

Abstract
Background:
Inherited cardiomyopathy associates with a range of phenotype, mediated by genetic and non-genetic factors. Non-inherited cardiomyopathy also displays varying progression and outcomes. Expression of cardiomyopathy genes is under the regulatory control of promoters and enhancers, and human genetic variation in promoters and enhancers may contribute to this variability.
Methods:
We superimposed epigenomic profiling from hearts and cardiomyocytes, including promoter-capture chromatin conformation information, to identify enhancers for two cardiomyopathy genes, MYH7 and LMNA. Enhancer function was validated in human cardiomyocytes derived from induced pluripotent stem cells. We also conducted a genome-wide search to ascertain genomic variation in enhancers positioned to alter cardiac expression and correlated one of these variants to cardiomyopathy progression using biobank data.
Results:
Multiple enhancers were identified and validated for LMNA and MYH7, including a key enhancer that regulates the switch from MYH6 expression to MYH7 expression. Deletion of this enhancer resulted in a dose-dependent increase in MYH6 and faster contractile rate in engineered heart tissues. We searched for genomic variation in enhancer sequences across the genome, with focus on nucleotide changes that create or interrupt transcription factor binding sites. rs875908 disrupts a TBX5 binding motif and maps to an enhancer region 2KB from the transcriptional start site of MYH7. Gene editing to remove the enhancer harboring this variant markedly reduced MYH7 expression in human cardiomyocytes. Using biobank-derived data, rs875908 associated with longitudinal echocardiographic features with cardiomyopathy.
Conclusions:
Enhancers regulate cardiomyopathy gene expression, and genomic variation within these enhancer regions associates with cardiomyopathic progression over time. This integrated approach identified noncoding modifiers of cardiomyopathy and is applicable to other cardiac genes.
Keywords: myosin, lamin A/C, enhancer, regulatory variants, epigenomics, engineered heart tissue, modifier
INTRODUCTION
Protein coding mutations in over 100 genes have been linked to autosomal dominant cardiomyopathy, which leads to heart failure and significant burden.1–3 A well-recognized clinical feature of inherited and non-inherited cardiomyopathy is its variable phenotypic expression. Genetic cardiomyopathy demonstrates an age-dependent penetrance, variable expressivity, and variable clinical presentations, even in patients sharing identical primary mutations.4, 5 Protein coding variants have been described as altering the phenotypic expression of primary cardiomyopathy-causing mutations.5–7 However, the contribution of noncoding variation as modifiers of the clinical presentation of cardiomyopathy has been less well investigated.
Noncoding regions of the genome harbor important regulatory sequences that control the expression of genes through both distal enhancers and proximal gene promoters.8 ChIP-seq, ATAC-seq, and CAGE-seq can mark genomic regions as having regulatory function, but do not provide information on their gene target. Chromatin conformation assays evaluate genomic three-dimensional organization and link enhancers to their target genes. However, as enhancer function is dependent on tissue-specific transcription factors, assays for enhancer function or targets require the context of relevant tissues/cells.
To define the contribution of noncoding variation, we evaluated the regulatory regions for two commonly mutated cardiopathy genes, MYH7 and LMNA. Mutations in MYH7 are a common cause of hypertrophic cardiomyopathy while mutations in LMNA are a common cause of dilated cardiomyopathy with arrhythmias.4, 9 MYH7 maps in tandem with MYH6 on human chromosome 14. MYH7 encodes β-myosin heavy chain (MHC), which is the major left ventricular myosin heavy chain in the adult human. MYH6 encodes α-MHC and is the major myosin heavy chain in the developing human ventricle and adult atrium. Mice display an entirely different myosin expression pattern where the adult murine ventricular myocardium is dominated by α-MHC, further underscoring the importance of studying MYH7 regulatory regions in human systems.
We used an integrative analysis that relied on >20 heart enhancer function and enhancer target datasets to identify MYH7 and LMNA left ventricle enhancer regions. We confirmed the activity of these regions using reporter assays and CRISPr-mediated deletion in human cardiomyocytes derived from induced pluripotent stem cells (IPSC-CMs). These regulatory regions contained sequence variants within transcription factor binding sites that altered enhancer function. Extending this strategy genome-wide, we identified an enhancer modifying variant upstream of MYH7. This common variant correlated with MYH7 expression in the GTEx eQTL dataset. Finally, we identified this variant also correlated with a more dilated left ventricle over time. These findings link noncoding enhancer variation to cardiomyopathy phenotypes and provide direct evidence of the importance of genetic background.
METHODS
Transparency and Openness Statement.
All code and scripts used in this manuscript are available at this link https://github.com/sdkearns/Find_Regulatory_Variants.
Human Subject Approvals.
Participants in Northwestern’s biobank provided informed consent under approval from Northwestern University’s Institutional Review Board.
Epigenetic Dataset Downloads, Visualization, and Candidate Selection.
Epigenetic datasets were identified from the Encode data repository or GEO (Table I in the Supplement). A UCSC genome browser session containing all tracks used for left ventricle enhancer identification is available by searching “Gacita_et_al_LV_Enhancer_Tracks” in UCSC’s public sessions repository. Candidate enhancers were identified by evaluating Hi-C interaction regions or Hi-C proximal regions that also had H3K27Ac and ATAC-seq signal. We prioritized regions with an out of phase organization of H3K27Ac and open chromatin signal, which likely represents a nucleosome-depleted region flanked by marked histones. Hi-C interaction regions meeting these criteria that also have high H3K4me3 signal and overlap the promoter of a target gene transcript were excluded due to their likely promoter function. Of the remaining candidates, we prioritized regions that were marked by additional epigenetic data, including CTCF, p300, GATA4, TBX5 and NKX2.5 binding.
Enhancer Region Cloning.
Candidate enhancer regions were ligated into luciferase plasmids using a Gateway cloning strategy.
Luciferase Reporter Assay.
HL-1 cardiomyocytes (Millipore Sigma Cat#SCC065) were cultured on fibronectin coated flasks in Claycomb media with 10% HL-1 qualified FBS as previously described.10 The firefly luciferase signal from each well was recorded from three separate replicates and internally normalized to Renilla luciferase signal. Each enhancer construct was tested in a minimum of two separate wells on three separate days. Transfection efficiency was optimized using flow cytometry and averaged 10% for the experiments.
Induced pluripotent stem cell (iPSC)-derived cardiomyocytes (IPSC-CMs) were generated according to standard protocols.11 At approximately day 10 of differentiation, cardiomyocytes were re-plated on to white clear-bottom 96-well plates at 40,000 cells per well. The media was changed every two days and cells began to beat as a syncytium day 14–16. On day 18, cardiomyocytes were transfected with Lipofecamine3000 (Thermo Fisher) according to manufacturer’s instructions. All wells were transfected with 0.2μl of 0.15μM enhancer firefly luciferase plasmid, 5ng of pRL-SV40 (Promega), 0.15μl Lipofecamine3000, and 0.2μl of P3000 in 10μl of Opti-MEM. Forty-eight hours after transfection, the luciferase assay was performed with the Dual-Glo luciferase assay kit (Promega) according to manufacturer’s instructions. Firefly luciferase signal was read using 96-well plate reader and signals were internally normalized to the same well’s Renilla luciferase signal. Signals from candidate enhancer luciferase plasmids were compared to the signal from a 500bp desert region in cells from the same differentiation. Each enhancer construct was tested in 8 separate wells on at least three separate cardiomyocyte differentiations. Table II in the Supplement describes the regions tested.
CRISPr Enhancer Deletion in IPSCs.
To delete enhancer regions, guides targeting the 5’ and 3’ end of enhancer regions were designed (Table III in the Supplement) and used as described in the detailed methods. The genotypes of edited cells are provided in Table IV in the Supplement and off target assessment is in Table V in the Supplement.
Engineered Heart Tissue Generation and Measurement of Contractile Properties.
Engineered heart tissues (EHTs) were generated according to previously published methods.12 IPSC-CMs were differentiated as previously described and when beating cells were present (~day 10), cells were washed with PBS and digested with TrypLE (Thermo). One million cells per EHT were centrifuged at 500g for 5min and resuspended in 65μl of EHT media (CDM3,11 containing 10% of heat-inactivated FBS, 2μM thiazovivin, 33μg/mL aprotinin, and 5U/mL penicillin/streptomycin), 25μl of 25mg/mL fibrinogen and 10μl of Matrigel (Corning). 100μl of this EHT mix was added to 3μl of 100U/mL thrombin and mixed. The whole mixture was pipetted between PDMS posts (EHT Technologies) in an EHT mold created from 2% agarose and a Teflon spacer in a 24-well Nunc plate (Thermo Fisher). Fibrin gel was allowed to polymerize for 2 hours and then 200μl of CDM3 was added to the EHT to help detach it from the mold. After 30min, the PDMS posts were lifted from the mold and the EHT was placed into a new 24 well plate containing 1.6 mL of RPMI containing B27 supplement (Thermo Fisher) and 33μg/mL aprotinin. Media was changed every other day until further processing. After 20 days of culture, videos of EHT contraction were taken on a KEYENCE BZ-X microscope at 50fps with 4×4 pixel binning. Videos were imported into Fiji and analyzed with MUSCLEMOTION macro with default settings.13 The contraction parameters for each contraction were averaged to give an EHT level measurement.
Regulatory Variants Computational Pipeline.
