

Abstract
The main protease (3CL Mpro) from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes COVID-19, is an essential enzyme for viral replication with no human counterpart, making it an attractive drug target. To date, no small-molecule clinical drugs are available that specifically inhibit SARS-CoV-2 Mpro. To aid rational drug design, we determined a neutron structure of Mpro in complex with the α-ketoamide inhibitor telaprevir at near-physiological (22 °C) temperature. We directly observed protonation states in the inhibitor complex and compared them with those in the ligand-free Mpro, revealing modulation of the active-site protonation states upon telaprevir binding. We suggest that binding of other α-ketoamide covalent inhibitors can lead to the same protonation state changes in the Mpro active site. Thus, by studying the protonation state changes induced by inhibitors, we provide crucial insights to help guide rational drug design, allowing precise tailoring of inhibitors to manipulate the electrostatic environment of SARS-CoV-2 Mpro.
Introduction
The number of confirmed COVID-19 cases worldwide is has surpassed one hundred million, while the number of deaths is relentlessly approaching a grim milestone of three million. Sadly, this deadly disease caused by the novel coronavirus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2)1−4 has become one of the leading causes of death on the planet in 2020, according to the World Health Organization (www.who.int). Although several vaccines have been developed5−7 to slow the spread of SARS-CoV-2, the need for therapeutic intervention options, including small-molecule drugs that inhibit essential steps in the viral replication cycle, cannot be overstated.8−12 Small-molecule clinical drugs have shown tremendous success in treating people infected with human immunodeficiency virus,13,14 hepatitis C,15,16 and influenza17,18 viruses, and an RNA polymerase inhibitor remdesivir has been recently approved for the treatment of COVID-19 by the US Food and Drug Administration.19
SARS-CoV-2, a single-stranded, positive-sense RNA virus with a genome comprising ∼30k nucleotides, belongs to the genus β-coronavirus of the family Coronaviridae.20 A vital step in the viral replication cycle is the cleavage of two polyproteins, pp1a and pp1ab, encoded by the viral replicase gene into individual functional viral proteins.20,21 Each polyprotein is mainly processed, or hydrolyzed, by a chymotrypsin-like protease, 3CL Mpro or main protease, that belongs to the class of cysteine protease enzymes.22,23 The functional main protease (hereafter referred to as Mpro) is essential for SARS-CoV-2 proliferation as the production of infectious virions depends on the enzymatic activity of Mpro. Hence, SARS-CoV-2 Mpro is undeniably a crucial target for designing specific small-molecule protease inhibitors24−29 and for potential repurposing of known clinical drugs.30−35 Though no clinical drugs are available for use against SARS-CoV-2 Mpro, several protease inhibitors have been designed to inhibit the very closely related SARS-CoV Mpro36−39 that shares 96% of amino acid sequence identity with the SARS-CoV-2 enzyme, has a similar catalytic efficiency, and an almost identical three-dimensional structure.25,27,40,41
Two identical protomers of SARS-CoV-2 Mpro, each with a molecular mass of ∼34 kDa, create the catalytically active homodimeric enzyme through non-covalent interactions (Figure 1A). Each protomer consists of three structural/functional domains—catalytic domains I (residues 8–101) and II (residues 102–184) and α-helical domain III (residues 201–303) crucial for protein dimerization (Figure 1B). Studies have shown that the monomeric enzyme is catalytically inactive, as was demonstrated for SARS-CoV Mpro.42,43 The active-site cavity is a shallow cleft located on the protein surface between domains I and II. There are six substrate-binding subsites, named S1′ through S5, that can bind either substrate residues or chemical groups of inhibitors in positions P1′ through P5. Peptide bond cleavage is carried out at the base of the well-defined subsite S1, where the non-canonical catalytic dyad composed of Cys145 and His41 is located. Catalysis is believed to be assisted by a water molecule positioned at the protein interior side of subsite S2 and hydrogen-bonded to the catalytic His41, His164, and Asp187.25,27,34,40 Scissile peptide bond cleavage begins through a nucleophilic attack by the Cys145 thiolate on the substrate carbonyl carbon. The negatively charged oxygen of the resultant hemithioketal intermediate is stabilized by a canonical oxyanion hole formed by the main chain amide NH groups of Gly143, Ser144, and Cys145.44 The hemithioketal intermediate is converted into a covalent acyl intermediate through the spontaneous cleavage of the scissile bond with the N-terminal portion of the substrate, and the acyl intermediate is then hydrolyzed by an incoming water molecule. Interestingly, subsites S2 and S4 need to be carved out by the substrate or inhibitor groups P2 and P4, respectively, that push protein residues away from their positions in the ligand-free enzyme.45 Conversely, subsites S1′, S3, and S5 are fully exposed to the bulk solvent.
Figure 1.

Joint X-ray/neutron structure of SARS-CoV-2 3CL Mpro and binding of hepatitis C clinical protease inhibitor telaprevir. (A) Catalytically active dimer is shown in a surface representation, with telaprevir shown in a ball-and-stick representation. (B) One enzyme protomer is shown in a cartoon representation colored according to the domain structure—domain I is yellow, domain II is blue, and domain III is dark pink. (C) Active-site cavity with the covalently bound telaprevir. H atoms are shown in gray, whereas D atoms are colored orange. (D) Chemical diagram of telaprevir with the ketone warhead shown in red. Telaprevir orientation matches that in panel C.
Our recent neutron crystallographic study of the ligand-free SARS-CoV-2 Mpro provided direct visualization of hydrogen (H) atom locations and hydrogen bonding interactions throughout the enzyme.46 The catalytic dyad was observed in a zwitterionic form in the enzyme without substrate or an inhibitor. The zwitterion comprises a deprotonated, negatively charged Cys145 thiolate and a doubly protonated, positively charged His41 imidazolium situated ∼3.8 Å apart from each other. Neutrons are unique probes of light atoms such as H as they are scattered by atomic nuclei instead of electron clouds, which interact with X-rays. Therefore, neutron crystallography can be used to accurately determine the positions of H and its heavier isotope deuterium (D) atoms in biomacromolecules.47,48 Deuterium has a potent neutron scattering length of 6.671 fm (https://www.ncnr.nist.gov/resources/n-lengths/), allowing its detection in protein structures even at moderate resolutions of 2.5–2.6 Å.49−51 Also, cold neutrons used in neutron crystallographic experiments with wavelengths in the range of 2–5 Å cause no radiation damage to biological samples; therefore, neutron diffraction data can be collected at near-physiological (room) temperature avoiding possible artifacts produced during cryo-cooling of protein crystals necessary for the mitigation of radiation damage in X-ray cryo-crystallography.45,48,52
Structure-guided and computer-assisted drug design approaches require a detailed atomic picture of a target biomacromolecule, which is generally obtained using X-ray cryo-crystallography. However, half of the atoms, namely, H atoms, are often overlooked in X-ray structures because they usually cannot be located with confidence unless X-ray data extend to sub-Å resolution. Even then, the most interesting and functionally relevant H atoms are typically invisible in electron density maps at ultrahigh resolutions because they are mobile and electron-poor, often participating in highly polarized chemical bonds.53−55 Detailed structures of the ligand-free and ligand-bound drug targets are essential to steer drug design efforts in the right direction. Knowledge of where H atoms relocate due to inhibitor binding can provide critical information on how protonation states and thus electric charges are modulated in the protein active-site cavity, improving rational drug design. For example, molecular dynamics simulations have predicted that protonation states in the SARS-CoV-2 Mpro active-site cavity may be altered when an inhibitor binds,56 but experimental evidence has been lacking.
