

Abstract
The autoimmune rheumatic diseases (ARDs) are characterised by a pathological triad composed of autoimmunity/inflammation, microangiopathy and aberrant tissue remodelling. Disease terms such as idiopathic inflammatory myopathy (IIM), scleroderma/systemic sclerosis (SSc), and systemic lupus erythematosus (SLE) are helpful clinically but disguise the considerable overlap that exists within these ‘distinct’ disorders. This is perhaps best demonstrated by inflammatory myopathy, which can be present in SSc or SLE, but can itself be absent in clinically amyopathic IIM. Archetypal clinical manifestations of ARD (such as Raynaud’s phenomenon) are frequently present, albeit with varying prominence, within each of these diseases. This is certainly the case for inflammatory myositis, which has long been recognised as an important clinical feature of both SSc and SLE. Progress in elucidating the clinicoserological spectrum of autoimmune rheumatic diseases has identified autoantibody specificities that are strongly associated with ‘overlap’ disease and the presence of inflammatory myositis in SSc and SLE. In this review, we shall describe the prevalence, burden, prognostic value and management considerations of IIM in the context of both SSc and SLE. A major emphasis on the value of autoantibodies shall highlight the value of these tools in predicting the future occurrence of inflammatory myositis in both SSc and SLE. Where applicable, unmet research needs shall be highlighted. The review emphasises the importance of myopathy as a common feature across all the ARDs, and highlights specific antibody specificities that are strongly associated with myopathy in the context of SLE and SSc.
Keywords: Polymyositis, dermatomyositis, Systemic sclerosis, Systemic Lupus Erythematosus, Clinical phenotype, Classification, Overlap syndromes
Introduction
The autoimmune rheumatic diseases (ARDs) are characterised by a pathological triad composed of autoimmunity/inflammation, microangiopathy and aberrant tissue remodelling. Disease entities such as idiopathic inflammatory myopathy (IIM), scleroderma/systemic sclerosis (SSc) and systemic lupus erythematosus (SLE) are helpful clinically but disguise the considerable overlap that exists within these ‘distinct’ disorders. Archetypal clinical manifestations of ARD, such as Raynaud’s phenomenon (RP), occur across each of these diseases, albeit varying in prevalence. The prominence of specific clinical features helps to determine both the clinical diagnosis and disease classification for the purposes of clinical research. This is certainly the case for inflammatory myositis, which is the pathological hallmark of IIM (although not an essential feature for diagnosis) and has long been recognised as an important clinical feature of SSc and SLE. In this review, we shall describe the prevalence, burden, prognostic value and management considerations of inflammatory myopathy in the context of SLE and SSc. Progress in elucidating the clinicoserological spectrum of autoimmune rheumatic diseases has revealed antibody specificities that are strongly associated with ‘overlap’ disease. The role of autoantibodies in predicting the future occurrence of inflammatory myositis in SSc and SLE shall be discussed. Where applicable, unmet research needs shall be highlighted.
Search Strategy and Selection Criteria
References for the present review were identified through a search in PubMed (18th April 2020) using the following search terms: ((myositis) or (myopathy)) AND ((lupus) OR (scleroderma) OR (systemic sclerosis)). No language or date restrictions were placed on the search. Of the 3897 studies identified, the titles and abstracts were reviewed for inclusion in the review on the basis of their relevance and originality. Additional cited manuscripts where identified through searches of the authors’ own files.
Systemic sclerosis
Clinical and pathological overlap between IIM and SSc-spectrum disorders
The most common clinical manifestations of SSc relate more to dysregulated fibrosis (in the skin (‘scleroderma’) and other organs) and vasculopathy (manifesting as RP, ischaemic digital ulceration, pulmonary arterial hypertension and scleroderma renal crisis), than overt inflammation. Nonetheless, inflammation in the muscles (and joints) are not uncommon features of SSc. Similarly, vascular injury (a pathological hallmark of SSc) is a cardinal feature of IIM, leading to an early proposal for the term ‘angiomyositis’ to describe IIM at the turn of the 20th century [1]. The aetiopathogenesis of SSc and tissue injury in the skin and muscle of IIM share many key features, that include endothelial injury, perivascular inflammatory cell infiltrate, reduction in capillary density and peri-fascicular ischaemia [2–5]. Clinical features such as RP occur in ~50% of patients with IIM and morphological abnormalities of the nailfold capillaries are common [6]. In contrast to the progressive microangiopathy of SSc, which is generally associated with poorer clinical outcomes [7], there can be resolution of nailfold capillaries (NC) abnormalities in IIM (particularly dermatomyositis [DM]), which can be associated with favourable clinical outcomes [6, 8–10]. Putative important molecular pathways, such as those relating to Vascular Endothelial Growth Factor (VEGF) - a factor strongly implicated in the pathogenesis of SSc-related vasculopathy - are also over-expressed IIM (and may be associated with the direct production of aminoacyl-tRNA synthetases in IIM) [11, 12]. Myositis-specific antibodies (particularly anti-synthetase antibodies) are not infrequently identified in IIM patients with a strong SSc phenotype (or even SSc patients without features of IIM), highlighting the strong clinical and pathological associations between these conditions [13, 14]. Impaired blood flow in microcirculation of the skeletal muscles may also be a contributing factor to the pathophysiology of muscle disease in SSc as was demonstrated in a novel muscle MRI study. [15]. Sometimes, evidence of muscle inflammation is an incidental finding on routine biochemistry (Figure 1), although enquiry about muscle symptoms and muscle strength assessment can be easily overlooked by clinicians whose focus is directed to other organ systems. In other patients, an overt symptomatic inflammatory myopathy is more clinically manifest.
Figure 1. Asymptomatic low-grade myopathy in overlap syndrome with anti-Ku antibodies.
