

SUMMARY
Recently, we showed that synthetic anion transporters DSC4P-1 and SA-3 had activity related to cancer cell death. They were found to increase intracellular chloride and sodium ion concentrations. They were also found to induce apoptosis (DSC4P-1) and both induce apoptosis and inhibit autophagy (SA-3). However, determinants underlying these phenomenological findings were not elucidated. The absence of mechanistic understanding has limited the development of yet-improved systems. Here, we show that three synthetic anion transporters, DSC4P-1, SA-3, and 8FC4P, induce osmotic stress in cells by increasing intracellular ion concentrations. This triggers the generation of reactive oxygen species via a sequential process and promotes caspase-dependent apoptosis. In addition, two of the transporters, SA-3 and 8FC4P, induce autophagy by increasing the cytosolic calcium ion concentration promoted by osmotic stress. However, they eventually inhibit the autophagy process as a result of their ability to disrupt lysosome function through a transporter-mediated decrease in a lysosomal chloride ion concentration and an increase in the lysosomal pH.
Graphical Abstract
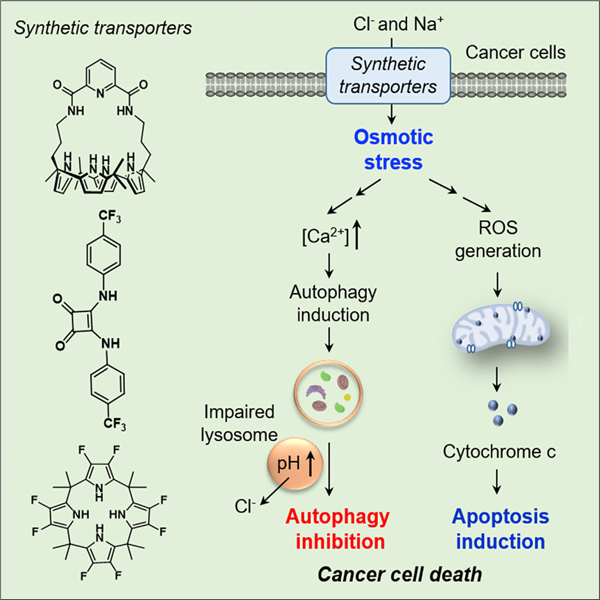
Park et al. show that three synthetic ion transporters, SA-3, 8FC4P, and DSC4P-1, promote apoptosis by increasing intracellular sodium and chloride concentrations in cells and consequently inducing osmotic stress. In addition, two of the transporters, SA-3 and 8FC4P, induce autophagy by increasing the cytosolic calcium ion concentration promoted by osmotic stress. However, they eventually inhibit the autophagy process because of their ability to disrupt lysosome function through a transporter-mediated decrease in lysosomal chloride ion concentration and an increase in the lysosomal pH.
INTRODUCTION
Autophagy is a self-degradative process that is associated with the maintenance of cellular homeostasis and cell survival under conditions of nutrient deficiency.1 During the autophagy process, cytoplasmic constituents are sequestered by phagophores, which are further expanded to form autophagosomes. Subsequently, the autophagosomes fuse with lysosomes to generate autolysosomes. Cytoplasmic constituents inside autolysosomes are degraded by lysosomal enzymes for their recycling.2,3 Multiple previous studies have shown that the level of autophagy in cancer cells is typically higher than that in normal cells.4 As a consequence, agents with autophagy-disrupting activity have potential for the use of anticancer drugs.5 Also, of prime importance in the context of anticancer drug development is apoptosis or programed cell death. This is a physiological process that removes damaged or unwanted cells so as to maintain tissue homeostasis.6 Tumor cells are resistant to apoptotic cell death via multiple anti-apoptotic processes,7 and thus many attempts have been made to develop potent apoptosis inducers that can be used as anticancer agents.8 One promising approach involves perturbing cellular ion concentrations.
Under normal physiological conditions, cellular ion concentrations are tightly regulated by the action of a variety of ion transporters and channels.9 However, perturbations in the intracellular concentrations of ions, such as chloride,10,11 calcium,12 and potassium ions,13 are closely correlated with the onset of apoptosis.14 Because cancer cells have evolved mechanisms to resist engagement of apoptotic cell death,7 channel blockers15 and ionophores16,17 (which induce apoptosis by disturbing the ion homeostasis of cells) have been developed as potential anticancer agents.7 In particular, small-molecule-based synthetic ion carriers or transporters that have the ability to perturb intracellular ion concentrations have attracted attention because of their potential as therapeutic agents.18–20
To date, several transporter systems capable of promoting apoptosis have been reported. For example, the naturally occurring tripyrrolic system prodigiosin and its analogs have been suggested to induce cancer cell death by changing intracellular pH gradients through HCl transport.21,22 In 2014, we showed that the diamide-strapped calix[4]pyrrole-1 (DSCP4–1) (Figure 1) promoted an influx of Cl− and Na+ into cells and consequently enhanced cell death via caspase-dependent apoptosis.23 More recently, we reported that squaramide-3 (SA-3) (Figure 1) enhanced apoptosis by increasing intracellular ion concentrations and blocked autophagy by increasing the lysosomal pH and disrupting lysosome function.24 However, the specific chemical and biochemical determinants of how these and other transporters might affect apoptosis and autophagy remain largely unknown. Acquiring such mechanistic insights is deemed critical if yet-improved transporters are to be designed.
Figure 1. Transmembrane Ion Transport in Liposomes.
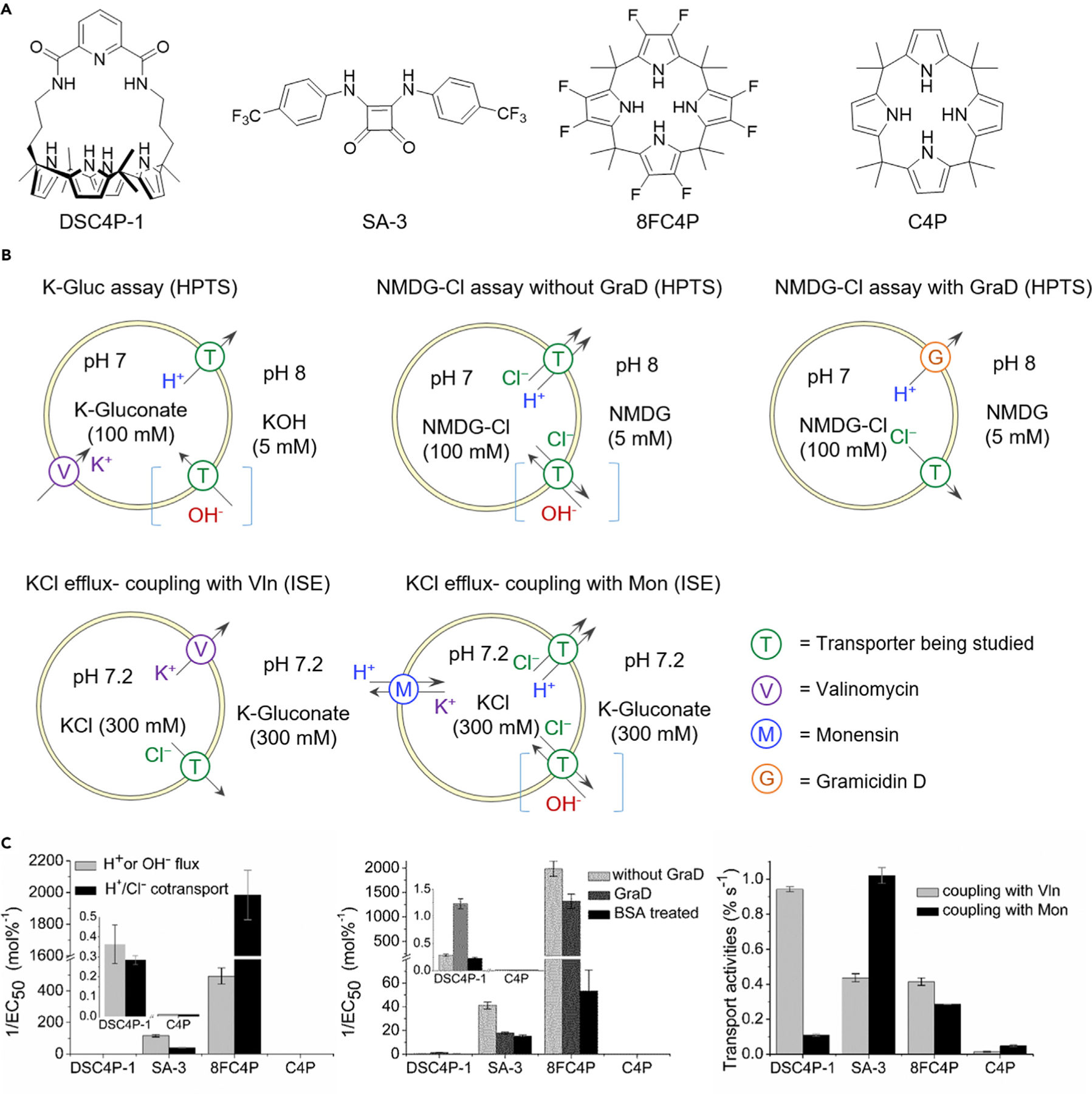
(A) Chemical structures of the synthetic ion transporters used in this study.
(B) Liposome-based assays used in this study; they were monitored by HPTS-fluorescence or chloride-ISE measurements.
(C) Bar charts summarizing transmembrane ion-transport results, expressed as the reciprocal of EC50 values for the K-Gluc and NMDG-Cl assays (standard errors from Hill analysis) and initial rate for the KCl efflux assay (mean ± SD; n = 2 or 3).
We have now elucidated mechanisms of synthetic ion transporters (DSC4P-1, SA-3, 8FC4P, and C4P) (Figure 1) and wish to report how specifically they influence apoptosis and autophagy. As detailed below, we have found that three transporters, DSC4P-1, SA-3, and 8FC4P, increase intracellular Cl− and Na+ concentrations, an event that induces osmotic stress in the cells. This osmotic stress leads to production of reactive oxygen species (ROS) through sequential processes and induces caspase-dependent apoptosis. However, C4P, which lacks ion-transport activity in cells, does not affect apoptosis. Two of the transporters with apoptosis-inducing activity, namely SA-3 and 8FC4P, induce autophagy early on by increasing the cytosolic calcium ion concentration promoted by osmotic stress. However, they eventually suppress the autophagy process by disrupting lysosome function through a decrease in the lysosomal Cl− concentration and an increase in the lysosomal pH. As a consequence, DSC4P-1 promotes cancer cell death by inducing apoptosis, while SA-3 and 8FC4P enhance cancer cell death by inducing apoptosis as well as suppressing autophagy. The control system C4P, which lacks ion-transport activity in cells, neither affects apoptosis nor influences autophagy. To our knowledge, this is the first mechanistic study of how synthetic ion transporters influence apoptosis and autophagy.
RESULTS AND DISCUSSION
Transmembrane Ion-Transport Activities in Liposomes
The ion-transport ability of four compounds (DSC4P-1, SA-3, 8FC4P, and C4P) was initially assessed by several 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposome membrane-based assays. Previously, we demonstrated that 8FC4P can function as a chloride anion transporter by using a chloride-nitrate exchange assay.25 In the current study, we employed a number of recently developed assays26 to provide further insights into the anion-transport mechanisms of 8FC4P, as well as DSC4P-1, SA-3, and C4P. To understand the mechanism of H+ or the equivalent OH− transport by the synthetic transporters, we used an assay based on measuring the intravesicular pH with the fluorescent indicator 8-hydroxypyrene-1,3,6-trisulfonic acid (HPTS) in conjunction with the potassium gluconate (K-Gluc) assay (Figure 1B) to monitor so-called electrogenic H+/OH− transport. An electrogenic process will result in a net flow of charge across a membrane, in this case either H+ efflux or OH− influx. Here, the assumption is that gluconate is not transported because of its high hydrophilicity and bulkiness and that an added K+ ionophore, valinomycin, provides a counterion pathway for electrogenic K+ transport. Results from the K-Gluc assay were compared with data from another complementary HPTS assay (N-methyl-D-glucamine chloride [NMDG-Cl] assay) (Figure 1B) devised to test for H+/Cl− symport or Cl−/OH− antiport; these are electroneutral processes with no charge buildup across the membrane. Dose-dependent experiments and Hill analyses were performed to quantify ion-transport activity (detailed in the Supplemental Information). As shown in Figure 1C, both DSC4P-1 and SA-3 proved active in the two assays. On the basis of these assays, SA-3 was found to be a better transporter than DSC4P-1. 8FC4P was more active in both assays. Whereas DSC4P-1 and SA-3 proved slightly better at H+ or OH− flux than H+/Cl− co-transport, 8FC4P was found to be more effective at H+/Cl− co-transport than H+ or OH– flux. C4P, the parent macrocycle, from which 8FC4P is formally derived, has been reported to be a membrane transport agent for cesium chloride as an ion pair;27 however, it displayed no transport activity even at 100 mol % concentration in all cases. Thus, C4P was considered to be a good control system for the biological studies detailed below.
