

SUMMARY
RTK/RAS/RAF pathway alterations (RPAs) are a hallmark of lung adenocarcinoma (LUAD). In this study, we use whole-genome sequencing (WGS) of 85 cases found to be RPA(–) by previous studies from The Cancer Genome Atlas (TCGA) to characterize the minority of LUADs lacking apparent alterations in this pathway. We show that WGS analysis uncovers RPA(+) in 28 (33%) of the 85 samples. Among the remaining 57 cases, we observe focal deletions targeting the promoter or transcription start site of STK11 (n = 7) or KEAP1 (n = 3), and promoter mutations associated with the increased expression of ILF2 (n = 6). We also identify complex structural variations associated with high-level copy number amplifications. Moreover, an enrichment of focal deletions is found in TP53 mutant cases. Our results indicate that RPA(–) cases demonstrate tumor suppressor deletions and genome instability, but lack unique or recurrent genetic lesions compensating for the lack of RPAs. Larger WGS studies of RPA(–) cases are required to understand this important LUAD subset.
In Brief
Carrot-Zhang et al. perform whole-genome characterization of lung adenocarcinomas (LUADs) lacking RTK/RAS/RAF pathway alterations (RPAs) and identify mutations or structural variants in both coding and non-coding spaces that define a unique entity of RPA(–) LUADs and potentially explain the underlying biology of this disease.
Graphical Abstract
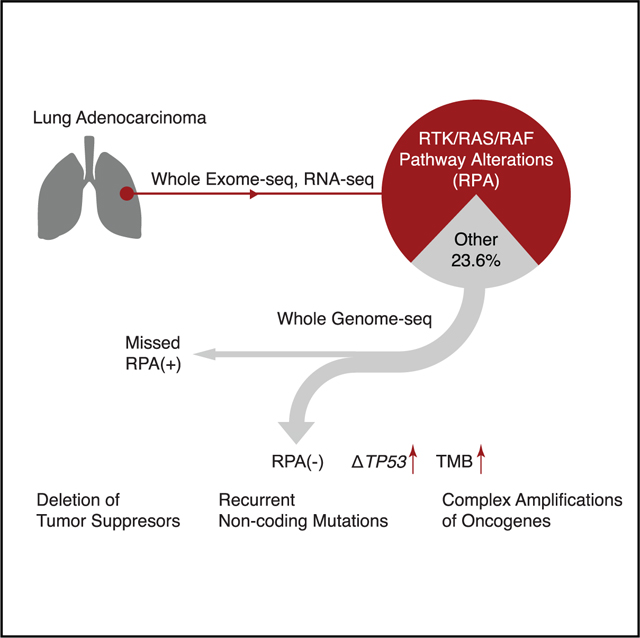
INTRODUCTION
Lung adenocarcinoma (LUAD) is the most common lung malignancy and a leading cause of cancer death in the United States (Siegel et al., 2019). Most LUADs are driven by constitutive activation of mitogen-activated protein kinase (MAPK) signaling, which is, in turn, a consequence of alterations in receptor tyrosine kinases (RTKs), downstream RAS/RAF/MEK cascade proteins, and their regulators (Desai et al., 2014). Comprehensive studies using whole-exome (WES) and/or transcriptome sequencing (RNA-seq) have identified RTK/RAS/RAF pathway driver alterations in 70%–80% of LUADs (Campbell et al., 2016; Cancer Genome Atlas Research Network, 2014; Imielinski et al., 2012). Because driver alterations in RTK/RAS/RAF pathway genes such as EGFR, BRAF, ALK, RET, ROS1, and KRAS can be targeted by small-molecule inhibitors that significantly prolong survival, therapeutic decision making in LUADs is routinely directed by testing for many but not all RTK/RAS/RAF alterations (Herbst et al., 2018).
The remaining 20%–30% of cases, which we refer to as RTK/RAS/RAF pathway alteration-negative, or RPA(–), LUADs, pose a major clinical challenge in precision thoracic oncology (Campbell et al., 2016). A key question is whether these RPA(–) LUADs represent a distinct RTK/RAS/RAF-independent entity that is associated with a unique evolutionary path and therapeutic sensitivities, or whether they have been mislabeled as RPA(–) due to technical factors (e.g., sample quality, limitations in the genomic profiling technologies). Specifically, apparent RPA(–) LUADs may harbor pathogenic variants in RTK/RAS/RAF pathway genes that are not adequately detected by WES, RNA-seq, or targeted gene panels such as those commonly used in large research studies or in clinical laboratories (Vinagre et al., 2013; Weischenfeldt et al., 2017).
We postulated that a more comprehensive analysis of candidate RPA(–) LUADs using whole-genome sequencing (WGS) in addition to WES and RNA-seq may illuminate more precisely the basic biology and also influence clinical management. We, therefore, performed WGS on LUAD samples from The Cancer Genome Atlas (TCGA) cohort that had appeared in previous analyses to lack an RTK/RAS/RAF pathway-activating alteration (Campbell et al., 2016). Since WGS is particularly effective in the identification of non-coding and structural genomic alterations, we hypothesized that analysis of those types of genome variation may reveal key features of RPA(–) LUAD biology.
RESULTS
Identification of RPA(–) LUADs
A previously published TCGA study of lung adenocarcinoma identified 383/501 (76%) of LUAD cases as RTK/RAS/RAF alteration positive, or RPA(+), using WES and RNA-seq (Table S1) (Campbell et al., 2016). Samples with activating mutations in KRAS, EGFR, BRAF, ERBB2, MET, RIT1, NRAS, RAF1, HRAS, ARAF, MAP2K1, or SOS1; loss of function mutations in NF1 or RASA1; fusions in ALK, ROS1, RET, MET, or NTRK2; and amplification of EGFR, ERBB2, KRAS, MET, FGFR1, or MAPK1 were classified as RPA(+) by WES and RNA-seq, which we henceforth designate as RPA(+)E. Among the remaining 118 LUADs, we performed WGS for 85 tumor-normal pairs (Figures 1A, 1B, and S1A) at an average of 73.5-fold (±7.9 SD) and 37.0-fold (±5.8 SD) coverage for tumor and matched normal samples, respectively, followed by somatic single-nucleotide variant (SNV), indel, copy number, and structural variant (SV) analysis (method details).