We recently obtained room-temperature X-ray structures of the SARS-CoV-2 Mpro in complex with hepatitis C virus clinical protease inhibitors boceprevir, narlaprevir, and telaprevir, establishing their mode of binding and mechanism of action.57 Using in vitro enzyme inhibition assays, we determined that these clinical drugs designed against hepatitis C virus NS3/4A protease inhibited the SARS-CoV-2 Mpro activity in the micromolar range, with an IC50 value of 18 μM for telaprevir.57 Here, we report a room-temperature (22 °C) neutron structure of the SARS-CoV-2 Mpro in complex with telaprevir (Mpro–telaprevir) determined at 2.4 Å resolution and refined jointly with a room-temperature X-ray data set collected from the same crystal to 2.0 Å resolution, thereby improving the accuracy of the experimental model (Table S1).58 We chose this complex because it produces crystals of morphology and size amenable for neutron diffraction (Figure S1). Telaprevir represents a promising class of covalent protease inhibitors called α-ketoamides, while also possessing chemical groups in positions P1′ through P5 (Figure 1C,D).27 Locations of D atoms in Mpro were accurately determined and compared to those observed in our previous neutron structure46 of the ligand-free enzyme, which was crystallized at the same pD value. We discovered that protonation states of key histidine residues (His41, His163, and His164) in the SARS-CoV-2 Mpro active-site cavity are altered after telaprevir binds, revealing different electric charges between the ligand-free and ligand-bound states of the enzyme but conserving the net electric charge of +1 in the active-site cavity. These results are crucial for continued efforts in structure-guided and computational drug design, emphasizing the essential nature of a full atomic-level visualization of protein structure, function, and enzyme inhibition.
Results
Protonation States in Mpro–Telaprevir
The reactive carbonyl warhead of the telaprevir α-ketoamide group (Figure 1D) is attacked by the Cys145 thiolate nucleophile to generate a reversible hemithioketal. Like other α-ketoamide inhibitors,27,34,57 the nucleophilic attack is stereospecific, generating only the S-enantiomer, in which the newly formed hydroxyl group of the hemithioketal faces toward His41 (Figure 2A). The distance between the hemithioketal oxygen and His41 Nε2 is 2.6 Å, implicating a strong hydrogen bond. In our neutron structure of the ligand-free SARS-CoV-2 Mpro, we observed that the Cys145–His41 catalytic dyad adopts a charge-separated state.46 In Mpro–telaprevir, the nuclear density clearly shows that the hemithioketal hydroxyl is protonated (Figure 2B), whereas His41 is neutral. A reasonable explanation of this observation is that the hemithioketal oxygen is protonated by His41 through a direct proton transfer either in concert with the Cys145 nucleophilic attack on the carbonyl warhead of telaprevir or following the hemithioketal S–C bond formation. Interestingly, the hydroxyl D atom is not positioned on the straight line connecting the hemithioketal oxygen and His41 Nε2 atoms, but instead the O–D···Nε2 angle is 129° (Figure 2C), indicating that this hydrogen bond is distorted from the ideal geometry expected for strong hydrogen bonds.59
Figure 2.
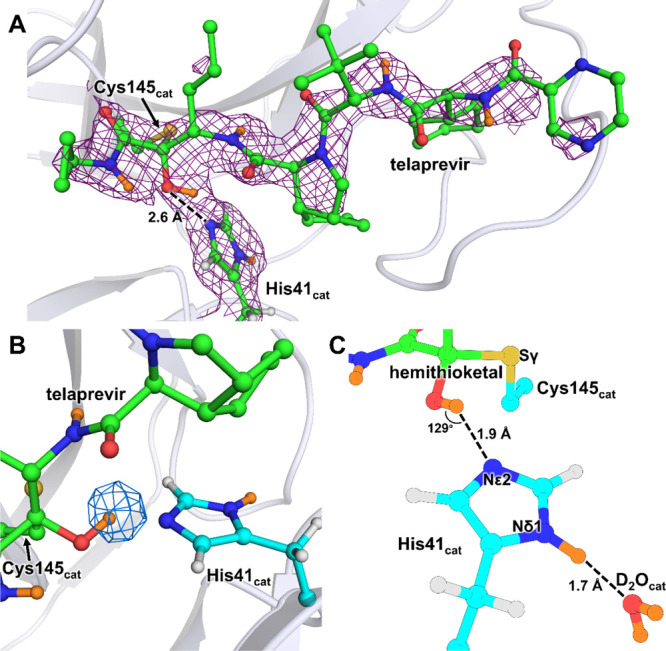
SARS-CoV-2 Mpro catalytic dyad forms a hemithioketal with telaprevir that possesses a protonated hydroxyl. (A) 2FO – FC nuclear density map of telaprevir and the catalytic His41 contoured at a level of 1.0 σ (violet mesh) in the SARS-CoV-2 Mpro active-site cavity. (B) Omit nuclear density map contoured at 3.5 σ level (blue mesh) clearly indicates that the hydroxyl of the hemithioketal is protonated. (C) Hydrogen bonding interactions involving His41 are shown in detail. H atoms on telaprevir are omitted for clarity, whereas D atoms are colored orange.
In Mpro–telaprevir, the His41 imidazole Nδ1 forms a 2.7 Å hydrogen bond with a conserved water molecule (Nδ1–D···OD2O distance of 1.7 Å) (Figure 2C), which is nearly identical to that observed in the ligand-free Mpro. This water is also hydrogen bonded with His164 and Asp187, but it is 3.5 Å away from the main chain ND of His41 compared to 3.2 Å in the ligand-free Mpro. Thus, the latter hydrogen bond is not present when telaprevir binds. Moreover, in the inhibitor complex, His164 is neutral, having lost the Nε2 D atom that was observed in ligand-free Mpro. This leads to a slight increase in Nε2His164···Oγ1Thr175 distance of 0.2 Å. Nevertheless, the hydrogen bond between His164 and Thr175 is effectively abolished upon telaprevir binding because the hydroxyl of Thr175 remains rotated toward and donates its D in a hydrogen bond with the main chain carbonyl of Asp176 (Figure S2). Consequently, in our Mpro–telaprevir structure, the conserved water molecule adjacent to His41, His164, and Asp187 is no longer surrounded by positively charged histidine side chains, significantly altering the electrostatics around the catalytic site.
Throughout the enzyme, several additional protonation state changes occur upon inhibitor binding. Surface residues His64 and His80 in domain I become neutral in Mpro–telaprevir through deprotonation of their Nδ1 and Nε2 atoms, respectively. Conversely, Glu288 of domain III located near Lys5 of the N-terminal loop gains a D atom on its side chain, which thus is converted from a negatively charged carboxylate into a neutral carboxylic acid when telaprevir binds. These three surface residues are over 20 Å away from the catalytic Cys145, and their electrostatic effect on the enzyme active site is unclear.
