

Abstract
Hydrogen sulfide (H2S), an important gaseous signalling molecule in the human body, has been shown to be involved in many physiological processes such as angiogenesis. Since the biological activities of H2S are known to be significantly affected by the dose and exposure duration, the development of H2S delivery systems that enable control of H2S release is critical for exploring its therapeutic potential. Here, we prepared polymeric micelles with different H2S release profiles, which were prepared from amphiphilic block copolymers consisting of a hydrophilic poly(N-acryloyl morpholine) segment and a hydrophobic segment containing H2S-releasing anethole dithiolethione (ADT) groups. The thermodynamic stability of the micelles was modulated by altering the ADT content of the polymers. The micelles with higher thermodynamic stability showed significantly slower H2S release. Furthermore, the sustained H2S release from the micelles enhanced migration and tube formation in human umbilical vein cells (HUVECs) and induced vascularlization in the in ovo chick chorioallantoic membrane (CAM) assay.
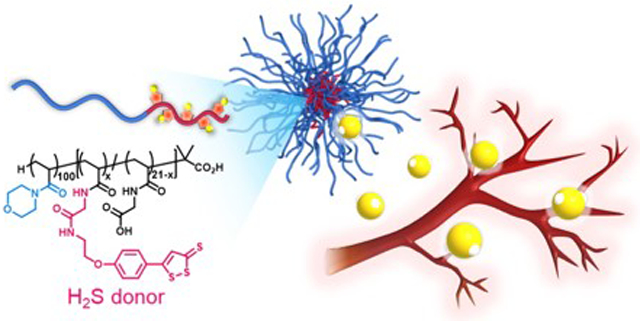
Graphical Abstract
H2S-releasing polymeric micelles promote angiogenesis
Introduction
In the human body, hydrogen sulfide (H2S) plays important roles as a endogenous signalling molecule in the cardiovascular, immune and nervous systems.1 Of particular interest are its roles in angiogenesis,2 the process of new blood vessel formation from pre-existing ones, which is a vital process during growth and development.3 The significance of endogenous H2S in angiogenesis was shown by impaired microvessel formation in aortic rings and wound healing in cystathionine-γ-lyase (CSE)-deficient mice.4 Therefore, H2S has been linked to pathophysiological conditions such as ischemia injury and diabetes underscoring its therapeutic potential. Indeed, exogenously delivered H2S, in the form of the CSE substrate propargylcysteine, has been shown to induce angiogenesis in a mouse model of hindlimb ischemia and a rat model of myocardial ischemia.5 Furthermore, H2S was also shown to promote angiogenesis after hindlimb ischemia in rats6 and improve wound-healing in type 2 diabetic mice.7 Further studies have shown that H2S stimulates angiogenesis by activating the VEGF receptor and ATP-sensitive potassium (KATP) channels in endothelial cells.5,8,9
Due to difficulties in handling and dosing of H2S gas, most studies employ sodium salts of H2S, i.e., Na2S or NaHS, that generate H2S upon dissolution in water.4,10 However, because of their instantaneous H2S release, which is unlikely to be relevant to the physiological H2S generation, these compounds are not suited for studying the biological functions of H2S as well as exploring its pharmaceutical applications.6 Instead, approaches using substrates for H2S-producing enzymes5 or small organic molecules that serve as prodrugs of H2S (H2S donors) are more viable.11-13 Especially the latter allows for design of diverse classes of organosulfur compounds with tailored H2S release kinetics in response to endogenous factors such as hydrolysis, reaction with reducing agents like cysteine or enzymatic digestion. However, their biological effects must be evaluated carefully as these low molecular weight organic compounds and their decomposition products can cause side effects that are not due to H2S released from these molecules.14, 15
The use of polymeric materials has emerged as an alternative approach that enables exogenous administration of H2S in a controlled manner with minimal side effects. In our previously studies, we showed that chemically incorporating the H2S donor anethole dithiolethione (ADT, Scheme 1A) into polymers, a poly(ethylene glycol) (PEG-ADT, Scheme 1B) conjugate14 and polymeric micelles of an amphiphilic block copolymer consisting of a hydrophilic poly(ethylene glycol) (PEG) segment and a hydrophobic ADT-containing segment (PEG-PADT, Scheme 1C), led to altered intracellular trafficking thereby reducing toxic side effects of the small ADT donor.16 These polymeric H2S donors showed a slower and continuous H2S release compared to ADT. Furthermore, the advantage of the polymeric micelle approach has been confirmed in an in vitro ischemia model where micelles were better in protecting cardiomyocytes from apoptosis.17 The benefits of polymeric H2S donors have also been recognized by others.18,19 Recently, reports have appeared tuning H2S release by controlling mobility in the micelle core20 and by shape-controlled micellar structures.21
Scheme 1.
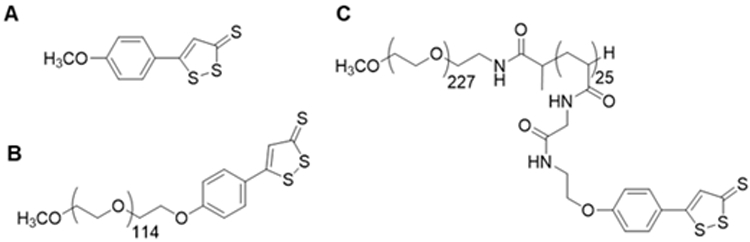
Structures of (A) ADT, (B) PEG-ADT conjugate and (C) micelle-forming PEG-PADT block copolymer.
Here, we present polymeric micelles with different H2S release profiles, which were prepared from amphiphilic block copolymers consisting of a hydrophilic poly (N-acryloyl morpholine) segment and a hydrophobic segment containing H2S-releasing anethole dithiolethione (ADT) groups. By changing the ADT content in the polymers, the thermodynamic stability and H2S release profiles from the micelles were modulated. In addition, the proangiogenic effects of the polymeric micelles with different H2S release profiles was evaluated in the in vitro human umbilical vein endothelial cells (HUVECs) migration and tube formation assays as well as the in ovo chick chorioallantoic membrane (CAM) assay.
Experimental
Instrumentation
1H NMR spectra were measured on a Bruker DPX400 NMR instrument at room temperature. For each sample a total of 32 scans was collected, and the D1 was set to 10 s. Chemical shifts are referenced to the residual undeuterated solvent signal at 2.50 ppm (DMSO-d6) or 7.26 ppm (CDCl3).
