

Abstract
Nucleophilic aromatic substitution (SNAr) is routinely used to install 19F− and 18F− in aromatic molecules, but is typically limited to electron-deficient arenes due to kinetic barriers associated with C–F bond formation. Here we demonstrate that a polarity-reversed photoredox-catalysed arene deoxyfluorination operating via cation radical-accelerated nucleophilic aromatic substitution (CRA-SNAr) enables the fluorination of electron-rich arenes with 19F− and 18F− under mild conditions, thus complementing the traditional arene polarity requirements necessary for SNAr-based fluorination. The utility of our radiofluorination strategy is highlighted by short reaction times, compatibility with multiple nucleofuges, and high radiofluorination yields, especially that of an important cancer positron emission tomography (PET) agent [18F]5-fluorouracil ([18F]FU). Taken together, our fluorination approach enables the development of fluorinated and radiofluorinated compounds that can be difficult to access by classical SNAr strategies, with the potential for use in the synthesis and discovery of PET radiopharmaceuticals.
Graphical Abstract

Introduction
General methods for site-selective and facile installation of fluorine (19F) and its radioisotope (18F) in simple and complex (hetero)aromatic molecules are highly desirable. The ubiquity and value of the resulting (radio)fluorinated molecules are well-documented in market pharmaceuticals1,2 and agrochemicals.3 Furthermore, 18F is an important radioisotope used in the synthesis of radiopharmaceuticals for positron emission tomography (PET)4,5 imaging. Nucleophilic aromatic substitution (SNAr) and transition metal catalysis have traditionally been used to construct arene C–F bonds with 19F− and 18F− in a site-selective manner (Figures 1A and 1B).6,7 Stepwise nucleophilic aromatic substitution (SNAr) methods are effective, but typically constrained to arenes possessing ortho and/or para electron-withdrawing substituents, which can stabilize a negatively-charged Meisenheimer complex upon nucleophile attack. However, a stepwise process may be inoperative in heteroarenes as SNAr occurs by a concerted mechanism via direct substitution at an sp2 carbon8,9, and rates of nucleofuge extrusion diminish upon the introduction of electron-rich groups.9 Despite its limitations, SNAr remains the method of choice for synthesizing 18F-labeled arenes due to its usability, with post-SNAr functional group interconversions used to modify arene electronics. To overcome the substrate-centered restrictions of SNAr, transition metal-catalysed methods have been developed, relying on palladium, copper or silver catalysis alongside aryl (pseudo)halides,10,11 boron electrophiles,12,13 or activated nucleofuges14,15 for successful C–19F and C–18F bond formation.4,7,16 However, the use of specialized ligands and high catalyst loadings can be potential roadblocks for its widespread adoption, especially in the context of radiofluorination.
Figure. 1. Strategies for arene fluorination.

Nucleophilic arene fluorination via (a) SNAr, (b) transition metal catalysis, (c) deoxyfluorination, and (d) cation radical accelerated SNAr strategies.
The direct fluorination of C–O bonds (Figure 1C) has recently gained attention as a method for interconversion of an electron-rich functional group (i.e. a phenol) into a fluorine atom. Concerted SNAr methods have been developed by both the Ritter9,17 and Sanford18,19 labs in which fluoride trapping at the nucleofuge (uronium or fluorosulfonate) leads to intramolecular fluoride delivery from either tetrahedral fluoroimidazoline (C-1) or trigonal pyramidal difluorosulfonate (C-2) intermediates. In both methods, nucleofuge assistance enables facile deoxyfluorination. Despite these successes, the application of deoxyfluorination methods to radiofluorination is still limited by high reaction temperatures and an activated uronium nucleofuge that is prone to hydrolysis with electron-rich phenols.9,20 With these challenges in mind, we sought to develop a strategy that would allow for site-selective arene deoxyfluorination on electron neutral to electron-rich (hetero)aromatics under mild conditions.
We recently disclosed photoredox-catalysed cation radical accelerated SNAr transformations which selectively aminate21 and cyanate22 methoxy- and benzyloxy- nucleofuges at the C–O bonds via arene radical cation intermediates. We envisioned that this reactivity could be translated to fluorination by both 19F and 18F (Figure 1D) and took inspiration from our recent report on arene C–H radiofluorination, in which we used caesium fluoride (CsF), a commonly used nucleophilic fluorine source, in tandem with tetrabutylammonium bisulfate (TBAHSO4), a phase transfer reagent, to install fluoride in several (hetero)arenes.23 In transferring this reaction towards C-O fluorination, initial attempts using fluoride as a nucleophile were unsuccessful when using 4-phenylanisole, either providing returned starting material or observable substrate de-methylation. We hypothesize that the increased acidity of O-methyl C–H bonds upon oxidation of the aryl methyl ether may lead to unproductive deprotonation under basic conditions.24 Thus, we decided to explore alternative nucleofuges that would circumvent the problem of acidic alkyl C–H bonds with minimal alteration of arene electronics.
Here, we demonstrate a strategy for the photoredox-catalysed, nucleophilic deoxyfluorination of phenol derivatives with 19F− and 18F−, with general methods established for unsymmetrical diaryl ethers. This approach enables selective arene fluorination via the preferential oxidization of the more electron-rich aromatic ring and only targets the phenoxy nucleofuge in the presence of other oxygen-based species within the molecule. The utility of our method is best highlighted by our radiofluorination protocol, which requires short reaction times, has good compatibility with multiple nucleofuges, and furnishes the desired 18F-labeled products in high radiofluorination yields (RCY).
Results
Enabling deoxyfluorination of electron-rich arenes with aryloxy nucleofuges via CRA-SNAr.
