

Abstract
Lymphomas with central nervous system (CNS) involvement confer a worse prognosis than those without CNS involvement, and patients currently have limited treatment options. T cells genetically engineered with CD19-targeted chimeric antigen receptors (CAR) are effective against B-cell malignancies and show tremendous potential in the treatment of systemic lymphoma. We aimed to leverage this strategy toward a more effective therapy for patients with lymphoma with CNS disease. NOD-scid IL2Rgammanull (NSG) mice with CNS and/or systemic lymphoma were treated with CD19-CAR T cells via intracerebroventricular (ICV) or intravenous (IV) injection. CAR T cells isolated post-treatment were rigorously examined for phenotype, gene expression, and function. We observed that CAR T cells infused ICV, but not IV, completely and durably eradicated both CNS and systemic lymphoma. CAR T cells delivered ICV migrated efficiently to the periphery, homed to systemic tumors, and expanded in vivo, leading to complete elimination of disease and resistance to tumor re-challenge. Mechanistic studies indicated that ICV-delivered CAR T cells are conditioned by exposure to cerebrospinal fluid in the ICV environment for superior anti-lymphoma activity and memory function compared with IV-delivered CAR T cells. Further analysis suggested that manipulating cellular metabolism or pre-activating therapeutic CAR T cells with antigen ex vivo may improve the efficacy of CAR T cells in vivo. Our demonstration that ICV-delivered CD19-CAR T cells had activity against CNS and systemic lymphoma could offer a valuable new strategy for treatment of B-cell malignancies with CNS involvement.
Keywords: CAR T cells, CNS lymphoma, chimeric antigen receptor, cerebroventricular, adoptive cellular therapy
Introduction
Primary central nervous system (CNS) lymphoma represents 2%–3% of all primary brain tumors and 1–2% of all lymphoma diagnoses (1). Systemic lymphoma with secondary CNS involvement occurs in 10–30% of patients with diffuse large B cell lymphoma (2). Surgery is not usually an option to treat primary or secondary CNS lymphoma due to the tendency of CNS lymphoma to be near critical structures and multi-centric. Although new therapeutic approaches have improved survival, the disease still poses a challenge to manage and is associated with a poor prognosis (3–5). Thus, it is critical to identify novel therapeutic modalities to treat CNS lymphoma.
Chimeric antigen receptor (CAR) T cells targeting CD19 have produced tremendous clinical success in treating several B cell lineage malignancies, but thus far have not been used specifically to treat CNS lymphoma. In all CD19-CAR T cell trials to date, T-cell products have been administered intravenously (IV). CD19-CAR T cells can traffic to the cerebrospinal fluid (CSF) following systemic administration (6–9), where they may mediate activity against CNS lymphoma. Although neurotoxicity is a common and potentially life-threatening toxicity of CD19-CAR T cells, there is no evidence indicating that the presence of CAR T cells in CSF is directly related to neurotoxicity (10). Therefore, the goals of this project were to identify ways to limit the toxicity that results from systemic infusion of high doses of CAR T cells (11–13) and to improve activity against CNS lymphoma.
Recent studies show that regional intraventricular (ICV) delivery of rituximab for CNS lymphoma is well-tolerated (14–16). In addition, ICV delivery of CAR T cells is safe and effective in patients with glioblastoma and in preclinical animal studies of glioblastoma and brain-metastatic solid tumors (17–19). Therefore, we hypothesized that ICV delivery of CD19-CAR T cells could hold promise as an effective therapeutic approach for both primary CNS lymphoma and systemic lymphoma with CNS involvement. Here we have demonstrated that a single dose of ICV-delivered CD19-CAR T cells completely eradicated both CNS and systemic lymphoma in a NOD-scid IL2Rgammanull (NSG) mouse model. Further, our mechanistic studies established that CD19-CAR T cells exposed to CSF in the ICV environment exhibited memory features and enhanced anti-lymphoma activity when compared with systemically administered CD19-CAR T cells.
Materials and Methods
Cell Lines
Daudi (Burkitt lymphoma) and Jeko (mantle cell lymphoma) cells were obtained in 2017 from ATCC and maintained in RPMI 1640 (Irvine Scientific) supplemented with 10% heat-inactivated FCS (Hyclone), 2mM L-glutamine (Irvine Scientific), and 25mM HEPES (Irvine Scientific) (complete medium). The cells were passaged twice a week up to 10 passage after thawing. Both cell lines are reauthenticated after thawing by CD19 positive expression analyzed with flow cytometry after staining with antibody against CD19 (Catalog No IM1285U; Beckman Coulter,). Mycoplasma testing was done regularly in house using the Myco alert Mycoplasma detection kit (Lonza, LT07–318). To generate tumor cell lines expressing enhanced green fluorescent protein (EGFP) and firefly luciferase (ffluc), Daudi/Jeko cells were transduced with a lentiviral vector encoding EGFP-ffluc at a multiplicity of infection (MOI) of 3 in complete medium. After expansion in complete medium for 14 days, GFP+ cells were sorted by FACS for >98% purity and expanded for in vivo experiments. OKT3–2A-Hygro_pEK (gift from Andrew Raubitschek, City of Hope National Medical Center) contains the anti-human-CD3ϵ immunoglobulin gene, the 2A peptide sequence, and the hygromycin resistance gene within the pEK vector (20). EBV-transformed LCLs were made from peripheral blood mononuclear cells (PBMCs) as previously described (21). OKT3-expressing LCL cells in experiments using the rapid expansion method (REM) (22) were generated by electroporating allogeneic LCLs with OKT3–2A-Hygromycin_pEK plasmid using the Amaxa Nucleofector I (Lonza), program T-20 according to the manufacturer’s instructions. Resulting cells were grown and passaged for 2 months in complete medium supplemented with 0.4mg/mL hygromycin (Stratagene).
Antibodies and Flow Cytometry
Fluorochrome-conjugated antibodies against CD3 (clone Leu-4), CD4 (clone SK3), CD8 (clone RPA-T8), CD25 (clone M-A251), CD107a (clone H4A3), CD28 (clone CD28.2), CD62L (clone SK11), CD127 (clone HIL-7R-M21), CD161 (clone DX12), and streptavidin-PE (Catalog No. 554061) were obtained from BD Biosciences. Antibodies against KLRG (clone 2F1), Tim3 (clone F38–2E2), and EGFR (clone AY13) were purchased from BioLegend. The antibody against LAG3 (Catalog No. LS-C130398) was from Lifespan Biosciences. Biotinylated cetuximab (Erbitux) was generated from Erbitux purchased from the City of Hope pharmacy. Briefly, 200 mg Erbitux was buffer exchanged to PBS (D-PBS, pH 7.5 ± 0.1) using a MidGee Hoop Cartridge (UFP-30-E-H42LA). The material (2 mg/mL) was modified with Sulfo-NHS-LC-biotin (20:1) in a 1-hour room temperature reaction and diafiltered to remove excess biotin. The biotinylated Erbitux was then buffer exchanged to PBS and frozen in 20% glycerol. Product purity was confirmed on NuPAGE Novex Bis-Tris gels with or without SDS reduction. For cell-surface phenotyping experiments, cells were stained with optimized antibody panels for 20 minutes at 4°C followed by two washes with PBS. IOTestBeta Mark TCR Vβ Repertoire Kits were obtained from Beckman Coulter and analysis was performed according to the kit instructions. Data acquisition for all experiments involving flow cytometry was performed on a MACSquant (Miltenyi) and analyzed using FCS Express Version 3 software.
