

SUMMARY
BACKGROUND:
Treatment for TB is lengthy and toxic, and new regimens are needed.
METHODS:
Participants with pulmonary drug-susceptible TB (DS-TB) were randomised to receive: 200 mg pretomanid (Pa, PMD) daily, 400 mg moxifloxacin (M) and 1500 mg pyrazinamide (Z) for 6 months (6Pa200MZ) or 4 months (4Pa200MZ); 100 mg pretomanid daily for 4 months in the same combination (4Pa100MZ); or standard DS-TB treatment for 6 months. The primary outcome was treatment failure or relapse at 12 months post-randomisation. The non-inferiority margin for between-group differences was 12.0%. Recruitment was paused following three deaths and not resumed.
RESULTS:
Respectively 4/47 (8.5%), 11/57 (19.3%), 14/52 (26.9%) and 1/53 (1.9%) DS-TB outcomes were unfavourable in patients on 6Pa200MZ, 4Pa200MZ, 4Pa100MZ and controls. There was a 6.6% (95% CI −2.2% to 15.4%) difference per protocol and 9.9% (95%CI −4.1% to 23.9%) modified intention-to-treat difference in unfavourable responses between the control and 6Pa200MZ arms. Grade 3+ adverse events affected 68/203 (33.5%) receiving experimental regimens, and 19/68 (27.9%) on control. Ten of 203 (4.9%) participants on experimental arms and 2/68 (2.9%) controls died.
CONCLUSION:
PaMZ regimens did not achieve non-inferiority in this under-powered trial. An ongoing evaluation of PMD remains a priority.
Keywords: tuberculosis, drug resistance, TB treatment, TB-HIV
Abstract
CONTEXTE :
Le traitement de la TB est long et toxique et de nouveaux protocoles sont requis.
MÉTHODES :
Des patients atteints de TB pulmonaire sensible aux médicaments (DS-TB) ont été randomisés pour recevoir : 200 mg de prétomanide (Pa, PMD) quotidien, 400 mg de moxifloxacine (M) et 1500 mg de pyrazinamide (Z) pendant 6 mois (6Pa200MZ) ou pendant 4 mois (4Pa200MZ) ; 100 mg de PMD quotidien pendant 4 mois dans la même combinaison (4Pa100MZ) ; ou un traitement standard de DS-TB pendant 6 mois. Le résultat principal a été l’échec du traitement ou la rechute à 12 mois après la randomisation. La marge de non infériorité pour les différences entre groupes a été de 12,0%. Le recrutement a été mis en pause à la suite de trois décès et n’a pas été repris.
RÉSULTATS :
Quatre sur 47 (8,5%), 11 sur 57 (19,3%), 14 sur 52 (26,9%) et 1 sur 53 (1,9%) des résultats de DS-TB ont été défavorables pour 6Pa200MZ, 4Pa200MZ, 4Pa100MZ, et les témoins. Il y a eu une différence de 6,6% (IC 95% −2,2 à 15,4) par protocole et 9,9% (IC 95% −4,1 à 23,9) en réponse défavorable entre les témoins et 6Pa200MZ. Des effets secondaires de grade 3+ ont affecté 68 patients sur 203 (33,5%) recevant des protocoles expérimentaux et 19 sur 68 (27,9%) on control. Dix sur 203 (4,9%) participants en bras expérimental et 2 sur 68 (2,9%) on control sont décédés.
CONCLUSION :
Les protocoles Pa200MZ n’ont pas atteint la non infériorité dans cet essai sous-puissant. La priorité reste l’évaluation du PMD.
TB is the leading cause of death from an infectious disease globally.1 TB treatment for drug-susceptible and -resistant disease is hindered by its long duration and toxicity.2 Pretomanid (PMD, Pa) is a member of the nitroimadazole drug class3 that has demonstrated significant bactericidal and sterilising activity against Mycobacterium tuberculosis.4,5 PMD has recently been approved by the US Food and Drug Administration (FDA) as part of a 6-month regimen in combination with bedaquiline (BDQ) and linezolid for the treatment of extensively drug-resistant TB (XDR-TB) and treatment-intolerant or non-responsive drug-resistant TB (DR-TB).
PMD, moxifloxacin (MFX, M) and pyrazinamide (PZA, Z) were studied in a Phase 2 clinical trial in combination, with promising results for 8-week bactericidal activity.6 The STAND (Shortening Treatment by Advancing Novel Drugs) trial investigated the efficacy and safety of PaMZ for the treatment of both drug-susceptible (DS) and rifampicin-resistant (RR) pulmonary TB.
METHODS
Study design
The study was designed as a partially randomised, open-label, non-inferiority Phase 3 clinical trial comparing three experimental treatment regimens against standard TB treatment for pulmonary DS-TB. RR-TB cases were allocated to a separate treatment arm without randomisation (Clinicaltrials.gov number NCT02342886). There were 27 sites across South Africa, Tanzania, the Philippines, Kenya, Malaysia, Uganda, Thailand and Ukraine. The trial protocol, laboratory manual and statistical analysis plan are available at https://www.tballiance.org/portfolio/trial/5091. A full list of ethics committee approvals is included in Supplementary Data (Clinicaltrials.gov number NCT02342886).