The pipeline relies on the bedtools tool to sequentially filter the starting variant list for variants that overlap regions with epigenetic evidence of enhancer modifying potential.14 The epigenetic datasets were all derived from IPSC-CMs and are listed in Table I in the Supplement. In datasets where multiple replicates were available, we created a superset representing all peaks found. The pipeline finds variants that are predicted to disrupt or create transcription factor binding sites. In order to use find new transcription factor binding sites created by variants, we used the GATK FastaAlternativeReferenceMaker to insert SNP variants into the reference genome.15 We then used Homer’s scanMotifGenomeWide.pl to search for GATA4 and TBX5 sites in the alternative reference and kept only sites that were new.16 In the case of multi-allelic variants, one alternative allele was chosen at random. These additional sites were used in the pipeline alongside sites present in the unchanged reference. We executed this pipeline on variants that passed all quality filters from the gnomAD v.2.1 release.
Association of Enhancer Variant with Phenotypic Data.
Phenotypic measurements of heart function and whole genome sequencing data were accessed as in.17 Individual measures were obtained for left ventricular internal diameter-diastole (LVIDd) and left ventricular posterior wall thickness during diastole (LVPWd) from echocardiogram reports and spanned as much as 14 years of echocardiogram data. The diagnosis of heart failure was determined by ICD9 diagnosis codes 425 and all sub-codes, and ICD10 diagnostic codes I42 and all sub-codes. Trajectory analysis of echo measurements was conducted as in.17 Briefly, we used PROC TRAJ in SAS 9.4,18 which uses a likelihood function to assign a each individual a phenotypic cluster and probability of belonging to that cluster. An individual’s variant status was regressed against cluster probability and was controlled for genetic ancestry (PC1–3) and sex in R.
Statistical Methods.
Statistical evaluation was performed using GraphPad Prism. Significance was set a priori at p < 0.05. Non parametric one-way ANOVA with Dunn’s multiple comparison correction was used to compare enhancer activity from individual plasmid in iPSC-CMs. One-way ANOVA with Dunnett’s multiple comparison testing was used to evaluate expression in edited iPSC-CMs and EHTs. Unpaired t test was used to compare expression from reference vs alternative alleles. Statistical tests are indicated for each data set in the legend.
RESULTS
Integrated epigenetic analysis identifies candidate enhancer regions for two cardiomyopathy genes.
To find putative modifying regulatory variants associated with cardiomyopathy we characterized the regulatory landscape of two of the most frequently involved genes in inherited cardiomyopathy, LMNA and MYH7. While mutations in these two genes cause cardiomyopathy, the genes differ in expression patterns, with MYH7 expression demonstrating high level and tissue-restricted expression and LMNA having lower expression and in nearly all cell types. To identify candidate enhancer regions in the human left ventricle, we overlaid multiple datasets beginning with promoter-capture Hi-C data from IPSC-CMs which identified genomic regions predicted to interact with promoters.19 These regions were then interrogated for signals from human left ventricle-derived H3K27Ac ChIP-seq and ATAC-seq, as well as ChIP-seq for the cardiac transcription factors GATA4, TBX3/5, and NKX2.5 (complete list shown in Table I in the Supplement). Intersection of these datasets identified two candidate enhancer clusters for MYH7 and three for LMNA (Figure 1). MYH7 cluster 1 overlaps the MYH6 promoter, consistent with their co-regulation.20 MYH7 cluster 2 is ~7kb upstream of MYH7 and is marked by H3K27Ac, CTCF, ATAC signal, transcription factor binding and relatively low H3K4me3 marks. Although the promoter capture Hi-C data suggested many interactions, the integrated analysis focused on three potential enhancer clusters for LMNA. Cluster 1 was located > 100kb from the LMNA gene within the ARHGEF2 gene, while LMNA cluster 2 was located directly upstream of LMNA. Cluster 3 mapped to the large first intron of LMNA, overlapping the second exon. Similar to the MYH7 sites, the LMNA sites showed H3K27Ac and CTCF marks and open chromatin enrichment. The low H3K4me3 signals differentiated these sites from promoter regions. No enhancer clusters crossed topological associating domain (TAD) boundaries defined by human left ventricle chromatin capture analysis.21
Figure 1. Integrated epigenomic analysis identifies candidate enhancer regions for MYH7 and LMNA.
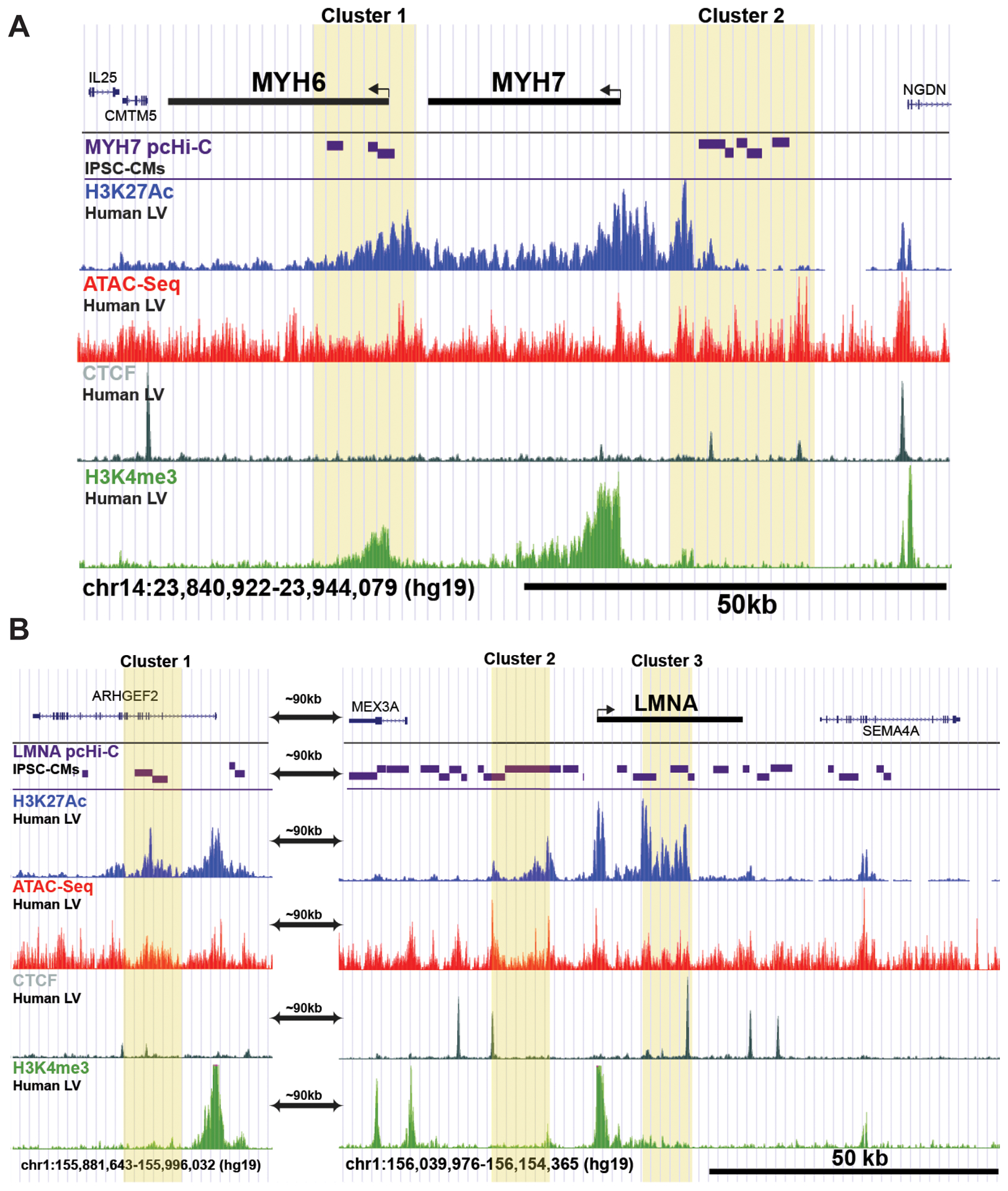
MYH7 encodes β-myosin heavy chain (MHC), the major contractile protein in the human left ventricle; mutations in MYH7 are a leading cause of inherited cardiomyopathy. Mutations in LMNA, which encodes lamin A/C also contribute to inherited cardiomyopathies. We intersected enhancer data from multiple sources to identify regulatory regions around these genes. Candidate enhancer regions are shown in yellow. A. The MYH6/7 genes are in near two clusters of candidate enhancers, highlighted in yellow boxes. B. Integrated epigenomic analysis identified three candidate enhancer clusters at the LMNA locus. The labels on the left indicate the data and cell/tissue source (full source listing is found in Table I in the Supplement). pcHi-C, promoter capture Hi-C. LV, left ventricle. IPSC-CMs, IPSC-derived cardiomyocytes.
Candidate enhancers display regulatory activity in cardiomyocytes.