Protonation States in Subsite S1
Important ionizable residues of subsite S1 are shown in Figure 3A. Subsite S1 is bordered by residues 140–144 spanning the oxyanion hole, His163, Glu166, and the N-terminal Ser1′ of the second enzyme protomer. Subsite S1 is selective for Gln at substrate position P1. Inhibitors possessing sub-μM affinity to Mpro typically mimic Gln by introducing a lactam functionality as their P1 substituent in order to engage His163 in a hydrogen bond.39 Telaprevir features a hydrophobic norvaline substituent in P1 position that cannot make hydrogen bonds in subsite S1. Compared to the ligand-free structure, inhibitor binding leads to the recruitment of a water molecule into subsite S1. This water molecule is positioned between telaprevir’s P1 norvaline and His163 and is hydrogen-bonded with the His163 imidazole. The weak electron and nuclear densities for this water molecule and its high B factor (∼65 Å–2) indicate its mobility. Despite the unfavorable norvaline P1 moiety, His163’s protonation state has changed such that the imidazole is doubly protonated and positively charged in Mpro–telaprevir compared to that in the ligand-free enzyme, where the Nδ1 not facing the S1 subsite was found to be protonated.46 This is significant because His163 protonation explains the apparent ability of the P1 lactam carbonyl to act as a hydrogen-bond acceptor with the His163 imidazole observed in several previous X-ray structures of other Mpro inhibitors.26,27,34 Notably, the superposition of Mpro–telaprevir with the X-ray structure of inhibitor 13b complex (PDB ID 6Y2F)27 reveals that the water molecule position observed in our neutron structure mimics the carbonyl oxygen of inhibitor 13b (Figure S3).
Figure 3.

(A) Ionizable residues of the active site and the S1 subsite of the Mpro–telaprevir complex. A 2FO – FC nuclear density map of the Ser1′ N-terminus and histidines 41, 163, 164, and 172 is shown contoured at 1.0 σ level (violet mesh). All distances are shown in Å with telaprevir (green carbon atoms) being transparent for clarity. (B) Superposition of Mpro–telaprevir (cyan carbon atoms) and apo-Mpro (pink carbon atoms, PDB ID 7JUN) neutron structures showing the S1 subsite. Distances are in Å.
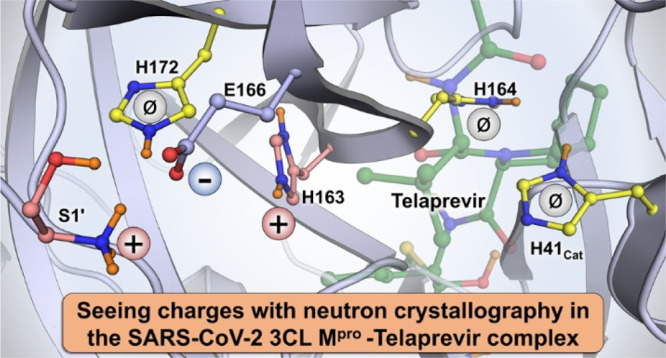
Similar to the ligand-free enzyme, the N-terminal amine is found to be protonated and positively charged (ND3+) in Mpro–telaprevir, where D atoms form hydrogen bonds with the Phe140 main chain carbonyl, the Glu166 carboxylate, and a D2O molecule. Proper positioning of Ser1′ from the second Mpro protomer is required for the catalytic function of this enzyme.25−27,31,33,34 However, the Glu166 carboxylate side chain is rotated by almost 90° relative to its conformation in the ligand-free enzyme, moving it 0.5 Å toward His172 (Figure 3B). Thus, the Oε1Glu166···Nε2His172 distance is reduced from 3.3 to 2.8 Å, shortening the D···O distance from 2.5 to 2.0 Å upon telaprevir binding and possibly considerably strengthening this hydrogen bond. Furthermore, Oε1Glu166 in the telaprevir complex is 1.2 Å closer to Nε2His163 than in ligand-free Mpro, in agreement with His163 becoming protonated in the Mpro–telaprevir complex.
Discussion
Crystallographic structures are essential for making well-educated predictions when constructing inhibitors utilizing computer-assisted and structure-guided drug-design approaches. It is crucial to determine the locations of heavy atoms and the lighter hydrogen atoms that govern the protonation states of amino acid residues and inhibitor substituents.55 The presence or absence of hydrogen atoms on ionizable chemical groups, for example, imidazole of histidine, amine of lysine or N-terminus, or carboxylate of aspartate or glutamate, governs their electric charges and thereby regulates the electrostatics in the vicinity of these groups. Detecting hydrogen atoms in a drug target and a bound ligand is undoubtedly essential to establish protonation states and hydrogen bonding interactions and decipher how protonation states may change due to ligand binding. The potent scattering of neutrons from hydrogen and its isotope deuterium enables neutron crystallography to directly and accurately map hydrogen atoms in biological macromolecules and bound ligands.46,49−51,60,61 In this work, we have successfully determined a room-temperature neutron structure of SARS-CoV-2 Mpro in complex with the hepatitis C virus clinical protease inhibitor, telaprevir, from a 0.5 mm3 crystal using a partially deuterated enzyme. This structure has permitted us to perform a detailed analysis of D atom positions and compare the protonation states in the active-site cavity between the ligand-bound and ligand-free states of the Mpro with broad implications for drug design.
In the SARS-CoV-2 Mpro–telaprevir neutron structure, we observed protonation of the hemithioketal hydroxyl, likely occurring through a proton transfer from His41 that was found doubly protonated in our previous neutron structure of the ligand-free enzyme (Figure 2). The hemithioketal hydroxyl makes a short, but distorted from the ideal geometry, hydrogen bond with the neutral His41 in Mpro–telaprevir. Stronger hydrogen bonds would possibly form when the hemithioketal or hemithioacetal oxygen from a warhead carbonyl is directed into the oxyanion hole upon a covalent attack on the inhibitor, as was observed in the structures of some Mpro inhibitors,26,34 which may enhance inhibitor binding affinity. The neutral His41 position is stabilized by an additional hydrogen bond with the catalytic water molecule, whose position is held by hydrogen bonds with Asp187 and His164 (Figure 3A). His164, positioned deeper in the protein interior and observed as doubly protonated in the ligand-free enzyme, has lost its Nε2 D atom resulting in the imidazole ring moving slightly away from its hydrogen bond partner Thr175 upon telaprevir binding (Figure S3). Intriguingly, His163 located in the subsite S1 has gained a proton on its Nε2 atom that faces the bulk solvent in the ligand-free enzyme and P1 norvaline in the telaprevir-bound complex (Figure 3A). Moreover, His163 protonation, evidently driven by inhibitor binding, induces a conformational change of the Glu166 side chain that brings it closer to the His163 imidazolium while significantly shortening its hydrogen bond with His172 and retaining the hydrogen bond with the N-terminal ammonium of the second protomer (Figure 3B). This observation agrees with His163 becoming positively charged, attracting the negatively charged Glu166. The establishment of hydrogen bond interactions between His163 and the inhibitor lactam ring in several X-ray structures of other inhibitor complexes implies that His163 is protonated in these complexes as well.25−27,34,62 Three additional residues, His64, His80, and Glu288, located on the surface of Mpro alter their protonation states when telaprevir binds, all carrying neutral changes in Mpro–telaprevir, whereas the histidines are positively charged, and the glutamate is negatively charged in the ligand-free enzyme.46 Because these residues are located far away (>20 Å) from the catalytic site, their role in inhibitor binding is unclear.