Lentivirus Vector Construction and CAR T cell generation
The CD19-CAR:CD28:ζ/EGFRt-epHIV7 (23) contains: 1) VH and VL gene segments of the CD19-specific FMC63 monoclonal antibody (mAb), IgG4 hinge with mutations at two sites (L235E; N297Q), CD28 costimulatory molecule, and CD3ζ chain; 2) the ribosomal skip T2A and truncated human EGFR (EGFRt) sequence (20) as depicted in Supplemental Figure 1. The BAFF-R-CAR:CD28:ζ/EGFRt-epHIV7 (24) contains: 1) BAFFR-specific VH and VL gene segments, IgG4 hinge with mutations at two sites (L235E; N297Q), CD28 costimulatory molecule, and CD3ζ chain; 2) the ribosomal skip T2A and truncated human EGFR (EGFRt) sequence (20). Leukapheresis products for CAR T–cell manufacturing were obtained from healthy donors using protocols approved by the City of Hope Institutional Review Board, which allows access to de-identified discard kits containing 50–100mL of leukapheresis product that was leftover in the tubing/filters from leukapheresis procedures on normal healthy donors. For experiments in Figures 1–4, a different healthy donor leukapheresis product was used for each experiment. Naïve and central memory T cells (Tn/mem) defined as CD62L+CD14- were isolated on a Miltenyi AutoMACS (Miltenyi Biotec, Inc.). Briefly, peripheral blood mononuclear cells were incubated with anti-CD14 microbeads (Catalog No. 130-019-101; Miltenyi Biotec, Inc.) for 30 minutes at room temperature and CD14+ cells were immediately depleted using the AutoMACS. The unlabeled negative cellular fraction was labeled with the CD62L-specific mAb (clone DREG-56; BD Biosciences) that was biotinylated as previously described (City of Hope Center for Biomedicine and Genetics), followed by incubation with anti-biotin microbeads for 30 minutes at room temperature. CD62L+ cells were purified with positive selection on the AutoMACS. Following selection, isolated Tn/mem cells were activated, transduced, and maintained as previously described (25–27) and non-transduced T cells were used as control (mock) T cells. Where indicated, CAR T cells were cultured in human CSF (Lee Biosolutions) or modified (60mg/dL glucose; 2.8 mEq/L potassium) RPMI (Invitrogen) for 48 hours at 37°C. To generate EGFP-ffluc–expressing CD19-CAR T cells, T cells were activated and co-transduced with CD19-CAR lentiviral vector at MOI 1 and EGFP-ffluc lentiviral vector at MOI 2. EGFR+ cells were purified with EGFR EasySep (Catalog No. 18309; StemCell Technologies, Inc.) following the manufacturer’s instructions, resulting in 60–70%GFP+EGFR+ double positive cells. To prime CAR T cells in vitro, 2×106 CD19-CAR T cells were co-cultured with irradiated (8000rads) Daudi cells at 10:1 (CAR:tumor) ratio in a 24-well plate for 1 hour at 37°C prior to infusion into mice.
Orthotopic brain and systemic lymphoma xenograft model
All animal experiments were performed under protocols approved by the City of Hope Institutional Animal Care and Use Committee using mice that were bred at our institutional animal facility. Six- to ten-week old NOD/Scid IL2RγCnull mice were injected intracranially and/or subcutaneously with EGFP-ffluc-Daudi or -Jeko cells. Mice were treated with CAR T cells or non-transduced mock cells by ICV (19,28) or IV infusion. Anesthetized mice were imaged using a Xenogen IVIS 100 series system (Xenogen). Photons from ffLuc+ tumor xenografts were quantified using the software program Living Image (Xenogen), and the bioluminescence signal was measured as total photon flux normalized for exposure time and surface area, expressed in units of photons per second per cm2 per steradian.
Reverse transcriptase (RT) quantitative PCR (qPCR) and single cell RNA-seq analysis
CAR T cells were cultured in RPMI or human CSF for 48 hours and RNA was extracted with RNeasy Kits (Qiagen) according to the manufacturer’s instructions. RT-qPCR was performed using the RT2 Profiler PCR Array (SABiosciences) designed with 48 customized genes involved with CAR T cell differentiation, survival, metabolism, and apoptosis (29,30) (Supplemental Table 1) and commercial primers provided by SABiosciences on CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Four control genes were used including housekeeping genes PPIA (Peptidylprolyl isomerase A) and RPL13A (Ribosomal protein L13a), control for reverse transcription, and positive PCR control. All samples passed the RNA quality control, PCR array reproducibility, RT efficiency, and genomic DNA contamination according to the manufacturer’s instructions. Data from duplicates were normalized by PPIA and analyzed by ΔΔCt method using RT2 Profiler PCR Array Web portal software (http://www.sabiosciences.com). Hierarchical clustering analysis was performed in Cluster 3.0 using the average linkage clustering method, with genes demonstrating an absolute fold change >1.3. Results were visualized in Treeview 1.1.6r4.
For single cell RNA-seq analysis, cells harvested from bone marrow of mice treated with CAR T cells were resuspended at 700–1200 cells/μl in 0.04% BSA (Sigma) in PBS. Single-cell RNA libraries were prepared according to the Chromium™ Single Cell 3’ Reagent Kits v3 User Guide using the Chromium single cell Controller instrument (10x Genomics). Libraries were sequenced with a Hiseq 2500 (Illumina) with a depth of 50k-100k reads per cell. Raw sequencing data were processed using the 10x Genomics Cell Ranger pipeline (version 3.0) and aligned to hg19 and mm10 genome. Data normalization was done using “Seurat” package v3.0, with library scaling and log2 transformation. All samples were processed by 10x instrument in the same batch, thus no batch removal was done. Cells with mitochondrial read rate >10% and <200 detectable genes were considered low quality and were not used in the analysis. Normalized and scaled data were clustered using the top significant principal components of highly variable genes and visualized by the t-distributed stochastic neighbor embedding (t-SNE) algorithm. Cluster-specific markers were identified to generate heatmap. The dot plots and feature plots in the identified cell clusters were generated for specific T-cell populations.
In vitro T-cell functional assays
For cytokine analysis, CD19-CAR T cells (105) that were expanded in either human CSF as described previously or complete medium were co-cultured in 96-well tissue culture plates with 105 Daudi cells in complete medium at 37°C. Supernatants were harvested 18 hours post-stimulation and analyzed in triplicate by cytometric bead array using the Bio-Plex Human Cytokine 17-Plex Panel (Bio-Rad Laboratories). For degranulation assays, CAR T cells (105) were co-cultured with Daudi cells (105) in medium containing Golgi plug (Catalog No. 555029; BD Biosciences) at 1μL/mL and 50μL/200μL CD107a-specific antibody for 6 hours at 37°C. Degranulation was determined by CD107a positivity using multicolor flow cytometry (see Antibodies and flow cytometry above).
NBDG uptake assay
NBDG fluorescence displays excitation/emission maxima of ~465/540 nm and can be visualized using a flow cytometer with a green/FITC channel. Spleens were collected from tumor-bearing mice following CAR T cell treatment and stimulated using the REM (22) for 2 cycles (14 days/cycle). Propagated cells were stained with an APC-conjugated human CD62L antibody (clone SK11; BD Biosciences,) and cultured in 100 μM 2-NBDG (Catalog No. N13195; Fisher Scientific) in PBS for 10 and 30 minutes at 37°C. The cells were analyzed with the MACSQuant after two washes with PBS.
