

Abstract

The use of high-valent antimony–oxo porphyrins as visible-light photocatalysts operating via direct hydrogen atom transfer has been demonstrated. Computational analysis indicates that the triplet excited state of these complexes shows an oxyl radical behavior, while the SbV center remains in a high-valent oxidation state, serving uniquely to carry the oxo moiety and activate the coordinated ligands. This porphyrin-based system has been exploited upon irradiation to catalyze C–H to C–C bond conversion via the addition of hydrogen donors (ethers and aldehydes) onto Michael acceptors in a redox-neutral fashion without the need of any external oxidant. Laser flash photolysis experiments confirmed that the triplet excited state of the photocatalyst triggers the desired C–H cleavage.
Keywords: C−C bond formation, hydrogen atom transfer, photocatalysis, porphyrins, radical reactions
Introduction
The development of eco-sustainable synthetic methodologies has recently received a fundamental contribution by visible-light photocatalysis.1 A photocatalyst (PC) can indeed activate the chosen organic substrate via single electron transfer (SET),1,2 hydrogen atom transfer (HAT),3 or energy transfer (EnT).4 These different modes of action typically require dedicated PCs (e.g. PCSET, PCHAT, or PCEnT, respectively). A single PC, however, may promote multiple pathways, as in the case of Eosin Y (a xanthene dye),5 known PCEnT for singlet oxygen (1O2) generation, which has been recently adopted as PCSET6,7 and PCHAT.8
As part of our ongoing research activity, we are interested in exploring the inherent advantages of photocatalyzed HAT in redox-neutral C–C bond forming reactions.3 However, finding new candidates to trigger this chemistry under visible light irradiation is a tough task, and only a handful of colored PCsHAT have been reported so far, notably Eosin Y,8 5,7,12,14-pentacenetetrone,9 and the uranyl cation.10 Inspired by the chameleonic behavior of Eosin Y, we reasoned that other classes of catalysts specifically designed for SET or EnT might work as visible light absorbing PCHAT as well. In particular, we decided to explore the class of porphyrin derivatives, widely employed as dyes for artificial photosynthesis11 and in many other fields.12,13 Porphyrins are commonly adopted as PCEnT for 1O2 generation14 in photodynamic therapy,15 albeit applications in organic synthesis are now also recognized.16,17 Thus, while free-base porphyrins act (in analogy with other PCSET) either through a reductive18 or an oxidative19 quenching mechanism, when the porphyrin ring is coordinated to a suitable metal center, the thus-formed metalloporphyrin complex may show a different reactivity, thanks to the axial ligand sphere.
Given their low toxicity and powerful oxidation ability under photoexcitation, we selected a class of antimony dihydroxo complexes [Sb(tap)(OH)2]+X– (tap = tetraarylporphyrin; X typically a halide ion or the hydroxide anion) for our purpose.20 These derivatives have been previously exploited to promote the photooxygenation of alkenes21 by using water as both the oxygen and electron donor in the presence of oxidative quenchers (e.g. methylviologen21a−21d or potassium hexachloroplatinate(IV);21e,21fFigure 1a). Intriguingly, they are known to give an antimony–oxo complex [Sb(tap)(=O)(OH)] upon deprotonation with a base (pKa = 9.7 for the tetraphenylporphyrin derivative).22 Accordingly, because currently known PCsHAT are characterized by the presence of a X=O moiety (X: carbon or metal) in their structures, we reasoned that these stable porphyrin complexes may trigger the desired HAT reactivity3 upon light exposure. In fact, antimony–oxo porphyrins have been reported to induce the conversion of ethanol to acetaldehyde (Figure 1b, HAT proposed as the rate determining step) when exposed to visible light (546 nm).23 An oxyl-radical reactivity of the triplet excited state was later postulated in the aerobic photocatalyzed conversion of alcohols/aldehydes to carboxylic acids under solar light irradiation (Figure 1b).24
Figure 1.
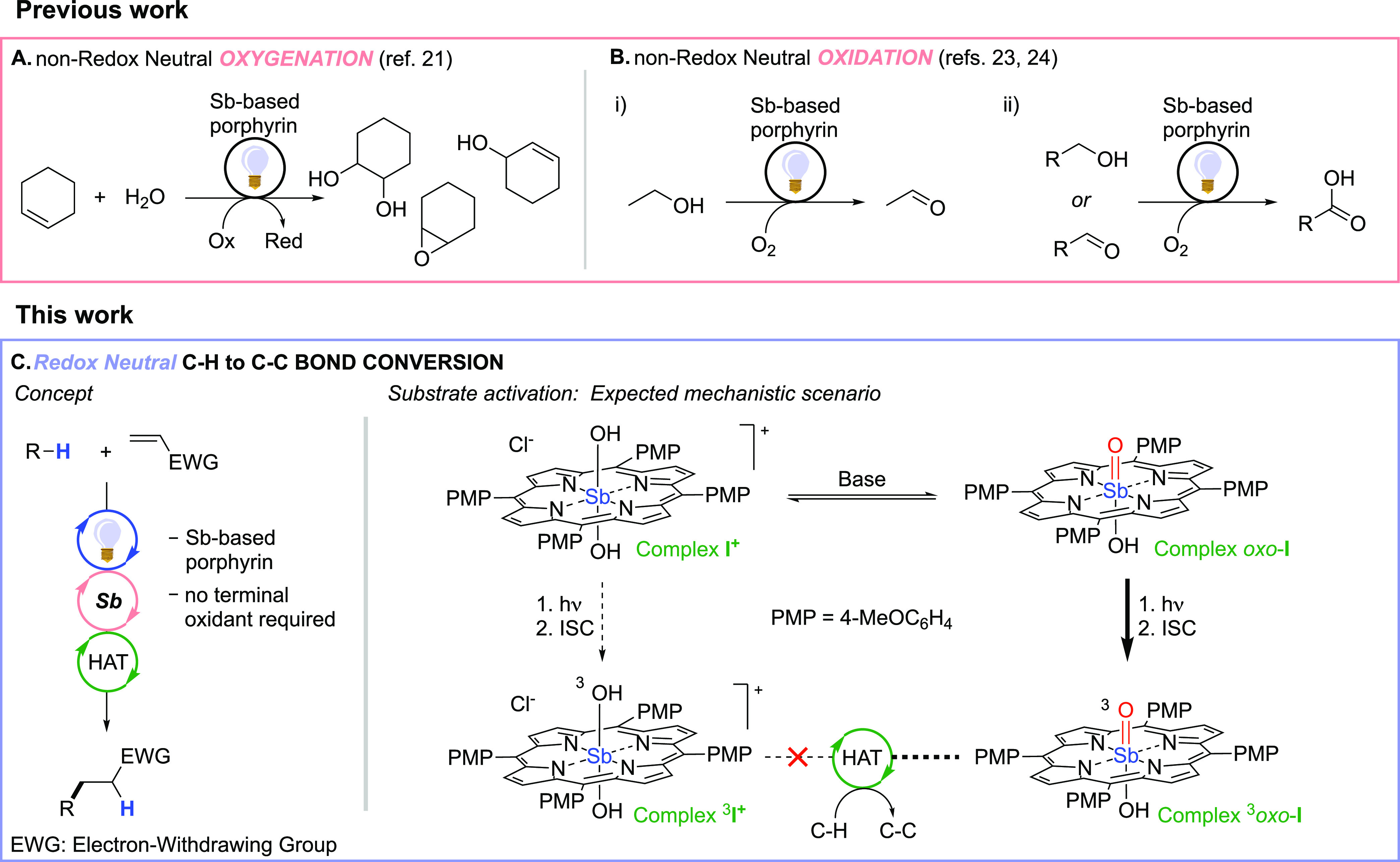
Previous examples of the use of antimony-based porphyrins as photocatalysts in non-redox neutral oxygenations (part a) and oxidations (part b), along with the new concept proposed in this work for the redox neutral C–H to C–C bond conversion and the expected mechanistic scenario (part c).
