

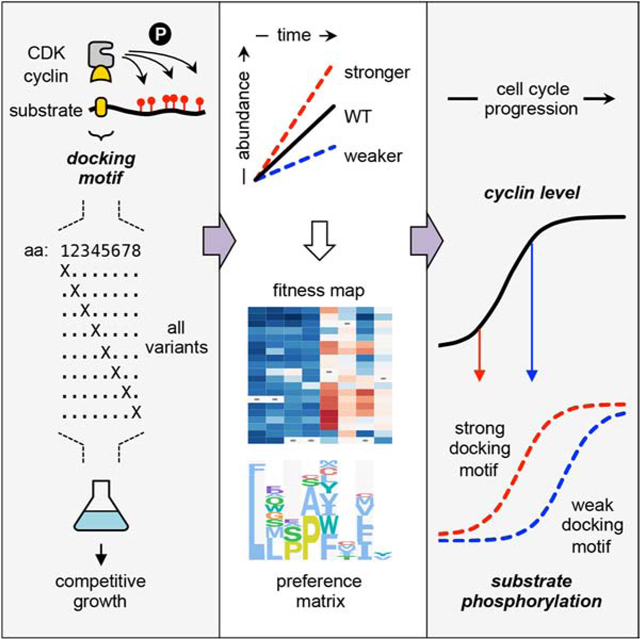
SUMMARY
Many protein-modifying enzymes recognize their substrates via docking motifs, but the range of functionally permissible motif sequences is often poorly defined. During eukaryotic cell division, cyclin-specific docking motifs help cyclin-dependent kinases (CDKs) phosphorylate different substrates at different stages, thus enforcing a temporally-ordered series of events. In budding yeast, CDK substrates with Leu/Pro-rich (LP) docking motifs are recognized by Cln1/2 cyclins in late G1 phase, yet the key sequence features of these motifs were unknown. Here we comprehensively analyzed LP motif requirements in vivo by combining a competitive growth assay with deep mutational scanning. We quantified the impact of all single-residue replacements in five different LP motifs, using six distinct G1 cyclins from diverse fungi including medical and agricultural pathogens. The results uncover substantial tolerance for deviations from the consensus sequence, plus requirements at some positions that are contingent on the favorability of other motif residues. They also reveal the basis for variations in functional potency among wild-type motifs, and allow derivation of a quantitative matrix that predicts the strength of other candidate motif sequences. Finally, we find that variation in docking motif potency can advance or delay the time at which CDK substrate phosphorylation occurs, and thereby control the temporal ordering of cell cycle regulation. The overall results provide a general method for surveying viable docking motif sequences and quantifying their potency in vivo, and they reveal how variations in docking strength can tune the degree and timing of regulatory modifications.
eTOC Blurb
CDKs use cyclin-mediated docking interactions to recognize substrates, but the docking motifs lack a quantitative description of how sequence variations affect recognition. Bandyopadhyay et al combine deep mutational scanning with an in vivo competitive growth assay to quantify the functional strength of motif variants and define recognition rules.
Graphical Abstract
INTRODUCTION
Protein function can be controlled by numerous post-translational modifications. Enzymes that catalyze these modifications must choose their substrates from among an excess of potential candidates in the proteome. A prevalent example is protein phosphorylation, which in eukaryotic cells is governed by hundreds of protein kinases. For these kinases to perform distinct functions, they cannot all phosphorylate the same substrates. Instead, several mechanisms allow them to be selective [1, 2]. These include the recognition of preferred phospho-acceptor residues and their surrounding sequence context by the enzyme’s catalytic site, plus the physical association and/or co-localization of the kinase with its substrates. Among the latter type of mechanism, several kinases (or their partner proteins) recognize “docking motifs” in their substrates, which are short linear motifs (SLiMs) that are separate from the phosphorylated residues, often in disordered regions [1–3]. Because such motifs are recognized by only a subset of kinases, they enhance the specificity of phosphorylation. Similar mechanisms regulate catalysis by phosphatases [4–6] and other protein modifications such as ubiquitination [7]. Although these general concepts are well established, for many docking motifs there is a lack of comprehensive information about the range of possible sequences that can be recognized and distinguished from the thousands of other competing peptide sequences in vivo. Hence, there remains a pressing need for defining the molecular rules that govern which substrates will be engaged by a given enzyme and how efficiently they will be modified.
Cyclin-dependent kinases (CDKs) are the central regulators of eukaryotic cell division [8]. All eukaryotes have multiple cyclin-CDK complexes that function at different stages of division. In the budding yeast Saccharomyces cerevisiae, a single CDK enzyme (Cdk1) associates with multiple different cyclins, which impart distinct properties to the enzyme complex as the cell cycle proceeds [9, 10]. Several of these cyclins recognize unique docking motifs that promote substrate phosphorylation in a cyclin-specific manner [11–17]. Early in the yeast cell cycle, a docking sequence called an “LP” motif (for its enrichment in Leu and Pro residues) is recognized specifically by late-G1 cyclins (Cln1 and Cln2), and not by the early-G1 cyclin (Cln3) or S- and M-phase cyclins (Clb1-Clb6). These LP docking interactions increase the efficiency, specificity, and multiplicity of substrate phosphorylation [11, 12, 18]. Nevertheless, fundamental issues remain poorly understood, including the key sequence features of LP motifs, their prevalence in the proteome, and the degree of conservation of LP docking among distinct organisms and cyclin subtypes.
Functional LP motifs likely exist in multiple Cln2-CDK substrates, as their phosphorylation in vitro can be inhibited by competing LP motif peptides or by Cln2 mutations that disrupt LP motif recognition [12, 18]. Yet for only three proteins has it been shown that their phosphorylation depends on a specific LP motif: Sic1 (a CDK inhibitor), Ste5 (a MAPK scaffold protein), and Ste20 (a PAK family kinase) [11, 12, 18]. While these share a minimal LxxP sequence, the key features of LP motifs remain undefined, which hinders their identification in other substrates and obscures insight into cyclin specificity. Moreover, in addition to Cln3 and Cln1/2 cyclin types, some budding yeasts contain a third type of G1 cyclin, Ccn1, which also recognizes LP motifs [18]. In contrast, most non-yeast fungi have only a single G1 cyclin (hereafter, “Cln”), which diverged prior to the expansion of the budding yeast genes into three types [19]. Thus, it is unclear if LP docking is an ancient function, present throughout fungi, or if it evolved more recently only in yeasts.
In this study we used the example of LP docking by G1 cyclins as a model to elucidate the functional consequences of SLiM sequence variation in vivo. In particular, we describe a comprehensive analysis of LP motifs, involving systematic mutational scanning and an in vivo competitive growth assay, which quantified the functional impact of all substitutions throughout the linear sequence. These efforts revealed the key features that control LP motif recognition, and how sequence variations modulate the strength and timing of CDK regulation. Similar variations in SLiM potency are likely to tune substrate recognition by other protein modifying enzymes.
RESULTS
LP motif recognition is broadly conserved among fungal G1 cyclins.
Before analyzing the key features of LP motifs, we first investigated the extent to which their recognition was conserved among fungal G1 cyclins, so that we could eventually compare sequence preferences for several distinct cyclin subtypes. Previously, we found that LP motifs are recognized by two types of G1 cyclins from budding yeasts, Cln1/2 and Ccn1, but not by a third type, Cln3 [18]. Unlike budding yeasts, most other fungi have only one type of G1 cyclin (Figure 1A) [19]. Thus, we tested if they can recognize LP motifs, using our prior in vivo phosphorylation assay. The foreign cyclins were expressed from a galactose-inducible promoter (PGAL1) in S. cerevisiae, with their N-termini fused to GST plus one half of a leucine zipper sequence; this latter feature allows the cyclins to engage a control substrate (with the partner half zipper), so that their function can be measured independent of LP docking [18, 20]. We chose representative cyclins from major subdivisions of Ascomycota and Basidiomycota (Figure 1A–B), the two most species-rich fungal phyla. Each was tested for the ability to drive phosphorylation of a substrate with no docking sequence, with an LP motif, or with a half leucine zipper (Figure 1B). As seen previously [18, 20], of two S. cerevisiae cyclins, only Cln2 and not Cln3 promoted phosphorylation of the substrate with the LP motif. Remarkably, most other cyclins behaved like Cln2, in that they could use the LP motif to promote substrate phosphorylation. The sole exception was Puc1 from the fission yeast S. pombe, but it also showed weak expression and functioned poorly with the leucine zipper substrate. In contrast, Puc1 from another fission yeast, S. japonicus, was able to drive phosphorylation of the LP motif substrate, and hence the S. pombe cyclin was an outlier.
Figure 1. LP docking conservation among fungal G1 cyclins.

(A) Phylogeny of representative fungal genera and G1 cyclin types found in each subdivision.
(B) Fungal G1 cyclins were expressed in S. cerevisiae as fusions to GST plus a half leucine zipper (GST-[lz]), along with HA-tagged substrates with no docking site, or an LP docking motif, or the partner half leucine zipper. Reduced mobility indicates phosphorylation.
(C) Top, Cln1/2-CDK inhibits pheromone-induced MAPK activation and G1 arrest. Bottom, Ste20Ste5PM chimeras contain a plasma membrane-binding domain (PM) and flanking CDK sites from Ste5 [11]; a functional LP docking motif allows these sites to be phosphorylated, which inhibits membrane binding by the PM domain and thus blocks signaling. Chimeras are tested in STE5–8A strains to prevent the CDK from inhibiting the native Ste5 protein [11, 22].
