

Abstract
Chronic alcohol consumption is accompanied by intestinal inflammation. However, little is known about how alterations to the intestinal immune system and sphingolipids contribute to pathogenesis of alcoholic liver disease (ALD). We used WT mice, RORγt-deficient mice, sphingosine kinase-deficient mice and local gut anti-inflammatory, 5-aminosalicyclic acid (5-ASA)-treated mice in a chronic-binge ethanol feeding model. Targeted lipidomics assessed the sphingolipids in gut and liver samples. Gut immune cell populations, the amounts of sphingolipids, and the level of liver injury were examined. Results: Alcohol intake induces a pro-inflammatory shift in immune cell populations in the gut, including an increase in Th17 cells. Using RORγt-deficient mice, we found that Th17 cells are required for alcohol-associated gut inflammation and the development of ALD. Treatment with 5-ASA decreases alcohol-induced liver injury and reverses gut inflammation by the suppression of CD4+/RORγt+/IL-17A+ cells. Increased Th17 cells were due to upregulation of SK1 activity and RORγt activation. We found that S1P/S1PR1 signaling are required for the development of Th17 cell-mediated ALD. Importantly, in vivo intervention blocking of S1P/S1PR1 signaling markedly attenuated alcohol-induced liver inflammation, steatosis, and damage. Conclusion: Gut inflammation is a functional alteration of immune cells in ALD. Reducing gut Th17 cells leads to reduced liver damage. S1P signaling was crucial in the pathogenesis of ALD in a Th17 cell dependent manner. Furthermore, our findings suggest that compounds that reduce gut inflammation locally may represent a unique targeted approach in the treatment of ALD.
Keywords: S1P, Th17 cells, gut inflammation, alcoholic steatohepatitis, 5-ASA
Introduction
Recent evidence has pointed to the intestine as a key site that becomes altered in alcoholic liver disease (ALD)(1). In the inflamed gut, CD4 T cells are the main IL-17-secreting cells. Th17 cell differentiation is mediated by the transcription factor, retinoid-related orphan receptor gamma t (RORγt)(2). It has been proposed that infiltration with IL-17A plays a pivotal role in the inflammation associated with ALD(3, 4). However, the local effects of alcohol on most immune cell populations in the gut and the function of gut CD4+IL17A+/RORγt+ cells in the pathogenesis of ALD remains unclear.
The development and maturation of the gut immune system are influenced by dietary lipids including sphingolipids. Some sphingolipids, such as ceramide, favor the development of T regulatory cells (Tregs) by the activation of PP2A(5). In contrast, we demonstrated that S1P, a bioactive sphingolipid, is necessary for Th17 cell migration to inflamed gut(6). Dysregulated sphingolipid metabolites have been implicated in numerous diseases, including fatty liver disease and colitis. The levels of sphingosine kinase 1 (SK1) and its product S1P are increased in patients with colitis(7). SK1 gene expression and synthesis of S1P occur in response to inflammatory stimuli (8). It has been reported that S1P is capable of promoting neutrophil infiltration into the crypts and lamina propria of the colon via ligation of its receptor S1PR1(9). However, the significance of S1P signaling in Th17 cell-mediated gut inflammation in ALD has yet to be evaluated.
Given that targeting intestinal barrier function may be an effective strategy in the prevention/treatment of ALD, we considered using a locally active, gut-specific anti-inflammatory agent. Mesalamine (5-ASA), a first-line maintenance therapy for inflammatory bowel disease (IBD), is a salicylic acid derivative with anti-inflammatory properties that acts locally in the gut with minimal systemic absorption and side effects(10, 11). As ALD is characterized by increased intestinal inflammation and altered permeability, we hypothesized that 5-ASA might have beneficial effects in ALD and may help elucidate roles of gut Th17 cells in this disease.
In this study, we show that the gut immune system is an important modulator of ALD. ALD is accompanied by a pro-inflammatory shift in lamina propria (LP) immune cell polarity, especially Th17 immune cells, in response to an intestinal barrier defect. We further report evidence that sphingolipids impart changes in the intestinal immune system and CD4 T cell homeostasis; notably, an increase of Th17 cells. Targeting gut immune dysregulation may lead to new classes of potentially effective, safe therapies for ALD.
Materials and Methods
Animals and treatments
C57BL/6 WT, RORγt−/−, SK1−/− and SK2−/− mice were obtained from Jackson Laboratory. All animal procedures were approved by the University of Louisville Institutional Animal Care and Use Committee. We used the binge-on-chronic NIAAA (Gao) model with 8-week-old mice. Details of other methods used in this study are described in the Supplemental Materials and Methods.
RESULTS
Alcohol feeding induces the accumulation of CD4+RORγt+/Th17 cells in the intestine
To examine how the gut immune system is affected in ALD, we fed an alcohol (alcohol feeding, AF) or an isocaloric control (pair feeding, PF) diet to male and female C57BL/6 mice. We investigated if gut immune cells are altered by alcohol feeding. After AF, we noted a significant reduction in the proportion of conventional CD8αβ + T cells in the intraepithelial lymphocytes (IEL) and the lamina propria lymphocytes (LPL) of ileum (Figure S1A) and CD4+ T cells in the IEL and LPL of colon (Figure S1B) in AF mice. However, natural CD8αα+ T cells and γδ T cells were less affected in the IEL of the ileum (Figure S1A and S1C). In the LPL of the large intestine (LI-LPL) and the small intestine (SI-LPL), the proportion and number of Th17 cells was significantly increased on the AF diet when compared to the PF diet (Figure 1A and Figure S1D). Th17 cell differentiation is mediated by RORγt. We also found an increase in the proportion and number of RORγt-expressing CD4 T cells in SI-LPL and LI-LPL in AF mice (Figure 1A and Figure S1E). This is consistent with the increase in IL-17A protein, and IL-17A and Rorc mRNA levels in LI-LPL in AF mice (Figure 1B). Furthermore, we detected a slight reduction of Th1 and a significant increase of CD8+ IL-17A+ T cells in the LPL (Figures S1F-S1G). In addition, there was a significant reduction in the proportion of innate lymphoid cells and Th22, and similar proportion of Tregs in AF mice (Figure S1H and S1I). Collectively, these results demonstrate a pro-inflammatory shift in some adaptive and innate T cell populations, and a significant increase in Th17 cells in the intestine of alcohol-fed mice.
Figure 1. ALD is associated with a pro-inflammatory shift in intestinal immune cells.
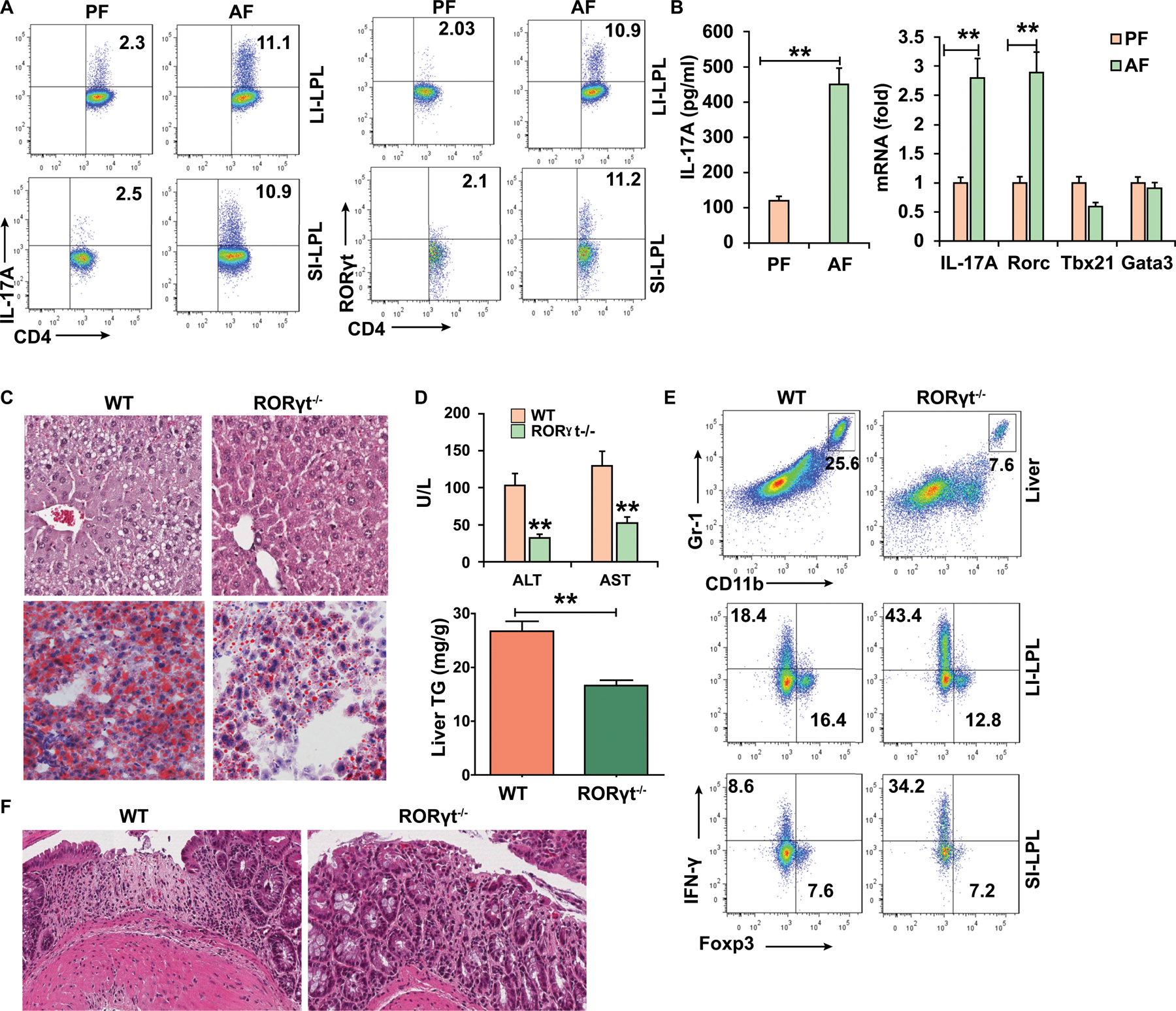
WT mice (A-B) and RORγt−/− (C-F) mice were fed control (pair-fed, PF) or alcohol (alcohol-fed, AF) diet and sacrificed 15–22 days later.
