

Abstract
Localized apoptosis of osteocytes, the tissue-resident cells within bone, occurs with fatigue microdamage and activates bone resorption. Osteoclasts appear to target and remove dying osteocytes, resorbing damaged bone matrix as well. Osteocyte apoptosis similarly activates bone resorption with estrogen loss and in disuse. Apoptotic osteocytes trigger viable neighbor (ie, bystander) osteocytes to produce RANKL, the cytokine required for osteoclast activation. Signals from apoptotic osteocytes that trigger this bystander RANKL expression remain obscure. Studying signaling among osteocytes has been hampered by lack of in vitro systems that model the limited communication among osteocytes in vivo (ie, via gap junctions on cell processes and/or paracrine signals through thin pericellular fluid spaces around osteocytes). Here, we used a novel multiscale fluidic device (the Macro-micro-nano, or Mμn) that reproduces these key anatomical features. Osteocytes in discrete compartments of the device communicate only via these limited pathways, which allows assessment of their roles in triggering osteocytes RANKL expression. Apoptosis of MLOY-4 osteocytes in the Mμn device caused increased osteocyte RANKL expression in the neighboring compartment, consistent with in vivo findings. This RANKL upregulation in bystander osteocytes was prevented by blocking Pannexin 1 channels as well as its ATP receptor. ATP alone caused comparable RANKL upregulation in bystander osteocytes. Finally, blocking Connexin 43 gap junctions did not abolish osteocyte RANKL upregulation, but did alter the distribution of RANKL expressing bystander osteocytes. These findings point to extracellular ATP, released from apoptotic osteocytes via Panx1 channels, as a major signal for triggering bystander osteocyte RANKL expression and activating bone remodeling.
Keywords: INTERCELLULAR COMMUNICATION, NANOFLUIDICS, BYSTANDER SIGNALING, EXTRACELLULAR ATP, OSTEOCYTES
Introduction
Recent studies have implicated apoptosis of osteocytes, the permanent resident cells within adult bone, as a key step in triggering osteoclastic bone resorption due to focal microdamage, estrogen loss, and disuse.(1–4) Despite differences in the causes of osteocyte death associated with these diverse bone remodeling stimuli, apoptosis in each instance was found to be required for recruitment and differentiation of myeloid (monocyte/macrophage) progenitor cells into osteoclasts, the specialized multinucleated phagocytes that remove apoptotic cell debris and resorb surrounding bone matrix in the process. Understanding the mechanism by which dying osteocytes recruit osteoclast progenitors from marrow to bone has been challenging. Osteocytes at baseline in adult bone do not express significant levels of receptor activator of nuclear factor κB ligand (RANKL), the obligate cytokine required for osteoclast recruitment and differentiation.(5) Furthermore, apoptotic cells do not carry out sustained production or release of any cytokine. Recent in vivo studies showed that apoptotic osteocytes induce their neighboring viable osteocytes (ie, bystander cells) to upregulate expression of RANKL.(6–8) However, this observation raises the broader question of which signals apoptotic cells might use to trigger RANKL production in bystander osteocytes.
Studies over the last decade revealed that cells undergoing apoptosis produce both “find-me” signals to attract specialized phagocytic cells to remove the cell and tissue debris and “eat-me” signals that mark the debris for engulfment. Among the key chemoattractant find-me signals released during early apoptosis are triphosphate nucleotides (ATP and UTP), lysophosphatidylcholine (lysoPC), and the chemokine CX3CL1.(9–11) Recently, Cheung and colleagues(6) in our laboratory provided evidence implicating ATP as a potential bystander signal produced by apoptotic osteocytes. They showed that osteocyte RANKL upregulation and bone remodeling following fatigue in vivo did not occur in mice genetically deficient in the membrane channel protein pannexin-1 (Panx1), although osteocyte apoptosis occurred normally. Panx1 is responsible for both low-level ATP release from cells in normal signaling and also for the large bolus of ATP released during apoptosis.(12) This locally high purinergic release at cell death acts as a direct find-me signal to macrophages and also a signal to bystander cells.(13) Whether such ATP, with or without additional signals, triggers bystander signaling from apoptotic osteocytes is not yet known.
Osteocytes, the majority cell population in bone (>90% of bone cells), pose unique problems when analyzing intercellular communication pathways, both in vivo and in vitro. Osteocytes in vivo are entombed within mineralized bone matrix, their cell bodies in matrix spaces termed lacunae and their numerous dendritic processes within tiny canals (canaliculi) of <1 μm diameter. Osteocytes communicate with each other and with bone surface cells by two pathways: via gap junctions formed where cell processes contact each other and via paracrine signals that move through the narrow proteoglycan-filled pericellular fluid space in the lacunar canalicular system (LCS) (see Schaffler and colleagues(14) for detailed discussion). Conventional monolayer cell culture techniques cannot capture key connectivity relationships and transport constraints experienced by authentic osteocytes in vivo. Three-dimensional cultures of osteocyte-like cells in collagen gel have been used to demonstrate that large mechanical injury stimulates RANKL production.(15,16) However, this approach could not distinguish among the potential factors (apoptosis, other cell injury, other factors in the system) responsible for activating RANKL expression. Moreover, the high water content of collagen gels allows open and unconstrained diffusion of potential signals, unlike the anatomic constraints seen in canaliculi.(17) To address this problem, McCutcheon and colleagues(18) in our laboratory developed a multiscale fluidic device (the Macro-micro-nano, or Mμn) that allows osteocytes to grow and connect via small channels with dimensions similar to those of the canalicular system in vitro. The system also allows for the spatial separation of apoptotic and non-apoptotic osteocyte populations, so communication between the two in this model occurs only by gap junctions on dendritic processes or by diffusion of signals through the extracellular fluid spaces around processes in the channels, analogous to the situation in vivo.
In the current studies, we established that this novel in vitro model system can replicate the osteocyte apoptosis and bystander signaling behavior we previously observed in vivo. Specifically, we determined whether RANKL upregulation in bystander cells operates through an apoptosis-dependent release of signal through Panx1 channels and activation of purinergic receptors. We further tested whether ATP is an essential trigger to turning on osteocyte RANKL expression. Last, we assessed how gap junctions between osteocytes contribute to the extent to which upregulated RANKL signaling spreads to bystander osteocytes.
Materials and Methods
Mμn device
The Mμn fluidic device (Fig. 1A,B) consists of two identical cell compartments for culturing cells (area/compartment = 10 mm2, volume = 0.5 μL), separated by a 100-μm-wide barrier penetrated by an array of nanochannels less than 1 μm diameter, similar to dimensions of osteocyte canaliculi in bone.(19,20) Each cell compartment has separate inlet and outlet ports for initial seeding of cells and to deliver culture medium and experimental agents. Equal media flow rates and pressures are maintained in both compartments throughout the experiment to prevent net convection between the two compartments. Consequently, any gradients of solute established by introduction of a test molecule into one chamber will be based only on solute concentration and diffusive properties, which in turn reflect molecular size. The device application also incorporates a specialized rapid heating element in one cell compartment to induce apoptosis exclusively in that compartment, and uses a heat sink to prevent thermal stress from spreading to the adjacent (bystander) compartment.
Fig. 1.
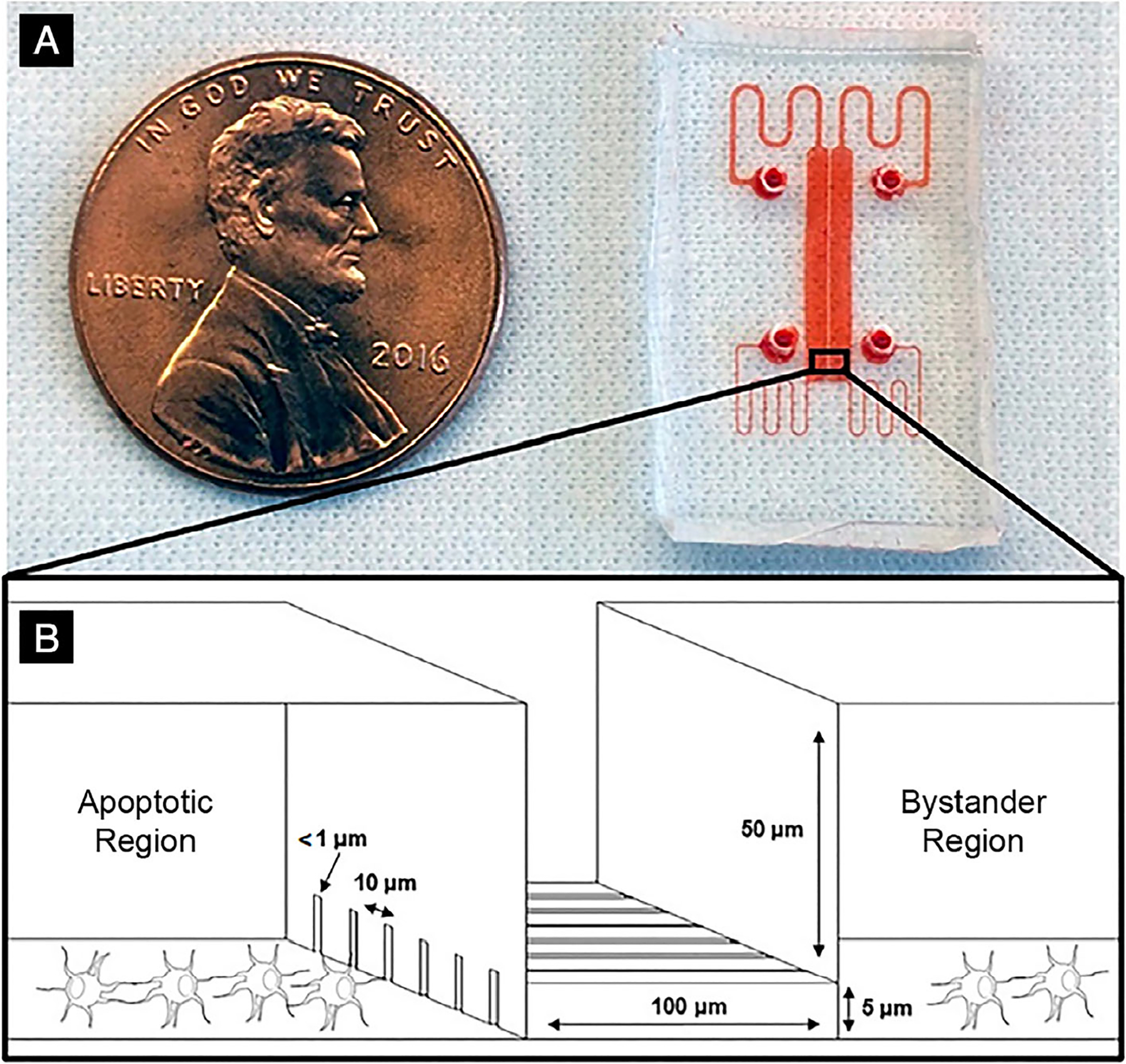
Mμn device. (A) The Mμn device, with fluid chambers and conduits labeled in red dye; (B) a 3-dimensional illustration of the nanochannel array interface in the center with rectangular seeding chambers on either side.