In an attempt to identify alternative nucleofuges that can be easily made in a single step from phenol derivatives, we surveyed a number of candidates. Initial attempts to employ carbonate or thiocarbonate nucleofuges were met with modest success (Figure 2A; 22– 23% yields). We next turned our attention to aryloxy groups, as they have been utilized as nucleofuges in traditional SNAr for electron-poor aromatics6,25 and display higher rates of expulsion relative to alkoxides.6 An important consideration upon adoption of aryloxy groups is arene site selectivity as substitution may occur on either aromatic ring. We envisioned that photoinduced electron transfer from an unsymmetrical biaryl ether to a photoexcited acridinium would generate the arene radical cation (Figure 2B). This electrophilic species has two potential sites of functionalization, but the formation of the cation radical on the arene with the lower oxidation potential is thermodynamically preferred.26 Preliminary studies of electron-poor aryloxy nucleofuges gave increased yields of the desired fluoroarene product 2 (Figure 2A, 23–79% yields) with the 4-chlorophenoxy adduct giving the best results (79% yield). Despite the fact that the dicyanophenoxy nucleofuge can be forged directly via SNAr from the phenol, we elected to carry on with the 4-chlorophenoxy nucleofuge as the diaryl ether starting material is easily prepared from the phenol by an Ullman coupling.
Figure 2. Nucleofuge scope and a rationale for nucleofuge site-selectivity (a).

Preliminary nucleofuge scope for the synthesis of 2. Yields reported as GC yields using 1,2-dichlorobenzene as an internal standard. (b) Selective arene oxidation dictates fluorination site selectivity.
After extensive optimization (Table 1) we found that using an acridinium-based photooxidant (1•BF4), CsF, and TBAHSO4 with a biphasic dichloromethane/water solvent system, provided 4-phenylfluorobenzene in excellent yield (79%) with minimal formation of 4-chloro-fluorobenzene (entry 1). This reaction could be performed under air or N2 without significant difference. At the outset, it was determined that 427 nm LEDs were interchangeable with 455 nm LEDs (See Supplementary Table 1), and most of the optimization was performed with 455 nm LEDs due to their higher commercial availability. Exchanging CsF for KF (entry 2) had little impact on the yield, however subsequent studies demonstrated better reproducibility for a range of substrates employing CsF (vide infra). The amount of phase transfer reagent used was important as yields diminished with lower TBAHSO4 loadings (entries 3 and 4). Decreasing fluoride stoichiometry was detrimental but increased nucleophile did not improve the yield (entries 5 to 7). Finally, photocatalyst 1•BF4 and irradiation were required as minimal (entry 8) and no conversion (entry 9) were observed with their exclusion.
Table 1.
Reaction optimization for arene deoxyfluorination
| entry | deviation from standard conditions | yield (%)a |
|---|---|---|
| 1 | none | 79b |
| 2 | KF (5.0 equiv) | 78b |
| 3 | TBAHSO4 (0.5 equiv) | 73c |
| 4 | TBAHSO4 (0.25 equiv) | 53c |
| 5 | TBAHSO4 (0.5 equiv), DCM:H2O (10:1), KF (5.0 equiv) | 48c |
| 6 | TBAHSO4 (0.5 equiv), DCM:H2O (10:1), KF (10.0 equiv) | 67c |
| 7 | TBAHSO4 (0.5 equiv), DCM:H2O (10:1), CsF (3.0 equiv) | 52c |
| 8 | No photocatalyst | N.D.c |
| 9 | No irradiation | 9 |
| 10 | TBAHSO4 (0.5 equiv), DCM:H2O (2.7:1), CsF (10.0 equiv) | 35c |
| 11 | TBAHSO4 (0.5 equiv), DCM:H2O (1:1), CsF (10.0 equiv) | 23c |
| 12 | TBAHSO4 (0.5 equiv), DCM:t-BuOH (10:1), CsF (10.0 equiv) | 5c |
| 13 | TBAHSO4 (0.5 equiv), DCM:t-BuOH:H2O (19:1:2), CsF (10.0 equiv) | 67c |
| 14 | TBAHSO4 (0.5 equiv), DCM:t-BuOH:H2O (100:1:9), CsF (10.0 equiv) | 71c |
| 15 | w/o TBAHSO4, DCM:t-BuOH:H2O (100:1:9), CsF (10.0 equiv) | 22c |
| 16 | w/o TBAHSO4, DCM:H2O (10:1) TBAF•H2O (10.0 equiv) | 9c |
| 17 | KHF2 (10.0 equiv) instead of CsF | 27c |

GC yields using 1,2 dichlorobenzene as an internal standard;
427 nm LEDs;
455 nm LEDs. See Supplementary Table 1 for full optimization details.
One important aspect of our transformation is the use of a biphasic solvent system, which is counterintuitive given fluoride’s reduced nucleophilicity in the presence of water.27 Our initial hypothesis for using water was to solubilize CsF and form TBAF from TBAHSO4. Small amounts of water are necessary for successful deoxyfluorination, however increasing the ratio of water to DCE led to a significant drop in the reaction efficiencies (entries 10 & 11). We were curious to examine if water was acting mainly as a hydrogen-bond donor in this reaction and investigated alcohol solvents such as t-BuOH in place of water, as the hydrogen-bonding stability of t-BuOH has been invoked in enhancing the nucleophilicity of alkali metal fluorides.28,29 This change did not prove advantageous and a significant loss of reactivity was observed (entry 12).30 However, when water was reintroduced as a co-solvent to t-BuOH and DCE, significant yield increases were observed (entries 13 & 14). Furthermore, when TBAHSO4 was removed or if TBAF trihydrate were used, yields suffered (entries 15 & 16). The latter result is surprising, but it may suggest that TBAF is decomposing quickly under the reaction conditions.
While it would be advantageous to use pure TBAF, it is prone to Hofmann elimination and unstable at room temperature, decomposing to tributylamine and bifluoride above room temperature31; MeCN solutions of TBAF are only stable for hours.32 Unfortunately, potassium bifluoride is an inefficient fluoride source in this case, resulting in significantly decreased yield (27%, entry 17) observed relative to CsF (79%).
Taken together, these observations suggest that anhydrous TBAF could be the active fluoride source during the reaction, formed in small quantities via phase transfer between aqueous CsF and the TBAHSO4 in the organic layer. This likely prevents high amounts of Hofmann elimination from occurring. Alternatively, the key species could be an activated fluoride benefiting from hydrogen bond-stabilizing interactions.33 Given the practicality and accessibility of alkali metal fluorides and tetraalkylammonium phase transfer reagents, we decided to move forward with this set of conditions.
Substrate scope for CRA-SNAr with 19F.