We also quantified the Cl− > H+/OH− selectivity of these transporters by using a modified NMDG-Cl assay. This assay involves the use of the proton channel gramicidin D (GraD) and vesicles treated with bovine serum albumin (BSA, 1 mol % with respect to lipid) to remove free-fatty-acid impurities from the vesicle membranes (Figure 1B).27 The results (Figure 1C; Table 1) revealed that 8FC4P had the highest selectivity (SBSA/G = 25) for Cl− > H+ in BSA-treated vesicles. However, selectivity (SG ~ 1) was completely lost in the untreated vesicles, indicating significant H+ flux in the presence of free fatty acid, as was also shown in the K-Gluc assay. Only DSC4P-1 displayed a moderate selectivity for chloride in both the untreated and BSA-treated experiments.
Table 1.
Summary of Transmembrane Transport Results of Transporters Used in This Study
| Compound | K-Gluc Assay | NMDG-CI Assay | Selectivity | k (mM S−1)a | Electrogenic Factord | ||||
|---|---|---|---|---|---|---|---|---|---|
| Without GraD | With GraD | BSA Treated | SGb | SBSA/Gc | With Vln | With Mon | |||
| EC50 (mol %) | EC50 (mol %) | EC50 (mol %) | EC50 (mol %) | ||||||
| DSC4P-1 | 2.76 | 3.52 | 0.812 | 4.42 | 4.3 | 5.4 | 2.83 | 0.328 | 8.6 |
| SA-3 | 8.67E–3 | 0.024 | 0.056 | 0.067 | 0.43 | 1.2 | 1.31 | 3.07 | 0.43 |
| 8FC4P | 2.03E–3e | 5.04E–4 | 7.58E–4 | 0.019 | 0.66 | 25 | 1.24 | 0.856 | 1.5 |
| C4P | >100f | >100f | >100f | >100f | – | – | 0.042 | 0.143 | 0.29 |
Hill analysis was performed to obtain the effective concentration to achieve 50% flux (EC50) at 200 s for the K-Gluc and NMDG-Cl assays for each carrier, shown as carrier/lipid molar percentages. The initial rate for KCl efflux assay was derived by nonlinear curve-fitting analysis with the single exponential function. See the Supplemental Information for details of the Hill analysis and initial rate determinations. Vln, valinomycin; Mon, monensin.
All initial rate (k) values of [Cl−] efflux were obtained with 1 mol % loading (ionophore/lipid molar percentage) of compound.
SG = EC50(without GraD)/EC50(with GraD); selectivity factor of Cl− > H+ in untreated vesicles (i.e., in the presence of free fatty acid).
SBSA/G = EC50(BSA treated)/EC50(with GraD); selectivity factor of Cl− > H+ in BSA-treated free-fatty-acid-removed vesicles.
Electrogenic factor = k(with Vln)/k(with Mon).
Using the corrected EC50 value.
Negligible transport at 100 mol % concentration.
We next sought to confirm the mechanism of transport of DSC4P-1. We did this by using cationophore coupling assays to determine whether the receptor-mediated ion transport occurs in an electrogenic or electroneutral fashion or in a nonspecific manner. Ionophore-induced Cl− efflux was measured by an ion-selective electrode (ISE) from KCl-loaded vesicles suspended in an inert external potassium gluconate solution, coupling with either valinomycin (an electrogenic K+ transporter) or monensin (an electroneutral M+/H+ transporter). On this basis, and as shown in Figure 1C, DSC4P-1 appeared to be more effective (by a factor of ~9; see Table 1) at mediating electrogenic Cl– transport than electroneutral H+/Cl− co-transport (or OH−/Cl− exchange).28 In contrast, SA-3 coupled much better to monensin than to valinomycin, supporting the notion that SA-3 acts as an electroneutral cotransporter rather than a chloride uniporter. 8FC4P coupled to both valinomycin and monensin, leading to the conclusion that 8FC4P is a nonspecific anionophore that can function as both a chloride uniporter and an HCl cotransporter. Taken in concert, these model studies thus serve to underscore that DSC4P-1, SA-3, and 8FC4P all function as ionophores but with distinctively different mechanisms. Our preceding findings23,24 lead us to postulate that all these chloride transporters (DSC4P-1, SA-3, and 8FC4P) can increase cytosolic chloride concentration and thus are likely to induce apoptosis. In contrast, only the electroneutral HCl cotransporters (SA-3 and 8FC4P) can decrease lysosomal H+ and Cl− concentration, leading us to suggest that they may act to disrupt autophagy. As described below, detailed cell biological studies were thus undertaken to confirm or refute the validity of these hypotheses.
Cellular Ion-Transport Activities of Synthetic Transporters
We then examined the cellular ion-transport activities of DSC4P-1, SA-3, 8FC4P, and C4P by determining the changes in concentration of several ions inside cells after treatment with each substance. Cancer cells (HeLa, human cervical cancer cells; A549, human lung adenocarcinoma epithelial cells), along with normal cells (FRT, Fischer rat thyroid epithelial cells), were individually incubated with DSC4P-1, SA-3, 8FC4P, and C4P and then treated with the chloride-ion-sensitive probe MQAE (N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide).29 Intracellular chloride ion concentrations were determined with Stern-Volmer plots for quenching of MQAE by Cl− (Figure S25A).29 The results revealed that the transporter 8FC4P promoted an increase in chloride ion concentrations in normal and cancer cells (Figures 2A and S25B). Similar effects were seen in cells treated with DSC4P-1 and SA-3.23,24 Specifically, treatment of cells with 20 µM SA-3 or 20 µM 8FC4P led to an increase in the intracellular chloride ion concentration from ca. 20 to 35–45 mM. DSC4P-1 displayed less effective chloride transport activity than SA-3 and 8FC4P because treatment with 20 µM DSC4P-1 caused an increase in the intracellular chloride ion concentration to ca. 30 mM. In contrast, the control system, C4P, did not induce a significant change in the intracellular chloride ion concentration.
Figure 2. Synthetic Ion Transporters Increase Intracellular Chloride and Sodium Ion Concentrations and Induce Apoptosis.
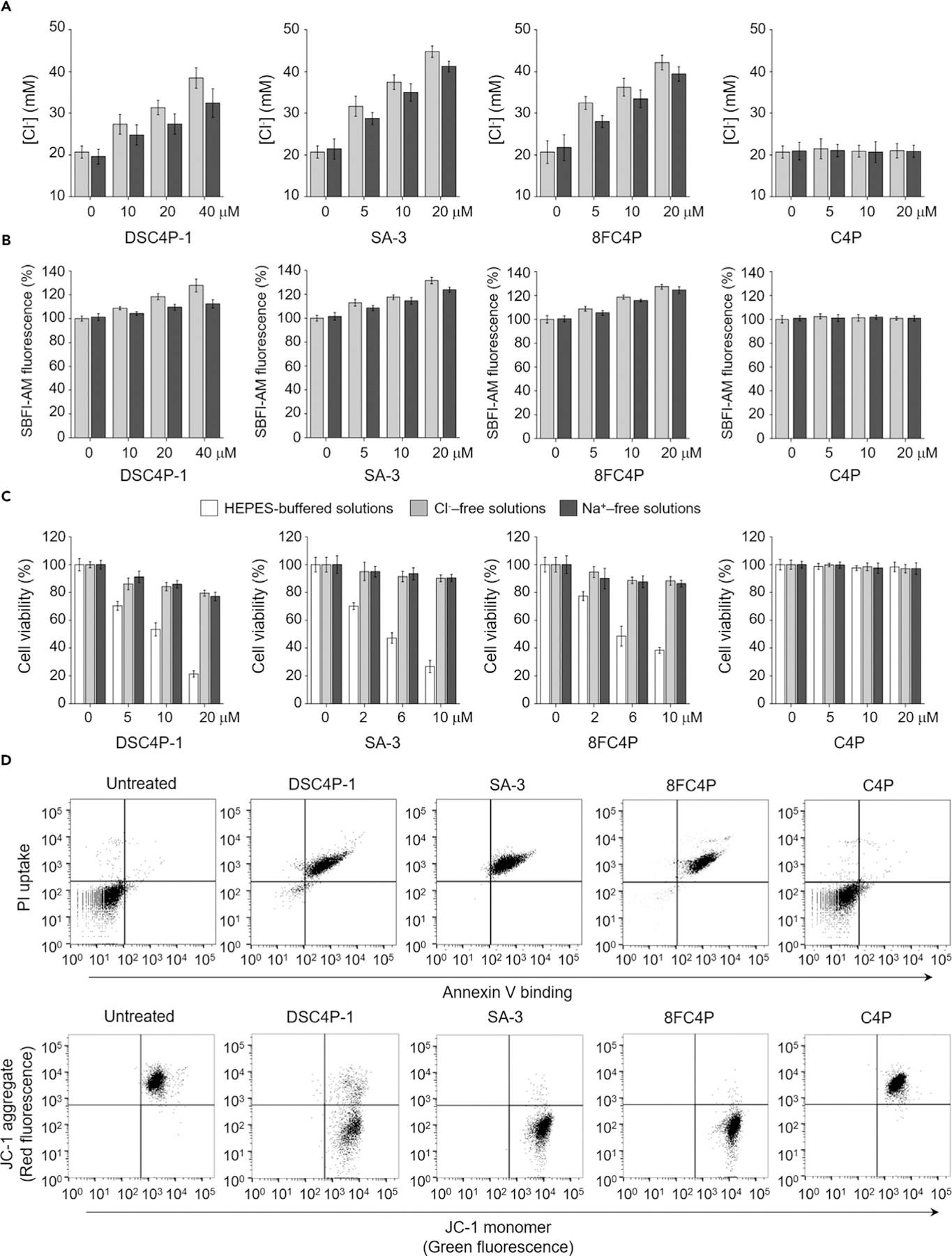
(A) HeLa cells, treated for 2 h with various concentrations of the indicated compounds in the absence (gray bars) and presence (black bars) of 1 mM amiloride, were incubated with 5 mM MQAE for 0.5 h. Intracellular chloride ion concentrations were determined by Stern-Volmer plots shown in Figure S25A (mean ± SD; n = 3).
(B) HeLa cells, treated with 10 µM SBFI-AM for 2 h, were incubated with various concentrations of the indicated agents for 2 h in the absence (gray bars) and presence (black bars) of 1 mM amiloride. SBFI-AM fluorescence was measured for determining changes in the intracellular sodium ion concentrations (mean ± SD; n = 3).
(C) HeLa cells were incubated for 18 h with various concentrations of the indicated agents in HEPES-buffered solutions or Cl−- or Na+-free solutions. Cell death was measured by an MTT assay (mean ± SD; n = 3).
(D) Top: flow cytometry of HeLa cells treated with 40 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, or 40 µM C4P for 24 h and stained with a mixture of fluorescein-annexin V and PI. Untreated cells are shown as a negative control. Bottom: flow cytometry of HeLa cells treated with 10 µM 8FC4P, 20 µM C4P, 10 µM SA-3, and 40 µM DSC4P-1 for 24 h and stained with JC-1. The results are shown as dot plots of red fluorescence (FL2, JC-1 aggregate) versus green fluorescence (FL1, JC-1 monomer).
Under physiological conditions, receptor-mediated into-cell chloride anion transport was expected to be correlated with cation uptake. Cell studies were thus carried out with the sodium fluorescent probe SBFI-AM (sodium-binding benzofuran isophthalate acetoxymethyl ester). It was found that 8FC4P and SA-3 increased the sodium ion concentration in normal and cancer cells with similar efficacy and that both were more efficient than DSC4P-1 (Figures 2B and S25C).23,24 As expected, C4P produced no change in the intracellular sodium ion concentration. To test whether sodium ions enter cells directly as a result of the transport by 8FC4P, SA-3, and DSC4P-1 or via cellular sodium channels, we examined the effect of a sodium channel blocker amiloride on the sodium influx elicited by synthetic transporters. The results revealed that the chloride- and sodium-ion-transport activities of DSC4P-1, SA-3, and 8FC4P were substantially reduced in the presence of the sodium-channel blocker amiloride (Figures 2A, 2B, and S25).23 Thus, it is likely that they act mainly as chloride anion transporters and that the sodium counterions enter cells concomitant with the chloride anions through cellular sodium channels. However, the intracellular potassium ion concentrations measured by a potassium-specific fluorescent probe, PBFI-AM (potassium-binding benzofuran isophthalate acetoxymethyl ester), were found to be unchanged after incubation of normal and cancer cells with DSC4P-1, SA-3, 8FC4P, and C4P (Figure S26).