The Mμn device was created by nanofabrication using a high-resolution two-step photolithography process.(18) From an initial template, the Mμn devices were cast in polydimethylsiloxane (PDMS, Sylgard 184; Dow Corning, Midland, MI, USA), bonded to #1 glass coverslips by ozone plasma treatment and sterilized with 70% ethanol, followed by three rinses with sterile deionized water. In the development studies for the device, osteocytes seeded in the Mμn device compartments attached and extended dendritic processes into the channels; cell viability within the device at 7 days was >90%.(18) Details on solute partitioning and gradients between cell compartments in the Mμn device as well as validation of apoptosis induction and its limitation to a single cell compartment are discussed in McCutcheon and colleagues.(18)
Osteocyte cell culture
MLO-Y4 osteocyte-like cells (RRID:CVCL_M098; Kerafast, Boston, MA, USA) were cultured in α-MEM (Gibco, Grand Island, NY, USA; 32571–036) supplemented by 2.5% heat-inactivated calf serum (Gibco; 16000036), 2.5% heat-inactivated fetal bovine serum (Gibco; 10082147), and 100 U/mL penicillin-streptomycin (Gibco; 15140122). Stock cultures were maintained in plates coated with 150 μg/mL rat tail Collagen I (Corning, Inc., Corning, NY, USA; CB-40236) and subcultured every 4 days.
For osteocyte culture in the Mμn device, cell compartments were coated with 30 μg/mL fibronectin (Gibco; F1141) in PBS (Gibco; 10010023) for 1 hour at room temperature, after which osteocyte suspensions at 1 × 106 cells/mL were seeded in each compartment using a syringe pump (NE-1600; New Era Pump Systems, Farmingdale, NY, USA) at 100 μL/min. Loading was performed under flow, but all experiments, unless otherwise noted, were performed under static conditions. Medium was replaced every 24 to 48 hours with undiluted osteocyte conditioned medium collected from our maintenance cultures every 4 days, and experiments were initiated at 7 days of culture, as has been demonstrated to be sufficient time post-seeding to obtain an osteocyte-like phenotype, namely formation of thin dendritic processes in our system.
RANKL reporter osteocytes
We created fluorescent RANKL reporter osteocytes to rapidly assess changes in RANKL expression on a per-cell basis. A cDNA construct containing a murine RANKL promoter upstream of an mCherry reporter and puromycin resistance gene (GeneCopoeia, Rockville, MD, USA; #MPRM21927-LvPM02, vector #pEZX-LvPM02) was introduced into competent Escherichia coli (New England Biolabs, Ipswich, MA, USA; C2987I) to increase yield. Plasmid DNA was then purified (Midiprep; Qiagen, Valencia, CA, USA; K210014) and used to produce lentiviral particles (LVP creation sourced to Vector Builder, Cyagen Biosciences, Santa Clara, CA, USA), using 293 T packaging cells (titer >108 particles/mL). MLO-Y4 cells were grown to 80% to 90% confluence and transduced with RANKL reporter LVPs at a multiplicity of infection (MOI) of 10. Stably transduced colonies were selected by resistance to 1 μg/mL puromycin (Gibco; A1113802) over 14 days. Monoclonal cell lines were selected with cloning rings (Millipore Sigma, St. Louis, MO, USA; TR-1004) and subcultured. The reporter line used for Mμn device experiments was expanded from the colony with the lowest mean fluorescence intensity (RANKL signal) per cell at baseline as determined by fluorescence microscopy using a Nikon Eclipse inverted microscope (Nikon, Tokyo, Japan) and analysis with NIH ImageJ software (v.1.51; Bethesda, MD, USA; https://imagej.nih.gov/ij/). In our cell cultures, approximately 20% of reporter osteocytes showed low-level fluorescence, consistent with the observation that some MLO-Y4 cells express RANKL.(21) Increased RANKL expression by osteocytes in Mμn device experiments (RANKL+ osteocytes) was defined as the increase of mean fluorescence intensity per cell ≥50% from maximum intensity measured at baseline. Fluorescence measurements for RANKL reporter studies were taken at 1-s exposure time.
Osteocyte process ingrowth and gap junction formation in the Mμn
Osteocytes were assessed after 7 days in culture using actin staining to demonstrate dendritic processes. Cells were fixed in the Mμn device using 4% formaldehyde in PBS and then permeabilized in 0.1% TritonX-100 for 5 min by manual perfusion of the chamber with a 1-mL syringe. After rinsing in PBS, cells were stained with 5 U/mL AlexaFluor488-labeled phalloidin (Life Technologies, Inc., Grand Island, NY, USA; A12379) in PBS containing 1% bovine serum albumin (BSA). Cells were then rinsed with PBS and glycerol was added to the cell seeding compartments via the input/outlet ports. Actin staining was visualized by confocal microscopy.
Gap junction communication via osteocyte processes in the nanochannel array was assessed using a modification of the parachute assay(22) in which dye-loaded “donor” cells are added (parachuted) on top of non-labeled cells; transfer of dye from the donor to existing (acceptor) cells occurs through formation of gap junctions. Osteocytes were cultured in both compartments of a Mμn device as described under osteocyte cell culture. Cells from a separate culture were labeled with 10μM Calcein AM (Thermo Fisher Scientific, Waltham, MA, USA; C3100MP) and lipophilic, non–gap junction transferrable DiI (10μM; Thermo Fisher Scientific; D282) as detailed elsewhere,(22) then trypsinized and resuspended in medium. The dye-loaded donor cells were added to one compartment of the Mμn device. After 90 min, the presence of calcein dye in cells on the opposite compartment of the device was assessed by fluorescence microscopy. To block dye transfer through gap junction channels, cells in both chambers were treated with 18α-glycyrrhetinic acid (AGA, 30μM; Sigma-Aldrich, St. Louis, MO, USA; G8503) for 60 min prior to the initiation of and during the parachute experiment. Experiments were repeated in triplicate.
Induction of osteocyte apoptosis and expression of RANKL in bystander osteocytes
RANKL reporter osteocytes were seeded in both compartments of Mμn devices as described under osteocyte cell culture. One hour prior to apoptosis induction in one compartment, the medium in both compartments was replaced with medium containing 5μM CellEvent, a caspase cleavage dye that fluoresces when cells undergo apoptosis (Thermo Fisher Scientific; C10423). Apoptosis was induced in one of the Mμn compartments by short-duration heating (60°C for 30 s), which caused apoptosis of the majority of osteocytes within that compartment after 12 hours, similar to what has been shown for surgically induced heat stress in bone in vivo.(18,23,24) In our development studies, we found no significant increase in apoptosis in the adjacent (bystander) compartment.(18) To inhibit apoptosis, the pan-caspase inhibitor QVD (Sigma-Aldrich; SML0063, 10μM)(25,26) was added only to stressed (apoptosis-induced) compartments following the protocol described by Kennedy and colleagues.(27,28)
Numbers of apoptotic or RANKL-expressing osteocytes were assessed in control and QVD-treated devices by fluorescence microscopy in both the challenge and non-challenge compartments at 1, 6, and 12 hours after apoptosis induction. Numbers of RANKL-expressing osteocytes in bystander compartments were measured as a function of distance from the channel array to assess whether a gradient response was observed like that seen in vivo.(27,28) In addition, mRNA expression for RANKL and osteoprotegerin (OPG)(29) was assessed from triplicate experiments using RT-PCR, to confirm results from the microscopy-based studies. RT-qPCR was performed by the manufacturer’s protocol (Thermo Fisher Scientific; Fast SYBR Green Cells-to-Ct, 4402956). Sequences for RANKL and OPG primers are detailed in Cheung and colleagues(6) and the GAPDH sequence was from Tsujita and colleagues.(30) In brief, at 12 hours post-heating, cells were rinsed three times with PBS then extracted separately from each side of the Mμn. Cells were trypsinized, recovered manually from the Mμn by 1-mL syringe, centrifuged at 200g and rinsed in cold PBS three times, then lysed at room temperature for 5 min to obtain mRNA extracts. RNA concentration was measured by spectrophotometer (Nanodrop; Thermo Fisher Scientific) after cell lysis to quantify concentration and quality. After RT reaction cDNA was diluted such that 2 ng/4 μL cDNA was used per 20 μL total volume of RT-qPCR reaction. PCR was performed with the following forward/reverse primers (5′ to 3′):
RANKL:ACGCAGATTTGCAGGACTCG / GGGCCACATCCAACCATGAG
OPG:TGGACCAAAGTGAATGCCG / CTGCTCTGTGGTGAGGTTCG
GAPDH:GACATGCCGCCTGGAGAAAC / AGCCCAGGATGCCCTTTAGT
Gene expression normalized to unheated control was determined by ΔΔCt, using GAPDH as a housekeeping gene. Unless otherwise indicated, n = 9 devices were used for each condition with groups of three devices pooled to obtain sufficient mRNA for quantification. Experiments were run in triplicate.