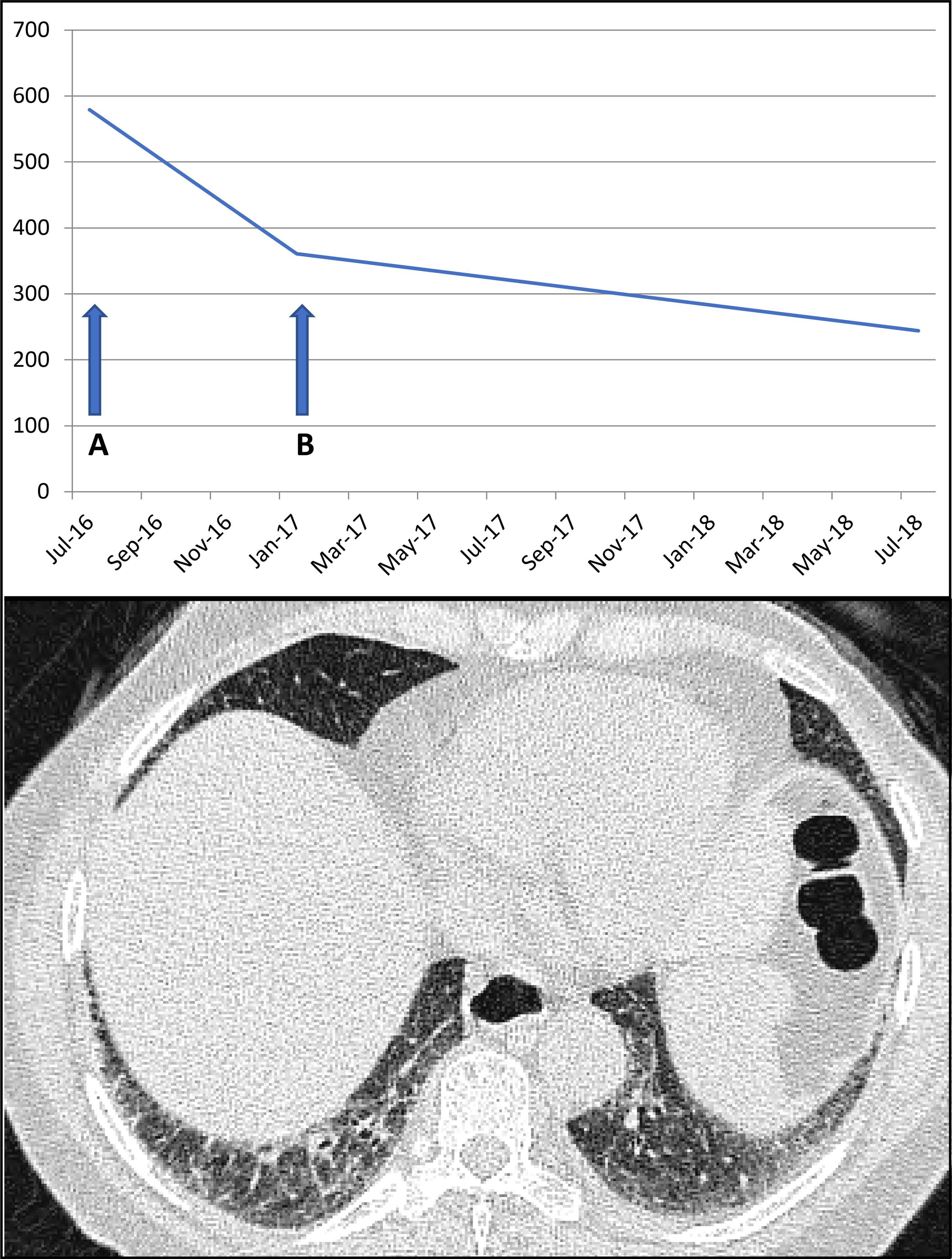
A 67yr old female developed inflammatory arthritis, Raynaud’s, diffuse skin thickening (modified Rodnan Skin Score 18/51) with positive Anti-Ku antibodies. There were no myopathic symptoms/signs, but moderately elevated creatine kinase was identified on routine biochemistry. The arthritis was treated with steroids (a) with mycophenolate mofetil added as steroid sparing agent (b), leading to reduction in CpK to normal range. Asymptomatic non-specific interstitial pneumonia was present on CT thorax.
Burden of myopathy in SSc-spectrum disorders
The prevalence of myopathy in SSc is greatly influenced by the definition (and objective testing) applied. Early attempts to assess muscle disease in SSc struggled to differentiate between a primary myopathy and disuse muscle atrophy secondary to general debility, associated arthropathy and skin disease [16]. There is a high prevalence of reduced muscle strength and endurance in SSc compared with age-matched controls; not all of which can be attributed to myositis [17]. Generalised fatigue is one associated factor that may contribute to muscle weakness in SSc [17]. Objective evidence of inflammatory myopathy can also be present in the absence of overt patient-reported symptoms of muscle weakness; possibly owing to a more insidious onset compared with IIM [16].
The typical distribution of proximal myopathy can mirror that of IIM, although distal weakness can also accompany skin involvement of the hands and forearms [16]. It was initially thought the myopathy of SSc was not particularly strongly influenced by disease subset [18], although subsequent studies have identified a paucity of IIM in the context of anti-centromere antibodies [19] and an association between myopathy and diffuse cutaneous SSc (dcSSc). Large registry analyses have identified objective evidence of muscle weakness in around a quarter of SSc patients in the EUSTAR database, although only a third of these (8% of the total) had an elevated creatine kinase [20]. The Canadian Scleroderma Research Group (CSRG), applying different definitions of myopathy, have reported evidence of myositis in ~13% of SSc patients [21]. The point prevalence of elevated creatinine kinase in SSc patients in the CSRG was 5.6%, with approximately 10% of patients having history of myositis/myopathy [22]. In the CSRG registry, a raised creatine kinase was associated with being male (~1/4 of patients), younger age, dcSSc, tendon friction rubs, higher skin score and a FVC <70% [22]. Alongside U1-RNP, patients with a raised creatine kinase were more likely to carry anti-topoisomerase antibodies [22]. Patients reported muscle pain and/or weakness is present in around 1/3 of patients with dcSSc, but there is objective evidence of muscle weakness and/or a raised creatine kinase is only present in around 10% of patients with dcSSc [20, 23, 24]. Muscle involvement is approximately twice as common in dcSSc compared with limited cutaneous SSc (for example, creatine kinase is elevated in 11.3%of dcSSc vs 4.4% in lcSSc) [24]. Sometimes, evidence of a low-grade myopathy in SSc can be identified on routine serum biochemistry in the absence of any muscle weakness (Figure 1). In other cases of SSc, the myopathy is more clinically overt, often with a correspondingly significantly elevated muscle enzyme level(Figure 2).
Figure 2. SSc associated myopathy with concomitant organising pneumonia in association with anti-PM-Scl antibodies.
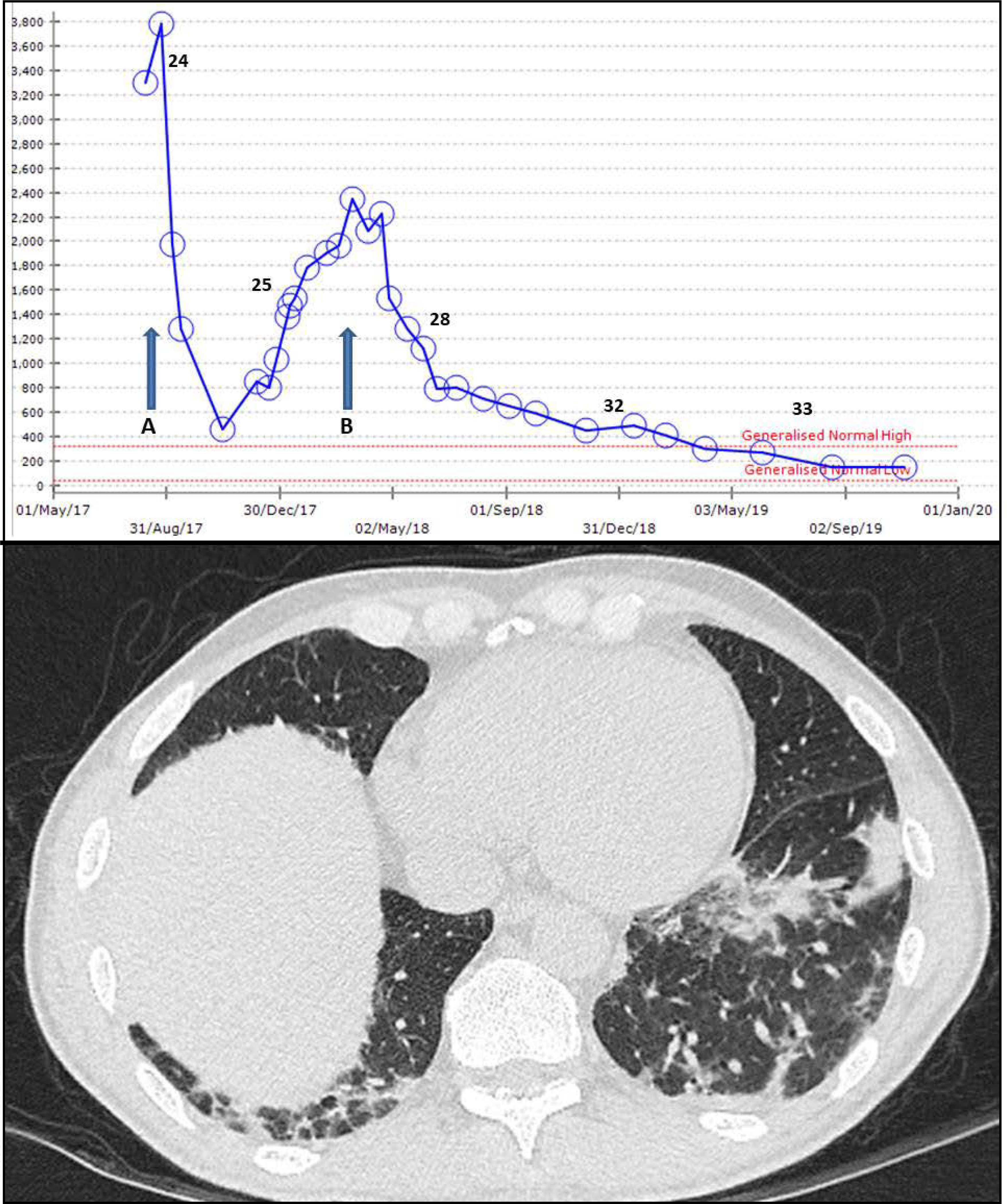
A 55yr old male with Raynaud’s, sclerodactyly and antibodies to PmScl-75, Pm-Scl 100 and Ro-52 developed acute proximal myopathy with raised CpK. Initial response to 6 IV cyclophosphamide infusions (a) was not maintained with mycophenolate mofetil (MMF). Rituximab was started (b) alongside ongoing MMF. CT thorax identified patchy organising pneumonia.