With the optimized conditions in hand, we sought to extend our method to a range of (hetero)aromatic systems (Figure 3). 4-Chlorophenoxy-substituted biphenyls were competent substrates as 2 to 5 furnished the respective fluorinated arenes in moderate to good yield. Cross-coupling handles for nickel catalysis were also compatible with our method, as (4-chlorophenoxy)phenyl pivalate and 4-chlorophenoxy-4-chlorobenzene were converted to their fluorinated analogs (6 & 7) in moderate to good yield. Electron-rich bis-oxygenated arenes are typically poor substrates for SNAr and have limited reactivity under uronium-17 and fluorosulfonate-18 based deoxyfluorination conditions. However, ethylguaiacol analogs were successfully fluorinated selectively at the phenoxy group using our method to furnish 8 and 9, with 4-substitution of the ethyl moiety relative to the nucleofuge furnishing higher yields than the respective 3-substituted isomer. Halogenated aromatic biaryl ethers were converted to their respective aryl fluorides (10 to 13) in moderate to good yields. Electron-withdrawing substituents such as carbonyls, nitriles, and ethylacryolyl groups were tolerated under reaction conditions, providing the anticipated fluorinated arenes (14 to 18). Dioxygenated benzyl alcohol and phenyl 2-butanone were also successfully converted to the respective desired fluorinated products 19 and 20, however a precipitous drop in yield was observed for the 4-substituted mono-oxygenated protected benzyl alcohol (21). This result thus prompted a brief examination of compatible functional groups. Arylsulfonamide (22), pyridine (23), and pyrimidine (24) substrates were fluorinated in moderate to good yield, while cyclohexyl (25), N-carboxybenzyl (Cbz)-protected piperidine (26), and tetrahydropyran (27) were converted to their fluorinated analogs albeit in modest levels of reaction efficiency. Tri-oxygenated aromatics are typically too electron rich to be fluorinated by traditional SNAr strategies, thus the facile synthesis of fluorinated 28 and 29 from their phenyloxy counterparts demonstrates the complementarity of our arene substitution method. Heterocycles were also compatible as competent substrates for this transformation as carbazole (30), pyridines (31 to 34), and pyrimidine (35) were successfully fluorinated. The synthesis of 35 is particularly interesting as it can be deprotected under mild acidic conditions34 to reveal 5-fluorouracil, an important antineoplastic drug typically used as first-line treatment of late-stage colorectal cancers.35 Bioactive molecules such as the antidiabetic nateglinide (36) and its diastereomer (37) were successfully obtained alongside fluorinated phenylalanines (38 to 40) and O-methyl tyrosines (41 and 42). Bisprotection of the amine as a phthalimide appeared to improve the yield, suggesting that unproductive pathways involving the monoprotected amine may be responsible for the observed decreases in yield. Additionally, substitution of the phenoxy nucleofuge relative to the glycine group appears important as a higher yield was observed for 42 than 41.
Figure 3. 19F fluorination of electron-rich aromatics.
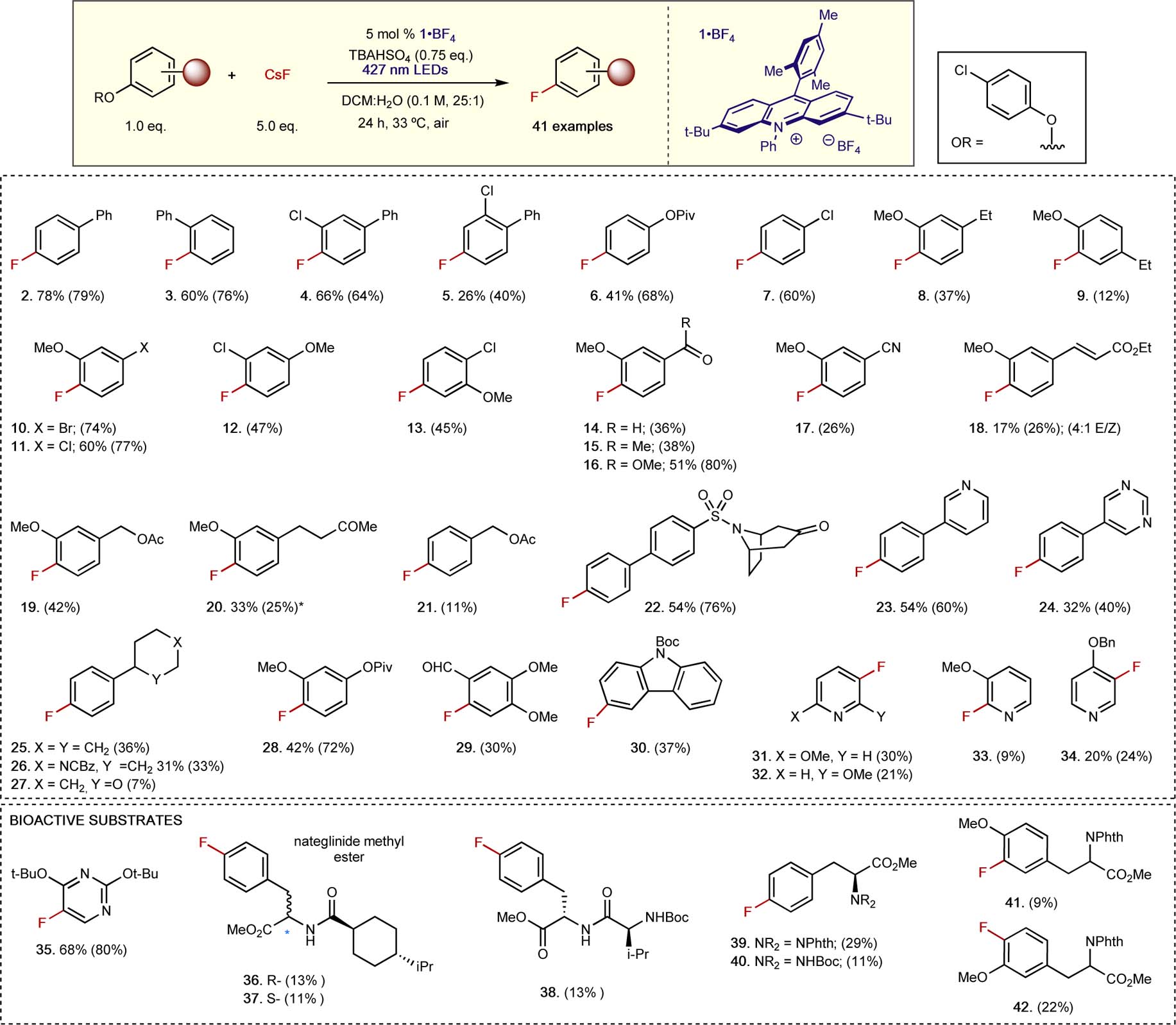
19F-NMR yields relative to an internal standard (fluorobenzene) are denoted by bracket; *NMR/isolated yield discrepancy due to excess remaining solvent for volatile product.