Size Exclusion Chromatography (SEC) was done on a Tosoh 8020 instrument equipped with a photo diode array (PDA) detector operating at a wavelength of 310 nm and a differential refractometer operating at 50°C. Polymer solutions (15 μL) were injected onto a KD-803 (Shodex) column kept at 50°C and eluted with 0.1 M LiCl solution in DMF. The number average molecular weight (Mn), weight average molecular weight (Mw), and dispersity (Mw/Mn) were calculated from the elution profiles of a PEG standard.
UV/Vis spectra were measured either with a Nanodrop 2000 or with a Tecan infinite M200 instrument in transparent polystyrene 96 well plates.
Fluorescence spectroscopy.
Fluorescence intensities were measured on a Tecan infinite M200 instrument in black polystyrene 96 well plate.
Dynamic light scattering (DLS) data were acquired on an Otsuka ELSZ-2 instrument in 4.5 mL disposable poly(methyl methacrylate) cuvettes. The diameter of the micelles in nm and the polydispersity index were calculated by the cumulant method.
Atomic force microscopy (AFM) images were acquired on a Seiko SPI300 instrument in the dynamic mode using a Seiko SI-DF20 Si probe. The micelle solutions were placed onto a fresh mica surface and water was removed by air blower before measurements.
Cell concentration was measured with the countess automated cell counter (Invitrogen) with disposable cell counting chamber slides using the trypan blue method.
An Olympus MVX10 macro zoom microscope was used to obtain bright field images of the Matrigels on the CAMs and the fluorescent images of HUVECs in the in vitro migration and tube formation assays.
An Olympus FV1000-D confocal laser scanning fluorescent microscope (CLSFM) was used to obtain fluorescent images of intracellular H2S release from the micelles in HUVECs.
Materials
Chemistry.
N,N'-dicyclohexylcarbodiimide (DCC), diethyl ether (Et2O), N,N-dimethylformamide (DMF) (super dehydrated and peptide grade), sodium hydrogen sulfate (NaHSO4), lithium chloride (LiCl), phosphorus pentoxide (P4O10), acetic acid (CH3COOH), n-hexane, ninhydrin, triphenylphosphine (PPh3), trifluoroacetic acid (TFA), azobisisobutyronitrile (AIBN), diisopropyl azodicarboxylate (DIAD), dry tetrahydrofran (THF) and anetholtrithione (ADT) were purchased from Wako Pure Chemical Industries (Osaka, Japan). 2-(tert-butoxycarbonylamino)-1-ethanol and benzyldimethyltetradecylammonium chloride (BTDACl) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Triethylamine (Et3N), ethyl acetate (EtOAc), N-methylpyrrolidone (NMP), potassium hydroxide (KOH), 3A molecular sieves and calcium hydride (CaH2) were purchased from Nacalai Tesque (Kyoto, Japan), 1-ethylpiperidine hypophosphite (EPHP), N-hydroxysuccinimide (NHS), alumina (Al2O3), polyethylene oxide (PEG) GPC standard, triisopropyl silane (TIS) and phenylmethanesulfonyl fluoride (PMSF) were purchased from Sigma-Aldrich (Missouri, USA). Deuterated chloroform (CDCl3) and dimethylsulfoxide (DMSO-d6) were purchased from Cambridge Isotope Laboratories (Massachusetts, USA). Dialysis tubing with a molecular weight cut off (MWCO) 2 kDa was purchased from Spectrum Labs. Sephadex LH-20 was purchased from GE Healthcare (Little Chalfont, UK). WSP-1 was purchased from Cayman Chemical (Michigan, USA). All chemicals were used as received unless indicated otherwise. Et3N was refluxed for 2 h over ninhydrin, filtered and distilled from KOH pellets under argon. 1,4-Dioxane was distilled from CaH2 and stored over molecular sieves in the presence of Xpell pellets to prevent peroxide formation. 3A molecular sieves were heated in a vacuum oven at 200°C for 6 h to remove water. AIBN was recrystallized from MeOH and kept at −20°C until use. CH2Cl2 was dried over molecular sieves overnight before use. Acryloyl morpholine was passed through a plug of Al2O3 to remove inhibitor. ADT-OH22 and PEG-Gly-ester-ADT14 were prepared following literature procedures.
Biology.
1-(4,5-Dimethyl-2-thiazolyl)-3,5-diphenylformazan (MTT) was purchased from Tokyo Chemical Industry (Tokyo, Japan). Slide-A-Lyzer G2 dialysis cassettes (MWCO 2 kD), acetoxymethyl calcein (calcein AM), Dulbecco's phosphate-buffered saline (D-PBS), Hank's balanced salt solution (HBSS) were purchased from Life Technologies Corporation (Tokyo, Japan). Normal human umbilical vein endothelial cells (HUVECs), endothelial cell growth medium EGM-2 MV SingleQuot kit supplements and growth factors and reagent pack subculture reagents containing trypsin/EDTA, trypsin neutralizing solution (TNS), and HEPES buffered saline (HEPES-BSS) were purchased from Lonza (New Jersey, USA). Water used for preparation of sterile micelle solutions and cell experiments was purchased from Otsuka Pharmaceutical Factory. μ-Slide angiogenesis for the tube formation assay and the culture-inserts 2 well for self-insertion were purchased from Ibidi (Martinsried, Germany). The μ-Angiogenesis activation assay kit was purchased from Millipore (Massachusetts, USA). Fibrinogen and thrombin from human plasma were purchased from Sigma-Aldrich (Missouri, USA). Recombinant human vascular endothelial growth factor-A121 (VEGF121), 16% paraformaldehyde solution (methanol free), and 25% glutaraldehyde solution were purchased from Wako (Osaka, Japan). Triple-well glass base dish, 24 and 96 well plates, culture flasks, Falcon tubes, dishes were purchased from Iwaki (Tokyo, Japan). CellTracker Green (5-chloromethylfluorescein diacetate, CMFDA) was purchased from ThermoFisher Scientific (Massachusetts, USA).
Synthesis of the PAM-PADT block copolymers
The PAM-PADT block copolymers were prepared as outlined in Scheme 2. Block copolymers 6a and 6b were prepared by RAFT polymerization following our previous report with modifications.23
Scheme 2.