Study participants
Participants were adults (≥18 years), and had sputum 1+ or greater (International Union Against Tuberculosis and Lung Disease/WHO scale). Baseline alanine aminotransferase (ALT) or aspartate aminotransferase (AST) of ≥3 times the upper limit of normal (xULN), or a total bilirubin >2xULN (other liver tests normal) or >1.5xULN (other liver tests abnormal) were exclusion criteria. HIV-positive participants with CD4+ counts <100 cells/mm3 or WHO Stage 4 disease were excluded. The inclusion and exclusion criteria can be found in Section 5 of the Supplementary Data.
Randomisation and study treatments
Participants with DS-TB were randomised to one of four treatment arms using online randomisation software in a 1:1:1:1 ratio: 200 mg PMD, 400 mg MFX and 1500 mg PZA daily for either 6 months (6Pa200MZ) or 4 months (4Pa200MZ); 100mg pretomanid for 4 months in the same combination daily (4Pa100MZ); or isoniazid (INH, H), rifampicin (RIF, R), PZA and ethambutol (EMB, E) daily for 8 weeks, followed by INH and RIF (HR) daily for 18 weeks (2HRZE/4HR) as detailed in the Supplementary Data Section 7.2. All participants with RR-TB were assigned to receive the 6Pa200MZ regimen.
The method of randomisation used for the DS-TB patients was minimisation with random element. Minimisation factors were centre, HIV status and presence or absence of cavities on local chest X-ray.
Laboratory methods
Rapid molecular testing was used to assess INH, RIF, fluoroquinolone (FQ) and PZA susceptibility at screening (GenoType MTBDRplus [Hain Lifescience, Nehren, Germany], Xpert® MTB/RIF [Cepheid, Sunnyvale, CA, USA], GenoType MTBDRsl [Hain Lifescience] and pncA genotyping). Mycobacterial liquid culture was performed using MGIT™ (BD, Franklin Lakes, NJ, USA), and phenotypic drug susceptibility testing (DST) for streptomycin (SM), INH, RIF, EMB, PZA and MFX was confirmed using MGIT. The Laboratory Manual has been included in the Supplementary Data. DS-TB cases were susceptible to HRZ and FQs. Participants with RR-TB were confirmed as RIF-resistant, PZA-susceptible and FQ-susceptible, and either INH-susceptible or INH-resistant.
Study procedures
Chest X-rays were performed during screening. At each study visit, sputum samples were obtained, a physical examination performed and information on adverse events (AEs) collected. Electrocardiograms were performed at pre-defined visits and at the site doctor’s discretion (see Section 8 in the Supplementary Data for the protocol visit schedule). AEs were graded according to the Division of Microbiology and Infectious Diseases (National Institutes of Health, Bethesda, MD, USA) criteria (available at https://www.niaid.nih.gov/sites/default/files/dmidadulttox.pdf) with an assessment of relationship to study drugs made by the investigator. AEs were considered “treatment-emergent” or “on-treatment” if they occurred in the period from first dose of trial drug up to 14 days after last dose. Follow-up was for 24 months after randomisation (see Section 13 in the Supplementary Data).
Study outcomes
The primary efficacy, “unfavourable” outcome, was the proportion of participants with bacteriologically or clinically defined treatment failure or relapse 12 months after randomisation (from 50 to 54 weeks). A “favourable” outcome was defined as having a negative culture status (defined as two consecutive negative culture results at least 1 week apart with no intervening positive result) at 12 months if not already classified as having an unfavourable outcome. Details are provided in Table 1. While patients favourable or unfavourable under modified intention to treat (mITT) may be excluded from the per-protocol (PP) analysis, unfavourable patients cannot become favourable under PP and vice versa. Relapse was declared if positive cultures after the end of treatment were considered identical to the baseline sample by whole-genome sequencing (WGS) (<20 single nucleotide polymorphisms difference), or if WGS was not available. Recurrence was considered to be re-infection if the Mycobacterium tuberculosis strain was different by >100 SNPs from the baseline strain.
Table 1.
Trial analysis populations and related unfavourable outcomes: list of definitions applied at end-point review for patients in the trial by analysis population *
| Modified intention-to-treat population |
|---|
All randomised patients included, except:
Unfavourable outcome definitions
|
| Per protocol population |
All randomised patients included, except:
Unfavourable outcome definitions:
|
* Relapse was declared if positive cultures after the end of treatment were considered identical to the baseline sample using WGS (difference of <20 SNPs), or if WGS was not available. Recurrence was considered to be re-infection if the M. tuberculosis strain was different by >100 SNPs from the baseline strain.
† Unless already declared as unfavourable.
WGS = whole-genome sequencing; SNP = single-nucleotide polymorphism.
Secondary efficacy endpoints included time to an unfavourable outcome, time to culture-negative status, and proportions of treatment failure or relapse 24 months after randomisation. The primary safety outcome was the proportion of participants with one or more Grade 3 or 4 AEs.
Study oversight
An independent data safety monitoring committee (DSMC) of clinicians and statisticians reviewed unblinded data and oversaw the conduct of the trial.
Statistical analysis and sample size
A sample size of 300 participants recruited to each DS-TB treatment arm was calculated to provide 90% power to show non-inferiority, with a margin of 12% in the upper boundary of the two-sided 95% Wald confidence interval (CI) for the proportion with unfavourable outcome and a one-sided significance level of 2.5%.