Rationale for deoxyfluorination site-selectivity
During our studies, the mass balance for low-yielding reactions is typically unreacted starting material, although we observed the formation of 4-chlorofluorobenzene (6%−19%), which arises from competitive deoxyfluorination, for several of our substrates (5, 14, 21, 34 and 40) (Figure 4). Because the formation of 4-chlorofluorobenzene is minimal for more electron-rich substrates, we hypothesized that differences in the thermodynamic stability of resultant arene cation radicals may be responsible for deoxyfluorination bias. For example, a 0.27 V difference in Ep/2 between the arene portions of A-1 results in highly selective deoxyfluorination whereas a separation of 0.06 V between the two halves of B-1 leads to an almost equal mixture of 21 and 7 (Figure 4A and 4B). Conversely, when the more electron-rich dioxygenated arene C-1 is used, a high degree of deoxyfluorination bias returns (Figure 4C). Furthermore, changing the nucleofuge to the more electron-deficient 4-cyanobenzene (D-1) results in the complete selectivity of deoxyfluorination on the more electron-rich aromatic ring albeit with decreased yield (Figure 4D). Therefore, deoxyfluorination site selectivity appears to be influenced by differences in Ep/2, thus supporting our initial hypothesis that the more electron-rich arene possesses a greater degree of partial positive charge in the cation radical and is preferentially fluorinated. We anticipate that this reactivity pattern can be used to guide future attempts at improving site selectivity.
Figure 4. Rationale for Arene Selectivity.

Greater disparities between the arene oxidation potentials lead to enhanced deoxyfluorination regioselectivity (a, c, d) whereas a lack of selectivity is observed when there is no difference in half peak potential (Ep/2) (b). Ep/2 is measured versus a saturated calomel electrode (SCE).
Developing CRA-SNAr deoxyfluorination with [18F]TBAF.
Given the success of the site-selective C–19F bond formation via our deoxyfluorination method, we began developing an analogous method for radiofluorination. The key difference between 19F− and 18F− fluorination protocols is the change of reagent stoichiometry; in 19F fluorination, fluoride is used in excess, whereas 18F fluorination employs the radioisotope as limiting reagent. As a starting point, we elected to adapt the conditions delineated in our method for arene C-H radiofluorination, in which we used an acridinium photocatalyst, [18F]-tetrabutylammonium fluoride ([18F]TBAF) and a 450 nm laser irradiation source,23 to the current transformation. After extensive optimization (Table 2), we found that adding excess tetrabutylammonium bicarbonate (TBAHCO3) and running the reaction for 30 min in a DCE:MeCN: t-BuOH solvent mixture gave [18F]2 in excellent RCY (entry 1). The solvent mixture was chosen as it provided general substrate solubility. We hypothesize that the excess TBAHCO3 helps to minimize the decomposition of [18F]TBAF via Hofmann elimination and buffers the system to a basic pH, which has been shown to improve [18F]fluoride nucleophilicity.36 In the absence of TBAHCO3, a significant drop in yield was observed (entry 2). Water proved deleterious to this reaction, as no product was observed when 100 μL of H2O was added to the reaction mixture. Since [18F]TBAF is usually synthesized with minimal H2O present,4 our results suggest that t-BuOH is forming beneficial adducts with [18F]TBAF30 and that excessive water mitigates 18F− nucleophilicity, which mirrors the drop in 19F-deoxyfluorination when H2O concentration increases. Next, we examined the impact of substrate and photocatalyst equivalency. A five-fold decrease of substrate did not lead to any appreciable loss of RCY (entry 4), however a larger loss was observed when a 25-fold drop in substrate was implemented (entry 5). Interestingly, a five-fold reduction in the amount of 1 led to a greater RCY loss than substrate reduction did (entry 6). This observation suggests that successful deoxyradiofluorination relies more on maintaining an appreciable amount of photocatalyst than substrate concentration.
Table 2.
Optimization for arene deoxyradiofluorination
| entry | deviation from standard conditions | RCY (%) |
|---|---|---|
| 1 | none | 94.6 ± 0.4 (n=3) |
| 2 | w/o TBAHCO3 | 75.2 |
| 3 | w/o TBAHCO3, + H2O (100 μL) | N.D. |
| 4 | S2 (0.01 mmol), w/o TBAHCO3 | 71.7 |
| 5 | S2 (0.002 mmol), w/o TBAHCO3 | 34.3 |
| 6 | S2 (0.01 mmol), 1 (0.3 mg), w/o TBAHCO3 | 24.3 |
| 7 | blue LEDs | 83.2 |
| 8 | No photocatalyst | N.D.b |
| 9 | No irradiation | 2.6 |

All radiochemical yields (RCYs) are calculated by HPLC isolation starting from azeotropically dried [18F]TBAF, and represent one experiment unless otherwise noted;
See Supplementary Tables 2–7 for full optimization details.
Replacing the 450 nm laser with 455 nm LEDs resulted in a minor RCY drop (entry 7), but a good RCY was obtained with 455 LEDs, indicating that the 450 nm laser can be replaced, albeit at the expense of a slight loss in RCY. Finally, control experiments (entries 8 and 9) show that both are crucial for radiodeoxyfluorination; minimal to no reaction is observed in their absence.