Next, using a luciferase reporter assay, we tested the regulatory potential of the candidate enhancer regions identified in the MYH7 and LMNA gene loci. We used promoter-capture high throughput sequencing capture (Hi-C) data to define the boundaries of individual enhancers within clusters. Because of size, some enhancers were divided into smaller regions. Four of five MYH7 candidate enhancer regions showed significant activity in IPSC-CMs compared to a negative control genomic desert region (Figure 2A and Figure I in the Supplement). MYH7-C3, which is ~7kb upstream of MYH7 had the strongest signal, consistent with its abundant H3K27Ac ChIP-seq marks. MYH7-C2, which overlaps the MYH6 promoter, was active but with lower activity. The MYH7-C2 region also displayed enhancer properties in mouse atrial HL-1 cardiomyocytes, consistent with its role in MYH6 expression in atria (Figure II in the Supplement). The VISTA browser revealed that both of these regions also showed cardiac enhancer activity in vivo in mouse embryos.22 MYH7-C4, located further upstream than MYH7-C3, also demonstrated significant enhancer activity in IPSC-CM reporter assays. MYH7-C4 demonstrated a lower magnitude of enhancer activity, indicating that it is weaker than MYH7-C3 is IPSC-derived cardiomyocytes. For LMNA, five of six candidate enhancer regions showed significant activity in IPSC-CMs (Figure 2B). LMNA enhancer activity was generally lower than MYH7 enhancer activity, consistent with lower LMNA gene expression in IPSC-CMs and in hearts. LMNA-C5, located at the 3’ end of LMNA’s large first intron, had the highest activity. This region shows low H3K4me3 signal, consistent with its role as an enhancer and not a promoter. LMNA-C3 showed modest activity IPSC-CMs and greater activity in HL-1s and does appear as an enhancer in mouse embryonic hearts in the VISTA dataset.22
Figure 2. Enhancer activity in IPSC-Derived Cardiomyocytes (IPSC-CMs).
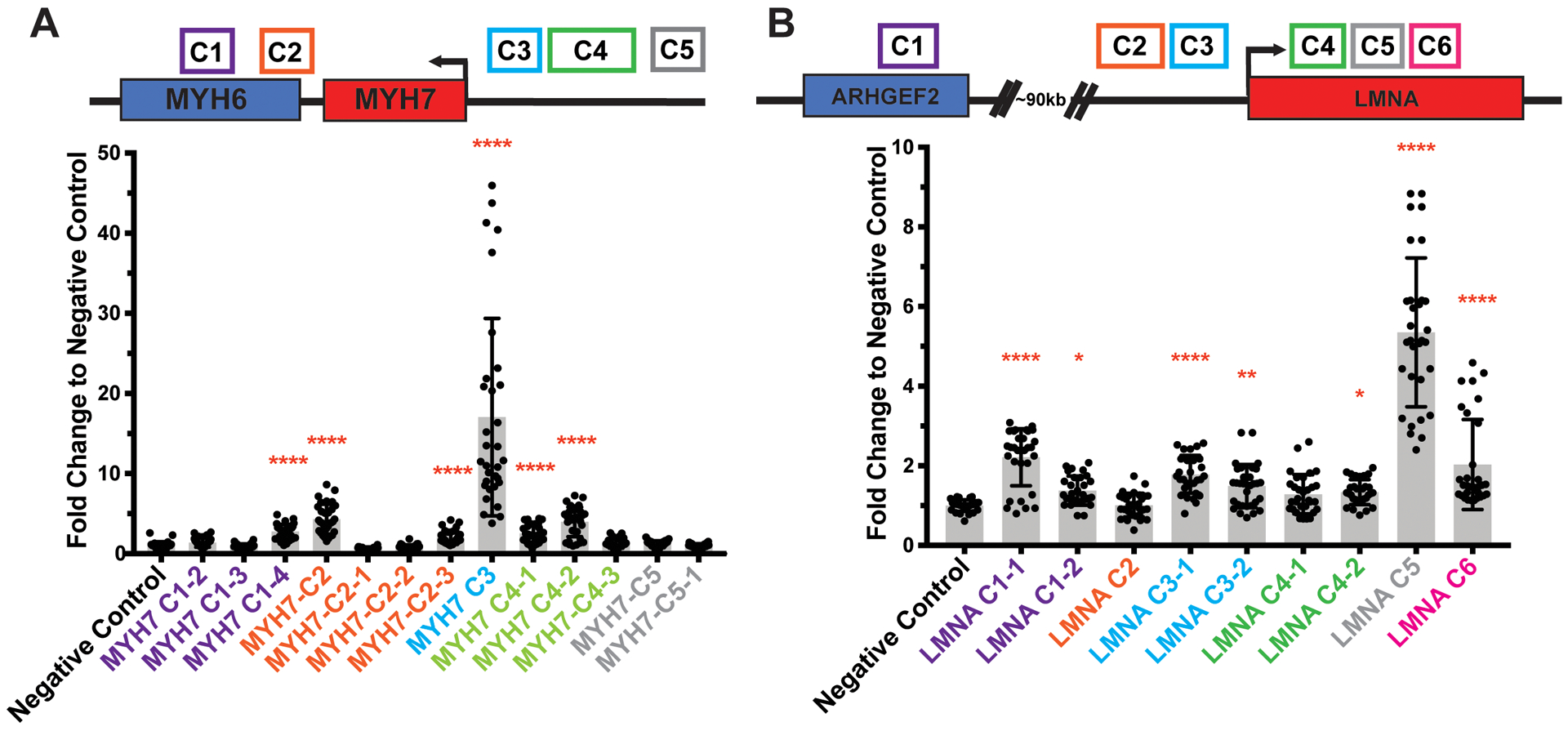
A luciferase reporter assay was used to test for enhancer activity in IPSC-CMs. The position of candidate enhancers is shown along the top in colored boxes. The clusters in Figure 1 were evaluated as smaller regions. A. Regions from 4 of 5 candidate enhancer regions demonstrated activity in IPSC-CMs, with the highest activity for MYH7 C3. B. Five of six candidate enhancer regions for LMNA showed activity in IPSC-CMs, with the highest being LMNA-C5. These data indicate the candidate MYH7 and LMNA enhancers have activity in human cardiomyocyte-like cells. Data is displayed as fold change to a negative control 500bp genomic desert region with mean ±SD. Expression was measured in an average of 32 assays per construct over four independent differentiations. Significance vs negative control determined by nonparametric one-way ANOVA with Dunn’s multiple comparisons correction. *<0.03, **<0.0021, ****<0.0001.
Loss of the MYH7-C3 enhancer shifts from MYH7 to MYH6 expression, altering protein levels and accelerating contractile rate in engineered heart tissues.
To test if candidate enhancers are required for target gene expression, we deleted regions of interest from the MYH6/7 locus in iPSCs using gene editing. We focused on MYH7-C3 and MYH7-C4 regions because they had significant activity in reporter assays. We employed a dual cutting CRISPr-Cas9 strategy to remove the candidate enhancer regions (Figure III in the Supplement). PCR genotyping confirmed the expected heterozygous and homozygous deletion in independent lines (Figure III in the Supplement). All edited cells passed karyotypic and off-target quality control testing (Figure IV in the Supplement). We differentiated enhancer-deleted iPSCs into cardiomyocytes and measured MYH7 and MYH6 mRNA expression using qPCR. MYH7-C3+/− and −/− cells had a significant decrease in MYH7 expression and increase in MYH6 expression, with dose-dependency (Figure 3A, Figure VI in the Supplement). We evaluated protein expression and found MYH7-C3+/− and −/− IPSC-CMs demonstrated a significant increase in the α-MHC to β-MHC protein ratio (Figure 3E, F). In general, MYH6 and MYH7 RNA changes were correlated with α-MHC to β-MHC ratio changes in IPSC-CMs (Figure VI in the Supplement). Deletion of the MYH7-C4 region had no significant impact on MYH7 or MYH6 mRNA or protein levels, indicating not all upstream regions impact gene and protein expression (Figure 3D and E). To ensure comparable maturity and purity, MYH7 and MYH6 gene expression measurements were normalized using a panel of cardiomyocyte genes. Additionally, there were no significant differences between genotypes in IPSC-CM purity as measured by cardiac troponin T (cTnT) flow cytometry (Figure III in the Supplement). α-MHC, encoded by MYH6, hydrolyzes ATP at a higher rate than MYH7, which leads to a faster rate of contraction.23 We evaluated the contractile properties of engineered heart tissues (EHTs) generated from MYH7-C3 deleted cardiomyocytes and unedited controls. EHTs deleted for MYH7-C3 showed a faster time to peak contraction and shorter relaxation time measurements, consistent with an faster rate of contraction and relaxation (Figure 3F). Average contraction amplitude was reduced in MYH7-C3 deleted EHTs (Figure VII in the Supplement), which might reflect a greater energetic cost associated with increased α-MHC expression. Therefore, deletion of MYH7-C3 decreases MYH7 and increases MYH6, which results in a faster contraction rate typical of α-MHC.
Figure 3. Deletion of the MYH7-C3 enhancer increases MYH6 and reduces MYH7 mRNA and protein and produces faster contraction and relaxation in engineered heart tissues.