(D) PGAL1-cyclin strains harboring Ste20Ste5PM chimeras with either a functional LP motif (LP_Ste5) or a control sequence (nonLP) were treated with pheromone and tested for phosphorylation of MAPKs (Fus3 and Kss1). Below, quantified results (mean ± SEM, n = 4).
(E) Phylogeny of fungal G1 cyclins [19] compared with LP docking abilities (check marks). Colored boxes denote three groups analyzed in panel F.
(F)Surface residue conservation for three cyclin groups, overlaid onto a model for the Cln2-CDK complex (see Methods). The CDK is grey. Cyclin coloring reflects conservation: red, 100% identical; white, 90% identical; blue, 0% identical. The patch of strongest conservation includes residues previously implicated in LP docking [18]. Also see Figure S1.
The conservation of LP motif recognition was confirmed via a separate assay that is based on the ability of Cln2 to inhibit signaling in the yeast pheromone response pathway [21, 22] (Figure 1C, top). The ordinary target of this inhibition is the scaffold protein Ste5 [22], but the regulatory effects can be transferred to a chimeric signaling protein, Ste20Ste5PM (Figure 1C, bottom; [11]). The localization and signaling activity of this Ste20Ste5PM chimera can be inhibited by CDK phosphorylation if it contains a functional LP motif [11], making it a useful context for assaying motifs and their recognition by cyclins. Using this assay, we found that all of the distantly related G1 cyclins could inhibit activation of Fus3, the main pheromone pathway MAPK (Figure 1D). Inhibition was weaker for some cyclins (e.g., from A. nidulans or S. japonicus), but all showed dependence on a functional LP motif. (The N. crassa cyclin was partly inhibitory even without an LP motif, implying a non-specific effect that we did not pursue.) Collectively, these results indicate that LP docking is an ancient property of fungal G1 cyclins, but this function was lost by Cln3 cyclins after they diverged from the Cln1/2 group (Figure 1E). Indeed, a patch of surface residues in the cyclin, near those previously implicated in LP motif recognition [18], is more markedly conserved within groups that recognize LP motifs than within the Cln3 group (Figures 1F, S1).
Extended LP motif sequences impart variation in cyclin recognition.
The sequence features defining a functional LP motif are poorly characterized. Of three motifs known to function in the context of the native protein, those from Sic1 and Ste5 share a 4-residue sequence, LLPP (Figure 2A); however, the key residues in this stretch and the role of flanking sequence were unknown, and they only partly resemble the sequence in Ste20 (LDDP). To begin defining required features, we mutated the Ste5 motif in blocks of four residues around the LLPP sequence (Figure 2B). Mutations on the N-terminal side had no effect, whereas mutation of the LLPP or the next four C-terminal residues disrupted recognition by both Cln1/2 and Ccn1, reminiscent of prior results with the Ste20 motif [11]. Further evidence implicated these C-terminal residues in differential recognition by distinct cyclins. Namely, despite a shared LLPP sequence, the Ste5 motif was recognized by both Cln1/2 and Ccn1, whereas the Sic1 motif was recognized only by Cln1/2 (Figure 2C). These behaviors could imply that Cln1/2 and Ccn1 have distinct preferences in the C-terminal part of the motif, or that Ccn1 is inherently more dependent on that region. In either case, they suggest a basis by which substrates might be engaged to different extents by different cyclins. Indeed, in binding assays, Ccn1 co-precipitated with only a subset of Cln2 partners (Figure S2). Therefore, binding sites for both Cln1/2 and Ccn1 extend beyond the core LxxP motif, and the two groups are differentially sensitive to this flanking sequence.
Figure 2. Extended LP motif sequences impart variation in cyclin recognition.

(A) LP motif sequences used throughout this study.
(B) Four blocks of 4 residues in the Ste5 LP motif region were mutated (to AAAA) and tested for effects on substrate phosphorylation (as in Figure 1B) induced by Cln2 or Ccn1 cyclins.
(C) Cln1/2 and Ccn1 cyclins were tested for phosphorylation of substrates with LP motifs from Ste5 or Sic1. Also see Figure S2.
(D) PGAL1-cyclin strains harboring Ste20Ste5PM chimeras with different LP motifs were tested for pheromone-induced MAPK phosphorylation. Below, quantified results (mean ± SEM, n = 4).
To study additional motifs, we used the pheromone response inhibition assay (as in Figure 1C–D) to compare Ste20Ste5PM chimeras with six different docking sequences. These included a nonfunctional control (nonLP), plus LP motifs from Ste5, Sic1, and Ste20, and an LLPP-containing motif from another Cln1/2-regulated substrate, Whi5 (Figure 2A). A final LP sequence was from Lam5, which we identified in a recent (unpublished) screen for proteins that bind wild-type Cln2 but not a docking-defective mutant; the responsible docking motif in Lam5 was of interest here because it contains further variations in both the LxxP and adjacent sequences (Figure 2A). All five LP motifs allowed Cln2 to inhibit signaling, although inhibition was noticeably weaker with the Sic1 motif (Figure 2D). With Ccn1, three motifs allowed strong inhibition (Ste5, Ste20, and Lam5), whereas the Whi5 motif was less potent and the Sic1 motif was nearly indistinguishable from the control. Thus, differences in both motif and cyclin can contribute to variation in the strength of CDK regulation.
A competitive growth assay for docking motif potency.
We wished to systematically interrogate the features of LP motifs that control their recognition and potency. To facilitate this, we designed an in vivo competitive growth assay in which LP motifs provide a selective advantage (Figure 3A). Namely, pheromone signaling ordinarily causes arrest of cell division (and loss of cyclin expression), but this arrest can be blocked by ectopic expression of cyclins that inhibit pheromone signaling. Hence, this inhibitory activity can provide a growth advantage in pheromone-treated cultures. Moreover, because this inhibition depends on the cyclin-LP interaction, strong LP motifs should be more advantageous than weak motifs. Thus, using the Ste20Ste5PM chimeras, we compared the relative enrichment or depletion of the five wild-type LP motifs described earlier (Figure 3B). A mixed population of cells harboring these constructs was treated first with galactose (to induce PGAL1-cyclin expression) and then with pheromone for various times. The population frequency of each motif was measured via deep sequencing of plasmid DNAs recovered at each time point.
Figure 3. Competitive growth assay for the functional potency of cyclins and LP motifs.

(A) Schematic of the competitive growth assay, using Ste20Ste5PM chimeras with a mixture of either wild-type (WT) LP motifs or libraries of randomized codon mutations.
(B)Sequences of five LP motifs and the control (nonLP) motif; residues are colored where ≥ 50% are identical (blue) or similar (yellow).
(C) Examples of competitive growth results showing changes in frequency of LP motifs after pheromone treatment, in cells ± PGAL1-ScCLN2. Plots show averages of two independent experiments. Also see Figure S3A.
(D) Experiments in panel C were performed in eight PGAL1-cyclin strains and the control strain (none). Bars show the ratio (log2) of the change in frequency for each motif compared to the nonLP motif (at 44 hr), normalized to the control strain; values show averages of two independent experiments.
(E) Example showing changes in frequencies of Ste5 LP motif variants (in PGAL1-ScCLN2 cells). The dashed black line is WT. Green and red show all WT synonyms and termination codons, respectively. Orange and blue show examples of missense synonyms with mild (Pro3 to Ser) or strong (Leu1 to Ser) defects; all six Ser codons are shown to illustrate that phenotypes are similar regardless of codon sequence.
(F) Fitness score distributions for all nucleotide variants of two LP motifs (Ste5 and Sic1) in two PGAL1-cyclin strains. Green, WT synonyms; Red, terminators; grey, missense mutations. Also see Figure S3B.
(G) Correlation of fitness scores for all amino acid variants between two independent replicate experiments. Also see Figure S3C.
Without a PGAL1-cyclin construct, none of the LP motifs had a selective advantage, but in strains expressing PGAL1-ScCLN2, they showed clear differences in enrichment and depletion (Figure 3C). The most potent were those from Ste5 and Lam5, which became enriched over the control motif by roughly 7-fold after 20 hours and 50-fold after 44 hours. The next most potent was the motif from Ste20, followed by those from Whi5 and Sic1. A similar rank order was observed with G1 cyclins from seven other fungi (Figure 3D), although those more distant from ScCLN2 were less able to utilize the weaker motifs. These results reveal a range of functional potencies among different LP motifs that can lead to quantifiable growth differences, with a hierarchy of preference that is conserved among divergent G1 cyclins. Strikingly, the strongest and weakest motifs contain the same core LLPP sequence, and hence the flanking residues must make important contributions to potency.
Comprehensive analysis of LP motif sequence requirements.
We exploited the competitive growth assay to analyze the determinants of LP motif potency in a comprehensive manner. First, the wild-type motifs in the Ste20Ste5PM chimeras were replaced with sequences in which individual codons were fully randomized (i.e., to all 64 codons), thus creating libraries with all possible substitutions at each amino acid position (Figure 3A–ii). This was done for 8 consecutive codons in each motif. Then, the libraries were transformed into PGAL1-cyclin strains and analyzed by the competitive growth assay. Changes in population frequency for all mutants were analyzed using software [23] that calculates fitness scores based on their rate of depletion (negative scores) or enrichment (positive scores) relative to the wild-type motif (defined as zero).