Radiofluorination reactions are typically conducted on a short timescale due to a relatively short 18F half-life (t1/2 ~ 110 min). We were interested to examine the impact of time on this transformation, especially since our C-H radiofluorination protocol required extended reaction times to observe yield increases. Thus, we were surprised to see that significant product formation (62.7% RCY) occurs after only 2 minutes and displays yield growth until 20 min (Extended Data Figure 1). However, the lag in conversion after 2 min probably arises from competitive oxidation of the nucleofuge (4-chlorophenol) by 1.37 It is important to note that while 20 mins was optimal for S2, other substrates required slightly longer reaction times, thus we chose 30 minutes as the standard for substrate scope studies. Overall, this reactivity is different from previous deoxyfluorination strategies as the ground state stabilities between the arene and nucleofuge are similar, but selective single electron arene oxidation enables fluoride trapping at the desired aromatic ring.
Nucleofuge scope for deoxyradiofluorination beyond 4-chlorophenoxy ethers.
While the 4-chlorophenoxy group is the optimal nucleofuge for our system, it is important to note that other electron-poor aryloxy groups explored earlier are also compatible with our radiofluorination method (Figure 5). A greater range of compatible carbonate and thiocarbonate leaving groups all provided [18F]2 in moderate to good RCY (24.1% to 57.1% RCYs). However, not all nucleofuges were useful leaving groups as acetate, mesylate, tosylate and methyl ether nucleofuges demonstrated minimal or unsuccessful radiofluorination. Nevertheless, these results demonstrate that nucleofuge tolerance is more general with arene deoxyradiofluorination, and these competent alternatives should be considered if the 4-chlorophenoxy congener is synthetically difficult or time-consuming to access. For the remainder of this study, we opted to use the 4-chlorophenoxy group due to its greater nucleofuge efficacy for both deoxyfluorination methods.
Figure 5. Nucleofuge scope for the synthesis of [18F]4-fluorobiphenyl ([18F]-2).

All radiochemical yields (RCYs) are calculated by HPLC isolation starting from azeotropically dried [18F]TBAF, decay corrected and represent one experiment unless otherwise noted. *See Supplementary Table S63 for full optimization details.
Substrate scope for CRA-SNAr with 18F.
While the success of our 19F−-fluorination method appears to be substrate-dependent, greater generality is observed for radiofluorination (Figure 6) as the 18F-labeled congeners are isolated with moderate to excellent RCY. We hypothesize that the relatively higher arene concentration and increased stability of [18F]TBAF enables efficient [18F]F− capture while minimizing substrate sensitivity observed in our 19F-deoxyfluorination conditions. Consequently, we are able to obtain purified 18F-labeled electron-rich arenes, ranging from simple aromatics ([18F]2 to [18F]34) to molecules with demonstrated bioactivity ([18F]35 to [18F]42). One notable highlight of our deoxyradiofluorination method is the highly efficient synthesis of ([18F]35) in excellent RCY; [18F]35 also can be easily deprotected to known radiotracer 18F-5-fluorouracil ([18F]35–1)38,39 under acidic conditions. This radiopharmaceutical is thus synthesized in two steps with an overall RCY of 82.4 ± 4% and a molar activity of 74.7 ± 14.1 GBq/μmol (2.02 ± 0.37 Ci/μmol). This result is complementary to existing routes involving spirocylic hypervalent iodoniums40 or stoichiometric aryl nickel complexes34, and enables facile production of [18F]35–1. We anticipate that this result should accelerate the availability of high molar activity 18F-FU34 and enable downstream radiochemical synthesis of 18F-FU pro(radio)drugs with extended biological lifetimes and safety profiles.41–43 The former is especially important as PET radiotracers often have molar activities of 37 GBq/μmol (1.0 Ci/μmol).44
Figure 6. Reaction scope of 18F fluorination of aromatics.
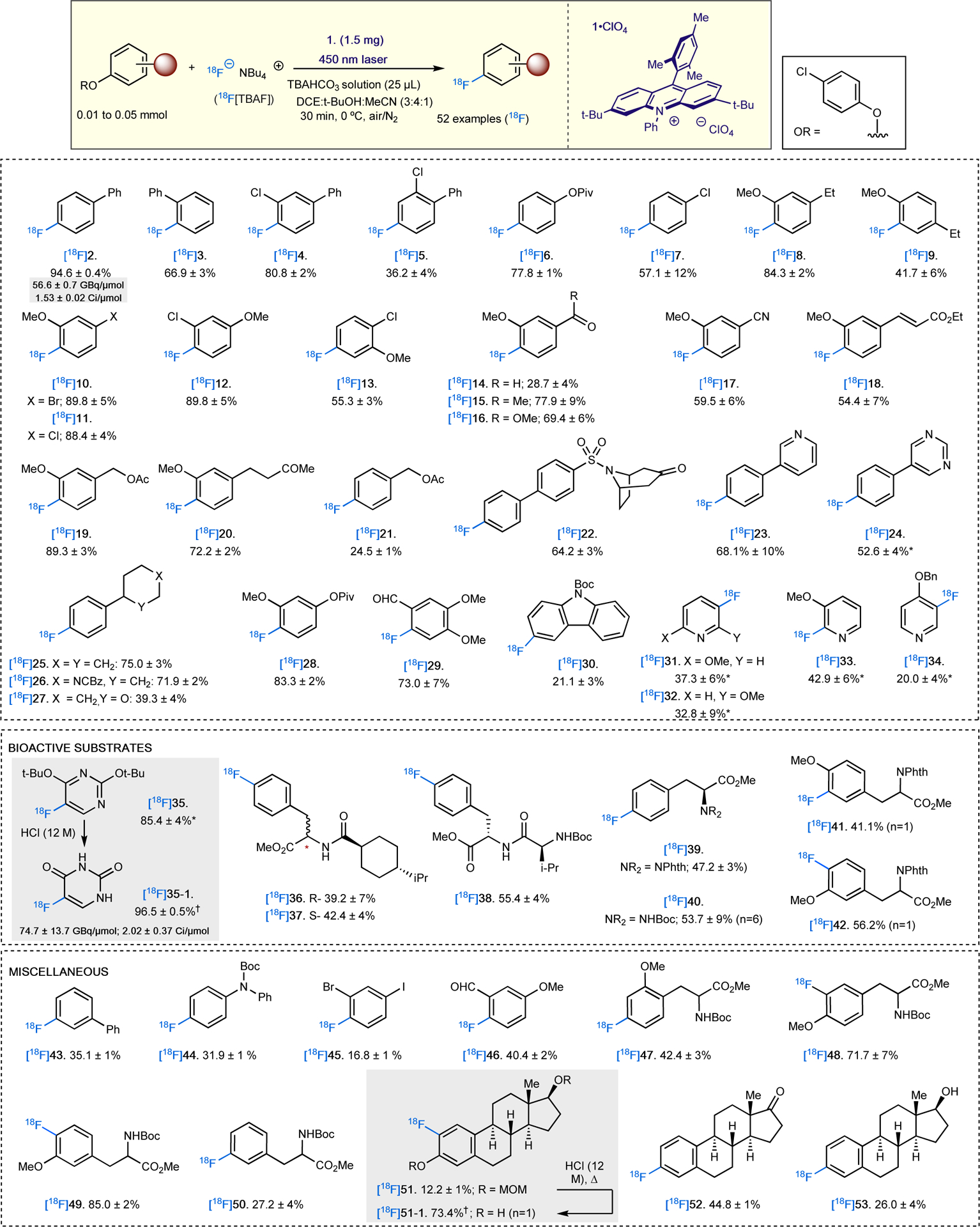
All radiochemical yields (RCYs) are calculated by HPLC isolation starting from azeotropically dried [18F]TBAF, decay corrected, and averaged over 3 experiments unless otherwise noted. *N2 atmosphere used instead of air; †Deprotection RCY.
