

Abstract
Although palladium-catalyzed cross-coupling of aryl halides and reactive pseudohalides has revolutionized the way organic molecules are constructed today across various fields of chemistry, comparatively less progress has been made in the palladium-catalyzed cross-coupling of less reactive C–O electrophiles. This is despite the fact that the use of phenols and phenol derivatives as bench-stable cross-coupling partners has been well-recognized to bring about major advantages over aryl halides, such as (1) natural abundance of phenols, (2) avoidance of toxic halides, (3) orthogonal cross-coupling conditions, (4) prefunctionalization of phenolic substrates by electrophilic substitution or C–H functionalization, (5) ready availability of phenols from a different pool of precursors than aryl halides. In this review, we present an overview of recent advances made in the field of palladium-catalyzed cross-coupling of C–O electrophiles with a focus on (1) catalytic systems, (2) reaction type, and (3) class of C–O coupling partners. Although the field has been historically dominated by nickel catalysis, it is now evident that the use of more versatile, more functional group tolerant and highly active palladium catalysts supported by appropriately designed ancillary ligands enables the cross-coupling with improved substrate scope and generality, and likely represents a practical solution to the broadly applicable cross-coupling of various C–O bonds across diverse chemical disciplines. The review covers the period through June 2020.
Graphical Abstract

We present an overview of recent advances made in the field of palladium-catalyzed cross-coupling of C–O electrophiles with a focus on (1) catalytic systems, (2) reaction type, and (3) class of C–O coupling partners. Although the field has been historically dominated by nickel catalysis, it is now evident that the use of more versatile, more functional group tolerant and highly active palladium catalysts likely represents a practical solution to the broadly applicable cross-coupling of various C–O bonds across diverse chemical disciplines.
1. Introduction
Transition-metal-catalyzed cross-couplings have transformed the arsenal of catalytic technologies accessible to practitioners of organic synthesis.1,2 The 2010 Nobel Prize in Chemistry well highlighted the practical value of Pd-catalyzed cross-couplings, applications of which have spurred long-lasting advances in the fields ranging from biotechnology and medicine to plastics, advanced materials and electronic devices, which are now present in our everyday life.3,4
In these reactions, aryl halides represent the most common class of electrophiles. Notable advances in the design of electron-rich, bulky ligands, such as trialkylphosphines, dialkylbiarylphosphines, and NHCs (NHC = N-heterocyclic carbene), enable now routine cross-coupling of less reactive aryl chlorides (cf. bromides or iodides).1–4 In contrast, routine application of highly desirable C–O electrophiles in palladium-catalyzed cross-couplings is predominantly limited to the highly active, yet unstable and expensive, triflates. This is despite the fact that there are several major benefits of using phenols and derivates as cross-coupling partners over conventional aryl halides, such as (1) phenols are naturally abundant, (2) toxic halides are avoided, (3) cross-coupling conditions are often orthogonal, (4) there is significant potential for prefunctionalization of phenolic substrates by electrophilic substitution or C–H functionalization, (5) phenols are readily available from a different pool of precursors than aryl halides,5 which together with other emerging methods permit for a major expansion of the cross-coupling toolbox for practitioners of organic synthesis.6–8
Pioneered by Wenkert and co-workers in the late 70s, the cross-coupling of less reactive C–O electrophiles has been dominated by nickel catalysis, with remarkable contributions by several research groups.6 Among many attractive classes of less reactive C–O electrophiles in cross-coupling, such as (1) sulfonates, (2) sulfamates, (3) phosphates, (4) esters, (5) ethers, and (6) phenols, that have been accomplished with Ni-catalysis, Pd-catalyzed cross-couplings are limited to sulfonates, phosphates and sulfamates, with examples of Pd-catalyzed cross-couplings of esters, ethers and phenols yet to be reported.1–4 Since this relative reactivity trend is closely related to the ease of oxidative addition of the C–O bond,9 the use of electron-rich ligands on palladium enables the cross-coupling of less reactive C–O electrophiles, which in some cases already matches the reactivity of Ni catalysts, while providing much superior functional group tolerance.1–4,6,8,9
The recent studies make it evident that the use of more versatile, more functional group tolerant and highly active palladium catalysts supported by appropriately designed ancillary ligands permit the cross-coupling of less reactive C–O electrophiles with improved substrate scope and generality, and likely represents a practical solution to the broadly applicable cross-coupling of various C–O bonds across diverse chemical disciplines.
In this review, we present an overview of recent advances made in the field of palladium-catalyzed cross-coupling of C–O electrophiles with a focus on (1) catalytic systems, (2) reaction type, and (3) class of C–O coupling partners. Throughout the review, the most representative or the most general examples are discussed in more detail. The cross-coupling of triflates is not covered as are not Kumada cross-couplings because these methods involve highly reactive and less air- and moisture-stable electrophiles and/or nucleophiles and have been the topic of previous reviews.10 Note that Negishi cross-couplings feature significantly higher functional group tolerance and have been included in the review.1–3 The review covers the period through June 2020. We hope that this review will stimulate future progress in the development of catalytic systems and application of versatile Pd-catalyzed cross-coupling reactions of less reactive C–O electrophiles by a range of interested chemists.
At the same time, several potential areas for future improvements in the C–O cross-coupling methodology can be noted, including (1) the advantage of in situ activation protocols of phenols and alcohols to obviate a separate synthetic step (cf. commercially available halides), (2) in some cases generation of large molecular weight side products that might not be soluble, and thus easily removed by aqueous wash, (3) some phenols are synthesized from diazonium salts, which could open up new opportunities for the use of lignin as a renewable source of phenols, (4) some C–O activators, e.g. SO2F2 have potential toxicity concerns.
Figure 1 summarizes development timeline that can be compared with the evolution of Pd-catalyzed cross-coupling of halides,2,3 catalytic cycle, and classes of C–O electrophiles that thus far have proved amenable to efficient Pd-catalyzed cross-couplings as well as the relative order of reactivity of C–O electrophiles. In general, the development of Pd-catalyzed C–O cross-couplings has been achieved by two major directions: (1) ancillary ligand development, and (2) design of more reactive yet stable C–O electrophiles. The relative order of reactivity is closely related to the ease of oxidative insertion of Pd(0) to the C–O bond.9 The reactivity order has been typically determined by competition studies. It should also be noted that some catalytic systems are limited to activated electrophiles that feature electron-withdrawing groups on the aryl electrophile, facilitating oxidative addition of Pd to C–O bonds. Figure 2 presents major classes of ligands that have enabled Pd-catalyzed cross-coupling of C–O electrophiles.
Figure 1.

(A) Pd-catalyzed cross-coupling of C–O electrophiles; (B) Catalytic cycle; (C) Relative order of reactivity of C–O electrophiles amenable to Pd-catalyzed cross-coupling protocols.
Figure 2.

Structures of ancillary ligands frequently used in the Pd-catalyzed cross-coupling of bench-stable C–O electrophiles.
2. Palladium-Catalyzed Suzuki–Miyaura Couplings of C–O Electrophiles
Since its discovery in 1979, Suzuki-Miyaura cross-coupling has witnessed an astounding progress and this technology has now become one of the most frequently executed synthetic methods due to its broad functional group tolerance, mild reaction conditions, operational simplicity, high stability, low toxicity, and commercially availability of organoboron reagents and catalysts.11–12
In 1995, during their study of metal-catalyzed homocoupling reactions of aryl mesylates, Percec and co-workers disclosed the first example of a palladium-catalyzed Suzuki-Miyaura cross-coupling of mesylates and arenesulfonates (Scheme 1).13 Although relatively low yields of activated mesylates and p-fluorobenzensulfonates were obtained, this study is very significant because it showed for the first time that Pd catalysis supported by simple bidentate phosphines, such as dppf and dppp can be used to activate bench-stable mesylates and arenesulfonates as reactive C–O electrophiles in the Suzuki-Miyaura cross-coupling.
Scheme 1.

Pd-catalyzed Suzuki–Miyaura cross-coupling of activated mesylates and arenesulfonates reported by Percec in 1995.
2.1. Suzuki–Miyaura Cross-Couplings of Aryl Mesylates
Subsequently, major progress in the cross-coupling of aryl mesylates has been achieved through ancillary ligand design. It is worth noting that mesylates are among the most desirable sulfonate derivatives in the Pd-catalyzed cross-coupling of activated C–O electrophiles due to high-atom economy as well as cheap and benign activating group.
In 2008, Kwong and co-workers reported a general protocol for the cross-coupling of aryl mesylates and organoboron nucleophiles using Pd(OAc)2 as a palladium source and their electron-rich, indole-based CM-Phos as a ligand (Scheme 2A).14 Under the optimized conditions, both unactivated and activated aryl mesylates as well as heteroaryl mesylates could be successfully used as cross-coupling partners. Furthermore, common organoboron nucleophiles, such as arylboronic acids, pinacolboronic esters, and trifluoroborate salts afforded the cross-coupled products in high yields. A noteworthy feature of the Kwong’s protocol is broad functional group tolerance in that functional groups such as formyl, ketone, ester, cyanide, some of which that could be problematic using Ni catalysis,1–4,6,8,9 were readily compatible during the course of the C–O cross-couplings.
Scheme 2.

Pd-catalyzed Suzuki–Miyaura cross-coupling of aryl mesylates reported by (A) Kwong, and (B) Buchwald.
One year later, Buchwald and co-workers achieved a similar cross-coupling of aryl mesylates with boronic acids using their dialkylbiarylphosphine ligand BrettPhos instead of CM-Phos (Scheme 2B). The combination of K3PO4 and tBuOH was found to be the most effective in terms of base and solvent, respectively.15 Interestingly, t-AmOH afforded comparable results to t-BuOH, and was opted as the optimal solvent due to its higher boiling point. In terms of scope, the protocols by Kwong and Buchwald are complementary and both should be screened when optimizing the Pd-catalyzed Suzuki-Miyaura cross-coupling of aryl mesylates.
Further examples of Pd-catalyzed Suzuki–Miyaura cross-coupling of aryl mesylates using CM-Phos, XPhos, benzimidazolyl phosphine and SPhos based catalyst systems have also been reported16–19 Importantly, the hemilabile phosphine ligand PCy-PhMezole-Phos based on benzimidazolyl scaffold (cf. indolyl) developed by Kwong and co-workers was more effective than its analogue CM-Phos, permitting the cross-coupling at 0.5 mol% palladium loading.18
In addition, Shao and co-workers reported a NHC-based system using their imidazole-supported [(IPr)Pd(1-Me-im)Cl2] catalyst for the cross-coupling of aryl mesylates.20 In particular this system represented the first example in which a well-defined Pd–NHC complex was utilized as catalyst for the Suzuki-Miyaura cross-coupling of aryl sulfonates. The synthesis of biaryls was achieved by the coupling of aryl mesylates and boronic acids in morpholine at 120 °C in the presence of K3PO4. This catalyst system was also extended to the Suzuki cross-coupling of aryl tosylates and 4-fluorobenzenesulfonates.
2.2. Suzuki–Miyaura Cross-Couplings of Aryl Tosylates
Although tosylates are less atom economic than mesylates, historically, their use is more established in organic synthesis and catalysis due to ease of preparation and favourable crystalline properties of the products. It was not until 2003 that the first general palladium-catalyzed Suzuki-Miyaura of aryl tosylates was reported by Buchwald and co-workers utilizing Pd(OAc)2 and XPhos as the catalyst system (Scheme 3A).21 A wide range of aryl, heteroaryl tosylates were compatible with this protocol and enabled the cross-coupling with various aryl and heteroaryl boronic acids, including extremely hindered 2,4,6-triisopropylphenylboronic acid. However, the cross-coupling of electron-rich tosylates was not explored.
Scheme 3.

Pd-catalyzed Suzuki–Miyaura cross-coupling of tosylates reported by (A) Buchwald, (B) Lakshman, (C) Skrydstrup, and (D) Fu.
In 2004, Lakshman and co-workers reported the cross-coupling of 2’-deoxyguanosine O6-tosylates nucleosides with arylboronic acids to obtain 2-amino-6-arylpurine 2’-deoxynucleosides using Pd(PPh3)4 as a catalyst (Scheme 3B).22 The very mild conditions of this protocol provide a clear advantage of using Pd over other metals in the cross-coupling of medicinally-relevant nucleosides by C–O bond disconnection.
In 2008, Skrydstrup and co-workers disclosed general reaction conditions for the Suzuki–Miyaura coupling of vinyl tosylates to provide styrene derivatives (Scheme 3C).23 The key to the success of this reaction was using electron-rich ferrocenyl-based palladacycle precatalyst, SK-CC02-A (nor = norbornyl). With this method, a gem-difluorovinyl group could also be installed using 2,2-difluorovinyl tosylate (F2C=CH-OTs) as the electrophilic partner.
Despite the great advances in the Suzuki-Miyaura cross-coupling in C(sp2)–C(sp2) bond formation, utilizing this reaction to construct C(sp3)–C(sp3) bonds has been rarely reported due to facile β-hydride elimination. In 2002, Fu and co-workers reported that Suzuki-Miyaura coupling of alkyl sulfonates could be achieved by employing a combination of Pd(OAc)2 and Pt-Bu2Me (Scheme 3D).24 A wide range of alkyl tosylates featuring acetals, silyl ethers, esters, tertiary amides, ketones, nitriles, olefins and tertiary alcohols were coupled with alkyl or aryl 9-BBN in good yields. This important study demonstrated the viability of Pd-catalyzed C–O cross-coupling of alkyl sulfonates to construct not only biaryls but also C(sp3)–C(sp3) bonds. Studies on the mechanism showed inversion of configuration during oxidative addition, while no change of configuration occurred during reductive elimination.
Other examples Pd-catalyzed Suzuki-Miyaura cross-coupling of aryl tosylates using dialkylbiarylphosphines, such as BrettPhos, SPhos, CM-Phos, XPhos, DavePhos and trialkylphoshines, such as PCy3 have been reported.15,25–41 In particular, in comparison with XPhos, BrettPhos showed improved performance as substrates which were not compatible with XPhos were well-tolerated using BrettPhos, such as electron-rich aryl tosylates, vinyl tosylates, 2-heteroarylboronic acids, alkyl- and vinylboronic acids.15
In addition, NHC-systems with morpholine as a throw-away ligand, and indanone-based systems have also been reported as efficient catalysts for the Suzuki-Miyaura cross-coupling of aryl tosylates.42–44
2.3. Suzuki–Miyaura Cross-Couplings of Various Aryl Sulfonates
In addition to mesyates and tosylates, several other arenesulfonates have been employed as coupling partners in the Pd-catalyzed Suzuki-Miyaura cross-coupling of C–O electrophiles, including benzenesulfonates (–SO2Ph),45 nosylates (–SO2C6H4-p-NO2),46 and pentafluorobenzene-sulfonates (–SO2C6F5, PFB). The most useful are pentafluorobenzenesulfonates, which due to the electronic activation of the arene permit for exceptionally mild cross-coupling compatible with numerous functional groups using Pd(PPh3)2Cl2 catalyst system at room temperature with ample substrate scope as demonstrated by Shashikanth (Scheme 4).47
Scheme 4.

Pd-catalyzed Suzuki–Miyaura cross-coupling of aryl pentafluorobenzenesulfonates reported by Shashikanth.
2.4. Suzuki–Miyaura Cross-Couplings of Aryl Fluorosulfonates and Aryl Nonaflates
Aryl fluorosulfonates (–SO2F) represent an attractive emerging class of electrophilic coupling partners that recently have attracted major attention in metal-catalyzed cross-couplings due to high atom-economy and high reactivity owing to the electronegativity of fluorine atom.48–52
In 2015, Hanley and co-workers reported a detailed study of the Suzuki–Miyaura cross-coupling of aryl fluorosulfonates with boronic acid using both nickel and palladium catalyst systems (Scheme 5).53 With Pd(OAc)2 and inexpensive PPh3 as a catalyst system, aryl fluorosulfonates featuring both electron-donating and electron-withdrawing groups were smoothly cross-coupled with aryl and heteroaryl boronic acids under mild conditions at 60 °C in good to excellent yields. The authors conducted a series of intermolecular competition experiments to elucidate the relative order of reactivity of aryl fluorosulfonates and other common electrophiles. They found the following order of reactivity under their reaction conditions: I > Br > OTf ≅ OFs ≫ Cl, OTs, OMs. Thus, the reactivity of fluorosulfonates is similar to that of triflates, while the fluorosulfonate group presents several advantages, such as higher stability to hydrolysis, availability in bulk of the activating sulfuryl fluoride (SO2F2), mild and economic conditions for activation of phenols.
Scheme 5.

Pd-catalyzed Suzuki–Miyaura cross-coupling of aryl fluorosulfonates reported by Hanley.
Other catalyst systems have been reported for the Pd-catalyzed Suzuki-Miyaura cross-coupling of aryl fluorosulfonates, including ligand-free conditions,54–56 and [Pd-PEPPSI-IPr] for the assembly of polysubstituted pyridines.57
In 2016, Kumar and co-workers reported palladium catalyzed the Suzuki-Miyaura cross-coupling of 4-methyl-7-nonafluorobutyl-sulfonyloxy coumarins under microwave irradiation conditions (Scheme 6).58 Bidentate dppp was found to be the optimal ligand due to moderate bite angle, while TBAF·3H2O was identified as the key base additive serving also as a solvating agent. Both aryl and heteroaryl boronic acid gave the desired biaryl products in excellent yields.
Scheme 6.

Pd-catalyzed Suzuki–Miyaura cross-coupling of aryl nonaflates reported by Kumar.
Other examples of Pd-catalyzed Suzuki-Miyaura cross-coupling of nonaflates using SPhos, PPh3 and ligandless conditions have been reported.59–61
It is worthwhile to note that the use of nonaflates is advantageous over triflates due to significantly higher hydrolytic stability; typically, the substrates can be readily purified by flash chromatography, while the perfluoroalkyl activating group permits for more facile oxidative addition of Pd(0) to the C–O bond.9
2.5. Suzuki–Miyaura Cross-Couplings of Aryl Imidazolyl Sulfonates
Imidazolylsulfonates represent an attractive development in using benign, air- and hydrolytically-stable sulfonyl-activating groups for the cross-coupling of C–O electrophiles.
In 2009, Albaneze-Walker and co-workers reported the first Suzuki-Miyaura cross-coupling of aryl imidazolylsulfonates (Scheme 7).62 They found that the combination of Pd(dppf)Cl2 and DMAc gave the best results at 60 °C, however, several other catalysts and solvents were also capable of promoting the cross-coupling in comparable yields. Importantly, they established that the reactivity of imidazolylsulfonates was similarto that of triflates, while tosylates were recovered unchanged under their reaction conditions. However, the scope of this coupling was limited to a few examples.
Scheme 7.

