

Abstract
Ring opening metathesis polymerization (ROMP) is widely considered an excellent living polymerization technique that proceeds rapidly under ambient conditions and is highly functional group tolerant when performed in organic solvents. However, achieving the same level of success in aqueous media has proved to be challenging, often requiring an organic co-solvent or a very low pH to obtain fast initiation and high monomer conversion. The ability to efficiently conduct ROMP under neutral pH aqueous conditions would mark an important step towards utilizing aqueous ROMP with acid-sensitive functional groups or within a biological setting. Herein we describe our efforts to optimize ROMP in an aqueous environment under neutral pH conditions. Specifically, we found that the presence of excess chloride in solution as well as relatively small changes in pH near physiological conditions have a profound effect on molecular weight control, polymerization rate and overall monomer conversion. Additionally, we have applied our optimized conditions to polymerize a broad scope of water-soluble monomers and used this methodology to produce nanostructures via ring opening metathesis polymerization induced self-assembly (ROMPISA) under neutral pH aqueous conditions.
Graphical Abstract

Introduction
The development of ring opening metathesis polymerization (ROMP) under aqueous conditions is an attractive endeavor for several diverse fields. From the perspective of green chemistry, eliminating organic solvents improves the sustainability and environmental impact of ROMP. Furthermore, metathesis is a bioorthogonal transformation that could be exploited for novel biological applications.1–4 Indeed, cross metathesis (CM) and ring closing metathesis (RCM) have been investigated extensively within this context.2,5–14 However, utilizing ROMP within a biological setting remains underdeveloped.15
ROMP in neat water presents unique challenges when compared to polymerization in organic solvents. Early work by Grubbs, studying a water-soluble variant of the Grubbs 1st generation catalyst (G1), observed rapid catalyst decomposition and deactivation which was caused by the presence of hydroxide ions in solution.16–19 They demonstrated that lowering the solution pH is a successful strategy to minimize the hydroxide concentration and maintain an active catalyst, allowing polymerization to occur rapidly and in a living manner (Scheme 1A). They surmised this was due to degradation of the ruthenium catalyst in which the chloride ligands are displaced by water or hydroxide anions in solution.16–18 Later examples using water-soluble Grubbs 2nd and 3rd generation (G2 and G3)19–22 as well as Hoveyda-Grubbs 2nd generation (HG2) type catalysts13,23–25 also demonstrated the importance of having low pH for ROMP to proceed efficiently, with pH values ranging from pH 1.5 – 4. Recent work from O’Reilly et al 26–29 has harnessed this strategy to synthesize polynorbornene based nanostructures in water via ROMP induced self-assembly (ROMPISA). While significant advances have been made to conduct ROMP under aqueous conditions, the low pH generally required for polymerizations to occur could be detrimental to acid sensitive monomers or to relevant biologics such as peptides, proteins and cells.
Scheme 1.

Previous work exploring pH and chloride concentration effects on metathesis in water (A, B) and the work described in this manuscript (C). Structure of water-soluble Hoveyda-Grubbs type catalyst used in this study (AquaMet, Right).
Our research seeks to synthesize cell-and protein-polymer conjugates via a ROMP graft-from approach for novel therapeutic biologics.15,30 However, in order for ROMP to take place in the presence of relevant biologics such as cells and proteins, an improved methodology for ROMP at physiological conditions (i.e. neutral pH, room temperature and no organic co-solvents) must be realized. These conditions must achieve high monomer conversion, allow a broad monomer scope and maintain an active ruthenium chain-end for block copolymer synthesis.
To this end, we were inspired by the work of Matsuo and coworkers who demonstrated that the addition of a chloride source was vital to maintain the activity of a water soluble HG2 catalyst for RCM in water near neutral pH (Scheme 1B).31 They proposed that excess chloride in solution shifts the equilibrium towards the active dichloro catalyst and away from a deactivated catalyst species in which a chloride ligand has been displaced by water or hydroxide anion. Indeed, previous work from Grubbs also hinted at the role of added chloride in solution for shifting the equilibrium away from solvolysis of chloride ligand on a water soluble Grubbs 1st generation catalyst.18
We hypothesized that the knowledge gained by Matsuo and Grubbs could be applied toward the optimization of ROMP under neutral aqueous conditions. Herein, we explore how variables such as chloride concentration and pH can be optimized to enhance the efficiency of ROMP in an aqueous environment (Scheme 1C).