Apoptosis-induced RANKL expression in bystander osteocytes: Inhibition of pannexin 1, P2X7, and connexins
RANKL reporter osteocytes were cultured in both compartments of Mμn devices for 7 days. One hour prior to apoptosis induction, the cultures received medium containing inhibitors of pannexin-1 channels Probenecid (Sigma Aldrich; P861, 500μM)(31) and 10panx1 (GenScript Corporation, Piscataway, NJ, USA; 100μM),(32) P2X7, the ATP receptor typically associated with Panx1(33) (Brilliant Blue G [BBG]; Sigma-Aldrich; B0770, 10μM), or connexin43 gap junctions(34) (18 α-glycyrrhetinic acid [AGA], 30μM). In all studies, osteocyte apoptosis, RANKL-expressing osteocytes, and mRNA were measured as described above. All inhibitors implemented have IC50 values well below the concentrations we used and have precedent in cell culture and in vivo studies.
Purinergic stimulation of RANKL expression in bystander osteocytes in the absence of apoptosis
During apoptosis, caspase-3–driven opening of Panx1 channels results in release of a large bolus of potential signals, including ATP, into the extracellular space.(13,35) To test whether the candidate purinergic stimuli would trigger RANKL expression in viable bystander osteocytes in the absence of apoptosis, RANKL reporter osteocytes were seeded on one side of the Mμn device and cultured as described under osteocyte cell culture; no cells were cultured in the opposing compartment. At day 7, medium alone was flowed (10 μL/min) into the osteocyte-containing compartment while medium containing 20nM or 1μM ATP (Sigma/FLAAS; Millipore Sigma-Aldrich) or 1μM Adenosine (Sigma-Aldrich; A4036) was flowed into the compartment with no cells. Flow was stopped after 10 min, establishing a separation of purine-supplemented versus non-supplemented medium across the channel array. RANKL expression was assessed by fluorescence at 1, 6, and 12 hours and RANKL and OPG gene expression changes were assessed at 12 hours by RT-qPCR.
Inhibition of RANKL upregulation in bystander osteocytes by scavenging ATP
RANKL reporter osteocytes were cultured in both compartments of Mμn devices for 7 days. One hour prior to apoptosis induction, the cultures received medium containing 0.5 U/mL apyrase (Sigma-Aldrich; A6535). Osteocyte apoptosis, RANKL-expressing osteocytes, and mRNA were measured. Experiments were repeated in nine devices. At 12 hours post-apoptosis induction, extracellular fluid was extracted in parallel from both sides of the Mμn simultaneously by syringe pump at 20 μL/min. ATP content of the extracellular fluid was quantified by firefly luciferase bioluminescence assay (Thermo Fisher Scientific; A22066).
Statistical analyses
Data were analyzed by one-way ANOVA with post hoc multiple comparison performed using Tukey’s test (MATLAB; MathWorks, Natick, MA, USA) for groups of three or more and Student’s (unpaired) t test for comparison between two groups. Data are shown as mean ± standard deviation (SD).
Results
Osteocyte process ingrowth and gap junction formation in the Mμn
Osteocytes grew well-differentiated processes into the nanochannel array of the Mμn device, with processes from cells in both compartments extending into channels (Fig. 2A). By 7 days in culture, approximately 60% (60.2% ± 8.5%; n = 8 devices) of the nanochannels were occupied by osteocyte dendritic processes. In acute studies, dye transfer from the prelabeled donor cells parachuted onto osteocytes was completely inhibited by AGA gap junction blockade. Moreover, dye transfer from donor osteocytes to osteocytes on the opposite sides of the channels was inhibited by AGA treatment, establishing gap junctional communication between via processes within the channels (Fig. 2A–D) of the Mμn system.
Fig. 2.
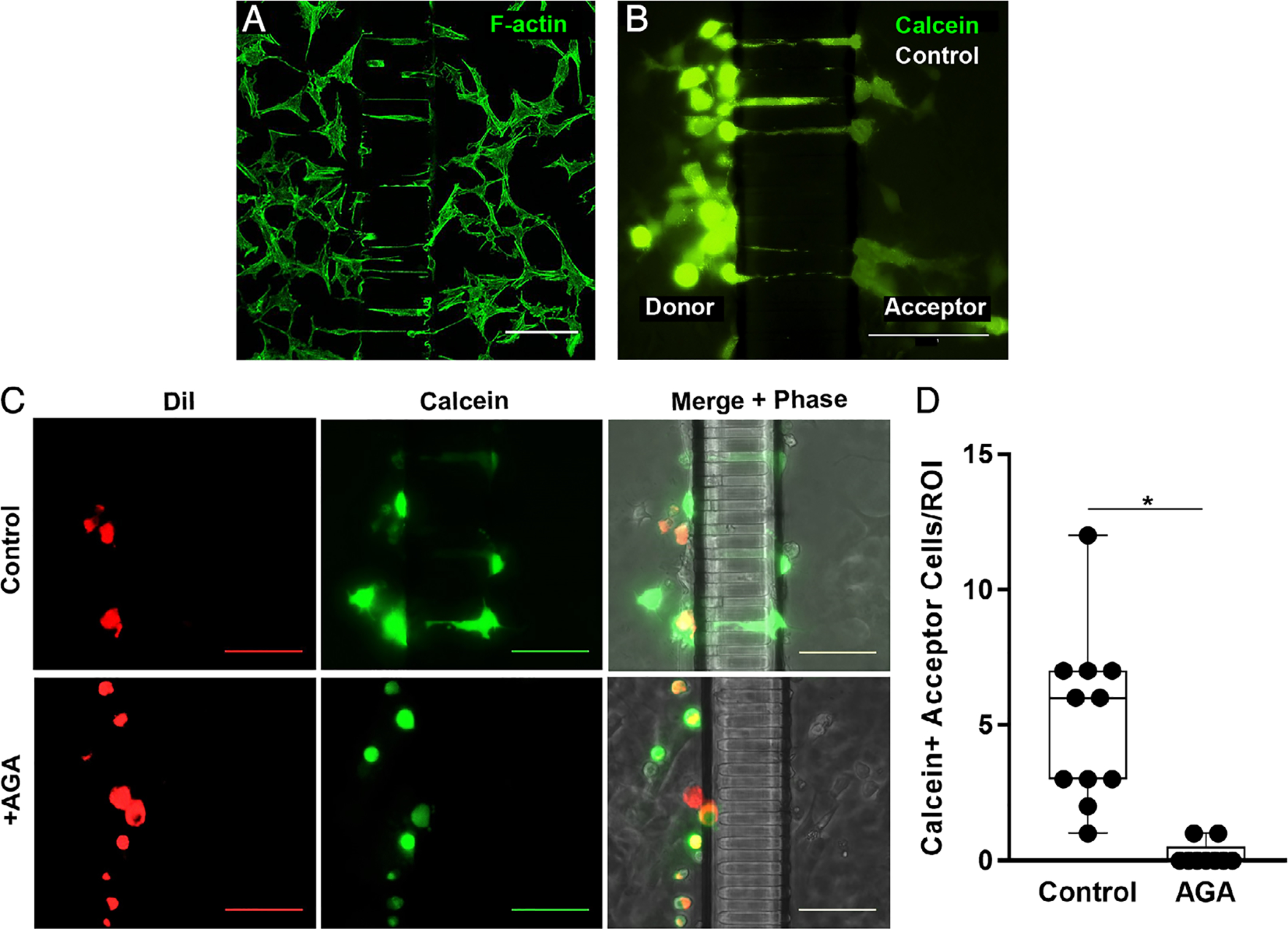
Osteocyte process ingrowth and gap junction functionality. (A) Confocal photomicrograph showing MLOY-4 osteocyte dendritic processes growing into the nanochannel array at 7 days culture in the Mμn device, visualized by AlexaFluor488-labeled phalloidin staining for F-Actin. (B) Parachute dye transfer assay image, showing Calcein-AM (green) transfer from donor compartment osteocytes to acceptor osteocytes in the “bystander” compartment of Mμn device. (C) Fluorescence and corresponding phase-contrast micrographs showing dye transfer across the channel array in osteocytes cultured under control conditions (top row), while no transfer to the acceptor side of the Mμn occurred when the connexin 43 blocker AGA was added, confirming gap junction communication between compartments. MLO-Y4 cells, labeled with gap junction permeable dye Calcein-AM (green) and gap junction impermeable membrane dye DiI (red), were parachuted onto one side of a previously MLO-Y4–seeded Mμn. Calcein-only labeled cells indicate gap junctional transfer from parachuted cells (donors) to cells on same side of the channel array, as well as across the channel array (acceptors) via first and second order network connections. (All scale bars =100 μm). (D) Number of Calcein-AM positive (Calcein+) cells on the acceptor side of the Mμn under control conditions and with AGA. (ROI = 100 μm on either side of channel array). n = 11 devices, *p = .0002, unpaired t test.