Figure 1. Identification of RPA(–) LUADs.

(A) Identification of 118 RPA(–)E LUAD cases from the 501 TCGA LUAD cohort, defined by WES or RNA-seq analysis. Eighty-five of the 118 samples were sent for WGS. The RTK/RAS/RAF pathway alterations used to define the RPA(+) or RPA(–) cases are listed in Table S1.
(B) WGS uncovered genomic alterations in the RTK/RAS/RAF pathway in 28/85 samples; 57/85 samples remain as RPA(–) after WGS analysis.
(C) Visualization of sequencing reads covering a KRAS p.G12C mutation in WGS (upper panel) and WES (lower panel) for sample (TCGA-55–7574). Both read depth and the number of reads supporting the mutation are higher in WGS than in WES.
(D) An example of EGFR amplification coupled with EGFR overexpression in TCGA-50–5939. In the second panel, purity-adjusted copy number and SV junctions (red lines) support a BFBC event underlying the amplification. Lower panels indicate WGS read depth and gene location in the region. CN, copy number.
(E) Example of a RASA1 simple deletion spanning from exon 21 to the end of the gene (12 kbp) coupled with RASA1 loss of expression in TCGA-55–8614.
See also Figure S1.
Our multi-step analysis schema is shown in Figure S1. In the first step, we re-analyzed the 85 RPA(–)E cases to determine whether there was truly no evidence of coding mutations in the RTK/RAS/RAF pathway. Surprisingly, we found that 20/85 cases harbored KRAS hotspot mutations, with 8 samples showing the recently targetable p.G12C mutation (Canon et al., 2019; Ostrem et al., 2013). Re-examination of the WES data for those samples confirmed the mutation calls for 16/20 samples, but the read support was insufficient to enable high-confidence variant calling without invoking the WGS information (Figures 1C and S1B). The poor WES coverage for those KRAS mutations was likely due to low capture efficiency (Clark et al., 2011) for the first coding exon of KRAS, which contains codons 12 and 13. In addition, the samples with KRAS mutations missed by WES showed lower tumor purity than did KRAS-mutated samples identified by WES (Figure S1C; Table S1), confirming that high sequencing depths were necessary for detecting the mutations reliably.
Among the 85 samples, we also identified 8 cases with somatic SV and copy-number alterations (SCNAs) in known RTK/RAS/RAF pathway members. Among those alterations, we found complex SVs driving high-level amplification and overexpression of oncogenes, including EGFR (n = 1) and MAPK1 (n = 3). Further classification of complex SVs showed that the EGFR amplification was driven by a breakage-fusion-bridge cycle (BFBC) (Figure 1D). We also found deletions coupled with the loss of heterozygosity (LOH), resulting in the decreased expression of RASA1 (n = 1) or NF1 (n = 1) (Figures 1B and 1E; Table S2), both of which negatively regulate RTK/RAS/RAF signaling. The focal deletion (379-bp length) in NF1 affected only a single exon that is not well represented in WES or RNA-seq, highlighting the advantage of WGS for the identification of focal SV events (Table S2). We also identified one case with ARAF amplification and one case with NRG1 fusion, which are alterations previously shown to activate RTK/RAS/RAF signaling in LUADs (Fernandez-Cuesta et al., 2014; Imielinski et al., 2014). Interestingly, we found amplification and overexpression of SOS1 in one case (TCGA-62–8399). Although SOS1 mutations have been shown to activate RTK/RAS/RAF signaling (Cai et al., 2019), the role played in RTK/RAS/RAF activation by the amplification described here is unclear.
Overall, we identified 28 (33%) additional RPA(+) cases among the 85 RPA(–)E that had undergone WGS (Figure S1A). We labeled the remaining 57 cases as RPA(–)G LUADs because they lacked an RTK/RAS/RAF pathway alteration identified by WGS (as well as WES and RNA-seq). The remainder of the results reported here focus on that subset.
Recurrent coding alterations in RPA(–)G LUADs
Having identified the 57 RPA(–)G LUAD cases, we sought to define their protein-coding driver alteration landscape. Among the coding sequences of 14,987 known protein-coding genes with a median expression RSEM ≥ 1 in all of the TCGA LUAD tumors, we analyzed protein-altering indels and SNVs across 57 RPA(–)G LUADs to identify recurrently mutated genes. The algorithm uses a gamma Poisson regression background model (Imielinski et al., 2017) correcting for known covariates of LUAD mutation density (e.g., chromatin state, replication timing, GC content; method details). We found four tumor suppressor genes—TP53, STK11, KEAP1, and SMARCA4—significantly mutated in the RPA(–)G samples (false discovery rate [FDR] < 0.1; Figure 2A). GISTIC analysis (Mermel et al., 2011) of WGS-derived SCNAs identified significantly deleted regions harboring SETD2 and significantly amplified regions harboring NKX2–1, KAT6A, CCNE1, MDM2, MYC, MCL1, and MYCL (FDR < 0.1; Figure 2A). Of note, we did not identify novel genes that were significantly mutated or amplified/deleted in the RTK/RAS/RAF pathway; in other words, we were unable to identify new putative driver alterations in an obvious candidate member gene of the RTK/RAS/RAF pathway, possibly owing to the limited sample size.
Figure 2. Recurrent coding alterations in RPA(–)G LUADs.

(A) Overview of genomic alterations in 57 RPA(–)G LUADs. Genes significantly mutated (*) or significantly amplified/deleted in the RPA(–)G samples are listed.
(B and C) Example of KEAP1 (3 kbp length) (B) and (C) STK11 (8 kbp length) simple, homozygous deletion (CN = 0), resulting in loss of expression. The distribution of the KEAP1 or STK11 expression is plotted based on the full TCGA LUAD cohort.