Immunohistochemistry staining (IHC)
CAR T cells propagated following REM (22) as described above (see NBDG uptake assay) were stained with mitochondrial selective probe 100 nM MitoTracker (Green) (ThermoFisher Scientific) diluted in complete medium for 30 minutes at 37°C. 1×105 cells were embedded onto slides by cytospin, fixed with 4% PFA, and imaged using an LSM880 confocal microscope.
Statistical Analysis
Analyses were performed using Prism (GraphPad Software, Inc.) or SAS 9.4. For normally distributed endpoints, raw or after logarithm transformation (e.g. flux), Student’s t test was applied to comparisons between two independent and one-way ANOVA models were applied to comparisons among 3 or more independent groups. The non-parametric Mann-Whitney test was applied to group comparisons for non-normally distributed data. Linear mixed models were applied to repeated measures (e.g. flux) from the same subject to account for the variance-covariance structure. For survival data, Kaplan-Meier method and Log-rank test were applied to survival function estimation and group comparisons. P values were corrected for multiple comparisons by Holm’s method. P-values of ≤0.05 were considered statistically significant.
Results
ICV-administered CAR T cells exhibit potent antitumor activity against CNS lymphoma
Administration of CD19-CAR T cells in all clinical settings to date has been via IV delivery. We tested the hypothesis that CD19-CAR T cells delivered by ICV infusion would result in superior CAR T cell efficacy against CNS lymphoma. We first evaluated tumor growth and survival outcomes comparing IV and ICV delivery of CD19-CAR T cells to treat CNS lymphoma. To model CNS lymphoma (Figure 1A), we implanted Daudi lymphoma cells engineered to express EGFP and ffluc into the right forebrain of NSG mice. We treated mice 5 days post-engraftment with either IV or ICV delivery of CD19-CAR T cells engineered using our clinical manufacturing platform in which Tn/mem cells are enriched prior to CAR-transduction (25–27). When determining the CAR T cell dose, we worked under the assumption that ICV-delivered CAR T cells, but not IV-delivered CAR T cells, would have direct access to CNS tumors (31), and achieving comparable therapeutic effects would require different cell doses. Therefore, in these experiments, we infused 3×106 CAR T cells IV and 1.3×106 CAR T cells ICV. We also treated tumor-bearing mice with mock-transduced T cells to control for potential xenoreactivity in our model. Tumors in mice receiving mock T cells delivered either IV or ICV progressed first in the brain and then systemically (Figure 1B). By contrast, tumors in mice that received 1.3 ×106 ICV-delivered CD19-CAR T cells were undetectable by bioluminescent imaging (BLI) by day 14, suggesting that ICV-delivered CD19-CAR T cells mediated rapid anti-CNS lymphoma activity. Although a high dose of IV-delivered CD19-CAR T cells (3×106) also displayed anti-tumor activity, the kinetics of response were delayed; mice treated IV had significantly higher lymphoma burden at day 14 (P<0.01) and did not have complete tumor eradication until day 21 (Figure 1B).
Figure 1. ICV infused CAR T cells eradicate CNS lymphoma more efficiently than IV infused CAR T cells.

(A) In our models of CNS lymphoma, NSG mice were injected intracranially (IC) with either EGFP+ffluc+ Daudi lymphoma cells or Jeko MCL cells. Following tumor engraftment, Tn/mem-derived CD19- or BAFF-R-CAR T cells were administered IV or ICV. For the CD19-CAR T cell model, 3×106 cells and 1.3×106 cells were administered IV and ICV, respectively. For the BAFF-R-CAR T cell model, equal number of cells (1×106) were administered for both IV and ICV conditions. (B) For the CD19-CAR T cell model, tumor burden, as measured in Flux (photons/sec) by bioluminescent imaging, was evaluated once weekly. N=5 mice per group in one experiment. Linear mixed models were used to compare logarithm transformed flux of mice that received ICV versus IV CAR T cells over time; **P<0.01. (C) For the BAFF-R-CAR T cell model, Flux (photons/sec) was determined by measuring bioluminescence once per week. N=5 mice per group in one experiment. Linear mixed models were used to compare logarithm transformed flux of mice that received ICV versus IV CAR T cells over time; **P<0.01. Survival of mice treated with ICV- versus IV-delivered BAFF-R-CAR T cells was analyzed by Log-rank test; **P<0.01. (D-E) 105 days post-CD19-CAR T cell treatment, blood was collected and analyzed for expression of human CD45, CD8, CD4, and CAR (Erbitux) by flow cytometry. Data from a representative mouse for ICV- and IV-delivered CD19-CAR T cells is shown in (D). Percentages (mean ±SD) of human T cells (hCD45+GFP−) and CD4+ and CD8+ cells for all mice are quantified in (E). Gating was set based on isotype-matched monoclonal antibodies or streptavidin. Mean± SDs from multiple mice/measurements are depicted.
To demonstrate that this observation was not limited to a specific tumor or CAR, we repeated our experiments using a different tumor and CAR model. B cell activation factor receptor (BAFF-R) is commonly expressed on B-cell malignancies including mantle cell lymphoma (MCL). We engrafted Jeko MCL cells engineered to express EGFP and ffluc into the right forebrain of NSG mice and treated with second generation BAFF-R CAR T cells (24) derived from Tn/mem cells. Consistent with our CD19-CAR T cell data, ICV-delivered BAFF-R-CAR T cells exhibited significantly improved activity against CNS lymphoma on day 20 (P<0.01) and prolonged mouse survival when compared with IV-delivered BAFF-R-CAR T cells (Figure 1C) (P<0.01).
To understand the effect of route of delivery on CAR T cell persistence, mice treated with CD19-CAR T cells in Figure 1B were euthanized at the same point (day 105) and CAR T cells in the blood were examined. BAFF-R-CAR T cells administered ICV were detectable 20 days after CAR T cell infusion in peripheral blood obtained through retro-orbital bleeding, indicating that ICV-delivered CAR T cells can migrate out of the CNS (Figure 1D-E, Supplemental Figure 2).
ICV-administered CAR T cells completely eradicate both CNS and systemic lymphoma
Because we detected ICV-delivered CAR T cells in the periphery in two lymphoma/CAR models (Figure 1D, Supplemental Figure 2), we investigated whether ICV-administered CAR T cells could eliminate systemic lymphoma. We established a dual model of CNS and systemic lymphoma by engrafting Daudi lymphoma cells in the brain as described above and subcutaneously (SC) in the right flank of the same mouse (Figure 2A). While ICV-delivered mock-transduced T cells did not affect tumor growth compared to untreated controls, ICV-delivered CD19-CAR T cells induced significant regression of both CNS and systemic lymphoma (P<0.001), with tumors at both sites undetectable by 21 days post-infusion (Figure 2B). Strikingly, mice treated with ICV-delivered CD19-CAR T cells remained tumor free for >300 days (Figure 2B). In contrast, mice treated with an equal number of IV-delivered CD19-CAR T cells showed a significantly delayed response, with only 3 of 5 mice achieving tumor eradication at 43 days post-CAR T cell infusion. Moreover, all mice treated IV died before day 200 from relapse of both CNS and systemic lymphoma (Figure 2B) (P<0.01), suggesting that IV-delivered CAR T cells failed to completely eradicate either form of lymphoma. Similarly, ICV-delivery of CD19-CAR T cells modestly improved survival of mice bearing CNS and systemic Jeko tumors relative to ICV-delivered mock T cells (Supplemental Figure 3A). However, there was no statistical difference in survival between IV- and ICV-delivered CD19-CAR T cells (P>0.05), which is likely due to high systemic tumor burden in this model (Supplemental Figure 3A). Together, these data indicate that ICV-delivered CAR T cells can migrate out of the CNS and have improved activity against systemic lymphoma when compared to IV-delivered CAR T cells.