For the present work, we started looking at antimony-based porphyrins from a new perspective. While previous reports described nonredox neutral approaches for oxygenations/oxidations requiring the use of external oxidants,21,23,24 we hereby document the competency of this class of photocatalysts in the redox neutral C–H to C–C bond conversion under oxidant-free conditions. We also explain which species is competent in the HAT step and the features of the photocatalyst in the deactivated form, generated upon cleavage of the relevant C–H bond. Because of its intense and broad absorption bands in the visible region,25,26 we focused our attention on the tetra(p-methoxyphenyl)-substituted metalloporphyrin I+ (chloride salt, PMP = 4-MeOC6H4; Figure 1c).
Results and Discussion
Computational Analysis
At the onset of our project, we undertook a computational analysis intended to describe the different forms of the employed photocatalyst possibly involved in the process and their competency, when appropriate, in triggering the desired HAT step. Thus, previous computational investigations on the reactivity of high-valent Fe- and Mn-based porphyrins highlighted that hydrocarbon activation may occur via hydrogen abstraction by either the metal-hydroxo or the metal–oxo group.27 Accordingly, we initially considered both the cationic complex I+ and the corresponding oxo-derivative obtained via deprotonation (tagged as oxo-I; no charge present; Figure 1c). We adopted a theoretical approach based on density functional theory (DFT) simulations at the ωB97XD/def2SVP level of theory (see Supporting Information for details), well-known to correctly handle the modeling of metal complexes.28
We investigated the structures of I+ and oxo-I in the gas phase, and the corresponding optimized structures are reported in the Supporting Information (see Figure S1), clearly showing the presence of a significantly shorter Sb–O bond in oxo-I (Sb=O: 1.84 Å vs Sb–OH: 2.00 Å), indicative of a double bond character. Next, we performed a time-dependent (TD) DFT study to simulate the UV–vis spectrum of both I+ and oxo-I, with the final aim to collect information about the relevant electronic transitions connected with excitation of the chromophore and the involved orbitals. Notably, the TD-DFT simulations performed in acetonitrile on the previously optimized structures of I+ and oxo-I indicate that these two forms show very similar spectra, and a close agreement between the simulated and the experimental spectrum of oxo-I was found, further confirming the accuracy of the computational method used (Figure S2). Careful inspection of the orbitals implicated in these transitions reveals that they mainly involve the displacement of electronic density within the aromatic π-system of the tetrapyrrole ring without a significant contribution of the Sb-center and its ligands. These results are consistent with the experimental data typically obtained for high-valent antimony porphyrins, which can be classified as normal type metalloporphyrin complexes with regular electronic absorption spectra dominated by intraligand π–π* transitions of the porphyrin macrocycle.29
Because it is well-known that HAT reactivity is typical of triplet excited states30 and previous studies demonstrated that the photoreactivity of Sb-based porphyrins involves this spin manifold,21,23,24 we also optimized the structures of the lowest lying triplet state of 3I+ and 3oxo-I because they could be populated upon intersystem crossing (ISC) from the first formed singlet excited state(s). At variance with the above, 3I+ and 3oxo-I show markedly different features in terms of the electronic structure. In 3oxo-I, inspection of the two singly occupied molecular orbitals (SOMOs) reveals that one electron is located on the π-system of the porphyrin ring, while the second one is centered on the oxygen atom of the oxo group, as we postulated. On the other hand, both SOMOs in 3I+ show electronic density only at the porphyrin ring and the p-methoxyphenyl substituents (see Figure S3). This difference is further corroborated by the spin density plots reported in Figure 2, where a contribution by the oxo ligand is apparent only for 3oxo-I, highlighting the presence of an unpaired electron on this site (red circle in Figure 2b). Quite interestingly, no spin localization is present at the antimony core in the analyzed triplets, with the central Sb atom remaining in the fully oxidized state. For the sake of comparison, we extended the latter analysis to the case of the parent tetraphenylporphyrin complex [Sb(tpp)(OH)2]+ (Ia+; tpp = tetraphenylporphyrin). Thus, the lowest lying triplet state 3oxo-Ia showed a strictly related electronic configuration to 3oxo-I, sharing the same features observed above in terms of the SOMO location (Figure S3) and spin density plot (Figure 2c).
Figure 2.