Pd-catalyzed Suzuki–Miyaura cross-coupling of aryl imidazolylsulfonates reported by Albaneze-Walker.
Furthermore, Nájera and co-workers reported Suzuki-Miyaura cross-coupling of aryl imidazolylsulfonates with aryl and alkenylpotassium trifluoroborates using their oxime-derived palladacycle catalysts under phosphine-free conditions.63–65 Under their conditions, aryl imidazolylsulfonates featuring electron-donating, electron-withdrawing and sterically hindered substituents were successfully coupled in water.65
2.6. Suzuki–Miyaura Cross-Couplings of Aryl Sulfamates
Palladium-catalyzed Suzuki-Miyaura cross-coupling of aryl sulfamates at room temperature has been achieved by Hazari and co-workers using XPhos-ligated precatalyst based on (η3-1-t-Bu-ind)Pd(L)Cl scaffold developed by their group (Scheme 8).66 A mixture of toluene and methanol was found to be the optimal solvent mixture due to higher solubility of boronic acid and base as well as the role of methanol in precatalyst activation to Pd(0). Under the developed conditions, naphthyl, phenyl and substituted phenyl sulfamates were successfully reacted with a variety of boronic acids to afford biaryl products in excellent yields. The high reactivity of the 1-t-Bu-ind supported precatalyst could be a result of preventing the formation of off-cycle Pd(I) allyl dimers.66
Scheme 8.

Pd-catalyzed Suzuki–Miyaura cross-coupling of aryl sulfamates reported by Hazari.
Further examples of Pd-catalyzed Suzuki-Miyaura cross-coupling of aryl sulfamates using Buchwald’s 2nd generation palladacycles, XPhos-Pd-G2, and NHC-Pd catalysts with morpholine and imidazole throw-away ligands have been reported.67–69 It is worth noting that the 1-t-Bu-ind supported precatalyst Pd-XPhos66 appears to outperform the palladacycle XPhos-Pd-G2 in terms of substrate scope.67
2.7. Suzuki–Miyaura Cross-Coupling of Vinyl Phosphates and Aryl Phosphonium Salts
As early as in 1999, Coudert and co-workers disclosed Pd(PPh3)4-catalyzed Suzuki-Miyaura cross-coupling of vinyl phosphates in DME and water under reflux conditions to yield nitrogen-containing heterocycles (Scheme 9A).70
Scheme 9.

Pd-catalyzed Suzuki–Miyaura cross-coupling of (A) vinyl phosphates reported by Coudert, and (B) aryl phosphonium salts reported by Kang.
In 2008, Kang and co-workers in a very significant study reported Pd-catalyzed direct cross-coupling of tautomerizable heterocycles using in situ prepared phosphonium salts (Scheme 9B).71 The products were obtained by activation/cross-coupling sequence in a one-pot fashion via a heterocycle-phosphonium salt intermediate, showing analogy to preactivated phosphates. Under the optimized conditions, using Pd(PPh3)4 catalyst system, a remarkable range of electron-deficient tautomerizable heterocycles was cross-coupled with aryl boronic acids in good to excellent yields. Importantly, 6-arylpurine ribonucleoside was also obtained in a single step from unactivated and unprotected inosine, again highlighting the advantages of using Pd catalysis in the synthesis of biorelevant biaryls.
Other examples of the Pd-catalyzed Suzuki-Miyaura cross-coupling of aryl phosphates using Pd(PPh3)4, Pd(PPh3)Cl2 and Pd(dppf)Cl2 catalyst systems have been reported.72–80
3. Palladium-Catalyzed Buchwald–Hartwig Couplings of C–O Electrophiles
Buchwald-Hartwig amination is now one of the most direct and powerful methods to construct aryl C–N bonds in organic synthesis.81–83 The first Buchwald-Hartwig amination of arenesulfonates was reported by Hartwig and co-workers in 1998 using aryl tosylates as substrates (Scheme 10).84 The reactions were performed in toluene at 110 °C with 2 mol% of Pd(dba)2 or Pd(OAc)2 and 3 mol% of ferrocenyl-based phosphine ligands, dtbpf or Josiphos. In this initial report, the authors concluded that a strong base, such as NaOt-Bu, is required to obtain high yields. Typically, strong bases are required for Buchwald-Hartwig amination, however, it is also interesting to note that much weaker bulky phenolate bases (e.g. NaOC6H2-2,4,6-t-Bu) are effective under some conditions.
Scheme 10.

Pd-catalyzed amination of aryl tosylates reported by Hartwig in 1998.
3.1. Buchwald–Hartwig Cross-Couplings of Aryl Mesylates
In 2008, Kwong reported the first general method for the Pd-catalyzed amination of aryl mesylates using their indole-based CM-Phos ligand (Scheme 11A).85 Impressively, the catalytic system was successfully applied to the cross-coupling of electronically-deactivated mesylates with cyclic and acyclic amines, indoles, pyrroles and carbazoles at as low as 0.5 mol% palladium loading. The cross-coupling could also be performed under solventless conditions or in water as a benign solvent.
Scheme 11.

Pd-catalyzed Buchwald–Hartwig cross-coupling of aryl mesylates reported by (A) Kwong, and (B) Buchwald.
Shortly thereafter, Buchwald and co-workers disclosed their conditions for the Pd-catalyzed cross-coupling of aryl mesylates with anilines using a combination of BrettPhos and BrettPhos Palladacycle G1 as the catalyst system (Scheme 11B).86 The protocol permitted amination of deactivated mesylates and anilines with good compatibility for electron-withdrawing and electron-donating groups and excellent functional group tolerance in high yields.
The Pd-catalyzed cross-coupling of aryl mesylates with amides and monoarylation of 1° alkyl amines has been reported with t-BuBrettPhos and Mor-DalPhos ligands, respectively.87–88
3.2. Buchwald–Hartwig Cross-Couplings of Aryl Tosylates
Following their seminal study (Scheme 10), Hartwig and co-workers developed a general method for the Pd-catalyzed amination of tosylates at room temperature (Scheme 12A).89 A combination of Pd[P(o-tol)3]2 and electron-rich Josiphos-type ligand, CyPF-t-Bu, formed a highly reactive catalyst system for this cross-coupling. An impressive range of amines, including 1° alkylamines and anilines, were coupled with both electronically-deactivated and heteroaryl tosylates to smoothly furnish the cross-coupling products under mild conditions. Most of the reactions were performed with 0.1 mol% catalyst loading, thus demonstrating the high efficiency of this catalyst system.
Scheme 12.

Pd-catalyzed Buchwald–Hartwig cross-coupling of aryl tosylates reported by (A-B) Hartwig, and (C) Stradiotto.
Later, Hartwig and co-workers reported the coupling of tosylates with ammonia to give 1° arylamines using the same catalyst system (Scheme 12B).90 Interestingly, they found that sterically-hindered aryl tosylates cross-coupled in higher yields than less sterically hindered tosylates due to more facile O–S bond cleavage in unhindered substrates. However, note that the scope of tosylates is limited to ortho-substituted aryl tosylates.
In 2010, Stradiotto and co-workers reported the Pd-catalyzed amination of tosylates with hydrazine to give aryl hydrazines, isolated as hydrazones, using [Pd(cin)Cl]2 and Mor-DalPhos catalyst system under mild conditions (Scheme 12C).91 A variety of ortho-, meta-, and para-substituted tosylates could be cross-coupled with hydrazine using this protocol to give the corresponding hydrazone products, which were isolated after condensation with benzaldehyde in good to excellent yields.
Further examples of Pd-catalyzed amination of aryl tosylates using XPhos, CyPF-t-Bu, DavePhos, dipf, MorDal-Phos, dppf, CM-Phos have been reported.22,27,92–100 Specifically, the P,N-ligand MorDal-Phos appears to be more efficient in the cross-coupling of aryl tosylates with ammonia than the CyPF-t-Bu system,90 and could selectively catalyze monoarylation of ammonia with sterically non-hindered tosylates in high yields at room temperature.95
Moreover, César and co-workers reported an electron-rich Pd-NHC system, Pd-PEPPSI-IPr(NMe2)2, for the amination of aryl tosylates.101f In particular, it is worth noting that the latter system represents a rare example of well-defined Pd(II)–NHC complex that is successful for the Buchwald-Hartwig cross-coupling of aryl tosylates. This reaction proceeded well in alcoholic solvents in the presence of K3PO4, while other solvents and bases had a deleterious effect. Electron-rich and deficient tosylates were aminated in high to excellent yields; however, a slightly higher catalyst loading was required compared with the Hartwig system.89
3.3. Buchwald–Hartwig Cross-Couplings of Aryl Fluorosulfonates
In consideration of the unique features of fluorosulfonate activating group, in 2016, Hanley and co-workers reported the Pd-catalyzed amination of aryl fluorosulfonates and anilines using CpPd(cin)/Xantphos catalyst system (Scheme 13).49 Both electron-rich and sterically-hindered fluorosulfonates gave the corresponding amination products in excellent yields with only 1.0 mol% palladium loading. Remarkably, deployment of this protocol permitted the transformation of phenol to 2° arylamines directly in a one-pot fashion.
Scheme 13.

Pd-catalyzed Buchwald–Hartwig cross-coupling of aryl fluorosulfonates reported by Hanley.
In addition, Pd-catalyzed amination of aryl fluorosulfonates using PPh3 and dppp systems has been reported.102–103
3.4. Buchwald–Hartwig Cross-Couplings of Aryl Benzenesulfonates
The Pd-catalyzed amination of aryl benzenesulfonates was reported by Buchwald and co-workers in 2003 (Scheme 14).92 In general, benzenzesulfonates (–SO2Ph) are slightly more reactive than tosylates (–SO2-4-Tol), although the inductive effect of the 4-Me group is mild. The conditions using Pd(OAc)2, XPhos and a catalytic amount of benzeneboronic acid to activate the Pd(II) precatalyst were found optimal for this cross-coupling. A variety of substrates such as aliphatic amines, anilines, diarylamines, benzophenone imine, pyrrolidinone, primary amides, N-methyl formamide, and N-Boc amides were cross-coupled with various electronically-deactivated benzenesulfonates in high yields. It is noteworthy that this catalyst system also allowed for the cross-coupling of amides with benzenesulfonates by C–O bond cleavage for the first time.
Scheme 14.

Pd-catalyzed Buchwald–Hartwig cross-coupling of aryl benzenesulfonates reported by Buchwald.
3.5. Buchwald–Hartwig Cross-Couplings of Aryl Nonaflates
Buchwald and co-workers reported the Pd-catalyzed amination of aryl nonaflates using the combination of Pd2dba3 and DavePhos at 80 °C in 2003 (Scheme 15A).104 This methodology offers access to various 2° amines, including the very sterically hindered tetra-ortho-substituted amines in high to excellent yields. However, high temperature was required to achieve good yields
Scheme 15.

Pd-catalyzed Buchwald–Hartwig cross-coupling of aryl nonaflates reported by (A-B) Buchwald, and (C) Dunn.
The Buchwald group has also found that the use of more sterically-hindered JohnPhos ligand instead of DavePhos allowed for the amination of deactivated aryl nonaflates at room temperature (Scheme 15B).104
In 2011, Dunn and co-workers reported the Pd-catalyzed amination of aryl nonaflates with 1° sulfonamides using t-BuXPhos as ligand (Scheme 15C).105 This protocol is notable for its excellent functional group tolerance, including cyano, nitro, ester, aldehyde, ketone, chloride, carbamate and phenol groups, which were all well tolerated under the reaction conditions. Sterically-hindered substrates, such as 2,6-disubstituted aryl nonaflates are not compatible with this reaction. The authors proposed that reductive elimination was the rate-limiting step for this cross-coupling.
In addition, the Pd-catalyzed cross-coupling of aryl nonaflates using Xantphos and QUINAP catalyst systems have been reported.106–108
3.6. Buchwald–Hartwig Cross-Couplings of Aryl Imidazolyl Sulfonates
The Pd-catalyzed amination of aryl imidazolylsulfonates was reported by Ackermann and co-workers in 2011 using Pd(OAc)2/BINAP catalyst system (Scheme 16).109 Despite the fact that the cross-couplings were performed at 105 °C with 10 mol% palladium loading, a variety of functional groups, such as fluoro, cyano, nitro, ester, ketone as well as chloride were all well tolerated under the reaction conditions.
Scheme 16.

Pd-catalyzed Buchwald–Hartwig cross-coupling of aryl imidazolyl sulfonates reported by Ackermann.
4. Palladium-Catalyzed Heck Couplings of C–O Electrophiles
Since its discovery in 1968, the Heck reaction has been established as one of the most powerful catalytic transformations for the synthesis of olefins.110–111 The use of alkenes as coupling partners allows for the preparation of functionalized olefins that are key intermediates in the assembly of small molecules, pharmaceuticals, agrochemicals and natural products in academia and industry.112–114
4.1. Heck Cross-Couplings of Vinyl Tosylates and Mesylates
The first example of a Pd-catalyzed Heck cross-coupling of C–O electrophiles was reported by Fu and co-workers at Schering-Plough in 2002 using a combination of Pd(OAc)2 and PPh3 for the synthesis of conjugated olefins derived from β-diketones (Scheme 17).115
Scheme 17.

Pd-catalyzed Heck cross-coupling of vinyl tosylates reported by Fu in 2002.
Later, in 2005, Skrydstrup and co-workers extended this protocol and disclosed the regioselective Heck coupling of vinyl mesylates and tosylates catalyzed by Pd2dba3/dppf catalytic system after isolation of the intermediate vinyl OMs electrophiles (Scheme 18).116
Scheme 18.

Pd-catalyzed Heck cross-coupling of vinyl mesylates reported by Skrydstrup.
In 2006, the Pd-catalyzed Heck cross-coupling of unactivated vinyl tosylates with electron-deficient alkenes was achieved by the same group using 5 mol% PdCl2(cod) and 10 mol% P(t-Bu)3·HBF4 in DMF at 100 °C (Scheme 19A).117 LiCI was identified as the key additive, presumably to trap the cationic Pd(II) intermediate. In general, high yields were obtained using trisubstituted cyclic alkenyl tosylates as substrates. In contrast, the expected products were not obtained when bulky 1-t-Butylvinyl tosylate and related bulky tosylates were used as substrates (Scheme 19B). Interestingly, in these cases, the 1,2-migration products were isolated in good to excellent yields. A possible mechanism involves oxidative addition, β-hydride elimination to give alkyne, and 1,2-migration.117
Scheme 19.

Pd-catalyzed Heck cross-coupling of vinyl tosylates reported by Skrydstrup.
Further examples of Pd-catalyzed Heck cross-couplings of mesylates and tosylates have been reported by Djakovitch and Skrydstrup groups.118–119 In particular, a protocol for the Mizoroki-Heck coupling of heteroaromatic tosylates using dppf as ligand is noteworthy;119 however, these Heck reactions of C–O electrophiles are significantly less developed than that of the corresponding halides.
4.2. Heck Cross-Couplings of Nonaflates
In 2018, Lassaletta and co-workers reported a highly diastereo- and enantioselective, Pd-catalyzed Heck cross-coupling of aryl nonaflates and 2,3-dihydrofuran or N-Boc-2,3-dihydropyrrole (Scheme 20A).120 High yields and excellent enantioselectivity were achieved using (R)-DM-BINAP class of ligands. Furthermore, Josiphos-type ligand, SL-J002-1, permitted a highly diastereo- and enantioselective coupling of aryl nonaflates and electron-rich olefins under the similar reaction conditions (Scheme 20B).
Scheme 20.

Pd-catalyzed asymmetric Heck cross-coupling of aryl nonaflates reported by Lassaletta.
Additional examples of using aryl nonaflates as electrophiles in the Pd-catalyzed Heck cross-coupling were reported by the Reissig, Peng and Valdés groups.121–123
4.3. Heck Cross-Couplings of Vinyl Phosphates
Building on their observation of 1,2-migration during the Heck coupling of tosylates (Scheme 19B),117 in 2007, Skrydstrup and co-workers reported a detailed study on the synthesis of dienes via Pd-catalyzed Heck cross-coupling of vinyl phosphates and electron-deficient alkenes (Scheme 21).124 In this divergent cross-coupling protocol, the typical Heck olefins were obtained as main products in high yields when XPhos was used as the ligand (Scheme 21A). In contrast, 1,2-migration was the main reaction pathway when P(tBu)3·HBF4 was used as a ligand in combination with α-quaternary vinyl phosphates (Scheme 21B).
Scheme 21.

Pd-catalyzed Heck cross-coupling of vinyl phosphates reported by Skrydstrup.
Furthermore, Sasaki and co-workers reported a rare intramolecular Heck cross-coupling of vinyl phosphates using Pd(PPh3)4.125
5. Palladium-Catalyzed Negishi Couplings of C–O Electrophiles
Since its discovery in 1977, the Negishi cross-coupling has become an effective and powerful way to construct C(sp2)–C(sp2) and C(sp2)–C(sp3) carbon-carbon bonds under mild conditions and with high chemoselectivity.126
In 1991, Roth and co-workers reported the seminal Negishi cross-coupling of aryl fluorosulfonates with aryl and heteroarylzinc reagents using Pd(PPh3)4 in the presence of LiCI in THF at 50 °C (Scheme 22).48 Electron-neutral, electron-rich and heteroaryl fluorosulfonates afforded good to high yields of the biaryl products, while electronically-deactivated fluorosulfonates gave moderate yields under these conditions.
Scheme 22.