Figure 2. CD19-CAR T cells administered by ICV infusion can completely eradicate both CNS and systemic lymphoma.

(A) NSG mice were injected subcutaneously (SC) with 3×106 EGFP+ ffluc+ Daudi lymphoma cells and 14 days later injected intracranially (IC) with 0.2×106 EGFP+ ffluc+ Daudi lymphoma cells. Five days following IC tumor inoculation, 2×106 mock-transduced T cells or CD19-CAR T cells were administered either ICV or IV. (B) Flux (photons/sec) was determined by measuring bioluminescence once weekly. N=5 mice per group. Linear mixed models were used to compare logarithm transformed flux of ICV- and IV-treated mice over time; **P<0.01, ***P<0.001. Survival of mice was analyzed by Log-rank test; **P<0.01. (C-E) At the time of euthanasia (day 334 for the ICV group and between days 79–180 for the IV group) blood was collected and analyzed for expression of human CD45, CD8, CD4, CAR (EGFR) and central memory receptors by flow cytometry. Representative data from two separate experiments are presented. Percentages (mean ±SD) of human T cells in the blood (D) and T cells expressing CD62L, CD127, CD28, and CAR+ CD4 and CD8 T cell subsets (E) are presented. ICV and IV were compared by Student’s t test. **P<0.01. (F) Human T cells harvested from mouse spleens 334 days post ICV and 180 days post IV CAR T cell infusion were re-stimulated with REM method containing OKT3, irradiated PBMC, and LCL for 14 days and their TCR repertoire was analyzed. Percent of TCR Vβ expression in CD3 positive cells is depicted.
ICV-administered CAR T cells exhibit memory phenotype and function with a broad TCR vβ repertoire
To determine whether there were phenotypic differences between ICV- and IV-delivered CD19-CAR T cells post-infusion that might explain the difference in potency, we analyzed CAR T cells isolated from peripheral blood of mice at the study endpoint by flow cytometry. Blood samples were collected at euthanasia, which occurred on day 334 for mice treated ICV and between days 79–180 for mice treated IV; the latter group were euthanized earlier due to tumor progression. Although mice treated ICV had undetectable tumor burden at the time of blood collection, we detected CAR T cells (CD3+; human CD45+; CAR+) in the blood (Figure 2C-D). ICV-infused CAR T cells may exhibit a high degree of homeostatic proliferation and persistence, which are characteristics of memory T cells. When we compared the persisting CD19-CAR T cells, we found that ICV-delivered cells expressed significantly higher levels of CD28 compared with IV-delivered cells (P=0.01), even following a longer in vivo engraftment (Figure 2E), which may have contributed to disease elimination and CAR T-cell persistence. Consistent with the Daudi lymphoma model, CD19-CAR T cells were detected in peripheral blood of the Jeko MCL model 20 days post-infusion. However, the trafficking and/or persistence in this model appears to be dependent on presence of tumor as CD19-CAR T cells were detected only if there was concurrent brain or systemic lymphoma (Supplemental Figure 3B).
To understand whether anti-lymphoma activity was due to clonal or polyclonal CAR T cell expansion, human T cells harvested at the study endpoint were re-stimulated for 14 days using REM, followed by TCR repertoire analysis. The pre-infusion product, and both the ICV- and IV-delivered CAR T cells all displayed broad TCR Vβ usage (Figure 2F, Supplemental Figure 4), indicating that the CAR T cells mediating anti-lymphoma activity were polyclonal.
Antigen-primed CAR T cells potently eradicate systemic lymphoma and resist tumor re-challenge
To explain the observation that ICV-infused CAR T cells more effectively eradicated both CNS and systemic lymphoma in our dual lymphoma model (Figure 2), we hypothesized that ICV-infused CAR T cells may have been primed by exposure to tumor antigen in the CNS, resulting in potent activity against systemic lymphoma. To assess the contribution of antigen priming on the potency of ICV-delivered CAR T cells, we designed an in vitro priming model in which CAR T cells were co-cultured with irradiated tumor cells for 1 hour before ICV-injection into mice bearing only systemic lymphoma. Mice treated with in vitro-primed CD19-CAR T cells delivered ICV had significant regression of systemic lymphoma when compared to mice treated with unprimed CD19-CAR T cells by day 21 (P<0.01), leading to a significant survival advantage (P<0.05) (Figure 3A) and supporting the hypothesis that priming may further improve antitumor activity and persistence of ICV-delivered CAR T cells. When surviving mice were subsequently re-challenged with new systemic lymphoma 133 days post-CAR T cell infusion, primed and unprimed CAR T cells delivered ICV were equally efficient at rejecting systemic lymphoma (Figure 3A, Supplemental Figure 5A).
Figure 3. Antigen-primed CAR T cells possess better effector memory function and can resist tumor re-challenge.

NSG mice were inoculated with subcutaneous injections of 3×106 Daudi lymphoma cells in the right flank 19 days prior to CAR T cell treatment. On the day of CAR T cell infusion, 2×106 CD19-CAR T cells were primed with irradiated Daudi cells at 10:1 (CAR:tumor) ratio for 1 hour prior to infusion by (A) ICV- or (B) IV-delivery. Mock T cells were infused into control mice. Flux (photons/sec) was determined by measuring bioluminescence by live imaging once per week. 135 days post-CAR T cell infusion, surviving mice were re-challenged (dotted lines) with Daudi lymphoma cells by subcutaneous injection. Naïve mice were inoculated with Daudi lymphoma cells at re-challenge as controls. Mean ±SDs from 5 mice per group are presented. Linear mixed models were used to compare flux (logarithm transformed) among different groups over time and log-rank test was used to compare survival functions among the groups. If a mouse died during imaging processes with no prior sign of tumor progression, then it was considered an anesthesia-related death and the mouse was excluded from survival analysis. *P<0.05; **P<0.01; ***P<0.001. (C) Blood was collected retro-orbitally 133 days post-CAR T cell treatment and analyzed for expression of human CD45, CD3, and CAR (EGFR) by flow cytometry. No mice survived in the unprimed CD19-CAR IV group. Significance was determined by one-way ANOVA test. ***P<0.001.
To determine if the enhanced CAR T-cell function we observed following antigen priming was independent of exposure to CSF, we treated mice bearing systemic lymphoma with primed or unprimed CD19-CAR T cells IV. Consistent with previous observations, primed CAR T cells delivered IV induced superior tumor regression when compared with unprimed CAR T cells (P<0.001) on day 28 (Figure 3B). All mice that received unprimed CD19-CAR T cells IV died before day 113. In contrast, 3 of 5 mice that received primed CAR T cells IV survived >135 days, and all surviving mice resisted tumor re-challenge (Figure 3B, Supplemental Figure 5B). When we examined the peripheral blood of mice 133 days post-CAR T-cell infusion, we observed significantly higher levels of CAR T cells in mice treated with primed ICV-delivered CAR T cells when compared to mice treated with either unprimed ICV-delivered or primed IV-delivered CAR T cells (Figure 3C) (P<0.001). Overall, this data supports the notion that T-cell activation of ICV-delivered CAR T cells by exposure to lymphoma in the brain (i.e. priming) does contribute in part to enhanced CAR T cell persistence and potency against systemic lymphoma.