Results and Discussion
Polymerization Optimization.
We chose to use a commercially available, water soluble HG2 catalyst (AquaMet) as it obviates the need for difficult catalyst synthesis32 and is well-defined for precise catalyst loading.15,19,33 Furthermore, it has been successfully used for ROMPISA in neat water by the Gianneschi group.34 To form a baseline comparison to previous work, monomer NB-PEG (0.1 M) was polymerized in non-degassed deionized water for 1 hour and reached 80% conversion (Table 1, entry 1, Figure S10) with a Mn of 37.8 kDa (targeted Mn = 24.7 kDa) and Ɖ = 1.49. While the molecular weight control and polymer dispersity differ considerably from that obtained by Gianneschi in pure water, we note that factors such as the variable pH of neat water and monomer concentration can play a significant role in polymerization initiation and propagation under aqueous conditions, highlighting the need for a better understanding of optimal conditions to perform ROMP in aqueous media.17,35
Table 1.
ROMP of monomer NB-PEG in buffered water with varying [Cl-]
 | ||||||
|---|---|---|---|---|---|---|
| Entrya,b | Aqueous Media | [salt] | pH | % Conversionf | Mn (kDa)c,d | Ɖ |
| 1 | diH2O | 0 mM | 4.6 | 80 | 37.8 | 1.49 |
| 2 | 50 mM K+ P.B. | 0 mM | 7.4 | 36 | 7.4e | 1.41 |
| 3 | 50 mM K+ P.B. | 10 mM MgCl2 | 7.4 | 50 | 12.6 | 1.69 |
| 4 | 50 mM K+ P.B. | 50 mM MgCl2 | 7.4 | 90 | 27.5 | 1.82 |
| 5 | 50 mM K+ P.B. | 10 mM NaCl | 7.4 | 32 | 9.5 | 1.74 |
| 6 | 50 mM K+ P.B. | 50 mM NaCl | 7.4 | 76 | 19.7 | 1.71 |
| 7 | 50 mM K+ P.B. | 100 mM NaCl | 7.4 | 93 | 26.8 | 1.86 |
| 8 | 10 mM K+ P.B. | 50 mM NaCl | 7.4 | 77 | 28.2 | 1.89 |
| 9 | 50 mM K+ P.B. | 100 mM NaCl | 6.5 | >99 | 40.0 | 1.87 |
| 10 | 50 mM K+ P.B. | 100 mM NaCl | 8.0 | 16 | 7.5 | 2.66 |
| 11 | 50 mM MOPS | 0 mM | 7.4 | 28 | 5.4e | 1.66 |
| 12 | 50 mM MOPS | 50 mM MgCl2 | 7.4 | 93 | 22.8 | 1.82 |
| 13 | 50 mM HEPES | 0 mM | 7.4 | 30 | 5.7e | 1.70 |
| 14 | 50 mM HEPES | 50 mM MgCl2 | 7.4 | 89 | 25.8 | 1.89 |
Polymerizations were conducted at room temperature open to air for one hour.
[NB-PEG] = 0.1 M, [AquaMet] : [NB-PEG] = 1 : 50.
The molecular weights of polymers were determined by SEC-GPC in THF using polystyrene standards.
Targeted Mn = 24.7 kDa.
A bimodal polymer distribution observed by SEC-GPC. The Mn for the highest intensity refractive index peak is reported.
% conversion was determined by 1H NMR spectroscopy comparing the relative integrations of polymer olefinic peaks and total olefin from monomer and polymer.
In an effort to maintain pH values relevant to physiological conditions, polymerizations were conducted in buffered aqueous media in which the pH and chloride concentration were carefully controlled. In 50 mM potassium phosphate buffer (K+ P.B.) at pH 7.4 with no added chloride (Table 1, entry 2), only 36% monomer conversion was achieved. Gel permeation chromatography (GPC) displayed a bimodal polymer distribution (Figure 1) which could be indicative of multiple catalytic species present in solution with different activities. Gratifyingly, with the addition of 100 mM NaCl (Table 1, entry 7) a single peak was observed by GPC (Figure 1) corresponding to 93% monomer conversion and an experimental Mn 26.8 kDa, which is very close to the targeted molecular weight of Mn 24.7 kDa.
Figure 1.