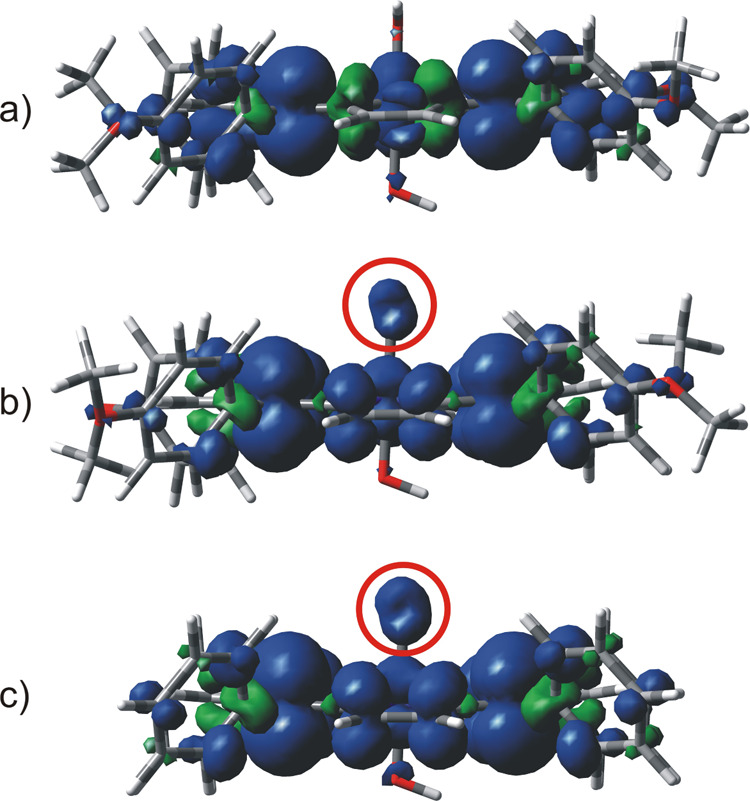
Spin density plots of the lowest lying triplet state of (a) 3I+, (b) 3oxo-I, and (c) 3oxo-Ia at the UωB97XD/def2SVP level of theory in the gas phase (side view).
Next, we computationally studied the deactivated form of the photocatalyst, namely complex I• (Figure 3), formed from 3oxo-I through a HAT step. It is worth mentioning here that this species is a neutral doublet (2I•); its optimized structure can be found in Figure S1, while the corresponding spin density plot is reported in Figure 3. Also in this case, no spin localization can be observed at the Sb-center, which remains in the SbV oxidation state and the π-system of the porphyrin ring is the only responsible acceptor site for the extra electron present in complex I•. In other words, the redox isomeric π-radical anion form of the reduced antimony porphyrin complex and not a SbIV radical intermediate is involved in the process,22 which is consistent with a ligand-to-ligand charge transfer character of the reactive triplet excited state of 3oxo-I, as also reflected by the spin-density plot of the photoactivated complex (see Figure 2b above).
Figure 3.
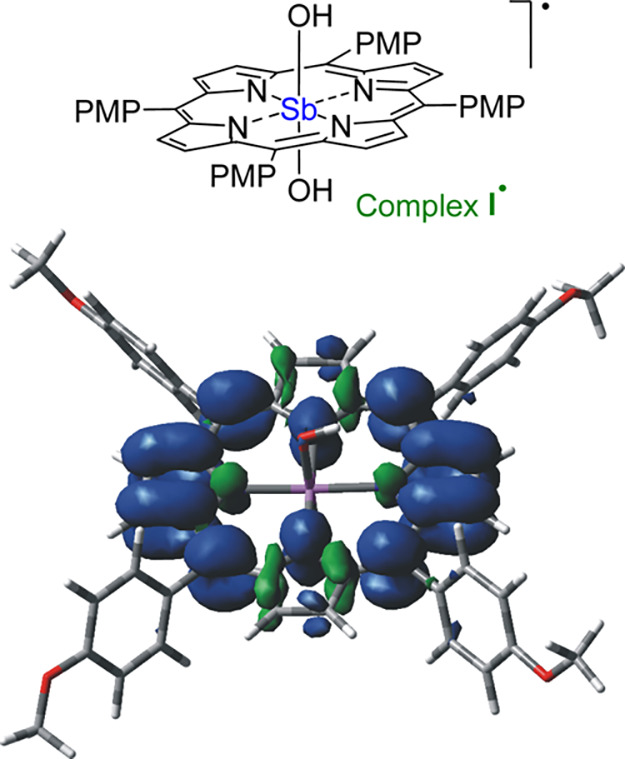
Spin density plot for complex 2I• as from the calculations at the UωB97XD/def2SVP level of theory in the gas phase. PMP = p-methoxyphenyl.
Experimental Data on the Use of Antimony–Oxo Porphyrins as PCHAT
In view of these encouraging computational results, we decided to test the potential of antimony porphyrins as visible-light PCHAT to promote the redox-neutral C–H to C–C conversion via a homolytic cleavage in organic substrates (Section 2.1 in the Supporting Information). As depicted in Table 1, we initially studied the addition of tetrahydrofuran (THF; 1a) onto dimethyl maleate (2a) in the presence of complex I+, under simulated sunlight irradiation (SolarBox equipped with a 1.5 kW Xe Lamp, light intensity 500 W·m–2). When an acetonitrile solution of 2a (0.05 M), 1a (0.5 M, 10 equiv) and complex I+ (1 mol %) in a 1 mm optical path cuvette was irradiated for 48 h, no consumption of olefin was observed, and the desired succinate 3 was not detected (entry 1). However, shifting to MeCN/H2O 95:5 along with the addition of 1 mol % NaOH led to the formation of 3 (60% gas chromatography (GC) yield, based on 67% consumption of 2a, entry 2). Varying the catalyst loading (in the 0.2–2 mol % range) confirmed that 1 mol % was the optimal choice (entries 3–5). Increasing the amount of water had a detrimental effect (entry 6), while the presence of oxygen completely inhibited the process (entry 7). Blank experiments demonstrated that both light and the catalyst are necessary (entries 8–9). To further promote the reactivity, we decided to use the more electrophilic dimethyl fumarate (2b)31 as the radical trap, which allowed the preparation of 3 in 77% yield after 24 h irradiation (entry 10). On the other hand, if complex I+ was replaced by the analogous tetraphenylporphyrin complex Ia+, a slightly diminished yield of 3 (66%) was observed (entry 11). In analogy to what observed in entry 1 for I+, no product 3 was detected when using complex Ia+ if base was omitted (entry 12). The result from entry 10 gave the opportunity to evaluate the effect of the light source by choosing different wavelengths, according to the absorption bands in the UV–vis spectrum of oxo-I (Figure S2).
