfonamide (118 to 119, Figure 13b).93 Nucleophilic attack of the benzyne by the pendant sulfonamide nitrogen gives the zwitterion LXVII. The sulfonyl group then migrates to the carbanionic center, a process involving a four-center reaction made possible by access to d-orbital space around the third-row sulfur atom.
The second reaction exploits an unprecedented pseudo-1,3-dipolar cycloaddition of thioamides with HDDA benzyne. The conversion of 51 to the benzothiazoline 120 is exemplary (Figure 13c).94 Initial nucleophilic attack by the sulfur atom forms the iminium zwitterion LXVIIIa. In an unusual elementary event, the positively charged nitrogen is suggested to then be attacked by the anionic arene carbon atom to yield the CF3-stabilzed nitrogen ylide LXVIIIb. All of the thioamides that participated in this pseudo-1,3-dipolar reaction contained an electron withdrawing substituent on the thiono carbon atom. Intramolecular proton transfer extrudes ethylene and accounts for the product 120.
4.9. Introduction of C–N and C–H bonds (Figure 14)
Figure 14.
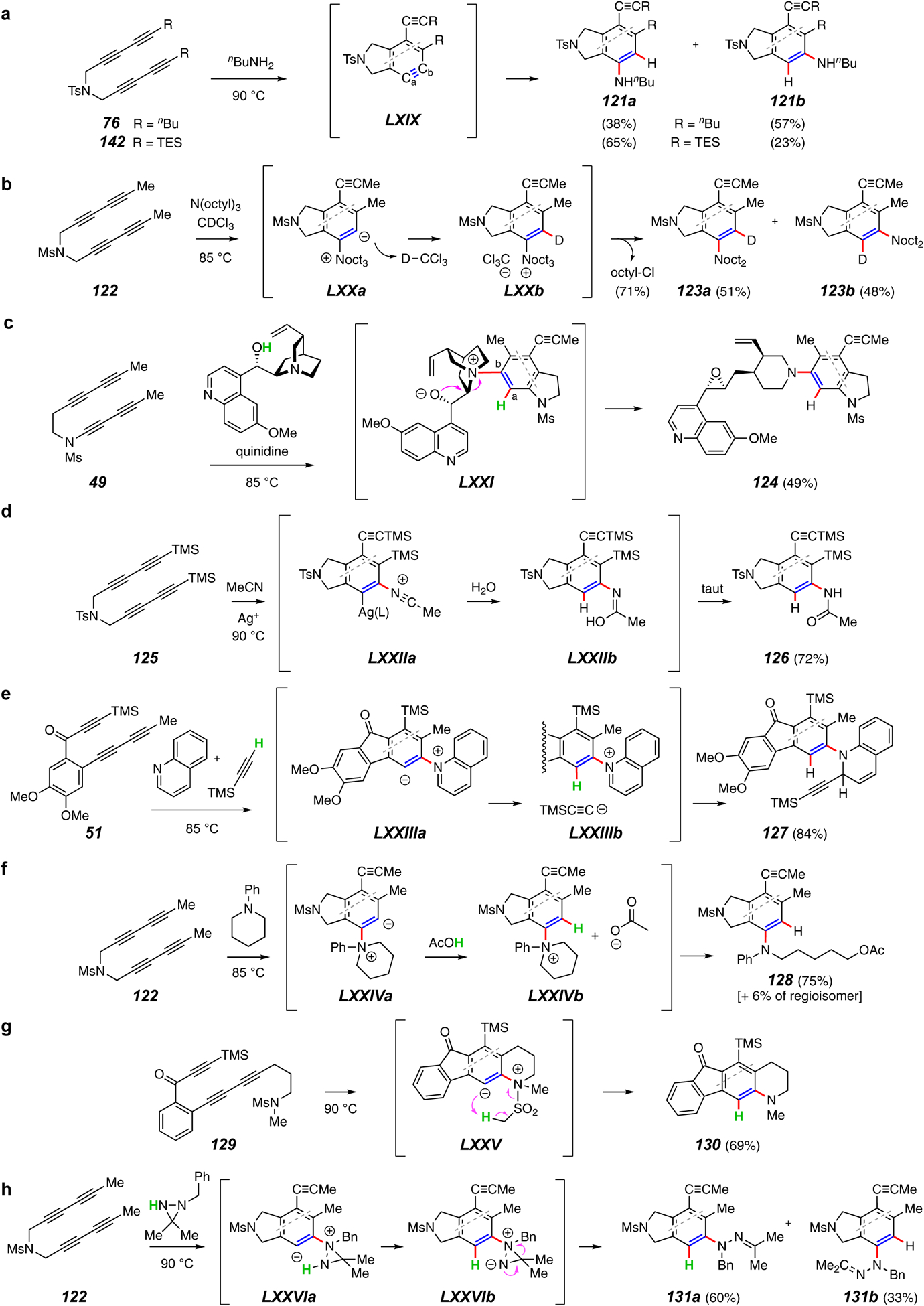
Trapping reactions forming a C–N and a C–H bond.
In contrast to the above category, there is a wide variety of reaction types that make both a C–N and a C–H bond during a trapping reaction. The simple nucleophilic attack of a primary or secondary amine is very common. The examples shown in Figure 14a come from a report that examined in considerable depth the factors that impact the sense and extent of regioselectivity of the addition of amines. For example, n-butylamine adds to the benzyne produced from the tetrayne 76 to give the isomeric adducts 121a and 121b (R = nBu) in a 1:1.5 ratio.90 Isoindolinynes such as LXIX show relatively little distortion and often lead to mixtures of isomers arising from competitive attack at the Ca- vs. the Cb-benzyne atoms. Steric interactions between the attacking nucleophile and the substituents in the 3- and 6-positions ortho to the benzyne can then be more influential in directing the incoming trapping agent. When an analogous reaction was performed with the TES-substituted tetrayne 142, the larger TES group in LXIX (R = TES) guided the attack more toward Ca to give the major product 121a (R = TES). When 142 was trapped by the bulkier tertiary amine Et3N, the added steric bulk of that nucleophile directed the attack to Ca, giving, solely, the diethylamino analog of 121a (R = TES) and showing that the steric interactions can completely dominate over the electronic bias imposed by the silyl substituent in the benzyne. An additional directing effect in benzyne LXIX (R = TES) stems from the presence of a trialkylsilyl group adjacent to the benzyne, a substituent known to electronically enhance nucleophilic attack at the adjacent rather than remote benzyne carbon.95,96,97,98 As is often the case in evaluating selectivity of a given reaction outcome, whether before (prediction) or after (rationalization) the fact, when multiple opposing directing effects are in play90 – here steric vs. electronic vs. distortion – it is not possible to judge which combination will dominate without a more extensive and systematic evaluation of substrate substituent effects (e.g., here, changing the sizes of the trialkylsilyl group).
The mechanism by which tertiary amines trap a HDDA benzyne has been investigated (Figure 14b).99 Shown here is the reaction of trioctylamine with the tetrayne substrate 122, which gives an ca. 1:1 mixture of products 123a and 124b. When performed in CDCl3 solution, these products are essentially fully deuterated, showing that a solvent molecule (and not a proton from the β-position of one of the octyl groups) is the source of the hydrogen atom in the product (cf. LXXa to LXXb). The fate of the departed octyl group was also identified; 1-chlorooctane was produced in nearly stoichiometric amount through dealkylation of LXXb [and the elimination product 1-octene was not observed (in situ NMR or GCMS)].
In contrast to isoindolinynes such as LXIX , the indolinyne derived from the unsymmetrical tetrayne 4933 (not shown) engages the tertiary amine in quinidine highly regioselectively at its Ca to produce, following proton transfer from the adjacent hydroxyl group, the alkoxyammonium ion LXXI.58 Collapse to the epoxyamine leads to formation of 124. A large number of other reactions between HDDA-generated benzynes and multifunctional natural products100 were also revealed in this study, which, among other things, revealed a noteworthy high level of kinetic selectivity among multiple potential reaction types.58
The conversion of tetrayne 125 to the acetanilide derivative 126 (Figure 14d) is achieved by Ag(I)-catalyzed addition of acetonitrile, the solvent.101 This Ritter-like reaction is viewed to proceed through the nitrilium ion LXXIIa. Water in the reaction mixture then adds to furnish the amide enol LXXIIb before final tautomerization to the product.
Multicomponent trapping reactions offer an attractive route for introducing structural complexity into HDDA products, some of which involve creation of C–N and C–H bonds (e.g., Figures 14e and 14f102). In the first, nucleophilic attack of the benzyne from 51 by aromatic heterocycles of the pyridine class, shown here with quinoline, produces the quinolinium-containing zwitterion LXXIIIa. The anionic character of this species is apparently sufficiently basic to deprotonate terminal alkynes also present in the reaction mixture as the third component. The resulting ion pair LXXIIIb then collapses to the product 127. In the second example, substrate 122, when heated in the presence of the cyclic tertiary amine N-phenylpiperidine and acetic acid, gives the 5-acetoxypentylaniline derivative 128. This can be rationalized by initial formation of the ammonium zwitterion LXXIVa [faster, incidentally, than the addition of acetic acid (cf. Figure 12f)], which is then protonated by AcOH to give the ammonium acetate LXXIVb. Nucleophilic ring-opening then completes the formation of 128. A wide assortment of cyclic amines as well as other protic nucleophiles function in these capacities as well.
The methanesulfonamide substrate 129 is an alkyl analog of the arylsulfonamide 118 shown above in Figure 13b. However, the fate of the Ms-containing zwitterion LXXV (Figure 14g) is different from that of the NTs analog.93 Rather than sulfonyl migration to the anionic center, LXXV apparently undergoes internal proton transfer with extrusion of sultene to produce the N-methylated tetrahydroquinoline derivative 130.
Finally, an unusual trapping reaction between diaziridines and an HDDA benzyne has been reported (Figure 14h).103 Following the HDDA cyclization of, e.g., 122, a nucleophilic diaziridine nitrogen attacks the aryne to give adduct LXXVIa. Proton transfer and electrocyclic ring opening forms the new C–H bond and yields the hydrazone product 131a. Notably, the more hindered but more electron rich nitrogen of mono-N-alkylated diaziridines adds preferentially. This likely reflects the expectation that the distance between the nucleophilic nitrogen and electrophilic carbon atoms is quite long in the TS leading to zwitterions such as LXXVIa. Again, a mixture of regioisomeric adducts (cf., 131b) was formed from this class of benzyne intermediate.
4.10. Introduction of C–H and C–H bonds (Figure 15)
Figure 15.
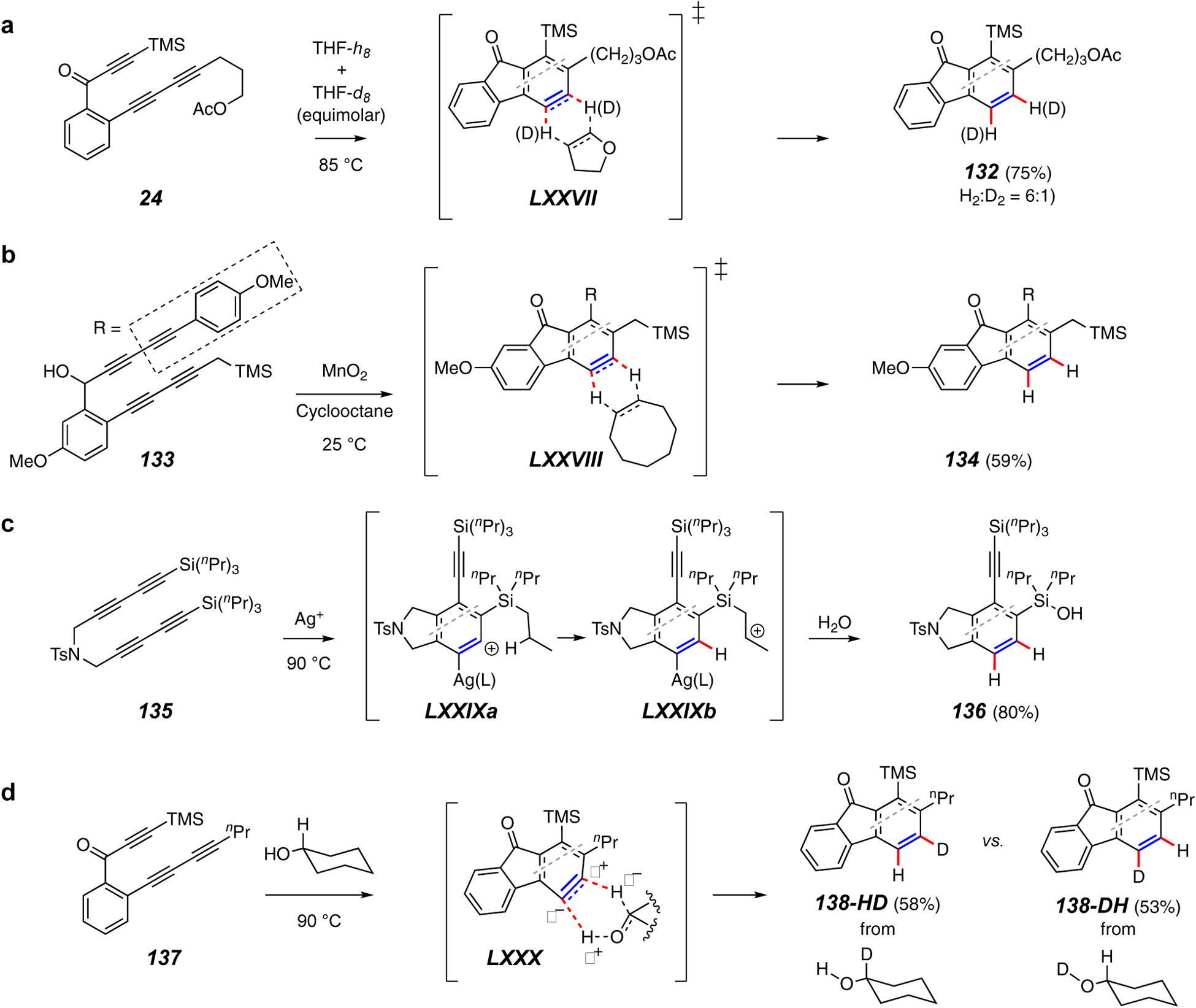
Trapping reactions forming two C–H bonds.