Induction of osteocyte apoptosis and expression of RANKL in bystander osteocytes
Osteocyte apoptosis in the stressed compartment of the Mμn device was increased approximately fivefold to sevenfold over osteocytes in the non-stressed (bystander) compartment at 12 hours after induction, consistent with our earlier development studies.(18) Apoptosis in the bystander compartment of the device was low and similar to control osteocyte cultures (<15%) (Fig. 3A,B). QVD treatment prevented the increase in osteocyte apoptosis in the stressed side of the Mμn device. Apoptosis data are summarized in Fig. 3. Extracellular ATP levels in the medium as measured by firefly bioluminescence were increased by 245% ± 40% (n = 3 devices, p = .0288) over baseline at 12 hours post–apoptosis induction (the time of peak apoptosis in this system), consistent with a large release of ATP from dying cells (Fig. 4A).
Fig. 3.
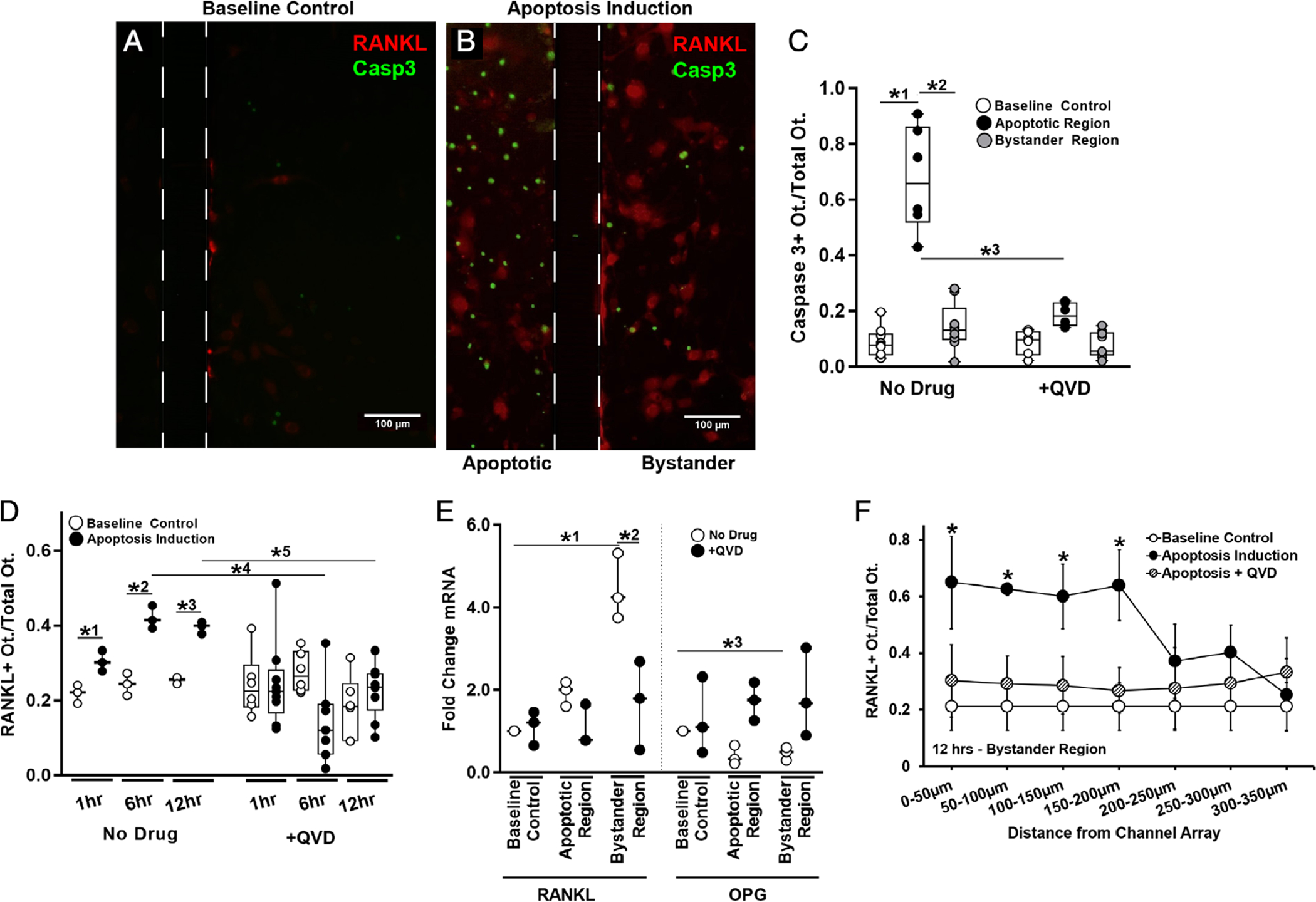
Osteocyte apoptosis and RANKL expression in the Mμn device. (A) Fluorescence photomicrograph of baseline control conditions for MLO-Y4 osteocytes cultured in the Mμn system, showing a few weakly fluorescent mCherry RANKL expressing osteocytes (red fluorescence). Boundaries of the nanochannel array outlined with a dotted line. Scale bar = 100 μm. (B) Fluorescence photomicrograph showing large numbers of apoptotic osteocytes (green fluorescence, shown using caspase 3 cleavage dye) at 12 hours after induction; apoptosis is effectively restricted to within the induction compartment of the Mμn device (on the left). The right side of the panel shows the opposite (non-stressed) compartment of the Mμn device, with large numbers of red fluorescing RANKL-expressing reporter osteocytes and a few apoptotic cells. (C) Quantification of the fraction of caspase 3–positive apoptotic cells before (baseline control) and in the apoptotic and bystander regions after apoptosis induction in the Mμn device. Data to the right are apoptosis data following addition of the apoptosis inhibitor QVD (10μM) 1 hour prior to apoptosis induction. Note that whereas almost 70% of osteocytes in the induction chamber undergo apoptosis (*1p = .0001 versus baseline control cells); there was no significant increase in osteocyte apoptosis in the bystander (non-stressed) compartment (*2p = .0001 versus apoptotic region and p = .32 versus baseline control), demonstrating that osteocyte apoptosis in the Mμn device is contained and separate. (D) Number of RANKL-expressing osteocytes in the bystander compartment was increased modestly at 1 hour (*1p = .01) and nearly twofold at 6 and 12 hours post-apoptosis induction (*2p = .0001 and *3p = .002, respectively). Inhibiting apoptosis with QVD prevented the increases in osteocyte RANKL expression in the bystander compartment at 6 and 12 hours (*4p = .001 and *5p = .003, respectively). n = 3 devices for control and n = 6 for QVD-treated groups. (E) With apoptosis induction, there was a more than fourfold increase in RANKL mRNA and a ~25% reduction in OPG mRNA in bystander osteocytes (*1p = .0001 for RANKL and *3p = .004 for OPG). Inhibiting apoptosis prevented the increase in RANKL gene expression (*2p < .001 versus apoptotic no drug) and the decrease in OPG mRNA level (p > .8 vs baseline for each condition), n = 3 devices per sample, tested in triplicate. (F) A high number of RANKL-expressing osteocytes extended ~200 μm from the nanochannel array/apoptotic signal source. Expression levels were low and homogeneous in the absence of apoptosis induced in the neighboring chamber and when treated with QVD. Comparison versus baseline control (*p = .043 at 0 to 50 μm, p = .0001 at 50 to 100 μm, and p = .02 at 100 to 150 μm and 150–200 μm), n = 9 devices. Data are shown as mean ± SD.
Fig. 4.