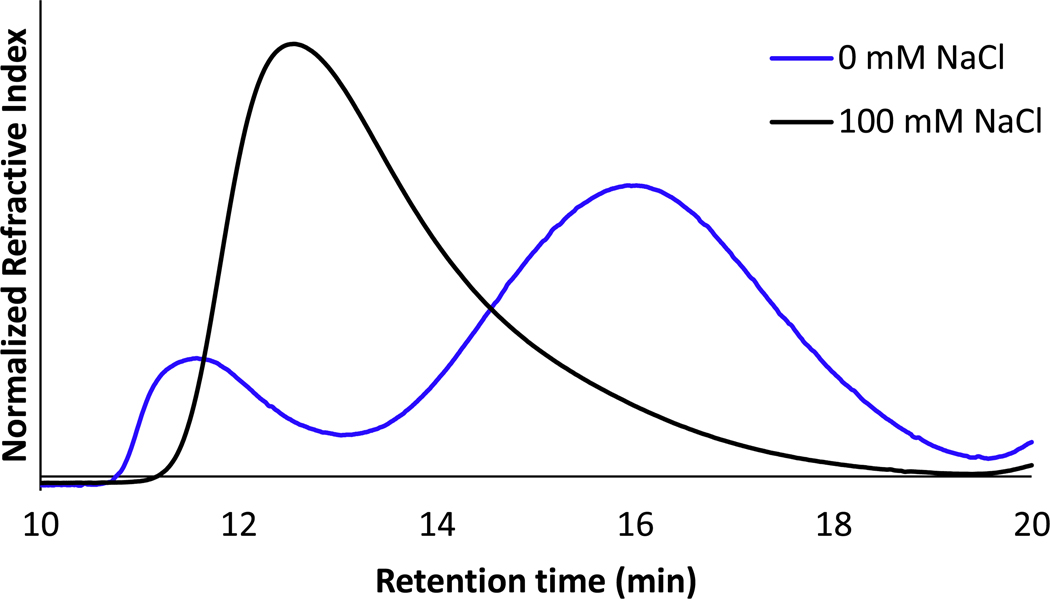
SEC-GPC trace of poly(NB-PEG) polymerized in 50 mM K+ P.B., pH 7.4 with and without the addition of 100 mM NaCl.
Information from Figure 1 clearly demonstrates that the addition of chloride improves the efficiency of ROMP in water at physiologically relevant pH. We then began to probe the role that varying concentrations of chloride played on monomer conversion at neutral pH. With the addition of chloride via either MgCl2 (Table 1, entry 3 – 4) or NaCl (Table 1, entry 5 – 10, Figure S11 – S13), we observed a chloride concentration dependent increase in monomer conversion. Importantly, we note that the chloride source is irrelevant as long as the pH is carefully controlled. This is particularly important in the case of MgCl2. We observed a significant decrease in buffer pH when a concentrated MgCl2 stock solution is added, which would manifest as higher catalytic activity.
When the pH is controlled, monomer conversion appears to be solely dependent on overall chloride concentration. A 100 mM chloride concentration provided the highest monomer conversion with an experimental Mn very close to the targeted molecular weight (Mn 24.7 kDa; [AquaMet] : [NB-PEG] = 1 : 50) (Table 1, entries 4 and 7), indicating this methodology provides good molecular weight control. Unfortunately, higher chloride concentrations caused the catalyst to precipitate out of solution. While lowering the buffer concentration does not appear to affect the overall monomer conversion, surprisingly a loss of molecular weight control was observed (Table 1, entry 8). However, common buffers used for biological systems such as MOPS and HEPES did not have any appreciable effect on monomer conversion or molecular weight control at a given concentration and pH (Table 1, entry 11 – 14, Figure S16 – S19).
The pH of the aqueous solution plays a crucial role in monomer conversion, as expected (Figure S3). Good monomer conversion (93%) can be achieved at pH 7.4 (Table 1 entry 7). While decreasing to pH 6.5 leads to near quantitative monomer conversion (Table 1, entry 9, Figure S14), interestingly, this came at the expense of molecular weight control. However, a significant drop in monomer conversion to 16% occurred at pH 8.0 (Table 1, entry 10, Figure S15). This corroborates literature reports that a high hydroxide concentration can cause catalyst decomposition and deactivation.16,31 Collectively, these results demonstrate that relatively small changes in pH can lead to dramatically different polymerization outcomes.