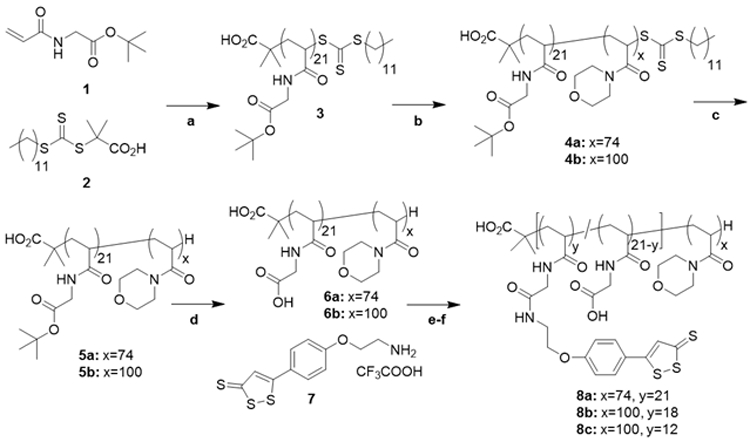
Synthesis of the PAM-PADT diblock copolymers.
(a) AIBN, 1,4-dioxane, 70°C, (b) acryloyl morpholine, AIBN, 1,4-dioxane, 70°C, (c) EPHP, AIBN, 1,4-dioxane, 95°C, (d) TFA/H2O, (e) NHS, DMAP, DMF, 25°C, (f) Et3N, DMF, 25°C. For explanation of the used abbreviations see the materials section.
Synthesis of polymer 3.
Monomer 1 (185.3 mg, 1.0 mmol), 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid 2 (14.7 mg, 0.04 mmol) and AIBN (1.32 mg, 0.008 mmol) were dissolved in 1,4-dioxane resulting in a final monomer concentration of 2 M. The clear solution was degassed by five argon freeze-pump-thaw cycles and placed in an oil bath at 70°C. After 24 h the reaction was stopped by cooling in liquid nitrogen and opening to air. The clear yellow solution was added to 200 mL hexane to precipitate the polymer. The polymer was recovered by filtration, washed with hexane and dried under vacuum to yield 169.1 mg (84%) of the homopolymer as a yellow solid. The 1H NMR spectrum in CDCl3 is shown in Figure S1 supporting information. The degree of polymerization was calculated to be 21.
Synthesis of polymers 4a and 4b.
As an example, the synthesis of 4b is described. Acryloyl morpholine (568 mg, 4 mmol), polymer 3 (171.2 mg, 0.04 mmol) and AIBN (0.66 mg, 0.004 mmol) were dissolved in 1,4-dioxane resulting in a final monomer concentration of 2 M. The clear solution was degassed by five argon freeze-pump-thaw cycles and placed in an oil bath at 70°C. After 24 h the reaction was stopped by cooling in liquid nitrogen and opening to air. The clear yellow solution was added to 200 mL cold Et2O to precipitate the polymer. The polymer was recovered by filtration, washed with Et2O and dried under vacuum to yield 614 mg (82%) of the block copolymer as a yellow solid. The 1H NMR spectrum in CDCl3 is shown in Figure S2 supporting information. The degree of polymerization was calculated to be 100.
Synthesis of polymers 5a and 5b.
As an example, the synthesis of polymer 5b is described. Polymer 4b (384.2 mg, 0.02 mmol), 1-ethylpiperidine hypophosphite (234.1 mg, 1.3 mmol) and AIBN (2.2 mg, 0.013 mmol) were dissolved in 1.4 mL 1,4-dioxane. The clear colourless solution was degassed by five argon freeze-pump-thaw cycles and placed in an oil bath at 95°C. After 24 h the reaction was stopped by cooling in liquid nitrogen and opening to air. The clear solution was diluted with water causing the precipitation of a small amount of solid that was removed by filtering through a 0.8 μm syringe filter. The clear solution was dialyzed (2 kDa dialysis tubing) against 4 L milliQ water for 3 d with regularly replacing the water to remove 1-ethylpiperidine hypophosphite-related impurities. The polymer was recovered by lyophilization to yield end group-removed polymer 5b (334.0 mg, 88%) as a white solid.
Synthesis of polymers 6a and 6b.
As an example, the synthesis of 6b is described. Polymer 5b (276 mg) was dissolved in 3.0 mL of TFA/H2O (9:1) and stirred at room temperature. After 24 h the clear solution was evaporated by passing a stream of nitrogen and the residue dried under reduced pressure. The residue was dissolved in water and lyophilized to yield polymer 6b (219 mg, 84%) as a white solid.
Synthesis of 7.
This compound was prepared in two steps with improved overall yield compared to that reported14 employing a Mitsunobu reaction (Scheme S1 supporting information). Under argon flow 560 mg (2.1 mmol) of PPh3 in a dried Schlenk tube was dissolved in 20 mL THF. While maintaining an argon flow the solution was put in a NaCl/ice bath and cooled to −20°C. After 5 min at −20 °C 440 μL (2.2 mmol) of DIAD was added under argon. After 15 min 400 mg (1.8 mmol) of ADT-OH was added to the formed yellow suspension followed by 290 mg (1.8 mmol) of 2-(tert-butoxycarbonylamino)-1-ethanol dissolved in 0.5 mL dry THF. The mixture was kept for another hour at −20°C and stirred overnight at room temperature. The mixture was concentrated under reduced pressure and purified by Al2O3 column chromatography eluting with EtOAc/hexane 1:1. Fractions containing product were concentrated and the solid triturated with 50 mL of hexane. After decanting the hexane and drying this yielded 489 mg (1.3 mmol, 74%) of an orange solid. The 1H NMR spectrum in CDCl3 is shown in Figure S3 supporting information.
Deprotection was carried out following our previous report14 with the difference that a solution of Boc-protected amine was added to a solution of TIS in TFA: 118 mg (0.3 mmol) was dissolved in 6 mL CH2Cl2 and added dropwise to a mixture of 1 mL TIS in 3 mL CH2Cl2 that had been cooled at 0°C for 10 min. After everything was added the resulting mixture was stirred for 1 h at 0°C before warming to room temperature. After 1 h TLC (SiO2, EtOAc/hexane 1:1) showed the absence of the starting material. The reaction mixture was then placed in a water bath at 30°C and the TFA, TIS and CH2Cl2 were evaporated by gently passing a flow of air. To the oily residue was added 2-3 mL CH2Cl2 and again evaporated three time total. To the orange solid was added 25 mL Et2O and the suspension was stirred for 10 min and the fine solid isolated by filtration on a glass filter. The solid was washed on the filter with Et2O (2 x 25 mL) and CH2Cl2 (8 x 2 mL) and finally dissolved in 10 mL MeOH. After removing MeOH on the rotavapor and drying under vacuum this yielded 90.1 mg, (0.23 mmol, 75%) of the amine as an orange solid. The 1H NMR is shown in Figure S4 supporting information.
Synthesis of the PAM-PADT polymers 8a-c.