(D) Expression comparison of samples with loss-of-function alterations in STK11 and (E) KEAP1 to other RPA(–)G LUAD samples. p values are calculated from Mann-Whitney U tests. Boxplots show median, interquartile range, and 1.5 times the interquartile range.
See also Figure S1.
Our SV analysis (method details) identified simple, focal deletions (<100 kbp) targeting STK11 and KEAP1, coupled with LOH, leading to the decreased expression of these genes (Figures 2B–2E; Table S2). In two cases (TCGA-86–7711 and TCGA-50–6592), the focal deletions targeted the promoter/transcription start site of KEAP1 without altering the coding regions but resulted in the reduced expression of KEAP1 (Figure 2B; Table S2). The identification of focal deletions within the gene bodies of STK11, SMARCA4, and TP53 was reinforced by concordant exon-skip-ping junctions in the corresponding RNA-seq data, suggesting that those transcriptomic variants were driven by DNA alterations and not by alternative splicing (Figures S2A–S2C). Alterations in STK11 in KRAS-driven LUADs have been shown to be associated with immune exclusion and a poor response to immunotherapies (Skoulidis et al., 2018). We found that loss-of-function events in STK11 were anti-correlated with the computationally estimated fraction of leukocytes in the tumor (Figure S2D). We then combined the 28 additional RPA(+)G cases identified from our cohort with 40 RPA(+)G cases that have WGS data from a previously published TCGA study (Imielinski et al., 2017) (Figure S1A). We found STK11 focal deletions in 4/68 RPA(+)G samples, but found no significant difference in STK11 deletion frequency between RPA(+)G and RPA(–)G (p = 0.22, Fisher’s exact test), suggesting that this event may be equally prevalent in both LUAD types.
As opposed to 411 RPA(+) cases from the full TCGA LUAD cohort (including the 28 cases rescued from the RPA(–)E category by our WGS analysis), we found a significant enrichment of TP53 mutations (p = 5.5 × 10−4, odds ratio [OR] = 2.97, Fisher’s exact test), KEAP1 mutations (p = 4.3 × 10−4, OR = 3.06), and SMARCA4 (p = 3.4 × 10−4, OR = 4.16) in the RPA(–)G cases (Figure S2E) for genes listed in Figure 2A. When expanding the analysis to 239 COSMIC cancer genes, we found an additional enrichment of mutations in NRG1 (p = 1.2 × 10−5, OR = 12.0), ESR1 (p = 4.5 × 10−4, OR = 11.4), BLM (p = 1.1 ×10−3, OR = 12.4), and FOXO3 (p = 2.3 × 10−3, OR = 9.29; Figures S2E and S2F). The RPA(–)G samples also showed significantly higher tumor mutation burden (TMB) in a linear regression model controlled for tumor purity (Figure S2G). We did not find additional differences between RPA(+)G and RPA(–)G LUADs in their molecular/clinical features (leukocyte fraction, genome doubling, degree of aneuploidy, age of diagnosis, and genetic ancestry) (Carrot-Zhang et al., 2020). We found that recent smokers (last smoking year <15) were enriched in the RPA(–)G group compared to the RPA(+) group, although mutagen activity associated with tobacco smoking activity (COSMIC SNV signature 4) was not different between the RPA(–)G group and the RPA(+)G group, controlling for tumor purity (Figure S2H).
Recurrent non-coding alterations in RPA(–)G LUADs
We next asked whether the RPA(–)G cases harbored novel SNVs or indels outside the coding genome. We focused the search on regions nominated by a recent TCGA ATAC-seq (assay for transposase-accessible chromatin using sequencing) study that identified regions of open chromatin, and required that the region be identified in at least 2/44 LUAD samples subject to ATAC-seq (Corces et al., 2018). Open chromatin regions are associated with active promoters, enhancers, and transcription factor-binding sites, and thus may be targets of positive somatic selection (Khurana et al., 2016). When we analysed 60,572 total mutations in 139,841 LUAD-specific open chromatin regions (2.7% of the genome) using gamma Poisson regression with known covariates of LUAD passenger mutation density (method details), we found 3 loci that were nominally enriched (FDR < 0.25) in SNVs or indels, located near ILF2, CUL2, and TSN (Figure 3A). Applying the intuition that expressed and dosage-sensitive genes may be targets of noncoding alteration, we examined genes that were consistently expressed across TCGA LUAD (median RSEM > 10) and recurrently amplified in RPA(–)G samples (see method details). This analysis yielded ILF2 as the sole significant peak (FDR < 0.1; Figure 3B).
Figure 3. Identification of ILF2 promoter mutations in RPA(–)G LUADs.

(A) Three genes with non-coding mutations nominated through recurrence analysis across LUAD-related ATAC peaks (left). Red dots indicate loci with FDR < 0.25.
(B) Same as (A), but restricted to ATAC-seq peaks in genes with RSEM ≥ 10 across TCGA LUAD and recurrently amplified in RPA(–)G samples. Red dots indicate FDR < 0.1.
(C) Among 57 RPA(–)G samples, 6 SNVs are observed in the promoter region of ILF2; all are located within ATAC-seq peaks.
(D) Expression comparison of RPA(–)G samples with ILF2 promoter mutations and amplifications. p values are calculated from linear regression analysis correlating expression, adjusting for the local copy number of ILF2 and purity. Boxplot shows median, interquartile range, and 1.5 times the interquartile range. See also Figure S3.