Table 1. Investigation on the Photocatalyzed Addition of THF (1a) Onto Electron-Poor Olefins (2) in the Presence of Antimony-Based Porphyrin Complexesa.

| entry | olefin | Sb-based porphyrin | variation from optimized conditions | consumption (%) | yield (%)b |
|---|---|---|---|---|---|
| 1 | 2a | I+ | no base (NaOH) | 0 | n.d. |
| 2 | 2a | I+ | none | 67 | 60 |
| 3 | 2a | I+ | catalyst, base loading: 0.2 mol % | 19 | 11 |
| 4 | 2a | I+ | catalyst, base loading: 0.4 mol % | 18 | 15 |
| 5 | 2a | I+ | catalyst, base loading: 2.0 mol % | 32 | 28 |
| 6 | 2a | I+ | MeCN/H2O 1:1 | 90 | 43 |
| 7 | 2a | I+ | air-equilibrated conditions | 15 | traces |
| 8 | 2a | I+ | in the dark | 0 | n.d. |
| 9 | 2a | I+ | no photocatalyst | 0 | n.d. |
| 10 | 2b | I+ | 24 h irradiation | 100 | 77 |
| 11 | 2b | Ia+ | 24 h irradiation | 100 | 66 |
| 12 | 2b | Ia+ | no base (NaOH) | 0 | n.d. |
| 13 | 2b | I+ | irradiation wavelength: 366 nm | 0 | n.d. |
| 14 | 2b | I+ | irradiation wavelength: 405 nm | 80 | 70 |
| 15 | 2b | I+ | irradiation wavelength: 455 nm | 53 | 70 |
| 16 | 2b | I+ | irradiation wavelength: 589 nm | 0 | n.d. |
Conditions: reaction performed in a 1 mm cuvette on a 300 μL nitrogen-purged solution containing 1a (10 equiv), 2 (0.05 M) and complex I+ or Ia+ (1 × 10–4 to 1 × 10–3 M, 0.2–2.0 mol %); the corresponding oxo-I or oxo-Ia complexes are generated in situ in the presence of NaOH in the chosen reaction medium (see Supporting Information, method B).
GC yields referred to the consumption of the limiting reagent (2), using n-dodecane as the internal standard; n.d.: not detected.
Thus, irradiation of complex oxo-I with ten 15 W phosphor-coated lamps (λEM centered at 366 nm) under the optimized conditions did not lead to any appreciable consumption of 2b (entry 13). The situation changed dramatically, however, when employing monochromatic visible-light LEDs (1 W) with emission centered at 405 or 455 nm because we consistently observed the formation of product 3 in 70% yield, albeit in the former case a higher conversion of 2b (80 vs 53%) was found (compare entries 14 and 15). Furthermore, we also tested a sodium vapour lamp (emission centered at 589 nm), but the olefin remained untouched (entry 16).
Next, we tested the catalytic system on a bigger scale (0.05 mmol), making use of more powerful visible light irradiation sources (405 or 455 nm LEDs, 18 W) to extend the optimized protocol to different reaction partners (Table 2). Gratifyingly, we found excellent yields in most of the tested cases and, in general terms, the reactions proceeded faster under 405 nm than 455 nm irradiation. Thus, the addition of 1a onto 2b afforded product 3 in 91 and 44% yield, respectively, upon irradiation for 48 h, albeit only a partial conversion of the starting materials was observed. Similarly, 1a could be smoothly added onto 2-cyclohexylidenemalononitrile (2c) and 2-benzylidenemalononitrile (2d) to give 4 and 5, respectively, in yields up to 82%. Finally, the feasibility of a C(sp2)–H cleavage was verified by adopting heptaldehyde 1b as the substrate. Also in this case, compounds 6 and 7 were obtained in good yields from the reaction with 2b and 2d, respectively. On the other hand, no product formation was observed when the challenging alkylation of 2b with cyclohexane 1c as the H-donor was attempted (data not shown).
Table 2. Investigation on the Photocatalyzed Addition of Different Hydrogen Donors (1a,b) Onto Electron-Poor Olefins 2b-d in the Presence of Antimony–Porphyrin Complex I+a,b.


1a: tetrahydrofuran; 1b: heptaldehyde; 2b: dimethyl fumarate; 2c: 2-cyclohexylidenemalononitrile; and 2d: 2-benzylidenemalononitrile.
Conditions: reaction performed in a 1 dram vial on a 1 mL nitrogen-purged solution containing 1 (10 equiv for 1a; 1 equiv for 1b), 2 (0.05 M) and oxo-I (5 × 10–4 M, 1.0 mol %) in MeCN/H2O 95:5 (see Supporting Information, method A). GC yields referred to the consumption of the limiting reagent (2), using n-dodecane as the internal standard. Brsm: based on remaining starting materials.
Mechanistic Investigation
The radical nature of the process can be inferred from its inhibition in the presence of TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl; Scheme 1a). Building on this result, we then performed isotopic labelling experiments to have insights into the mechanistic scenario and the results are gathered in Scheme 1 (see also Sections 2.2 and 2.3 in the Supporting Information). When the reaction between THF and 2b was performed by adopting an equimolar mixture of 1a and perdeuterated 1a-d8 (5 equiv each), a preferential activation of the former occurred, with formation of 3 and 3-d7 in a 3.5:1 ratio (Scheme 1b) according to GC–MS analysis (see Supporting Information for details). Next, we repeated the experiment in the presence of a deuterated medium, to verify if the solvent had a role. When the experiment was run in a CD3CN/H2O mixture, no deuterium was incorporated in the obtained product, while a significant deuteration of the 3-position of 3 was observed when adopting CH3CN/D2O as the medium (Scheme 1c).
Scheme 1. Chemical Quenching Experiments (Part a) and Isotopic Labeling Studies in the Reaction between 1a and 2b: Cross-Over Experiment (Part b); Use of the Deuterated Media (Part c).

