

Abstract
Neonatal diabetes (ND) appears during the first months of life and is caused by a single gene mutation. It is heterogenous and very different compared to other forms of multi-factorial or polygenic diabetes. Clinically, this form is extremely severe, however, early genetic diagnosis is pivotal for successful therapy. A large palette of genes is demonstrated to be a cause of ND, however, the mechanisms of permanent hyperglycemia are different. This review will give an overview of more frequent genetic mutations causing ND, including the function of the mutated genes and the specific therapy for certain sub-forms.
Keywords: Classification, Mutations, Neonatal diabetes (ND), Syndromes
Introduction
Most of the patients with diabetes mellitus (DM) in childhood suffer from DM type 1 (DMT1) that is of autoimmune etiology [1,2]. This form of diabetes appears after the first 6 months of life, and reaches the highest incidence in the 9-14 years age group, with the rising incidence in the age group before 4 years of age [3]. The persisting hyper-glycemia is due to autoimmune destruction of insulin producing β-cells in the endocrine pancreas leading to insulin deficiency. It is considered a polygenic disease involving mostly DQ and DR human leukocyte antigen (HLA) genes conferring susceptibility or resistance toward the disease [4, 5, 6, 7, 8]. Diabetes mellitus type 1 accounts for more than 95.0% of cases in childhood [7,9,10]. During the last few decades, obesity in childhood has caused increase of DM type 2 (DMT2) in children, which is induced by an interplay of genetic and environmental factors. Genetic factors involved in DMT2 are still not precisely elucidated. A number of different gene polymorphisms affecting β-cell function causes impaired insulin secretion or insulin resistance [11,12]. The prevalence of DMT2 in childhood and adolescence varies in different countries, reaching up to 30.0% of all cases with DM in regions with the highest childhood obesity rates [1].
Monogenic diabetes (MD) caused by mutations of a single gene is a separate form of DM with a distinct etiology, clinical presentation, therapy and outcome. Monogenic diabetes is a heterogenous group of sub-forms of diabetes caused by mutations in different, highly penetrant genes that are pivotal for pancreas development, sensing of the level of glucose for insulin secretion, cellular metabolism, cell membrane depolarization control, or insulin synthesis/excretion [13, 14, 15, 16, 17, 18, 19]. Monogenic diabetes is not common, but it does account for 1.0-6.0% of pediatric diabetes cases [20].
Generally, based upon the detected mutation, timing of presentation, and affected genes, MD can be classified as follows [19]: 1) neonatal diabetes (ND) (occurring before 6 months), 2) syndromic ND associated with extra pancreatic features, and 3) autosomal dominant familial hyperglycemia or diabetes or maturity onset diabetes of the young (MODY). This review will give an overview of mutations causing diabetes in the neonatal period that includes ND and syndromic diabetes.
Genes Involved in Monogenic Diabetes. Mutations in about 40 different genes are recognized as a cause of MD. These encompass genes involved in different processes that could compromise insulin secretion and action such as: reduction of β-cell number or pancreatic aplasia, impaired β-cell development, glucose sensing or metabolism, failure to depolarize cellular membrane and expulsion of insulin in circulation, failure of insulin synthesis, increased destruction of β-cells including immune-based destruction, increased apoptosis, and endoplasmatic reticulum stress, as well as other mechanisms that need to be elucidated [21, 22, 23]. Each of these steps in insulin secretion and action is under the control of a particular gene [19,24] (Tables 1 and 2).
Table 1.
Gene mutations in transient neonatal diabetes mellitus.
| Mechanism of β-Cell Dysfunction | Gene Mutation | Chromosome Locus | Inheritance | Additional Features | Therapy |
|---|---|---|---|---|---|
| Reduced β-cell development | ZAC (IPLAG1)/ HYMA1 | 6q24 | imprinting; AD | macroglossia; umbilical hernia | insulin |
| ZEP57 | 6p22.1 | AR | insulin | ||
| HNF1B | 17q21.3 | AD | pancreatic cysts hypoplasia; renal | ||
| Failure membrane to depolarize | KCNJ11a (Kir6.2) | 11p15.1 | AD; de novo | low developmental birth weight; delay; DEND | sulfonylurea |
| Failure channel to close KATP | ABCC8a (SUR1) | 11p15.1 | AD; AR; de novo | low birth weight | sulfonylurea |
| Abnormal β-cell function | INSa (proinsulin) | 11p15.5 | AR | low birth weight | insulin |
AD: autosomal dominant; AR: autosomal recessive; DEND: developmental delay, epilepsy, DM; KATP: ATP-dependent potassium channel.
a These mutations can also be found in permanent neonatal diabetes mellitus.
Table 2.
Gene mutations in permanent neonatal diabetes mellitus.
| Mechanism of β-Cell Dysfunction | Gene Mutation | Chromosome Locus | Inheritance | Additional Features | Therapy |
|---|---|---|---|---|---|
| Failure membrane to depolarize | KCNJ11 (Kir6.2) | 11p15.1 | AD; de novo | low developmental birth weight; delay; DEND | sulfonylurea |
| Failure KATP channel to close | ABCC8 (SUR1) | 11p15.1 | de novo; AD; AR | low birth weight | sulfonylurea |
| Abnormal function β-cell | INS (proinsulin) | 11p15.5 | de novo; AD; AR | low birthweight | insulin |
| Abnormal sensing glucose | GCK | 7p15-13 | AR | ||
| Abnormal development pancreatic | PDX1 | 13.q12.1 | AR | pancreatic afenesis steatorrhea |
AD: autosomal dominant; AR: autosomal recessive; DEND: developmental delay, epilepsy, DM.
Most of the recent molecular analyses in MD have been performed by Sanger sequencing using specific primers, or targeted next generation sequencing (NGS) of involved exons and searching for multiple gene mutations [25, 26, 27, 28]. We have reviewed most of the recent guidelines for the diagnosis and treatment of all forms of ND, as well as numerous original articles describing characterizations of ND caused by different mutations.
Neonatal Diabetes. Although the neonatal period encompasses only the first 4 weeks of postnatal life, the term ND, by convention, is used for the DM that appears in neonates and infants up to 6 months of age, although some forms might appear up to 9-12 months of age [20,24]. It was first described in 1852 and has been reported since then in different countries and ethnicities around the world [24,28, 29, 30, 31, 32, 33]. If the hyperglycemia appears in the infant of 6-12 months, the distinction of the ND form early onset of DMT1 is necessary [34]. However, most of the patients do not display autoimmunity at this early age and belong to the monogenic “neonatal diabetes” [35]. The MODY mutations are present at birth, and they cause the appearance of diabetes later in life. Neonatal diabetes is rare, it appears, according to different authors, in one case per 100,000, 300,000 or even per 500,000 live births [19,23,36,37]. It can be classified as: transient ND mellitus (TNDM), and permanent ND mellitus (PNDM) [19,24,34,38,41]. Aguillar-Bryan et al. [24] have collected more than 195 patients from different studies and calculated that TNDM is more frequent, affecting 57.0% (111/ 195) of all neonates with ND.
