

Abstract
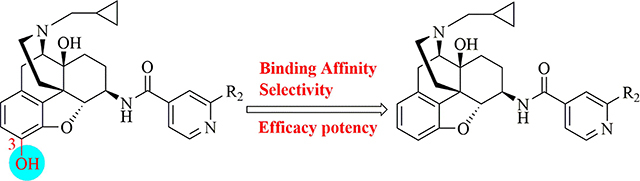
In the present study, the role of 3-hydroxy group of a series of epoxymorphinan derivatives in their binding affinity and selectivity profiles toward the opioid receptors (ORs) has been investigated. It was found that the 3-hydroxy group was crucial for the binding affinity of these derivatives for all three ORs due to the fact that all the analogues 1a-e exhibited significantly higher binding affinities compared to their counterpart 3-dehydroxy ones 6a-e. Meanwhile most compounds carrying the 3-hydroxy group possessed similar selectivity profiles for the kappa opioid receptor over the mu opioid receptor as their corresponding 3-dehydroxy derivatives. [35S]-GTPγS functional assay results indicated that the 3-hydroxy group of these epoxymorphinan derivatives was important for maintaining their potency on the ORs with various effects. Further molecular modeling studies helped comprehend the remarkably different binding affinity and functional profiles between compound 1c (NCP) and its 3-dehydroxy analogue 6c.
Keywords: 3-Hydroxy group, Opioid receptors, Binding affinity, Selectivity, Molecular docking
Graphical Abstract
1. Introduction
The opioid receptors (ORs) belong to G-protein-coupled receptors (GPCRs) and generally fall into four subtypes: μ opioid receptor (MOR), κ opioid receptor (KOR), δ opioid receptor (DOR), and the nociceptin/orphanin FQ (N/OFQ) peptide (NOP) receptor [1, 2]. ORs couple to Gi/Go G-proteins, generating the efflux of K+, closing voltage-gated Ca2+ channels and inhibiting adenylyl cyclase to reduce formation of cyclic adenosine monophosphate (cAMP) [3]. It has been extensively reported that activation of different types of ORs may yield various pharmacological behaviors [4]. As a result, a large number of full agonists, partial agonists and antagonists targeting ORs have been applied to treat different diseases or used in pharmacological research. For example, the MOR agonists, including morphine have been the frontline medication in the management of moderate to severe pain for a long time [5]. Naloxone and naltrexone, two primary MOR antagonists, have been utilized in the treatment of opioid use disorders [6]. Nalbuphine, a KOR agonist and MOR partial agonist, is the only approved non-scheduled opioid analgesic in the U.S [7]. Norbinaltorphimine (nor-BNI), behaved as a KOR antagonist in animal models, and has demonstrated antidepressant and antipanic-like activities [8]. As a highly selective DOR antagonist, naltrindole (NTI) was found to significantly attenuate the discriminative stimulus properties of cocaine in rats [9], and showed promise in the treatment of stress disorders [10]. However, these OR ligands are also accompanied by a battery of potential side effects which have compromised their applications [11]. Therefore, novel small molecules targeting ORs with minimal side effects are still highly desirable for various clinical applications.
In the past decade, we have been constructing an opioid ligands library, with small molecules featuring diversified heterocyclic substituents introduced onto the 4,5-epoxymorphinan skeleton [4, 12–21], and have explored the potential therapeutic applications of these new opioid ligands in preclinical studies [6, 22–28]. In an attempt to develop peripheral selective MOR ligands as therapeutic agents toward opioid-induced constipation (OIC) [12, 14, 15, 21–23, 25, 28], we obtained compounds 1a-e which were preliminarily identified as KOR/MOR dual selective ligands with subnanomolar binding affinities through the competitive radioligand binding assays, among which 1b and 1c exhibited moderate KOR selectivity over the MOR (Ki ratio, μ/κ: 3.5 and 9.62, respectively) [15] (Fig. 1).
Fig. 1.

Chemical structures of compounds 1a-e.
It has been well-accepted that the 4,5-epoxymorphinan skeleton embeds a prevalent para-hydroxyphenylethylamine moiety that corresponds to the N-terminal tyrosine residue in the endogenous opioid peptides, e.g. enkephalins, endorphins and dynorphins (Fig. 2) [29] and such a chemical structural feature should be critical for opioid receptor recognition in general, which was derived from previous studies of opioid ligands that were mainly from medicinal chemists’ perspective [30–37].
Fig. 2.

General structure of 4,5-epoxymorphinan containing the para-hydroxyphenylethylamine moiety.
It is noteworthy that nalfurafine (TRK-820), approved for the treatment of uremic pruritus in Japan, also bears the 4,5-epoxymorphinan skeleton while acting as a selective KOR agonist [38]. It has been reported that the 3-hydroxy, an essential functional group in the critical para-hydroxyphenylethylamine moiety, of nalfurafine derivatives, bearing the 4,5-epoxymorphinan skeleton or the decahydro(iminoethano)phenanthrene skeleton, has been proven indispensable for maintaining binding affinities for the opioid receptors [33, 39, 40]. Meanwhile, apparently such an observation mainly came from the structure-activity relationship (SAR) studies of nalfurafine derivatives with very limited information from structural biology due to technical limitation previously.
With vast progresses achieved in modern technology, a large number of GPCR crystal structures have been resolved in the last two decades. More recently, the crystal complexes of all three major ORs (i.e. MOR, KOR, and DOR) at both active and inactive states have been obtained [37, 41–45], which allow us to re-examine the roles of some “indispensable” moieties in opioid ligands from the angle of structural biology and possibly to revisit many taken-for-granted concepts and conclusions in the field. In 2018, the crystal structure of human KOR in complex with a potent 4,5-epoxymorphinan opioid agonist and an active-state-stabilizing nanobody was reported [37]. In this study, binding affinities of nalfurafine for the KOR and MOR were determined respectively, with KOR Ki = 0.32 nM and MOR Ki = 4.20 nM, and μ/κ Ki ratio at 13. Meanwhile, removal of the 3-hydroxy group in nalfurafine yielded compound CT-18 (Fig. 3). It was found that binding affinities of CT-18 for both the KOR and MOR (for KOR, Ki = 1.50 nM; for MOR, Ki = 533 nM; μ/κ: 355) decreased compared to those of nalfurafine while its MOR binding affinity appeared to be affected much more significantly than for the KOR [37]. Thus removing 3-hydroxy group seemed more favorable for opioid ligand selectivity for the KOR over the MOR. Such observation motivated us to reconsider the role of 3-hydroxy group for its influence on binding affinity and particularly on the selectivity profiles to the three ORs.
Fig. 3.

Chemical structures of nalfurafine and compound CT-18
Moreover, another example from a previous study demonstrated that chemical modification on the 3-hydroxy group of levorphanol with the aminothiazole moiety was well tolerant, and a highly potent KOR selective ligand ATPM was generated which the selectivity for the KOR over the MOR was reversed [46] (Fig. 4). These results further suggested that 3-hydroxy group may not be as critical as a required pharmacophore in at least some epoxymorphinan derivatives.
Fig. 4.

Chemical structures of levorphanol and compound ATPM
Take together, it seems that certain modifications at the 3-position may be acceptable from both perspectives of structural biology and medicinal chemistry. Therefore, we believe that it is imperative to re-investigate the influence of 3-hydroxy group of epoxymorphinan derivatives on especially KOR selectivity over the MOR. More specifically, we implemented the current study in our epoxymorphinan derivatives 1a-e as KOR/MOR dual selective ligands by removing their 3-hydroxy group and verifying such an operation on ligand selectivity and functional profiles toward opioid receptors. Herein, we report the synthesis of the newly designed compounds 6a-e, their SAR and molecular modeling study results.
2. Results and discussion
2.1. Chemistry
The chemical synthesis of compounds 1a-e has been reported [15]. The synthetic route of compounds 6a-e is outlined in Scheme 1. The syntheses of compounds 6a-e was performed in a similar way to that of a previous report [47] and to that of 1a-e with the removal of 3-hydroxy group first: the tetrazolyl ether 2 was obtained by reacting the starting material naltrexone with 5-chloro-1-phenyl-1H-tetrazole. Catalytic hydrogenation of compound 2 in glacial acetic acid provided the 3-desoxy-naltrexone 3 with a yield of 82%. The intermediate 3 was undergone a stereoselective reductive amination to give the 6β-dibenzyl protected compound 4. Subsequent deprotection of the dibenzyl group of compound 4 under catalytic hydrogenation condition afforded the key intermediate 5. Finally, the desired compounds 6a-e were prepared through the amide coupling reaction of 5 with substituted pyridylcarboxylic acids and subsequent salt formation with reasonable yields. All the target compounds were fully characterized.
Scheme 1.

Synthesis of the target compounds 6a-e.
2.2. In vitro radioligand binding assays and [35S]-GTPγS functional assays
To determine the binding affinity and selectivity profiles of the new compounds 6a-e on the three opioid receptors, the in vitro competitive radioligand binding assays were conducted using Chinese hamster ovary (CHO) cells overexpressing monoclonal opioid receptors. The KOR and DOR were labeled with [3H]diprenorphine, while the MOR was labeled with [3H]naloxone, respectively.