Across all conditions tested, a relatively high polymer dispersity was observed. In fact, polymer dispersity appears to increase as the chloride concentration and conversion increases. The broad polymer dispersity is likely due to the gradual decomposition of the propagating catalytic species on the polymer chain end, which is prolonged by increasing chloride concentration. This leads to a mixture of polymer chain ends with catalytically active species capable of propagation and chain extension and polymer chain ends that propagate more slowly or have prematurely terminated altogether. A broad polymer dispersity is commonly observed in the literature for ROMP in an aqueous environment, even under acidic conditions.17,20,21,26,36
Catalyst Decomposition Studies.
We believed the differences observed in monomer conversion under different conditions were likely due to the stability of AquaMet under those conditions. Therefore, the effects of pH and chloride concentration on catalyst AquaMet were monitored by UV-vis spectroscopy. In the presence of 100 mM NaCl, a peak at 376 nm, which is characteristic of the HG2 complex,31,37,38 showed a slow decrease in intensity (Figure 2B). In the absence of NaCl (Figure 2A), a peak at ~360 nm, rapidly decreases over the course of 100 minutes. This shift in λmax has been attributed to the partial dissociation of chloride from the metal center.31 The generation of a new peak at ~260 nm suggests other ruthenium complexes are also being generated in solution.
Figure 2.
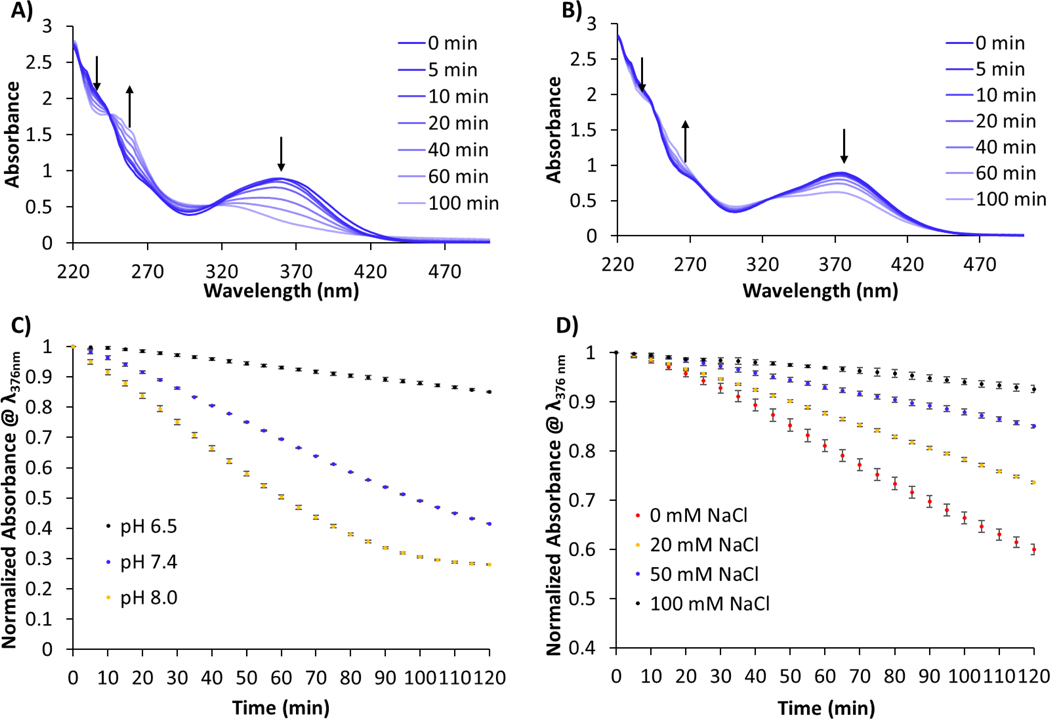
UV-vis of AquaMet (100 µM) in 50 mM K+ P.B., pH 7.4 A) without NaCl and B) with 100 mM NaCl. Arrows indicate the depletion of the peak at 376 nm which is diagnostic of HG2 catalyst. C) The decomposition of catalyst in 50 mM K+ P.B, 50 mM NaCl was followed as a function of pH (pH 6.5, 7.4 and 8.0). D) The decomposition of catalyst in 50 mM K+ P.B, pH 6.5 was followed as a function of NaCl concentration (0, 20, 50 and 100 mM NaCl).