We have observed that the binding of a covalent inhibitor can modulate the protonation states of histidine residues in the active-site cavity of SARS-CoV-2 Mpro. His41, His163, and His164 have different protonation states relative to those in the ligand-free enzyme (Table 1). His41 and His164 lose hydrogens to become uncharged and neutral, whereas His163 gains a proton to become positively charged when telaprevir binds. Cys145 is also uncharged as it is covalently bonded to telaprevir. Nevertheless, the overall electric charge of the active-site cavity of +1 is maintained upon inhibitor binding, even though the electrostatics of the ligand-binding cavity differ significantly between the ligand-free and ligand-bound enzyme. It is not unreasonable to suggest that the same protonation state changes of the active-site histidine residues may occur upon binding of other α-ketoamide inhibitors. There are 54 possible combinations of protonation states in the active site of ligand-free Mpro with His41, His163, His164, and Cys145 in consideration. Mutual exclusivity of telaprevir’s hemithioketal and His41 Nε2 states reduce this value by 2 to 52 in the telaprevir complex, but combinational possibilities would multiply if, unlike telaprevir, an inhibitor has additional chemical groups ionizable at physiological pH. Furthermore, we note that the exact protonation state combinations of the ionizable residues that we observed in the active-site cavities of ligand-free46 and ketoamide inhibitor-bound SARS-CoV-2 Mpro have not been predicted by molecular simulations,56 emphasizing the critical importance of experimentally determining the locations of H atoms. As a result, the design of covalent inhibitors against SARS-CoV-2 Mpro should consider the observed protonation state changes of the ionizable residues in the active-site cavity triggered by inhibitor binding.
Table 1. Comparison of the Protonation States and the Corresponding Electric Charges of the Ionizable Residues in the SARS-CoV-2 Mpro Active Site Observed in the Neutron Structures of the Ligand-Free and Telaprevir-Bound Enzyme.
| Mpro ligand-free |
Mpro–telaprevir |
|||
|---|---|---|---|---|
| (PDB ID 7JUN) |
(PDB ID 7LB7) |
|||
| residue | charge | species | charge | species |
| Cys145cat | –1 | thiolate (S–) | 0 | S–C–OD (hemithioketal) |
| His41cat | +1 | Nδ1–D, Nε2–D | 0 | Nδ1–D |
| His163 | 0 | Nδ1–D | +1 | Nδ1–D, Nε2–D |
| His164 | +1 | Nδ1–D, Nε2–D | 0 | Nδ1–D |
| His172 | 0 | Nε2–D | 0 | Nε2–D |
| net charge | +1 | +1 | ||
Conclusions
Enzymes rely upon the transfer and movement of hydrogen atoms to carry out their function. We have shown that the active-site cleft of SARS-CoV-2 Mpro contains three histidine residues that can change their protonation states upon an inhibitor binding, revealing how Mpro can tune its electrostatics to accommodate inhibitor and substrate binding into the active-site cleft. We observed that inhibitor binding results in a cascade of protonation/deprotonation (Table 1) and conformational events that remodel the SARS-CoV-2 Mpro active-site cavity as follows:
-
(1)
Upon hemithioketal formation, a proton is transferred from doubly protonated His41 to the hemithioketal hydroxyl to retain the overall neutral charge of the catalytic site.
-
(2)
His164 loses a proton from the Nε2 atom, becoming neutral, and moves away from Thr175 side-chain hydroxyl, which in effect eliminates the hydrogen bond between these two residues.
-
(3)
His163 obtains a proton to become doubly protonated, recruiting a loosely bound water molecule to Nε2. The positively charged imidazolium cation of His163 attracts the negatively charged carboxylate of Glu166 that changes its conformation to move 1.2 Å closer to His163 and concomitantly strengthens its hydrogen bond with His172.
-
(4)
His172 remains singly protonated on Nε2, with no charge because Nδ1 is locked in a hydrogen bond with the main chain NH of Gly138.
-
(5)
The overall +1 electric charge of the active-site cavity is retained.
By considering how the protonation states and electric charges of the ionizable histidine residues in the active site can be altered by inhibitor binding in structure-assisted and computational drug design, protease inhibitors can be improved to specifically target the Mpro enzyme from SARS-CoV-2.
Methods
General Information
Protein purification columns were purchased from Cytiva (Piscataway, New Jersey, USA). Crystallization reagents were purchased from Hampton Research (Aliso Viejo, California, USA). Crystallographic supplies were purchased from MiTeGen (Ithaca, New York, USA) and Vitrocom (Mountain Lakes, New Jersey, USA). Telaprevir was purchased from BioVision Inc. (Milpitas, CA, USA). A detailed protocol for hydrogenated enzyme expression, purification, and crystallization of Mpro to grow neutron diffraction quality crystals has been published elsewhere.63
Cloning, Expression, and Purification of Partially Deuterated SARS-CoV-2 Mpro
The codon-optimized sequence of Mpro from SARS-CoV-2 was cloned into the pD451-SR vector harboring kanamycin resistance (ATUM, Newark, CA) and transformed into chemically competent Escherichia coli BL21 (DE3) cells. Before producing deuterated Mpro, a frozen glycerol stock was first revived in H2O minimal medium.64 Unlabeled glucose (0.5% w/v) and kanamycin (100 mg/mL) were used in this and all other media. After initial growth in H2O minimal medium, the cells were adapted stepwise to minimal medium containing increasing percentages (50, 75, and 100%) of D2O. The final D2O-adapted preculture was used to inoculate the bioreactor vessel to an initial volume of 2.8 L. Following inoculation, the BioFlo 310 bioreactor controller console (Eppendorf, Enfield, CT) was set to maintain the temperature (30 °C) and the dissolved oxygen level (>30%). The pD was kept above 7.3 by the controlled addition of sodium deuteroxide solution in D2O (10% w/w). Once the initial glycerol was exhausted, the culture was fed with a solution containing 20% (w/v) unlabeled glucose, 0.2% (w/v) MgSO4, and kanamycin (100 μg/mL). At an OD600 of ∼8.8, isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 0.5 mM to induce protein expression. The cells were collected ∼13.5 h later by centrifugation at 6000g for 40 min. After removing the supernatant, the wet cell paste was harvested and stored at −80 °C until further use. The protein was purified according to the published procedure.45,63 Upstream of the Mpro, the N-terminus codes for a maltose-binding protein, followed by the protease autoprocessing site SAVLQ↓SGFRK (arrow indicates the autocleavage site), which corresponds to the cleavage position between NSP4 and NSP5 in the viral polyprotein. Downstream to the Mpro, the C-terminus codes for the human rhinovirus 3C (HRV-3C) protease cleavage site (SGVTFQ↓GP), which is connected to a 6× His tag. The N-terminal flanking sequence is autoprocessed in vivo during expression, whereas the C-terminal flanking sequence is removed upon in vitro treatment with HRV-3C protease (MilliporeSigma, St. Louis, MO).