ICV-delivered CAR T cells show superior proliferative potential compared with IV-delivered CAR T cells
Because antigen priming played a role in the potency of both IV- and ICV-delivered CAR T cells against systemic lymphoma, we next assessed whether CAR T cells would traffic to systemic tumors and proliferate in the absence of priming. To track CAR T cells in vivo, CD19-CAR T cells were transduced to express EGFP and ffluc (Figure 4A; Supplemental Figure 6A) and administered ICV or IV into mice bearing systemic tumors. Both ICV- and IV-delivered CAR T cells migrated to and efficiently infiltrated the systemic tumors as assessed by BLI (Figure 4B). Interestingly, the bioluminescent signals were significantly higher in ICV- versus IV-treated mice at day 35 (P<0.001) (Figure 4C), indicating greater in vivo expansion of ICV-delivered CAR T cells. This expansion was dependent on engagement of the CAR as mock T cells did not expand in vivo (Supplemental Figure 6B) and tumor volumes were not controlled by mock T cells (Supplemental Figure 6C). When we examined CAR T-cell persistence, we observed that the ratio of CAR+ to CAR– human CD45+ cells increased over time for both ICV- and IV-treated groups (Figure 4D-E), suggesting that CAR+ cells were preferentially persisting. However, levels of human CD45+ cells (CAR+ or CAR–) diminished over time, with the IV-delivered cells disappearing more rapidly than the ICV-delivered cells (P<0.05) (Figure 4D-E). Thus, while both ICV- and IV-delivered CAR T cells can migrate to systemic lymphoma, ICV-delivered cells have superior capacity for proliferation and persistence.
Figure 4. ICV-infused CAR T cells exhibit similar trafficking, but superior proliferation and persistence potential when compared with IV-delivered CAR T cells.

(A) CD19-CAR T cells were transduced with EGFP-ffluc and expanded in vitro. NSG mice were implanted subcutaneously with Daudi lymphoma in the right flank. Nineteen days after tumor engraftment, 2×106 EGFP+ffluc+ CAR T cells were administered ICV or IV and CAR T cell proliferation was determined by measuring bioluminescence by live imaging every other day (B-C). The same scale was used for each time point. (D-E) Blood was collected at different time points post-CAR T cell infusion, and T cell (human CD45+) and CAR+ T cell (EGFR+) levels were detected by flow cytometry. Mean ±SDs from 5 mice per group are presented. Significance was determined with the Mann-Whitney test; *P<0.05; ***P<0.001. Experiments were repeated twice and representative data are presented.
CSF conditions CAR T cells for enhanced memory and reduced exhaustion
Exposure to CSF may enhance CAR T-cell potency
We observed that both ICV- and IV-delivered CAR T cells have similar homing potential to systemic tumors even in the absence of antigen priming (Figure 4). We therefore explored why ICV delivery yielded CAR T cells possessing superior anti-lymphoma capacity. We hypothesized that exposure to CSF upon ICV administration may modify CAR T cells to enhance their function. Glucose and potassium, which are known to be involved in T-cell memory and function (32–36), are found at lower levels in CSF (60mg/dL of glucose; 2.8 mEq/L of potassium) compared with serum (90mg/dL of glucose; 4.5 mEq/L of potassium) (37). To investigate if exposure to CSF affects CAR T-cell gene expression, we cultured CD19-CAR T cells in regular RPMI (Invitrogen) (200 mg/dL of glucose; 5.3 mEq/L of potassium) or CSF from healthy human donors for 48 hours and analyzed for expression of genes associated with memory, effector function, and metabolism. Across 4 different donors, we consistently observed elevated levels (>1.3-fold change) of genes related to survival and memory function, including BCL2, BCL2L11, KLF2, and IL7Rα (CD127), and decreased expression (>1.3-fold change) of effector differentiation genes like PRF1, IFNγ, GZMB, and TBX21 in CAR T cells cultured in CSF when compared to RPMI (Figure 5A). Of note, numerous other genes regulating glucose metabolism, including CPT1A and Pgam-1 (33), were downregulated in CSF cultures. These data support the hypothesis that CAR T cells cultured in CSF underwent metabolic reprogramming to favor memory formation. Consistent with the transcriptional profile suggesting that CSF maintains CAR T cells in a less differentiated state compared with RPMI (Figure 5A), CAR T cells cultured in CSF produced significantly lower levels of effector cell-related Th1 and T cell-inhibitory Th2 cytokines upon in vitro stimulation with Daudi lymphoma cells compared with RPMI-cultured CAR T cells (Figure 5B).
Figure 5. CSF conditions CAR T cells for enhanced memory and reduced exhaustion.

(A) CD19-CAR T cells were cultured in RPMI or human CSF for 48 hours and subjected to RT-PCR array analysis. Fold-changes of genes in CSF over RPMI of >1.3 are shown. N=4 donors. (B) CAR T cells cultured in CSF or RPMI for 48 hours were stimulated overnight with Daudi lymphoma cells. Cytokines released into the supernatant were measured with cytometric bead array using the Bio-Plex Human Cytokine 17-Plex Panel. Mean ±SDs from 6 replicates are presented. CSF and RPMI (logarithm transformation) were compared by Student’s t test; **P<0.01; ***P<0.001. (C) CAR T cells were activated, transduced, and expanded for 13 days in modified RPMI (60mg/dL glucose; 2.8 mEq/L potassium) or in regular RPMI (200 mg/dL glucose; 5.3 mEq/L potassium). CAR T cells cultured in modified RPMI were subsequently cultured in regular RPMI (Switched) for 14 days. Percentages (mean ±SD) of CD28 positive cells in CAR-gated populations from N=4 different human donors are presented. Linear mixed models were used to compare the three conditions based on repeated measures from the same donor; *P<0.05; **P<0.01; ***P<0.001. (D) CAR T cells were cultured in modified or regular RPMI, stimulated with Daudi cells for 6 hours, and assessed for degranulation. Percentages (mean ±SD) of CD107+ cells in CAR gated populations from N=4 different human donors are presented.
To control for differences between CSF and regular RPMI, we performed experiments with modified RPMI medium that mimics the levels of glucose and potassium in CSF (60mg/dL glucose; 2.8 mEq/L potassium). CD19-CAR T cells generated in modified RPMI showed lower fold-expansion than cells generated in regular RPMI (Supplemental Figure 7A), which may be a mechanism that prevents differentiation and maintains memory. In support of this, CD19-CAR T cells expanded in modified RPMI expressed elevated levels of memory marker CD28 compared to cells generated in regular RPMI (P<0.001) (Figure 5C) and activation marker CD25 (P<0.01), but did not show change in the exhaustion markers LAG3, Tim3, and KLRG (Supplemental Figure 7B-C). Remarkably, these cells sustained significantly higher levels of CD28 when compared to cells cultured in regular RPMI even after cultures were switched from modified to regular RPMI medium for 14 days (Figure 5C). Moreover, these cells maintained effector capabilities, as indicated by comparable CD107a expression (i.e. degranulation) upon co-culture with Daudi target cells (Figure 5D). Overall, our data suggest that the improved function of ICV-delivered CAR T cells may be attributed to reprogramming to memory T cells in the CSF environment.