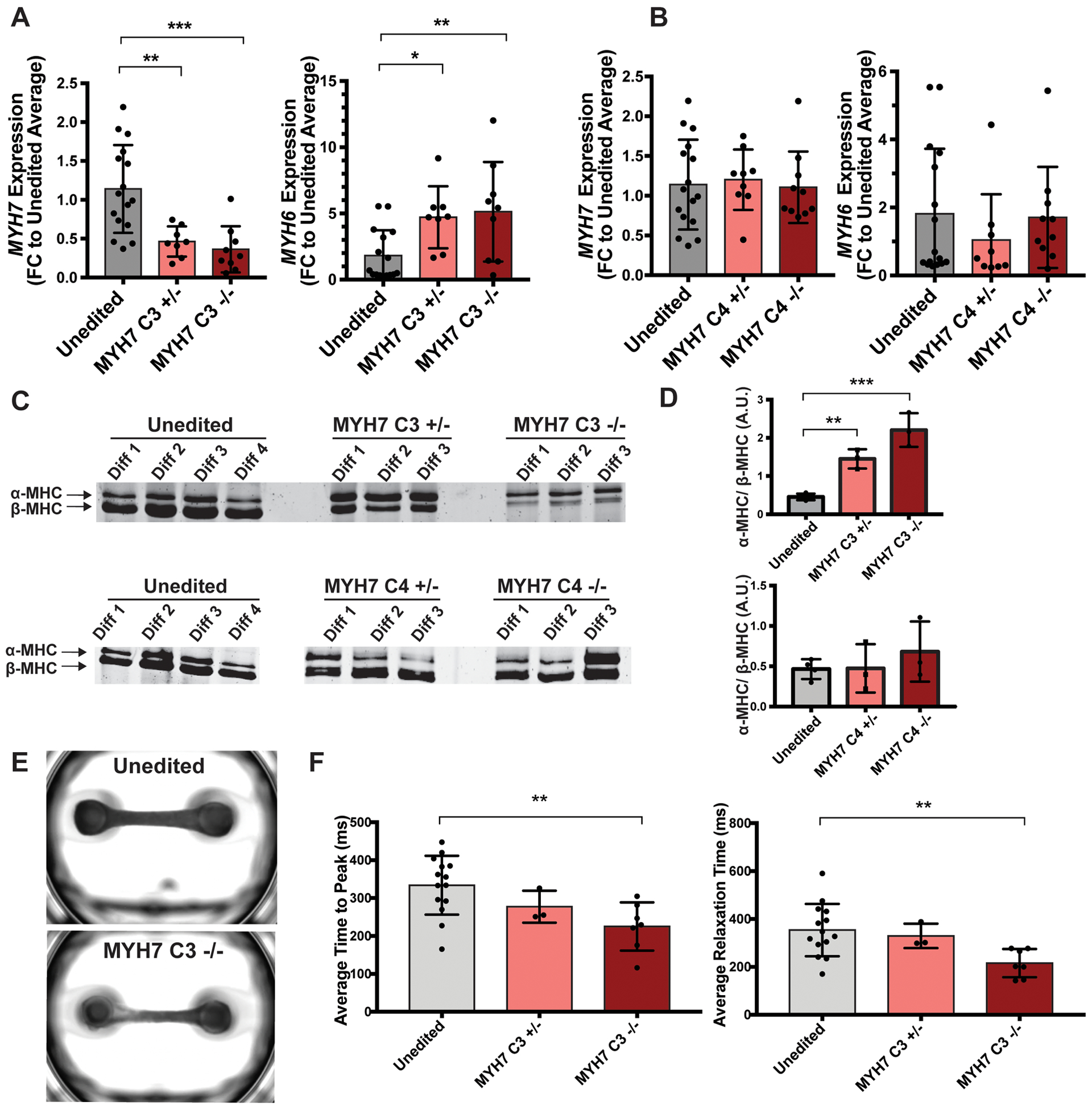
A. Gene editing was used to delete the MYH7-C3 enhancer heterozygously (+/−) or homozygously (−/−). MYH6 and MYH7 mRNA expression was assayed by qPCR and showed a dose-dependent increase in MYH6 expression and reduction in MYH7 expression. Therefore, the MYH7-C3 enhancer is required for MYH7 expression (unedited n=16, MYH7 C3+/− n=8, MYH7 C3−/− n=9.) B. Deletion of the MYH7-C4 enhancer had little effect, demonstrating a specificity of these findings to MYH7-C3 (unedited n=16, MYH7 C4+/− n=9, MYH7 C4−/− n=10.) C. α-MHC and β-MHC protein ratios were quantified using SDS-PAGE. D. Quantification of α-MHC/β-MHC protein ratios demonstrating correlation with the differences seen at the RNA level. E. Representative images of engineered heart tissues (EHTs) containing unedited or MYH7-C3 homozygous deleted IPSC-CMs. F. Average time to peak (top) and average relaxation times (bottom) measurements of EHT contractions containing unedited or MYH7-C3 deleted cells showed a decrease in time to peak contraction and relaxation in MYH7-C3 deleted EHTs, consistent with the shift from MYH7/β-MHC to MYH6/α-MHC and the known faster ATPase cycle for α-MHC. Each point represents the average time to peak measurement of a single EHT across multiple contractions (unedited n=14, MYH7 C3+/− n=3, MYH7 C3−/− n=7.) All data shown as mean ±SD. * determined by one-way ANOVA with Dunnett’s multiple comparisons correction. *<0.03, **<0.0021, ***<0.0002.
Active cardiac enhancers harbor genetic variants in transcription factor binding sites.
We next queried the MYH7 enhancers for naturally occurring sequence variants using the gnomAD database and selecting those that overlapped cardiac transcription factor binding motifs and/or were correlated with MYH7 expression in the GTEx eQTL dataset.24, 25 We identified six unique variants within MYH7 enhancers that overlapped transcription factor binding motifs and were within or nearby ChIP-seq peaks showing transcription factor binding in cardiac cells (Figure 4A, B, top). For each variant, we compared luciferase signals from plasmids carrying the reference or alternative allele in IPSC-CMs. The variant rs373958405 which is upstream of MYH6 disrupts a highly conserved site in the NKX2.5 binding motif, and plasmids encoding this variant demonstrated significantly reduced signals in IPSC-CMs compared to the reference allele when assessed by luciferase assay (Figure 4A, top right). Within MYH7-C3, we identified rs7149564 which disrupted a less conserved site in the NKX2.5 motif, and consequently, showed a more modest trending reduction in luciferase signal (Figure 4B, bottom left). A nearby variant (chr14_23912371_C), also in MYH7-C3, creates a TCF21 motif and correlated with higher luciferase activity, relative to reference. Within MYH7-C4, rs116554832, which overlapped a highly conserved site within a TBX5 motif, resulted in a reduction in luciferase signal (Figure 4B, bottom middle). A second C4 variant, rs10873105, correlated with MYH7 expression in GTEx skeletal muscle expression. This variant generates a Hox10 motif and causes an increased signal in luciferase reporter assays (Figure 4B, bottom right). These enhancer modifying variants (EMVs) are positioned to regulate cardiac function.
Figure 4. Genomic variation in MYH7 enhancer regions.
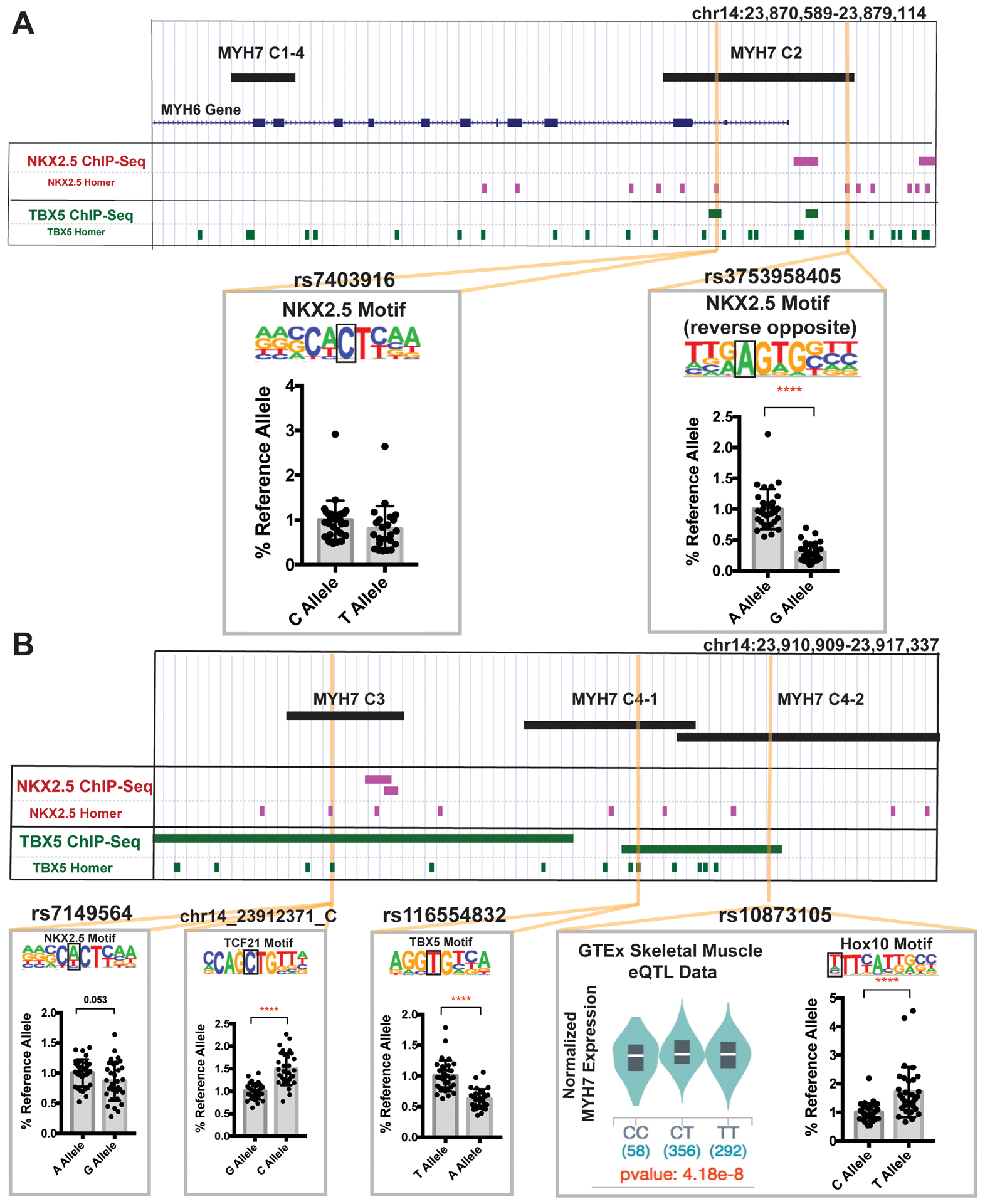
A. We queried the human genetic variation database gnomAD for naturally occurring human sequence variants in MYH7 enhancers. We selected variants overlapping cardiac transcription factor binding motifs, and/or correlating with MYH7 expression in the GTEx eQTL dataset. rs7403916 and rs373958405 fall within MYH7 C2 and disrupt NKX2.5 motifs. These variants were evaluated for reporter activity in IPSC-CMs and rs373958405 demonstrated reduced activity compared to the reference allele, indicating this variant may reduce expression by disrupting the enhancer activity of MYH7-C2. B. MYH7-C3 contains rs7149564 and chr14_23912371_C. rs7149564 disrupts an NKX2.5 motif and produced a trending reduction in activity in IPSC-CMs. chr14_23912371_C generates a TCF21 motif and produced an increased IPSC-CM luciferase signal. MYH7-C4 contains rs116554832 and rs10873105. rs116554832 disrupts a TBX5 motif and reduced activity in IPSC-CMs. rs10873105 is correlated with MYH7 expression in GTEx skeletal muscle data and creates a Hox10 motif. This variant generated increased activity in IPSC-CMs. These data indicate that sequence variants in transcription factor binding sites within enhancer regions can alter enhancer function and potentially affect MYH7 gene expression. All data shown as mean ± SD. Data was derived from an average of 30 different assays per condition, from across least 3 separate differentiations. Significance determined by unpaired t-test. ****<0.0001.