Another point worth to be investigated relates to the activation mechanism of H-donors by the excited porphyrin complex. Previous studies indicated that the triplet excited state of Sb-based tetraarylporphyrins is able to oxidize monoelectronically the hydroxide anion to the hydroxyl radical HO•,21c a potent oxidant able to trigger an indirect photocatalytic HAT process.3b However, this pathway has been reported to occur in the presence of a large excess of the base (500-fold),21c while an equimolar amount of NaOH with respect to antimony porphyrin is added under our conditions to form oxo-I. Nonetheless, with the final aim to safely exclude a role for this indirect pathway and to further elucidate the nature of the light-mediated primary steps, we carried out excited state quenching experiments with THF (1a) as the H-donor (Section 2.4 in the Supporting Information). Upon addition of increasing amounts of 1a to samples of the active photocatalyst oxo-I, no indication of the singlet excited state quenching was observed, as monitored by fluorescence quantum yield measurements (data not shown). On the other hand, a nanosecond laser flash photolysis study of the triplet excited state manifold clearly confirmed that a dynamic bimolecular quenching process between the photocatalyst and 1a was taking place.
Time-resolved spectra of the photoexcited catalyst oxo-I in the absence of a quencher are shown in Figure 4 (see also Figure S9 for a longer timescale). Bleaching of the ground state absorption spectrum giving rise to a negative Soret-band signal between 400 and 440 nm is accompanied by an arising transient signal with a peak around 460 nm, which is characteristic for the triplet excited state of regular tetraarylporphyrin metal complexes.32,33 In addition, the triplet–triplet absorption spectrum of the antimony–oxo complex in the given solvent mixture displays a second conspicuous signal with a maximum at 510 nm (Figure 4), which decays with a lifetime of (8.0 ± 0.1) μs, in agreement with a significant charge-transfer character of the triplet excited state manifold of 3oxo-I.
Figure 4.
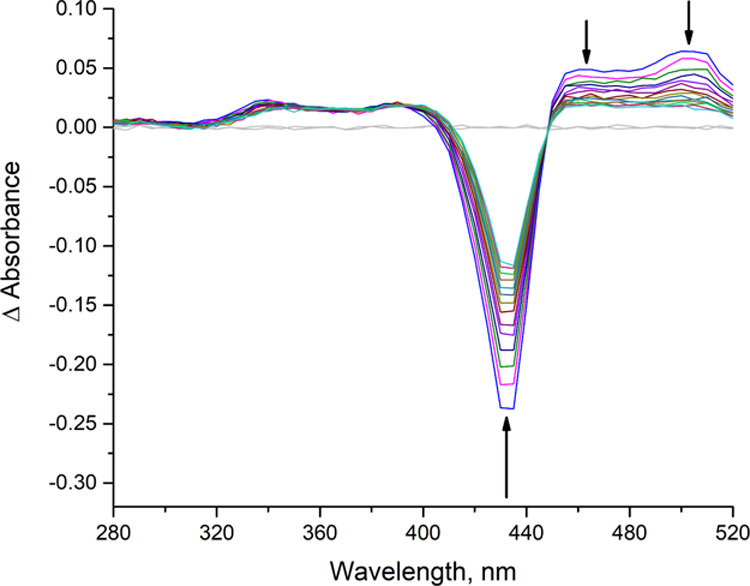
Transient differential absorption spectra obtained upon nanosecond flash photolysis (532 nm) of oxo-I in degassed CH3CN/H2O 95:5 showing the spectral variations occurring within the first 17 μs after the laser flash. The flat lines in gray represent the pre-pulse signals.
The deactivation rate of the lowest triplet excited state of oxo-I significantly increases upon addition of 1a, clearly acting as a quencher (Figure 5). The corresponding Stern–Volmer plot is linear in the tested concentration range (up to 1 M THF) and yields a slope of KSV = (0.26 ± 0.01) M–1 at 298 K in agreement with a dynamic bimolecular process. From the corresponding lifetime data, a quenching rate constant of kQ = (3.3 ± 0.2) × 104 M–1·s–1 was obtained.
Figure 5.

Left side: time absorption profiles recorded at 510 nm, monitoring the monoexponential decay process of the lowest excited triplet state of oxo-I in degassed CH3CN/H2O 95:5 in the absence (blue line) and presence (red line) of THF. Right side: Photokinetic data analysis indicates that bimolecular quenching of the oxo-I triplet state manifold by the hydrogen atom donor THF occurs.
Based on the above reported computational and experimental evidence, we propose that the present photocatalytic process occurs as depicted in Scheme 2 for the case of the tetra(p-methoxyphenyl)-substituted metalloporphyrin. Thus, dihydroxo complex I+ is not active as PCHAT, however, upon addition of a stoichiometric amount of base (aqueous NaOH), the antimony–oxo functionality of oxo-I is generated in situ. Following visible light absorption and ISC, the triplet excited state (3oxo-I) endowed with a partial oxyl radical character is populated, which confirms previous speculations present in the literature.24 Notably, the same behavior has been confirmed for the parent 3oxo-Ia derivative as well. According to excited state quenching experiments, 3oxo-I is prone to abstract a hydrogen atom from 1 to give the C-centered radical 8•, which is then trapped by 2 to afford 9•. Finally, the desired Giese adduct is generated upon back-HAT or stepwise electron/proton transfer. This last step consists in the closure of the photocatalytic cycle, which also restores the active form of the photocatalyst without the need of terminal oxidants. Furthermore, according to Scheme 1c, we suggest that a ligand exchange involving a protic solvent may occur at the I• stage to give I•-d1, explaining the partial deuteration observed when a deuterated protic medium was employed.
Scheme 2. Proposed Mechanism. EWG: Electron-Withdrawing Group.

The hereby reported use of an oxo-porphyrin derivative to promote a redox-neutral process occurring via visible-light photocatalyzed HAT has not been previously explored. Key to the success of this chemistry is the adoption of a stable metal–oxo porphyrin complex, containing a high-valent oxidation state element of the p-block, notably antimony, for several reasons. First of all, SbV can coordinate two additional ligands (the hydroxyl groups) other than porphyrin, which is a prerequisite to install the axial oxo functionality implicated in the excited state reactivity. On top of that, high-valent antimony is superior to other elements because it exerts a peculiar electron-withdrawing electronic effect and imposes a strong polarization to the attached axial ligands and the porphyrin macrocycle, accordingly.34 This property is expected to have a crucial role in both controlling the pKa of the attached hydroxyl ligands and the reactivity in the triplet excited state.