The promoter region of ILF2 (p = 2.7 3 10−6, coefficient = 2, gamma Poisson regression) was mutated in 6/57 cases (Figure 3C). We used the FunSeq2 method to annotate the sequence motifs bound by transcription factors (Fu et al., 2014). All 6 mutations lay in the “sensitive” and “ultrasensitive” (i.e., highly conserved) regions of the genome (Khurana et al., 2013). One mutation (chr1:153643633, G → A) was predicted to disrupt a HOXB6 motif and another mutation (chr1:153643690, G → T) was predicted to disrupt an NR3C1 motif. We did not observe any mutational signature enriched in the 6 mutations. Moreover, RPA(–)G cases harboring ILF2 promoter mutations showed increased expression of ILF2, compared to ILF2-wild-type cases (Figure 3D) or cases harboring other mutations within the ±10-kbp window of the promoter region (Figures S3A and S3B), after controlling for the local copy number of ILF2 and tumor purity. However, non-coding mutations near CUL2 did not affect CUL2 expression, and intronic mutations in TSN showed a non-significant trend toward an increase in TSN expression (p = 0.066). ILF2 is located in chr1q21.3, which is frequently amplified in LUADs. In myeloma, increases in ILF2 expression through amplification have been shown to promote tolerance of genomic instability and drive resistance to DNA-damaging therapies, through dysregulation of RNA splicing and DNA damage response pathways (Marchesini et al., 2017). Consistent with that role of ILF2, we found ILF2 expression to be associated with increased SV burden (p = 0.01, coefficient = 0.9, negative binomial regression).
Complex SV patterns in RPA(–)G LUADs
By integrating read depth changes with rearrangement breakpoint locations to generate junction-balanced genome graphs (method details), we analyzed genome graphs (Hadi et al., 2020) of the 57 RPA(–)G cases to identify complex patterns of structural variation. Analysis of subgraphs (https://github.com/mskilab/gGnome) in those graphs identified multiple instances of complex amplicons (18 double minute, 5 BFBC, 5 tyfonas, 52 pyrgo), as well as simple duplications (mean = 16.3 per sample; Figure 4A). Tyfonas are recently identified SV patterns comprising hundreds of high junction copy number (JCN) and fold-back inversion junctions (Hadi et al., 2020) that are enriched in cancers such as dedifferentiated liposarcomas and acral melanomas. As an example, we show an amplification of NKX2–1 driven by tyfonas in Figure S4A. Pyrgo, which comprises “towers” of low copy duplication junctions (Hadi et al., 2020), were also found to drive the amplification of LUAD loci, including NKX2–1 (Figure S4B).
Figure 4. Classification of SVs in RPA(–)G LUADs.

(A) Identification of simple and complex SV events. Upper panel: SVs resulting in copy-number gain (double minute, BFBC, tyfonas, pyrgo, simple duplication). Lower panel: SVs resulting in copy-number loss (chromothripsis, rigma, chromoplexy, templated insertion chain, simple deletion). Key indicates the range of event count of SV types observed in each sample.
(B) Expression quantile of genes located in SV types with copy-number gain.
(C) Simple deletion count is more significantly enriched in the TP53 mutant RPA(–)G samples than in the TP53-wild-type RPA(–)G samples. p value is obtained from Mann-Whitney U test. Violin plots reflect kernel density estimations.
(D and E) Example of a double minute in TCGA-55–5899 spanning 3 genes (D), and (E) 2 of which (UBL3 and LIG4) showed marked overexpression relative to RNA-seq data for the full TCGA LUAD cohort. See also Figure S4.
Genes located inside double minute, BFBC, and tyfonas events were markedly enriched in expression outlier genes (p < 1 × 10−16, Mann-Whitney U test) relative to genes involved in pyrgo and simple duplication events (Figure 4B), suggesting that the former events were retained in the cancer cell due to the growth-promoting effects of altered gene expression. Although none of these complex SV types were correlated with TP53 mutations, which are thought to generate genomic instability, there was a significantly higher incidence of simple deletions observed in the TP53 mutant cases (p = 1 × 10−4, Mann-Whitney U test; Figure 4C). That association held true when including the 68 RPA(+)G samples and controlling for purity and RPA status (p = 2 × 10−4, coefficient = 12, linear regression).
Double minutes were the most common complex SV type seen in the RPA(–)G cases (12/57 samples; Figure 4A). Like extrachromosomal circular DNA segments, double minutes do not segregate symmetrically; thus, their dosage per cell is exquisitely responsive to selection pressure (Verhaak et al., 2019; Wu et al., 2019). As a result, at least one of the genes in any given double minute likely contributes to tumor development. To leverage this intuition, we focused on a relatively small double minute identified in case TCGA-55–5899 (Figure 4D). We found that this double minute fused and amplified multiple focal regions on chromosome 13 spanning 1.0 Mbp and resulted in the high-level gain (>10 copies) of 3 intact genes (UBL3, SOX21, and LIG4). Two of these, UBL3 and LIG4, were overexpressed in TCGA-55–5899 relative to the full LUAD cohort with RNA-seq data (Figure 4E). Because we did not observe any genes to be amplified and overexpressed in more than one RPA(–)G case (Table S3), larger numbers of cases would have to be analyzed to gain an understanding of the possible role that genes amplified by double minutes play in driving RPA(–) LUADs.
DISCUSSION
Although large-scale genomic studies have shown LUADs to be molecularly heterogeneous, the majority of LUAD cases share the common feature of RTK/RAS/RAF signaling activation through a genetic driver (Desai et al., 2014; Swanton and Govindan, 2016). A key question, however, is whether RPA(–) LUADs—which, by definition, lack RTK/RAS/RAF drivers—represent a biologically distinct entity. Our results suggest that they are heterogeneous but that they do share common biological features, including a high frequency of TP53 mutations and high mutation burden. Those features tend to distinguish RPA(–) LUADs from their RPA(+) counterparts.
A key confounder when we try to define RPA(–) LUADs as a distinct entity is the technical limitation of reliably detecting RTK/RAS/RAF pathway genomic lesions in impure tumor samples. Strikingly, 28/85 cases in the present study that were previously found to be negative for any RTK/RAS/RAF lesion by WES and RNA-seq pipelines were subsequently shown by our WGS analysis to harbor a somatic RTK/RAS/RAF driver. The high prevalence of overlooked KRAS mutations is explained in part by low tumor purity and/or decreased probe affinity for the GC-rich exons in KRAS during WES library preparation (Clark et al., 2011). The recent discovery of small molecules with activity against KRAS p.G12C LUAD (Canon et al., 2019; Ostrem et al., 2013) highlights the importance of the precise identification of mutations at that locus. However, many of the challenges could be overcome by higher read depth (e.g., >40 × minimal on-target coverage), which is now routinely achieved by some clinical-grade target capture assays (Goodman et al., 2017; Zehir et al., 2017). Nevertheless, the high rate of missed RTK/RAS/RAF lesions in our WGS cohort tells a cautionary tale: false-negative calls should always be considered even when working with high-quality datasets.