As shown in Table 1, all the compounds carrying 3-hydroxy group, i.e. 1a-e, possessed subnanomolar binding affinity for the KOR and subnanomolar to one-digit nanomolar binding affinity for the MOR, while they bound to the DOR with relatively low affinity of Ki values at two- digit nanomolar to submicromolar level. Moreover, the 3-dehydroxy compounds 6a-e exhibited lower binding affinity for the KOR, with Ki values at the one- to two-digit nanomolar level. They also bound to the MOR and DOR with Ki values at the two-digit nanomolar level and the micromolar level, respectively, which were substantially lower in affinity compared to their 3-hydroxylated counterparts. These results clearly demonstrated that the 3-hydroxy was crucial for the affinities of these epoxymorphinan ligands in binding to all three opioid receptors. In addition, the effects of different substituents varying in size and electronic natures at the 2’-position of the pyridine ring on binding affinity for the KOR and MOR were examined. It seemed that electron-withdrawing groups were more favorable than electron-donating groups for KOR binding affinity though not significantly. Among these compounds, compound 1c (NCP) was found to display the highest binding affinity for the KOR, and displayed the highest selectivity for the KOR over the MOR (μ/κ = 9.62) as well as very high selectivity for the KOR over the DOR (δ/κ = 579). Overall, these compounds exhibited higher binding affinities for the KOR and MOR than for the DOR. Based on our previous studies [5, 12, 15, 21], it has been demonstrated that the binding pocket in both the KOR and MOR can form aromatic stacking, hydrophobic and hydrogen bonding interaction with these ligands containing a 4’-pyridyl moiety.
Table 1.
The Radioligand Binding Affinity to the KOR, MOR, and DOR and Selectivity Profiles of Compounds 1a-e and 6a-e.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Compounds | R1 | R2 | Ki (nM) | Selectivity | |||
| KOR | MOR | DOR | μ/κ | δ/κ | |||
| 1a | OH | Cl | 0.15 ± 0.04b | 0.10 ± 0.04b | 602.20 ± 22.30b | 0.67 | 4015 |
| 6a | H | Cl | 10.46 ± 1.38 | 18.84 ± 3.01 | 2123.66 ± 101.18 | 1.8 | 203 |
| 1b | OH | Br | 0.18 ± 0.03b | 0.63 ± 0.18b | 173.70 ± 134.90b | 3.5 | 965 |
| 6b | H | Br | 7.44 ± 0.7 | 24.51 ± 3.23 | 2086.15 ± 71.59 | 3.3 | 280 |
| 1c (NCP) | OH | CN | 0.13 ± 0.01b | 1.25 ± 0.55b | 75.30 ± 7.00b | 9.6 | 579 |
| 6c | H | CN | 6.13 ± 0.74 | 58.27 ± 5.16 | 3375.73 ± 743.96 | 9.5 | 551 |
| 1d | OH | CH3 | 0.58 ± 0.12b | 0.39 ± 0.20b | 90.10 ± 17.10b | 0.67 | 155 |
| 6d | H | CH3 | 96.07 ± 14.84 | 39.03 ± 2.56 | 3649.72 ± 158.17 | 0.41 | 38.0 |
| 1e | OH | OCH3 | 0.46 ± 0.04b | 0.60 ± 0.23b | 160.40 ± 6.60b | 1.3 | 349 |
| 6e | H | OCH3 | 86.91 ± 9.22 | 45.63 ± 4.59 | 5900.42 ± 300.78 | 0.53 | 67.9 |
The values are the mean ± SEM of at least three independent experiments.
Data have been reported in Reference [15], and are presented here for comparison purposes.
The selectivity of these compounds for the KOR over the MOR and DOR was also compared. In general, compounds carrying electron-withdrawing groups, e.g. CN and Br, exhibited higher selectivity for the KOR over the MOR and DOR than compounds with electron-donating groups (CH3 and OCH3). As discussed above, compounds 1c and 6c with the cyano group at 2’-position of the 4’-pyridyl ring possessed the highest selectivity for the KOR over the MOR and DOR overall. Interestingly, the selectivity profile for the KOR over the MOR was relatively uninfluenced by the bearing of 3-hydroxy group (e.g. 1b vs 6b, 1c vs 6c, and 1d vs 6d), suggesting that the impact of removing 3-hydroxy group on the binding affinities was similar for the KOR and MOR because of the nearly same degree of selectivity profile shift between these two sets of compounds. This observation was somewhat different from the previous report [37]. On the other hand, the selectivity profiles for the KOR over the DOR of all the 3-hydroxy group bearing analogues were relatively higher than those of the corresponding 3-dehydroxy analogues, revealing that the 3-hydroxy group may exert noteworthy influence on the selectivity profiles for the KOR over the DOR.
Compounds 1b and 1c (NCP) together with their 3-dehydroxy analogues 6b and 6c were chosen to further determine their potencies and relative efficacies at the KOR, MOR, and DOR. [35S]-GTPγS functional assays were conducted on KOR-, MOR- and DOR-expressed CHO cells. EC50 and Emax values are measures of potency and efficacy, respectively. Emax values are expressed as % of the maximal response produced by the full agonists U50,488H, DAMGO, and DPDPE at the KOR, MOR, and DOR, respectively (Table 2). As shown in Table 2, compounds 1b and 1c (NCP) acted as full agonists of the KOR with % Emax values of 98.76% and 97.14%, and exhibited subnanomolar potency with EC50 values of 0.34 nM and 0.28 nM, respectively. Their counterparts without a 3-hydroxy group (6b and 6c) also demonstrated full agonism of KOR (% Emax = 99.27% and 100.20%, respectively) but with much lower potency (EC50 = 81.88 nM and 92.00 nM, respectively). Apparently, the 3-hydroxy group did not significantly affect the efficacy at the KOR whereas its removal led to a dramatic decrease in KOR potency. Additionally, compound 1b and 1c (NCP) behaved as MOR partial agonists with moderate efficacy (1b, % Emax = 61.29%; 1c, % Emax = 58.14%), while 6b and 6c acted as MOR partial agonists with low efficacy (6b, % Emax = 33.62%; 6c, % Emax = 26.48%). Moreover, compounds 1b and 1c (NCP) acted as DOR partial agonists with moderate efficacy (1b, % Emax = 57.34%; 1c, % Emax = 55.68%), while 6b and 6c exhibited full agonism to DOR (6b, % Emax = 145.10%; 6c, % Emax = 113.90%) but with much lower potency. Combined together, these results indicated that the 3-hydroxy group of these epoxymorphinan derivatives appeared to have different influences on activation profiles of the three ORs.
Table 2.
[35S]-GTPγS Functional Assay Results of Compounds 1b, 1c, 6b, and 6c.a
| Compounds | KOR [35S]-GTPγS binding | MOR [35S]-GTPγS binding | DOR [35S]-GTPγS binding | |||
|---|---|---|---|---|---|---|
| EC50 (nM) | % Emax of U50,488H | EC50 (nM) | % Emax of DAMGO | EC50 (nM) | % Emax of DPDPE | |
| 1b | 0.34 ± 0.03 | 98.76 ± 1.52 | 0.42 ± 0.05 | 61.29 ± 1.11 | 15.79 ± 2.96 | 57.34 ± 2.48 |
| 6b | 81.88 ± 9.09 | 99.27 ± 2.79 | 290.90 ± 41.99 | 33.62 ± 3.92 | 2675 ± 689 | 145.10 ± 12.31 |
| 1c (NCP) | 0.28 ± 0.03 | 97.14 ± 1.50 | 1.82 ± 0.28 | 58.14 ± 1.48 | 29.88 ± 3.99 | 55.68 ± 1.71 |
| 6c | 92.00 ± 8.47 | 100.20 ± 2.37 | 338.43 ± 31.10 | 26.48 ± 3.73 | 1850 ± 152 | 113.90 ± 3.10 |
The values are the mean ± SEM of at least three independent experiments.
2.3. Molecular modeling study
From the binding and functional assays results, it was found that NCP (Fig. 5a) possessed about 50-fold higher binding affinities for both the KOR and MOR than those of compound 6c (Fig. 5b). In addition, both NCP and 6c acted as KOR full agonists and MOR partial agonists, with NCP showing higher efficacy than compound 6c on the MOR. Notably, both of NCP and 6c contained a ‘message’ moiety (epoxymorphinan moiety) and an ‘address’ moiety (2-cyanopyridyl moiety), which were similar to other epoxymorphinan derivatives [4, 20, 21]. The only difference between the chemical structures of NCP and compound 6c was the 3-hydroxy group on their phenyl ring.
Fig. 5.

The chemical structures of NCP (a) and compound 6c (b). The structures with notions were derived from the optimized NCP_KORactive and compound 6c_KORactive complexes.
To investigate why NCP and compound 6c with such similar chemical structures exhibited significantly different binding affinity and functional profiles at the KOR and MOR, NCP was docked into the active KOR and MOR crystal structures, and compound 6c was docked into the active KOR and the inactive MOR ones. The docking solution with the highest CHEM-PLP score was merged into the respective receptor to afford the ligand-receptor complex. Subsequently, energy minimization was further conducted on the four ligand-receptor complexes (NCP_KORactive, NCP_MORactive, compound 6c_KORactive, and compound 6c_MORinactive, Fig. S1). Furthermore, 100 ns molecular dynamics (MD) simulations were conducted on the four ligand-receptor complexes and their binding modes from the MD simulations of each complex evaluated by the root-mean-square deviation (RMSD, Fig. S2) were shown in Fig. 6.
Fig. 6.