Reactions that create two C–H bonds amount to a net reduction of a benzyne to its benzene analog. Tetrahydrofuran was the first molecule observed to transfer two hydrogen atoms to a thermally generated benzyne; namely, the benzyne shown in IV in Figure 2c was reduced by gaining two hydrogens when it was created in THF solution.10 Viewed at the time as hydrogen atom abstraction events, the net redox reaction with THF was subsequently demonstrated to involve a concerted transfer of two hydrogen atoms as depicted in LXXVII (Figure 15a).104 One key experiment involved heating 24 in an equimolar mixture of THF-d8 and THF-h8. Product 132 was formed as a 6:1 ratio of di-protio and di-deuterio isotopomers, but none of the mono-H/mono-D analog was detectable. This is consistent with a single-encounter event like that depicted in TS LXXVII; the relatively small kinetic isotope effect for simultaneous cleavage of two C–H bonds reflects the relatively early transition state for this highly exergonic (DFT) process.
Two vicinal hydrogen atoms will also transfer from a saturated, cyclic hydrocarbon, as depicted for the transformation of 133 to 134 in Figure 15b. This key step in the Lee synthesis of selaginpulvilins C and D23 was carried out by oxidizing a precursor propargylic alcohol, which rapidly cyclized at ambient temperature in cyclooctane solvent. In competition experiments,104 cyclooctane (and cyclopentane) reduces HDDA benzynes faster than cyclohexane, consistent with the energetically easier access to the more closely eclipsed geometry computed for a concerted transition structure like that shown in LXXVIII.
Another instance in which silver(I) has promoted unusual reactivity is shown in Figure 15c.70 The benzyne-silver complex LXXIXa is argued to be sufficiently electrophilic to accept hydride donation from one of the pendant n-propyl groups in substrate 135 to produce the carbocation LXXIXb, stabilized by the β-silicon atom. Water intervention and extrusion of propene results in the net-hydrogenated silanol product 136.
Alcohols are also capable of reducing benzynes in a process reminiscent of a Cannizzaro reaction (e.g., 137 to 138, Figure 15d).88 A tandem experimental and computational study revealed the ability of primary and secondary alcohols to donate two H atoms in a concerted process similar to the cyclic hydrocarbons. Labeling studies using isomeric mono-deuterated cyclohexanols demonstrated that the process was both concerted and regiospecific, as shown in Figure 15d. Computation of TS geometries for addition of cyclohexanol to benzyne shows a geometry like that shown in LXXX. This is consistent with the deuterium labeling pattern in products 138-HD and 138-DH and indicates that the CH methine hydrogen atom is transferring as a hydride-like nucleophile and the OH as a proton-like electrophile.
4.11. Introduction of C–H and C–Halogen bonds (Figure 16)
Figure 16.
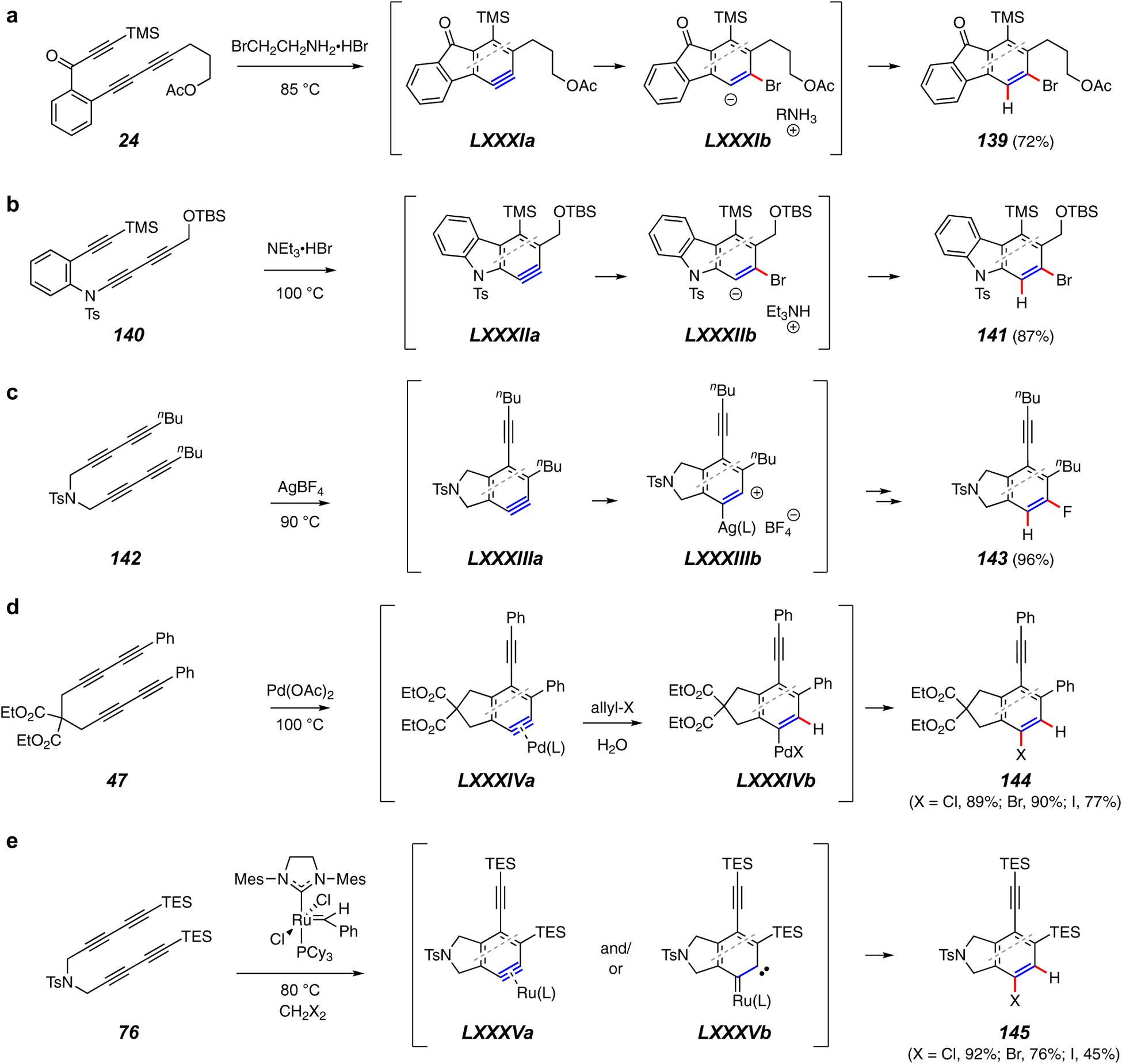
Trapping reactions forming a C–H bond and a C–Hal bond.
The use of a trapping reagent that produces both C–H and C–halogen bonds yields products with useful handles for further synthetic manipulation. Perhaps the simplest way to accomplish this is through the net addition of a mineral acid. Although the use of HBr had been demonstrated in a number of publications, the reagents of choice here are ammonium halide salts. For example, triynes 24 or 140 give the bromides 139 or 141 when heated in the presence of primary or tertiary ammonium bromides (Figures 16a20 or 16b24). The soft bromide ion nucleophilically adds to the HDDA benzyne LXXXIa or LXXXIIa and protonation of the ion pair LXXXIb or LXXXIIb leads to the product. The level of regioselectivity of nucleophilic addition to each of these benzynes is known to be high.
In one of the Lee group’s earliest studies of Ag(I)-enabled chemistry, they developed a method for the net addition of HF across the benzyne using AgBF4 (Figure 16c).75 Addition of fluoride is a rare outcome for aryne chemistry – consider the thousands of reactions that use the Kobayashi fluoride-induced aryne generation21 in which F– does not further engage the reactive intermediate. Presumably this reflects the reluctance of the soft, polarizable aryne bond to accept a hard, compact nucleophile. The silver-benzyne complex LXXXIIIb, formed following generation of benzyne LXXXIIIa from 142, presumably accepts a fluoride ion from the (softer) BF4– counterion. Protonation – perhaps occurring during workup – yields the product 143.
Reactions catalyzed by palladium (Figure 16d)105 and ruthenium (Figure 16e)25 that result in the net addition of HCl, HBr, and HI to an HDDA benzyne have been reported. In the conversion of, e.g., 47 to 144, allyl halides serve as the halogen source whereas in the reaction of, e.g., 76 to 145, dihalomethanes serve that role. In both reactions initial formation of an intermediate benzyne-metal complex (cf. LXXXIVa or LXXVa/LXXXVb) is proposed. In the Pd-variant, a water molecule was demonstrated (D2O) to be the proton source.
4.12. Introduction of C–H and C–S bonds (Figure 17)
Figure 17.
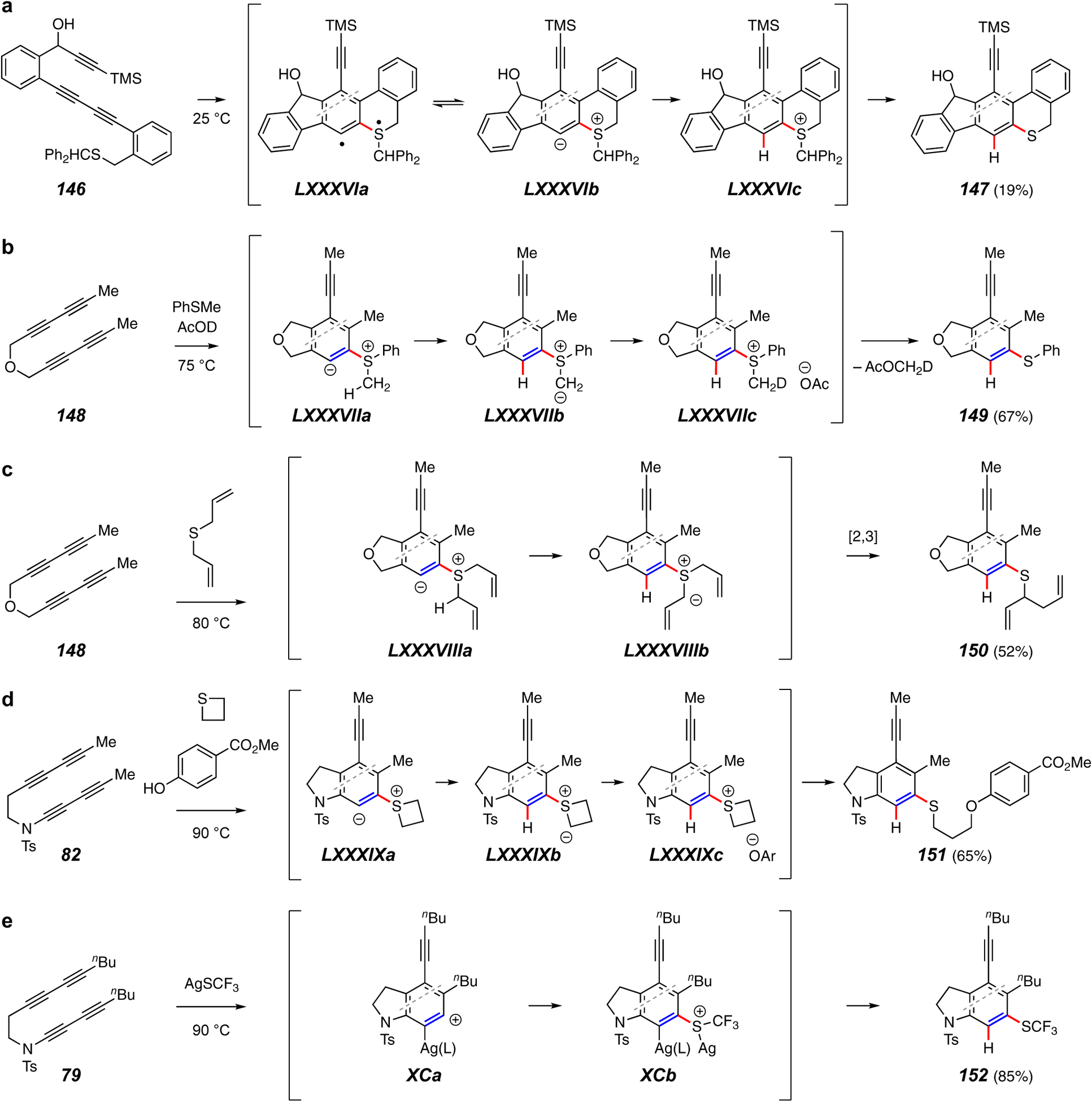
Trapping reactions that form a C–H bond and a C–S bond.