Eight of the 28 rescued RPA(+)G cases in our WGS cohort harbored cryptic SV lesions (protein-coding fusions, SCNAs), which represent alternative mutational mechanisms for (in)activating RTK/RAS/RAF genes (e.g., EGFR BFBC, focal deletions of NF1 and RASA1). WGS is naturally adapted to detect such complex or subtle structural alterations (Hadi et al., 2020). We leveraged that capability to identify a spectrum of SV patterns among the 57 RPA(–)G cases. Notably, 9/85 (11%) samples in the cohort harbored focal deletions in STK11 that were undetected by WES. Alterations in genes such as STK11 and KEAP1 have come into focus as possible prognostic and/or predictive biomarkers in patients with lung cancer (Arbour et al., 2018; Skoulidis et al., 2018); the inclusion of full genomic capture probe sets for those genes may become necessary in the near future to identify accurately samples with alterations. Double minutes, the most prevalent SV type among complex SVs in the RPA(–)G cases have recently been implicated in genomic plasticity, oncogene selection, and chromatin evolution (Verhaak et al., 2019). Further studies analyzing larger cohorts will be valuable for dissecting the role that they may play in driving RPA(–) LUAD biology in greater detail.
If the 57 RPA(–)G cases identified in this study represent a distinct biological entity of RTK/RAS/RAF-independent LUADs, then what pathway drives them to proliferate? Do RPA(–) LUADs show distinct genetic or therapeutic vulnerabilities? Although our analyses have nominated candidate drivers in LUAD (e.g., ILF2), it is unclear how tumors harboring an alteration in such genes would phenocopy the proliferative effect of RTK/RAS/RAF alteration. Perhaps the frequent SV-driven loss of tumor suppressors (e.g., STK11) or amplification of genes operating downstream of RTK/RAS/RAF signaling (e.g., MYC), which we observed in our RPA(–)G LUADs, can cooperate to fill this missing role (Sears et al., 2000). It is also possible that a small subset of RPA(–) tumors are still RTK/RAS/RAF driven but activate the pathway through epigenetic dysregulation or genetic alterations in genes that are less frequently altered in LUAD, and therefore are not detected by the statistical methods used in this study. Alternatively, the biology of RPA(–) LUADs may resemble that of other cancer types in which RTK/RAS/RAF alterations are rarely seen and are marked by early TP53 loss and high TMB; those tumor subtypes may take a different evolutionary path that is less dependent on the sustained proliferative signaling that RTK/RAS/RAF activation provides (Chen et al., 2019; Drosten et al., 2014; Salgueiro et al., 2020). Such a path may accumulate key genetic alterations in a different order than RPA(+) tumors (Lee et al., 2019), begin in an alternate cell of origin, or undergo lineage switching in the course of evolution.
Our findings suggest that RPA(–) LUAD is likely to represent a heterogeneous entity and that the WGS of much larger cohorts of RPA(–) and RPA(+) LUADs would be necessary to fully address the nature of their underlying biology.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for data or code generated in this study should be directed to and will be fulfilled by the Lead Contact, Marcin Imielinski (mai9037@med.cornell.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Whole-genome BAM files of 85 RPA(–) samples (https://portal.gdc.cancer.gov/repository?facetTab=files&filters=%7B%22op%22%3A%22and%22%2C%22content%22%3A%5B%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22cases.project.program.name%22%2C%22value%22%3A%5B%22TCGA%22%5D%7D%7D%2C%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22cases.project.project_id%22%2C%22value%22%3A%5B%22TCGA-LUAD%22%5D%7D%7D%2C%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22files.experimental_strategy%22%2C%22value%22%3A%5B%22WGS%22%5D%7D%7D%5D%7D) and raw mutation/SV calls generated during this study are released to Genomic Data Commons (https://gdc.cancer.gov/about-data/publications/TCGALUAD). The controlled data access is covered in dbGaP with the accession number dbGaP: phs000178 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000178.v10.p8). All software packages used in the study are available to the public and links to sources can be found in the Key resources table and Method details. Other analysis scripts are available upon request. Experimental model and subject details
KEY RESOURCES TABLE
In this study, we used tumor and matched normal samples collected by the Cancer Genome Atlas (TCGA) Research Network with informed consent under their local Institutional Institutional Review Boards. DNA samples from 85 lung cancer cases were processed by whole-genome sequencing, with associated clinicopathologic data collected by the TCGA.
METHOD DETAILS
Sample selection and whole-genome sequencing
Eighty-five samples were selected for WGS, among 118 previously whole-exome sequenced TCGA LUAD samples that were negative for 1) activating mutations in KRAS, EGFR, BRAF, ERBB2, MET, RIT1, NRAS, RAF1, HRAS, ARAF, MAP2K1 and SOS1; 2) loss-of-function mutations in NF1 and RASA1; 3) fusions in ALK, ROS1, RET, MET and NTRK2; and 4) amplification in EGFR, ERBB2, KRAS, MET, FGFR1 and MAPK1. The same criteria were applied to re-identify RPA(–) samples in the WGS analysis, except that overexpression (defined as a z-score greater than 1.96 for gene expression among the full TCGA LUAD samples) was additionally required to qualify an amplification as an oncogenic driving event.
DNA was sequenced using the Illumina HiSeq platform. Paired-end sequencing reads were aligned to hg19 using BWA (v0.6.2) (Li and Durbin, 2009) aln and processed through NovoSort (v1.03.01) to mark PCR duplicates (http://www.novocraft.com/products/novosort/), then through GATK (v3.4) (McKenna et al., 2010) for indel realignment (jointly for the normal and tumor samples). Base quality scores were recalibrated with GATK.