The numbers represent Myositis Muscle Dysfucntion Scores (range 0–40 with higher numbers indicative of better strength).
The first major study in 1978 to prospectively examine and classify myopathy in SSc included 24 patients with SSc (the majority of whom it seems had a diffuse cutaneous disease). Twenty three of 24 (95.8%) of this cohort had one or more myopathic features (raised muscle enzymes, objective weakness on muscle testing, abnormal EMG and/or abnormal biopsy when performed) [25]. The majority (19/24) were perceived to have a ‘simple myopathy’ which they defined as a myopathy with modest rise in muscle enzymes (creatine kinase ~140% of normal) and often not necessitating treatment with corticosteroids [25]. The remaining patients had a ‘complicated myopathy’ which they defined as patients with significant weakness, abnormal electromyography and significantly elevated muscle enzymes (creatine kinase ~1700% of normal) [25]. While this classification of myopathy dichotomized muscle disease in SSc, it became apparent in a subsequent study in 2009 focused on histological features of 35 patients with SSc-myopathy that muscle disease was far more heterogeneous than previously thought. Muscle biopsy features ranged from inflammation, atrophy, necrosis, fibrosis, and microangiopathy [26]. Scleroderma specific associations in this study also identified the diffuse subset in 26 (74%) of cases and that myopathy was an early feature of SSc, typically occurring within 5 years from diagnosis (Figure 1 and 2) [26]. A case-control study comparing SSc patients with and without an associated myopathy (matched for disease subset, age, gender) identified an increased frequency of reduced FVC, heart involvement and scleroderma renal crisis in patients versus controls [27]. Sudden cardiac death in the setting of muscle disease in SSc has also been reported highlighting the importance of early identification of these patients [28]. Associated cardiac disease has been highlighted as a challenge in children with SSc-associated myopathy [29].
Investigation of myopathy in SSc-spectrum disorders
Role of muscle enzymes
SSc patients with a raised creatine kinase or clinical evidence of myopathy have higher Health Assessment Questionnaire Disability Index (HAQ-DI) scores [17, 22, 30, 31] Myopathy is an independent risk factor for worse outcomes such as disability even after controlling for potential confounders such as age, restrictive lung disease, or GI disease [32]. Both assessment of muscle strength and laboratory evidence of muscle inflammation are important modes of investigation in SSc associated myopathy. An analysis from the Pittsburgh cohort identified proximal muscle weakness as an important determinant of future death on univariate analysis and Cox proportional hazards analysis [33]. Furthermore, in this study, aldolase was suggested as having a stronger association with death than creatinine kinase [33]. A separate French study also demonstrated that elevated muscle enzymes are a useful predictor of future myopathy in SSc (9/137 over 4 years) and plasma aldolase may be superior to creatinine kinase in this regard [34]. Raised creatine kinase and/or those with a past history myopathy, has also been associated with higher mortality [22]. Therefore, based on these studies, the assessment of muscle strength, in particular in the proximal distribution and muscle enzymes (both CK and aldolase) can be easy and important tools to determine the presence of a myopathy.
Since there can be multiple other aetiologies for elevated muscle enzymes and proximal muscle weakness, to comprehensively assess for the presence of a myopathy in SSc patients should ideally include additional studies such as electromyography, MRI of the muscles, or muscle biopsy when available. EMG results supporting a myopathic process is usually identified in patients with SSc associated myopathy [26]. Similarly, symmetric muscle oedema will be classically seen in T2 STIR images typical of myositis.
Histological assessment of myopathy in SSc
Muscle biopsy can be a helpful diagnostic and management tool in SSc associated myopathy, particularly if the diagnosis is unclear. In a study of 42 SSc patients with a myopathy, muscle histopathology was studied to identify whether unique subsets existed in SSc muscle disease [35]. Distinct histopathological categories were identified that included polymyositis (5%, often associated with early disease), acute denervation (7%) and muscle fibrosis (7%, often associated with anti-PM-Scl antibodies) [35]. On follow-up study of this cohort, the histopathological subset that was identified with predominant fibrosis on muscle biopsy, or a fibrosing myopathy, was found to a distinct clinical phenotype. The presence of fibrosing myopathy on muscle biopsy was associated with diffuse cutaneous disease, African-American race, a lower FVC and a higher mortality (5/8, 62,5%) when compared to those with an inflammatory myopathy [36]. Interestingly, patients with a fibrosing myopathy also tended to have lower muscle enzymes when compared to those with an inflammatory myopathy, and patients died of cardiac complications (i.e. non-ischemic cardiomyopathy). The predominance of fibrosis on muscle biopsy was also reported in another case series of 35 SSc patients when compared to those with an inflammatory myopathy (80% vs 32%, p<0.05) [37].
The treatment of SSc associated myopathy is not well defined. The recently published data classify SSc associated myopathy based on muscle histopathology, but it is not known if this is predictive of outcomes or should be better utilised to guide specific personalised treatment approaches in SSc-associated myopathy. While it is clear that those with a clear overlap myositis will respond to immunosuppression, it is not clear whether those with significant fibrosis on muscle biopsy will have the same response. In fact, in one case series, a lymphocytic infiltrate on muscle biopsy (identified in 2/3 of cases) was associated with a more favourable response to corticosteroids (90% vs 38% response rate) [26]. Future studies should examine the value of histological analysis of myopathy in predicting treatment response and outcomes in the context of SSc.
The clinical relevance of autoantibodies in SSc-associated myopathy
Autoantibodies have proved reliable prognostic markers for a number of disease manifestations of SSc. This is also true of SSc-associated myopathy. In the aforementioned case-control study, anti-centromere antibodies were protective against SSc-myopathy (odds ratio, OR, 0.11), whereas the presence of antibodies targeting PM-Scl (OR 5.0) and U1-RNP (OR 6.9) were both strongly associated with SSc-associated myopathy [27]. Despite the aforementioned reported association between myopathy and dcSSc, antibodies targeting RNA polymerase III (4% of patients) and topoisomerase (9%) are not strongly associated with SSc-associated myositis [38]. Unsurprisingly, the antibodies most commonly associated with lcSSc (anti-centromere and anti-Th/To) are also weakly associated with SSc-myositis (1% and 6% of patients respectively) [38].