Pd-catalyzed Negishi cross-coupling of aryl fluorosulfonates reported by Roth in 1991.
5.1. Negishi Cross-Couplings of Alkyl Mesylates
In 2006, Organ and co-workers reported a method for the Pd-catalyzed cross-coupling of alkyl mesylates and alkyl and arylzinc reagents at room temperature (Scheme 23).127 The key to the success was using their easily prepared, air-stable, well-defined [Pd-PEPPSI-IPr] precatalyst. Under the developed conditions, excellent yields were achieved with both alkyl and arylzinc reagents; however, aryl mesylates are not suitable substrates for this cross-coupling.
Scheme 23.
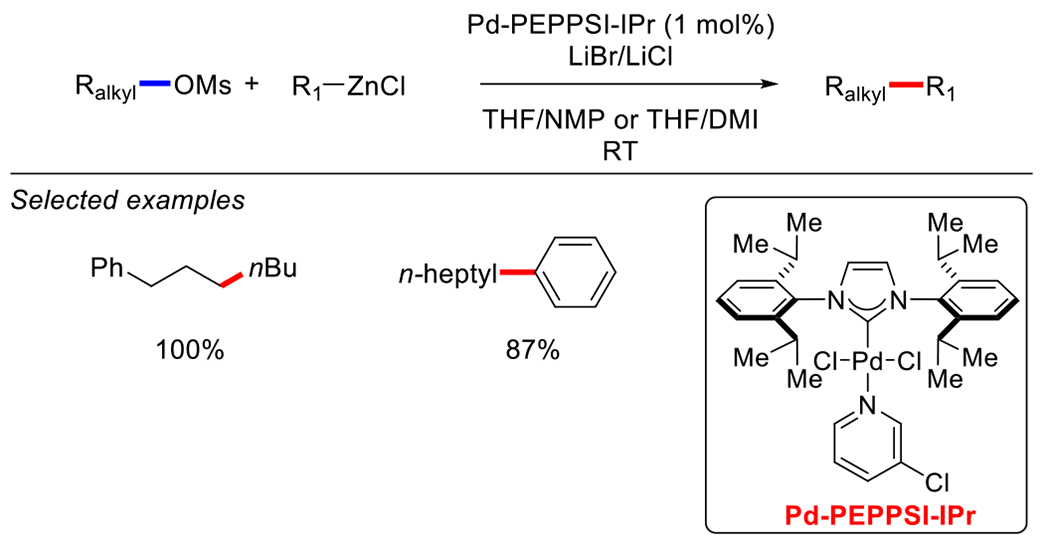
Pd-catalyzed Negishi cross-coupling of aryl mesylates reported by Organ.
5.2. Negishi Cross-Couplings of Vinyl Tosylates
The cross-coupling of 4-tosylcoumarines with a variety of organozinc reagents was reported by Yang and co-workers using the Pd(PPh3)4 catalytic system (Scheme 24).128 High yields were obtained using aryl, benzyl, and heteroarylzinc reagents. Furthermore, it was found that 3-cyanopropylzinc bromides could be successfully employed due to coordination of the cyano group to the metal center. Unfortunately, this methodology was restricted to coumarin derivatives.
Scheme 24.

Pd-catalyzed Negishi cross-coupling of vinyl tosylates reported by Yang.
Other examples of Pd-catalyzed Negisihi cross-coupling of tosylates using [Pd-PEPPSI-IPr], Pd2dba3/PCy3 and Pd(OAc)2 catalytic systems have been reported.127,129–130 Importantly, [Pd-PEPPSI-IPr] allowed for the coupling of alkyl tosylates with alkyl and arylzinc reagents in promising yields.127
5.3. Negishi Cross-Couplings of Aryl Nonaflates
The Pd-catalyzed Negishi cross-coupling of aryl nonaflates was disclosed by Knochel and co-workers in 1998 using the combination of Pd(dba)2/dppf (Scheme 25).131 The reactions were performed at 55–60 °C in THF at 2 mol% palladium loading. The reaction afforded a variety of polyfunctionalized terphenyls in excellent yields.
Scheme 25.

Pd-catalyzed Negishi cross-coupling of aryl nonaflates reported by Knochel.
Furthermore, Rossi and co-workers reported Negishi cross-coupling of aryl nonaflates under similar reaction conditions.132
5.4. Negishi Cross-Couplings of Aryl Fluorosulfonates
Following the early study by Roth (Scheme 22), more recently, Schoenebeck and co-workers reported the Pd-catalyzed synthesis of polysubstituted arenes via selective Negishi cross-coupling between aryl fluorosulfonates and organozinc bromides catalyzed by their well-defined Pd(I) dimer, [Pd(Pt-Bu3)I]2 (Scheme 26).133 The reactions were performed in a mixture of NMP/THF at room temperature at 5 mol% palladium loading. Impressively, full selectivity for the cross-coupling of fluorosulfonates in the presence of C–Cl bonds was achieved, irrespective of the electronic and steric demands of the fluorosulfonate substrates. The authors also demonstrated that trisubstituted arenes could be synthesized in one-pot by sequential cross-couplings with the same catalyst in excellent yields.
Scheme 26.

Pd-catalyzed Negishi cross-coupling of aryl fluorosulfonates reported by Schoenebeck.
5.5. Negishi Cross-Couplings of Vinyl Phosphates
In 2007, Skrydstrup and co-workers reported the Pd-catalyzed Negishi coupling of enol phosphates using Pd2dba3/dppf catalyst system (Scheme 27A).134 The reactions proceed well with both alkyl and arylzinc bromides in THF at 70 °C. It should be pointed out that the addition of LiCI (5 equiv), presumably to increase the concentration of the anionic Pd(0), was required as organozinc bromides were not reactive under the reaction conditions.
Scheme 27.

Pd-catalyzed cross-coupling of vinyl phosphates with (A) organozinc reagents reported by Skrydstrup, (B-C) organoaluminum and organomanganese reagents by Oshima.
In a related method, Oshima and co-workers disclosed the cross-coupling between enol phosphates and organoaluminium reagents catalyzed by Pd(PPh3)4 (Scheme 27B).135 The reaction provides an efficient method to convert ketones into substituted olefins. Alkyl, alkenyl and alkynyl organoaluminum reagents could be employed as coupling partners to give the corresponding products in good to excellent yields. The same group reported a related cross-coupling of vinyl phosphates with organomanganese reagents catalyzed by Pd(PPh3)4 (Scheme 27C).136
Further examples of Pd-catalyzed Negishi-type cross-couplings of vinyl phosphates using PPh3 and PPF-t-Bu based systems have been reported by Oshima and Skrydstrup.137–139
6. Palladium-Catalyzed Sonogashira Couplings of C–O Electrophiles
At present, the palladium-catalyzed Sonogashira cross-coupling represents the most straightforward and synthetically useful method for the synthesis of substituted alkynes.140
In 1998, Nicolaou and co-workers reported a seminal example of the cross-coupling of enol phosphates with trimethylsilylacetylene, affording the desired product in 84% yield in the presence of 10 mol% Pd(PPh3)4 and 10 mol% of Cul as the catalyst system (Scheme 28).141 This report set the stage for the development of new catalytic systems for the Pd-catalyzed Sonogashira cross-coupling of C–O electrophiles, and to date this class of reactions is among the best developed in the field.
Scheme 28.

Pd-catalyzed Sonogashira cross-coupling of vinyl phosphates reported by Nicolaou in 1998.
6.1. Sonogashira Cross-Couplings of Aryl Mesylates
In 2010, Kwong and co-workers established the first general method for the Pd-catalyzed, Cu-free Sonogashira cross-coupling of aryl mesylates using 2 mol% Pd(OAc)2 in combination with 6 mol% of their CM-Phos ligand in t-BuOH at 110 °C (Scheme 29).142 Importantly, electronically-deactivated and heteroaryl mesylates were capable coupling partners under these conditions.
Scheme 29.

Pd-catalyzed Sonogashira cross-coupling of aryl mesylates reported by Kwong.
6.2. Sonogashira Cross-Couplings of Aryl and Vinyl Tosylates
In 2001, Braga and co-workers disclosed a mild protocol for the Pd/Cu-co-catalyzed synthesis of β-chalcogeneno-enynes (S, Se) via the Sonogashira cross-coupling of vinylic tosylates using 3 mol% Pd(PPh3)2Cl2 in the presence of 17 mol% Cul (Scheme 30A).143 Relatively complex enynes containing selenium and sulfur were prepared in good to excellent yields using this method. Much lower yields were observed in the absence of either palladium or copper.
Scheme 30.

Pd-catalyzed Sonogashira cross-coupling of aryl tosylates reported by (A) Braga, (B) Buchwald, and (C) Nazaré.
In contrast, in 2003, Buchwald and co-workers reported a Pd-catalyzed, Cu-free method for the Sonogashira cross-coupling of electron-deficient aryl tosylates using their XPhos as ligand (Scheme 30B).144 However, this protocol also required the use of propionitrile as a solvent and slow alkyne addition, which limits its application.
In 2010, Nazaré and co-workers reported the first general method for the Pd-catalyzed, Cu-free Sonogashira cross-coupling of aryl and heteroaryl tosylates using Pd(TFA)2/CyPF-t-Bu catalytic system (Scheme 30C).145 Importantly, Cu was not required to promote the cross-coupling under these conditions. Furthermore, tosylates featuring both electron-withdrawing and deactivating, electron-rich functions afforded the desired alkynes in high yields. The method is characterized by excellent functional group tolerance in that functionalities, such as nitriles, ketones, methyl esters, aldehydes, hydroxy groups, and even amino group are well compatible under this catalyst system, demonstrating the generality of the protocol.
Further examples of Pd-catalyzed Sonogashira cross-coupling of tosylates using CM-Phos, PPh3, BrettPhos, and DPEPhos based catalytic systems have been reported.26,130,142,146–148 Moreover, Shen and co-workers reported a Pd(II)–NHC system based on bulky IPr* ligand for the Sonogashira cross-coupling of aryl tosylates under Cu-free conditions.149
6.3. Sonogashira Cross-Couplings of Aryl Nonanflates
In 2003, Maleczka Jr. and co-workers reported the Pd/Cu-co-catalyzed cross-coupling of aryl nonaflates with alkynes using a combination of Pd(PPh3)2Cl2 and CuCl at room temperature (Scheme 31).150 Polymethylhydrosiloxane and CsF were found to be necessary additives in this reaction to facilitate the formation of an alkynyl siloxane intermediate. The method affords functionalized alkynes in good to excellent yields under mild conditions; tolerance to aryl bromides should be noted (see Scheme 31).
Scheme 31.

Pd-catalyzed Sonogashira cross-coupling of aryl nonaflates reported by Maleczka, Jr.
An additional example of Pd-catalyzed cross-coupling of nonanflates has been reported by the same group.151
6.4. Sonogashira Cross-Couplings of Aryl Imidazolyl Sulfonates
Williams and co-workers reported a Pd-catalyzed, Cu-free Sonogashira cross-coupling of aryl/heteroaryl imidazolylsulfonates in the presence of 10 mol% palladium and 20 mol% XPhos (Scheme 32).152 These reactions proceeded quite smoothly in DMSO at 65 °C owing to the high stability and electronic activating effect of the imidazolylsulfonate group, affording the corresponding alkynes in high yields.
Scheme 32.

Pd-catalyzed Sonogashira cross-coupling of aryl imidazolyl sulfonates reported by Williams.
Moreover, Nájera reported a microwave-promoted Sonogashira cross-coupling of aryl imidazolylsulfonates catalyzed by their oxime palladacycle and SPhos combination.153
6.5. Sonogashira Cross-Couplings of Vinyl Phosphates and Phosphonium Salts
Following their study on the Suzuki cross-coupling of vinyl phosphates (Scheme 9B), in 2010, Kang and co-workers disclosed a Pd/Cu-co-catalyzed Sonogashira cross-coupling of vinyl phosphonium salts (Scheme 33).154 These reactions proceed via a formal C–H/C–OH bond activation, enabling C(sp2)-C(sp) bond formation in various tautomerizable heterocycles. This class of dehydrative cross-couplings affords a broad range of heteroaryl alkynes in good to excellent yields. Cu is the required additive as the reactions stopped at low conversions in the absence of copper.
Scheme 33.

Pd-catalyzed Sonogashira cross-coupling of vinyl phosphates reported by Kang.
In addition, Aldrich and co-workers reported a Pd-catalyzed, Cu-free Sonogashira cross-coupling of tautomerizable heterocycles via phosphonium salts using CyJohnPos as a ligand.155
7. Palladium-Catalyzed Stille Couplings of C–O Electrophiles
While Stille cross-couplings are among the most effective methods for the formation of carbon-carbon bonds, the toxicity of tin rendered this class of reactions less appealing. Nevertheless, numerous examples of Stille cross-coupling are still performed in both academic and industrial laboratories, especially in cases when other organometallic nucleophiles are less effective.156
In 1991, Scott and co-workers reported the cross-coupling of vinyl mesylates with organostannanes in the presence of 5 mol% Pd(PPh3)4 in THF at 80 °C (Scheme 34A).157 Generally high yields were achieved when vinylstannanes were used; however, arylstannanes were not suitable coupling partners under these conditions, resulting in low yields.
Scheme 34.

Pd-catalyzed Stille cross-coupling of vinyl phosphates reported by (A) Scott, and (B) Roth in 1991.
In the same year, Roth and co-workers reported the Stille cross-coupling of aryl fluorosulfonates using 5 mol% of Pd(PPh3)2Cl2 in DMF at room temperature (Scheme 34B).48 Fluorosulfonates are typically more reactive than mesylates due to facile Pd(0) insertion into the C–O bond.9 Accordingly, Roth demonstrated that both vinyl and arylstannanes performed well in the cross-coupling. Remarkably, the conditions were compatible with a C(sp2)–Br bond, showing excellent functional group tolerance of the protocol.
7.1. Stille Cross-Couplings of Aryl Mesylates
In 2010, Buchwald and co-workers reported a general method for the Stille cross-coupling of aryl mesylates (Scheme 35).158 A combination of Pd(OAc)2 and XPhos was found to be the optimum catalyst system for this cross-coupling. This protocol is carried out in refluxing t-BuOH in the presence of CsF, and exhibits broad scope and excellent functional group tolerance. Electronically-deactivated, electron-deficient and heteroaryl mesylates underwent smooth cross-coupling in good yields. Furthermore, acidic N–H bonds, such as in 2° amides are tolerated in this reaction. Sterically hindered biaryls can also be obtained by this method.
Scheme 35.

Pd-catalyzed Stille cross-coupling of aryl mesylates reported by Buchwald.
7.2. Stille Cross-Couplings of Aryl Tosylates
In 1997, Sasaki and co-workers investigated the synthesis of 6-functionalized 2-aminopurine nucleosides prepared by the Pd-catalyzed Stille cross-coupling of guanosine 6-O-tosylates and vinyl-tributylstannanes using 20 mol% of Pd(PPh3)4 in refluxing dioxane (Scheme 36, cf. Scheme 3B by Lakshman and co-workers using more practical organoboranes).159 The method afforded quantitative yields when the amino group at the 2-position was unprotected, and lower yields when the amino group was converted to the isobutyryl amide.
Scheme 36.

Pd-catalyzed Stille cross-coupling of heteroaryl tosylates reported by Sasaki.
Further examples of Pd(PPh3)4 and Pd(PPh3)2Cl2 catalyst systems for the Stille cross-coupling of tosylates have been reported.160–161
7.3. Stille Cross-Couplings of Aryl Nonaflates
In 1998, Knochel and co-workers reported a single example of Pd-catalyzed Stille cross-coupling of aryl nonaflates to afford biaryls using phenyl tributylstannane in the presence of Pd(dba)2/dppf in DMF (Scheme 37A).131 The cross-coupling afforded the desired product in 82% yield; however, the nonaflate was electronically-activated by the presence of an ester group at the 4-position.
Scheme 37.

Pd-catalyzed Stille cross-coupling of aryl nonaflates reported by (A) Knochel, and (B) Swager.
In 2008, Swager and co-workers demonstrated that norbornene derivatives can be efficiently synthesized by the Stille cross-coupling between aryl di-nonaflates and heteroarylstannanes using Pd(PPh3)2Cl2 in 71–91% yields (Scheme 37B).162 These products are of interest as monomers for conductive polymers.
Furthermore, Braun and co-workers reported Stille cross-coupling of aryl nonaflates using Pd(PPh3)4 as catalyst.163
7.4. Stille Cross-Couplings of Aryl Fluorobenzenesulfonates
In another interesting development, Badone and co-workers reported the Pd-catalyzed Stille cross-coupling of aryl 4-fluoro-benzenesulfonates with organostannanes using Pd(OAc)2/dppp or Pd(OAc)2/Ph2PMe catalyst systems (Scheme 38).164 The use of more electron-deficient 4-fluoro-benzenesulfonates permits to achieve higher yields than with benzenesulfonates or tosylates. This protocol works well with electronically-activated substrates, while electron-rich electrophiles gave lower yields. A range of organostannanes, including alkyl, vinyl, allyl, and aryl performed well as coupling partners under the developed reaction conditions.
Scheme 38.

Pd-catalyzed Stille cross-coupling of aryl 4-fluorobenzensulfonates reported by Badone.
7.5. Stille Cross-Couplings of Vinyl Phosphates
In 1997, Nicolaou and co-workers reported a general method for the functionalization of lactones to cyclic enol ethers via Pd-catalyzed Stille cross-coupling of ketene acetal phosphates with vinylstannanes (Scheme 39A).165 These reactions were conducted using 5 mol% of Pd(PPh3)4 in refluxing THF. This protocol allows to obtain dienes of medium and large ring systems (6-16-membered rings) in good to excellent yields. In addition, the cross-coupling of a ketene acetal phosphate with hexamethylditin afforded the desired vinyl trimethylstannane in 75% yield, which could be used as a nucleophile in the subsequent cross-coupling.
Scheme 39.