The binding modes of NCP_KORactive, NCP_MORactive, compound 6c_KORactive, and compound 6c_MORinactive complexes after 100 ns MD simulations. The receptors were shown as cartoon models, the active KOR in light-pink, the active MOR in light-blue, and the inactive MOR in light-green. NCP, compound 6c, and key amino acid residues were shown as stick models. Carbon atoms: NCP in cyan; compound 6c in yellow; key amino acid residues of the KOR in magenta, and the MOR in orange. The dashed lines in yellow represented potential water-mediated hydrogen bonds. The key residues at the allosteric site of the MOR were shown as stick and ball models in magentas. The key residues at the allosteric site of the KOR were shown as stick and ball models in orange. The water molecules were shown as sphere models in red.
As shown in Fig. 6, NCP and compound 6c exhibited similar interactions with the orthosteric sites in the NCP_KORactive, NCP_MORactive, compound 6c_KORactive, and compound 6c_MORinactive complexes, which was the same to the molecular docking results (Fig. S1). The orthosteric sites of the KOR and MOR accommodated the rigid and bulky 4, 5-epoxymorphinan moiety of NCP and compound 6c. Three types of interactions between the key residues located at the orthosteric site and the two ligands were established. First, residues M3.36, W6.48, I6.51, and H6.52 formed hydrophobic interactions with the 4, 5-epoxymorphinan moiety. Second, D3.32 formed ionic interaction with the piperidine quaternary ammonium nitrogen atom of the ligands’ 4, 5-epoxymorphinan nucleus. Third, a hydrogen bonding interaction was observed between Y3.33 and the dihydrofuran oxygen atom of the ligands. However, the interactions between compound 6c and the allosteric site in compound 6c_MORinactive complex were different from those of NCP in NCP_KORactive and NCP_MORactive complexes and compound 6c in compound 6c_KORactive complex. The binding subdomain of the 2-cyanopyridyl moiety of compound 6c in compound 6c_MORinactive complex showed a significant change after 100 ns MD simulations (Fig. 6) compared with that from molecular docking operation (Fig. S1). As shown in Fig. 6, the 2-cyanopyridyl moiety of NCP and compound 6c was adopted in a subdomain of the allosteric site of the KOR and MOR, formed by residues Q2.60, WECL1, and L(KOR)/I(MOR)3.29. These residues seemed to establish hydrophobic interactions with the pyridyl ring of NCP. Similar interactions were also formed in compound 6c_KORactive complex. While in compound 6c_MORinactive complex, the 2-cyanopyridyl moiety of compound 6c bound with another subdomain of the allosteric site of the MOR, formed by residues V6.55 and W7.35. To be noted, the binding modes of NCP in the KOR and MOR and compound 6c in the KOR, were almost identical to those of the KOR agonist MP1104 in the active KOR and the MOR agonist BU72 in the active MOR (Fig. S3) [37, 42]. Additionally, our previous molecular modeling studies revealed that the ‘address’ moiety of an epoxymorphinan derivative interacted with the subdomain formed by residues Q2.60, WECL1, and I3.29 of the allosteric site of the MOR may exhibit positive allosteric modulation to the ‘message’ moiety and induce the ligand to display agonism profile to the MOR. On the contrary, the ‘address’ moiety of an epoxymorphinan derivative bound with the subdomain formed by residues V6.55 and W7.35 of the allosteric site of the MOR may demonstrate negative allosteric modulation to the ‘message’ moiety and leads to the ligand acting as an antagonist to the MOR [20, 48]. Therefore, we speculated that these findings might explain why NCP and compound 6c exhibited similar agonism activities at the KOR as that of MP1104 and why NCP possessed a different activation profile from that of compound 6c at the MOR.
In addition, as observed in the crystal structures of the active KOR and MOR, the 3-hydroxy group of the phenolic moiety of the original ligands, MP1104 and BU72, may interact with residues Y3.33, K5.39, and H6.52 through water-mediated hydrogen bonds [37, 42]. Similarly, as shown in the binding mode of NCP_KORactive complex after 100 ns MD simulations (Fig. 6), these water-mediated hydrogen bonds were also established between the 3-hydroxy group of NCP and residues Y3.33, K5.39, and H6.52. However, in the compound 6c_KORactive complex, due to the lack of the 3-hydroxy group, compound 6c did not form any water-mediated hydrogen bonds with residues Y3.33, K5.39, and H6.52. These similar observations could also be found in the NCP_MORactive and compound 6c_MORinactive complexes, in which NCP formed the water-mediated hydrogen-bonding interactions with residues Y3.33, K5.39, and H6.52 through its 3-hydroxy group while compound 6c did not. More importantly, it can be concluded from Table S1 that the water-mediated hydrogen-bonding interactions could strengthen the interactions between NCP and residues Y3.33, K5.39, and H6.52 compared with that in the compound 6c_KORactive and compound 6c_MORinactive complexes. Therefore, the lack of water-mediated hydrogen bonding interactions due to removal of the 3-hydroxy group may lead to decrease in binding affinities for the KOR and MOR, which provided a computational explanation to the lower binding affinities of compound 6c to the KOR and MOR compared to those of NCP.
3. Conclusion
In summary, we have investigated the role of the 3-hydroxy group of the epoxymorphinan derivatives in the binding affinity and selectivity profiles toward the opioid receptors for a series of compounds from our library. All the 3-dehydroxy analogues 6a-e demonstrated reduced binding affinities for the ORs compared to compounds 1a-e containing the 3-hydroxy group, providing further evidence that the 3-hydroxy is critical for the affinity of epoxymorphinan derivatives binding to all three ORs. Moreover, most compounds carrying the 3-hydroxy group possessed similar selectivity profiles for the KOR over the MOR as those of their corresponding 3-dehydroxy analogues, suggesting that the 3-hydroxy group may not be as influential to ligand selectivity profiles between the two receptors as expected. Further [35S]-GTPγS functional assay results indicated that the 3-hydroxy group of these epoxymorphinan derivatives played a vital role in the potency to activate the opioid receptors, and demonstrated different degrees of influence on maximal activation profiles at the three ORs. Binding modes from the MD simulations showed that the 3-dehydroxy compound 6c and NCP exhibited similar interactions with the orthosteric sites, while the interactions between compound 6c and the allosteric site in compound 6c_MORinactive complex were different from those of NCP in NCP_KORactive and NCP_MORactive complexes and compound 6c in compound 6c_KORactive complex. Moreover, NCP formed water-mediated hydrogen bonds with residues Y3.33, K5.39, and H6.52 in both complexes through its 3-hydroxy group, while the 3-dehydroxy compound 6c did not. These findings furnished possible explanations for the different in vitro binding affinity profiles and functional behaviors between compounds 1c (NCP) and 6c in the KOR and MOR.
4. Experimental
4.1. Chemistry
All nonaqueous reactions were carried out under a pre-dried nitrogen gas atmosphere. All solvents and reagents were purchased from commercial suppliers, and were used as received without further purification. Melting points were measured on a MPA100 OptiMelt automated melting point apparatus without correction. IR spectra were recorded on Thermo Scientific Nicolet iS10 FT-IR Spectrometer. Analytical thin-layer chromatography (TLC) analyses were carried out on Analtech Uniplate F254 plates and flash column chromatography (FCC) was performed over silica gel (230−400 mesh, Merck). 1H (400 MHz) and 13C (100 MHz) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Ultrashield 400 Plus spectrometer. Chemical shifts were expressed in δ units (ppm), using TMS as an internal standard, and J values were reported in hertz (Hz). Mass spectra were obtained on an Applied BioSystems 3200 Q trap with a turbo V source for Turbolon Spray. Analytical reversed-phase high performance liquid chromatography (HPLC) was performed on a Varian ProStar 210 system using Agilent Microsorb-MV 100–5 C18 column (250 × 4.6 mm). All analyses were conducted at an ambient temperature with a flow rate of 0.5 mL/min. HPLC eluent condition: acetonitrile/water (with 0.1% trifluoroacetic acid), acetonitrile increased from 40% to 100% in gradient within 20 min of test. The UV detector was set up at 210 nm. The injection volume was 5 μL. The purities of final compounds were calculated as the percentage peak area of the analyzed compound, and retention time (Rt) was presented in minutes. The purity of all newly synthesized compounds was identified as ≥ 95%.