As an example, the synthesis of 8c is described: 70.8 mg (4.2 μmol, 0.088 mmol COOH groups), 15.0 mg (0.13 mmol) of NHS were dried overnight under vacuum at 60°C before dissolving in 2.1 mL of DMF. To the solution was added 21.5 mg (0.10 mmol) of DCC and 1.0 mg (0.008 mmol) DMAP in 400 μL of DMF. The reaction mixture was stirred for 24 h at room temperature during which time the solution became turbid due to the formation of dicyclohexl urea. To the mixture was then added 17.9 mg (0.047 mmol, 0.5 eq) of 7 followed by 13 μL (0.093 mmol) of Et3N. After 24 h acetic acid was added (1 mL) and the mixture filtered over a plug of glass wool. The residue was washed with DMF (4 x 0.5 mL) and the orange filtrate diluted with water and dialyzed against 500 mL of water (MWCO 2 kDa) for 3 days with regularly replacing the water. After lyophilizing this yielded an orange solid. The crude polymer was dissolved in DMF and purified by size exclusion chromatograph on Sephadex LH20 eluting with DMF. Fractions containing polymer were combined and diluted with 50 mL Et2O to precipitate the polymer. The 1H NMR is shown in Figure S5 supporting information.
Micelle Formation
The micelle solutions were prepared in a laminar cell culture hood by adding solutions of the polymers in 200 μL NMP (25 mg/mL) drop wise to 1800 μL milliQ water under vigorous stirring at RT. After stirring for 30 min, the solution was transferred to a slide-a-lyzer G2 (MWCO 2 kDa) and dialyzed against 5 L of milliQ water for 1 day with replacing the water two times. Micelle solutions were then sterile filtered under laminar flow. The micelle concentration, after diluting 1:1 with DMF, was calculated from the absorbance at 436 nm and a standard curve of ADT in the same solvent mixture.
Critical micelle concentration (CMC) measurement
Micelle solutions at different concentrations (2 μL) were placed onto a parafilm-coated surface and the contact angle was measured using a Kyowa Interface Science Drop Master DM 300 instrument. From a plot of the contact angle versus polymer concentration the critical micelle concentration was calculated. Measurements were done in triplicate.
Cell culture
HUVECs were cultured in endothelial cell growth medium EGM-2 supplemented with 2% FBS, 0.04% hydrocortisone, 0.4% hFGF, 0.1% VEGF, 0.1% R3-IGF-1, 0.1% ascorbic acid, 0.1% hEGF, 0.1% GA-1000, and 0.1% heparin in 5% CO2 inside an incubator at 37°C. Cells were subcultured when reaching 70-80% confluency. Cells were washed with HEPES-BBS and 2 mL of trypsin/EDTA was added. Periodically, the trypsinization process was checked under the microscope and after 2-3 min the trypsin was neutralized by adding 4 mL of TNS. The cell suspension was centrifuged at 1200 rpm for 5 min and after removing the supernatant resuspended in 3 mL of medium. This cell suspension was then diluted to the concentration needed for the experiment.
Preparation of HUVEC cell lysate for the H2S release experiments
HUVECs were detached and the cell suspension was centrifuged at 500 rpm for 5 min. After washing the cell pellet with cold PBS, the cells were resuspended in PBS supplemented with 1 mM PMSF (Final concentration: 1.4 × 106 cells/mL). The cell suspension was sonicated for 1 min with a Taitec VP-050N ultrasonic homogenizer and centrifuged. The supernatant was collected and stored at −80°C until use.
H2S release from the micelles in HUVEC lysate with the WSP-1 H2S fluorescent detection probe
Cell lysate was diluted with PBS at a volume ratio of 1:4 and mixed with WSP-1 and BTDACl (Final concentrations: 10 μM for WSP-1 and 2.5 μM for BTDACl). This solution was mixed with the micelle solutions in a 96 well plate (Final concentration of ADT groups: 100 μM). The fluorescence intensity was measured as function of time on the plate reader (λex = 465 nm, λem = 515 nm).
H2S release of the micelles in HUVECs
HUVECs were seeded in a 96 well plate at 7.5 x 103 cells/well and cultured for 2 d. After removing the medium, cells were incubated with 25 μM WSP-1/PBS at room temperature for 30 min. After the WSP-1 solution was removed, cells were cultured in medium containing the micelles 8a, 8b and 8c (ADT concentration: 50 μM). At the indicated time points, the fluorescence intensity was measured (λex=465 nm, λem=515 nm) using the bottom reading mode.
Intracellular H2S detection by CLSFM
HUVECs were seeded into a triple-well glass base dish at 1 × 104 cells/well and cultured for 24 h in a CO2 incubator. The medium was replaced with 100 μL fresh medium containing ADT or the micelles (Final ADT concentration: 100 μM). After culturing for 6 h, the cells were washed with FBS-free medium and incubated in 100 μL FBS-free medium containing 10 μM WSP-1 and 2.5 μM BTDACl for 30 min at 37°C. The cells were washed with PBS and observed by CLSFM.
MTT assay
HUVECs were seeded in a 96 well plate at 1 × 104 cells/well and cultured for 24 h. After replacing the medium with 100 μL of fresh medium containing ADT or the micelles at different concentrations, cells were cultured for 24 h. The medium was replaced with 100 μL of fresh medium containing 0.5 mg/mL MTT. After culturing for 4 h, 100 μL/well of 0.1 g/mL sodium dodecyl sulfate in 0.01 M HCl (aq) was added to lyse the cells and solubilize the formazan crystals. The absorbance at 570 nm was measured and the cell viability is expressed as the percentage of the absorbance for cells that had not received any treatment.
Migration Assay
HUVECs at 70-85% confluency were stained with Cell Tracker solution (1 μg/mL) in HBSS for 15 min at 37°C. Cells were detached by trypsinization, resuspended in EBM-2 medium containing 2% FBS and seeded at 3.5 × 104 cells (70 μL medium) in an ibidi Culture-Insert 2 well, which is placed in a 24-well plate. After culturing for 24 h, the culture-inserts were removed to create a confluent cell monolayer with a cell-free gap of 500 μm. To each well was added 300 μL medium containing VEGF121, ADT or the micelles (Final concentrations: 100 ng/mL for VEGF121, 100 μM for ADT or ADT groups of the micelles). Cells were cultured in a CO2 incubator and observed under a fluorescence microscope at the indicated time points. The MetaMorph software was used to analyze the images and to calculate the rate of cell migration.24
Tube formation assay
Frozen solutions of fibrinogen and thrombin were thawed inside an icebox while cooling the μ-Slide Angiogenesis plates inside the fridge. To the wells on the cold μ-Slide Angiogenesis plate was then added 5.5 μL of fibrinogen solution followed by 5.5 μL thrombin solution. The resulting fibrinogen-thrombin solution mixture was gently mixed by pipetting. The μ-Slide Angiogenesis plate was then placed inside the cell incubator at 37˚C for 1 h to form the fibrin gel. To each well was added 45 μL of a HUVEC cell suspension (2.3 × 106 cells/mL) and 5 μL of either water (NT), VEGF121, ADT or the micelle solutions (Final concentrations: 100 ng/ mL for VEGF121, 330 μM for ADT or ADT groups of the micelles). After culturing for 24 h, the cells were stained with calcein AM and observed under a macro zoom fluorescence microscope.