Pd-catalyzed Stille cross-coupling of vinyl phosphates reported by (A) Nicolaou, and (B) Coudert.
In 1999, Coudert and co-workers reported the Pd-catalyzed Stille cross-coupling of vinyl phosphates for the synthesis of 3-substituted-4H-1,4-benzoxazines (Scheme 39B).166 A broad range of vinyl, aryl and heteroaryl organostannanes could be coupled by this mild protocol in the presence of 5 mol% of Pd(PPh3)4 in refluxing THF.
Further studies on the Pd-catalyzed Stille cross-coupling of phosphates using Pd(PPh3)4 systems have been reported.141,167–170
8. Palladium-Catalyzed Hiyama Couplings of C–O Electrophiles
The Hiyama cross-coupling between organic electrophiles and organosilanes has been developed into an attractive cross-coupling manifold due to low toxicity, superb functional group tolerance and availability of organosilicon reagents, which in several cases offer advantages even over organoboranes (Suzuki cross-coupling).171
In 2002, Denmark reported a stereospecific Hiyama cross-coupling of aryl nonaflates with (E)- and (Z)-heptenyl-dimethylsilanols catalyzed by a combination of PdBr2 and JohnPhos at room temperature (Scheme 40).172 Importantly, both activated and nonactivated nonaflates could be used as substrates in this seminal study giving the desired products in 88–97% yield with high retention of configuration. The use of fluoride and water were found to be critical to tune the reactivity in these protocols.
Scheme 40.
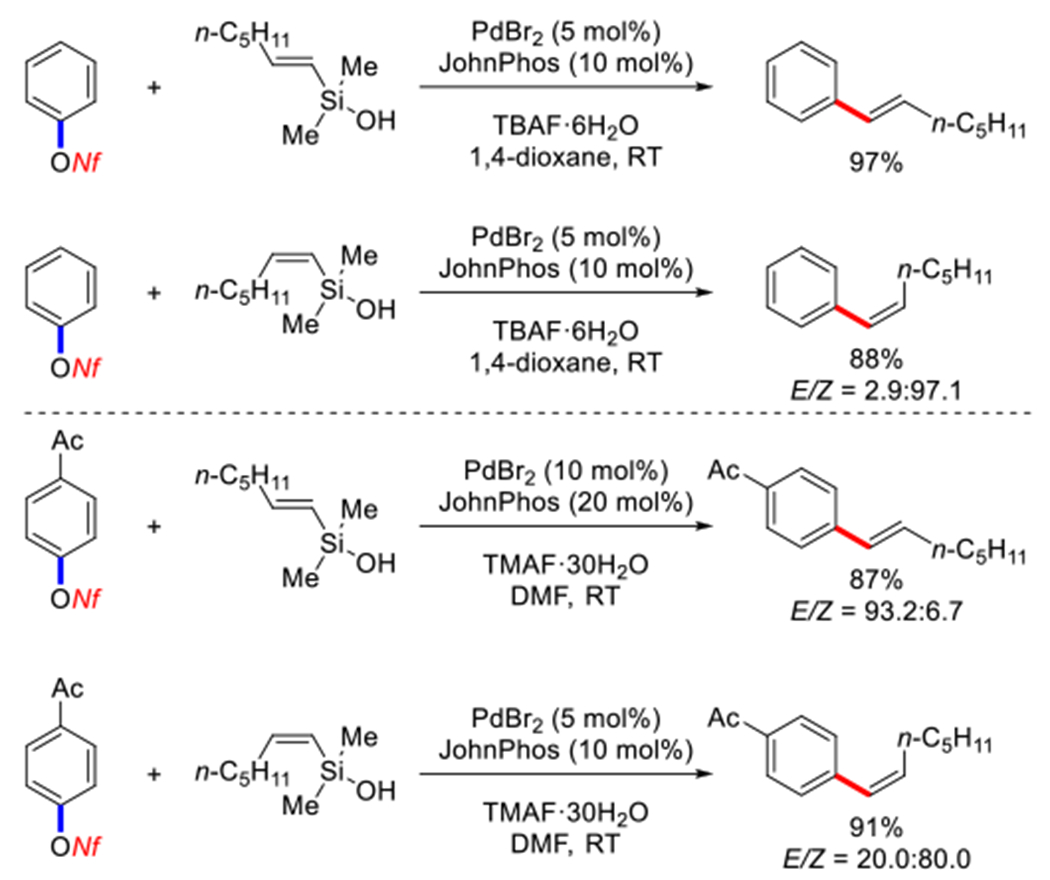
Pd-catalyzed Hiyama-Denmark cross-coupling of aryl nonaflates reported by Demark in 2002.
8.1. Hiyama Cross-Couplings of Aryl Mesylates
In 2008, Wu and co-workers reported the first general method for the Pd-catalyzed Hiyama cross-coupling between aryl mesylates and arylsilanes using a catalytic system based on 4 mol% of Pd(OAc)2 and 10 mol% of XPhos in THF/tBuOH at 90 °C (Scheme 41).173 TBAF was identified as an efficient additive. Trimethoxyarylsilanes and triethoxyarylsilanes containing electron-withdrawing and electron-donating groups at the para or ortho positions were cross-coupled with electronically-deactivated aryl mesylates to give the corresponding biaryls in generally excellent yields. Interestingly, competition experiments demonstrated than aryl chlorides are more reactive than aryl mesylates under these conditions.
Scheme 41.

Pd-catalyzed Hiyama cross-coupling of aryl mesylates reported by Wu.
In 2009, Kwong and co-workers reported the Pd-catalyzed Hiyama cross-coupling of aryl mesylates using their CM-Phos as a ligand.174
8.2. Hiyama Cross-Couplings of Aryl Tosylates
In 2008, Wu and co-workers reported a general method for the cross-coupling of aryl tosylates with organosilanes using the same catalyst system as identified for the cross-coupling of mesylates (Scheme 42 cf. Scheme 41).175 Moreover, aryl benzenesulfonates and vinyl tosylates could also be coupled under these conditions.
Scheme 42.
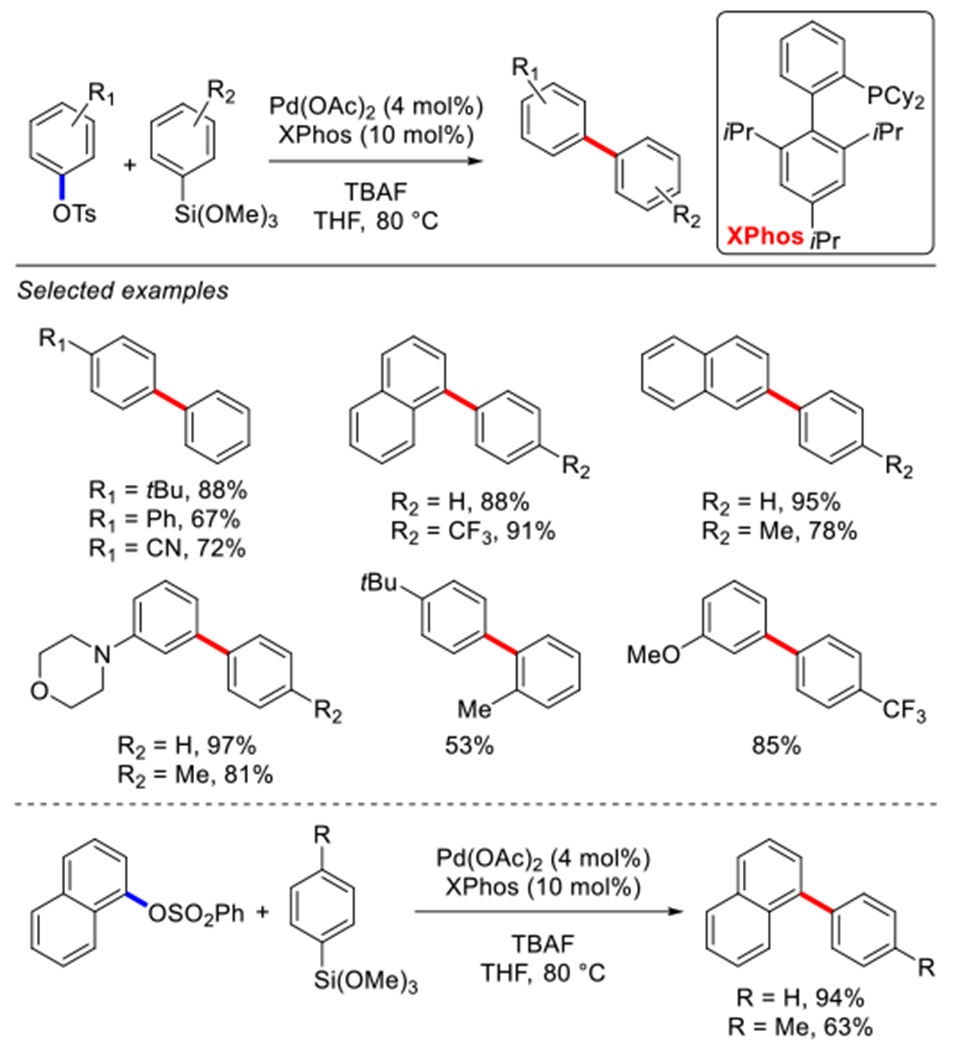
Pd-catalyzed Hiyama cross-coupling of aryl tosylates reported by Wu.
In 2016, Nakao and co-workers reported a co-operative Pd/Cu catalysis system for the cross-coupling of aryl tosylates and organosilanes using a heterobidentate ArPCy2 ligand.176
8.3. Hiyama Cross-Couplings of Aryl Sulfamates
In 2016, Hazari and co-workers reported the Pd-catalyzed Hiyama cross-coupling of aryl sulfamates with aryl silanolates using Pd-RuPhos catalyst based on (η3-1-t-Bu-ind)Pd(L)Cl system developed by their group (Scheme 43).66 Importantly, external base is not required for this cross-coupling. The use of additional 5 mol% of RuPhos resulted in significantly improved yields due to preventing the catalyst decomposition. The scope of this reaction is broad with a variety of substituted aryl and heteroaryl sulfamates compatible with the reaction conditions, including 2-Np, Ar and quinolinyl. However, 1-Np sulfamates are not tolerated in this protocol due to increased steric hindrance.
Scheme 43.

Pd-catalyzed Hiyama cross-coupling of aryl sulfamates reported by Hazari.
8.4. Hiyama Cross-Couplings of Aryl Imidazolyl Sulfonates
In 2010, Williams and co-workers reported the cross-coupling of aryl imidazolylsulfonates with HOMSi reagents (HOMSi = o-hydroxymethylphenyldimethylsilyl) catalyzed by 5 mol% Pd(dppf)Cl2 in DMSO at 65 °C (Scheme 44).152 The method works well for both electron-poor and electronically-deactivated substrates. However, lower yields were observed for ortho-substituted electrophiles.
Scheme 44.

Pd-catalyzed Hiyama cross-coupling of aryl imidazolyl sulfonates reported by Williams.
9. Palladium-Catalyzed α-Arylations using C–O Electrophiles
The α-arylation of carbonyl compounds, wherein aryl halides are cross-coupled with enolates in the presence of a transition-metal-catalyst represents a highly valued process due to the prevalence of α-aryl carbonyl moieties as basic building blocks for the synthesis of biologically active molecules.177–178
In 1999, Hartwig and co-workers demonstrated the first example of Pd-catalyzed α-arylation of C–O electrophiles and reported cross-coupling of propiophenone with aryl tosylate as an electrophile using catalyst system based on Pd(OAc)2/Josiphos type ligand in THF at 70 °C (Scheme 45).179
Scheme 45.

Pd-catalyzed a-arylation of ketones with aryl tosylates reported by Hartwig in 1999.
9.1. α–Arylation Reactions with Aryl Mesylates
In 2013, Stradiotto and co-workers reported a general method for the α-monoarylation of ketones with aryl mesylates using their Mor-DalPhos ligand and [Pd(cin)Cl]2 as palladium source (Scheme 46).88 Both cyclic and acyclic dialkyl-ketones were monoarylated in high yields to selectively afford benzyl methyl ketone products. However, electron-deficient and ortho-substituted aryl mesylates were not suitable coupling partners in this protocol.
Scheme 46.

Pd-catalyzed α-arylation of ketones with aryl mesylates reported by Stradiotto.
In 2016, Kwongand co-workers reported the α-monoarylation of aryl- and heteroarylketones using a catalyst system based on Pd(OAc)2 and a 4,7-dimethyl-derivative of their indolyl phosphine CM-Phos.180 Importantly, this methodology tolerates a wide range of substrates, including arylketones and heteroaromatic ketones (cf. alkylketones).88
9.2. α–Arylation Reactions with Aryl Tosylates
In 2003, Buchwald and co-workers reported the α-arylation of 1,3-cyclopentanedione with an activated aryl tosylate in the presence of 2 mol% Pd(OAc)2 and 5 mol% XPhos (Scheme 47A).21 The monoarylation product was isolated in 95% yield.
Scheme 47.

Pd-catalyzed α-arylation of ketones with aryl tosylates reported by (A) Buchwald, and (B) Stradiotto.
In 2011, Stradiotto and co-workers reported the challenging α-monoarylation of acetone using aryl tosylates as C–O electrophiles (Scheme 47B).181 The method works best with electron-rich aryl tosylates. The use of [Pd(cin)Cl]2, Mor-DalPhos and Cs2CO3 was identified as the optimum catalyst system for this reaction. A striking feature of this protocol is that valuable α-monoarylated acetones were generated in high to excellent yields.
9.3. α–Arylation Reactions with Aryl Benzenesulfonates
In 2003, the Buchwald group reported the α-arylation of ketones and esters with aryl benzenesulfonates using Pd(OAc)2 and XPhos catalyst system (Scheme 48).21 The developed conditions mirror the protocol for the cross-coupling of aryl tosylates by the same group (Scheme 47A). Ketones and esters were arylated in good to high yields in a Tol/t-BuOH at 110 °C.
Scheme 48.

Pd-catalyzed α-arylation of ketones with aryl benzenesulfonates reported by Buchwald.
9.4. α–Arylation Reactions with Aryl Imidazolyl Sulfonates
In 2012, Ackermann and co-workers reported the α-monoarylation of acetone with aryl imidazolylsulfonates in the presence of Pd(OAc)2 and XantPhos in dioxane (Scheme 49).182 The substrate scope was further extended to alkyl-methyl ketones, aryl-methyl ketones, and di-alkyl ketones. In all cases, the reactions proceed in good to excellent yields to give the monoarylated products with high selectivity. This protocol complements the study with aryl mesylates (cf. Scheme 46) using more electronically-activated and also benign imidazolylsulfonates.
Scheme 49.

Pd-catalyzed α-arylations of ketones with aryl imidazolyl sulfonates reported by Ackermann.
10. Palladium-Catalyzed Direct C–H Arylations of C–O Electrophiles
The direct C–H arylation is one the most attractive methods to create biaryl motifs.183 Extensive advances have been made in this reaction manifold as a more environmentally and economically viable strategy to the traditional cross-coupling in recent years.184
10.1. Direct Arylation Reactions with Aryl Mesylates
Ackermann and co-workers have made major progress in the field of direct arylation of C–O electrophiles (Schemes 50–52). In 2009, they reported the direct arylation of benzoxazole with activated aryl mesylates using Pd(OAc)2 and XPhos catalyst system in the presence of tBuCO2H (Scheme 50A cf. Scheme 51).185
Scheme 50.

Pd-catalyzed direct C–H arylation with aryl mesylates reported by (A) Ackermann, (B) Kwong, and (C) Kalyani.
Scheme 52.

Pd-catalyzed C–H arylations with aryl imidazolyl sulfonates reported by Ackermann.
Scheme 51.

Pd-catalyzed direct C–H arylation with aryl tosylates reported by Ackermann.
In 2010, Kwong and co-workers disclosed a general method for the direct arylation of benzoxazoles with aryl mesylates (Scheme 50B).186 They identified Pd(OAc)2 and CM-Phos as effective catalyst system. Interestingly, additional acid is not required in this protocol. A broad range of electronically-differentiated mesylates afforded the cross-coupling products in good to excellent yields. The method was further extended to other heterocycles, such as caffeine, triazole and benzothiazole, which were smoothly arylated using this method.
In 2014, Kalyani and co-workers reported an impressive method for the intramolecular direct arylation of arenes and heteroarenes with aryl mesylates as C–O electrophiles (Scheme 50C).187 They found a combination of Pd(OAc)2 and dcype as the optimal catalyst system for this transformation with Rb2CO3 and CsOPiv as stoichiometric additives. The use of alternative conditions was not reported. Diverse heterocycles could be synthesized by this protocol in good to excellent yields. Notably, the method was further extended to the direct arylation of benzoxazoles and related heterocycles by a sequential, one-pot mesylation and arylation of phenols in excellent yields.
Further examples of Pd-catalyzed direct arylation with aryl mesylates using XPhos, CM-Phos, SPhos, and XPhos based catalytic systems have been reported.188–191
10.2. Direct Arylation Reactions with Aryl Tosylates
In 2009, Ackermann and co-workers reported the first general method for the direct arylation of heterocycles with tosylates as C–O electrophiles (Scheme 51).184 A catalyst system based on Pd(OAc)2 and XPhos in the presence of tBuCO2H additive was found to be effective for this transformation. Electronically-diverse tosylates were coupled with various heterocycles, including benzoxazole, oxazole, caffeine, and 1,2,3-triazole to furnish biaryl motifs in good to high yields, demonstrating the generality of the protocol.
Additional studies of Pd-catalyzed direct arylation with aryl tosylates using XPhos, CyJohnPhos, and dcype ligands have been reported.188,191–193
10.3. Direct Arylation Reactions with Aryl Imidazolyl Sulfonates
In 2010, the Ackermann group disclosed a general method for the Pd-catalyzed direct arylation using aryl imidazolylsulfonates (Scheme 52).194 The reaction was accomplished with Pd(OAc)2 and dppe in the presence of Cs2CO3 additive. Ortho-, meta-, and para-substituted aryl imidazolylsulfonates could be coupled with various benzoxazoles and oxazoles in high yields.
10.4. Arylation Reactions with Aryl Nonaflates
In 2016, Manabe and co-workers reported an intramolecular direct C–H arylation of aryl nonaflates to yield fluoranthenes (Scheme 53).195 A combination of Pd2dba3 and SPhos with 1-Ad-CO2H as additive was identified as the optimal catalyst system. Electronically-diverse fluoranthenes could be prepared by this protocol in good to high yields.
Scheme 53.

Pd-catalyzed C–H arylations with aryl nonaflates reported by Manabe.
Another example of Pd-catalyzed intermolecular C–H arylation of aryl nonaflates using PCy3 has been reported.196
10.5. Arylation Reactions with Vinyl Phosphates
In 2010, Ackermann and co-workers reported the direct arylation of benzoxazoles with vinyl phosphates (Scheme 54).194 Interestingly, the catalyst system reflects the system developed for the direct arylation of aryl imidazolylsulfonates (cf. Scheme 52). Following this protocol, various alkenylated benzoxazoles could be prepared in good yields with vinyl phosphates as coupling partners.
Scheme 54.