DSC4P-1, SA-3, and 8FC4P did not affect the level of the calcium ion concentration in normal cells, as measured by the calcium fluorescent probe Fluo-4 NM (Figure S27A).23,24 However, they were found to increase the calcium ion concentrations in cancer cells (Figures S27B and S27C). When the cancer cells were co-treated with each of the three synthetic transporters and 2-aminoethoxydiphenyl borate (2-APB), an inhibitor of an inositol 1,4,5-trisphosphate receptor (IP3R), which is an intracellular Ca2+-release channel located on the endoplasmic reticulum (ER),30 the calcium ion concentrations were not increased (Figures S27B and S27C). On this basis, we conclude that the increase in calcium ions in the cancer cells seen in the presence of the synthetic transporters does not originate from the extracellular media but is the result of the release of calcium ions from the ER through IP3R, as detailed further below. Importantly, DSC4P-1, SA-3, and 8FC4P function as chloride anion transporters, an effect that is correlated with channel-mediated sodium cation co-transport but not with the delivery of either extracellular potassium or calcium ions into the cytosol.
Synthetic Ion Transporters Induce Apoptosis in Cancer Cells
We previously showed that synthetic transporters DSC4P-1 and SA-3 promoted cancer cell death by increasing intracellular ion concentrations.23,24 To allow for valid comparisons, we examined the effects of 8FC4P and C4P on cell viability by incubating several cancer cell lines (HeLa cells, A549 cells, PLC/PRF/5 [Alexander hepatoma] cells, HepG2 [human hepatoblastoma carcinoma] cells, Capan-1 [human pancreatic adenocarcinoma] cells, and HCT116 [human colorectal carcinoma] cells) with each substance. On the basis of the results of an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, 8FC4P was found to induce cancer cell death effectively with half maximum inhibitory concentration (IC50) values similar to that of SA-3 (i.e., 3–5 µM) (Figure S28D). Both of these transporters proved more active than DSC4P-1 (IC50 values of 10–15 µM) (Figure S28D), a compound with moderate ion-transport activity (vide supra). The control system C4P, which proved ineffective as an ion transporter both in cells and in the model transport studies, displayed little cytotoxicity. The findings provide support for the suggestion that, at least for the present series of compounds, there is a strong correlation between the ability to act as an ion transporter and to promote cancer cell death.
To test further whether the observed synthetic transporter-induced cell death was caused by an increase in the intracellular concentrations of chloride and sodium ions, we determined the effect of extracellular Cl− and Na+ on transporter-induced cytotoxicity. HeLa and A549 cells were individually incubated with DSC4P-1, SA-3, 8FC4P, and C4P in buffers containing both chloride and sodium ions (HEPES-buffered solutions) or in analogous buffers depleted in either chloride anion (Cl−-free solutions) or sodium cations (Na+-free solutions).24 MTT assays revealed that the cell-death activities of DSC4P-1, SA-3, and 8FC4P were greatly attenuated in both Cl−-free and Na+-free solutions (Figures 2C and S29). The cells were also incubated with synthetic transporters in the presence of various non-toxic concentrations of amiloride. A decrease in cell-death activity was observed in cells treated with DSC4P-1, SA-3, or 8FC4P in conjunction with amiloride (Figure S30). Taken together, the findings lead us to conclude that the cell death promoted by DSC4P-1, SA-3, and 8FC4P results from an increase in intracellular chloride and sodium ion concentrations and that suppression of sodium ion co-transport reduces activity.
Previously, we demonstrated that DSC4P-1 and SA-3 induce apoptotic cancer cell death.23,24 Thus, the ability of 8FC4P to induce apoptosis was examined. HeLa cells were incubated with 8FC4P, along with DSC4P-1, SA-3, and C4P, and then treated with a mixture of fluorescein-annexin V and propidium iodide (PI). Flow-cytometry analyses revealed that cells treated with 8FC4P displayed positive annexin V binding and PI uptake, phenomena that were also seen in cells treated with DSC4P-1 and SA-3 but not for the cells treated with C4P (Figure 2D). Analysis of the cell size by flow cytometry revealed that cells treated with 8FC4P, DSC4P-1, or SA-3 were characterized by a considerable degree of cell shrinkage; however, this was not the case for cells incubated with C4P (Figure S31A).
The loss of mitochondrial membrane potential, a hallmark of apoptosis, was examined with a membrane potential-sensitive probe, JC-1.31 Flow-cytometry analysis revealed a considerable decrease in the red fluorescence in the case of cells treated with 8FC4P, DSC4P-1, or SA-3 (Figure 2D).32 No such decrease was seen for cells treated with C4P. Moreover, an increase in DNA fragmentation was observed in cells treated with 8FC4P, DSC4P-1, or SA-3 (Figure S31B). Collectively, the findings provide support for the suggestion that cells treated with the synthetic transporters 8FC4P, DSC4P-1, and SA-3 undergo apoptosis but that those treated with C4P do not.
Synthetic Transporters Induce Osmotic Stress in Cells
Next, we sought to elucidate the cellular mechanisms by determining how the synthetic transporters 8FC4P, DSC4P-1, and SA-3 promote apoptosis. On the basis of the above findings, namely that these substances increase intracellular ion concentrations, we assumed that their mode of action might involve osmotic stress-induced apoptotic cell death.33,34 It is known that a sudden alteration in the solute concentration around cells induces osmotic stress and, within a few minutes, causes a change in the cell volume that can persist up to 2.5–3 h.35,36 Under conditions of high extracellular solute concentrations (hyperosmotic conditions), water molecules exit from cells, leading to a decrease in the cell volume (cell shrinkage). However, at low external solute concentrations (hyposmotic conditions), water molecules flow into cells, leading to an increase in the cell volume (cell swelling). Osmotic stresses that induce a change in cell size eventually promote apoptotic cell death.34
To test the ability of the present series of synthetic transporters to induce osmotic stress, we individually incubated HeLa cells with DSC4P-1, SA-3, 8FC4P, and C4P (as a control), along with a known osmotic stress inducer, sucrose.37 The cell size was then measured over a 1.5 h period by confocal microscopy. As shown in Figure 3A and Videos S1, S2, S3, S4, S5, and S6, the cell size increased after short-term incubation with DSC4P-1, SA-3, or 8FC4P before recovering back to the original size within 1 h. These changes were also seen in cells treated with sucrose. However, cells treated with C4P, as well as untreated cells, did not display any change in cell size. The cell-size changes induced by the present series of synthetic transporters were found to be correlated with cellular ion-transport activity. Specifically, SA-3 and 8FC4P with good cellular ion-transport activity induced greater changes in the cell size than DSC4P-1, a system with moderate cellular ion-transport activity. The overall pattern of cell-size changes promoted by DSC4P-1, SA-3, and 8FC4P is typical of those for cells undergoing hyposmotic stress.35
Figure 3. Synthetic Transporters Induce Osmotic Stress in Cells.
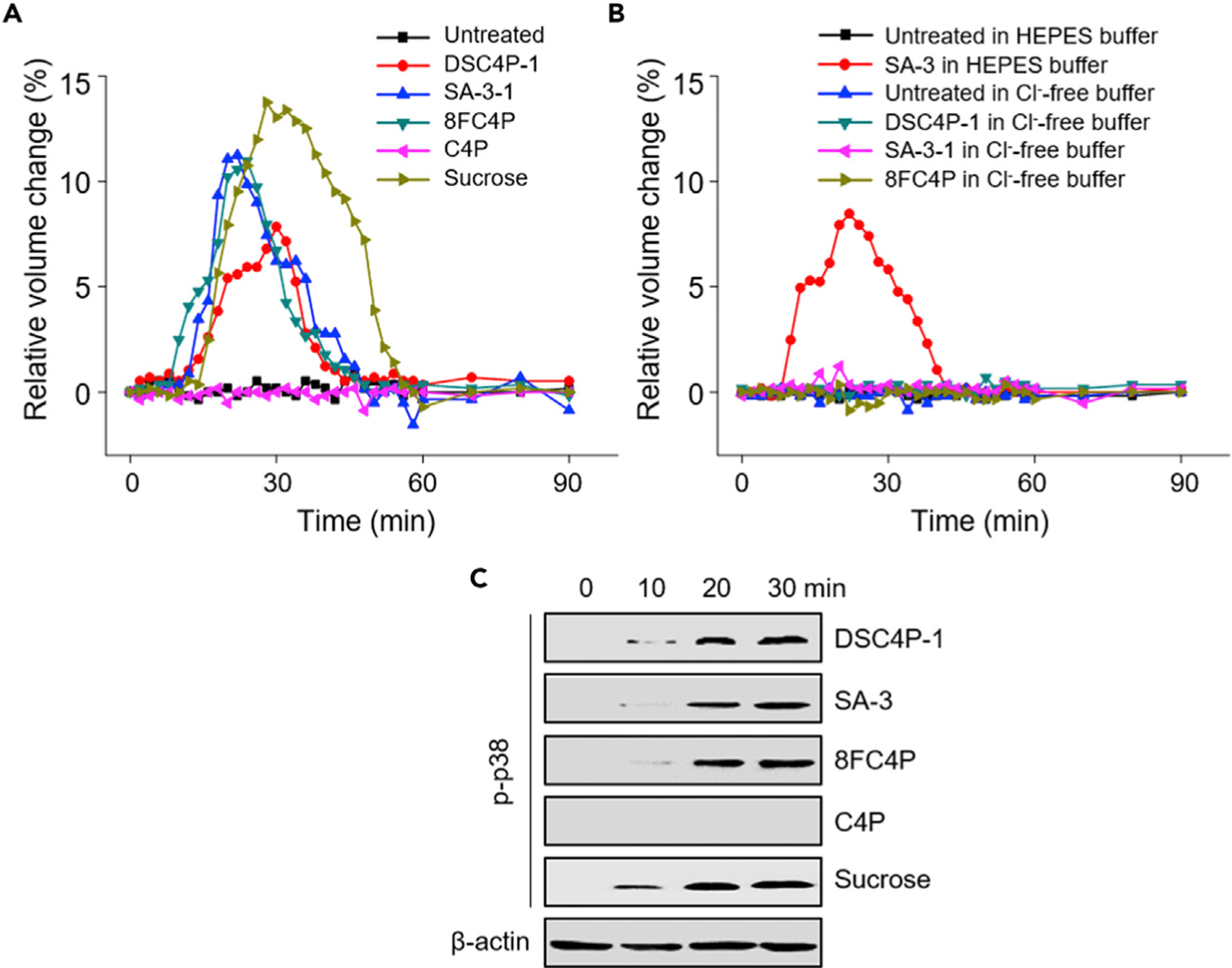
(A and B) HeLa cells were treated with 40 µM DSC4P-1, 20 µM SA-3, 20 µM 8FC4P, 40 µM C4P, or 100 mM sucrose in cell-culture media (A) and with 40 µM DSC4P-1, 20 µM SA-3, or 20 µM 8FC4P in HEPES-buffered solutions or Cl−-free solutions (B) over a 1.5 h period. Cell size was analyzed with ImageJ software. Experiments were repeated three times and gave similar results.
(C) HeLa cells were treated with 40 µM DSC4P-1, 20 µM SA-3, 20 µM 8FC4P, 40 µM C4P, or 100 mM sucrose for the indicated times. Immunoblotting was conducted with the phospho-p38 antibody. β-Actin was used as a loading control.
To determine the effect of chloride ions on the observed cell size changes, we also measured the cell size after treatment of cells with DSC4P-1, SA-3, and 8FC4P in Cl−-free HEPES buffer. The size of cells treated with DSC4P-1, SA-3, or 8FC4P in Cl−-free HEPES buffer was unchanged (Figure 3B; Videos S7, S8, S9, S10, S11, and S12). These findings are consistent with the suggestion that transporter-induced ion influx into cells triggers osmotic stress.
Multiple previous studies have shown that induction of osmotic stress in cells leads to activation of p38 (see Figure 6A).38,39 This typically occurs within a short time window and is ascribed to the stress-induced activation of the guanine nucleotide exchange factor, Brx, followed by the formation of its osmosensing complex with the c-Jun NH2-terminal kinase interacting protein 4 (JIP4).40,41 Thus, increased levels of phosphorylated p38 are taken as an indication of osmotic stress induction.42 On this basis, we tested whether the synthetic transporters of this study had the ability to activate p38. The results of western blot analyses using a phospho-p38 antibody revealed that treatment of DSC4P-1, SA-3, and 8FC4P, as well as sucrose, led to an increase in the levels of phosphorylated p38 after a 10–20 min incubation period (Figure 3C). As expected, C4P had no effect on p38 activation. Collectively, the findings provide support for the hypothesis that because DSC4P-1, SA-3, and 8FC4P act to increase the intracellular ion concentrations rapidly (Figure S32), they induce (hypo)osmotic stress.