(A) Intracellular staining of IL-17A and RORγt in CD4+ T cells in the lamina propia from large intestine (LI-LPL) or small intestine (SI-LPL).
(B) ELISA analysis of IL-17A in the supernatant of colonic LPLs stimulated with IL-23 (left) and real-time PCR analysis of IL-17A and Rorc mRNA in colonic LPLs (right).
(C-F) WT mice or RORγt−/− mice were fed alcohol diet and sacrificed 15 days later.
(C) HE and Oil red O staining of liver tissue.
(D) Serum levels of ALT and AST and hepatic triglycerides.
(E) Frequencies of immature myeloid cells (CD11b+Gr-1+) in the liver and Th1 and Tregs cells in the gut LPLs.
(F) Large intestine was stained by H&E.
Data in all panels are presented as Mean ± SEM. n=11 * P<0.05; ** P<0.01
CD4+RORγt+/ Th17 cells promote the development of ALD
To define the importance of increased gut RORγt/Th17 cells in ALD, we fed ethanol or control diet to WT and RORγt knockout (RORγt−/−) mice. Of note, some of RORγt−/− mice fed the alcohol diet rapidly died due to a deleterious effect of T-cell lymphomas(12). However, in contrast to WT mice, RORγt−/− mice with no lymphomas showed significant amelioration of the morphological features of ALD including significant attenuation of liver injury, steatosis, and hepatic triglyceride concentrations (Figures 1C and 1D). Moreover, the RORγt−/− mice had less immature myeloid cell accumulation in the liver than WT mice (Figure 1E). We also found a higher proportion of Th1 cells (Figure 1E) and decreased inflammatory infiltrates (Figure 1F) in the gut of the RORγt−/− mice fed alcohol than in WT mice. Clearly, RORγt-deficient mice develop less evidence of alcoholic liver disease and the increase of IL-17+/RORγt+ CD4+ T cells is strongly associated with the development of ALD.
Anti-inflammatory 5-ASA alleviates alcohol-associated gut inflammation and subsequent liver disease
Since alcohol consumption is associated with a pro-inflammatory shift in gut immune populations, we reasoned that therapies aimed at targeting gut inflammation such as mesalamine (5-ASA) may have a role in the treatment of ALD. AF mice treated with 5-ASA had gradual weight loss, a ratio of liver to body weight and food intake similar to AF control mice (Figures S2A-S2B). However, 5-ASA treatment prevented alcohol-induced liver damage and steatosis (Figures 2A–2B). 5-ASA-treated mice also had reduced serum ALT and AST (Figure 2C). Alcohol feeding significantly upregulated several serum lipid markers including TG, VLDL and TC/HDL but only in untreated AF mice, not in 5-ASA treated animals (Figure 2D). To reflect the clinical scenario in which most patients present with alcoholic hepatitis superimposed on established ALD, mice were first fed the ethanol or control diet for 2 weeks to allow development of ALD and then orally treated with 50 mg/kg/d 5-ASA for the last 2 weeks of feeding, while still maintaining the alcohol diet. Treatment of established ALD with 5-ASA prevented progression of alcohol-induced liver damage (ALT increase) and steatosis (Figures S2C and S2D). We also found that 5-ASA treated mice showed an overt reduction of liver fibrosis, as indicated by Sirius red staining (Figure 2E) and by the reduced expression of markers of fibrogenesis, including Tgfb1, alpha-smooth muscle actin (α-SMA), fibronectin (FBN) and pro-Col1a1 (Figure 2F). To confirm the effect of 5-ASA on alcoholic liver fibrosis, we analyzed the serum markers of liver fibrosis, including tissue inhibitor of matrix metalloproteinases 1 (TIMP-1) and hyaluronic acid. 5-ASA treatment significantly decreased these markers compared to PBS in AF mice (Figure 2G).
Figure 2. 5-ASA administration protects mice from ALD.
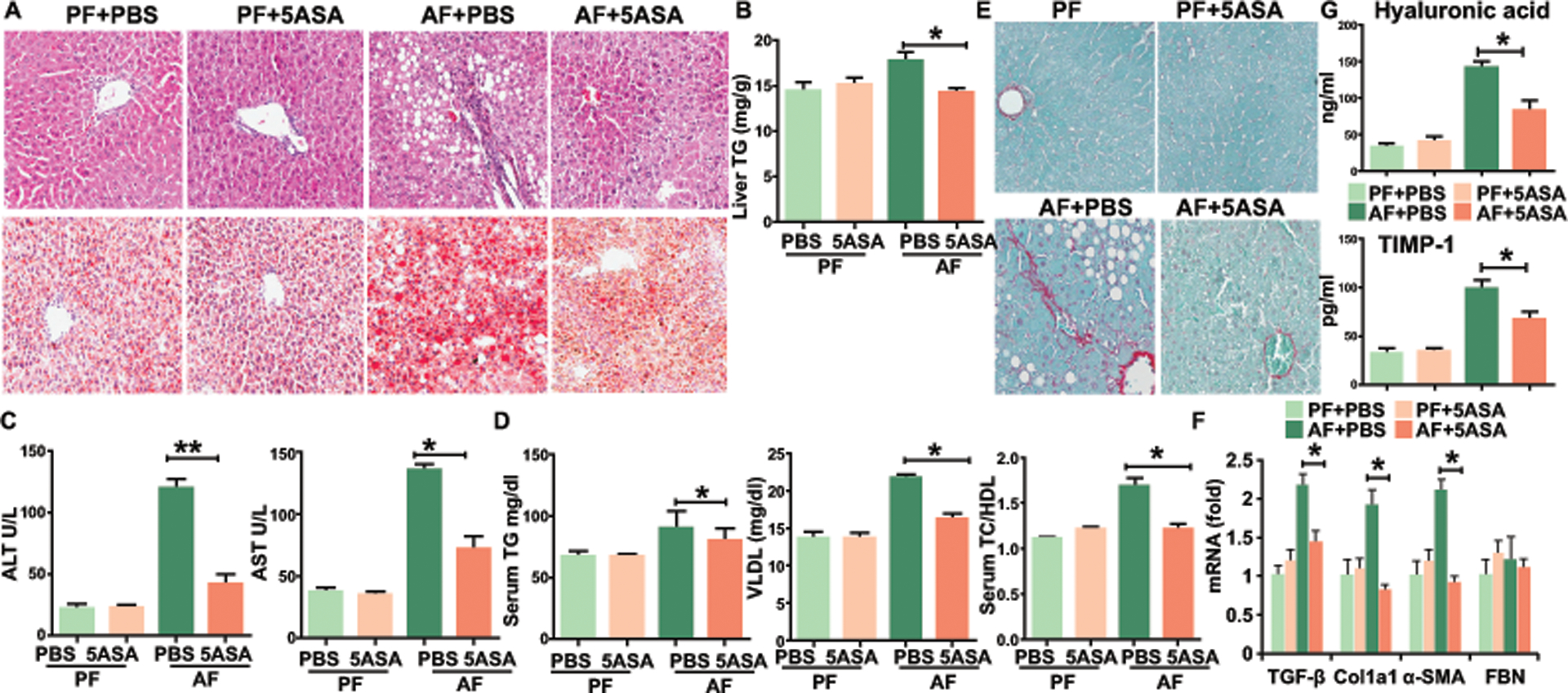
C57BL/6 mice on a pair-fed or binge ethanol diet, with 5-ASA oral supplementation (50 mg/kg/day) or vehicle for 15 days (A-D) or for 28 days (E-G).
(A) H&E and Oil red O staining of liver tissue.
(B) Hepatic triglycerides.
(C) Serum levels of ALT and AST.
(D) Serum levels of lipids were analyzed by piccolo lipid pane plus.
(E) Fibrosis was evaluated by Sirius Red staining.
(F) Real-time PCR analysis of the expression of indicated genes in liver fibrosis.
(G) Serum markers of liver fibrosis, including hyaluronic acid and TIMP-1.