All five LP motif libraries were analyzed multiple times in strains expressing ScCLN2 and KwCCN1, plus in additional trials with four other cyclins. Several general properties indicated that the behaviors of mutant variants were logical and reproducible: (a) Wild-type synonyms mimicked the original wild-type sequence (Figure 3E), with scores clustered around zero (Figures 3F, S3B), confirming that fitness levels reflected amino acid sequence irrespective of nucleotide sequence. Similarly, changes in fitness due to missense mutations were consistent regardless of the specific codon (Figure 3E). (b) Termination codons were the most favorable, yielding the highest scores (Figure 3E–F, S3B), which is expected because they eliminate the protein required for the growth arrest signal. (c) Individual variants showed reproducible behavior in independent experiments (Figures 3G, S3C). (d) The largest negative scores were observed with the strongest motifs (e.g., Ste5, Lam5; Figure S3B–C), because they are furthest from non-functional (and so have the “most to lose”). By comparison, negative scores were smaller for weak motifs (e.g., Sic1, Whi5; Figure S3B–C), especially when combined with cyclins that poorly recognize the wild-type sequence (e.g., KwCCN1 combined with the Sic1 motif).
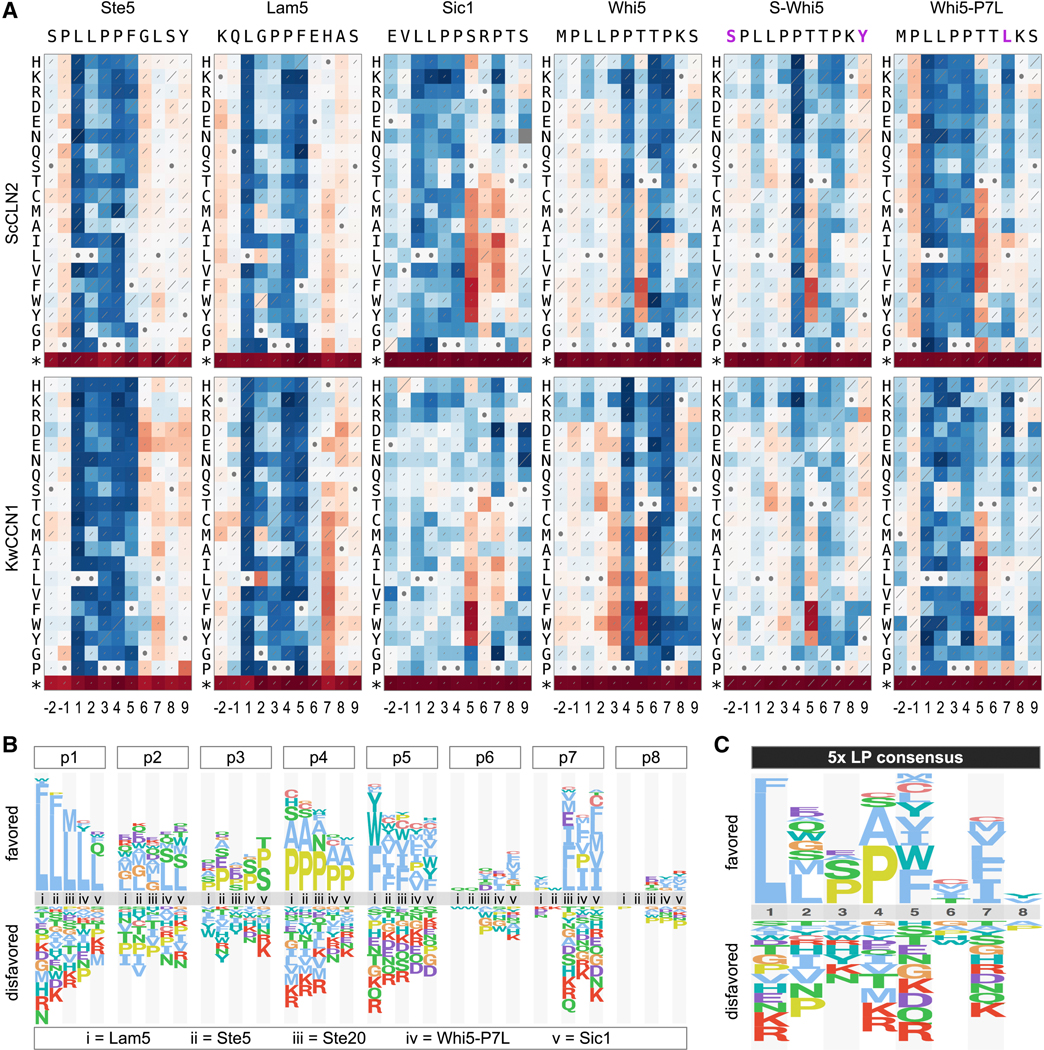
Key features of each motif were revealed by plotting results as heat maps and sequence logos (Figures 4A–B). The Whi5 motif was unusual, as will be discussed later. For the other four motifs, several notable features emerged. (i.) Consistent with the invariant LxxP signature in all known motifs, there were strong preferences for Leu and Pro at positions p1 and p4, although Ala was tolerated well at p4, and even some mutations at p1 (Phe or Met) were not always fully inactivating. (ii.) More substitutions were tolerated in the intervening positions, p2 and p3, with a clear hierarchy among the p2 residues found in wild-type motifs (i.e., Leu > Gly > Asp). (iii.) Despite a preference for Leu at p2, chemically similar residues such as Ile and Val were more disruptive than many dissimilar residues, such as polar or charged groups. (iv.) At p5 and p7 there were broader preferences for non-polar side chains, with Phe usually the most favored at p5. Notably, the preference at p7 was relatively mild with the strong motifs (Ste5, Lam5) and stronger with the weaker motifs (Ste20, Sic1). (v.) Positions p6 and p8 showed minimal selectivity, although a mild bias for polar groups at p6 was evident with the Ste5 motif (Figures 4A, S4). (vi.) With the Sic1 motif, mutations at p1 to p4 caused reduced fitness with ScCLN2 but not with other cyclins (Figures 4A, S4), reflecting the fact that the wild-type motif is already nearly non-functional for these other cyclins. Conversely, the Sic1 motif was strengthened (and hence recognized by all cyclins) by non-polar substitutions at p5 or p7, indicating that unfavorable residues at these positions explain its weakness.
Figure 4. Comprehensive analysis of LP motif sequence preferences.

(A) Fitness effects of all single-residue substitutions in five motifs. Red and blue indicate better and worse performance, respectively, relative to the wild-type motif; each panel is scaled to its own maximum and minimum. Diagonal lines depict standard errors, scaled such that the highest value covers the entire diagonal. Circles denote wild-type residues. Data represent three independent experiments. See Figure S4 for analyses with four other cyclins.
(B) Sequence logos showing relative preferences of ScCLN2 along motifs in panel A.
(C) Number of inactivating mutations (defined as normalized scores within 15% of minimum; see Methods) at each position in 4 “typical” motifs (i.e., excluding Whi5). Also see Figure S5.
(D) Pearson correlation coefficients (r) for all pair-wise comparisons of cyclins, calculated from the raw fitness score matrices for all 5 motifs.
Calculating the number of inactivating mutations (Figures 4C, S5A) showed that p1 and p4 were the least tolerant of substitutions, followed by p5; the results at p2, p3, and p7 were more context-dependent, with p7 being less tolerant in the weaker motifs (Ste20, Sic1). A similar hierarchy was evident in measurements of functional inequality (Figure S5B), in which positions ranged from uniquely selective (p1, p4), to broadly selective (p2, p3, p5, p7) to nonselective (p6, p8). Importantly, the requirements were highly similar for all other cyclins (Figure S4), and we found no qualitative distinctions. Instead, sequence preferences were strongly correlated for all pair-wise comparisons of cyclins (Figure 4D), and in all cases the most favored residues fit the sequence LLPPΦxΦ (where Φ is hydrophobic).
Atypical behavior of the Whi5 sequence reveals overlapping motifs.
Unlike other motifs, the Whi5 motif was not strongly impaired by mutations at p1 to p3 (Figure 4A). Moreover, it showed an extreme bias for threonine at p6, a position that was not selective in other motifs. We considered two possible explanations for this atypical behavior. First, different flanking sequences might alter the reliance on positions within the 8-residue LP motif. Because in most libraries the motifs were inserted as 11-residue segments with otherwise identical surrounding sequence (see Methods), any such context effects would be limited to two preceding residues (positions p-2 and p-1) and one following residue (p9). Second, the required threonine at p6 is part of a TP dipeptide, a potential phosphorylation site. The resulting phospho-Thr moiety might improve cyclin binding, or it might be recognized by the Cks1 subunit of the cyclin-CDK complex [24, 25]; an overlapping Cks1 binding site could explain reduced reliance on the p1-p3 residues usually required for cyclin binding.