Our radiofluorination method can also be applied to several arenes that demonstrate minimal conversion under our 19F-fluorination conditions. Simple arenes such [18F]3-fluoro-1,1’-biphenyl ([18F]43), [18F]4-fluoro-N-Boc-phenylaniline ([18F]44), [18F]3-bromo-4-iodo-fluorobenzene ([18F]45) and [18F]2-fluoro-5-methoxybenzaldehyde. ([18F]46) were obtained in moderate to good RCY; conversions for these substrates when using 19F were usually <5%. Radiofluorinated N-Boc-O-methyltyrosines ([18F]47 to [18F]49) and phenylalanine ([18F]50) that demonstrated poor yields under regular fluorination conditions due to the sensitivity of the NHBoc moiety were successfully fluorinated in moderate to excellent RCY, demonstrating a greater substrate compatibility for the radiofluorination method. One limitation of our current method are unprotected amines, which are oxidized by the acridinium photocatalyst. However, deactivating the amine with common protecting groups (e.g. NHBoc) limits this unproductive pathway. Additionally, we also show that aryl bromides and iodides are tolerated under the current system, albeit with limited examples.
Products [18F]43, [18F]47 and [18F]50 are especially interesting as deoxyradiofluorination at the meta position relative to the most electron-donating group is observed, thus suggesting that this transformation is not limited to substrates with para or ortho electron-rich substituents. Finally, estrogens bis-methoxymethyl (MOM)-protected [18F]2-fluoroestradiol ([18F]51) and its deprotected form ([18F]51–1), [18F]deoxy-3-fluoroestrone ([18F]52), and [18F]deoxy-3-fluoroestradiol ([18F]53) were obtained in moderate to good RCY. These 18F-labeled steroids are potential alternatives to PET radiotracer [18F]16α-fluoroestradiol ([18F]FES) which is used for quantifying the expression of estrogen and progesterone receptors in primary and metastatic breast cancers.46
CONCLUSIONS
Nucleophilic aromatic substitution (SNAr) is a powerful method for substrate-dependent functionalizations, but it has historically relied on electron-withdrawing groups to stabilize charge buildup in Meisenheimer complexes or neighboring heteroatoms (e.g. nitrogen in pyridine) to accelerate concerted substitution. Our photoredox-catalysed arene deoxy(radio)fluorination method reverses the polarity demand associated with SNAr as it operates by CRA-SNAr. Consequently, selective (radio)fluorination of electron-rich arenes with CsF/KF and [18F]TBAF is obtained under mild conditions. Site-selectivity is dictated by arene electronics, thus more electron-rich aromatic species can be selectivity fluorinated over the less oxidizable arene nucleofuge. (Thio)carbonate and acyl nucleofuges can also replace the 4-chlorophenoxy group, but demonstrate lower deoxy(radio)fluorination efficiency. The utility of our radiofluorination strategy is highlighted by short reaction times, compatibility with multiple nucleofuges, and the high-yielding syntheses of [18F]5-fluorouracil, an important PET imaging agent for colon cancer.
Taken together, our fluorination approach enables the development of fluorinated and radiofluorinated compounds that are typically difficult to access by classical SNAr or deoxyfluorination strategies, and we anticipate that the ease of our method, coupled with facile access to substrate precursors, will allow for rapid access to new aromatic 18F-labeled PET tracers of use in neurology, oncology, and other disease diagnosis applications. Insights gained from this study should also aid in the discovery and translation of photoredox-catalysed arene functionalizations to radiochemistry.
Methods
General procedure for nucleophilic deoxyfluorination
To a clean, dry 2 dram vial containing a Teflon-coated magnetic stir bar was added 76.0 mg of cesium fluoride (0.500 mmol, 5 equiv) under an inert atmosphere. The vial was then removed from the inert atmosphere and a series of reagents were added: 0.0050 mmol of 1 (0.05 equiv), tetrabutylammonium hydrogensulfate (0.075 mmol, 0.75 equiv), and arene (0.100 mmol, 1.0 equiv). The reagent mixture was then dissolved in DCM (1.2 mL, 0.083 M) and DI water (0.05 mL, 0.5 M) was subsequently added. The vial was then sealed with a Teflon-lined septum screw cap and stirred rapidly for approximately 1 minute. The vial was positioned on a stir plate approximately 3–4 cm from a Kessil PR160–427 LED Photoredox Light supplying blue light (λ = 427 nm) and irradiated for a designated time. The crude reaction mixture was then concentrated in vacuo and purified by flash chromatography or reverse-phase flash liquid chromatography. Isolation yields were obtained on a 0.20 mmol scale while 19F NMR yields are measured on a 0.10 mmol scale, using an equimolar amount of fluorobenzene (0.10 mmol) as an internal standard.