4.1.1. Preparation of Target Compounds 6a-e.
4.1.1.1. 17-Cyclopropylmethyl-4,5α-epoxy-14β-hydroxy-3-[(1-phenyl-1H-tetrazol-5-yl)oxy]-m orphinan-6-one (2)
To a mixture of naltrexone (1.30 g, 3.81 mmol) and potassium carbonate (1.16 g, 8.38 mmol) in dry DMF (10 mL, dried over 4 Å molecular sieves) was added 5-chloro-1-phenyl-1H-tetrazole (756 mg, 4.19 mmol) in portions. The resulting mixture was stirred at room temperature for 6 h. After completion of the reaction (monitored by TLC), DMF was removed under reduced pressure. The crude residue was extracted with DCM, and the combined organic layers were washed with water and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to give a brown solid. The crude product was washed with methanol to afford the pure intermediate 2 as a white solid. Yield: 97%. Mp: 151.1–151.8°C. 1H NMR (400 MHz, CDCl3): δ 7.89–7.87 (m, 2H), 7.61–7.57 (m, 2H), 7.52–7.47 (m, 2H), 7.18 (d, J = 8.3 Hz, 1H), 6.76 (d, J = 8.3 Hz, 1H), 4.76 (brs, 1H), 3.14 (d, J = 19.0 Hz, 1H), 3.12–3.03 (m, 1H), 2.95–2.87 (m, 1H), 2.73–2.40 (m, 4H), 2.32 (dt, J = 14.6, 3.0 Hz, 1H), 2.22 (brs, 1H), 1.99 (brs, 1H), 1.66–1.58 (m, 3H), 0.95 (brs, 1H), 0.61 (d, J = 6.3 Hz, 2H), 0.22 (brs, 2H). 13C NMR (100 MHz, CDCl3): 207.40, 159.44, 146.61, 135.77, 133.12, 131.44, 131.22, 129.71 (Ph-C × 2), 129.40, 122.45 (Ph-C × 2), 121.19, 119.87, 91.24, 69.96, 61.81, 59.20, 50.91, 43.40, 36.10, 31.30, 30.70, 23.00, 9.37, 4.06, 3.81. HRMS (ESI) m/z: 486.2049 [M+H]+, 508.1864 [M+Na]+, C27H27N5O4 (485.2063).
4.1.1.2. 17-Cyclopropylmethyl-4,5α-epoxy-14β-hydroxymorphinan-6-one (3)
A mixture of compound 2 (1.87 g, 3.85 mmol), 10% Pd/C (675 mg), and 7 mL of glacial acetic acid was hydrogenated (H2, 52 psi) at 40 °C for 10 h. The reaction mixture was filtered through Celite and the Celite was washed with DCM (3 times). The filtrate was concentrated to give a light brown oil. The oil was treated with 4 N NaOH (6 mL) and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was further purified by column chromatography (MeOH/DCM = 1/100) to provide the pure compound 3 as a white solid. Yield: 82%. Mp: 143.2–143.9 °C. 1H NMR (400 MHz, CDCl3): δ 7.05 (td, J = 7.8, 1.1 Hz, 1H), 6.73 (d, J = 7.9, 1H), 6.67 (d, J = 7.6, 1H), 4.63 (s,1H), 3.19 (d, J = 5.9 Hz, 1H), 3.10 (d, J = 18.8 Hz, 1H), 3.03 (td, J = 14.5, 5.0 Hz, 1H), 2.69 (dd, J = 12.0, 4.8 Hz, 1H), 2.62 (dd, J = 18.8, 6.0 Hz, 1H), 2.46–2.38 (m, 3H), 2.29 (d, J = 14.5 Hz, 1H), 2.12 (td, J = 12.0, 3.5 Hz, 1H), 1.87 (dt, J = 13.2, 4.0 Hz, 1H), 1.66–1.58 (m, 1H), 1.54–1.51 (m, 1H), 0.92–0.82 (m, 1H), 0.57–0.53 (m, 2H), 0.16–0.12 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 209.08, 157.40, 133.16, 129.18, 127.88, 118.66, 108.05, 89.85, 70.17, 62.08, 59.25, 50.11, 43.53, 36.20, 31.39, 30.82, 23.33, 9.43, 3.99, 3.84. HRMS (ESI) m/z: 326.1728 [M+H]+, C20H23NO3 (325.1678).
4.1.1.3. 17-Cyclopropylmethyl-4,5α-epoxy-6β-dibenzylamino-14β-hydroxymorphinan (4)
To a mixture of 3 (400 mg, 1.23 mmol), benzoic acid (165 mg, 1.35 mmol), and a trace amount of p-toluenesulfonic acid (p-TsOH, 20 mg) in anhydrous toluene (120 mL) and ethanol (30 mL) was added dibenzylamine (255 mg, 1.29 mmol) under N2 atmosphere. The reaction mixture was heated to reflux, employing a Dean-Stark trap for removal of generated water. The solvent was removed from the trap until the volume of the reaction mixture reduced to ca. 30 mL over 8 h. After cooling down to room temperature, additional anhydrous toluene (120 mL) and ethanol (30 mL) was added and the mixture was refluxed overnight. Next day, the solvent was removed from the trap to ca. 30 mL left again and the mixture was cooled to ambient temperature. Molecular sieves (3 g, 4 Å) and anhydrous ethanol (30 mL) were added followed by the addition of sodium cyanoborohydride (62 mg, 0.99 mmol). The reaction mixture was stirred at room temperature for 24 h under N2 atmosphere. After completion of the reaction (monitored by TLC), the reaction mixture was filtered through Celite and the filtrate was concentrated under reduced pressure. The residue was treated with 3% ammonia water and extracted with ethyl acetate (three times). The combined organic layers were washed with brine and dried over Na2SO4. Removal of ethyl acetate under reduced pressure gave a brown oil which was further purified by column chromatography (16%−18% ethyl acetate/hexane) to deliver compound 4 as a white solid. Yield: 72%. Mp: 119.8–120.4 °C. 1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 7.4 Hz, 4H), 7.27–7.24 (m, 4H), 7.16 (t, J = 7.3 Hz, 2H), 6.92 (t, J = 7.7, 1H), 6.53 (d, J = 7.6, 1H), 6.48 (d, J = 7.8, 1H), 4.69 (d, J = 7.8 Hz, 1H), 3.89 (d, J = 14.2 Hz, 2H), 3.65 (d, J = 14.2 Hz, 2H), 3.03–2.97 (m, 2H), 2.61 (dd, J = 11.8, 5.5 Hz, 1H), 2.57–2.51 (m, 2H), 2.38–2.30 (m, 2H), 2.21 (td, J = 12.4, 5.0 Hz, 1H), 2.10–1.94 (m, 2H), 1.65–1.54 (m, 2H), 1.42 (dd, J = 12.4, 2.2 Hz, 1H), 1.20 (td, J = 13.0, 2.8 Hz, 1H), 0.90–0.78 (m, 1H), 0.55–0.47 (m, 2H), 0.14–0.07 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 156.55, 140.53, 133.19, 130.74, 128.62, 128.43, 128.16, 126.69, 117.59,108.39, 91.00, 70.47, 62.64, 59.53, 59.37, 54.59, 47.09, 44.07, 30.96, 30.73, 23.46, 19.00, 9.61, 4.02, 3.98. IR (Diamond, cm−1) νmax: 3390, 3025, 2910, 2835, 1629, 1493, 1454, 1374, 1245, 1136, 1049, 925, 731, 697. HRMS (ESI) m/z: 507.3032 [M+H]+, C34H38 N2O2 (506.2933).
4.1.1.4. 17-Cyclopropylmethyl-4,5α-epoxy-6β-amino-14β-hydroxymorphinan hydrochloride (5·2HCl)
To a suspension of intermediate 4 (750 mg, 1.48 mmol) in 12 mL of anhydrous methanol was added concentrated HCl (0.3 mL, pH = 2). Then, 10% Pd/C (262 mg) was added and the resulting mixture was hydrogenated at room temperature under 65 psi pressure for 4 days. After completion of the reaction (by TLC monitoring), the reaction mixture was filtered through Celite and the Celite was washed with methanol. The combined filtrate was concentrated to afford the key intermediate 5 as an off-white solid. Yield: 81%. Mp: > 250 °C. This compound could be used in the next step without any further purification. A small quantity of 5 was recrystallized using methanol/ether to get a pure product for characterization. 1H NMR (400 MHz, DMSO-d6): δ 9.01 (brs, 1H, exchangeable), 8.58 (brs, 3H, exchangeable), 7.21 (t, J = 7.8 Hz, 1H), 6.86 (d, J = 7.7, 1H), 6.80 (d, J = 7.9, 1H), 6.56 (brs, 1H), 4.77 (d, J = 7.3 Hz, 1H), 4.13 (brs, 1H, exchangeable), 3.45 (d, J = 19.8 Hz, 1H), 3.39–3.34 (m, 1H), 3.19–3.12 (m, 1H), 2.05 (d, J = 10.3 Hz, 1H), 2.92–2.88 (m, 1H), 2.73 (brs, 1H), 2.48–2.44 (m, 2H), 2.02 (q, J = 12.8 Hz, 1H), 1.88 (d, J = 13.9 Hz, 1H), 1.77 (d, J = 9.8 Hz, 1H), 1.43 (d, J = 10.8 Hz, 1H), 1.28 (t, J = 13.4 Hz, 1H), 1.10–1.07 (m, 1H), 0.69–0.65 (m, 1H), 0.62–0.58 (m, 1H), 0.56–0.50 (m, 1H), 0.43–0.39 (m, 1H). 13C NMR (100 MHz, DMSO-d6): 155.13, 131.44, 129.71, 127.69, 119.33, 109.01, 88.22, 69.30, 61.10, 56.66, 52.34, 46.03, 44.96, 28.67, 27.07, 23.62, 21.31, 5.68, 5.09, 2.63. IR (Diamond, cm−1) νmax: 3342, 3207, 2967, 2832, 1630, 1610, 1514, 1455, 1389, 1338, 1225, 1161, 1056, 909, 784. HRMS (ESI) m/z: 327.2082 [M+H]+, C20H26N2O2 (326.1994).