To preserve the overall type I error, a hierarchical approach to the analysis was adopted. The first comparison was 6Pa200MZ vs. the control arm. Comparison between the control and the 4-month arms would only be made if this was found to be non-inferior. Non-inferiority would only be demonstrated if indicated in both the mITT and PP analyses. Table 1 lists the definitions for mITT and PP populations in the trial. Participants who received at least one dose of trial medication were included in the safety analysis.
RESULTS
Study participants
The recruitment disposition of the participants in the trial is shown in Figure 1 with 271 of planned 1,200 patients randomised and 13 RR-TB patients allocated. In total, 234 of 271 (86.3%) participants were included in mITT and 209 of 271 participants (77.1%) in the PP population. The majority of participants were enrolled in South Africa (218/284, 76.8%). The first patient was enrolled on 27 January 2015 and last patient completed on 29 November 2017. Within the DS-TB mITT population and PP population, respectively 3/4 and 26/30 participants excluded from the analysis were lost to follow-up or withdrawn. In total, 234/271 (86.3%) participants were included in mITT and 209/271 participants (77.1%) in the PP population.
Figure 1.
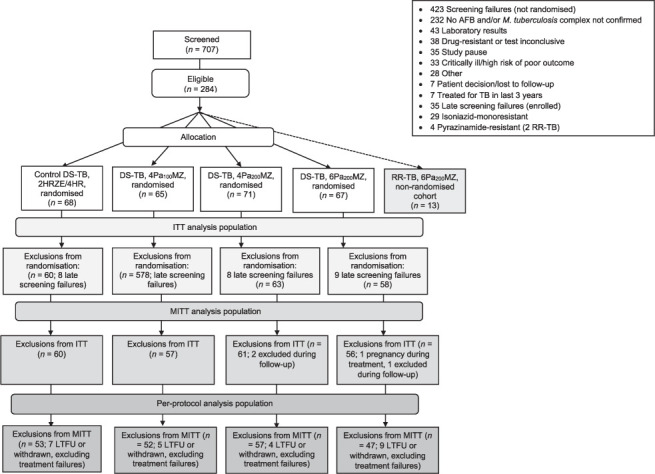
CONSORT diagram, indicating randomisations and exclusions. Patients with RR-TB were not randomised and allocated to receive 6MPa200Z. Pa 200 mg daily, M 400 mg and Z 1500 mg for 6 months (6Pa200MZ) or 4 months (4Pa200MZ); Pa 100 mg daily for 4 months in the same combination (4Pa100MZ). Late identification of drug resistance and withdrawal of consent were the most common reasons for exclusion during follow-up. AFB = acid-fast bacilli; RR-TB = rifampicin-resistant TB; DS-TB = drug-susceptible TB; H = isoniazid; R = rifampicin; Z = pyrazinamide; E = ethambutol; Pa = pretomanid; M = moxifloxacin; ITT= intention-to-treat; MITT = modified ITT; LTFU = lost to follow-up. CONSORT = Consolidated Standards of Reporting Trials.
The first DSMC review (8 months after enrolment began) recommended a pause in trial enrolment following three deaths associated with hepatotoxicity on the experimental regimen arms. While these deaths raised concern about the contribution of the experimental regimen, it was noted that some of the deaths involved delays in the recognition and management of hepatotoxicity, and administration of concomitant potentially hepatotoxic medications. Patients already enrolled in the trial continued their allocated treatment without interruption. A full review of the safety data relating to the trial, and the earlier Phase 2 study,6 was undertaken and included external specialists in hepatotoxicity and the DSMC. No conclusive evidence was found supporting an unduly increased risk for severe drug-induced liver injury (DILI) on the experimental arms. The DSMC subsequently recommended resuming enrolment into the trial with additional safety monitoring in place. Nevertheless, the sponsor decided instead to pursue a Phase 3 clinical trial of a combination of BDQ with the Pa200MZ regimen that had demonstrated very promising bactericidal activity.7
Primary outcome
The baseline characteristics of the participants are presented in Table 2. The primary efficacy results are presented in Table 3. In the PP analysis, 4/47 (8.5%) participants had an unfavourable outcome at 12 months on 6Pa200MZ arm compared to 1/53 (1.9%) on the control arm. The absolute difference in unfavourable outcomes was 6.6% (95% CI −2.2% to 15.4%). There were 11/57 (19.3%) patients with an unfavourable outcome on the 4Pa200MZ arm (absolute difference 17.4%, 95% CI −6.5% to 28.3) and 14/52 (26.9%) on 4Pa100MZ (absolute difference 25.0%, 95% CI −12.4 to 37.6). Of 11 assessable RR-TB participants receiving 6Pa200MZ one patient had an unfavourable outcome in the mITT analysis (withdrawn due to AE); in the PP population, none of the 10 participants had an unfavourable outcome.
Table 2.