Genome-wide evaluation of enhancer modifying variants identifies variants controlling the activity of cardiac enhancers.
Since we identified EMVs within MYH7 enhancers, we next sought other regulatory variants applying this strategy genome-wide. We created a computational filtering pipeline to use publicly available data from IPSC-CMs to identify variants within enhancer regions that alter transcription factor binding, with focus on transcription factors important for the heart (Figure 5A). We benchmarked this pipeline using variant sets from GTEx and gnomAD.24, 25 As expected, eQTLs in heart tissues were more likely to be found using this strategy (Figure 5B). Rare variants were also more likely to survive the filtering steps of this pipeline, consistent with transcription factor binding sites within enhancer regions being under greater evolutionary constraint and less subject to change (Figure 5C). We executed this pipeline on the variants in gnomAD variants and identified 1,747 variants with EMV potential. Ninety-four of these variants mapped to orthologous regions in the mouse that had been tested in the VISTA database (Figure VIII in the Supplement), and 56 (60%) showed activity in the developing mouse heart. We selected five variants for experimental testing by choosing variants near five genes important for normal heart function (MICAL2, MYH6, NPPA, TNNT2, and GATA4).26–29 A detailed map of each genomic regions is shown in Figures IX–XV in the Supplement. The candidate enhancers from each of these genes was first tested for luciferase activity in IPSC-CMs, and four of the five variants were active (Figure 5D). We then compared expression from the reference and alternative alleles in IPSC-CMs. The alternative allele for MYH6 and GATA4 showed significantly reduced function, demonstrating this pipeline has the capacity to identify EMVs for cardiac genes.
Figure 5. Computational pipeline to identify enhancer modifying variants.
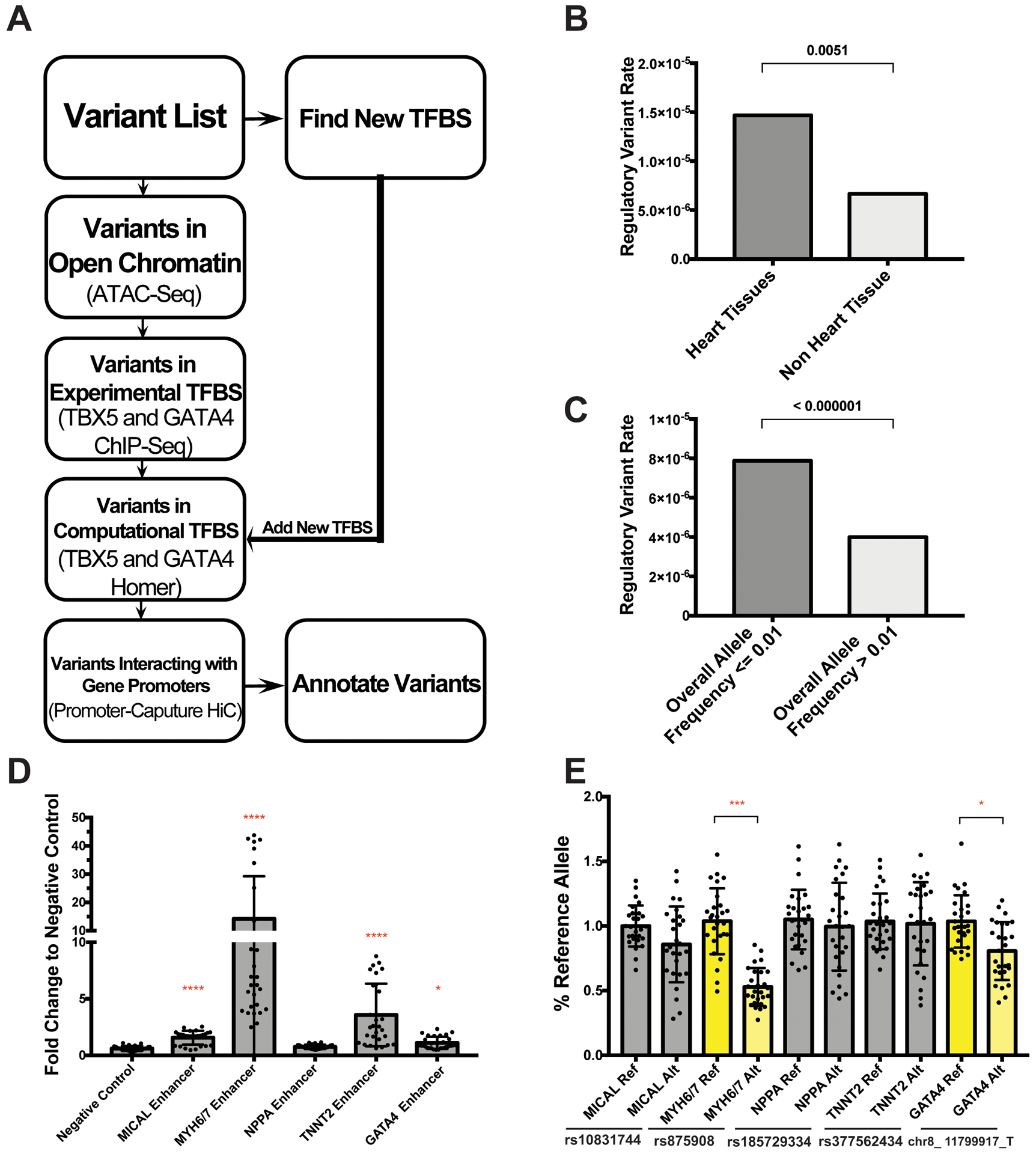
A. Schematic showing the pipeline filtering steps used to identify enhancer modifying variants (EMVs) genome-wide. B. This strategy disproportionately identified significant GTEx eQTLs from heart tissues versus non-heart tissues, and disproportionately identified rare alleles, indicating both tissue specificity and sequence conservation. C. Significance determined in B & C by Fisher’s exact test. D. Candidate enhancers for five genes were selected for testing in the luciferase reporter assay in IPSC-CMs. Four of five of these candidate regions validated as functional enhancer regions with signal greater than the negative control region (as tested in an average of 32 assays using four separate differentiations). Significance vs negative control was determined by nonparametric one-way ANOVA with Dunn’s multiple comparisons correction. E. We then tested the reference and alternate alleles to assess whether the alternative variant shifted activity. The enhancer modifying variant in MYH6/7 and in GATA4 significantly changed expression (highlighted in yellow) (data derived from an average of 27 assays per condition, from four separate differentiations). Significance determined by unpaired t-test. All data shown as mean ± SD. *<0.03, ***<0.0002, ****<0.0001.
A variant ~2kb upstream of MYH7 correlates with cardiomyopathic features in longitudinal echocardiographic imaging.
We next determined the potential of EMVs to act as modifiers of cardiomyopathy. rs875908, which was predicted to regulate MYH6 by the computational pipeline, is an EMV located ~2kb upstream of the transcriptional start site of MYH7 (Figure 6A). The region harboring this variant showed significant luciferase activity (MYH6/7 enhancer in Figure 5D). We deleted the region harboring this variant, MYH7-C6, in iPSCs (Figure III in the Supplement). Heterozygous removal of this region in IPSC-CMs caused a reduction in MYH7 expression but no change in MYH6 expression in IPSC-CMs (Figure 6B). Homozygous deletion of this region showed an approximately 100 fold reduction in MYH7 expression levels and a qualitative, but no significant increase in MYH6 levels (Figure 6B). Homozygous deleted cells also showed a significant increase in the α/β-MHC protein ratio (Figure 6C, D). The rs875908 variant was identified because it is bound by GATA4 and TBX5 sites and is predicted to disrupt a TBX5 motif (Figure 7A). GTEx eQTL data show this variant correlates with MYH7 expression in skeletal muscle with trending significance for expression in left ventricle (Figure 7B).
Figure 6. Deletion of the C6 enhancer region alters MYH6/7 expression.