Identification of somatic mutations and SCNAs
Somatic SNVs were called by MuTect (v1.1.7) (Cibulskis et al., 2013), Strelka (v1.0.14) (Saunders et al., 2012) and LoFreq (v2.1.3a) (Wilm et al., 2012). Somatic indels were called by Strelka, Pindel (v0.2.5) (Ye et al., 2009) and Scalpel (v0.5.3) (Narzisi et al., 2014). Variants called by only one of the three callers were filtered out. Final VCFs were formatted to pass the EBI validator (v 0.4.3) (https://vcftools.github.io/perl_module.html). Variants were annotated for their effect (non-synonymous coding, nonsense, etc.) using snpEff (Cingolani et al., 2012b) based on human genome annotations from ENSEMBL. We further annotated the variants using snpEff (Cingolani et al., 2012b), snpSift (Cingolani et al., 2012a) and GATK VariantAnnotator module with information from COSMIC (Sondka et al., 2018), 1000 Genomes Project (Auton et al., 2015), ExAC (Karczewski et al., 2017), CIViC (Griffith et al., 2017), and UniProt (UniProt Consortium, 2019). Non-coding mutations were further annotated by Funseq2 (Fu et al., 2014). Mutational signatures were analyzed using SignatureAnalyzer (Kim et al., 2016).
Data on genetic ancestry, genome double, aneuploidy, leukocyte fraction and other clinical features were downloaded from the Genomic Data Commons (https://portal.gdc.cancer.gov/). GISTIC 2.0 (Mermel et al., 2011) was used to identify significant SCNAs using copy number segments generated by Titan (Ha et al., 2014). High-level amplification was defined by log2-transformed copy number ratios > 1. To calculate the allelic fraction of KRAS mutations in WGS and WES, we applied a custom script counting reads supporting the altered alleles and the reference alleles, respectively (Carrot-Zhang and Majewski, 2017). Reads with based quality and mapping quality lower than 30 were removed.
Identification of structural variations and genome graph reconstructions
Somatic aberrant junctions (i.e., pairs of strand-specific disconnected loci that form neo-adjacencies in the cancer genome) were identified by SvABA v1.1.3 (Wala et al., 2018), using the default setting for tumor-normal pairs. We then used JaBbA, a junction balance analysis (Hadi et al., 2020) to reconstruct a genome graph for each sample through the application of a maximum likelihood model to high-resolution binned (200bp) normalized read depth and unfiltered SvABA junctions (after exclusion of small (< 1kbp) deletion-like junctions). The read depth input to JaBbA was calculated as the ratio between tumor and normal sample’s WGS read counts in all 200bp genomic bins, corrected for guanine/cytosine content and 100-mer mappability. Subsequently, we used Circular Binary Segmentation (Olshen et al., 2004) with alpha parameter at 1×10−5 to derive a primary segmentation for each sample. The segmentation was later combined with SvABA-identified aberrant junctions to build the genome graph. The affine mapping between read depth signal and integer copy number is dictated by the hyperparameters ploidy and purity. We used the published purity and ploidy values from the GDC (https://portal.gdc.cancer.gov/). Any sample missing from that resource was supplemented by Sequenza (Favero et al., 2015), based on the allelic read counts at germline heterozygous sites using Samtools (Li et al., 2009). A total of 13 types of simple and complex SV events were annotated and visualized using gGnome (https://github.com/mskilab/gGnome), with the same default parameters as described in Hadi et al. (2020). SV burden per sample was defined by the number of junctions of simple SVs in that sample. Genome graphs drawn in copy number ~genomic coordinates plots are made with gTrack (https://github.com/mskilab/gTrack) and gGnome (https://github.com/mskilab/gGnome).
Oncoprints, mutation barplots, and expression quantiles
Genomic alterations affecting genes in the cohort were plotted with ComplexHeatmap (Gu et al., 2016) with the aforementioned definitions for SNVs and CNAs. Tumor mutation burden was calculated by dividing the total number of SNVs in the eligible mutation calling region proposed in (Li, 2014) by the total width of these regions (2429.397 Mbp). Expression quantiles and density plots are made with the gene’s RSEM (RNA-seq by Expectation Maximization) values of the full set of 507 LUAD RNA-seq in TCGA. Mutation barplots (“loliplot”) are made with trackViewer package (http://bioconductor.org/packages/release/bioc/html/trackViewer.html).
Differential alteration frequencies between RPA(–)G and RPA(+)G
The frequency of genomic alterations in various genes were compared between RPA(–)G and RPA(+)G using Fisher’s exact tests with false discovery rate (FDR) threshold below 0.1. To maximize statistical power, we only considered the variant types that can be detected both through WES and WGS, and compared the frequency of alteration in the RPA(–)G group (N = 57) to the rest of all the TCGA LUAD samples with WES-based variant calls from the PanCanAtlas (N = 411, https://api.gdc.cancer.gov/data/1c8cfe5f-e52d-41ba-94da-f15ea1337efc). Same method was used for clinical or molecular features, including smoking history, age of diagnosis, leukocyte fraction, genome doubling, degree of aneuploidy, genetic ancestry, primary disease stage (data available through GDC) using Fisher’s exact test or Wilcoxon’s Rank test. TMB comparison was performed based on WES-defined TMB values using linear regression controlling for tumor purity.
SCISSOR analysis
RNA-seq BAM files for TCGA LUAD samples were used as input to SCISSOR (Choi et al., 2021) for analysis of structural changes in RNA transcripts. Briefly, SCISSOR is a statistical method for unsupervised screening of a range of structural alterations in RNA-seq data including alternative splicing, intron retention, de novo splice sites, intra-/intergenic deletions, and alternative transcription start/termination. For each gene under consideration, SCISSOR aims to identify anomalous shapes in expression profile by considering aligned short read data through a base-level read pileup. To identify such profile shape changes, it quantifies the level of abnormality of each sample using a projection depth approach (Dang and Serfling, 2010; Donoho and Gasko, 1992) and then uses the statistics from the cohort to detect and rank shape outliers. Using reference exon coordinates derived from UCSC hg19 known genes, we constructed base-resolution read coverage data including intronic parts from the BAM files. For TP53, STK11, and SMARCA4, SCISSOR identified shape changes where the aligned coverage shape is significantly different from the majority of samples with a significance level 1×10−4. This process confirmed the effects of focal, exon deletions on RNA transcripts.