Sulfides, soft nucleophiles, are quite effective trapping agents. This often leads to products in which adjacent C–S and C–H bonds are introduced at the benzyne carbon atoms. The first example was in the intramolecular setting of the conversion of 146 to 147 (Figure 17a).14 Intermediates LXXXVIa-c were invoked. Subsequent studies have made it clear that proton transfer from an alkyl group within an intermediate such as the zwitterion LXXXVIb often generates a sulfur ylide (examples below), likely involved in this reaction as well. An ylide intermediate is supported by a number of experiments, for example the labeling reaction shown in Figure 17b.106 In the conversion of 148 to 149, the zwitterion LXXXVIIa gives the ylide LXXXVIIb, which is then protonated by AcOD also present in the reaction mixture. The acetate in the ion pair LXXXVIIc then produces the monodeuterio-methyl acetate, detected by in situ 1H NMR analysis. Notably, the soft sulfur in thioanisole adds to the benzyne in preference to the addition of acetic acid (cf. Figure 12f), even though the latter is a competent trapping agent.
Diallyl sulfide adds to the benzyne from 148 to produce the zwitterion and ylide LXXXVIIIa and LXXXVIIIb, respectively (Figure 17c).106 A [2.3]-sigmatropic rearrangement produces the arylsulfide product 150. Use of cyclic sulfides allows for three-component reactions (Figure 17d).106 For example, 82 gives 151 when heated in the presence of thietane and various phenols. Intermediates LXXXIXa-c explain that outcome.
Finally, in another silver-catalyzed trapping of HDDA benzynes with a fluorine-containing reagent, the substrate 79 gives rise to the trifluoromethyl sulfide 152 when heated in the presence of AgSCF3 (Figure 17e).75 Intermediates XCa and XCb were offered as rationale.
4.13. Introduction of a C–Hal and a C–Hal or a C–O bond (Figure 18)
Figure 18.
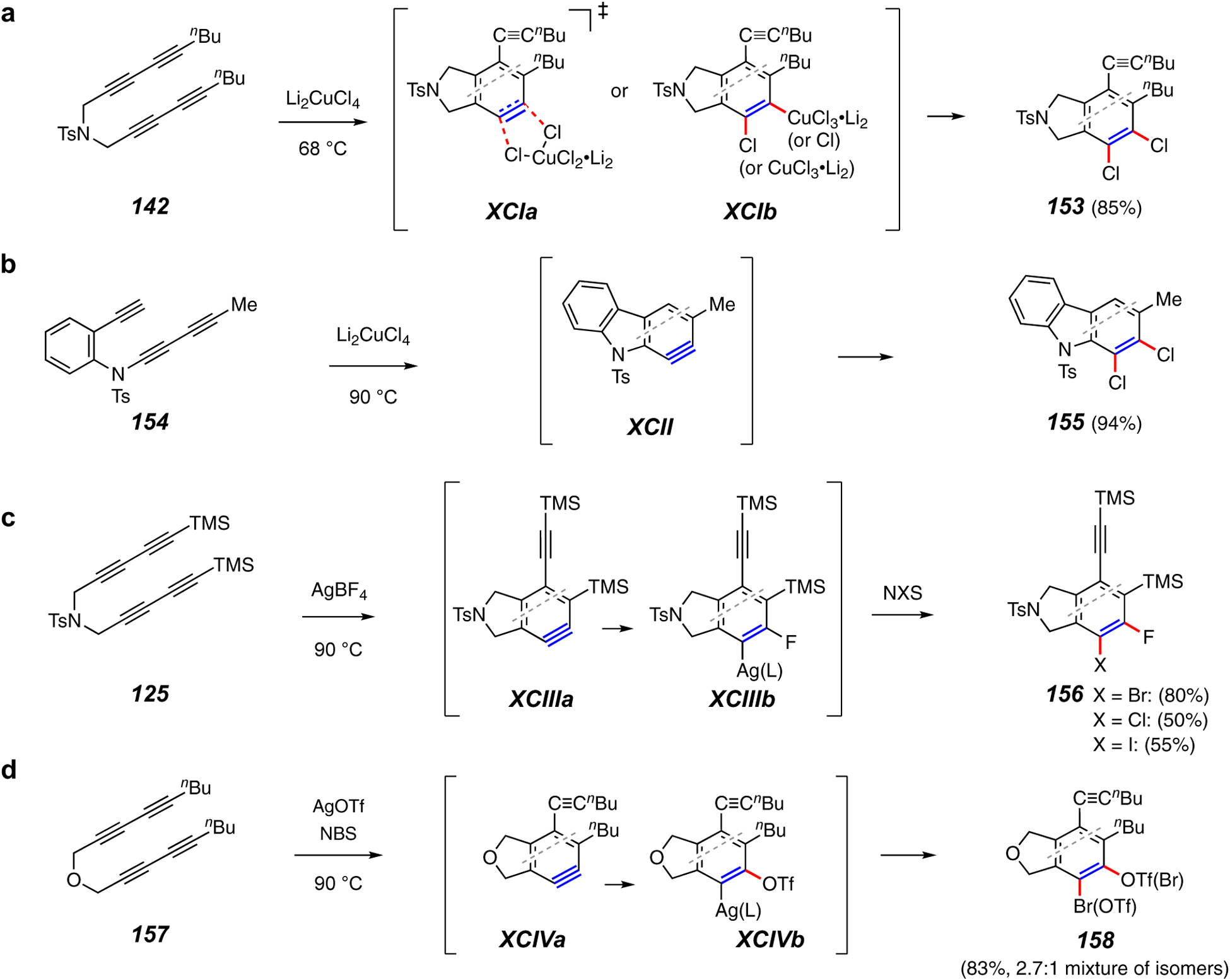
Trapping reactions that form a C–Hal bond as well as a (second) C–Hal or a C–O bond.
Dihalogenation represents an example of an HDDA trapping reaction in which compatibility of the polyyne substrate with the trapping reagent envisioned for capturing the benzyne needs to be taken into consideration. For example, elemental dihalogens would react with the alkynes in the substrate before there was any appreciable formation of an HDDA benzyne. Dilithium tetrachlorocuprate (Li2CuCl4) was identified as one solution to this limitation.107 The tetrayne 142 gave the 1,2-dichlorobenzene derivative 153 (Figure 18a). An associated mechanistic study in which the substrate contained a tethered trapping moiety, which served as an internal clock reaction, allowed determination of the kinetic order of the post-rate limiting trapping step by varying the concentration of Li2CuCl4. The capture is first order in Li2CuCl4; however, this leaves open the question of the detailed mechanism of the reaction. A direct transfer of both chlorine atoms through a TS such as XCIa or an intermediate chlorocuprate species such as XCIb would both explain the outcome. A related example proceeds through the carbazolyne XCII (154 to 155, Figure 18b).24
Additional variants of silver(I)-promoted reactions lead to either the fluorohalobenzene derivatives 156 (Figure 18c)75 or o-halotriflates such as 158 (Figure 18d).89 In the first reaction (125 to 156), a stoichiometric amount of AgBF4 gave adduct XCIIIb by reaction with the initial benzyne XCIIIa. Subsequent addition of an N-halosuccinimide gave the product dihalides. This represents a rare example in which trapping of an aryne gives a stable intermediate species that then reacts further upon later addition of another reagent. In the second reaction, both silver triflate and an N-halosuccinimide (NBS in the example of 157 to 158) are present from the outset. Here the organosilver species XCIVb (from XCIVa) is halogenated in situ.
4.14. Introduction of miscellaneous bond pairs (Figure 19)
Figure 19.
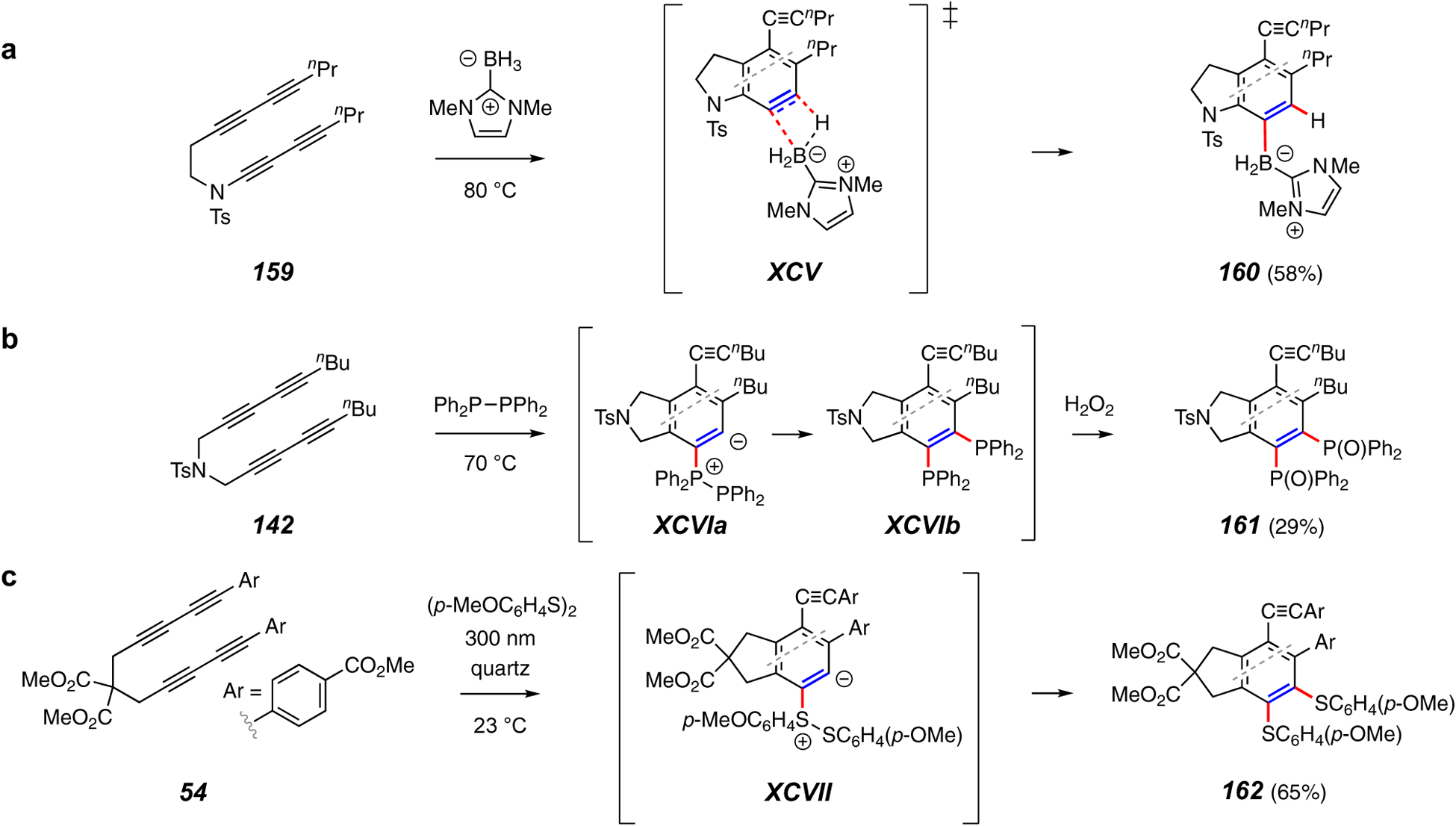
Trapping reactions that form (a) a C–H and a C–B bond, (b) two C–P bonds, and (c) two C–S bonds.
In this last subsection, several “one-off” examples of some unusual transformations, each fueled by the high potential energy of the reactive benzyne intermediate, are presented. The first of these is a net hydroboration of the HDDA benzyne resulting in the conversion of 159 to 160 (Figure 19a).108 As with the dihalogens discussed above, hydroboranes as trapping agents would be expected to rapidly consume the substrate alkynes, precluding benzyne formation. In contrast, the indicated carbene-borane complex effects this net hydroboration transformation. The mechanism of this reaction is not obvious. The sense of the (complete) regioselectivity observed for this reaction suggests that the process involves a step in which nucleophilic attack by hydride leads the way. A concerted TS such as XCV can be formulated, although it is not clear if the boron atom can sustain this type of bonding and bond reorganization.