Transient Neonatal Diabetes Mellitus. Transient ND mellitus appears in the first days or weeks after birth, usually in newborns with intrauterine growth retardation (IUGR). After introduction of low doses of insulin therapy, it resolves, usually approximately up to 4 months of age, but its duration could be extended up to 18 months [35, 36, 37, 38]. However, during adolescence or in the young adult period, relapses can occur in about 50.0% of patients, when it resembles DMT2 [36, 37, 38]. Genes involved in TNDM are provided in Table 1.
Table 3.
Mutations in syndromic neonatal diabetes mellitus.
| Mechanism | Gene Mutation | Chromosome Locus | Inheritance | Additional Features |
|---|---|---|---|---|
| Fanconi-Bickel syndrome | SCL2A2 (GLUT2) | 3q26.1-26.3 | AR | hypergalatosemia; liver dysfunction |
| Roger syndrome | SCL19A2 | 1q23.3 | AR | thiamine-sensorinueral responsive deafness megaloblastic anemia; |
| Abnormal development pancreatic | RFX6 | 6q22.1 | AR | intestinal atresia + bladder agenesis |
| GATA6 | 18q11-q11.2 | AD | pancreatic biliary abnormalities agenesis; heart defects; | |
| GATA4 | 8p23.1 | AD | pancreatic agenesis + heart defects | |
| GLIS3 | 9p24.3-p23 | AR | congenital hepatic fibrosis; hypothyridism; renal cysts glaucoma; | |
| NEURG3 | 10q21.3 | AR | malabsorbtive diarrhea | |
| NEUROD1 | 2q32 | AR | cerebellar deafness hypoplasia; visual impairment; | |
| PAX6 | 11.p13 | AR | microphtalmia; brain malformations | |
| MNX1 | 7q36.3 | AR | developmental imperforate anus delay; sacral agenesis; | |
| MNX2-2 | 20p11.22 | AR | developmental deafness delay; hypotonia; short stature; | |
| PTF1 | 10.p12.2 | AR | pancreatic hypoplasia; cerebellar hypoplasia | |
| Destruction of β-cells | ||||
| Wolcott-Rallison syndrome | EIF2AK3 | 2p11.2 | de novo or AD | skeletal dysplasia; liver dysfunction |
| IER3IP1 | 18q21.2 | AR | microcephaly; lisencephaly; enceph-alopathy | |
| IPEX syndrome | FOXP3 | Xp11.23-p13.3 | X-recessive linked; | autoimmune autoimmune enteropathy; hypothyroidism; eczema |
| Wolfram (DIDMOAD)syndrome a | WFS1 | 4p16.1 | AR | optic atrophy; DM; DI |
| WFS1 | 4p16.1 | AD | congenital cataracts; deafness |
AR: autosomal recessive; AD: autosomal dominant.
a Also known as Wolfram syndrome (see text).
The genetic origin of TNDM has been established for approximately 90.0% of patients with TNDM. The major genetic change causing the disease is abnormal imprinting at chromosome 6q24, which appears in approximately 70.0% of patients with TNDM [36,39,40, 41, 42, 43]. Usually, for this chromosome region, only alleles inherited from the father are expressed, whereas mother’s alleles are imprinted. Overexpression of paternal genes in this region can happen through uniparental isodisomy or inheritance of the duplication of the region from the father [44,45]. On the other hand, defects in maternal methylation of the 6q24 region can cause activation of the maternal alleles [46,47]. The methylation defect might be inherited or can appear sporadically. The 6q24 region is rich in imprinted genes. Only a few of them have been studied in detail, such as the ZAC and HYMA1 genes. The regulator of the methylation of this chromosome region is the ZFP57 gene. ZAC (zinc finger protein that regulates apoptosis and cell cycle arrest) is a multifunctional transcription factor and coactivator of p53 and coactivator or corepressor of some nuclear hormone receptors [48]. ZAC has been described as a tumor suppressor gene and its overexpression in cell lines has shown decreased rate of cell replication, increased apoptosis and cell mitosis G1 arrest [49]. Thus, when over-expressed, it would reduce growth of the β-cell mass, possibly through increase of the peroxisome proliferator-activated receptor γ (PPARγ) expression, which is an insulin sensitizer, during embryogenesis and slow down the β-cell proliferation. The function of HYMAI (hydatiform mole-associated and imprinted also called PLAGL1) has still to be elucidated. These methylation defects have the following effects: decreased cell replication, increased apoptosis, delayed maturation of pancreatic islets, and decreased β-cell mass that cause impaired insulin secretion in utero and after birth. DNA multiple methylation defects can also appear and they are usually caused by mutation in the ZFP57 gene when TNDM is only a part of the complex clinical picture [49]. Most of these facts have been confirmed in the mouse model [50].
Approximately 30.0% of children with TNDM have additional features such as umbilical hernia or macroglossia [43]. Insulin therapy is necessary, however, the dose is quickly tapered-down and not necessary after 12 weeks [19]. The relapse is usually common during puberty, it appears in 50.0-60.0% of patients and is presented as DMT2. Due to some residual degree of endogenous insulin secretion, many patients are successfully treated with sulfonylurea [51,52]. If the cause of the disease is duplication of the parental 6q24 region, the genetic risk for future children should be discussed with the family.
Transient ND mellitus can also be caused by mutations of the ABCC8 and KCNJ11 genes, and their function will be discussed with PNDM, as these two mutations can cause both TNDM and PNDM. Transient ND mellitus has rarely been described in association with HNF1 β mutations, causing pancreatic hypoplasia and associated prenatal cystic kidney [53], autosomal recessive insulin gene mutations [54], or homozygous glucokinase (GCK) mutation inherited from both parents in consanguineous families (Table 1). These genes and their mutations will be discussed in the section on PNDM.
Permanent Neonatal Diabetes Mellitus. Permanent ND mellitus has a very similar clinical presentation as TNDM, and can be distinguished only on the basis of the gene mutations, especially in hyperglycemic children with a low birth weight [55]. This diabetes is permanent, remission does not occur. About 10 genes are involved in the etiology of PNDM causing abnormal pancreatic development, increased apopotosis, reduction of β-cell mass and β-cell dysfunction. However, the most common mutations are in the KCNJ11 and ABCC8 genes, which account for approximately 30.0% of all PNDM cases, INS gene mutations accounting for 12.0%, and glucokinase (GCK gene) mutations. All of these genes are involved in glucose sensing and insulin secretion [19,24,55, 56, 57]. The function of these genes has been elucidated in Table 2 (Figure 1).
Figure1.