Recurrence analysis of genomic alterations
A gamma-Poisson regression framework (fishHook) that takes into account different confounders that affect the mutation count in a population was used (Imielinski et al., 2017). fishHook allows for defining the genomic region of interest, called the hypotheses set, to be used for recurrence analyses. For coding mutation analysis, models were fitted with gene bodies as the hypotheses set. Non-synonymous, missense and truncating mutations were tested separately. We also corrected for multiple hypotheses on 47 genes identified by prior genomic studies on LUAD (Campbell et al., 2016; Cancer Genome Atlas Research Network, 2014; Imielinski et al., 2012). For non-coding mutation analysis, lung-specific ATAC-seq peaks (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG) defining open chromatin regions (Corces et al., 2018) were used as the hypotheses set. Lift over was used to map hg38 coordinates to hg19. ATAC peaks occurring at least 2 of 44 LUAD samples were used, and peaks within 100bp distance were merged. The model was fitted with the following covariates of the neutral mutation density: 1) Fraction of heterochromatic regions in each query interval. Heterochromatin annotation obtained from chromHMM on A549 cell line from Epigenomics Roadmap (Kundaje et al., 2015); 2) Gene expression data for LUAD for A549 cell line; 3) GC content in reference genome; 4) Replication timing from normal human epidermal keratinocytes; 5) DNA accessibility annotation from DNase-seq for A549 cell line. Besides genome-wide hypotheses, we also tested within the subset of ATAC peaks that overlaps recurrently amplified regions (Campbell et al., 2016) and with putative target gene median RSEM > 10 among LUAD samples.
Associations between non-coding mutation and target gene expression are evaluated through fitting an ordinary linear model to log RSEM values with the non-coding mutation presence and amplification status of ILF2 gene.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests are carried out using R (v3.6.1) and Bioconductor (v3.10) and listed within the figure legends and Results. Fisher’s exact tests are executed with “fisher.test” function, Wilcoxon’s Rank test with “wilcox.test,” ordinary linear models with “lm.”
Supplementary Material
Highlights.
Whole-genome sequencing of LUAD(–) RPA reveals complex structural variations
RPA(–) LUADs harbor focal deletions in tumor suppressors
RPA(–) LUADs show elevated TMB and TP53 mutation frequency
ILF2 promoter mutations are recurrent in RPA(–) LUADs
ACKNOWLEDGMENTS
We thank the US National Cancer Institute for funding through U24 grants CA210999, CA210974, CA211006, CA210949, CA210978, CA210952, CA210989, CA210957, CA210990, CA211000, CA210950, CA210969, and CA210988. J.C.-Z. holds a CIHR Banting fellowship. M.I., X.Y., and A.D. were also partially supported by M.I.’s Burroughs Wellcome Fund Career Award for Medical Scientists and startup funds from the Weill Cornell Medicine Department of Pathology and Laboratory Medicine. We are grateful for advice from numerous colleagues at our respective institutions, TCGA and GDAN collaborators, and the GDC technical support team.
Footnotes
CONSORTIA
The members of the TCGA Research Network are Kanika Arora, Minita Shah, Jennifer Shelton, Reanne Bowlby, Verena Friedl, Mary Goldman, Brian Craft, David I. Heiman, Iman Hajirasouliha, Camir Ricketts, Pavana Anur, Kami E. Chiotti, Samantha J. Caesar[HYPHEN]Johnson, John A. Demchok, Martin L. Ferguson, Anab Kemal, Roy Tarnuzzer, Zhining Wang, Liming Yang, Paul T. Spellman, Benjamin Raphael, Rehan Akbani, Jingchun Zhu, Steven J.M. Jones, and Hui Shen.
DECLARATION OF INTERESTS
M.M. is listed as the inventor on the patent for the EGFR mutation analysis for lung cancer diagnosis licensed to LabCorp; holds research support from Bayer, Janssen, and Ono; and serves as scientific advisory board chair for OrigiMed. P.W.L. serves on the scientific advisory boards of AnchorDx and Progenity. D.J.K. has research support from Revolution Medicines and Genentech and is a consultant to AADi.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108707.