Data in all panels are presented as Mean ± SEM. n>15 * P<0.05; ** P<0.01
5-ASA improves intestine inflammation in AF mice
To understand the mechanisms by which 5-ASA can exert effects on ALD, we examined the effects of 5-ASA on systemic and local immune function during alcohol feeding. 5-ASA treatment showed no effects on immune cell populations and cytokine section in the spleen (Figures S2E), or on circulating immune cell polarity in the blood (Figures S2F). Consistent with minor systemic effects on immune cell function, we could identify only traces of 5-ASA compound in the serum of mice using of high-performance liquid chromatography (HPLC). Instead, 5-ASA was concentrated in the colon and small bowel (Figures S2G). Interestingly, 5-ASA has less effects on the percentages of CD8+ IFNγ+ T cells, Th1, NK and NKT cells in the liver (Figure S2H). The results are in agreement with previous literature demonstrating poor systemic absorption of 5-ASA upon oral administration (10) and highlight the relative specificity of our gut anti-inflammatory therapy. 5-ASA treatment strongly prevented increased accumulation of F4/80-positive macrophages and immature myeloid cells (Figure 3A) and reduced the infiltration of neutrophils into the liver (Figure 3B) compared with untreated AF mice. The levels of TNF-α, COX2, S100A8, S100A9, and CCL20 mRNA in the liver were also decreased in 5-ASA-treated mice (Figure 3C).
Figure 3. 5-ASA improves liver and gut inflammation in mice during alcohol feeding.
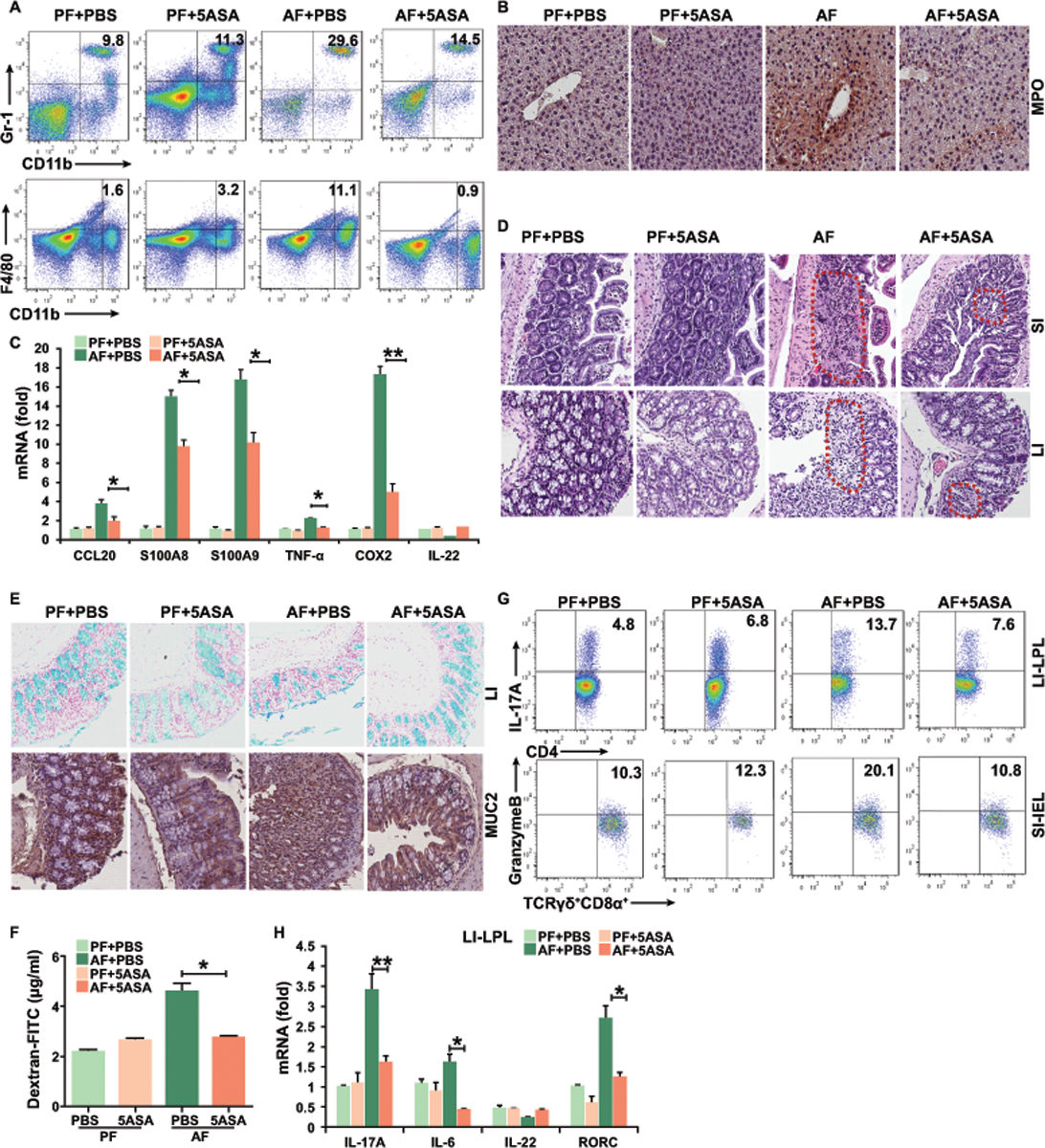
(A) Frequencies of immature myeloid cells (CD11b+Gr-1+) and macrophages (CD11b+F4/80+) in the liver.
(B) Immunohistochemistry staining of myeloperoxidase (MPO) in the liver.
(C) Real-time PCR analysis of the expression of indicated genes in the liver.
(D) H&E staining of small and large intestine.
(E) Alcian Blue staining and immunohistochemistry staining of mucin 2 of colonic tissue.
(F) Plasma levels of Dextran-FITC for gut permeability.
(G) Intracellular staining of IL-17A+ CD4+ T cells in LPL of colon or granzyme B+ γδ+ T cells in IEL of small intestine.
(H) Real-time PCR analysis of the expression of indicated genes in the colonic LPL.
Data in all panels are presented as Mean ± SEM. n>15 * P<0.05; ** P<0.01
We next assessed whether the attenuated liver disease was due to a decreased inflammatory environment in the gut of 5-ASA-treated mice. 5-ASA treatment prevented inflammatory infiltrates in the gut (Figure 3D). 5-ASA-treated mice also had more colon goblet cells and increased expression of mucin compared to AF untreated mice (Figure 3E). RT-PCR analysis showed that the expression of tight junction zonula occludens 1 (ZO-1) and mucin 2 as well as 5a/c were greatly enhanced by 5-ASA treatment (Figure S3A). Indeed, 5-ASA treatment dramatically prevented the translocation of fluorescence-labeled dextran into the plasma (Figure 3F). Of note, 5-ASA treatment showed a significant reduction in Th17 cells and granzymeB+ γδ+T cells in the colon (Figure 3G) and small intestine (Figures S3B). This is consistent with the decrease in IL-6, IL17A and RORγt mRNA levels in the colonic LPL of AF mice after treatment (Figure 3H). However, Th1 cells, Tregs, CD8αβ+ T cells and γδ+T cells in IEL or LPL in the small intestine of 5-ASA-treated AF mice remained similar to those in untreated AF mice (Figures S3C and S3D). These data indicate that 5-ASA treatment caused an overall reversal of the local pro-inflammatory immune shift in the gut of AF mice.
5-ASA targets sphingolipid metabolism to regulate adaptive gut immunity in ALD
To determine if the effects of 5-ASA were mediated through anti-inflammatory actions that require adaptive immune cells like Th17 cells, we treated 7-week-old AF Rag1−/− mice with 5-ASA. Treatment of AF Rag1−/− mice with 5-ASA had a slight, but not significant, effect on liver damage and steatosis or liver inflammation (Figures S4A-S4C). Though we did identify a significant increase in the expression of tight junction and mucin genes (Figure S4D), 5-ASA treatment showed no significant effects on innate immune cell populations or cytokine secretion in the liver and gut (Figure S4E). Clearly, these data suggest that the beneficial effects of 5-ASA require components of the adaptive immune system. To further pinpoint the effect of 5-ASA on intestinal cells, we fed alcohol with 5-ASA to RORγt−/− mice. Interestingly, similar to the Rag1−/− mice, treatment with 5-ASA had no additional effects on the improvement of liver injury in the RORγt−/− mice (Figures S4F-S4G). Thus, the beneficial effects of 5-ASA require a Th17 cell-mediated adaptive immune response.