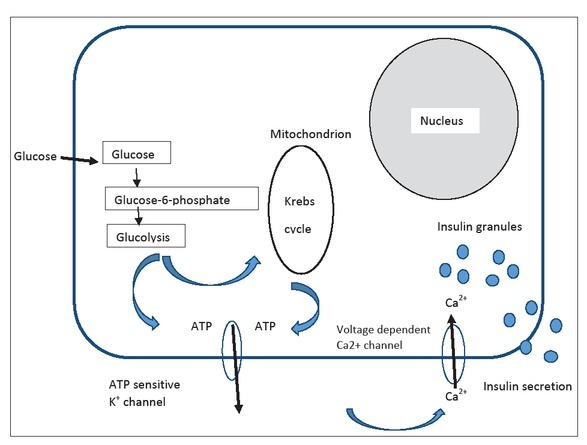
Normal insulin secretion process after glucose enters the β-cell is presented. KCNJ11 and ABCC8 gene mutations influence the ATP channel causing inability of insulin expulsion.
Channelopathies. KCNJ11 and ABCC8 gene mutations are responsible for approximately 40.0% of permanent or transient hyperglycemia (NDM) cases [56,58]. Most of them (approximately 60.0%) occur de novo. They act through increase of activity of ATP-sensitive potassium channels located on the β-cell membrane [35,59,60]. Glucose enters the cell facilitated by the Glut1 transporter, and the enzyme glucokinase (GCK) converts it to glucose-6-phosphate that is then transferred into mitochondria.
There, increased metabolic activity induces an increase of ATP/ ADP ratio that results in a closure of the membrane KATP channel. As a result, the β-cell membrane gets depolarized, followed by influx of Ca++ into the cell, which triggers insulin secretion and expulsion from the β-cell [57,59] (Figure 1). The KATP channel is a key component of the glucose stimulated insulin secretion pathway. It is composed of four Kir6.2 subunits that compose the K+ conducting pore encoded by the KCNJ11 gene. Four SUR1 regulatory subunits encoded by the ABCC8 gene are located at the external site of the pore and regulate the channel activity. Dominant activating mutations in KCNJ11 or ABCC8 cause a permanently opened KATP channel, irrespective of the glucose level, which decreases the channel sensitivity toward ATP, disables the membrane depolarization and prevents insulin secretion causing NDM [57, 58, 59]. More than 205 different mutations of the KCNJ11 and 748 of the ABCC8 genes have been reported [59, 60, 61, 62, 63, 64, 65] Many additional polymorphisms with significance to be elucidated also have been referred [65]. Mutations in the KCNJ11 gene are usually located at the N or C terminus below the plasma membrane, around the inhibitory ATP binding site, and they reduce affinity for ATP. The ABCC8 gene mutations, on the other hand, are located throughout the entire molecule, however, the mechanism of the channel activation is poorly understood although it has been suggested that SUR1 acts to antagonize the Mg-dependent stimulatory action on Kir6.2 [61,64]. Mutations could be point mutations, e.g. missense, nonsense, frameshift, splicing mutations and deletions [65]. Depending upon the mutations, different numbers of permanently opened KATP channels and their different sensitivity for flux of ATP occurs, thus causing PNDM or TNDM. A complex interplay between Kir6.2 and SUR1 subunits, also involving Mg++ has been described [66]. KCNJ11 and SUR1 gene mutations occur de novo in 80.0% of patients without family history, causing isolated PNDM. The remaining cases are familial, and they are always dominantly transferred. KCNJ11 gene mutations cause PNDM in 90.0% of patients who carry it, whereas the ABCC8 gene mutations causes PNDM in less than 40.0% of patients. The remaining 10.0% and >60.0% of mutations, respectively cause TNDM [65]. Both KCNJ11 and ABCC8 genes are located on chromosome 11p, 4.5 kb apart. KCNJ11 encodes for the 390-amino acid Kir6.2 protein, and ABCC8 consists of 39 exons. It encodes for the SUR1 (sulphonyl-urea receptor) protein that consists of 1582 amino acids [65].
Permanent ND mellitus due to KCNJ11 and ABCC8 gene mutations usually have an acute clinical presentation in neonates at the age from several days of age up to 26 weeks after birth, who are born with a lower birth weight, but not as low as in TNDM [67]. Extreme hyperglycemia, severe dehydration, ketoacidosis hypoinsulinemia, and immediate insulin dependency are the common set of symptoms [19,24,68]. However, milder forms have been described in certain mutations [24,60]. Due to the confirmed expression of KATP channels in muscle cells and neurons, approximately 20.0-30.0% of patients have associated neurological features, which are usually associated with certain mutations such as V59M or R210 C of the KCNJ11 gene [65,69, 70, 71, 72]. Some of the children have PNDM, muscle weakness and hypotonia associated with epilepsy when this is termed DEND (developmental delay, epilepsy, ND) syndrome [71]. Others have different level of developmental delay, lower IQ, may have attention deficit hyperactivity disorder (ADHD), autism, anxiety, hypotonia, muscule weakness, balance problems, learning disabilities, and are termed iDEND (intrermediate DEND) syndrome. The V59M mutation has mostly been blamed for iDEND. In general, patients have lower academic achievements [72, 73, 74]. There are some data confirming that mutations that increase channel activity less than 15-fold are associated with both PNDM and TNDM, whereas, if the channel activity is more than 15-fold higher, DEND syndrome occurs [24]. Breakthrough in the treatment of children with PNDM occurred when sulfonylurea was shown to be a successful therapy that normalizes the function of the KATP channel [75,76] but also improves, to some extent, the developmental issues if given early enough [77]. Beyond 90.0% of patients with PNDM can be transferred from insulin to sulfonylurea and treated long-term with careful tapering of the doses that provides a very stable, long-term glycemic control [76,78]. However, in the review by Aguilar-Bryan and Bryan [24], 22 different KCNJ11 gene mutations had a good response to sulfonylurea, whereas seven did not, whereas 20 ABCC8 gene mutations had a good response vs. three mutations where the response did not occur. In the patients who did not react favorably to sulfonylurea, therapy with insulin was necessary [24]. Therefore, genetic testing is mandatory for individualized treatment, improved outcome, and a better quality of life (QoL) [58,79, 80, 81].
Insulin gene mutations. Dominant mutations of the insulin gene (INS) are the second most frequent cause of PNDM [82,83]. Mutated genes usually appear de novo in 80.0% of cases, and cause disturbances in the folding of the proinsulin and/or insulin protein that becomes nonfunctional, but additionally causes endoplasmic reticulum stress due to protein accumulation inducing apoptosis of the β-cell [56]. No other tissues or organs are affected. A significant portion of mutations are de novo, usually dominant, and affect the disulfide bonds in the insulin molecule [84]. Diabetes is typically insulin-deficient, affected newborns have a low birth weight, hyperglycemia usually occurs during the second month of life. It can occasionally appear after the age of 6 months, and therefore, in all babies with negative anti β-cell antibodies, molecular testing for insulin gene mutations is necessary. Insulin gene mutations causing PNDM can also be recessive, causing delayed fetal growth, low birth weight, and severe early presentation after birth [82,83]. All newborns with a mutation of the insulin gene require insulin therapy.