4.1.1.5. General Procedure for the Preparation of Target Compounds 6a-e
To a solution of substituted pyridylcarboxylic acids (2 equiv) in anhydrous DMF (2 mL) were added 4 Å molecular sieves, EDCI (1.5 equiv), HOBt (1.5 equiv), and Et3N (5.0 equiv) at 0 °C (ice-water bath) under N2 atmosphere. After stirring for 1 h, a solution of 5 (hydrochloride salt, 1.0 equiv) in DMF (1 mL) was added dropwise. The resultant mixture was allowed to warm to room temperature and was stirred for 4–5 days. Upon completion of the reaction, the mixture was then filtered through Celite. The filtrate was concentrated under reduced pressure to remove DMF. The crude residue was purified by column chromatography (MeOH/DCM) to provide the desired free bases of 6a-e. After confirmation by 1H NMR, the free bases were converted into their hydrochloride salts 6a-e. In this regard, the free base (1 equiv) was dissolved in MeOH (0.5 to 1 mL) and cooled in an ice-water bath. Next, HCl in methanol solution (1.25 M, 4 equiv) was added dropwise and the resultant mixture was stirred for 1 h. Finally, ether (25 mL) was added and the mixture was stirred at room temperature overnight. Next day, the precipitate was collected by filtration and dried to obtain the desired hydrochloride salt (6a-e), which was then used for further analysis and biological assays.
17-Cyclopropylmethyl-4,5α-epoxy-14β-hydroxy-6β-{[4′-(2′-chloropyridyl)]carboxami do}morphinan hydrochloride (6a)
Free base of 6a was synthesized following the general procedure by reacting 2-chloroisonicotinic acid (79 mg, 0.50 mmol) with 5 (100 mg, 0.25 mmol). Yellow sticky solid. Yield: 56%. The free base was then converted into its hydrochloride salt 6a. Off-white solid. Yield: 79%. Mp: > 250 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.13 (d, J = 8.2 Hz, 1H), 8.94 (brs, 1H, exchangeable), 8.58 (d, J = 5.1 Hz, 1H), 7.93 (s, 1H), 7.82 (dd, J = 5.1, 1.3 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 6.86 (d, J = 7.7, 1H), 6.76 (d, J = 7.8, 1H), 6.29 (s, 1H, exchangeable), 4.80 (d, J = 7.7 Hz, 1H), 3.94 (d, J = 5.1 Hz, 1H), 3.72–3.64 (m, 1H), 3.46 (d, J = 19.9 Hz, 1H), 3.39–3.32 (m, 1H), 3.20 (dd, J = 19.7, 5.6 Hz, 1H), 3.05 (d, J = 7.4 Hz, 1H), 2.90–2.86 (m, 1H), 2.46 (d, J = 8.6 Hz, 2H), 1.99–1.89 (m, 1H), 1.82 (d, J = 13.8 Hz, 1H), 1.60–1.54 (m, 1H), 1.46 (d, J = 8.8 Hz, 1H), 1.43–1.36 (m, 1H), 1.10–1.07 (m, 1H), 0.70–0.66 (m, 1H), 0.63–0.57 (m, 1H), 0.55–0.51 (m, 1H), 0.45–0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 162.63, 155.82, 151.00, 150.83, 144.56, 131.33, 129.59, 128.38, 121.94, 120.93, 118.93, 109.07, 90.06, 69.64, 61.55, 56.74, 51.40, 46.27, 44.98, 29.37, 27.25, 23.77, 23.44, 5.76, 5.18, 2.70. IR (Diamond, cm−1) νmax: 3083, 1652, 1540, 1459, 1363, 1333, 1117, 1031, 909, 751. HRMS (ESI) m/z: 466.1901 [M+H]+, C26H28ClN3O3 (465.1819). HPLC purity: 99.80%. Rt: 7.888 min.
17-Cyclopropylmethyl-4,5α-epoxy-14β-hydroxy-6β-{[4′-(2′-bromopyridyl)]carboxami do}morphinan (6b)
Free base of 6b was synthesized following the general procedure by reacting 2-bromoisonicotinic acid (81 mg, 0.40 mmol) with 5 (80 mg, 0.20 mmol). Light brown solid. Yield: 62%. The free base was then converted into its hydrochloride salt 6b. Off-white solid. Yield: 81%. Mp: > 250 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.13 (d, J = 8.2 Hz, 1H), 8.94 (brs, 1H, exchangeable), 8.56 (d, J = 5.0 Hz, 1H), 8.07 (d, J = 0.6 Hz, 1H), 7.84 (dd, J = 5.1, 1.4 Hz, 1H), 7.19 (t, J = 7.8 Hz, 1H), 6.86 (d, J = 7.7 Hz, 1H), 6.76 (d, J = 7.8 Hz, 1H), 6.30 (s, 1H, exchangeable), 4.80 (d, J = 7.8 Hz, 1H), 3.94 (d, J = 5.2 Hz, 1H), 3.71–3.63 (m, 1H), 3.46 (d, J = 19.8 Hz, 1H), 3.41–3.32 (m, 1H), 3.20 (dd, J = 19.8, 5.8 Hz, 1H), 3.09–3.02 (m, 1H), 2.90–2.86 (m, 1H), 2.46 (d, J = 8.8 Hz, 2H), 1.98–1.89 (m, 1H), 1.82 (d, J = 13.9 Hz, 1H), 1.58–1.54 (m, 1H), 1.46 (d, J = 9.0 Hz, 1H), 1.42–1.36 (m, 1H), 1.11–1.07 (m, 1H), 0.72–0.66 (m, 1H), 0.63–0.57 (m, 1H), 0.55–0.49 (m, 1H), 0.45–0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 162.50, 155.82, 151.34, 144.15, 141.95, 131.33, 129.60, 128.30, 125.55, 121.24, 118.94, 109.08, 90.06, 69.64, 61.55, 56.74, 51.40, 46.27, 44.98, 29.37, 27.25, 23.76, 23.43, 5.76, 5.18, 2.70. IR (Diamond, cm−1) νmax: 3082, 2937, 1650, 1533, 1493, 1456, 1332, 1252, 1133, 1029, 907, 733. HRMS (ESI) m/z: 510.1388 [M+H]+, C26H28BrN3O3 (509.1314). HPLC purity: 99.21%. Rt: 8.038 min.
17-Cyclopropylmethyl-4,5α-epoxy-14β-hydroxy-6β-{[4′-(2′-cyanopyridyl)]carboxami do}morphinan (6c)
Free base of 6c was synthesized following the general procedure by reacting 2-cyanoisonicotinic acid (59 mg, 0.40 mmol) with 5 (80 mg, 0.20 mmol). Off-white solid. Yield: 41%. The free base was then converted into its hydrochloride salt 6c. Off-white solid. Yield: 86%. Mp: > 250 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.24 (d, J = 8.2 Hz, 1H), 8.93 (dd, J = 5.0, 0.4 Hz, 2H, exchangeable for 1H), 8.43 (d, J = 0.7 Hz, 1H), 8.13 (dd, J = 5.1, 1.6 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 6.86 (d, J = 7.7, 1H), 6.77 (d, J = 7.8, 1H), 6.29 (s, 1H, exchangeable), 4.80 (d, J = 7.8 Hz, 1H), 3.93 (d, J = 5.2Hz, 1H), 3.74–3.65 (m, 1H), 3.46 (d, J = 19.9 Hz, 1H), 3.40–3.33 (m, 1H), 3.21 (dd, J = 19.9, 5.7 Hz, 1H), 3.06 (d, J = 7.3 Hz, 1H), 2.90–2.85 (m, 1H), 2.46 (d, J = 8.8 Hz, 2H), 1.99–1.90 (m, 1H), 1.82 (d, J = 13.9 Hz, 1H), 1.60–1.55 (m, 1H), 1.47 (d, J = 9.0 Hz, 1H), 1.44–1.38 (m, 1H), 1.11–1.07 (m, 1H), 0.70–0.66 (m, 1H), 0.64–0.57 (m, 1H), 0.55–0.49 (m, 1H), 0.45–0.40 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 162.30, 155.80, 152.21, 142.45, 133.30, 131.33, 129.63, 128.26, 126.53, 125.35, 118.97, 117.26, 109.09, 90.05, 69.63, 61.58, 56.75, 51.49, 46.27, 44.98, 29.35, 27.25, 23.76, 23.41, 5.75, 5.17, 2.69. IR (Diamond, cm−1) νmax: 3185, 3082, 1660, 1630, 1552, 1458, 1384, 1254, 1169, 1132, 1050, 917, 782. HRMS (ESI) m/z: 457.2245 [M+H]+, C27H28N4O3 (456.2161). HPLC purity: 100%. Rt: 7.404 min.