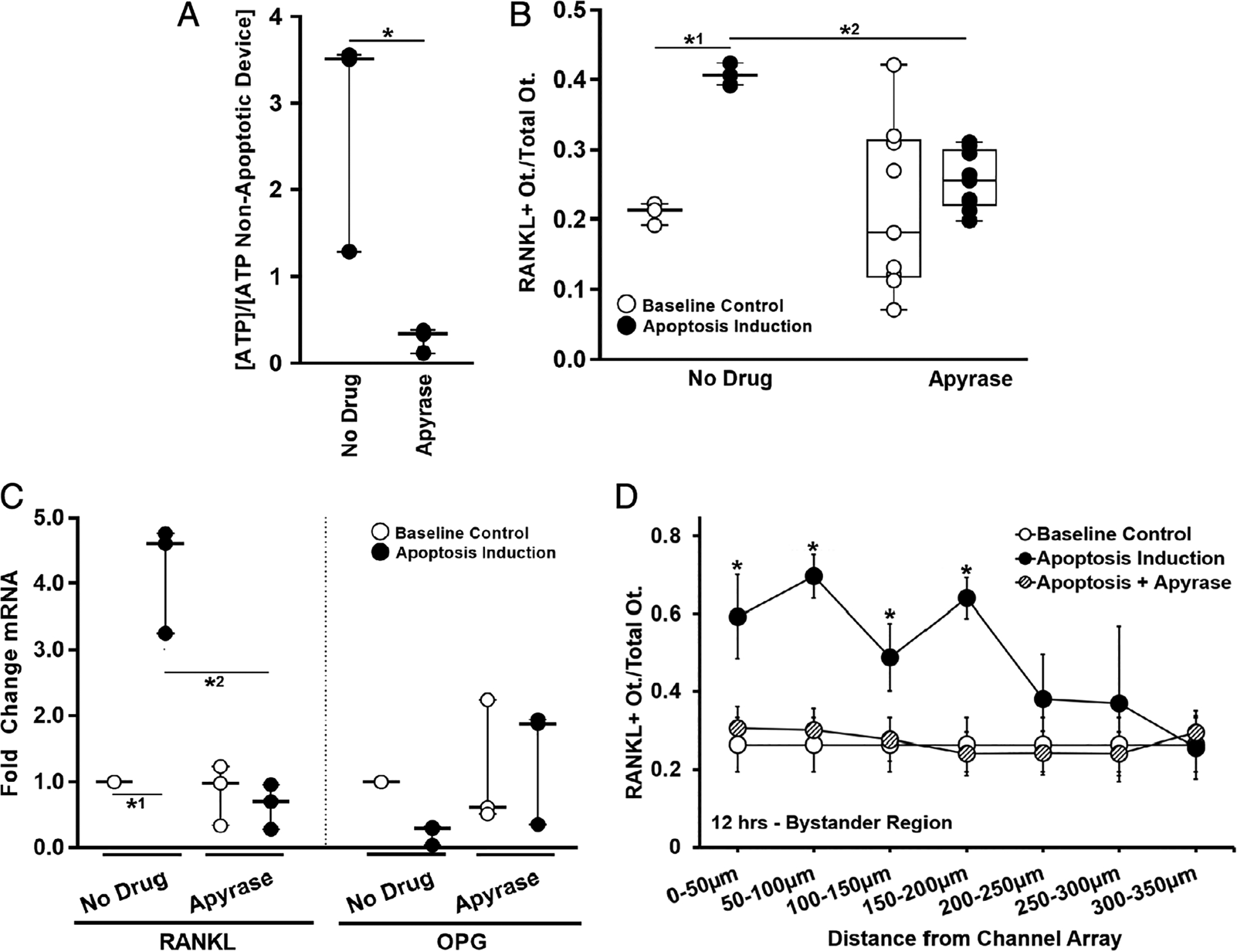
Effect of removing ATP on bystander osteocyte RANKL expression. (A) Induction of apoptosis induced sustained elevation in extracellular ATP in the Mμn (12 hours post-apoptosis induction, n = 3 devices, *p = .029); this increase was no longer seen when apyrase was added to the medium to hydrolyze the extracellular ATP. (B) The increased numbers of RANKL-expressing osteocytes expected after apoptosis induction (*1p = .031 versus control) was prevented by the addition of apyrase (p = .825 versus baseline control, *2p = .043 versus apoptotic control), n = 3 devices for control and n = 9 devices for apyrase treatment. (C) Scavenging the extracellular ATP with apyrase also prevented the changes in RANKL and OPG gene expression in bystander osteocytes caused by apoptosis of neighboring cells. (RANKL/apyrase p = .999 versus baseline control. *2p < .0001 versus no drug apoptotic control). n = 3 samples (three devices per sample), tested in triplicate. (D) Effect of apyrase on distribution of RANKL-expressing osteocytes as a function of the distance from the nanochannel array/apoptotic signal source, showing that hydrolyzing the ATP prevented increases in osteocyte RANKL expression through the entire bystander compartment (n = 9 devices, significance only for apoptosis induction-no apyrase versus baseline control [*p = .0001 at 0 to 50 μm, p = .0001 at 50 to 100 μm, p = .022 at 100 to 150 μm, and p < .0001 at 150 to 200 μm]). Data are shown as mean ± SD.
Apoptosis caused a significant (approximately twofold) increase in the number of RANKL-expressing osteocytes in the bystander compartment of the Mμn device at 6 and 12 hours (40.2% ± 8.9% at 12 hours post-apoptosis compared to 26.0% ± 7.8% at baseline) as shown in Fig. 3C. RANKL gene expression assessed by RT-PCR mirrored this increase in the bystander compartment and showed that OPG, the decoy receptor for RANKL, expression decreased significantly (Fig. 3D). Inhibition of osteocyte apoptosis in the stressed compartment prevented increase in RANKL-expressing osteocytes and RANKL gene expression in the bystander compartment (Fig. 3E). Finally, the number of cells expressing RANKL in the bystander compartment was highest close to the nanochannel array and declined with increasing distance from the array (Fig. 3F).
Apoptosis-induced RANKL expression in bystander osteocytes: the roles of pannexin 1/purinergic receptor signaling and gap junctions
Blocking Panx1 (probenecid and 10-panx1) after induction of osteocyte apoptosis prevented the expected increase in both RANKL-expressing cell numbers and gene expression in bystander osteocytes. Similarly, blocking the P2X7 receptor with BBG inhibited the upregulation of RANKL expression in bystander osteocytes. In contrast, blocking Cx43 channels did not alter the overall upregulation of RANKL expression in bystander osteocytes. However, AGA altered the distribution (spread) of RANKL-expressing osteocytes, as indicated by the significant difference in RANKL between control and treated osteocytes at distances >150 to 200 μm from the nanochannel array/apoptotic osteocytes. Data are summarized in Fig. 5A–C. It was also shown that elimination of extracellular ATP by apyrase inhibited RANKL upregulation in bystander osteocytes in our fluorescent reporter system and at the transcript level (Fig. 4B–D).
Fig. 5.
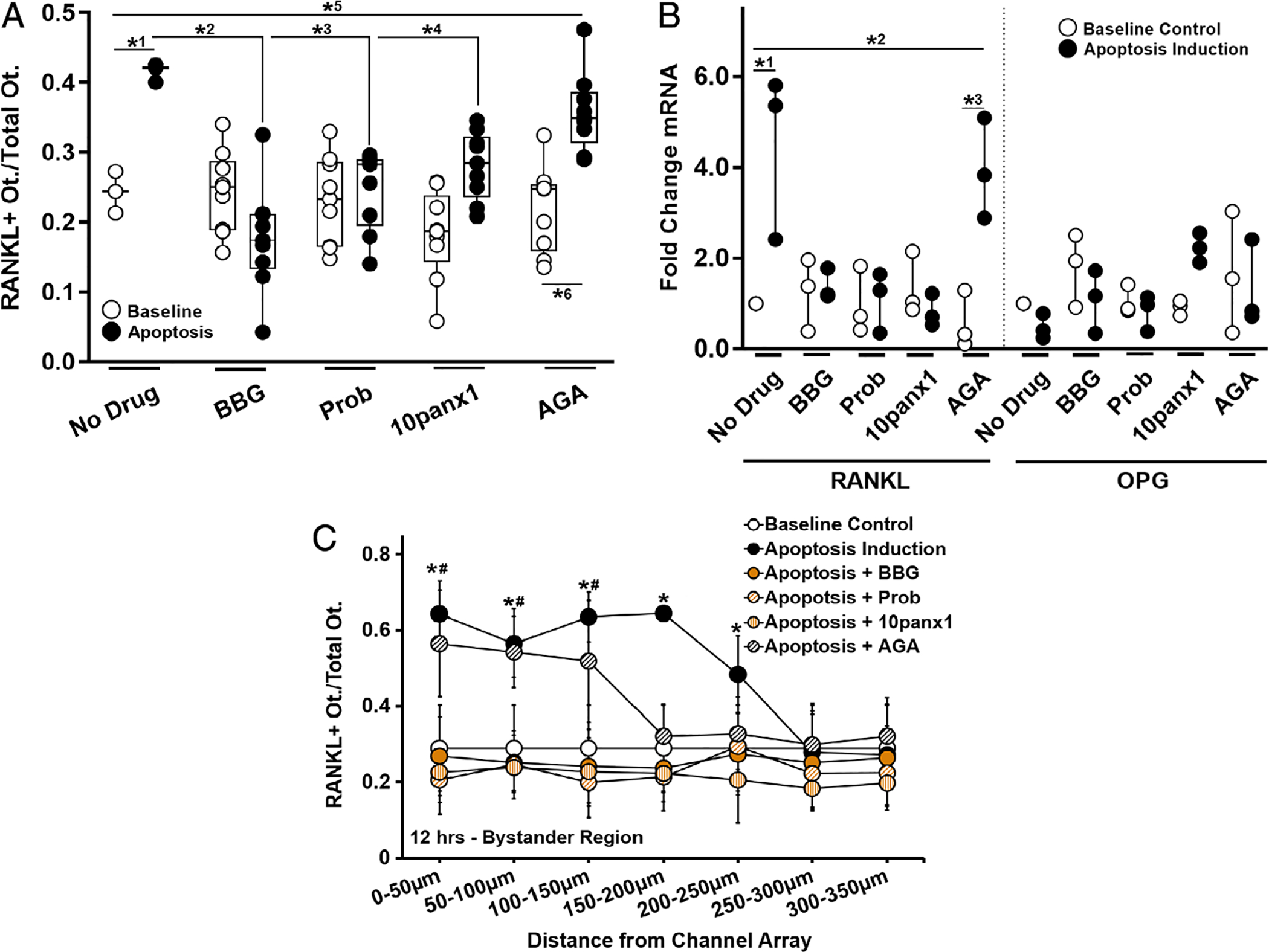
Effect of Panx1, P2X7, and Cx43 inhibition on bystander osteocyte RANKL expression. (A) Increased numbers of RANKL-expressing bystander osteocytes after apoptosis was induced in the neighboring chamber (*1p = .024) was prevented by inhibition of P2X7 by BBG (10μM) (*2p = .0001) or by blocking Panx1 using probenecid (Prob, 500μM) (*3p = .003) or biomimetic peptide 10panx1 (100μM) (*4p = .038). Blocking Cx43 (AGA, 30μM) did not prevent the increase in RANKL-expressing osteocytes in the bystander compartment (*5p = .008 versus baseline control and *6p < .001 versus AGA treated non-apoptotic control), n = 3 devices for controls and n = 9 devices for treatment groups. (B) The effects of channel inhibitors on RANKL and OPG mRNA expression in bystander osteocytes with and without apoptosis induction of apoptosis in the neighboring chamber. Increased in RANKL gene expression was blocked by BBG, Prob, and 10panx1 (all p = .999 versus baseline, but not by blocking Cx43 with AGA; *2p = .011 versus baseline control and *3p = .004 versus non-apoptotic AGA-treated group), n = 3 samples (three devices per sample), tested in triplicate. (C) The effects of channel inhibitors on the distribution of RANKL-expressing osteocytes as a function of the distance from the nanochannel array/apoptotic signal source. Note that whereas probenecid, 10panx1, and BBG prevented RANKL upregulation in all regions, blocking Cx43 (AGA) altered the extent of the spread from the nanochannel array/apoptotic signal source (n = 9 devices), and this effect was significant up to 150 to 200 μm away from the nanochannel array/apoptotic signal source. All other treatments did not differ from baseline control or among themselves. Apoptosis induction-no drug control versus baseline control (*p = .004 at 0 to 50 μm, p = .01 at 50 to 100 μm, p < .001 at 100 to 150 μm, p < .0001 at 150 to 200 μm, and p = .041 at 200 to 250 μm). Apoptosis induction–AGA-treated versus baseline control (#p < .001 at 0 to 50 μm, p < .0001 at 50 to 100 μm, and p = .002 at 100 to 150 μm). Data are shown as mean ± SD.