Preparation of the Matrigels with VEGF and the H2S donors for the CAM assay
PBS solutions of the micelles 8a-c, PEG-Gly-ester-ADT and VEGF121 (5 μL) were mixed with cold growth factor reduced Matrigel (30 μL) and kept at 37°C for 30 min to form the Matrigels (Final concentrations: 11 μg/mL for VEGF121, 400 μM for ADT or ADT groups of the micelles).
Chick Chorioallantoic Membrane (CAM) Assay.
On embryonic day 1, the eggs were cleaned with tissue paper wetted with 70% ethanol (aq) and put inside the incubator at 37.6°C. On day 3 the eggs were candled with a LED light source to locate the air sac. The egg was then punctured at this side with an egg hole puncher and through the hole about 2-3 mL albumen was removed using a 21 G x 11/2 plastic syringe.25 The hole was closed with Opsite tape (Smith and Nephew). Then a square window of about 2 cm x 2 cm was cut in the centre of the egg and the shell and inner shell membrane carefully removed with a pincer. The cut window was covered with Opsite and the area above the window cut away with scissors and covered with Tegaderm tape (3M). The eggs were then placed back inside the incubator. At day 9, eggs were candled and those containing a live embryo (60-77%) were used for the experiment. The Tegaderm covering the window on the egg was cut away and the Matrigels placed onto the CAM using sterilized tweezers. After resealing the window with Tegaderm tape the egg was put back in the incubator. On day 11, the CAM was fixed by adding 2 mL of fixation solution containing methanol-free paraformaldehyde (16 w/v%) solution and glutaraldehyde (25 w/v%) solution, in 20 mM PBS pH7.4 at room temperature for 1 d. The CAM was taken out using a pincer and scalpel, washed with PBS and placed inside a 6 well plate. The images were collected using a macro zoom microscope and analysed by blind test using the reported method.26
Results and discussion
Synthesis of ADT-containing diblock copolymers (PAM-PADT)
The amphiphilic PAM-PADT block copolymers consisting of a hydrophilic poly (N-acryloyl morpholine) (PAM) segment and a hydrophobic ADT-containing (PADT) segment were synthesized as outlined in Scheme 2. We first prepared block copolymers consisting of a carboxylic acid-containing block and PAM block using the reversible addition fragmentation transfer (RAFT) polymerization technique. RAFT polymerization of tert-butyl glycine acrylamide 1 using thiocarbonate 2 as the chain transfer agent (CTA) gave homopolymer 3. This polymer had a dispersity of 1.06 as determined by gel permeation chromatography (GPC). The homopolymer 3 was then used as a macro CTA to polymerize N-acryloyl morpholine to yield diblock copolymers 4a and 4b. The dispersity of both polymers was 1.07 and 1.12 respectively, showing the successful synthesis of the diblock copolymers with narrow size distributions.
Then, the CTA end-group of polymers 4a and 4b was removed by radical-induced reduction using 1-ethylpiperidine hypophosphite as the hydrogen source.27 Successful thiocarbonate group removal was confirmed by the absence of the absorbance at 310 nm as measured by the PDA detector connected to the GPC instrument. After removing the tert-butyl esters groups with trifluoroacetic acid, the carboxylic acid-containing polymers 6a and 6b were first activated by preparing the corresponding N-hydroxysuccinimide esters and used to conjugate with the amine linker-modified ADT 7 to yield the PAM-PADT polymers (8a-c).
We obtained the polymers with different ADT contents by changing the molar feed ratio of ADT (7) to -COOH groups of polymers 6a and 6b (21 per polymer). The number of ADT groups per polymer (x) of 8a, 8b and 8c were 21, 18 and 12, respectively, as determined from the 1H NMR spectra (Table 1).
Table 1.
PAM-PADT polymers and their micelles.
| Polymer | Feed ratio | DLSb | CMCc | ||
|---|---|---|---|---|---|
| 7/COOH | xa | Dh [nm] | PDI | [pM] | |
| 8a | 23/21 | 21 | 34 | 0.24 | 245 ± 8 |
| 8b | 17/21 | 18 | 35 | 0.27 | 328±13d |
| 8c | 12/21 | 12 | 37 | 0.25 | 390±21e |
Number of ADT groups per polymer as determined by 1H NMR
Z-average hydrodynamic diameter (Dh) and polydispersity index (PDI=μ2Γ−2) of the micelles as determined by dynamic light scattering (DLS) after fitting the correlation function using the cumulant method.
Critical micelle concentration (CMC) as determined by the surface tension method.
p = 0.0031 vs. 8a.
p = 0.035 vs. 8b.
Micelle preparation and characterization
The micelles were prepared by dispersing the PAM-PADT diblock copolymers (8a-c) in water. Due to the hydrophobic nature of the ADT groups, the block copolymers self-assembled in water to form micelles with an average hydrodynamic diameter (Dh) of 34-37 nm as measured by dynamic light scattering (DLS) as shown in Table 1. Furthermore, the spherical structures of the micelles were confirmed by atomic force microscopy (AFM) as shown in Figure 1. Average size distribution of the micelles as well as cross-section curves can be found in Table S1 and Figure S6 (supporting information).
Figure 1.

Morphology of the micelles (A) 8a, (B) 8b and (B) 8c by atomic force microscopy (AFM). The micelles were adsorbed onto a fresh mica surface and observed by AFM after removing water by air blower. The images show the height image with the height profile from 0 to 4.87 nm indicated below each image.
To see how the ADT content of the polymers affects the thermodynamic stability of the micelles, the critical micelle concentration (CMC), the polymer concentration above which micelles form, were determined by the surface tension method.16 As shown in Table 1, the CMC values follow the order: 8a < 8b < 8c. This result shows that the micelles prepared from the polymers with a higher ADT content (i.e., 8a) has a higher thermodynamic stability, which is likely to be due to the increased hydrophobicity of the PADT segment.