Figure 6. Proposed Mechanism of Action Leading to Apoptosis Induction and Autophagy Disruption for the Synthetic Ion Transporters in This Study.
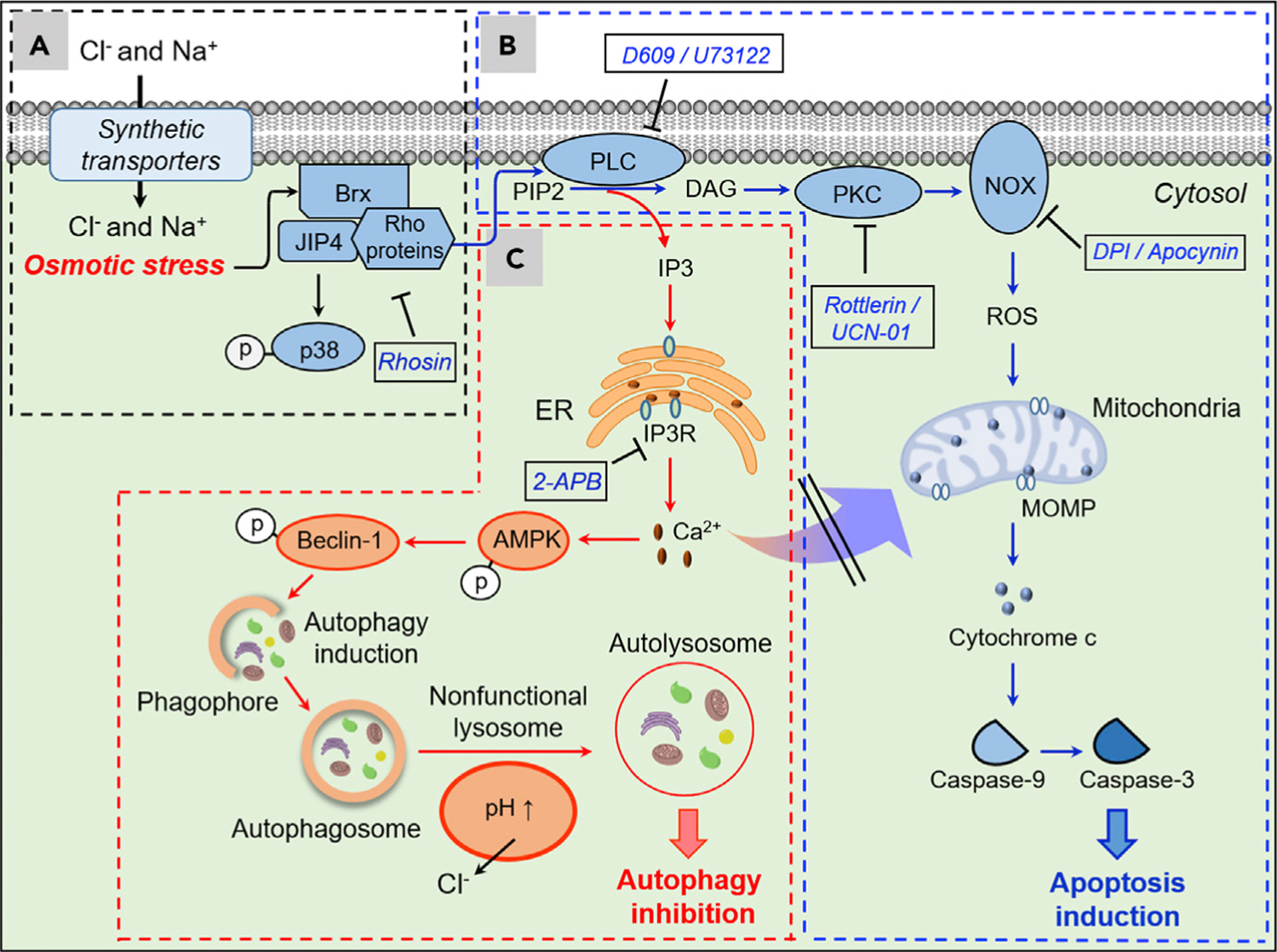
(A) Synthetic transporters DSC4P-1, SA-3, and 8FC4P increase intracellular Cl− and Na+ concentrations, an event that induces osmotic stress in cells. The induced osmotic stress leads to p38 activation presumably through sequential stimulation of Brx and JIP4.
(B) Rho proteins activated by transporter-induced osmotic stress stimulate PLC, which catalyzes cleavage of PIP2 into DAG and IP3. The DAG produced in this way activates PKC, which in turn stimulates NOX to generate ROS. This induces MOMP, thus promoting caspase-dependent apoptosis.
(C) IP3 generated by PLC, which is activated by osmotic stress induced by SA-3 and 8FC4P, leads to an increase in the cytosolic calcium ion concentration through activation of IP3R. This stimulates AMPK and beclin-1 and induces autophagy. However, SA-3 and 8FC4P also disrupt lysosome function by decreasing the lysosomal Cl− concentrations and increasing the lysosomal pH. As a consequence, these transporters eventually block the autophagy process. However, the cytosolic calcium ions increased by action of IP3 do not affect apoptosis. Substances in the box are inhibitors of indicated proteins.
Osmotic Stress Induced by Synthetic Transporters Promotes Apoptosis
Next, we investigated how the osmotic stress induced by DSC4P-1, SA-3, and 8FC4P promotes apoptosis. It is known that when cells are under osmotic stress, Rho-type small G (Rho) proteins are stimulated, presumably through Brx activation (see Figure 6).41 This occurs in addition to p38 activation. The stimulation of Rho proteins in turn activates phospholipase C (PLC), which is responsible for cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-bisphosphate (IP3).43–45 We thus examined whether DSC4P-1, SA-3, and 8FC4P would induce activation of PLC by measuring the PLC activity with a commercial PLC activity assay kit. The results revealed that the PLC activity in cells treated with DSC4P-1, SA-3, or 8FC4P was increased in a concentration-dependent manner; however, little increase was detected in the case of cells treated with C4P (Figure 4A). Moreover, in the presence of either rhosin (an inhibitor of Rho proteins)46 or D609 (a PLC inhibitor),47 the PLC activity in the cells treated with DSC4P-1, SA-3, or 8FC4P was reduced to the basal level (Figure 4A). These findings are taken as further evidence that the osmotic stress in cells treated with the synthetic ion transporters of this study leads to an enhancement in PLC activation through stimulation of Rho proteins.
Figure 4. Osmotic Stress Induced by Synthetic Transporters Promotes ROS Generation.
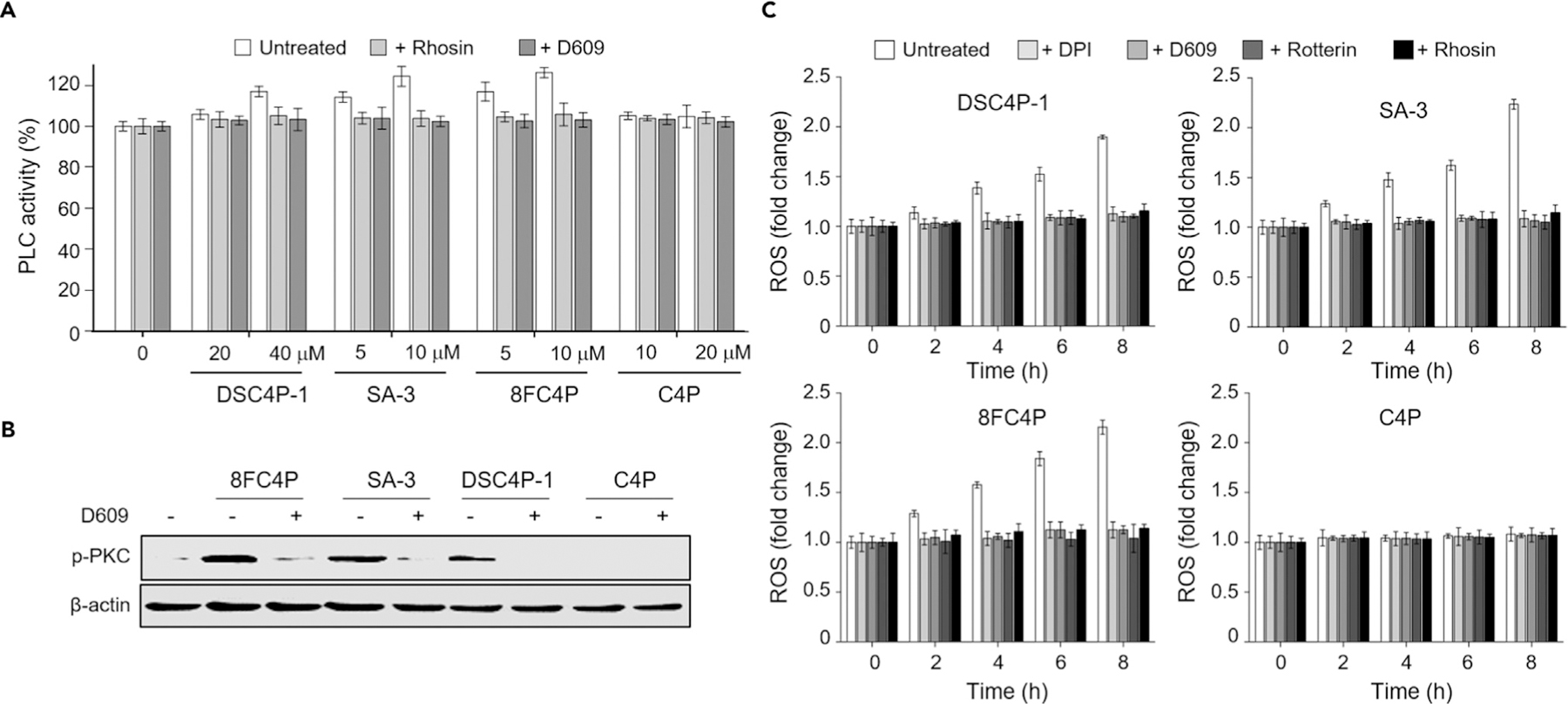
(A) HeLa cells were treated with the indicated concentrations of 8FC4P, SA-3, DSC4P-1, and C4P for 3 h in the absence and presence of either 10 µM rhosin or 10 µM D609. Cell lysates were used to detect PLC activity using a PLC activity assay kit (mean ± SD; n = 3).
(B) HeLa cells, treated with 10 µM 8FC4P, 10 µmM SA-3, 40 µM DSC4P-1, or 40 µM C4P in the absence and presence of 10 µM D609 for 6 h, were subjected to western blotting using the phospho-PKC antibody.
(C) HeLa cells were incubated for 8 h with 20 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, or 20 µM C4P in the absence and presence of each of the indicated inhibitors (10 µM DPI, 10 µM D605, 10 µM rottlerin, and 10 µM rhosin). Treated cells were incubated with 10 µM PF1 for 1 h. The fluorescence intensity of PF1 was measured with a microplate reader (mean ± SD; n = 3).
DAG, produced by activated PLC, is known to stimulate protein kinase C (PKC) and generate phosphorylated PKC through direct binding of DAG to PKC.48–50 Therefore, we sought to determine whether treatment of cells with DSC4P-1, SA-3, and 8FC4P would lead to activation of PKC. The results of western blot analyses revealed that DSC4P-1, SA-3, and 8FC4P significantly increased the level of phospho-PKC, whereas C4P did not (Figure 4B). In contrast, when cells were co-treated with each synthetic transporter and a PLC inhibitor (D609), the level of phospho-PKC was greatly reduced (Figure 4B). These findings are consistent with the notion that DSC4P-1, SA-3, and 8FC4P serve to activate PKC by stimulating PLC.
Multiple previous studies have shown that activated PKC stimulates NADPH oxidase (NOX).51,52 NOX is responsible for the generation of ROS, which are known to induce apoptotic cell death.51 Therefore, we examined the ability of the synthetic ion transporters DSC4P-1, SA-3, and 8FC4P to generate NOX-mediated ROS. Cell studies using the ROS fluorescent probe PF124 revealed that the levels of ROS in cells treated with DSC4P-1, SA-3, or 8FC4P gradually increased in a time-dependent manner (Figure 4C). In contrast, C4P did not affect the level of ROS in cells. ROS production was almost completely abrogated in cells treated with DSC4P-1, SA-3, or 8FC4P in the presence of each inhibitor of Rho proteins (rhosin), PLC (D609, U73122),53 PKC (rottlerin, UCN-01),54,55 and NOX (diphenyleneiodonium (DPI), apocynin),56,57 all of which are inhibitors with no effects on ROS generation, cytochrome c release from mitochondria into the cytosol, caspase activation, or cell viability (Figures 4C and S33). These findings are taken as a clear indication that ROS are generated upon treatment with the synthetic ion transporters of this study and that this occurs via the sequential activation of Rho proteins PLC, PKC, and NOX. This cascade is initially triggered by ion-transport-induced osmotic stress.