The creation of two C–P bonds has been achieved with tetraphenyl diphosphane (142 to 161, Figure 19b).109 A DFT analysis suggested that the intermediate XCVIa can progress directly to the bis-phosphine XCVIb in a concerted event. A similar reaction with a diaryldisulfane gives the bis-sulfide 162 (Figure 19c).60 The contrasting behavior between this reagent, which presumably produces the zwitterion XCVII, and that of the analogous higher chalcogenides of selenium and tellurium (cf. Figure 10c) is notable. Finally, note that in this instance the substrate 54 was cyclized photochemically, a process discussed in more detail next in section 5.1.
5. STRATEGY-LEVEL DEVELOPMENTS FOR THE HDDA REACTION
There have been several notable conceptual developments that demonstrate important new dimensions of HDDA chemistry. The specific transformations that demonstrate these new strategic advances draw largely from the same pool of substrate tethers and trapping reactions described in Sections 2–4 above. An overview of each development with illustrative examples is presented in Sections 5.1–5.4.
5.1. The Photochemical HDDA Reaction (Figure 20)
Figure 20.
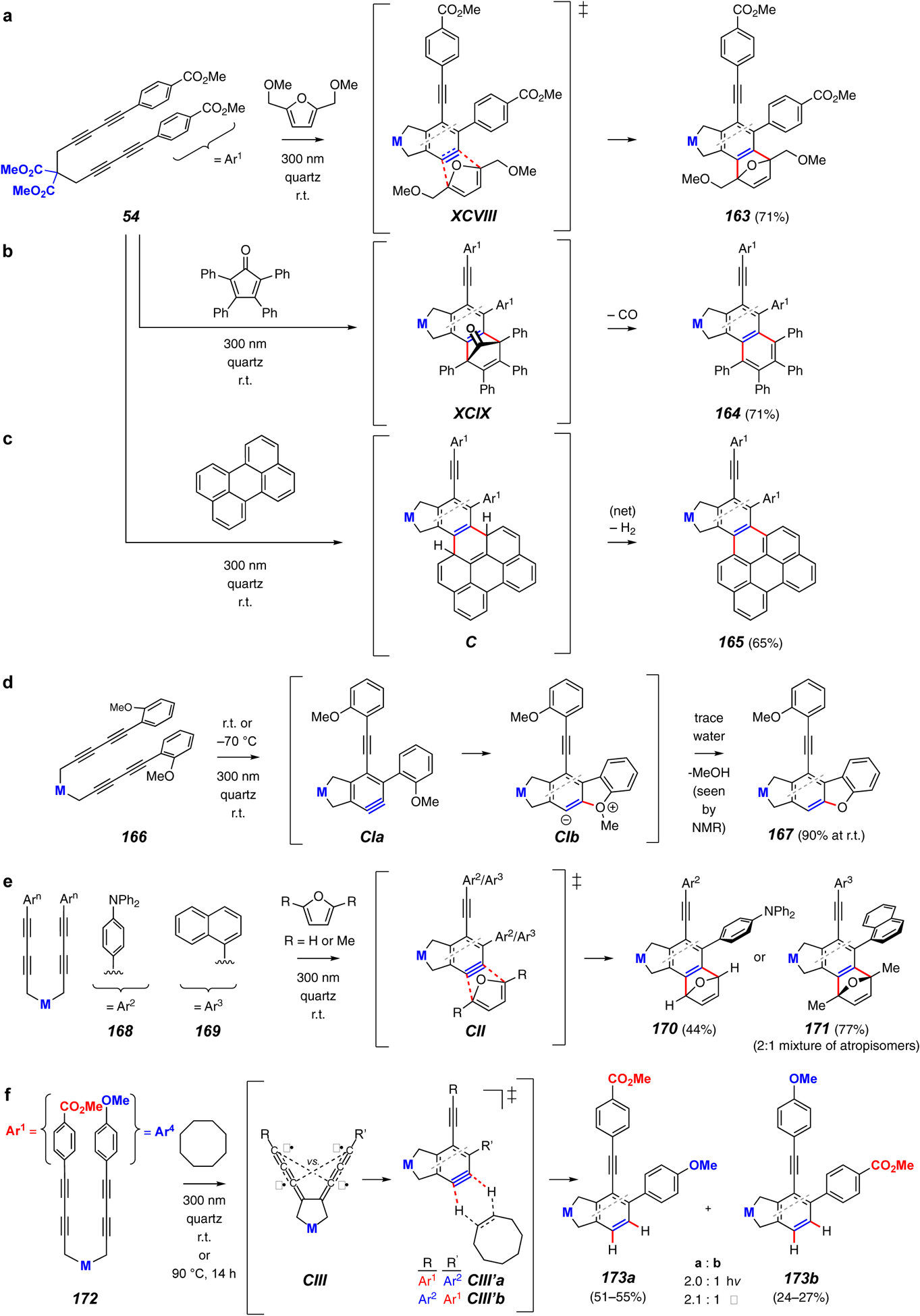
Examples of the photochemical HDDA (hv-HDDA) reaction.
The photochemical HDDA reaction, reported in 2017, uses a photon rather than thermal energy to initiate the HDDA cyclization.60 To date the only class of substrate reported to undergo this transformation is a series of tetraynes bearing two terminal aromatic substituents, as seen from each of the examples in Figure 20. Not surprisingly, substrate 54 can be trapped with a furan derivative to produce 163 (via XCVIII, Figure 20a). More informative are the reactions in Figures 20b and 20c in which the intermediate [4+2] adducts were not isolated but lost carbon monoxide (cf. XCIX) and dihydrogen (cf. C) to give the naphthalene and naphthoperylene derivatives 164 and 165, respectively. These examples show that those finishing events occur even at ambient temperature, a fact not discernable from the thermal variant of these types of HDDA trapping reactions.
Although most reactions were performed, out of convenience, at ambient temperature, in the case of the substrate 166, bearing an internal trapping moiety in the form of the o-methoxyphenyl substituent, the reaction was shown to proceed also at ca. −70 °C to give 167 (Figure 20d). In this experiment the byproduct methanol was observed, suggesting that a small amount of water in the reaction mixture was providing the proton to zwitterion CIb (from cyclization of CIa) as well as the nucleophile to subsequently accept the electrophilic methyl group.
Electron rich aryls (cf. 166, 168, and 169) also can be cyclized photochemically. The last two (Figure 20e) are demonstrated by the oft-used furan trapping (cf. CII) to produce 170 or 171, respectively. The latter, where 2,5-dimethylfuran was the trapping agent, was observed by NMR analysis to be a mixture of atropisomers because of hindered biaryl rotation on the NMR time scale.
Finally, important mechanistic information was obtained from study of the reaction of the unsymmetrically substituted tetrayne substrate 172 (Figure 20f). This can be cyclized either thermally (half-life of 12 hours at 75 °C) or photochemically. Under thermal conditions, two isomeric benzynes, CIII’a and CIII’b, are formed and give rise to products 173a and 173b in a 2.1:1 ratio, respectively. The trapping agent here was cyclooctane (cf. Figure 15b), which made the products easily discernable by NOE experiments. When the same reaction was performed photochemically, the same ratio (within error) of products was observed. This result suggests that the mechanisms leading to formation of the isomeric benzynes converge through a common intermediate, regardless of the means of energization. This is consistent with the view that benzyne formation occurs by a stepwise pathway through a common biradical similar to CIII, which then bifurcates during ring closure to either CIII’a or CIII’b.24,28,30,32 Further, it suggests that the electronic spin characteristics of this biradical are the same, whether formed by initial thermal or photochemical activation.
5.2. The Domino HDDA Reaction (Figure 21)
Figure 21.
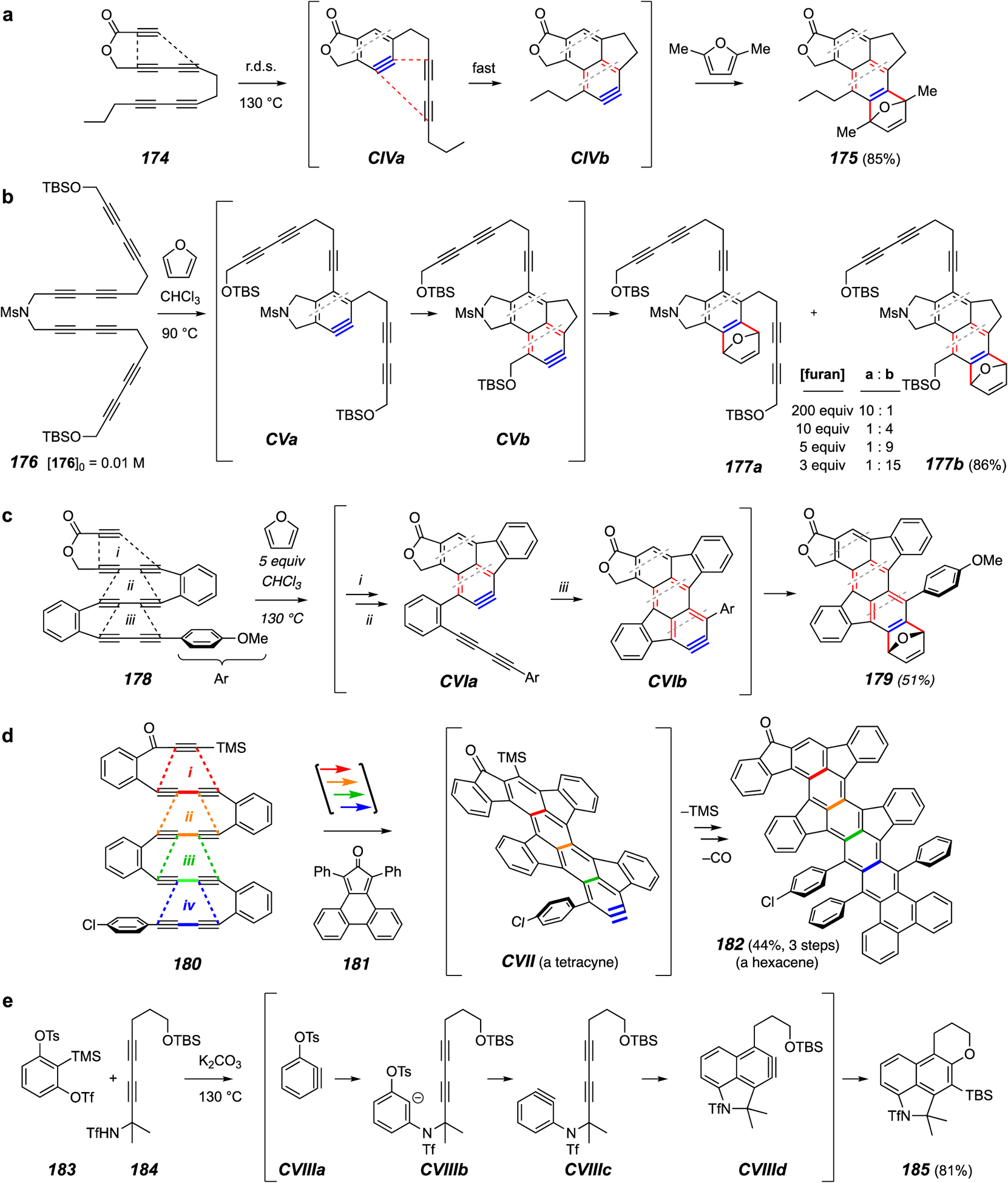
Examples of the domino HDDA reaction. (a) Benzyne to naphthyne. (b) Evidence for the intermediacy of a benzyne. (c) Benzyne to naphthyne to anthracyne. (d) Benzyne to naphthyne to anthracyne to tetracyne. (e) A domino cascade involving benzynes generated by both classical and HDDA processes.
If one is good, two (or more) is (often) better. That idea led researchers to hypothesize that an appropriate linear array of multiple tethered diynes would support not one but a series of intramolecular cyclizations wherein each successive aryne would become the diynophile for the next diyne in line – a domino HDDA process.13 In short, this process initially generates one benzyne that then undergoes subsequent intramolecular HDDA cyclizations with tethered diynes to produce increasingly large members of the acyne family.
The concept was first demonstrated using the pentayne 174 (Figure 21a).110 This was designed to include a three-atom tether between the first diynophile (the top ynoate) and its proximal 1,3-diyne but with only two atoms (CH2CH2 in 174) separating the pair of 1,3-diynes. As planned, the initial cyclization produced the benzyne CIVa, which proceeded to engage the second diyne, now five atoms away from the proximal benzyne carbon, to give the naphthyne CIVb. This was trapped by in situ 2,5-dimethylfuran to give essentially nothing but the adduct 175. Numerous examples of naphthyne trapping were shown to be effective.
The results shown in Figure 21b demonstrate that the process is sequential. That is, the intermediate benzyne CVa, produced by initial cycloisomerization of the central tetrayne in substrate 176, has a finite lifetime. This benzyne is trapped relatively efficiently when the reaction is performed in furan as solvent. As the concentration of furan is systematically lowered, there is a longer lifetime for CVa, giving ample opportunity for the second cyclization to occur to produce the naphthyne CVb as revealed by the formation of increasing amounts of the naphthalene product 177b.
The conversion of heptayne 178 to the anthracene derivative 179 (Figure 21c) shows that the two-atom linker between adjacent pairs of diynes also can be a 1,2-disubstituted arene moiety. This particular transformation involves three successive HDDA cyclizations, the last (step iii) converting the naphthyne CVIa to the anthracyne CVIb.