The approach is remarkably different from that of iron- and manganese–oxo porphyrins known to promote hydroxylation and halogenation reactions in alkanes via a thermal HAT step.35 The latter behavior may be attributed to the presence of a partially filled d shell in the metal center, endowing the complex with a radicaloid character. These complexes are characterized by several low-lying, close-in-energy electronic states that mix together, giving rise to the so-called “redox mesomerism” phenomenon.36 Thus, these metal–oxo porphyrins trigger a thermal homolytic C–H cleavage, and the resulting C-centered radical can be used for C–X bond formation through a rebound mechanism,35 with the concomitant reduction of the complex (the oxidation state at M goes from n+ to (n – 2)+). Accordingly, these complexes have been conveniently exploited as catalysts in net-oxidative processes, where the presence of a terminal oxidant is mandatory for the formation of the oxo-species. This peculiar characteristic makes these complexes unsuitable for other types of chemistry, such as the redox-neutral process presented herein.
Conclusions
Starting off from computational investigation, we experimentally demonstrated that a thermally unreactive SbV–oxo porphyrin can be exploited to trigger a redox-neutral C–H to C–C bond conversion based on a photocatalytic HAT step occurring under visible light irradiation. We also proved the radical nature of the process via inhibition experiments performed in the presence of TEMPO, while the involvement of a HAT step is corroborated by the observed kinetic isotopic effect. Meticulous spectroscopic investigation allowed to identify the nature of the activation step and to determine the quenching constant value, as well.
Finally, according to our computational analysis, the SbV center remains in the high-valent oxidation state under the conditions explored, serving uniquely to carry the oxo moiety and to activate the coordinated ligands. Thus, antimony has a spectator role in the key steps of the photocatalytic cycle, suggesting that other high-valent porphyrin complexes featuring an oxo ligand may be envisaged as PCHAT. Experiments in this direction are ongoing in our laboratories.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c02250.
Experimental details about the used materials; sample preparation; mechanistic experiments; and computational data (including optimized structures) (PDF)
Author Contributions
G.K. and D.R. conceived the project and supervised the activities. G.K developed and provided the catalyst, M.E. performed photophysical experiments, L.C. conducted the reactivity tests, and D.R. performed the computational work. M.F. helped writing the manuscript and contributed to the interpretation of experimental data. All authors have given approval to the final version of the manuscript.
L.C. and D.R. thank the MIUR for financial support (SIR Project “Organic Synthesis via Visible Light Photocatalytic Hydrogen Transfer”; Code: RBSI145Y9R). G.K. gratefully acknowledges the contributions of the COST Action CA15106 “C–H Activation in Organic Synthesis”. Calculations were carried out at the CINECA Supercomputer Center (Italy), with computer time granted by ISCRA projects (code: HP10C30YAJ).
The authors declare no competing financial interest.
Supplementary Material
References
- a Fagnoni M.; Protti S.; Ravelli D.. Photoorganocatalysis in Organic Synthesis; World Scientific Publishing: Singapore, 2019. [Google Scholar]; b Stephenson C.; Yoon T.; MacMillan D. W. C.. Visible Light Photocatalysis in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018. [Google Scholar]; c König B.Chemical Photocatalysis; De Gruyter: Berlin, Boston, 2013. [Google Scholar]
- For selected reviews, see:; a Ravelli D.; Protti S.; Fagnoni M. Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. 10.1021/acs.chemrev.5b00662. [DOI] [PubMed] [Google Scholar]; b Sideri I. K.; Voutyritsa E.; Kokotos C. G. Photoorganocatalysis, Small Organic Molecules and Light in the Service of Organic Synthesis: The Awakening of a Sleeping Giant. Org. Biomol. Chem. 2018, 16, 4596–4614. 10.1039/c8ob00725j. [DOI] [PubMed] [Google Scholar]
- a Protti S.; Fagnoni M.; Ravelli D. Photocatalytic C-H Activation by Hydrogen-Atom Transfer in Synthesis. ChemCatChem 2015, 7, 1516–1523. 10.1002/cctc.201500125. [DOI] [Google Scholar]; b Capaldo L.; Ravelli D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2056–2071. 10.1002/ejoc.201601485. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Knör G. The Concept of Photochemical Enzyme Models – State of the Art. Coord. Chem. Rev. 2016, 325, 102–115. 10.1016/j.ccr.2016.06.006. [DOI] [Google Scholar]
- Strieth-Kalthoff F.; James M. J.; Teders M.; Pitzer L.; Glorius F. Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions. Chem. Soc. Rev. 2018, 47, 7190–7202. 10.1039/c8cs00054a. [DOI] [PubMed] [Google Scholar]
- Xiao W.-J.; Hu X.-Q.; Chen J.-R.. Oxygen Heterocycles: Eosin Derivatives. In Photoorganocatalysis in Organic Synthesis; Fagnoni M., Protti S., Ravelli D., Eds.; World Scientific Publishing: Singapore, 2019. [Google Scholar]
- Ravelli D.; Fagnoni M. Dyes as Visible Light Photoredox Organocatalysts. ChemCatChem 2012, 4, 169–171. 10.1002/cctc.201100363. [DOI] [Google Scholar]