17-Cyclopropylmethyl-4,5α-epoxy-14β-hydroxy-6β-{[4′-(2′-methylpyridyl)]carboxami do}morphinan (6d)
Free base of 6d was synthesized following the general procedure by reacting 2-methylisonicotinic acid (55 mg, 0.40 mmol) with 5 (80 mg, 0.20 mmol). Yellow sticky solid. Yield: 79%. The free base was then converted into its hydrochloride salt 6d. Off-white solid. Yield: 71%. Mp: decomposed at 234.4°C. 1H NMR (400 MHz, DMSO-d6): δ 9.42 (d, J = 7.6 Hz, 1H), 8.97 (brs, 1H, exchangeable), 8.83 (d, J = 5.7 Hz, 1H), 8.15 (s, 1H), 8.04 (d, J = 5.2 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 6.86 (d, J = 7.7, 1H), 6.76 (d, J = 7.9, 1H), 6.37 (brs, 1H, exchangeable), 4.85 (d, J = 7.8 Hz, 1H), 3.97 (d, J = 4.8 Hz, 1H), 3.74–3.66 (m, 1H), 3.46 (d, J = 19.9 Hz, 1H), 3.41–3.31 (m, 1H), 3.20 (dd, J = 19.7, 5.6 Hz, 1H), 3.06 (d, J = 8.3 Hz, 1H), 2.92–2.88 (m, 1H), 2.74 (s, 3H), 2.45–2.42 (m, 2H), 2.02–1.92 (m, 1H), 1.85 (d, J = 13.5 Hz, 1H), 1.58–1.55 (m, 1H), 1.45 (d, J = 9.8 Hz, 1H), 1.43–1.37 (m, 1H), 1.11–1.07 (m, 1H), 0.70–0.66 (m, 1H), 0.64–0.59 (m, 1H), 0.56–0.51 (m, 1H), 0.44–0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 162.63, 156.05, 155.81, 131.38, 131.25, 129.59, 128.32, 124.24, 120.90, 118.96 (2 × C), 109.05, 90.00, 69.64, 61.49, 56.74, 51.57, 46.28, 45.00, 29.38, 27.24, 23.78, 23.44, 20.88, 5.79, 5.19, 2.71. IR (Diamond, cm−1) νmax: 3363, 3118, 1660, 1624, 1552, 1454, 1425, 1333, 1278, 1128, 1029, 920, 749. HRMS (ESI) m/z: 446.2452 [M+H]+, C27H31N3O3 (445.2365). HPLC purity: 96.99%. Rt: 7.058 min.
17-Cyclopropylmethyl-4,5α-epoxy-14β-hydroxy-6β-{[4′-(2′-methoxypyridyl)]carboxa mido}morphinan (6e)
Free base of 6e was synthesized following the general procedure by reacting 2-methoxyisonicotinic acid (77 mg, 0.50 mmol) with 5 (100 mg, 0.25 mmol). Yellow-orange solid. Yield: 67%. The free base was then converted into its hydrochloride salt 6e. White solid. Yield: 91%. Mp: decomposed at 235.0°C. 1H NMR (400 MHz, DMSO-d6): δ 8.95 (d, J = 7.9 Hz, 2H, exchangeable), 8.30 (dd, J = 5.3, 0.4 Hz, 1H), 7.39 (dd, J = 5.3, 1.1 Hz, 1H), 7.23 (s, 1H), 7.19 (t, J = 7.8 Hz, 1H), 6.85 (d, J = 7.7, 1H), 6.76 (d, J = 7.9, 1H), 4.81 (d, J = 7.8 Hz, 1H), 3.94 (d, J = 4.5 Hz, 1H), 3.90 (s, 3H), 3.71–3.63 (m, 1H), 3.46 (d, J = 19.9 Hz, 1H), 3.39–3.32 (m, 1H), 3.20 (dd, J = 19.7, 5.9 Hz, 1H), 3.05 (d, J = 6.6 Hz, 1H), 2.90–2.85 (m, 1H), 2.45 (d, J = 8.7 Hz, 2H), 1.98–1.88 (m, 1H), 1.81 (d, J = 13.5 Hz, 1H), 1.57–1.52 (m, 1H), 1.46 (d, J = 9.0 Hz, 1H), 1.42–1.36 (m, 1H), 1.11–1.07 (m, 1H), 0.72–0.66 (m, 1H), 0.63–0.56 (m, 1H), 0.55–0.49 (m, 1H), 0.45–0.39 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.23, 163.74, 155.88, 147.65, 144.49, 131.37, 129.57, 128.41, 118.90, 114.75, 109.07, 108.44, 90.14, 69.69, 61.49, 56.74, 53.71, 51.23, 46.30, 45.00, 29.41, 27.27, 23.80, 23.58, 5.82, 5.22, 2.73. IR (Diamond, cm−1) νmax: 3407, 3114, 1640, 1504, 1457, 1425, 1386, 1299, 1126, 1030, 916, 751. HRMS (ESI) m/z: 462.2382 [M+H]+, C27H31N3O4 (461.2315). HPLC purity: 97.10%. Rt: 7.694 min.
4.2. Biological Evaluation
The free base of naltrexone was provided through NIDA Drug Supply Program. All drugs and test compounds were dissolved in sterile-filtered distilled/deionized water. All other reagents and radioligands were purchased from either Sigma-Aldrich or Perkin-Elmer.
4.2.1. In Vitro Competitive Radioligand Binding Assay
The competition binding assay was conducted using monoclonal mice opioid receptor expressed in CHO cell lines (monoclonal human δ opioid receptor was used in the DOR assay). In this assay, 30 μg of membrane protein was incubated with the corresponding radioligand in the presence of different concentrations of test compounds in TME buffer (50 mM Tris, 3 mM MgCl2, and 0.2 mM EGTA, pH 7.4) for 1.5 h at 30 °C. The bound radioligand was separated by filtration using the Brandel harvester. Specific (i.e., opioid receptor-related) binding to the KOR, MOR, and DOR was determined as the difference in binding obtained in the absence and presence of 5 μM of U50,488, DAMGO, and SNC80, respectively. Relative affinity values (IC50) were determined by fitting displacement binding inhibition values by non-linear regression using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA), where %inhibition value was calculated as follows: %inhibition = 100% - (binding in the presence of tested compound - nonspecific binding)/specific binding × 100%. The IC50 values were converted to Ki values using the Cheng-Prusoff equation: Ki = IC50/[1 + ([L*]/KD)], where [L*] is the concentration of the radioligand and KD is the KD of the radioligand.[49]
4.2.2. In Vitro [35S]-GTPγS Functional Assay
[35S]-GTPγS functional assays were conducted in the rat MOR, human KOR, mouse DOR cell membranes. Membrane proteins (10 μg) were incubated with varying concentrations of drugs, GDP (15 μM), and 80 pM [35S]-GTPγS in assay buffer for 1 h at 30 °C. Nonspecific binding was determined with 10 μM unlabeled GTPγS. U50,488H (5 μM), DAMGO (5 μM), and DPDPE (5 μM) were included in the assay for a maximally effective concentration of a full agonist for the KOR, MOR, and DOR, respectively. The Emax values for receptors were calculated as relative to net full agonist-stimulated [35S]-GTPγS binding, which is defined as (net-stimulated binding by ligand/net-stimulated binding by maximally effective concentration of a full agonist) × 100%. By using the equation Ke = [Ant]/DR-1, where [Ant] is the concentration of antagonist and DR is the ratio of the EC50 values of the full agonist in the presence and absence of antagonist, Ke values in the competitive antagonism studies were determined.
4.3. Molecular Modeling Study
The PDB files of crystal complexes of the agonist MP1104 with KOR (PDB ID: 6B73) [37], the agonist BU72 with MOR (PDB ID: 5C1M),[42] and the antagonist β-FNA with MOR (PDB ID: 4DKL) [41] were downloaded from Protein Data Bank (http://www.rcsb.org). Prior to conducting the molecular docking, the three receptors were firstly prepared. The monomers of the KOR and MOR, and the three crystallographic water molecules involved in the water-mediated hydrogen bonding interactions between residues Y3.33, K5.39, and H6.52 and the original ligands (MP1104, BU72, and β-FNA) in the three crystal structures were remained. While the three original ligands, the active-state-stabilizing nanobody in 6B73, the G protein-mimetic camelid-antibody fragment in 5C1M, the T4 lysozyme fragment in 4DKL, solvent molecules, and other water molecules in the three crystal structures were removed. Subsequently, hydrogen atoms were added to each receptor and the two ligands, NCP and compound 6c, were sketched by applying SYBYL-X 2.1 (Tripos Inc., St Louis). Moreover, the two compounds were assigned with Gasteiger-Hückel charges and energy minimized under the Tripos force field (TFF) [50]. After preparing the structures of the ligands and the receptors, GOLD 5.6 [51] was applied to dock the two ligands, NCP and compound 6c, into the above three receptors. In the process of docking study, the binding sites of the KOR and MOR were defined by the atoms within 10 Å of the γ-carbon atom of D3.32 in the three receptors [52] and two distance constraints were applied. One is the distance between the piperidine quaternary ammonium nitrogen atom of the ligands’ 4, 5-epoxymorphinan nucleus and D3.32. The other one is the distance between the ligands’ dihydrofuran oxygen atom and the phenolic oxygen atom of Y3.33. In addition, 50 docking solutions were obtained after molecular docking study. Except for above setting parameters, default parameters were applied for the other docking procedure. The docking solution with the highest CHEM-PLP score was selected as the optimal docking pose of each compound. Subsequently, the optimal docking poses were merged into their respective receptors to obtain ligand-receptor complexes (NCP_KORactive, NCP_MORactive, compound 6c_KORactive, and compound 6c_MORinactive).
After molecular docking study, each ligand-receptor complex was firstly inserted into the POPC lipid membrane bilayers, solvated in a rectangle box by the TIP3 water molecules, surrounded with sodium and chloride ions in the concentration of 0.15 M by applying the CHARMM-GUI website service [53] and then further conducted 100 ns molecular dynamics (MD) simulations on each system by Amber14.0 [54] to obtain reliable and stable ligand-receptor complexes. In the process of the MD simulations, periodic boundary conditions were applied. The long-range electrostatic interactions were calculated by Particle Mesh Ewald (PME) method [55]. The cut-off of the non-bonded van der Waals interactions was set at 10 Å. The constant temperatures at 310 K was controlled by the Langevin thermostat. After 100 ns MD simulation, the root-mean-square deviations (RMSD) of the backbone atoms of the amino acid residues and ligand in each complex during the 100 ns MD simulations were computed to evaluate the equilibrium of the system (Fig. S2).