The most complex substrate examined was the nonayne 180 (Figure 21d). This proceeded through four consecutive events to the tetracyne CVII, which was trapped with several agents; shown here is the use of the cyclopentadienone derivative 181. The initial adduct was desilylated and heated to 180 °C to eject carbon monoxide and produce the dibenzohexacene derivative 182. This was shown to have a significantly twisted architecture. The highly conjugated molecules available from these domino HDDA cyclizations have interesting photonic and electronic properties, potentially valuable for use in various electronic devices.
Finally, the domino strategy can be exported into other settings. This was demonstrated by the reaction shown in Figure 21e.111 As shown by researchers in the Li lab, reactant 183 serves as the equivalent of 1,2-bisbenzyne.112 When exposed to carbonate and an appropriate nucleophile, this initially forms the benzyne CVIIIa, which then engages the nucleophile, here the conjugate base of the triflamide 184. Net Sn2’ displacement of the tosylate leaving group in CVIIIb gives the second benzyne CVIIIc. This now engages the pendant 1,3-diyne in the HDDA event to give the penultimate intermediate, the naphthyne CVIIId. Internal trapping with the tethered TBS ether produces 185, completing this cascade that marries classical Kobayashi-like with HDDA benzyne formation.
5.3. The Aza-HDDA Reaction (Figure 22)
Figure 22.
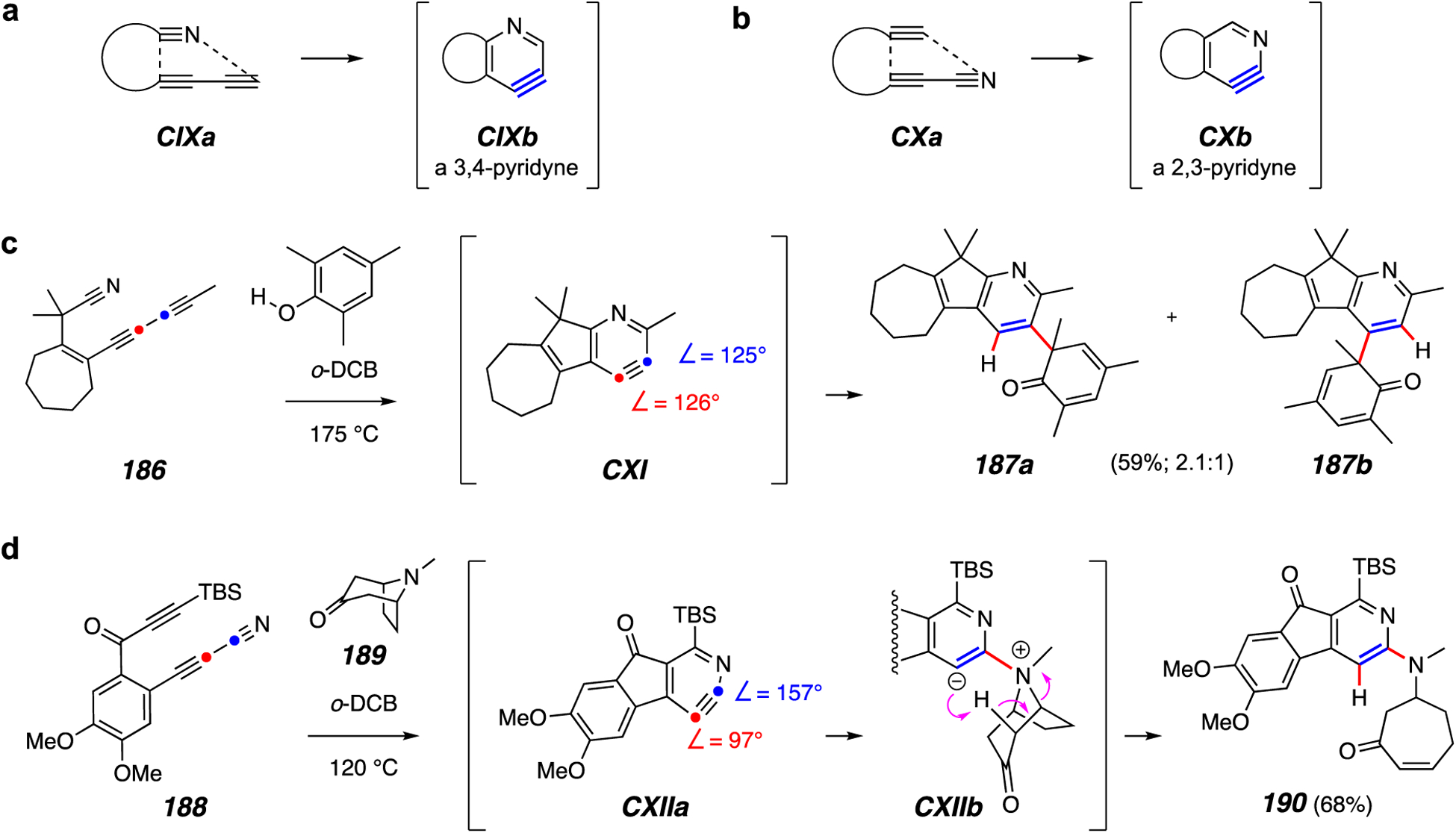
Aza-HDDA reactions. (a) Generic formation of a 3,4-pyridyne. (b) Generic formation of a 2,3-pyridyne. (c) A relatively unselective trapping of a 3,4-pyridyne. (d) A highly selective trapping of a 2,3-pyridyne.
Replacement of one of the two terminal carbon atoms in a triyne substrate with a nitrogen atom opens the possibility to effect an analogous cycloisomerization to form a pyridyne intermediate. This aza-HDDA reaction has been recently reduced to practice (Figure 22).113 This represents a fundamentally novel approach towards the construction of highly functionalized pyridines from acyclic precursors. Based on the original location of the nitrile, either a 3,4-pyridyne (CIXa to CIXb, Figure 22a) or a 2,3-pyridyne (CXa to CXb, Figure 22b) is formed.
DFT studies of these types of isomeric pyridynes indicate that they have dramatically different levels of geometric distortion. This was shown experimentally by comparing the behavior of these isomeric pyridynes, which exhibit quite different selectivity upon reaction with a nucleophilic trapping agent. For example, substrate 186, having the nitrile as a diynophile, gives the dienone 187, but as a mixture of both 187a and 187b via the relatively undistorted pyridyne CXI (Figure 22c). In contrast, 188, having an aza-diyne and the precursor to the 2,3-pyridiyne CXIIa, reacts with, for example, tropinone (189) to give the amine-trapped product 190 (via zwitterion CXIIb, Figure 22d). This was the only regioisomer observed; eight other trapping reactions of 2,3-pyridiynes also showed complete regioselectivity, including one with 2,4,6-trimethylphenol.
In many respects the aza-HDDA reaction behaves analogously to the all-carbon HDDA transformation – many of the same traps and tethers can be used. However, the aza variant has several limitations. The aza-HDDA cycloisomerization reaction is considerably slower, with some substrates requiring a reaction temperature in excess of 200 °C. This is consistent with the fact that the bond energy of a C–N triple bond is considerably higher than that of a C–C triple bond. Moreover, product yields of many examples are below 50% and substrate synthesis is not as straightforward as for analogous triynes. Despite a more limited scope, the aza-HDDA variant still represents a novel and potentially useful process for creating certain highly substituted pyridine derivatives.
5.4. Reagent-induced Net HDDA Reactions (Figure 23)
Figure 23.
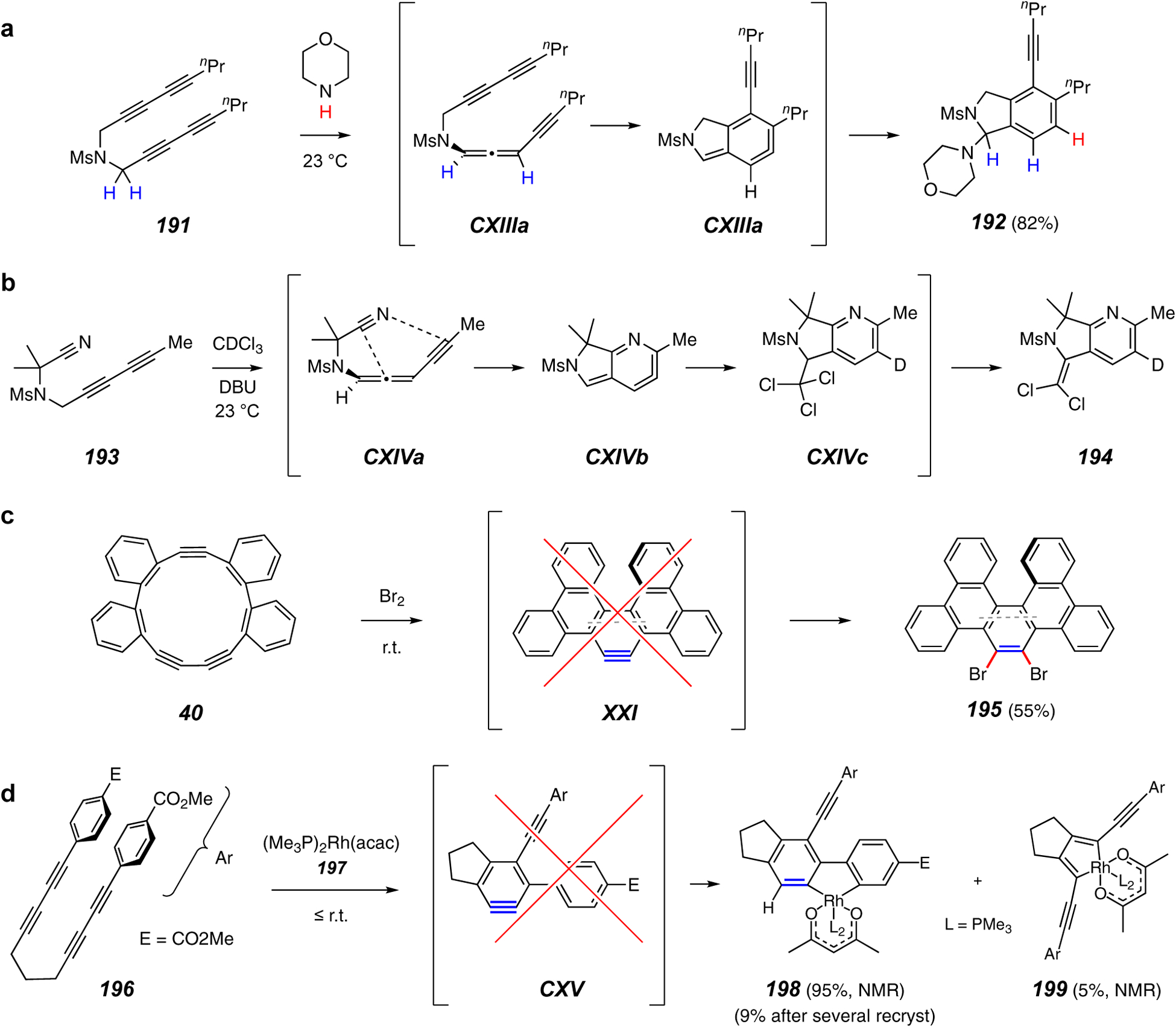
Reactions of polyyne substrates that proceed under conditions that would not promote appreciable levels of HDDA benzynes, indicating that the co-reactant is promoting cyclization by engaging the substrate prior to the thermal HDDA cycloisomerization.
Several reactions have been reported to give products that are HDDA-like, but that do not proceed by way of a benzyne. These are shown in Figure 23. In the first example, the tetrayne 191 was observed to react at ambient temperature in the presence of secondary amines such as morpholine to produce the isoindoline product 192.114 Mechanistic studies revealed that the process proceeds by an initial, base-catalyzed isomerization to the allenyne CXIIIa that then cyclizes to the strained allene CXIIIb. This analog of the known, parent α,3-dehydrotoluene115 is then captured by the amine.116 This process was dubbed the pentadehydro-Diels-Alder (PDDA) reaction, signifying that the cyclization occurs via an acyclic intermediate, CXIIIa, in which five, not six, of the atoms are sp-hybridized.
In a related reaction, the cyanodiyne 193 was converted to the pyridine derivative 194 under the action of DBU in deuterochloroform (Figure 23b). This was the first instance in which the energetically more demanding cyano group participated in an HDDA-related cyclization. This aza-PDDA reaction likely proceeds via intermediates CXIVa-c.
As shown earlier in Figure 5l, the cyclic triyne 40 requires heating for many hours at 70 °C to generate the benzyne XXI. Shown in Figure 23c is the reaction of 40 with bromine, which rapidly (<1 h) at room temperature generates the dibrominated product 195.53 This strongly suggests that XXI is not involved in this “bromine-induced transannular cyclization.”53
Finally, the hydrometallation product 198 was generated highly efficiently (accompanied by a small amount of the isomeric rhodacycle 199) when the potential HDDA tetrayne substrate 196 was exposed to a stoichiometric amount of the Rh(I) complex 197.117 Based on the known rate of reaction of tetraynes structurally very similar to 196 [e.g., 172 (cf. Figure 20f) cyclizes thermally with a half-life of ca. 4 h at 90 °C60], it can be expected that 196 would require elevated temperature to cyclize to the HDDA benzyne CXV at any reasonable rate. Therefore, it is most probable that Rh(I) is inducing cyclization leading, ultimately, to the new benzenoid ring in 198 by a pathway that does not include the free benzyne as an intermediate.