Anti-PM-Scl
It is over 40 years since Wolfe et al. identified antibodies targeting a novel antigen present in a high proportion of patients with myositis (but not other diseases), that they termed anti-PM-1 [39]. Subsequent work noted an association between this antibody and a strong SSc clinical phenotype (Raynaud’s phenomenon (RP), sclerodactyly, proximal scleroderma, telangiectases and interstitial lung disease) in over half of patients, resulting in the re-deignation of this antibody as anti-PM-Scl [40]. The strong clinical SSc phenotype (particularly Raynaud’s, ILD and sclerodactyly), in comparison with other IIMs has been confirmed on subsequent studies [41] (Figure 2). Overall, antibodies targeting PM-Scl are identified in between 2.5–10.6% of cases of SSc, and typically associated with either the lcSSc subtype (88%) and/or overlap syndromes [38, 42]. The 75kDa subunit is the major antigen target of anti-PM-Scl antibodies, although there is some evidence that patients carrying antibodies targeting the 100kDa subunit have a higher creatine kinase elevation [42]. Overall, a raised creatine kinase is present in around 35% of patients carrying anti-PM-Scl (compared with 6–8% of all SSc patients) [42]. Muscle weakness is only present in around 1/3 of patients with anti-PM-Scl at presentation but over 90% will develop myopathy at some stage during the disease course, typically affecting the upper limbs more often than in other forms of IIM [41]. Antibodies to PM-Scl have the strongest association with myopathy overall (58% prevalence of myopathic features in SSc patients), followed by antibodies to U1-RNP (27%, see later) and U3-RNP (18% with myopathy in a cohort of whom ~1/3 were African-American) [38, 43].
Overall, features of SSc (such as RP) are present in ~40% of patients with a diagnosis of IIM carrying anti-PM-Scl antibodies [43]. This rises to 78%−100% when the clinical features of unselected anti-PM-Scl positive patients are reported, allowing more of those whose clinical phenotype may be more consistent with SSc to be included [41, 44]. One cohort of IIM patients carrying anti-PM-Scl, noted that 30% of patients also fulfilled classification criteria for SSc with clinical features such as RP (78%), sclerodactyly (66%), telangiectases (66%), gastro-oesophageal reflux disease (61%), puffy hands (39%) and calcinosis cutis (39%) more commonly identified in PM-Scl positive patients compared with patients with anti-synthetase syndrome, DM or immune-mediated necrotising myopathy [41]. It was noted that even when overt features of IIM or SSc did not allow a diagnosis to be made in patients carrying PM-Scl antibodies, there were often still features of SSc-spectrum disorders such as ILD [45] (see Figure 2 for example).
Anti-Ku antibodies
A rarer antibody that occurs in patients with polymyositis-scleroderma overlap are anti-Ku antibodies (targeting DNA binding proteins) [46]. Anti-Ku antibodies are occasionally identified in SLE, rheumatoid arthritis, Sjogren’s syndrome or as part of an undifferentiated connective tissue disease, sometimes in conjunction with other autoantibodies (Figure 1). They are present in only 1–2% of patients with isolated clinical features of SSc or SLE, but found in 33–55% of patients with SLE-SSc-polymyositis overlap syndromes [46]. A recent study that combined several large SSc registries identified anti-Ku antibodies in 1.1% of SSc patients (24/2140), around half of whom (13/24) had no other antibodies [47]. SSc patients with single-specificity anti-Ku antibodies had higher prevalence of ILD (58% vs 34%) and raised creatine kinase levels (>3× normal) at both baseline (11% vs 1%) and during follow-up (10% vs 2%) [47]. Patients with anti-Ku fall within 2 distinct clusters comprising those with a strong SLE phenotype characterise by raised dsDNA (associated with glomerulonephritis) or those with a stronger SSc/IIM phenotype characterised by a raised creatine kinase (associated with ILD) [48, 49]. Lung disease in patients with Ku autoantibodies have also been reported to have more corticosteroid resistant disease [50].
Anti-RuvBL1/2 antibodies
The most recently identified SSc-specific antibody to be associated with myositis in SSc is anti-RUV-BL 1/2 [51]. Anti-RuvBL1/2 are identified in ~2% of SSc cases and associated with dcSSc in overlap with myositis [51]. In contrast to, anti-PM-Scl and anti-Ku, anti-RuvBL1/2 antibodies are associated with an older age of onset, male sex and diffuse skin involvement [51].
Other causes of myopathy in SSc-spectrum disorders
Whilst no longer typically included in the modern management of SSc, there have been a number of reports of myositis occurring following D-penicillamine use in rheumatic disease such as SSc [52, 53]. An early report of the clinical experience of D penicillamine in SSc noted mortality from cardiomyopathy in ~10% of cases, although this was attributed to the disease rather than the intervention [54]. Other forms of myopathy (e.g. neuromuscular disease) should also be considered in the work of SSc patients with myopathic features.
Systemic lupus erythematosus
Burden of myopathy in SLE
Musculoskeletal manifestations in SLE have a major impact on health-related quality of life. The most common musculoskeletal manifestations of SLE are inflammatory arthritis and fibromyalgia; each of which could impact on the assessment myopathy in SLE depending on the definition applied [55]. While myositis is a recognised manifestation of SLE, it is less well studied compared with myositis in other rheumatic diseases such as SSc. The first major assessment of myopathy in SLE examined the records of 228 patients with SLE, identifying prominent muscle involvement in 18 (8%) of the cohort [56]. Consistent with the aforementioned study of SSc, this preliminary work suggested serum aldolase may be a better marker than creatine kinase for assessing muscle involvement in SLE [56]. This early work also noted that up to 20% of patients carried antibodies to either U1-RNP and/or PM-Scl (then termed PM-1) [56], which may partly account for early observations that that myopathy in SLE seldom occurred in the presence of renal involvement [56]. Indeed, early reports of lupus myositis consistently reported an association with a more favourable disease course [57]. Whilst some of the earliest studies suggested SLE was associated with a milder myopathy, a prospectively identified cohort study which used the Bohan and Peter criteria, found very similarly elevated creatine kinase and objective muscle weakness between IIM and SLE myositis groups [58]. This study also suggested patients with SLE myositis where more likely to be female and younger at disease onset, than patients with other forms of IIM [58]. A more recent large retrospective study evaluated the prevalence of myositis in 1718 SLE cases (including 451 paediatric patients) identified during a 9 year period [59]. They found an overall prevalence of myopathy in SLE of around 6%, similar to earlier studies [59]. Of the 108 SLE myositis patients in this cohort, myositis was a presenting feature in 64% (as seen in the case described in Figure 3) and myositis preceded the diagnosis of SLE in 15% [59]. In those where it was not a presenting feature, myositis occurred an average of 5.25 years after SLE diagnosis. Applying a more stringent definition of myopathy (requiring the presence of objective muscle weakness, raised creatine kinase and abnormal electromyography) another recent cross-sectional study of over 1700 SLE patients identified a point prevalence of myopathy of only 2.6% [60]. The prevalence of myositis may be higher in paediatric populations with one estimate of 31% within a US cohort in Alabama (based on either symptoms, objective weakness, raised muscle enzymes and/or MRI findings) [61].
Figure 3. Myositis in newly diagnosed SLE.