Crystallization
Initial protein crystallization conditions were discovered by screening conducted at the Hauptman-Woodward Medical Research Institute (HWI).65 Crystal aggregates were reproduced using the sitting drop vapor diffusion method using 25% PEG3350, 0.1 M [bis(2-hydroxyethyl)amino]tris(hydroxymethyl)methane (Bis-Tris) pH 6.5 in 20 μL drops with 1:1 ratio of the protein/well solution. Aggregates were transformed into microseeds using Hampton Research Seed Beads. For partially deuterated Mpro–telaprevir co-crystallization, freshly purified deuterated Mpro in 20 mM Tris, 150 mM NaCl, and 1 mM tris(2-carboxyethyl)phosphine, pH 8.0 was concentrated to ∼10.3 mg/mL and mixed with telaprevir, from 60 mM stocks in 100% dimethyl sulfoxide, in a 1:5 molar ratio. After room-temperature incubation for 30 min, precipitation in the sample was removed via centrifugation and filtration through a 0.2 μm centrifugal filter. Crystallization was achieved in a Hampton 9-well plate and sandwich box setup with 50 μL drops of protein mixed with 18% PEG3350 and 0.1 M Bis-Tris pH 6.5 at 1:1 ratio seeded with 0.2 μL of microseeds at a 1:200 dilution. After 28 days of incubation at 14 °C, the crystal used for neutron diffraction data collection grew to the final dimensions of ∼1.5 × 0.7 × 0.5 mm (∼0.5 mm3) (Figure S1). The crystal was mounted in a fused quartz capillary accompanied by 20% PEG3350 prepared with 100% D2O and allowed to H/D exchange for 2 weeks before starting the neutron data collection. The pH in the crystallization drop at the time of crystal mounting was measured by a microelectrode to be 6.6, corresponding to a final pD of 7.0 (pD = pH + 0.4); these are identical conditions to those previously used to determine the neutron structure of the ligand-free enzyme.46
Neutron Diffraction Data Collection
The Mpro–telaprevir crystal was screened for diffraction quality using a broad-bandpass Laue configuration using neutrons from 2.8 to 10 Å with the IMAGINE instrument at the High Flux Isotope Reactor (HFIR) at Oak Ridge National Laboratory.66−68 The full neutron diffraction data set was then collected using the macromolecular neutron diffractometer instrument at the Spallation Neutron Source (SNS).69−71 The crystal was held stationary at room temperature, and diffraction data were collected for 20 h using all neutrons between 2 and 4.16 Å. The next 20 h diffraction image was collected after crystal rotation by Δϕ = 10°. A total of 21 data frames were collected in the final neutron data set. Diffraction data were reduced using the Mantid package, with integration carried out using three-dimensional TOF profile fitting.72,73 Wavelength normalization of the Laue data was performed using the Lauenorm program from the Lauegen suite.74,75 The neutron data collection statistics are shown in Table S1.
X-ray Diffraction Data Collection
The room-temperature X-ray diffraction data set was collected from the same crystal following the neutron data collection on a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator and Osmic VariMax optics. The diffraction images were collected using an Eiger R 4M hybrid photon-counting detector. Diffraction data were integrated using the CrysAlis Pro software suite (Rigaku Inc., The Woodlands, TX). Diffraction data were then reduced and scaled using the Aimless76 program from the CCP4 suite;77 molecular replacement using PDB code 6XQS(57) was then performed with Molrep from the CCP4 program suite. The protein structure was first refined against the X-ray data using phenix.refine from the Phenix78 suite of programs to obtain an accurate model for the subsequent X-ray/neutron joint refinement. The X-ray data collection statistics are shown in Table S1.
Joint X-ray/Neutron Refinement
The joint X-ray/neutron refinement of Mpro-telaprevir was performed using nCNS,79 and the structure was manipulated in Coot.80 After initial rigid-body refinement, several cycles of positional, atomic displacement parameter, and occupancy refinement were performed. The structure was checked for the correctness of side-chain conformations, hydrogen bonding, and orientations of D2O water molecules built based on the mFO – DFC difference neutron scattering length density maps. The 2mFO – DFC and mFO – DFC neutron scattering length density maps were then examined to determine the correct orientations of hydroxyl (Ser, Thr, Tyr), thiol (Cys), and ammonium (Lys) groups, and protonation states of the enzyme residues. The protonation states of some disordered side chains could not be obtained directly and remained ambiguous. All water molecules were refined as D2O. Initially, water oxygen atoms were positioned according to their electron density peaks and then were shifted slightly in accordance with the neutron scattering length density maps. Because Mpro used for this study was partially deuterated, all H positions in the protein were modeled as D atoms. In telaprevir, only the labile H atoms were modeled as Ds. The occupancies of D atoms were refined individually within the range of −0.56 (pure H) to 1.00 (pure D) because the neutron scattering length of H is −0.56 times that of D. Before depositing the neutron structure to the PDB, a script was run that converts a record for the coordinates of a D atom into two records corresponding to an H and a D partially occupying the same site, both with positive partial occupancies that add up to unity. The percent D at a specific site is calculated according to the following formula: % D = {occupancy(D) + 0.56}/1.56.
Acknowledgments
This research was supported by the DOE Office of Science through the National Virtual Biotechnology Laboratory (NVBL), a consortium of DOE national laboratories focused on response to COVID-19, with funding provided by the Coronavirus CARES Act. This research used resources at the SNS and the HFIR, which are DOE Office of Science User Facilities operated by the Oak Ridge National Laboratory (ORNL). The Office of Biological and Environmental Research supported research at ORNL’s Center for Structural Molecular Biology (CSMB), a DOE Office of Science User Facility. This research used resources of the Spallation Neutron Source Second Target Station Project at ORNL. ORNL is managed by UT-Battelle LLC for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. L.C. acknowledges support by the NIH (R01-GM071939). We thank Dr. Hugh M. O’Neill from ORNL for assistance during expression of the partially deuterated protein.
Glossary
Abbreviations
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- 3CL Mpro
chymotrypsin-like main protease
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00058.
Neutron and X-ray data collection statistics; photo of 0.5 mm3 crystal of SARS-CoV-2 Mpro–telaprevir complex; superposition of Mpro–telaprevir neutron structure with the neutron structure of ligand-free Mpro (PDB ID 7JUN); and superposition of Mpro–telaprevir neutron structure with the X-ray structure of Mpro in complex with inhibitor 13b (PDB ID 6Y2F) (PDF)
Author Contributions
L.C. and A.K. conceived the study. A.K., G.P., and Q.Z. designed and cloned the gene. G.P., K.L.W., and Q.Z. performed expression of the partially deuterated protein. D.W.K. and A.K. crystallized the protein. A.K. and D.W.K. collected the X-ray diffraction data. L.C. collected and reduced the neutron diffraction data. A.K., D.W.K., and L.C. refined the joint X-ray/neutron structure. D.W.K., L.C., and A.K. wrote the paper with help from all co-authors.
The authors declare no competing financial interest.
Notes
Coordinates and structure factors for the SARS-CoV-2 3CL Mpro–telaprevir complex have been deposited in the PDB with the accession code 7LB7. Authors will release the atomic coordinates upon article publication. Any other relevant data are available from the corresponding authors upon reasonable request.