Glucokinase mutations. Glucokinase is the enzyme that regulates adequate glucose-stimulated insulin secretion from the β-cell, and is termed a sensor for glucose [85]. In humans, the threshold of glycemia for insulin release is set to approximaltely 5 mmol/L. More than 200 mutations have been detected in the GCK gene. However, most of them being autosomal dominant mutations, if in heterozygous form, cause MODY2, e.g. mild hyperglycemia due to the inappropriate sensing of glucose level. They are usually detected later in childhood or during young adult life. However, several homozygous inactivating mutations are described in PNDM [86,87]. Recessive mutations, when in homozygous or compound homozygous states, cause complete lack of insulin secretion and PNDM, especially in consanguineous families. Parents usually have a history of low glucose tolerance or untreated mild diabetes. This form of diabetes accounts for only 2.0-3.0% of all patients with PNDM [88]. These neonates have low body mass due to insulin deficiency and are treated with insulin. Mutations of other genes causing ND are rare, and are given in Table 1.
Syndromic Neonatal Diabetes Mellitus. Syndromic ND mellitus (SNDM) should be considered after more common forms (KATP channelopathies) and other more frequent mutations are excluded. The number of genes discovered inducing SNDM is increasing with the newer molecular techniques. Most cases of SNDM belong to syndromes with associated extra-pancreatic features affecting different organs or functions. All are very rare, some with only a few patients described. All require therapy with insulin, and other therapeutic procedures for associated problems. Major characteristics have been presented in Table 2. Only some will be mentioned briefly.
Eukaryotic initiation factor 2α kinase 3 (EIF2AK3) mutations in homozygotes or compound heterozygotes cause Wolcott-Rallison syndrome. It is the most common form of PNDM in consanguineous families. Clinical presentation is complex, and comprises additional features such as multiple epiphyseal dysplasia, growth retardation, occasionally associated with learning difficulties, epilepsy, hepatic and/or renal dysfunction, abnormalities of the cardiovascular system and dysfunction of the exocrine pancreas [89,90]. The location of this gene is at chromosome 2p12 and it is involved in the regulation of protein-folding in the endoplasmic reticulum [90,91]. If mutated, it causes endoplasmic stress and initiates apoptosis in many tissues including β-cells. About 20 different mutations have so far been reported, children present with ketoacidosis and insulin therapy is mandatory [24,91,92].
Mutations in the WFS1 gene cause Wolfram syndrome. It is a very complex syndrome, also known as DID-MOAD, symptoms consisting of DM, diabetes insipidus, optic atrophy, deafness as well as neurodegeneration. It appears in 1:160,000-1:770,000 individuals. Although, on average it appears at 6 years of age, neonatal cases have been described with insulin-dependence, and additional features during childhood. The β-cell degeneration due to irregular folding of the protein wolframin, induces endoplasmic stress in different organs [93, 94, 95]. Mutations can be autosomal recessive, however, several dominant mutations have also been described [96]. Insulin therapy is mandatory, however, it should be accompanied by a complex involvement of ophtalmologists, nephrologists, otolaryngologists and neurologists. Life-span is shortened [97].
Immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome is caused by mutations of the fork-head box protein 3 (FOXP3) gene located on the short arm of the X chromosome (Xp11.23). It encodes FOXP3 protein that is crucial for the function of regulatory T-cells, thus the diabetes is of an autoimmune nature and general disorder of immunity. More than 70 different mutations have been described. The most common mutation is the substitution of the Pro339 by alanine or other amino acids [98]. Clinical presentation involves enteropathy, autoimmune diabetes, immunodeficiency, severe infections; 65.0% of patients survive. Treatment with immunosuppressive agents is recommended [99].
Utility of Testing for Neonatal Diabetes. Having in mind all previously mentioned forms of PNDM, and many others that are extremely rare, there is a reasonable algorithm of testing that should be performed when NDM occurs. The first approach is to test for 6q24 abnormalities which cause 68.0% of TNDM. If it is negative, further testing for the KCNJ11 gene should follow (10.0% of patients with NDM carry it), and if it is normal, tests for ABCC8 gene mutations should be performed, as they are the cause of ND in 9.0% of patients. Novel methods provide simultaneous testing for many genes involved in monogenic diabetes including both NDM and MODY. Targeted NGS has been successfully applied [26] as well as the combination of NGS and methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA), assay for the detection of both transient and permanent NDM [100].
Molecular diagnosis of NDM is of the utmost importance as it provides the appropriate selection of therapy [77, 78, 79], which is in concordance with precision medicine [101]. It is also economically valuable as it provides the opportunity for transfer to oral therapy in certain patients [102,103], improving the QoL, and decreasing the cost of treatment [104]. On the other hand, knowing the molecular mechanisms of hyperglycemia provides new research for novel therapies [105], and gives insight into the mechanisms of more common forms of diabetes [106].
Conclusions. Neonatal diabetes is monogenic, and genetically polymorphic. Due to the severity of onset, danger of an unfavorable outcome and uncertain future, NDM should be genetically characterized as soon as possible through international platforms for countries with funding problems for genetic testing. The importance of this testing is providing a precise diagnosis, precision medicine, individual therapy and best possible outcome. However, genetic diagnosis has also taught endocrinologists of many physiological pathways in the β-cell function, genetic control of glucose homeostasis and interplay of involved genes in control of other organs.