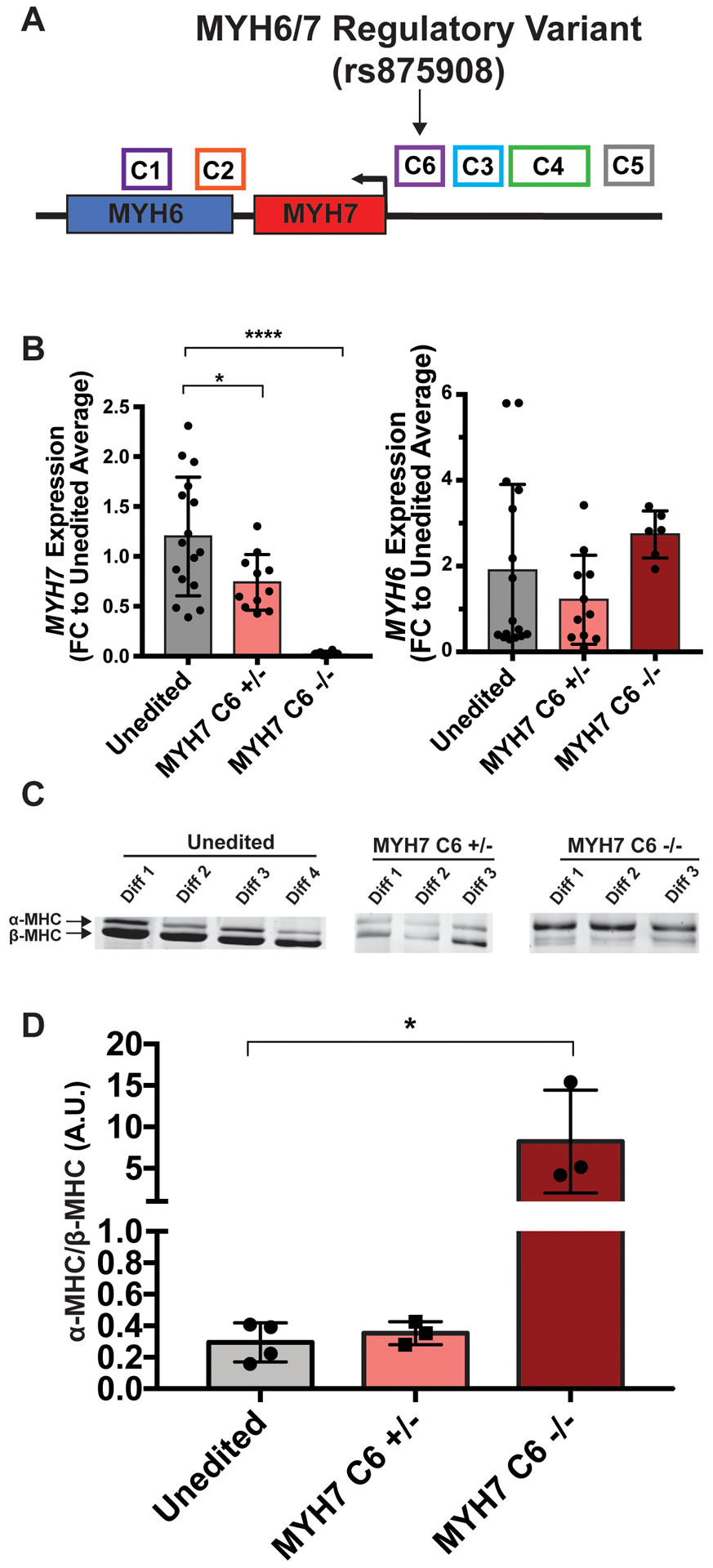
A. Schematic demonstrating the location of the MYH6/7-C6 enhancer region; the position of rs875908 is indicated. B. IPSC-CM MYH6 and MYH7 expression levels in cells deleted heterozygously or homozygously for the C6 enhancer region containing rs875908. MYH6/7 levels were assayed by qPCR, and showed a dose-dependent reduction in MYH7 expression. Therefore, rs875908, is within an enhancer region required for strong MYH7 expression in human cardiomyocyte like cells (unedited n=16, MYH7-C6+/− n=11, MYH7-C6−/− n=6). C. SDS-PAGE analysis of myosin heavy chain protein (MHC) isoforms in MYH7-C6 +/− and −/− cells. D. Quantitation of α-MHC/β-MHC ratios indicates that RNA differences correlate with changes in protein level. Significance determined by one-way ANOVA with Dunnett’s multiple comparisons correction. *<0.03, ****<0.0001.
Figure 7. Correlation of MYH6/7 rs875908 EMV with MYH7 mRNA cardiac expression and longitudinal shift in left ventricular dimensions over time in human cardiomyopathy patients.
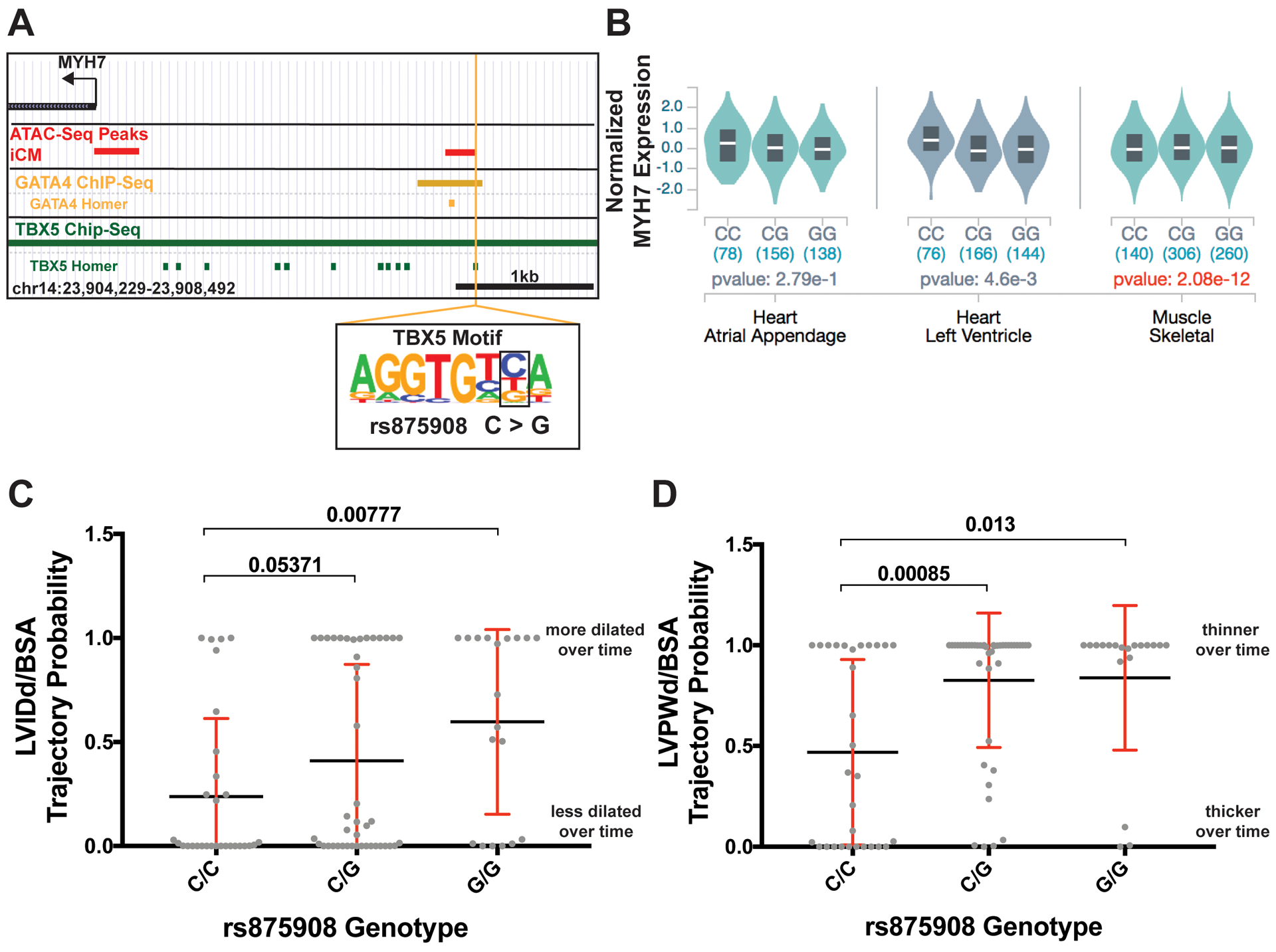
A. The location of the MYH6/7 regulatory variant is shown; it is bound by both GATA4 and TBX5 signals. The variant disrupts a site within the TBX5 transcription factor motif. B. eQTL data from the GTEx project shows that the variant genotype correlates with MYH7 expression across multiple striated muscle samples; the larger number of skeletal muscle samples yields genome wide significance in expression levels. C. Association of variant status with LVIDd/BSA over time in cardiomyopathy patients whose data was derived from the electronic data warehouse from Northwestern Medicine. D. Association of variant genotype with LVPWd/BSA over time in in cardiomyopathy cases. These data indicate that rs875908 associated with changes of left ventricular morphology including a more dilated left ventricle and thinner posterior wall. Significance determined using a linear regression model corrected for genetic ancestry and sex. LVIDd/BSA, left ventricular internal diameter during diastole corrected for body surface area. LVPWd/BSA, left ventricular posterior wall thickness during diastole corrected for body surface area.