Supplementary Material
References
- Hussain A.; Yadav S.; Hadda V.; Suri T. M.; Tiwari P.; Mittal S.; Madan K.; Mohan A. Covid-19: a comprehensive review of a formidable foe and the road ahead. Expert Rev. Respir. Med. 2020, 14, 869–879. 10.1080/17476348.2020.1782198. [DOI] [PubMed] [Google Scholar]
- Rastogi Y. R.; Sharma A.; Nagraik R.; Aygün A.; Şen F. The novel coronavirus 2019-nCoV: its evolution and transmission into humans causing global COVID-19 pandemic. Int. J. Environ. Sci. Technol. 2020, 17, 4381–4388. 10.1007/s13762-020-02781-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfi M.; Hamblin M. R.; Rezaei N. COVID-19: transmission, prevention, and potential therapeutic opportunities. Clin. Chim. Acta 2020, 508, 254–266. 10.1016/j.cca.2020.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmy Y. A.; Fawzy M.; Elaswad A.; Sobieh A.; Kenney S. P.; Shehata A. A. The COVID-19 pandemic: a comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. J. Clin. Med. 2020, 9, 1225. 10.3390/jcm9041225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. What Pfizer’s landmark COVID vaccine results mean for the pandemic. Nature 2020, 10.1038/d41586-020-03166-8. [DOI] [PubMed] [Google Scholar]
- Voysey M.; Clemens S. A. C.; Madhi S. A.; Weckx L. Y.; Folegatti P. M.; Aley P. K.; Angus B.; Baillie V. L.; Barnabas S. L.; Bhorat Q. E. l.; Bibi S.; Briner C.; Cicconi P.; Collins A. M.; Colin-Jones R.; Cutland C. L.; Darton T. C.; Dheda K.; Duncan C. A.; Emary K.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomized controlled trails in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. 10.1016/S0140-6736(20)32661-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. COVID vaccine excitement builds as Moderna reports third positive result. Nature 2020, 587, 337–338. 10.1038/d41586-020-03248-7. [DOI] [PubMed] [Google Scholar]
- Liu C.; Zhou Q.; Li Y.; Garner L. V.; Watkins S. P.; Carter L. J.; Smoot J.; Gregg A. C.; Daniels A. D.; Jervey S.; Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020, 6, 315–331. 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T.; Meenakshisundaram S.; Manickam M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discovery Today 2020, 25, 668–688. 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar C.; Mondal M.; Islam M. T.; Martorell M.; Docea A. O.; Maroyi A.; Sharifi-Rad J.; Calina D. Potential therapeutic options for COVID-19: current status, challenges, and future perspectives. Front. Pharmacol. 2020, 11, 572870. 10.3389/fphar.2020.572870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A.; Chan J. F. W.; Azhar E. I.; Hui D. S. C.; Yuen K.-Y. Coronaviruses—drug discovery and therapeutic options. Nat. Rev. 2016, 15, 327–347. 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil C.; Ginex T.; Maestro I.; Nozal V.; Barrado-Gil L.; Cuesta-Geijo M. Á.; Urquiza J.; Ramírez D.; Alonso C.; Campillo N. E.; Martinez A. COVID-19: drug targets and potential treatments. J. Med. Chem. 2020, 63, 12359–12386. 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- Engelman A.; Cherepanov P. The structural biology of HIV-1: mechanistic and therapeutic insights. Nat. Rev. 2012, 10, 279–290. 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D.-Y.; Wu H.-Y.; Yarla N. S.; Xu B.; Ding J.; Lu T.-R. HAART in HIV/AIDS treatments: future trends. Infect. Disord.: Drug Targets 2018, 18, 15–22. 10.2174/1871526517666170505122800. [DOI] [PubMed] [Google Scholar]
- Soriano V.; Vispo E.; Poveda E.; Labarga P.; Martin-Carbonero L.; Fernandez-Montero J. V.; Barreiro P. Directly acting antivirals against hepatitis C virus. J. Antimicrob. Chemother. 2011, 66, 1673–1686. 10.1093/jac/dkr215. [DOI] [PubMed] [Google Scholar]
- Shahid I.; AlMalki W. H.; Hassan S.; Hafeez M. H. Real-world challenges for hepatitis C virus medications: a critical review. Crit. Rev. Microbiol. 2017, 44, 143–160. 10.1080/1040841x.2017.1329277. [DOI] [PubMed] [Google Scholar]
- Webster R. G.; Govorkova E. A. Continuing challenges in influenza. Ann. N.Y. Acad. Sci. 2014, 1323, 115–139. 10.1111/nyas.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toots M.; Plemper R. K. Next-generation direct-acting influenza therapeutics. Transl. Res. 2020, 220, 33–42. 10.1016/j.trsl.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman R. T.; Roth J. S.; Brimacombe K. R.; Simeonov A.; Shen M.; Patnaik S.; Hall M. D. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. 10.1021/acscentsci.0c00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.; Zhao S.; Yu B.; Chen Y.-M.; Wang W.; Song Z.-G.; Hu Y.; Tao Z.-W.; Tian J.-H.; Pei Y.-Y.; Yuan M.-L.; Zhang Y.-L.; Dai F.-H.; Liu Y.; Wang Q.-M.; Zheng J.-J.; Xu L.; Holmes E. C.; Zhang Y.-Z. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Zhao S.; Teng T.; Abdalla A. E.; Zhu W.; Xie L.; Wang Y.; Guo X. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 2020, 12, 244. 10.3390/v12020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A. E.; Snijder E. J. Viral cysteine proteases. Perspect. Drug Discovery Des. 1996, 6, 64–86. 10.1007/bf02174046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L. Viral proteases. Chem. Rev. 2002, 102, 4609–4626. 10.1021/cr010184f. [DOI] [PubMed] [Google Scholar]
- Douangamath A.; Fearon D.; Gehrtz P.; Krojer T.; Lukacik P.; Owen C. D.; Resnick E.; Strain-Damerell C.; Aimon A.; Abranyi-Balogh P.; Brandao-Neto J.; Carbery A.; Davison G.; Dias A.; Downes T. D.; Dunnett L.; Fairhead M.; Firth J. D.; Jones S. P.; Keeley A.; Keseru G. M.; Klein H. F.; Martin M. P.; Noble M. E. M.; O’Brien P.; Powell A.; Reddi R. N.; Skyner R.; Snee M.; Waring M. J.; Wild C.; London N.; von Delft F.; Walsh M. A. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; Duan Y.; Yu J.; Wang L.; Yang K.; Liu F.; Jiang R.; Yang X.; You T.; Liu X.; Yang X.; Bai F.; Liu H.; Liu X.; Guddat L. W.; Xu W.; Xiao G.; Qin C.; Shi Z.; Jiang H.; Rao Z.; Yang H. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Dai W.; Zhang B.; Jiang X.-M.; Su H.; Li J.; Zhao Y.; Xie X.; Jin Z.; Peng J.; Liu F.; Li C.; Li Y.; Bai F.; Wang H.; Cheng X.; Cen X.; Hu S.; Yang X.; Wang J.; Liu X.; Xiao G.; Jiang H.; Rao Z.; Zhang L.