Footnotes
Declaration of Interest. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Chiang JL, Maahs DM, Garvey KC, Hood KK, Laffel LM, Weinzimer SA. Type 1 diabetes in children and adolescents: A position statement by th American Diabetes Association. Diabetes Care. 2018;41(9):2026–2044. doi: 10.2337/dci18-0023. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tosur M, Geyer SM, Rodriguez H, Libman I, Baidal DA, Redondo MJ; Type 1 Diabetes TrialNet Study Group. Ethnic differences in progression of islet autoimmunity and type 1 diabetes in relatives at risk. Diabetologia. 2018;61(9):2043–2053. doi: 10.1007/s00125-018-4660-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patterson CC, Dahlquist G, Soltész G, Green A; EURODIAB ACE Study Group. Variation and trends in the incidence of childhood diabetes in Europe. Lancet. 2000;355(9207):873–876. [PubMed] [Google Scholar]
- 4.Roark CL, Anderson KM, Simon LJ, Schuyler RP, Aubrey MT, Freed BM. Multiple HLA epitopes contribute to type 1 diabetes susceptibility. Diabetes. 2014;63(1):323–331. doi: 10.2337/db13-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kocova M, Blagoevska M, Bogoevski M, Konstantinova M, Dorman J, Trucco M. HLA class II molecular typing in an European Slavic population with a low incidence of insulin-dependent diabetes mellitus. Tissue Antigens. 1995;45(3):216–219. doi: 10.1111/j.1399-0039.1995.tb02442.x. [DOI] [PubMed] [Google Scholar]
- 6.Platz P, Jakobsen BK, Morling N, Ryder LP, Svejgaard A, Thomsen M. HLA-D and -DR antigens in genetic analysis of insulin dependent diabetes mellitus. Diabetologia. 1981;21(2):108–115. doi: 10.1007/BF00251276. et al. [DOI] [PubMed] [Google Scholar]
- 7.Pociot F, Lenmark A. Genetic risk factors for type 1 diabetes. Lancet. 2016;387(10035):2331–2339. doi: 10.1016/S0140-6736(16)30582-7. [DOI] [PubMed] [Google Scholar]
- 8.Noble JA, Valdes AM. Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diab Rep. 2011;11(6):532–542. doi: 10.1007/s11892-011-0223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patterson CC, Harjutsalo V, Rosenbauer J, Neu A, Cinek O, Skrivarhaug T. Trends and cyclical variation in the incidence of childhood type 1 diabetes in 26 European centres in the 25 year period 19892013: A mul-ticentre prospective registration study. Diabetologia. 2019;62(3):408–417. doi: 10.1007/s00125-018-4763-3. et al. [DOI] [PubMed] [Google Scholar]
- 10.Qiu YH, Deng FY, Li MJ, Lei SF. Identification of novel risk genes associated with type 1 diabetes mellitus using a genome-wide gene-based association analysis. J Diabetes Investig. 2014;5(6):649–656. doi: 10.1111/jdi.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: Pathogenesis and treatment. Lancet. 2008;371(9631):2153–2156. doi: 10.1016/S0140-6736(08)60932-0. [DOI] [PubMed] [Google Scholar]
- 12.Unger RH. Reinventing type 2 diabetes: Pathogenesis, treatment, and prevention. JAMA. 2008;299(10):1185–1187. doi: 10.1001/jama.299.10.1185. [DOI] [PubMed] [Google Scholar]
- 13.Fendler W, Borowiec M, Baranowska-Jazwiecka A, Szadkowska A, Skala-Zamorowska E, Deja G. Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia. 2012;55(10):2631–2635. doi: 10.1007/s00125-012-2621-2. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irgens HU, Molnes J, Johansen BB, Rindal M, Skivarhaug T, Undlien D. Prevalence of monogenic diabetes in the population-based Norwegian Childhood Diabetes Registry. Diabetologia. 2013;56(7):1512–1519. doi: 10.1007/s00125-013-2916-y. et al. [DOI] [PubMed] [Google Scholar]
- 15.Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, Dolan LM. Prevalence, characteristics, and clinical diagnosis of maturity onset diabetes of the young due to mutations HNF1A, HNF4A, and glucokinase results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab. 2013;98(10):40554062. doi: 10.1210/jc.2013-1279. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johanson BB, Irgens HU, Molnes J, Sztromwasser P, Aukrust I, Juliuson PB. Targeted next generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia. 2017;60(4):625–635. doi: 10.1007/s00125-016-4167-1. et al. [DOI] [PubMed] [Google Scholar]
- 17.Delvecchio M, Mozzillo E, Salzano G, Iafusco D, Frontino G, Patera PI. Monogenic diabetes accounts for 6.3 of cases referred to 15 Italian pediatric diabetes centers during 2007 to 2012. J Clin Endocrinol Metab. 2017;102(6):1826–1834. doi: 10.1210/jc.2016-2490. et al. [DOI] [PubMed] [Google Scholar]
- 18.Shepherd M, Shields B, Hammersley S, Hudson M, McDonald T, Colclough K. Systematic population screening, using biomarkers and genetic testing identifies 2.5% of the UK pediatric diabetes population with monogenic diabetes. Diabetes Care. 2016;39(11):1879–1888. doi: 10.2337/dc16-0645. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hattersley AT, Greenley AW, Polak M, Rubio-Cabezas O, Njolstad PR, Mlynarski W. ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19(Suppl 27):47–63. doi: 10.1111/pedi.12772. et al. [DOI] [PubMed] [Google Scholar]
- 20.Nansseu JRN, Ngo-Um SS, Balti EV. Incidence, prevalence and genetic determinants of neonatal diabetes mellitus: A systematic review and meta-analysis protocol. Syst Rev. 2016;5(1):188. doi: 10.1186/s13643-016-0369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jennings RE, Berry AA, Strutt JP, Gerrard DT, Hanley NA. Human pancreas development. Development. 2015;142(18):3126–3137. doi: 10.1242/dev.120063. [DOI] [PubMed] [Google Scholar]
- 22.Servitja JM, Ferrer J. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia. 2004;47(4):597–613. doi: 10.1007/s00125-004-1368-9. [DOI] [PubMed] [Google Scholar]
- 23.Polak M, Cavé H. Neonatal diabetes mellitus: A disease linked to multiple mechanisms. Orphanet J Rare Dis. 2007;2:12. doi: 10.1186/1750-1172-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguilar-Bryan L, Bryan J. Neonatal diabetes mellitus. Endocr Rev. 2008;29(3):265–291. doi: 10.1210/er.2007-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petruzelkova L, Dusatkova P, Cinek O, Sumnik Z, Pruhova S, Hardsky O. Substantial proportion of MODY among multiplex families participating in a Type 1 diabetes prediction programme. Diabetic Med. 2016;33(12):1712–1716. doi: 10.1111/dme.13043. et al. [DOI] [PubMed] [Google Scholar]