To address these possibilities, we extended the systematic analysis of four motifs to include all 11 unique residues, and in parallel we analyzed two variant forms of the Whi5 sequence (Figure 5A). In one variant, S-Whi5, we altered residues flanking the 8-residue Whi5 motif to match those flanking the Ste5 motif, to test their influence. In the other, called Whi5-P7L, we made a Pro to Leu substitution at p7, to simultaneously disrupt the potential TP phosphorylation site and match the preference seen in other motifs. For all motifs, mutations at flanking positions (p-2, p-1, and p9) had minimal effects (Figure 5A). Furthermore, the altered flanking sequence in the S-Whi5 motif had no notable impact, as it retained the atypical behavior of the wild-type Whi5 motif. In contrast, the Whi5-P7L motif displayed radically altered behavior (Figure 5A): it showed increased dependence on p1-p3 and strongly reduced selectivity at p6. Thus, when the potential TP phosphorylation site was disrupted, the sequence reverted to typical LP motif preferences. These results support the notion that the Whi5 sequence contains an overlapping Cks1 site, which can promote CDK regulation of the substrate even when mutations (i.e., at p1 to p3) disrupt cyclin recognition. In this light it is noteworthy that the wild-type Whi5 motif showed a strong bias for proline at p4 (Figures 4A, 5A), which could be required by Cks1 or by the kinase that phosphorylates the TP site (either the CDK or the pheromone-activated MAPK; see Discussion).
Figure 5. Analysis of atypical and consensus motif preferences.
(A) Heat maps (as in Figure 4A) from mutational scanning of 11-residue sequences for four wild-type motifs plus two variants of the Whi5 motif (S-Whi5 and Whi5-P7L; magenta letters indicate sequence changes). Data represent two independent experiments.
(B) Comparison of individual position preferences in five “typical” motifs.
(C) Sequence logo showing the consensus preferences of the motifs in panel B.
A quantitative LP motif matrix predicts the potency of other candidate sequences.
To distill results from multiple motifs into consensus preferences, we generated sequence logos that compared the biases at each position from five motifs with typical behavior (Figure 5B). These plots highlighted the consistently strong selectivity at p1 (Leu), p4 (Pro or Ala), and p5 (non-polar), along with the context-dependent selectivity for non-polar residues at p7 evident in weaker motifs. These quantitative preference scores constitute position-specific scoring matrices (PSSMs; see Methods) from each motif, which we averaged to generate a “consensus” PSSM (Figures 5C, S6A). We then used this consensus PSSM to assess which amino acid properties best explain the preferences at individual positions, by testing for correlations to a prior set of 28 physicochemical descriptors [26]. The strongest correlations were seen at p2, p5, and p7 (Figure S6B). Positions p5 and p7 correlated best with the descriptor of “hydrophobicity”, in accord with their noted preference for nonpolar residues. Position p2 correlated best with a complex descriptor (“steric hindrance / P[helix]”) that combines a preference for two properties – low steric hindrance and high α-helical propensity. This fits with the observation that, among chemically similar residues, proximally-branched side chains were disfavored (e.g., Leu > Ile/Val, Gln > Asn, Ser > Thr). The poorest correlations were at the most selective positions, p1 and p4, presumably reflecting a stringent requirement for a unique side-chain property. To assess whether these results depend on the specific method of generating the PSSM, we derived an alternative PSSM using distinct mathematical operations and found that the pattern of physicochemical correlations was similar (Figures S6B–D).
The preference for Pro residues at p3 and p4 raised the possibility that the LP motif might adopt a polyproline II helical structure. Because such helices show two-fold pseudosymmetry, peptide ligands with this conformation can often present the key side chains equally well in N-to-C or C-to-N orientation [27]. Thus, we compared four LP motifs in both forward and reverse orientation (Figure S6E), and in all cases the forward orientation was strongly preferred. This finding helps constrain searches for candidate LP motifs in other proteins to a single orientation.
To further define permissible LP motifs, we tested additional candidates from proteins phosphorylated by Cln2-CDK [12, 18, 24, 28] or found to co-precipitate with Cln2 in a separate (unpublished) project. Within each protein, we located all LxxP motifs in predicted disordered regions, and selected 14 examples with varying degrees of matches to the favored residues at other positions. When tested by the competitive growth assay, they showed a broad range of potency, from stronger than Ste5 to indistinguishable from the nonLP control (Figure 6A), and their rank order was largely similar among different cyclins (Figure 6B). We then used the consensus PSSMs to calculate a predicted score for each motif, which correlated well with the observed results (Figures 6C, S6F). Because all motifs contained a core LxxP sequence, their variation in potency must be due to the other positions. Thus, a consensus PSSM derived from the systematic analyses can effectively predict the functional potency of candidate motifs.
Figure 6. Observed and predicted potencies of new motifs.

(A) Frequency changes during competitive growth of 14 candidate LP motifs, plus Ste5 and nonLP controls, in cells with PGAL1-ScCLN2 or no cyclin. Plots show averages of two independent experiments.
(B) Panel A experiments were performed with eight PGAL1-cyclin strains. Heat map colors indicate the ratio (log2) of the change in frequency for each motif compared to the nonLP control (at 20 hr), normalized to the results in the no-cyclin strain; two independent experiments were averaged. Motif sequences are colored where >50% are identical (red) or similar (blue).
(C) Observed vs. predicted potency of new motifs (excluding Ste5 and nonLP controls). Observed potency is the log2 ratio value (as in panel B) for the ScCLN2 strain, at 20 or 44 hr. Predicted scores were derived from the preference score consensus PSSM (see Figures 5C and S6A) by calculating the sum of PSSM values for the residues at p1-p8 of each motif.
Also see Data S1.
Finally, we used an in silico method [29] to search the yeast proteome for matches to the consensus PSSMs within predicted disordered regions. Dozens of sequences scored at least as strongly as the Ste20 motif, and hundreds scored at least strongly as the Sic1 motif (see Data S1), raising the possibility that a vast network of functionally consequential LP motifs are recognized by Cln1/2-CDK in vivo.
Disfavored residues in the Sic1 motif tune the timing of its CDK-mediated degradation.
The LP motif from Sic1 showed weak potency with all cyclins, presumably because, despite its ideal LLPP sequence, it lacks favored residues at p5 and p7. Indeed, replacement of either position with hydrophobic residues strengthened the motif (see Figures 4A, 5A). Curiously, the absence of favored residues at p5 and p7 is generally conserved among Sic1 orthologs (Figure 7A), raising the possibility that a weak LP motif in Sic1 is beneficial. In the native protein, this motif initiates a cascade of CDK phosphorylation reactions that ultimately trigger ubiquitin-mediated degradation of Sic1 and entry into S phase [30, 31]. Thus, we hypothesized that the submaximal potency of this motif helps set the timing of Sic1 degradation.
Figure 7. A weak Sic1 motif delays its CDK-mediated degradation.

(A) Sic1 LP sequences from ten budding yeasts; residues are colored where ≥ 50% are identical or similar. Preferred residues are at top; arrows emphasize the conserved absence of favorites at p5 and p7.
(B) Fluorescence intensities of Sic1-GFP variants, relative to the time when nuclear exit of Whi5-mCherry is 50% complete. Dark lines, mean; shaded bands, SEM. The “llpp” mutant substitutes VLLPP with AAAAA.
(C) Degradation time and duration (see Methods) for Sic1-GFP variants. Circles, individual cells; lines, mean and 95% CI.
To test this view, we used a fluorescent reporter construct to monitor Sic1 degradation by live cell microscopy [14] (Figure 7B). Here, GFP is fused to a Sic1 N-terminal fragment (residues 1–215) that includes multiple CDK sites and the LP motif (other docking sites for later S-phase cyclins were removed). We changed the p5 and p7 positions of the LP motif to Phe and Ile, respectively, which yielded the strongest enhancements in the competitive growth assays. In native Sic1, these mutations correspond to S141F and P143I. Remarkably, each single mutation substantially accelerated the timing of Sic1-GFP degradation (Figure 7B–C), to a degree that was even greater than the difference between the wild-type motif and a defective motif (llpp). The effect was slightly stronger for S141F than for P143I (Figure 7B–C), which agrees with their relative strengths in the competitive growth assay, and the double mutation caused no further effect. The advanced timing caused the majority of Sic1-GFP to be degraded within 10 minutes after the cell cycle commitment point, or “Start” (Figure 7B); this is comparable to the earliest timing observed previously by adding an optimized di-phosphodegron [14]. Notably, this timing precedes the degradation of full-length, wild-type Sic1 and the consequent release of Clb-CDK activity [14, 18, 30, 31], and hence it is presumably driven solely by Cln1/2-CDK. The mutations only affected the time when degradation was initiated, and not the duration of time needed to complete degradation (Figure 7C). We conclude that the disfavored residues in the Sic1 LP motif provide a beneficial tuning effect that delays the initiation of Sic1 degradation by Cln1/2-CDK until a time closer to the appearance of Clb-CDK activity, thus permitting the rapid two-stage phosphorylation relay documented for Sic1 [30, 31].
DISCUSSION
This study provides new insights into mechanisms of substrate selection by cyclin-CDK complexes and the recognition of LP docking motifs by G1 cyclins. By combining systematic mutagenesis and in vivo competition assays, we uncovered the key sequence features that dictate LP motif function and potency. The maximally preferred features define a 7-residue consensus motif, LLPPΦxΦ, yet we found substantial tolerance for deviation from this consensus, as well as complex contingency relationships between different positions in the motif. Moreover, varying degrees of plasticity at several key positions allow motifs to vary in functional potency, which provide a mechanism for tuning the sensitivity to CDK activity and the cell cycle timing of substrate phosphorylation. These findings carry general implications for the regulatory functions of docking motifs, and the method has potential to be extended broadly to other SLiM-based interactions.