Pd-catalyzed C–H arylations with vinyl phosphates reported by Ackermann.
11. Palladium-Catalyzed Carbonylative Couplings of C–O Electrophiles
The transition-metal-catalyzed carbonylation has been an active area of catalysis and provided a straightforward way for the synthesis of many polar carbonyl derivatives utilizing inexpensive CO.197
In 1998, Sugi and co-workers disclosed the first example of a Pd-catalyzed carbonylative cross-coupling of C–O electrophiles using activated aryl tosylates and PdCl2/dppp system at 10 bar CO (Scheme 55A).198 This procedure worked well for electronically-activated aryl tosylates, while lower yields were obtained with other substrates.
Scheme 55.

Pd-catalyzed carbonylative cross-coupling of aryl tosylates and vinyl phosphates reported by (A) Sugi, and (B) Nicolaou.
In the same year, Nicolaou and co-workers reported Pd-catalyzed carbonylation of lactam ketene aminal phosphates (Scheme 55B).141 These reactions were performed under mild conditions using a combination of 10 mol% Pd(OAc)2 and 20 mol% PPh3 at 1 atm CO in methanol. Good to excellent yields were obtained with various lactams featuring small to large rings (5-16 membered rings).
11.1. Carbonylative Cross-Couplings of Aryl Mesylates
In 2008, Buchwald reported the first general and mild protocol for the carbonylation of aryl mesylates to furnish esters using 2 mol% of Pd(OAc)2 and 2.2 mol% of dcypp at 1 atm CO pressure at 80–110 °C (Scheme 56).199 This process tolerates electron-rich, electron-poor and sterically-hindered mesylates, affording the ester products in high yields. The method is further noteworthy for its functional group tolerance, including ketones, acetals and heterocycles. Importantly, primary and secondary alcohols could be used as nucleophiles giving the corresponding ester products in high yields.
Scheme 56.

Pd-catalyzed carbonylative cross-coupling of aryl mesylates reported by Buchwald.
11.2. Cabonylative Cross-Couplings of Aryl Tosylates
The Buchwald group reported that aryl tosylates could also undergo the Pd-catalyzed carbonylative cross-coupling following the same procedure as for aryl mesylates (Scheme 57 cf. Scheme 56).199 As an advantage, a broader range of heterocyclic tosylate substrates is compatible with this protocol.
Scheme 57.

Pd-catalyzed carbonylative cross-coupling of aryl tosylates reported by Buchwald.
11.3. Carbonylative Cross-Couplings of Aryl Nonaflates
In 2012, Beller and co-workers reported an important method for the direct carbonylation of phenols via in situ formed aryl nonaflates (Scheme 58).200 A broad range of esters was formed using Pd-dimer, [Pd(cin)Cl]2, and dppf as the catalyst system. Methanol, primary and secondary alcohols, as well as electronically-diverse phenols were coupled to give the corresponding esters in high yields.
Scheme 58.

Pd-catalyzed carbonylative cross-coupling of aryl nonaflates reported by Beller.
11.4. Carbonylative Cross-Couplings of Aryl Pentafluorobenzenesulfonates
In 2015, Shashikanth and co-workers reported a remarkably mild protocol for the Pd-catalyzed carbonylation of aryl pentafluorobenzenesulfonates using 3 mol% Pd(dppf)Cl2 as catalyst and Co2(CO)8 as carbon monoxide source at room temperature (Scheme 59A–B).201 Both oxygen (Scheme 59A) and nitrogen (Scheme 59B) nucleophiles are compatible with this method, affording esters, carboxylic acids, and amides with excellent functional group tolerance.
Scheme 59.

Pd-catalyzed carbonylative (A) esterification, and (B) amidation of aryl pentafluorobenzenesulfonates by Shashikanth.
11.5. Carbonylative Cross-Couplings of Aryl Fluorosulfonates
In 2017, Qin and co-workers reported a one-pot protocol for the carbonylation of phenols via aryl fluorosulfonates (Scheme 60A).202 The method employs Pd(OAc)2 and dppp catalyst system at 1 atm CO pressure, affording carboxylic acids and esters in high yields.
Scheme 60.

Pd-catalyzed carbonylative (A) esterification, and (B) amidation of aryl fluorosulfonates reported by Qin.
In 2018, they disclosed a similar system for the carbonylative amidation of phenols at 1 atm CO pressure (Scheme 60B).103
11.6. Carbonylative Cross-Couplings of Aryl Fluorobenzensulfonates
One of the advantages of 4-fluorobenzenesulfonates is the activating effect of the fluorine on C–O activation, while preserving high stability of the sulfonate group.
In 2006, Rivera and co-workers reported a protocol for the carbonylative esterification of 4-fluorobenzenesulfonates catalyzed by Pd(OAc)2 and a Josiphos-type ligand at 135 °C (Scheme 61).203 Despite high temperature, the method is quite robust and tolerates a variety of functional groups affording esters in high yields.
Scheme 61.
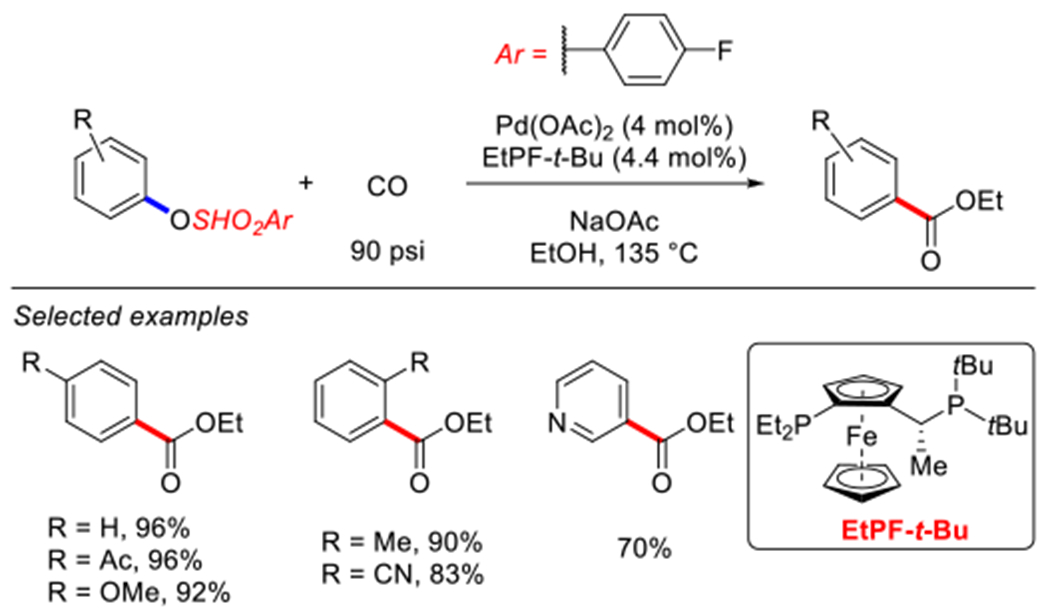
Pd-catalyzed carbonylative cross-coupling of aryl 4-fluorobenzensulfonates reported by Rivera.
11.7. Carbonylative Cross-Couplings of Aryl Imidazolyl Sulfonates
In 2009, Albaneze-Walkers and co-workers reported a single example of carbonylative esterification of 2-Np-imidazolylsulfonate using Pd(BINAP)Cl2 in methanol at 60 °C (Scheme 62).62
Scheme 62.

Pd-catalyzed carbonylative cross-coupling of aryl imidazolyl sulfonates reported by Albaneze-Walker.
11.8. Carbonylative Cross-Couplings of Vinyl Phosphates
In 2009, Larhed and co-workers reported the carbonylative amidation of vinyl phosphates to give acrylamides under microwave irradiation conditions (Scheme 63).204 They found that a combination of Hermann’s palladacycle (cataCXium C) and tBu3P allowed to use Mo(CO)6 as an attractive solid source of CO. In this protocol, alkyl alkenyl phosphates could be coupled with alkyl amines in good yields, while the coupling of aryl alkenyl phosphates or anilines afforded lower yields of the amide products.
Scheme 63.

Pd-catalyzed carbonylative cross-coupling of vinyl phosphates reported by Larhed.
Another method for the Pd-catalyzed carbonylation of vinyl phosphates using PPh3 has been reported.205
12. Palladium-Catalyzed Decarboxylative Couplings of C–O Electrophiles
The transition-metal-catalyzed decarboxylative coupling taking advantage of the availability, ubiquity and orthogonal properties of carboxylic acids is a widely attractive strategy for the construction of carbon-carbon and carbon-heteroatom bonds in catalysis.206
12.1. Decarboxylative Cross-Couplings of Aryl Mesylates
In 2013, Gooßen and co-workers reported the Pd-catalyzed cross-coupling of aryl mesylates with aryl and heteroaryl potassium carboxylates using a dual catalytic system of 5 mol% Pd(dba)2 and 5 mol% Cu2O in the presence of an imidazolyl phosphine ligand under microwave irradiation conditions (Scheme 64).207 The harsh conditions necessary for decarboxylation should be noted. 2-Np tosylate, activated aryl and heteroaryl carboxylates were identified as good substrates for this protocol; however, more sterically-hindered 1-Np as well as Ar tosylates afforded the products in lower yields. Importantly, the authors demonstrated that alkenyl mesylates are also viable C–O electrophiles and could be cross-coupled with various aromatic carboxylates to give substituted olefins.
Scheme 64.

Pd-catalyzed decarboxylative cross-coupling of aryl mesylates reported by Gooßen.
12.2. Decarboxylative Cross-Couplings of Aryl Tosylates
In 2010, the Gooßen group reported the Pd-catalyzed decarboxylative cross-coupling of aromatic potassium carboxylates and aromatic tosylates using a dual Pd/Cu catalyst system (Scheme 65).208. These cross-coupling reactions were performed using a combination of Pd(acac)2 and Cu2O either under microwave irradiation conditions or at 170 °C in NMP. XPhos was identified as the most effective ligand. Under the optimized reactions conditions, tosylates featuring various electronically-differentiated groups cross-coupled with aryl and heteroaryl carboxylates in high to excellent yields. Importantly, this protocol was shown to be compatible with a broad range of functional groups, such as aldehydes, nitro, esters, ketone, and tertiary amino groups, showing good functional group tolerance despite high temperatures required for decarboxylation.
Scheme 65.
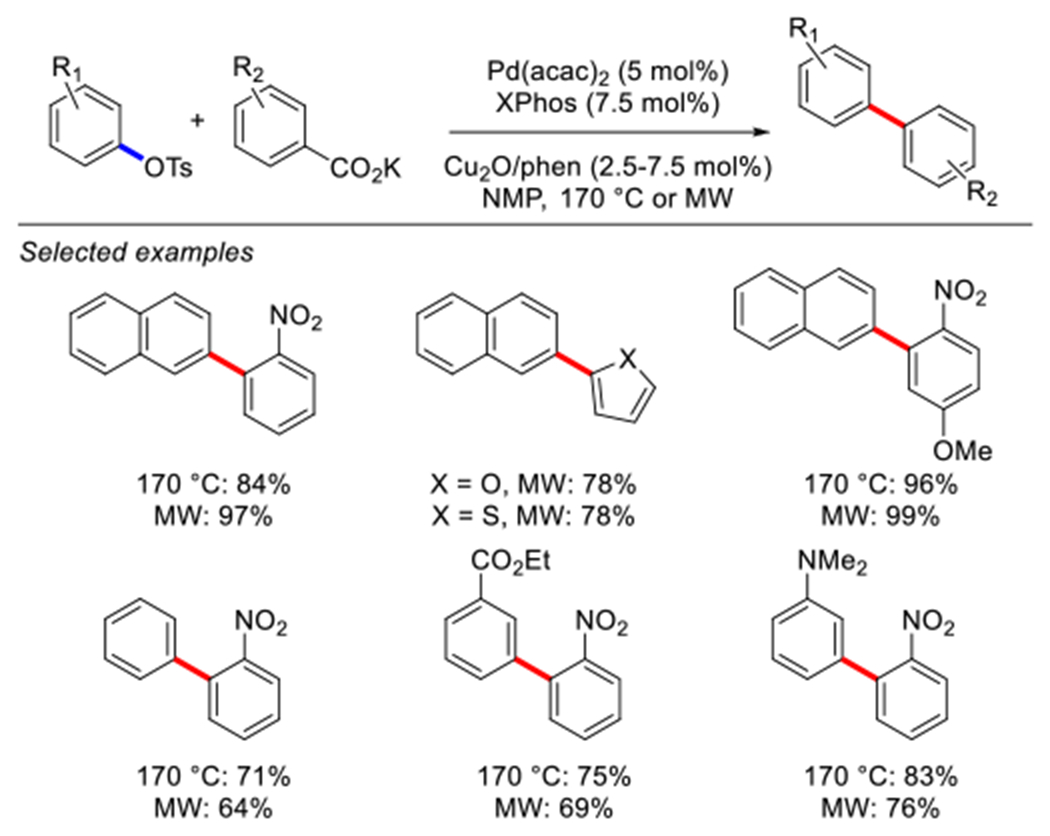
Pd-catalyzed decarboxylative cross-coupling of aryl tosylates reported bv Gooßen.
13. Palladium-Catalyzed Cyanation of C–O Electrophiles
The direct cyanation by C–O cross-coupling establishes an attractive method to convert phenols to aromatic nitriles as a practical alternative to Sandmeyer type reactions of anilines. More generally, the replacement of phenol to nitrile (Section 13), boron (Section 14), phosphorus (Section 15), arene (Section 16) or thiophenol (Section 16) represent a fundamental type of C–O to C–X (X = C, B, P, H, S) interconversion, wherein the phenol group can be utilized as a traceless directing group or a less conventional electrophile derived from an orthogonal pool of synthetic precursors.
In 2000, Hanack and co-workers reported the first example of cyanation of aryl nonafluorosulfonates using Pd(PPh3)4 as catalyst and Zn(CN)2 as cyanide source (Scheme 66).209 It was noteworthy that two- and hexa-fold cyanations to afford phthalonitrile and hexacyanotribenzylene were readily conducted under these conditions in 83% and 32% yield, respectively.
Scheme 66.

Pd-catalyzed cyanation of aryl nonaflates reported by Hanack in 2000.
13.1. Cyanations of Aryl Mesylates
In 2010, Kwong reported a general protocol for the Pd-catalyzed cyanation of aryl mesylates using 2 mol% Pd(OAc)2 and 8 mol% of their CM-Phos ligand as a catalyst system (Scheme 67).210 Notably, they deployed a green cyanating reagent, K4[Fe(CN)6]·3H2O, and most of the reactions could be performed in water. Using this environmentally-friendly method, a broad range of activated, deactivated and heteroaryl mesylates were converted to benzonitriles in good to excellent yields.
Scheme 67.

Pd-catalyzed cyanation of aryl mesylates reported by Kwong.
Another example of Pd-catalyzed cyanation of aryl mesylates using hemilabile, binaphthyl-type ligands and K4[Fe(CN)6]·3H2O has been reported by Xie and co-workers.211
13.2. Cyanations of Aryl Tosylates
Kwong and co-workers reported that their protocol developed for cyanation of mesylates could be used for the cyanation of aryl tosylates (Scheme 68 cf. Scheme 67).210 Aryl and heteroaryl tosylates featuring various functional groups, such as ester, amino, aldehyde, and amide were well-tolerated under the reaction conditions.
Scheme 68.

Pd-catalyzed cyanation of aryl tosylates reported by Kwong.
Furthermore, cyanation of aryl tosylates in H2O/dioxane mixture using XPhos-SO3Na based system has been reported.212
13.3. Cyanations of Aryl Fluorosulfonates
In 2018, in another important development, Qin and co-workers demonstrated that phenols can be converted to benzonitriles in a one-pot process via in situ formed fluorosulfonates (Scheme 69).213 The authors identified a combination of Pd(OAc)2 and PhJohnPhos as the optimal system for this one-pot activation/cross-coupling protocol. Electron-deficient phenols afforded benzonitriles in high yields, while electronically-deactivated substrates afforded the cross-coupling products in lower yields.
Scheme 69.
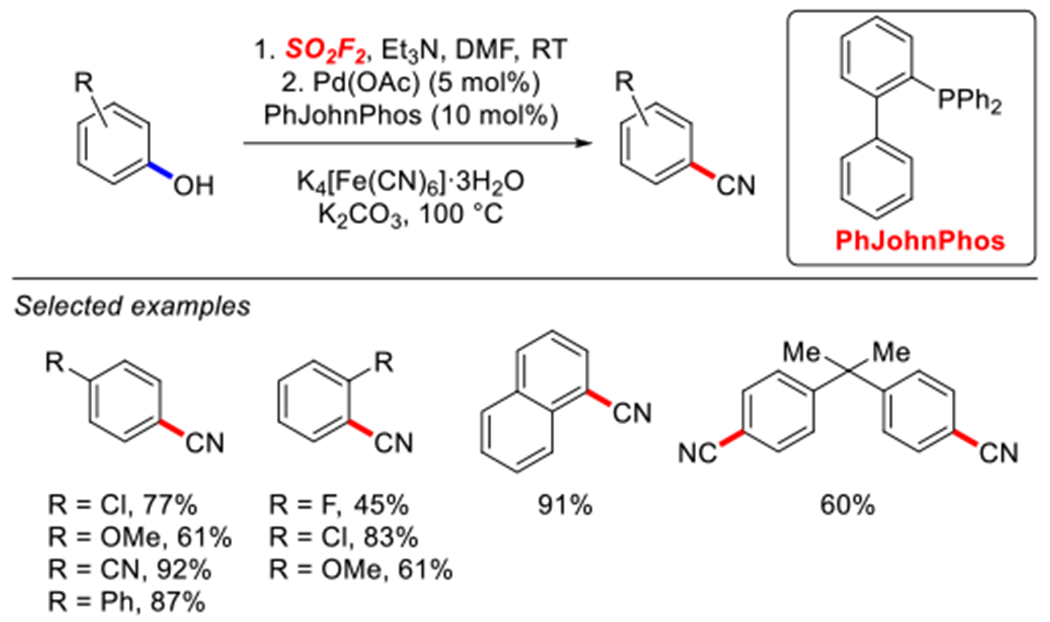
Pd-catalyzed cyanation of aryl fluorosulfonates reported by Qin.
13.4. Cyanations of Aryl Benzenesulfonates
In 2010, Huang and co-workers reported the Pd-catalyzed of aryl benzenesulfonates using Pd(OAc)2 and water-soluble XPhos-SO3Na (Scheme 70).212 In general, similar yields to the cross-coupling of aryl tosylates were obtained across the substrates examined.
Scheme 70.