Effector T cell entry into the intestinal mucosa is a complex event regulated by selective expression of intestinal homing receptors on the T cell surface and corresponding ligands within the intestinal mucosa. We recently demonstrated that the recruitment of Th17 cells to the intestinal mucosa was due to the release of S1P by IECs in the gut(6). We also found dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity(13). S1P is mainly generated by the action of sphingosine kinase (SK1 and SK2)(7). Therefore, we assessed whether SK and S1P levels were affected in alcohol-induced injury. SK1 and SK2 mRNA levels significantly increased in the gut and liver after AF when compared to PF controls (Figure 4A). AF also induces the higher levels of SK1 protein in the gut compared with PF (Figure S5A). Next, sphingolipid metabolites including sphingosine, S1P, sphinganine, dihydrosphingosine 1-phosphate (DHS1P), sphingomyelin and ceramide concentrations were measured in the liver and gut of control and alcohol-fed mice. Alcohol exposure was associated with a significant increase in liver levels of S1P, DHS1P and ceramides, including C14:0–18:1 and C22:0–26:0 ceramides (Figure 4B). However, the levels of sphingosine, sphinganine and sphingomyelin with fatty acids (FAs) of varying chain length were not increased in the liver (Figure S5B). In the gut, sphingosine and sphinganine were both significantly increased in alcohol-fed mice (Figure 4C). Interestingly, most of the gut ceramides and sphingomyelin were not changed (Figure S5C), however, gut C20:0, C24:0, and C24:1 ceramides were significantly elevated after AF (Figure 4C). Similarly, gut C20:0, C22:0, C24:0, and C24:1 sphingomyelin were also increased in alcohol-fed mice (Figure 4C). Next, we conducted a thorough analysis of sphingolipids including the specific acyl-chain ceramides and sphingomyelin in the serum and the expression profiling of SK1 in the liver in samples from patients with moderate and severe alcoholic hepatitis (AH). The levels of C16:0, C18:0, C24:1 and C26:1 ceramides were significantly elevated in moderate and severe AH patients (Figure 4D). The increased levels of C18:1, and C14:0 ceramides and decreased levels of C24:0 and C26:0 ceramides were found in severe AH patients, but not in moderate AH patients (Figure 4D). With regard to sphingomyelin, the opposite phenomenon was observed: significant decreases were observed in C16:0 and C18:0 sphingomyelin in moderate and severe AH patients (Figure 4D). However, the levels of C22:0, C24:0, and C26:0 sphingomyelin were significantly decreased in severe AH patients, but not in moderate AH patients (Figure 4D). Although the levels of sphingosine, S1P, sphinganine and DHS1P remained unchanged in patients’ serum (Figure S5D), we found a significant increase in SK1 expression in the livers of AH patients compared to healthy controls (Figure 4E). Because the Stat3 pathway has been implicated as an essential regulator of S1P and IL-17A and links to acute inflammation and colitis(7, 14), activation of Stat3 was next examined. Indeed, nuclear accumulation of phosphorylated Stat3 was strongly stimulated in colon mucosa by AF compared to PF, as shown by western blotting and confocal analysis (Figure 4F). Importantly, the levels of SK expression in the liver and gut (Figure 4A and Figure S5A), the levels of liver S1P, DHS1P, and ceramides (Figure 4B), and the levels of gut sphinosine and sphinganine (Figure 4C) in ALD mice were strongly suppressed by 5-ASA treatment. In vivo administration of 5-ASA also prevented Stat3 activation induced by AF (Figure 4F). Our FACS data further showed that S1PR1 is highly expressed in CD4+ T cells in the gut from AF mice compared to PF mice (Figure S5E). Clearly, the SK1/S1P-S1PR1 axis may regulate the migration of CD4+IL-17+cells in ALD. 5-ASA may target the migration and activation of Th17 cells via S1P/S1PR1-Stat3 signaling.
Figure 4. Activation of sphingosine kinase and metabolism of sphingolipids in ALD.
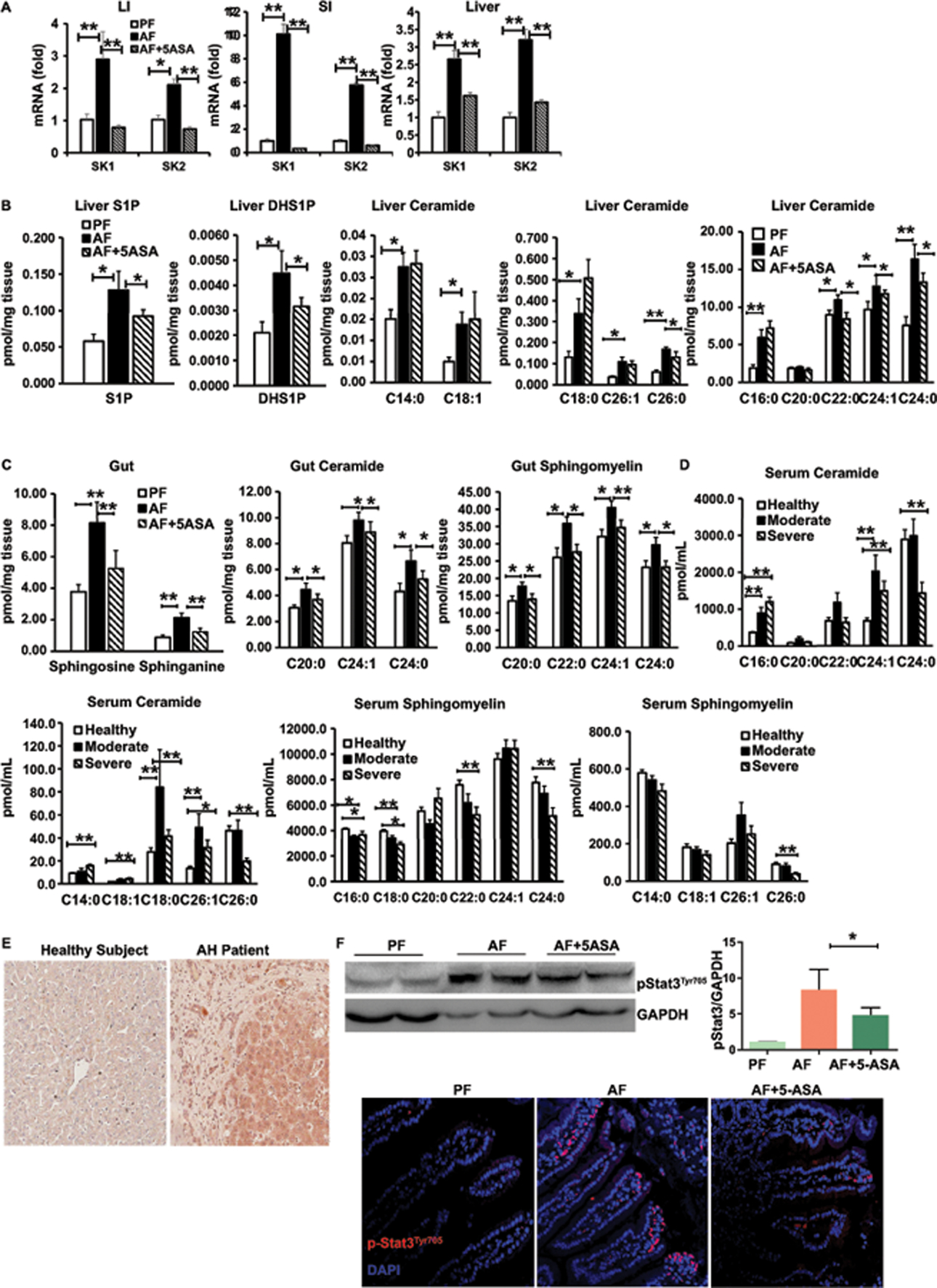
(A) Real-time PCR analysis of the expression of sphingosine kinase in the colon, small intestine and liver.
(B) Levels of S1P, DHS1P and acyl-chain ceramides in the liver.
(C) Levels of sphingosine, sphinganine, acyl-chain ceramides and sphingomyelin in the gut.
(D) Levels of acyl-chain ceramides and sphingomyelin in the serum of moderate, severe alcoholic hepatitis (AH) patients or healthy donors (normal liver). n=13
(E) Immunohistochemistry staining analysis of SK1 expression in the liver of AH patients and healthy control.
(F) Levels of pStat3 were evaluated using immunoblotting and immunofluorescence in the colon of mice.
Data from mice are presented as Mean ± SEM. n=8 * P<0.05; ** P<0.01
SK1 deficiency ameliorates alcoholic liver steatosis and injury as well as gut inflammation
Next, we investigated the role of SK1 in ALD using mice with a genetic deficiency of SK1. WT mice and SK1−/− mice had similar weight loss and ratio of liver/body weight after alcohol feeding (Figures S6A-S6B). Deletion of SK1 remarkably attenuated alcohol-induced liver damage and steatosis (Figures 5A–5C). Moreover, deficiency of SK1 prevented the accumulation of macrophages and immature myeloid cells, infiltration of neutrophils and the levels of Th1 and Th17 cells in the liver (Figures 5D and 5E). Furthermore, deficiency of SK1 decreased the levels of the proinflammatory genes and increased the level of IL-10 compared with WT mice (Figure 5F). Taken together, these data support the crucial role of SK1 in the pathogenesis of alcohol-induced liver inflammation.
Figure 5. Deficiency of SK1 attenuates alcoholic liver inflammation, steatosis, and damage.