The rates of catalyst decomposition were compared as a function of pH and chloride concentration. Figure 2C clearly shows the effect of pH on maintaining catalyst fidelity. Across all pH values, the peak at 376 nm decreases over time. However, at pH 6.5 catalyst decomposition is much slower than at pH 7.4 which is subsequently slower than decomposition of AquaMet at pH 8.0. Salt concentration also has a profound effect on catalyst stability, with the rate of catalyst decomposition decreasing with increasing salt concentration up to 100 mM NaCl (Figure 2D). Collectively, this data is in good agreement with the parameters observed for efficient monomer conversion seen in Table 1. However, decomposition of AquaMet appears to occur across all pH values and chloride concentrations studied. Therefore, when the catalyst is attached to the end of a polymer chain, gradual decomposition of the propagating ruthenium complex likely leads to the observed broad polymer dispersity.
Effects on Polymerization Rate.
Next, we looked at how factors such as pH and chloride concentration affected the polymerization rate. Monomer NB-PEG was polymerized in buffered D2O at various pH and salt concentrations (Figure 3). Interestingly, pH and chloride concentration produce different outcomes with regards to monomer conversion. In the case of changing pH in the presence of 100 mM NaCl, polymerization rate increases as the pH decreases, as expected (Figure 3A). Overall conversion did not seem to be significantly affected in the case of pH 6.5 and pH 7.4. However, conducting the polymerization in media at pH 8.0 caused a dramatic decline in both the polymerization rate and overall monomer conversion. This seems to indicate a critical hydroxide concentration exists in which complete deactivation of the chain-end ruthenium complex will occur at the onset of polymerization.
Figure 3.

A) Conversion of NB-PEG in 50 mM K+ P.B. in D2O and 100 mM NaCl at different pH. B) Conversion of NB-PEG in 50 mM K+ P.B, pH 7.4 and various NaCl concentrations. For all polymerizations, [AquaMet] : [NB-PEG] = 1 : 50.
Changing the chloride concentration in the media seems to affect not only the rate of polymerization but also has a significant effect on overall monomer conversion (Figure 3B). For example, in buffered D2O at pH 7.4 and in the presence of 100 mM NaCl, monomer conversion plateaus at approximately 90% conversion, whereas at 50 mM NaCl and 20 mM NaCl, monomer conversion plateaus at approximately 70% and 45% respectively. In the absence of NaCl, negligible levels of polymerization are observed under these conditions.
Collectively, our data and previous work from the literature suggests that during ROMP in an aqueous environment each propagating polymer chain contains one of several possible ruthenium complexes with varying levels of catalytic activity which ultimately affects the rate of polymerization and overall monomer conversion (Scheme 2).18,31 Furthermore, these chain-end complexes can further decompose over the course of polymerization, leading to polymer chain ends with premature termination and a broad molecular weight distribution. Recent work by Fogg and colleagues have demonstrated that HG2 complexes, in which the chloride ligands are displaced by hydroxide anions (HG2-OH), still display some catalytic activity, albeit minimal.39 These HG2-OH complexes will then further decompose into metathesis incompetent complexes. It is a reasonable assumption that these ruthenium decomposition products also form on the ruthenium metal center of the propagating polymer chain.
Scheme 2.

Proposed effects of excess chloride on ROMP in an aqueous environment at neutral pH. Increasing the concentration of chloride anions maintains the catalyst for a longer time in the active dichloro species, whereby a reduction in chloride ions leads to a more rapid decomposition into mono-and dihydroxyl species that decrease activity.
However, the presence of excess chloride in solution appears to have a significant effect on not only the decomposition of these complexes but also the initial distribution. The bimodal distribution in the GPC trace for polymerizations in which no excess chloride is present (Figure 1) suggests the catalyst immediately begins to decompose prior to polymer initiation and propagation. Conducting the polymerization in the presence of excess chloride appears to shift the distribution of ruthenium complexes towards the most active complex, presumably the dichloro complex. Additionally, excess chloride delays catalyst decomposition to the deactivated complex, preventing premature termination of monomer consumption.
We can conclude, however, that the most active form of the catalyst exists on the polymer chain end for a relatively short period of time at neutral pH and at all chloride concentrations. This is evidenced by all polymerizations appearing to plateau after ~10 minutes. This observation seems inconsistent with the data from Figure 2. AquaMet catalyst appeared to persist over the course of 100 minutes under neutral conditions and in the presence of NaCl. However, this discrepancy could be due to differences in stability of the ruthenium alkylidene on the propagating polymer chain versus the ruthenium benzylidene in the absence of monomer.