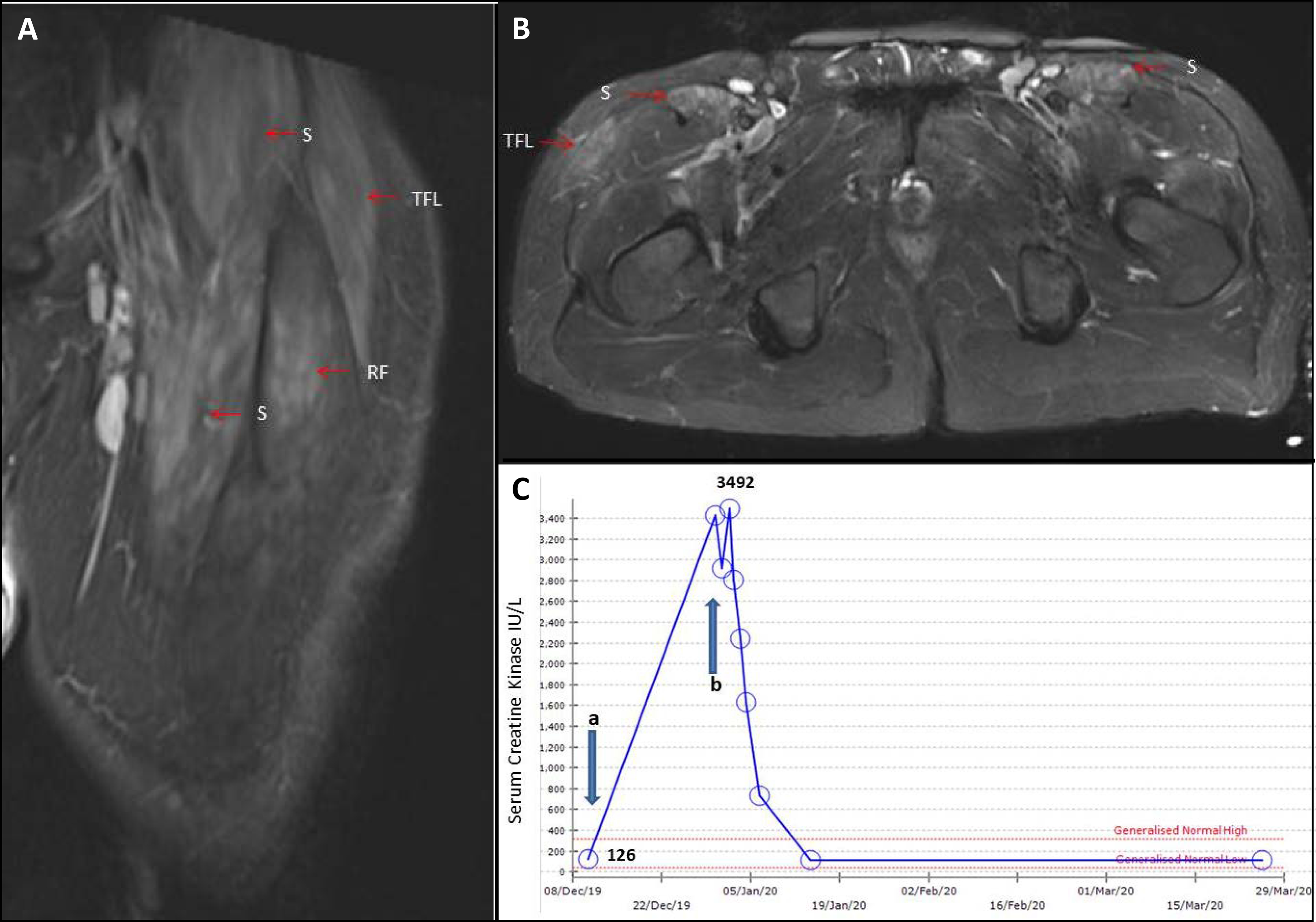
A 49yr old male presented with arthralgia, malaise and plasma viscosity 2.22 (NR <1.74). He developed fever, myalgia, proximal weakness, synovitis and neutropenia. The CpK rose from normal (a) to 3,400 (b). He had positive ds-DNA (59.9IU/ML) and low complement. MRI revealed symmetrical oedema within sartorius (S), tensor fascia lata (TFL) and rectus femoris (RF). Treatments with pulsed IV methylprednisolone and mycophenolate mofetil resulted in resolution of symptoms and normalisation of creatine kinase, ds-DNA and complements.
The distribution of myopathy can differ in SLE in comparison with other forms of IIM. Early studies of lupus myopathy noted a predilection for disuse muscle atrophy in close proximity to affected joint inflammation, that was distinct from a true inflammatory myopathy [62]. Orbital myositis is a specific rare manifestation of SLE [63, 64], although impaired extraocular muscle movement can also occur secondary to focal tenosynovitis [65].
Clinical and serological characteristics associated with SLE myopathy
The enormous overlap across the ARDs is again highlighted by the observation that almost half of SLE-associated IIM patients had at least 2 features of SSc, with 28% of patients fulfilling classification criteria for SSc (many carrying U1-RNP or myositis-associated antibodies); once again emphasising the futility of considering these disorders truly distinct [59]. An association with black ethnicity and paediatric-onset SLE has also been noted [59]. A number of studies have also demonstrated an association between myositis and other specific SLE manifestations including arthritis (up to 94% in one study [59]), leukopenia, thrombocytopenia, hypocomplementaemia, muco-cutaneous and pulmonary disease [59, 60]. A negative association has been observed with the presence of renal disease [56]. High SLE disease activity, as measured by the SLE disease activity index (SLEDAI) has also been shown to be independently associated with presence of myositis; suggesting as seen in case 3, that myositis is often present in conjunction with other manifestations of active SLE [60].
As with SSc-associated myopathy, serological studies can help predict the likelihood of myopathy in SLE, particularly anti-U1-RNP (found in up to 77% of SLE myositis cases) and raised dsDNA antibodies (as seen in Figure 3) [60]. Patients with anti-Ku antibodies with a strong SLE phenotype characterised by raised dsDNA (often associated with glomerulonephritis) appear to be less likely to develop myopathy than patients with stronger SSc clinical phenotype [48, 49]. Interestingly one study also found the presence of anti-phospholipid antibodies in 71% of SLE myositis patients [59]. However only 20% of the cohort had clinical antiphospholipid syndrome, which is similar to the prevalence in the SLE population as a whole.
Histological findings in SLE
Abnormal muscle histology is common in SLE. Studies examining muscle biopsies in SLE have identified 2 major histological patterns. A lymphocytic vasculitis is often associated with a raised ESR, arthritis and sicca symptoms (often with antibodies to Ro/La) whereas a true inflammatory myositis is typically associated with a raised creatinine kinase, proximal myopathy and U1-RNP antibodies [66]. Several studies have also described cases of necrotising myositis on biopsy [59, 60].
Other causes of myopathy in SLE
High cumulative exposure to corticosteroids was thought to be a common cause of chronic myopathy in SLE, prior to the emergence of effective steroid-sparing treatment strategies [56, 62]. Hydroxychloroquine myopathy is another potential explanation for acute myositis in the context of SLE [67]. Myopathy is thought to occur in around 1–1.2% of people treated with antimalarial therapy and the risk is considered to be higher with chloroquine than hydroxychloroquine [68, 69]. Unilateral myositis (typically lower limb) in association with fever and adjacent joint inflammation, should prompt consideration of pyomyositis in SLE, particularly in patients receiving corticosteroids [70].
Mixed Connective Tissue Disease
No clinical diagnosis better encapsulates the considerable overlap that exists across the ARDs than the disease entity known as mixed connective tissue disease (MCTD). As the name implies, MCTD is the archetypal overlap syndrome and the presence of an (often low grade) steroid-responsive inflammatory myopathy in overlap with features of other ARDs represents one of the defining characteristics of patients carrying antibodies targeting U1-RNP [71]. It was the identification of autoantibodies targeting ribonucleoproteins in patients with a clinical phenotype comprising features of SLE, SSc and polymyositis, that resulted in the term MCTD [72]. Subsequent efforts to identify and characterise the clinical phenotype of patients carrying what was subsequently characterised to be anti-U1RNP antibodies, consistently identified a high prevalence of Raynaud’s phenomenon, swollen hands, inflammatory arthralgia and myopathy [73]. Whereas these antibodies were found to occur in ~15% of patients with a clinical diagnosis of SSc (of whom around 27% have an associated myopathy) and a smaller proportion of patients with SLE (10%), they were virtually never found within cohorts of patients with a clinical diagnosis of isolated IIM, highlighting the overlap features with myopathy that characterises the presence of these antibodies [38, 73]. MCTD is particularly strongly associated with the co-existence of myositis with clinical features of SSc such as RP and of SLE such as inflammatory arthralgia and mucocutaneous lesions [71]. The aforementioned early study examining myopathy in SSc, noted the presence of anti-U1RNP antibodies in half of the patients designated as having a ‘complicated myopathy’ with muscle enzyme levels and EMG findings that more closely resembled that of IIM controls [25]. Furthermore as previously mentioned Bittencourt et al. found 77% of SLE patients with myositis has u1-RNP antibodies [59].