Our systematic method of analyzing motif requirements is analogous to in vitro approaches such as peptide spot arrays [32, 33], phage display [34, 35], or spectrally coded beads [36]. A beneficial aspect of our approach is that it operates under conditions present in living cells, including numerous competing interactions, and hence it intrinsically highlights variations in binding strength that are functionally consequential in vivo. Furthermore, because it monitors enrichment or depletion of all sequences in the population, the quality of data is similar for both functional and nonfunctional motifs, in contrast to capture-based approaches (such as phage display) that provide only indirect inferences about non-binding sequences. Thus, the approach offers a valuable complement to in vitro methods, and one that is inexpensive and technically straightforward. It should be readily generalizable to other domain-SLiM interactions, either in their native contexts or as substitutes for the cyclin-motif interaction in our synthetic system.
Our comprehensive analyses defined the core requirements common to all motifs and cyclins, while also uncovering context-dependent features. In particular, the preference for a hydrophobic residue at p7 was partly masked in strong motifs but highly pronounced in weaker motifs. Similar contingency relationships have been observed for other SLiMs. For example, the phosphatase PP2A B56 subunit can recognize an LxxIxE motif if there is a favored residue at either one of two key positions (p1-Leu or p4-Ile) [4], and APC/C degrons can deviate from the classical consensus requirements when the non-consensus positions are optimal [7]. In all of these cases, positions that are highly flexible in one peptide context can be highly constrained in another. Collectively, they emphasize that SLiM consensus sequences, while a useful shorthand, are not adequate to describe the complexity of functional motif space. Instead, a transition toward quantitative matrices of motif preferences will be required for system-level understanding of docking-based networks. Comprehensive methods to analyze specificity determinants will be essential for achieving these goals.
We found that cyclin docking strength can tune the magnitude and timing of CDK regulation, which is likely applicable to other eukaryotic cyclins. In particular, strengthening the LP motif in Sic1 accelerated its degradation. In G1, Sic1 retards S-phase entry by blocking Clb-CDK activity until the S-phase cyclin Clb5 has accumulated and Cln1/2-CDK activity has peaked [37, 38]; at this stage, multi-site phosphorylation of Sic1 is initiated by Cln1/2-CDK and then is accelerated by the resulting release of Clb5-CDK activity, leading to Sic1 degradation and G1/S transition [30, 31, 39, 40]. Our findings suggest that a weak Sic1 LP motif helps delay the ability of Cln1/2 to initiate these events until Clb5-CDK complexes accumulate and are poised for release from Sic1 inhibition. More generally, they emphasize that maximal SLiM binding affinity is not necessarily optimal. This view is reinforced by prior examples in which modulation of SLiM affinity could tune the balance of competing substrate modifications [4, 6]. Relatedly, the preferred sequence context for CDK phosphorylation is absent from most CDK-regulated sites [41]. The fact that submaximal sequences can be functional, and even beneficial, impedes bioinformatic efforts to identify viable motifs and cautions against demanding matches to a maximal consensus. This further accentuates the need for comprehensive quantification of motif preferences, to define both favored residues and prohibited substitutions for filtering out nonfunctional sequences.
Surprisingly, the Whi5 sequence harbors two overlapping motifs: an LP motif for cyclin binding and a potential phospho-Thr site for binding Cks1. Both types of motifs can promote substrate engagement and processive phosphorylation by CDKs [24]. Because our experiments involved both cyclin expression and pheromone treatment, the kinase that phosphorylates the TP site could be either the CDK or the pheromone-activated MAPK; indeed, they can regulate shared targets [42]. We note that the dual-motif behavior of the Whi5 sequence in the context of the Ste20Ste5PM chimera might not operate in the context of native Whi5. Yet it is also noteworthy that Whi5 orthologs show strong conservation of a 10-residue sequence (LLPPTTPKSR) that spans both motifs, plus C-terminal basic residues preferred by CDKs [43, 44]. Their overlap might also allow phosphorylation to modulate cyclin binding.
The capacity of G1 cyclins for LP docking and their motif preferences are conserved throughout fungi, including diverse phyla that diverged roughly 0.5 billion years ago. Apart from yeast systems, fungal G1 cyclins have not been studied extensively, except for recent work in Aspergillus [45]. In yeasts, in addition to cell cycle entry, Cln1/2-related cyclins promote polarized morphogenesis [46–49], and LP docking can facilitate this role [18]. Hence, their counterparts might help drive the polarized tip growth of hyphae in filamentous fungi, or filamentous growth in fission yeast [50]. Because fungal G1 cyclins evolved from a B-type cyclin precursor after fungi and animals diverged [19], LP docking likely arose specifically in fungi. Consequently, this function could present a target for anti-fungal therapeutics.
Finally, although LP docking by fungal G1 cyclins is widely conserved, the individual motifs are not. As discussed previously [51, 52], because SLiM-binding domains recognize dozens of targets, they face greater constraints to genetic drift than do the individual motifs, which can be rapidly acquired and discarded by target proteins. Furthermore, the key function of a particular SLiM interaction might often be achieved equally well by targeting any one of several proteins in the same pathway or complex, allowing evolutionary drift of which protein harbors the SLiM. Indeed, this notion is illustrated by our ability to synthetically switch the CDK regulatory target in the pheromone pathway from Ste5 to Ste20 [11]. Therefore, additional efforts to measure how genetic substitutions alter SLiM potency will help illuminate how evolution sculpts regulatory networks. Such SLiM variants can provide a spectrum of docking strengths for tuning the timing and specificity of their regulatory effects, and we expect this principle will apply to many mechanisms of docking-mediated enzyme recruitment.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Peter Pryciak (peter.pryciak@umassmed.edu).
Materials availability
Plasmids and strains generated for this study are available from the lead contact by request.
Data and Code Availability
Datasets generated during this study are available at Mendeley Data (http://dx.doi.org/10.17632/b3czpfhjrm.1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains and growth conditions
Standard procedures were used for growth and genetic manipulations of yeast [53, 54]. Unless indicated otherwise, cells were grown at 30˚C in yeast extract/peptone medium with 2% glucose (YPD) or in synthetic complete medium (SC) with 2% glucose and/or raffinose. Strains and plasmids used in this study are listed in Tables S1 and S2, respectively.
Fungal G1 cyclin genes were amplified by PCR from genomic DNA samples and ligated into episomal or integrating PGAL1-GST vectors. For genes with introns (AnCLN, NcCLN, BdCLN, CcCLN, CnCLN), exon sequences were amplified separately and then joined together to create a pseudo-cDNA.
Strains used for mobility-shift assays of substrate phosphorylation harbored plasmid-borne PGAL1-GST-cyclin expression constructs with C-terminal truncations (ΔC) that remove destabilizing sequences, which allowed maximal expression and accumulation of comparable protein levels for most cyclins. Full-length cyclins were used for pheromone-inhibition experiments and experiments involving longer-term expression, such as for in vivo selection of LP motif variants, because galactose-induced expression of the truncated forms inhibited growth. These full-length PGAL1-GST-cyclin constructs were integrated at the HIS3 locus. Strains for microscopy expressed Whi5-mCherry from the native WHI5 locus, and Sic1-GFP reporter constructs were inserted in single copy at the URA3 locus, as described previously [14].
METHOD DETAILS
Phosphorylation, binding, and signaling assays
Mobility-shift assays to monitor cyclin-induced phosphorylation in vivo were performed as described previously [11, 18, 20]. Briefly, cells harboring PGAL1-GST-cyclin constructs and HA-tagged CDK substrates were induced with 2% galactose for 2.5 hr, to induce cyclin expression. Two mL of these cultures were harvested by centrifugation, and then the pellets were frozen in liquid nitrogen and stored at −80˚C. As before [18] all substrates were all based on the Ste5 N-terminus: (i) the “LP dock” substrate is Ste5 residues 1–337; (ii) the “no dock” substrate is Ste5 1–337 with its LLPP motif mutated to AAAA; (iii) the “leu zipper” substrate is Ste5 1–260 fused to a half leucine zipper sequence (E34[N]). Substrate phosphorylation was visualized by reduced mobility in SDS-PAGE, and detected by anti-HA immunoblotting.
GST co-precipitation assays were performed as described previously [11, 18, 20]. Briefly, 10-mL cultures harboring PGAL1-GST fusion proteins and V5-tagged cyclins were treated with 2% galactose for 1.5 hr. Extracts were prepared by glass bead lysis in a nonionic detergent buffer, and then GST fusions and co-bound proteins were captured on glutathione-sepharose beads. Proteins in input and bound fractions were detected by anti-V5 and anti-GST immunoblots.
To assay the ability of fungal cyclins to inhibit pheromone signaling and MAPK activation, ste20Δ STE5–8A strains were transformed with plasmids that encode derivatives of a chimeric signaling protein, Ste20Ste5PM [11], with different LP motifs; the STE5–8A allele prevents the CDK from inhibiting Ste5 [11, 22], and thus the CDK must phosphorylate the Ste20Ste5PM chimera in order to block pheromone signaling. These strains also contained integrated PGAL1-GST-cyclin constructs, as indicated in the figures. The transformants were grown to exponential phase in 2% raffinose media, and then cyclin expression was induced by adding 2% galactose for 75 minutes. Then, cells were treated with pheromone (50 nM) for 15 minutes. Two mL of these cultures were harvested by centrifugation, and then the pellets were frozen in liquid nitrogen and stored at −80˚C. MAPK activation was detected by immunoblots using anti-p44/42 antibodies.