- Hari D. P.; König B. Synthetic Applications of Eosin Y in Photoredox Catalysis. Chem. Commun. 2014, 50, 6688–6699. 10.1039/c4cc00751d. [DOI] [PubMed] [Google Scholar]
- Fan X.-Z.; Rong J.-W.; Wu H.-L.; Zhou Q.; Deng H.-P.; Tan J. D.; Xue C.-W.; Wu L.-Z.; Tao H.-R.; Wu J. Eosin Y as a Direct Hydrogen-Atom Transfer Photocatalyst for the Functionalization of C–H Bonds. Angew. Chem., Int. Ed. 2018, 57, 8514–8518. 10.1002/anie.201803220. [DOI] [PubMed] [Google Scholar]
- Kamijo S.; Kamijo K.; Maruoka K.; Murafuji T. Aryl Ketone Catalyzed Radical Allylation of C(sp3)–H Bonds under Photoirradiation. Org. Lett. 2016, 18, 6516–6519. 10.1021/acs.orglett.6b03586. [DOI] [PubMed] [Google Scholar]
- Capaldo L.; Merli D.; Fagnoni M.; Ravelli D. Visible Light Uranyl Photocatalysis: Direct C–H to C–C Bond Conversion. ACS Catal. 2019, 9, 3054–3058. 10.1021/acscatal.9b00287. [DOI] [Google Scholar]
- a Gust D.; Moore T. A.; Moore A. L. Solar Fuels via Artificial Photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]; b Fukuzumi S.; Ohkubo K.; Suenobu T. Long-Lived Charge Separation and Applications in Artificial Photosynthesis. Acc. Chem. Res. 2014, 47, 1455–1464. 10.1021/ar400200u. [DOI] [PubMed] [Google Scholar]; c Knör G. Recent Progress in Homogeneous Multielectron Transfer Photocatalysis and Artificial Photosynthetic Solar Energy Conversion. Coord. Chem. Rev. 2015, 304–305, 102–108. 10.1016/j.ccr.2014.09.013. [DOI] [Google Scholar]
- Kadish K. M., Smith K. M., Guilard R.. Handbook of Porphyrin Science; World Scientific Publishing: Singapore, 2010. [Google Scholar]
- a Sarma T.; Panda P. K. Annulated Isomeric, Expanded, and Contracted Porphyrins. Chem. Rev. 2017, 117, 2785–2838. 10.1021/acs.chemrev.6b00411. [DOI] [PubMed] [Google Scholar]; b Lu H.; Kobayashi N. Optically Active Porphyrin and Phthalocyanine Systems. Chem. Rev. 2016, 116, 6184–6261. 10.1021/acs.chemrev.5b00588. [DOI] [PubMed] [Google Scholar]
- Baptista M. S.; Cadet J.; Di Mascio P.; Ghogare A. A.; Greer A.; Hamblin M. R.; Lorente C.; Nunez S. C.; Ribeiro M. S.; Thomas A. H.; Vignoni M.; Yoshimura T. M. Type I and Type II Photosensitized Oxidation Reactions: Guidelines and Mechanistic Pathways. Photochem. Photobiol. 2017, 93, 912–919. 10.1111/php.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Abrahamse H.; Hamblin M. R. New Photosensitizers for Photodynamic Therapy. Biochem. J. 2016, 473, 347–364. 10.1042/bj20150942. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Protti S.; Albini A.; Viswanathan R.; Greer A. Targeting Photochemical Scalpels or Lancets in the Photodynamic Therapy Field-The Photochemist’s Role. Photochem. Photobiol. 2017, 93, 1139–1153. 10.1111/php.12766. [DOI] [PubMed] [Google Scholar]; c Lanzilotto A.; Kyropoulou M.; Constable E. C.; Housecroft C. E.; Meier W. P.; Palivan C. G. Porphyrin-Polymer Nanocompartments: Singlet Oxygen Generation and Antimicrobial Activity. J. Biol. Inorg Chem. 2018, 23, 109–122. 10.1007/s00775-017-1514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García H.; Remiro-Buenamañana S.. Nitrogen Heterocycles: Porphyrins. In Photoorganocatalysis in Organic Synthesis; Fagnoni M., Protti S., Ravelli D., Eds.; World Scientific Publishing: Singapore, 2019. [Google Scholar]
- a Alberti M.; Orfanopoulos M. Recent Mechanistic Insights in the Singlet Oxygen Ene Reaction. Synlett 2010, 999–1026. [Google Scholar]; b Walaszek D. J.; Rybicka-Jasińska K.; Smoleń S.; Karczewski M.; Gryko D. Mechanistic Insights into Enantioselective C–H Photooxygenation of Aldehydes via Enamine Catalysis. Adv. Synth. Catal. 2015, 357, 2061–2070. 10.1002/adsc.201500056. [DOI] [Google Scholar]; c Mojarrad A. G.; Zakavi S. Simple Low Cost Porphyrinic Photosensitizers for Large Scale Chemoselective Oxidation of Sulfides to Sulfoxides under Green Conditions: Targeted Protonation of Porphyrins. Catal. Sci. Technol. 2018, 8, 768–781. 10.1039/c7cy02308a. [DOI] [Google Scholar]; d Fukuzumi S.; Lee Y. M.; Nam W. Photocatalytic Oxygenation Reactions Using Water and Dioxygen. ChemSusChem 2019, 12, 3931–3940. 10.1002/cssc.201901276. [DOI] [PubMed] [Google Scholar]
- Rybicka-Jasińska K.; Shan W.; Zawada K.; Kadish KM; Gryko D. Porphyrins as Photoredox Catalysts: Experimental and Theoretical Studies. J. Am. Chem. Soc. 2016, 138, 15451–15458. [DOI] [PubMed] [Google Scholar]
- Rybicka-Jasińska K.; König B.; Gryko D. Porphyrin-Catalyzed Photochemical C-H Arylation of Heteroarenes. Eur. J. Org. Chem. 2017, 2104–2107. [Google Scholar]
- Shiragami T.; Matsumoto J.; Inoue H.; Yasuda M. Antimony Porphyrin Complexes as Visible-Light Driven Photocatalyst. J. Photochem. Photobiol., C 2005, 6, 227–248. 10.1016/j.jphotochemrev.2005.12.001. [DOI] [Google Scholar]