H2S release from the micelles in presence of HUVEC’s cell lysate
The effect of the thermodynamic stability of the PAM-PADT micelles on their H2S release profiles was evaluated. In our previous report, we showed that H2S release from ADT was induced by cell lysate from mouse macrophages16 and rat cardiomoycytes.17 In this study, we measured H2S release from the micelles in cell lysate from HUVECs using the WSP-1 H2S detection probe.28 Interestingly, the H2S release profiles differed significantly between the micelles. As shown in Figure 2, the order is 8a < 8b < 8c, suggesting that the micelles with a higher stability (i.e., lower CMC) show the slowest H2S release.
Figure 2.
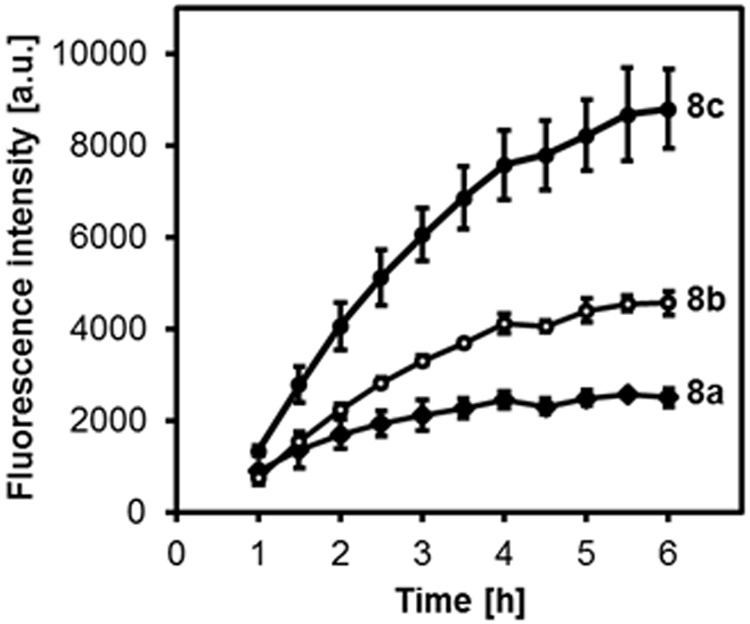
H2S release profiles of the micelles 8a, 8b and 8c. The micelles (ADT concentration: 100 μM) were added to HUVEC cell lysate containing the WSP-1 H2S detection fluorescent probe (10 μM). λex= 465 nm, λem= 515 nm. n=3.
We previously showed that ADT-based H2S donors release H2S in cell lysate but not fetal bovine serum or glutathione-containing PBS.14 This implies that H2S release is induced by intracellular components, presumably enzymes as was recently reported for rat liver microsomes.29 Since the ADT groups, which are located within the micellar core, are unlikely to be accessible for enzymes, it is reasonable to assume that micelles partially dissociate in the presence of cell lysate, resulting in H2S release. This hypothesis is not contradicted by the result in Figure 2 showing that the micelles with a higher CMC (8c), i.e. low thermodynamic stability, exhibits faster H2S release.
H2S release from the micelles in HUVECs
We compared the H2S release profiles of the micelles 8a, 8b and 8c in live cells. HUVECs were pretreated with the WSP-1 H2S detection probe and cultured in the presence of micelles. The fluorescence intensity within the cells was measured by a plate reader using the bottom reading mode for real time monitoring of H2S release in HUVECs. As shown in Figure 3, the H2S release follows the same trend as that observed in cell lysate.
Figure 3.
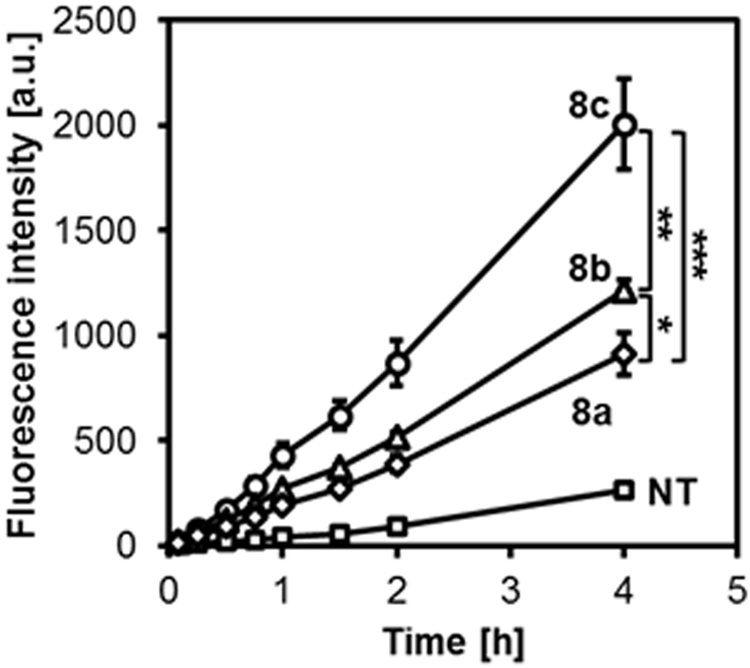
H2S release in HUVECs. Cells in a 96 well plate were pretreated with the WSP-1 H2S detection probe in PBS for 30 min and cultured in medium without (NT, squares) or with micelles 8a (rhombus), 8b (triangles) and 8c (circles). ADT concentration: 50 μM. The fluorescence intensity was monitored using the bottom reading mode (λex=465 nm, λem=515 nm). *p < 0.05, **p < 0.01,***p < 0.001 (n=3).
To show that H2S release occurred intracellularly we used the WSP-1 H2S detection probe to visualize intracellular H2S by confocal laser scanning fluorescence microscopy (CLSFM). HUVECs were treated with the different H2S donors (100 μM) for 6 h at 37°C and observed by CLSFM. We observed increased fluorescence within cells treated with the small H2S donor ADT (Figure 4B) as well as the micelles (Figure 4C-E) compared to the non-treated cells (Figure 4A). These CLSFM images clearly show that ADT as well as the micelles can release H2S inside HUVECs. Furthermore, in the case of ADT, due to its low water solubility, small crystallites of ADT appeared, as indicated by the black spots in the DIC image in Figure 4B.
Figure 4.

H2S release from the micelles in HUVECs. The cells were treated with (A) NT (PBS), (B) ADT, (C) micelles 8a, (D) 8b and (E) 8c for 6 h. Cells were washed with PBS and treated with the WSP-1 probe to detect H2S within cells. ADT concentration: 100 μM. Scale bars: 50 μm.