Distinct gene signatures of in vivo persisting ICV and IV CAR T cells
To quantitatively dissect differences in expression of memory, differentiation, and immune checkpoint markers, we harvested CAR T cells delivered ICV or IV from mice bearing both CNS and systemic lymphoma and performed single cell RNA-sequencing. Human T cells were isolated from mouse bone marrow 68 days post-CAR T cell infusion. Clustering analysis identified 5 clusters; t-SNE plots demonstrated that CAR T cells harvested from ICV-treated mice are enriched in cluster 3, while CAR T cells from IV-treated mice are enriched in cluster 1 (Figure 6A). In agreement with in vitro CSF-treated cells (Figure 5), in vivo persisting ICV-delivered CAR T cell-enriched cluster 3 are characterized by higher levels of hallmark memory genes (KLF2, BCL2L11, BCL2, IL7R, CD27, and CD28) and lower levels of differentiation and effector genes (PDCD1, EOMES, FOS, TBX21, IFNG, PRF1, GZMH, and GZMB) when compared with IV-delivered CAR T cell-enriched cluster 1 (Figure 6B). Overall gene expression patterns, as shown in the heatmap in Figure 6C, display distinct gene signatures between the clusters enriched for ICV- and IV-delivered CAR T cells.
Figure 6. ICV- and IV-delivered CAR T cells that persist in vivo have distinct gene signatures.
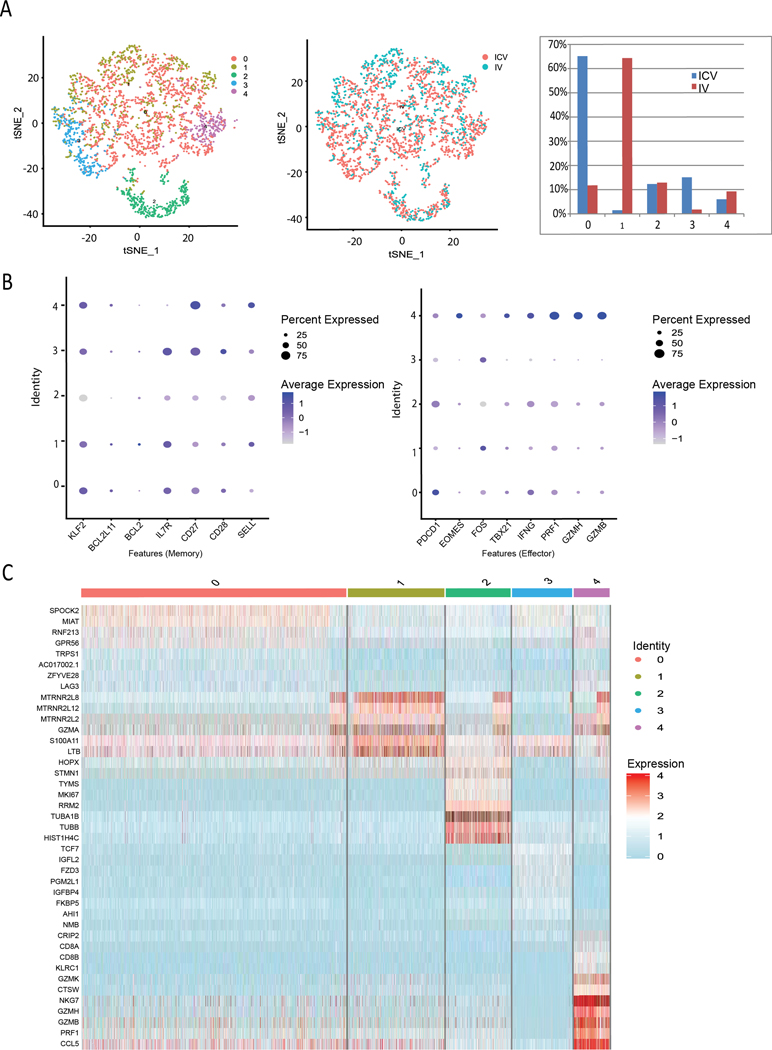
T cells were harvested from the bone marrow of mice 68 days post ICV or IV CAR T cell infusion and the samples from one mouse in each group were subjected to single cell RNA-seq (scRNA-seq) analysis. The subsequent analysis of human gene data was performed using “Seurat” package v3.0 and R scripts. (A) t-distributed stochastic neighbor embedding (tSNE) visualization plot of scRNA-seq data and (B) dot plots and feature plots in the identified cell clusters were generated for specific T cell populations. ICV delivered cells were enriched in cluster 3, while IV delivered cells were enriched in cluster 1. (C) Cluster specific markers were identified to generate a heatmap.
Enhanced memory and reduced exhaustion of ICV-delivered CAR T cells
To investigate if the gene expression patterns that distinguished ICV- from IV-delivered CAR T cells (Figure 6) resulted in enhanced memory and/or reduced exhaustion, we performed an in vitro serial re-stimulation with CAR T cells harvested from the spleens of mice at the study endpoint. T cells were stimulated with REM for 2 cycles (14 days/cycle), which included both OKT3 and CD19-mediated antigen stimulation (Figure 7A). CAR T cells from ICV-treated mice expanded 9.1±0.9 fold from REM1 to REM2, whereas CAR T cells from IV-treated mice had either lower expansion or reduction in overall numbers (1.7±1.6 fold) (Figure 7B), suggesting that ICV-delivered CAR T cells resisted T cell activation-induced cell death (38,39) and had superior expansion potential. Despite 334 days of in vivo incubation and repeated in vitro stimulation, CAR T cells from ICV-treated mice maintained higher levels of CD127, CD62L, and CD161—markers that represent central memory T cells and memory stem cells with high efflux capacity (40)—when compared with CAR T cells from IV-treated mice harvested after only 180 days of in vivo engraftment (Figure 7C).
Figure 7. ICV administered CD19-CAR T cells exhibit memory phenotype and function.

(A) T cells harvested from two representative mouse spleens (334 days post-infusion for ICV mice and 180 days post-infusion for IV mice) were stimulated with OKT3 and feeder cells, including peripheral blood mononuclear cells and LCL cells, for 2 cycles (14 days/cycle). (B) Percentages of CAR T cells (CD3+EGFR+) and fold change following first and second REM stimulation are presented. (C) CAR T cells were analyzed for expression of CD161, CD127, and CD62L following the second REM stimulation. (D) Uptake of glucose analog NBDG after 30 minutes was analyzed by flow cytometry. (E) CD62L+ and CD62L- cell populations were assessed for the ability to uptake NBDG overtime. Percentages (mean ±SD) of NBDG+ cells are presented. Linear mixed models were used to compare CD62+ and CD62L over time; **P<0.01. (F) Mitochondria of T cells following the second REM stimulation were stained with 100 nM MitoTracker (Green) and imaged using an LSM880 confocal microscope. Images were taken at 63X; scale bars=5 μm.
Low glycolytic activity represents an intrinsic quality of CAR T cells associated with clinical response (41). To functionally assess the glycolytic status of the ICV- and IV-delivered CAR T cells harvested from mice, we examined their capacity for uptake of the glucose analog NBDG. We observed that CD62L+ cells, which are enriched in those isolated from ICV-treated mice (Figure 7C), exhibited significantly lower (P<0.01) NBDG uptake than CD62L- cells (Figure 7D-E), supporting the memory-like behavior of these CD62L+ cells (33,41). To meet differing energy requirements, effector T cells activate mitochondrial fission and subsequent fragmentation, while memory T cells inhibit mitochondrial fission, leading to mitochondrial elongation (42,43). In line with these reported observations, T cells from ICV-treated mice maintained fused mitochondrial networks that appeared elongated, while T cells from IV-treated mice had punctate mitochondria (Figure 7F).