To ascertain whether rs875908 correlated with cardiac outcomes, we evaluated trajectory probabilities of left ventricular dimensions over time using genomic and echocardiographic information derived from the Northwestern biobank. This approach assigns a probability of maintaining an echocardiographic change overtime.17 The rs875908-G allele correlates with a more dilated left ventricle over time in participants selected with cardiomyopathy diagnosis codes (Figure 7C). This correlation was not observed when using clinical data from non-selected biobank participants (Figure V in the Supplement). The rs875908-G allele also correlates with a thinner left ventricle posterior wall thickness at end-diastole (LVPWd) over time in those with cardiomyopathy diagnostic codes (Figure 7B). Variant association with left ventricular wall thickness was also present with all subjects, but with a weaker signal (Figure V in the Supplement). The cardiomyopathy diagnostic codes in this cohort were for dilated cardiomyopathy. In dilated cardiomyopathy, a thinner wall over time translates to a more diseased heart. These data support that the EMV rs875908 correlates with a more severe dilated cardiomyopathy phenotype and demonstrates the pipeline identifies EMVs on a genome-wide scale and that some of these EMVs have clinical correlates.
DISCUSSION
Cardiomyopathy gene enhancers.
We integrated epigenomic data to uncover candidate enhancers for a highly expressed and tissue restricted locus like the MYH6/7 genes. We also showed that this approach can be used on more ubiquitously expressed genes like LMNA, a gene also important for cardiomyopathy. This data integration has the power to identify regulatory regions remote from the gene of interest and uncover human genetic variation that alters the activity of these regions.
An MYH7/6 Super-Enhancer.
Promoter capture Hi-C data from human cardiomyocytes19 indicates that the MYH7 and MYH6 gene promoters contact each other within 3-dimensional space. Further, an enhancer cluster positioned ~7kb upstream of MYH7 also interacts with the MYH7 gene promoter. Since multiple individual parts of this enhancer cluster have activity in human cardiomyocytes, it is likely this cluster represents a super-enhancer.30 Super-enhancers are known to regulate genes critical for cell identity.31 We showed that deletion of the MYH7-C3 enhancer region reduced MYH7 expression in IPSC-CMs, and, correspondingly, deletion of the MYH7-C3 enhancer increased MYH6 expression resulting in an αMHC/βMHC ratio and a faster rate of contraction in EHTs. Deletion of the MYH7-C3 promoter reciprocally shifts expression, lowering MYH7 and increasing MYH6, akin to what has been described after thyroid hormone exposure and approaching the pattern seen in the developing rodent ventricle.32–34 The faster rate of contraction/relaxation after deleting MYH7-C3 is distinct from what occurs in hypertrophic cardiomyopathy EHTs, which better reflect the relaxation defects seen in hypertrophic cardiomyopathy.35, 36 Importantly, deletion of a nearby region (the C4 region) had no effect on MYH6 or MYH7 expression. Thus, the observed expression changes are not the result of nonspecific disruption of this highly active locus.
These data favor a model where the MYH6 and MYH7 promoter regions form a 3-dimensional complex with the super-enhancer upstream of MYH7 (Figure 8). In this model, the super-enhancer, containing C3, plus additional MYH7-specific enhancer regions induce MYH7 expression, which is critical during heart development, and this same region may be employed in heart failure. The concomitant increase in MYH6 expression may be due to an inhibitory function in C3 or an independent mechanism that compensates for reduced MYH7 expression. These findings are reminiscent of the murine Scn5A-Scn10A locus, a region important for regulating electrical control of the heart.37
Figure 8. Schematic diagram of the MYH6/7 locus during cardiac development.
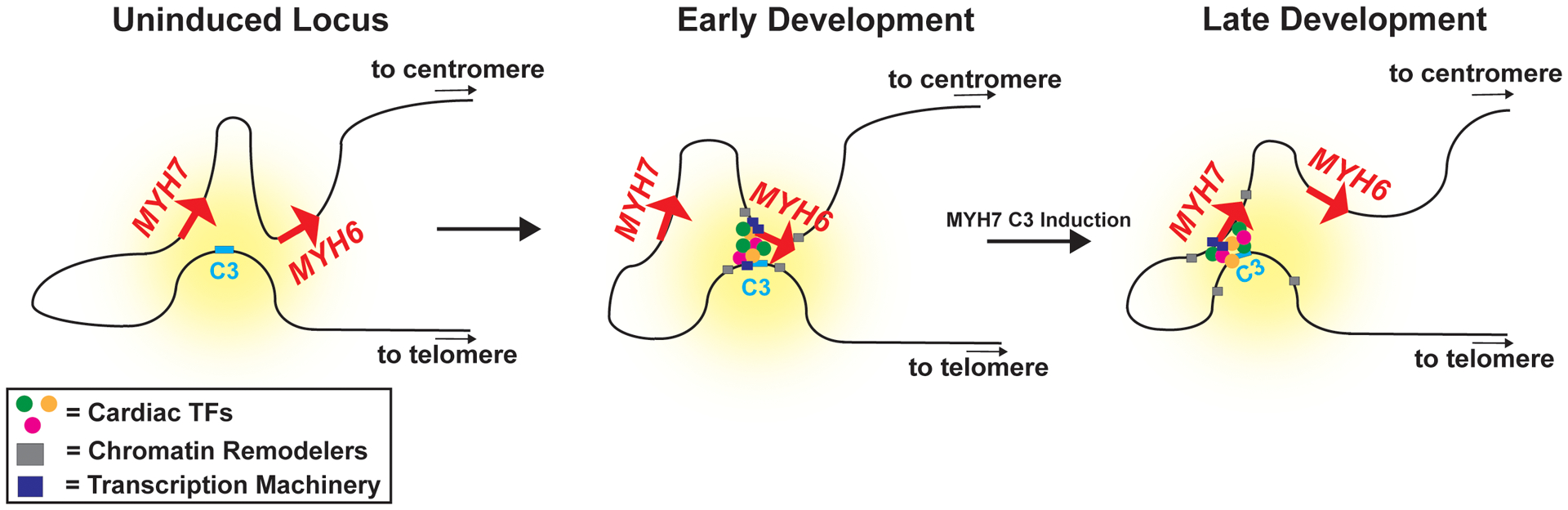
At the uninduced locus, the MYH7 and MYH6 promoter regions form a complex with a super-enhancer containing the MYH7-C3 enhancer. The effect of the super-enhancer is depicted as the yellow hue. In early development, this super-enhancer is primarily in contact with the MYH6 promoter, which recruits cardiac transcription factors, chromatin remodelers, and transcription machinery to drive MYH6 expression. During development, activation of the MYH7-C3 region reorganizes the complex and the MYH7 promoter preferentially contacts the regulatory regions. Through competition for transcriptional machinery or through an independent separate mechanism, the MYH6 gene is downregulated. Cells lacking the MYH7-C3 region can established the chromatin structure of the locus but are unable to switch interactions to the MYH7 promoter, causing an upregulation of MYH6 and a downregulation of MYH7. This model highlights the importance of an MYH7 enhancer which is an attractive therapeutic target. TFs, transcription factors.
Integrated genomics to identify EMVs.
The computational pipeline identified rs875908, a common variant with MAF ranging from 35% to 47% in various populations, as a candidate EMV for cardiomyopathy. This variant correlated with altered MYH7 expression and with a more severe dilated cardiomyopathy phenotype over time, as marked by a more dilated, thinner walled ventricle. The MYH6/7 ratio is known to shift during heart failure, with end stage hearts exhibiting an increase in MYH7 and a decrease in MYH6. With prolonged shift of myosin expression, or a specific magnitude of shift, this change in myosin expression may actually contribute to heart failure.38 Supporting this, the MYH6/7 ratio has previously been implicated in heart failure phenotypes.39 A distinct contributory mechanism could involve variants within MYH6/7 enhancers, variants in linkage disequilibrium or even pathogenic coding mutations. Varied expression of pathogenic MYH7 mutations has been shown to affect cardiomyopathy phenotypes.40, 41 A region related to the C6 enhancer, containing the EMV rs875908, was previously deleted in a mouse. Mice missing this C6 orthologous region had reduced MYH7/β-MHC but no change in MYH6/α-MHC42, similar to what was shown here in human cells. This study measured MYH7 expression in the mouse embryonic heart, which differs from the human developing and mature heart. Consistent with the human genetic findings, mouse hearts lacking this enhancer region demonstrated reduced fractional shortening and higher amounts of myofiber disarray, which additionally support the functionality of this region.
A pipeline for EMVs.
As deep sequencing data of intergenic regions becomes more available, the importance of noncoding annotation of disease genes will become vital and permit the integration of this information into clinical care. We demonstrated here a robust pipeline to identify genetic variants positioned to alter gene expression. We identified >1,700 putative EMVs in the gnomAD database, which were linked to multiple genes important for cardiac function like TNNT2, NPPA, GJA5, and MEF2A. As anticipated, many of the predicted EMVs were infrequent in the population, further supporting the functional role of this type of expression-altering change. However, we did identify EMVs at higher population frequency; higher frequency EMVs, because of their prevalence, are more likely to show population level clinical correlates, such as what was detected using electronic health record data. Targeted assessment of EMVs annotated by specific epigenetic marks can have clinical utility.
Supplementary Material
Clinical Perspective.
What is new?
Genetic variation in the coding regions of genes like MYH7 is known to cause inherited cardiomyopathy and heart failure.
We now identified genetic variation in the noncoding regions of MYH7 which regulates expression of beta myosin heavy chain, the major motor protein in the heart.
One genetic variant, rs875906, correlated with cardiomyopathy features derived from biobank and electronic health record information.
What are the clinical implications?
Molecular markers like epigenetic footprints and chromatin conformation can be used to find the regulatory regions for cardiac genes, including genes that cause cardiomyopathy.
Genetic variation in these regulatory regions can alter expression of cardiac genes and change heart function.
ACKNOWLEDGEMENTS
We thank Dr. David Barefield and Dr. Mattia Quattrocelli for insight and support. We thank Patrick Page for technical support.