Bolus-type ATP stimulation triggers osteocyte RANKL expression in the absence of apoptosis
Addition of ATP to one Mμn compartment containing no cells caused upregulation of osteocyte expression of RANKL in the adjacent (bystander) compartment. RANKL-expressing osteocyte number was increased approximately twofold over baseline at 12 hours after ATP exposure (Fig. 6A,B). RANKL expression was not detectably increased at 1 hour after ATP exposure and was increased moderately at 6 hours (data not shown). Both 20nM and 1μM ATP yielded similar increases in RANKL-positive osteocytes, although RANKL expression was less pronounced at longer distance (250 to 300 μm) from the source at the lower concentration (Fig. 6C). RANKL gene expression was increased by ATP exposure (approximately fourfold versus unstimulated control) whereas OPG gene expression decreased more than 50% from baseline. Adenosine had no effect on either RANKL or OPG expression.
Fig. 6.
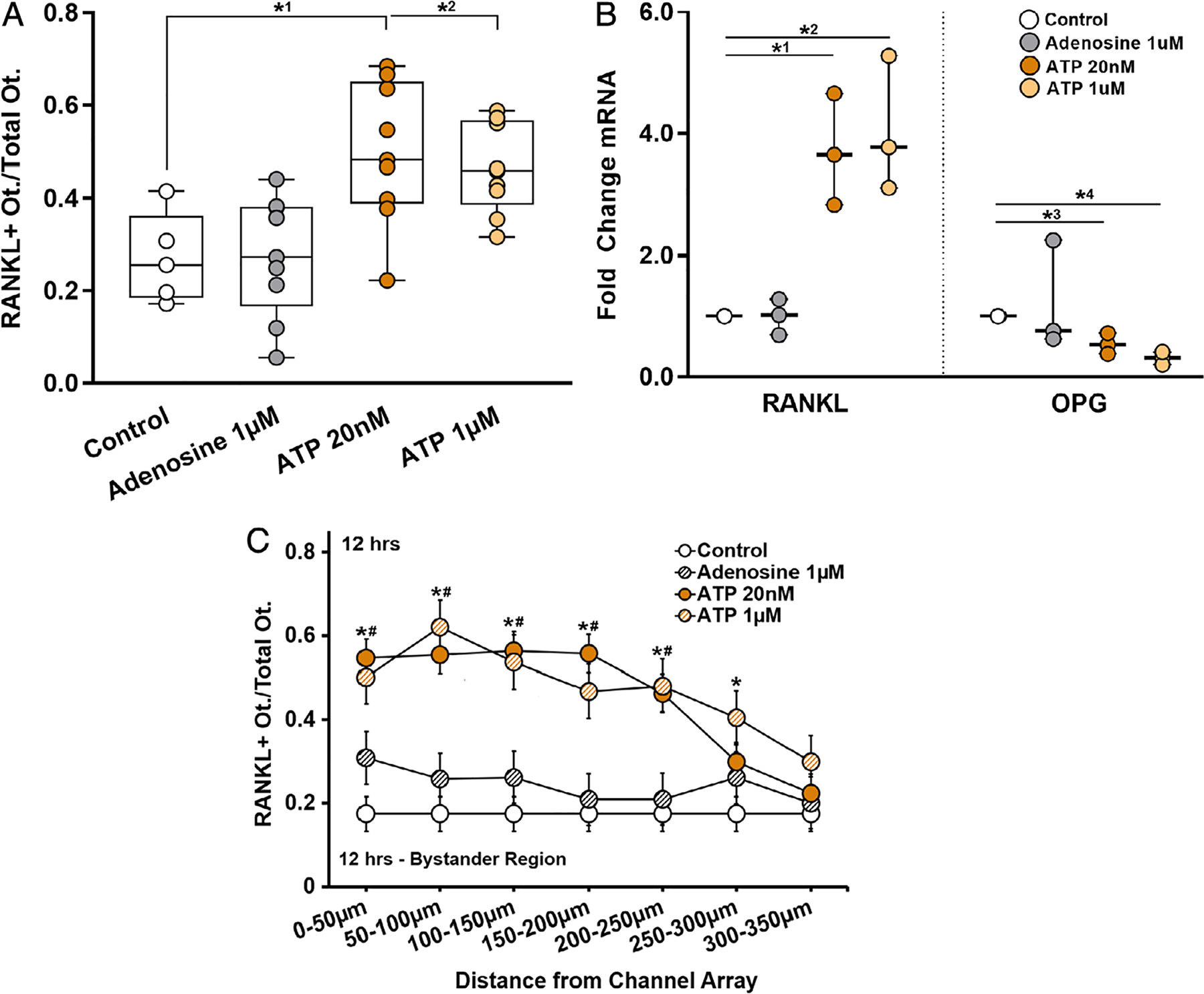
Purinergic stimulation induced changes on osteocyte RANKL expression. (A) ATP at 20nM and 1μM both similarly increased the number of RANKL-expressing osteocytes in the bystander compartment nearly twofold compared to control (*1p = .0133 and *2p = .044, respectively); adenosine treatment had no effect. n = 5 to 9 devices. (B) RANKL mRNA was markedly increased with exposure to ATP at 20nM or 1μM (*1p = .008 and *2p = .004) and OPG expression significantly decreased (*3p = .007 and *4p < .001, respectively), n = 3 samples (3 devices per sample), tested in triplicate; again adenosine had no effect. (C) The distribution of RANKL-expressing cells as a function of distance from the ATP source (nanochannel array) was similar to that observed when bystander osteocyte RANKL was triggered by apoptosis of neighboring cells; see Fig. 3. Both ATP 20nM* and 1μM# treatment groups were significantly different compared to untreated controls up to 300 μm from the ATP source (n = 9 devices), p = .001 at 0 to 50 μm, p = .0001 at 50 to 100 μm, p = .002 at 100 to 150 μm, p = .001 at 150 to 200 μm, p = .002 at 200 to 250 μm, and p = .011 at 250 to 300 μm). Adenosine had no effect. Data are shown as mean ± SD.
Discussion
The current studies show that apoptotic MLO-Y4 osteocytic cells in vitro activate RANKL production in bystander cells, analogous to behavior of osteocytes in vivo. Specifically, bystander signaling was dependent upon the apoptosis-dependent release of signal(s) through Panx1 channels and the activation of its P2 receptor. We also discovered that ATP, which is known to be released in large amounts through Panx1 channels, during apoptosis, is a key trigger to activate osteocyte RANKL expression, as evidenced by lack of RANKL upregulation post-apoptosis with extracellular ATP scavenging. Last, we found that blockade of gap junctions between osteocytes does not prevent bystander cell induction of RANKL expression, although it does reduce the extent of spread of bystander RANKL induction by cells in the neighboring chamber. This finding indicates that the Panx1/P2XR complex and gap junctions play complementary roles, with the former providing the mechanism whereby extracellular spread of ATP mediates the induction of the RANKL gene expression program triggered by dying cells, and the latter extending the intercellular spread of the osteoclastogenic activation signal. Such dual roles of gap junctions and Panx1 channels are prominent in the spread of intercellular Ca2+ waves(36) and in a variety of bystander phenomena.(37) Gap junctions are formed by the pairing of connexon subunits, or hemichannels, and unpaired hemichannels have been proposed to provide a route for ATP release that would support intercellular signaling, similar to Panx1 channels. Whether hemichannels contribute to the extent of bystander gene expression induction remains to be determined, but the pharmacological blockade of the spread of gene expression and apoptosis by specific inhibitors clearly establishes the major role played by Panx1 and P2 receptors in this phenomenon.