Because NOX-mediated ROS generation leads to induction of apoptosis,52 we examined whether DSC4P-1, SA-3, and 8FC4P change the levels of proapoptotic (tBid and Bim) and antiapoptotic (Bcl-2 and Bcl-xL) proteins. The results of western blot analysis of cells treated with DSC4P-1, SA-3, or 8FC4P in the presence of cell-permeable pan-caspase inhibitor (Z-VAD-FMK)24 showed that whereas the levels of antiapoptotic proteins were reduced in treated cells, the levels of proapoptotic proteins were increased (Figure 5A).58–61 However, C4P did not alter the levels of proapoptotic and antiapoptotic proteins in cells. On the basis of these findings, we further examined whether DSC4P-1, SA-3, and 8FC4P activate Bax by determining the induction of mitochondrial outer membrane permeabilization (MOMP). Immunocytochemistry analysis with a Bax (active monomer) antibody revealed that cells treated with DSC4P-1, SA-3, or 8FC4P, but not with C4P, displayed an increased level of active Bax, which co-localized with MitoTracker red (Figure 5B). However, this phenomenon was not observed in cells co-treated with SA-3 and DPI, leading us to conclude that MOMP was induced by ROS generated by NOX (Figure 5B).
Figure 5. Osmotic Stress Induced by Synthetic Transporters Promotes Caspase Activation.
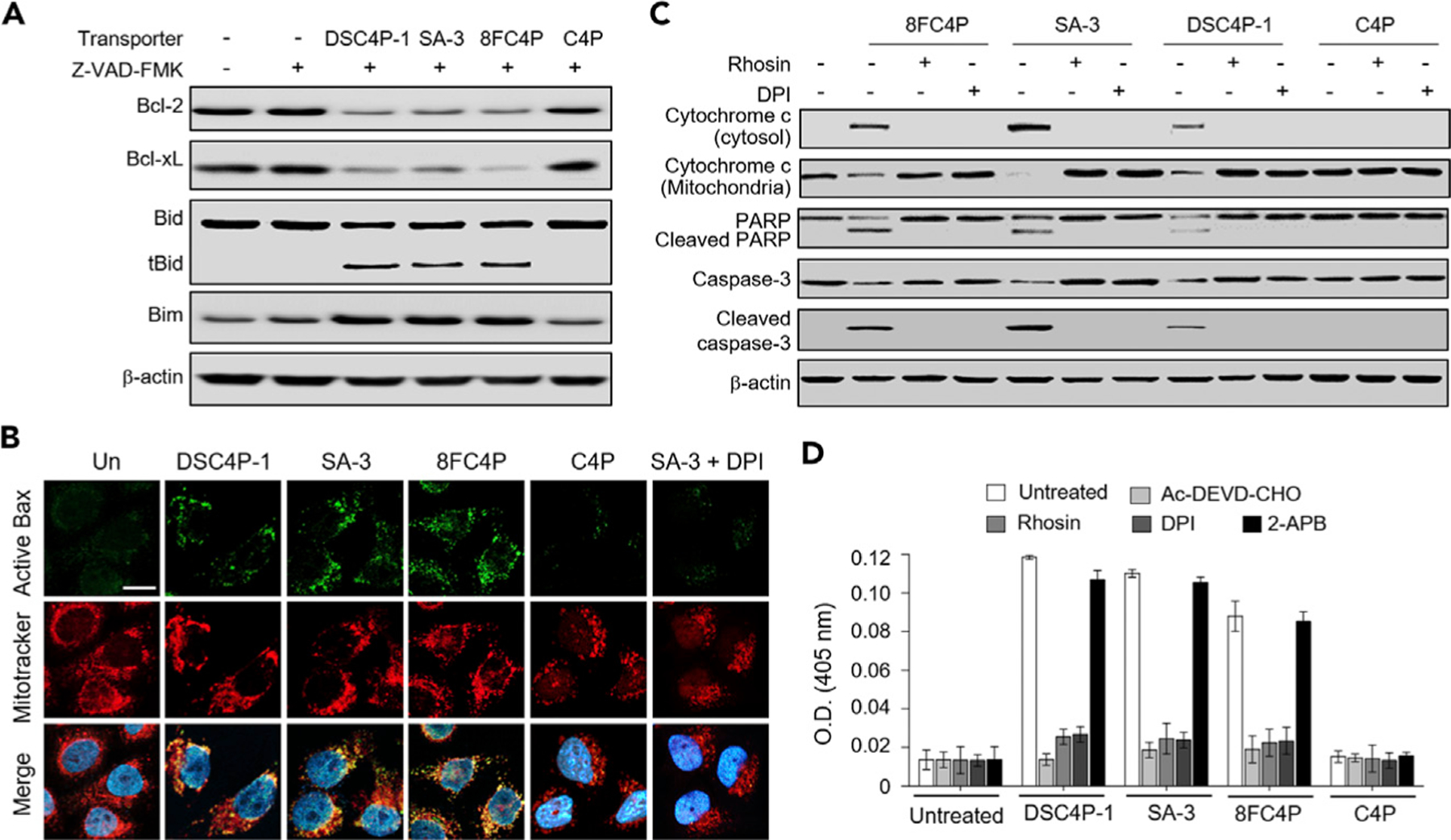
(A) HeLa cells, treated with 40 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, and 40 µM C4P in the absence and presence of 20 µM Z-VAD-FMK for 8 h, were subjected to western blotting with the corresponding antibodies.
(B) HeLa cells, treated with 40 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, and 40 µM C4P, or 10 µM SA-3 + 10 µM DPI for 12 h, were incubated with MitoTracker red and then immunostained with active Bax antibody (green). DAPI was used to stain the nuclei (scale bar, 10 µm).
(C) HeLa cells, treated with 10 µM 8FC4P, 10 µM SA-3, 40 µM DSC4P-1, or 40 µM C4P in the absence and presence of either 10 µM DPI or 10 µM rhosin for 18 h, were subjected to western blotting with the corresponding antibodies.
(D) HeLa cells were treated for 18 hr with 40 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, or 40 µM C4P. The caspase activities of the cell lysates were determined with 200 µM Ac-DEVD-pNA in theabsence and presence of 20 µM Ac-DEVD-CHO (mean ± SD; n = 3). In addition, HeLa cells were treated for 18 h with 40 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, or 40 µM C4P in the presence of each inhibitor (10 µM rhosin, 10 µM DPI, and 10 µM 2-APB). The caspase activities of the cell lysates were determined with 200 µM Ac-DEVD-pNA.
When MOMP is induced in cells, cytochrome c is released from the mitochondria into the cytosol, where it interacts with Apaf-1 and activates caspase-9, which in turn activates caspase-3, thereby inducing apoptosis via caspase activation.62,63 The results of western blotting revealed that cytochrome c is released from mitochondria into the cytosol and that generation of cleaved caspase-3 takes place after incubation with DSC4P-1, SA-3, or 8FC4P, but not with C4P (Figure 5C). Additional experiments aimed at evaluating the cleavage of the cellular caspase substrate poly(ADP-ribose) polymerase (PARP) revealed that the cleaved product of PARP was generated in cells treated with DSC4P-1, SA-3, or 8FC4P. On the other hand, co-treatment of each transporter with each inhibitor of Rho proteins (rhosin), PLC (U73122), PKC (UCN-01), and NOX (DPI, apocynin) led to a remarkable decrease in cytochrome c release as well as the levels of cleaved caspase-3 and PARP (Figures 5C and S34B). The finding provides further support for the suggestion that the cytochrome c release and procaspase-3 cleavage mediated by DSC4P-1, SA-3, and 8FC4P result from osmotic stress and subsequently generated ROS.
To determine whether the synthetic transporters of the present study induce caspase activation, we determined the caspase activities of lysates of cells treated with DSC4P-1, SA-3, and 8FC4P, as well as C4P, by using a colorimetric peptide substrate for caspases, Ac-DEVD-pNA (p-nitroaniline [pNA]). The results revealed that the caspase activity in cells treated with DSC4P-1, SA-3, or 8FC4P was increased (Figure 5D). This increase was blocked when Ac-DEVD-CHO, a known inhibitor of caspases, was added to the lysates of cells treated with DSC4P-1, SA-3, or 8FC4P. In addition, almost no caspase activity was detected when each inhibitor of Rho proteins (rhosin), PLC (U73122), PKC (UCN-01), and NOX (DPI, apocynin) was co-treated with DSC4P-1, SA-3, or 8FC4P (Figures 5D and S34C).
Taken together, the findings provide evidence in support of the conclusion that the present synthetic transporters induce osmotic stress in cells by increasing intracellular ion concentrations. This then leads to the generation of ROS via the sequential activation of Rho proteins, PLC, PKC, and NOX, which then promotes caspase-dependent apoptosis (Figures 6A and 6B).
Synthetic Transporters SA-3 and 8FC4P Inhibit the Autophagy Process by Disrupting Lysosome Function
As a complement to our study of apoptosis, we investigated the effects of synthetic transporters on autophagy. As described above, the osmotic stress induced by the present synthetic transporters was found to stimulate PLC, an enzyme that cleaves PIP2 into DAG and IP3. Previous studies have led to an appreciation that the IP3 produced by PLC serves to activate IP3R, which is an intracellular Ca2+-release channel of the ER (Figure 6C).64,65 When IP3R is activated by IP3, Ca2+ ions are released from the ER into the cytosol. The increased cytosolic Ca2+ concentration stimulates the AMP-activated protein kinase (AMPK).66 These events also activate beclin-1, which plays a central role in autophagy by enhancing the formation of phagophores.67,68 We thus tested whether the synthetic transporters of the present study would affect the cytosolic Ca2+ concentration and whether they would induce activation of AMPK and beclin-1.
HeLa cells were individually incubated with DSC4P-1, SA-3, 8FC4P, and C4P in the absence and presence of inhibitors of PLC (D609, U73122) and IP3R (2-APB). The cytosolic Ca2+ ion levels were then measured with the probe Fluo-4 NW. Activation of AMPK and beclin-1 was determined via western blot analysis using antibodies against phosphorylated AMPK and beclin-1, respectively. It was found that 8FC4P and SA-3 significantly increased the cytosolic Ca2+ concentrations, whereas DSC4P-1 only increased the Ca2+ levels slightly and C4P engendered almost no change in the concentrations (Figure 7A). Treatment of 8FC4P and SA-3 was found to increase the levels of phosphorylated AMPK and beclin-1, whereas DSC4P-1 and C4P did not (Figure 7B). When inhibitors of PLC and IP3R were used in conjunction with DSC4P-1, SA-3, or 8FC4P, the cytosolic Ca2+ concentrations in the treated cells were decreased to almost basal levels (Figure 7A). The levels of phosphorylated AMPK and beclin-1 were likewise greatly reduced when 2-APB, D609, or U73122 was co-treated with SA-3 and 8FC4P (Figures 7B and S35). It is worth mentioning that PLC inhibitors (D609 and U73122) themselves do not affect autophagy (Figure S36). The findings are taken as evidence that both SA-3 and 8FC4P increase the cytosolic Ca2+ concentration by releasing Ca2+ ions from the ER into the cytosol through IP3R, thus stimulating AMPK and beclin-1. However, the less effective transporter DSC4P-1 does not activate AMPK and beclin-1 appreciably. This finding is in line with the fact that DSC4P-1 produces only a slight increase in the cytosolic Ca2+ concentration. It should be emphasized that the Ca2+ ion increase seen in the cytosol of cancer cells in the presence of SA-3 and 8FC4P originates from the ER and not from the extracellular media.
Figure 7. Synthetic Ion Transporters Disrupt Lysosome Function.