6. ADDITIONS MADE DURING MANUSCRIPT REVISION (Figure 24)
Several additions, including a summary of recently published reports, are presented in Figure 24. In a few instances where relevant, these have been referred to (called out) by a comment at an appropriate location in earlier discussion.
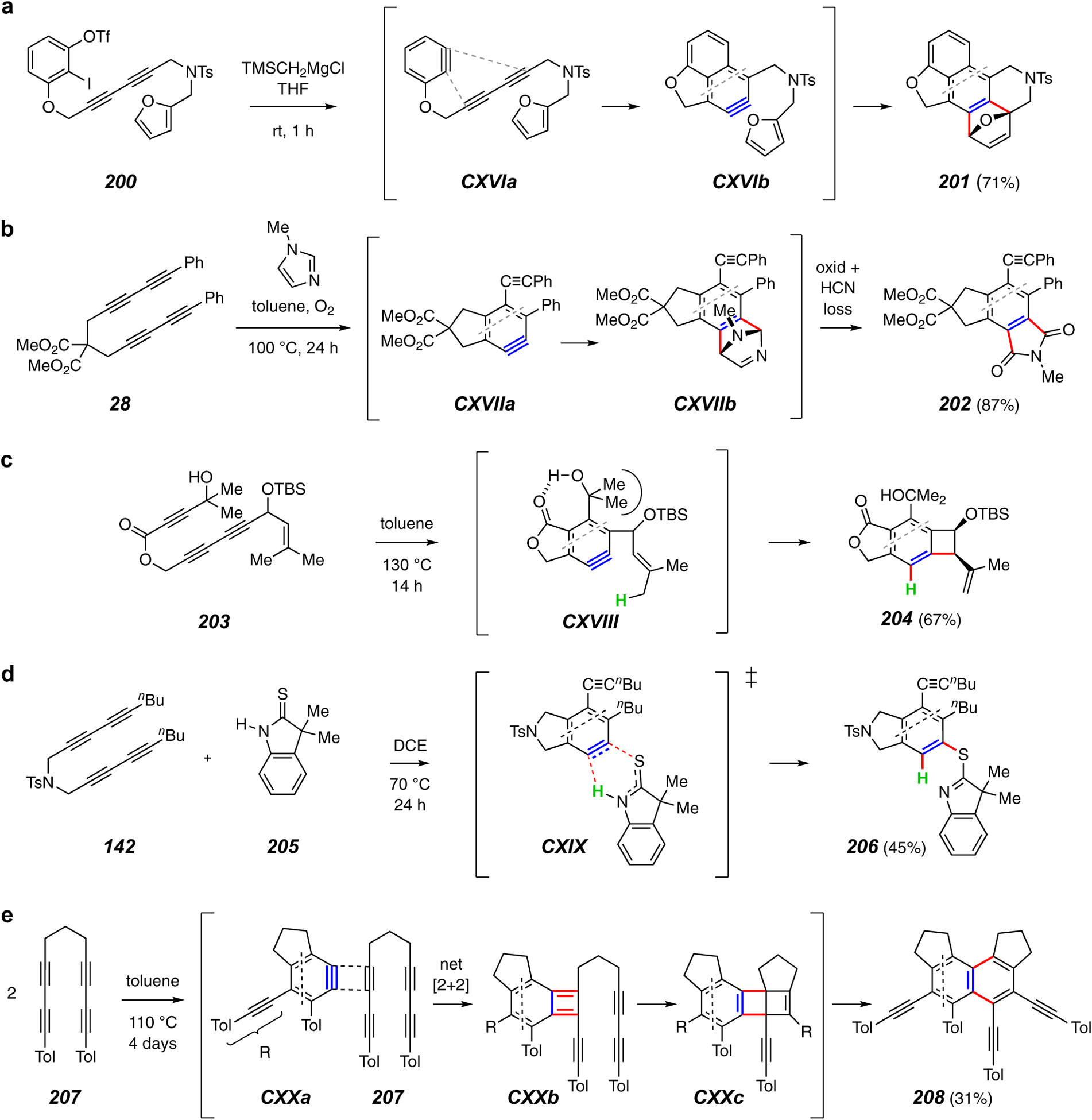
Yoshida, Hosoya, and coworkers devised the interesting cascade reaction shown in Figure 24a.118 This involves initial generation of the benzyne CXVIa from substrate 200, which then functions as the diynophile to engage the tethered diyne. The resulting naphthyne CXVIb is then trapped by the tethered furan to produce the polycyclic structure 201. Hu et al. have described trapping reactions with a series of N-substituted imidazoles (Figure 24b).119 For example, the benzyne CXVIIa from tetrayne 28 adds to 1-methylimidazole in the presence of oxygen to give the imide 202. The exact sequence of HCN extrusion and oxidation of the suggested initial Diels-Alder adduct CXVIIb was not delineated. Xia, Lee, and their coworkers reported the cyclization of 203 to give the strained benzocyclobutene derivative 204 (Figure 24c).120 This demonstrates how even subtle changes in structure can impact reaction outcomes. The alcohol-containing substrate 203 gave a higher yield of Alder-ene product than any of a series of similar substrates that lacked a hydroxyl group. The hydrogen bond in the benzyne CXVIII is argued to induce bond-angle strain, increasing the proximity of the alkene and benzyne in CXVIII. This is an example of a phenomenon that the purely thermal HDDA reaction rendered detectable; it likely would be invisible, if not inoperative, in the presence of the reagents present in virtually all of the methods for generating benzynes by classical methods.
In recent work, researchers in the Yao and Tan labs have described the reaction between HDDA benzynes and the thiolactam 205 (Figure 24d).121 For example, heating 142 with this oxindole analog gave the thioimino ether product 206 via a process that can be rationalized by the transition structure CXIX.
As mentioned earlier, it is often the case that if no trapping agent is present when an HDDA benzyne is formed, an intractable array of products is often formed. However, certain substrate polyynes are exceptional; instead they form structurally complex, unsymmetrical dimers (cf. Figure 5j).52 Researchers in the Engels and Marder groups have reported an interesting example of this dimerization process. As shown in Figure 24e, the tetrayne 207, when heated in toluene solution, gave the naphthalene derivative 210 as the major product.122 It is notable that the initial benzyne CXXa was trapped by Diels-Alder reaction with a toluene solvent molecule when the initial concentration of 207 was ca. 0.012 M. Increasing the [207]0 to 0.16 M allowed for a more competitive intervention of a second molecule of the tetrayne to proceed, via benzocyclobutadiene CXXb and Dewar naphthalene CXXc, to the dimer adduct 208. This study also demonstrates another feature common to a number of HDDA reactions. The identification of additional minor products (not shown here) allowed several other energetically viable pathways to be delineated. It is often the case that the practice of ‘digging through’ minor products, while of arguable preparative value, expands our awareness and understanding of entirely new mechanistic pathways.
Finally, capitalizing on the photo-HDDA60 reaction, investigators in the Marder, Mitric, and Brixner labs have recently reported their use of femtosecond transient absorption spectroscopy to provide the first direct evidence for the intermediacy of a benzyne in an HDDA reaction.123 A body of data from the excitation and decay of species produced by irradiation of a solution of substrate 207 was interpreted to support the existence of a steady state concentration of the benzyne CXXa in this fundamentally important study.
7. CONCLUSION
In conclusion, the hexadehydro-Diels-Alder reaction provides a valuable (and intriguing) route for the preparation of complex benzenoids by way of a benzyne intermediate. A wide variety of structurally diverse species are accessible due to the large number of tethers and trapping agents that are compatible with the purely thermal reaction conditions (i.e., in the absence of other reagents). Examples of new tethers and new trapping reagents or reactions will continue to emerge as increasing numbers of investigators contemplate the opportunities provided by this approach to forming benzynes by a protocol that is quite complementary to and distinct from other aryne-generating methods. Studies of HDDA reactions have also provided new mechanistic understanding and insights about benzyne reactivity, and many additional such results are embedded in the primary publications cited here. Finally, new avenues are also being opened by strategic developments such as the domino-, photochemical-, and aza-HDDA reactions, which adds further versatility to the synthetic utility. Future new strategic advances can also be anticipated.
ACKNOWLEDGEMENTS
Aspects of this research are supported in the corresponding author’s laboratory by grants from the Institute of General Medical Sciences of the United States Department of Health and Human Services (R35 GM127097) and the United States National Science Foundation (CHE-1665389).
Biographies
Thomas R. Hoye is currently a Distinguished Professor of Science and Engineering at the University of Minnesota (UMN). Dr. Hoye received his Bachelor of Science and Master of Science degrees in chemistry from Bucknell University in 1972 under the tutelage of Professor Harold W. Heine and the Ph.D. in chemistry from Harvard University in 1976, where he carried out his thesis research in the laboratory of Professor Robert B. Woodward. He is about to embark on his 45th year of teaching at the UMN, where he leads a research group engaged in studies of synthetic, mechanistic, structural, and spectroscopic organic, medicinal, natural product, and polymer chemistries.
Lucas Fluegel received his Bachelor of Science degree in 2020, having completed studies as a UMN Honors Program student majoring in chemistry with a minor in Spanish Studies. In the Fall of 2020 he was matriculated into the doctoral program in chemical and biological sciences at Scripps Research, Florida.