Baseline characteristics (mITT population)
| DS-TB | RR-TB | |||||
|---|---|---|---|---|---|---|
| Total* (n = 234) n (%) | HRZE (n = 60) n (%) | 4Pa100MZ† (n = 57) n (%) | 4Pa200MZ† (n = 61) n (%) | 6Pa200MZ† (n = 56) n (%) | 6Pa200MZ† (n = 11) n (%) | |
| Male sex | 167 (71.4) | 42 (70.0) | 46 (80.7) | 42 (68.9) | 37 (66.1) | 5 (45.5) |
| Age, years, median (min–max) | 34.0 (18.0–77.0) | 32.5 (19.0–69.0) | 37.0 (18.0–60.0) | 37.0 (18.0–77.0) | 31.0 (18.0–64.0) | 28.0 (20.0–43.0) |
| Weight, kg, median (min–max) | 53.0 (32.2–137.8) | 54.9 (34.0–107.5) | 51.3 (37.0–137.8) | 53.0 (35.6–81.4) | 52.6 (32.2–82.0) | 55.7 (43.1–74.0) |
| Black | 164 (70.1) | 41 (68.3) | 41 (71.9) | 43 (70.5) | 39 (69.6) | 8 (72.7) |
| Smoking history | ||||||
| Never | 81 (34.6) | 16 (26.7) | 23 (40.4) | 20 (32.8) | 22 (39.3) | 6 (54.5) |
| Past | 41 (17.5) | 14 (23.3) | 7 (12.3) | 14 (23.0) | 6 (10.7) | 2 (18.1) |
| Current | 107 (45.7) | 29 (48.3) | 24 (42.1) | 27 (44.3) | 27 (48.2) | 3 (27.3) |
| Missing | 5 (2.1) | 1 (1.7) | 3 (5.3) | 0 (0.0) | 1 (1.8) | 0 (0.0) |
| HIV-positive | 58 (24.8) | 15 (25.0) | 13 (22.8) | 13 (21.3) | 17 (30.4) | 6 (54.5) |
| Resistant to INH | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 5 (45.4) |
| CXR cavitation present | 170 (72.6) | 41 (68.3) | 40 (70.2) | 43 (70.5) | 46 (82.1) | 10 (90.9) |
| Baseline TTP ≥ median | 125 (53.4) | 28 (46.7) | 28 (49.1) | 34 (55.7) | 35 (62.5) | 11 (100.0) |
* Includes control arm but does not include the (non-randomised) RR-TB patients.
† 200 mg pretomanid (Pa, PMD) daily, 400 mg moxifloxacin (M) and 1500 mg pyrazinamide (Z) for 6 months (6Pa200MZ) or 4 months (4Pa200MZ); 100 mg pretomanid daily for 4 months in the same combination (4Pa100MZ).
mITT = modified intention-to-treat; DS-TB = drug-susceptible TB; RR-TB = rifampicin-resistant TB; H, INH = isoniazid; R = rifampicin; Z = pyrazinamide; E = ethambutol; M = moxifloxacin; Pa = pretomanid; CXR = chest X-ray; TTP = time to positive result (in liquid culture).
Table 3.
Primary efficacy outcomes
| DS-TB | RR-TB | |||||
|---|---|---|---|---|---|---|
| Total* | HRZE | 4Pa100MZ† | 4Pa200MZ† | 6Pa200MZ† | 6Pa200MZ† | |
| Modified intention-to-treat analysis | ||||||
| Total randomised | 271 | 68 | 65 | 71 | 67 | 13 |
| Unassessable | 37 | 8 | 8 | 10 | 11 | 2 |
| Assessable | 234 | 60 | 57 | 61 | 56 | 11 |
| Unfavourable, n (%) | 55 (23.5) | 8 (13.3) | 19 (33.3) | 15 (24.6) | 13 (23.2) | 1 (9.1) |
| Adverse event | 17 | 2 | 3 | 3 | 9 | 1 |
| Withdrawal (culture-positive)‡ | 16 | 0 | 9 | 6 | 1 | 0 |
| Confirmed relapse | 4 | 0 | 2 | 1 | 1 | 0 |
| Death (not violent or accidental)§ | 4 | 0 | 1 | 1 | 2 | 0 |
| Non-adherence to study protocol | 6 | 4 | 1 | 1 | 0 | 0 |
| Physician decision | 4 | 2 | 0 | 2 | 0 | 0 |
| Death (TB-related)¶ | 2 | 0 | 1 | 1 | 0 | 0 |
| Clinical deterioration in follow-up | 1 | 0 | 1 | 0 | 0 | 0 |
| Lost to follow-up (during treatment) | 1 | 0 | 1 | 0 | 0 | 0 |
| Favourable, n (%) (95% CI) | 179 (76.5) | 52 (86.7) | 38 (66.7) | 46 (75.4) | 43 (76.8) | 10 (90.9) |
| (70.5 to 81.8) | (75.4 to 94.1) | (52.9 to 78.6) | (62.7 to 85.5) | (63.6 to 87.0) | (58.7 to 99.8) | |
| Difference from HRZE in unfavourable, % (95% CI) | — | — | 20.0 (−5.0 to 35.0) | 11.3 (−2.6 to 25.1) | 9.9 (−4.1 to 23.9) | — |
| Per-protocol analysis | ||||||
| Total randomised | 271 | 68 | 65 | 71 | 67 | 13 |
| Unassessable | 62 | 15 | 13 | 14 | 20 | 3 |
| Assessable | 209 | 53 | 52 | 57 | 47 | 10 |
| Unfavourable, n (%) | 30 (14.4) | 1 (1.9) | 14 (26.9) | 11 (19.3) | 4 (8.5) | 0 (0.0) |
| Withdrawal (culture-positive)‡ | 16 | 0 | 9 | 6 | 1 | — |
| Confirmed relapse | 4 | 0 | 2 | 1 | 1 | — |
| Death (not violent or accidental)§ | 4 | 0 | 1 | 1 | 2 | — |
| Death (TB-related)¶ | 2 | 0 | 1 | 1 | 0 | — |
| Treatment failure | 2 | 1 | 0 | 1 | 0 | — |
| Other | 2 | 0 | 1 | 1 | 0 | — |
| Favourable, n (%) (95% CI) | 179 (85.6) | 52 (98.1) | 38 (73.1) | 46 (80.7) | 43 (91.5) | 10 (100.0) |
| (80.1 to 90.1) | (89.9 to 100.0) | (59.0 to 88.4) | (68.1 to 90.0) | (79.6 to 97.6) | (69.2 to 100.0) | |
| Difference from HRZE in unfavourable, % (95% CI) | — | — | 25.0 (−12.4 to 37.6) | 17.4 (−6.5 to 28.3) | 6.6 (−2.2 to 15.4) | — |
* Total includes control arm but does not include the (non-randomised) MDR-TB patients.