Pd-catalyzed cyanation of aryl benzenesulfonates reported by Huang.
13.5. Cyanations of Aryl Imidazolyl Sulfonates
In 2014, Clarke reported a single example of the Pd-catalyzed cyanation of an ortho-substituted aryl imidazolylsulfonate (Scheme 71).214 Extensive optimization revealed 1 mol% Pd(TFA)2 and 3 mol% PPh3 as the optimal catalyst system for this reaction.
Scheme 71.

Pd-catalyzed cyanation of aryl imidazolylsulfonates reported by Clarke.
14. Palladium-Catalyzed Miyaura Borylations of C–O Electrophiles
Miyaura borylation provides an attractive access to aryl boronates from phenols under mild conditions; however, these C–O cross-coupling strategies are not well developed at present.
In 2005, Occhiato and co-workers reported an early example of the Pd-catalyzed borylation of vinyl phosphates for the synthesis of tetrahydropyridine-2-boronate (Scheme 72).215 The conditions using Pd(PPh3)2Cl2 with an additional PPh3 worked well to afford the desired six-membered product in 63% yield. Interestingly, the corresponding five-membered pyrrolidine derivative was unreactive and afforded only the olefin reduction product.
Scheme 72.

Pd-catalyzed borylation of vinyl phosphate reported by Occhiato in 2005.
14.1. Borylations of Aryl Mesylates
In 2011, Kwong and co-workers reported a general method for the Pd-catalyzed borylation of aryl mesylates (Scheme 73).216 They established that a combination of Pd(OAc)2 and their electron-rich indolyl-based MeO-CM-Phos ligand as the most effective system for this transformation. The functional group tolerance, excellent yields, and compatibility with various aryl and heteroaryl mesylates in this protocol are noteworthy.
Scheme 73.

Pd-catalyzed borylation of aryl mesylates reported by Kwong.
14.2. Borylations of Aryl Tosylates
The Kwong group reported Miyaura borylation of aryl tosylates following the same reaction conditions as they developed for the cross-coupling of mesylates (Scheme 74 cf. Scheme 73).216 Importantly, the palladium loading could be reduced to 1 mol% achieving good to high yields in most substrates examined.
Scheme 74.

Pd-catalyzed borylation of aryl tosylates reported by Kwong.
Moreover, the Pd-catalyzed borylation of aryl tosylates using pinacolborane and dtbpf ligand has been reported by Murata.217
14.3. Borylations of Aryl Benzenesulfonates
In 2011, Murata and co-workers reported Miyaura borylation of aryl benzenesulfonates using dialkoxyboranes and a catalytic system based on Pd(dba)2, dtbpf and stoichiometric Bu4NI (Scheme 75).217 Aryl benzenesulfonates featuring various functional groups, such as cyano, ketone, ester afforded the borylated products in good to high yields. In the same study, the authors have also examined aryl pyrazolylsulfonates and aryl 4-trifluorbenzenesulfonates under the same catalytic conditions, with the latter providing significant improvements in case of less reactive C–O electrophiles.
Scheme 75.

Pd-catalyzed borylation of aryl benzenesulfonates reported by Murata.
15. Palladium-Catalyzed Hirao Phosphorylations of C–O Electrophiles
The Hirao cross-coupling has been leveraged to provide access to C–P bonds with key applications as ligands, biological probes and flame retardants using a range of organic coupling partners.
In 1999, Lipshutz and co-workers reported the first phosphorylation of aryl nonaflates using 5 mol% Pd(PPhs)4 and diphenylphosphine-borane as the coupling partner (Scheme 76).218 The protocol was used to prepare BH3-stabilized triarylphosphines in good to excellent yields. Chemoselectivity with respect to aryl chlorides, which remained unreacted under these conditions, is noteworthy feature of this method.
Scheme 76.

Pd-catalyzed phosphorylation of aryl nonaflates reported by Lipshutz in 1999.
15.1. Phosphorylations of Aryl Mesylates
In 2015, Kwong and co-workers reported the first general method for the palladium-catalyzed Hirao cross-coupling of aryl mesylates (Scheme 77).219 The reactions were performed using 0.5 mol% Pd(OAc)2 and 2 mol% CM-Phos with electronically-activated mesylates as C–O electrophiles, while less activated substrates required 1.5 mol% palladium loading. Dialkyl phosphites and phenylphosphinates could be applied as phosphorylating partners. This transformation showed broad substrate scope. Importantly, medicinally-relevant substrates, such as 17β-estradiol and 6-hydroxyflavone afforded the phosphorylated products in high yields.
Scheme 77.

Pd-catalyzed phosphorylation of aryl mesylates reported by Kwong.
15.2. Phosphorylations of Aryl Tosylates
In 2019, the Kwong group has also demonstrated that aryl tosylates undergo the Pd-catalyzed Hirao cross-coupling using the same catalyst system as for aryl mesylates (Scheme 78 cf. Scheme 77).219 It is worth noting that some cross-couplings of tosylates could be performed at 0.25 mol% loading.
Scheme 78.

Pd-catalyzed phosphorylation of aryl tosylates reported by Kwong.
15.3. Phosphorylations of Aryl Nonaflates
In 2016, Lassaletta and co-workers reported the synthesis of axially chiral P,N-ligands via phosphorylation of aryl nonaflates (Scheme 79).220 This cross-coupling was catalyzed by Pd(dba)2 and a Josiphos-type bidentate ligands using TMS activated diaryl or dialkylphosphines. The axially chiral products were formed in high yields and with excellent enantioselectivity in most cases.
Scheme 79.

Pd-catalyzed phosphorylation of aryl nonaflates reported by Lassaletta.
Other examples of palladium-catalyzed phosphorylation of aryl nonaflates using PPh3 and dppe based systems have been reported.221–223
15.4. Phosphorylations of Aryl Imidazolyl Sulfonates
In 2009, Wu and co-workers reported an efficient protocol for the Hirao cross-coupling of aryl imidazolylsulfonates with H-phosphonate diesters (Scheme 80).224 The reaction was catalyzed by 5 mol% Pd(OAc)2 and 5 mol% dppp. A broad range of electronically-diverse imidazolylsulfonates afforded the desired arylphosphonates in good to excellent yields using diethyl or diisopropyl phosphite as the cross-coupling partners.
Scheme 80.

Pd-catalyzed phosphorylation of aryl imidazolyl sulfonates reported by Wu.
16. Palladium-Catalyzed Reductions of C–O Electrophiles
Palladium-catalyzed reduction permits for de-functionalization of nucleophilic phenols to neutral arenes under mild conditions.
In 1987, Greene and co-workers reported reduction of vinyl phosphates to olefins using Pd(PPh3)4 a catalyst in the presence of Al(C2H5)3 a as reductant (Scheme 81).225 This mild method (RT, DCE), afforded olefins in high yields and with good stereoselectivity.
Scheme 81.

Pd-catalyzed reduction of vinyl phosphates reported by Greene in 1987.
16.1. Reductions of Aryl Mesylates
In 1990, Cabri and co-workers reported the reduction of aryl mesylates using Pd(OAc)2/dppb catalyst system in the presence of triethylammonium formate as a reductant (Scheme 82).226 Under these reaction conditions, electronically-activated aryl mesylates, 1-naphthyl mesylate and 1-hydroxy-9,10-anthraquinone mesylate were reduced to the corresponding hydrocarbons in high yields.
Scheme 82.

Pd-catalyzed reduction of aryl mesylates reported by Cabri.
16.2. Reductions of Aryl Tosylates
In 2014, the Kwong group reported the first general method for the Pd-catalyzed reduction of aryl tosylates (Scheme 83).227 The combination of Pd(OAc)2 and CM-Phos using i-PrOH as the reductant was identified as the most effective system for this transformation. The catalyst loading could be reduced to 0.5 mol% in case of electronically-activated tosylates. Furthermore, several sensitive functional groups, including nitriles, ketones, and esters, were well compatible with this protocol. Interestingly, labelling studies indicated that the hydride source is the C–H bond of isopropanol rather than the O–H moiety.
Scheme 83.

Pd-catalyzed reduction of aryl tosylates reported by Kwong.
16.3. Reductions of Aryl Fluorosulfonates
In 2017, Qin and co-workers reported another efficient method for the direct reduction of phenols via in situ formed aryl fluorosulfonates (Scheme 84).228 The reduction was performed using Pd(OAc)2/ dppp system in the presence of HCO2H as a reductant at very mild room temperature conditions. A broad range of phenols was compatible with this method, irrespective of the electronic substitution of the substrates. Moreover, polyphenols have also been found effective substrates for this reaction.
Scheme 84.

Pd-catalyzed reduction of aryl fluorosulfonates reported by Qin.
16.4. Reductions of Aryl Benzenesulfonates
The reduction of aryl benzenesulfonates to areneswas reported by Cabri and co-workers using Pd(OAc)2 and dppb catalyst system in the presence of HCO2H (Scheme 85).226 In general, comparable yields to the reduction of aryl tosylates were obtained (cf. Scheme 82).
Scheme 85.

Pd-catalyzed reduction of aryl benzenesulfonates reported by Cabri.
16.5. Reductions of Aryl Nonaflates
In 1999, Lipshutz and co-workers reported a protocol for the reduction of aryl nonaflates to arenes using Pd(PPh3)4 catalyst and dimethylamine-borane as a reductant (Scheme 86).229 Notably, the method is compatible with a broad range of functional groups, including olefins, amides, esters, aryl chlorides and even bromides.
Scheme 86.

Pd-catalyzed reduction of aryl nonaflates reported by Lipshutz.
16.5. Reductions of Vinyl Phosphates
In 2008, Sasaki and co-workers reported the synthesis of enol ethers and enecarbamates via Pd-catalyzed reduction of vinyl phosphates using Pd(PPh3)4 as a catalyst and Me2PhSiH as a reductant (Scheme 87).230 Alkenyl phosphates were reduced with excellent chemoselectivity, affording the cross-coupling products in high yields. Furthermore, the protocol was applied as a key step to the synthesis of two alkaloids, lennoxamine and chilenine.
Scheme 87.

Pd-catalyzed reduction of vinyl phosphates reported by Sasaki.
17. Palladium-Catalyzed Thiolations of C–O Electrophiles
There are only a few examples of Pd-catalyzed thiolation of C–O electrophiles at present.
17.1. Thiolation of Aryl Tosylates
In 2006, Hartwig and co-workers reported a single example of the cross-coupling between an aryl tosylate and 1-octanethiol (Scheme 88).231 The desired product was obtained in 86% yield using a catalyst system based on 2 mol% Pd(OAc)2 and 2 mol% CyPF-t-Bu. However, the reaction was not observed using benzenethiol as nucleophile.
Scheme 88.

Pd-catalyzed thiolation of aryl tosylates reported by Hartwig.
17.2. Thiolation of Vinyl Phosphates
In 2003, Kocienski and co-workers reported the synthesis of α-benzenesulfanyl enol ethers via Pd-catalyzed cross-coupling between in situ formed vinyl phosphates and sodium thiophenolate (Scheme 89).232 The reaction afforded moderate to good yields using Pd(PPhs)4 as a catalyst in refluxing THF.
Scheme 89.

Pd-catalyzed thiolation of vinyl phosphates reported by Kocienski.
18. Conclusions
In summary, major advances have been made in the palladium-catalyzed cross-coupling of bench-stable C–O electrophiles. Two major research directions involved (1) the development of more reactive catalyst system, mostly enabled by the design of electron-rich ancillary ligands, and (2) the establishment of more effective air- and moisture-stable activating groups for C–O electrophiles. The excellent functional group tolerance of palladium-catalyzed cross-couplings established across numerous reactions, catalyst systems and C–O electrophiles, compares very favourably with other metals, including more electropositive Ni, and bodes well for the routine application of the palladium-catalyzed C–O cross-couplings of bench-stable electrophiles across various areas of catalysis (cf. Figure 1A).
Among the key developments in this area were (1) the design of dialkylbiarylphosphines, in particular XPhos, and indolyl-phosphines, in particular, CM-Phos, by Buchwald and Kwong groups, respectively, as well as MorDal-Phos by Stradiotto group, and the development of several general catalytic procedures involving diverse C–O electrophiles, some at low catalyst loading; (2) establishment of robust protocols for in situ activation of phenols by Beller, Qin, Kalyani and Hanley groups using diverse activating groups; (3) expansion of the palladium-catalyzed cross-coupling to less reactive C–O electrophiles, such as sulfamates by the groups of Hazari, Molander, and Shao; (4) application of the C–O cross-coupling to the synthesis of biologically-relevant targets by the groups of Lakshman, Kang and Sasaki; and (5) expansion of the C–O cross-coupling manifold of bench-stable electrophiles to novel nucleophiles, including acidic arenes and carboxylates by the Ackermann and Gooßen groups, respectively.
Despite remarkable progress, several challenges remain to be addressed in this field, including: (1) only few protocols for the Pd-catalyzed cross-coupling of less reactive C–O electrophiles have been reported; (2) there is an urgent need for the development of mild conditions at low temperature and low catalyst loading that would be of wide interest to industrial chemists; (3) only few novel transformations have been reported; (4) green, sustainable protocols using C–O electrophiles, including flow synthesis, have not received appropriate attention. It would also be interesting to probe the reactivity of less activated electrophiles, such as O-acyl groups including acetates233 that potentially could make the Pd-catalysed manifold more attractive owing to the synthetic and economic advantages. Another interesting point would involve a more detailed investigation of the behaviour of anions in R–Pd(II)–OX intermediates after oxidative addition, which should behave differently from the halide-type R–Pd(II)–X intermediates. We believe that the major advancements in this area will be accomplished through further studies on ancillary ligand design and will be closely tied to the mechanistic understanding of C–O cross-couplings.
Overall, the studies described in this review represent exciting developments that are likely to facilitate general applications of the C–O catalysis platform.
Acknowledgements
We thank Rutgers University, the NSF (CAREER CHE-1650766), and the NIH (1R35GM133326) for generous financial support of our studies in catalysis and inert bond activation.
Biographies
BIOGRAPHIES

Tongliang Zhou was born and raised in Hubei Province, P.R. of China, and obtained his B.Sc. degree from Wuhan University in 2013. After a short stay in industry, he joined the group of Prof. Ping Xu at Peking University, and obtained his M.Sc. degree in 2018 in medicinal chemistry. Currently, he is a Ph.D. student in the group of Prof. Michal Szostak at Rutgers University. His research interests are focused on transition-metal-catalysis, catalyst development, C–N and C–O activation reactions.