Monomer Scope.
With the optimized conditions in hand, we next wanted to examine the scope of monomers that can be used for ROMP in physiologically relevant aqueous conditions (Table 2, Table S1). To that end, monomers based on norbornene (Table 2, entries 1 – 6) and oxanorbornene (Table 2, entries 7 – 12) scaffolds were tested. Monomers modified with a polyethylene glycol oligomer (NB-PEG and oNB-PEG), a sulfobetaine (NB-Sulfo and oNB-Sulfo) and a tertiary amine (NB-NMe2 and oNB-NMe2) were compared for their conversion under optimized conditions.
Table 2.
Monomer scope for ROMP under physiological conditions
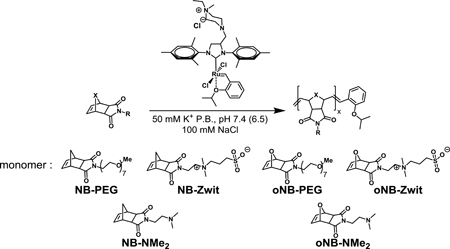 | |||
|---|---|---|---|
| Entrya,b | Monomer | pH | % Conversionc |
| 1 | NB-PEG | 7.4 | 93 |
| 2 | NB-PEG | 6.5 | >99 |
| 3 | NB-Sulfo | 7.4 | 83 |
| 4 | NB-Sulfo | 6.5 | >99 |
| 5 | NB-NMe2 | 7.4 | 0 |
| 6 | NB-NMe2 | 7.4 | 0 |
| 7 | oNB-PEG | 7.4 | 35 |
| 8 | oNB-PEG | 6.5 | 64 |
| 9 | oNB-Sulfo | 7.4 | 10 |
| 10 | oNB-Sulfo | 6.5 | 22 |
| 11 | oNB-NMe2 | 7.4 | 0 |
| 12 | oNB-NMe2 | 6.5 | 0 |
Polymerizations were conducted at room temperature open to air for one hour.
[monomer] = 0.1 M, [AquaMet] : [monomer] = 1 : 50.
% conversion was determined by 1H NMR spectroscopy comparing the relative integrations of polymer olefinic peaks and total olefin from monomer and polymer.
A drop in conversion from 93% to 83% was observed when comparing the conversion of NB-PEG to NB-Sulfo at pH 7.4 (Table 2, entries 1 and 3). This could be due to sulfate groups on the monomer inhibiting olefin coordination and metathesis. Decreasing the pH to 6.5, increased conversion of both monomers to >99% (Table 2, entries 2 and 4). NB-NMe2 did not polymerize at either pH 7.5 or pH 6.5 (Table 2, entries 5 and 6). Previous work from Foster et al achieved full conversion of this monomer at very low pH (pH 2) in which the tertiary amine is completely ionized.26 Under our conditions, the lack of monomer conversion is likely due to coordination of the un-ionized tertiary amine on the monomer to the metal center, inhibiting olefin approach and catalyst turnover. However, we acknowledge that other amine-triggered decomposition pathways may also contribute to the observed catalyst deactivation.40 Interestingly, oxanorbornene based monomers had a much lower conversion than their corresponding norbornene counterparts (Table 2, entries 7 – 12). Conceivably, this could be due to the coordination of the bridging oxygen atom on the oxanorbornene monomer inhibiting productive olefin coordination and metathesis while concomitant catalyst decomposition prevents significant monomer conversion. While decreasing the pH to 6.5 improved conversion, quantitative conversion was not observed.
We next examined the extent to which monomers formed block copolymers using our methodology. Inspired by recent work from the Gianneschi and O’Reilly labs,26,28,34 we looked to utilize aqueous ROMPISA to generate nanostructures near physiological conditions. Based on our optimization experiments, we chose to use pH 6.5 as our reaction medium to ensure optimal fidelity of the ruthenium chain end and thus increase the efficiency for chain extension. NB-PEG was used as the corona-forming monomer and polymerized in 50 mM K+ P.B., pH 6.5, 100 mM NaCl for 25 minutes (Figure 4A). The polymer was then chain extended with NB-OMe as the core-forming monomer for an additional 60 minutes to synthesize the final block copolymer. While chain extension with NB-OMe increased the molecular weight from Mn 11.1 kDa to Mn 25.4 kDa, the GPC trace suggests inefficient reinitiation, consistent with decomposition of the ruthenium chain end (Figure 4B). Dynamic light scattering (DLS) measurements confirms that the resulting polymer solution contains nanostructures with a Dh of 105 nm (Figure 4C). Nanostructures can also be formed using NB-Sulfo as the corona-forming monomer, expanding the current scope of ROMPISA monomers (See Figure S1).