† 200 mg pretomanid (Pa, PMD) daily, 400 mg moxifloxacin (M) and 1500 mg pyrazinamide (Z) for 6 months (6Pa200MZ) or 4 months (4Pa200MZ); 100 mg pretomanid daily for 4 months in the same combination (4Pa100MZ).
‡ Patients who failed to achieve sustained culture-negative status and were withdrawn based on clinical assessment of treatment failure.
§ During treatment.
¶ During follow-up.
DS-TB = drug-susceptible TB; RR-TB = rifampicin-resistant TB; H = isoniazid; R = rifampicin; Z = pyrazinamide; E = ethambutol; M = moxifloxacin; Pa = pretomanid; CI = confidence interval; MDR-TB = multidrug-resistant TB.
Secondary endpoints
Figure 2 contains Kaplan-Meier curves for time to culture-negative status and time to unfavourable outcome by treatment arm for DS-TB participants. There were 31/57 (54.3%) culture-negative patients at 8 weeks on control, compared to respectively 35/53 (66.0%), 36/58 (62.1%) and 33/54 (61.1%) on the 6Pa200MZ, 4Pa200MZ, and 4Pa100MZ arms in the mITT population (Supplementary Tables S13.9 and S13.10).
Figure 2.
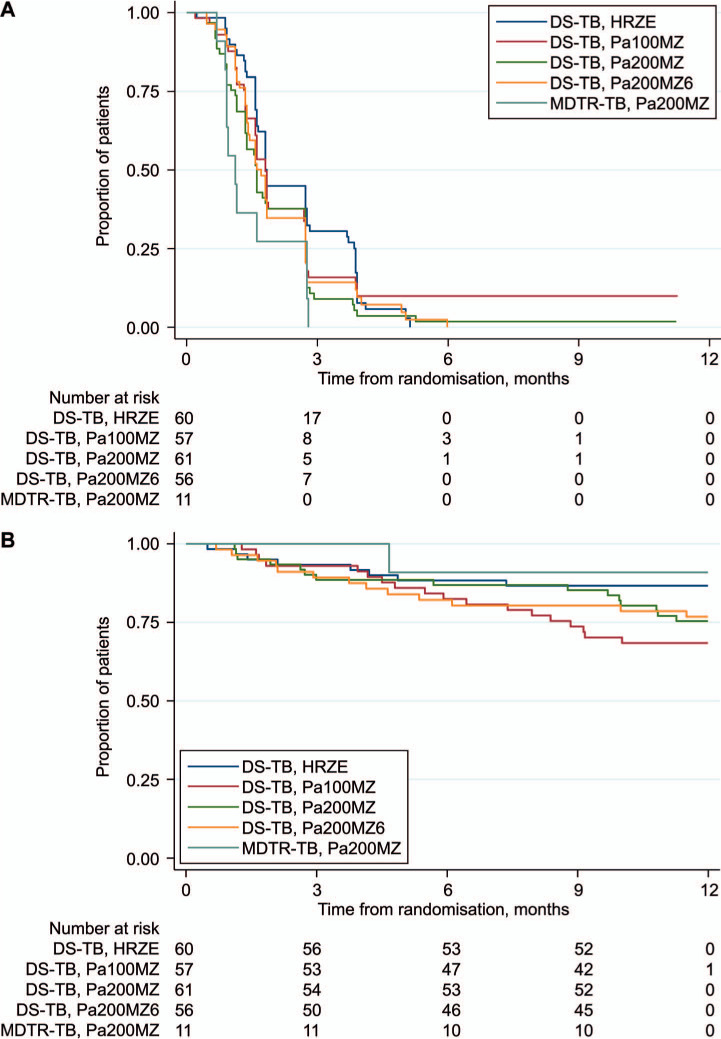
KM curves: A) time to first culture-negative status by trial treatment arm; and B) time to an unfavourable outcome by treatment arm in the modified intention-to-treat analysis.
At 24 months, respectively 14/55 (25.0%), 16/57 (28.0%), 19/54 (35.0%) and 10/56 (17.9%) assessable DS-TB participants who had received 6Pa200MZ, 4Pa200MZ, 4Pa100MZ and control had an unfavourable outcome in the mITT population. Patients receiving 6Pa200MZ had 4.8% (95% CI − 6.48 to 15.97) more unfavourable outcomes than controls in the PP and 7.6% (95% CI −7.7 to 22.9) more in the mITT analysis (Supplementary Tables S13.5 and S13.6).
Subgroup analyses
There were 14/43 (32.6%) HIV-positive participants with an unfavourable outcome on the experimental arms in the mITT analysis compared to 2/15 (13.3%) receiving the control regimen. More details can be found in Section 14 of the Supplementary Data.