-K.; Xu Y.; Yang H.; Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R. L.; Kania R. S.; Brothers M. A.; Davies J. F.; Ferre R. A.; Gajiwala K. S.; He M.; Hogan R. J.; Kozminski K.; Li L. Y.; Lockner J. W.; Lou J.; Marra M. T.; Mitchell L. J.; Murray B. W.; Nieman J. A.; Noell S.; Planken S. P.; Rowe T.; Ryan K.; Smith G. J. III; Solowiej J. E.; Steppan C. M.; Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. S.; Ma X. R.; Ma Y.; Alugubelli Y. R.; Scott D. A.; Vatansever E. C.; Drelich A. K.; Sankaran B.; Geng Z. Z.; Blankenship L. R.; Ward H. E.; Sheng Y. J.; Hsu J. C.; Kratch K. C.; Zhao B.; Hayatshahi H. S.; Liu J.; Li P.; Fierke C. A.; Tseng C. T. K.; Xu S.; Liu W. R. A Quick Route to Multiple Highly Potent SARS-CoV-2 Main Protease Inhibitors. ChemMedChem 2020, 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerlemans R.; Ruiz-Moreno A. J.; Cong Y.; Dinesh Kumar N.; Velasco-Velazquez M. A.; Neochoritis C. G.; Smith J.; Reggiori F.; Groves M. R.; Dömling A. Repurposing the HCV NS3–4A Protease Drug Boceprevir as COVID-19 Therapeutics. RSC Med. Chem. 2021, 10.1039/D0MD00367K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva L.; Yuan S.; Yin X.; Martin-Sancho L.; Matsunaga N.; Pache L.; Burgstaller-Muehlbacher S.; De Jesus P. D.; Teriete P.; Hull M. V.; Chang M. W.; Chan J. F.-W.; Cao J.; Poon V. K.-M.; Herbert K. M.; Cheng K.; Nguyen T.-T. H.; Rubanov A.; Pu Y.; Nguyen C.; Choi A.; Rathnasinghe R.; Schotsaert M.; Miorin L.; Dejosez M.; Zwaka T. P.; Sit K.-Y.; Martinez-Sobrido L.; Liu W.-C.; White K. M.; Chapman M. E.; Lendy E. K.; Glynne R. J.; Albrecht R.; Ruppin E.; Mesecar A. D.; Johnson J. R.; Benner C.; Sun R.; Schultz P. G.; Su A. I.; García-Sastre A.; Chatterjee A. K.; Yuen K.-Y.; Chanda S. K. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Zhao Y.; Sun Y.; Zhang B.; Wang H.; Wu Y.; Zhu Y.; Zhu C.; Hu T.; Du X.; Duan Y.; Yu J.; Yang X.; Yang X.; Yang K.; Liu X.; Guddat L. W.; Xiao G.; Zhang L.; Yang H.; Rao Z. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- Fu L.; Ye F.; Feng Y.; Yu F.; Wang Q.; Wu Y.; Zhao C.; Sun H.; Huang B.; Niu P.; Song H.; Shi Y.; Li X.; Tan W.; Qi J.; Gao G. F. Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Hu Y.; Townsend J. A.; Lagarias P. I.; Marty M. T.; Kolocouris A.; Wang J. Ebselen, disulfiram, carmofur, PX-12, tideglusib, and shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. 10.1021/acsptsci.0c00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno S.; Thanigaimalai P.; Yamamoto T.; Nakada K.; Kakiuchi R.; Takayama K.; Yamazaki Y.; Yakushiji F.; Akaji K.; Kiso Y.; Kawasaki Y.; Chen S.-E.; Freire E.; Hayashi Y. Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety. Bioorg. Med. Chem. 2013, 21, 412–424. 10.1016/j.bmc.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J.; Grum-Tokars V.; Zhou Y.; Turlington M.; Saldanha S. A.; Chase P.; Eggler A.; Dawson E. S.; Baez-Santos Y. M.; Tomar S.; Mielech A. M.; Baker S. C.; Lindsley C. W.; Hodder P.; Mesecar A.; Stauffer S. R. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridine-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013, 56, 534–546. 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T.; Manickam M.; Namasivayam V.; Hayashi Y.; Jung S.-H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Liang C.; Xin L.; Ren X.; Tian L.; Ju X.; Li H.; Wang Y.; Zhao Q.; Liu H.; Cao W.; Xie X.; Zhang D.; Wang Y.; Jian Y. The development of coronavirus 3C-like protease (3CLpro) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. 10.1016/j.ejmech.2020.112711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K.; Ziebuhr J.; Wadhwani P.; Mesters J. R.; Hilgenfeld R. Coronavirus main protease (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Solowiej J.; Thomson J. A.; Ryan K.; Luo C.; He M.; Lou J.; Murray B. W. Steady-state and pre-steady-state kinetic evaluation of severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro cysteine protease: development of an ion-pair model for catalysis. Biochemistry 2008, 47, 2617–2630. 10.1021/bi702107v. [DOI] [PubMed] [Google Scholar]
- Hsu W.-C.; Chang H.-C.; Chou C.-Y.; Tsai P.-J.; Lin P.-I.; Chang G.-G. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. 2005, 280, 22741–22748. 10.1074/jbc.m502556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P.-Y.; Chou C.-Y.; Chang H.-C.; Hsu W.-C.; Chang G.-G. Correlation between dissociation and catalysis of SARS-CoV main protease. Arch. Biochem. Biophys. 2008, 472, 34–42. 10.1016/j.abb.2008.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménard R.; Storer A. C. Oxyanion hole interactions in serine and cysteine protease. Biol. Chem. Hoppe Seyler 1992, 373, 393–400. 10.1515/bchm3.1992.373.2.393. [DOI] [PubMed] [Google Scholar]
- Kneller D. W.; Phillips G.; O’Neill H. M.; Jedrzejczak R.; Stols L.; Langan P.; Joachimiak A.; Coates L.; Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202. 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W.; Phillips G.; Weiss K. L.; Pant S.; Zhang Q.; O’Neill H. M.; Coates L.; Kovalevsky A. Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 2020, 295, 17365–17373. 10.1074/jbc.ac120.016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura N.; Podjarny A.. Neutron Protein Crystallography; Oxford University Press, 2011; p 250. [Google Scholar]
- Neutron Crystallography in Structural Biology; Moody P. C. E., Ed.; Methods in Enzymology; Academic Press, 2020; Vol. 634, p 389. [DOI] [PubMed] [Google Scholar]
- Gerlits O.; Weiss K. L.; Blakeley M. P.; Veglia G.; Taylor S. S.; Kovalevsky A. Zooming in on protons: neutron structure of protein kinase A trapped in a product complex. Sci. Adv. 2019, 5, eaav0482 10.1126/sciadv.aav0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langan P. S.; Close D. W.; Coates L.; Rocha R. C.; Ghosh K.; Kiss C.; Waldo G.; Freyer J.; Kovalevsky A.; Bradbury A. R. M. Evolution and characterization of a new reversibly photoswitching chromogenic protein, Dathail. J. Mol. Biol. 2016, 428, 1776–1789. 10.1016/j.jmb.2016.02.029. [DOI] [PubMed] [Google Scholar]
- Banco M. T.; Mishra V.; Ostermann A.; Schrader T. E.; Evans G. B.; Kovalevsky A.; Ronning D. R. Neutron structures of the Helicobacter pylori 5’-methylthioadenosine nucleosidase highlight proton sharing and protonation states. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 13756–13761. 10.1073/pnas.1609718113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garman E. F. Radiation damage in macromolecular crystallography: what is it and why should we care?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 339–351. 10.1107/s0907444910008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley M. P.; Mitschler A.; Hazemann I.; Meilleur F.; Myles D. A. A.; Podjarny A. Comparison of hydrogen determination with X-ray and neutron crystallography in a human aldose reductase-inhibitor complex. Eur. Biophys. J. 2006, 35, 577–583. 10.1007/s00249-006-0064-8. [DOI] [PubMed] [Google Scholar]
- Gardberg A. S.; Del Castillo A. R.; Weiss K. L.; Meilleur F.; Blakeley M. P.; Myles D. A. A. Unambiguous determination of H-atom positions: comparing results from neutron and high-resolution X-ray crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 558–567. 10.1107/s0907444910005494. [DOI] [PubMed] [Google Scholar]