Bystander signaling is a well-established mechanism by which apoptotic cells recruit phagocytic cells such as macrophages to remove dying cells and often their associated matrix in nonskeletal tissues.(37) Dying cells have limited ability to produce large amounts of find-me signals for extended periods of time, making recruitment of phagocytic cells from remote sites problematic. Consequently, apoptotic cells also produce signals that induce non-apoptotic neighboring cells in the surrounding “penumbra” region to continue to produce phagocyte chemoattractants and other regulators as well. Bystander signaling greatly expands the capability, both in intensity and duration of the response, to recruit phagocytic cells to a given site and has been extensively investigated in cases of tissue repair due to focal injury, especially in cardiac and neural tissues.(37)
Growing evidence from in vivo studies supports the concept that bone resorption falls within the paradigm of bystander signaling. Resorption is carried out by specialized phagocytic cells (osteoclasts) and was shown to be dependent on the apoptosis of resident cells (osteocytes) in experimental models representing three major instances of bone remodeling: microdamage, estrogen loss, and disuse.(1–4) Moreover, Kennedy and colleagues(5) in our laboratory explicitly demonstrated that apoptosis-dependent upregulation of the essential pro-osteoclastogenic cytokine RANKL occurred in bystander osteocytes. Thus, bone appears to have co-opted this essential response cascade used by dying cells to signal their viable neighbors to “call for help”.(6–8)
Cheung and colleagues(6) provided a critical insight into the potential nature of the signals that come from apoptotic osteocytes to trigger RANKL production in bystander osteocytes. They found that the expected upregulation of RANKL in bystander osteocytes and activation of bone remodeling following fatigue did not occur in Panx1-deficient mice, although osteocyte apoptosis occurred normally. Pharmacological blocking of the purine receptor that typically associates with Panx1 similarly prevented the upregulation of osteocyte RANKL. These data suggested that release of purinergic signals via the Panx1 axis may be key to the activation of RANKL expression in bystander osteocytes, yet it was undetermined if ATP was the obligate ligand.
Pannexins are vertebrate membrane proteins related to the invertebrate innexins.(38) However, unlike the vertebrate family of connexin gap junction proteins, pannexins are non-junctional; ie, they are obligate hemichannels that do not couple with complementary channels on adjacent cells.(37) Pannexins have been shown to function as large transmembrane channels connecting the intracellular and extracellular space, allowing the passage of small molecules. In particular, Panx1 is the major channel responsible for ATP release from cells for both normal regulatory signaling and also for the large ATP release that occurs during apoptosis. There is a caspase-dependent terminal opening of Panx1 channels during apoptosis that results in a large bolus release of ATP from the dying cells.(13,39) This locally high ATP release is a core find-me signal to attract professional phagocytic cells (ie, macrophages) to the site of apoptosis to remove dying cells. This ATP also serves as a critical signal to activate or even cause additional cell death bystander cells.(37) In the current studies we found that acute ATP exposure turned on osteocyte RANKL expression, consistent with previous studies showing that ATP can stimulate RANKL expression.(40,41) We further discovered that with osteocyte apoptosis, ATP was the essential signal driving the increase in bystander osteocyte RANKL expression.
Defining the mechanisms of bystander signaling among osteocytes has posed substantial experimental difficulties, given the complex architectural environment of bone and its consequent constraints on osteocyte communication. Osteocytes in vivo live within an extremely spatially restricted environment. Communication among osteocytes is limited to gap junctions on their numerous cell processes and to paracrine signals that move through the tiny proteoglycan-filled pericellular fluid space around osteocyte cell bodies (pericellular space <1000 nm wide) and their processes (pericellular space <100 nm wide).(14) The model system used in this study was designed to replicate as effectively as possible the distinctive environment of osteocytes in vivo in addition to crucial elements of the osteocyte phenotype. Consequently, an initial goal of this study was to test whether the system could reproduce bystander signaling behavior that we had observed in previous in vivo studies.
The Mμn microfluidic culture device, which allowed osteocytic cells to communicate through nanochannels with dimensions similar to those of canaliculi in vivo, avoided limitations that rendered conventional monolayer or open multicompartment cell culture systems (eg, Boyden chamber, transwell plates) insufficient to model the essential relationships among osteocytes. Furthermore, the system enforced spatial separation of apoptotic and non-apoptotic osteocytes, with communication between the two populations limited only to gap junctions on dendritic processes or movement of extracellular signals through the small fluid spaces around processes in the channels, analogous to the situation in vivo. McCutcheon and colleagues(18) further showed that solute transport limits between compartments of the Mμn device are similar to those of the LCS in vivo. In this system, MLO-Y4 osteocytes grew dendritic processes into the channels and established junctional communication with cells on opposite sides of the channel array. Moreover, this model system was able to replicate essential features of bystander signaling between apoptotic and non-apoptotic osteocytes previously observed in vivo. Specifically, (i) osteocyte apoptosis in one location triggered upregulation of osteocyte RANKL expression within the opposite bystander compartment(27,28); and (ii) this triggering of osteocyte RANKL expression in non-apoptotic osteocytes by neighboring apoptotic osteocytes was dependent on presence of extracellular ATP, functional Panx1 channels, and the purinergic P2X7 receptor.(6) Having established that the compartmented design could replicate osteocyte behavior in vivo, we were able to show that RANKL upregulation in non-apoptotic osteocytes could be replicated by delivery of ATP alone in the absence of inducing (apoptotic) cells and to show that gap junctions did not play a major role in regulation osteocyte apoptosis or in the upregulation of RANKL in non-apoptotic bystander osteocytes.
We chose to use MLO-Y4 osteocyte-like cells in our studies for their well-established ability to elaborate consistent dendritic processes and networks with gap junctions, which is key to our model and studies. These cells can be reliably transfected and maintained throughout the experiments, which was essential for our RANKL reporter studies. Primary osteocytes could not be used for these studies because they do not divide and thus cannot be used for a stable reporter system. One difference between MLO-Y4 osteocytes and mature osteocytes in vivo is that some MLO-Y4 osteocytes can show high constitutive RANKL expression, whereas in mature osteocytes in vivo do not.(27,28) In the current studies, MLO-Y4 osteocytes were near confluence and thus little RANKL expression was detected. Moreover, the current studies tested for RANKL upregulation by measuring the number of cells strongly expressing this ligand. The increased number of RANKL-expressing osteocytes observed in response to apoptosis of their neighbors is consistent with the recruitment of bystander cells for the analogous situation of osteocyte apoptosis and response in vivo.
RANKL upregulation in bystander MLO-Y4 osteocytes replicated our previous findings for osteocytes in vivo, showing a complete dependency on apoptosis of cells in the opposing cell compartment as well as on both Panx1 and P2 receptors. The pan-caspase inhibitor QVD used to block apoptosis was the same as that used in vivo to demonstrate the requirement for apoptosis in initiating bone resorption in response to the three major remodeling stimuli: microdamage,(1,2,28) disuse,(4,42) and estrogen withdrawal.(3,43,44) Similarly, the approach we used to block P2X7R (the antagonist BBG) in this study was the same as used by us in vivo.(6) To confirm the dependence of RANKL upregulation upon Panx1, we used both probenecid and a Panx1-specific blocking peptide 10panx1 to block Panx1 in this study—with effectively identical results for both. Functionally, probenecid itself is considered to be a specific Panx1 blocker, but in theory might potentially also act on Cx43 hemichannels. That potential is mitigated by the use of the Panx1 inhibitory mimetic peptide 10panx1. Last, and critically, the absence of RANKL response when Panx1 channels were blocked indicates that this response is channel-dependent, and not due to a loss of cell membrane integrity of the dying osteocytes that would allow ATP and damage-associated molecular patterns (DAMPs) (eg, alarmins) to leak out of cells. Rather, our data support the idea that a “bolus”-type release of ATP from the terminal opening of Panx1 channels during apoptosis is key to activating bone resorption and remodeling.
Although inhibition of both Panx1 and P2X7R were able to block RANKL upregulation completely, inhibition of Cx43-based channels with AGA had little effect in RANKL upregulation. The only consequence of AGA inhibition was a more rapid decline in high RANKL-expressing cells with distance from the apoptotic cell compartment, suggesting that the principal role played by gap junction channels is on the spread of apoptosis-derived signal rather than in triggering RANKL response of bystanders. Indeed, previous studies revealed that the amount of osteoclastic resorption at bone microdamage sites correlated with the extent of osteocyte apoptosis and bystander RANKL signaling, which spreads for several hundred microns on either side of a microcrack—well beyond the few microns of bone that is actually damaged.(28) In focal tissue injury in brain, retina, and myocardium, the spread of apoptotic and surrounding cell responses in the “penumbra” around these in these focal injuries has been shown to be governed by Cx43-based gap junctions and/or hemichannels.(45–48) Given biological parallels between soft tissue focal injury and bone microdamage and the connectivity of osteocytes via gap junctions on their processes, we speculate that that gap junctions play a similar role in regulating the spread of the osteocyte apoptosis and in turn the related bystander signaling.