Michal Szostak received his Ph.D. from the University of Kansas with Prof. Jeffrey Aubé in 2009. After postdoctoral stints at Princeton University with Prof. David MacMillan and University of Manchester with Prof. David Procter, in 2014, he joined the faculty at Rutgers University. His research group is focused on the development of new catalytic methods based on transition-metal-catalysis, amide bonds, N–C, O–C, C–H bond activation, Pd–NHC catalysis, twisted amides and synthetic methodology. He is the author of over 170 peer refereed publications.
Footnotes
Publisher's Disclaimer: This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication.
Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.For general reviews, see:; (a) Molander GA, Wolfe JP and Larhed M, Science of Synthesis: Cross-Coupling and Heck-Type Reactions, 1st ed., Thieme, 2013; [Google Scholar]; (b) de Meijere A, Bräse S and Oestreich M, Metal-Catalyzed Cross-Coupling Reactions and More, 1st ed., Wiley, 2014; [Google Scholar]; (c) Colacot TJ, New Trends in Cross-Coupling, 1st ed., The Royal Society of Chemistry, 2015. [Google Scholar]
- 2.For perspectives on the historical importance of Pd-catalyzed cross-couplings, see:; (a) Wu XF, Anbarasan P, Neumann H and Beller M, Angew. Chem. Int. Ed, 2010, 49, 9047; [DOI] [PubMed] [Google Scholar]; (b) Johansson-Seechurn CCC, Kitching MO, Colacot TJ and Snieckus V, Angew. Chem. Int. Ed, 2012, 51, 5062. [DOI] [PubMed] [Google Scholar]
- 3.For further reviews on Pd-catalyzed cross-couplings, see:; (a) Torborg C and Beller M, Adv. Synth. Catal, 2009, 351, 3027; [Google Scholar]; (b) Beller M and Blaser HU, Organometallics as Catalysts in the Fine Chemicals Industry, Springer, 2012; [Google Scholar]; (c) Magano J and Dunetz JR, Chem. Rev, 2011, 111, 2177; [DOI] [PubMed] [Google Scholar]; (d) Busacca CA, Fandrick DR, Song JJ and Senanayake CH, Adv. Synth. Catal, 2011, 353, 1825; [Google Scholar]; (e) Crawley ML and Trost BM, Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, Wiley, 2012; [Google Scholar]; (f) Molnar A, Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments, Wiley, 2013; [Google Scholar]; (g) Dorel R, Grugel CP and Haydl AM. Angew. Chem. Int. Ed, 2019, 58, 17118. [DOI] [PubMed] [Google Scholar]
- 4.For recent reviews, see:; (a) Gildner PG and Colacot TJ, Organometallics, 2015, 34, 5497; [Google Scholar]; (b) Campeau LC and Hazari N, Organometallics, 2019, 38, 3; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Beletskaya IP, Alonso F and Tyurin V, Coord. Chem. Rev, 2019, 385, 137. [Google Scholar]
- 5.(a) Rappoport Z, The Chemistry of Phenols, Wiley, 2003; [Google Scholar]; (b) Warren S, Chemistry of the Carbonyl Group, Wiley, 1991; For further perspectives, see; [Google Scholar]; (c) Gooßen LJ, Gooßen K and Stanciu C, Angew. Chem. Int. Ed, 2009, 48, 3569; [DOI] [PubMed] [Google Scholar]; (d) Knappke CEI and Jacobi von Wangelin A, Angew. Chem. Int. Ed, 2010, 49, 3568. [DOI] [PubMed] [Google Scholar]
- 6.For the seminal study by Wenkert, see:; (a) Wenkert E, Michelotti EL and Swindell CS, J. Am. Chem. Soc, 1979, 101, 2246; For reviews on Ni-catalyzed C–O activation, see; [Google Scholar]; (b) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK and Percec V, Chem. Rev, 2011, 111, 1346; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yamaguchi J, Muto K and Itami K, Eur. J. Org. Chem, 2013, 19; [Google Scholar]; (d) Cornella J, Zarate C and Martin R, Chem. Soc. Rev, 2014, 43, 8081; [DOI] [PubMed] [Google Scholar]; (e) Tobisu M and Chatani N, Acc. Chem. Res, 2015, 48, 1717; [DOI] [PubMed] [Google Scholar]; (f) Tollefson EJ, Hanna LE and Jarvo ER, Acc. Chem. Res, 2015, 48, 2344; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhang YF and Shi ZJ, Acc. Chem. Res, 2019, 52, 161. [DOI] [PubMed] [Google Scholar]
- 7.For a review on Fe-catalyzed C–O activation, see:; Bisz E and Szostak M, ChemSusChem, 2017, 10, 3964. [DOI] [PubMed] [Google Scholar]
- 8.For a recent review on Pd-catalyzed activation of ester C–O bonds, see:; Shi S, Nolan SP and Szostak M, Acc. Chem. Res, 2018, 51, 2589. [DOI] [PubMed] [Google Scholar]
- 9.Hartwig JF, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 2010. [Google Scholar]
- 10.(a) Li WN and Wang ZL, RSC Adv, 2013, 3, 25565; [Google Scholar]; (b) So CM and Kwong FY, Chem. Soc. Rev, 2011, 40, 4963; [DOI] [PubMed] [Google Scholar]; (c) Nelson DJ, Cazin CSJ and Nolan SP, Phys. Sci. Rev, 2016, Vol. 3, 20160018; [Google Scholar]; (d)see, refs. 1–3.
- 11.Miyaura N, Yamada K and Suzuki A, Tetrahedron Lett, 1979, 20, 3437. [Google Scholar]
- 12.Miyaura N and Suzuki A, Chem. Rev, 1995, 95, 2457. [Google Scholar]
- 13.Percec V, Bae J-Y and Hill DH, J. Org. Chem, 1995, 60, 1060. [Google Scholar]
- 14.So CM, Lau CP and Kwong FY, Angew. Chem., Int. Ed, 2008, 47, 8059. [DOI] [PubMed] [Google Scholar]
- 15.Bhayana B, Fors BP and Buchwald SL, Org. Lett, 2009, 11, 3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chow WK, So CM, Lau CP and Kwong FY, J. Org. Chem, 2010, 75, 5109. [DOI] [PubMed] [Google Scholar]
- 17.Molander GA and Beaumard F, Org. Lett, 2011, 13, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung KH, So CM, Wong SM, Luk CH, Zhou ZY, Lau CP and Kwong FY, Chem. Commun, 2012, 48, 1967. [DOI] [PubMed] [Google Scholar]
- 19.Molander GA and Shin I, Org. Lett, 2012, 14, 3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z-Y, Chen G-Q and Shao L-X, J. Org. Chem, 2012, 77, 6608. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen HN, Huang X and Buchwald SL, J. Am. Chem. Soc, 2003, 125, 11818. [DOI] [PubMed] [Google Scholar]
- 22.Gunda P, Russon LM and Lakshman MK, Angew. Chem., Int. Ed, 2004, 43, 6372. [DOI] [PubMed] [Google Scholar]
- 23.Gøgsig TM, Søbjerg LS, Lindhardt AT, Jensen KL and Skrydstrup T, J. Org. Chem, 2008, 73, 3404. [DOI] [PubMed] [Google Scholar]
- 24.Netherton MR and Fu GC, Angew. Chem., Int. Ed, 2002, 41, 3910. [DOI] [PubMed] [Google Scholar]
- 25.Fang Y, Zhang L, Li J, Jin X, Yuan M, Li R, Wu R and Fang J, Org. Lett, 2015, 17, 798. [DOI] [PubMed] [Google Scholar]
- 26.Quan Z-J, Jing F-Q, Zhang Z, Da Y-X and Wang X-C, Eur. J. Org. Chem, 2013, 7175. [Google Scholar]
- 27.Yuen OY, Pang WH, Chen X, Chen Z, Kwong FY and So CM, Synlett, 2019, 30, 731. [Google Scholar]
- 28.Yang J-F, Liu S-J, Zheng J-F and Zhou J-R, Eur. J. Org. Chem, 2012, 6248. [Google Scholar]
- 29.So CM, Lau CP, Chan ASC and Kwong FY, J. Org. Chem, 2008, 73, 7731. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, Meng T and Wu J, J. Org. Chem, 2007, 72, 9346. [DOI] [PubMed] [Google Scholar]
- 31.Wu J, Zhu Q, Wang L, Fathi R and Yang Z, J. Org. Chem, 2003, 68, 670. [DOI] [PubMed] [Google Scholar]
- 32.Wu J, Zhang L and Xia HG, Tetrahedron Lett, 2006, 47, 1525. [Google Scholar]
- 33.Luo Y and Wu J, Tetrahedron Lett, 2009, 50, 2103. [Google Scholar]
- 34.Wang Z, Fan R and Wu J, Adv. Synth. Catal, 2007, 349, 1943. [Google Scholar]
- 35.Zhang L, Meng T, Fan R and Wu J, J. Org. Chem, 2007, 72, 7279. [DOI] [PubMed] [Google Scholar]
- 36.Baxter JM, Steinhuebel D, Palucki M and Davies IW, Org. Lett, 2005, 7, 215. [DOI] [PubMed] [Google Scholar]
- 37.Nakatsuji H, Nishikado H, Ueno K and Tanabe Y, Org. Lett, 2009, 11, 4258. [DOI] [PubMed] [Google Scholar]
- 38.Jacks TE, Belmont DT, Briggs CA, Horne NM, Kanter GD, Karrick GL, Krikke JJ, McCabe RJ, Mustakis JG, Nanninga TN, Risedorph GS, Seamans RE, Skeenan RE, Winkle DD and Zennie TM , Org. Process Res. Dev, 2004, 8, 201. [Google Scholar]
- 39.Lakshman MK, Thomson PF, Nuqui MA, Hilmer JH, Sevova N and Boggess B, Org. Lett, 2002, 4, 1479. [DOI] [PubMed] [Google Scholar]
- 40.Lakshman MK, Gunda P and Pradhan P, J. Org. Chem, 2005, 70, 10329. [DOI] [PubMed] [Google Scholar]
- 41.Kang SB, De Clercq E and Lakshman MK, J. Org. Chem, 2007, 72, 5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X-X, Xu B-B, Song W-T, Sun K-X and Lu J-M, Org. Biomol. Chem, 2015, 13, 4925. [DOI] [PubMed] [Google Scholar]
- 43.Abe T, Mino T, Watanabe K, Yagishita F and Sakamoto M, Eur. J. Org. Chem, 2014, 3909. [DOI] [PubMed] [Google Scholar]
- 44.Waheed M and Ahmed N, Synthesis, 2017, 49, 4372. [Google Scholar]
- 45.Wang Q, Dai Z, Di X, Huang Q, Wang Y and Zhu J, Mol. Divers, 2019, DOI: 10.1007/s11030-019-10001-4. [DOI] [PubMed] [Google Scholar]
- 46.Dikova A, Cheval NP, Blanc A, Weibel J-M and Pale P, Tetrahedron, 2016, 72, 1960. [Google Scholar]
- 47.Joseph JT, Sajith AM, Ningegowda RC, Nagaraj A, Rangappa KS and Shashikanth S, Tetrahedron Lett, 2015, 56, 5106. [Google Scholar]
- 48.Roth GP and Fuller CE, J. Org. Chem, 1991, 56, 3493. [Google Scholar]
- 49.Hanley PS, Clark TP, Krasovskiy AL, Ober MS, O’Brien JP and Staton TS, ACS Catal, 2016, 6, 3515. [Google Scholar]
- 50.Domino K, Veryser C, Wahlqvist BA, Gaardbo C, Neumann KT, Daasbjerg K, De Borggraeve WM and Skrydstrup T, Angew. Chem., Int. Ed, 2018, 57, 6858. [DOI] [PubMed] [Google Scholar]
- 51.Ma C, Zhao C-Q, Xu X-T, Li Z-M, Wang X-Y, Zhang K and Mei T-S, Org. Lett, 2019, 21, 2464. [DOI] [PubMed] [Google Scholar]
- 52.Lekkala R, Lekkala R, Moku B, Rakesh KP and Qin HL, Org. Chem. Front, 2019, 6, 3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hanley PS, Ober MS, Krasovskiy AL, Whiteker GT and Kruper WJ, ACS Catal, 2015, 5, 5041. [Google Scholar]
- 54.Liang Q, Xing P, Huang Z, Dong J, Sharpless KB, Li X and Jiang B, Org. Lett, 2015, 17, 1942. [DOI] [PubMed] [Google Scholar]
- 55.Li X, Ma Y, Hu Q, Jiang B, Wu Q and Yuan Z, Catal. Commun, 2018, 117, 57. [Google Scholar]
- 56.Li X, Zhang H, Feng F, Hu Q and Yuan Z, ChemistrySelect, 2018, 3, 12287. [Google Scholar]
- 57.Zhang E, Tang J, Li S, Wu P, Moses JE and Sharpless KB, Chem. Eur. J, 2016, 22, 5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nibin Joy M, Yadav Bodke D, AbdulKhader KK, Sajith AM, Venkatesh T and Ajeesh Kumar AK, J. Fluor. Chem, 2016, 182, 109. [Google Scholar]
- 59.Yamaguchi M, Kimura T, Shinohara N and Manabe K, Molecules, 2013, 18, 15207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hommes P, Reissig H-U, Asian J. Org. Chem, 2016, 5, 1033. [Google Scholar]
- 61.Blettner CG, König WA, Stenzel W and Schotten T, J. Org. Chem, 1999, 64, 3885. [Google Scholar]
- 62.Albaneze-Walker J, Raju R, Vance JA, Goodman AJ, Reeder MR, Liao J, Maust MT, Irish PA, Espino P and Andrews DR, Org. Lett, 2009, 11, 1463. [DOI] [PubMed] [Google Scholar]
- 63.Cívicos JF, Gholinejad M, Alonso DA and Nájera C, Chem. Lett, 2011, 40, 907. [Google Scholar]
- 64.Cívicos JF, Alonso DA and Nájera C, Eur. J. Org. Chem, 2012, 3670. [Google Scholar]
- 65.Cívicos JF, Alonso DA and Nájera C, Adv. Synth. Catal, 2012, 354, 2771. [Google Scholar]
- 66.Melvin PR, Hazari N, Beromi MM, Shah HP and Williams MJ, Org. Lett, 2016, 18, 5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Molander GA and Shin I, Org. Lett, 2013, 15, 2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Z-Y, Ma Q-N, Li R-H and Shao L-X, Org. Biomol. Chem, 2013, 11, 7899. [DOI] [PubMed] [Google Scholar]
- 69.Wang X-X, Luo M-J and Lu J-M, Org. Biomol. Chem, 2015, 13, 11438. [DOI] [PubMed] [Google Scholar]
- 70.Lepifre F, Buon C, Rabot R, Bouyssou P and Coudert G, Tetrahedron Lett,1999, 40, 6373. [Google Scholar]
- 71.Kang FA, Sui Z and Murray WV, J. Am. Chem. Soc, 2008, 130, 11300. [DOI] [PubMed] [Google Scholar]
- 72.Sasaki M, Fuwa H, Ishikawa M and Tachibana K, Org. Lett, 1999, 1, 1075. [Google Scholar]
- 73.Lepifre F, Clavier S, Bouyssou P and Coudert G, Tetrahedron, 2001, 57, 6969. [Google Scholar]
- 74.Campbell IB, Guo J, Jones E and Steel PG, Org. Biomol. Chem, 2004, 2, 2725. [DOI] [PubMed] [Google Scholar]
- 75.Larsen US, Martiny L and Begtrup M, Tetrahedron Lett, 2005, 46, 4261. [Google Scholar]
- 76.Fuwa H, Ebine M, Bourdelais AJ, Baden DG and Sasaki M, J. Am. Chem. Soc, 2006, 128, 16989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mousset D, Gillaizeau I, Sabatié A, Bouyssou P and Coudert G, J. Org. Chem, 2006, 71, 5993. [DOI] [PubMed] [Google Scholar]
- 78.Pedzisa L, Vaughn IW and Pongdee R, Tetrahedron Lett, 2008, 49, 4142. [Google Scholar]
- 79.Li S-M, Huang J, Chen G-J and Han F-S, Chem. Commun, 2011, 47, 12840. [DOI] [PubMed] [Google Scholar]
- 80.Kosugi M, Kameyama M and Migita T, Chem. Lett, 1983, 12, 927. [Google Scholar]
- 81.Guram AS, Rennels RA and Buchwald SL, Angew. Chem., Int. Ed, 1995, 34, 1348. [Google Scholar]
- 82.Louie J and Hartwig JF, Tetrahedron Lett, 1995, 36, 3609. [Google Scholar]
- 83.Ingoglia BT, Wagen CC and Buchwald SL, Tetrahedron, 2019, 75, 4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hamann BC and Hartwig JF, J. Am. Chem. Soc, 1998, 120, 7369. [Google Scholar]
- 85.So CM, Zhou Z, Lau CP and Kwong FY, Angew. Chem., Int. Ed, 2008, 47, 6402. [DOI] [PubMed] [Google Scholar]
- 86.Fors BP, Watson DA, Biscoe MR and Buchwald SL, J. Am. Chem. Soc, 2008, 130, 13552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang W and Du D-M, Org. Lett, 2010, 12, 2350. [DOI] [PubMed] [Google Scholar]
- 88.Alsabeh PG and Stradiotto M, Angew. Chem., Int. Ed, 2013, 52, 7242. [DOI] [PubMed] [Google Scholar]
- 89.Ogata T and Hartwig JF, J. Am. Chem. Soc, 2008, 130, 13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vo GD and Hartwig JF, J. Am. Chem. Soc, 2009, 131, 11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lundgren RJ and Stradiotto M, Angew. Chem., Int. Ed, 2010, 49, 8686. [DOI] [PubMed] [Google Scholar]
- 92.Huang X, Anderson KW, Zim D, Jiang L, Klapars A and Buchwald SL, J. Am. Chem. Soc, 2003, 125, 6653. [DOI] [PubMed] [Google Scholar]
- 93.Roy AH and Hartwig JF, J. Am. Chem. Soc, 2003, 125, 8704. [DOI] [PubMed] [Google Scholar]
- 94.Klapars A, Campos KR, Chen CY and Volante RP, Org. Lett, 2005, 7, 1185. [DOI] [PubMed] [Google Scholar]
- 95.Lundgren RJ, Peters BD, Alsabeh PG and Stradiotto M, Angew. Chem., Int. Ed, 2010, 49, 4071. [DOI] [PubMed] [Google Scholar]
- 96.Mantel MLH, Lindhardt AT, Lupp D and Skrydstrup T, Chem. Eur. J, 2010, 16, 5437. [DOI] [PubMed] [Google Scholar]
- 97.Choy PY, Chung KH, Yang Q, So CM, Sun RW-Y, Kwong FY, Chem. Asian J, 2018, 13, 2465. [DOI] [PubMed] [Google Scholar]
- 98.Yang Q, Choy PY, Zha Q, Leung MP, Chan HS, So CM, Wong W-T and Kwong FY, J. Org. Chem, 2018, 83, 11369. [DOI] [PubMed] [Google Scholar]