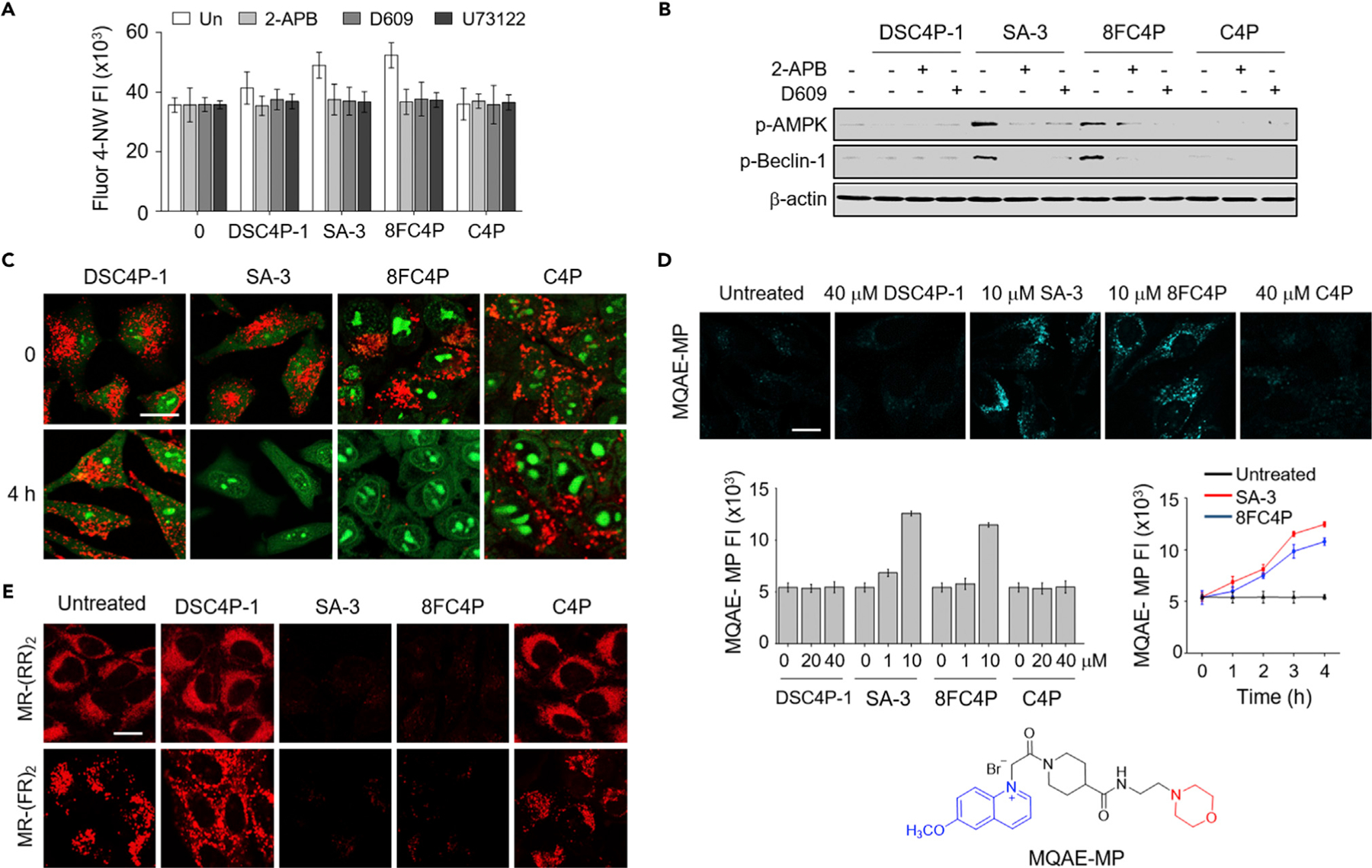
(A) HeLa cells, treated with 10 µM Fluo-4 NW for 1 h, were incubated with 20 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, or 20 µM C4P in the absence and presence of 10 µM 2-APB, 10 µM D609, or 5 µM U73122 for 6 h. Fluo-4 fluorescence was measured with a microplate reader (mean ± SD; n = 3).
(B) HeLa cells, treated with 20 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, or 20 µM C4P in the absence and presence of either 10 µM 2-APB or 10 µM D609 for 8 h, were subjected to immunoblotting with the corresponding antibodies.
(C) HeLa cells, treated with 20 µM DSC4P-1, 10 µM SA-3 10 µM 8FC4P, or 20 µM C4P for 4 h, were incubated with 100 nM acridine orange for 30 min. Cell images were obtained by confocal microscopy (scale bar, 10 µm) (see Figure S33 for more cell images).
(D) HeLa cells, treated with the indicated concentrations of 8FC4P, SA-3, DSC4P-1, and C4P for 4 h, were incubated with 5 mM MQAE-MP for 30 min. Cell images were obtained by confocal microscopy (scale bar, 10 µm) (see Figure S34 for more cell images). Left graph: HeLa cells were incubated with 8FC4P, SA-3, DSC4P-1, and C4P for 4 h and then treated with 5 mM MQAE-MP for 30 min. The graph shows the fluorescence intensity of MQAE-MP in cells, as analyzed by ZEN 2011 software (mean ± SD; n = 3). Right graph: HeLa cells were treated with 10 µM SA-3 or 10 µM 8FC4P over a 4 h period. Every hour, the treated cells were incubated with 5 mM MQAE-MP. The graph shows the fluorescence intensity of MQAE-MP in cells, which was analyzed by ZEN 2011 software (mean ± SD; n = 3).
(E) HeLa cells were incubated for 8 h with 20 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, or 20 µM C4P and then treated with MR-(RR)2 or MR-(FR)2 for 4 h. Cell images were obtained by confocal fluorescence microscopy (scale bar, 10 µm).
In addition to a role that cytosolic Ca2+ ions play in the induction of autophagy, these ions are also known to be associated with caspase-dependent apoptosis.69 To test whether the cytosolic Ca2+ ion concentration increased upon treatment with the present synthetic transporters affects apoptosis, we treated cancer cells with each of the synthetic transporters in the absence and presence of the IP3R inhibitor 2-APB. Caspase activity was then measured with Ac-DEVD-pNA. It was found that the caspase activity of cells treated with DSC4P-1, SA-3, or 8FC4P was almost unchanged in the presence of 2-APB (Figure 5D). Collectively, these findings lead us to suggest that 8FC4P and SA-3 promote autophagy by activating AMPK and beclin-1 without inducing Ca2+-mediated apoptosis and that DSC4P-1 does not enhance Ca2+-mediated autophagy (Figure 6C). Slight increases in cytosolic Ca2+ ions are known to induce autophagy, whereas an overload in Ca2+ promotes apoptosis.70,71 We thus consider it likely that the cytosolic Ca2+ concentration increase induced by 8FC4P and SA-3 is sufficient to induce autophagy but not apoptosis.
Our previous study showed that SA-3 disrupted lysosome function by increasing the lysosomal pH, consequently inhibiting the autophagy process.24 Since most lysosomal enzymes are stable and active in the acidic pH range (pH % 5),72 the increase in the lysosomal pH induced by SA-3 was expected to lead to disruptions in lysosome function.73 On the other hand, if lysosomal functions essential for the autophagy process are disrupted, autophagy should be inhibited; this should be true even under conditions that might otherwise induce autophagy (e.g., conditions that enhance the phagophore formation). Accordingly, we examined whether the present synthetic transporters would affect lysosome function by initially measuring the lysosomal pH. Toward this end, cells were incubated with DSC4P-1, SA-3, 8FC4P, or C4P and then treated with acridine orange, a stain routinely used to detect acidic vesicles, including lysosomes.74 The results of cell image analysis revealed that 8FC4P increased the lysosomal pH as effectively as SA-3, as judged from the observed decrease in the red fluorescence arising from acridine orange (Figures 7C and S37). In marked contrast, the extent of red fluorescence remained unchanged in cells treated with DSC4P-1 and C4P.
Multiple previous studies have shown that the lysosomal pH is correlated with the lysosomal Cl− concentration regulated by the lysosomal chloride transport protein ClC-7.72,75
Higher lysosomal pH values are correlated with relatively low lysosomal Cl− concentrations. Since the lysosomal chloride concentration is higher (~ 80 mM) than the cytosolic chloride concentration (5–40 mM),76 a lysosomal membrane-permeable synthetic chloride ion transporter can potentially disrupt the autophagy process by promoting the transfer of Cl− and therefore H+ out of the lysosome. To determine whether treatment with the present synthetic ion transporters leads to decreases in the lysosomal Cl− concentration, we incubated cells with each transporter and then treated them with a lysosomal chloride-ion-selective fluorescent probe, MQAE-MP (Figure 7D).77 On the basis of the extent of fluorescence quenching of MQAE-MP, incubation with 8FC4P and SA-3 serves to decrease the lysosomal Cl− concentrations (Figures 7D and S38). In contrast, DSC4P-1 had almost no effect on the lysosomal Cl− levels.
Transporters 8FC4P and SA-3 were both found to promote chloride-anion-mediated H+ co-transport (or OH−-for-Cl− exchange) in the model membrane studies and to induce an increase in the lysosomal pH in cells (vide supra). It was thus considered likely that these two transporters could affect lysosomal enzyme activities that are optimal in the acidic pH range (pH % 5). To test this possibility, we determined the effect of the present synthetic ion transporters on the activity of the lysosomal enzymes cathepsins B and L by using cell-permeable cathepsin B and L probes, MR-(RR)2 and MR-(FR)2, respectively.24 Treatment of cells with 8FC4P and SA-3 led to a noticeable decrease in the activity levels of cathepsins B and L, as judged from the diminished red fluorescence derived from MR-(RR)2 and MR-(FR)2 (Figure 7E). However, DSC4P and C4P showed no effect on the activity of either cathepsin B or L. Taken together, these findings support the contention that 8FC4P and SA-3, but not DSC4P and C4P, disrupt lysosomal function by decreasing the lysosomal Cl− anion concentrations.
Because lysosome function is critical for the autophagy process, transporters with an ability to disrupt lysosome function should block the autophagy process. To test this proposal, we studied the fluorescence intensity of a tandem mRFP-EGFP-LC3 fusion protein (mRFP, monomeric red fluorescence protein; EGFP, enhanced green fluorescence protein). It is known that both RFP and GFP fluorescence signals are observed in autophagosomes, but only RFP signals are detected in acidic autolysosomes.78 Cells stably expressing the mRFP-EGFP-LC3 fusion protein were incubated with DSC4P-1, SA-3, 8FC4P, or C4P, along with bafilomycin A1 (BfA1) and torin-1 as controls for autophagy inhibition and induction, respectively.24 Confocal fluorescence microscopy image analysis of cells treated with 8FC4P revealed a conspicuous increase in yellow vesicles, reflecting a merger of the GFP and RFP fluorescence emission (Figures 8A and S39). This phenomenon was also observed in SA-3- or BfA1-treated cells. In contrast, cells treated with torin-1 exhibited predominantly red fluorescent vesicles, reflecting its ability to induce autophagy. Finally, cells treated with DSC4P-1 and C4P displayed fluorescence patterns similar to those of untreated cells (Figures 8A and S39), a finding we take as indicating that they do not affect autophagy.
Figure 8. Synthetic Ion Transporters Disrupt Autophagy.
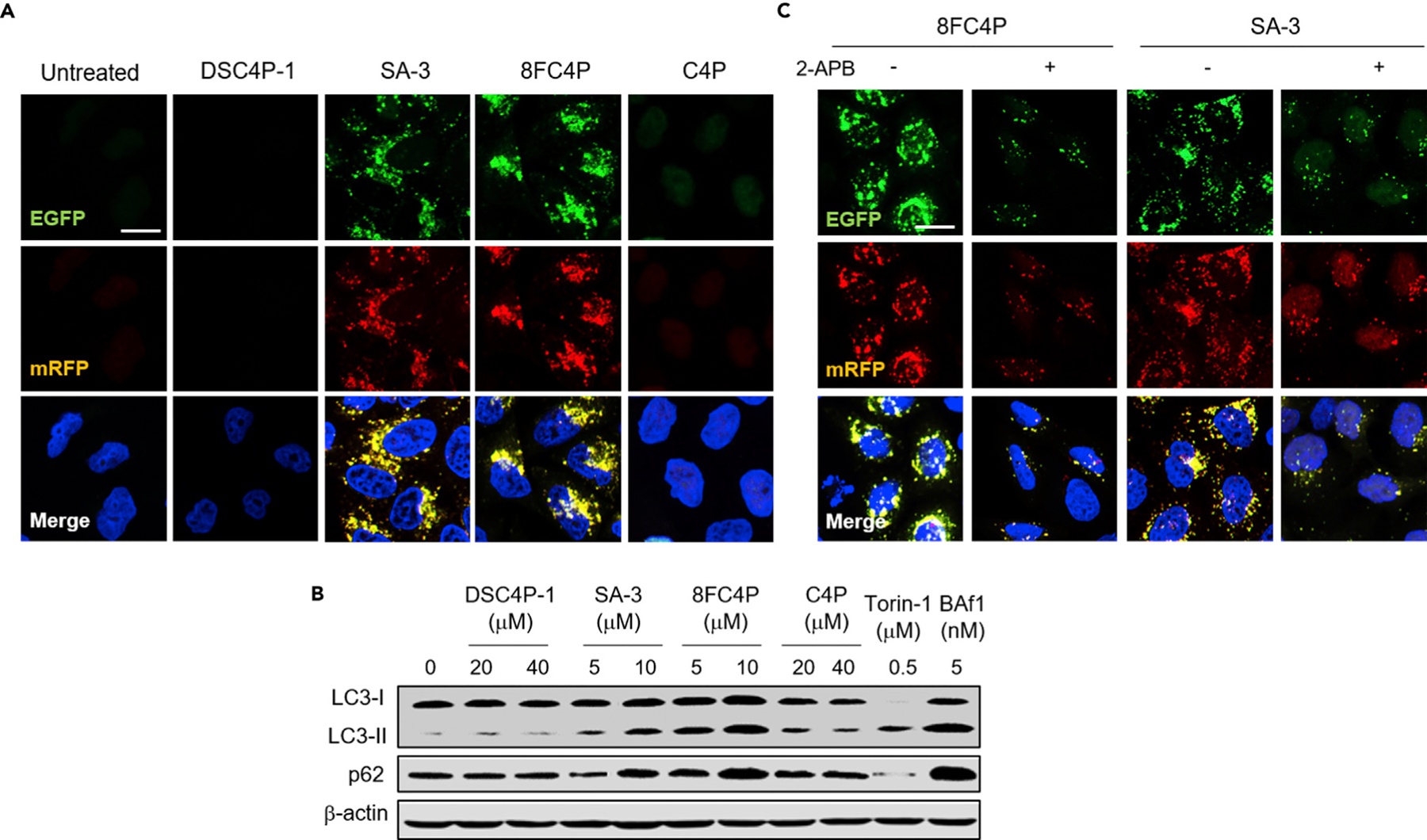
(A) HeLa cells stably expressing mRFP-EGFP-LC3 fusion protein were treated with 20 µM DSC4P-1, 10 µM SA-3, 10 µM 8FC4P, or 20 µM C4P for 12 h. Cell images were obtained by confocal microscopy (scale bar, 10 µm) (see Figure S35 for more cell images).