- 26.Ellard S, Lango Allen H, De Franco E, Flanagan SE, Hysenaj G, Colclough K. Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia. 2013;56(9):1958–1963. doi: 10.1007/s00125-013-2962-5. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bansal V, Gassenhuber J, Phillips T, Oliveira G, Harbaugh R, Villarasa N. Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC Medicine. 2017;15(1):213. doi: 10.1186/s12916-017-0977-3. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Globa E, Zelinska N, Mackay DJG, Temple K, Houghton JAL, Hattersley AT. Neonatal diabetes in Ukraine: Incidence, genetic, clinical phenotype and treatment. J Pediatr Endocrinol Metab. 2015;28(11-12):1279–1286. doi: 10.1515/jpem-2015-0170. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubio-Cabezas O, Ellard S. Diabetes mellitus in neonates and infants: Genetic heterogeneity, clinical approach to diagnosis, and therpeutic options. Horm Res Paediatr. 2013;80(3):137–146. doi: 10.1159/000354219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nansseu JR, Ngo-Um SS, Balti EV. Incidence, prevalence and genetic determinants of neonatal diabetes mellitus: A systematic review and meta-analysis protocol. Syst Rev. 2016;5(1):188. doi: 10.1186/s13643-016-0369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagashima K, Tanaka D, Inagaaki N. Epidemiology, clinical chracteristics, and genetic etiology of neonatal diabetes in Japan. Pediatr Int. 2017;59(2):129–133. doi: 10.1111/ped.13199. [DOI] [PubMed] [Google Scholar]
- 32.Huopio H, Miettinen PJ, Ilonen J, Nykanen P, Veijola R, Keskinen P. Clinical, genetic, and biochemical characteristics of early-onset diabetes in the Finnish population. J Clin Endocrinol Metab. 2016;101(8):3018–3026. doi: 10.1210/jc.2015-4296. et al. [DOI] [PubMed] [Google Scholar]
- 33.Cao B, Gong C, Wu D, Lu C, Liu F, Liu X. Genetic analysis and follow-up of 25 neonatal diabetes mellitus patients in China. J Diabetes Res. 2016;2016:6314368. doi: 10.1155/2016/6314368. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blanco Lemelman M, Letourneau L, Greeley SAW. Neonatal diabetes mellitus: An update on diagnosis and managment. Clin Perinatol. 2018;45(1):41–59. doi: 10.1016/j.clp.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russo L, Iafusco D, Brescianini S, Nocerino V, Bizzarri C, Toni S. Permanent diabetes mellitus during the first year of life: Multiple gene screening in 54 patients. Diabetologia. 2011;54(7):1693–1701. doi: 10.1007/s00125-011-2094-8. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Mühlendah KE, Herkenhof H. Long-term course of neonatal diabetes. N Engl J Med. 1995;333(11):704–708. doi: 10.1056/NEJM199509143331105. [DOI] [PubMed] [Google Scholar]
- 37.Polak M, Cavé H. Neonatal diabetes mellitus: A disease linked to multiple mechanisms. Orphanet J Rare Dis. 2007;2:12. doi: 10.1186/1750-1172-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fösel S. Transient and permanent neonatal diabetes. Eur J Pediatr. 1995;154(12):944–948. doi: 10.1007/BF01958635. [DOI] [PubMed] [Google Scholar]
- 39.Temple IK, Shield JPH. Transient neonatal diabetes, a disorder of imprinting. J Med Genet. 2002;39(12):872–875. doi: 10.1136/jmg.39.12.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naylor RN, Greeley SAW, Bell IGI, Philipson LH. Genetics and pathophysiology of neonatal diabetes mellitus. J Diabetes Investig. 2011;2(3):158–169. doi: 10.1111/j.2040-1124.2011.00106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Temple IK, Gardner RJ, Mackay DJ, Barber JC, Robinson DO, Shield JP. Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes. 2000;49(8):1359–1366. doi: 10.2337/diabetes.49.8.1359. [DOI] [PubMed] [Google Scholar]
- 42.Gardner RJ, Mackay DJ, Mungall AL, Polychronakos C, Siebert R, Shield JP. An imprinted locus associated with transient neonatal diabetes mellitus. Hum Mol Genet. 2000;9(4):589–596. doi: 10.1093/hmg/9.4.589. et al. [DOI] [PubMed] [Google Scholar]
- 43.Docherty LE, Kabwama S, Lehman A, Hawke E, Harrison L, Flanagan SF. Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype-phenotype correlation in an international cohort of patients. Diabetologia. 2013;56(4):758–762. doi: 10.1007/s00125-013-2832-1. et al. [DOI] [PubMed] [Google Scholar]
- 44.Hermann R, Laine AP, Johansson C, Niederland T, TokarskaL Dziatkowiak H. Transient but not permanent neonatal diabetes mellitus is associated with paternal isodisomy of chromosome 6. Pediatrics. 2000;105(1):49–52. doi: 10.1542/peds.105.1.49. et al. [DOI] [PubMed] [Google Scholar]
- 45.Whiteford ML, Narendra A, White MP, Cooke A, Wilkinson AG, Robertson KJ. Paternal uniparental disomy for chromosome 6 causes transient diabetes mellitus. J Med Genet. 1997;34(2):167–168. doi: 10.1136/jmg.34.2.167. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mackay D, Bens S, Perez de Nanclares G, Siebert R Temple K. Clinical utility gene card for: Transient neonatal diabetes mellitus, 6q24-related. Eur J Hum Genet. 2014;22(9) doi: 10.1038/ejhg.2014.27. Epub 2014 Feb 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mackay DJ, Boonen SE, Clayton-Smith J, Good-ship J, Hahnemann JMD, Kant SG. A maternal hypo-methylation syndrome presenting as transient neonatal diabetes mellitus. Hum Genet. 2006;120(2):262–269. doi: 10.1007/s00439-006-0205-2. et al. [DOI] [PubMed] [Google Scholar]
- 48.Varrault A, Ciani E, Apiou F, Bilanges A, Hofmann A, Pantaloni C. hZAC encodes a zink finger protein with antiproliferative properties and maps to a chromosomal region frequently lost in cancer. Natl Acad Sci USA. 1998;95(15):8835–8840. doi: 10.1073/pnas.95.15.8835. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Touati A, Errea-Dorronsoro J, Nouri S, Halleb Y, Pereda A, Mahdhaoui N. Transient neonatal diabetes mellitus and hypomethylation at additional imprinted loci: Novel ZFP57 mutation and review of the literature. Acta Diabetol. 2019;56(3):301–307. doi: 10.1007/s00592-018-1239-3. et al. [DOI] [PubMed] [Google Scholar]
- 50.Ma D, Shield JPH, Dean W, Leclerc I, Knauf C, Burcelin R. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest. 2004;114(3):339–348. doi: 10.1172/JCI19876. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sovik O, Aagenaes O, Eide SA, Mackay D, Temple IK, Molven A. Familial occurrence of neonatal diabetes with duplications in chromosome 6q24: Treatment with and 40-yr follow-up. Pediatr Diabetes. 2012;1(3):155–162. doi: 10.1111/j.1399-5448.2011.00776.x. et al. [DOI] [PubMed] [Google Scholar]
- 52.Carmody D, Beca FA, Bell CD, Hwang JD, Dickens JT, Devine NA. Role of noninsulin therapies alone or in combination in chromosome 6q24-related transient neonatal diabetes: Sulfonylurea improves but does not always normalize insulin secretion. Diabetes Care. 2015;38(6):e86–e87. doi: 10.2337/dc14-3056. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism. J Clin Endocrinol Metab. 2004;89(6):2905–2908. doi: 10.1210/jc.2003-031828. et al. [DOI] [PubMed] [Google Scholar]