Conclusions
SSc, SLE and IIM are each part of a unified clinicoserological spectrum of disease (Figure 4). Consistent with the considerable overlap of clinical features across the ARDs and shared aetiopathogenic pathways, it is unsurprising that features of inflammatory myopathy are common in both SLE and SSc. The exclusion of myopathic features in classification criteria for both SSc and SLE should not dissuade clinicians from making a clinical diagnosis of either SLE or SSc, where the overriding clinical features indicate the presence of one of these disorders.
Figure 4. The clinicoserological spectrum of overlap CTD.
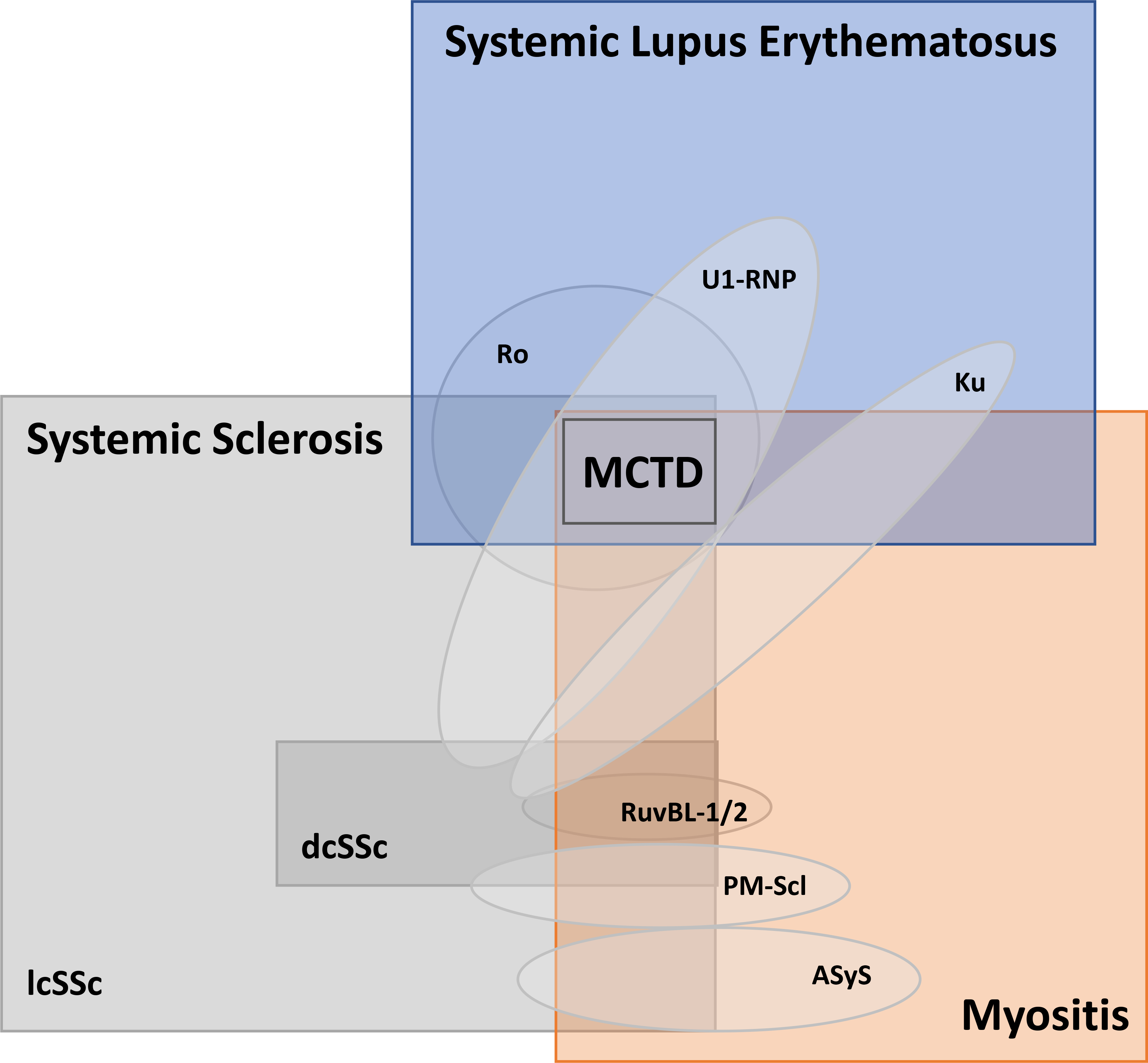
The square boxes reflect the clinical diagnosis with the oval shapes representing the common clinical characteristics (and relative frequency) of relevant antibody specificities. For example, anti-Ku antibodies, meanwhile, can be found in SSc, SLE, overlap SLE/myositis or overlap SSc/myositis. The Mixed Connective Tissue Disease (MCTD) is frequently diagnosed in all patients with anti-U1 RNP antibodies, although only a minority of patients with this antibody will truly have overlap features of SLE, SSc and myositis.
Similarly, clinicians need to be alert to the possibility of inflammatory myopathy in both SSc and SLE, and it is prudent routinely enquire about myopathic symptoms and incorporate an assessment of muscle strength during physical examination (and consider assessing muscle enzymes in routine serum biochemistry), particularly early in the disease course and during periods of increased disease activity. Open muscle biopsy should be considered in patients with elevated muscle enzymes and/or objective muscle weakness, particularly if there is doubt as to the cause (e.g. disease-related or iatrogenic) or if myopathy is refractory to immunomodulatory treatments (when alternative genetic or metabolic forms of myopathy may need to be excluded). The identification and characterisation of antibodies targeting PM-Scl, U1-RNP, Ku, RuvBL-1/2 and others has provided considerable insight into the clinicoserological relationship between myopathy and ARDs, in addition to providing a valuable prognostic marker to alert clinicians to the possibility of future muscle involvement in ARDs.
Table 1.
Summary of clinicoserological spectrum of myopathy in the context of SSc and SLE with respect to major overlap antibody specificities
| Clinical features | ||||||
|---|---|---|---|---|---|---|
| Diffuse cutaneous disease | Limited cutaneous disease | Interstitial lung disease | Inflammatory joint disease | Mucocutaneous lupus features | ||
| Antibody | PM-Scl | + | ++ | + | + | + |
| U1-RNP | + | ++ | + | ++ | ++ | |
| Ku | − | + | + | ++ | ++ | |
| RuvBL1/2 | ++ | + | + | − | -- | |
Acknowledgments
Sources of support: JJP is supported by NIAMS K23 AR073927.