(B) HeLa cells, treated with the indicated concentrations of 8FC4P, C4P, DSC4P-1, or SA-3 for 12 h, were subjected to immunoblotting with LC3 and p62 antibodies.
(C) HeLa cells were treated with 10 µM SA-3 or 10 µM 8FC4P in the absence and presence of 10 µM 2-APB for 12 h, and the resulting cell images were obtained by confocal microscopy (scale bar, 10 µm).
The LC3-II and p62 levels were also analyzed. Typically, increased levels of LC3-II and decreased levels of p62 are observed during autophagy induction.5 In contrast, increased levels of both LC3-II and p62 are considered indicative of disruptions in autophagic flux. Analysis of LC3-II and p62 levels in cells treated separately with DSC4P-1, SA-3, 8FC4P, and C4P revealed that both SA-3 and 8FC4P acted to increase the levels of both LC3-II and p62 (Figure 8B). This was also true for cells treated with BfA1. In contrast, the LC3-II and p62 levels in cells treated with DSC4P-1 and C4P were similar to those in untreated cells (Figure 8B). As expected, cells treated with torin-1 were characterized by an increase in the LC3-II level and a decrease in the p62 level. Collectively, these findings are interpreted in terms of 8FC4P and SA-3 acting as autophagy inhibitors, but not DSC4P-1 or C4P.
As discussed above, both SA-3 and 8FC4P activate AMPK and beclin-1 by increasing the cytosolic Ca2+ concentration, thereby inducing the autophagy process. We also found that these two transporters eventually inhibit autophagy by disrupting lysosome function. On this basis, we surmised that if the autophagy induction promoted by SA-3 and 8FC4P is blocked by an IP3R inhibitor, then both the number and size of puncta of autophagosomes and/or nonfunctional autolysosomes in cells should be decreased in relation to those in cells treated with the synthetic transporters alone. To test this postulate, we individually treated cells stably expressing mRFP-EGFP-LC3 with SA-3 and 8FC4P in the absence and presence of 2-APB, an IP3R inhibitor that reduces the cytosolic Ca2+ concentration and thus suppresses activation of AMPK and beclin-1. As expected, cells treated with SA-3 or 8FC4P alone exhibited both larger-sized and a greater number of puncta than those co-treated with 2-APB and each transporter (Figure 8C). Also, larger puncta and a greater number of puncta were observed in cells co-treated with torin-1 as an autophagy inducer and BfA1 as a lysosome function disruptor than in cells treated with BfA1 alone (Figure S40).79 These combined findings provide support for the notion that SA-3 and 8FC4P induce autophagy by increasing the cytosolic Ca2+ concentration and that they eventually inhibit the autophagy process because they disrupt lysosome function. Particularly noteworthy is the distinct correlation between the effects on autophagy and the model membrane studies. Specifically, the electroneutral HCl cotransporters (8FC4P and SA-3) were found to regulate lysosomal functions, whereas the electrogenic chloride transporter (DSC4P-1) was found to have little discernible effect.
Conclusions
To date, considerable effort has been devoted to developing artificial ion transporters that have anticancer activity.23,24,80–83 However, most of this effort has been empirical in nature. Understanding the cellular mechanisms underlying cancer cell death induced by artificial transporters could allow for further, more rational designs of active transporters. In the present study, we analyzed in detail three synthetic ion transporters, namely DSC4P-1, SA-3, and 8FC4P. These systems, but not the control C4P, increase intracellular Cl− and Na+ concentrations. This leads to (hypo)osmotic stress in cells and the production of ROS via sequential processes, which in turn induce caspase-dependent apoptosis. C4P does not affect apoptosis. A particularly noteworthy finding to emerge from the present study is that two of the synthetic ion transporters, SA-3 and 8FC4P, increase cytosolic calcium ion concentrations and activate AMPK and beclin-1, which are critical for autophagy induction. However, SA-3 and 8FC4P also disrupt lysosome functions essential for the autophagy process by decreasing the lysosomal Cl− concentrations and increasing the lysosomal pH. As a consequence, SA-384 and 8FC4P eventually block the autophagy process. Taken together, these results underscore subtle differences in what on the surface may appear to be ostensibly similar transport function and effects. In particular, DSC4P-1 promotes cancer cell death by inducing apoptosis, whereas 8FC4P and SA-3 enhance cancer cell death by inducing apoptosis and suppressing autophagy. To our knowledge, this is the first mechanistic study of how synthetic ion transporters affect apoptosis and autophagy. It thus sets the stage for further advances in the design of potential ion-transport-based drug leads.
Supplementary Material
HIGHLIGHTS.
The transport and biological properties of a series of anionophores were studied
Synthetic ion transporters induce osmotic stress in cells
SA-3 and 8FC4P promote cell death by inducing apoptosis and inhibiting autophagy
DSC4P-1 enhances cell death by inducing apoptosis without affecting autophagy
The Bigger Picture.
New approaches to treating cancer remain at the forefront of necessity in spite of considerable clinical progress. Among the attractive strategies currently being explored at the early stages of drug discovery is the use of ion transporters that can disrupt the normal ion balance across cell membranes. Recently, a number of synthetic ion transporters have been described. Several with so-called drug-like properties have been shown to enhance chloride fluxes and to induce apoptosis, a process also known as programed cell death. Select agents also show promise for inhibiting autophagy, a key cellular degradation process that is critical to maintaining a healthy cellular environment. Understanding how synthetic ion transporters affect these two cellular processes may prove critical to elucidating their mode of action. The present study thus serves to highlight the potential of ion transporters as possible drug leads and underscore the complexities associated with their mode of action.
ACKNOWLEDGMENTS
This paper is dedicated to the memory of our friend and colleague Professor Vladi-mír Král. This study was financially supported by a grant from the National Creative Research Initiative Program (2010–0018272) in Korea. The work in Austin was supported by the National Institutes of Health (RO1 GM 103790 to J.L.S.) and the Robert A. Welch Foundation (F-0018 to J.L.S.). P.A.G. thanks the Australian Research Council (DP180100612), the University of Sydney, the Royal Society, the Wolfson Foundation, and the Engineering and Physical Sciences Research Council (EP/J009687/1) for funding.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chempr.2019.05.001.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES AND NOTES
- 1.Mizushima N, and Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- 2.Glick D, Barth S, and Macleod KF (2010). Autophagy: cellular and molecular mechanisms. J. Pathol 221, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eskelinen EL, and Saftig P (2009). Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 1793, 664–673. [DOI] [PubMed] [Google Scholar]
- 4.White E (2012). Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baek KH, Park J, and Shin I (2012). Autophagy regulating small molecules and their therapeutic applications. Chem. Soc. Rev 41, 3245–3263. [DOI] [PubMed] [Google Scholar]
- 6.Newmeyer DD, and Ferguson-Miller S (2003). Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112, 481–490. [DOI] [PubMed] [Google Scholar]
- 7.Igney FH, and Krammer PH (2002). Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer 2, 277–288. [DOI] [PubMed] [Google Scholar]
- 8.Ko SK, Kim J, Na DC, Park S, Park SH, Hyun JY, Baek KH, Kim ND, Kim NK, Park YN, et al. (2015). A small molecule inhibitor of ATPase activity of HSP70 induces apoptosis and has antitumor activities. Chem. Biol 22, 391–403. [DOI] [PubMed] [Google Scholar]
- 9.Gould D (1998). Hum. Physiol: from cells to systems, [Third edition]. J. Adv. Nurs 28, 680–682. [Google Scholar]
- 10.Yu L, Jiang XH, Zhou Z, Tsang LL, Yu MK, Chung YW, Zhang XH, Wang AM, Tang H, and Chan HC (2011). A protective mechanism against antibiotic-induced ototoxicity: role of prestin. PLoS One 6, e17322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsukimoto M, Harada H, Ikari A, and Takagi K (2005). Involvement of chloride in apoptotic cell death induced by activation of ATP-sensitive P2X7 purinoceptor. J. Biol. Chem 280, 2653–2658. [DOI] [PubMed] [Google Scholar]
- 12.Lee JM, Davis FM, Roberts-Thomson SJ, and Monteith GR (2011). Ion channels and transporters in cancer. 4. Remodeling of Ca(2+) signaling in tumorigenesis: role of Ca(2+) transport. Am. J. Physiol. Cell Physiol 301, C969–C976. [DOI] [PubMed] [Google Scholar]
- 13.Remillard CV, and Yuan JX-J (2004). Activation of K+ channels: an essential pathway in programmed cell death. Am. J. Physiol. Lung Cell. Mol. Physiol 286, L49–L67. [DOI] [PubMed] [Google Scholar]
- 14.Yu SP, Canzoniero LMT, and Choi DW (2001). Ion homeostasis and apoptosis. Curr. Opin. Cell Biol 13, 405–411. [DOI] [PubMed] [Google Scholar]
- 15.Arcangeli A, Crociani O, Lastraioli E, Masi A, Pillozzi S, and Becchetti A (2009). Targeting ion channels in cancer: a novel frontier in antineoplastic therapy. Curr. Med. Chem 16, 66–93. [DOI] [PubMed] [Google Scholar]
- 16.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, and Lander ES (2009). Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding WQ, Liu B, Vaught JL, Yamauchi H, and Lind SE (2005). Anticancer activity of the antibiotic clioquinol. Cancer Res 65, 3389–3395. [DOI] [PubMed] [Google Scholar]
- 18.Olschewski A, Papp R, Nagaraj C, and Olschewski H (2014). Ion channels and transporters as therapeutic targets in the pulmonary circulation. Pharmacol. Ther 144, 349–368. [DOI] [PubMed] [Google Scholar]
- 19.Fürstner A (2003). Chemistry and biology of roseophilin and the prodigiosin alkaloids: a survey of the last 2500 years. Angew. Chem. Int. Ed 42, 3582–3603. [DOI] [PubMed] [Google Scholar]
- 20.Busschaert N, Wenzel M, Light ME, Iglesias-Hernández P, Pérez-Tomás R, and Gale PA (2011). Structure–activity relationships in tripodal transmembrane anion transporters: the effect of fluorination. J. Am. Chem. Soc 133, 14136–14148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato T, Konno H, Tanaka Y, Kataoka T, Nagai K, Wasserman HH, and Ohkuma S (1998). Prodigiosins as a new group of H+/Cl− symporters that uncouple proton translocators. J. Biol. Chem 273, 21455–21462. [DOI] [PubMed] [Google Scholar]
- 22.Shen B, Li X, Wang F, Yao X, and Yang D (2012). A synthetic chloride channel restores chloride conductance in human cystic fibrosis epithelial cells. PLoS One 7, e34694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ko SK, Kim SK, Share A, Lynch VM, Park J, Namkung W, Van Rossom W, Busschaert N, Gale PA, Sessler JL, et al. (2014). Synthetic ion transporters can induce apoptosis by facilitating chloride anion transport into cells. Nat. Chem 6, 885–892. [DOI] [PubMed] [Google Scholar]
- 24.Busschaert N, Park SH, Baek KH, Choi YP, Park J, Howe ENW, Hiscock JR, Karagiannidis LE, Marques I, Félix V, et al. (2017). A synthetic ion transporter that disrupts autophagy and induces apoptosis by perturbing cellular chloride concentrations. Nat. Chem 9, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gale PA, Tong CC, Haynes CJE, Adeosun O, Gross DE, Karnas E, Sedenberg EM, Quesada R, and Sessler JL (2010). Octafluorocalix[4]pyrrole: a chloride/bicarbonate antiport agent. J. Am. Chem. Soc 132, 3240–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X, Howe ENW, and Gale PA (2018). Supramolecular transmembrane anion transport: new assays and insights. Acc. Chem. Res 51, 1870–1879. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, and Gale PA (2016). Small-molecule uncoupling protein mimics: synthetic anion receptors as fatty acid-activated proton transporters. J. Am. Chem. Soc 138, 16508–16514. [DOI] [PubMed] [Google Scholar]