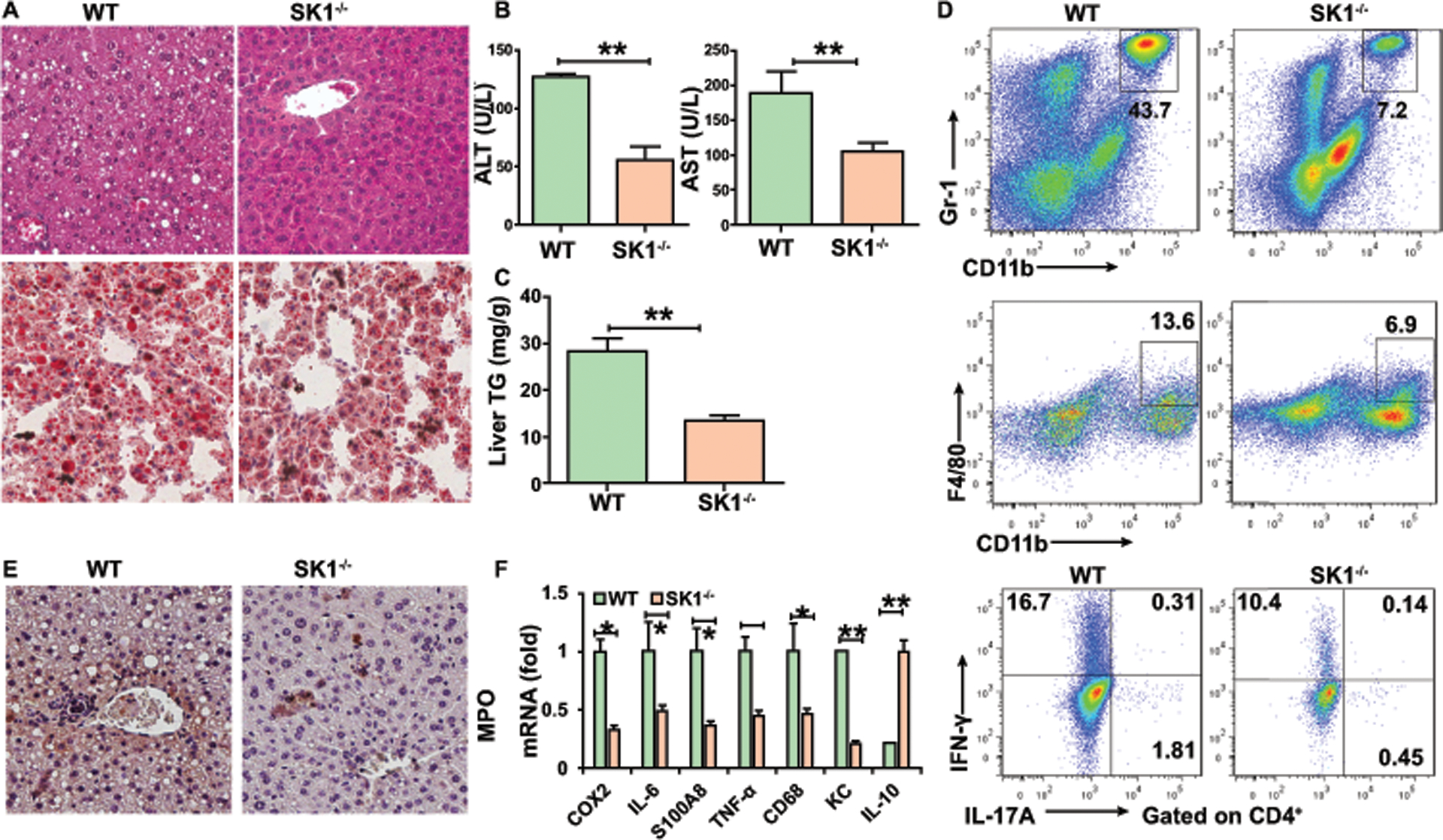
WT or SK1−/− mice were fed alcohol diet and sacrificed 15 days later.
(A) H&E staining and Oil red O staining of liver.
(B) Serum ALT and AST.
(C) Hepatic triglycerides.
(D) Frequencies of immature myeloid cells, macrophages, Th1, and Th17 cells in the liver.
(E) Immunohistochemistry staining of myeloperoxidase (MPO) in the liver.
(F) Real-time PCR analysis of the relative expression of indicated genes in the liver.
Data in all panels are presented as Mean ± SEM. n=11 * P<0.05; ** P<0.01
We next determined if deletion of SK1 influenced intestinal Th17 cell accumulation and function. In comparison to AF WT mice, AF SK1−/− mice had a reduced inflammatory infiltrate in the small intestine and colon (Figure 6A), more colon goblet cells (Figure 6B), and higher expression of mucin (Figure 6B) which is consistent with lower intestinal permeability (Figure 6C) in SK1−/− mice compared with AF WT mice. Furthermore, AF SK1−/− mice had higher expression of TJ protein and mucin and decreased amounts of COX-2, RORC, RORA, IL-23, and IL-17A mRNA in the LPL of the colon (Figure 6D). ELISA analysis confirmed that cytokines IL-17A and IL-23 were reduced in the LPL from AF SK1−/− mice compared with AF WT mice (Figure 6E). Moreover, deficiency of SK1 significantly attenuated the accumulation of Th17 cells in gut LPLs after AF compared to WT mice (Figure 6F). Our data showed no differences of Th1, CD8αβ+ T cells, TCRγδ T cells, and CD4+ T cells as well as Treg cells (Figure 6F and Figures S6C-S6F) in the guts of AF SK1−/− mice compared to the AF WT mice. Furthermore, we found significantly lower levels of S1P and reduced nuclear accumulation of phosphorylated STAT3 in the colon of SK1−/− mice (Figures 6G–6H). Taken together, these data demonstrated a critical role of the SK1/S1P-Th17 axis in the pathogenesis of alcohol-induced gut inflammation.
Figure 6. Deficiency of SK1 attenuates the gut inflammation and accumulation of Th17 cells in ALD.
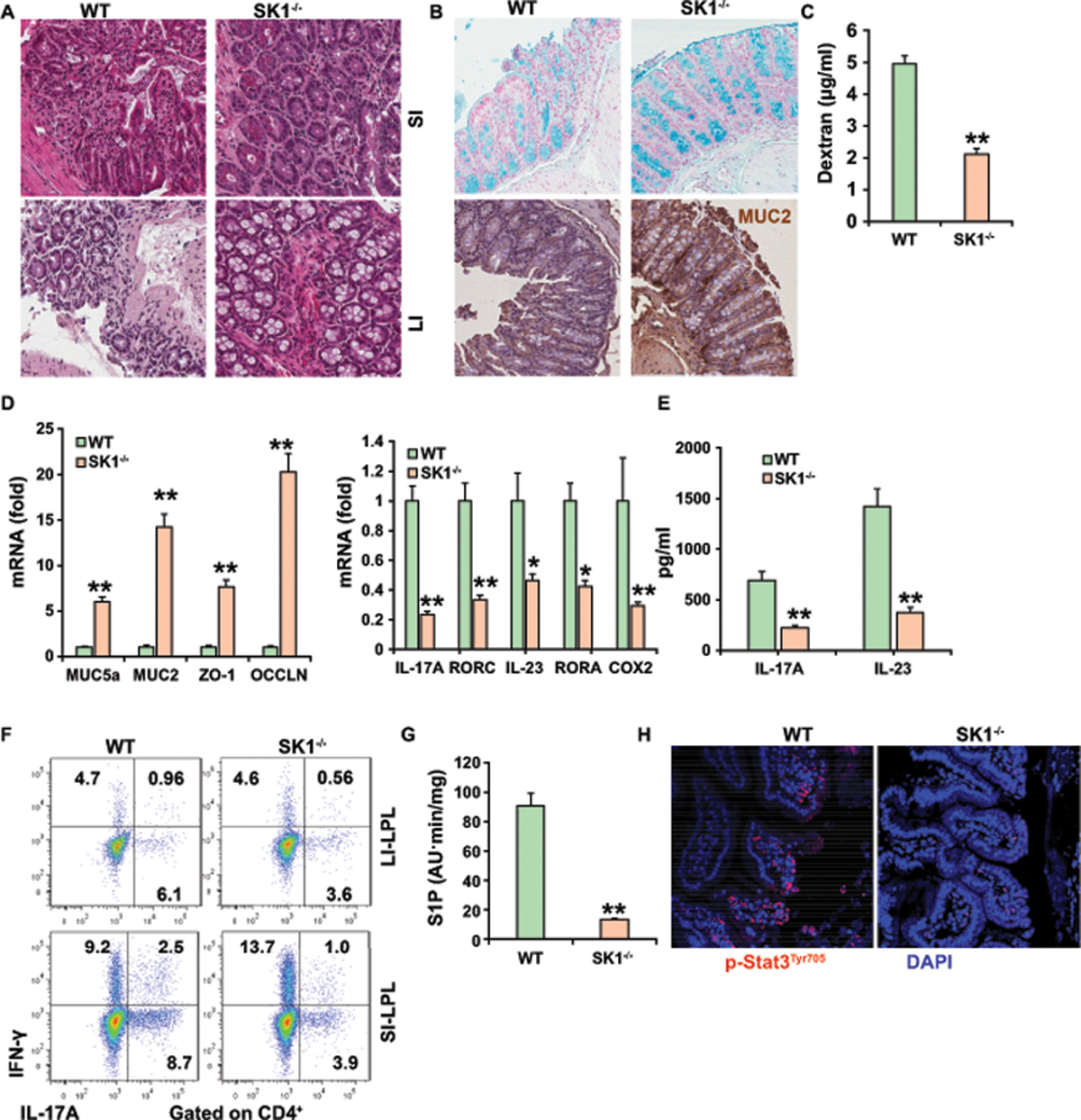
(A) H&E staining of small and large intestine.
(B) Alcian Blue staining and immunohistochemistry staining of mucin 2 in colonic tissue.
(C) Plasma levels of Dextran-FITC for gut permeability.
(D) Real-time PCR analysis of the expression of indicated genes in the colon (left) and IL-17A and Rorc mRNAs (right) in colonic LPLs.
(E) ELISA analysis of IL-17A or IL-23 in the supernatant of colonic LPLs.
(F) Intracellular staining of IL-17A+ and IFN-γ+ CD4+ T cells from gut LPL.
(G) Relative levels of S1P in the ileum of alcohol-fed mice.
(H) Immunofluorescence was used to evaluate the expression of pSTAT3 in the ileum.