Footnotes
Conflicts of Interest: None of the authors report any conflicts of interest relevant to the content of this work
References
- 1.Lépine R, Lépine (R.) Polymyosite (derinatomyosite; angiomyosite. Rev. de Med., Paris, 1901. 21: p. 426–8. [Google Scholar]
- 2.Dalakas MC and Hohlfeld R, Polymyositis and dermatomyositis. Lancet, 2003. 362(9388): p. 971–82. [DOI] [PubMed] [Google Scholar]
- 3.Casademont J, et al. , Relationship between capillary and muscle damage in dermatomyositis. Int J Dermatol, 1990. 29(2): p. 117–20. [DOI] [PubMed] [Google Scholar]
- 4.Gitiaux C, et al. , Whole microvascular unit deletions in dermatomyositis. Ann Rheum Dis, 2013. 72(3): p. 445–52. [DOI] [PubMed] [Google Scholar]
- 5.Bosello S, et al. , Characterization of inflammatory cell infiltrate of scleroderma skin: B cells and skin score progression. Arthritis Res Ther, 2018. 20(1): p. 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganczarczyk ML, Lee P, and Armstrong SK, Nailfold capillary microscopy in polymyositis and dermatomyositis. Arthritis Rheum, 1988. 31(1): p. 116–9. [DOI] [PubMed] [Google Scholar]
- 7.Paxton D and Pauling JD, Does nailfold capillaroscopy help predict future outcomes in systemic sclerosis? A systematic literature review. Semin Arthritis Rheum, 2018. 48(3): p. 482–494. [DOI] [PubMed] [Google Scholar]
- 8.Leteurtre E, et al. , [Vascular manifestations of dermatomyositis and polymyositis. Clinical, capillaroscopic and histological aspects]. Rev Med Interne, 1994. 15(12): p. 800–7. [DOI] [PubMed] [Google Scholar]
- 9.Manfredi A, et al. , Nailfold capillaroscopic changes in dermatomyositis and polymyositis. Clin Rheumatol, 2015. 34(2): p. 279–84. [DOI] [PubMed] [Google Scholar]
- 10.Kubo S, et al. , Significance of nailfold videocapillaroscopy in patients with idiopathic inflammatory myopathies. Rheumatology (Oxford), 2019. 58(1): p. 120–130. [DOI] [PubMed] [Google Scholar]
- 11.Grundtman C, et al. , Vascular endothelial growth factor is highly expressed in muscle tissue of patients with polymyositis and patients with dermatomyositis. Arthritis Rheum, 2008. 58(10): p. 3224–38. [DOI] [PubMed] [Google Scholar]
- 12.Williams TF, et al. , Secreted Threonyl-tRNA synthetase stimulates endothelial cell migration and angiogenesis. Sci Rep, 2013. 3: p. 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pauling JD, et al. , Presence of anti-eukaryotic initiation factor-2B, anti-RuvBL1/2 and anti-synthetase antibodies in patients with anti-nuclear antibody negative systemic sclerosis. Rheumatology (Oxford), 2018. 57(4): p. 712–717. [DOI] [PubMed] [Google Scholar]
- 14.Maundrell A, Proudman S, and Limaye V, Prevalence of other connective tissue diseases in idiopathic inflammatory myopathies. Rheumatol Int, 2019. 39(10): p. 1777–1781. [DOI] [PubMed] [Google Scholar]
- 15.Partovi S, et al. , Impaired skeletal muscle microcirculation in systemic sclerosis. Arthritis Res Ther, 2012. 14(5): p. R209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medsger TA Jr., et al. , Skeletal muscle involvement in progressive systemic sclerosis (scleroderma). Arthritis Rheum, 1968. 11(4): p. 554–68. [DOI] [PubMed] [Google Scholar]
- 17.Justo AC, et al. , Muscle function in women with systemic sclerosis: Association with fatigue and general physical function. Clin Biomech (Bristol, Avon), 2017. 47: p. 33–39. [DOI] [PubMed] [Google Scholar]
- 18.Ringel RA, et al. , Muscle involvement in the scleroderma syndromes. Arch Intern Med, 1990. 150(12): p. 2550–2. [PubMed] [Google Scholar]
- 19.Mimura Y, et al. , Clinical and laboratory features of scleroderma patients developing skeletal myopathy. Clin Rheumatol, 2005. 24(2): p. 99–102. [DOI] [PubMed] [Google Scholar]
- 20.Avouac J, et al. , Characteristics of joint involvement and relationships with systemic inflammation in systemic sclerosis: results from the EULAR Scleroderma Trial and Research Group (EUSTAR) database. J Rheumatol, 2010. 37(7): p. 1488–501. [DOI] [PubMed] [Google Scholar]
- 21.Muangchan C, et al. , The 15% rule in scleroderma: the frequency of severe organ complications in systemic sclerosis. A systematic review. J Rheumatol, 2013. 40(9): p. 1545–56. [DOI] [PubMed] [Google Scholar]
- 22.Jung M, et al. , Myopathy is a poor prognostic feature in systemic sclerosis: results from the Canadian Scleroderma Research Group (CSRG) cohort. Scand J Rheumatol, 2014. 43(3): p. 217–20. [DOI] [PubMed] [Google Scholar]
- 23.Clements PJ, et al. , High-dose versus low-dose D-penicillamine in early diffuse systemic sclerosis: analysis of a two-year, double-blind, randomized, controlled clinical trial. Arthritis Rheum, 1999. 42(6): p. 1194–203. [DOI] [PubMed] [Google Scholar]
- 24.Walker UA, et al. , Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Ann Rheum Dis, 2007. 66(6): p. 754–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clements PJ, et al. , Muscle disease in progressive systemic sclerosis: diagnostic and therapeutic considerations. Arthritis Rheum, 1978. 21(1): p. 62–71. [DOI] [PubMed] [Google Scholar]
- 26.Ranque B, et al. , A descriptive and prognostic study of systemic sclerosis-associated myopathies. Ann Rheum Dis, 2009. 68(9): p. 1474–7. [DOI] [PubMed] [Google Scholar]
- 27.Ranque B, et al. , Myopathies related to systemic sclerosis: a case-control study of associated clinical and immunological features. Scand J Rheumatol, 2010. 39(6): p. 498–505. [DOI] [PubMed] [Google Scholar]
- 28.Follansbee WP, Zerbe TR, and Medsger TA Jr., Cardiac and skeletal muscle disease in systemic sclerosis (scleroderma): a high risk association. Am Heart J, 1993. 125(1): p. 194–203. [DOI] [PubMed] [Google Scholar]
- 29.Quartier P, et al. , Severe cardiac involvement in children with systemic sclerosis and myositis. J Rheumatol, 2002. 29(8): p. 1767–73. [PubMed] [Google Scholar]
- 30.Jaeger VK, et al. , Functional disability and its predictors in systemic sclerosis: a study from the DeSScipher project within the EUSTAR group. Rheumatology (Oxford), 2018. 57(3): p. 441–450. [DOI] [PubMed] [Google Scholar]
- 31.Clements PJ, et al. , Correlates of the disability index of the health assessment questionnaire: a measure of functional impairment in systemic sclerosis. Arthritis Rheum, 1999. 42(11): p. 2372–80. [DOI] [PubMed] [Google Scholar]
- 32.Paik JJ, et al. , Independent Association of Severity of Muscle Weakness With Disability as Measured by the Health Assessment Questionnaire Disability Index in Scleroderma. Arthritis Care Res (Hoboken), 2016. 68(11): p. 1695–1703. [DOI] [PubMed] [Google Scholar]
- 33.Altman RD, et al. , Predictors of survival in systemic sclerosis (scleroderma). Arthritis Rheum, 1991. 34(4): p. 403–13. [DOI] [PubMed] [Google Scholar]