- Bax B.; Chung C.-W.; Edge C. Getting the chemistry right: protonation, tautomers and the importance of H atoms in biological chemistry. Acta Crystallogr., Sect. D: Struct. Biol. 2017, 73, 131–140. 10.1107/s2059798316020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova A.; Lynch D. L.; Daidone I.; Zanetti-Polzi L.; Smith M. D.; Chipot C.; Kneller D. W.; Kovalevsky A.; Coates L.; Golosov A. A.; Dickson C. J.; Velez-Vega C.; Duca J. S.; Vermaas J. V.; Pang Y. T.; Acharya A.; Parks J. M.; Smith J. C.; Gumbart J. C. Inhibitor binding influences the protonation states of histidines in SARS-CoV-2 main protease. Chem. Sci. 2021, 12, 1513–1527. 10.1039/d0sc04942e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W.; Galanie S.; Phillips G.; O’Neill H. M.; Coates L.; Kovalevsky A. Malleability of SARS-CoV-2 3CL Mpro active site cavity facilitates binding of clinical antivirals. Structure 2020, 28, 1313–1320. 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Mustyakimov M.; Afonine P. V.; Langan P. Generalized X-ray and Neutron Crystallographic Analysis: More Accurate and Complete Structures for Biological Macromolecules. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 567–573. 10.1107/s0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski S. J. What is the covalency of hydrogen bonding?. Chem. Rev. 2011, 111, 2597–2625. 10.1021/cr800346f. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Serpersu E. H.; Cuneo M. J. A low-barrier hydrogen bond mediates antibiotic resistance in a noncanonical catalytic triad. Sci. Adv. 2018, 4, eaas8667 10.1126/sciadv.aas8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalevsky A.; Aggarwal M.; Velazquez H.; Cuneo M. J.; Blakeley M. P.; Weiss K. L.; Smith J. C.; Fisher S. Z.; McKenna R. “To be or not to be” protonated: atomic details of human carbonic anhydrase-clinical drug complexes by neutron crystallography and simulation. Structure 2018, 26, 383–390.e3. 10.1016/j.str.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Rathnayake A. D.; Zheng J.; Kim Y.; Perera K. D.; Mackin S.; Meyerholz D. K.; Kashipathy M. M.; Battaile K. P.; Lovell S.; Perlman S.; Groutas W. C.; Chang K.-O. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med. 2020, 12, eabc5332 10.1126/scitranslmed.abc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W.; Phillips G.; Kovalevsky A.; Coates L. Room-temperature neutron and X-ray data collection of 3CL Mpro from SARS-CoV-2. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2020, 76, 483–487. 10.1107/s2053230x20011814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törnkvist M.; Larsson G.; Enfors S.-O. Protein release and foaming in Escherichia coli cultures grown in minimal medium. Bioprocess Eng. 1996, 15, 231–237. 10.1007/bf02391583. [DOI] [Google Scholar]
- Luft J. R.; Collins R. J.; Fehrman N. A.; Lauricella A. M.; Veatch C. K.; DeTitta G. T. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J. Struct. Biol. 2003, 142, 170–179. 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- Meilleur F.; Kovalevsky A.; Myles D. A. A. IMAGINE: The neutron protein crystallography beamline at the high flux isotope reactor. Methods Enzymol. 2020, 634, 69–85. 10.1016/bs.mie.2019.11.016. [DOI] [PubMed] [Google Scholar]
- Meilleur F.; Coates L.; Cuneo M.; Kovalevsky A.; Myles D. The neutron macromolecular crystallography instruments at Oak Ridge National Laboratory: Advances, challenges, and opportunities. Crystals 2018, 8, 388. 10.3390/cryst8100388. [DOI] [Google Scholar]
- Meilleur F.; Munshi P.; Robertson L.; Stoica A. D.; Crow L.; Kovalevsky A.; Koritsanszky T.; Chakoumakos B. C.; Blessing R.; Myles D. A. A. The IMAGINE instrument: first neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 2157–2160. 10.1107/s0907444913019604. [DOI] [PubMed] [Google Scholar]
- Coates L.; Cao H. B.; Chakoumakos B. C.; Frontzek M. D.; Hoffmann C.; Kovalevsky A. Y.; Liu Y.; Meilleur F.; dos Santos A. M.; Myles D. A. A.; Wang X. P.; Ye F. A suite-level review of the neutron single-crystal diffraction instruments at Oak Ridge National Laboratory. Rev. Sci. Instrum. 2018, 89, 092802. 10.1063/1.5030896. [DOI] [PubMed] [Google Scholar]
- Coates L.; Sullivan B. The macromolecular neutron diffractometer at the spallation neutron source. Methods Enzymol. 2020, 634, 87–99. 10.1016/bs.mie.2019.11.020. [DOI] [PubMed] [Google Scholar]
- Coates L.; Cuneo M. J.; Frost M. J.; He J.; Weiss K. L.; Tomanicek S. J.; McFeeters H.; Vandavasi V. G.; Langan P.; Iverson E. B. The macromolecular neutron diffractometer MaNDi at the Spallation Neutron Source. J. Appl. Crystallogr. 2015, 48, 1302–1306. 10.1107/s1600576715011243. [DOI] [Google Scholar]
- Sullivan B.; Archibald R.; Langan P. S.; Dobbek H.; Bommer M.; McFeeters R. L.; Coates L.; Wang X.; Gallmeier F.; Carpenter J. M.; Lynch V.; Langan P. Improving the accuracy and resolution of neutron crystallographic data by three-dimensional profile fitting of Bragg peaks in reciprocal space. Acta Crystallogr., Sect. D: Struct. Biol. 2018, 74, 1085–1095. 10.1107/s2059798318013347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold O.; Bilheux J. C.; Borreguero J. M.; Buts A.; Campbell S. I.; Chapon L.; Doucet M.; Draper N.; Ferraz Leal R.; Gigg M. A.; Lynch V. E.; Markvardsen A.; Mikkelson D. J.; Mikkelson R. L.; Miller R.; Palmen K.; Parker P.; Passos G.; Perring T. G.; Peterson P. F.; Ren S.; Reuter M. A.; Savici A. T.; Taylor J. W.; Taylor R. J.; Tolchenov R.; Zhou W.; Zikovsky J. Mantid—Data analysis and visualization package for neutron scattering and μ SR experiments. Nucl. Instrum. Methods Phys. Res., Sect. A 2014, 764, 156–166. 10.1016/j.nima.2014.07.029. [DOI] [Google Scholar]
- Campbell J. W.; Hao Q.; Harding M. M.; Nguti N. D.; Wilkinson C. LAUEGEN version 6.0 and INTLDM. J. Appl. Crystallogr. 1998, 31, 496–502. 10.1107/s0021889897016683. [DOI] [Google Scholar]
- Helliwell J. R.; Habash J.; Cruickshank D. W. J.; Harding M. M.; Greenhough T. J.; Campbell J. W.; Clifton I. J.; Elder M.; Machin P. A.; Papiz M. Z.; Zurek S. The recording and analysis of synchrotron X-radiation Laue diffraction photographs. J. Appl. Crystallogr. 1989, 22, 483–497. 10.1107/s0021889889006564. [DOI] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/s0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustyakimov M.; Langan P.. Copyright C-06, 104 Patch for CNS; nCNS an Open Source Distribution Patch for CNS for Macromolecular Structure Refinement; Los Alamos National Security: Los Alamos, NM, USA, 2007. [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.