Data in all panels are presented as Mean ± SEM. n=11 * P<0.05; ** P<0.01
SK2 deficiency exacerbates alcohol-induced liver injury as well as gut Th17 accumulation
Given that sphingosine is phosphorylated by both of SK1 and SK2 to produce S1P, we investigated the role of SK2 in ALD. Unexpectedly, in contrast to WT mice, SK2−/− mice had more severe alcohol-induced liver damage and steatosis (Figures 7A and 7B). Moreover, deficiency of SK2 induced the accumulation of immature myeloid cells (Figure 7C) and upregulation of the proinflammatory genes mRNA in the livers in AF mice compared with AF WT controls (Figure S7A). Furthermore, histopathological analysis revealed more severely damaged colon mucosa with more extensive infiltration of inflammatory cells, fewer colon goblet cells, and reduced expression of mucin (Figures 7D–7E and Figures S7B-S7C) in AF SK2−/− mice in comparison to AF WT mice. The colons of SK2−/− mice also contained higher levels of IL-17A and Rorc mRNA in LI-LPL after AF (Figure 7E). We consistently observed a significant increase in the gut permeability and in the proportions of Th17 cells and CD4+ RORγt+ cells, but not Th1 and Tregs cells in the LPL of gut of SK2−/− mice after AF (Figure 7F and Figures S7D-S7E). Surprisingly, the levels of SK1 (Figure 7E), S1P (Figure S7F), and pStat3 (Figures S7G and S7H), in the gut were markedly increased in SK2−/− mice compared to littermates during AF, indicating that the increases in Th17 cells and severity of gut inflammation in SK2−/− mice are likely due to upregulation of SK1 and S1P. Consistent with our previous data, we also observed decreased liver injury and steatosis of AF mice in 5-ASA-treated SK2−/− mice (Figures 7A–7C). 5-ASA also decreased gut inflammation (Figures 7D), the level of IL-17A and SK1 mRNA (Figure 7E) in SK2−/− mice compared with untreated SK2−/− mice, which further confirmed that 5-ASA reduces Th17 cells via SK1-dependent mechanisms in ALD.
Figure 7. Deficiency of SK2 promotes liver injury in ALD.
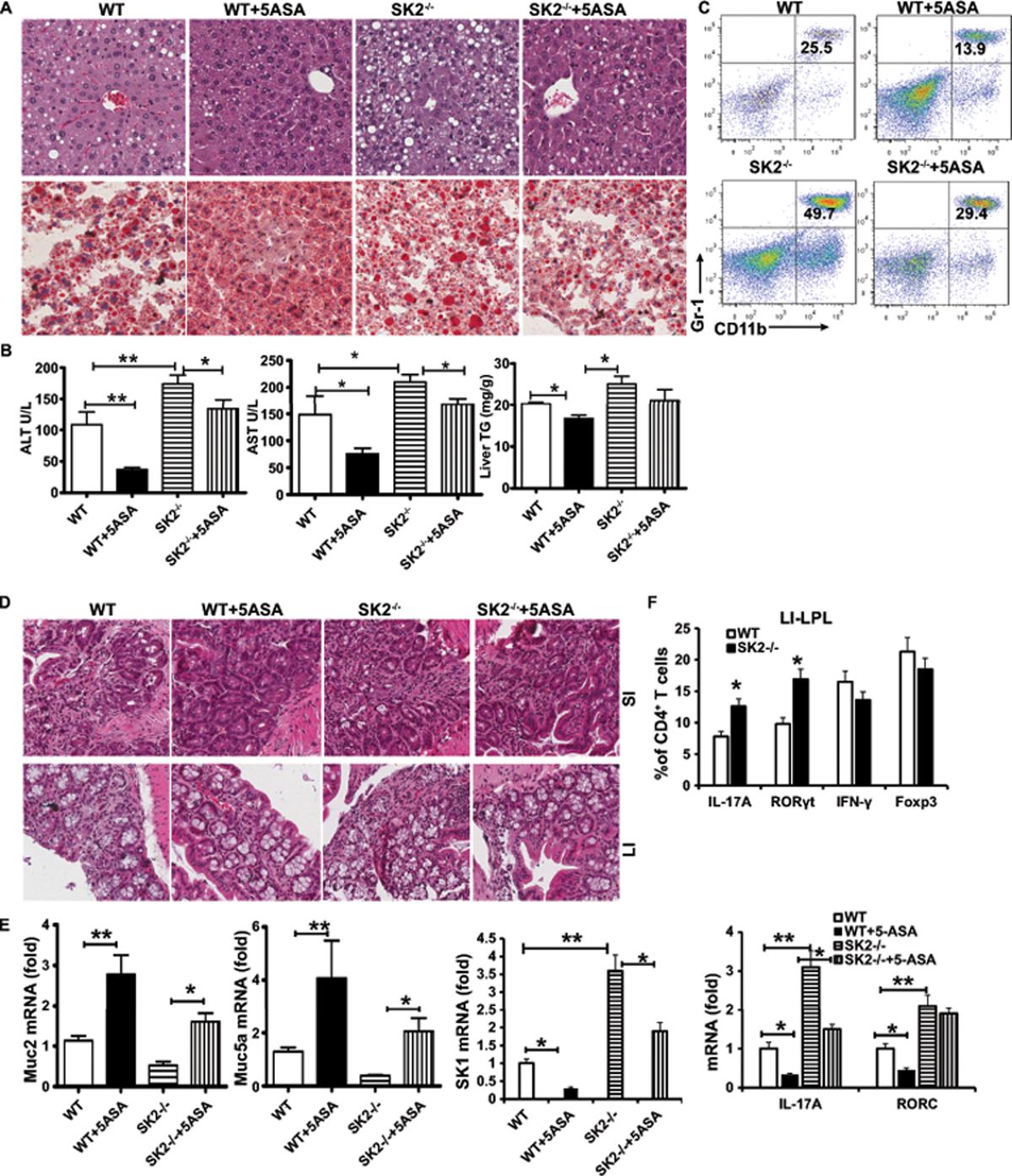
WT or SK2−/− mice on a chronic-binge ethanol diet, with/without 5-ASA oral supplementation (50 mg/kg/day) or vehicle for 15 days.
(A) H&E staining and Oil red O staining of liver.
(B) The levels of serum ALT, AST and hepatic triglycerides.
(C) Frequencies of immature myeloid cells (CD11b+Gr-1+) in the liver.
(D) H&E staining of small and large intestine.
(E) Real-time PCR analysis of the levels of Muc2, Muc5a, IL-17A, Rorc and SK1 mRNA in the colon.
(F) Intracellular staining of IL-17A, IFN-γ, Foxp3 or RORγt in CD4+ T cells from colonic LPL.
Data in all panels are presented as Mean ± SEM. n=7 * P<0.05; ** P<0.01
Pharmacologic intervention via inhibition of S1P-S1PR1 signaling ameliorates ALD development and progression.
To evaluate whether pharmacological inhibition of SK1 has a protective effect in ALD, we took advantage of an SK1 inhibitor, SKI-178, to treat mice along with or without 5-ASA and found that SKI-178 alone slightly ameliorated alcohol-induced liver steatosis and damage (Figures 8A–8B). Administration of SKI-178 also slightly decreased the accumulation of immature myeloid cells and neutrophils (Figure 8C) and the levels of Cox2 and TNF-α mRNA (Figure 8D) in the liver. However, the combined treatment with SK1 and 5-ASA significantly prevented progression of alcohol-induced liver damage and steatosis, expression of inflammatory genes, and accumulation of immature myeloid cells and neutrophils (Figures 8A–8D). In the liver, the level of S1P was significantly decreased in SKI-178 treated AF mice when compared with untreated AF mice (Figure S8A). We observed an increase in the levels of C16:0-ceramide and a simultaneous decrease in the levels of C22-C24.1-ceramide in the liver of SKI-178 treated AF relative to untreated AF mice (Figure S8B). Accordingly, in the liver of SKI-178 treated AF mice, the total ceramide levels were not significantly different from untreated AF mice (Figure S8B). These results indicate that the sphingosine kinase 1 (SK1) is necessary to regulate the S1P levels, but not bulk ceramide levels in the liver. Presumably, several ceramidases (acid, neutral, alkaline), which can catalyze the degradation of ceramide, or other ceramide synthases (CerS 1–6), function to control metabolic levels of ceramide in cells. These data demonstrated that administration of SKI-178 and 5-ASA together could achieve protection from ALD similar to that observed in SK1−/− mice (Figures 5–6).
Figure 8. Pharmacologic intervention via inhibition of S1P/SPR1 signaling ameliorates ALD progression.