- 99.Xie X, Ni G, Ma F, Ding L, Xu S, Zhang Z, Synlett 2011, 7, 955. [Google Scholar]
- 100.Alsabeh PG, Lundgren RJ, McDonald R, Johansson Seechurn CCC, Colacot TJ and Stradiotto M, Chem. Eur. J, 2013, 19, 2131. [DOI] [PubMed] [Google Scholar]
- 101.Zhang Y, Lavigne G and César V, J. Org. Chem, 2015, 80, 7666. [DOI] [PubMed] [Google Scholar]
- 102.Lim T, Byun S and Kim BM, Asian J. Org. Chem, 2017, 6, 1222. [Google Scholar]
- 103.Fang W-Y, Huang Y-M, Leng J and Qin H-L, Asian J. Org. Chem, 2018, 7, 751. [Google Scholar]
- 104.Anderson KW, Mendez-Perez M, Priego J and Buchwald SL, J. Org. Chem, 2003, 68, 9563. [DOI] [PubMed] [Google Scholar]
- 105.Shekhar S, Dunn TB, Kotecki BJ, Montavon DK and Cullen SC, J. Org. Chem, 2011, 76, 4552. [DOI] [PubMed] [Google Scholar]
- 106.Joy MN, Bodke YD, Khader KKA, Ali Padusha MS, Sajith AM and Muralidharan A, RSC Adv, 2014, 4, 19766. [Google Scholar]
- 107.Ramírez-López P, Ros A, Romero-Arenas A, Iglesias-Sigüenza J, Fernández R and Lassaletta JM, J. Am. Chem. Soc, 2016, 138 , 12053. [DOI] [PubMed] [Google Scholar]
- 108.Kawaguchi K, Nakano K and Nozaki K, J. Org. Chem, 2007, 72, 5119. [DOI] [PubMed] [Google Scholar]
- 109.Ackermann L, Sandmann R and Song WF, Org. Lett, 2011, 13, 1784. [DOI] [PubMed] [Google Scholar]
- 110.Heck RF and Nolley JP Jr., J. Org. Chem, 1972, 37, 2320. [Google Scholar]
- 111.Heck Org RF. React, 1982, 27, 345. [Google Scholar]
- 112.Beller M and Blaser HU, Top. Organomet. Chem, 2012, 42, 1. [Google Scholar]
- 113.Dounay AB, Overman LE, Chem. Rev, 2003, 103, 2945. [DOI] [PubMed] [Google Scholar]
- 114.Magano J, Dunetz JR, Chem. Rev, 2011, 111, 2177. [DOI] [PubMed] [Google Scholar]
- 115.Fu X, Zhang S, Yin J, McAllister TL, Jiang SA, Tann C-H, Thiruvengadam TK and Zhang F, Tetrahedron Lett, 2002, 43, 573. [Google Scholar]
- 116.Hansen AL and Skrydstrup T, Org. Lett, 2005, 7, 5585. [DOI] [PubMed] [Google Scholar]
- 117.Hansen AL, Ebran J-P, Ahlquist M, Norrby P-O and Skrydstrup T, Angew. Chem., Int. Ed, 2006, 45, 3349. [DOI] [PubMed] [Google Scholar]
- 118.Noël S. b., Pinel C and Djakovitch L, Org. Biomol. Chem, 2006, 4, 3760. [DOI] [PubMed] [Google Scholar]
- 119.Gøgsig TM, Lindhardt AT, Dekhane M, Grouleff J and Skrydstrup T, Chem. Eur. J, 2009, 15, 5950. [DOI] [PubMed] [Google Scholar]
- 120.Carmona JA, Hornillos V, Ramírez-Lopez P, Ros A, Iglesias-Sigüenza J, Gomez-Bengoa E, Fernandez R and Lassaletta JM, J. Am. Chem. Soc, 2018, 140, 11067. [DOI] [PubMed] [Google Scholar]
- 121.Aulenta F and Reissig H-U, Synlett, 2006, 18, 2993. [Google Scholar]
- 122.Peng AY, Chen BT, Wang Z, Wang B, Mo XB, Wang YY and Chen PJ, J. Fluorine Chem, 2011, 132, 982. [Google Scholar]
- 123.Barluenga J, Florentino L, Aznar F and Valdés C, Org. Lett, 2011, 13, 510. [DOI] [PubMed] [Google Scholar]
- 124.Ebran JP, Hansen AL, Gøgsig TM and Skrydstrup T, J. Am. Chem. Soc, 2007, 129, 6931. [DOI] [PubMed] [Google Scholar]
- 125.Fuwa H and Sasaki M, Chem. Commun, 2007, 2876. [DOI] [PubMed] [Google Scholar]
- 126.Negishi E, Hu Q, Huang Z, Qian M, Wang G, Aldrichimica Acta, 2005, 38, 71. [Google Scholar]
- 127.Organ MG, Avola S, Dubovyk I, Hadei N, Kantchev EA, O’Brien CJ and Valente C, Chem. Eur. J, 2006, 12, 4749. [DOI] [PubMed] [Google Scholar]
- 128.Wu J, Liao Y and Yang Z, J. Org. Chem, 2001, 66, 3642. [DOI] [PubMed] [Google Scholar]
- 129.Zhou J and Fu GC, J. Am. Chem. Soc, 2003, 125, 12527. [DOI] [PubMed] [Google Scholar]
- 130.Gao W, Luo Y, Ding Q, Peng Y and Wu J, Tetrahedron Lett, 2010, 51, 136. [Google Scholar]
- 131.Rottlander M and Knochel P, J. Org. Chem, 1998, 63, 203. [DOI] [PubMed] [Google Scholar]
- 132.Rossi R, Carpita A, Bellina F, Stabile P and Mannina L, Tetrahedron, 2003, 59, 2067. [Google Scholar]
- 133.Mendel M, Kalvet I, Hupperich D, Magnin G, and Schoenebeck F, Angew. Chem., Int. Ed, 2020, 59, 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hansen AL, Ebran J-P, Gøgsig TM and Skrydstrup T, J. Org. Chem, 2007, 72, 6464. [DOI] [PubMed] [Google Scholar]
- 135.Takai K, Oshima K and Nozaki H, Tetrahedron Lett, 1980, 21, 2531. [Google Scholar]
- 136.Fugami K, Oshima K and Utimoto K, Chem. Lett, 1987, 16, 2203. [Google Scholar]
- 137.Sato M, Takai K, Oshima K and Nozaki H, Tetrahedron Lett, 1981, 22, 1609. [Google Scholar]
- 138.Takai K, Sato M, Oshima K and Nozaki H, Bull. Chem. Soc. Jpn, 1984, 57, 108. [Google Scholar]
- 139.Lindhardt AT, Gogsig TM and Skrydstrup T, J. Org. Chem, 2009, 74, 135. [DOI] [PubMed] [Google Scholar]
- 140.Doucet H and Hierso J-C, Angew. Chem., Int. Ed, 2007, 46, 834. [DOI] [PubMed] [Google Scholar]
- 141.Nicolaou KC, Shi G-Q, Namoto K, Bernal F, Chem. Commun, 1998, 1757. [Google Scholar]
- 142.Choy PY, Chow WK, So CM, Lau CP and Kwong FY, Chem. Eur. J, 2010, 16, 9982. [DOI] [PubMed] [Google Scholar]
- 143.Braga AL, Emmerich DJ, Silveira CC, Martins TLC and Rodrigues OED, Synlett, 2001, 369. [Google Scholar]
- 144.Gelman D and Buchwald SL, Angew. Chem., Int. Ed, 2003, 42, 5993. [DOI] [PubMed] [Google Scholar]
- 145.R’kyek O, Halland N, Lindenschmidt A, Alonso J, Lindemann P, Urmann Ma. and Nazaré M, Chem. Eur. J, 2010, 16, 9986. [DOI] [PubMed] [Google Scholar]
- 146.Fu X, Zhang S, Yin J and Schumacher D, Tetrahedron Lett, 2002, 43, 6673. [Google Scholar]
- 147.Luo Y and Wu J, Tetrahedron, 2009, 65, 6810. [Google Scholar]
- 148.Nishihara Y, Ogawa D, Noyori S and Iwasaki M, Chem. Lett, 2012, 41, 1503. [Google Scholar]
- 149.Lu CH, Wang L, Yang F, Wu R and Shen W, RSC Adv, 2014, 4, 30447. [Google Scholar]
- 150.Gallagher WP and Maleczka RE Jr., J. Org. Chem, 2003, 68, 6775. [DOI] [PubMed] [Google Scholar]
- 151.Gallagher WP and Maleczka RE Jr., Synlett, 2003, 537. [Google Scholar]
- 152.Shirbin SJ, Boughton BA, Zammit SC, Zanatta SD, Marcuccio SM, Hutton CA and Williams SJ, Tetrahedron Lett, 2010, 51, 2971. [Google Scholar]
- 153.Cívicos JF, Alonso DA and Nájera C, Adv. Synth. Catal, 2013, 355, 203. [Google Scholar]
- 154.Kang F-A, Lanter JC, Cai C, Sui Z and Murray WV, Chem. Commun, 2010, 46, 1347. [DOI] [PubMed] [Google Scholar]
- 155.Shi C and Aldrich CC, Org. Lett, 2010, 12, 2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Espinet P and Echavarren AM, Angew. Chem., Int. Ed, 2004, 43, 4704. [DOI] [PubMed] [Google Scholar]
- 157.Hettrick CM, Kling JK and Scott WJ, J. Org. Chem, 1991, 56, 1489. [Google Scholar]
- 158.Naber JR, Fors BP, Wu X, Gunn JT and Buchwald SL, Heterocycles, 2010, 80, 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nagatsugi F, Uemura K, Nakashima S, Maeda M and Sasaki S, Tetrahedron, 1997, 53, 3035. [Google Scholar]
- 160.Nagatsugi F, Uemura K, Nakashima S, Maeda M and Sasaki S, Tetrahedron Lett, 1995, 36, 421. [PubMed] [Google Scholar]
- 161.Mathieu J and Marsura A, Synth. Commun, 2003, 33, 409. [Google Scholar]
- 162.Kang HA, Bronstein HE and Swager TM, Macromolecules, 2008, 41, 5540. [Google Scholar]
- 163.Braun M, Sigloch M and Cremer J, Adv. Synth. Catal, 2007, 349, 337. [Google Scholar]
- 164.Badone D, Cecchi R and Guzzi U, J. Org. Chem, 1992, 57, 6321. [Google Scholar]
- 165.Nicolaou KC, Shi G-Q, Gunzner JL, Gärtner P and Yang Z, J. Am. Chem. Soc, 1997, 119, 5467. [Google Scholar]
- 166.Buon C, Bouyssou P and Coudert G, Tetrahedron Lett, 1999, 40, 701. [Google Scholar]
- 167.Jiang J, DeVita RJ, Doss GA, Goulet MT and Wyvratt MJ, J. Am. Chem. Soc, 1999, 121, 593. [Google Scholar]
- 168.Claveau E, Gillaizeau I, Blu J, Bruel A and Coudert G, J. Org. Chem, 2007, 72, 4832. [DOI] [PubMed] [Google Scholar]
- 169.Simas ABC, de Sales DL and Pais KC, Tetrahedron Lett, 2009, 50, 6977. [Google Scholar]
- 170.Steel PG and Woods TM, Synthesis, 2009, 3897. [Google Scholar]
- 171.Denmark SE and Sweis RF, Acc. Chem. Res, 2002, 35, 835. [DOI] [PubMed] [Google Scholar]
- 172.Denmark SE and Sweis RF, Org. Lett, 2002, 4, 3771. [DOI] [PubMed] [Google Scholar]
- 173.Zhang L, Qing J, Yang P and Wu J, Org. Lett, 2008, 10, 4971. [DOI] [PubMed] [Google Scholar]
- 174.So CM, Lee HW, Lau CP and Kwong FY, Org. Lett, 2009, 11, 317. [DOI] [PubMed] [Google Scholar]
- 175.Zhang L and Wu J, J. Am. Chem. Soc, 2008, 130, 12250. [DOI] [PubMed] [Google Scholar]
- 176.Ohgi A, Semba K, Hiyama T and Nakao Y, Chem. Lett, 2016, 45, 973. [Google Scholar]
- 177.Culkin DA and Hartwig JF, Acc. Chem. Res, 2003, 36, 234. [DOI] [PubMed] [Google Scholar]
- 178.Johansson CCC and Colacot TJ, Angew. Chem., Int. Ed, 2010, 49, 676. [DOI] [PubMed] [Google Scholar]
- 179.Kawatsura M and Hartwig JF, J. Am. Chem. Soc, 1999, 121, 1473. [Google Scholar]
- 180.Fu WC, So CM, Yuen OY, Lee IT and Kwong FY, Org. Lett, 2016, 18, 1872. [DOI] [PubMed] [Google Scholar]
- 181.Hesp KD, Lundgren RJ and Stradiotto M, J. Am. Chem. Soc, 2011, 133, 5194. [DOI] [PubMed] [Google Scholar]
- 182.Ackermann L and Mehta VP, Chem. Eur. J, 2012, 18, 10230. [DOI] [PubMed] [Google Scholar]
- 183.McGlacken GP and Bateman LM, Chem. Soc. Rev, 2009, 38, 2447. [DOI] [PubMed] [Google Scholar]
- 184.Ackermann L, Vicente R and Kapdi A, Angew. Chem., Int. Ed, 2009, 48, 9792. [DOI] [PubMed] [Google Scholar]
- 185.Ackermann L, Althammer A and Fenner S, Angew. Chem., Int. Ed, 2009, 48, 201. [DOI] [PubMed] [Google Scholar]
- 186.So CM, Lau CP and Kwong FY, Chem. Eur. J, 2011, 17, 761. [DOI] [PubMed] [Google Scholar]
- 187.Ferguson DM, Rudolph SR and Kalyani D, ACS Catal, 2014, 4, 2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ackermann L and Fenner S, Chem. Commun, 2011, 47, 430. [DOI] [PubMed] [Google Scholar]
- 189.Lee DS, Choy PY, So CM, Wang J, Lau CP and Kwong FY, RSC Adv, 2012, 2, 9179. [Google Scholar]
- 190.Chang JWW, Chia EY, Chai CLL and Seayad J, Org. Biomol. Chem, 2012, 10, 2289. [DOI] [PubMed] [Google Scholar]
- 191.Yi Z, Aschenaki Y, Daley R, Davick S, Schnaith A, Wander R and Kalyani D, J. Org. Chem, 2017, 82, 6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fan S, Yang J and Zhang X, Org. Lett, 2011, 13, 4374. [DOI] [PubMed] [Google Scholar]
- 193.Nervig CS, Waller PJ and Kalyani D, Org. Lett, 2012, 14, 4838. [DOI] [PubMed] [Google Scholar]
- 194.Ackermann L, Barfüsser S and Pospech J, Org. Lett, 2010, 12, 724. [DOI] [PubMed] [Google Scholar]
- 195.Yamaguchi M, Higuchi M, Tazawa K and Manabe K, J. Org. Chem, 2016, 81, 3967. [DOI] [PubMed] [Google Scholar]
- 196.Manabe K and Kimura T, Org. Lett, 2013, 15, 374. [DOI] [PubMed] [Google Scholar]
- 197.Brennführer A, Neumann H and Beller M, Angew. Chem., Int. Ed, 2009, 48, 4114. [DOI] [PubMed] [Google Scholar]
- 198.Kubota Y, Nakada S and Sugi Y, Synlett, 1998, 183. [Google Scholar]
- 199.Munday RH, Martinelli JR and Buchwald SL, J. Am. Chem. Soc, 2008, 130, 2754. [DOI] [PubMed] [Google Scholar]
- 200.Wu X-F, Neumann H and Beller M, Chem. Eur. J, 2012, 13, 3831. [DOI] [PubMed] [Google Scholar]
- 201.Joseph JT, Sajith AM, Ningegowda RC and Shashikanth S, Adv. Synth. Catal, 2017, 359, 419. [Google Scholar]
- 202.Fang W-Y, Leng J and Qin H-L, Chem. Asian J, 2017, 12, 2323. [DOI] [PubMed] [Google Scholar]
- 203.Cai C, Rivera NR, Balsells J, Sidler RR, McWilliams JC, Shultz CS and Sun Y, Org. Lett, 2006, 8, 5161. [DOI] [PubMed] [Google Scholar]
- 204.Lagerlund O, Mantel MLH and Larhed M, Tetrahedron, 2009, 65, 7646. [Google Scholar]
- 205.Occhiato EG, Scarpi D and Prandi C, Heterocycles, 2010, 80, 697. [Google Scholar]
- 206.Rodriguez N and Gooßen LJ, Chem. Soc. Rev, 2011, 40, 5030. [DOI] [PubMed] [Google Scholar]
- 207.Song B, Knauber T and Gooßen LJ, Angew. Chem., Int. Ed, 2013, 52, 2954. [DOI] [PubMed] [Google Scholar]
- 208.Gooßen LJ, Rodríguez N, Lange PP and Linder C, Angew. Chem., Int. Ed, 2010, 49, 1111. [DOI] [PubMed] [Google Scholar]
- 209.Krempl H, Mattmer R and Hanack M, Synthesis, 2000, 1705. [Google Scholar]
- 210.Yeung PY, So CM, Lau CP and Kwong FY, Angew. Chem., Int. Ed, 2010, 49, 8918. [DOI] [PubMed] [Google Scholar]
- 211.Tu Y, Zhang Y, Xu S, Zhang Z and Xie X, Synlett, 2014, 2938. [Google Scholar]
- 212.Zhang JL, Chen XR, Hu TJ, Zhang YA, Xu KL, Yu YP and Huang J, Catal. Lett, 2010, 139, 56. [Google Scholar]
- 213.Zhao C, Fang W-Y, Rakesh KP and Qin H-L, Org. Chem. Front, 2018, 5, 1835. [Google Scholar]
- 214.Leckie SM, Harkness GJ and Clarke ML, Chem. Commun, 2014, 50, 11511. [DOI] [PubMed] [Google Scholar]
- 215.Occhiato EG, Galbo FL and Guarna A, J. Org. Chem, 2005, 70, 7324. [DOI] [PubMed] [Google Scholar]
- 216.Chow WK, So CM, Lau CP and Kwong FY, Chem. Eur. J, 2011, 17, 6913. [DOI] [PubMed] [Google Scholar]
- 217.Murata M, Oda T, Sogabe Y, Tone H, Namikoshi T and Watanabe S, Chem. Lett, 2011, 40, 962. [Google Scholar]
- 218.Lipshutz BH, Buzard DJ and Yun CS, Tetrahedron Lett, 1999, 40, 201. [Google Scholar]
- 219.Fu WC, So CM and Kwong FY, Org. Lett, 2015, 17, 5906. [DOI] [PubMed] [Google Scholar]
- 220.Ramírez-López P, Ros A, Estepa B, Fernández R, Fiser B, Gómez-Bengoa E and Lassaletta JM, ACS Catal, 2016, 6, 3955. [Google Scholar]
- 221.Kwong FY and Chan KS, Organometallics, 2001, 20, 2570. [Google Scholar]
- 222.Wang Y, Lai CW, Kwong FY, Jia W and Chan KS, Tetrahedron, 2004, 60, 9433. [Google Scholar]
- 223.Botman PNM, Fraanje J, Goubitz K, Peschar R, Verhoeven JW, van Maarseveen JH and Hiemstra H, Adv. Synth. Catal, 2004, 346, 743. [Google Scholar]
- 224.Luo Y and Wu J, Organometallics, 2009, 28, 6823. [Google Scholar]
- 225.Charbonnier F, Moyano A and Greene AE, J. Org. Chem, 1987, 52, 2303. [Google Scholar]
- 226.Cabri W, De Bernadinis S, Francalanci F and Penco S, J. Org. Chem, 1990, 55, 350. [Google Scholar]
- 227.Chow WK, So CM, Lau CP and Kwong FY, Org. Chem. Front, 2014, 1, 464. [Google Scholar]
- 228.Wang X-Y, Leng J, Wang S-M, Asiri AM, Marwani HM and Qin H-L, Tetrahedron Lett, 2017, 58, 2340. [Google Scholar]
- 229.Lipshutz BH, Buzard DJ and Vivian RW, Tetrahedron Lett, 1999, 40, 6871. [Google Scholar]
- 230.Fuwa H and Sasaki M, Heterocycles, 2008, 76, 521. [Google Scholar]
- 231.Fernμndez-Rodríguez MA, Shen Q and Hartwig JF, J. Am. Chem. Soc, 2006, 128, 2180. [DOI] [PubMed] [Google Scholar]
- 232.Milne JE and Kocienski PJ, Synthesis 2003, 584. [Google Scholar]
- 233.Becica J, Gaube G, Sabbers WA and Leitch DC, Dalton Trans, 2020, doi: 10.1039/D0DT01119C [DOI] [PubMed] [Google Scholar]