- a Inoue H.; Sumitani M.; Sekita A.; Hida M. Photochemical Epoxidation of Alkenes by Visible Light in a Redox System Involving Tetraphenylporphyrinantimony(V) and Water. J. Chem. Soc., Chem. Commun. 1987, 1681–1682. [Google Scholar]; b Inoue H.; Okamoto T.; Komiyama M.; Hida M. Photochemical Epoxidation of Alkene by Visible Light in a Redox System Involving Metalloporphyrins and Water. J. Photochem. Photobiol., A 1992, 65, 221–227. 10.1016/1010-6030(92)85047-x. [DOI] [Google Scholar]; c Takagi S.; Okamoto T.; Shiragami T.; Inoue H. Photochemical Electron Transfer from Hydroxide Ion to the Excited Triplet State of Tetraphenylporphyrinatoantimony(V) upon Visible Light Irradiation in Aqueous Acetonitrile. Chem. Lett. 1993, 793–796. 10.1246/cl.1993.793. [DOI] [Google Scholar]; d Inoue H.; Okamoto T.; Kameo Y.; Sumitani M.; Fujiwara A.; Ishibashi D.; Hida M. Photochemical Epoxidation of Cyclohexene Sensitized by Tetraphenylporphyrinatoantimony(V) in the Presence of Water Acting Both as an Electron and an Oxygen Donor. J. Chem. Soc., Perkin Trans. 1 1994, 105–111. [Google Scholar]; e Shiragami T.; Kubomura K.; Ishibashi D.; Inoue H. Efficient Photochemical Oxygenation of Cyclohexene with Water as an Oxygen Donor Sensitized by Dimethoxy-Coordinated Tetraphenylporphyrinatoantimony(V). J. Am. Chem. Soc. 1996, 118, 6311–6312. 10.1021/ja960020u. [DOI] [Google Scholar]; f Takagi S.; Suzuki M.; Shiragami T.; Inoue H. Photochemical P-450 Oxygenation of Cyclohexene with Water Sensitized by Dihydroxy-Coordinated (Tetraphenylporphyrinato)Antimony(V) Hexafluorophosphate. J. Am. Chem. Soc. 1997, 119, 8712–8713. 10.1021/ja971371r. [DOI] [Google Scholar]
- Knör G.; Vogler A.; Roffia S.; Paolucci F.; Balzani V. Switchable Photoreduction Pathways of Antimony(V) Tetraphenylporphyrin. A Potential Multielectron Transfer Photosensitizer. Chem. Commun. 1996, 1643–1644. 10.1039/cc9960001643. [DOI] [Google Scholar]
- Knör G. Bionic Catalyst Design: A Photochemical Approach to Artificial Enzyme Function. ChemBioChem 2001, 2, 593–596. . [DOI] [PubMed] [Google Scholar]
- Hajimohammadi M.; Schwarzinger C.; Knör G. Controlled Multistep Oxidation of Alcohols and Aldehydes to Carboxylic Acids Using Air, Sunlight and a Robust Metalloporphyrin Sensitizer with a PH-Switchable Photoreactivity. RSC Adv. 2012, 2, 3257–3260. 10.1039/c2ra01076c. [DOI] [Google Scholar]
- Ertl M.; Wöβ E.; Knör G. Antimony porphyrins as red-light powered photocatalysts for solar fuel production from halide solutions in the presence of air. Photochem. Photobiol. Sci. 2015, 14, 1826–1830. 10.1039/c5pp00238a. [DOI] [PubMed] [Google Scholar]
- An analogous trend has been observed for strictly-related SbIII–porphyrin complexes, see:Knör G.; Vogler A. Photochemistry and photophysics of antimony(III) hyper porphyrins: activation of dioxygen induced by a reactive sp excited state. Inorg. Chem. 1994, 33, 314–318. [Google Scholar]
- Silaghi-Dumitrescu R. High-Valent Metalloporphyrins in Hydrocarbon Activation: Metal(v)-Oxo or Metal(v)-Hydroxo?. New J. Chem. 2010, 34, 1830–1833. 10.1039/c0nj00297f. [DOI] [Google Scholar]
- a Qi S.-C.; Hayashi J.-i.; Zhang L. Recent Application of Calculations of Metal Complexes Based on Density Functional Theory. RSC Adv. 2016, 6, 77375–77395. 10.1039/c6ra16168e. [DOI] [Google Scholar]; b Minenkov Y.; Singstad Å.; Occhipinti G.; Jensen V. R. The Accuracy of DFT-Optimized Geometries of Functional Transition Metal Compounds: A Validation Study of Catalysts for Olefin Metathesis and Other Reactions in the Homogeneous Phase. Dalton Trans. 2012, 41, 5526–5541. 10.1039/c2dt12232d. [DOI] [PubMed] [Google Scholar]
- Knör G. Reductive Fluorescence Quenching of the Photoexcited Dihydroxy Antimony(V) Tetraphenylporphine Cation in Acetonitrile Solution. Chem. Phys. Lett. 2000, 330, 383–388. 10.1016/s0009-2614(00)01093-9. [DOI] [Google Scholar]
- Waele V. D.; Poizat O.; Fagnoni M.; Bagno A.; Ravelli D. Unraveling the Key Features of the Reactive State of Decatungstate Anion in Hydrogen Atom Transfer (HAT) Photocatalysis. ACS Catal. 2016, 6, 7174–7182. 10.1021/acscatal.6b01984. [DOI] [Google Scholar]
- Allgäuer D. S.; Mayr H. Electrophilicities of 1,2-Disubstituted Ethylenes. Eur. J. Org. Chem. 2014, 2956–2963. 10.1002/ejoc.201301779. [DOI] [Google Scholar]
- Gouterman M., Rentzepis P. M., Straub K. D.. Porphyrins: Excited States and Dynamics; ACS Symposium Series; American Chemical Society: Washington, DC, 1986; Vol. 321. [Google Scholar]
- Beddard G. S.; Carlin S.; Harris L.; Porter G.; Tredwell C. J. Quenching of Chlorophyll Fluorescence by Nitrobenzene. Photochem. Photobiol. 1978, 27, 433–438. 10.1111/j.1751-1097.1978.tb07625.x. [DOI] [Google Scholar]
- Furhop J. H.; Kadish K. M.; Davis D. G. Redox behavior of metallo octaethylporhyrins. J. Am. Chem. Soc. 1973, 95, 5140–5147. 10.1021/ja00797a008. [DOI] [PubMed] [Google Scholar]
- a Song W. J.; Ryu Y. O.; Song R.; Nam W. Oxoiron(IV) Porphyrin π-Cation Radical Complexes with a Chameleon Behavior in Cytochrome P450 Model Reactions. J. Biol. Inorg Chem. 2005, 10, 294–304. 10.1007/s00775-005-0641-9. [DOI] [PubMed] [Google Scholar]; b Nam W. High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions. Acc. Chem. Res. 2007, 40, 522–531. 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; c Liu W.; Groves J. T. Manganese Catalyzed C–H Halogenation. Acc. Chem. Res. 2015, 48, 1727–1735. 10.1021/acs.accounts.5b00062. [DOI] [PubMed] [Google Scholar]; d Milan M.; Salamone M.; Costas M.; Bietti M. The Quest for Selectivity in Hydrogen Atom Transfer Based Aliphatic C–H Bond Oxygenation. Acc. Chem. Res. 2018, 51, 1984–1995. 10.1021/acs.accounts.8b00231. [DOI] [PubMed] [Google Scholar]
- Ogliaro F.; de Visser S. P.; Groves J. T.; Shaik S. Chameleon States: High-Valent Metal-Oxo Species of Cytochrome P450 and Its Ruthenium Analogue. Angew. Chem., Int. Ed. 2001, 40, 2874–2878. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.