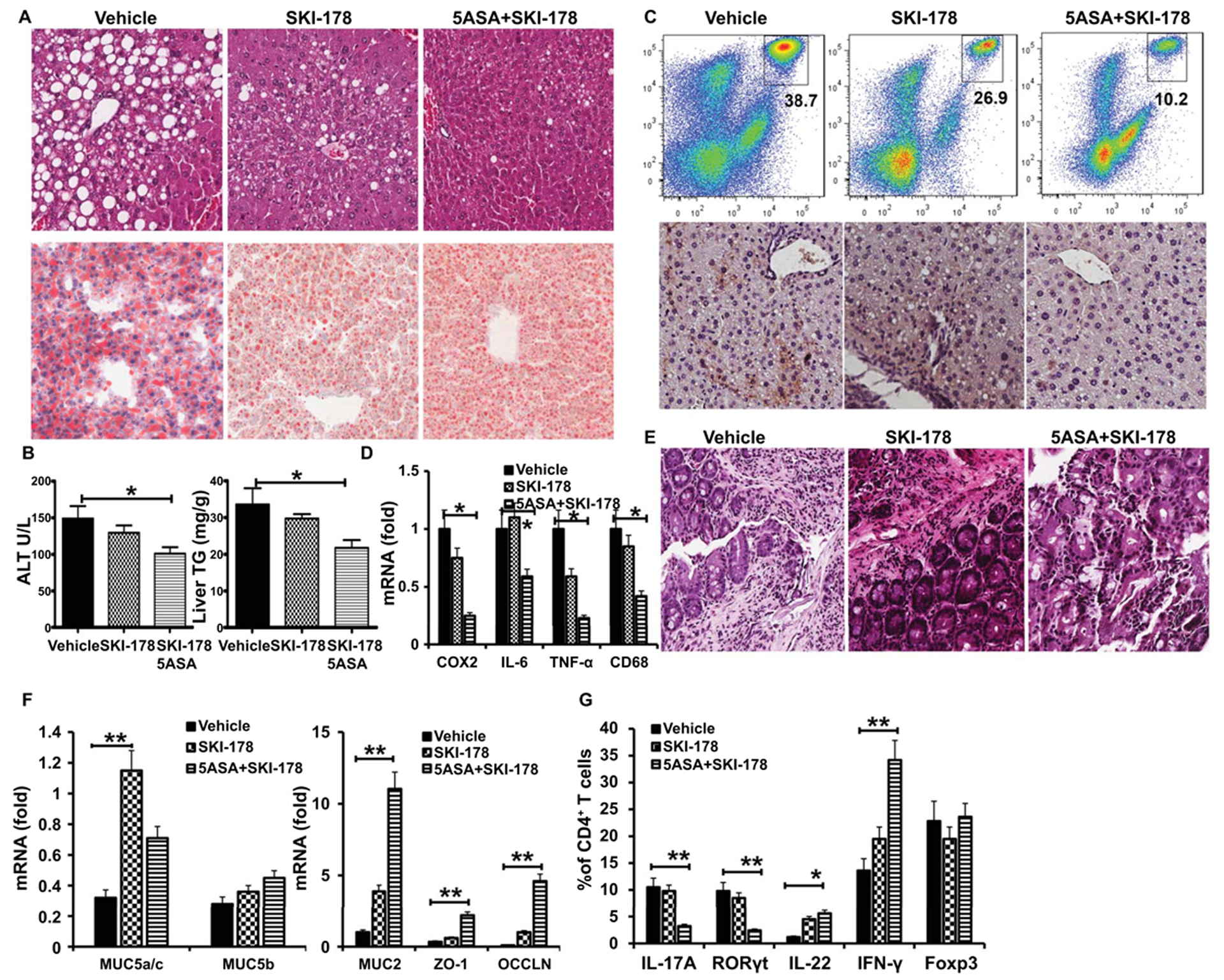
(A) H&E staining and Oil red O staining of liver.
(B) Serum levels of ALT and hepatic triglycerides.
(C) Frequencies of immature myeloid cells (Top) and immunohistochemistry analysis of the expression of myeloperoxidase (MPO) (bottom) in the liver.
(D) Real-time PCR analysis of the expression of indicated genes in the liver.
(E) H&E staining of large intestine.
(F) Real-time PCR analysis of the expression of indicated genes in the colon.
(G) The proportion of IL-17A, IFN-γ, Foxp3 or RoRγt in CD4+ T cells from colonic LPL.
Data in all panels are presented as Mean ± SEM. n=7 * P<0.05; ** P<0.01
We next evaluated the effect of dual inhibitors on Th17 cells in the gut. We found that the administration of 5-ASA in combination with SKI-178 significantly inhibited progression of alcohol-induced gut inflammation (Figure 8E), which was associated with substantially increased genes encoding TJ protein and mucin in the gut compared to AF mice with single treatment (Figure 8F). FACS analysis for RORγt and IL-17A in mouse gut sections revealed a significant increase in CD4+ cells in the gut after alcohol feeding that was unchanged by giving SKI-178 alone, but was modestly reduced when 5-ASA was administered with SKI-178 (Figure 8G). Interestingly, the administration of SKI-178 and 5-ASA together remarkably enhanced the expression of IFN-γ in CD4+ cells but did not affect the proportion of CD4+Foxp3+ cells and CD4+ IL-22+ in LPL from LI (Figure 8G).
The activity of S1P signaling is tightly regulated by FTY720, a natural antagonist of S1PR1. Next, we asked whether blockage of S1PR1 can ameliorate alcohol-induced liver injury by preventing the recruitment of Th17 cells to gut. At the conclusion of the experiment, the mice received the combination treatment of FTY720 with 5-ASA, but not FTY720 alone, which resulted in a substantially improved histological picture of ALD: lower levels of serum ALT, less infiltration of myeloid cells, and less expression of inflammatory genes compared with alcohol-fed, vehicle or FTY720-treated mice (Figures S9A-S9F). Interestingly, the administration of FTY720 alone or the combination treatment of FTY720 and 5-ASA markedly blocked the migration of CD4+ T cells to liver (Figures S9G).
Discussion
ALD is tightly linked to gut inflammation in humans as well as in experimental models(15). We have identified the gut immune system as an active orchestrator and therapeutic target in ALD. Specifically, we demonstrated that inhibition of gut inflammation with a gut-specific anti-inflammatory agent, 5-ASA, during alcohol feeding can alter liver steatosis. We further found that the bioactive lipid, S1P, drives the pathogenesis of alcohol-induced gut inflammation and that the pathogenic effect of S1P is mediated by Th17 cells specifically. We demonstrated, for the first time to our knowledge, that SK1 was involved in Th17 cells accumulating in the gut of alcohol fed mice and AH patients, and that a deficiency in RORγt and SK1, but not in SK2, consistently prevented alcoholic gut inflammation and significantly attenuated alcohol-induced steatosis and liver damage in experimental ALD. Pharmacologic inhibition of S1P-S1PR1 signaling greatly protected against alcohol-induced steatosis and liver injury in experimental ALD.
Previous work has shown that AF reduces the expression of IL-22 in type 3 innate lymphoid cells in the colons of AF mice(16). We consistently saw that the pro-inflammatory shift in immune cell populations observed in the gut was associated with obvious inflammatory histological changes in AF mice, which are characterized by increased IL-17A-producing CD4+ and CD8+ T cells as well as increased RORγt+ CD4+ T cells in the LP. In AF RORγt−/− mice, we observed overall improvements in gut barrier function and inflammation. These findings implicate local intestinal IL-17A production as one of the critical pathogenic mediators of intestinal permeability in experimental ALD. The development of Th17 cells is influenced by gut microbiota (17). Some commensal bacteria, such as segmented filamentous bacteria (SFB), and bacterial metabolites, like bile acids, are necessary for gut Th17 cell development (18, 19). We observed the number of Th17 cells is slightly elevated, but not significantly, in the liver by alcohol feeding. Therefore, additional work is needed to explore if gut metabolites (e, g. bile acid) can regulate the differentiation/accumulation of Th17 cells in the liver by RORγt in ALD (20).
We further showed that inhibition of gut inflammation with a gut-specific anti-inflammatory agent, 5-ASA, during alcohol feeding can attenuate liver injury. Treatment with 5-ASA has beneficial effects on the intactness of the gut epithelial barrier in IBD models(21, 22). We observed similar beneficial effects of 5-ASA on gut barrier function during alcohol feeding that are linked to reduced levels of inflammatory cytokines, such as TNF-α and IL-17A. Improvements seen with 5-ASA treatment were found to be dependent on adaptive and gut immune systems due to minimal effects seen in ALD 5-ASA-fed Rag1−/− mice and RORγt−/− mice. The observed direct effect of 5-ASA in vivo on IL-17A production highlights potential crosstalk between intestinal epithelial cells and gut immune cells in mediating the effects of 5-ASA on the alcohol-induced liver injury. These experiments indicate the relative specificity of gut anti-inflammatory therapy, such as sulfasalazine, colon-specific prodrug for 5-ASA, might also attenuate the development of ALD.
Abundant evidence reveals that gut inflammation is associated with enhanced leukocyte trafficking to the gut mucosa and altered expression of adhesion molecules. The α4 integrins (α4β1 and α4β7,) and chemokines (CCL25/CCR9) play a regulatory role in lymphocyte homing and recruitment to the inflamed intestine(23, 24). S1P, through ligand binging of its receptor S1PR1 in the blood and lymph(25), is also essential for naïve lymphocytes to preferentially migrate into the gut (26). S1P is mainly generated via ceramide by the action of sphingosine kinase (SK1 and SK2). We found that the expression of SK1 and SK2 was increased in experimental ALD, which we believe to be novel. More importantly, we also found elevated SK1 in human AH samples compared with healthy human donor liver samples. We have confirmed that hepatic SIP levels are increased following alcohol exposure in mice. Reported increases in ceramide levels from different models of nonalcoholic fatty liver have led to suggestions that sphingolipids are important in fatty liver disease development, although the mechanistic link between hepatic steatosis and ceramide is unclear(27–29). In this study, SK1−/− mice had significantly attenuated alcohol-induced steatosis and liver damage after alcohol feeding. Importantly, these mice showed reduced immune cell infiltrates in the liver and gut during alcohol feeding, including reductions in IL-17A-producing CD4+ not only in the gut, but also in the liver. Ethanol-induced S1P seems to be due to augmented expression and activity of SK. However, these two isoforms of SK, SK1 and SK2, have differences in subcellular localization, i.e., cytosol for SK1 and nucleus for SK2, as well as different physiological functions(30, 31). Although the absence of SK1 was protective against DSS-induced colitis, SK2 depletion resulted in enhanced proliferation and proinflammatory cytokine production and thus, IBD progression(7, 32). Indeed, our data showed that knockout mice for SK2 were more susceptible to developing severely damaged and necrotic colon mucosa with increased expression of SK1 and extensive infiltration of inflammatory Th17 cells. The severity of colitis in SK2−/− mice correlated with NFκB activation, IL-6 and TNFα formation, STAT3 activation, and S1PR1 expression. The associated increase in SK1 and S1P in SK2−/− mice was attributed to loss of SK2-mediated HDA1/2C inhibition, thereby increasing the induction of c-Jun and its target gene SK1(7). We have shown that nuclear accumulation of phosphorylated STAT3 was strongly stimulated in colonic mucosa by chronic alcohol feeding (Figure 4F). Phosphorylated STAT3 was also increased and suppressed by 5-ASA in the colons of SK2−/− mice (Figures S7G and 7H). The Stat3 pathway has been implicated as an essential regulator of S1P and IL-17A and NFκB/IL-6/STAT3/S1PR1 amplification loop links to acute inflammation and colitis (33). Our future study will explore if the potential mechanism to control SK1 expression by 5-ASA is through the regulation of STAT3 in ALD. More in-depth mechanistic approaches will be required to fully understand the mechanism of SK1/S1P-mediated activation of RORγt signaling, including studies in mice with CD4 T cell-specific deletion of RORγt, or with hepatocyte/IEC-specific deletion of SK1, to dissect the precise roles of SK1 and Th17 cells in ALD in vivo.