Discussion
While CAR T-cell therapy has shown great promise in the treatment of hematologic diseases, the generation of durable responses against tumor relapse remains an obstacle. We here propose that ICV-delivery of CD19-CAR T cells holds superior potential to treat CNS and systemic lymphoma, which supports the conclusions of two recently published studies that demonstrate that ICV-delivered CAR T cells more effectively control primary and metastatic brain tumors (31,44). In the present study, ICV-delivered CAR T cells potently eradicated both CNS and systemic lymphoma, leading to lymphoma elimination, in conditions where equal or higher doses of IV-delivered CAR T cells did not yield disease elimination (Figure 1–2). Furthermore, CAR T cells isolated from ICV-treated mice showed increased phenotypic and functional memory characteristics (Figure 2, 6-7), which we were able to recapitulate by in vitro culture with CSF (Figure 5). These findings suggest that CAR T cells are conditioned by exposure to CSF in the ICV environment to promote anti-tumor activity and memory formation.
Importantly, this study examined how persistence of CAR T cells is affected by route of administration. CAR T cells delivered ICV were detectable in peripheral blood of mice >300 days post-infusion, even without detectable lymphoma (Figure 2). We acknowledge that the possibility of xenoreactivity of CAR T cells is a potential limitation of our model. However, our studies were well controlled with equal dosing of non-transduced (mock) T cells, which demonstrated no anti-lymphoma activity. Moreover, we followed the duration of response to ICV-delivered CAR T cells for over 11 months (334 days) and did not observe any evidence of toxicity due to graft-versus-host disease (GVHD), which would indicate xenoreactivity. Absence of GVHD suggests that there is limited engagement of the endogenous TCR in our model and, therefore, xenoreactivity likely does not affect our conclusions. In line with these observations, a recent study by Mulazzani et al (45) showed that intracerebral infusion of CAR T cells exhibits superior anti-tumor efficacy and persistence compared to IV delivery, where xenoreactivity of CAR T cells is ruled out using mock T cells. These data support our observation that differences in outcomes in mice treated with ICV and IV delivery reflected differences in capacity for anti-tumor activity promoted by the two routes of delivery. The long-term persistence of memory CAR T cells following ICV administration in absence of antigen, also observed by Mulazzani et al (45), could be due to homeostatic mechanisms driven by factors such as cytokines that are species cross-reactive, such as IL-7 (46,47).
ICV-delivered CAR T cells exhibited a higher degree of memory phenotype than did IV-delivered CAR T cells (Figure 6–7), and an elongated mitochondrial morphology consistent with maintaining memory T-cell persistence (43) (Figure 7). With t-SNE clustering and underlying data output, single cell RNA-seq datasets revealed distinct gene signatures between ICV- and IV-delivered CAR T cells that supported evolving memory features of ICV-delivered CAR T cells but advanced differentiation signatures of IV-delivered CAR T cells (Figure 6).
We propose that exposure of CAR T cells to CSF within the ICV environment leads to a metabolic reprogramming that favors the formation of memory. Proper engagement of metabolic pathways is critical to fulfilling the nutrient demands of immune cells (33,35,36,48–50) and inhibition of glycolysis enhances memory T-cell formation (33,36). Moreover, elevated extracellular potassium is believed to suppress T-cell function through the PP2A pathways (51). Compared with serum, CSF contains lower levels of glucose and potassium and CAR T cells cultured in CSF or modified RPMI compared to RPMI with glucose and potassium levels similar to serum had changes in gene expression that promoted a memory phenotype (Figure 5).
In keeping with clinical observations that reactivating CAR T cells with antigen further augments their efficacy (7), our data showed that antigen-primed CAR T cells possessed significantly enhanced anti-tumor activity, expansion, and persistence regardless of route of administration when compared with unprimed CAR T cells (Figure 3). These data indicate that antigen priming of CAR T cells contributes to improved anti-tumor activity, independent of the CSF microenvironment.
Collectively, the data presented here provide rationale to clinically evaluate ICV delivery of CAR T cells to treat primary CNS lymphoma and systemic lymphoma with CNS involvement, as we demonstrated that a single dose of ICV-delivered CAR T cells has the potential to cure both primary CNS and systemic lymphoma in mice. Unlike classical T-cell differentiation patterns, where enhanced effector function is associated with impaired memory and persistence, ICV-infused CAR T cells successfully acquired complete effector function for anti-tumor activity while maintaining memory function for long-term immune surveillance and resistance to tumor re-challenge. These differences in potency and longevity should be evaluated in other systemic tumor types where CNS involvement is common and may represent a generalizable strategy to improve CAR T-cell therapy.
Supplementary Material
Synopsis:
In the clinic, CD19-CAR T cells are administered IV and are not used specifically to treat CNS lymphoma. The authors show a single ICV infusion of CD19-CAR T cells completely eradicated both CNS and systemic lymphoma in mice.
Acknowledgments
This paper is dedicated to the memory of Tia Palermo whose life and illness inspired this work. We thank Dr. Brian Armstrong in the Light Microscopy Digital Imaging Core at City of Hope for technical assistance.
Financial support: Small Animal Imaging Core supported by the National Cancer Institute of the National Institutes of Health (P30CA033572); Toni Stephenson Lymphoma Center (PI: L.W.K.); Leukemia and Lymphoma Society Mantle cell lymphoma Research Initiative (SCOR 7000-18; PI: L.W.K.; Project Leaders: S.J.F.; X.W.; L.E.B.); Department of Defense (CA170783, PI: L.W.K.). The study was also supported by Borstein family foundation (X.W.).
Footnotes
Conflict of Interest Statement: The authors declare they have no competing interests.