In summary, results of these studies point to extracellular ATP release from dying osteocytes via Panx1 channels and working through ATP receptor-gated (P2) channels as the major molecular signal for triggering bystander osteocytes to turn on RANKL expression. This, in turn, activates osteoclast recruitment and bone remodeling. That this RANKL response was prevented when Panx1 channels were blocked further indicates that this response is channel-dependent, and thus loss of cell membrane integrity in dying osteocytes is not implicated in this acute ATP release and the bystander osteocyte RANKL response it triggers. The precise molecular mechanisms through which ATP can activate osteocyte RANKL signaling is not yet known and requires further study. In a broader biological context, the large ATP release that occurs during apoptosis provides a critical find-me signal that activates and attracts to sites of dying cells and activates neighboring bystander cells to coordinate other local cell responses. This activation of bystander cell signaling is a well-established mechanism by which acutely released signals from apoptotic cells (ie, ATP) trigger surviving neighbors to produce the regulatory molecular mechanism needed to recruit phagocytic cells. The results of the current studies suggest that bone appears to rely on this fundamental mechanism to activate osteoclastic resorption, such that bystander osteocytes are triggered to turn on RANKL expression in response to acute ATP release from nearby apoptotic osteocytes. Those bystander osteocytes, in turn, produce the RANKL responsible for recruiting specialized phagocytic cells, ie, osteoclasts, that target bone tissue and dying cells within for resorption.
Acknowledgments
This study was supported by grants from the National Institute of Arthritis, Musculoskeletal and Skin Diseases (AR041210, AR070547) and National Institute of Aging (AG056397), the National Eye Institute (EY026752), the National Science Foundation (CBET0939511), and the CUNY Advance Science Research Center. We thank Tanya Singh (CCNY) for technical assistance with these studies and Dr. Randy Stout for guidance on reporter plasmid development.
Footnotes
Disclosures
The authors have no conflicts to disclose.
References
- 1.Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res. 2000;15:60–7. [DOI] [PubMed] [Google Scholar]
- 2.Noble B, Alini M, Richards R. Bone microdamage and cell apoptosis. Eur Cell Mater. 2003;6:46–55. [DOI] [PubMed] [Google Scholar]
- 3.Tomkinson A, Gevers E, Wit J, Reeve J, Noble B. The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res. 1998;13:1243–50. [DOI] [PubMed] [Google Scholar]
- 4.Bakker A, Klein-Nulend J, Burger E. Shear stress inhibits while disuse promotes osteocyte apoptosis. Biochem Biophys Res Commun. 2004;320:1163–8. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50:1115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung WY, Fritton JC, Morgan SA, et al. Pannexin-1 and P2X7-receptor are required for apoptotic osteocytes in fatigued bone to trigger RANKL production in neighboring bystander osteocytes. J Bone Miner Res. 2016;31:890–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB. Osteocyte apoptosis controls activation of Intracortical Resorption in response to bone fatigue. J Bone Miner Res. 2009;24:597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong J, O’Brien CA. Osteocyte RANKL: new insights into the control of bone remodeling. J Bone Miner Res. 2012;27:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peter C, Waibel M, Radu CG, et al. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem. 2008;283:5296–305. [DOI] [PubMed] [Google Scholar]
- 12.Yanguas SC, Willebrords J, Johnstone SR, et al. Pannexin1 as mediator of inflammation and cell death. Biochim Biophys Acta Mol Cell Res. 2017;1864:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chekeni FB, Elliott MR, Sandilos JK, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaffler MB, Cheung W-Y, Majeska R, Kennedy O. Osteocytes: master orchestrators of bone. Calcif Tissue Int. 2014;94:5–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosaku K, J HT, Hidehiko H, Kalervo VH. Bone marrow cell differentiation induced by mechanically damaged osteocytes in 3D gel-embedded culture. J Bone Miner Res. 2006;21:616–25. [DOI] [PubMed] [Google Scholar]
- 16.Mulcahy LE, Taylor D, Lee TC, Duffy GP. RANKL and OPG activity is regulated by injury size in networks of osteocyte-like cells. Bone. 2011;48:182–8. [DOI] [PubMed] [Google Scholar]
- 17.Anderson EJ, Knothe Tate ML. Idealization of pericellular fluid space geometry and dimension results in a profound underprediction of nano-microscale stresses imparted by fluid drag on osteocytes. J Biomech. 2008;41:1736–46. [DOI] [PubMed] [Google Scholar]
- 18.McCutcheon S, Majeska R, Schaffler M, Vazquez M. A multiscale fluidic device for the study of dendrite-mediated cell to cell communication. Biomed Microdevices. 2017;19:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burger EH, Klein-Nulend J. Mechanotransduction in bone-role of the lacuno-canalicular network. FASEB J. 1999;13(Suppl):S101–12. [PubMed] [Google Scholar]
- 20.You LD, Weinbaum S, Cowin SC, Schaffler MB. Ultrastructure of the osteocyte process and its pericellular matrix. Anat Rec A Discov Mol Cell Evol Biol. 2004;278:505–13. [DOI] [PubMed] [Google Scholar]
- 21.Zhao S, Zhang YK, Harris S, Ahuja SS, Bonewald LF. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J Bone Miner Res. 2002;17:2068–79. [DOI] [PubMed] [Google Scholar]
- 22.Yellowley CE, Li Z, Zhou Z, Jacobs CR, Donahue HJ. Functional gap junctions between osteocytic and osteoblastic cells. J Bone Miner Res. 2000;15:209–17. [DOI] [PubMed] [Google Scholar]
- 23.Dolan E, Haugh M, Tallon D, Casey C, McNamara L. Heat-shock-induced cellular responses to temperature elevations occurring during orthopaedic cutting. J R Soc Interface. 2012;9:3503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dolan EB, Haugh MG, Voisin MC, Tallon D, McNamara LM. Thermally induced osteocyte damage initiates a remodelling signaling cascade. PLoS One. 2015;10:e0119652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McStay G, Salvesen G, Green D. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15:322. [DOI] [PubMed] [Google Scholar]
- 26.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026. [DOI] [PubMed] [Google Scholar]
- 27.Kennedy OD, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone. 2014;64:132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy OD, Lendhey M, Mauer P, Philip A, Basta-Pljakic J, Schaffler MB. Microdamage induced by in vivo Reference Point Indentation in mice is repaired by osteocyte-apoptosis mediated remodeling. Bone. 2017;95:192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyce BF, Xing L. The Rankl/rank/Opg pathway. Curr Osteoporos Rep. 2007;5:98–104. [DOI] [PubMed] [Google Scholar]
- 30.Tsujita Y, Muraski J, Shiraishi I, et al. Nuclear targeting of Akt antagonizes aspects of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2006;103:11946–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silverman W, Locovei S, Dahl G. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am J Physiol Cell Physiol. 2008;295:C761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iglesias R, Locovei S, Roque A, et al. P2X7 receptor-Pannexin1 complex: pharmacology and signaling. Am J Physiol Cell Physiol. 2008;295(3):C752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang L-H, Mackenzie AB, North RA, Surprenant A. Brilliant blue G selectively blocks ATP-gated rat P2X(7) receptors. Mol Pharmacol. 2000;58:82. [PubMed] [Google Scholar]
- 35.Sandilos JK, Chiu YH, Chekeni FB, et al. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012;287:11303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54:716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spray DC, Hanstein R, Lopez-Quintero SV, Stout RF Jr, Suadicani SO, Thi MM. Gap junctions and bystander effects: good Samaritans and executioners. Wiley Interdiscip Rev Membr Transp Signal. 2013;2:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baranova A, Ivanov D, Petrash N, et al. The mammalian pannexin family is homologous to the invertebrate innexin gap junction proteins. Genomics. 2004;83:706–16. [DOI] [PubMed] [Google Scholar]
- 39.Medina C, Ravichandran K. Do not let death do us part:’find-me’ signals in communication between dying cells and the phagocytes. Cell Death Differ. 2016;23:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallagher J. ATP P2 receptors and regulation of bone effector cells. J Musculoskelet Neuronal Interact. 2004;4:125. [PubMed] [Google Scholar]
- 41.Komori T. Cell death in chondrocytes, osteoblasts, and osteocytes. Int J Mol Sci. 2016;17:2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cabahug-Zuckerman P, Frikha-Benayed D, Majeska RJ, et al. Osteocyte apoptosis caused by hindlimb unloading is required to trigger osteocyte RANKL production and subsequent resorption of cortical and trabecular bone in mice femurs. J Bone Miner Res. 2016;31:1356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kameda T, Mano H, Yuasa T, et al. Estrogen inhibits bone resorption by directly inducing apoptosis of the bone-resorbing osteoclasts. J Exp Med. 1997;186:489–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emerton K, Hu B, Woo AA, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2010;46:577–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chew SS, Johnson CS, Green CR, Danesh-Meyer HV. Role of connexin43 in central nervous system injury. Exp Neurol. 2010;225:250–61. [DOI] [PubMed] [Google Scholar]
- 46.Davidson JO, Drury PP, Green CR, Nicholson LF, Bennet L, Gunn AJ. Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS One. 2014;9:e96558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.García-Dorado D, Rodríguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia–reperfusion. Cardiovasc Res. 2004;61:386–401. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Ma A, Zhu W, et al. The role of connexin 43 and hemichannels correlated with the astrocytic death following ischemia/reperfusion insult. Cell Mol Neurobiol. 2013;33:401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]