Whole cell lysates and Immunoblotting
Whole cell lysates were prepared from frozen cell pellets by lysis in trichloroacetic acid as described previously [20]. Protein concentrations were measured by a bicinchoninic acid assay (Thermo Scientific # 23225), and equal amounts (10 or 20 μg) were loaded per lane. Proteins were resolved by SDS–PAGE and transferred to PVDF membranes (Immobilon-P; Millipore) in a submerged tank. Membranes were blocked (1 hr, room temperature) in TTBS (0.2% Tween-20, 20 mM TRIS-HCl, 500 mM NaCl, pH 7.5) containing 5% non-fat milk, and then probed with antibodies in the equivalent solution. Primary antibodies were mouse anti-HA (1:1000, Covance #MMS101R), mouse anti-V5 (1:5000, Invitrogen #46–0705) mouse anti-phospho-p44/42 (1:1000, Cell Signaling Technology #9101), mouse anti-GST (1:1000, Santa Cruz Biotechnologies #sc-138), rabbit anti-myc (1:200, Santa Cruz Biotechnologies #sc-789), and rabbit anti-G6PDH (1:100000, Sigma #A9521). HRP-conjugated secondary antibodies were goat anti-rabbit (1:3000, Jackson ImmunoResearch #111–035-144), and goat anti-mouse (1:3000, BioRad #170–6516). Enhanced chemilluminescent detection used a BioRad Clarity substrate (#170–5060). Densitometry was performed using ImageJ software.
Library construction and competitive growth assay
The construction of mutant libraries and the performance of bulk growth competition assays were based on procedures described previously [55]. Variant LP motifs were tested in the context of a chimeric Ste20Ste5PM signaling protein [11], in which the membrane-binding domain from Ste5 and its flanking CDK phosphorylation sites replace the native membrane-binding domain in Ste20, and LP docking motifs are placed at the N-terminus. An initial parent construct (pPP4364) harbored the LP motif region from Ste20 on a 108-bp SpeI-BglII fragment that includes native Ste20 residues 80–109; in a later derivative (pPP4365), internal restriction sites were introduced within this region so that a smaller piece of the Ste20 LP motif sequence region (residues 86–96) could be replaced with 45-bp AatII-NheI fragments that contain 11 unique codons between the AatII and NheI sites (the products of which contain the descriptor “G25” in Table 2). To make libraries of point mutants, pPP4364 was digested with SpeI and BglII (for Ste20 libraries), or pPP4365 was digested with AatII and NheI (for all other libraries). These digested vectors were ligated to annealed pairs of oligonucleotides (with single-stranded overhangs suitable for ligation with the vector) in which single codons were randomized (NNN) to generate all 64 possibilities. The ligation products were transformed into E. coli and plated on LB+Amp plates. Colonies (> 5000 per library) were harvested by adding LB+Amp liquid and gently agitating with a glass rod spreader, and then plasmid DNA was prepared from the suspension of pooled colonies. The isolated DNAs were checked by Sanger sequencing to verify that all four nucleotides were comparably represented at each position in the randomized codon.
For competitive growth assays, a solution was prepared that mixed together equimolar amounts of each individual codon library, plus a fraction (4–6%) of the wild-type plasmid. This pool was transformed into PGAL1-cyclin yeast strains. After the transformation procedure, an aliquot (10%) was reserved, dilutions of which were spread on –URA plates; after 2 days incubation, colonies were counted to ensure that the number of transformants (usually > 10,000) exceeded the number of mutant variants in the library by at least 10-fold. The remainder of the transformation mixture was inoculated directly into 20 mL of –URA/glucose liquid medium and incubated at 30˚C overnight (15–16 hr). Then, the cells were pelleted, washed five times with –URA/raffinose, and suspended in 50 mL of –URA/raffinose. This culture was incubated at 30˚C for 48 hours, during which it was diluted back with fresh medium three times (at 22 hr, 31 hr, and 43 hr) to prevent saturation. The culture was then treated with 2% galactose for 75 minutes (in a volume of 70 mL) to induce cyclin expression. At this time (t = 0), an aliquot (40 mL) was harvested, and the remaining culture was treated with pheromone (500 nM) and returned to incubation. Cells were diluted with fresh medium (including galactose and pheromone) after the first 8 hours and subsequently after every 12 hours to maintain an OD660 below 1 (in a volume of 50 mL). Aliquots (20 mL) were harvested at 20, 32 and 44 hours. Harvested cells were collected by centrifugation (5 min., 5000 rpm, 4˚C), washed with 25 mL sterile water, resuspended in 1 mL sterile water, and transferred to 1.5 mL microcentrifuge tubes. These suspensions were centrifuged, the supernatants were aspirated, and the pellets were frozen using liquid nitrogen and stored at −80˚C.
DNA preparation and deep sequencing.
Isolation and sequencing of DNA from competitive growth assays followed previously described methods [55]. DNA was purified from yeast cells using the Zymo Research ZR Plasmid Miniprep Kit (#D4015). Frozen cell pellets were thawed and suspended in 200 μL of solution P1, and then were lysed using Zymolase (0.2 units/μL; Zymo Research #E1005) for 1.5 hours at 37˚C, before proceeding with the remaining purification steps. The purified DNA was digested with NotI to improve the efficiency of the subsequent PCR. A test PCR (20 μL) was performed to confirm equal abundance of the template in all samples and to optimize the number of cycles for the final PCR (50 μL), which was 17 cycles for all results reported here. The forward primer included a P5 sequence (for binding the Illumina flow cell) followed by a Illumina sequencing primer binding site, a 6-nt bar code, and an upstream plasmid-annealing sequence; the reverse primer included a P7 sequence (for binding the Illumina flow cell) followed by a 6-nt i7-index sequence, an i7 sequencing primer binding site, and a downstream plasmid-annealing sequence.
Aliquots (5 μL) of the PCR products were run in 1.2% agarose gels to confirm the presence of the desired product, and the remainders were purified using Zymo Spin I columns (Zymo Research #C1003–250) and eluted in 10 mM TRIS-HCl, pH 8. The concentration of the eluted products was measured using nanodrop, and a mixture was prepared containing equal amounts of each DNA product. This final mixture was then re-purified using another Zymo Spin I column, and the DNA concentration was determined by qPCR using a KAPA Library Quantification Kit (Kapa Biosystems #KK4824). Samples were diluted to 20 nM, and 50 μL portions were submitted to the UMMS Deep Sequencing Core Facility for sequencing on an Illumina MiSeq instrument (single-end read, 100 bp, plus i7-Index sequencing when relevant).
Sequencing data were de-multiplexed by using bar code and index identifiers. Time-dependent changes in the frequency of individual sequence variants were analyzed using Enrich2 software [23]. Fitness scores calculated by Enrich2 describe the rate of change of mutant variants compared to the wild-type sequence, expressed as a natural log: i.e., (fitness score) = ln[(change in frequency of mutant variant) ÷ (change in frequency of wild-type)]. Overall, the relative fitness scores were similar regardless of whether they were calculated from the full time course (0, 20, 32, 44 hr) or just the initial stage (0–20 hr); all scores reported here were calculated from the 0–20 hr stage.
With only one exception, fitness results reported for each library incorporate all nucleotide variants. The exception is the Ste20 motif library, which showed an unusual behavior in which a minority of nucleotide variants became excessively enriched during the selection, in a manner that was not shared by their synonymous codons. The basis of this behavior remains unknown, but because it was not observed with other libraries it likely involves sequence differences between the two parent vectors that received the randomized oligonucleotides (pPP4364 and pPP4365; described above). To prevent the fitness score calculations from being skewed by these spurious variants, the Ste20 library data were filtered as follows. After initial fitness scores were calculated (by Enrich2) for all nucleotide variants, we used a custom Python script to identify the least-fit (i.e., lowest-scoring) synonym for each amino acid variant and create an output file with their raw counts. Then, the Enrich2 analysis was repeated using the counts data for only this set of least-fit synonyms.
Source data containing raw fitness scores are available at Mendeley Data (see Key Resources Table), including all nucleotide and amino acid variants, for all 8-residue and 11-residue motif mutagenesis results.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-HA | Covance | Cat# MMS101R |
| Mouse anti-V5 | Invitrogen | Cat# 46–0705 |
| Mouse anti-phospho-p44/42 | Cell Signaling Technology | Cat# 9101 |
| Mouse anti-GST | Santa Cruz Biotechnology | Cat# sc-138 |
| Rabbit anti-myc | Santa Cruz Biotechnology | Cat# sc-789 |
| Rabbit anti-G6PDH | Sigma | Cat# A9521 |
| Goat anti-mouse, HRP-conjugated | Bio-Rad | Cat# 170–6516 |
| Goat anti-rabbit, HRP-conjugated | Jackson ImmunoResearch | Cat# 111–035-144 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| α factor peptide (WHWLQLKPGQMPY) | Tufts University Core Facility | N/A |
| Critical Commercial Assays | ||
| BCA Protein Assay Kit | Thermo Scientific | Cat# 23225 |
| Clarity Western ECL Substrate | Bio-Rad | Cat# 170–5060 |
| ZR Plasmid Miniprep Kit | Zymo Research | Cat# D4015 |
| Zymo Spin I Columns | Zymo Research | Cat# C1003 |
| KAPA Library Quantification Kit | Kapa Biosystems | Cat# KK4824 |
| Deposited Data | ||
| Source data for western blots, competitive growth assays, and quantitative microscopy | Mendeley Data | http://dx.doi.org/10.17632/b3czpfhjrm.1 |
| Experimental Models: Organisms/Strains | ||
| See Table S2 for yeast strains (S. cerevisiae) | This paper | N/A |
| Oligonucleotides | ||
| P5/barcode forward primers: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTnnnnnnGGCCGACTAGTCGATGATGACAAT | Eurofins Genomics | N/A |
| P7 reverse primer: CAAGCAGAAGACGGCATACGAGATAGTCATTGTCTGTAGGAGTTTCCATCATGAGAT | Eurofins Genomics | N/A |
| P7/i7-Index reverse primers: CAAGCAGAAGACGGCATACGAGATnnnnnnGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGTCTGTAGGAGTTTCCATCATGAGAT | Eurofins Genomics | N/A |
| Recombinant DNA | ||
| See Table S3 for plasmids | This paper | N/A |
| Software and Algorithms | ||
| ImageJ (western blot image quantification) | https://imagej.nih.gov/ij/ | N/A |
| UCSF Chimera (3D structure annotation) | [56] | N/A |
| Enrich2 (deep mutational scanning data analysis) | [23] | N/A |
| PSSMSearch (proteome-wide motif search) | [29] | N/A |
| Other | ||
| Zymolyase | Zymo Research | Cat# E1005 |
Fitness score transformations and PSSM derivation.