Furthermore, we investigated whether the micelles can be internalized by HUVECs. The micelles were prepared from cyanine3 (Cy3)-labeled polymers 8b and 8c (Figure S7, supporting information). As shown in Figure S8 (supporting information), HUVECs treated with the micelles showed bright fluorescence, supporting that the micelles were taken up by cells and thereafter released H2S intracellularly.
Pro-angiogenic effects of the micelles in the HUVEC migration and tube formation assays
H2S is known to induce angiogenesis by stimulating endothelial cell migration and tubulogenesis.5,8 We therefore tested the proangiogenic activity of the micelles by the gap closure migration and tube formation assays using HUVECs.
The effect of ADT and the micelles on migration of HUVECs was evaluated in the gap closure migration assay where a confluent cell monolayer with an artificial cell free gap (width: 500 μm) was first prepared and cell migration into the gap was monitored. As shown in Figure 5 and S9A-E in the supporting information, the micelles enhanced migration into the cell free gap and about 75% of the gap surface was covered over the course of 9 h. Note that the effect on cell migration is very similar with that of the growth factor VEGF121.
Figure 5.

The gap closure migration assay. HUVECs were seeded in the ibidi Culture-Insert 2 Well to create a confluent cell monolayer with an artificial gap. After removing the insert, cells were cultured in culture medium containing (A) NT (water), (B) VEGF121 (100 ng/mL), (C) ADT, (D) micelles 8a, (E) 8b and (F) 8c. ADT concentration 100 μM. Top panels: the cell free gap at 0 h, Bottom panels: the same area after 9 h. Scale bar: 500 μm.
Compared to the micelles, ADT showed less migratory activity. This may be related to its higher toxicity compared to micelles in HUVECs as shown in Figure S10 in the supporting information. Furthermore, the images obtained at different time points were analysed to determine the rate of cell migration (Figure S9F in the supporting information). Compared to non-treatment (NT) and ADT, the micelles showed a higher cell migration rate. These results show that micelles efficiently stimulate migration of HUVECs compared to ADT. There was no obvious effect of the difference in H2S release profiles of the micelles on cell migration.
We then tested tubulogenic activity of the micelles in the in vitro tube formation assay. Cells were seeded on fibrin gels and cultured for 24 h in medium containing VEGF121, ADT and micelles. As shown in Figure 6B, in the presence of VEGF121, cells have migrated and aligned themselves to form tubular structures, as indicated by the white arrows, which were not observed for the non-treated cells (Figure 6A). The micelles showed formation of capillary tubes (Figures 6D-F) indicating the tubulogenic effect of H2S released from the micelles. Interestingly, the number of capillary-like tubes formed by the micelles was in the order of 8a < 8b < 8c as shown by a quantitative analysis (Figure S11 supporting information).This suggests that the tubulogenesis of HUVECs is affected by the H2S release rate. The micelles with a faster H2S release rate stimulate endothelial cell tube formation more efficiently.
Figure 6.

Tube formation assay. Representative images of HUVECs treated with (A) NT (water), (B) VEGF121, (C) ADT, (D) micelles 8a, (E) 8b and (F) 8c. HUVECs were seeded on fibrin gel and treated with 100 ng/mL VEGF121, ADT and micelles (ADT concentration: 330 μM) for 24 h. The cells were stained with calcein AM and observed under a macro zoom fluorescence microscope. Arrows indicate capillary tubes. Scale bar: 500 μm.
Pro-angiogenic effect of the micelles in the in ovo chick chorioallantoic membrane assay
To evaluate the pro-angiogenic effects of the micelles in a more realistic model, we used the in ovo chick chorioallantoic membrane (CAM) assay.30 The CAM is a vascularized membrane that forms around day 4 to 5 of embryonic development that covers the chick embryo. During embryo development, the CAM capillary endothelium develops into a dense network of arteries and veins in the mesodermal layer. From this network, the capillary plexus is formed, i.e., branching network of blood vessels, that serves as the respiratory organ until the time of hatching.31,32 Due to its unique capillary plexus formation, CAM has been used to study the pro-angiogenic effect of drugs including Na2S.4,33,34
We placed growth factor reduced Matrigels containing VEGF121 and the micelles on the CAMs on embryonic day 9. The Matrigels containing ADT could not be prepared due to its low aqueous solubility. Instead, we used a PEG-Gly-ester-ADT conjugate having ADT linked via a hydrolysable ester bond as a prodrug of ADT-OH.14 As shown in our previous report, this conjugate is rapidly hydrolyzed in aqueous solutions to release ADT-OH. Although ADT-OH lacks the methyl group, this compound is also known to release H2S similar to ADT. On embryonic day 12, the CAM area around the Matrigels was taken out and observed under a macro zoom microscope (Figure 7A-F).
Figure 7.

Pro-angiogenic effect in the CAM assay. Photographs of CAMs treated with Matrigel containing (A) NT (PBS), (B) VEGF121 (11 μg/mL), (C) ADT-OH (PEG-Gly-ester-ADT), (D) micelles 8a, (E) 8b and (F) 8c. ADT concentration: 400 μM. The location where the gel was applied has been indicated by the circled area. Scale bar: 2 mm. (G) Semi-quantitative scoring. * p < 0.001 versus NT, # p < 0.001, ## p < 0.05 versus VEGF, • p < 0.001, •• p < 0.05 versus ADT-OH. † p < 0.05 versus 8a. n= 8-10.
Compared to the non-treated CAMs (NT), the CAM treated with all formulations increased the number of blood vessels that are sprouting out from the area where Matrigels were placed. We further used the reported semi-quantitative scoring system to compare the pro-angiogenic effect of the different formulations.26 As can be seen in Figure 7G, the micelles showed significantly stronger pro-angiogenic effects compared to NT (p < 0.001) while no statistically significant effects was observed for ADT-OH. All micelles were more effective in inducing angiogenesis compared to ADT-OH, and the micelles 8b and 8c showed a significantly stronger effect than VEGF121. These data clearly show the advantage of using the micelles for promoting vascularization on the CAMs. Furthermore, although the difference between the micelles 8b and 8c was not significant, the proangiogenic activities of both micelles 8b and 8c were significantly stronger compared to 8a (p < 0.05) showing that the H2S-release profile affects vascularization in the CAM assay (Figure 7).