Supplementary Material
Highlights.
3-Hydroxy was crucial for the epoxymorphinan ligands in binding to all three opioid receptors.
The impact of removing 3-hydroxy group on the binding affinities seemed similar for the KOR and MOR.
Molecular modeling studies elucidated the remarkably different binding affinity and functional profiles between compound 1c and its 3-dehydroxy analogue 6c.
Acknowledgements
The authors are grateful to NIDA Drug Supply Program for providing the free base of naltrexone. This work was partially supported by NIH/NIDA Grants R01DA024022, R01DA044855 and UG3DA050311 (Y.Z.); and R01DA041359, P30DA013429 and R21DA045274 (L.-Y.L.-C.) The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health.
Footnotes
Conflict of interest
The authors declare no competing financial interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Brownstein MJ, A brief history of opiates, opioid peptides, and opioid receptors, Proceedings of the National Academy of Sciences 90(12) (1993) 5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Valentino RJ, Volkow ND, Untangling the complexity of opioid receptor function, Neuropsychopharmacology 43(13) (2018) 2514–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Azzam AAH, McDonald J, Lambert DG, Hot topics in opioid pharmacology: mixed and biased opioids, Br. J. Anaesth 122(6) (2019) e136–e145. [DOI] [PubMed] [Google Scholar]
- [4].Ma H, Obeng S, Wang H, Zheng Y, Li M, Jali AM, Stevens DL, Dewey WL, Selley DE, Zhang Y, Application of Bivalent Bioisostere Concept on Design and Discovery of Potent Opioid Receptor Modulators, J. Med. Chem 62(24) (2019) 11399–11415. [DOI] [PubMed] [Google Scholar]
- [5].Yuan Y, Elbegdorj O, Chen J, Akubathini SK, Beletskaya IO, Selley DE, Zhang Y, Structure selectivity relationship studies of 17-cyclopropylmethyl-3,14beta-dihydroxy-4,5alpha-epoxy-6beta-[(4’-pyridyl)carboxa mido]morphinan derivatives toward the development of the mu opioid receptor antagonists, Bioorg. Med. Chem. Lett 21(18) (2011) 5625–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Obeng S, Yuan Y, Jali A, Selley DE, Zhang Y, In vitro and in vivo functional profile characterization of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3-carboxamido) morphinan (NAQ) as a low efficacy mu opioid receptor modulator, Eur. J. Pharmacol 827 (2018) 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].De Souza EB, Schmidt WK, Kuhar MJ, Nalbuphine: an autoradiographic opioid receptor binding profile in the central nervous system of an agonist/antagonist analgesic, J. Pharmacol. Exp. Ther 244(1) (1988) 391. [PubMed] [Google Scholar]
- [8].Maraschin JC, Almeida CB, Rangel MP, Roncon CM, Sestile CC, Zangrossi H, Graeff FG, Audi EA, Participation of dorsal periaqueductal gray 5-HT1A receptors in the panicolytic-like effect of the κ-opioid receptor antagonist Nor-BNI, Behav. Brain Res. 327 (2017) 75–82. [DOI] [PubMed] [Google Scholar]
- [9].Suzuki T, Mori T, Funada M, Misawa M, Nagase H, Attenuation of the discriminative stimulus properties of cocaine by δ-opioid receptor antagonists, Eur. J. Pharmacol 263(1) (1994) 207–211. [DOI] [PubMed] [Google Scholar]
- [10].Chavkin C, Cohen JH, Land BB, Repeated Administration of Norbinaltorphimine Produces Cumulative Kappa Opioid Receptor Inactivation, Front. Pharmacol 10 (2019) 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stefanucci A, Dimmito MP, Macedonio G, Ciarlo L, Pieretti S, Novellino E, Lei W, Barlow D, Houseknecht KL, Streicher JM, Mollica A, Potent, Efficacious, and Stable Cyclic Opioid Peptides with Long Lasting Antinociceptive Effect after Peripheral Administration, J. Med. Chem 63(5) (2020) 2673–2687. [DOI] [PubMed] [Google Scholar]
- [12].Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y, Design, Synthesis, and Biological Evaluation of 6α- and 6β-N-Heterocyclic Substituted Naltrexamine Derivatives as μ Opioid Receptor Selective Antagonists, J. Med. Chem 52(5) (2009) 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li G, Aschenbach LCK, He H, Selley DE, Zhang Y, 14-O-Heterocyclic-substituted naltrexone derivatives as non-peptide mu opioid receptor selective antagonists: Design, synthesis, and biological studies, Bioorg. Med. Chem. Lett 19(6) (2009) 1825–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yuan Y, Elbegdorj O, Chen J, Akubathini SK, Beletskaya IO, Selley DE, Zhang Y, Structure selectivity relationship studies of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6 β -[(4 ′ -pyridyl)carboxamido]morphinan derivatives toward the development of the mu opioid receptor antagonists, Bioorg. Med. Chem. Lett 21(18) (2011) 5625–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yuan Y, Elbegdorj O, Chen J, Akubathini SK, Zhang F, Stevens DL, Beletskaya IO, Scoggins KL, Zhang Z, Gerk PM, Selley DE, Akbarali HI, Dewey WL, Zhang Y, Design, Synthesis, and Biological Evaluation of 17-Cyclopropylmethyl-3,14 β -dihydroxy-4,5 α -epoxy-6 β -[(4 ′ -pyridyl)carboxamido]morphinan Derivatives as Peripheral Selective μ Opioid Receptor Agents, J. Med. Chem 55(22) (2012) 10118–10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yuan Y, Zaidi SA, Elbegdorj O, Aschenbach LCK, Li G, Stevens DL, Scoggins KL, Dewey WL, Selley DE, Zhang Y, Design, Synthesis, and Biological Evaluation of 14-Heteroaromatic-Substituted Naltrexone Derivatives: Pharmacological Profile Switch from Mu Opioid Receptor Selectivity to Mu/Kappa Opioid Receptor Dual Selectivity, J. Med. Chem 56(22) (2013) 9156–9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yuan Y, Elbegdorj O, Beletskaya IO, Selley DE, Zhang Y, Structure activity relationship studies of 17-cyclopropylmethyl-3,14 β -dihydroxy-4,5 α -epoxy-6 α -(isoquinoline-3 ′ -carboxamido)morphinan (NAQ) analogues as potent opioid receptor ligands: Preliminary results on the role of electronic characteristics for affinity and function, Bioorg. Med. Chem. Lett 23(18) (2013) 5045–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang Y, Elbegdorj O, Yuan Y, Beletskaya IO, Selley DE, Opioid receptor selectivity profile change via isosterism for 14-O-substituted naltrexone derivatives, Bioorg. Med. Chem. Lett 23(13) (2013) 3719–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yuan Y, Zaidi SA, Stevens DL, Scoggins KL, Mosier PD, Kellogg GE, Dewey WL, Selley DE, Zhang Y, Design, syntheses, and pharmacological characterization of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5 α -epoxy-6 α -(isoquinoline-3 ′ -carboxamido)morphinan analogues as opioid receptor ligands, Biorg. Med. Chem 23(8) (2015) 1701–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Obeng S, Wang H, Jali A, Stevens DL, Akbarali HI, Dewey WL, Selley DE, Zhang Y, Structure–Activity Relationship Studies of 6α- and 6β-Indolylacetamidonaltrexamine Derivatives as Bitopic Mu Opioid Receptor Modulators and Elaboration of the “Message-Address Concept” To Comprehend Their Functional Conversion, ACS Chem. Neurosci 10(3) (2019) 1075–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zheng Y, Obeng S, Wang H, Jali AM, Peddibhotla B, Williams DA, Zou C, Stevens DL, Dewey WL, Akbarali HI, Selley DE, Zhang Y, Design, Synthesis, and Biological Evaluation of the Third Generation 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6 β -[(4 ′ -pyridyl)carboxamido]morphinan (NAP) Derivatives as μ/κ Opioid Receptor Dual Selective Ligands, J. Med. Chem 62(2) (2019) 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yuan Y, Li G, He H, Stevens DL, Kozak P, Scoggins KL, Mitra P, Gerk PM, Selley DE, Dewey WL, Zhang Y, Characterization of 6α- and 6β-N-Heterocyclic Substituted Naltrexamine Derivatives as Novel Leads to Development of Mu Opioid Receptor Selective Antagonists, ACS Chem. Neurosci 2(7) (2011) 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yuan Y, Stevens DL, Braithwaite A, Scoggins KL, Bilsky EJ, Akbarali HI, Dewey WL, Zhang Y, 6β-N-Heterocyclic substituted naltrexamine derivative NAP as a potential lead to develop peripheral mu opioid receptor selective antagonists, Bioorg. Med. Chem. Lett 22(14) (2012) 4731–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Williams DA, Zheng Y, David BG, Yuan Y, Zaidi SA, Stevens DL, Scoggins KL, Selley DE, Dewey WL, Akbarali HI, Zhang Y, 6β-N-Heterocyclic Substituted