Figure 4.

A) Synthesis of p(NB-PEG_B_NB-OMe) in 50 mM K+ P.B., pH 6.5, 100 mM NaCl. B) GPC refractive index traces of 1st block: p(NB-PEG) and 2nd block p(NB-PEG_b_NB-OMe) in THF. C) DLS of p(NB-PEG_b_NB-OMe) in PBS.
Conclusion
Herein, we have demonstrated optimized conditions for ROMP in water under physiologically relevant conditions. Crucially, the addition of NaCl up to 100 mM appears to enhance the persistence of the active dichloro ruthenium complex on the polymer chain end over the course of the polymerization. This is likely due to a shift in equilibrium away from HG2-OH complexes with little to no metathesis reactivity and towards the active dichloro species. Increasing the chloride concentration has a profound effect on the polymerization in an aqueous environment, providing excellent monomer conversions and polymers with a well-controlled molecular weight. These results can be achieved near physiologically relevant pH whereas the previous literature for ROMP in an aqueous environment have required media at a very low pH. Additionally, we have shown this can be accomplished at room temperature and without the use of organic co-solvents. We believe these findings will be especially useful for conducting ROMP in biological settings such as in the presence of proteins and cells which require the use of physiological buffers.
Supplementary Material
Acknowledgments
The authors acknowledge funding from the National Science foundation (1808031) and the National Institutes of Health (R21 EB024874).
References
- 1.Sabatino V and Ward TR, Beilstein Journal of Organic Chemistry, 2019, 15, 445–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binder JB and Raines RT, Current Opinion in Chemical Biology, 2008, 12, 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomasek J and Schatz J, Green Chemistry, 2013, 15, 2317. [Google Scholar]
- 4.Burtscher D and Grela K, Angewandte Chemie International Edition, 2009, 48, 442–454. [DOI] [PubMed] [Google Scholar]
- 5.Lin YA, Chalker JM, Floyd N, Bernardes GJL and Davis BG, Journal of the American Chemical Society, 2008, 130, 9642–9643. [DOI] [PubMed] [Google Scholar]
- 6.Chalker JM, Lin YA, Boutureira O and Davis BG, Chemical Communications, 2009, 3714. [DOI] [PubMed] [Google Scholar]
- 7.Lin YA, Chalker JM and Davis BG, Journal of the American Chemical Society, 2010, 132, 16805–16811. [DOI] [PubMed] [Google Scholar]
- 8.Pelegri-O’Day EM, Matsumoto NM, Tamshen K, Raftery ED, Lau UY and Maynard HD, Bioconjugate Chemistry, 2018, 29, 3739–3745. [DOI] [PubMed] [Google Scholar]
- 9.Ai H, Shen W, Brustad E and Schultz PG, Angewandte Chemie International Edition, 2010, 49, 935–937. [DOI] [PubMed] [Google Scholar]
- 10.Sabatino V, Rebelein JG and Ward TR, Journal of the American Chemical Society, 2019, 141, 17048–17052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Timmer BJJ and Ramström O, Chemistry – A European Journal, 2019, 25, 14408–14413. [DOI] [PubMed] [Google Scholar]
- 12.Messina MS and Maynard HD, Materials Chemistry Frontiers, 2020, 4, 1040–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masuda S, Tsuda S and Yoshiya T, Organic & Biomolecular Chemistry, 2018, 16, 9364–9367. [DOI] [PubMed] [Google Scholar]
- 14.Bhushan B, Lin YA, Bak M, Phanumartwiwath A, Yang N, Bilyard MK, Tanaka T, Hudson KL, Lercher L, Stegmann M, Mohammed S and Davis BG, Journal of the American Chemical Society, 2018, 140, 14599–14603. [DOI] [PubMed] [Google Scholar]