Safety analysis
In the experimental arms, 68/203 (33.5%) participants with DS-TB experienced one or more Grade 3 or higher AE (G3 + AE) while on treatment vs. 19/68 (27.9%) participants in the control arm (Table 4 and Supplementary Table S16.2). Six of 71 (8.5%) DS-TB participants on the 4Pa200MZ arm, 6/65 (9.2%) on the 4Pa100MZ arm, 11/67 (16.4%) on the 6Pa200MZ arm and 4/68 (5.9%) participants receiving the control regimen discontinued treatment due to an AE: most commonly related to increased liver enzymes.
Table 4.
Safety data *
| DS-TB | RR-TB | |||||
|---|---|---|---|---|---|---|
| Total† (n = 271) n (%) | HRZE (n = 68) n (%) | 4Pa100MZ‡ (n = 65) n (%) | 4Pa200MZ‡ (n = 71) n (%) | 6Pa200MZ‡ (n = 67) n (%) | 6Pa200MZ‡ (n = 13) n (%) | |
| Grade 3+ AEs | ||||||
| On treatment§ | 87 (32.1) | 19 (27.9) | 25 (38.5) | 21 (29.6) | 22 (32.8) | 3 (23.1) |
| Post-treatment | 21 (7.7) | 3 (4.4) | 4 (6.2) | 11 (15.5) | 3 (4.5) | 0 |
| Patients with at least one treatment-emergent AE¶ leading to early discontinuation from study drug | 27 (10.0) | 4 (5.9) | 6 (9.2) | 6 (8.5) | 11 (16.4) | 0 |
| Top 5 treatment-emergent AEs reported for ≥5% patients by preferred term | ||||||
| Hyperuricaemia | 77 (28.4) | 22 (32.4) | 20 (30.8) | 21 (29.6) | 14 (20.9) | 2 (15.4) |
| Arthralgia | 73 (26.9) | 15 (22.1) | 15 (23.1) | 24 (33.8) | 19 (28.4) | 4 (30.1) |
| Aspartate aminotransferase increased | 61 (22.5) | 17 (25.0) | 15 (23.1) | 11 (15.5) | 18 (26.9) | 1 (7.7) |
| ALT increased | 53 (19.6) | 11 (16.2) | 13 (20.0) | 11 (15.5) | 18 (26.9) | 1 (7.7) |
| Blood uric acid increased | 46 (17.0) | 8 (11.8) | 15 (23.1) | 11 (15.5) | 11 (16.4) | 0 (0.0) |
| Patients with ≥1 SAE | 22 (8.1) | 3 (4.4) | 3 (4.6) | 8 (11.3) | 8 (11.9) | 3 (23.1) |
| Liver-related SAEs | 9 (3.3) | 0 (0.0) | 1 (1.5) | 4 (5.6) | 4 (6.0) | 0 (0.0) |
| Peak ALT result | ||||||
| >3xULN | 40 (14.8) | 5 (7.4) | 9 (13.8) | 12 (16.9) | 14 (20.9) | 1 (7.7) |
| >5xULN | 27 (9.9) | 4 (5.9) | 4 (6.2) | 9 (12.6) | 10 (14.9) | 1 (7.7) |
| >10xULN | 16 (5.9) | 2 (2.9) | 4 (6.2) | 5 (7.0) | 5 (7.5) | 1 (7.7) |
| Mean change from baseline in QTcF (95% CI)# | — | 9.2 (5.4–12.9) | 13.3 (10.0–16.6) | 17.6 (13.8–21.5) | 18.3 (15.1–21.5) | 13.7 (2.5–23.8) |
| Deaths | 12 (4.4) | 2 (2.9) | 4 (6.2) | 3 (4.2) | 3 (4.5) | 1 (7.7) |
* All participants who received one or more doses of study drug were included in the safety analysis.
† Includes control arm but does not include the (non-randomised) MDR-TB patients.
‡ 200 mg pretomanid (Pa, PMD) daily, 400 mg moxifloxacin (M) and 1500 mg pyrazinamide (Z) for 6 months (6Pa200MZ) or 4 months (4Pa200MZ); 100 mg pretomanid daily for 4 months in the same combination (4Pa100MZ).
§ Period from first dose of trial drug up to 14 days after last dose.
¶ AEs occurring between first dose of study medication and up to 14 days after last dose.
# Mean change from baseline in QTcB interval across visits for readings taken on or after the first administration of trial drug up to and including 14 days after the last administration of trial drug.
DS-TB = drug-susceptible TB; RR-TB = rifampicin-resistant TB; H = isoniazid; R = rifampicin; Z = pyrazinamide; E = ethambutol; M = moxifloxacin; Pa = pretomanid; AE = adverse event; ALT =alanine aminotransferase; SAE = serious adverse event; ULN = upper limit of normal; CI = confidence interval.
At least one liver-related treatment-emergent AE (TEAE) was reported among 61/216 (28.2%) participants with DS-TB receiving the experimental regimens: 24/67 (35.8%) on 6Pa200MZ, 17/71 (23.9%) on 4Pa200MZ, and 19/65 (29.2%) on 4Pa100MZ. In the control arm, 21/68 (30.9%) participants had at least one-liver related TEAE. One case met the criteria for Hy’s Law8 on the control arm; 23/203 (11.3%) participants receiving experimental regimens had a peak ALT of >5xULN, compared to 4/68 (5.9%) participants receiving standard TB treatment.