General procedure for the preparation of [18F]TBAF
[18F]Fluoride was produced via the 18O(p,n)18F reaction by proton irradiation (40 μA, 45 min) of an [18O]H2O containing target in a GE PETTrace cyclotron. The aqueous solution of [18F]fluoride was delivered in to a hot cell and passed through a QMA cartridge (water preconditioning) to trap the [18F]fluoride. The [18F]fluoride was eluted into a 5 mL V-vial which sealed with a Teflon-lined septum screw cap with 600 μL solution of 70 μL tetrabutylammonium bicarbonate (TBAHCO3) solution (20%, w/w), 53 μL of H2O and 477 μL of MeCN. This solution was azeotropically dried with MeCN (1 mL × 3) under a stream of Argon at 100 °C and then placed under vacuum for 3 min. The resulting residue was diluted with ~1 mL anhydrous MeCN to obtain the [18F]TBAF solution which was used for the labeling reaction. In our procedure, approximately 2.2 Ci 18F was trapped on QMA and ~2.1 Ci was eluted in to the V-vial and 1.6–1.8 Ci [18F]TBAF was obtained after drying.
General procedure for nucleophilic deoxyradiofluorination
Substrate (0.01 or 0.05mmol) and photocatalyst (1.5 mg) were weighed into a 1.5 mL Eppendorf tube and transferred (with solvent when the substrate is liquid or oil) into a 5 mL V-vial via pipette. DCE (300 μL), anhydrous MeCN (45–65 μL), t-BuOH(400 μL) and 25 μL of TBAHCO3 in MeCN solution (~60 mg/mL) were sequentially added to the V-vial. Then a 10–30 μL aliquot of [18F]TBAF in MeCN (typically 0.37–1.11 GBq (10–30 mCi)) was added to the reaction vial via pipette. The reaction V-vial was then fixed on an iron support and cooled using an ice bath. A needle connected to an air/N2 filled balloon was inserted to the bottom of the V-vial and the reaction medium was continuously sparged with air/N2 for the entire reaction time. The reaction was then irradiated top-down with an optic fiber of an OEM diode laser (MDL-D-450, 450 nm, 3.5W after fiber coupling) (Supplementary Fig. 3) or a A160WE Tuna Blue Kessil LED lamp (Supplementary Fig. 4) for 30 min and 18F activity was recorded at the end of the reaction. The resulting solution was diluted and evenly mixed with MeCN (500 μL). An aliquot of the reaction mixture (typically 0.011–0.037 GBq (300–1000 μCi)) was taken for radio-HPLC analysis. The activity injected into the HPLC was measured (this activity was denoted by α) and the time was recorded. The fraction corresponding to radiolabeled product was collected and the activity was measured (this activity was denoted by β) and the time was recorded. The decay-corrected β could be calculated from the recorded isolation time of each substrate. The decay-corrected radiochemical yield (RCY) was obtained by dividing the decay-corrected β by α. Quality control (QC) was run separately to ensure the purity of isolated radiolabeled compounds. Co-injection of the purified 18F-labeled compound with commercial or synthesized 19F standards via HPLC was used to confirm the identity of the isolated radiolabeled compound.
Radio-HPLC analysis and characterization for 18F-radiolabeled arenes
All 18F-labelling reactions were performed according to the general procedure for arene radiofluorination. The starting activity ([18F]TBAF), injected and collected activities, isolation time and decay corrected activity for each reaction are summarized for each substrate. The radiolabeling of each substrate was run in triplicate unless specified otherwise. All 18F-labeled products were analyzed according to the general HPLC conditions. QC testing was run separately after collection using a different HPLC condition, which is listed respectively for each substrate in Supplementary Methods Section 1.6.6. The red HPLC traces in the spectra were obtained with a UV signal at 212 nm. The black HPLC traces represent the radio signal. The desired radiofluorinated products were confirmed via co-injection of the purified -18F-labeled species and an authentic 19F-labeled standard.
Extended Data
Extended Data Figure 1. Time dependence studies for deoxyradiofluorination with arene S2.

All radiochemical yields (RCYs) are calculated by HPLC isolation starting from azeotropically dried [18F]TBAF, decay corrected and represent one experiment unless otherwise noted. See Supplemental Table S5 for more details.
Supplementary Material
Acknowledgements:
Financial support was provided in part by the National Institutes of Health (NIBIB) 5R01EB014354 (Z.L.) and by the UNC Department of Radiology, Biomedical Research Imaging Center, and UNC Lineberger Comprehensive Cancer Center (start-up fund to Z.L.). UNC LCCC pilot grant, N.E.S.T. and V.A.P are grateful for NSF Graduate Research Fellowships. A.L. thanks the American Australian Association for a Chevron Fellowship. W.C. thanks Dr. Gerald T. Bida for assistance with cyclotron operation.
Abbreviations
- Mes
mesityl
- t-Bu
tert-butyl
- Boc
tert-butyloxycarbonyl
Footnotes
Competing Interests: The authors declare no competing interests.