References
- 1.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol 2013;15 Suppl 2:ii1–56 doi 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomita N, Kodama F, Kanamori H, Motomura S, Ishigatsubo Y. Secondary central nervous system lymphoma. Int J Hematol 2006;84(2):128–35 doi 10.1532/ijh97.06091. [DOI] [PubMed] [Google Scholar]
- 3.Cai QQ, Hu LY, Geng QR, Chen J, Lu ZH, Rao HL, et al. New risk factors and new tendency for central nervous system relapse in patients with diffuse large B-cell lymphoma: a retrospective study. Chin J Cancer 2016;35(1):87 doi 10.1186/s40880-016-0150-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Besien K, Gisselbrecht C, Pfreundschuh M, Zucca E. Secondary lymphomas of the central nervous system: risk, prophylaxis and treatment. Leuk Lymphoma 2008;49 Suppl 1:52–8 doi 10.1080/10428190802311458. [DOI] [PubMed] [Google Scholar]
- 5.Maciocia P, Badat M, Cheesman S, D’Sa S, Joshi R, Lambert J, et al. Treatment of diffuse large B-cell lymphoma with secondary central nervous system involvement: encouraging efficacy using CNS-penetrating R-IDARAM chemotherapy. Br J Haematol 2016;172(4):545–53 doi 10.1111/bjh.13867. [DOI] [PubMed] [Google Scholar]
- 6.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385(9967):517–28 doi 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abramson JS, McGree B, Noyes S, Plummer S, Wong C, Chen YB, et al. Anti-CD19 CAR T Cells in CNS Diffuse Large-B-Cell Lymphoma. The New England journal of medicine 2017;377(8):783–4 doi 10.1056/NEJMc1704610. [DOI] [PubMed] [Google Scholar]
- 8.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015;33(6):540–9 doi 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. The New England journal of medicine 2013;368(16):1509–18 doi 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer discovery 2018;8(8):958–71 doi 10.1158/2159-8290.cd-17-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer discovery 2017;7(12):1404–19 doi 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 2016;127(26):3312–20 doi 10.1182/blood-2016-02-629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hay KA, Hanafi L-A, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017;130(21):2295–306 doi 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadoch C, Li J, Wong VS, Chen L, Cha S, Munster P, et al. Complement activation and intraventricular rituximab distribution in recurrent central nervous system lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20(4):1029–41 doi 10.1158/1078-0432.ccr-13-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubenstein JL, Fridlyand J, Abrey L, Shen A, Karch J, Wang E, et al. Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J Clin Oncol 2007;25(11):1350–6 doi 10.1200/jco.2006.09.7311. [DOI] [PubMed] [Google Scholar]
- 16.Rubenstein JL, Li J, Chen L, Advani R, Drappatz J, Gerstner E, et al. Multicenter phase 1 trial of intraventricular immunochemotherapy in recurrent CNS lymphoma. Blood 2013;121(5):745–51 doi 10.1182/blood-2012-07-440974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. The New England journal of medicine 2016;375(26):2561–9 doi 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(1):95–105 doi 10.1158/1078-0432.ccr-17-2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Molecular therapy : the journal of the American Society of Gene Therapy 2018;26(1):31–44 doi 10.1016/j.ymthe.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011;118(5):1255–63 10.1182/blood-2011-02-337360 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelloquin F, Lamelin JP, Lenoir GM. Human B lymphocytes immortalization by Epstein-Barr virus in the presence of cyclosporin A. In Vitro Cell Dev Biol 1986;22(12):689–94 doi 10.1007/bf02621085. [DOI] [PubMed] [Google Scholar]
- 22.Crossland KD, Lee VK, Chen W, Riddell SR, Greenberg PD, Cheever MA. T cells from tumor-immune mice nonspecifically expanded in vitro with anti-CD3 plus IL-2 retain specific function in vitro and can eradicate disseminated leukemia in vivo. Journal of immunology (Baltimore, Md : 1950) 1991;146(12):4414–20. [PubMed] [Google Scholar]
- 23.Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Molecular therapy : the journal of the American Society of Gene Therapy 2015;23(4):757–68 doi 10.1038/mt.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin H, Dong Z, Wang X, Cheng WA, Wen F, Xue W, et al. CAR T cells targeting BAFF-R can overcome CD19 antigen loss in B cell malignancies. Sci Transl Med 2019;11(511) doi 10.1126/scitranslmed.aaw9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang WC, et al. Phenotypic and Functional Attributes of Lentivirus-modified CD19-specific Human CD8+ Central Memory T Cells Manufactured at Clinical Scale. Journal of immunotherapy (Hagerstown, Md : 1997) 2012;35(9):689–701 doi 10.1097/CJI.0b013e318270dec7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khaled SK, Blanchard S, Wang X, Wagner J, Naranjo A, Simpson J, et al. Adult Patients with ALL Treated with CD62L+ T Naïve/Memory-Enriched T Cells Expressing a CD19-CAR Mediate Potent Antitumor Activity with a Low Toxicity Profile. Blood 2018;132(Suppl 1):4016 doi 10.1182/blood-2018-99-119883. [DOI] [Google Scholar]
- 27.Popplewell L, Wang X, Blanchard S, Wagner J, Naranjo A, Arencibia A, et al. CD19-CAR Therapy Using Naive/Memory or Central Memory T Cells Integrated into the Autologous Stem Cell Transplant Regimen for Patients with B-NHL. Blood 2018;132(Suppl 1):610 doi 10.1182/blood-2018-99-119650. [DOI] [Google Scholar]
- 28.Stastny MJ, Brown CE, Ruel C, Jensen MC. Medulloblastomas expressing IL13Ralpha2 are targets for IL13-zetakine+ cytolytic T cells. Journal of pediatric hematology/oncology 2007;29(10):669–77 doi 10.1097/MPH.0b013e3181468c68. [DOI] [PubMed] [Google Scholar]
- 29.Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood 2011;117(3):808–14 doi 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. Journal of immunology (Baltimore, Md : 1950) 2005;175(9):5895–903. [DOI] [PubMed] [Google Scholar]
- 31.Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med 2020;26(5):720–31 doi 10.1038/s41591-020-0827-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Frontiers in immunology 2015;6:1 doi 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. The Journal of clinical investigation 2013;123(10):4479–88 doi 10.1172/jci69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature 2017;548(7669):537–42 doi 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gurusamy D, Clever D, Eil R, Restifo NP. Novel “Elements” of Immune Suppression within the Tumor Microenvironment. Cancer immunology research 2017;5(6):426–33 doi 10.1158/2326-6066.cir-17-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI insight 2017;2(23) doi 10.1172/jci.insight.95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guyton AC, Hall JE. Textbook of medical physiology (11th edition). Edinburgh : Elsevier Saunders; Oxford: : Elsevier Science [distributor], 2006.; 2005. [Google Scholar]
- 38.Youngblood B, Davis CW, Ahmed R. Making memories that last a lifetime: heritable functions of self-renewing memory CD8 T cells. International immunology 2010;22(10):797–803 doi 10.1093/intimm/dxq437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature reviews Immunology 2015;15(8):486–99 doi 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR. A distinct subset of self-renewing human memory CD8+ T cells survives cytotoxic chemotherapy. Immunity 2009;31(5):834–44 doi 10.1016/j.immuni.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nature medicine 2018;24(5):563–71 doi 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liesa M, Shirihai OS. Mitochondrial Networking in T Cell Memory. Cell 2016;166(1):9–10 doi 10.1016/j.cell.2016.06.035. [DOI] [PubMed] [Google Scholar]
- 43.Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016;166(1):63–76 doi 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med 2020;26(5):712–9 doi 10.1038/s41591-020-0821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mulazzani M, Frassle SP, von Mucke-Heim I, Langer S, Zhou X, Ishikawa-Ankerhold H, et al. Long-term in vivo microscopy of CAR T cell dynamics during eradication of CNS lymphoma in mice. Proceedings of the National Academy of Sciences of the United States of America 2019;116(48):24275–84 doi 10.1073/pnas.1903854116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity 2008;29(6):848–62 doi 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 47.Barata JT, Silva A, Abecasis M, Carlesso N, Cumano A, Cardoso AA. Molecular and functional evidence for activity of murine IL-7 on human lymphocytes. Experimental hematology 2006;34(9):1133–42 doi 10.1016/j.exphem.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 48.Irving M, Vuillefroy de Silly R, Scholten K, Dilek N, Coukos G. Engineering Chimeric Antigen Receptor T-Cells for Racing in Solid Tumors: Don’t Forget the Fuel. Frontiers in immunology 2017;8:267 doi 10.3389/fimmu.2017.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nature communications 2015;6:6692 doi 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016;537(7621):539–43 doi 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conforti L. Potassium channels of T lymphocytes take center stage in the fight against cancer. Journal for immunotherapy of cancer 2017;5:2 doi 10.1186/s40425-016-0202-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.