There were 12 deaths among participants with DS-TB and 1 death among those with RR-TB (Table 5). Two deaths on the 4Pa200MZ arm and one death on the 4Pa100MZ arm were attributed to hepatotoxicity and assessed as possibly related to the study treatment; delay in recognition of DILI and withdrawing drug were identified as contributing factors (see Section 16.2 of the Supplementary Data for more details).
Table 5.
Deaths in the trial
| Treatment group*† | Trial day (post-randomisation) | Trial status | Cause of death | Relationship of adverse event(s) to trial drug‡ |
|---|---|---|---|---|
| DS-TB 4Pa100MZ | Day 482 | Follow-up | Fell down from bulldozer at work | Not related |
| Day 305 | Follow-up | Haematemesis | Not related | |
| Day 39 | On treatment | Fulminant liver failure | Possibly related | |
| Day 436 | Follow-up | Massive haemoptysis | Not related | |
| DS-TB 4Pa200MZ | Day 343 | Follow-up | Sepsis | Not related |
| Day 28 | On treatment | Hepatotoxicity | Possibly related | |
| Day 34 | On treatment | Liver failure with hepatic encephalopathy | Possibly related | |
| DS-TB 6Pa200MZ | Day 163 | Follow-up | Natural causes (unknown) | Unlikely related |
| Day 114 | On treatment | Post-mortem chest X-rays indicated pneumothorax | Not related | |
| Day 469 | Follow-up | Poorly differentiated squamous cell carcinoma of the anal canal | Not related | |
| DS-TB 2HRZE/4HR | Day 576 | Follow-up | Left lobar pneumonia | Not related |
| Day 707 | Follow-up | Lower respiratory tract infection | Not related | |
| MDR-TB 6Pa200MZ | Day 34 | On treatment | Metastatic lung cancer | Not related |
* All participants who received one or more doses of trial drug included in the analysis.
† 200 mg pretomanid (Pa, PMD) daily, 400 mg moxifloxacin (M) and 1500 mg pyrazinamide (Z) for 6 months (6Pa200MZ) or 4 months (4Pa200MZ); 100 mg pretomanid daily for 4 months in the same combination (4Pa100MZ).
‡ As per site investigator assessment.
DS-TB = drug-susceptible TB; H = isoniazid; R = rifampicin; Z = pyrazinamide; E = ethambutol; M = moxifloxacin; Pa = pretomanid; MDR-TB = multidrug-resistant TB.
DISCUSSION
It was not possible to assess whether the 6Pa200MZ regimen was non-inferior in terms of efficacy compared to the standard TB treatment regimen for the treatment of DS-TB because the trial was stopped early after Phase 2 trial data suggested that the addition of BDQ was associated with greater efficacy. Experimental arms had higher proportions of treatment failure or relapse than the standard TB regimen; however, because of the small numbers these results are inconclusive.
In the safety analysis, a higher proportion of TEAE and serious AEs was observed in the experimental arms than in the standard TB regimen among DS-TB participants, and were most commonly hepatic in nature. Overall, 11% of DS-TB participants receiving experimental regimens and 6% of participants allocated to the standard TB regimen demonstrated a peak ALT ≥5xULN. This is higher than the proportion reported in the REMoxTB trial for the standard regimen (3% for ALT ≥5xULN, and 6% for peak ALT ≥3xULN9), and suggests that the inclusion criteria for this trial could have recruited patients with higher risk of hepatoxicity (for example, lower permitted haemoglobin and CD4+count at baseline).
In the Phase 2 study that preceded this trial, there was little difference observed in liver-related withdrawals between the experimental arms and standard TB treatment: eight participants were withdrawn because of elevated aminotransferases on each of the two experimental arms (100 mg and 200 mg PMD arms), and six participants were withdrawn for the same reason from the standard TB regimen.6 However, the numbers of patients with peak ALT/AST ≥3xULN on the experimental arms in the Phase 2 study were higher than the number of liver-related withdrawals, at respectively 10/60 (16.7%) and 14/88 (15.9%) on the 100 mg and 200 mg (DS-TB and DRTB) PMD arms, compared to 7/59 (11.8%) on standard treatment. The higher incidence of clinically significant peak ALT/AST elevations seen in the current trial compared to the Phase 2 study may be due to differences in the patient population recruited, which included more HIV-positive participants with lower CD4+ counts and a lower permitted haemoglobin at baseline. Previous work has indicated that comorbidities and less favourable baseline physiological parameters are associated with higher rates of significant hepatotoxicity with TB treatment.10–12 The REMoxTB study recruitment was more restrictive than STAND, and it was hoped that this would represent the wider population of TB patients and enhance the generalisability of the results.
This report highlights the need to better understand the relationship between TB treatment and hepatotoxicity.13 Although likely to be multifactorial, oxidative stress on hepatocytes could be a major factor.14 Individual responses can be hard to predict, for example some participants can tolerate the same regimen when reintroduced for unknown reasons.15 We need to focus on the pathological basis of TB treatment DILI to improve prediction of potentially hepatotoxic regimens.
Study limitations were predominantly related to the early halt in recruitment. Phase 2 and 3 clinical trials are not powered to make statistical analyses relating to safety, and this is especially true when halted early. Analysis of data using small patient numbers can over- or under-estimate efficacy and/or safety16 associated with a certain intervention, although there were approximately 60 DS-TB participants per arm. Furthermore, very few TB-HIV co-infected patients were participants in the trial.