REFERENCES
- Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, Lito P, Berger M, Solit DB, Hellmann M, et al. (2018). Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 24, 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR; 1000 Genomes Project Consortium (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Choi PS, Gelbard M, and Meyerson M. (2019). Identification and Characterization of Oncogenic SOS1 Mutations in Lung Adenocarcinoma. Mol. Cancer Res. 17, 1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et al. ; Cancer Genome Atlas Research Network (2016). Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. [DOI] [PubMed] [Google Scholar]
- Carrot-Zhang J, and Majewski J. (2017). LoLoPicker: detecting low allelic-fraction variants from low-quality cancer samples. Oncotarget 8, 37032–37040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrot-Zhang J, Chambwe N, Damrauer JS, Knijnenburg TA, Robertson AG, Yau C, Zhou W, Berger AC, Huang K-L, Newberg JY, et al. ; Cancer Genome Atlas Analysis Network (2020). Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell 37, 639–654.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Carrot-Zhang J, Zhao Y, Hu H, Freeman SS, Yu S, Ha G, Taylor AM, Berger AC, Westlake L, et al. (2019). Genomic and immune profiling of pre-invasive lung adenocarcinoma. Nat. Commun. 10, 5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HT, Jo H, Zhao X, Hoadley KA, Newman S, Holt J, Hayward MC, Love MI, Marron JS, and Hayes DN (2021). SCISSOR: a framework for identifying structural changes in RNA transcripts. Nat. Commun. 12, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, and Lu X. (2012a). Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012b). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Chen R, Lam HYK, Karczewski KJ, Chen R, Euskirchen G, Butte AJ, and Snyder M. (2011). Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 29, 908–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. ; Cancer Genome Atlas Analysis Network (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang X, and Serfling R. (2010). Nonparametric depth-based multivariate outlier identifiers, and masking robustness properties. J. Stat. Plan. Inference 140, 198–213. [Google Scholar]
- Desai TJ, Brownfield DG, and Krasnow MA (2014). Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoho DL, and Gasko M. (1992). Breakdown Properties of Location Estimates Based on Halfspace Depth and Projected Outlyingness. Ann. Stat. 20, 1803–1827. [Google Scholar]
- Drosten M, Sum EYM, Lechuga CG, Simón-Carrasco L, Jacob HKC, García-Medina R, Huang S, Beijersbergen RL, Bernards R, and Barbacid M. (2014). Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc. Natl. Acad. Sci. USA 111, 15155–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, and Eklund AC (2015). Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 26, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Cuesta L, Plenker D, Osada H, Sun R, Menon R, Leenders F, Ortiz-Cuaran S, Peifer M, Bos M, Daßler J, et al. (2014). CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 4, 415–422. [DOI] [PubMed] [Google Scholar]
- Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, Khurana E, and Gerstein M. (2014). FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 15, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, and Kurzrock R. (2017). Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 16, 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, Ainscough BJ, Ramirez CA, Rieke DT, Kujan L, et al. (2017). CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 49, 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Ha G, Roth A, Khattra J, Ho J, Yap D, Prentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, et al. (2014). TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 24, 1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadi K, Yao X, Behr JM, Deshpande A, Xanthopoulakis C, Tian H, Kudman S, Rosiene J, Darmofal M, DeRose J, et al. (2020). Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 183, 197–210.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Morgensztern D, and Boshoff C. (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. [DOI] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. (2012). Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Greulich H, Kaplan B, Araujo L, Amann J, Horn L, Schiller J, Villalona-Calero MA, Meyerson M, and Carbone DP (2014). Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J. Clin. Invest. 124, 1582–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Guo G, and Meyerson M. (2017). Insertions and Deletions Target Lineage-Defining Genes in Human Cancers. Cell 168, 460–472.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha KE, Cummings BB, et al. ; The Exome Aggregation Consortium (2017). The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 45 (D1), D840–D845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, et al. ; 1000 Genomes Project Consortium (2013). Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342, 1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Chakravarty D, Demichelis F, Rubin MA, and Gerstein M. (2016). Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 17, 93–108. [DOI] [PubMed] [Google Scholar]
- Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, Rosenberg JE, Van Allen EM, D’Andrea A, and Getz G. (2016). Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 48, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. ; Roadmap Epigenomics Consortium (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ-K, Park S, Park H, Kim S, Lee J, Lee J, Youk J, Yi K, An Y, Park IK, et al. (2019). Tracing Oncogene Rearrangements in the Mutational History of Lung Adenocarcinoma. Cell 177, 1842–1857.e21. [DOI] [PubMed] [Google Scholar]
- Li H. (2014). Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30, 2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini M, Ogoti Y, Fiorini E, Aktas Samur A, Nezi L, D’Anca M, Storti P, Samur MK, Ganan-Gomez I, Fulciniti MT, et al. (2017). ILF2 Is a Regulator of RNA Splicing and DNA Damage Response in 1q21- Amplified Multiple Myeloma. Cancer Cell 32, 88–100.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G. (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narzisi G, O’Rawe JA, Iossifov I, Fang H, Lee Y-H, Wang Z, Wu Y, Lyon GJ, Wigler M, and Schatz MC (2014). Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat. Methods 11, 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, and Wigler M. (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572. [DOI] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgueiro L, Buccitelli C, Rowald K, Somogyi K, Kandala S, Korbel JO, and Sotillo R. (2020). Acquisition of chromosome instability is a mechanism to evade oncogene addiction. EMBO Mol. Med. 12, e10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, and Nevins JR (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 14, 2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A. (2019). Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34. [DOI] [PubMed] [Google Scholar]
- Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. (2018). STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 8, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton C, and Govindan R. (2016). Clinical Implications of Genomic Discoveries in Lung Cancer. N. Engl. J. Med. 374, 1864–1873. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47 (D1), D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RGW, Bafna V, and Mischel PS (2019). Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat. Rev. Cancer 19, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, et al. (2013). Frequency of TERT promoter mutations in human cancers. Nat. Commun. 4, 2185. [DOI] [PubMed] [Google Scholar]
- Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, Schumacher S, Li Y, Weischenfeldt J, Yao X, et al. (2018). SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res. 28, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Dubash T, Drainas AP, Mardin BR, Chen Y, Stütz AM, Waszak SM, Bosco G, Halvorsen AR, Raeder B, et al. (2017). Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 49, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, and Nagarajan N. (2012). LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, et al. (2019). Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole-genome BAM files of 85 RPA(–) samples (https://portal.gdc.cancer.gov/repository?facetTab=files&filters=%7B%22op%22%3A%22and%22%2C%22content%22%3A%5B%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22cases.project.program.name%22%2C%22value%22%3A%5B%22TCGA%22%5D%7D%7D%2C%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22cases.project.project_id%22%2C%22value%22%3A%5B%22TCGA-LUAD%22%5D%7D%7D%2C%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22files.experimental_strategy%22%2C%22value%22%3A%5B%22WGS%22%5D%7D%7D%5D%7D) and raw mutation/SV calls generated during this study are released to Genomic Data Commons (https://gdc.cancer.gov/about-data/publications/TCGALUAD). The controlled data access is covered in dbGaP with the accession number dbGaP: phs000178 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000178.v10.p8). All software packages used in the study are available to the public and links to sources can be found in the Key resources table and Method details. Other analysis scripts are available upon request. Experimental model and subject details
KEY RESOURCES TABLE
In this study, we used tumor and matched normal samples collected by the Cancer Genome Atlas (TCGA) Research Network with informed consent under their local Institutional Institutional Review Boards. DNA samples from 85 lung cancer cases were processed by whole-genome sequencing, with associated clinicopathologic data collected by the TCGA.