Conclusions
We prepared amphiphilic block copolymers composed of a hydrophilic poly(N-acryloyl morpholine) segment and a hydrophobic segment bearing different numbers of ADT groups, using the RAFT polymerization technique. The polymers assembled in water to form spherical micelles. The thermodynamic stability of the micelles, which was determined by critical micelle concentration, was enhanced by increasing the number of ADT groups. The micelles with higher thermodynamic stability showed slower H2S release in cell lysate of HUVECs. The micelles showed stronger pro-angiogenic effects in the in vitro migration and tube formation assay compared to the small H2S donor ADT. Furthermore, the micelles efficiently induced vascularization in the in ovo CAM assay. The micelle approach would have promise in promoting angiogenesis by delivering H2S in a controlled manner.
Supplementary Material
Acknowledgements
This work was supported by Center for Molecular Analysis of Disease Pathways (CMADP), National Institute of General Medical Sciences of the National Institutes of Health, Award Number P20GM103638.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: 1H NMR spectra, MTT assay data, AFM cross section curves and size distribution, cellular uptake fluorescent micelles, rates of cell migration and quantitative analysis capillary-like tube formation in HUVECs. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Kimura H, Molecules, 2014, 19, 16146–16157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mistry RK and Brewer AC, Free Radical Biology and Medicine, 2017, 108, 500–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmeliet P, Nature Medicine, 2003, 9, 653–660. [DOI] [PubMed] [Google Scholar]
- 4.Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R and Szabó C, Proceedings of the National Academy of Sciences, 2009, 106, 21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kan J, Guo W, Huang C, Bao G, Zhu Y and Zhu YZ, Antioxidants & Redox Signaling, 2014, 20, 2303–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y and Zhu YC, Antioxidants & Redox Signaling, 2010, 12, 1065–1077. [DOI] [PubMed] [Google Scholar]
- 7.Liu F, Chen D-D, Sun X, Xie H-H, Yuan H, Jia W and Chen AF, Diabetes, 2014, 63, 1763–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai W-J, Wang M-J, Moore PK, Jin H-M, Yao T and Zhu Y-C, Cardiovascular Research, 2007, 76, 29–40. [DOI] [PubMed] [Google Scholar]
- 9.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE and Snyder SH, Circulation Research, 2011, 109, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan S, Pardue S, Shen X, Alexander JS, Orr AW and Kevil CG, Redox Biology, 2016, 9, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Ji X, Ji K and Wang B, Acta Pharmaceutica Sinica B, 2015, 5, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szabo C and Papapetropoulos A, Pharmacological Reviews, 2017, 69, 497–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Powell CR, Dillon KM and Matson JB, Biochemical Pharmacology, 2018, 149, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasegawa U and van der Vlies AJ, Bioconjugate Chemistry, 2014, 25, 1290–1300. [DOI] [PubMed] [Google Scholar]
- 15.Yuan S, Patel RP and Kevil CG, American Journal of Physiology-Lung Cellular and Molecular Physiology, 2015, 308, L403–L415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasegawa U and van der Vlies AJ, MedChemComm, 2015, 6, 273–276. [Google Scholar]
- 17.Takatani-Nakase T, Katayama M, Matsui C, Hanaoka K, van der Vlies AJ, Takahashi K, Nakase I and Hasegawa U, Molecular BioSystems, 2017, 13, 1705–1708. [DOI] [PubMed] [Google Scholar]
- 18.Connal LA, Journal of Materials Chemistry B, 2018, 6, 7122–7128. [DOI] [PubMed] [Google Scholar]
- 19.Urquhart MC, Ercole F, Whittaker MR, Boyd BJ, Davis TP and Quinn JF, Polymer Chemistry, 2018, 9, 4431–4439. [Google Scholar]
- 20.Foster JC, Carrazzone RJ, Spear NJ, Radzinski SC, Arrington KJ and Matson JB, Macromolecules, 2019, 52, 1104–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu SH, Esser L, Khor SY, Senyschyn D, Veldhuis NA, Whittaker MR, Ercole F, Davis TP and Quinn JF, Journal of Polymer Science Part A: Polymer Chemistry, 2019, 57, 1982–1993. [Google Scholar]
- 22.Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P and Moore PK, Free Radical Biology and Medicine, 2007, 42, 706–719. [DOI] [PubMed] [Google Scholar]
- 23.Hasegawa U, Tateishi N, Uyama H and van der Vlies AJ, Macromolecular Bioscience, 2015, 15, 1512–1522. [DOI] [PubMed] [Google Scholar]
- 24.van der Vlies AJ, Morisaki M, Neng HI, Hansen EM and Hasegawa U, Bioconjugate Chemistry, 2019, 30, 861–870. [DOI] [PubMed] [Google Scholar]
- 25.Samkoe KS, Clancy AA, Karotki A, Wilson BC and Cramb DT, Journal of Biomedical Optics, 2007, 12, 1–14. [DOI] [PubMed] [Google Scholar]
- 26.Ribatti D, Nico B, Vacca A and Presta M, Nature Protocols, 2006, 1, 85–91. [DOI] [PubMed] [Google Scholar]
- 27.Hasegawa U, van der Vlies AJ, Simeoni E, Wandrey C and Hubbell JA, Journal of the American Chemical Society, 2010, 132, 18273–18280. [DOI] [PubMed] [Google Scholar]
- 28.Peng B, Chen W, Liu C, Rosser EW, Pacheco A, Zhao Y, Aguilar HC and Xian M, Chemistry – A European Journal, 2014, 20, 1010–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dulac M, Nagarathinan C, Dansette PM, Mansuy D and Boucher J-L, Drug Metabolism and Disposition, 2018, 46, 1390–1395. [DOI] [PubMed] [Google Scholar]
- 30.Mary R and Gurmit S, Current Drug Targets - Cardiovascular & Hematological Disorders, 2003, 3, 155–185. [DOI] [PubMed] [Google Scholar]
- 31.Ribatti D, Nico B, Vacca A, Roncali L, Burri PH and Djonov V, The Anatomical Record, 2001, 264, 317–324. [DOI] [PubMed] [Google Scholar]
- 32.Dimitropoulou C, Malkusch W, Fait E, Maragoudakis ME and Konerding MA, Angiogenesis, 1998, 2, 255–263. [DOI] [PubMed] [Google Scholar]
- 33.Melkonian G, Munoz N, Chung J, Tong C, Marr R and Talbot P, Journal of Experimental Zoology, 2002, 292, 241–254. [DOI] [PubMed] [Google Scholar]
- 34.Vázquez F, Hastings G, Ortega M-A, Lane TF, Oikemus S, Lombardo M and Iruela-Arispe ML, Journal of Biological Chemistry, 1999, 274, 23349–23357 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.