Footnotes
The authors declare no competing financial interests.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrev.9b00560
REFERENCES
- (1).Diels O; Alder K Synthesen in der hydroaromatischen Reihe. Justus Liebigs Ann. Chem 1928, 460, 98–122. [Google Scholar]
- (2).Wessig P; Müller G The dehydro-Diels-Alder reaction. Chem. Rev 2008, 108, 2051–2063. [DOI] [PubMed] [Google Scholar]
- (3).Johnson RP Dehydropericyclic routes to reactive intermediates. J. Phys. Org. Chem 2010, 23, 283–292. [Google Scholar]
- (4).Li W; Zhou L; Zhang J Recent progress in dehydro(genative) Diels–Alder reaction. Chem. Eur. J 2016, 22, 1558–1571. [DOI] [PubMed] [Google Scholar]
- (5).Baire B; Niu D; Willoughby PH; Woods BP Synthesis of complex benzenoids via the intermediate generation of o-benzynes through the hexadehydro-Diels-Alder reaction. Nature Protoc. 2013, 8, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Holden C; Greaney MF The hexadehydro-Diels-Alder reaction: a new chapter in aryne chemistry. Angew. Chem. Int. Ed 2014, 53, 5746–5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Diamond OJ; Marder TB Methodology and applications of the hexadehydro-Diels–Alder (HDDA) reaction. Org. Chem. Front 2017, 4, 891–910. [Google Scholar]
- (8).Miyawaki K; Suzuki R; Kawano T; Ueda I Cycloaromatization of a non-conjugated polyenyne system: synthesis of 5H-benzo[d]fluoreno[3,2-b]pyrans via diradicals generated from 1-[2-{4-(2-alkoxymethylphenyl)butan-1,3-dinyl}]phenylpentan-2,4-diyn −1-ols and trapping evidence for the 1,2-didehydrobenzene diradical. Tetrahedron Lett. 1997, 38, 3943–3946. [Google Scholar]
- (9).Bradley AZ; Johnson RP Thermolysis of 1,3,8-nonatriyne: evidence for intramolecular [2+4] cycloaromatization to a benzyne intermediate. J. Am. Chem. Soc 1997, 119, 9917–9918. [Google Scholar]
- (10).Tsui JA; Sterenberg BT A metal-templated 4 + 2 cycloaddition reaction of an alkyne and a diyne to form a 1,2-aryne. Organometallics 2009, 28, 4906–4908. [Google Scholar]
- (11).Miyawaki K; Kawano T; Ueda I Multiple cycloaromatization of novel aromatic enediynes bearing a triggering device on the terminal acetylene carbon. Tetrahedron Lett. 1998, 39, 6923–6926. [Google Scholar]
- (12).Ueda I; Sakurai Y; Kawano T; Wada Y; Futai M An unprecedented arylcarbene formation in thermal reaction of non-conjugated aromatic enetetraynes and DNA strand cleavage. Tetrahedron Lett. 1999, 40, 319–322. [Google Scholar]
- (13).Miyawaki K; Kawano T; Ueda I Domino thermal radical cycloaromatization of non-conjugated aromatic hexa- and heptaynes: synthesis of fluoranthene and benzo[a]rubicene skeletons. Tetrahedron Lett. 2000, 41, 1447–1451. [Google Scholar]
- (14).Miyawaki K; Ueno F; Ueda I Synthesis of indeno[1,2-b]phenanthrene-type heterocycles by cycloaromatization of acyclic non-conjugated benzotetraynes. Heterocycles 2001, 54, 887–900. [Google Scholar]
- (15).Miyawaki K; Kawano. T.; Ueda. I. Synthesis and properties of functionalized [6] helicenes by the thermal domino radical cycloaromatization of acyclic polyynes. Polycyclic Aromatic Compounds 2001, 19, 133–154. [Google Scholar]
- (16).Kawano T; Inai H; Miyawaki K; Ueda I Synthesis of indenothiophenone derivatives by cycloaromatization of non-conjugated thienyl tetraynes. Tetrahedron Lett. 2005, 46, 1233–1236. [Google Scholar]
- (17).Kawano T; Inai H; Miyawaki K; Ueda I Effect of water molecules on the cycloaromatization of non-conjugated aromatic tetraynes. Bull. Chem. Soc. Jpn 2006, 79, 944–949. [Google Scholar]
- (18).Torikai K; Otsuka Y; Nishimura M; Sumida M; Kawai T; Sekiguchi K; Ueda I Synthesis and DNA cleaving activity of water-soluble non-conjugated thienyl tetraynes. Bioorganic Med. Chem 2008, 16, 5441–5451. [DOI] [PubMed] [Google Scholar]
- (19).Nicolaou KC; Smith AL; Yue EW Chemistry and biology of natural and designed enediynes. Proc. Natl. Acad. Sci. USA 1993, 90, 5881–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hoye TR; Baire B; Niu D; Willoughby PH; Woods BP The hexadehydro-Diels-Alder reaction. Nature 2012, 490, 208–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Himeshima Y; Sonoda T; Kobayashi H Fluoride-induced 1,2-elimination of o-trimethylsilylphenyl triflate to benzyne under mild conditions. Chem. Lett 1983, 12, 1211–1214. [Google Scholar]
- (22).Woods BP; Baire B; Hoye TR Rates of hexadehydro-Diels-Alder (HDDA) cyclizations: impact of the linker structure. Org. Lett 2014, 16, 4578–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Karmakar R; Lee D Total synthesis of selaginpulvilin C and D relying on in situ formation of arynes and their hydrogenation. Org. Lett 2016, 18, 6105–6107. [DOI] [PubMed] [Google Scholar]
- (24).Wang T; Hoye TR Hexadehydro-Diels-Alder (HDDA)-enabled carbazolyne chemistry: single step, de novo construction of the pyranocarbazole core of alkaloids of the murraya koenigii (curry tree) family. J. Am. Chem. Soc 2016, 138, 13870–13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Karmakar R; Wang K-P; Yun SY; Mamidipalli P; Lee D Hydrohalogenative aromatization of multiynes promoted by ruthenium alkylidene complexes. Org. Biomol. Chem 2016, 14, 4782–4788. [DOI] [PubMed] [Google Scholar]
- (26).Wang T; Niu D; Hoye TR The hexadehydro-Diels-Alder cycloisomerization reaction proceeds by a stepwise mechanism. J. Am. Chem. Soc 2016, 138, 7832–7835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cahill KJ; Ajaz A; Johnson RP New thermal routes to ortho-benzyne. Aust. J. Chem 2010, 63, 1007–1012. [Google Scholar]
- (28).Ajaz A; Bradley AZ; Burrell RC; Li WHH; Daoust KJ; Bovee LB; Dirico KJ; Johnson RP Concerted vs stepwise mechanisms in dehydro-Diels-Alder reactions. J. Org. Chem 2011, 76, 9320–9328. [DOI] [PubMed] [Google Scholar]
- (29).Skraba-Joiner SL; Johnson RP; Agarwal J Dehydropericyclic reactions: symmetry-controlled routes to strained reactive intermediates. J. Org. Chem 2015, 80, 11779–11787. [DOI] [PubMed] [Google Scholar]
- (30).Marell DJ; Furan LR; Woods BP; Lei X; Bendelsmith AJ; Cramer CJ; Hoye TR; Kuwata KT Mechanism of the intramolecular hexadehydro-Diels-Alder reaction. J. Org. Chem 2015, 80, 11744–11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yu P; Yang Z; Liang Y; Hong X; Li Y; Houk KN Distortion-controlled reactivity and molecular dynamics of dehydro-Diels-Alder reactions. J. Am. Chem. Soc 2016, 138, 8247–8252. [DOI] [PubMed] [Google Scholar]
- (32).Liang Y; Hong X; Yu P; Houk KN Why alkynyl substituents dramatically accelerate hexadehydro-Diels-Alder (HDDA) reactions: stepwise mechanisms of HDDA cycloadditions. Org. Lett 2014, 16, 5702–5705. [DOI] [PubMed] [Google Scholar]
- (33).Chen M; He CQ; Houk KN Mechanism and regioselectivity of an unsymmetrical hexadehydro-Diels-Alder (HDDA) reaction. J. Org. Chem 2019, 84, 1959–1963. [DOI] [PubMed] [Google Scholar]
- (34).Ghorai S; Lee D Aryne-based multicomponent coupling reactions. Synlett 2020, 31, 750–771. [Google Scholar]
- (35).Luu Nguyen Q; Baire B; Hoye TR Competition between classical and hexadehydro-Diels-Alder (HDDA) reactions of HDDA triynes with furan. Tetrahedron Lett. 2015, 56, 3265–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wertjes WC; Southgate EH; Sarlah D Recent advances in chemical dearomatization of nonactivated arenes. Chem. Soc. Rev 2018, 47, 7996–8017. [DOI] [PubMed] [Google Scholar]
- (37).Pogula VD; Wang T; Hoye TR Intramolecular [4 + 2] trapping of a hexadehydro-Diels-Alder (HDDA) benzyne by tethered arenes. Org. Lett 2015, 17, 856–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang Y; Hoye TR Intramolecular capture of HDDA-Derived benzynes: (i) 6- to 12-membered ring formation, (ii) internally (vis-á-vis remotely) tethered traps, and (iii) role of the rate of trapping by the benzynophile. Org. Lett 2018, 20, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Karmakar R; Le A; Xie P; Xia Y; Lee D Reactivity of arynes for arene dearomatization. Org. Lett 2018, 20, 4168–4172. [DOI] [PubMed] [Google Scholar]
- (40).Mitake A; Nagai R; Sekine A; Takano H; Sugimura N; Kanyiva KS; Shibata T Consecutive HDDA and TDDA reactions of silicon-tethered tetraynes for the synthesis of dibenzosilole-fused polycyclic compounds and their unique reactivity. Chem. Sci 2019, 10, 6715–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chen J; Baire B; Hoye TR Cycloaddition reactions of azide, furan, and pyrrole units with benzynes generated by the hexadehydro-Diels-Alder (HDDA) reaction. Heterocycles 2014, 88, 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang M-X; Shan W; Chen Z; Yin J; Yu G-A; Liu SH Diels-Alder Reactions of Arynes in Situ Generated from DA Reaction between Bis-1,3-Diynes and Alkynes. Tetrahedron Lett 2015, 56, 6833–6838. [Google Scholar]
- (43).Liu B; Hu Q; Yang F; Zheng X; Hu Y Fused multifunctionalized bridge aromatic hydrocarbons from in situ-generated arynes and anthracene derivatives. Chinese Chem. Lett 2020, 31, 1305–1308. [Google Scholar]
- (44).Meng X; Cheng D; Lv S; Ma J; Hu Y Synthesis of a type of bridged heterocycles via tetraynes. Heterocycles 2017, 94, 2111–2124. [Google Scholar]
- (45).Xu F; Hershey KW; Holmes RJ; Hoye TR Blue-emitting arylalkynyl naphthalene derivatives via a hexadehydro-Diels-Alder cascade reaction. J. Am. Chem. Soc 2016, 138, 12739–12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Xu F; Xiao X; Hoye TR Reactions of HDDA-derived benzynes with perylenes: rapid construction of polycyclic aromatic compounds. Org. Lett 2016, 18, 5636–5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ghorai S; Lin Y; Xia Y; Wink DJ; Lee D Silver-catalyzed selective multicomponent coupling reactions of arynes with nitriles and isonitriles. Org. Lett 2020, 22, 642–647. [DOI] [PubMed] [Google Scholar]
- (48).Liu B; Mao C; Hu Q; Yao L; Hu Y Direct methylation and carbonylation of in situ generated arynes via HDDA-Wittig coupling. Org. Chem. Front 2019, 6, 2788–2791. [Google Scholar]
- (49).Hamura T; Ibusuki Y; Sato K; Matsumoto T; Osamura Y; Suzuki K Strain-induced regioselectivities in reactions of benzyne possessing a fused four-membered ring. Org. Lett 2003, 5, 3551–3554. [DOI] [PubMed] [Google Scholar]
- (50).Garr AN; Luo D; Brown N; Cramer CJ; Buszek KR; VanderVelde D Experimental and theoretical investigations into the unusual regioselectivity of 4,5-, 5,6-, and 6,7-indole aryne cycloadditions. Org. Lett 2010, 12, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cheong PH-Y; Paton RS; Bronner SM; Im G-YJ; Garg NK; Houk KN Indolyne and aryne distortions and nucleophilic regioselectivites. J. Am. Chem. Soc 2010, 132, 1267–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Xiao X; Woods BP; Xiu W; Hoye TR Benzocyclobutadienes: an unusual mode of access reveals unusual modes of reactivity. Angew. Chem. Int. Ed 2018, 57, 9901–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Nobusue S; Yamane H; Miyoshi H; Tobe Y [4.2](2,2’)(2,2’)Biphenylophanetriyne: a twisted biphenylophane with a highly distorted diacetylene bridge. Org. Lett 2014, 16, 1940–1943. [DOI] [PubMed] [Google Scholar]
- (54).Heaney H; Jablonski JM Reactions of arynes in the synthesis of 2H-chromens. Chem. Commun 1968, 1139–1139. [Google Scholar]
- (55).Meng X; Lv S; Cheng D; Hu Q; Ma J; Liu B; Hu Y Fused multifunctionalized chromenes from tetraynes and α,β-unsaturated aldehydes. Chem. - A Eur. J 2017, 23, 6264–6271. [DOI] [PubMed] [Google Scholar]
- (56).Wang T; Oswood CJ; Hoye TR Trapping of hexadehydro-Diels-Alder benzynes with exocyclic, conjugated enals as a route to fused spirocyclic benzopyran motifs. Synlett 2017, 28, 2933–2935. [Google Scholar]
- (57).Hu Y; Hu Y; Hu Q; Ma J; Lv S; Liu B; Wang S Direct access to fused salicylaldehydes and salicylketones from tetraynes. Chem. - A Eur. J 2017, 23, 4065–4072. [DOI] [PubMed] [Google Scholar]
- (58).Ross SP; Hoye TR Reactions of hexadehydro-Diels-Alder benzynes with structurally complex multifunctional natural products. Nature Chem. 2017, 9, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Arora S; Zhang J; Pogula V; Hoye TR Reactions of Thermally generated benzynes with six-membered N-heteroaromatics: pathway and product diversity. Chem. Sci 2019, 10, 9069–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Xu F; Xiao X; Hoye TR Photochemical hexadehydro-Diels-Alder reaction. J. Am. Chem. Soc 2017, 139, 8400–8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Arora S; Sneddon DS; Hoye TR Reactions of HDDA benzynes with C,N-diarylimines (ArCH=NAr’). Eur. J. Org. Chem 2020, 2379–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ghorai S; Lin Y; Xia Y; Wink DJ; Lee D Silver-catalyzed annulation of arynes with nitriles for synthesis of structurally diverse quinazolines. Org. Lett 2020, 22, 626–630. [DOI] [PubMed] [Google Scholar]
- (63).Zheng X; Liu B; Yang F; Hu Q; Yao L; Hu Y Access to benzoxazepines and fully substituted indoles via HDDA coupling. Org. Lett 2020, 22, 956–959. [DOI] [PubMed] [Google Scholar]