- 34.Toledano C, et al. , Aldolase predicts subsequent myopathy occurrence in systemic sclerosis. Arthritis Res Ther, 2012. 14(3): p. R152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paik JJ, et al. , Spectrum of Muscle Histopathologic Findings in Forty-Two Scleroderma Patients With Weakness. Arthritis Care Res (Hoboken), 2015. 67(10): p. 1416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paik JJ, et al. , Association of Fibrosing Myopathy in Systemic Sclerosis and Higher Mortality. Arthritis Care Res (Hoboken), 2017. 69(11): p. 1764–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corallo C, et al. , Histopathological findings in systemic sclerosis-related myopathy: fibrosis and microangiopathy with lack of cellular inflammation. Ther Adv Musculoskelet Dis, 2017. 9(1): p. 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steen VD, Autoantibodies in systemic sclerosis. Semin Arthritis Rheum, 2005. 35(1): p. 35–42. [DOI] [PubMed] [Google Scholar]
- 39.Wolfe JF, Adelstein E, and Sharp GC, Antinuclear antibody with distinct specificity for polymyositis. J Clin Invest, 1977. 59(1): p. 176–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reichlin M, et al. , Antibodies to a nuclear/nucleolar antigen in patients with polymyositis overlap syndromes. J Clin Immunol, 1984. 4(1): p. 40–4. [DOI] [PubMed] [Google Scholar]
- 41.De Lorenzo R, et al. , Muscular and extramuscular clinical features of patients with anti-PM/Scl autoantibodies. Neurology, 2018. 90(23): p. e2068–e2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanke K, et al. , Antibodies against PM/Scl-75 and PM/Scl-100 are independent markers for different subsets of systemic sclerosis patients. Arthritis Res Ther, 2009. 11(1): p. R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marie I, et al. , Long-term outcome of patients with polymyositis/ dermatomyositis and anti-PM-Scl antibody. Br J Dermatol, 2010. 162(2): p. 337–44. [DOI] [PubMed] [Google Scholar]
- 44.Marguerie C, et al. , The clinical and immunogenetic features of patients with autoantibodies to the nucleolar antigen PM-Scl. Medicine (Baltimore), 1992. 71(6): p. 327–36. [DOI] [PubMed] [Google Scholar]
- 45.Schnitz W, et al. , Anti-PM/Scl autoantibodies in patients without clinical polymyositis or scleroderma. J Rheumatol, 1996. 23(10): p. 1729–33. [PubMed] [Google Scholar]
- 46.Mimori T, et al. , Characterization of a high molecular weight acidic nuclear protein recognized by autoantibodies in sera from patients with polymyositis-scleroderma overlap. J Clin Invest, 1981. 68(3): p. 611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoa S, et al. , Single-specificity anti-Ku antibodies in an international cohort of 2140 systemic sclerosis subjects: clinical associations. Medicine (Baltimore), 2016. 95(35): p. e4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spielmann L, et al. , Anti-Ku syndrome with elevated CK and anti-Ku syndrome with anti-dsDNA are two distinct entities with different outcomes. Ann Rheum Dis, 2019. 78(8): p. 1101–1106. [DOI] [PubMed] [Google Scholar]
- 49.Ogawa-Momohara M, Muro Y, and Akiyama M, Overlap of systemic lupus erythematosus and myositis is rare in anti-Ku antibody-positive patients. Ann Rheum Dis, 2019. [DOI] [PubMed] [Google Scholar]
- 50.Rigolet A, et al. , Inflammatory myopathies with anti-Ku antibodies: a prognosis dependent on associated lung disease. Medicine (Baltimore), 2012. 91(2): p. 95–102. [DOI] [PubMed] [Google Scholar]
- 51.Kaji K, et al. , Autoantibodies to RuvBL1 and RuvBL2: a novel systemic sclerosis-related antibody associated with diffuse cutaneous and skeletal muscle involvement. Arthritis Care Res (Hoboken), 2014. 66(4): p. 575–84. [DOI] [PubMed] [Google Scholar]
- 52.Lund HI and Nielsen M, Penicillamine-induced dermatomyositis. A case history. Scand J Rheumatol, 1983. 12(4): p. 350–2. [DOI] [PubMed] [Google Scholar]
- 53.Leden I and Libelius R, Penicillamine-induced polymyositis. Scand J Rheumatol, 1985. 14(1): p. 90–3. [DOI] [PubMed] [Google Scholar]
- 54.Jayson MI, et al. , Penicillamine therapy in systemic sclerosis. Proc R Soc Med, 1977. 70 Suppl 3: p. 82–8. [PMC free article] [PubMed] [Google Scholar]
- 55.Piga M, et al. , Musculoskeletal manifestations as determinants of quality of life impairment in patients with systemic lupus erythematosus. Lupus, 2018. 27(2): p. 190–198. [DOI] [PubMed] [Google Scholar]
- 56.Tsokos GC, Moutsopoulos HM, and Steinberg AD, Muscle involvement in systemic lupus erythematosus. JAMA, 1981. 246(7): p. 766–8. [PubMed] [Google Scholar]
- 57.Foote RA, Kimbrough SM, and Stevens JC, Lupus myositis. Muscle Nerve, 1982. 5(1): p. 65–8. [DOI] [PubMed] [Google Scholar]
- 58.Garton MJ and Isenberg DA, Clinical features of lupus myositis versus idiopathic myositis: a review of 30 cases. Br J Rheumatol, 1997. 36(10): p. 1067–74. [DOI] [PubMed] [Google Scholar]
- 59.Bitencourt N, et al. , Inflammatory myositis in systemic lupus erythematosus. Lupus, 2020: p. 961203320918021. [DOI] [PubMed] [Google Scholar]
- 60.Liang Y, et al. , Associated Variables of Myositis in Systemic Lupus Erythematosus: A Cross-Sectional Study. Med Sci Monit, 2017. 23: p. 2543–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Record JL, Beukelman T, and Cron RQ, High prevalence of myositis in a southeastern United States pediatric systemic lupus erythematosus cohort. Pediatr Rheumatol Online J, 2011. 9: p. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isenber DA and Snaith ML, Muscle Disease in systemic lupus erythematosus: a study of its nature, frequency and cause. J Rheumatol, 1981. 8(6): p. 917–24. [PubMed] [Google Scholar]
- 63.Jenkins PO, et al. , Systemic Lupus Erythematosus Presenting as Orbital Myositis. Neuroophthalmology, 2014. 38(5): p. 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serop S, et al. , Orbital myositis secondary to systemic lupus erythematosus. Acta Ophthalmol (Copenh), 1994. 72(4): p. 520–3. [DOI] [PubMed] [Google Scholar]
- 65.Alonso-Valdivielso JL, et al. , Acquired Brown’s syndrome in a patient with systemic lupus erythematosus. Ann Rheum Dis, 1993. 52(1): p. 63–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lim KL, et al. , Muscle biopsy abnormalities in systemic lupus erythematosus: correlation with clinical and laboratory parameters. Ann Rheum Dis, 1994. 53(3): p. 178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whisnant JP, et al. , Chloroquine Neuromyopathy. Proc Staff Meet Mayo Clin, 1963. 38: p. 501–13. [PubMed] [Google Scholar]
- 68.Posada C, et al. , Chloroquine-induced myopathy. Lupus, 2011. 20(7): p. 773–4. [DOI] [PubMed] [Google Scholar]
- 69.Casado E, et al. , Antimalarial myopathy: an underdiagnosed complication? Prospective longitudinal study of 119 patients. Ann Rheum Dis, 2006. 65(3): p. 385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meesiri S, Pyomyositis in a patient with systemic lupus erythaematosus and a review of the literature. BMJ Case Rep, 2016. 2016: p. 101136/bcr-2016-214809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jonsson J and Norberg R, Symptomatology and diagnosis in connective tissue disease. II. Evaluations and follow-up examinations in consequence of a speckled antinuclear immunofluorescence pattern. Scand J Rheumatol, 1978. 7(4): p. 229–36. [DOI] [PubMed] [Google Scholar]
- 72.Sharp GC, et al. , Mixed connective tissue disease--an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med, 1972. 52(2): p. 148–59. [DOI] [PubMed] [Google Scholar]
- 73.Cohen ML, et al. , Clinical significance of antibodies to ribonucleoprotein. Ann Rheum Dis, 1979. 38(1): p. 74–8. [DOI] [PMC free article] [PubMed] [Google Scholar]