- 54.Garin I, Edghill EL, Akerman I, Rubio-Cabezas O, Rica I, Locke JM. Recessive mutations in INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci USA. 2010;107(7):3105–3110. doi: 10.1073/pnas.0910533107. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Besser REJ, Flanagan SE, Mackay DGJ, Temple IK, Shepherd MH, Shields BM. Prematurity and genetic testing for neonatal diabetes. Pediatrics. 2016;138(3):10. doi: 10.1542/peds.2015-3926. et al. 1542/peds.2015-3926e20153926. Epub 2016 Aug 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoy J, Edghill EL, Flanagan SF, Ye H, Paz VP, Pluzhnikov A. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA. 2007;104(38):15040–15044. doi: 10.1073/pnas.0707291104. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.John SA, Weiss JN, Xie LH, Ribalet B. Molecular mechanism for ATP-dependent closure of the K+ channel Kir6.2. J Physiol. 2003;552(Pt 1):23–34. doi: 10.1113/jphysiol.2003.048843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Franco E, Flanagan SF, Houghton JAL, Lango Allen H, Mackay DJG, Temple IK. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet. 2015;386(9997):957–963. doi: 10.1016/S0140-6736(15)60098-8. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tinker A, Aziz Q, Li Y, Specterman M. ATP-Sensitive potassium channels and their physiological and pathophysiological roles. Compr Physiol. 2008;8(4):1463–1511. doi: 10.1002/cphy.c170048. [DOI] [PubMed] [Google Scholar]
- 60.Gloyn AL, Pearson ER, Antcliff LF, Proks P, Bruining GJ, Slingerland AS. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir 6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350(18):1838–1849. doi: 10.1056/NEJMoa032922. et al. [DOI] [PubMed] [Google Scholar]
- 61.Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Eng J Med. 2006;355(5):456–466. doi: 10.1056/NEJMoa055068. et al. [DOI] [PubMed] [Google Scholar]
- 62.Achroft FM. ATP-sensitive potassium channelopathies: Focus on insulin secretion. J Clin Invest. 2005;115(8):2047–2058. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flanagan SF, Edghill EL, Glyon AL, Ellard S, Gattersley AT. Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia. 2006;49(6):1190–1197. doi: 10.1007/s00125-006-0246-z. [DOI] [PubMed] [Google Scholar]
- 64.Vaxillaire M, Populaire C, Busiah K, Cave H, Gloyn AL, Hattersley AT. Kir 6.2 mutations are a common cause of permanent neonatal diabetes in a large cohort of French patients. Diabetes. 2004;53(10):2719–2722. doi: 10.2337/diabetes.53.10.2719. et al. [DOI] [PubMed] [Google Scholar]
- 65.De Franco E, Saint Martin C, Brusgaard K, Knight Johnson AE, Aguilar-Bryan L, Bowman P. Upadate of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat. 2020;41(5):884–905. doi: 10.1002/humu.23995. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Proks P, Arnold AL, Bruining J, Girard C, Flanagan SE, Larkin B. A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes. Hum Mol Genet. 2006;15(11):1793–1800. doi: 10.1093/hmg/ddl101. et al. [DOI] [PubMed] [Google Scholar]
- 67.Flanagan SF, Patch AM, Mackay DJ, Edghill EL, Gloyn AL, Robinson D. Mutations in ATP sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007;56(7):1930–1937. doi: 10.2337/db07-0043. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Letoumeau LR, Carmody D, Wriblewski K, Denson AM, Sanyoura M, Rochelle N. Diabetes presentation in infancy: High risk of diabetic ketoacidosis. Diabetes Care. 2017;40(10):e147–e148. doi: 10.2337/dc17-1145. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dahl A, Kumar S. Recent advances in neonatal diabetes. Diabetes, metabolic syndrome and obesity: Targets and therapy. Diabetes Metab Syndr Obes. 2020;13:355–364. doi: 10.2147/DMSO.S198932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gloyn AL, Diatloff-Zito C, Edghill EL, Bellanné-Chantelot C, Nivot S, Coutan R. KCNJ11 activating mutations are associated with developmental delay, epilepsy and neonatal diabetes syndrome and other neurological features. Eur J Hum Genet. 2006;14(7):824–830. doi: 10.1038/sj.ejhg.5201629. et al. [DOI] [PubMed] [Google Scholar]
- 71.Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: New clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54(9):2503–2513. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- 72.Bowman P, Broadbridge E, Knight BA, Pettit L, Flanagan SE, Reville M. Psychiatric morbidity in children with KCNJ 11 neonatal diabetes. Diabet Med. 2016;33(10):1387–1391. doi: 10.1111/dme.13135. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Clark RH, McTaggart JS, Webster R, Knight BA, Pettit L, Flanagan SE. Muscle disfunction caused by a KATP channel mutation in neonatal diabetes is neuronal in origin. Science. 2010;329(5990):458–461. doi: 10.1126/science.1186146. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carmody D, Pastore AN, Landmeier KA, Letourneau LR, Martin R, Hwang JL. Patients with KC-NJ11-related diabetes frequently have neuropsychological impairment compared with sibling controls. Diabet Med. 2016;33(10):1380–1386. doi: 10.1111/dme.13159. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pearson ER, Flechtner I, Njolstad PR, Malecki MT, Flanagan SF, Larkin B. Switching from insulin to oral sulfonylureas in paients with diabetes due to 6.2 mutations. N Engl J Med. 2006;355(5):467–477. doi: 10.1056/NEJMoa061759. et al. [DOI] [PubMed] [Google Scholar]
- 76.Rafiq M, Flanagen SE, Patch AM, Shields BM, Ellard S, Hattersley AT. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31(2):204–209. doi: 10.2337/dc07-1785. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beltrand J, Elie C, Busiah K, Fournier E, Boddaert N, Bahi-Buisson N. GlidKir Study Group. Erratum. Sulfonylurea therapy benefits neurological and psychomotor functions in patients with neonatal diabetes owing to potassium channel mutations. Diabetes Care. 2015;38:2033–2041. doi: 10.2337/dc15-0837. et al. [DOI] [PubMed] [Google Scholar]
- 78.Bowman O, Sulen A, Barbetti F, Beltrand J, Svalastoga P, Codner E. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: An international cohort study. Lancet Diabetes Endocrinol. 2018;6(8):637–646. doi: 10.1016/S2213-8587(18)30106-2. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Torbjornsdotter T, Marosvari-Barna T, Henckel E, Corrias E, Norgren M, Janson A. Successful treatment of a cohort of infants with neonatal diabetes using insulin pumps including data on genetics and estimated incidence. Acta Paediatr. 2020;109(6):1131–1137. doi: 10.1111/apa.15100. [DOI] [PubMed] [Google Scholar]