To create sequence logos of sequence preferences, we transformed the fitness score data generated by Enrich2, which is expressed relative to the WT motif sequence, into a preference metric that expresses the bias for each residue relative to the set of all possible residues. First, the raw fitness score for each amino acid variant was normalized to the lowest (= 0) and highest (= 1) scores in a given motif array: (normalized score) = (raw score – minimum) ÷ (mean terminator score – minimum). Then, the normalized scores were converted to a frequency metric by dividing each by the sum of all scores at the same position: (frequency) = (normalized score) ÷ (column sum). Finally, the frequency metric was converted to a preference metric by subtracting 0.05, so that a neutral preference is represented by zero, favored residues are positive, and disfavored residues are negative: (preference score) = (frequency – 0.05). These preference scores were used to generate sequence logos via a web-based tool (http://slim.icr.ac.uk/visualisation/index.html). Source data files containing these calculations are available at Mendeley Data.
To assess the number of inactivating mutations, we used the normalized scores, which allow the function of all sequence variants in all motifs to be compared relative to a uniform scale from non-functional (= 0) to maximally functional (= 1). From the normalized scores for each motif, we calculated the number of values at each position that were below a minimum threshold (20%, 15%, or 10%) above the minimum value (zero). Source data files containing these calculations are available at Mendeley Data. Note that, because the Sic1 motif is weak, residues that are slightly better than the worst residue at a given position are noticeably closer to the wild-type residue in heat maps and logos, but they are still at the very low end of the uniform normalized scale, and hence they score as “inactivating” because they fall below the minimum threshold.
To assess the similarities in motif sequence preferences for different cyclins, we used the raw fitness scores to calculate Pearson correlation coefficients (r) for all pair-wise comparisons of cyclins. This calculation was performed individually for each of the 5 original motifs, as well as collectively for the set of all 5 motifs (as shown in Figure 4D). Source data files containing these calculations are available at Mendeley Data.
To calculate the consensus PSSM, the individual preference score matrices for 5 motifs (Ste5, Lam5, Ste20, Sic1, and Whi5-P7L) were averaged. To assess whether the specific mathematical operations used to create this “Preference” PSSM influence its predictive utility, we also generated an alternative PSSM (“Difference” PSSM), in which the scores were not first normalized or converted to a frequency. Instead, we simply calculated the difference between the raw fitness score for each amino acid and the average score of all (non-terminator) residues at the same position: (difference score) = (raw score – column average). As above, to calculate the consensus PSSM, the individual PSSMs for 5 motifs were averaged. Source data files containing both types of calculations are available at Mendeley Data. The two methods of PSSM calculation gave similar patterns of correlation with physico-chemical descriptors and with the observed scores of tested candidate LP motifs, although they yielded some differences in the rank order of motifs identified in the in silico search of the yeast proteome. Hence, results using each method are included (in Figure S6 and Table S1). In general, we note that the Difference PSSM performs slightly better at predicting the rank order of empirically-tested motifs with detectable function, whereas the Preference PSSM more accurately represents inactivating substitutions and hence is better at filtering out non-functional motifs.
Structural and sequence conservation analysis.
To display the pattern of cyclin sequence conservation on the domain surface, we used a predicted model for the structure of ScCln2 that we generated previously [18]; this model includes ScCln2 residues 1–376, from which we removed a variable insert region unique to Cln1/2 cyclins (residues 134–150) in order to improve the quality of the model. This ScCln2 model was oriented relative to the CDK by 3D alignment with a known cyclin-CDK crystal structure (PDB 5HQ0), using UCSF Chimera software [56]. Then, sequence alignments of the indicated groups of cyclins were superimposed upon the ScCln2-CDK model and colored according to the conservation criteria described in the Figure 1F legend.
To compare patterns of surface residue conservation among different groups of cyclins, we first performed sequence alignments of large numbers of fungal G1 cyclins, and then calculated the amino acid frequency at each position for two groups: Cln3 cyclins (which do not recognize LP motifs) and non-Cln3 cyclins (i.e., Cln1/2, Ccn1, Puc1, and Cln, which do recognize LP motifs). Separately, we used the model for ScCln2 (described above) to extract surface exposure values at each position; these were calculated as the proportion of the amino acid that is accessible in the structure, using the program DSSP [57]. Finally, to identify surface-exposed positions with strong differential conservation, we calculated a weighted difference in amino acid frequency: (weighted Δ aa freq.) = [(non-Cln3 aa freq.)^2 – (Cln3 aa freq.)^2]*(surface exposure). All three metric types are displayed in Figure S1A.
Live cell microscopy assays.
Time-lapse fluorescence microscopy of Whi5-mCherry localization and Sic1-GFP intensity was measured by methods that have been described in detail previously [14], using cells that were grown in SC/glucose medium, placed on agarose pads, and imaged at 30˚C. Plots showing mean Sic1-GFP intensity were expressed relative to the time at which the nuclear Whi5-mCherry signal declined by 50% in the same cells, and the Sic1-GFP degradation time in individual cells was defined as the point when its signal declined by 50%. Duration of Sic1-GFP signal was defined as the time from the start of its decline until the time it reached background levels.
QUANTIFICATION AND STATISTICAL ANALYSIS
For competitive growth assays of LP motif mutant variants, fitness scores and standard errors were calculated by Enrich2 software [23]. Other statistical analyses, including calculation of means, range, SEM, Pearson’s correlation (r), coefficient of determination (R2), and Gini index, were performed using Microsoft Excel. The numbers of biological replicates are described in the Figure Legends.
Supplementary Material
A: Results of in silico search using the “Preference” PSSM.
B: Results of in silico search using the “Difference” PSSM.
C: Comparison of rank order and p-values of hits from the two distinct searches.
D: Cross-reference of search hits with a prior dataset of Cdk1 phosphorylation sites.
Highlights.