In the studies presented here, the fact that the S1P/S1PR1 axis is essential for lymphocyte egress added a new potential target for blocking Th17 migration to the inflamed gut in ALD. In terms of IBD, FTY720 ameliorated experimental colitis arising as a result of chemical induction, T-cell transfer, and IL-10 deficiency, suggesting that it may be a potential candidate for ALD treatment(34, 35). Indeed, our studies showed that SKI-178 or FTY720 interferes with S1P signaling and blocks the response of Th17 cells in ALD mice. Our findings suggest that the combination of 5-ASA with pharmacological blockade of S1P/S1PR1 signaling may prevent the accumulation of Th17 cells not only in the gut, but also in the liver, and attenuate the development of ALD to the same extent as a global deficiency of SK1 or RORγt.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH R21AA025724, R21AI128206 and R01 DK115406 (Z.D.), NIH R01AA023190 (W.F.) and NIH P50AA024337, P20GM113226, U01AA026936 (CJ.M.). We thank Dr. J. Ainsworth and Marion McClain for editorial assistance.
Footnotes
Conflicts of Interest
The authors disclose no conflicts of interest.
Reference
- 1.Albillos A, Gottardi A, Rescigno M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol 2019. [DOI] [PubMed] [Google Scholar]
- 2.Xiao S, Yosef N, Yang JF, Wang YH, Zhou L, Zhu C, Wu C, et al. Small-Molecule ROR gamma t Antagonists Inhibit T Helper 17 Cell Transcriptional Network by Divergent Mechanisms. Immunity 2014;40:477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gyongyosi B, Cho Y, Lowe P, Calenda CD, Iracheta-Vellve A, Satishchandran A, Ambade A, et al. Alcohol-induced IL-17A production in Paneth cells amplifies endoplasmic reticulum stress, apoptosis, and inflammasome-IL-18 activation in the proximal small intestine in mice. Mucosal Immunol 2019;12:930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin F, Taylor NJ, Su H, Huang X, Hussain MJ, Abeles RD, Blackmore L, et al. Alcohol dehydrogenase-specific T-cell responses are associated with alcohol consumption in patients with alcohol-related cirrhosis. Hepatology 2013;58:314–324. [DOI] [PubMed] [Google Scholar]
- 5.Apostolidis SA, Rodriguez-Rodriguez N, Suarez-Fueyo A, Dioufa N, Ozcan E, Crispin JC, Tsokos MG, et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nature Immunology 2016;17:556–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng ZB, Mu JY, Tseng M, Wattenberg B, Zhuang XY, Egilmez NK, Wang QL, et al. Enterobacteria-secreted particles induce production of exosome-like S1P-containing particles by intestinal epithelium to drive Th17-mediated tumorigenesis. Nature Communications 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, et al. Sphingosine-1-Phosphate Links Persistent STAT3 Activation, Chronic Intestinal Inflammation, and Development of Colitis-Associated Cancer. Cancer Cell 2013;23:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heaver SL, Johnson EL, Ley RE. Sphingolipids in host-microbial interactions. Current Opinion in Microbiology 2018;43:92–99. [DOI] [PubMed] [Google Scholar]
- 9.Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, Obeid LM. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. Faseb Journal 2009;23:143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, Metzger D, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. Journal of Experimental Medicine 2005;201:1205–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiPaolo MC, Merrett MN, Crotty B, Jewell DP. 5-aminosalicylic acid inhibits the impaired epithelial barrier function induced by gamma interferon. Gut 1996;38:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ueda E, Kurebayashi S, Sakaue M, Backlund M, Koller B, Jetten AM. High incidence of T-cell lymphomas in mice deficient in the retinoid-related orphan receptor ROR gamma. Cancer Research 2002;62:901–909. [PubMed] [Google Scholar]
- 13.Deaciuc IV, Nikolova-Karakashian M, Fortunato F, Lee EY, Hill DB, McClain CJ. Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity. Alcohol Clin Exp Res 2000;24:1557–1565. [PubMed] [Google Scholar]
- 14.Krebs CF, Paust HJ, Krohn S, Koyro T, Brix SR, Riedel JH, Bartsch P, et al. Autoimmune Renal Disease Is Exacerbated by S1P-Receptor-1-Dependent Intestinal Th17 Cell Migration to the Kidney. Immunity 2016;45:1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo G, Mandrekar P. A Recent Perspective on Alcohol, Immunity, and Host Defense. Alcoholism-Clinical and Experimental Research 2009;33:220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendrikx T, Duan Y, Wang YH, Oh JH, Alexander LM, Huang W, Starkel P, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut 2019;68:1504–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sommer F, Backhed F. The gut microbiota--masters of host development and physiology. Nat Rev Microbiol 2013;11:227–238. [DOI] [PubMed] [Google Scholar]
- 18.Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009;31:677–689. [DOI] [PubMed] [Google Scholar]
- 19.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, U Karaoz, D Wei, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009;139:485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, Ha S, et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019;576:143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown JB, Lee G, Managlia E, Grimm GR, Dirisina R, Goretsky T, Cheresh P, et al. Mesalamine inhibits epithelial beta-catenin activation in chronic ulcerative colitis. Gastroenterology 2010;138:595–605, 605 e591–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu XC, Mei Q, Xu JM, Hu J. Balsalazine decreases intestinal mucosal permeability of dextran sulfate sodium-induced colitis in mice. Acta Pharmacol Sin 2009;30:987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luda KM, Joeris T, Persson EK, Rivollier A, Demiri M, Sitnik KM, Pool L, et al. IRF8 Transcription-Factor-Dependent Classical Dendritic Cells Are Essential for Intestinal T Cell Homeostasis. Immunity 2016;44:860–874. [DOI] [PubMed] [Google Scholar]
- 24.Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, Schady D, et al. Critical Role for the Microbiota in CX(3)CR1(+) Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity 2018;49:151–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004;427:355–360. [DOI] [PubMed] [Google Scholar]
- 26.Murphy CT, Nally K, Shanahan F, Melgar S. Shining a Light on Intestinal Traffic. Clinical & Developmental Immunology 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Summers SA. Sphingolipids and insulin resistance: the five Ws. Curr Opin Lipidol 2010;21:128–135. [DOI] [PubMed] [Google Scholar]
- 28.Park JW, Park WJ, Kuperman Y, Boura-Halfon S, Pewzner-Jung Y, Futerman AH. Ablation of very long acyl chain sphingolipids causes hepatic insulin resistance in mice due to altered detergent-resistant membranes. Hepatology 2013;57:525–532. [DOI] [PubMed] [Google Scholar]
- 29.Meikle PJ, Summers SA. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat Rev Endocrinol 2017;13:79–91. [DOI] [PubMed] [Google Scholar]
- 30.Lai WQ, Irwan AW, Goh HH, Melendez AJ, McInnes IB, Leung BP. Distinct roles of sphingosine kinase 1 and 2 in murine collagen-induced arthritis. J Immunol 2009;183:2097–2103. [DOI] [PubMed] [Google Scholar]
- 31.Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. Journal of Biological Chemistry 2003;278:46832–46839. [DOI] [PubMed] [Google Scholar]
- 32.Samy ET, Meyer CA, Caplazi P, Langrish CL, Lora JM, Bluethmann H, Peng SL. Cutting edge: Modulation of intestinal autoimmunity and IL-2 signaling by sphingosine kinase 2 independent of sphingosine 1-phosphate. Journal of Immunology 2007;179:5644–5648. [DOI] [PubMed] [Google Scholar]
- 33.Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010;465:1084–U1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizushima T, Ito T, Kishi D, Kai Y, Tamagawa H, Nezu R, Kiyono H, et al. Therapeutic effects of a new lymphocyte homing reagent FTY720 in interleukin-10 gene-deficient mice with colitis. Inflammatory Bowel Diseases 2004;10:182–192. [DOI] [PubMed] [Google Scholar]
- 35.Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4(+)CD25(+) regulatory T cells. Journal of Immunology 2007;178:2458–2468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.