In this Phase 3 trial, the results for the experimental regimens compared to standard TB treatment in a Phase 2 study did not appear to translate into non-inferior outcomes in either the 6-month or 4-month treatment arms. However, the study failed to reach an adequate sample size, and this limits conclusions relating to the efficacy and safety data. An ongoing evaluation of the PMD safety profile remains a priority.
Acknowledgments
The authors thank all the participants for their participation in the study; the nursing and laboratory staff; all those who advised, volunteered, or otherwise supported community engagement around the STAND clinical trial sites; and the members of the DSMC for their oversight of the safety of the trial. STAND was sponsored by TB Alliance with support from the UK Department for International Development, UK Department of Health (London, UK), Bill and Melinda Gates Foundation (Seattle, WA, USA), US Agency for International Development (Washington DC, USA), Directorate General for International Cooperation of the Netherlands (Amsterdam, The Netherlands), Irish Aid (Dublin, Ireland), Australia Department of Foreign Affairs and Trade (Canberra ACT, Australia) and the Federal Ministry for Education and Research of Germany (Berlin, Germany) through KfW (Kreditanstalt für Wiederaufbau) and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH; Bethesda, MD, USA) under Award Numbers UM1 AI068634 and UM1 AI068636. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. AMC and AJN are supported by Medical Research Council Grant: MC_UU_12023/27 Tuberculosis Treatment Trials.
The sponsor of the study was involved in study design, data collection, data analysis, data interpretation, and writing of this report. The trial sites had substantial previous experience in running clinical trials; laboratory staff were blinded to the treatment arms associated with patient samples; monitoring visits were carried out at the sites to ensure accurate data collection relating to both efficacy and safety findings; and all data collected during the course of the trial was included in the analyses (with populations defined as per the Methods section of the manuscript).The first author (CDT) had full access to all the data in the study and had final responsibility for submission.
Qualified researchers may contact the corresponding author (CDT) to obtain specific de-identified clinical trial data with the permission of TB Alliance.
Conflicts of interest: none declared.
References
- 1.World Health Organization. Geneva, Switzerland: WHO; 2019. Global tuberculosis report, 2019. WHO/CDS/TB/2019.15. [Google Scholar]
- 2.Furin J, Cox H, Pai M. Tuberculosis. Lancet. 2019;393:1642–1656. doi: 10.1016/S0140-6736(19)30308-3. [DOI] [PubMed] [Google Scholar]
- 3.Stover CK, et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 4.Hu Y, Coates ARM, Mitchison DA. Comparison of the sterilising activities of the nitroimidazopyran PA-824 and moxifloxacin against persisting Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 2008;12:69–73. [PubMed] [Google Scholar]
- 5.Prideaux B, et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med. 2015;21:1223–1227. doi: 10.1038/nm.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson R, et al. Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: A phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pul. Lancet. 2015;385:1738–1748. doi: 10.1016/S0140-6736(14)62002-X. [DOI] [PubMed] [Google Scholar]
- 7.Tweed CD, et al. Bedaquiline, moxifloxacin, pretomanid, and pyrazinamide during the first 8 weeks of treatment of patients with drug-susceptible or drug-resistant pulmonary tuberculosis: a multicentre, open-label, partially randomised, phase 2b trial. Lancet Respir Med. 2019;7:1048–1058. doi: 10.1016/S2213-2600(19)30366-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15:241–243. doi: 10.1002/pds.1211. [DOI] [PubMed] [Google Scholar]
- 9.Tweed CD, et al. Liver toxicity associated with tuberculosis chemotherapy in the REMoxTB study. BMC Infect Dis. 2018;18:46. doi: 10.1186/s12879-018-3230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosford JD, et al. Hepatotoxicity from antituberculous therapy in the elderly: A systematic review. Tuberculosis. 2014;95:112–122. doi: 10.1016/j.tube.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yimer G, et al. Evaluation of patterns of liver toxicity in patients on antiretroviral and anti-tuberculosis drugs: a prospective four arm observational study in ethiopian patients. PLoS One. 2014;9:e94271. doi: 10.1371/journal.pone.0094271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tweed CD, et al. Toxicity associated with tuberculosis chemotherapy in the REMoxTB study. BMC Infect Dis. 2018;18:317. doi: 10.1186/s12879-018-3230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saukkonen JJ, Powell K, Jereb JA. Monitoring for tuberculosis drug hepatotoxicity: moving from opinion to evidence. Am J Respir Crit Care Med. 2012;185:598–599. doi: 10.1164/rccm.201112-2174ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yew WW, et al. Oxidative stress and first-line antituberculosis drug-induced hepatotoxicity. Antimicrob Agents Chemother. 2018;62:e02637–02617. doi: 10.1128/AAC.02637-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma SK, et al. Safety of 3 different reintroduction regimens of antituberculosis drugs after development of antituberculosis treatment-induced hepatotoxicity. Clin Infect Dis. 2010;50:833–839. doi: 10.1086/650576. [DOI] [PubMed] [Google Scholar]
- 16.Bassler D, et al. Stopping randomized trials early for benefit and estimation of treatment effects systematic review and meta-regression analysis. JAMA. 2010;303(12):1180–1187. doi: 10.1001/jama.2010.310. [DOI] [PubMed] [Google Scholar]