In vivo assay quantifies the functional impact of hundreds of docking motif variants
Hierarchical and contingent residue preferences define a spectrum of motif strengths
A quantitative matrix surpasses a simple consensus for predicting motif potency
Variation in docking motif potency modulates the degree and timing of CDK regulation
ACKNOWLEDGEMENTS
We are indebted to Dan Bolon, Julia Flynn, and David Mavor for advice and guidance with deep mutational scanning methods. We also thank David Mavor and Alan Rubin for assistance with deep sequencing data processing and Enrich2 software. In addition, we thank Matt Winters and Brian Gruessner for technical help, Charlie Specht and Nick Rhind for fungal DNA samples, Nick Rhind for helpful discussions, and Dan McCollum and Jenny Benanti for feedback on the manuscript. This work was supported by grants from the NIH (R01 GM057769) to P.M.P, by ERC Consolidator Grant 649124 and Estonian Science Agency grant PRG550 to M.L., and from CRUK (Senior Cancer Research Fellowship C68484/A28159) to N.E.D.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and three tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Miller CJ, and Turk BE (2018). Homing in: Mechanisms of Substrate Targeting by Protein Kinases. Trends Biochem Sci 43, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ubersax JA, and Ferrell JE Jr. (2007). Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol 8, 530–541. [DOI] [PubMed] [Google Scholar]
- 3.Remenyi A, Good MC, and Lim WA (2006). Docking interactions in protein kinase and phosphatase networks. Curr Opin Struct Biol 16, 676–685. [DOI] [PubMed] [Google Scholar]
- 4.Hertz EPT, Kruse T, Davey NE, Lopez-Mendez B, Sigurethsson JO, Montoya G, Olsen JV, and Nilsson J. (2016). A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol Cell 63, 686–695. [DOI] [PubMed] [Google Scholar]
- 5.Kataria M, Mouilleron S, Seo MH, Corbi-Verge C, Kim PM, and Uhlmann F. (2018). A PxL motif promotes timely cell cycle substrate dephosphorylation by the Cdc14 phosphatase. Nat Struct Mol Biol 25, 1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy J, Li H, Hogan PG, and Cyert MS (2007). A conserved docking site modulates substrate affinity for calcineurin, signaling output, and in vivo function. Mol Cell 25, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davey NE, and Morgan DO (2016). Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol Cell 64, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan DO (2007). The Cell Cycle: Principles of Control, (London: New Science Press; ). [Google Scholar]
- 9.Bloom J, and Cross FR (2007). Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol 8, 149–160. [DOI] [PubMed] [Google Scholar]
- 10.Ord M, and Loog M. (2019). How the cell cycle clock ticks. Mol Biol Cell 30, 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhaduri S, and Pryciak PM (2011). Cyclin-specific docking motifs promote phosphorylation of yeast signaling proteins by G1/S Cdk complexes. Curr Biol 21, 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kõivomägi M, Valk E, Venta R, Iofik A, Lepiku M, Morgan DO, and Loog M. (2011). Dynamics of Cdk1 Substrate Specificity during the Cell Cycle. Mol Cell 42, 610–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loog M, and Morgan DO (2005). Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature 434, 104–108. [DOI] [PubMed] [Google Scholar]
- 14.Ord M, Moll K, Agerova A, Kivi R, Faustova I, Venta R, Valk E, and Loog M. (2019). Multisite phosphorylation code of CDK. Nat Struct Mol Biol 26, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ord M, Venta R, Moll K, Valk E, and Loog M. (2019). Cyclin-Specific Docking Mechanisms Reveal the Complexity of M-CDK Function in the Cell Cycle. Mol Cell 75, 76–89 e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulman BA, Lindstrom DL, and Harlow E. (1998). Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc Natl Acad Sci U S A 95, 10453–10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilmes GM, Archambault V, Austin RJ, Jacobson MD, Bell SP, and Cross FR (2004). Interaction of the S-phase cyclin Clb5 with an “RXL” docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev 18, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhaduri S, Valk E, Winters MJ, Gruessner B, Loog M, and Pryciak PM (2015). A docking interface in the cyclin Cln2 promotes multi-site phosphorylation of substrates and timely cell-cycle entry. Curr Biol 25, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medina EM, Turner JJ, Gordan R, Skotheim JM, and Buchler NE (2016). Punctuated evolution and transitional hybrid network in an ancestral cell cycle of fungi. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pope PA, Bhaduri S, and Pryciak PM (2014). Regulation of cyclin-substrate docking by a G1 arrest signaling pathway and the Cdk inhibitor Far1. Curr Biol 24, 1390–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oehlen LJ, and Cross FR (1994). G1 cyclins CLN1 and CLN2 repress the mating factor response pathway at Start in the yeast cell cycle. Genes Dev 8, 1058–1070. [DOI] [PubMed] [Google Scholar]
- 22.Strickfaden SC, Winters MJ, Ben-Ari G, Lamson RE, Tyers M, and Pryciak PM (2007). A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell 128, 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubin AF, Gelman H, Lucas N, Bajjalieh SM, Papenfuss AT, Speed TP, and Fowler DM (2017). A statistical framework for analyzing deep mutational scanning data. Genome Biol 18, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kõivomägi M, Ord M, Iofik A, Valk E, Venta R, Faustova I, Kivi R, Balog ER, Rubin SM, and Loog M. (2013). Multisite phosphorylation networks as signal processors for Cdk1. Nat Struct Mol Biol 20, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGrath DA, Balog ER, Koivomagi M, Lucena R, Mai MV, Hirschi A, Kellogg DR, Loog M, and Rubin SM (2013). Cks confers specificity to phosphorylation-dependent CDK signaling pathways. Nat Struct Mol Biol 20, 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abriata LA, Palzkill T, and Dal Peraro M. (2015). How structural and physicochemical determinants shape sequence constraints in a functional enzyme. PLoS One 10, e0118684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zarrinpar A, Bhattacharyya RP, and Lim WA (2003). The structure and function ofproline recognition domains. Sci STKE 2003, RE8. [DOI] [PubMed] [Google Scholar]
- 28.McCusker D, Denison C, Anderson S, Egelhofer TA, Yates JR 3rd, Gygi SP, and Kellogg DR (2007). Cdk1 coordinates cell-surface growth with the cell cycle. Nat Cell Biol 9, 506–515. [DOI] [PubMed] [Google Scholar]
- 29.Krystkowiak I, Manguy J, and Davey NE (2018). PSSMSearch: a server for modeling, visualization, proteome-wide discovery and annotation of protein motif specificity determinants. Nucleic Acids Res 46, W235–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kõivomägi M, Valk E, Venta R, Iofik A, Lepiku M, Balog ER, Rubin SM, Morgan DO, and Loog M. (2011). Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 480, 128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X, Lau KY, Sevim V, and Tang C. (2013). Design principles of the yeast G1/S switch. PLoS Biol 11, e1001673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frank R. (2002). The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports--principles and applications. J Immunol Methods 267, 13–26. [DOI] [PubMed] [Google Scholar]
- 33.Turk BE, and Cantley LC (2003). Peptide libraries: at the crossroads of proteomics and bioinformatics. Curr Opin Chem Biol 7, 84–90. [DOI] [PubMed] [Google Scholar]
- 34.Davey NE, Seo MH, Yadav VK, Jeon J, Nim S, Krystkowiak I, Blikstad C, Dong D, Markova N, Kim PM, et al. (2017). Discovery of short linear motif-mediated interactions through phage display of intrinsically disordered regions of the human proteome. FEBS J 284, 485–498. [DOI] [PubMed] [Google Scholar]
- 35.Tong AH, Drees B, Nardelli G, Bader GD, Brannetti B, Castagnoli L, Evangelista M, Ferracuti S, Nelson B, Paoluzi S, et al. (2002). A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295, 321–324. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen HQ, Roy J, Harink B, Damle NP, Latorraca NR, Baxter BC, Brower K, Longwell SA, Kortemme T, Thorn KS, et al. (2019). Quantitative mapping of protein-peptide affinity landscapes using spectrally encoded beads. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider BL, Yang QH, and Futcher AB (1996). Linkage of replication to start by the Cdk inhibitor Sic1. Science 272, 560–562. [DOI] [PubMed] [Google Scholar]
- 38.Schwob E, Bohm T, Mendenhall MD, and Nasmyth K. (1994). The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79, 233–244. [DOI] [PubMed] [Google Scholar]
- 39.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, and Tyers M. (2001). Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414, 514–521. [DOI] [PubMed] [Google Scholar]
- 40.Verma R, Feldman RM, and Deshaies RJ (1997). SIC1 is ubiquitinated in vitro by a pathway that requires CDC4, CDC34, and cyclin/CDK activities. Mol Biol Cell 8, 1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, and Morgan DO (2009). Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Repetto MV, Winters MJ, Bush A, Reiter W, Hollenstein DM, Ammerer G, Pryciak PM, and Colman-Lerner A. (2018). CDK and MAPK Synergistically Regulate Signaling Dynamics via a Shared Multi-site Phosphorylation Region on the Scaffold Protein Ste5. Mol Cell 69, 938–952 e936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinna LA, and Ruzzene M. (1996). How do protein kinases recognize their substrates? Biochim Biophys Acta 1314, 191–225. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki K, Sako K, Akiyama K, Isoda M, Senoo C, Nakajo N, and Sagata N. (2015). Identification of non-Ser/Thr-Pro consensus motifs for Cdk1 and their roles in mitotic regulation of C2H2 zinc finger proteins and Ect2. Sci Rep 5, 7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paolillo V, Jenkinson CB, Horio T, and Oakley BR (2018). Cyclins in aspergilli: Phylogenetic and functional analyses of group I cyclins. Stud Mycol 91, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hungerbuehler AK, Philippsen P, and Gladfelter AS (2007). Limited functional redundancy and oscillation of cyclins in multinucleated Ashbya gossypii fungal cells. Eukaryot Cell 6, 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lew DJ, and Reed SI (1993). Morphogenesis in the yeast cell cycle: regulation by Cdc28 and cyclins. J Cell Biol 120, 1305–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loeb JD, Sepulveda-Becerra M, Hazan I, and Liu H. (1999). A G1 cyclin is necessary for maintenance of filamentous growth in Candida albicans. Mol Cell Biol 19, 4019–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng X, and Wang Y. (2004). Hgc1, a novel hypha-specific G1 cyclin-related protein regulates Candida albicans hyphal morphogenesis. EMBO J 23, 1845–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kinnaer C, Dudin O, and Martin SG (2019). Yeast-to-hypha transition of Schizosaccharomyces japonicus in response to environmental stimuli. Mol Biol Cell 30, 975–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldman A, Roy J, Bodenmiller B, Wanka S, Landry CR, Aebersold R, and Cyert MS (2014). The calcineurin signaling network evolves via conserved kinase-phosphatase modules that transcend substrate identity. Mol Cell 55, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davey NE, Cyert MS, and Moses AM (2015). Short linear motifs - ex nihilo evolution of protein regulation. Cell Commun Signal 13, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rothstein R. (1991). Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol 194, 281–301. [DOI] [PubMed] [Google Scholar]
- 54.Sherman F. (2002). Getting started with yeast. Methods Enzymol 350, 3–41. [DOI] [PubMed] [Google Scholar]
- 55.Hietpas R, Roscoe B, Jiang L, and Bolon DN (2012). Fitness analyses of all possible point mutations for regions of genes in yeast. Nat Protoc 7, 1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 57.Kabsch W, and Sander C. (1983). Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Results of in silico search using the “Preference” PSSM.
B: Results of in silico search using the “Difference” PSSM.
C: Comparison of rank order and p-values of hits from the two distinct searches.
D: Cross-reference of search hits with a prior dataset of Cdk1 phosphorylation sites.
Data Availability Statement
Datasets generated during this study are available at Mendeley Data (http://dx.doi.org/10.17632/b3czpfhjrm.1).