- 15.Isarov SA and Pokorski JK, ACS Macro Letters, 2015, 4, 969–973. [DOI] [PubMed] [Google Scholar]
- 16.Lynn DM, Mohr B and Grubbs RH, Journal of the American Chemical Society, 1998, 120, 1627–1628. [Google Scholar]
- 17.Lynn DM, Mohr B, Grubbs RH, Henling LM and Day MW, Journal of the American Chemical Society, 2000, 122, 6601–6609. [Google Scholar]
- 18.Lynn DM and Grubbs RH, Journal of the American Chemical Society, 2001, 123, 3187–3193. [DOI] [PubMed] [Google Scholar]
- 19.Gallivan JP, Jordan JP and Grubbs RH, Tetrahedron Letters, 2005, 46, 2577–2580. [Google Scholar]
- 20.Breitenkamp K and Emrick T, Journal of Polymer Science Part A: Polymer Chemistry, 2005, 43, 5715–5721. [Google Scholar]
- 21.Samanta D, Kratz K, Zhang X and Emrick T, Macromolecules, 2008, 41, 530–532. [Google Scholar]
- 22.Trnka TM and Grubbs RH, Accounts of Chemical Research, 2001, 34, 18–29. [DOI] [PubMed] [Google Scholar]
- 23.Mayer C, Gillingham DG, Ward TR and Hilvert D, Chemical Communications, 2011, 47, 12068. [DOI] [PubMed] [Google Scholar]
- 24.Sauer DF, Himiyama T, Tachikawa K, Fukumoto K, Onoda A, Mizohata E, Inoue T, Bocola M, Schwaneberg U, Hayashi T and Okuda J, ACS Catalysis, 2015, 5, 7519–7522. [Google Scholar]
- 25.Sauer DF, Gotzen S and Okuda J, Organic & Biomolecular Chemistry, 2016, 14, 9174–9183. [DOI] [PubMed] [Google Scholar]
- 26.Foster JC, Varlas S, Blackman LD, Arkinstall LA and O’Reilly RK, Angewandte Chemie International Edition, 2018, 57, 10672–10676. [DOI] [PubMed] [Google Scholar]
- 27.Varlas S, Foster JC and O’Reilly RK, Chemical Communications, 2019, 55, 9066–9071. [DOI] [PubMed] [Google Scholar]
- 28.Varlas S, Foster JC, Arkinstall LA, Jones JR, Keogh R, Mathers RT and O’Reilly RK, ACS Macro Letters, 2019, 8, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varlas S, Keogh R, Xie Y, Horswell SL, Foster JC and O’Reilly RK, Journal of the American Chemical Society, 2019, 141, 20234–20248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Church DC and Pokorski JK, Angewandte Chemie International Edition, , DOI: 10.1002/anie.202005148. [DOI] [Google Scholar]
- 31.Matsuo T, Yoshida T, Fujii A, Kawahara K and Hirota S, Organometallics, 2013, 32, 5313–5319. [Google Scholar]
- 32.Skowerski K, Szczepaniak G, Wierzbicka C, Gułajski Ł, Bieniek M and Grela K, Catalysis Science & Technology, 2012, 2, 2424. [Google Scholar]
- 33.Hong SH and Grubbs RH, Journal of the American Chemical Society, 2006, 128, 3508–3509. [DOI] [PubMed] [Google Scholar]
- 34.Wright DB, Touve MA, Thompson MP and Gianneschi NC, ACS Macro Letters, 2018, 7, 401–405. [DOI] [PubMed] [Google Scholar]
- 35.Muhlebach A, Bernhard P, Buhler N, Karlen T and Ludi A, J Mol Catal, 1994, 90, 143–156. [Google Scholar]
- 36.Dunbar MA, Balof SL, Roberts AN, Valente EJ and Schanz H-J, Organometallics, 2011, 30, 199–203. [Google Scholar]
- 37.Matsuo T, Imai C, Yoshida T, Saito T, Hayashi T and Hirota S, Chemical Communications, 2012, 48, 1662. [DOI] [PubMed] [Google Scholar]
- 38.Mingotaud A-F, Mingotaud C and Moussa W, Journal of Polymer Science Part A: Polymer Chemistry, 2008, 46, 2833–2844. [Google Scholar]
- 39.Goudreault AY, Walden DM, Nascimento DL, Botti AG, Steinmann SN, Michel C and Fogg DE, ACS Catalysis, 2020, 10, 3838–3843. [Google Scholar]
- 40.Ireland BJ, Dobigny BT and Fogg DE, ACS Catalysis, 2015, 5, 4690–4698. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.