Data Availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information or from the authors upon reasonable request.45
References:
- 1.Wang J et al. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 114, 2432–2506 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y et al. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 116, 422–518 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Fujiwara T & O’Hagan D Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem 167, 16–29 (2014). [Google Scholar]
- 4.Preshlock S, Tredwell M & Gouverneur V 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev 116, 719–766 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Coenen HH et al. Fluorine-18 radiopharmaceuticals beyond [18F]FDG for use in oncology and neurosciences. Nucl. Med. Biol 37, 727–740 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Terrier F Modern Nucleophilic Aromatic Substitution: Terrier/Modern Nucleophilic Aromatic Substitution. (Wiley-VCH Verlag GmbH & Co. KGaA, 2013). doi: 10.1002/9783527656141. [DOI] [Google Scholar]
- 7.Deng X et al. Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed 58, 2580–2605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwan EE, Zeng Y, Besser HA & Jacobsen EN Concerted nucleophilic aromatic substitutions. Nat. Chem 10, 917–923 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann CN, Hooker JM & Ritter T Concerted nucleophilic aromatic substitution with 19F− and 18F−. Nature 534, 369–373 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Watson DA et al. Formation of ArF from LPdAr(F): Catalytic Conversion of Aryl Triflates to Aryl Fluorides. Science 325, 1661–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sather AC et al. A Fluorinated Ligand Enables Room-Temperature and Regioselective Pd-Catalyzed Fluorination of Aryl Triflates and Bromides. J. Am. Chem. Soc 137, 13433–13438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye Y, Schimler SD, Hanley PS & Sanford MS Cu(OTf)2-Mediated Fluorination of Aryltrifluoroborates with Potassium Fluoride. J. Am. Chem. Soc 135, 16292–16295 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Furuya T, Kaiser HM & Ritter T Palladium-Mediated Fluorination of Arylboronic Acids. Angew. Chem. Int. Ed 47, 5993–5996 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Ichiishi N et al. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett 16, 3224–3227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell MG & Ritter T Modern Carbon–Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev 115, 612–633 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Cardinale J et al. Carrier-effect on palladium-catalyzed, nucleophilic 18F-fluorination of aryl triflates. J. Label. Compd. Radiopharm 55, 450–453 (2012). [Google Scholar]
- 17.Tang P, Wang W & Ritter T Deoxyfluorination of Phenols. J. Am. Chem. Soc 133, 11482–11484 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schimler SD et al. Nucleophilic Deoxyfluorination of Phenols via Aryl Fluorosulfonate Intermediates. J. Am. Chem. Soc 139, 1452–1455 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Schimler SD, Froese RDJ, Bland DC & Sanford MS Reactions of Arylsulfonate Electrophiles with NMe4F: Mechanistic Insight, Reactivity, and Scope. J. Org. Chem 83, 11178–11190 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Beyzavi MH et al. 18F-Deoxyfluorination of Phenols via Ru π-Complexes. ACS Cent. Sci 3, 944–948 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tay NES & Nicewicz DA Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. J. Am. Chem. Soc 139, 16100–16104 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Holmberg-Douglas N & Nicewicz DA Arene Cyanation via Cation-Radical Accelerated-Nucleophilic Aromatic Substitution. Org. Lett 21, 7114–7118 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Chen W et al. Direct arene C–H fluorination with 18F− via organic photoredox catalysis. Science 364, 1170–1174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zweig Arnold., Hodgson WG & Jura WH The Oxidation of Methoxybenzenes. J. Am. Chem. Soc 86, 4124–4129 (1964). [Google Scholar]
- 25.Um I-H, Kim M-Y & Dust JM Medium effect (water versus MeCN) on reactivity and reaction pathways for the SNAr reaction of 1-aryloxy-2,4-dinitrobenzenes with cyclic secondary amines. Can. J. Chem 95, 1273–1279 (2017). [Google Scholar]
- 26.Schmittel M & Burghart A Understanding Reactivity Patterns of Radical Cations. Angew. Chem. Int. Ed. Engl 36, 2550–2589 (1997). [Google Scholar]
- 27.Nolte C, Ammer J & Mayr H Nucleofugality and Nucleophilicity of Fluoride in Protic Solvents. J. Org. Chem 77, 3325–3335 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Kim DW et al. A New Class of SN2 Reactions Catalyzed by Protic Solvents: Facile Fluorination for Isotopic Labeling of Diagnostic Molecules. J. Am. Chem. Soc 128, 16394–16397 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Kim DW et al. Facile Nucleophilic Fluorination Reactions Using tert-Alcohols as a Reaction Medium: Significantly Enhanced Reactivity of Alkali Metal Fluorides and Improved Selectivity. J. Org. Chem 73, 957–962 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Kim DW, Jeong H-J, Lim ST & Sohn M-H Tetrabutylammonium Tetra(tert-Butyl Alcohol)-Coordinated Fluoride as a Facile Fluoride Source. Angew. Chem. Int. Ed 47, 8404–8406 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Sharma RK & Fry JL Instability of anhydrous tetra-n-alkylammonium fluorides. J. Org. Chem 48, 2112–2114 (1983). [Google Scholar]
- 32.Sun H & DiMagno SG Anhydrous Tetrabutylammonium Fluoride. J. Am. Chem. Soc 127, 2050–2051 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Lee J-W et al. Hydrogen-bond promoted nucleophilic fluorination: concept, mechanism and applications in positron emission tomography. Chem. Soc. Rev 45, 4638–4650 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Hoover AJ et al. A Transmetalation Reaction Enables the Synthesis of [18F]5-Fluorouracil from [18F]Fluoride for Human PET Imaging. Organometallics 35, 1008–1014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webber EM, Kauffman TL, O’Connor E & Goddard KA Systematic review of the predictive effect of MSI status in colorectal cancer patients undergoing 5FU-based chemotherapy. BMC Cancer 15, 156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemaire CF et al. Fast Production of Highly Reactive No-Carrier-Added [18F]Fluoride for the Labeling of Radiopharmaceuticals. Angew. Chem. Int. Ed 49, 3161–3164 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Roth HG, Romero NA & Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 27, 714–723 (2016). [Google Scholar]
- 38.Fowler JS, Finn RD, Lambrecht RM & Wolf AP The Synthesis of 18F-5-Fluorouracil. VII. J. Nucl. Med 14, 63–64 (1973). [PubMed] [Google Scholar]
- 39.Saleem A et al. Modulation of fluorouracil tissue pharmacokinetics by eniluracil: in-vivo imaging of drug action. The Lancet 355, 2125–2131 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Rotstein BH, Stephenson NA, Vasdev N & Liang SH Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun 5, 4365 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Sulkes A, Benner SE & Canetta RM Uracil-ftorafur: an oral fluoropyrimidine active in colorectal cancer. J. Clin. Oncol 16, 3461–3475 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Álvarez P et al. 5-Fluorouracil derivatives: a patent review. Expert Opin. Ther. Pat 22, 107–123 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Li M, Liang Z, Sun X, Gong T & Zhang Z A Polymeric Prodrug of 5-Fluorouracil-1-Acetic Acid Using a Multi-Hydroxyl Polyethylene Glycol Derivative as the Drug Carrier. PLOS ONE 9, e112888 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sergeev M et al. Performing radiosynthesis in microvolumes to maximize molar activity of tracers for positron emission tomography. Commun. Chem 1, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun Y et al. The Preliminary Study of 16α-[18F]fluoroestradiol PET/CT in Assisting the Individualized Treatment Decisions of Breast Cancer Patients. PLOS ONE 10, e0116341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao GJ, Clark AS, Schubert EK & Mankoff DA 18F-Fluoroestradiol PET: Current Status and Potential Future Clinical Applications. J. Nucl. Med 57, 1269–1275 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.