- (64).Niu D; Hoye TR The aromatic ene reaction. Nature Chem 2014, 6, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Karmakar R; Mamidipalli P; Yun SY; Lee D Alder-ene reactions of arynes. Org. Lett 2013, 15, 1938–1941. [DOI] [PubMed] [Google Scholar]
- (66).Karmakar R Lee, D. Reactions of arynes promoted by silver ion. Chem. Soc. Rev 2016, 45, 4459–4470. [DOI] [PubMed] [Google Scholar]
- (67).Ghorai S; Lee D Silver-catalyzed cycloaddition reactions. In Silver Catalysis in Organic Synthesis; Li C-J, Bi X, Eds.; Wiley-VCH,: New York, 2018, pp 33–83. [Google Scholar]
- (68).Lee N-K; Yun SY; Mamidipalli P; Salzman RM; Lee D; Zhou T; Xia Y Hydroarylation of arynes catalyzed by silver for biaryl synthesis. J. Am. Chem. Soc 2014, 136, 4363–4368. [DOI] [PubMed] [Google Scholar]
- (69).Yun SY; Wang K-P; Lee N-K; Mamidipalli P; Lee D Alkane C-H insertion by aryne intermediates with a silver catalyst. J. Am. Chem. Soc 2013, 135, 4668–4671. [DOI] [PubMed] [Google Scholar]
- (70).Mamidipalli P; Yun SY; Wang K-P; Zhou T; Xia Y; Lee D Formal hydrogenation of arynes with silyl Cβ-H bonds as an active hydride source. Chem. Sci 2014, 5, 2362–2367. [Google Scholar]
- (71).Shen H; Xiao X; Haj MK; Willoughby PH; Hoye TR BF3-Promoted, carbene-like, C-H insertion reactions of benzynes. J. Am. Chem. Soc 2018, 140, 15616–15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Gupta S; Xie P; Xia Y; Lee D Reactivity and selectivity in the intermolecular Alder-ene reactions of arynes with functionalized alkenes. Org. Lett 2017, 19, 5162–5165. [DOI] [PubMed] [Google Scholar]
- (73).Gupta S; Xie P; Xia Y; Lee D Reactivity of arynes toward functionalized alkenes: intermolecular alder-ene vs. addition reactions. Org. Chem. Front 2018, 5, 2208–2213. [Google Scholar]
- (74).Zhang J; Niu D; Brinker VA; Hoye TR The phenol-ene reaction: Biaryl synthesis via trapping reactions between HDDA-generated benzynes and phenolics. Org. Lett 2016, 18, 5596–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Wang K-P; Yun SY; Mamidipalli P; Lee D Silver-mediated fluorination, trifluoromethylation, and trifluoromethylthiolation of arynes. Chem. Sci 2013, 4, 3205–3211. [Google Scholar]
- (76).Ghorai S; Lee D Synthesis of imides, imidates, amidines, and amides by intercepting the aryne-isocyanide adduct with weak nucleophiles. Org. Lett 2019, 21, 7390–7393. [DOI] [PubMed] [Google Scholar]
- (77).Xiao X; Wang T; Xu F; Hoye TR CuI-mediated bromoalkynylation and hydroalkynylation reactions of unsymmetrical benzynes: complementary modes of addition. Angew. Chem. Int. Ed 2018, 57, 16564–16568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Kerisit N; Toupet L; Larini P; Perrin L; Guillemin J-C; Trolez Y Straightforward synthesis of 5-bromopenta-2,4-diynenitrile and its reactivity towards terminal alkynes: a direct access to diene and benzofulvene scaffolds. Chem. - A Eur. J 2015, 21, 6042–6047. [DOI] [PubMed] [Google Scholar]
- (79).Palani V; Chen J; Hoye TR Reactions of hexadehydro-Diels-Alder (HDDA)-derived benzynes with thioamides: synthesis of dihydrobenzothiazino-heterocyclics. Org. Lett 2016, 18, 6312–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Hu Y; Ma J; Li L; Hu Q; Lv S; Liu B; Wang S Fused multifunctionalized dibenzoselenophenes from tetraynes. Chem. Commun 2017, 53, 1542–1545. [DOI] [PubMed] [Google Scholar]
- (81).Liu B; Hu Q; Wen Y; Fang B; Xu X; Hu Y Versatile dibenzothio[seleno]phenes via hexadehydro-Diels-Alder domino cyclization. Front. Chem 2019, 7, Article 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Woods BP; Hoye TR Differential scanning calorimetry (DSC) as a tool for probing the reactivity of polyynes relevant to hexadehydro-Diels-Alder (HDDA) cascades. Org. Lett 2014, 16, 6370–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Hoye TR; Baire B; Wang T Tactics for probing aryne reactivity: mechanistic studies of silicon-oxygen bond cleavage during the trapping of (HDDA-generated) benzynes by silyl ethers. Chem. Sci 2014, 5, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Shen H; Xiao X; Hoye TR Benzyne cascade reactions via benzoxetenonium ions and their rearrangements to o-quinone methides. Org. Lett 2019, 21, 1672–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Vandavasi JK; Hu W-P; Hsiao C-T; Senadi GC; Wang J-J A new approach for fused isoindolines via hexadehydro-Diels-Alder reaction (HDDA) by Fe(0) catalysis. RSC Adv. 2014, 4, 57547–57552. [Google Scholar]
- (86).Kimura H; Torikai K; Miyawaki K; Ueda I Scope of the thermal cyclization of nonconjugated ene-yne-nitrile system: a facile synthesis of cyanofluorenol derivatives. Chem. Lett 2008, 37, 662–663. [Google Scholar]
- (87).Smela MP; Hoye TR A traceless tether strategy for achieving formal intermolecular hexadehydro-Diels-Alder reactions. Org. Lett 2018, 20, 5502–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Willoughby PH; Niu D; Wang T; Haj MK; Cramer CJ; Hoye TR Mechanism of the reactions of alcohols with o-benzynes. J. Am. Chem. Soc 2014, 136, 13657–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Karmakar R; Ghorai S; Xia Y; Lee D Synthesis of phenolic compounds by trapping arynes with a hydroxy surrogate. Molecules 2015, 20, 15862–15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Karmakar R; Yun SY; Wang K-P; Lee D Regioselectivity in the nucleophile trapping of arynes: the electronic and steric effects of nucleophiles and substituents. Org. Lett 2014, 16, 6–9. [DOI] [PubMed] [Google Scholar]
- (91).Zhang J; Hoye TR Divergent reactivity during the trapping of benzynes by glycidol analogs: ring cleavage via pinacol-like rearrangements vs oxirane fragmentations. Org. Lett 2019, 21, 2615–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).E.g., Shi F; Waldo JP; Chen Y; Larock RC Benzyne click chemistry: synthesis of benzotriazoles from benzynes and azides. Org. Lett 2008, 10, 2409–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Wang Y; Zheng L; Hoye TR Sulfonamide-trapping reactions of thermally generated benzynes. Org. Lett 2018, 20, 7145–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Zhang J; Page ACS; Palani V; Chen J; Hoye TR Atypical mode of [3 + 2]-cycloaddition: pseudo-1,3-dipole behavior in reactions of electron-deficient thioamides with benzynes. Org. Lett 2018, 20, 5550–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Ikawa T; Nishiyama T; Shigeta T; Mohri S; Morita S; Takayanagi S; Terauchi Y; Morikawa Y; Takagi A; Ishikawa Y; et al. ortho-Selective nucleophilic addition of primary amines to silylbenzynes: synthesis of 2-silylanilines. Angew. Chem. Int. Ed 2011, 50, 5674–5677. [DOI] [PubMed] [Google Scholar]
- (96).Bronner SM; Mackey JL; Houk KN; Garg NK Steric effects compete with aryne distortion to control regioselectivities of nucleophilic additions to 3-silylarynes. J. Am. Chem. Soc 2012, 134, 13966–13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Ikawa T; Takagi A; Goto M Aoyama, Y.; Ishikawa, Y.; Itoh, Y.; Fujii, S.; Tokiwa, H.; Akai, S. Regiocomplementary cycloaddition reactions of boryl- and silylbenzynes with 1,3-dipoles: selective synthesis of benzo-fused azole derivatives. J. Org. Chem 2013, 78, 2965–2983. [DOI] [PubMed] [Google Scholar]
- (98).Medina JM; Mackey JL; Garg NK; Houk KNJ The role of aryne distortions, steric effects, and charges in regioselectivities of aryne reactions J. Am. Chem. Soc 2014, 136, 15798–15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Ross SP; Baire B; Hoye TR Mechanistic duality in tertiary amine additions to thermally generated hexadehydro-diels-alder benzynes. Org. Lett 2017, 19, 5705–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Tasker SZ; Cowfer AE; Hergenrother PJ Preparation of structurally diverse compounds from the natural product lycorine. Org. Lett 2018, 20, 5894–5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Ghorai S; Lee D Aryne formation via the hexadehydro Diels-Alder reaction and their Ritter-type transformations catalyzed by a cationic silver complex. Tetrahedron 2017, 73, 4062–4069. [Google Scholar]
- (102).Ross SP; Hoye TR Multiheterocyclic motifs via three-component reactions of benzynes, cyclic amines, and protic nucleophiles. Org. Lett 2018, 20, 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Arora S; Palani V; Hoye TR Reactions of diaziridines with benzynes give N-arylhydrazones. Org. Lett 2018, 20, 8082–8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Niu D; Willoughby PH; Woods BP; Baire B; Hoye TR Alkane desaturation by concerted double hydrogen atom transfer to benzyne. Nature 2013, 501, 531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Zhang H; Hu Q; Li L; Hu YY; Zhou P; Zhang X; Xie H; Yin F; Hu YY; Wang S Convenient one-step construction of yne-functionalized aryl halides through domino cyclization from tetraynes. Chem. Commun 2014, 50, 3335–3337. [DOI] [PubMed] [Google Scholar]
- (106).Chen J; Palani V; Hoye TR Reactions of HDDA-derived benzynes with sulfides: Mechanism, modes, and three-component reactions. J. Am. Chem. Soc 2016, 138, 4318–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Niu D; Wang T; Woods BP; Hoye TR Dichlorination of (hexadehydro-Diels-Alder generated) benzynes and a protocol for interrogating the kinetic order of bimolecular aryne trapping reactions. Org. Lett 2014, 16, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Watanabe T; Curran DP; Taniguchi T Hydroboration of arynes formed by hexadehydro-Diels-Alder cyclizations with N-heterocyclic carbene boranes. Org. Lett 2015, 17, 3450–3453. [DOI] [PubMed] [Google Scholar]
- (109).Okugawa Y; Hayashi Y; Kawauchi S; Hirano K; Miura M Diphosphination of arynes with diphosphines. Org. Lett 2018, 20, 3670–3673. [DOI] [PubMed] [Google Scholar]
- (110).Xiao X; Hoye TR The domino hexadehydro-Diels-Alder Reaction transforms polyynes to benzynes to naphthynes to anthracynes to tetracynes (and beyond?). Nature Chem. 2018, 10, 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Xiao X; Hoye TR One-pot, three-aryne cascade strategy for naphthalene formation from 1,3-diynes and 1,2-benzdiyne equivalents. J. Am. Chem. Soc 2019, 141, 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Shi J; Li Y; Li Y Aryne multifunctionalization with benzdiyne and benztriyne equivalents. Chem. Soc. Rev 2017, 46, 1707–1719. [DOI] [PubMed] [Google Scholar]
- (113).Thompson SK; Hoye TR The aza-hexadehydro-Diels-Alder reaction. J. Am. Chem. Soc 2019, 141, 19575–19580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Wang T; Naredla RR; Thompson SK; Hoye TR The pentadehydro-Diels-Alder reaction. Nature 2016, 532, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Logan CF; Ma JC; Chen P The photoelectron spectrum of the α,3-dehydrotoluene biradical. J. Am. Chem. Soc 1994, 116, 2137–2138. [Google Scholar]
- (116).Myers AG; Kuo EY; Finney NS Thermal generation of α,3-dehydrotoluene from (Z)-1,2,4-heptatrien-6-yne. J. Am. Chem. Soc 1989, 111, 8057–8059. [Google Scholar]
- (117).Sieck C; Tay MG; Thibault M-H; Edkins RM; Costuas K; Halet J-F; Batsanov AS; Haehnel M; Edkins K; Lorbach A; et al. Reductive coupling of diynes at rhodium gives fluorescent rhodacyclopentadienes or phosphorescent rhodium 2,2’-biphenyl complexes. Chem. Eur. J 2016, 22, 10523–10532. [DOI] [PubMed] [Google Scholar]
- (118).Yoshida S; Shimizu K; Uchida K; Hazama Y; Igawa K; Tomooka K; Hosoya T Construction of condensed polycyclic aromatic frameworks through intramolecular cycloaddition reactions involving arynes bearing an internal alkyne moiety. Chem. Eur. J 2017, 23, 15332–15335. [DOI] [PubMed] [Google Scholar]
- (119).Hu Q; Li L; Yin F; Zhang H; Hu Y; Liu B; Hu Y Fused multifunctionalized isoindole-1,3-diones via the coupled oxidation of imidazoles and tetraynes. RSC Adv. 2017, 7, 49810–49816. [Google Scholar]
- (120).Gupta S; Lin Y; Xia Y; Wink DJ; Lee D Alder-ene reactions driven by high steric strain and bond angle distortion to form benzocyclobutenes. Chem. Sci 2019, 10, 2212–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Saputra A; Fan F; Yao T; Chen J; Tan J Synthesis of 2-(arylthio)indolenines via chemoselective arylation of thio-oxindoles with arynes. Adv. Synth. Catal 2020, 362, 2683–2688. [Google Scholar]
- (122).Maier J; Deutsch M; Merz J; Ye Q; Diamond O; Schilling M-T; Friedrich A; Engels B; Marder TB Highly conjugated π-systems arising from cannibalistic hexadehydro-Diels-Alder couplings: cleavage of C-C single and triple bonds. Chem. Eur. J 2020, Early View. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Ma X; Maier J; Wenzel M; Friedrich A; Steffen A; Marder TB; Mitric R; Brixner T Direct observation of o-benzyne formation in photochemical hexadehydro-Diels–Alder (hv-HDDA) reactions. Chem. Sci 2020, 11, 9198–9208. [DOI] [PMC free article] [PubMed] [Google Scholar]