- 28.Clarke HJ, Howe ENW, Wu X, Sommer F, Yano M, Light ME, Kubik S, and Gale PA (2016). Transmembrane fluoride transport: direct measurement and selectivity studies. J. Am. Chem. Soc 138, 16515–16522. [DOI] [PubMed] [Google Scholar]
- 29.Verkman AS, Sellers MC, Chao AC, Leung T, and Ketcham R (1989). Synthesis and characterization of improved chloride-sensitive fluorescent indicators for biological applications. Anal. Biochem 178, 355–361. [DOI] [PubMed] [Google Scholar]
- 30.Mikoshiba K (2007). IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J. Neurochem 102, 1426–1446. [DOI] [PubMed] [Google Scholar]
- 31.Williams DR, Ko S-K, Park S, Lee M-R, and Shin I (2008). An apoptosis-inducing small molecule that binds to heat shock protein 70. Angew. Chem. Int. Ed 47, 7466–7469. [DOI] [PubMed] [Google Scholar]
- 32.Tzung SP, Kim KM, Basañez G, Giedt CD, Simon J, Zimmerberg J, Zhang KY, and Hockenbery DM (2001). Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat. Cell Biol 3, 183–191. [DOI] [PubMed] [Google Scholar]
- 33.Jäckle T, Hasel C, Melzner I, Brüderlein S, Jehle PM, and Möller P (2001). Sustained hyposmotic stress induces cell death: apoptosis by defeat. Am. J. Physiol. Cell Physiol 281, C1716–C1726. [DOI] [PubMed] [Google Scholar]
- 34.Ball BA (2008). Oxidative stress, osmotic stress and apoptosis: impacts on sperm function and preservation in the horse. Anim. Reprod. Sci 107, 257–267. [DOI] [PubMed] [Google Scholar]
- 35.Tilly BC, Gaestel M, Engel K, Edixhoven MJ, and de Jonge HR (1996). Hypo-osmotic cell swelling activates the p38 MAP kinase signalling cascade. FEBS Lett 395, 133–136. [DOI] [PubMed] [Google Scholar]
- 36.Kiehl TR, Shen D, Khattak SF, Jian Li Z, and Sharfstein ST (2011). Observations of cell size dynamics under osmotic stress. Cytometry A 79, 560–569. [DOI] [PubMed] [Google Scholar]
- 37.Andronic J, Shirakashi R, Pickel SU, Westerling KM, Klein T, Holm T, Sauer M, and Sukhorukov VL (2015). Hypotonic activation of the myo-inositol transporter SLC5A3 in HEK293 cells probed by cell volumetry, confocal and super-resolution microscopy. PLoS One 10, e0119990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou X, Naguro I, Ichijo H, and Watanabe K (2016). Mitogen-activated protein kinases as key players in osmotic stress signaling. Biochim. Biophys. Acta 1860, 2037–2052. [DOI] [PubMed] [Google Scholar]
- 39.Nandigama R, Padmasekar M, Wartenberg M, and Sauer H (2006). Feed forward cycle of hypotonic stress-induced ATP release, purinergic receptor activation, and growth stimulation of prostate cancer cells. J. Biol. Chem 281, 5686–5693. [DOI] [PubMed] [Google Scholar]
- 40.Kelkar N, Standen CL, and Davis RJ (2005). Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol. Cell. Biol 25, 2733–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kino T, Segars JH, and Chrousos GP (2010). The guanine nucleotide exchange factor Brx: a link between osmotic stress, inflammation and organ physiology and pathophysiology. Expert. Rev. Endocrinol. Metab 5, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagata Y, and Todokoro K (1999). Requirement of activation of JNK and p38 for environmental stress-induced erythroid differentiation and apoptosis and of inhibition of ERK for apoptosis. Blood 94, 853–863. [PubMed] [Google Scholar]
- 43.Hao JJ, Liu Y, Kruhlak M, Debell KE, Rellahan BL, and Shaw S (2009). Phospholipase C–mediated hydrolysis of PIP2 releases ERM proteins from lymphocyte membrane. J. Cell Biol 184, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suh PG, Park JI, Manzoli L, Cocco L, Peak JC, Katan M, Fukami K, Kataoka T, Yun S, and Ryu SH (2008). Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep 41, 415–434. [DOI] [PubMed] [Google Scholar]
- 45.Park D, Jhon DY, Lee CW, Lee KH, and Rhee SG (1993). Activation of phospholipase C isozymes by G protein beta gamma subunits. J. Biol. Chem 268, 4573–4576. [PubMed] [Google Scholar]
- 46.Shang X, Marchioni F, Sipes N, Evelyn CR, Jerabek-Willemsen M, Duhr S, Seibel W, Wortman M, and Zheng Y (2012). Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem. Biol 19, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh AT, Radeff JM, Kunnel JG, and Stern PH (2000). Phosphatidylcholine-specific phospholipase C inhibitor, tricyclodecan-9-yl xanthogenate (D609), increases phospholipase D-mediated phosphatidylcholine hydrolysis in UMR-106 osteoblastic osteosarcoma cells. Biochim. Biophys. Acta 1487, 201–208. [DOI] [PubMed] [Google Scholar]
- 48.Huang KP (1989). The mechanism of protein kinase C activation. Trends Neurosci 12, 425–432. [DOI] [PubMed] [Google Scholar]
- 49.Newton AC (1995). Protein kinase C: structure, function, and regulation. J. Biol. Chem 270, 28495–28498. [DOI] [PubMed] [Google Scholar]
- 50.Chang ZL, and Beezhold DH (1993). Protein kinase C activation in human monocytes: regulation of PKC isoforms. Immunology 80, 360–366. [PMC free article] [PubMed] [Google Scholar]
- 51.Lambeth JD (2004). NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol 4, 181–189. [DOI] [PubMed] [Google Scholar]
- 52.Jiang F, Zhang Y, and Dusting GJ (2011). NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev 63, 218–242. [DOI] [PubMed] [Google Scholar]
- 53.Bleasdale JE, and Fisher SK (1993). Use of U-73122 as an inhibitor of phospholipase C-dependent processes. Neuroprotocols 3, 125–133. [Google Scholar]
- 54.Gschwendt M, Müller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, and Marks F (1994). Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun 199, 93–98. [DOI] [PubMed] [Google Scholar]
- 55.Akinaga S, Gomi K, Morimoto M, Tamaoki T, and Okabe M (1991). Antitumor activity of UCN-01, a selective inhibitor of protein kinase C, in murine and human tumor models. Cancer Res 51, 4888–4892. [PubMed] [Google Scholar]
- 56.Altenhöfer S, Radermacher KA, Kleikers PW, Wingler K, and Schmidt HH (2015). Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid. Redox Signal 23, 406–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petrônio MS, Zeraik ML, Fonseca LM, and Ximenes VF (2013). Apocynin: chemical and biophysical properties of a NADPH oxidase inhibitor. Molecules 18, 2821–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herrera B, Fernández M, Alvarez AM, Roncero C, Benito M, Gil J, and Fabregat I (2001). Activation of caspases occurs downstream from radical oxygen species production, Bcl-xL down-regulation, and early cytochrome c release in apoptosis induced by transforming growth factor beta in rat fetal hepatocytes. Hepatology 34, 548–556. [DOI] [PubMed] [Google Scholar]
- 59.Rahmani M, Aust MM, Attkisson E, Williams DC Jr., Ferreira-Gonzalez A, and Grant S (2013). Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res 73, 1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacob SF, Würstle ML, Delgado ME, and Rehm M (2016). An analysis of the truncated Bid- and ROS-dependent spatial propagation of mitochondrial permeabilization waves during apoptosis. J. Biol. Chem 291, 4603–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bedard K, and Krause KH (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev 87, 245–313. [DOI] [PubMed] [Google Scholar]
- 62.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, and Wang X (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. [DOI] [PubMed] [Google Scholar]
- 63.Bratton SB, and Salvesen GS (2010). Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci 123, 3209–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schlossmann J, Ammendola A, Ashman K, Zong X, Huber A, Neubauer G, Wang GX, Allescher HD, Korth M, and Wilm M (2000). Regulation of intracellular calcium by a signaling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature 404, 197–201. [DOI] [PubMed] [Google Scholar]
- 65.Thillaiappan NB, Chavda AP, Tovey SC, Prole DL, and Taylor CW (2017). Ca2+ signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun 8, 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jeon SM (2016). Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med 48, e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He C, Zhu H, Li H, Zou MH, and Xie Z (2013). Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 62, 1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang R, Zeh HJ, Lotze MT, and Tang D (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Joseph SK, and Hajnóczky G (2007). IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 12, 951–968. [DOI] [PubMed] [Google Scholar]
- 70.Pinton P, Giorgi C, Siviero R, Zecchini E, and Rizzuto R (2008). Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhivotovsky B, and Orrenius S (2011). Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium 50, 211–221. [DOI] [PubMed] [Google Scholar]
- 72.Graves AR, Curran PK, Smith CL, and Mindell JA (2008). The Cl− /H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 453, 788–792. [DOI] [PubMed] [Google Scholar]
- 73.Xu H, and Ren D (2015). Lysosomal physiology. Annu. Rev. Physiol 77, 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palmgren MG (1991). Acridine orange as a probe for measuring pH gradients across membranes: mechanism and limitations. Anal. Biochem 192, 316–321. [DOI] [PubMed] [Google Scholar]
- 75.Hosogi S, Kusuzaki K, Inui T, Wang X, and Marunaka Y (2014). Cytosolic chloride ion is a key factor in lysosomal acidification and function of autophagy in human gastric cancer cell. J. Cell. Mol. Med 18, 1124–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stauber T, and Jentsch TJ (2013). Chloride in vesicular trafficking and function. Annu. Rev. Physiol 75, 453–477. [DOI] [PubMed] [Google Scholar]
- 77.Park SH, Hyun JY, and Shin I (2019). A lysosomal chloride ion-sensitive fluorescent probe for biological applications. Chem. Sci 10, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kimura S, Noda T, and Yoshimori T (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460. [DOI] [PubMed] [Google Scholar]
- 79.Yuan H, Tan B, and Gao SJ (2017). Tenovin-6 impairs autophagy by inhibiting autophagic flux. Cell Death Dis 8, e2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rodilla AM, Korrodi-Gregorio L, Hernando E, Manuel-Manresa P, Quesada R, Perez-Tomas R, and Soto-Cerrato V (2017). Synthetic tambjamine analogues induce mitochondrial swelling and lysosomal dysfunction leading to autophagy blockade and necrotic cell death in lung cancer. Biochem. Pharmacol 126, 23–33. [DOI] [PubMed] [Google Scholar]
- 81.Jowett LA, Howe ENW, Soto-Cerrato V, Van Rossom W, Pérez-Tomás R, and Gale PA (2017). Indole-based perenosins as highly potent HCl transporters and potential anticancer agents. Sci. Rep 7, 9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soto-Cerrato V, Manuel-Manresa P, Hernando E, Calabuig-Fariñas S, Martínez-Romero A, Fernández-Dueñas V, Sahlholm K, Knöpfel T, García-Valverde M, Rodilla AM, et al. (2015). Facilitated anion transport induces hyperpolarization of the cell membrane that triggers differentiation and cell death in cancer stem cells. J. Am. Chem. Soc 137, 15892–15898. [DOI] [PubMed] [Google Scholar]
- 83.Sessler JL, Eller LR, Cho WS, Nicolaou S, Aguilar A, Lee JT, Lynch VM, and Magda DJ (2005). Synthesis, anion-binding properties, and in vitro anticancer activity of prodigiosin analogues. Angew. Chem. Int. Ed 44, 5989–5992. [DOI] [PubMed] [Google Scholar]
- 84.Zhang S, Wang Y, Xie W, Howe ENW, Busschaert N, Sauvat A, Leduc M, Gomes-da-Silva LC, Chen G, Martins I, et al. (2019). Squaramide-based synthetic chloride transporters activate TFEB but block autophagic flux. Cell Death Dis 10, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.