FUNDING SOURCES
National Institutes of Health NIH HL128075, NIH HL142187, NIH HL141698, American Heart Association 18CDA34110460.
Nonstandard Abbreviations
- cTnT
cardiac troponin T
- EMV
enhancer modifying variant
- eQTL
expression quantitative trait loci
- GEO
gene expression omnibus
- GTEx
genotype tissue expression project
- Hi-C
high throughput sequencing capture
- IPSC-CMs
induced pluripotent stem cell-derived cardiomyocytes
- LVIDd
left ventricular internal diameter-diastole
- LVPWd
left ventricular posterior wall thickness during diastole
- MHC
myosin heavy chain
- pcHi-C
promoter capture Hi-C
Footnotes
CONFLICT OF INTEREST DISLOSURES:
The authors declare no competing interests related to the content of this work. EMM is or was a consultant to Amgen, Avidity, AstraZeneca, Cytokinetics, Pfizer, 4D Molecular Therapeutics, Exonics, Tenaya Therapeutics and is the founder of Ikaika Therapeutics, and these activities are outside the content of this work.
REFERENCES
- 1.McNally EM, Golbus JR and Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konno T, Chang S, Seidman JG and Seidman CE. Genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol. 2010;25:205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maron BJ, Rowin EJ and Maron MS. Global Burden of Hypertrophic Cardiomyopathy. JACC Heart Fail. 2018;6:376–378. [DOI] [PubMed] [Google Scholar]
- 4.McNally EM and Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017;121:731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marian AJ. Modifier genes for hypertrophic cardiomyopathy. Curr Opin Cardiol. 2002;17:242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barp A, Bello L, Politano L, Melacini P, Calore C, Polo A, Vianello S, Soraru G, Semplicini C, Pantic B, et al. Genetic Modifiers of Duchenne Muscular Dystrophy and Dilated Cardiomyopathy. PLoS One. 2015;10:e0141240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verdonschot JAJ, Robinson EL, James KN, Mohamed MW, Claes GRF, Casas K, Vanhoutte EK, Hazebroek MR, Kringlen G, Pasierb MM, et al. Mutations in PDLIM5 are rare in dilated cardiomyopathy but are emerging as potential disease modifiers. Mol Genet Genomic Med. 2020;8:e1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersson R and Sandelin A. Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet. 2020;21:71–87. [DOI] [PubMed] [Google Scholar]
- 9.Tanjore R, Rangaraju A, Vadapalli S, Remersu S, Narsimhan C and Nallari P. Genetic variations of beta-MYH7 in hypertrophic cardiomyopathy and dilated cardiomyopathy. Indian J Hum Genet. 2010;16:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Claycomb WC, Lanson NA, Jr., Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A and Izzo NJ, Jr. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaaf S, Eder A, Vollert I, Stohr A, Hansen A and Eschenhagen T. Generation of strip-format fibrin-based engineered heart tissue (EHT). Methods Mol Biol. 2014;1181:121–129. [DOI] [PubMed] [Google Scholar]
- 13.Sala L, van Meer BJ, Tertoolen LGJ, Bakkers J, Bellin M, Davis RP, Denning C, Dieben MAE, Eschenhagen T, Giacomelli E, et al. MUSCLEMOTION: A Versatile Open Software Tool to Quantify Cardiomyocyte and Cardiac Muscle Contraction In Vitro and In Vivo. Circ Res. 2018;122:e5–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quinlan AR and Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H and Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pottinger TD, Pesce LL, Gacita A, Montefiori L, Hodge N, Kearns S, Salamone IM, Pacheco JA, Rasmussen-Torvik LJ, Smith ME, et al. Trajectory analysis of cardiovascular phenotypes from biobank data uncovers novel genetic associations. bioRxiv. Preprint posted online May 11, 2020:2020.2005.2010.087130. [Google Scholar]
- 18.Jones BL and Nagin DS. Advances in Group-Based Trajectory Modeling and an SAS Procedure for Estimating Them. Sociological Methods & Research. 2007;35:542–571. [Google Scholar]
- 19.Montefiori LE, Sobreira DR, Sakabe NJ, Aneas I, Joslin AC, Hansen GT, Bozek G, Moskowitz IP, McNally EM and Nobrega MA. A promoter interaction map for cardiovascular disease genetics. Elife. 2018;7:e35788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morkin E Regulation of myosin heavy chain genes in the heart. Circulation. 1993;87:1451–1460. [DOI] [PubMed] [Google Scholar]
- 21.Leung D, Jung I, Rajagopal N, Schmitt A, Selvaraj S, Lee AY, Yen CA, Lin S, Lin Y, Qiu Y, et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature. 2015;518:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Visel A, Minovitsky S, Dubchak I and Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.VanBuren P, Harris DE, Alpert NR and Warshaw DM. Cardiac V1 and V3 myosins differ in their hydrolytic and mechanical activities in vitro. Circ Res. 1995;77:439–444. [DOI] [PubMed] [Google Scholar]
- 24.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv. 2020:531210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lundquist MR, Storaska AJ, Liu TC, Larsen SD, Evans T, Neubig RR and Jaffrey SR. Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell. 2014;156:563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houweling AC, van Borren MM, Moorman AF and Christoffels VM. Expression and regulation of the atrial natriuretic factor encoding gene Nppa during development and disease. Cardiovasc Res. 2005;67:583–593. [DOI] [PubMed] [Google Scholar]
- 28.Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S, Cowan J, Morales A, Parvatiyar MS and Potter JD. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2009;2:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yilbas AE, Hamilton A, Wang Y, Mach H, Lacroix N, Davis DR, Chen J and Li Q. Activation of GATA4 gene expression at the early stage of cardiac specification. Front Chem. 2014;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pott S and Lieb JD. What are super-enhancers? Nat Genet. 2015;47:8–12. [DOI] [PubMed] [Google Scholar]
- 31.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA and Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metzger JM, Wahr PA, Michele DE, Albayya F and Westfall MV. Effects of myosin heavy chain isoform switching on Ca2+-activated tension development in single adult cardiac myocytes. Circ Res. 1999;84:1310–1317. [DOI] [PubMed] [Google Scholar]
- 33.Cappelli V, Bottinelli R, Poggesi C, Moggio R and Reggiani C. Shortening velocity and myosin and myofibrillar ATPase activity related to myosin isoenzyme composition during postnatal development in rat myocardium. Circ Res. 1989;65:446–457. [DOI] [PubMed] [Google Scholar]
- 34.Rundell VL, Manaves V, Martin AF and de Tombe PP. Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol. 2005;288:H896–903. [DOI] [PubMed] [Google Scholar]
- 35.Prondzynski M, Lemoine MD, Zech AT, Horváth A, Di Mauro V, Koivumäki JT, Kresin N, Busch J, Krause T, Krämer E, et al. Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol Med. 2019;11:e11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eschenhagen T and Carrier L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes-a systematic review. Pflugers Arch. 2019;471:755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Man JCK, Mohan RA, Boogaard MVD, Hilvering CRE, Jenkins C, Wakker V, Bianchi V, Laat W, Barnett P, Boukens BJ, et al. An enhancer cluster controls gene activity and topology of the SCN5A-SCN10A locus in vivo. Nat Commun. 2019;10:4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakao K, Minobe W, Roden R, Bristow MR and Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100:2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abraham WT, Gilbert EM, Lowes BD, Minobe WA, Larrabee P, Roden RL, Dutcher D, Sederberg J, Lindenfeld JA, Wolfel EE, et al. Coordinate changes in Myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype. Mol Med. 2002;8:750–760. [PMC free article] [PubMed] [Google Scholar]
- 40.Tripathi S, Schultz I, Becker E, Montag J, Borchert B, Francino A, Navarro-Lopez F, Perrot A, Ozcelik C, Osterziel KJ, et al. Unequal allelic expression of wild-type and mutated beta-myosin in familial hypertrophic cardiomyopathy. Basic Res Cardiol. 2011;106:1041–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang J, Wakimoto H, Seidman JG and Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickel DE, Barozzi I, Zhu Y, Fukuda-Yuzawa Y, Osterwalder M, Mannion BJ, May D, Spurrell CH, Plajzer-Frick I, Pickle CS, et al. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat Commun. 2016;7:12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim EY, Barefield DY, Vo AH, Gacita AM, Schuster EJ, Wyatt EJ, Davis JL, Dong B, Sun C, Page P, et al. Distinct pathological signatures in human cellular models of myotonic dystrophy subtypes. JCI Insight. 2019;4:e122686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Q, Jiang C, Xu J, Zhao MT, Van Bortle K, Cheng X, Wang G, Chang HY, Wu JC and Snyder MP. Genome-Wide Temporal Profiling of Transcriptome and Open Chromatin of Early Cardiomyocyte Differentiation Derived From hiPSCs and hESCs. Circ Res. 2017;121:376–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, et al. Large-scale discovery of enhancers from human heart tissue. Nat Genet. 2011;44:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ang YS, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu JD, Spencer CI, et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell. 2016;167:1734–1749 e1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, t Hoen PA, Bakkers J, Barnett P, et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012;122:2519–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He A, Kong SW, Ma Q and Pu WT. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci U S A. 2011;108:5632–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gacita AM, Dellefave-Castillo L, Page PGT, Barefield DY, Wasserstrom JA, Puckelwartz MJ, Nobrega MA and McNally EM. Altered Enhancer and Promoter Usage Leads to Differential Gene Expression in the Normal and Failed Human Heart. Circ Heart Fail. 2020;13:e006926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.England J and Loughna S. Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci. 2013;70:1221–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.