- 80.Greeley SA, Zielinski MC, Poudel A, Ye H, Berry S, Taxy JB. Preservation of reduced numbers of insulin-positive cells in sulfonylurea-unresponsive KCNJ11-related diabetes. J Clin Endocrinol Metab. 2017;102(1):1–5. doi: 10.1210/jc.2016-2826. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taberner P, Flanagan SE, Mackay DJ, Ellard S, Taverna MJ, Ferraro M. Clinical and genetic features of Argentinian children with diabetes-onset before 12 months of age: Successful transfer from insulin to oral sulfonylurea. Diabetes Res Clin Pract. 2016;117:104–110. doi: 10.1016/j.diabres2016.04.005. [DOI] [PubMed] [Google Scholar]
- 82.Polak M, Dechaume A, Cavé H, Nimri R, Crosnier H, Sulmont V. French ND (neonatal diabetes) Study Group. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: A report from the French ND (neonatal diabetes) study group. Diabetes. 2008;57(4):1115–1119. doi: 10.2337/db07-1358. et al. [DOI] [PubMed] [Google Scholar]
- 83.Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B. Neonatal Diabetes International Collaborative Group. Insulin mutation screening in 1044 patients with diabetes: Mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57(4):1034–1042. doi: 10.2337/db07-1405. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fu J, Wang T, Li M, Xiao X. Identification of insulin gene variants in patients with neonatal diabetes in the Chinese population. J Diabetes Investig. 2020;11(3):578–584. doi: 10.1111/jdi.13156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matschinsky FM. Glucokinase, glucose homeostasis and diabetes mellitus. Curr Diab Rep. 2005;5(3):171–176. doi: 10.1007/s11892-005-0005-4. (Author: please see reference below) [DOI] [PubMed] [Google Scholar]
- 86.Lin DC, Huang CY, Ting WH, Lo FS, Lin CL, Yang HW. Mutations in glucokinase and other genes detected in neonatal and type 1B diabetes patient using whole exome sequencing may lead to diseasecausing changes in protein activity. Biochim Biophys Acta. 2019;1865(2):428–433. doi: 10.1016/j.bbadis.2018.11.013. et al. [DOI] [PubMed] [Google Scholar]
- 87.Njølstad PR, Søvik O, Cuesta-Muñoz A, Bjørkaug L, Massa O, Barbetti F. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344(21):1588–1592. doi: 10.1056/NEJM200105243442104. et al. [DOI] [PubMed] [Google Scholar]
- 88.Reis AF, Kannengiesser C, Jennane F, Manna TD, Cheurfa N, Oudin C. Two novel mutations in the EIF2AK3 gene in children with Wolcott-Rallison syndrome. Pediatr Diabetes. 2011;12(3 Pt 1):187–191. doi: 10.1111/j.1399-5448.2010.00679.x. et al. [DOI] [PubMed] [Google Scholar]
- 89.Habener JF, Kemp DM, Thomas MK. Minireview: Transcriptional regulation in pancreatic development. Endocrinology. 2005;146(3):1025–1034. doi: 10.1210/en.2004-1576. [DOI] [PubMed] [Google Scholar]
- 90.Thornton CM, Carson DJ, Stewart FJ. Autopsy findings in Wolcott-Rallison syndrome. Pediatr Pathol Lab Med. 1997;17(3):487–496. [PubMed] [Google Scholar]
- 91.Sümegi A, Hendrik Z, Gáll T, Felszeghy E, Szakszon K, Antal-Szalamas P. A novel splice site indel alteration in the EIF2AK3 gene is responsible for the first cases of Wolcott-Rallison syndrome in Hungary. BMC Med Genet. 2020;21(1):61. doi: 10.1186/s12881-020-0985-6. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Welters A, Meissner T, Konrad K, Freiberg C, Warnicke K, Judmaier S. Diabetes management in Wolcott-Rallison syndrome: analysis from the German/ Austrian DPV database. Orphanet J Rare Dis. 2020;15(1):100. doi: 10.1186/s13023-020-01359-y. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iver S, Korada M, Rainbow L, Kirk J, Brown RM, Shaw N. Wolcott-Rallison syndrome: A clinical and genetic study of three children, novel mutation in EIF2AK3 and a review of the literature. Acta Paediatr. 2004;93(9):1195–1201. et al. [PubMed] [Google Scholar]
- 94.Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet. 1995;346(8988):1458–1463. doi: 10.1016/s0140-6736(95)92473-6. [DOI] [PubMed] [Google Scholar]
- 95.Khanim F, Kirk J, Latif F, Barett TG. WFS1/ wolframin mutations, Wolfram syndrome, and associated diseases. Hum Muat. 2001;17(5):357–367. doi: 10.1002/humu.1110. [DOI] [PubMed] [Google Scholar]
- 96.Marshall BA, Permutt MA, Paciorkowski AR, Paciorkowsky AR, Hoekel J, Karzon R. Washington University Wolfram Study Group. Phenotypic characteristics of early Wolfram Sundrome. Orphanet J Rare Dis. 2013;8:64. doi: 10.1186/1750-1172-8-64. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rigoli L, Bramanti P, Di Bella C, De Luca F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr Res. 2018;83(5):921–929. doi: 10.1038/pr.2018.17. [DOI] [PubMed] [Google Scholar]
- 98.Bueno GE, Ruiz-Castañeda D, Martínez JR, Muñoz MR, Alascio PC. Natural history and clinical characteristics of 50 patients with Wolfram syndrome. Endocrine. 2018;61(3):440–446. doi: 10.1007/s12020-018-1608-2. [DOI] [PubMed] [Google Scholar]
- 99.Agakidis C, Agakidou E, Sarafidis K, Papoulidis I, Xinias I, Farmaki E. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome associated with a novel mutation of FOXP3 gene. Front Pediatr. 2019;7:20. doi: 10.3389/fped.2019.00020. eCollection 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX-like chidren. J Clin Immunol. 2008;28(5):581–587. doi: 10.1007/s10875-008-9196-1. [DOI] [PubMed] [Google Scholar]
- 101.Alkorta-Aranburu G, Sukhanova M, Carmody D, Hoffman T, Wysinger L, Keller-Ramey J. Improved molecular diagnosis of patients with neonatal diabetes using a combined next-generation sequencing and MS- approach. J Pediatr Endocrinol Metab. 2016;29(5):523–531. doi: 10.1515/jpem-2015-0341. et al. [DOI] [PubMed] [Google Scholar]
- 102.Letourneau LR, Greeley SAW. Precision medicine: Long-term treatment with sulfonylureas in patients with neonatal diabetes due to KKCNJ11 mutations. Curr Diab Rep. 2019;19(8):52. doi: 10.1007/s11892-019-1175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Naylor R. Economics of genetic testing for diabetes. Curr Diab Rep. 2019;19(5):23. doi: 10.1007/s11892-019-1140-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ma S, Viola R, Sui L, Cherubini V, Barbetti F, Egli D. β Cell replacement after gene editing of a neonatal diabetes causing mutation at the insulin locus. Stem Cell Reports. 2018;11(6):1407–1415. doi: 10.1016/j.stemcr.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang Y, Chan L. Monogenic diabetes: What it teaches us on the common forms of type 1 and type 2 diabetes. Endocr Rev. 2016;37(3):190–222. doi: 10.1210/er.2015-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]