Naltrexamine Derivative BNAP: A Peripherally Selective Mixed MOR/KOR Ligand, ACS Chem. Neurosci 7(8) (2016) 1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang Y, Williams DA, Zaidi SA, Yuan Y, Braithwaite A, Bilsky EJ, Dewey WL, Akbarali HI, Streicher JM, Selley DE, 17-Cyclopropylmethyl-3,14 β -dihydroxy-4,5 α -epoxy-6 β -(4 ′ -pyridylcarboxamido)morphinan (NAP) Modulating the Mu Opioid Receptor in a Biased Fashion, ACS Chem. Neurosci 7(3) (2016) 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zheng Y, Obeng S, Wang H, Stevens DL, Komla E, Selley DE, Dewey WL, Akbarali HI, Zhang Y, Methylation Products of 6β-N-Heterocyclic Substituted Naltrexamine Derivatives as Potential Peripheral Opioid Receptor Modulators, ACS Chem. Neurosci 9(12) (2018) 3028–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Obeng S, Jali A, Zheng Y, Wang H, Schwienteck KL, Chen C, Stevens DL, Akbarali HI, Dewey WL, Banks ML, Liu-Chen L-Y, Selley DE, Zhang Y, Characterization of 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-7-carboxamido)morp hinan (NAN) as a Novel Opioid Receptor Modulator for Opioid Use Disorder Treatment, ACS Chem. Neurosci 10(5) (2019) 2518–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zheng Y, Obeng S, Reinecke BA, Chen C, Phansalkar PS, Walentiny DM, Gerk PM, Liu-Chen L-Y, Selley DE, Beardsley PM, Zhang Y, Pharmacological characterization of 17-cyclopropylmethyl-3,14-dihydroxy-4,5-epoxy-6-[(3′-fluoro-4 ′-pyridyl)acetamido]morphinan (NFP) as a dual selective MOR/KOR ligand with potential applications in treating opioid use disorder, Eur. J. Pharmacol 865 (2019) 172812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kawai K, Hayakawa J, Miyamoto T, Imamura Y, Yamane S, Wakita H, Fujii H, Kawamura K, Matsuura H, Izumimoto N, Kobayashi R, Endo T, Nagase H, Design, synthesis, and structure–activity relationship of novel opioid κ-agonists, Biorg. Med. Chem 16(20) (2008) 9188–9201. [DOI] [PubMed] [Google Scholar]
- [30].Snyder SH, Childers SR, Opiate Receptors and Opioid Peptides, Annu. Rev. Neurosci. 2(1) (1979) 35–64. [DOI] [PubMed] [Google Scholar]
- [31].Misicka A, Lipkowski AW, Slaninova J, Davis P, Yamamura HI, Porreca F, Hruby VJ, The synthesis and opioid receptor binding affinities of analogues of dermorphin and its N-terminal tetrapeptide fragment with dibasic acids in position 2, Life Sci. 57(18) (1995) 1633–1640. [DOI] [PubMed] [Google Scholar]
- [32].Nagase H, Hayakawa J, Kawamura K, Kawai K, Takezawa Y, Matsuura H, Tajima C, Endo T, DISCOVERY OF A STRUCTURALLY NOVEL OPIOID K-AGONIST DERIVED FROM 4, 5-EPOXYMORPHINAN, Chem. Pharm. Bull. (Tokyo) 46(2) (1998) 366–369. [DOI] [PubMed] [Google Scholar]
- [33].Hiroshi N, Hideaki F, Essential Structure of the κ Opioid Receptor Agonist Nalfurafine for Binding to the κ Receptor, Curr. Pharm. Des 19(42) (2013) 7400–7414. [DOI] [PubMed] [Google Scholar]
- [34].Urbano M, Guerrero M, Rosen H, Roberts E, Antagonists of the kappa opioid receptor, Bioorg. Med. Chem. Lett 24(9) (2014) 2021–2032. [DOI] [PubMed] [Google Scholar]
- [35].O’Connor C, White KL, Doncescu N, Didenko T, Roth BL, Czaplicki G, Stevens RC, Wüthrich K, Milon A, NMR structure and dynamics of the agonist dynorphin peptide bound to the human kappa opioid receptor, Proceedings of the National Academy of Sciences 112(38) (2015) 11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fenalti G, Zatsepin NA, Betti C, Giguere P, Han GW, Ishchenko A, Liu W, Guillemyn K, Zhang H, James D, Wang D, Weierstall U, Spence JCH, Boutet S, Messerschmidt M, Williams GJ, Gati C, Yefanov OM, White TA, Oberthuer D, Metz M, Yoon CH, Barty A, Chapman HN, Basu S, Coe J, Conrad CE, Fromme R, Fromme P, Tourwé D, Schiller PW, Roth BL, Ballet S, Katritch V, Stevens RC, Cherezov V, Structural basis for bifunctional peptide recognition at human δ-opioid receptor, Nat. Struct. Mol. Biol 22(3) (2015) 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, Han GW, Lee M-Y, Pardon E, Steyaert J, Huang X-P, Strachan RT, Tribo AR, Pasternak GW, Carroll FI, Stevens RC, Cherezov V, Katritch V, Wacker D, Roth BL, Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor, Cell 172(1) (2018) 55–67.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.[] Endoh T, Tajima A, Izumimoto N, Suzuki T, Saitoh A, Suzuki T, Narita M, Kamei J, Tseng LF, Mizoguchi H, Nagase H, TRK-820, a Selective κ-Opioid Agonist, Produces Potent Antinociception in Cynomolgus Monkeys, The Japanese Journal of Pharmacology 85(3) (2001) 282–290. [DOI] [PubMed] [Google Scholar]
- [39].Nagase H, Imaide S, Hirayama S, Nemoto T, Fujii H, Essential structure of opioid κ receptor agonist nalfurafine for binding to the κ receptor 2: Synthesis of decahydro(iminoethano)phenanthrene derivatives and their pharmacologies, Bioorg. Med. Chem. Lett 22(15) (2012) 5071–5074. [DOI] [PubMed] [Google Scholar]
- [40].Fujii H, Imaide S, Hirayama S, Nemoto T, Gouda H, Hirono S, Nagase H, Essential structure of opioid κ receptor agonist nalfurafine for binding to the κ receptor 3: Synthesis of decahydro(iminoethano)phenanthrene derivatives with an oxygen functionality at the 3-position and their pharmacologies, Bioorg. Med. Chem. Lett 22(24) (2012) 7711–7714. [DOI] [PubMed] [Google Scholar]
- [41].Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S, Crystal structure of the μ-opioid receptor bound to a morphinan antagonist, Nature 485(7398) (2012) 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, Kobilka BK, Structural insights into μ-opioid receptor activation, Nature 524(7565) (2015) 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC, Structure of the human kappa-opioid receptor in complex with JDTic, Nature 485(7398) (2012) 327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Claff T, Yu J, Blais V, Patel N, Martin C, Wu L, Han GW, Holleran BJ, Van der Poorten O, White KL, Hanson MA, Sarret P, Gendron L, Cherezov V, Katritch V, Ballet S, Liu Z-J, Müller CE, Stevens RC, Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists, Science Advances 5(11) (2019) eaax9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK, Structure of the δ-opioid receptor bound to naltrindole, Nature 485(7398) (2012) 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang A, Li F, Ding C, Yao Q, Knapp BI, Bidlack JM, Neumeyer JL, Synthesis and Pharmacological Evaluation of 6,7-Indolo/Thiazolo-MorphinansFurther SAR of Levorphanol, J. Med. Chem 50(11) (2007) 2747–2751. [DOI] [PubMed] [Google Scholar]
- [47].Krassnig R, Schmidhammer H, A new and efficient synthesis of the μ-selective opioid antagonist cyprodime, Heterocycles 38(4) (1994) 877–881. [Google Scholar]
- [48].Wang H, Reinecke BA, Zhang Y, Computational insights into the molecular mechanisms of differentiated allosteric modulation at the mu opioid receptor by structurally similar bitopic modulators, J. Comput. Aided Mol. Des. 34(8) (2020) 879–895. [DOI] [PubMed] [Google Scholar]
- [49].Yung-Chi C, Prusoff WH, Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction, Biochem. Pharmacol 22(23) (1973) 3099–3108. [DOI] [PubMed] [Google Scholar]
- [50].Clark M, Cramer Iii RD, Van Opdenbosch N, Validation of the general purpose tripos 5.2 force field, J. Comput. Chem. 10(8) (1989) 982–1012. [Google Scholar]
- [51].Jones G, Willett P, Glen RC, Leach AR, Taylor R, Development and validation of a genetic algorithm for flexible docking11Edited by F. E. Cohen, J. Mol. Biol 267(3) (1997) 727–748. [DOI] [PubMed] [Google Scholar]
- [52].Ballesteros JA, Weinstein H, [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors, in: Sealfon SC (Ed.), Methods in Neurosciences, Academic Press; 1995, pp. 366–428. [Google Scholar]
- [53].Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, Klauda JB, Im W, CHARMM-GUI Membrane Builder toward realistic biological membrane simulations, J. Comput. Chem 35(27) (2014) 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].D.B. Case J; Betz R; Cerutti D; Cheatham T Iii; Darden T; Duke R; Giese T; Gohlke H; Goetz A, AMBER 2015. University of California, San Francisco, (2015). [Google Scholar]
- [55].Darden T, York D, Pedersen L, Particle mesh Ewald: An N.log(N) method for Ewald sums in large systems, The Journal of Chemical Physics 98(12) (1993) 10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.